
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 07 July 2021
Sec. Virus and Host
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.705129
This article is part of the Research TopicInfection and Control of Vector-Borne DiseasesView all 34 articles
 Triwibowo Ambar Garjito1*
Triwibowo Ambar Garjito1* Widiarti Widiarti1
Widiarti Widiarti1 Muhammad Choirul Hidajat1,2
Muhammad Choirul Hidajat1,2 Sri Wahyuni Handayani1Mujiyono Mujiyono1
Sri Wahyuni Handayani1Mujiyono Mujiyono1 Mega Tyas Prihatin1
Mega Tyas Prihatin1 Rosichon Ubaidillah3Mohammad Sudomo4Tri Baskoro Tunggul Satoto5
Rosichon Ubaidillah3Mohammad Sudomo4Tri Baskoro Tunggul Satoto5 Sylvie Manguin6
Sylvie Manguin6 Laurent Gavotte7
Laurent Gavotte7 Roger Frutos8*
Roger Frutos8*Currently, Aedes aegypti, the principal vector of dengue virus in Indonesia, has spread throughout the archipelago. Aedes albopictus is also present. Invasion and high adaptability of the Aedes mosquitoes to all of these areas are closely related to their ecology and biology. Between June 2016 and July 2017, larval and adult mosquito collections were conducted in 43 locations in 25 provinces of Indonesia using standardized sampling methods for dengue vector surveillance. The samples collected were analyzed for polymorphism and phylogenetic relationship using the mitochondrial cox1 gene and the nuclear ribosomal internal transcribed spacer 2 (ITS2). Almost all Ae. aegypti samples collected in this study (89%) belonged to the same haplotype. A similar situation is observed with the nuclear ITS2 marker. Populations of Ae. aegypti characterized few years ago were genetically different. A closely related observation was made with Aedes albopictus for which the current populations are different from those described earlier. Ae. aegypti populations were found to be highly homogenous all over Indonesia with all samples belonging to the same maternal lineage. Although difficult to demonstrate formally, there is a possibility of population replacement. Although to a lower extent, a similar conclusion was reached with Ae. albopictus.
Aedes aegypti is known as a major vector of dengue viruses (family Flaviviridae, genus Flavivirus, DENV) (Simmons et al., 2012; Kraemer et al., 2015), yellow fever virus (family Flaviviridae, genus Flavivirus, YF) (da Costa-da-Silva et al., 2005; Kraemer et al., 2015; Yohan et al., 2018), zika virus (family Flaviviridae, genus Flavivirus, ZIKV) (Ja, 2016; Olson et al., 2020), and chikungunya virus (family Togaviridae, genus Alphavirus, CHIKV) (Kraemer et al., 2015; Yohan et al., 2018). This mosquito species originates from the forest of Africa and, since the 18th century, has spread via transcontinental trade throughout tropical and subtropical regions (Gubler, 1997; da Costa-da-Silva et al., 2005; Gubler, 2011; Joyce et al., 2018; Tedjou et al., 2019). In Southeast Asia, Ae. aegypti was formally identified for the first time in Malaysia and Thailand (1907) at the early 20th century (Urdaneta-Marquez and Failloux, 2011; Parimittr et al., 2018). Ae. aegypti was formally identified in Indonesia in 1908 (Leicester, 1908). A local strain of Ae. aegypti, the Medan strain, was first reported and successfully colonized in a laboratory in the 1930s (Kuno, 2010). Currently, the species is reported to have spread throughout the archipelago (Setiati et al., 2006; IVRCRD, 2018).
Another dengue vector species is the Asian tiger mosquito, Aedes albopictus. This species has for long been considered as a secondary vector of several viruses (Paupy et al., 2009; Grard et al., 2014; Goubert et al., 2016; Mulyatno et al., 2018). Ae. albopictus originates in the forests of Southeast Asia, and has spread worlwide since the 1970s (Moncayo et al., 2004). According to the Global invasive species database (http://www.issg.org/database/), this species has been recorded as one of the worst invasive species in the world. Currently, Ae. albopictus can be found in Asia, Africa, Europe, North and South America, and many locations in the Pacific and Indian oceans except Antartica (Paupy et al., 2009; Kraemer et al., 2015). As an invasive species, Ae. albopictus plays a potential role in triggering a re-emergence of arboviruses transmission in many locations. Recently, this species played an important role in Dengue, Chikungunya, and Zika outbreaks in both endemic and invaded regions (Rezza et al., 2007; Teixeira et al., 2009; Paupy et al., 2010; McKenzie et al., 2019; Lai et al., 2020).
The invasion and adaptation to all of these areas are closely related to their ecology and biology. Ae. aegypti has high adaptability to urban and peridomestic areas, where it breeds in the vicinity of human dwellings in a variety of artificial and natural containers in urban and rural areas (Kusriastuti and Sutomo, 2005; Kraemer et al., 2015; IVRCRD, 2018). This species is also recognized as the most anthropophilic mosquito and has the ability to blood-feed repeatedly on humans almost on a daily basis (Ritchie, 2014). This behavior may have contributed to the capacity of Ae. aegypti to cause high epidemics of dengue fever in Indonesia. A total of 68,407 dengue cases (incidence: 78.85/100,000) with 493 deaths (case fatality rates (CFR): 0.72%) were reported in 2017 (MoH Indonesia, 2018; Harapan et al., 2019). Ae. albopictus displays a strong ecological plasticity and has shown a remarkable capacity to adapt to urban and sub-urbans under various climate conditions, displacing Ae. aegypti population in some areas. Ae. albopictus has now become a significant vector of CHIKV and DENV (Knudsen., 1995; Paupy et al., 2009; Thiberville et al., 2013; Ngoagouni et al., 2015).
While an effective multivalent dengue vaccine is still under research and not yet available, vector control and entomological surveillance are the only reliable means of prevention and control of dengue fever (Wilder-Smith et al., 2010; Thisyakorn and Thisyakorn, 2014; Chang et al., 2015; Parra et al., 2018). Updated information on the genetic diversity and evolutionary patterns among Ae. aegypti and Ae. albopictus populations is needed to provide clues for better understanding the origin, the structuration, and the distribution of populations. Moreover, this is also a prerequisite to define differences in vector competence and capacity to transmit dengue virus, in ecological adaptations and in resistance to insecticides (Gupta and Preet, 2014; Yohan et al., 2018; Naim et al., 2020). However, a comprehensive information about genetic diversity and structuration of populations of Ae. aegypti and Ae. albopictus in Indonesia is still missing.
Therefore, we investigated the genetic diversity, evolutionary relationship, and distribution of Ae. aegypti and Ae. albopictus mosquitoes collected in different locations and islands, from Western Sumatra (Aceh) to Eastern Indonesia (Papua) using the mitochondrial cox1 or COI gene and the internal transcribed spacer 2 (ITS2) of the ribosomal DNA as target sequences.
Larva, pupa, and adult mosquitoes were collected using standardized sampling methods for dengue vector surveillance (Focks and Special Program for Research and Training in Tropical Diseases (TDR), 2003; Kusriastuti and Sutomo, 2005; WHO, 2011; Ritchie, 2014; WHO, 2016; MoH Indonesia, 2018; Harapan et al., 2019). In each house, larvae and pupae from different containers were put in different plastic bags. All samples were then transported to a field laboratory. Larvae and pupae were reared in the field laboratory for 3 days until the emergence of adult mosquitoes. Mosquitoes were then morphologically identified, sorted according to locality, and preserved in 250 µl of RNAlater (Ambion-Thermo Fisher Scientific, Watham, USA). This large sampling campaign was conducted as part of a nationwide program supervised by the Ministry of Health. Mosquitoes were collected during this campaign as a cohort of samples for use in different projects. This explains why the samples were stored individually in RNAlater even though no virus detection was conducted in this work. They were then stored at −80°C until further analysis. Larvae which did not emerge after 3 days were preserved the same way as adult mosquito samples for further analysis. All mosquito samples were individual samples, and only female mosquitoes were used as samples.
