 Jasper Götting1
Jasper Götting1 Katrin Lazar2
Katrin Lazar2 Nicolás M. Suárez3
Nicolás M. Suárez3 Lars Steinbrück1Tabea Rabe2
Lars Steinbrück1Tabea Rabe2 Rangmar Goelz4
Rangmar Goelz4 Thomas F. Schulz1
Thomas F. Schulz1 Andrew J. Davison3
Andrew J. Davison3 Klaus Hamprecht2
Klaus Hamprecht2 Tina Ganzenmueller2*
Tina Ganzenmueller2*- 1Institute of Virology, Hannover Medical School, Hannover, Germany
- 2Institute for Medical Virology and Epidemiology of Viral Diseases, University Hospital Tuebingen, Tuebingen, Germany
- 3MRC-University of Glasgow Centre for Virus Research, Glasgow, United Kingdom
- 4Department of Neonatology, University Children’s Hospital, Tuebingen, Germany
Reactivation and shedding of human cytomegalovirus (HCMV) in breast milk during lactation is highly frequent in HCMV-seropositive mothers. This represents a key transmission route for postnatal HCMV infection and can lead to severe disease in preterm neonates. Little is known about HCMV strain composition or longitudinal intrahost viral population dynamics in breast milk from immunocompetent women. We performed HCMV-specific target enrichment and high-throughput sequencing of 38 breast milk samples obtained in Germany between days 10 and 60 postpartum from 15 mothers with HCMV DNA lactia, and assembled HCMV consensus sequences de novo. The genotype distribution and number of HCMV strains present in each sample were determined by quantifying genotype-specific sequence motifs in 12 hypervariable viral genes, revealing a wide range of genotypes (82/109) for these genes in the cohort and a unique, longitudinally stable strain composition in each mother. Reactivation of up to three distinct HCMV strains was detected in 8/15 of mothers, indicating that a representative subset of the woman’s HCMV reservoir might be locally reactivated early during lactation. As described previously, nucleotide diversity of samples with multiple strains was much higher than that of samples with single strains. Breast milk as a main source of postnatal mother-to-infant transmission may serve as a repository for viral diversity and thus play an essential role in the natural epidemiology of HCMV.
Introduction
Infections with human cytomegalovirus (HCMV; species Human betaherpesvirus 5), which is a large, enveloped virus from subfamily Betaherpesvirinae of family Herpesviridae, are highly prevalent in people worldwide and result in life-long viral persistence (Boppana and Fowler, 2007; Davison, 2010; Manicklal et al., 2013). HCMV infections are acquired during all stages of life via numerous transmission routes (vertically and by various bodily fluids), and mostly remain asymptomatic in immunocompetent individuals. However, HCMV infection can lead to severe complications in transplant recipients, congenitally infected children, and upon postnatal transmission (Balfour, 1979; Manicklal et al., 2013; Kotton et al., 2018; Krishna et al., 2019).
Postnatal HCMV infections of preterm or very low birth weight (VLBW) infants pose the risk of severe disease with around 4% fatality (Martins-Celini et al., 2016; Hamprecht and Goelz, 2017; Osterholm and Schleiss, 2020). A main source of postnatal HCMV infections is the feeding of raw breast milk (BM), which is considered to be the principal transmission route (Stagno et al., 1980; Doctor et al., 2005; Hamprecht et al., 2008). Local HCMV reactivation during lactation is observed in up to 97% of HCMV-IgG-seropositive mothers, lasting around three months postpartum, and viral DNA can be detected by PCR-based methods in milk whey (DNA lactia) (Hamprecht et al., 2001; Lazar et al., 2020). This unique reactivation mode at the onset of lactation is limited to a local reactivation in the mammary glands, whereas HCMV DNA is not detected in the mother’s blood during this period (Mosca et al., 2001; Numazaki et al., 2001; Hamprecht et al., 2003).
Given its role in the natural transmission of HCMV and the resulting health burden on preterm and VLBW infants, BM is an important material to study. However, detailed studies analyzing whole HCMV genomes from BM are few, and little is known about viral strain composition and diversity, or about the longitudinal population dynamics of HCMV in BM during lactation-induced reactivation (Hamprecht et al., 2008; Hamprecht and Goelz, 2017; Suárez et al., 2019a). The diversity of HCMV genomes (236 kbp) is an evolutionary result of the generation of hypervariable genes coupled with a general loss of genetic linkage due to recombination (Bradley et al., 2008; Sijmons et al., 2015; Lassalle et al., 2016). The advent of target-enrichment as a prelude to sequencing library preparation has enabled high-throughput sequencing (HTS) of HCMV genomes directly from clinical specimens with less bias and higher coverage compared to metagenomic and amplicon-based approaches (Depledge et al., 2011; Houldcroft et al., 2016; Houldcroft et al., 2017; Suárez et al., 2019a; Suárez et al., 2019b). However, most research on HCMV strain composition has focused on genotyping single hypervariable genes in the context of HCMV as an important transplant pathogen, which, due to the involvement of immunosuppression, reinfection, and donor/recipient serostatus, does not represent natural transmission situations (Görzer et al., 2010; Renzette et al., 2013; Hage et al., 2017).
In this study, we explore the strain diversity and longitudinal intrahost dynamics of HCMV in BM from a cohort of healthy, immunocompetent mothers. This was accomplished using HTS of HCMV genomes by target enrichment coupled with a recently developed genotyping method that discriminates short motifs in hypervariable genes and allows single-strain infections to be distinguished from multiple-strain infections (Suárez et al., 2019b).
Material and Methods
Cohort and BM Specimens
BM samples originated from the BlooMil study (Lazar et al., 2020; Rabe et al., 2020) and were analyzed in collaboration with the Department of Neonatology at the University Children’s Hospital, Tuebingen. Samples (~80 ml) from HCMV-seropositive mothers of mostly preterm infants were collected within four defined time periods: 10–15, 25–30, 40–45 and 55–60 days postpartum (T1–T4). No data were available on the site of sample collection (i.e. left or right breast). Additionally, women filled out questionnaires providing data on age and number of children. Women gave informed consent, and the study was approved by the local Ethics Committee of University Hospital Tuebingen (567/2017BO2).
