
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol., 13 April 2021
Sec. Microbes and Innate Immunity
Volume 11 - 2021 | https://doi.org/10.3389/fcimb.2021.629836
This article is part of the Research TopicMicrobial Modulation of Host Programmed Cell DeathView all 4 articles
 Siyuan Feng1,2†
Siyuan Feng1,2† Zhongsi Hong1†
Zhongsi Hong1† Guoliang Zhang3†
Guoliang Zhang3† Jiachen Li1,2
Jiachen Li1,2 Guo-Bao Tian1,2Haibo Zhou4*
Guo-Bao Tian1,2Haibo Zhou4* Xi Huang1,2,3,4,5*
Xi Huang1,2,3,4,5*Genome scale mutagenesis identifies many genes required for mycobacterial infectivity and survival, but their contributions and mechanisms of action within the host are poorly understood. Using CRISPR interference, we created a knockdown of ppe31Mm gene in Mycobacterium marinum (M. marinum), which reduced the resistance to acid medium. To further explore the function of PPE31, the ppe31 mutant strain was generated in M. marinum and Mycobacterium tuberculosis (M. tuberculosis), respectively. Macrophages infected with the ppe31Mm mutant strain caused a reduced inflammatory mediator expressions. In addition, macrophages infected with M. marinum Δppe31Mm had decreased host cell death dependent on JNK signaling. Consistent with these results, deletion of ppe31Mtb from M. tuberculosis increased the sensitivity to acid medium and reduced cell death in macrophages. Furthermore, we demonstrate that both ppe31 mutants from M. marinum and M. tuberculosis resulted in reduced survival in macrophages, and the survivability of M. marinum was deceased in zebrafish due to loss of ppe31Mm. Our findings confirm that PPE31 as a virulence associated factor that modulates innate immune responses to mycobacterial infection.
Several mycobacterial species are successful intracellular pathogens of humans and other animals, and can survive and replicate within host macrophages (Houben et al., 2006; Pieters, 2008). Mycobacterium tuberculosis, the leading infectious killer of humans worldwide, specifically has developed a plethora of strategies to escape killing by host defense mechanisms. Under acidic conditions, M. tuberculosis Type VII secretion systems can cause phagolysosomal rupture (Conrad et al., 2017) and damage to the phagosomal membrane facilitates bacterial translocation into the cytosol, a process that ultimately leads to macrophage necrosis (Abdallah et al., 2011; Houben et al., 2012). In addition, necrotic cells provide an adaptive environment for proliferation/survival (Behar et al., 2010). Virulence factor used by M. tuberculosis manipulated macrophage death pathways, which is one of strategies to evade host immune defenses. Although the evasion of intracellular host defense by M. tuberculosis is crucial for host cell necrosis and bacterial dissemination, the molecular mechanisms involved remains incompletely understood.
Members of the PE/PPE protein family, which are present primarily in slowly growing mycobacteria, have been associated with virulence (Tobin and Ramakrishnan, 2008). Most PE/PPE proteins are located in the cell wall and some have been shown to modulate host cell physiology by different mechanisms (Deng et al., 2015; Yang et al., 2017). For example, M. tuberculosis PPE38 dampens CD8+ T cell responses by inhibiting macrophage MHC Class I expression (Meng et al., 2017), suggesting a unique role of PE/PPE protein to facilitate mycobacteria to escape host immunity. PPE31 belongs to the PPE subfamily and is highly expressed in M. tuberculosis during acidic stress, Mg2+ starvation and antibiotic treatment, as well as in M. tuberculosis infected macrophages (Walters et al., 2006; Rohde et al., 2007; Liu et al., 2016). Furthermore, an M. tuberculosis ppe31 transposon mutant was attenuated virulence in a mouse infection model (Sassetti and Rubin, 2003), indicating a role for PPE31 in virulence; however, its precise function in pathogenesis is unknown.
In the current study, we showed that ppe31Mm (MMAR_2683), the M. marinum orthologue of ppe31Mtb (rv1807), was associated with mycobacterial survival in acid medium. The mutants for ppe31Mm reduced host cell death through JNK-dependent regulation of reactive oxygen species (ROS) signaling. we also confirm that deletion of ppe31Mtb in M. tuberculosis H37Rv decreased inflammatory cytokine expressions and reduced cell death. Consistent with these findings, PPE31 not only provides a survival advantage to M. marinum in macrophage, but also, was necessary for intracellular growth of M. tuberculosis. These findings identify PPE31 contributing to host cell death, and provide new insights into a mechanism by which PPE proteins alter host signaling to affect M. tuberculosis pathogenesis.
With the approval from Ethics Committee of Zhongshan School of Medicine on Laboratory Animal Care (reference number: 2016-159), Sun Yat-sen University, all the animal experiments were conducted based on the standard of the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
In order to perform microinjections, borosilicate needles were prepared. The needles were then connected to a Warner PLI-100A pump and handled using a Narishige MN-152 micromanipulator. All the injections were performed on zebrafish larvae previously anaesthetized with tricaine (Finquel; 0.02% in embryo water) and placed on a Petri dish containing a hardened solution of 1% agarose in egg water. M. marinum M or its isogenic strain containing pSMT3-mCherry was grown. The injection of M. marinum was performed at 36 h postfertilization with an injection of 300 colony-forming units (CFU) per embryo in the neural tube. After injection, embryos were transferred into fresh egg water and incubated at 28°C for 4 days before collection. Proper infection was controlled by fluorescent imaging before embryo dissociation.
M. marinum M wild-type strain and the modified vector, pPR27-GFP (ts oriM; sacB counter selection; GFP; GentR) was a gift from Prof. Qian Gao, Fudan University. M. marinum Δppe31Mm and M. marinum comp-Δppe31Mm strains were generated in our laboratory. Δppe31Mm was generated by means of a two-step gene replacement strategy using pPR27-GFP as described previously (Pelicic et al., 1997) (Figure S1). Primer sequences used are listed in Table S1.
For construction of the complementation strains, M. tuberculosis homolog ppe31 Mtb (rv1807) fused with a HA tag was first cloned into a pMV306-hsp (mycobacterial integration vector; integrates into the attB site; KanR; hsp60 promoter), and the recombinant plasmid pMV306-hsp-ppe31Mtb-HA was integrated into the chromosomes of the deletion strains.
To generate the ppe31Mtb knock-out in M. tuberculosis H37Rv strain, Δppe31Mtb was generated by phage specialized transduction as described previously (Jain et al., 2014). The deletion was confirmed by PCR analysis and sequencing (Figure S4).
The CRISPR interference system in M. marinum was constructed as previously described (Singh AK et al., 2016). Briefly, oligos targeted 3 sites of ppe31 gene were designed for the construction of sgRNAs. The oligos were annealed for the generation of double-stranded DNA inserted fragment, and the terminals were phosphorylated by T4 polynucleotide kinase. The inserted fragments were then ligated into BbsI digested pRH2521 to obtain pRH2521-sgRNA for the encoding of sgRNAs. pRH2521 harboring sgRNA targeting at gene of interest was electroporated into M. marinum with the expression of dCas9 encoded by pRH2502. Transformants were screened by hygromycin and kanamycin resistance agar plate.
For M. marinum knockdown strains, to induce the expression of the sgRNA and dCas9, different strains were grown to an OD=0.2, then the anhydrotetracycline (aTc) was added to the final concentration 200 ng/ml every 48h into 7H9-OADC medium containing hygromycin and kanamycin. Once the growth of each strain reached the OD600 = 0.5, the culture was centrifuged and the cell pellet was resuspended in a 7H9 medium (pH= 4.5) for 9 h in the presence or absence of aTc. After the treatment, ten-fold dilutions were spotted onto Middlebrook 7H10 agar containing hygromycin and kanamycin and bacteria numbers were counted after 7-9 days culture.
For M. marinum knockout strains, WT, Δppe31Mm or comp-Δppe31Mm strains were grown into optimal concentration (OD600 = 0.4) in 7H9 medium containing indicated antibiotics. Bacteria were then pelleted, washed three times with 7H9/Tween-80 and cells were resuspended in Middlebrook 7H9 of pH=4.5 for 9h. After treatment, the corresponding strains ten-fold dilutions were spotted onto Middlebrook 7H10 agar, and number bacteria were counted after 7-10 days culture.
For M. tuberculosis knockout strains, Δppe31Mtb or H37Rv strains were grown into optimal concentration (OD600 = 0.4) in 7H9 medium containing indicated antibiotic. Bacteria were then pelleted, washed three times with 7H9/Tween-80 and cells were resuspended in Middlebrook 7H9 of pH= 4.5 or pH= 5.5 for 7 days. After treatment, the corresponding strains ten-fold dilutions were spotted onto Middlebrook 7H10 agar, and bacterial enumeration was performed after 21 days post culture.
Bone marrow was isolated from the femurs and tibiae of 8 to 12-week old mice were used for the preparation of bone marrow derived macrophage (BMDMs). To induce differentiation, the cells were cultivated in DMEM containing 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 µg/ml streptomycin, and 30% L929 conditioned medium. In DMEM supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin, 100 µg/ml streptomycin (GIBCO, Invitrogen). Bone marrow isolated from the femurs and tibiae of 8 to 12-week old mice were used for the preparation of BMDMs. To induce differentiation, the cells were cultivated in DMEM containing 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin, 100 µg/ml streptomycin, and 30% L929 conditioned medium. Non-adherent cells were washed by PBS after 24 h and cultured for 7 days.