Whole DNA from each mosquito was individually extracted using a DNeasy® Blood & Tissue Kit (Qiagen, Hilden, Germany) according to the manufacturer’s standard protocol. ITS2 and cox1 (COI) were selected as target sequence because they are the most used sequences for phylogenetic and population structure analyses of mosquitoes. They are present in databases. They were also used in previous works on the genotyping of Aedes mosquitoes in Indonesia, allowing thus for comparison. The amplification of cox1 was conducted using the primers CI-N-2087 (5′-AAT TTC GGT CAG TTA ATA ATA TAG-3′ and TY-J-1460 (5′-TAC AAT TTA TCG CCT AAA CTT CAG CC-3′) as previously described (Rueanghiran, et al., 2011; Ngoagouni et al., 2015). The ITS2 sequence was amplified using the primers ITS2a (5′-TGT GAA CTG CAG GAC ACA T-3′) and ITS2b (5′-TAT GCT TAA ATT CAG GGG GT-3′). PCR reactions were carried out using the GoTaq® Green Master Mix (Promega, Madison, WI, USA). The conditions for PCR amplification of the cox1 gene were as follows: one cycle at 94°C for 1 min for initial denaturation, followed by five cycles at 94°C for 30 s, 45°C for 40 s, and 72°C for 1 min. This was then followed by 35 cycles at 94°C for 30 s, 44°C for 40 s, and 72°C for 1 min, and by a final extension step at 72°C for 10 min (Ngoagouni et al., 2015). PCR thermocycling conditions for ITS2 were as follows: 94°C for 10 min; followed by 40 cycles of denaturation at 94°C for 1 min, annealing at 56°C for 45 s, and elongation at 72°C for 1 min; followed by a final extension step at 72°C for 10 min. PCR products were electrophoresed in 1.5% agarose gel and visualized by SYBR® safe DNA gel stain (Invitrogen, Carlsbad, CA, USA) using a Biorad Molecular Image Gel Doc XR (Biorad Laboratories Inc, California, USA). A 100-bp DNA ladder was used for calculating the size of the PCR products. Amplicons were purified using Applied Biosystems ExoSAP-IT™ (Thermo Fisher Scientific, Vilnius, Lithuania). Cycle sequencing was performed using the primers listed above and an Applied Biosystems BigDye™ Terminator v.3.1 Cycle Sequencing Kit (Life Technologies Cooperation, Austin, TX, USA). To remove unincorporated BigDye® terminators and salts, cycle sequencing products were purified using a BigDye® Xterminator Purification Kit (Life technologies, Bedford, MA, USA). Sequence data were obtained using a DNA sequencer (Applied Biosystems® 3500 Genetic Analyzer) and analyzed using the Sequencing Analysis 6 program (Applied Biosystems). All sequences have been deposited in Genbank under the accession numbers MW280620 to MW280792, MW280794 to MW280797 and MW280800 to MW280818 for Ae. aegypti cox1 sequences. The accession numbers for Ae. aegypti ITS2 sequences range from MW288143 to MW288145 and MW290431 to MW290468. The accession numbers for Ae. albopictus cox1 sequences are MW280793, MW280798, MW280799, and MW283303 to MW283318. The accession numbers for Ae. albopictus ITS2 sequences are MW287155 to MW287157. The accession numbers of the cox1 and ITS2 sequences of the unknown species Aedes sp (sls25_Asp) are MW286812 and MW293720, respectively.
Sequences were analyzed for definition of haplotypes using DnaSP software v.6.12.03 (Rozas et al., 2017). The relationship between haplotypes, based on pairwise difference to generate a minimum spanning tree (MST) and minimum spanning network (MSN), was calculated and modeled using Network software and Hapstar v. 0.7. Multiple alignment and phylogenetic analysis were performed using the SaeView package (Gouy et al., 2010). Phylogenetic trees were built using maximum-likelihood (ML) with the general time reversible model with gama distributed with four discrete categories (GTR + I + G). The clade support was assessed via 500 bootstrap replicates.
Collections were conducted in 43 districts/municipalities in 25 dengue-endemic provinces in Indonesia (Supplementary Table 1 and Figure 1). These provinces were Aceh, Riau, Riau Islands, Jambi, Bangka-Belitung, Lampung, Banten, West Java, Central Java, Yogyakarta, East Java, West Kalimantan, South Kalimantan, Central Kalimantan, East Kalimantan, Bali, West Nusa Tenggara, East Nusa Tenggara, North Sulawesi, Central Sulawesi, South Sulawesi, Southeast Sulawesi, Maluku, North Maluku, and West Papua. Sampling of larva and adult mosquitoes was conducted as part of the 2nd year of the “Rikhus Vektora” project in July–August 2016 in 28 locations, the WHO project SEINO (#1611945) in September–October 2016 in six locations, and subsequently in nine locations as part of the 3rd year of “Rikhus Vektora” project in May–July 2017 (Figure 1). A total of 60,873 Ae. aegypti mosquitoes were collected: 2,184 adults, 54,251 larvae, and 4,438 adults obtained from reared larvae. With respect to Ae. albopictus, 16,223 mosquitoes were collected with the following breakdown: 4,957 adults, 9,638 larvae, and 1,628 adults obtained from reared larvae. From these samples 196 Ae. aegypti mosquitoes were sequenced: 34 adults and 162 adults obtained from reared larvae. For Ae. albopictus, 19 samples were sequenced, two adults and 17 adults obtained from reared larvae.
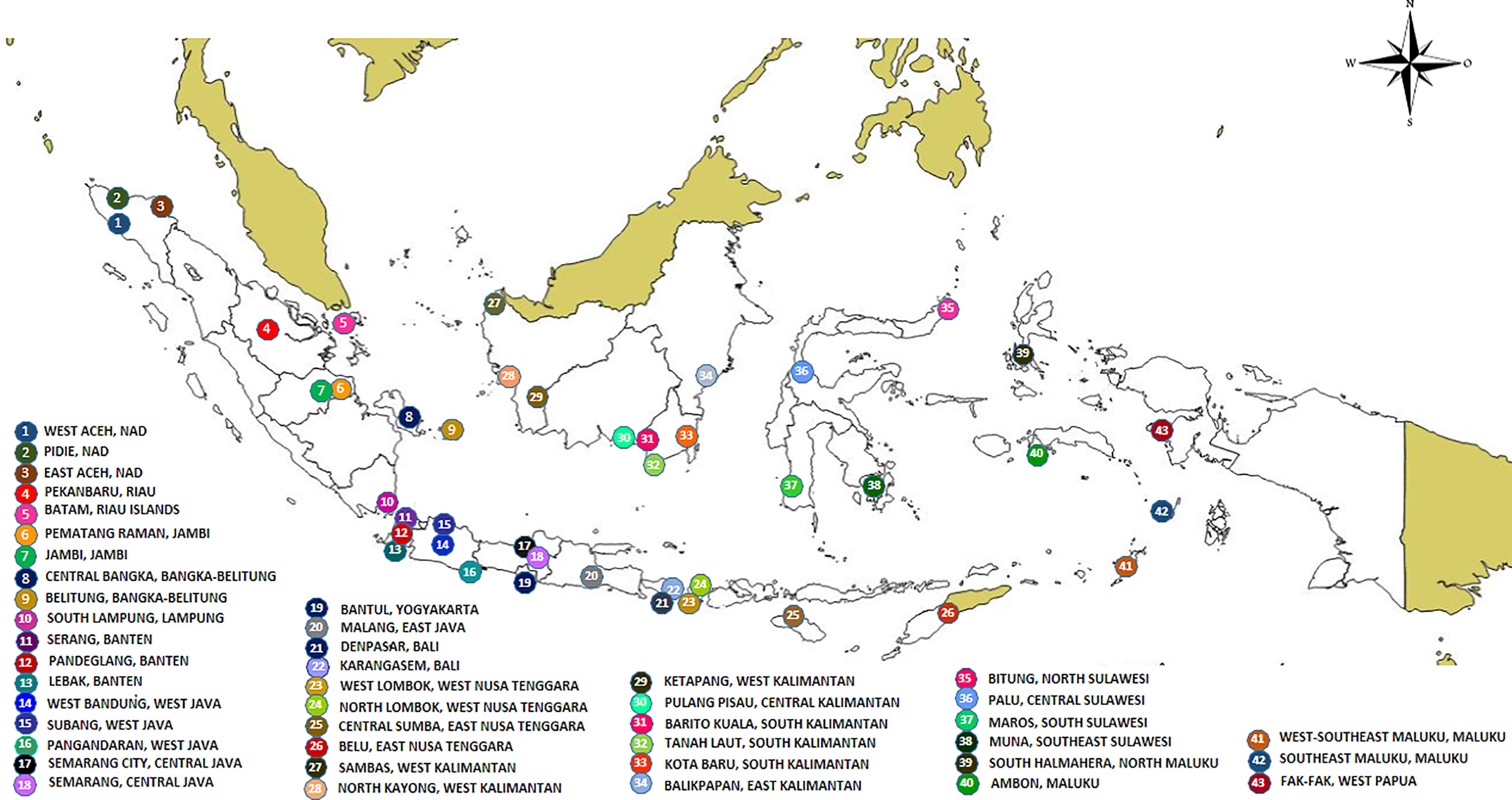
Figure 1 Map of sampling sites.
The samples collected in this work fell into three different branches, both for cox1 and ITS2 (Supplementary Figure 1). These branches corresponded to Ae. aegypti, Ae. albopictus, and another undetermined Aedes species. The latter sample was therefore named sls25_Asp (Supplementary Figure 1). The cox1 gene phylogeny showed the presence of two main clusters in Ae. aegypti, Cluster Aae1 and Cluster Aae2, with Cluster Aae1 being separated into two subclusters: Subcluster Aae1a and Subcluster Aae1b (Supplementary Table 1 and Supplementary Figure 1). These clusters were separated by very low bootstrap values indicating that the tree was not well structured and that the samples belonged to the same population (Supplementary Figure 1). However, with the cox1 gene from Ae. albopictus, three clusters could be identified with strong bootstraps (100), Cluster Aal1, Cluster Aal2, and Cluster Aal3 (Supplementary Table 2 and Supplementary Figure 1).