BM Preparation and DNA Extraction
BM was prepared and centrifuged as described previously (Lazar et al., 2020) to separate cellular, whey and fatty fractions. HCMV DNA loads of cell-free whey fraction specimens were determined by quantitative real-time PCR (CMV-R-Gene, bioMérieux, lower limit of detection = 600 copies/ml) on a LightCycler (Roche). Whey fraction samples were archived at –80°C. Based on viral load (>1000 copies/ml) and availability of longitudinal samples, 38 specimens from 15 mothers were selected for HTS. DNA was extracted from 400 µl archived BM whey fractions and eluted in 200 µl buffer using a QIAgen DNeasy Blood & Tissue kit on a QIAcube (Qiagen) according to the manufacturer’s instructions.
Library Preparation, HCMV-Specific Target Enrichment, and HTS
Library preparation and sequencing were performed as described previously, with minor modifications (Hage et al., 2017). DNA was fragmented by sonication, sequencing libraries were prepared (KAPA library preparation kit, KAPA Biosystems), and PCR amplification prior to target enrichment was carried out with adapter-specific primers (14 cycles). Up to 750 ng amplified DNA was target-enriched using HCMV-specific RNA baits (ELID 0659711, SureSelect XT, Agilent). HCMV-enriched libraries were indexed, amplified (14–16 cycles), multiplexed and sequenced on a MiSeq (Illumina) using a v3 Reagent Kit to generate 2 × 300 nucleotide paired-end reads.
De Novo Assembly and Phylogenetic Analysis
De novo assembly was performed using a pipeline incorporating fastp (adapter trimming and read quality filtering), Kraken2 (human read removal), SPAdes (contig assembly), and Qiagen CLC Genomics Workbench 10 (contig scaffolding and subsequent read mappings) (Nurk et al., 2013; Nurk et al., 2017; Chen et al., 2018; Wood et al., 2019). Duplicate read pairs stemming from the same DNA fragment were removed using Picard v2.3.0, and draft genomes were polished using GapFiller and Pilon with the deduplicated reads (Boetzer and Pirovano, 2012; Walker et al., 2014; Broad Institute, 2019). The polished draft genomes were inspected by read mapping of their corresponding read sets, annotated from HCMV strain Merlin (GenBank accession no. AY446894.2) using Geneious Prime 2020.0.3 (Biomatters), and trimmed of the inverted repeats at the genome termini. Complete, annotated HCMV consensus genomes from mothers with single-strain HCMV reactivations were deposited in GenBank (accession numbers MW528458 – MW528464), and their respective human-filtered read data sets were deposited in the European Nucleotide Archive (http://www.ebi.ac.uk/ena/data/view/PRJEB42695, sample accession numbers ERS5621307 – ERS5621320).
Genotype Analysis and Strain Enumeration
The quality of the libraries (fraction of human reads and number of unique HCMV reads) was assessed before genotype analysis (Table S1). Genotyping of 12 hypervariable HCMV genes (RL5A, RL6, RL12, RL13, UL1, UL9, UL11, UL73, UL74, UL120, UL146 and UL139) was achieved using the VATK pipeline to determine the number of reads containing conserved, genotype-specific sequence motifs of 20–31 bp in the trimmed, unfiltered read datasets, as described previously (Centre for Virus Research, 2019; Suárez et al., 2019b). The number of HCMV strains present in a sample was set as the maximum number of genotypes detected for ≥2 genes, requiring ≥25 reads containing the relevant motifs and representing ≥5% of the total number of reads identified for that gene. Genotype profiles were visualized in R using dplyr and ggplot2 3.3.0 (Wickham, 2016; R Core Team, 2019). The output was used to distinguish single-strain from multiple-strain reactivations.
Variant Analysis and Calculation of Intrahost Nucleotide Diversity
Single nucleotide polymorphisms (SNPs) were called after mapping reads to the relevant consensus genome or, in cases of longitudinal samples, after mapping the reads from each time-point to the sequence derived from the first time-point. SNPs fulfilling the following criteria were considered: read depth ≥25 (deduplicated data), average base quality ≥20, forward/reverse read balance 0.3–0.5, and variant frequency (the relative frequency of a SNP at this position) ≥2%. The SNP profiles were visualized in R as described above. This depiction facilitates the identification of the approximate abundance of minor viral populations (i.e. variant frequency <50%). The genome-wide intrahost nucleotide diversity value (π) was calculated as described previously (Nelson and Hughes, 2015). Briefly, diversity Di at each variable position i in the genome was calculated based on the frequency of the four nucleotides and the coverage of deduplicated reads mapping against the de novo assembly of each dataset (intra-sample diversity). The sum of the diversity Di of all variable positions in a genome was then divided by the length of that genome to obtain π.
Results
HTS of HCMV From BM
Thirty-eight HCMV DNA positive BM samples from 15 mothers (median age 33 [26–49] years; n=8 already had given birth to at least one other child) were analyzed (Table 1). Samples from at least two time-points were sequenced for each mother, and the full set of four samples was available for four women. As a result, it was possible to evaluate the longitudinal population dynamics for all participants (Table 1).
