 María Gabriela Libisch
María Gabriela Libisch Natalia Rego
Natalia Rego Carlos Robello
Carlos Robello- 1Laboratorio de Interacciones Hospedero Patógeno-UBM, Institut Pasteur de Montevideo, Montevideo, Uruguay
- 2Unidad de Bioinformática, Institut Pasteur de Montevideo, Montevideo, Uruguay
- 3Departamento de Bioquímica, Facultad de Medicina, Universidad de la República, Montevideo, Uruguay
Chagas Disease, caused by the protozoan parasite Trypanosoma cruzi, affects nearly eight million people in the world. T. cruzi is a complex taxon represented by different strains with particular characteristics, and it has the ability to infect and interact with almost any nucleated cell. The T. cruzi-host cell interactions will trigger molecular signaling cascades in the host cell that will depend on the particular cell type and T. cruzi strain, and also on many different experimental variables. In this review we collect data from multiple transcriptomic and functional studies performed in different infection models, in order to highlight key differences between works that in our opinion should be addressed when comparing and discussing results. In particular, we focus on changes in the respiratory chain and oxidative phosphorylation of host cells in response to infection, which depends on the experimental model of T. cruzi infection. Finally, we also discuss host cell responses which reiterate independently of the strain, cell type and experimental conditions.
Introduction
Chagas disease, discovered more than 100 years ago by the Brazilian physician Carlos Chagas, affects nearly eight million people mostly in rural areas of Latin America (Schmunis and Yadon, 2010). It is considered a neglected tropical disease (NTD) by the World Health Organization (WHO), affecting the very poorest in society, which receive insufficient treatments and preventive measures. Its causative agent, the protozoan parasite Trypanosoma cruzi, has a complex life cycle alternating between an hematophagous triatomine insect and a mammalian host, exhibiting different forms during its life cycle (Kollien and Schaub, 2000; Parodi-Talice et al., 2007). During a blood meal, the insect ingests from the host the infective and non-replicative form trypomastigotes. They migrate to the vector´s digestive tract where they transform into epimastigotes, a replicative and non-infective form, which in the hindgut differentiate into the infective and non-replicative metacyclic trypomastigotes. These forms are excreted with faeces of the insect vector and enter the mammalian hosts through the injured skin or the mucosa. Once the parasite reaches the bloodstream it can spread easily, because it has the ability to infect almost any nucleated cell. Intracellularly, the parasite undergoes differentiation into amastigotes, a replicative and also infective (Mortara et al., 1999) form. Inside the cell amastigotes multiply by binary fission and when they reach a high density, they transform into trypomastigotes that are released into the blood where they can invade new cells and propagate the infection. The premature lysis of infected host cells can also liberate amastigotes that will contribute to the intra-host propagation of the infection. Other forms of transmission in humans include non-tested blood transfusion, organ transplantation, vertical transmission and oral infection by ingestion of food contaminated by infected triatomine feces (Coura and Borges-Pereira, 2010).
Chagas disease presents two main forms, the acute and the chronic phase. The acute phase lasts between four and eight weeks after parasite inoculation and is asymptomatic in most cases, or could exhibit unspecific symptoms like fever, muscle pain, subcutaneous edema, hepatosplenomegaly and lymphadenopathy (Rassi et al., 2010). In the chronic phase 60-70% of the patients do not develop symptoms or detectable pathology, despite being seropositive for T. cruzi. The remaining 30-40% of the patients, after a period of 10 to 30 years, might develop cardiac (Chronic Chagas Cardiomyopathy, CCC), gastrointestinal (megacolon or megaesophagus), neurological pathologies, or mixed forms of the previous manifestations (Fernandes and Andrews, 2012; Coura, 2015; Lewis and Kelly, 2016).
The understanding of the host-pathogen interaction process involves the study, among others, of the changes in gene expression provoked by the parasite in the host cell. For this purpose different models are used, both in vitro (cell cultures) and in vivo (mainly mice) infections. The host cell infection shows considerable heterogeneity at each step of this process, and the specific result may vary with each unique combination of parasite strain, stage (trypomastigotes and/or amastigotes) and host cell type (Epting et al., 2010). Besides, many experimental variables can influence the final result, such as the protocol used to propagate and purify trypomastigotes (Libisch et al., 2020), the interaction time between the parasite and the host cell, the multiplicity of infection (MOI, defined as the ratio of parasites to host cells), the analyzed time, the technology used (qPCR, microarrays or massive sequencing) and the data analysis layout. In addition, it is well known that long lasting T. cruzi cultures can suffer a decrease in the infectivity capacity (Contreras et al., 1998). Figure 1 summarizes the main factors that can vary through the different studies, those which we consider most important are analyzed in this work.
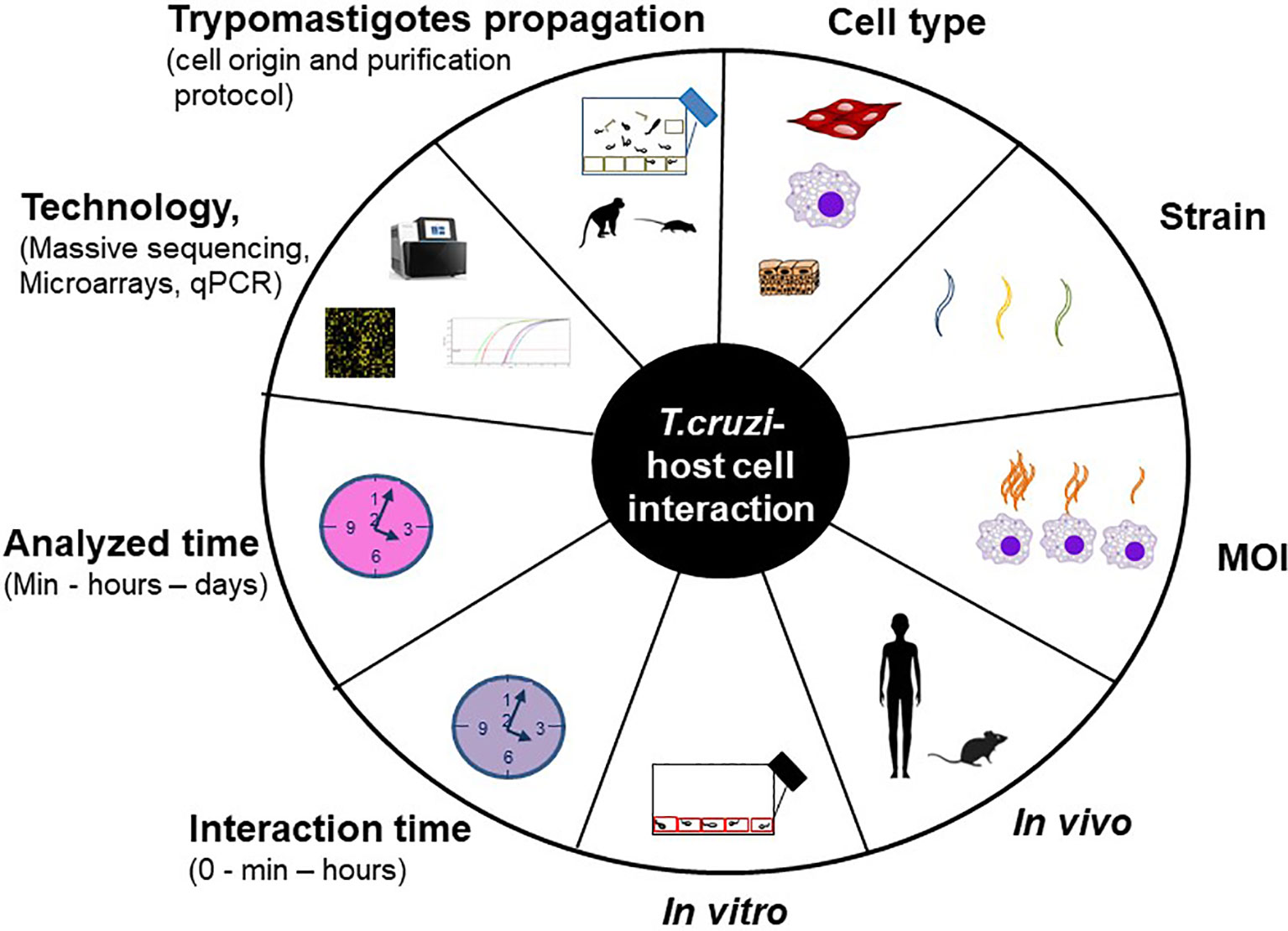
Figure 1 Experimental variables that impact on T. cruzi – host cell interaction studies.
T. cruzi Strain Variability
It has been shown that different T. cruzi strains exhibit biological differences, such as virulence, infectivity, pathogenicity, among others (Vago et al., 1996; Andrade et al., 1999; Vago et al., 2000; Buscaglia and Di Noia, 2003; Andrade et al., 2010). In an effort to understand this variability, the different strains have been classified into six main groups, known as discrete typing units called DTU (TcI to TcVI). This term was proposed to describe a set of strains that are genetically more related to each other than to any other strain. The first four DTUs are non-hybrid lineages while TcV and TcVI are hybrids of TcII and TcIII (Zingales et al., 2012). However, there exists a high genetic and biological diversity of T. cruzi strains even at the intra-DTU level (Berná et al., 2019) and it is known to impact the host response to infection. For example, by comparing the gene expression profiles of two T. cruzi TcVI strains that display contrasting virulence phenotypes (CL14 and CL Brener which present low and high in vivo virulence, respectively), Belew et al. (Belew et al., 2017) found a central role for genes encoding surface proteins associated with the differentiation from intracellular replicative amastigotes to infective trypomastigotes. In order to understand this phenotypic variability, it is important to consider the complex T. cruzi genome with its highly repetitive nature: 5–10% of the genome is composed of a 195 bp satellite sequence, and the remaining 40-45% of the repetitive DNA is composed of multi-gene families, tandem repeats and retrotransposable elements (El-Sayed et al., 2005b). The main multi-gene families are: transialidases (TS), mucins (heavily glycosylated proteins), the mucin-associated surface proteins (MASP), GP63, the families DGF-1 (dispersed gene family) and RHS (retrotransposon hotspot) proteins. Our group has found important differences in copy numbers for some of these protein families depending on the DTU (TCC/TcVI and Dm28c/TcI) (Berna et al., 2017; Berna et al., 2018). Wang et al. (2021) also showed important differences between two T. cruzi strains (Brazil/TcI and Y/TcII), where all gene families exhibit a broad range of sequence identity between their members, especially TS, MASP, mucins and RHS. In addition, a repeat-driven recombination and generation of antigenic diversity was recently proposed (Talavera-Lopez et al., 2021). These genomic features allow the parasites to display great plasticity and the ability to invade almost any type of nucleated cell. For sure, ongoing works will improve genome assemblies and gene annotations, which in turn will contribute to a better understanding about parasite virulence and plasticity, as well as will allow for a better assessment on the parasite proteins triggering the T. cruzi-host interaction.
