 Eriko Ohsaki
Eriko Ohsaki Keiji Ueda
Keiji Ueda- Division of Virology, Department of Microbiology and Immunology, Osaka University Graduate School of Medicine, Suita, Japan
Interactions between viruses and cellular factors are essential for viral replication or host defense. The DNA damage response (DDR) orchestrates a molecular network of cellular mechanisms that integrates cell cycle regulation and DNA repair or apoptosis. Numerous studies have revealed that the DDR is activated by virus infection, aberrant DNA structures generated by viral DNA replication, or the integration of retroviruses. Although the DDR is an essential function for maintaining the genomic integrity of cells, viruses may utilize this mechanism to build a convenient environment for themselves, and the resulting perturbation of the DDR has been shown to increase the risk of tumorigenesis. There have been many studies investigating the roles of the DDR in oncogenic viruses such as Epstein-Barr virus (EBV), human papillomavirus (HPV), hepatitis B virus (HBV), human T-cell leukemia virus type 1 (HTLV-1), and Kaposi’s sarcoma-associated herpesvirus (KSHV). This review summarizes current knowledge on the roles of DDR in the KSHV lifecycle.
Introduction
For the survival of organisms, the faithful transmission of genetic information from a parent cell to its daughter cells is essential. Such accurate transmission requires not only mechanisms for the faithful replication of DNA and segregation of chromosomes, but also mechanisms to prevent spontaneous and/or exogenously induced DNA damages. To accomplish all these goals, cells have monitoring systems that survey aberrant chromosomal structures. After sensing DNA damage, a DNA damage checkpoint coordinates with the cell-cycle regulation and repair systems. In response to DNA damage, the DNA damage response (DDR) controls cell cycle arrest to allow enough time for repair. When DNA damage is too severe to rescue, the apoptosis pathway is activated by the DDR.
KSHV is classified as a member of the Gammaherpesvirinae subfamily, which also includes Kaposi’s sarcoma and lymphoproliferative disorders such as primary effusion lymphoma (PEL) and multicentric Castleman’s disease (MCD), and KSHV is often associated with HIV infection (Cesarman and Knowles, 1995; Soulier et al., 1995). KSHV has two distinct lifecycles, a latent phase and lytic replication phase. During latency, the viral genome is maintained with limited gene expression in host cells (Sarid et al., 1998; Fakhari and Dittmer, 2002; Lieberman, 2013; Campbell et al., 2020). When latency is disrupted, the virus shifts to a lytic phase in which infectious progeny virions are produced (Sun et al., 1998; Purushothaman et al., 2015; Aneja and Yuan, 2017).
Affinity purification of DNA-binding proteins first demonstrated that several DDR proteins such as poly (ADP-ribose) polymerase 1 (PARP1) and MutS homolog 2/3/6 (MSH2/3/6) bind to the terminal repeat (TR) region (Ohsaki et al., 2004), and that MSH2/6, PARP1, DNA-dependent protein kinase (DNA-PK), and Ku70/80 bind to lytic DNA replication origins (ori-Lyt) (Wang et al., 2008). Recently, many studies have reported that DDR proteins are upregulated by viral replication and involved in the KSHV lifecycle, as described in a later section.
Viral infection causes global disruption of nuclear architecture and chromosomal aberration, and DDR is activated as a potent antiviral defense (Fortunato and Spector, 2003). In addition, structures of the viral genome, including linear double stranded DNA (dsDNA) (herpesviruses, adenoviruses), circular dsDNA (polyomaviruses, papillomaviruses), and RNA genomes which are reverse transcribed to linear dsDNA (retroviruses), can be recognized by DDR sensor proteins (Everett, 2006; Kerur et al., 2011; Lilley et al., 2011; Barber, 2014; Hau and Tsao, 2017; Kleinberger, 2020). On the other hand, viruses take advantage of the DDR pathway to modulate the cell cycle and hijack cellular proteins to support viral replication (Everett, 2006; Lilley et al., 2010; Weitzman and Fradet-Turcotte, 2018). Since DDR signaling pathways induce cell cycle arrest or apoptosis, which are negative effects on virus production, viruses have developed suppressive strategies against the DDR. Over the past two decades, numerous studies have reported the interactions between DDR signaling pathways and human tumor viruses including HPV, HTLV-1, HBV, HCV, EBV, and KSHV (Weitzman and Fradet-Turcotte, 2018).
Deregulation of DDRs because of the competition among such virus-host defense systems increases the risk of tumorigenesis. This is because various kinds of cell signaling networks maintaining homeostasis are perturbed. Therefore, an improved understanding of these relationships between viruses and DDR systems will help us to develop strategies for anti-tumorigenic and anti-viral therapies. In the first section of this review we summarize DDR signaling pathways, and in the second section we focus on the relationships between KSHV and DDR.
DNA Damage Response
DDR consists of DNA damage sensors followed by transducers, and effectors (Figure 1). All the DDR pathways including cell cycle checkpoints and DNA repair pathways are completed by proper signaling from sensors to transducers and to effectors.