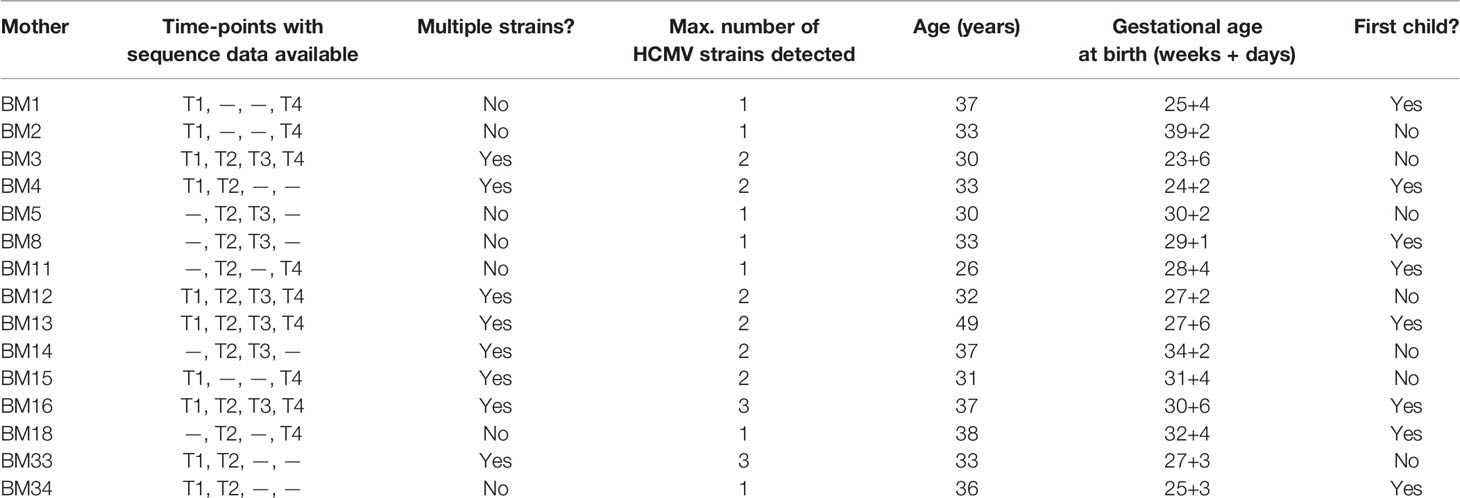
Table 1 Clinical information on the 15 mothers included in the study, sequenced time-points and number of detected HCMV strains.
The sequencing libraries prepared from milk whey samples had a median viral input load of 4.4 × 103 copies (9.8 × 102 to 2.6 × 105) per library (Table S1). HCMV-specific target enrichment and sequencing with an average MiSeq flow cell utilization of 12% yielded an excellent human-to-viral reads ratio and sufficient reads for the assembly of consensus HCMV genomes in all 38 cases. Even after removal of duplicate reads, the median average sequencing coverage depth of the assemblies was quite high, with 339 (15–3420) deduplicated reads per position. This coverage depth correlated very well (R2 = 0.71) with the viral input load during library preparation (Figure S1). Thus, human milk whey is an excellent specimen type for HCMV genome sequencing.
Genotype Diversity and Strain Enumeration
To determine HCMV strain diversity during natural reactivation in immunocompetent mothers, 12 hypervariable viral genes were genotyped in each sample dataset using a genotype-specific sequence motif search. A very wide genotype variety (82 out of the 109 possible genotypes) was observed within the cohort, and the intrahost genotype composition (viral haplotype) within each mother was unique. Thus, no two mothers had the same HCMV strain(s). Multiple-strain reactivations involving up to three different HCMV strains were detected in more than half of the mothers (8/15, 53%; Table 2), and each of the other seven women had reactivations involving a single strain only.
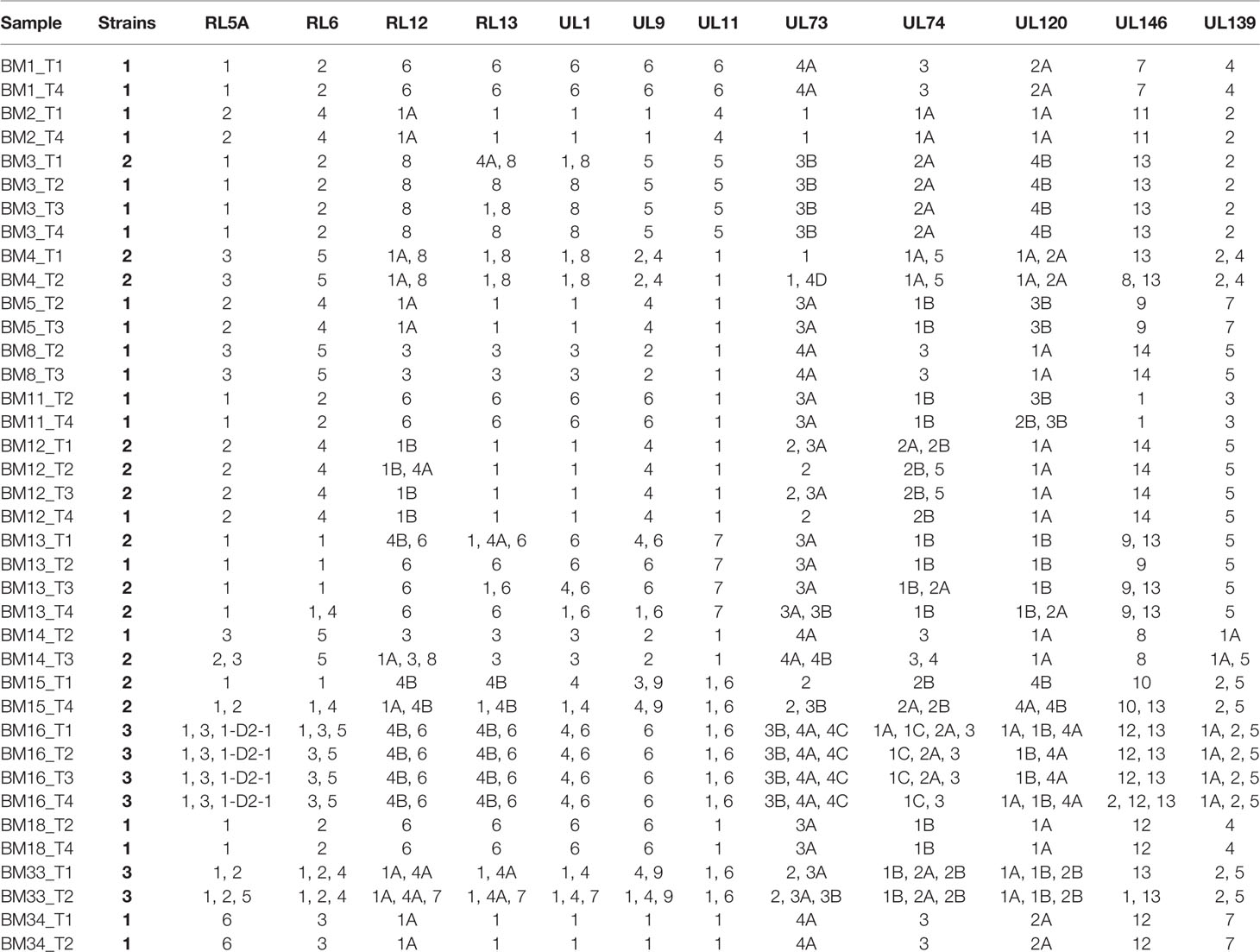
Table 2 Genotypes of hypervariable genes in BM specimens determined via genotype-specific sequence motifs.