We also want to highlight that the specific strain does not only impact T. cruzi host cell interaction studies, but also on infections with other trypanosomatids. For instance, for another intracellular trypanosomatid as Leishmania major, infection by different strains results in different levels of virulence (Markikou-Ouni et al., 2012). In the case of Trypanosoma brucei, an extracellular parasite, different strains also show variable virulence (Gitonga et al., 2017).
The Invasion Process
To succeed in the invasion process the parasites must attach to the host cell surface prior to entry. Most of the interactions between the parasite and the host cell occur through different proteins belonging to multigene families (Woolsey et al., 2003; El-Sayed et al., 2005a; De Pablos et al., 2011). In the case of mucins, they facilitate the adhesion of the trypomastigote to the host cell and its subsequent invasion and also participate in immune response evasion (Andrews and Whitlow, 1989). Transialidases (TS) transfer sialic acid from the host cell membrane to the parasite’s glycoproteins and favors the parasite interaction with the host cell during the invasion process (Woolsey and Burleigh, 2004; Mott et al., 2009). MASP proteins, present in the membrane of trypomastigotes, participate in the invasion of the host cell and in host immune evasion mechanisms (De Pablos et al., 2011; Dos Santos et al., 2012). Individual members of the protease GP63 family also play a role in the infection process (Kulkarni et al., 2009). There are fewer studies about the DGF-1 and RHS families, but members of the former are expressed on the trypomastigote surface and might be associated with the ability of T. cruzi to bind extracellular matrix proteins (Kawashita et al., 2009), and the RHS family may play a role in modulating the chromatin structure but also they may participate, possibly as an adjuvant, in the interaction with the mammalian host (Bernardo et al., 2020). After the attachment to the host cell, the parasite enters into the cell. The mechanisms and route of cell invasion vary greatly with the host cell type since T. cruzi trypomastigotes are capable of directly invading both professional and non-professional phagocytic cells. In macrophages, trypomastigotes induce their uptake by engaging Toll-like receptors (Maganto-Garcia et al., 2008). In the case of non-professional phagocytic cells, at least two major pathways have been characterized. The first relies upon calcium-mediated signaling at the surface for lysosomal trafficking, dependent upon actin polymerization and microtubules (Schenkman et al., 1991; Tardieux et al., 1994), while the second is a plasma membrane-mediated invagination involving PI3 kinase signaling and independent of actin polymerization (De Souza, 2002; Andrade and Andrews, 2005; Burleigh, 2005). The specific mechanism by which T. cruzi invades the host could also depend on the strain (Neira et al., 2002). In any case, the interaction between the parasite and the cell will trigger the transduction of different signals depending on the involved specific host cell receptors and the parasite surface molecules. As it is known, most of these signal transduction pathways will ultimately impact the activity of different transcription factors, resulting in changes in gene expression, which can be considered as a reprogramming of host cells.
Independently of the specific invasion process and the parasite stage (trypomastigotes or amastigotes), components of the host endosomal-lysosomal system such as early endosomes, late endosomes and lysosomes, participate in the formation of a parasitophorous vacuole with an acidic pH (Ley et al., 1990). In the vacuole, trypomastigotes release trans-sialidase, that will remove sialic acid residues from the membrane, making it sensitive to the action of Tc-Tox (a peptide that has homology with the factor 9 of the human complement, Andrews and Whitlow, 1989), which probably contributes to the disintegration of the parasitophorous vacuole (De Souza et al., 2010). The acidic pH favors the conversion of trypomastigotes into amastigotes and the escape to the cytoplasm forto the initiation of amastigotes replication (Ley et al., 1990). Regarding the invasion kinetics (the time the parasite takes to escape from the vacuole and start replication), there are some studies that analyze, with both specific DTUs and host cells, aspects of this process. Ley et al. found that two hours after interaction between human monocytes and parasites from the Y strain (trypomastigotes or amastigotes), 70% of the parasites are in disrupted vacuoles or free in the cytosol (Ley et al., 1990). Stecconi-Silva et al. found that there is a population of trypomastigotes of the G strain that leaves the vacuole at 7 to 8h and another set that leaves the vacuole between 12-15h post-invasion of HeLa and Vero cells (Stecconi-Silva et al., 2003). Dias et al. found that the JG and Col1.7G2 trypomastigotes escape from the vacuole at 8 to 12h post-invasion of primary neonatal cardiomyocytes cultures (Dias et al., 2017). Once free in the cytoplasm the parasites start replication to finally, 3-5 days post infection, lyse the cell.
Although many studies analyze the host cell response to T. cruzi infection, the comparison between them becomes very difficult due to significant differences in the incubation periods and analyzed times after infection, which encompass distinct biological events during the infection process. These differences together with the particular molecular interactions established by the specific combination of strain and cell type will impact the host transcriptomic and functional response in very different ways.
Are Transcriptomic Experiments Comparable to Each Other?
For in vivo gene profiling experiments mice are the animal model of choice for the majority of the studies. Despite the study of the immune response using mice models have yielded important insights into the workings of the human immune system, there are also significant differences in both innate and adaptive immunity including the balance of leukocyte subsets, Toll-like receptors, FcR, immunoglobulins subsets, the B and T cell signaling pathway components, cytokines and cytokine receptors, Th1/Th2 differentiation, Ag-presenting function of endothelial cells, among others (Mestas and Hughes, 2004). Regarding general metabolic features between mice and humans, mice have a higher mass-specific metabolic rate and therefore a weak capacity to maintain cellular homeostasis, while humans have a lower mass-specific metabolic rate and a strong capacity to maintain cellular homeostasis (Demetrius, 2004; Demetrius, 2005). All these differences show that there are important caveats to consider that could impact the in vitro results between experiments done with mice or human origin cells, and when it comes to extrapolating results from in vivo mice models to humans.
To perform transcriptomic host – T. cruzi experiments, propagation of parasites in order to perform the infection assays constitutes a variable that is not usually considered. One of the most common systems to do the parasite propagation is the in vitro infection of Vero (or other types of monkey-derived) cells (Duran-Rehbein et al., 2014). We recently demonstrated that when parasites are grown in a cell line genetically close to the target cells in the infection experiment, false-positive results may occur in the subsequent transcriptomic analysis (Libisch et al., 2020). This contamination happens because, in the trypomastigote RNA purification process, RNA coming from the cell line used to propagate the parasites is also extracted. This observation showed that particular attention must be paid both in the selection of the cell origin used for parasite propagation (the further away phylogenetically from the target cells, the better) and in the protocol used to purify the trypomastigotes in order to avoid this type of contamination. Another important aspect of the experimental conditions that could impact the final host response is the MOI, which not only can affect the percentage of infected host cells but also could modulate the type of host cell death, as has been observed in some bacterial infections (Lee et al., 2006; Willingham et al., 2007). Finally, the different technologies used to obtain the transcriptomic data (q-PCR, microarrays or massive sequencing) and the specific type of data analysis can impact the final results.
All these variables make it difficult to compare the different results, and not taking them into account can lead to incorrect conclusions. Table 1 shows different transcriptomic studies performed by diverse groups where they analyze the host cell response to T. cruzi infection. We summarize 11 in vivo and 18 in vitro studies. In the case of the in vivo studies six involved mice and five CCC patient samples. The number of parasites used to establish the mice infections were as few as 50 parasites to as many as 106 parasites, and the analyzed time varied between 3dpi to 8 months. In the case of the in vitro studies, the MOI varied between 1 to 80, and the experiments were done in cells derived from mice (4), humans (11) and rats (3). The host cell type more frequently analyzed in the in vitro studies were fibroblast (6) and cardiomyocytes (4) followed by epithelial HeLa cells (3), rat myoblast (3), human placenta (1) and endothelial cells (1). The cell lines more frequently used to propagate the parasites were cells derived from monkeys (Vero and LCMK2 lines). The interaction time varied between 0 to 60h and the analyzed time between 30min to 96hpi.