The sample sls25_Asp from Maros in South Sulawesi (site No 36 in Figure 1) was initially misidentified in the field as Culex quinquefasciatus. This sample was branching apart from Ae. aegypti and Ae. albopictus for the cox1 gene indicating that it was neither Ae. aegypti nor Ae. albopictus (Supplementary Figure 1). It was also different from Cx. quinquefasciatus which was used as outgroup. The phylogeny of the ITS2 sequences showed a similar result. The sample sls25_Asp was different from Ae. aegypti and Ae. albopictus (Supplementary Figure 2). The ITS2 sequence showed a best hit with an Aedes polynesiensis mosquito from Fidji (AY822662) with a percentage of identity of 88.24%, whereas the cox1 sequence displayed a best hit with an Ae. albopictus sample from Vietnam (HQ398902) with 91.95% identity. However, there was no cox1 sequence for Ae. polynesiensis in Genbank, and it was thus impossible to confirm if the cox1 gene would also link sls25_Asp to Ae. polynesiensis. A morphological analysis showed that the closest species, although still with morphological differences, was Aedes paullusi (data not shown). There is no cox1 or ITS2 records for Ae. paullusi in databases. The Ae. albopictus and Ae. polynesiensis hits for sls25_Asp cox1 and ITS2 sequences, respectively, might simply be default hits due of the lack of relevant sequences in the databases.
The cox1 sequences from 196 samples collected for this work were compared to the only other source of Ae. aegypti cox1 sequences from Indonesia, a 17-sample collection from 2013 in the North Coast of Central Java (Yohan et al., 2018). These samples, identified in the tree by their accession numbers from KP869121 to KP869126 and KP334259 to KP334269, make a completely separate cluster (Figure 2). The genetic distance between the sequences from this work and those reported by Yohan et al. (2018) ranged from 0.4 to 3.1%. With a mutation rate of the cox1 gene in insects ranging from 2.4% Mya−1 to 3.5% Mya−1 (Brower, 1994; Papadopoulou et al., 2010), the time needed for the accumulation of these mutations ranges from 114,285 to 167,000 years for a mutation rate of 2.4% Mya−1 and from 885,714 to 1,291,667 years for a mutation rate of 3.1% Mya−1. When blasting the cox1 sequences from this work against Genbank data, the sequences from previously Ae. aegypti identified from Indonesia in 2013 (Yohan et al., 2018) did not respond as best hits. However, best hits were obtained with the same score with a series of nine cox1 sequences of Ae. aegypti mosquitoes captured in Peru, Cambodia, Puerto Rico, India, Georgia, England, and Germany (Supplementary Table 3). Subcluster Aae2a showed two best hits both from Kenya (Supplementary Table 3). Subclusters Aae2b and Aae2d had only one corresponding best hit in Genbank from Mozambique and Haiti, respectively (Supplementary Table 4). Subcluster Aae2c showed five best hits with the same score from Egypt and Kenya (Supplementary Table 3). Interestingly, Subcluster Aae2e displayed four best hits with the same score, but only one was a wild-type mosquito captured in Haiti. The other three best hits corresponded to the reference strains reared in laboratory conditions of Liverpool and RED (Supplementary Table 3). Finally, each of the two individual samples diverging from Cluster 1, 46_Aae (IS1) and 28-1-Aae (IS2) displayed a different best hit. IS1 showed a best hit with a mosquito collected in Russia, whereas IS2 showed a best hit with Aedes aegypti formosus, which is considered an ancestral feral population from sub-Saharan Africa (Powell and Tabachnick, 2013; Gloria-Soria et al., 2016) (Supplementary Table 3). The breakdown into individual haplotypes showed that Cluster Aae1 comprised 176 samples out of 198 (89%) and 39 haplotypes out of 53 (73.6%) with two of them, H1 and H4, being the most represented (Supplementary Tables 1, 3 and Figure 3). H1 haplotype comprised 57 samples (32.85%) whereas the haplotype H4 contained 61 samples (34.6%) (Supplementary Table 1). Subcluster Aae1a comprised 16 haplotypes, including haplotype H1, and 75 samples, whereas Subcluster Aae1b contained haplotype H4 and 22 other haplotypes for a total of 102 samples (Supplementary Table 1).
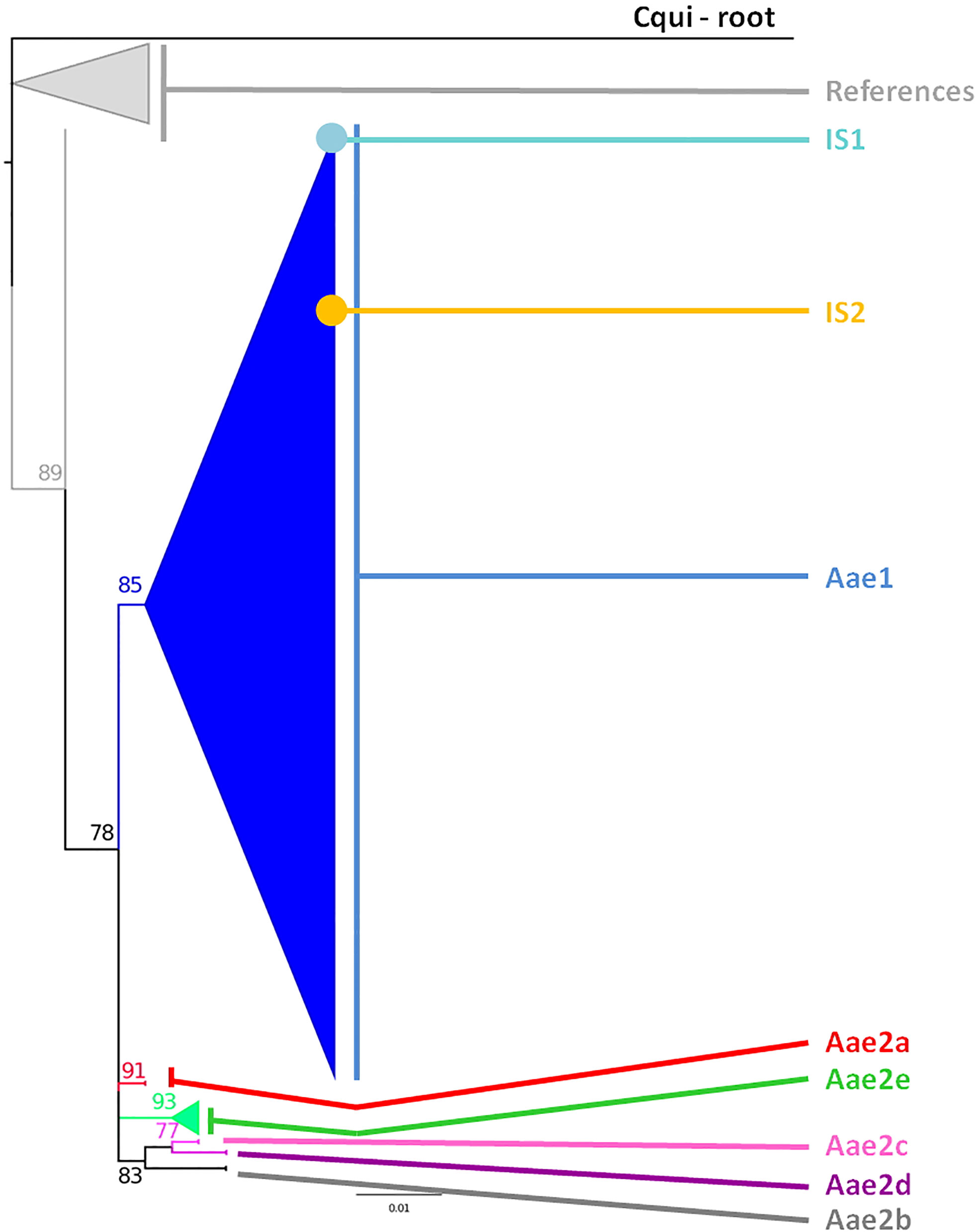
Figure 2 Phylogeny of the Aedes aegypti cox1 gene. The phylogenetic trees were built using maximum-likelihood (ML) with the general time reversible model with gama distributed with four discrete categories (GTR + I + G). The clade support was assessed via 500 bootstrap replicates. The tree was rooted using the Culex quinquefasciatus cox1 gene (MK265737) as outgroup. The color code is that of the cox1 subclusters shown in Supplementary Table 1: light gray: References, dark blue: Subcluster Aae1, red: Subcluster Aae2a, dark gray: Subcluster Aae2b, pink: Subcluster Aae2c, purple: Subcluster Aae2d, green: Subcluster Aae2e, light blue: individual sample 1, yellow: individual sample 2, black: root. “References” correspond to the Ae. aegypti cox1 sequences published by Yohan et al. (2018) from samples collected in 2013 whose accession numbers are KP334259 to KP334269 and KP869121 to KP89126.