Analysis of the intrahost genotype profile of the 12 analyzed viral genes among samples taken at longitudinal time-points for each mother revealed largely stable genotype compositions over time. Shifts in the relative proportions of individual genotypes occurred over time, but there were no drastic qualitative changes among the two or more samples obtained from each individual. This indicates that no additional genotypes possibly indicating superinfections or sequentially reactivated strains emerged at later time-points, but rather that a representative part of the latency reservoir of each host was reactivated early during lactation. In a few cases (e.g. BM15), additional genotypes seemed to have appeared at later time-points, but closer analysis revealed that these were already present at earlier time-points, but at levels below the defined reporting thresholds (Table S1). These genotyping results are summarized in Table 2, and the genotype profiles of all samples are shown in Figures 1 and 2. The genotype composition of a single sample containing two HCMV strains is illustrated in further detail in Figure S3.
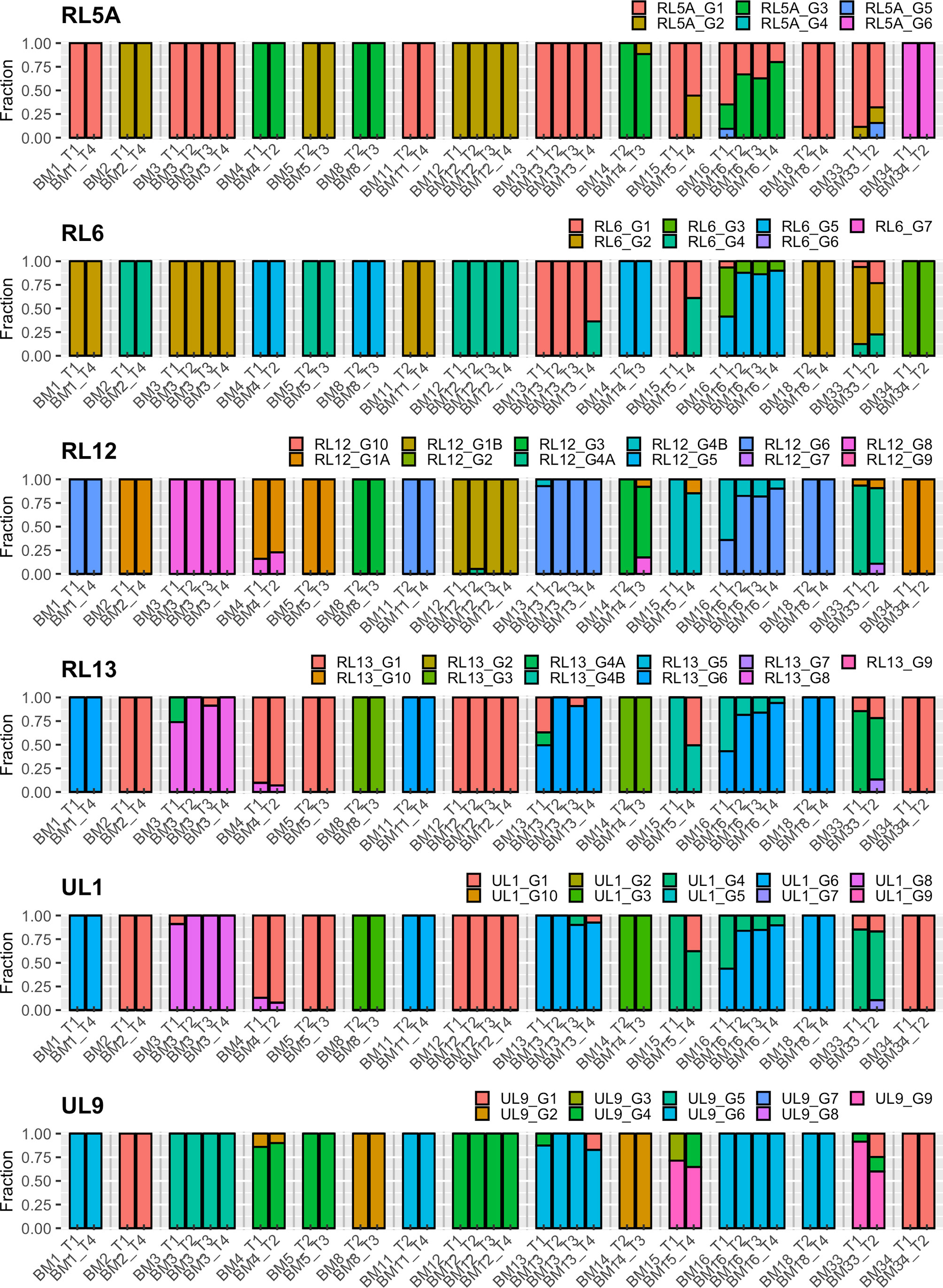
Figure 1 Genotype profile and relative abundance of HCMV genes RL5A – UL9 genotypes in the 38 BM specimens. Each color represents a different genotype of a hypervariable gene. Each bar represents a different sample, grouped by subject. The Y-axis indicates the fraction of genotypes in the total sequence reads for that gene in the sample.