BMDMs were grown in DMEM containing 10% FBS and 10% L929 conditioned medium. Cells were allowed to adhere in a 24-well plate for 24 h at 37°C at a density of 3×105 cells/well in 1 ml at 37°C under an atmosphere containing 5% CO2. For infections of Raw264.7 cells and BMDMs, mycobacteria were cultivated at 30°C (for M. marinum) or 37°C (for M. tuberculosis) in 7H9 supplemented with 10%OADC and 0.05% Tween 80 to reach mid-log phase growth. Bacterial culture pellets were re-suspended with 1 ml of PBS. Mycobacterium pellets were homogenized to generate single cell suspension, and the aggregates were removed by a short spin for 1 min at 1200 rpm. The mycobacterium suspensions were diluted by DMEM. BMDMs were then infected at the indicated multiplicity of infection (MOI). BMDMs were incubated for 1h at 30°C with 5% CO2 to induce phagocytosis. Extracellular bacteria were washed out with PBS for three times. To quantify the number of internalized bacteria, BMDMs were incubated for an additional 48 hours under the same condition. At indicated time point, BMDMs were lysed by 0.1% Triton X-100 in PBS and serial dilutions of the lysates were plated on 7H10-OADC. Colony forming units (CFU) were counted after 8–10 days of incubation at 30°C for M. marinum and after 21 days of incubation at 37°C for M. tuberculosis, respectively.
Total RNA was extracted as previously described (Wu et al., 2013). Quantitative real-time PCR was performed in Bio-Rad CFX96 real-time detection system. For mammalian cells, relative mRNA expression levels were calculated by normalization to β-actin. For mycobacterial cells, relative mRNA expression levels were calculated by normalization to sigA.
Cytokine expression was measured in culture supernatants harvested from M. tuberculosis-infected Raw264.7 macrophages at 24 h after infection. TNF and IL-6 levels were measured by ELISA kits (R&D Systems) according to the manufacturer’s instructions.
Cells were washed three times with ice-cold PBS and lysed by lysis buffer containing 1 mM phenylmethylsulfonyl fluoride, 1% (vol/vol) protease inhibitor cocktail (Sigma) and 1 mM DTT, followed by centrifugation at 12,000g for 5 minutes. Equal amounts (20 µg) of cell lysates were loaded for SDS-PAGE and transferred to PVDF membranes. Membranes were blocked by 5% BSA in PBST and incubated overnight with the respective primary antibodies at 4°C. Antibodies against phosphorylated MAPKs (CST) were diluted in 1: 1000 ratio. The membranes were incubated at room temperature for 1 h with appropriate HRP-conjugated secondary antibodies (1: 3000 dilution). The immunoblots were further visualized by reacting with Plus-ECL (PerkinElmer, Shelton, CA) according to the manufacturer’s protocol.
Immunostaining was conducted as previously described (Wang et al., 2013). Briefly, cells were seeded on coverslips, and treated, then fixed, thereafter permeabilized and blocked. Cells were then incubated with primary antibodies at 4°C overnight, and then with secondary antibodies for 1 h at room temperature. Additionally, nuclei were labeled by 4,6-diamidino-2-phenylindole (DAPI). Coverslips were mounted with ProLong Gold anti-fade reagent (Invitrogen), and images were captured by Olympus BX53 fluorescence microscope (Olympus Corporation, Tokyo, Japan).
Macrophages were stained with propidium iodide (PI) (KeyGEN BioTECH) for 10 minutes and sorted by flow cytometry (at least 8,000 cells acquired, BD Accuri C6). For TUNEL staining, cells were fixed by 4% paraformaldehyde overnight prior to staining. Detection of cell death was conducted under manufacturer’s instructions (Roche) and examined by fluorescence microscopy.
Cytosolic lactate dehydrogenase (LDH) release (OD490nm) was measured using CytoTox 96 assay (Promega) to monitor cytotoxicity. Lysing of infected cells with 1% Triton-X enabled maximum LDH release, while supernatants of control cells lysates were assessed for spontaneous LDH release. Measurement of cytotoxicity was carried out by calculating the percentage of LDH release net change as shown in the following formula: (test LDH release - spontaneous release)/(maximal release - spontaneous release) ×100.
The measurement of ROS levels in BMDMs were conducted as previously described (Wang et al., 2014). At the indicated time points after infection, cells were harvested and stained with 10μM CM-H2DCFDA (Invitrogen) for 30 minutes at 37°C in basic DMEM and then washed twice with PBS. Following workflow were analyzed by flow cytometry (at least 8,000 cells acquired, BD Accuri C6).
Statistical analysis was performed using Prism (version 6.0c; GraphPad Software). Statistical significance of paired comparison data was assessed by paired student’s t-test. Statistical significance of data with multiple confounding factors were assessed by analysis of variance (ANOVA). A p value of 0.05 or lower was considered of statistical significance.
According to previous studies, the ppe31 is upregulated in M. tuberculosis during acidic stress (Walters et al., 2006; Rohde et al., 2007; Goodsmith et al., 2015). We sought to probe the phenotype of PPE31 through knock down ppe31Mm gene in M. marinum by means of CRISPR interference (CRISPRi) (Singh AK et al., 2016). To do this, silencing efficiency was measured by real-time PCR and showed ~80% decreased expression of ppe31Mm (Figure 1A). We found that the decreased ppe31Mm expression significantly reduced the survival of M. marinum in 7H9 medium of low pH compared to the strain without induction of aTc (control) (Figure 1B). To further explore the function of ppe31Mm, we constructed a ppe31Mm deletion mutant (Δppe31Mm) in M. marinum by substituting the ppe31Mm gene with a cassette coding for hygromycin resistance (Figure S1) and used this strain to investigate the impact of the PPE31 on its survivability in acid medium, and we also constructed a complementation strain comp-Δppe31Mm, which integrated M. tuberculosis ppe31Mtb fused with a HA tag sequence into the chromosomes of M. marinum Δppe31Mm strain. WT, Δppe31Mm or comp-Δppe31Mm were cultured in 7H9 of pH4.5, the result showed that when compared to WT or comp-Δppe31Mm, M. marinum lack of ppe31Mm significantly reduced the resistance to acid medium (Figure 1C).
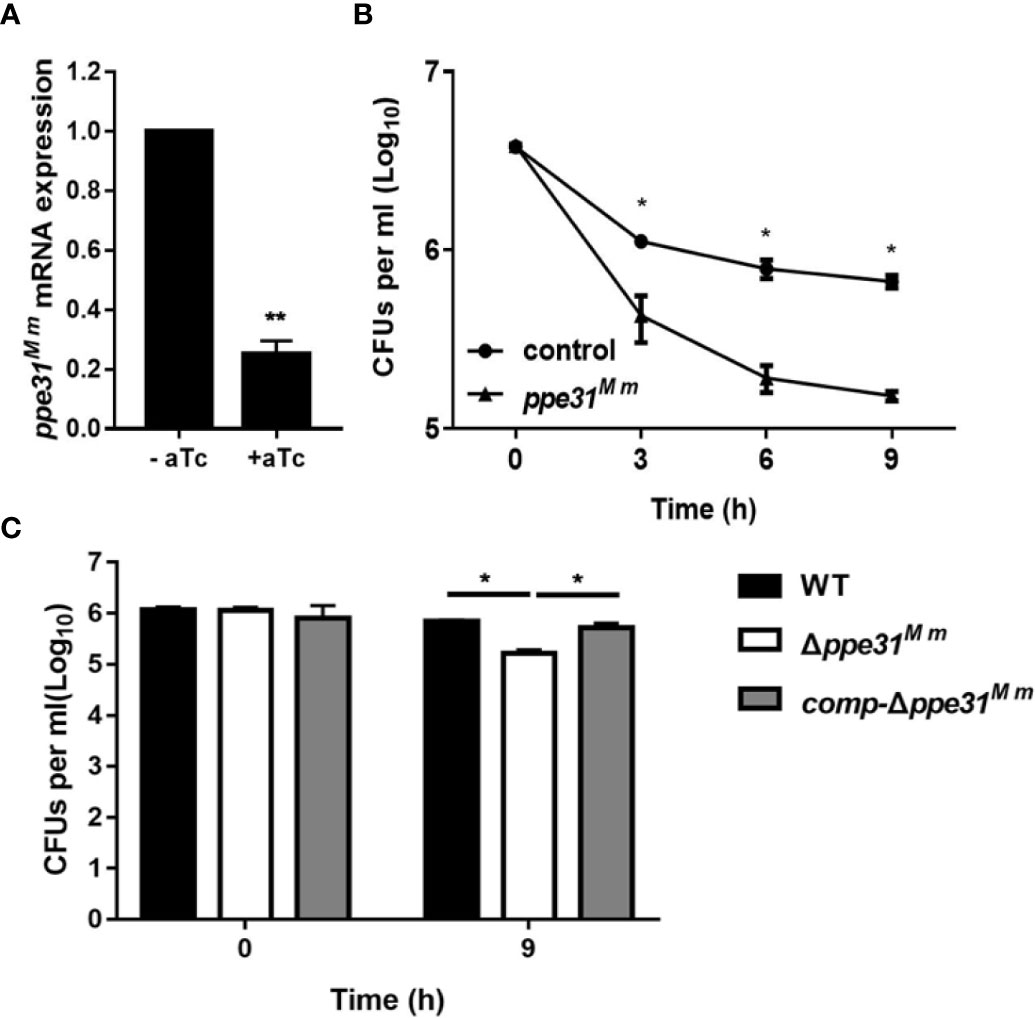
Figure 1 PPE31Mm was required for the resistance of M. marinum to acid medium in vitro. (A) CRISPR interference has been performed for knockdown of ppe31Mm. Expression of ppe31Mm determined by RT-qPCR with or without induction of aTc. (B) M. marinum contain dCas9 and sgRNA targeting ppe31Mm were exposed to 7H9 medium of pH4.5 with or without (control) induction of aTc. The bacterial survival was monitored by CFU counting at the indicated time. (C) WT, Δppe31Mm, or comp-Δppe31Mm were exposed to 7H9 medium of pH4.5 at 30°C. The bacterial survival was monitored by CFU counting for 9h. Data are shown as mean ± S.E.M. of three independent experiments. *p < 0.05, **p < 0.01.