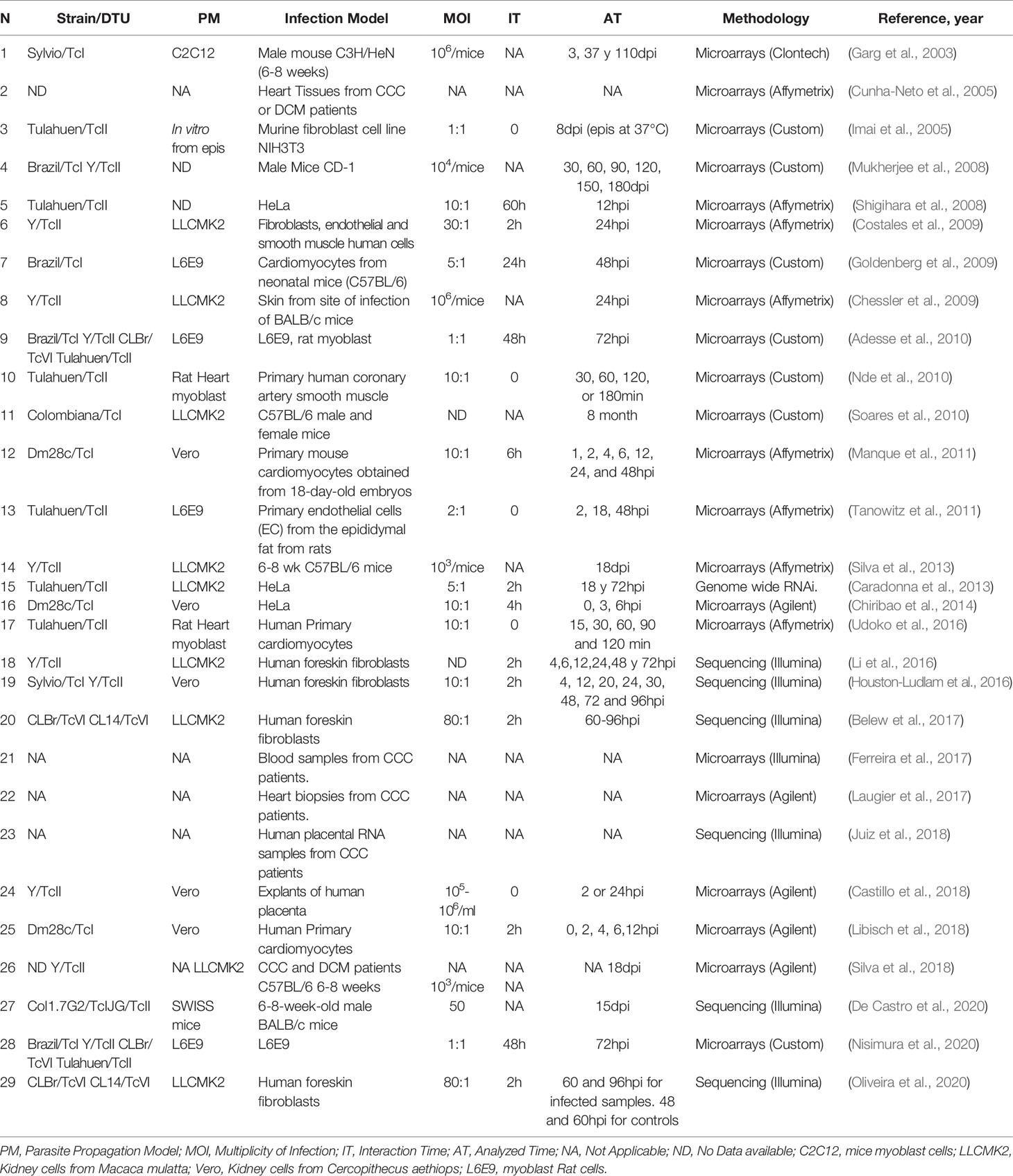
Table 1 Different T. cruzi-host cell interaction studies incorporating transcriptomics of the infected host cell.
All the variables mentioned above also affect transcriptional changes induced by other intracellular trypanosomes, as Leishmania (different infection models, the source and origin of the macrophages, parasite species, levels of infectivity, MOI, the used technology, data analysis, and interaction and analyzed times of infection) (Ashwin et al., 2018; Stamper et al., 2019) and probably they will also affect the transcriptional host changes caused by any intracellular microorganism.
How Is Host Cellular Respiration Affected by T. cruzi?
To exemplify the differences between the host responses to T. cruzi infection given the experiment design, we will focus the analysis on the respiratory chain and oxidative phosphorylation (OXPHOS) responses (Table 2). First, when comparing in vitro-based transcriptomic responses of human primary cardiomyocytes at early times post infection we found, using the Dm28c strain (TcI), upregulation of pathways related to energy metabolism (Libisch et al., 2018), while Udoko et al. (2016) using the Tulahuen strain (TcVI) did not found these changes. Concerning in vitro transcriptomic experiments performed in murine (instead of human) primary cardiomyocytes at early time post-infection, Manque et al. (Manque et al., 2011), using the Dm28c strain did not found significant changes in pathways related to energy metabolism, while Goldenberg et al. with the Brazil strain (TcI) observed a down-regulation of the electron transport activity GO term (Goldenberg et al., 2009), although in this last case the analysis was on late time post infection. These comparisons exemplify how transcriptomic in vitro results depend on the specific strain, the origin of the infected cells (human or mouse), as well as on the experimental interaction and analyzed times. It is important to highlight that in only one of these studies a functional validation of the OXPHOS response was performed (see below).
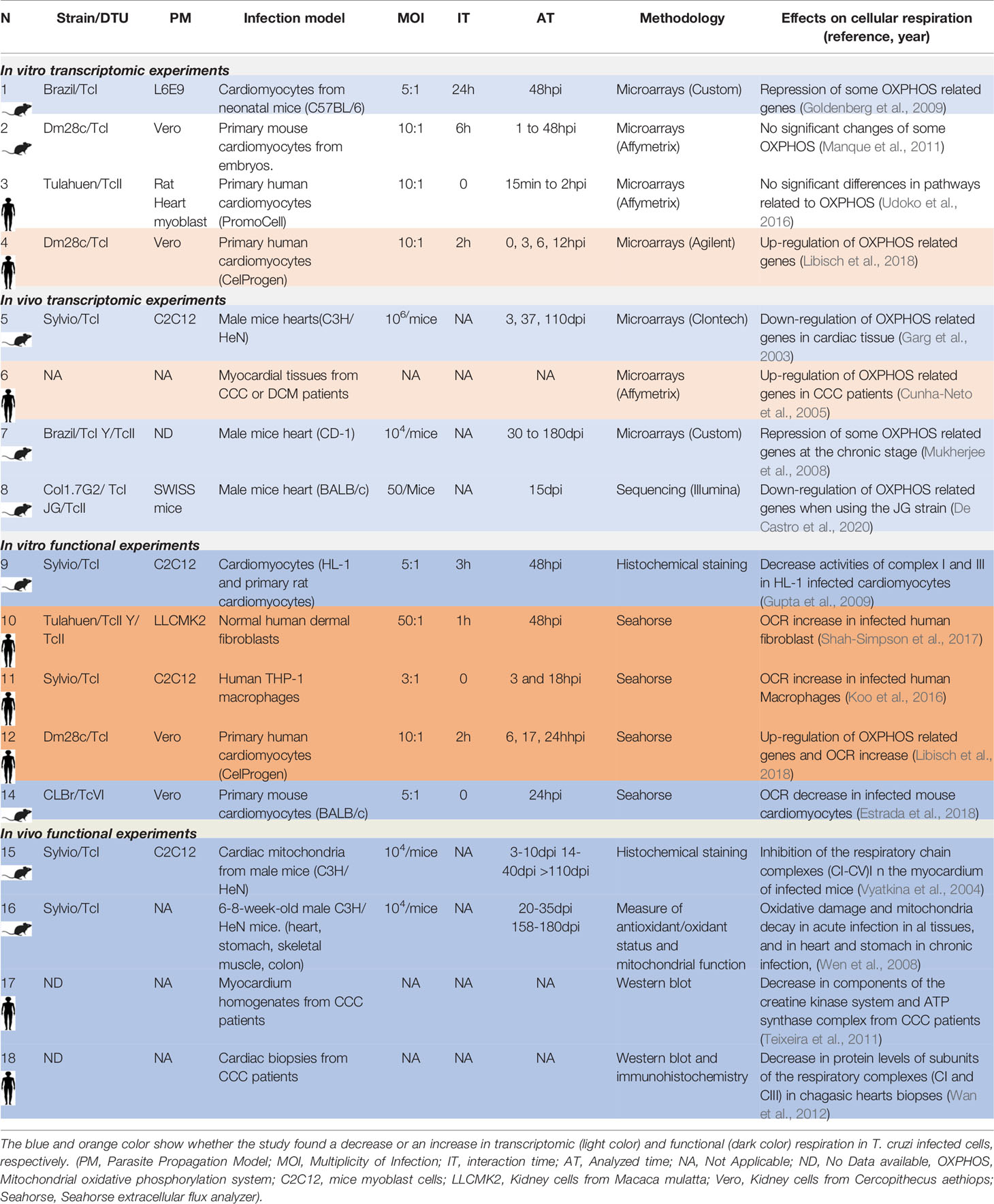
Table 2 Transcriptomic and functional results from different studies related to the host cell respiration response to T. cruzi infection.
By comparing in vivo-based transcriptomics data from mice heart infections, Garg et al. (2003) (3-110dpi) using the Sylvio strain (TcI), and Mukherjee et al. (2008) (3-180dpi) using the Brazil (TcI) and Y (TcII) strains, showed a decrease in the energy metabolism pathway. De Castro et al. (2020) analyzed the transcriptomic response of murine hearts infected with the Col1.7G2 (TcI), and JG (TcII) strains, 15dpi, and also found a decrease in pathways related to energy metabolism. Concerning samples from patients with Chronic Chagas Cardiomyopathy (CCC), the study from Cunha-Neto et al. (2005) showed the contrary, an increase in genes related to energy metabolism. These works showed that in vivo transcriptomic results could also depend on the origin of the infected cells (human or mouse). Of note, additional specific factors will also impact results, such as the genetic background and the immune status of each cell donor, which should be at least described.
It is important to note that transcriptional changes in response to infection do not imply a correlation with protein levels and/or activity, and hence to the functional response. Regulatory processes occurring after mRNA transcription such as the stability or differential degradation of the RNAs, post-transcriptional modifications, the accessibility to the translation machinery, translational and protein degradation regulation are important factors in controlling steady-state protein levels and specially, post-translational modifications are important factors in controlling protein activity (Vogel and Marcotte, 2012).