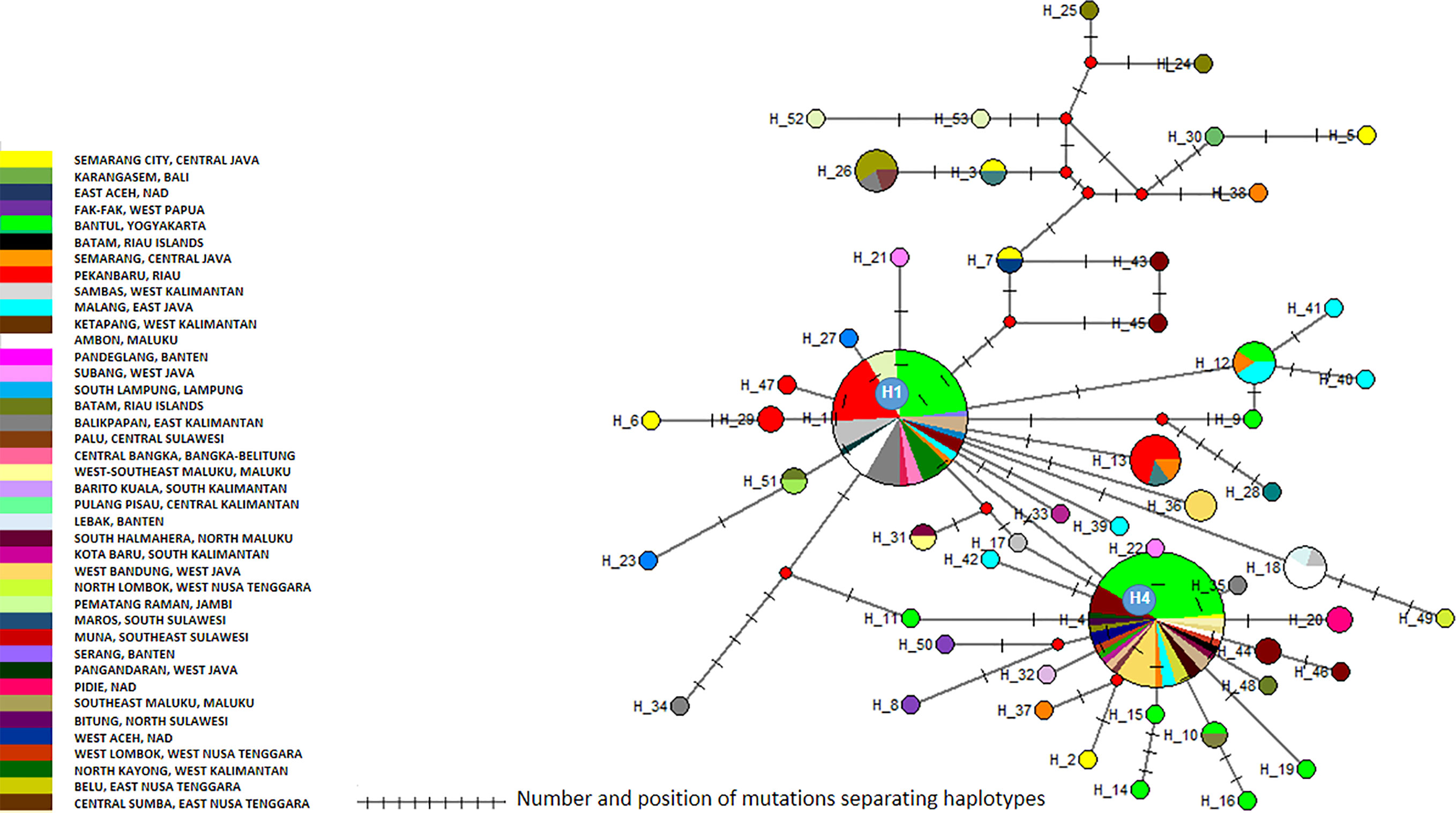
Figure 3 Network of Aedes aegypti cox1 haplotypes. The figure represents the frequency of each haplotype of the cox1 gene of Ae. aegypti in the regions sampled. The network represents the number of mutations between the haplotypes and their location.
Cluster Aae1 was, as expected, present everywhere with the exception of East Aceh and North Lombok (Supplementary Figure 3 and Supplementary Table 1). No correlation could be found between any cluster and any location. When considering the geographic distribution of the haplotypes, a lack of correlation was also observed (Supplementary Figure 4). Only a default correlation could be observed, i.e. rare haplotypes from a region with few samples. However, this is a sampling bias and is not significant.
The 40 sequences were distributed into two clusters and 21 haplotypes (Supplementary Table 1, Figure 4 and Supplementary Table 5). Cluster 1 is divided into four subclusters (1a to 1d), which displayed limited variations (Figure 5). As a consequence, Cluster 1 gathered 42 samples and 23 haplotypes representing all the sequence variations observed within this monophyletic group (Figure 5). The different haplotypes were closely related with a maximum relative distance of 11.54% (Supplementary Table 5). Cluster 2 comprised only four samples, each one corresponding to a different haplotype. They were very closely related with an overall variation of 0.51% (Supplementary Table 5).
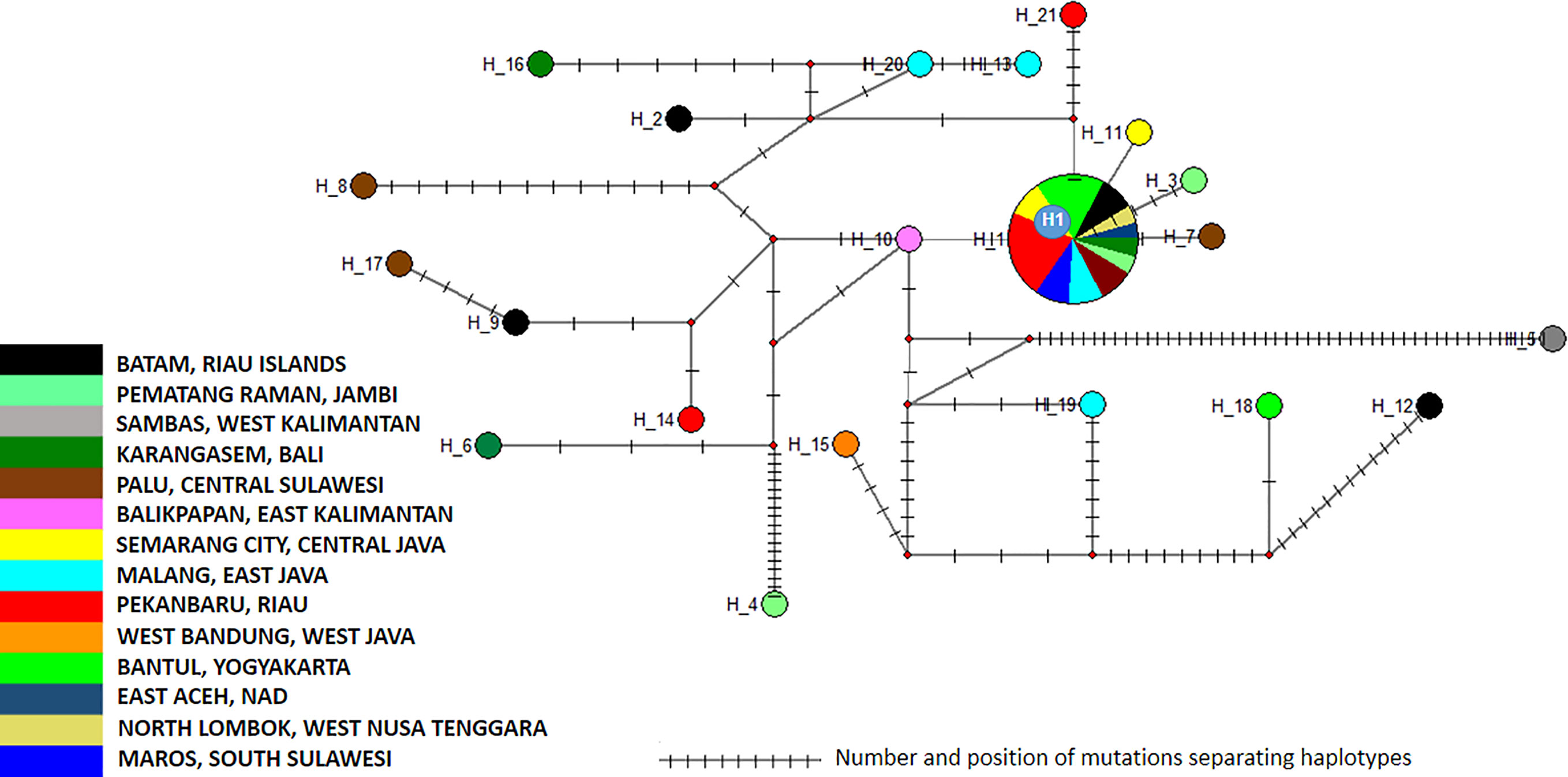
Figure 4 Network of Aedes aegypti ITS2 sequences. The figure represents the frequency of each haplotype of the ITS2 sequence of Ae. aegypti in the regions sampled. The network represents the number of mutations between the haplotypes and their location.

Figure 5 Phylogeny of the Aedes aegypti ITS2 sequences. The phylogenetic tree was built using maximum-likelihood (ML) with the general time reversible model with gama distributed with four discrete categories (GTR + I + G). The clade support was assessed via 500 bootstrap replicates. The tree was rooted using the Culex quinquefasciatus ITS2 sequence (HQ848572) as outgroup. The color code used is that of the ITS2 subclusters given in Supplementary Table 1.