PE/PPE family proteins, which are mainly located in the cell surface, interact with innate receptors in the phagocytes to modulate inflammatory mediators such as IL-6, IL-12p40 and TNF, and the generation of reactive oxygen species (ROS) (Cadieux et al., 2011; Deng et al., 2015; Deng et al., 2016). The expression of proinflammatory cytokines were examined and TNF and IL-6 mRNA expression was decreased in the BMDMs infected with Δppe31Mm strain compared to WT or comp-Δppe31Mm although all strains showed increased cytokine production with increased bacterial inocula (Figures 2A, B). These findings suggest a regulatory role for ppe31Mm in proinflammatory cytokine expression in macrophages infected with M. marinum.
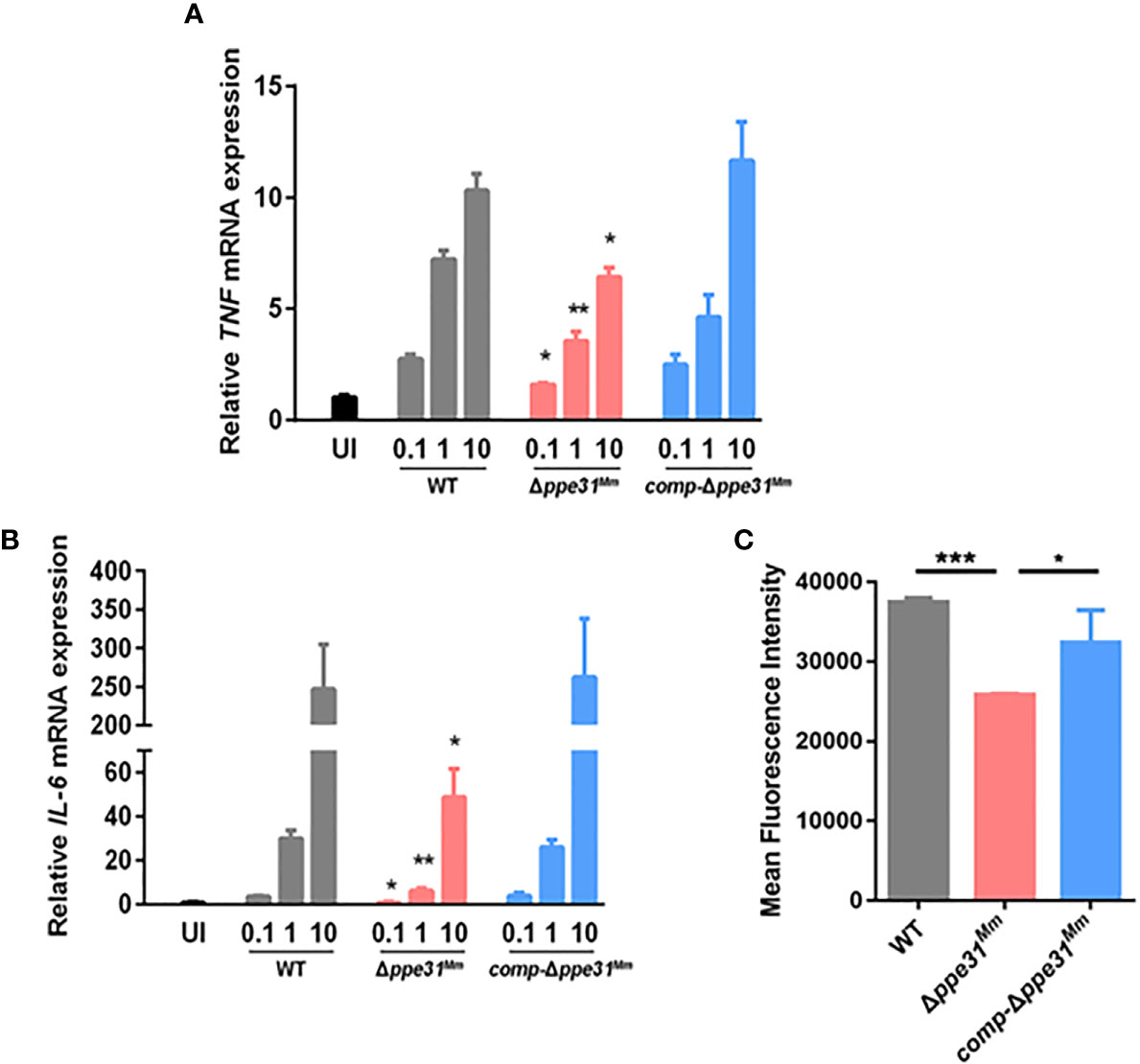
Figure 2 Δppe31Mm infection fails to partly induce the expression of proinflammatory cytokines and ROS generation in BMDMs. BMDMs were infected with WT, Δppe31Mm, or comp-Δppe31Mm at different MOIs (0.1, 1 and 10) for 6 h. The level expressions of inflammatory cytokines, TNF (A) and IL-6 (B) were assessed by RT-qPCR. (C) BMDMs were stimulated with WT, Δppe31Mm or comp-Δppe31Mm for 30 min. Cells were then labeled with DCFH-DA that were used detecting cytosolic ROS, and were analyzed for cytosolic ROS levels using flow cytometry. Data are shown as mean ± S.E.M. of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. WT, wild-type M. marinum; UI, uninfected.
Since proinflammatory cytokine production was strongly related with ROS generation, our hypothesis is that ROS levels differed between cells infected with WT, comp-Δppe31Mm and Δppe31Mm, strains of M. marinum. The production of ROS was measured by flow cytometry, using 2,7’-dichlorofluorescein-diacetate (DCFH-DA). Compared with BMDMs infected with WT or comp-Δppe31Mm strains, cells infected with Δppe31Mm displayed significantly decreased of intracellular DCFH-DA fluorescence (Figure 2A). Additionally, we explored whether there is a relationship between ROS generation and the inflammatory response. When we inhibited the generation of ROS by using DPI, a reactive oxygen species inhibitor, we observed that the expression of inflammatory cytokines was significantly reduced (Figure S2). Altogether, these data suggest that PPE31Mm is involved in the modulation of inflammatory mediators in BMDMs.
During infection, we observed that at 24h post-infection, morphological changes associated with cell death in WT or comp-Δppe31Mm-infected cells, but not in Δppe31Mm (Figure 3A), indicating that wild-type M. marinum disrupted the integrity of cell membrane, a process that is critical for necrosis. We next examined the cytotoxicity of Δppe31Mm, after infection with these three strains at an MOI of 10, a significant decrease in cell cytotoxicity at 24h and 36h post-infection was observed in Δppe31Mm-infected BMDMs, whereas WT and comp-Δppe31Mm-infected cells displayed higher cytotoxicity (Figure 3B). To determine whether PPE31 can regulate host cell death in macrophages, we further assessed whether necrosis played a role in cell death induced by M. marinum. Using propidium iodide (PI) staining, we found that ppe31Mm deficiency causes less cell death of infected BMDM cells at two different MOI (Figure 3C). To further examine the mechanism by which Δppe31Mm-infected cells induce cell death, we cultured BMDM cells infected with WT or Δppe31Mm (MOI = 10) in the presence or absence of the caspase inhibitor Z-VAD-FMK. We found that this inhibitor could not block WT-mediated cell death (Figure 3D). Collectively, these results suggest that Δppe31Mm is associated with decreased cell cytotoxicity and reduced caspase-independent cell death.