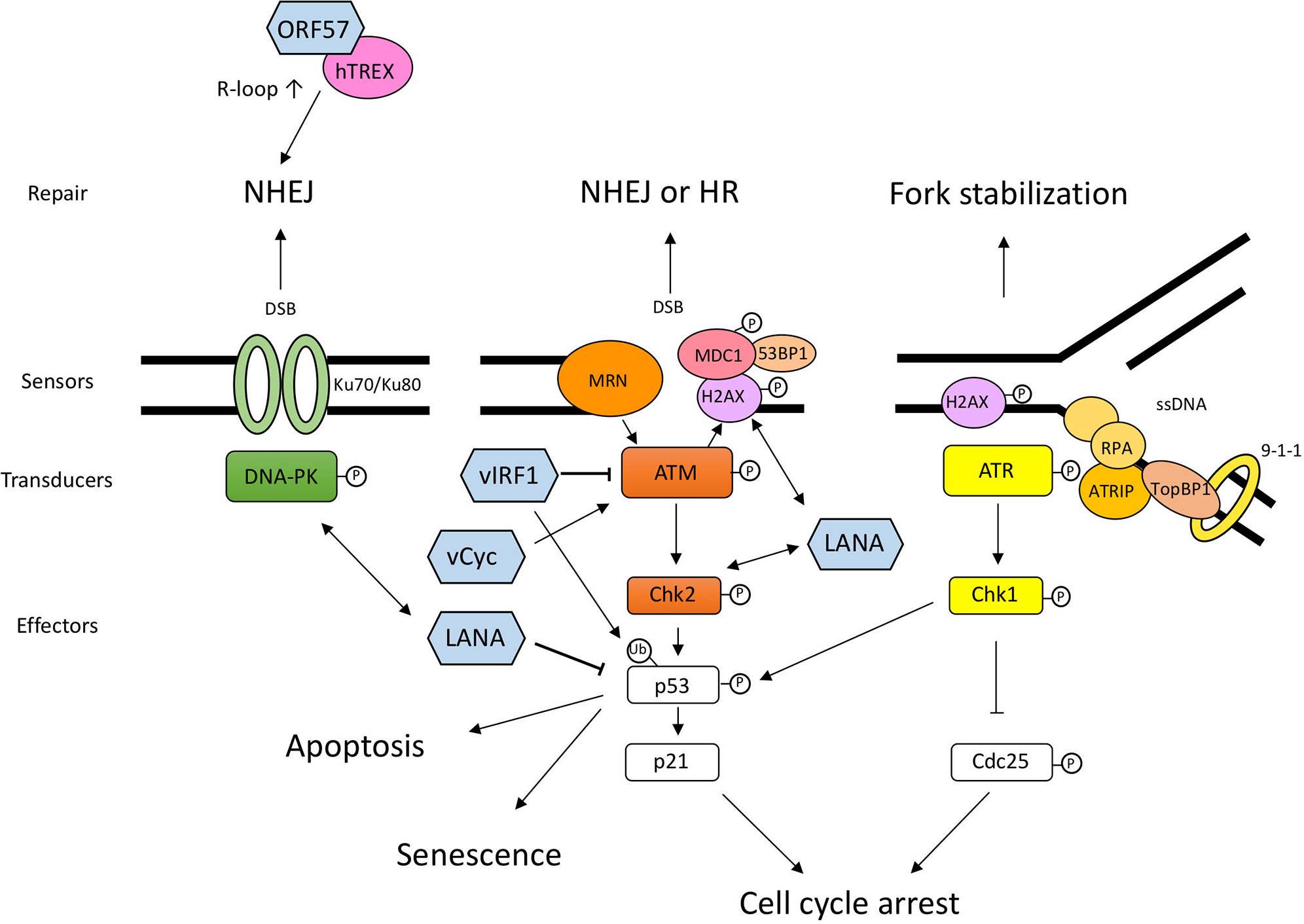
Figure 1 Schematic diagram of DDR pathways and the interplay between DDR and KSHV proteins. DNA damage sensors recognize the aberrant DNA structure and activate transducers, such as DNA-PK, ATM, and ATR. These transducers activate downstream effectors related to cell cycle arrest and DNA repair. When the DNA damage is too severe, cells undergo permanent cell-cycle arrest, senescence, or cell death. The viral proteins shown in this figure are not exhaustive but include the proteins mentioned in this article. LANA and v-cyclin lead to the activation of sensors and transducers via direct or indirect interaction. vIRF1 interacts with ATM and downregulates its kinase activity and ubiquitinates p53 to degrade it. ORF57 expression induces several NHEJ proteins and interacts with hTREX, and consequently transcribed mRNA becomes unstable and forms R-loops, which leads to genomic instability. The double-headed arrow shows the interaction between viral protein and DDR protein. DNA-PK, DNA-dependent protein kinase; ATM, Ataxia telangiectasia mutated; ATR, ATM and Rad3-related; MRN, Mre11-Rad50-NBS1 complex; Chk1, Checkpoint kinase 1; Chk2, Checkpoint kinase 2; MDC1, mediator of DNA damage checkpoint protein 1; ATRIP, ATR-interacting protein; RPA (replication protein A); TopBP1, DNA topoisomerase II binding protein 1.