Taking these aspects into consideration we looked for studies that had evaluated not only changes at the transcriptional level but also the functional response related to cellular respiration in host cells infected by T. cruzi. Looking at functional data related to cellular respiration in mouse and human in vitro infection systems, an increase in respiration of different infected human origin host cells was observed: we found that human primary cardiomyocytes infected with Dm28c (17-24hpi) increase respiration and mitochondrial biogenesis (Libisch et al., 2018). Shah-Simpson et al. also found both an increase in respiration and mitochondrial biogenesis of T. cruzi infected human dermal fibroblast (48hpi) (Shah-Simpson et al., 2017), which is maintained even when the respiration of T. cruzi is specifically inhibited. An increase in respiration (3-18hpi) was also described in human macrophages infected with Sylvio (TcI) by Koo et al. (Koo et al., 2016). In all three cases the respiration was evaluated by the quantification of the oxygen consumption rate (OCR), using the same methodology. On the contrary, Estrada et al. (Estrada et al., 2018) found a decrease in respiration in infected murine cardiomyocytes with the CLBr (TcVI) strain at similar analyzed times (24hpi). Gupta et al. (2009) also described a decrease in the respiration complexes I and III of HL1 mice cardiomyocytes infected with Sylvio (TcI) at 48hpi. These functional works showed again that results are closely related to the origin of infected cells (human or mouse). Regarding in vivo infections, Vyatkina et al. (2004) evaluated protein expression of different respiration complexes in cardiac mitochondria from infected mice with the Sylvio strain, finding a decreased expression in some of the complexes (3 to 110dpi). Wen et al. (2008) also found a decreased mitochondrial function in hearts, stomach, skeletal muscle and colon cells of acute mice infection, and also in hearts and stomach of chronic infections. Finally, there are very few studies that analyze the cellular respiration in cells or tissues derived from patients. For example, in a discrete group of CCC heart samples (five patients), Teixeira et al. (Teixeira et al., 2011) found a discrete decrease in the expression levels of the alpha subunit of the ATP synthase enzyme (complex V). Similarly, the work of Wan et al. (2012) in a group of eight CCC heart patients samples, showed a decrease in the Cyt B mitochondrial respiration protein (complex III) and ND1 protein (complex I). Given the diversity of cells present in the heart (cardiomyocytes, fibroblasts, endothelial cells), and in the case of the CCC additional infiltrates of macrophages and T cells, it would be of great importance to perform more studies in a large number of heart biopsies of CCC patients, as well as in cardiomyocytes purified from them.
As mentioned before, working with human or mouse models can have a great impact on the observed host response to infection. Finding an increase in the respiration of infected human cells but a decrease in the respiration in infected cells with a murine origin may be linked to the fact that mice have a basal metabolic rate per gram of body weight approximately seven times higher than that of a human. As a consequence, mice have a lower ability to maintain adequate cellular homeostasis against different types of stress such as reactive oxygen species (ROS) (Demetrius, 2004; Demetrius, 2005). During normal cellular respiration, ROS are usually generated (Scheibye-Knudsen et al., 2015), and particularly they increase in T. cruzi infected cardiomyocytes (Gupta et al., 2009; Libisch et al., 2018), and macrophages (Cardoni et al., 1997; Alvarez et al., 2011), and perhaps it is one of the reasons why cells of murine or human origin do not behave the same against infection, at least concerning energy metabolism. Besides there exists an established relationship between the immune system and mitochondrial bioenergetics (Breda et al., 2019), so it is possible that the differences in immune systems between mice and humans also impact on the mitochondrial respiration.
Does T. cruzi Induce Any Common Pathway Independently From Experimental Conditions?
With so many studies performed under so many different experimental variables, we found that activation of the PI3K/AKT pathway is a common response to T. cruzi infection. This pathway is an intracellular transduction signal that promotes proliferation, metabolism, cell survival, growth and angiogenesis in response to extracellular signals. Its key components are receptor tyrosine kinases (RTKs), phosphatidylinositol 3-kinase (PI3K), phosphatidylinositol-4,5-bisphosphate (PIP2), phosphatidylinositol-3,4,5-trisphosphate (PIP3) and the serine/threonine protein kinase B/AKT. There are three classes of PI3K. The substrates for class I enzymes are PIP2, which are predominantly found on the plasma membrane where they are phosphorylated to PIP3. The class I enzymes are further divided into two subclasses that differ in their structure and in the stimuli that activate them. The Class Ia enzymes (PI3K α, β, δ) are activated by tyrosine kinase receptors. The class Ib (PI3K γ) is activated primarily by G-protein receptors (Kima, 2016). In all the cases the PI3K class I phosphorylation provokes the AKT phosphorylation and activation. Results obtained by our laboratory in human primary cardiomyocytes and HeLa cells infected with the Dm28c strain show that the PI3K/AKT pathway is activated (Chiribao et al., 2014; Libisch et al., 2018). Wilkowsky et al. and Todorov et al. also found this pathway activated in human and mouse macrophages infected with a TcVI and a TcII strain, respectively (Todorov et al., 2000; Wilkowsky et al., 2001). Caradonna et al. found the PI3K/AKT pathway activated in infected human epithelial (HeLa) cells with the Tulahuen (TcII) strain and its activation was critical for parasite replication (Caradonna et al., 2013). Silva et al. found its activation in mouse cardiomyocytes infected with the Y (TcII) strain (Silva et al., 2018). Chuenkova et al. showed that infected human glial cells of the Schwann cell type with the Sylvio-TcI strain increase the PI3K pathway (Chuenkova and Pereiraperrin, 2009). Nagajyothi et al. also described the PI3K/AKT activation in murine adipocytes infected with the Tulahuen strain-TcII (Nagajyothi et al., 2008), as well as Wilkowsky et al. in murine fibroblasts infected with a TcVI strain (Wilkowsky et al., 2001). PI3K/AKT activation was also demonstrated in patients with CCC by Silva et al. and they also showed that the activation of this pathway in myeloid cells (macrophages) would restrict infection in the heart by protecting cardiomyocytes (Silva et al., 2018). In general, the role of the PI3K/AKT activation is related to its antiapoptotic capacity (Zhou et al., 2000; Yamaguchi and Wang, 2001) and its role in the survival and repair of different cell types has been associated with the ability of the parasite to persist in the host (Nagajyothi et al., 2012). The PI3K pathway could also be necessary for the T. cruzi invasion process of non-phagocytic cells as previously mentioned (Woolsey et al., 2003; Burleigh, 2005). In Table 3 we summarize the high number of works in which, independently of the T. cruzi strain, interaction and analyzed time, cell type and cell origin (human or mouse), activation of the PI3K pathway was found. This fact gives strong evidence of the crucial role of this pathway in the interaction of the host cell with the parasite. Chuenkova et al. showed that T. cruzi TSs are responsible for activating this pathway in infected Schwann cells (Chuenkova et al., 2001). De Melo et al. showed that a T. cruzi trans-sialidase is able to interact with the membrane receptor tyrosine kinase TrKA by promoting invasion of mammalian cells (neuronal, epithelial, and phagocytic cells) and at the same time allowing activation of the PI3K/AKT pathway (De Melo-Jorge and Pereiraperrin, 2007). It is important to consider that activation of PI3K/AKT was reported in infections produced by many different microorganisms. Diehl et al. reviewed different viral strategies which exploit the PI3K/AKT signaling pathway for effective viral replication (Diehl and Schaal, 2013). The activation of this pathway showed to be essential for Staphylococcus aureus internalization (Oviedo-Boyso et al., 2011) and also for Pseudomonas aeruginosa strain PAK internalization (Kierbel et al., 2005). It also played an important role in host defense for some bacterial infections (Yilmaz et al., 2004; Brown et al., 2011; Cremer et al., 2011). Finally, the PI3K signaling plays a role in the host response to Leishmania infections (Kima, 2016). Although this pathway appears to be essential in the host response to many types of infections, the particular molecules that activate this pathway are in most cases still unknown. In the case of T. cruzi, only TSs in trypomastigotes have been shown to activate this pathway in some mammalian cells (neuronal, epithelial, and phagocytic cells) through TrKA (De Melo-Jorge and Pereiraperrin, 2007). However, it remains to be elucidated which particular trans-sialidases and which particular enzyme of the class Ia PI3K family are involved. It has been shown that T. cruzi also activates the PI3Kγ, class Ib enzyme, in macrophages and hearts from CCC patients (Silva et al., 2018). Which particular molecules of T. cruzi and G-protein receptors are involved in this activation remains elusive. Understanding the specific molecules and host receptors that participate in the PI3K/AKT activation in different infection models will shed light on its modulation and could be a strategy to control T. cruzi infection, replication and/or appearance of clinical signs. Besides, the role of the PI3K/AKT pathway has been extensively studied in the context of cancer, and different drugs targeting PI3K have been developed and employed in clinical trials evaluations (Yang et al., 2019), bringing the option of considering them as repurposed drugs for host-directed therapies in Chagas and others infectious diseases.
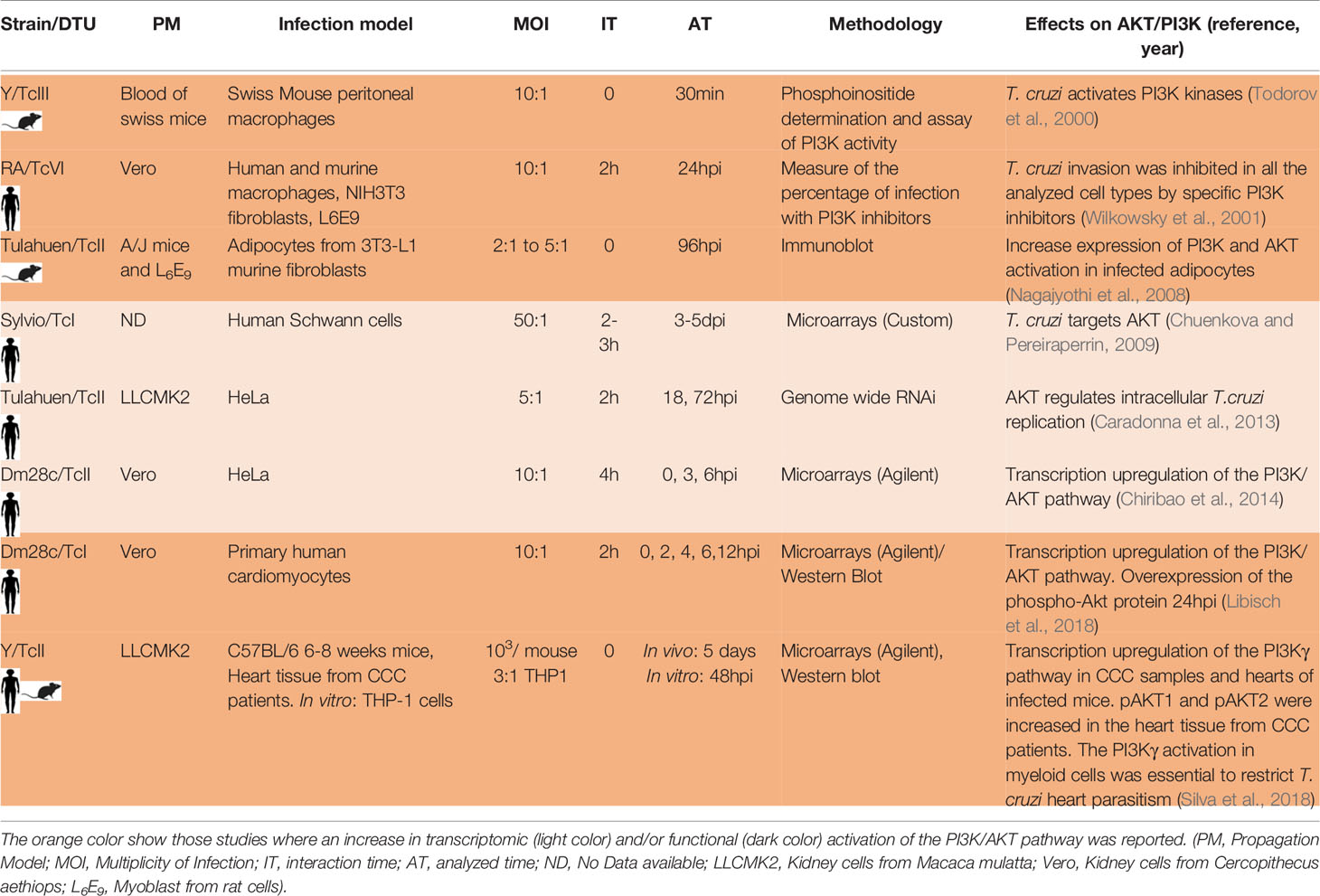
Table 3 Studies related to the host AKT/PI3K response to T. cruzi infection.