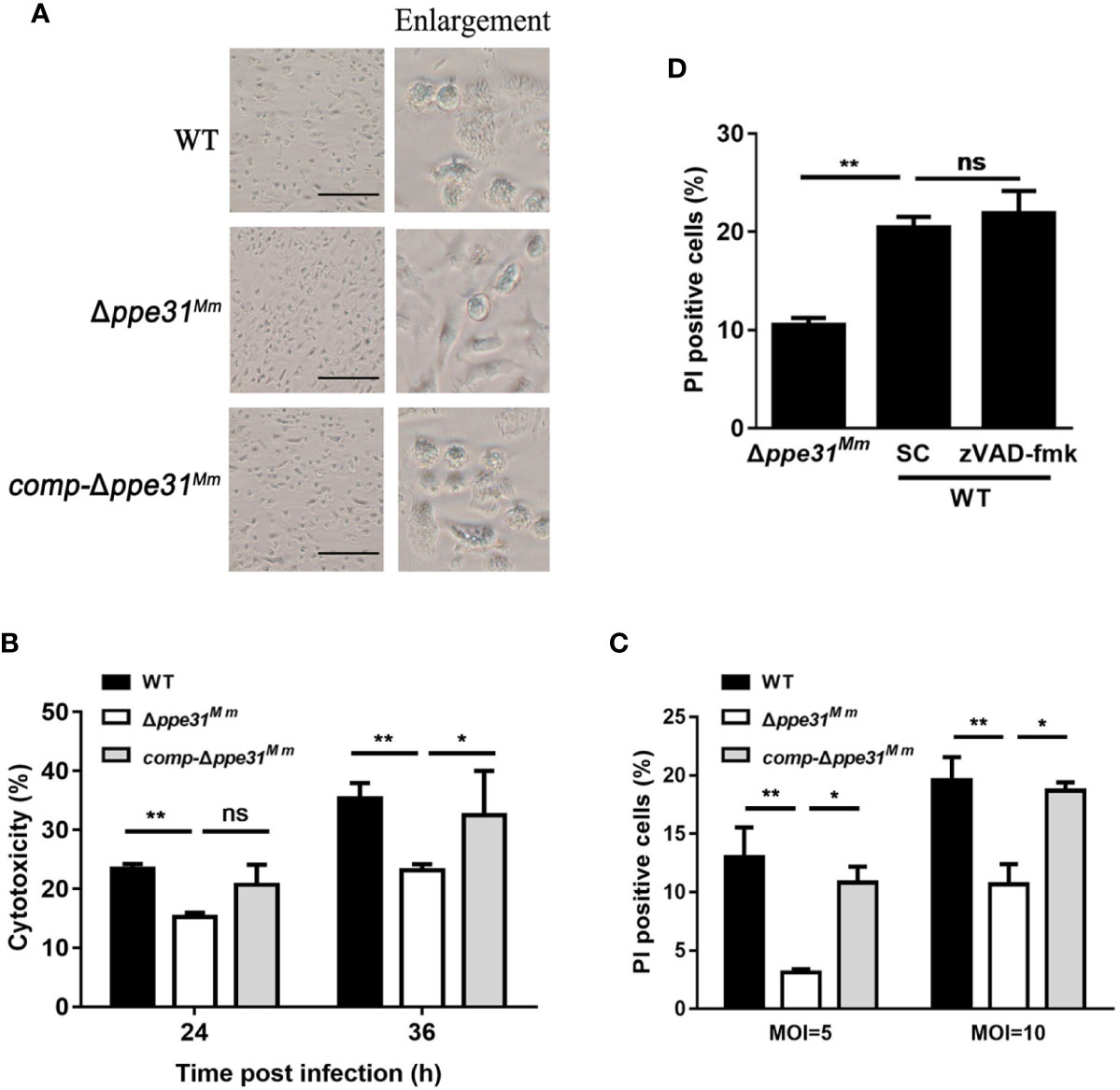
Figure 3 Macrophages infected with Δppe31Mm show reduced caspase- independent cell death. (A) BMDMs were infected with WT, Δppe31Mm, or comp-Δppe31Mm (MOI = 10) for 24h for analysis of morphological changes. Scale bar in 100 μm. (B) BMDMs were infected with WT, Δppe31Mm, or comp-Δppe31Mm (MOI = 10). The cytotoxicity was assessed by LDH release assay at indicated times. (C) BMDMs were infected with either WT, Δppe31Mm, or comp-Δppe31Mm (MOI = 10 or 5) for 24 h, and cell death was detected by PI staining and then examined by flow cytometry. (D) BMDMs were infected with WT, Δppe31Mm or comp-Δppe31Mm (MOI = 10) in the presence or absence of Z-VAD-FMK (20 μM), a caspase inhibitor, then cells were stained with PI and then examined by flow cytometry. Data are shown as mean ± S.E.M. of three independent experiments. *p < 0.05, **p < 0.01. WT, wild-type M. marinum; UI, uninfected; SC, solvent control (0.1% DMSO); ns, no significant.
MAPK signaling pathways are critical in oxidative stress-mediated cell death during mycobacterial infection (Kim et al., 2012). To investigate whether MAPK signaling pathways are involved in PPE31Mm-mediated cell death, BMDM were infected with WT, comp-Δppe31Mm or Δppe31Mm strains. We found the activation kinetics of phosphorylated p38 and ERK1/2 between cells infected with these three strains was no significant difference. On the contrary, a decrease in JNK/SAPK phosphorylation was observed in cells infected with M. marinum Δppe31Mm when compared with cells infected with WT or comp-Δppe31Mm (Figure 4A). These results indicate that JNK signaling may be involved in PPE31Mm mediated cell death.
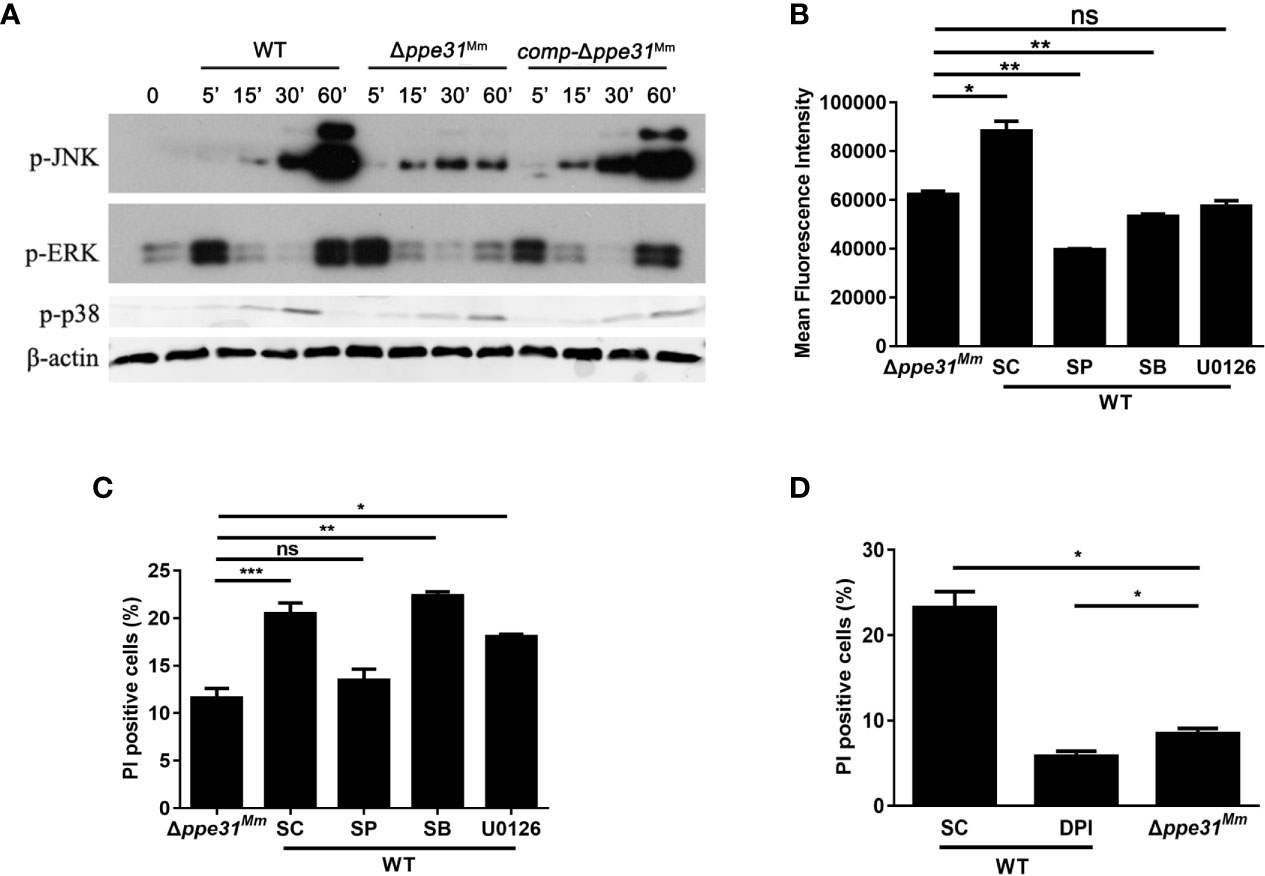
Figure 4 Infection with Δppe31Mm reduces cell death through JNK-dependent signaling. (A) BMDMs were infected with WT, Δppe31Mm or comp-Δppe31Mm (MOI = 10) for the indicated periods of time, and then subjected to Western blot analysis using antibodies raised to p-ERK1/2, p-p38, p-JNK, and β-actin. (B) BMDMs were pretreated with the following MAPK signaling pathways inhibitors U0126 (20 μM), SB203580 (SB; 10 μM), or SP600125 (SP; 20 μM) for 1h, and then infected with WT, Δppe31Mm or comp-Δppe31Mm for 30 min, Cells were then incubated with DCFH-DA and analyzed immediately for ROS generation by flow cytometry. (C) BMDMs were pretreated with U0126 (20 μM), SB (10 μM), or SP (20 μM) for 1h, and then infected with WT, Δppe31Mm or comp-Δppe31Mm for 24h, cells were stained with PI and then examined by flow cytometry. (D) BMDMs were infected with WT, Δppe31Mm or comp-Δppe31Mm (MOI = 10) in the presence or absence of DPI (10 μM). After 24h, cells were then stained with PI and analyzed immediately for cell death using flow cytometry. Data are shown as mean ± S.E.M. of three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001. WT, wild-type; M, marinum; UI, uninfected; SC, solvent control (0.1% DMSO); ns, no significant.