The analysis of the polymorphism of Ae. aegypti ITS2 haplotype in this study showed that cluster 1 was the dominant one (Supplementary Figure 5). Subcluster 1a displayed the most extensive distribution, covering Sumatra (East Aceh-Aceh, Pematang Raman-Jambi, Pekan Baru-Riau, South Lampung-Lampung), Java (West Bandung-West Java, Semarang-Central Java, Bantul-Yogyakarta, Malang-East Java), Kalimantan (Sambas-West Kalimantan, Balikpapan-East Kalimantan), Bali (Karangasem), West Nusa Tenggara (Lombok), and Sulawesi (Palu-Central Sulawesi, Maros-South Sulawesi) (Supplementary Figure 5). Subcluster 1a showed a best hit with mosquitoes collected in Russia, Sri Lanka. The other subclusters, namely 1b, 1c, and 1d, showed a limited distribution. Subcluster 1b was only found in the Sambas-West Kalimantan region, while Subcluster 1c was identified in two locations: Batam-Riau Islands and Bantul-Yogyakarta. Subcluster 1d was found in two locations, i.e. Pematang Raman-Jambi and Palu-Central Sulawesi (Supplementary Figure 5 and Supplementary Table 5). Cluster 2 had a more limited distribution. This cluster was found in Karangasem-Bali, Ambon-Maluku, Malang-East Java and Batam-Riau islands (Supplementary Figure 5 and Supplementary Table 5).
The 19 cox1 sequences of the Ae. albopictus samples collected in this work were compared to those released by Maynard et al. (2017) who collected samples in Jakarta in 2012, in Waingapu (Sumba) in 2013, and in Timika (Papua) in 2015. Sequences were also compared to those released by Battaglia et al. (2016) who established a worldwide classification of Ae. albopictus cox1 haplogroups (Battaglia et al., 2016). The COI sequences from Maynard et al. (2017) matched perfectly the haplogroups defined by Battaglia et al. (2016) and were distributed within two different haplogroups, A2a and A1b1a. The sequences obtained in this work did not correspond to the sequences reported by Maynard et al. (2017) and did not match any of the haplogroups defined by Battaglia et al. (2016) (Figure 6). Out of the three clusters identified within the Ae. albopictus sequences reported in this work, Cluster Aal1 was closer, although clearly different, to the haplogroups A2a; Cluster Aal3 was closer, although different, to the haplogroup A1ba1; and Cluster Aal2 was not close to any haplogroup. A total of 11 different haplotypes were found (Figure 7 and Supplementary Table 2). The genetic distance between the cox1 sequences reported in this work and those from Maynard et al. (2017) ranges from 0.4 to 1.3%, depending on the sample. The time needed to accumulate the number of mutations separating the samples from this work to those described by Maynard et al. (2017) ranges from 114,285 to 167,000 years for a mutation rate of 2.4% Mya−1 and from 371,428 to 542,000 years for a mutation rate of 3.5% Mya−1. Cluster Aal1 was found only in Central Kalimantan, whereas the other two clusters were spread over different provinces (Supplementary Figure 6). However, the sample size is too small to draw any significant conclusion on the phylogeography. When blasted on databases, Cluster Aal1 and Cluster Aal2 both displayed best hits with the same Ae. albopictus populations from The Philippines but with differing percentage of identity ranging from 99.51 to 99.85% (Supplementary Table 4). Cluster Aal3 showed best hits with invasive populations of Ae. albopictus found in D.R. Congo, China, Thailand, Greece, Brazil, and the USA with 99.65 to 99.84% of identity (Supplementary Table 4). With respect to ITS2, the number of sequences available (only three) was too small to draw a conclusion. Nevertheless, they corresponded to invasive populations of Ae. albopictus found in Italy, Georgia, Israel, or Sri Lanka (Supplementary Table 6).

Figure 6 Phylogeny of Aedes albopictus cox1 genes. The phylogenetic tree was built using maximum-likelihood (ML) with the general time reversible model with gama distributed with four discrete categories (GTR + I + G). The clade support was assessed via 500 bootstrap replicates. The tree was rooted using the Culex quinquefasciatus cox1 gene (MK265737) as outgroup. The color code used is that of the cox1 subclusters displayed in Supplementary Table 2. Gray: Subcluster Aal1, green: Subcluster Aal2, orange: Subcluster Aal3.
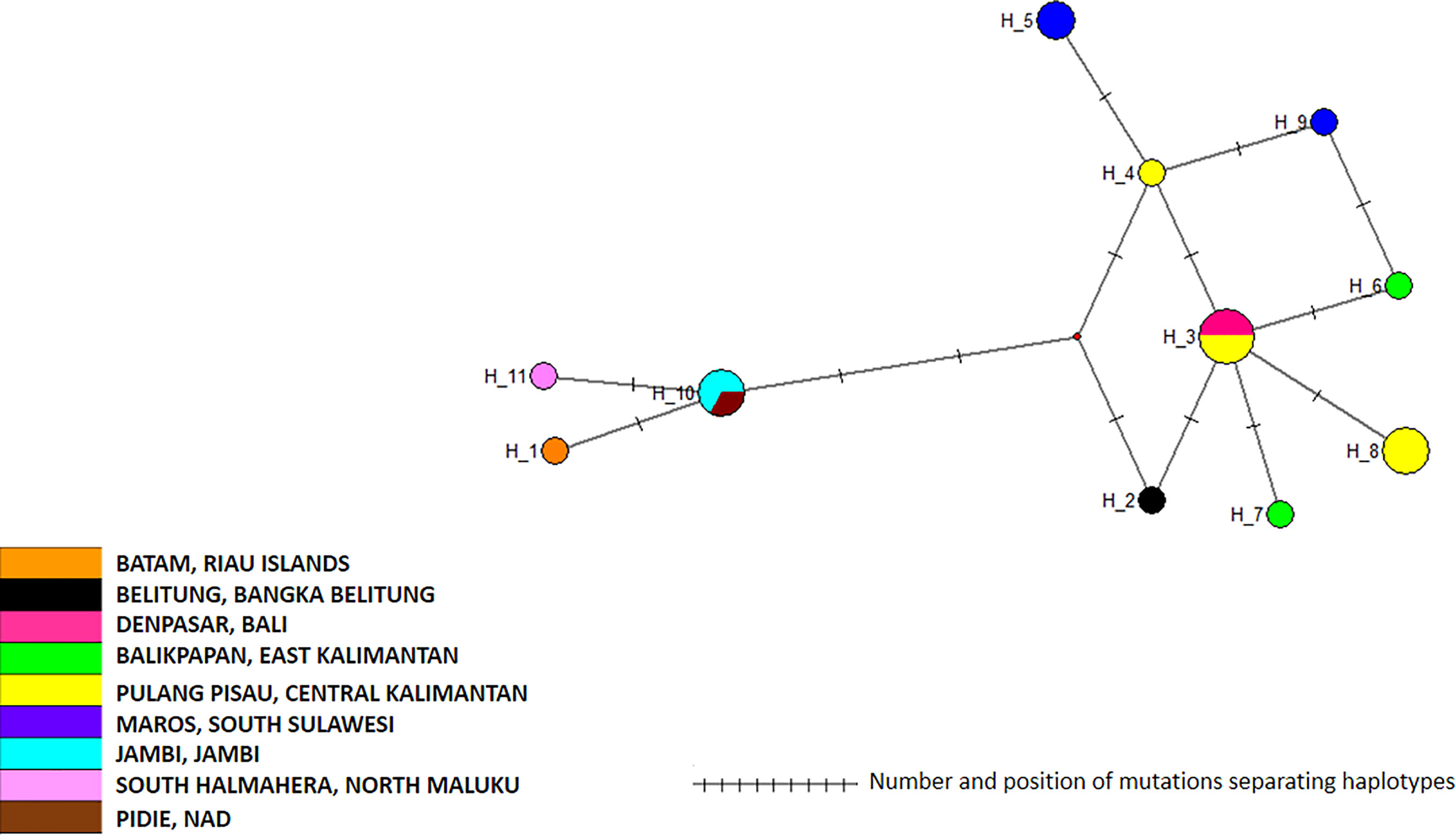
Figure 7 Network of Aedes albopictus cox1 haplotypes. The figure represents the frequency of each haplotype of the cox1 gene of Ae. albopictus in the regions sampled. The network represents the number of mutations between the haplotypes and their location on the gene sequence.
Dengue, which is the fastest spreading arbovirus disease worldwide, is also the first ranking vector-borne disease in Indonesia and, thus, a national health priority. In the absence of treatment and commercially available vaccine, vector management remains the only way to control the disease. However, in order to do so, the knowledge of vector population structure is an obligate prerequisite. The capacity of vectoring a given pathogen is not correlated with the species but instead with the population. Some populations of Ae. aegypti or Ae. albopictus are more prone to disseminate a given pathogen than others (Alto et al., 2008; Reinhold et al., 2018). Therefore, a species should be regarded as a metapopulation or the combination of cross-fertile genetically distinct populations displaying differing phenotypic traits (Garjito et al., 2020). Vector competence is one of these phenotypic traits, which in Aedes and other mosquitoes, was shown to be related to specific populations (Beerntsen et al., 2000; Severson and Behura, 2016) and not to the species per se. Deciphering the structure of the vector populations is thus essential.
Although dengue is the transmissible disease ranking number one in Indonesia, there have been very limited works on the analysis of the structure of the populations of Aedes with only one study on Ae. Aegypti (Yohan et al., 2018) and one on Ae. albopictus (Maynard et al., 2017). In both cases, the number of sampling sites was very limited. To these must be added studies aiming at assessing the stability of populations in the framework of a Wolbachia release program in Yogyakarta (Rašić et al., 2015). However, in this case, the genetic diversity was assessed with microsatellites and SNPs only on a sample from the city of Yogyakarta in South Central Java (Rašić et al., 2015). This work is, to our knowledge, the first one exploring the genetic diversity of Ae. aegypti and Ae. albopictus throughout Indonesia, a huge archipelago of more than 13,000 islands spanning 5,271 km from east to west and 2,210 km from north to south.