Concluding Remarks
T. cruzi-host transcriptomic and functional studies give important clues to understand the changes suffered by the host cell in response to infection. In this review we analyzed how multiple experimental variables can significantly affect the final transcriptomic and functional responses, highlighting the difficulties to compare different works and drawing general conclusions to host-cell infection response. Concerning molecular mechanisms of pathogenesis in Chagas Disease, there are still many questions to answer, but the main point will be to select systems that could represent in the best possible way the human molecular characteristics of the disease, with studies comparable to each other. We found important differences in the OXPHOS response to T. cruzi infection depending on the origin of the infected cell (human or mouse), highlighting in the in vitro studies opposite responses depending on this variable, both at the transcriptomic and functional level. We caution about not considering the genetic and immunological differences between mice and humans before comparing results, and also, we believe that the use of in vitro models that include primary human cells, human induced pluripotent stem cells and 3D cultures (Breyner et al., 2020) are recommended options. Finally, delving into the activation mechanisms of PI3K/AKT would open new perspectives in host-directed therapies in Chagas disease.
Author Contributions
MGL and CR contributed to conception and design of the study. MGL and NR organized the database. MGL wrote the first draft. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by: Research Council United Kingdom Grand Challenges Research Funder under grant agreement “A Global Network for Neglected Tropical Diseases” grant number MR/P027989/1; Agencia Nacional de Investigación e Innovación (UY) DCI-ALA/2011/023–502, “Contrato de apoyo a las políticas de innovación y cohesión territorial”, Fondo para la Convergencia Estructural del Mercado Común del Sur (FOCEM) 03/1.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
MGL and CR are members of the Sistema Nacional de Investigadores (ANII, Uruguay).
References
Adesse, D., Iacobas, D. A., Iacobas, S., Garzoni, L. R., Meirelles Mde, N., Tanowitz, H. B., et al. (2010). Transcriptomic Signatures of Alterations in a Myoblast Cell Line Infected With Four Distinct Strains of Trypanosoma Cruzi. Am. J. Trop. Med. Hyg. 82, 846–854. doi: 10.4269/ajtmh.2010.09-0399
Alvarez, M. N., Peluffo, G., Piacenza, L., Radi, R. (2011). Intraphagosomal Peroxynitrite as a Macrophage-Derived Cytotoxin Against Internalized Trypanosoma Cruzi: Consequences for Oxidative Killing and Role of Microbial Peroxiredoxins in Infectivity. J. Biol. Chem. 286, 6627–6640. doi: 10.1074/jbc.M110.167247
Andrade, L. O., Andrews, N. W. (2005). The Trypanosoma Cruzi-Host-Cell Interplay: Location, Invasion, Retention. Nat. Rev. Microbiol. 3, 819–823. doi: 10.1038/nrmicro1249
Andrade, L. O., Galvao, L. M., Meirelles Mde, N., Chiari, E., Pena, S. D., Macedo, A. M. (2010). Differential Tissue Tropism of Trypanosoma Cruzi Strains: An In Vitro Study. Mem. Inst. Oswaldo. Cruz. 105, 834–837. doi: 10.1590/s0074-02762010000600018
Andrade, L. O., Machado, C. R., Chiari, E., Pena, S. D., Macedo, A. M. (1999). Differential Tissue Distribution of Diverse Clones of Trypanosoma Cruzi in Infected Mice. Mol. Biochem. Parasitol. 100, 163–172. doi: 10.1016/s0166-6851(99)90035-x
Andrews, N. W., Whitlow, M. B. (1989). Secretion by Trypanosoma Cruzi of a Hemolysin Active at Low Ph. Mol. Biochem. Parasitol. 33, 249–256. doi: 10.1016/0166-6851(89)90086-8
Ashwin, H., Seifert, K., Forrester, S., Brown, N., Macdonald, S., James, S., et al. (2018). Tissue and Host Species-Specific Transcriptional Changes in Models of Experimental Visceral Leishmaniasis. Wellcome Open Res. 3, 135. doi: 10.12688/wellcomeopenres.14867.2
Belew, A. T., Junqueira, C., Rodrigues-Luiz, G. F., Valente, B. M., Oliveira, A. E. R., Polidoro, R. B., et al. (2017). Comparative Transcriptome Profiling of Virulent and Non-Virulent Trypanosoma Cruzi Underlines the Role of Surface Proteins During Infection. PloS Pathog. 13, e1006767. doi: 10.1371/journal.ppat.1006767PPATHOGENS-D-17-01304
Berna, L., Chiribao, M. L., Greif, G., Rodriguez, M., Alvarez-Valin, F., Robello, C. (2017). Transcriptomic Analysis Reveals Metabolic Switches and Surface Remodeling as Key Processes for Stage Transition in Trypanosoma Cruzi. PeerJ 5, e3017. doi: 10.7717/peerj.3017
Berná, L., Pita, S., Chiribao, M. L., Parodi-Talice, A., Alvarez-Valin, F., Robello, C. (2019). Biology of the Trypanosoma Cruzi Genome. In: DeSouza, W, eds. Biology of Trypanosoma cruzi (London, Intechopen Otd.). doi: 10.5772/intechopen.86144
Bernardo, W. P., Souza, R. T., Costa-Martins, A. G., Ferreira, E. R., Mortara, R. A., Teixeira, M. M. G., et al. (2020). Genomic Organization and Generation of Genetic Variability in the RHS (Retrotransposon Hot Spot) Protein Multigene Family in Trypanosoma Cruzi. Genes (Basel) 11. doi: 10.3390/genes11091085
Berna, L., Rodriguez, M., Chiribao, M. L., Parodi-Talice, A., Pita, S., Rijo, G., et al. (2018). Expanding an Expanded Genome: Long-Read Sequencing of Trypanosoma Cruzi. Microb. Genom. 4. doi: 10.1099/mgen.0.000177
Breda, C. N. S., Davanzo, G. G., Basso, P. J., Saraiva Camara, N. O., Moraes-Vieira, P. M. M. (2019). Mitochondria as Central Hub of the Immune System. Redox Biol. 26, 101255. doi: 10.1016/j.redox.2019.101255
Breyner, N. M., Hecht, M., Nitz, N., Rose, E., Carvalho, J. L. (2020). In Vitro Models for Investigation of the Host-Parasite Interface - Possible Applications in Acute Chagas Disease. Acta Trop. 202, 105262. doi: 10.1016/j.actatropica.2019.105262
Brown, J. B., Cheresh, P., Goretsky, T., Managlia, E., Grimm, G. R., Ryu, H., et al. (2011). Epithelial phosphatidylinositol-3-kinase Signaling is Required for Beta-Catenin Activation and Host Defense Against Citrobacter Rodentium Infection. Infect. Immun. 79, 1863–1872. doi: 10.1128/IAI.01025-10
Burleigh, B. A. (2005). Host Cell Signaling and Trypanosoma Cruzi Invasion: Do All Roads Lead to Lysosomes? Sci. STKE 2005:pe36. doi: 10.1126/stke.2932005pe36
Buscaglia, C. A., Di Noia, J. M. (2003). Trypanosoma Cruzi Clonal Diversity and the Epidemiology of Chagas’ Disease. Microbes Infect. 5, 419–427. doi: 10.1016/s1286-4579(03)00050-9
Caradonna, K. L., Engel, J. C., Jacobi, D., Lee, C. H., Burleigh, B. A. (2013). Host Metabolism Regulates Intracellular Growth of Trypanosoma Cruzi. Cell. Host. Microbe. 13, 108–117. doi: 10.1016/j.chom.2012.11.011
Cardoni, R. L., Antunez, M. I., Morales, C., Nantes, I. R. (1997). Release of Reactive Oxygen Species by Phagocytic Cells in Response to Live Parasites in Mice Infected With Trypanosoma Cruzi. Am. J. Trop. Med. Hyg. 56, 329–334. doi: 10.4269/ajtmh.1997.56.329
Castillo, C., Carrillo, I., Libisch, G., Juiz, N., Schijman, A., Robello, C., et al. (2018). Host-Parasite Interaction: Changes in Human Placental Gene Expression Induced by Trypanosoma Cruzi. Parasit. Vectors 11, 479. doi: 10.1186/s13071-018-2988-010.1186/s13071-018-2988-0
Chessler, A. D., Unnikrishnan, M., Bei, A. K., Daily, J. P., Burleigh, B. A. (2009). Trypanosoma Cruzi Triggers an Early Type I IFN Response In Vivo at the Site of Intradermal Infection. J. Immunol. 182, 2288–2296. doi: 10.4049/jimmunol.0800621
Chiribao, M. L., Libisch, G., Parodi-Talice, A., Robello, C. (2014). Early Trypanosoma Cruzi Infection Reprograms Human Epithelial Cells. BioMed. Res. Int. 2014, 439501. doi: 10.1155/2014/439501
Chuenkova, M. V., Furnari, F. B., Cavenee, W. K., Pereira, M. A. (2001). Trypanosoma Cruzi Trans-Sialidase: A Potent and Specific Survival Factor for Human Schwann Cells by Means of Phosphatidylinositol 3-Kinase/Akt Signaling. Proc. Natl. Acad. Sci. U. S. A. 98, 9936–9941. doi: 10.1073/pnas.161298398161298398