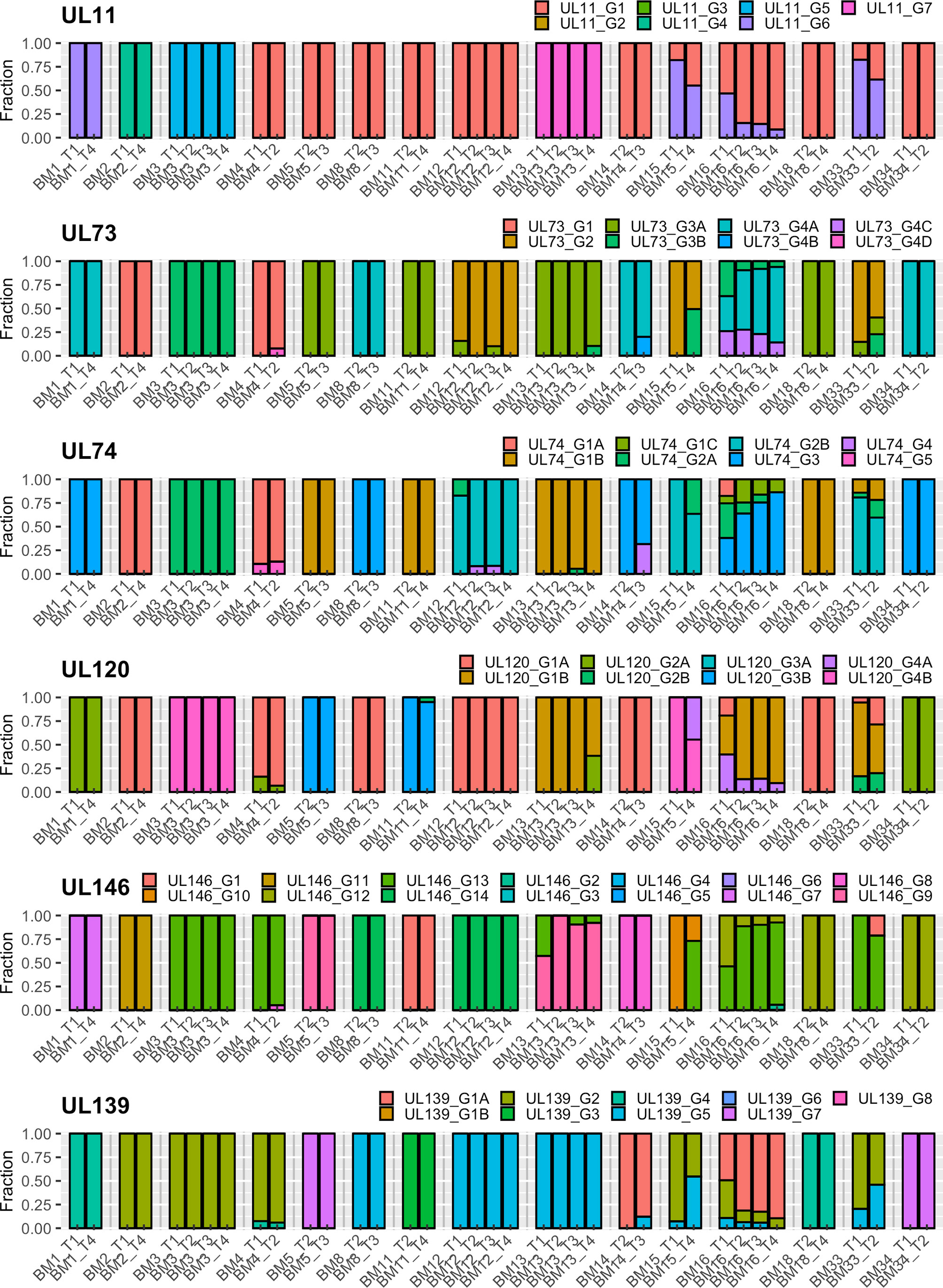
Figure 2 Genotype profile and relative abundance of HCMV genes UL11 – UL139 genotypes in the 38 BM specimens. Each color represents a different genotype of a hypervariable gene. Each bar represents a different sample, grouped by subject. The Y-axis indicates the fraction of genotypes in the total sequence reads for that gene in the sample.
Intrahost Diversity
Intrahost nucleotide diversity (π) was calculated separately for datasets representing single strains or multiple strains. Samples containing a single strain (n=20) had π values of 6.4 × 10–5 (± 2.1 × 10–4 SD), whereas the samples representing two (n=12) or three strains (n=6) exhibited higher π values of 1.2 × 10–3 (± 2.4 × 10–3 SD) or 5.2 × 10–3 (± 2.5 × 10–3 SD), respectively (Figure S2). These results can be ascribed to an average of 2074 ± 1470 (range: 271–5045) or 5976 ± 332 (range: 5596–6525) variants contributing to the diversity in datasets representing two or three strains, respectively. Single-strain datasets displayed only 361 ± 420 (range: 7–1336) variants (Figure S4).
Correlation of Age and Number of Children With Multiple-Strain Reactivation
No trends were observed between the presence of multiple-strain reactivation and whether a woman was below or above the median age of 33 (Fisher’s exact test, p = 1), or whether the child was the woman’s first (Fisher’s exact test, p = 0.62) (Figure S5).
Discussion
BM is an important focus because of its role in postnatal HCMV transmission and the health burden imposed by postnatal HCMV infection on preterm and VLBW infants. However, studies on HCMV population dynamics at the whole genome level in BM are rare (Hamprecht et al., 2008; Suárez et al., 2019a), especially with regard to the longitudinal intrahost diversity. Analysing BM from healthy mothers with HCMV reactivation opens an opportunity to study the natural epidemiology of HCMV strains and the diversity of the genome over time in immunocompetent adults, in contrast to investigations in transplanted or HIV-infected patients who display an entirely different risk pattern for multiple HCMV infections and reinfections. This is especially pertinent, given that immunocompetent persons generally do not reactivate HCMV with DNAemia levels sufficient to allow HTS of viral genomes.
In our hands, HCMV-specific target enrichment and HTS of HCMV genomes in BM specimens resulted in excellent human-to-viral read ratios and enabled the de novo assembly of HCMV consensus genomes despite relatively low viral input loads. This contrasts with our experience sequencing HCMV from human plasma samples (Dhingra et al., unpublished data), where the median unique coverage depth was five times lower. This difference was possibly due to the proportion of intact genomes from virions in mammary glands, which constitute the primary locus of HCMV reactivation (Maschmann et al., 2015).
To our knowledge, this is the first description of HCMV diversity in the BM of a cohort of predominantly European, immunocompetent women. Sequencing of viral populations from longitudinally collected specimens revealed a high level of genotypic complexity with stable intrahost population compositions. Multiple-strain reactivations with up to three strains were present in 8/15 mothers (53%). No correlation was detected between the presence of multiple-strain reactivation, a woman’s age or whether the child was the woman’s first. However, as the cohort was relatively small, epidemiological conclusions should be drawn cautiously.