To investigate the relationship between MAPK signaling pathway and PPE31Mm-mediated ROS generation and cell death, BMDMs were pretreated with specific inhibitors of JNK, p38, and MEK for 1 h prior to infection with WT or Δppe31Mm. All three inhibitors significantly decreased ROS generation, indicating that all three kinases in MAPK signaling pathways are involved in the regulation of ROS generation in response to mycobacterial infection (Figure 4B). In addition, inhibition of JNK and ERK signaling pathway, but not p38, reduced macrophage death infected with WT, and JNK signaling was dominant in PPE31Mm-mediated cell death (Figure 4C). We also examined the link between MAPK signaling pathways and the mRNA of inflammatory cytokines. Inhibition of all three kinases in MAPK signaling pathway, reduced PPE31Mm-induced mRNA of TNF and IL-6 in a dose-dependent manner (Figure S3).
Furthermore, we examined the relationship between ROS generation and macrophage survival. BMDMs were infected WT, comp-Δppe31Mm or Δppe31Mm, for 24 h in the presence or absence of DPI. Inhibition of ROS generation significantly decreased PPE31Mm-induced cell death (Figure 4D). Together, these data suggest that PPE31Mm modulates macrophage survival and inflammatory response in M. marinum infected cells through JNK-dependent regulation of ROS signaling.
Given the observation that M. marinum PPE31 contributed to control host immune response, we speculated that M. tuberculosis PPE31 may exhibit similar phenotype and functions. To do it, Δppe31Mtb was generated in M. tuberculosis H37Rv strain by recombineering (Figure S4). Next, M. tuberculosis H37Rv or Δppe31Mtb was cultured in 7H9 containing different gradient pH for 7 days. When compared to H37Rv, we found that, the survival rate of Δppe31Mtb was significantly decreased (Figure S5C). Additionally, assessment of transcript levels for TNF and IL-6 in infected macrophages showed that the expression of TNF and IL-6 was significantly decreased in Δppe31Mtb infected macrophages when compared to wild-type strains (Figures 5A, B and Figures S5A, B). Furthermore, RAW264.7 cells were infected with H37Rv and Δppe31Mtb and stained for genomic DNA fragmentation using the TUNEL assay. We found that ppe31Mtb deficiency in M. tuberculosis reduced host cell death during infection (Figure 5D). Notably, a decrease in cleaved PARP was observed in macrophages infected with Δppe31Mtb when compared with cells infected with H37Rv (Figure 5C).These results indicate that PPE31Mtb was also important and indispensable to regulate innate immune responses to M. tuberculosis infection.
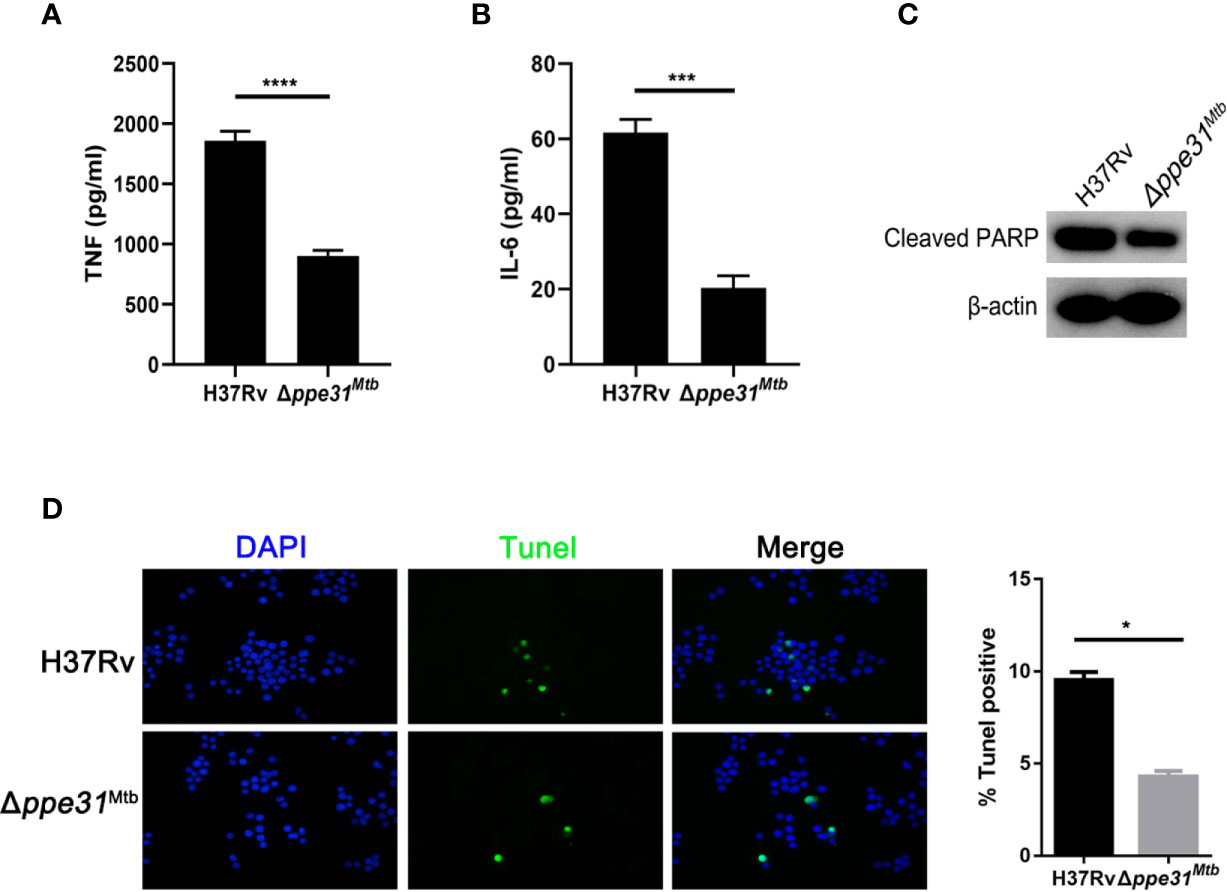
Figure 5 Mutants for ppe31Mtb decreases the inflammatory cytokine expressions and reduces host cell death. RAW264.7 cells were infected with H37Rv or Δppe31Mtb (MOIs=10) for 24 h. The supernatants were collected, sterile-filtered, and assayed for TNF (A) and IL-6 (B). (C) RAW264.7 were infected with H37Rv or Δppe31Mtb (MOI = 10) for 24h, and then subjected to Western blot analysis using antibodies raised to cleaved PARP and β-actin. (D) RAW264.7 were infected with H37Rv or Δppe31Mtb (MOI = 10) for 24h, and cell death was detected by TUNEL staining and then examined by confocal microscope. Data are shown as mean ± S.E.M. of three independent experiments. *p<0.05, ***p<0.001, ****p<0.00001. UI, uninfected; M, marker.
Based on the above data, we hypothesize that PPE31 is essential for mycobacteria survival in macrophage. To confirm this, RAW264.7 cells were infected with WT, Δppe31Mm or comp-Δppe31Mm strains of M. marinum and the surviving intracellular bacteria were assessed at 2, 4, 8, 24, and 48h after infection. We found deletion of ppe31Mm reduced the number of surviving mycobacteria at 48h in macrophages (Figure 6A). In addition, survival of H37Rv and Δppe31Mtb were assessed by CFU in macrophages. We found that ppe31Mtb deficiency significantly decreased its survival in macrophages when compared to H37Rv (Figure 6B), these results were consistent with the earlier observation in mice (Sassetti and Rubin, 2003). Furthermore, zebrafish larvae were infected with the WT or Δppe31Mm strains. A significant reduction in the virulence of Δppe31Mm was observed when compared with WT (Figure 6C).