Chuenkova, M. V., Pereiraperrin, M. (2009). Trypanosoma Cruzi Targets Akt in Host Cells as An Intracellular Antiapoptotic Strategy. Sci. Signal 2, ra74. doi: 10.1126/scisignal.2000374
Contreras, V. T., De Lima, A. R., Zorrilla, G. (1998). Trypanosoma Cruzi: Maintenance in Culture Modify Gene and Antigenic Expression of Metacyclic Trypomastigotes. Mem. Inst. Oswaldo Cruz 93, 753–760. doi: 10.1590/s0074-02761998000600011
Costales, J. A., Daily, J. P., Burleigh, B. A. (2009). Cytokine-Dependent and-Independent Gene Expression Changes and Cell Cycle Block Revealed in Trypanosoma Cruzi-Infected Host Cells by Comparative mRNA Profiling. BMC Genom. 10, 252. doi: 10.1186/1471-2164-10-252
Coura, J. R. (2015). The Main Sceneries of Chagas Disease Transmission. The Vectors, Blood and Oral Transmissions–a Comprehensive Review. Mem. Inst. Oswaldo Cruz 110, 277–282. doi: 10.1590/0074-0276140362
Coura, J. R., Borges-Pereira, J. (2010). Chagas Disease: 100 Years After its Discovery. A Systemic Review. Acta Trop. 115, 5–13. doi: 10.1016/j.actatropica.2010.03.008
Cremer, T. J., Butchar, J. P., Tridandapani, S. (2011). Francisella Subverts Innate Immune Signaling: Focus on Pi3k/Akt. Front. Microbiol. 5, 13. doi: 10.3389/fmicb.2011.00013
Cunha-Neto, E., Dzau, V. J., Allen, P. D., Stamatiou, D., Benvenutti, L., Higuchi, M. L., et al. (2005). Cardiac Gene Expression Profiling Provides Evidence for Cytokinopathy as a Molecular Mechanism in Chagas’ Disease Cardiomyopathy. Am. J. Pathol. 167, 305–313. doi: 10.1016/S0002-9440(10)62976-8
De Castro, T. B. R., Canesso, M. C. C., Boroni, M., Chame, D. F., Souza, D. L., De Toledo, N. E., et al. (2020). Differential Modulation of Mouse Heart Gene Expression by Infection With Two Trypanosoma Cruzi Strains: A Transcriptome Analysis. Front. Genet. 11, 1031. doi: 10.3389/fgene.2020.01031
De Melo-Jorge, M., Pereiraperrin, M. (2007). The Chagas’ Disease Parasite Trypanosoma Cruzi Exploits Nerve Growth Factor Receptor TrkA to Infect Mammalian Hosts. Cell. Host Microbe. 1, 251–261. doi: 10.1016/j.chom.2007.05.006
Demetrius, L. (2004). Caloric Restriction, Metabolic Rate, and Entropy. J. Gerontol. A Biol. Sci. Med. Sci. 59, B902–B915. doi: 10.1093/gerona/59.9.b902
Demetrius, L. (2005). Of Mice and Men. When it Comes to Studying Ageing and the Means to Slow it Down, Mice are Not Just Small Humans. EMBO Rep. 6, S39–S44. doi: 10.1038/sj.embor.7400422. Spec No.
De Pablos, L. M., Gonzalez, G. G., Solano Parada, J., Seco Hidalgo, V., Diaz Lozano, I. M., Gomez Samblas, M. M., et al. (2011). Differential Expression and Characterization of a Member of the Mucin-Associated Surface Protein Family Secreted by Trypanosoma Cruzi. Infect. Immun. 79, 3993–4001. doi: 10.1128/IAI.05329-11
De Souza, W. (2002). Basic Cell Biology of Trypanosoma Cruzi. Curr. Pharm. Des. 8, 269–285. doi: 10.2174/1381612023396276
De Souza, W., De Carvalho, T. M., Barrias, E. S. (2010). Review on Trypanosoma Cruzi: Host Cell Interaction. Int. J. Cell Biol. 2010. doi: 10.1155/2010/295394
Dias, P. P., Capila, R. F., Do Couto, N. F., Estrada, D., Gadelha, F. R., Radi, R., et al. (2017). Cardiomyocyte Oxidants Production may Signal to T. Cruzi Intracellular Development. PloS Negl. Trop. Dis. 11, e0005852. doi: 10.1371/journal.pntd.0005852PNTD-D-17-00252
Diehl, N., Schaal, H. (2013). Make Yourself at Home: Viral Hijacking of the PI3K/Akt Signaling Pathway. Viruses 5, 3192–3212. doi: 10.3390/v5123192
Dos Santos, S. L., Freitas, L. M., Lobo, F. P., Rodrigues-Luiz, G. F., Mendes, T. A., Oliveira, A. C., et al. (2012). The MASP Family of Trypanosoma Cruzi: Changes in Gene Expression and Antigenic Profile During the Acute Phase of Experimental Infection. PloS Negl. Trop. Dis. 6, e1779. doi: 10.1371/journal.pntd.0001779
Duran-Rehbein, G. A., Vargas-Zambrano, J. C., Cuellar, A., Puerta, C. J., Gonzalez, J. M. (2014). Mammalian Cellular Culture Models of Trypanosoma Cruzi Infection: A Review of the Published Literature. Parasite 21, 38. doi: 10.1051/parasite/2014040parasite140050
El-Sayed, N. M., Myler, P. J., Bartholomeu, D. C., Nilsson, D., Aggarwal, G., Tran, A. N., et al. (2005a). The Genome Sequence of Trypanosoma Cruzi, Etiologic Agent of Chagas Disease. Science 309, 409–415. doi: 10.1126/science.1112631
El-Sayed, N. M., Myler, P. J., Blandin, G., Berriman, M., Crabtree, J., Aggarwal, G., et al. (2005b). Comparative Genomics of Trypanosomatid Parasitic Protozoa. Science 309, 404–409. doi: 10.1126/science.1112181
Epting, C. L., Coates, B. M., Engman, D. M. (2010). Molecular Mechanisms of Host Cell Invasion by Trypanosoma Cruzi. Exp. Parasitol. 126, 283–291. doi: 10.1016/j.exppara.2010.06.023
Estrada, D., Specker, G., Martinez, A., Dias, P. P., Hissa, B., Andrade, L. O., et al. (2018). Cardiomyocyte Diffusible Redox Mediators Control Trypanosoma Cruzi Infection: Role of Parasite Mitochondrial Iron Superoxide Dismutase. Biochem. J. 475, 1235–1251. doi: 10.1042/BCJ20170698
Fernandes, M. C., Andrews, N. W. (2012). Host Cell Invasion by Trypanosoma Cruzi: A Unique Strategy That Promotes Persistence. FEMS Microbiol. Rev. 36, 734–747. doi: 10.1111/j.1574-6976.2012.00333.x
Ferreira, L. R., Ferreira, F. M., Nakaya, H. I., Deng, X., Candido, D. D., De Oliveira, L. C., et al. (2017). Blood Gene Signatures of Chagas Cardiomyopathy With or Without Ventricular Dysfunction. J. Infect. Dis. 215, 387–395. doi: 10.1093/infdis/jiw540
Garg, N., Popov, V. L., Papaconstantinou, J. (2003). Profiling Gene Transcription Reveals a Deficiency of Mitochondrial Oxidative Phosphorylation in Trypanosoma Cruzi-Infected Murine Hearts: Implications in Chagasic Myocarditis Development. Biochim. Biophys. Acta 1638, 106–120. doi: 10.1016/S0925-4439(03)00060-7
Gitonga, P. K., Ndung’u, K., Murilla, G. A., Thande, P. C., Wamwiri, F. N., Auma, J. E., et al. (2017). Differential Virulence and Tsetse Fly Transmissibility of Trypanosoma Congolense and Trypanosoma Brucei Strains. Onderstepoort J. Vet. Res. 84, e1–e10. doi: 10.4102/ojvr.v84i1.1412
Goldenberg, R. C., Iacobas, D. A., Iacobas, S., Rocha, L. L., Da Silva De Azevedo Fortes, F., Vairo, L., et al. (2009). Transcriptomic Alterations in Trypanosoma Cruzi-Infected Cardiac Myocytes. Microbes Infect. 11, 1140–1149. doi: 10.1016/j.micinf.2009.08.009
Gupta, S., Bhatia, V., Wen, J. J., Wu, Y., Huang, M. H., Garg, N. J. (2009). Trypanosoma Cruzi Infection Disturbs Mitochondrial Membrane Potential and ROS Production Rate in Cardiomyocytes. Free Radic. Biol. Med. 47, 1414–1421. doi: 10.1016/j.freeradbiomed.2009.08.008
Houston-Ludlam, G. A., Belew, A. T., El-Sayed, N. M. (2016). Comparative Transcriptome Profiling of Human Foreskin Fibroblasts Infected With the Sylvio and Y Strains of Trypanosoma Cruzi. PloS One 11, e0159197. doi: 10.1371/journal.pone.0159197
Imai, K., Mimori, T., Kawai, M., Koga, H. (2005). Microarray Analysis of Host Gene-Expression During Intracellular Nests Formation of Trypanosoma Cruzi Amastigotes. Microbiol. Immunol. 49, 623–631. doi: 10.1111/j.1348-0421.2005.tb03654.x
Juiz, N. A., Torrejon, I., Burgos, M., Torres, A. M. F., Duffy, T., Cayo, N. M., et al. (2018). Alterations in Placental Gene Expression of Pregnant Women With Chronic Chagas Disease. Am. J. Pathol. 188, 1345–1353. doi: 10.1016/j.ajpath.2018.02.011
Kawashita, S. Y., Da Silva, C. V., Mortara, R. A., Burleigh, B. A., Briones, M. R. (2009). Homology, Paralogy and Function of DGF-1, a Highly Dispersed Trypanosoma Cruzi Specific Gene Family and Its Implications for Information Entropy of its Encoded Proteins. Mol. Biochem. Parasitol. 165, 19–31. doi: 10.1016/j.molbiopara.2008.12.010