A previous study on HCMV strain diversity in BM specimens from HIV-positive mothers in Zambia found a similar number of different genotypes (89/109 genotypes) and reactivation of up to five strains in single individuals over a period of 4–16 weeks postpartum (Suárez et al., 2019a). The fraction of multiple-strain reactivations in our cohort was slightly lower, with 8/15 compared to 11/13 mothers in this category, and was more comparable to the 9/16 fraction observed in a cohort of transplant patients at Hanover Medical School, Germany (Hage et al., 2017). It is possible that HIV-related immunodeficiency in the Zambian cohort, as well as the generally higher seroprevalence of HCMV in developing countries, contributed to this difference (Ludwig and Hengel, 2009; Manicklal et al., 2013; Voigt et al., 2015).
HCMV strains in all 15 mothers from our cohort displayed a stable genotype composition with shifting abundance of subpopulations in multiple-strain samples but no novel emerging haplotypes that would indicate superinfection or reactivation of previously undetected strains. This is consistent with observations of two of the mothers in Zambia for whom longitudinal data were available, who also exhibited intrahost stability of HCMV populations in the initial months postpartum. Our estimates of the much lower level of intrahost diversity in single-strain reactivations in comparison with multiple-strain reactivations also corroborates the findings of previous studies in the blood compartment of immunocompromised patients (Cudini et al., 2019). This underlines the importance of controlling for the presence of multiple strains in studies of HCMV diversity in clinical specimens (Houldcroft et al., 2020; Suárez et al., 2020). Low levels of intrahost diversity in single-strain infections and reactivations stem from a small number of variants usually present at low frequency (Figure S4), a situation that makes diversity estimates very susceptible to outliers (Figure S2) (Suárez et al., 2019b).
Overall, the abundance of multiple strains reactivating in the mammary glands during lactation confirms the likelihood that BM serves not only as a major source of HCMV transmission but also as a key repository for viral diversity. We could not evaluate the transmission of HCMV to preterm infants, as sampling from neonates was not part of the study and infants under 32 weeks of gestational age were fed exclusively with short-term pasteurized BM, which would prevent transmission (Goelz et al., 2009; Klotz et al., 2018; Bapistella et al., 2019; Maschmann et al., 2019).
As HCMV reactivates strictly locally in the mammary glands during lactation and none of the women showed signs of systemic HCMV reactivation with DNAemia, strain composition and diversity among different compartments (blood and mammary glands) could not be analyzed (Lazar et al., 2020). Local reactivation during lactation is thought to be triggered by invasion of CD14+ monocytes (one site of HCMV latency) into the mammary glands (Maschmann et al., 2015). Thus, the viral populations and mixed strain reactivations that were detected likely have been present in the women prior to lactation. In the context of this reactivation mechanism the stable strain composition over time may indicate that a representative subset of the woman’s HCMV reservoir reactivates early and simultaneously during lactation.
In summary, our study provides insights into interhost and longitudinal intrahost variation of HCMV populations in BM after birth. It also demonstrates the feasibility of studying the natural diversity and transmission of HCMV in immunocompetent adults. Future work is necessary to elucidate HCMV strain dynamics during and after transmission to the infant.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, MW528458 – MW528464. https://www.ebi.ac.uk/ena, PRJEB42695, ERS5621307 – ERS5621320.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethics Committee of University Hospital Tuebingen, Tuebingen, Germany (No. 567/2017BO2). The patients/participants provided their written informed consent to participate in this study.
Author Contributions
JG conducted the sequencing, analyzed and interpreted the data and drafted the mansucript. LS conducted the sequencing and genome assembly. KL and TR collected samples and data, and extracted breast milk DNA. NS and AD developed the genotyping tools, conducted genome assembly and reviewed the manuscript. TS and RG contributed to conception of the study, and reviewed the manuscript. KH contributed to the study design and sample selection and reviewed the manuscript. TG designed and supervised the study, analyzed and interpreted data and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) in Collaborative Research Centre 900 (project Z1) and under Germany’s Excellence Strategy – EXC 2155 “RESIST” – Project ID 39087428. JG was supported by the graduate program “Infection Biology” of the Hanover Biomedical Research School (HBRS). KL was supported by “Sonderlinie Medizin: Verbundprojekt Freiburg-Tuebingen-Ulm” by the Ministry of Science and Art of Baden Wuerttemberg, grant number [KL 25280-0]. TR was supported by a medical thesis grant from IZKF Promotionskolleg, University Hospital Tuebingen [201802-23]. AD and NS were supported by Medical Research Council program grant MC_UU_12014/3. We acknowledge support by the Open Access Publishing Fund of the University of Tuebingen.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
First and foremost, we thank the mothers who participated in this study. We also thank Dr. Akshay Dhingra for guidance during the very early stages of the project and Karin Kollender, Andrea Baumeister and Susanne Rack for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.664247/full#supplementary-material
References
Balfour H. H. (1979). Cytomegalovirus: the troll of transplantation. Arch. Intern. Med. 139, 279–280. doi: 10.1001/archinte.139.3.279
Bapistella S., Hamprecht K., Thomas W., Speer C. P., Dietz K., Maschmann J., et al. (2019). Short-term Pasteurization of Breast Milk to Prevent Postnatal Cytomegalovirus Transmission in Very Preterm Infants. Clin. Infect. Dis. 69, 438–444. doi: 10.1093/cid/ciy945
Boetzer M., Pirovano W. (2012). Toward almost closed genomes with GapFiller. Genome Biol. 13, R56. doi: 10.1186/gb-2012-13-6-r56
Boppana S. B., Fowler K. B. (2007).Persistence in the population: epidemiology and transmisson. In: Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis (Cambridge: Cambridge University Press). Available at: http://www.ncbi.nlm.nih.gov/books/NBK47450/ (Accessed October 21, 2020).
Bradley A. J., Kovács I. J., Gatherer D., Dargan D. J., Alkharsah K. R., Chan P. K. S., et al. (2008). Genotypic analysis of two hypervariable human cytomegalovirus genes. J. Med. Virol. 80, 1615–1623. doi: 10.1002/jmv.21241
Broad Institute (2019). Picard toolkit (Broad Institute, GitHub repository). Available at: http://broadinstitute.github.io/picard/ (Accessed May 25, 2020).
Centre for Virus Research (2019). centre-for-virus-research/VATK (Centre for Virus). Available at: https://github.com/centre-for-virus-research/ (Accessed May 26, 2020).