A first conclusion from this work is the homogeneity of the Ae. aegypti populations found all over Indonesia. With respect to the cox1 sequences, all Ae. aegypti samples belonged to the same maternal lineage. Variations were observed and clusters were described, but they simply represent a polymorphism within a monophyletic population. All clusters identified correspond to co-circulating variants. The main difference is that one cluster, Cluster Aae2, comprised samples displaying a larger polymorphism. Cluster Aae1, and in particular the haplotypes H1 and H4, seemed to be populations with a very high colonizing and demographic potential. These two haplotypes represent each about 30% of the samples collected all over Indonesia. They represent indeed the very same population with very limited or no polymorphism at all with Cluster Aae1 making up 87% of all samples and being present everywhere in Indonesia. Considering the size and structure of Indonesia, an archipelago spanning from the Indian Ocean to the Pacific Ocean, this is unexpected. One would have instead expected patched populations differing from one island to the other. What is observed is exactly the contrary, the same population throughout the whole country.
Owing to the mutation rate of the cox1 gene in Ae. aegypti (Brower, 1994; Papadopoulou et al., 2010), the population described in this work cannot have evolved from the populations previously described. Even in the case of introgression, the maternal lineage remains the same. What is described in this case is a different maternal lineage. This indicates that the observed population is allogenic. The lack of BLAST best hits with the previously described Indonesian populations and the occurrence of BLAST best hits with populations from other parts of the world also support this allogenic origin. Two hypotheses must then be considered to explain the massive presence of this homogeneous and allogenic population of Ae. aegypti. The first hypothesis is that the population described in this work has been completely missed in previous studies. Previous sampling conducted in 2013 yielded genetically different populations (Yohan et al., 2018) but were very limited and covered a limited zone (Yohan et al., 2018). This result could then be considered a consequence of a sampling bias, all mosquitoes from the previous study (Yohan et al., 2018) having been captured in a specific area, i.e. Northern Coastal Central Java. However, considering the extension of Cluster Aae1 throughout Indonesia and its overwhelming presence among samples (87%), it seems unlikely that it would have been missed in the 2013 sampling campaign. Furthermore, Cluster Aae1 was found to be strongly present in this same Northern Coastal Central Java area. The second hypothesis is that this allogenic population might have invaded Indonesia after the 2013 sampling. This invasion would have been very fast then, within a maximum of three years between 2013, date of the former sampling, and 2016, date of our first sampling. However, owing to the very limited information available on the previous populations, it is not possible to formally demonstrate this replacement hypothesis. Nevertheless, the ITS2 marker showed a similar trend; and considering the high potential of nuclear DNA for recombination and variation and finding the same ITS2 cluster all over Indonesia confirm the presence of a set of genetically closely related populations in Indonesia with one specific population characterized by two very closely related, monophyletic maternal haplotypes, H1 and H4. These haplotypes seem to be highly invasive most likely due to higher demographic and adaptability potentials. This also suggests that assortative mating might occur, which restricts greatly intraspecies breeding with preexisting populations.
Ae. aegypti has been shown to be highly susceptible to satyrization by Ae. albopictus leading to the replacement of Ae. aegypti populations by Ae. albopictus ones, thus explaining in part the invasive potential of the latter (Bargielowski and Lounibos, 2016). However, what is observed in Indonesia does not match this model despite the presence of populations of Ae. albopictus described as invasive in other parts of the world. What is seen in Indonesia is instead a homogeneous population of Ae. aegypti occupying all the archipelago and outcompeting Ae. albopictus. As per Ae. aegypti, the populations of Ae. albopictus collected in Indonesia do not correspond to those previously described from 2012 to 2015 (Maynard et al., 2017). Unlike populations described by Maynard et al. (2017), they also do not correspond to the haplogroups designed on samples from 2013 (Battaglia et al., 2016). They are related but not the same. Owing to the rate of mutation of the cox1 gene in insects, 2.4% Mya−1 to 3.5% Mya−1 depending on the model (Brower, 1994; Papadopoulou et al., 2010), the variations observed are not compatible with an evolution of previous local populations and indicate an invasion by allogenic populations. However, just as per Ae. aegypti the very limited number of previous studies makes it difficult to formally demonstrate a replacement.
The domestication of Ae. aegypti and Ae. albopictus is a process closely linked to the development of the human society and in particular to long distance mobility, transportation of goods, and international trade (Paupy et al., 2009). The current expansion of the Aedes-borne diseases is by far a consequence of the global economy. Ae. aegypti and Ae. albopictus, like all living organisms, are structured in metapopulations, which differ slightly one from the other and due to massive international transportation are distributed all over the world within the areas suitable for the survival of these species. The mobility of these populations from one place to another is a stochastic event, which depends on the place of departure, the place of arrival, the genetic and physiological traits of the populations involved, the economic situation, and the commercial exchanges and routes at a given time. This is to our knowledge the first report of such a massive homogeneity of a population of Ae. aegypti over such a large area.
One limitation of this work is the lack of previous data at the scale of the whole country. Only two studies have been conducted prior to our work on the genotyping of Aedes mosquitoes in Indonesia. There is only one report on the genotyping of Ae. aegypti but only on few samples and only in one restricted area in northern coastal central Java (Yohan et al., 2018). This lack of previous data prevents us from fully analyzing and concluding on the putative replacement of populations that the phylogenetic analyses suggest. Regarding the analysis of Ae. albopictus, the same problem occurred with only one previous study (Maynard et al., 2017) thus preventing the conduct of a fully significant comparison. On another hand, a major limitation of this work is the limited number of Ae. albopictus samples and sequences of ITS2. This is due to a limitation in funding which prevented the conduct of a full scale sequencing program. Further analyses of Ae. albopictus diversity in Indonesia should be conducted to complete the current study.
A general consequence of our results is that populations are changing over time, even throughout a very large archipelago. Whatever the population, established or invasive, Ae. aegypti and Ae. albopictus mosquitoes will have to breed in the human environment. Then, the best way to prevent any population of vector from thriving is certainly to implement vector control at a very local level, at maximum at the community level, essentially by eliminating breeding sites using very simple and affordable means of control such as containers and garbage removal. The strategy of prevention of dengue transmission through community participation currently recommended in Indonesia will most likely be the most successful of all. This approach named 3M for “Menutup” for covering water containers, “Menguras” for cleaning water containers, and “Mengubur” for burying discarded containers, is implemented under the responsibility of families in each household with at least one person in charge of monitoring Aedes larvae in all water storage (Paupy et al., 2009; MoH Indonesia, 2015). This strategy shed light on what is most needed for the successful control of Aedes-borne diseases, not big science, big management or big strategies but simply information, education, people awareness and community-based management.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Specific permissions were not required for field Aedes larval collections. Oral consent to inspect the Aedes breeding places in the household was obtained from the homeowners and local government authorities. Formal approval to conduct these activities was granted by the Ethical Commission Board of the NIHRD, Ministry of Health, Indonesia (No. LB.02.01/5.2/KE.003/2016, January 13, 2016; and No. LB.02.01/5.2/KE.020/2017, February 6, 2017).
Conceptualization: TG, W, MH, RU, MS, and TS. Data curation: TG, SH, M, and MP. Investigation: TG, SH, M, and MP. Formal analysis: TG, MP, MH, RF, and LG. Resources: TG, MP, M, MH, and SH. Methodology: TG, RF, and LG. Project administration: MH and TG. Software: RF, LG, and TG. Supervision: SM and RF. Validation: RF, SM, TG, and LG. Writing—original draft: TG and RF. Writing—review and editing: RF, SM, and TG. All authors contributed to the article and approved the submitted version.
The research was supported by the Institute for Vector and Reservoir Control Research and Development, National Institute of Health Research and Development, Ministry of Health Indonesia under the project Rikhus Vektora 2016–2017 and as part of the WHO project SEINO1611945 in 2016. TG, RF, SM, and TS were supported in part by the PHC Nusantara projects Zika & Co and SOCIAL, co-funded by the French Ministry of Foreign Affairs and RISTEKDIKTI under the Indonesian Ministry of Education and Culture.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors would also like to thank the people in the study areas for cooperating during the entomological surveys, all field mosquito collectors and all technical team of the Rikhus Vektora project on disease vectors and reservoirs in 2016–2017. We also thank all team members of the WHO project SEINO 1611945 in 2016. Both projects were conducted under the supervision and coordination of the Institute for Vector and Reservoir Control Research and Development, National Institute of Health Research and Development, Ministry of Health, Republic of Indonesia.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.705129/full#supplementary-material
Supplementary Figure 1 | cox1 gene phylogeny of the collected samples. The phylogenetic trees were built using maximum-likelihood (ML) with the general time reversible model with gama distributed with four discrete categories (GTR + I + G). The clade support was assessed via 500 bootstrap replicates. The tree was rooted using the Culex quinquefasciatus cox1 gene (MK265737) as outgroup.
Supplementary Figure 2 | ITS2 phylogeny of the collected samples. The phylogenetic tree was built using maximum-likelihood (ML) with the general time reversible model with gama distributed with four discrete categories (GTR + I + G). The clade support was assessed via 500 bootstrap replicates. The tree was rooted using the Culex quinquefasciatus ITS2 sequence (HQ848572) as outgroup. The color code used is that of the ITS2 clusters of Ae. aegypti and Ae. albopictus displayed in Supplementary Tables 1 and 2, respectively. The color code for Ae. aegypti ITS2 clusters is: blue: Cluster 1a, red: Cluster 1b, orange: Cluster 1c.