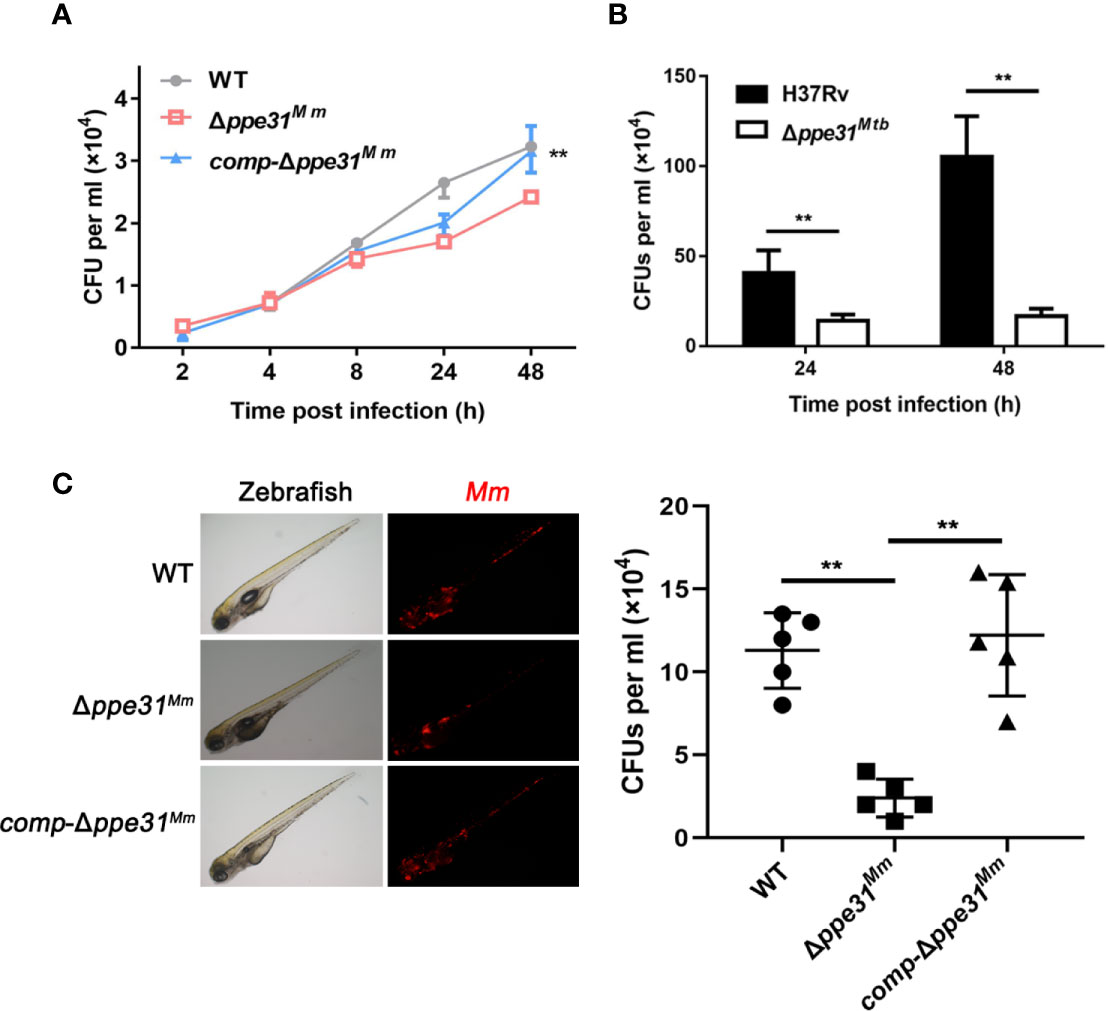
Figure 6 PPE31 promotes mycobacteria survival in macrophage and in zebrafish. (A) WT, Δppe31Mm or comp-Δppe31Mm-infected BMDMs were analyzed for number of surviving internalized bacilli by CFU counting at the indicated time. (B) H37Rv or Δppe31Mtb-infected RAW264.7 cells were analyzed for number of surviving internalized bacilli by CFU counting at the indicated time. (C) Bacterial survival advantage in the infected zebrafish. Three hundred CFU of M. marinum strains were injected to reach a comparable infection level at 4 dpi. Representative burden pictures and representative burden analysis derive from larvae collected at 4 dpi. Each point in represents 1 infected larva from a representative pool. Scale bar in 200 μm. Data are shown as mean ± S.E.M. of three independent experiments. **p < 0.01. WT, wild-type M. marinum; UI, uninfected.
Overall, these results demonstrated that PPE31 enhances the intracellular survivability of mycobacteria.
M. marinum is genetically related to M. tuberculosis (Tobin and Ramakrishnan, 2008). Both of them share the virulence related Type VII secretion systems ESX-1 and ESX-5 (Thi et al., 2013; Singh VK et al., 2016). ESX-1 secretion systems of pathogenic mycobacteria secrete the effector proteins, including ESAT-6, which are required for the disruption of the membrane integrity of the mycobacteria containing phagosome (Conrad et al., 2017; Augenstreich et al., 2017). This was followed by the translocation of mycobacteria to the cytosol of host cells (Simeone et al., 2012). Although disruption of ESX-5 has no effect on the translocation of mycobacteria from phagolysosome to cytosol, the effector proteins secreted by ESX-5 also induce a caspase-independent cell death after translocation has taken place (Abdallah et al., 2011). Intriguingly, accumulated evidences have shown that the ESX-5 substrates, PE/PPE family proteins, are involved in the evasion of host cell immune responses (Saini et al., 2016; Camassa et al., 2017).
The important environment factor of mycobacteria containing phagosome is the pH value. During M. tuberculosis infection, the pH value is ranging from 6.2 within the phagosome of immature macrophage to 4.5 in the phagolysosome following INF-γ activation (Machado et al., 2019). Here, we first demonstrated that PPE31 is required for survival under acidic condition in vitro. It is not surprising that ppe31Mtb partly complemented the phenotype of M. marinum lack of ppe31Mm, due to the following factors: (i) PPE31 of M. marinum is 71% identical to PPE31 of M. tuberculosis, (ii) M. marinum is closely related to M. tuberculosis in its pathogenicity as well as genetically. A previous study reported that PPE38 shared 73% identity in amino acid sequence to MMAR_3661, complemented the phenotype of ΔMMAR_3661 (Dong et al., 2012).
Next, we examined the role of PPE31Mm in immune regulation. We showed that a M. marinum Δppe31Mm provoked reduced expression of inflammatory cytokine and ROS generation during infection of phagocytic cells. Recent reports have emphasized that several PE/PPE family proteins are potent inducers of cytokines, including TNF and IL-6. For example, PE11-transformed M. smegmatis-infected mice induce high levels of TNF and necrotic cell death of macrophage (Deng et al., 2015; Singh P et al., 2016). TNF constitutes a critical host defense against tuberculosis, although excessive of TNF induces necrosis via mitochondrial reactive oxygen species in macrophages during M. marinum infection (Roca and Ramakrishnan, 2013). Based on the decreased cytokine production in the Δppe31Mm strain, we hypothesized that PPE31 may affect the survival of M. marinum-infected cells. Consistent with our hypothesis, we observed prominent signs of necrosis in macrophages infected with WT strain, such as lytic plasma membranes and lactate dehydrogenase release, but these were not seen in cells infected with a Δppe31Mm strain. Interestingly, this type of cell death was not inhibited by the inhibitor of caspase. These results suggested that PPE31 induced caspase-independent cell death during M. marinum infection.
Recent studies have shown that MAPK signaling pathways, especially JNK signaling, contribute to the induction of cell death by M. tuberculosis. Based on this, and given the closely relationship between ROS and cell death, our data comparing wild-type to Δppe31Mm show that JNK signaling is involved in macrophage necrosis in cells infected with M. marinum. M. tuberculosis Eis protein suppresses host immune defenses by negatively regulating cell death through JNK-dependent inhibition of ROS generation (Kim et al., 2012). Moreover, the Streptococcus pyogenes Toxin triggers a form of programmed necrosis dependent on JNK signaling in Streptococcus pyogenes infected cells (Chandrasekaran and Caparon, 2016). Our data demonstrated that phosphorylated JNK was also involved in PPE31Mm-induced ROS generation and cell death. Previous work has implicated mycobacterial factors in programmed necrosis via mitochondrial ROS (Miller et al., 2010; Mohanty et al., 2016). Our results demonstrate that DPI, an NADPH oxidase inhibitor, significantly decreased PPE31Mm-induced cell death. This supports the idea that phosphorylated JNK increases oxidative stress and contributes to the induction of host cell death during M. tuberculosis infection (Shin et al., 2010; Chandrasekaran and Caparon, 2016).
To strengthen the proposition that PPE31 is a virulence factor that modulates innate immune responses during mycobacterial infection, M. tuberculosis ppe31Mtb mutant was constructed by specialized-phage transduction. Given the relationship between M. marinum and M. tuberculosis genetically, our data reported that ppe31Mtb is also required for M. tuberculosis survival in acid medium. However, we did not observe macrophages undergoing a lytic death at early stages of infection. In contrast, we observed a decreased cleaved PARP, a maker of apoptosis, in macrophage infected with Δpppe31Mtb. Since several studies described that apoptosis is induced exclusively by virulent M. tuberculosis strains in marine macrophage (Derrick and Morris, 2007; Aguilo et al., 2014), indicating that the mechanism by which M. tuberculosis induces host cell death is different from M. marinum at early stage of infection. It should be noted that a delayed phagosomal rupture caused by M. tuberculosis infection was observed after 48 hours, but M. marinum escaped from phagosome within 2–4 hours (Abdallah et al., 2011).
It has been reported that PPE proteins form a heterodimer with PE proteins, and that secretion of PPE protein by ESX-5 is dependent on PE protein (Tundup et al., 2008; Tiwari et al., 2014; Tiwari et al., 2015; Chen et al., 2017), such as PE25/PPE41. Many different groups have shown that the expression of pe20 and ppe31 in M. tuberculosis are significantly increased under adverse stress (Walters et al., 2006; Rohde et al., 2007; Provvedi et al., 2009; Liu et al., 2016), indicating a close relationship between PE20 and PPE31. However, PE20 or its homologue gene is absent in M. marinum. Therefore, it is possible that PE20 is dispensable for mycobacterial virulence, at least in M. marinum. On the other hand, the substrate of ESX-5 was involved in the secretion phenotype of PE_PGRS proteins in M. marinum. Very recently, a study by Ates and colleagues reported that the secretion of ESX-5 substrates, including PPE-MPTR and PE_PGRS, was completely inhibited by genetic disruption of ppe38 and this phenotype has been already confirmed both in M. marinum and M. tuberculosis (Ates et al., 2018). In the light of these results linking PPE31 to ESX-5, it is plausible that PPE31, which might be transported by ESX-5, partly increased the cell envelope integrity in M. tuberculosis. Further studies will be necessary to address the relationship between PPE31 and ESX-5.