Kierbel, A., Gassama-Diagne, A., Mostov, K., Engel, J. N. (2005). The Phosphoinositol-3-Kinase-Protein Kinase B/Akt Pathway is Critical for Pseudomonas Aeruginosa Strain PAK Internalization. Mol. Biol. Cell 16, 2577–2585. doi: 10.1091/mbc.e04-08-0717
Kima, P. E. (2016). PI3K Signaling in Leishmania Infections. Cell Immunol. 309, 19–22. doi: 10.1016/j.cellimm.2016.09.004
Kollien, A. H., Schaub, G. A. (2000). The Development of Trypanosoma Cruzi in Triatominae. Parasitol. Today 16, 381–387. doi: 10.1016/s0169-4758(00)01724-5
Koo, S. J., Chowdhury, I. H., Szczesny, B., Wan, X., Garg, N. J. (2016). Macrophages Promote Oxidative Metabolism to Drive Nitric Oxide Generation in Response to Trypanosoma Cruzi. Infect. Immun. 84, 3527–3541. doi: 10.1128/IAI.00809-16
Kulkarni, M. M., Olson, C. L., Engman, D. M., Mcgwire, B. S. (2009). Trypanosoma Cruzi GP63 Proteins Undergo Stage-Specific Differential Posttranslational Modification and are Important for Host Cell Infection. Infect. Immun. 77, 2193–2200. doi: 10.1128/IAI.01542-08
Laugier, L., Frade, A. F., Ferreira, F. M., Baron, M. A., Teixeira, P. C., Cabantous, S., et al. (2017). Whole-Genome Cardiac DNA Methylation Fingerprint and Gene Expression Analysis Provide New Insights in the Pathogenesis of Chronic Chagas Disease Cardiomyopathy. Clin. Infect. Dis. 65, 1103–1111. doi: 10.1093/cid/cix506
Lee, J., Remold, H. G., Ieong, M. H., Kornfeld, H. (2006). Macrophage Apoptosis in Response to High Intracellular Burden of Mycobacterium Tuberculosis is Mediated by a Novel Caspase-Independent Pathway. J. Immunol. 176, 4267–4274. doi: 10.4049/jimmunol.176.7.4267
Lewis, M. D., Kelly, J. M. (2016). Putting Infection Dynamics at the Heart of Chagas Disease. Trends Parasitol. 32, 899–911. doi: 10.1016/j.pt.2016.08.009
Ley, V., Robbins, E. S., Nussenzweig, V., Andrews, N. W. (1990). The Exit of Trypanosoma Cruzi From the Phagosome is Inhibited by Raising the Ph of Acidic Compartments. J. Exp. Med. 171, 401–413. doi: 10.1084/jem.171.2.401
Libisch, M. G., Faral-Tello, Garg, N. J., Radi, R., Piacenza, L., Robello, C. (2018). Early Trypanosoma Cruzi Infection Triggers Mtorc1-Mediated Respiration Increase and Mitochondrial Biogenesis in Human Primary Cardiomyocytes. Front. Microbiol. 9, 1889. doi: 10.3389/fmicb.2018.01889
Libisch, M. G., Rego, N., Diaz-Viraque, F., Robello, C. (2020). Host-Pathogen Transcriptomics: Trypanosoma Cruzi as a Model for Studying RNA Contamination. J. Proteomics 223, 103804. doi: 10.1016/j.jprot.2020.103804
Li, Y., Shah-Simpson, S., Okrah, K., Belew, A. T., Choi, J., Caradonna, K. L., et al. (2016). Transcriptome Remodeling in Trypanosoma Cruzi and Human Cells During Intracellular Infection. PloS Pathog. 12, e1005511. doi: 10.1371/journal.ppat.1005511PPATHOGENS-D-15-00390
Maganto-Garcia, E., Punzon, C., Terhorst, C., Fresno, M. (2008). Rab5 Activation by Toll-like Receptor 2 is Required for Trypanosoma Cruzi Internalization and Replication in Macrophages. Traffic 9, 1299–1315. doi: 10.1111/j.1600-0854.2008.00760.x
Manque, P. A., Probst, C. M., Pereira, M. C., Rampazzo, R. C., Ozaki, L. S., Pavoni, D. P., et al. (2011). Trypanosoma Cruzi Infection Induces a Global Host Cell Response in Cardiomyocytes. Infect. Immun. 79, 1855–1862. doi: 10.1128/IAI.00643-10
Markikou-Ouni, W., Ben Achour-Chenik, Y., Meddeb-Garnaoui, A. (2012). Effects of Leishmania Major Clones Showing Different Levels of Virulence on Infectivity, Differentiation and Maturation of Human Dendritic Cells. Clin. Exp. Immunol. 169, 273–280. doi: 10.1111/j.1365-2249.2012.04611.x
Mestas, J., Hughes, C. C. (2004). Of Mice and Not Men: Differences Between Mouse and Human Immunology. J. Immunol. 172, 2731–2738. doi: 10.4049/jimmunol.172.5.2731
Mortara, R. A., Procopio, D. O., Barros, H. C., Verbisck, N. V., Andreoli, W. K., Silva, R. B., et al. (1999). Features of Host Cell Invasion by Different Infective Forms of Trypanosoma Cruzi. Mem. Inst. Oswaldo Cruz 94 Suppl 1, 135–137. doi: 10.1590/s0074-02761999000700014
Mott, A., Lenormand, G., Costales, J., Fredberg, J. J., Burleigh, B. A. (2009). Modulation of Host Cell Mechanics by Trypanosoma Cruzi. J. Cell Physiol. 218, 315–322. doi: 10.1002/jcp.21606
Mukherjee, S., Nagajyothi, F., Mukhopadhyay, A., Machado, F. S., Belbin, T. J., Campos De Carvalho, A., et al. (2008). Alterations in Myocardial Gene Expression Associated With Experimental Trypanosoma Cruzi Infection. Genomics 91, 423–432. doi: 10.1016/j.ygeno.2008.01.008
Nagajyothi, F., Desruisseaux, M. S., Thiruvur, N., Weiss, L. M., Braunstein, V. L., Albanese, C., et al. (2008). Trypanosoma Cruzi Infection of Cultured Adipocytes Results in an Inflammatory Phenotype. Obes. (Silver Spring) 16, 1992–1997. doi: 10.1038/oby.2008.331
Nagajyothi, F., Machado, F. S., Burleigh, B. A., Jelicks, L. A., Scherer, P. E., Mukherjee, S., et al. (2012). Mechanisms of Trypanosoma Cruzi Persistence in Chagas Disease. Cell Microbiol. 14, 634–643. doi: 10.1111/j.1462-5822.2012.01764.x
Nde, P. N., Johnson, C. A., Pratap, S., Cardenas, T. C., Kleshchenko, Y. Y., Furtak, V. A., et al. (2010). Gene Network Analysis During Early Infection of Human Coronary Artery Smooth Muscle Cells by Trypanosoma Cruzi and Its gp83 Ligand. Chem. Biodivers. 7, 1051–1064. doi: 10.1002/cbdv.200900320
Neira, I., Ferreira, A. T., Yoshida, N. (2002). Activation of Distinct Signal Transduction Pathways in Trypanosoma Cruzi Isolates With Differential Capacity to Invade Host Cells. Int. J. Parasitol. 32, 405–414. doi: 10.1016/s0020-7519(02)00004-8
Nisimura, L. M., Coelho, L. L., De Melo, T. G., Vieira, P. C., Victorino, P. H., Garzoni, L. R., et al. (2020). Trypanosoma Cruzi Promotes Transcriptomic Remodeling of the JAK/STAT Signaling and Cell Cycle Pathways in Myoblasts. Front. Cell Infect. Microbiol. 10, 255. doi: 10.3389/fcimb.2020.00255
Oliveira, A. E. R., Pereira, M. C. A., Belew, A. T., Ferreira, L. R. P., Pereira, L. M. N., Neves, E. G. A., et al. (2020). Gene Expression Network Analyses During Infection With Virulent and Avirulent Trypanosoma Cruzi Strains Unveil a Role for Fibroblasts in Neutrophil Recruitment and Activation. PloS Pathog. 16, e1008781. doi: 10.1371/journal.ppat.1008781
Oviedo-Boyso, J., Cortes-Vieyra, R., Huante-Mendoza, A., Yu, H. B., Valdez-Alarcon, J. J., Bravo-Patino, A., et al. (2011). The Phosphoinositide-3-kinase-Akt Signaling Pathway is Important for Staphylococcus Aureus Internalization by Endothelial Cells. Infect. Immun. 79, 4569–4577. doi: 10.1128/IAI.05303-11
Parodi-Talice, A., Monteiro-Goes, V., Arrambide, N., Avila, A. R., Duran, R., Correa, A., et al. (2007). Proteomic Analysis of Metacyclic Trypomastigotes Undergoing Trypanosoma Cruzi Metacyclogenesis. J. Mass Spectrom. 42, 1422–1432. doi: 10.1002/jms.1267
Rassi, A., Jr., Rassi, A., Marin-Neto, J. A. (2010). Chagas Disease. Lancet 375, 1388–1402. doi: 10.1016/S0140-6736(10)60061-X
Scheibye-Knudsen, M., Fang, E. F., Croteau, D. L., Wilson, D. M., 3rd, Bohr, V. A. (2015). Protecting the Mitochondrial Powerhouse. Trends Cell Biol. 25, 158–170. doi: 10.1016/j.tcb.2014.11.002
Schenkman, S., Robbins, E. S., Nussenzweig, V. (1991). Attachment of Trypanosoma Cruzi to Mammalian Cells Requires Parasite Energy, and Invasion can be Independent of the Target Cell Cytoskeleton. Infect. Immun. 59, 645–654. doi: 10.1128/iai.59.2.645-654.1991
Schmunis, G. A., Yadon, Z. E. (2010). Chagas Disease: A Latin American Health Problem Becoming a World Health Problem. Acta Trop. 115, 14–21. doi: 10.1016/j.actatropica.2009.11.003
Shah-Simpson, S., Lentini, G., Dumoulin, P. C., Burleigh, B. A. (2017). Modulation of Host Central Carbon Metabolism and In Situ Glucose Uptake by Intracellular Trypanosoma Cruzi Amastigotes. PloS Pathog. 13, e1006747. doi: 10.1371/journal.ppat.1006747