Supplementary Figure 3 | Geographic distribution of Aedes aegypti cox1 clusters.
Supplementary Figure 4 | Geographic distribution of Aedes aegypti cox1 haplotypes.
Supplementary Figure 5 | Geographic distribution of Aedes aegypti ITS2 sequences.
Supplementary Figure 6 | Geographic distribution of Aedes albopictus cox1 haplotypes.
Supplementary Table 1 | Specimens and sampling localities of Aedes aegypti.
Supplementary Table 2 | Specimens and sampling localities of Aedes albopictus.
Supplementary Table 3 | Polymorphism of Aedes aegypti cox1 haplotypes from Indonesia.
Supplementary Table 4 | Polymorphism of Aedes albopictus cox1 haplotypes from Indonesia.
Supplementary Table 5 | Polymorphism of Aedes aegypti ITS2 haplotypes from Indonesia.
Supplementary Table 6 | Polymorphism of Aedes albopictus ITS2 haplotypes from Indonesia.
Alto, B. W., Reiskind, M. H., Lounibos, L. P. (2008). Size Alters Susceptibility of Vectors to Dengue Virus Infection and Dissemination. Am. J. Trop. Med. Hyg. 79, 688–695. doi: 10.4269/ajtmh.2008.79.688
Bargielowski, I. E., Lounibos, L. P. (2016). Satyrization and Satyrization-Resistance in Competitive Displacements of Invasive Mosquito Species. Insect Sci. 23, 162–174. doi: 10.1111/1744-7917.12291
Battaglia, V., Gabrieli, P., Brandini, S., Capodiferro, M. R., Javier, P. A., Chen, X.-G., et al. (2016). The Worldwide Spread of the Tiger Mosquito as Revealed by Mitogenome Haplogroup Diversity. Front. Genet. 23, 208. doi: 10.3389/fgene.2016.00208
Beerntsen, B. T., James, A. A., Christensen, B. M. (2000). Genetics of Mosquito Vector Competence. Microbiol. Mol. Biol. Rev. 64, 115–137. doi: 10.1128/MMBR.64.1.115-137.2000
Brower, A. V. Z. (1994). Phylogeny of Heliconius Butterflies Inferred From Mitochondrial Dna Sequences (Lepidoptera: Nymphalidae). Mol. Phyl. Evol. 3, 159–174. doi: 10.1006/mpev.1994.1018
Chang, F. S., Tseng, Y. T., Hsu, P. S., Chen, C. D., Lian, I. B., Chao, D. Y. (2015). Re-Assess Vector Indices Threshold as an Early Warning Tool for Predicting Dengue Epidemic in a Dengue non-Endemic Country. PloS Negl. Trop. Dis. 9, 1–20. doi: 10.1371/journal.pntd.0004043
da Costa-da-Silva, A. L., Capurro, M. L., Bracco, J. E. (2005). Genetic Lineages in the Yellow Fever Mosquito Aedes (Stegomyia) aegypti (Diptera: Culicidae) From Peru. Mem. do Inst. Oswaldo. Cruz. 100, 539–544. doi: 10.1590/S0074-02762005000600007
Focks, D. A., Special Program for Research and Training in Tropical Diseases (TDR) (2003). Review of Entomological Sampling Methods and Indicators for Dengue Vectors Vol. 28 (Geneva, Switzerland: UNICEF, UNDP, World Bank, WHO Spec Program Res Train Trop Dis), 208–389.
Garjito, T. A., Hidajat, M. C., Kinansi, R. R., Setyaningsih, R., Anggraeni, Y. M., Mujiyanto, et al. (2020). Stegomyia Indices and Risk of Dengue Transmission: A Lack of Correlation. Front. Public Health 8, 328. doi: 10.3389/fpubh.2020.00328
Gloria-Soria, A., Ayala, D., Bheecarry, A., Calderon-Arguedas, O., Chadee, D. D., Chiappero, M., et al. (2016). Global Genetic Diversity of Aedes aegypti. Mol. Ecol. 25, 5377–5395. doi: 10.1111/mec.13866
Goubert, C., Minard, G., Vieira, C., Boulesteix, M. (2016). Population Genetics of the Asian Tiger Mosquito Aedes albopictus, an Invasive Vector of Human Diseases. Heredity. (Edinb). 117, 125–134. doi: 10.1038/hdy.2016.35
Gouy, M., Guindon, S., Gascuel, O. (2010). SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Grard, G., Caron, M., Mombo, I. M., Nkoghe, D., Mboui Ondo, S., Jiolle, D., et al. (2014). Zika Virus in Gabon (Central Africa)–2007: A New Threat From Aedes albopictus? PloS Negl. Trop. Dis. 8, e2681. doi: 10.1371/journal.pntd.0002681
Gubler, D. J. (1997). “Dengue and Dengue Hemorrhagic Fever: Its History and Resurgence as a Global Public Health Prolem,” in Dengue and Dengue Hemorrhagic Fever. Eds. Gubler, D., Kuno, G. (Wallingford, UK: CAB International), 1–22.
Gubler, D. J. (2011). Dengue, Urbanization and Globalization: The Unholy Trinity of the 21 St Century. Trop. Med. Health 39 (4 Suppl.), 3–11. doi: 10.2149/tmh.2011-S05
Gupta, S., Preet, S. (2014). Genetic Differentiation of Invasive Aedes albopictus by RAPD-PCR: Implications for Effective Vector Control. Parasitol. Res. 113, 2137–2142. doi: 10.1007/s00436-014-3864-2
Harapan, H., Michie, A., Mudatsir, M., Sasmono, R. T., Imrie, A. (2019). Epidemiology of Dengue Hemorrhagic Fever in Indonesia: Analysis of Five Decades Data From the National Disease Surveillance. BMC Res. Notes 12, 4–9. doi: 10.1186/s13104-019-4379-9
IVRCRD (2017). Manual on Mosquitoes Field Sampling Methods (in Bahasa Indonesia). 3rd edition (Indonesia: IVRCRD: NIHRD-MoH Indonesia, Salatiga).
IVRCRD (2018). Final Compilation Rikhus Vectora 2015-2018 (Indonesia: IVRCRD-Salatiga, NIHRD-MoH Indonesia), 80.
Ja, D. (2016). Etymologia: Aedes aegypti. Emerg. Infect. Dis. 22, 1807–1807. doi: 10.3201/eid2210.ET2210
Joyce, A. L., Torres, M. M., Torres, R., Moreno, M. (2018). Genetic Variability of the Aedes aegypti (Diptera: Culicidae) Mosquito in El Salvador, Vector of Dengue, Yellow Fever, Chikungunya and Zika. Parasit. Vectors 11, 1–14. doi: 10.1186/s13071-018-3226-5
Knudsen, A. B. (1995). Global Distribution and Continuing Spread of Aedes albopictus. Parassitologia 37, 91–97.
Kraemer, M., Sinka, M., Duda, K., Mylne, A., Shearer, F., Barker, C., et al. (2015). The Global Distribution of the Arbovirus Vectors Aedes aegypti and Ae. albopictus. Elife 4, e08347. doi: 10.7554/eLife.08347
Kuno, G. (2010). Early History of Laboratory Breeding of Aedes aegypti (Diptera: Culicidae) Focusing on the Origins and Use of Selected Strains. J. Med. Entomol. 47, 957–971. doi: 10.1603/ME10152
Kusriastuti, R., Sutomo, S. (2005). Evolution of Dengue Prevention and Control Programme in Indonesia DF/DHF Disease Burden. Dengue. Bull. 29, 1–7.
Lai, Z., Zhou, T., Zhou, J., Liu, S., Xu, Y., Gu, J., et al. (2020). Vertical Transmission of Zika Virus in Aedes albopictus. PloS Negl. Trop. Dis. 14, e0008776. doi: 10.1371/journal.pntd.0008776
Maynard, A. J., Ambrose, L., Cooper, R. D., Chow, W. K., Davis, J. B., Muzari, M. O., et al. (2017). Tiger on the Prowl: Invasion History and Spatio-Temporal Genetic Structure of the Asian Tiger Mosquito Aedes albopictus (Skuse 1894) in the Indo-Pacific. PloS Negl. Trop. Dis. 11, 1–27. doi: 10.1371/journal.pntd.0005546
McKenzie, B. A., Wilson, A. E., Zohdy, S. (2019). Aedes albopictus is a Competent Vector of Zika Virus: A Meta-Analysis. PloS One 14, e0216794. doi: 10.1371/journal.pone.0216794
MoH Indonesia. (2015). “Guidelines for Dengue Hemorrhagic Fever Control in Indonesia (in Bahasa Indonesia),” in Directorate General Disease Prevention and Control (Jakarta, Indonesia: Ministry of Health of Republic Indonesia).
MoH Indonesia (2018). “Dengue Hemorrhagic Fever Situation in Indonesia Tahun 2017,” in Center for Data and Information (MoH Republic of Indonesia).