Finally, we considered the role of PPE31 in intracellular survival of bacteria. Transposon mutagenesis previously showed that M. tuberculosis ppe31::tn caused significant attenuation in mice (Sassetti and Rubin, 2003). Our results showing shortened survival in macrophages of M. marinum ppe31Mm are consistent with this M. tuberculosis data in vivo. Moreover, M. tuberculosis Δppe31Mtb were also attenuated in macrophages. Several mechanisms could be involved in this observation; Firstly, TNF excess induced host cell necrosis is detrimental to bacterial clearance. Secondly, phagosome maturation is a vital strategy to destroy engulfed invading microorganisms. Finally, mycobacteria escape from the phagosome to access the cytosol. Then cytosolic mycobacteria are able to replicate and cause death. This suggests that an important role of PPE31 in mycobacterial survival during infection.
In summary, our results identify mycobacterial PPE31 as an important factor contributing to the modulation of host cell immune response. Our data show that this cell envelope localized protein modulates JNK signaling pathways in host cells to alter cytokine profiles and facilitate mycobacterial survival. Additional mechanisms by which PPE31 mediates the host-pathogen interaction will require further investigation.
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding authors.
The animal study was reviewed and approved by Ethics Committee of Zhongshan School of Medicine on Laboratory Animal Care (reference number: 2016-159), Sun Yat-sen University.
XH and HZ conceived the study and supervised global data analysis. SF, ZH, and GZ designed and performed experiments, analyzed data, and co-wrote paper. ZH and GZ performed construction of deletion of ppe31 gene in mycobacteria. JL performed the intracellular fitness experiment. G-BT contributed to data interpretation and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Science and Technology Key Projects for Major Infectious Diseases (2017ZX10302301-002), National Natural Science Foundation of China (31470877), Development Project of Foshan Fourth People’s Hospital (FSSYKF-2020003 and FSSYKF-2020017), and Guangzhou Science and Technology Planning Project (201704020226 and 201604020006).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2021.629836/full#supplementary-material
Supplementary Figure 1 | Construction of the ppe31Mm mutant. Schematic representation of the strategy used for the generation of ppe31Mm gene replacement mutant (A). Confirmation of ppe31 Mm disruption in mutant. PCR amplifications were performed using specific primers. The result shows the PCR amplification gives a product of 2.2kb for wildtype M. marinum and 1kb for Δppe31Mm (B).
Supplementary Figure 2 | PPE31 significantly increased the expression of inflammatory cytokines via reactive oxygen species in BMDMs. BMDMs were infected with WT, Δppe31Mm or comp-Δppe31Mm in the presence or absence in DPI (10μM). After 6h, the level of inflammatory cytokines was assessed by RT-qPCR for expressions of TNF-α and IL-6. WT, wild-type M. marinum, *p < 0.05, **p < 0.01. Data are shown as mean ± S.E.M. of three independent experiments. *p < 0.05, **p < 0.01.
Supplementary Figure 3 | PPE31 significantly increased the expression of inflammatory cytokines through JNK-dependent signaling. BMDMs were pretreated with U0126 (5, 10, 20 mM), SB203580 (SB; 1, 5, 10 mM), or SP600125 (SP; 5, 10, 20 mM) for 1h, and then infected with Δppe31Mm for1h, washed three times by PBS. After 6h, the level of inflammatory cytokines was assessed by real time PCR for expressions of TNF-α and IL-6.
Supplementary Figure 4 | Generation of ppe31Mtb gene replacement mutant. (A) Generation of M. tuberculosis ppe31Mtb gene replacement mutant. Schematic representation of the strategy used for the generation of ppe31Mtb gene replacement mutant. (B) Confirmation of ppe31Mtb disruption in mutant. PCR amplifications were performed using specific primers, using genomic DNA of H37Rv and Δppe31Mtb mutant. The result shows the PCR amplification with Rv ppe31-LL and Rv ppe31-RR pair, which gives a product of 3.2kb for H37Rv and 5.6kb for Δppe31.
Supplementary Figure 5 | RAW264.7 cells were infected with H37Rv or Δppe31Mtb (MOIs=10) for 6 h. The level expressions of inflammatory cytokines, TNF (A) and IL-6 (B) were assessed by RT-qPCR. The sensitivity of M. tuberculosis strains to acid condition. The bacteria grown to mid-log phase were collected by centrifugation and resuspended to the OD600 of 0.5 in 5 ml 7H9 (pH = 4.5 or 5.5). The results are percent survival post treatment with 7H9 medium (pH =4.5 or 5.5) for 7d (C). Data are shown as mean ± S.E.M. of three independent experiments. *p<0.05. UI, uninfected; M, marker.
Abdallah A. M., Bestebroer J., Savage N. D., de Punder K., van Zon M., Wilson L., et al. (2011). Mycobacterial secretion systems ESX-1 and ESX-5 play distinct roles in host cell death and inflammasome activation. J. Immunol. 187 (9), 4744–4753. doi: 10.4049/jimmunol.1101457
Aguilo N., Uranga S., Marinova D., Martin C., Pardo J. (2014). Bim is a crucial regulator of apoptosis induced by Mycobacterium tuberculosis. Cell Death Dis. 5, e1343. doi: 10.1038/cddis.2014.313
Ates L. S., Dippenaar A., Ummels R., Piersma S. R., van der Woude A. D., van der Kuij K., et al. (2018). Mutations in ppe38 block PE_PGRS secretion and increase virulence of Mycobacterium tuberculosis. Nat. Microbiol. 3 (2), 181–188. doi: 10.1038/s41564-017-0090-6
Augenstreich J., Arbues A., Simeone R., Haanappel E., Wegener A., Sayes F., et al. (2017). ESX-1 and phthiocerol dimycocerosates of Mycobacterium tuberculosis act in concert to cause phagosomal rupture and host cell apoptosis. Cell Microbiol. 19 (7). doi: 10.1111/cmi.12726
Behar S. M., Divangahi M., Remold H. G. (2010). Evasion of innate immunity by Mycobacterium tuberculosis: is death an exit strategy? Nat. Rev. Microbiol. 8 (9), 668–674. doi: 10.1038/nrmicro2387
Cadieux N., Parra M., Cohen H., Maric D., Morris S. L., Brennan M. J. (2011). Induction of cell death after localization to the host cell mitochondria by the Mycobacterium tuberculosis PE_PGRS33 protein. Microbiology 157 (Pt 3), 793–804. doi: 10.1099/mic.0.041996-0
Camassa S., Palucci I., Iantomasi R., Cubeddu T., Minerva M., De Maio F., et al. (2017). Impact of pe_pgrs33 gene polymorphisms on mycobacterium tuberculosis infection and pathogenesis. Front. Cell Infect. Microbiol. 7, 137. doi: 10.3389/fcimb.2017.00137
Chandrasekaran S., Caparon M. G. (2016). The NADase-negative variant of the Streptococcus pyogenes toxin NAD(+) glycohydrolase induces JNK1-mediated programmed cellular necrosis. MBio 7 (1), e02215–e02215. doi: 10.1128/mBio.02215-15
Chen X., Cheng H. F., Zhou J., Chan C. Y., Lau K. F., Tsui S. K., et al. (2017). Structural basis of the PE-PPE protein interaction in Mycobacterium tuberculosis. J. Bio Chem. 292 (41), 16880–16890. doi: 10.1074/jbc.M117.802645
Conrad W. H., Osman M. M., Shanahan J. K., Chu F., Takaki K. K., Cameron J., et al. (2017). Mycobacterial ESX-1 secretion system mediates host cell lysis through bacterium contact-dependent gross membrane disruptions. Proc. Natl. Acad. Sci. U. S. A. 114 (6), 1371–1376. doi: 10.1073/pnas.1620133114
Deng W., Zeng J., Xiang X., Li P., Xie J. (2015). PE11 (Rv1169c) selectively alters fatty acid components of Mycobacterium smegmatis and host cell interleukin-6 level accompanied with cell death. Front. Microbiol. 6, 613. doi: 10.3389/fmicb.2015.00613
Deng W., Yang W., Zeng J., Abdalla A. E., Xie J. (2016). Mycobacterium tuberculosis PPE32 promotes cytokines production and host cell apoptosis through caspase cascade accompanying with enhanced ER stress response. Oncotarget 7 (41), 67347–67359. doi: 10.18632/oncotarget.12030
Derrick S. C., Morris S. L. (2007). The ESAT6 protein of Mycobacterium tuberculosis induces apoptosis of macrophages by activating caspase expression. Cell Microbiol. 9 (6), 1547–1555. doi: 10.1111/j.1462-5822.2007.00892.x
Dong D. D., Wang D. C., Li M., Wang H., Yu J., Wang C., et al. (2012). PPE38 Modulates the Innate Immune Response and Is Required for Mycobacterium marinum Virulence. Infect. Immun. 80 (1), 43–54. doi: 10.1128/Iai.05249-11