Shigihara, T., Hashimoto, M., Shindo, N., Aoki, T. (2008). Transcriptome Profile of Trypanosoma Cruzi-Infected Cells: Simultaneous Up- and Down-Regulation of Proliferation Inhibitors and Promoters. Parasitol. Res. 102, 715–722. doi: 10.1007/s00436-007-0819-x
Silva, G. K., Costa, R. S., Silveira, T. N., Caetano, B. C., Horta, C. V., Gutierrez, F. R., et al. (2013). Apoptosis-Associated Speck-Like Protein Containing a Caspase Recruitment Domain Inflammasomes Mediate IL-1beta Response and Host Resistance to Trypanosoma Cruzi Infection. J. Immunol. 191, 3373–3383. doi: 10.4049/jimmunol.1203293
Silva, M. C., Davoli-Ferreira, M., Medina, T. S., Sesti-Costa, R., Silva, G. K., Lopes, C. D., et al. (2018). Canonical PI3Kgamma Signaling in Myeloid Cells Restricts Trypanosoma Cruzi Infection and Dampens Chagasic Myocarditis. Nat. Commun. 9, 1513. doi: 10.1038/s41467-018-03986-3
Soares, M. B., De Lima, R. S., Rocha, L. L., Vasconcelos, J. F., Rogatto, S. R., Dos Santos, R. R., et al. (2010). Gene Expression Changes Associated With Myocarditis and Fibrosis in Hearts of Mice With Chronic Chagasic Cardiomyopathy. J. Infect. Dis. 202, 416–426. doi: 10.1086/653481
Stamper, B. D., Davis, M., Scott-Collins, S., Tran, J., Ton, C., Simidyan, A., et al. (2019). Model-Based Evaluation of Gene Expression Changes in Response to Leishmania Infection. Gene Regul. Syst. Bio. 13, 1177625019828350. doi: 10.1177/1177625019828350
Stecconi-Silva, R. B., Andreoli, W. K., Mortara, R. A. (2003). Parameters Affecting Cellular Invasion and Escape From the Parasitophorous Vacuole by Different Infective Forms of Trypanosoma Cruzi. Mem. Inst. Oswaldo Cruz 98, 953–958. doi: 10.1590/s0074-02762003000700016
Talavera-Lopez, C., Messenger, L. A., Lewis, M. D., Yeo, M., Reis-Cunha, J. L., Matos, G. M., et al. (2021). Repeat-Driven Generation of Antigenic Diversity in a Major Human Pathogen, Trypanosoma Cruzi. Front. Cell Infect. Microbiol. 11, 614665. doi: 10.3389/fcimb.2021.614665
Tanowitz, H. B., Mukhopadhyay, A., Ashton, A. W., Lisanti, M. P., Machado, F. S., Weiss, L. M., et al. (2011). Microarray Analysis of the Mammalian Thromboxane receptor-Trypanosoma Cruzi Interaction. Cell Cycle 10, 1132–1143. doi: 10.4161/cc.10.7.15207
Tardieux, I., Nathanson, M. H., Andrews, N. W. (1994). Role in Host Cell Invasion of Trypanosoma Cruzi-Induced Cytosolic-Free Ca2+ Transients. J. Exp. Med. 179, 1017–1022. doi: 10.1084/jem.179.3.1017
Teixeira, P. C., Santos, R. H., Fiorelli, A. I., Bilate, A. M., Benvenuti, L. A., Stolf, N. A., et al. (2011). Selective Decrease of Components of the Creatine Kinase System and ATP Synthase Complex in Chronic Chagas Disease Cardiomyopathy. PloS Negl. Trop. Dis. 5. doi: 10.1371/journal.pntd.0001205PNTD-D-10-00158
Todorov, A. G., Einicker-Lamas, M., De Castro, S. L., Oliveira, M. M., Guilherme, A. (2000). Activation of Host Cell Phosphatidylinositol 3-Kinases by Trypanosoma Cruzi Infection. J. Biol. Chem. 275, 32182–32186. doi: 10.1074/jbc.M909440199
Udoko, A. N., Johnson, C. A., Dykan, A., Rachakonda, G., Villalta, F., Mandape, S. N., et al. (2016). Early Regulation of Profibrotic Genes in Primary Human Cardiac Myocytes by Trypanosoma Cruzi. PloS Negl. Trop. Dis. 10, e0003747. doi: 10.1371/journal.pntd.0003747PNTD-D-15-00537
Vago, A. R., Andrade, L. O., Leite, A. A., D’avila Reis, D., Macedo, A. M., Adad, S. J., et al. (2000). Genetic Characterization of Trypanosoma Cruzi Directly From Tissues of Patients With Chronic Chagas Disease: Differential Distribution of Genetic Types Into Diverse Organs. Am. J. Pathol. 156, 1805–1809. doi: 10.1016/s0002-9440(10)65052-3
Vago, A. R., Macedo, A. M., Oliveira, R. P., Andrade, L. O., Chiari, E., Galvao, L. M., et al. (1996). Kinetoplast DNA Signatures of Trypanosoma Cruzi Strains Obtained Directly From Infected Tissues. Am. J. Pathol. 149, 2153–2159.
Vogel, C., Marcotte, E. M. (2012). Insights Into the Regulation of Protein Abundance From Proteomic and Transcriptomic Analyses. Nat. Rev. Genet. 13, 227–232. doi: 10.1038/nrg3185
Vyatkina, G., Bhatia, V., Gerstner, A., Papaconstantinou, J., Garg, N. (2004). Impaired Mitochondrial Respiratory Chain and Bioenergetics During Chagasic Cardiomyopathy Development. Biochim. Biophys. Acta. 1689(2), 162–173. doi: 10.1016/j.bbadis.2004.03.005S0925443904000468
Wang, W., Peng, D., Baptista, R. P., Li, Y., Kissinger, J. C., Tarleton, R. L. (2021). Strain-Specific Genome Evolution in Trypanosoma Cruzi, the Agent of Chagas Disease. PloS Pathog. 17, e1009254. doi: 10.1371/journal.ppat.1009254
Wan, X., Gupta, S., Zago, M. P., Davidson, M. M., Dousset, P., Amoroso, A., et al. (2012). Defects of mtDNA Replication Impaired Mitochondrial Biogenesis During Trypanosoma Cruzi Infection in Human Cardiomyocytes and Chagasic Patients: The Role of Nrf1/2 and Antioxidant Response. J. Am. Heart Assoc. 1, e003855. doi: 10.1161/JAHA.112.003855jah3110
Wen, J. J., Dhiman, M., Whorton, E. B., Garg, N. J. (2008). Tissue-Specific Oxidative Imbalance and Mitochondrial Dysfunction During Trypanosoma Cruzi Infection in Mice. Microbes Infect. 10, 1201–1209. doi: 10.1016/j.micinf.2008.06.013
Wilkowsky, S. E., Barbieri, M. A., Stahl, P., Isola, E. L. (2001). Trypanosoma Cruzi: Phosphatidylinositol 3-Kinase and Protein Kinase B Activation is Associated With Parasite Invasion. Exp. Cell Res. 264, 211–218. doi: 10.1006/excr.2000.5123
Willingham, S. B., Bergstralh, D. T., O’connor, W., Morrison, A. C., Taxman, D. J., Duncan, J. A., et al. (2007). Microbial Pathogen-Induced Necrotic Cell Death Mediated by the Inflammasome Components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe 2, 147–159. doi: 10.1016/j.chom.2007.07.009
Woolsey, A. M., Burleigh, B. A. (2004). Host Cell Actin Polymerization is Required for Cellular Retention of Trypanosoma Cruzi and Early Association With Endosomal/Lysosomal Compartments. Cell Microbiol. 6, 829–838. doi: 10.1111/j.1462-5822.2004.00405.x
Woolsey, A. M., Sunwoo, L., Petersen, C. A., Brachmann, S. M., Cantley, L. C., Burleigh, B. A. (2003). Novel PI 3-Kinase-Dependent Mechanisms of Trypanosome Invasion and Vacuole Maturation. J. Cell Sci. 116, 3611–3622. doi: 10.1242/jcs.00666jcs.00666
Yamaguchi, H., Wang, H. G. (2001). The Protein Kinase PKB/Akt Regulates Cell Survival and Apoptosis by Inhibiting Bax Conformational Change. Oncogene 20, 7779–7786. doi: 10.1038/sj.onc.1204984
Yang, J., Nie, J., Ma, X., Wei, Y., Peng, Y., Wei, X. (2019). Targeting PI3K in Cancer: Mechanisms and Advances in Clinical Trials. Mol. Cancer 18, 26. doi: 10.1186/s12943-019-0954-x
Yilmaz, O., Jungas, T., Verbeke, P., Ojcius, D. M. (2004). Activation of the Phosphatidylinositol 3-Kinase/Akt Pathway Contributes to Survival of Primary Epithelial Cells Infected With the Periodontal Pathogen Porphyromonas Gingivalis. Infect. Immun. 72, 3743–3751. doi: 10.1128/iai.72.7.3743-3751.2004
Zhou, H., Li, X. M., Meinkoth, J., Pittman, R. N. (2000). Akt Regulates Cell Survival and Apoptosis at a Postmitochondrial Level. J. Cell Biol. 151, 483–494. doi: 10.1083/jcb.151.3.483
Keywords: Trypanosoma cruzi, Chagas disease, transcriptomics, respiratory chain, host-pathogen, Chagas cardiomyopathy, experimental variables
Citation: Libisch MG, Rego N and Robello C (2021) Transcriptional Studies on Trypanosoma cruzi – Host Cell Interactions: A Complex Puzzle of Variables. Front. Cell. Infect. Microbiol. 11:692134. doi: 10.3389/fcimb.2021.692134
Received: 07 April 2021; Accepted: 26 May 2021;
Published: 17 June 2021.
Edited by:
Rubem Figueiredo Sadok Menna-Barreto, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Santiago Martínez-Calvillo, National Autonomous University of Mexico, MexicoEdmundo C. Grisard, Federal University of Santa Catarina, Brazil
Copyright © 2021 Libisch, Rego and Robello. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos Robello, cm9iZWxsb0BwYXN0ZXVyLmVkdS51eQ==