Moncayo, A. C., Fernandez, Z., Ortiz, D., Diallo, M., Sall, A., Hartman, S., et al. (2004). Dengue Emergence and Adaptation to Peridomestic Mosquitoes. Emerg. Infect. Dis. 10, 1790–1796. doi: 10.3201/eid1010.030846
Mulyatno, K. C., Kotaki, T., Yotopranoto, S., Rohmah, E. A., Churotin, S., Sucipto, T. H., et al. (2018). Detection and Serotyping of Dengue Viruses in Aedes aegypti and Aedes albopictus (Diptera: Culicidae) Collected in Surabaya, Indonesia From 2008 to 2015. Jpn. J. Infect. Dis. 71, 58–61. doi: 10.7883/yoken.JJID.2017.117
Naim, D. M., Kamal, N. Z. M., Mahboob, S. (2020). Population Structure and Genetic Diversity of Aedes aegypti and Aedes albopictus in Penang as Revealed by Mitochondrial DNA Cytochrome Oxidase I. Saudi. J. Biol. Sci. 27, 953–967. doi: 10.1016/j.sjbs.2020.01.021
Ngoagouni, C., Kamgang, B., Nakouné, E., Paupy, C., Kazanji, M. (2015). Invasion of Aedes albopictus (Diptera: Culicidae) Into Central Africa: What Consequences for Emerging Diseases? Parasit. Vectors 8, 191. doi: 10.1186/s13071-015-0808-3
Olson, M. F., Ndeffo-Mbah, M. L., Juarez, J. G., Garcia-Luna, S., Martin, E., Borucki, M. K., et al. (2020). High Rate of non-Human Feeding by Aedes aegypti Reduces Zika Virus Transmission in South Texas. Viruses 12, 1–20. doi: 10.3390/v12040453
Papadopoulou, A., Anastasiou, I., Vogler, A. P. (2010). Revisiting the Insect Mitochondrial Molecular Clock: The Mid-Aegean Trench Calibration. Mol. Biol. Evol. 27, 1659–1672. doi: 10.1093/molbev/msq051
Parimittr, P., Chareonviriyaphap, T., Bangs, M. J., Arunyawat, U. (2018). Genetic Variation of Aedes aegypti Mosquitoes Across Thailand Based on Nuclear DNA Sequences. Agric. Nat. Resour. 52, 596–602. doi: 10.1016/j.anres.2018.11.021
Parra, M. C. P., Fávaro, E. A., Dibo, M. R., Mondini, A., Eiras, Á. E, Kroon, E. G., et al. (2018). Using Adult Aedes aegypti Females to Predict Areas at Risk for Dengue Transmission: A Spatial Case-Control Study. Acta Trop. 182, 43–53. doi: 10.1016/j.actatropica.2018.02.018
Paupy, C., Delatte, H., Bagny, L., Corbel, V., Fontenille, D. (2009). Aedes albopictus, an Arbovirus Vector: From the Darkness to the Light. Microbes Infect. 11, 1177–1185. doi: 10.1016/j.micinf.2009.05.005
Paupy, C., Ollomo, B., Kamgang, B., Moutailler, S., Rousset, D., Demanou, M., et al. (2010). Comparative Role of Aedes albopictus and Aedes aegypti in the Emergence of Dengue and Chikungunya in Central Africa. Vector. Borne. Zoonotic. Dis. 10, 259–266. doi: 10.1089/vbz.2009.0005
Powell, J. R., Tabachnick, W. J. (2013). History of Domestication and Spread of Aedes aegypti–a Review. Mem. Inst. Oswaldo. Cruz. 108 (Suppl 1), 11–17. doi: 10.1590/0074-0276130395
Rašić, G., Endersby-Harshman, N., Tantowijoyo, W., Goundar, A., White, V., Yang, Q., et al. (2015). Aedes aegypti has Spatially Structured and Seasonally Stable Populations in Yogyakarta, Indonesia. Parasit. Vectors 8, 1–12.
Reinhold, J. M., Lazzari, C. R., Lahondère, C. (2018). Effects of the Environmental Temperature on Aedes aegypti and Aedes albopictus Mosquitoes: A Review. Insects 9, 158. doi: 10.3390/insects9040158
Rezza, G., Nicoletti, L., Angelini, R., Romi, R., Finarelli, A. C., Panning, M., et al. (2007). Infection With Chikungunya Virus in Italy: An Outbreak in a Temperate Region. Lancet 370, 1840–1846. doi: 10.1016/S0140-6736(07)61779-6
Ritchie, S. A. (2014). “Dengue Vectors Bionomics : Why Aedes aegypti Such a Good Vector,” in Dengue and Dengue Hemorrhagic Fever, 2nd edition. Eds. Gubler, D. J., Ooi, E. E., Vasudevan, J. F. S. (Boston, USA: CAB International), 455–480.
Rozas, J., Ferrer-Mata, A., Sanchez-DelBarrio, J., Guirao-Rico, S., Librado, P., Ramos-Onsins, S., et al. (2017). DnaSP V6: DNA Sequence Polymorphism Analysis of Large Datasets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Rueanghiran, C., Apiwathnasorn, C., Sangthong, P., Samung, Y., Ruangsittichai, J. (2011). Utility of a Set of Conserved Mitochondrial Cytochrome Oxidase Subunit I Gene Primers for Mansonia Annulata Identification. Southeast. Asian J. Trop. Med. Publ. Health 42, 1381–1387.
Setiati, T. E., Wagenaar, J. F. P., de Kruif, M. D., Mairuhu, A. T. A., Van-gorp, E. C. M., Soemantri, A. (2006). Changing Epidemiology of Dengue Haemorrhagic Fever in Indonesia. Dengue. Bull. 30, 1–14.
Severson, D. W., Behura, S. K. (2016). Genome Investigations of Vector Competence in Aedes aegypti to Inform Novel Arbovirus Disease Control Approaches. Insects 7, 58. doi: 10.3390/insects7040058
Simmons, C. P., Farrar, J. J., Nguyen, V. V. C., Wills, B. (2012). Dengue. N. Engl. J. Med. 366, 1423–1432. doi: 10.1056/NEJMra1110265
Tedjou, A. N., Kamgang, B., Yougang, A. P., Njiokou, F., Wondji, C. S. (2019). Update on the Geographical Distribution and Prevalence of Aedes aegypti and Aedes albopictus (Diptera: Culicidae), Two Major Arbovirus Vectors in Cameroon. PloS Negl. Trop. Dis. 13, e0007137. doi: 10.1371/journal.pntd.0007137
Teixeira, M. G., Costa, M., Barreto, F., Barreto, M. L. (2009). Dengue: Twenty-Five Years Since Reemergence in Brazil Cad Saude Publica 25 (Suppl 1), S7–18. doi: 10.1590/s0102-311x2009001300002
Thiberville, S. D., Moyen, N., Dupuis-Maguiraga, L., Nougairede, A., Gould, E. A., Roques, P., et al. (2013). Chikungunya Fever: Epidemiology, Clinical Syndrome, Pathogenesis and Therapy. Antiviral Res. 99, 345–370. doi: 10.1016/j.antiviral.2013.06.009
Thisyakorn, U., Thisyakorn, C. (2014). Latest Developments and Future Directions in Dengue Vaccines. Ther. Adv. Vaccines 2, 3–9. doi: 10.1177/2051013613507862
Urdaneta-Marquez, L., Failloux, A.-B. (2011). Population Genetic Structure of Aedes aegypti, the Principal Vector of Dengue Viruses. Infect. Genet. Evol. 11, 253–261. doi: 10.1016/j.meegid.2010.11.020
WHO (2011). Comprehensive Guidelines for Prevention and Control of Dengue and Dengue Haemorrhagic Fever. Revised Eds (New Delhi, India: World Health Organization: Regional Office for South-East Asia).
WHO (2016). Entomological Surveillance for Aedes Spp. In the Context of Zika Virus: Interim Guidance for Entomologist Vol. 10 (Geneva, Switzerland: World Health Organization).
Wilder-Smith, A., Ooi, E. E., Vasudevan, S. G., Gubler, D. J. (2010). Update on Dengue: Epidemiology, Virus Evolution, Antiviral Drugs, and Vaccine Development. Curr. Infect. Dis. Rep. 12, 157–164. doi: 10.1007/s11908-010-0102-7
Keywords: Aedes aegypti, Aedes albopictus, Indonesia, cox1, internal transcribed spacer 2 (ITS2)
Citation: Garjito TA, Widiarti W, Hidajat MC, Handayani SW, Mujiyono M, Prihatin MT, Ubaidillah R, Sudomo M, Satoto TBT, Manguin S, Gavotte L and Frutos R (2021) Homogeneity and Possible Replacement of Populations of the Dengue Vectors Aedes aegypti and Aedes albopictus in Indonesia. Front. Cell. Infect. Microbiol. 11:705129. doi: 10.3389/fcimb.2021.705129
Received: 04 May 2021; Accepted: 10 June 2021;
Published: 07 July 2021.
Edited by:
Jun Feng, National Institute of Parasitic Diseases, ChinaReviewed by:
Padet Siriyasatien, Chulalongkorn University, ThailandCopyright © 2021 Garjito, Widiarti, Hidajat, Handayani, Mujiyono, Prihatin, Ubaidillah, Sudomo, Satoto, Manguin, Gavotte and Frutos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Triwibowo Ambar Garjito, dHJpd2lib3dvQGxpdGJhbmcua2Vta2VzLmdvLmlk; Roger Frutos, cm9nZXIuZnJ1dG9zQGNpcmFkLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.