Goodsmith N., Guo X. V., Vandal O. H., Vaubourgeix J., Wang R., Botella H., et al. (2015). Disruption of an M. tuberculosis membrane protein causes a magnesium-dependent cell division defect and failure to persist in mice. PloS Pathog. 11 (2), e1004645. doi: 10.1371/journal.ppat.1004645
Houben E. N., Nguyen L., Pieters J. (2006). Interaction of pathogenic mycobacteria with the host immune system. Curr. Opin. Microbiol. 9 (1), 76–85. doi: 10.1016/j.mib.2005.12.014
Houben D., Demangel C., van Ingen J., Perez J., Baldeon L., Abdallah A. M., et al. (2012). ESX-1-mediated translocation to the cytosol controls virulence of mycobacteria. Cell Microbiol. 14 (8), 1287–1298. doi: 10.1111/j.1462-5822.2012.01799.x
Jain P., Hsu T., Arai M., Biermann K., Thaler D. S., Nguyen A., et al. (2014). Specialized transduction designed for precise high-throughput unmarked deletions in Mycobacterium tuberculosis. MBio 5 (3), e01245–e01214. doi: 10.1128/mBio.01245-14
Kim K. H., An D. R., Song J., Yoon J. Y., Kim H. S., Yoon H. J., et al. (2012). Mycobacterium tuberculosis Eis protein initiates suppression of host immune responses by acetylation of DUSP16/MKP-7. Proc. Natl. Acad. Sci. U. S. A. 109 (20), 7729–7734. doi: 10.1073/pnas.1120251109
Liu Y., Tan S., Huang L., Abramovitch R. B., Rohde K. H., Zimmerman M. D., et al. (2016). Immune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J. Exp. Med. 213 (5), 809–825. doi: 10.1084/jem.20151248
Machado P., Bizarro C. V., Basso L. A. (2019). Resistance reversed in KatG mutants of Mycobacterium tuberculosis. Trends Microbiol. 27 (8), 655–656. doi: 10.1016/j.tim.2019.05.008
Meng L., Tong J., Wang H., Tao C., Wang Q., Niu C., et al. (2017). PPE38 protein of Mycobacterium tuberculosis inhibits macrophage MHC class I expression and dampens CD8(+) T cell responses. Front. Cell Infect. Microbiol. 7, 68. doi: 10.3389/fcimb.2017.00068
Miller J. L., Velmurugan K., Cowan M. J., Briken V. (2010). The type I NADH dehydrogenase of Mycobacterium tuberculosis counters phagosomal NOX2 activity to inhibit TNF-alpha-mediated host cell apoptosis. PloS Pathog. 6 (4), e1000864. doi: 10.1371/journal.ppat.1000864
Mohanty S., Dal Molin M., Ganguli G., Padhi A., Jena P., Selchow P., et al. (2016). Mycobacterium tuberculosis EsxO (Rv2346c) promotes bacillary survival by inducing oxidative stress mediated genomic instability in macrophages. Tuberculosis (Edinb.) 96, 44–57. doi: 10.1016/j.tube.2015.11.006
Pelicic V., Jackson M., Reyrat J. M., Jacobs W. R. Jr., Gicquel B., Guilhot C. (1997). Efficient allelic exchange and transposon mutagenesis in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 94 (20), 10955–10960. doi: 10.1073/pnas.94.20.10955
Pieters J. (2008). Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe 3 (6), 399–407. doi: 10.1016/j.chom.2008.05.006
Provvedi R., Boldrin F., Falciani F., Palu G., Manganelli R. (2009). Global transcriptional response to vancomycin in Mycobacterium tuberculosis. Microbiology 155 (Pt 4), 1093–1102. doi: 10.1099/mic.0.024802-0
Roca F. J., Ramakrishnan L. (2013). TNF dually mediates resistance and susceptibility to mycobacteria via mitochondrial reactive oxygen species. Cell 153 (3), 521–534. doi: 10.1016/j.cell.2013.03.022
Rohde K. H., Abramovitch R. B., Russell D. G. (2007). Mycobacterium tuberculosis invasion of macrophages: linking bacterial gene expression to environmental cues. Cell Host Microbe 2 (5), 352–364. doi: 10.1016/j.chom.2007.09.006
Saini N. K., Baena A., Ng T. W., Venkataswamy M. M., Kennedy S. C., Kunnath-Velayudhan S., et al. (2016). Suppression of autophagy and antigen presentation by Mycobacterium tuberculosis PE_PGRS47. Nat. Microbiol. 1 (9), 16133. doi: 10.1038/nmicrobiol.2016.133
Sassetti C. M., Rubin E. J. (2003). Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U. S. A. 100 (22), 12989–12994. doi: 10.1073/pnas.2134250100
Shin D. M., Jeon B. Y., Lee H. M., Jin H. S., Yuk J. M., Song C. H., et al. (2010). Mycobacterium tuberculosis eis regulates autophagy, inflammation, and cell death through redox-dependent signaling. PloS Pathog. 6 (12), e1001230. doi: 10.1371/journal.ppat.1001230
Simeone R., Bobard A., Lippmann J., Bitter W., Majlessi L., Brosch R., et al. (2012). Phagosomal rupture by Mycobacterium tuberculosis results in toxicity and host cell death. PloS Pathog. 8 (2), e1002507. doi: 10.1371/journal.ppat.1002507
Singh A. K., Carette X., Potluri L. P., Sharp J. D., Xu R., Prisic S., et al. (2016). Investigating essential gene function in Mycobacterium tuberculosis using an efficient CRISPR interference system. Nucleic Acids Res. 44 (18), e143. doi: 10.1093/nar/gkw625
Singh P., Rao R. N., Reddy J. R., Prasad R. B., Kotturu S. K., Ghosh S., et al. (2016). PE11, a PE/PPE family protein of Mycobacterium tuberculosis is involved in cell wall remodeling and virulence. Sci. Rep. 6, 21624. doi: 10.1038/srep21624
Singh V. K., Berry L., Bernut A., Singh S., Carrere-Kremer S., Viljoen A., et al. (2016). A unique PE_PGRS protein inhibiting host cell cytosolic defenses and sustaining full virulence of Mycobacterium marinum in multiple hosts. Cell Microbiol. 18 (11), 1489–1507. doi: 10.1111/cmi.12606
Thi E. P., Hong C. J., Sanghera G., Reiner N. E. (2013). Identification of the Mycobacterium tuberculosis protein PE-PGRS62 as a novel effector that functions to block phagosome maturation and inhibit iNOS expression. Cell Microbiol. 15 (5), 795–808. doi: 10.1111/cmi.12073
Tiwari B., Soory A., Raghunand T. R. (2014). An immunomodulatory role for the Mycobacterium tuberculosis region of difference 1 locus proteins PE35 ( Rv3872) and PPE68 ( Rv3873). FEBS J. 281 (6), 1556–1570. doi: 10.1111/febs.12723
Tiwari B., Ramakrishnan U. M., Raghunand T. R. (2015). The Mycobacterium tuberculosis protein pair PE9 (Rv1088)-PE10 (Rv1089) forms heterodimers and induces macrophage apoptosis through Toll-like receptor 4. Cell Microbiol. 17 (11), 1653–1669. doi: 10.1111/cmi.12462
Tobin D. M., Ramakrishnan L. (2008). Comparative pathogenesis of Mycobacterium marinum and Mycobacterium tuberculosis. Cell Microbiol. 10 (5), 1027–1039. doi: 10.1111/j.1462-5822.2008.01133.x
Tundup S., Pathak N., Ramanadham M., Mukhopadhyay S., Murthy K. J., Ehtesham N. Z., et al. (2008). The co-operonic PE25/PPE41 protein complex of Mycobacterium tuberculosis elicits increased humoral and cell mediated immune response. PloS One 3 (10), e3586. doi: 10.1371/journal.pone.0003586
Walters S. B., Dubnau E., Kolesnikova I., Laval F., Daffe M., Smith I. (2006). The Mycobacterium tuberculosis PhoPR two-component system regulates genes essential for virulence and complex lipid biosynthesis. Mol. Microbiol. 60 (2), 312–330. doi: 10.1111/j.1365-2958.2006.05102.x
Wang J., Yang K., Zhou L., Minhaowu, Wu Y., Zhu M., et al. (2013). MicroRNA-155 promotes autophagy to eliminate intracellular mycobacteria by targeting Rheb. PloS Pathog. 9 (10), e1003697. doi: 10.1371/journal.ppat.1003697
Wang J., Wu M., Wen J., Yang K., Li M., Zhan X., et al. (2014). MicroRNA-155 induction by Mycobacterium bovis BCG enhances ROS production through targeting SHIP1. Mol. Immunol. 62 (1), 29–36. doi: 10.1016/j.molimm.2014.05.012
Wu S., He L., Li Y., Wang T., Feng L., Jiang L., et al. (2013). miR-146a facilitates replication of dengue virus by dampening interferon induction by targeting TRAF6. J. Infect. 67 (4), 329–341. doi: 10.1016/j.jinf.2013.05.003
Keywords: virulence factors, PPE31, cell death, Mycobacterium tuberculosis, JNK signaling
Citation: Feng S, Hong Z, Zhang G, Li J, Tian G-B, Zhou H and Huang X (2021) Mycobacterium PPE31 Contributes to Host Cell Death. Front. Cell. Infect. Microbiol. 11:629836. doi: 10.3389/fcimb.2021.629836
Received: 16 November 2020; Accepted: 23 March 2021;
Published: 13 April 2021.
Edited by:
Yongqun Oliver He, University of Michigan, United StatesCopyright © 2021 Feng, Hong, Zhang, Li, Tian, Zhou and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xi Huang, aHVhbmd4aTZAbWFpbC5zeXN1LmVkdS5jbg==; Haibo Zhou, aGFpYm9femhvdTIwMDNAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.