Chen S., Zhou Y., Chen Y., Gu J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cudini J., Roy S., Houldcroft C. J., Bryant J. M., Depledge D. P., Tutill H., et al. (2019). Human cytomegalovirus haplotype reconstruction reveals high diversity due to superinfection and evidence of within-host recombination. PNAS 116, 5693–5698. doi: 10.1073/pnas.1818130116
Davison A. J. (2010). Herpesvirus systematics. Vet. Microbiol. 143, 52–69. doi: 10.1016/j.vetmic.2010.02.014
Depledge D. P., Palser A. L., Watson S. J., Lai I. Y.-C., Gray E. R., Grant P., et al. (2011). Specific capture and whole-genome sequencing of viruses from clinical samples. PloS One 6, e27805. doi: 10.1371/journal.pone.0027805
Doctor S., Friedman S., Dunn M. S., Asztalos E. V., Wylie L., Mazzulli T., et al. (2005). Cytomegalovirus transmission to extremely low-birthweight infants through breast milk. Acta Paediatr. 94, 53–58. doi: 10.1111/j.1651-2227.2005.tb01788.x
Goelz R., Hihn E., Hamprecht K., Dietz K., Jahn G., Poets C., et al. (2009). Effects of Different CMV-Heat-Inactivation-Methods on Growth Factors in Human Breast Milk. Pediatr. Res. 65, 458–461. doi: 10.1203/PDR.0b013e3181991f18
Görzer I., Guelly C., Trajanoski S., Puchhammer-Stöckl E. (2010). Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J. Virol. 84, 7195–7203. doi: 10.1128/JVI.00475-10
Hage E., Wilkie G. S., Linnenweber-Held S., Dhingra A., Suárez N. M., Schmidt J. J., et al. (2017). Characterization of Human Cytomegalovirus Genome Diversity in Immunocompromised Hosts by Whole-Genome Sequencing Directly From Clinical Specimens. J. Infect. Dis. 215, 1673–1683. doi: 10.1093/infdis/jix157
Hamprecht K., Goelz R. (2017). Postnatal Cytomegalovirus Infection Through Human Milk in Preterm Infants: Transmission, Clinical Presentation, and Prevention. Clin. Perinatol 44, 121–130. doi: 10.1016/j.clp.2016.11.012
Hamprecht K., Maschmann J., Vochem M., Dietz K., Speer C. P., Jahn G. (2001). Epidemiology of transmission of cytomegalovirus from mother to preterm infant by breastfeeding. Lancet 357, 513–518. doi: 10.1016/S0140-6736(00)04043-5
Hamprecht K., Maschmann J., Vochem M., Speer C. P., Jahn G. (2003). Transmission of Cytomegalovirus to Preterm Infants by Breast-Feeding. New Aspects CMV-Related Immunopathol 24, 43–52. doi: 10.1159/000073883
Hamprecht K., Maschmann J., Jahn G., Poets C. F., Goelz R. (2008). Cytomegalovirus transmission to preterm infants during lactation. J. Clin. Virol. 41, 198–205. doi: 10.1016/j.jcv.2007.12.005
Houldcroft C. J., Bryant J. M., Depledge D. P., Margetts B. K., Simmonds J., Nicolaou S., et al. (2016). Detection of Low Frequency Multi-Drug Resistance and Novel Putative Maribavir Resistance in Immunocompromised Pediatric Patients with Cytomegalovirus. Front. Microbiol. 7, 1317. doi: 10.3389/fmicb.2016.01317
Houldcroft C. J., Beale M. A., Breuer J. (2017). Clinical and biological insights from viral genome sequencing. Nat. Rev. Microbiol. 15, 183–192. doi: 10.1038/nrmicro.2016.182
Houldcroft C. J., Cudini J., Goldstein R. A., Breuer J. (2020). Reply to Jensen and Kowalik: Consideration of mixed infections is central to understanding HCMV intrahost diversity. Proc. Natl. Acad. Sci. U.S.A. 117, 818–819. doi: 10.1073/pnas.1918955117
Klotz D., Jansen S., Gebauer C., Fuchs H. (2018). Handling of Breast Milk by Neonatal Units: Large Differences in Current Practices and Beliefs. Front. Pediatr. 6, 235. doi: 10.3389/fped.2018.00235
Kotton C. N., Kumar D., Caliendo A. M., Huprikar S., Chou S., Danziger-Isakov L., et al. (2018). The Third International Consensus Guidelines on the Management of Cytomegalovirus in Solid-organ Transplantation. Transplantation 102, 900–931. doi: 10.1097/TP.0000000000002191
Krishna B. A., Wills M. R., Sinclair J. H. (2019). Advances in the treatment of cytomegalovirus. Br. Med. Bull. 131, 1–13. doi: 10.1093/bmb/ldz031
Lassalle F., Depledge D. P., Reeves M. B., Brown A. C., Christiansen M. T., Tutill H. J., et al. (2016). Islands of linkage in an ocean of pervasive recombination reveals two-speed evolution of human cytomegalovirus genomes. Virus Evol. 2, vew017. doi: 10.1093/ve/vew017
Lazar K., Rabe T., Goelz R., Hamprecht K. (2020). Human Cytomegalovirus Reactivation During Lactation: Impact of Antibody Kinetics and Neutralization in Blood and Breast Milk. Nutrients 12. doi: 10.3390/nu12020338
Ludwig A., Hengel H. (2009). Epidemiological impact and disease burden of congenital cytomegalovirus infection in Europe. Eurosurveillance 14, 19140. doi: 10.2807/ese.14.09.19140-en
Manicklal S., Emery V. C., Lazzarotto T., Boppana S. B., Gupta R. K. (2013). The “silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 26, 86–102. doi: 10.1128/CMR.00062-12
Martins-Celini F. P., Yamamoto A. Y., Passos D. M., do Nascimento S. D., Lima E. V., Di Giovanni C. M., et al. (2016). Incidence, Risk Factors, and Morbidity of Acquired Postnatal Cytomegalovirus Infection Among Preterm Infants Fed Maternal Milk in a Highly Seropositive Population. Clin. Infect. Dis. 63, 929–936. doi: 10.1093/cid/ciw394
Maschmann J., Goelz R., Witzel S., Strittmatter U., Steinmassl M., Jahn G., et al. (2015). Characterization of Human Breast Milk Leukocytes and Their Potential Role in Cytomegalovirus Transmission to Newborns. NEO 107, 213–219. doi: 10.1159/000371753
Maschmann J., Müller D., Lazar K., Goelz R., Hamprecht K. (2019). New short-term heat inactivation method of cytomegalovirus (CMV) in breast milk: impact on CMV inactivation, CMV antibodies and enzyme activities. Arch. Dis. Child Fetal Neonatal Ed. 104, F604–F608. doi: 10.1136/archdischild-2018-316117
Mosca F., Pugni L., Barbi M., Binda S. (2001). Transmission of cytomegalovirus. Lancet 357, 1800. doi: 10.1016/S0140-6736(00)04914-X
Nelson C. W., Hughes A. L. (2015). Within-Host Nucleotide Diversity of Virus Populations: Insights from Next-Generation Sequencing. Infect. Genet. Evol. 0, 1–7. doi: 10.1016/j.meegid.2014.11.026
Numazaki K., Chiba S., Asanuma H. (2001). Transmission of cytomegalovirus. Lancet 357, 1799–1800. doi: 10.1016/S0140-6736(00)04913-8
Nurk S., Bankevich A., Antipov D., Gurevich A., Korobeynikov A., Lapidus A., et al. (2013). “Assembling Genomes and Mini-metagenomes from Highly Chimeric Reads,” in Research in Computational Molecular Biology Lecture Notes in Computer Science. Eds. Deng M., Jiang R., Sun F., Zhang X. (Berlin, Heidelberg: Springer), 158–170. doi: 10.1007/978-3-642-37195-0_13
Nurk S., Meleshko D., Korobeynikov A., Pevzner P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Osterholm E. A., Schleiss M. R. (2020). Impact of breast milk-acquired cytomegalovirus infection in premature infants: Pathogenesis, prevention, and clinical consequences? Rev. Med. Virol. 30, e2117. doi: 10.1002/rmv.2117
R Core Team (2019). R: A Language and Environment for Statistical Computing (Vienna, Austria: R Foundation for Statistical Computing). Available at: https://www.R-project.org/.
Rabe T., Lazar K., Cambronero C., Goelz R., Hamprecht K. (2020). Human Cytomegalovirus (HCMV) Reactivation in the Mammary Gland Induces a Proinflammatory Cytokine Shift in Breast Milk. Microorganisms 8. doi: 10.3390/microorganisms8020289
Renzette N., Gibson L., Bhattacharjee B., Fisher D., Schleiss M. R., Jensen J. D., et al. (2013). Rapid Intrahost Evolution of Human Cytomegalovirus Is Shaped by Demography and Positive Selection. PloS Genet. 9. doi: 10.1371/journal.pgen.1003735
Sijmons S., Thys K., Mbong Ngwese M., Van Damme E., Dvorak J., Van Loock M., et al. (2015). High-Throughput Analysis of Human Cytomegalovirus Genome Diversity Highlights the Widespread Occurrence of Gene-Disrupting Mutations and Pervasive Recombination. J. Virol. 89, 7673–7695. doi: 10.1128/JVI.00578-15
Stagno S., Reynolds D. W., Pass R. F., Alford C. A. (1980). Breast milk and the risk of cytomegalovirus infection. N. Engl. J. Med. 302, 1073–1076. doi: 10.1056/NEJM198005083021908
Suárez N., Musonda K. G., Escriva E., Njenga M., Agbueze A., Camiolo S., et al. (2019a). Multiple-Strain Infections of Human Cytomegalovirus with High Genomic Diversity are Common In Breast Milk from HIV-Positive Women in Zambia. J. Infect. Dis. 220, 792–801. doi: 10.1093/infdis/jiz209
Suárez N. M., Wilkie G. S., Hage E., Camiolo S., Holton M., Hughes J., et al. (2019b). Human Cytomegalovirus Genomes Sequenced Directly From Clinical Material: Variation, Multiple-Strain Infection, Recombination, and Gene Loss. J. Infect. Dis. 220, 781–791. doi: 10.1093/infdis/jiz208
Suárez N. M., Blyth E., Li K., Ganzenmueller T., Camiolo S., Avdic S., et al. (2020). Whole-Genome Approach to Assessing Human Cytomegalovirus Dynamics in Transplant Patients Undergoing Antiviral Therapy. Front. Cell. Infect. Microbiol. 10, 267. doi: 10.3389/fcimb.2020.00267
Voigt S., Schaffrath Rosario A., Mankertz A. (2015). Cytomegalovirus Seroprevalence Among Children and Adolescents in Germany: Data From the German Health Interview and Examination Survey for Children and Adolescents (KiGGS), 2003–2006. Open Forum Infect. Dis. 3. doi: 10.1093/ofid/ofv193
Walker B. J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., et al. (2014). Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PloS One 9, e112963. doi: 10.1371/journal.pone.0112963
Wickham H. (2016). ggplot2: Elegant graphics for data analysis (New York: Springer-Verlag). Available at: https://ggplot2.tidyverse.org.
Keywords: cytomegalovirus, breast milk, genomes, strains, diversity, genotyping, immunocompetent, lactation
Citation: Götting J, Lazar K, Suárez NM, Steinbrück L, Rabe T, Goelz R, Schulz TF, Davison AJ, Hamprecht K and Ganzenmueller T (2021) Human Cytomegalovirus Genome Diversity in Longitudinally Collected Breast Milk Samples. Front. Cell. Infect. Microbiol. 11:664247. doi: 10.3389/fcimb.2021.664247
Received: 04 February 2021; Accepted: 22 March 2021;
Published: 16 April 2021.
Edited by:
Qiliang Cai, Fudan University, ChinaReviewed by:
Neelam Sharma-Walia, Rosalind Franklin University of Medicine and Science, United StatesJuliet Spencer, Texas Woman’s University, United States
Copyright © 2021 Götting, Lazar, Suárez, Steinbrück, Rabe, Goelz, Schulz, Davison, Hamprecht and Ganzenmueller. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tina Ganzenmueller, VGluYS5HYW56ZW5tdWVsbGVyQG1lZC51bmktdHVlYmluZ2VuLmRl