DNA Damage Sensors and Transducers
PIKKs: Transducers of DNA Damage Response
The cellular responses to DNA damage are mainly controlled by three phosphatidylinositol 3-kinase-like kinases (PIKKs): ATM (Ataxia telangiectasia mutated), ATR (ATM and Rad3-related), and DNA-PK (Cimprich and Cortez, 2008; Davis et al., 2014; Blackford and Jackson, 2017; Menolfi and Zha, 2020), which act as DNA damage transducers. ATM and DNA-PK are principally activated in response to double-strand breaks (DSBs). In contrast, ATR is activated by single-stranded DNA (ssDNA) during the S-phase to regulate the firing of replication origins and the stalled replication forks.
DNA Damage Sensing and Signaling
DSBs are recognized by the MRN complex, which is composed of Mre11, Rad50, and NBS1 and activates ATM (Lavin et al., 2015). MRN mediates cross-talk among the repair and checkpoint machinery. ATM phosphorylates downstream molecules, such as H2AX, a variant of the histone H2A protein family, and Chk2 (Checkpoint kinase 2) (Burma et al., 2001; Bartek and Lukas, 2003). Phosphorylated H2AX (γH2AX) is generated in chromatin near DSBs and recruits critical adaptor proteins such as MDC1 (mediator of DNA damage checkpoint protein 1) and 53BP1 (p53 binding protein 1) (Stewart et al., 2003; Xie et al., 2008; Kilic et al., 2019). Activated ATR and ATM phosphorylate the downstream targets Chk1 (Checkpoint kinase 1) and Chk2, which are key effectors in DDR (Bartek and Lukas, 2003; Smith et al., 2020).
Ku70/Ku80 are other sensor proteins recognizing DSBs and recruit the DNA-PK, and their main role is to facilitate non-homologous end joining (NHEJ), which is one of the DNA repair systems, as discussed in a later section (Jette and Lees-Miller, 2015; Chang et al., 2017). Following the sensing of DNA lesions, phosphorylation of transducer and effector molecules by PIKKs induces cell cycle arrest, DNA repair, and/or apoptosis or senescence.
Cell Cycle Checkpoints and DNA Repair Pathways
Activated Chk1 and Chk2 phosphorylate downstream targets such as Cdc25 and p53 followed by degradation of Cdc25 and cell cycle arrest (Donzelli and Draetta, 2003; Liu et al., 2020) or by activation of the p53-mediated signaling pathway for DNA repair, cell cycle arrest, and apoptosis (Bartek and Lukas, 2003; Williams and Schumacher, 2016). p21, the downstream target of p53, induces cell cycle arrest through inhibition of the cyclin E/Cdk2 complex (Waldman et al., 1995; Planas-Silva and Weinberg, 1997; Gire and Dulić, 2015).
PIKKs have roles in the recruitment of repair machineries via the phosphorylation of downstream elements. For DSB repair, two major pathways are used: NHEJ and homologous recombination (HR). The NHEJ pathway has largely four steps: recognition, resection, polymerization, and ligation of the DNA ends (Chang et al., 2017). In the NHEJ, Ku70/80 primarily recognizes DSBs, and DNA-PK is recruited and activated by Ku-bound DSB ends to promote NHEJ.
DSBs are also recognized by the MRN complex (Carson, 2003), which promotes ATM activation and resection of DSB ends to generate ssDNA overhangs. While NHEJ is active throughout interphase, HR is active only in the S and G2 phases, because a homologous chromosome is available in the S and G2 phases as a template for DNA repair. The relationships between HR proteins and viruses have been reported in EBV (Kudoh et al., 2009), HPV (Gillespie et al., 2012; Park et al., 2014), and HTLV-1 (Belgnaoui et al., 2010), but not in KSHV. Additional investigations will be needed to expand our knowledge of the relationships between KSHV and HR.
The mismatch repair system improves DNA replication fidelity by degrading an error-containing region of the newly synthesized strand and providing a chance for the DNA polymerase to correct errors (Li, 2008; Fishel, 2015; Gupta, 2019). MSH2, MSH3, and MSH6 participate in mismatch repair as heterodimers, i.e., MSH2/MSH6 or MSH2/MSH3. While MSH2/6 recognizes base pair mismatches and small insertion/deletions, MSH2/MSH3 recognizes various DNA mismatches, including DNA loops ranging from 1 to 14 nucleotides as well as longer insertion/deletion mismatches (Jiricny, 2006). Several studies have suggested that the mismatch repair pathway is involved in viral replication not only in KSHV (Ohsaki et al., 2004; Wang et al., 2008; Cha et al., 2010), but also in EBV (Daikoku et al., 2006), as described in a later section.
Poly (ADP-ribose) polymerase 1 (PARP1) is an ADP-ribosylating enzyme and a multifunctional nuclear enzyme that affects various aspects of cellular homeostasis, such as DNA repair, cell proliferation, apoptosis, and inflammation. PARP1 is another important SSB- and DSB-signaling protein and modifies both target proteins and PARP1 itself by poly (ADP) ribosylation (Ray Chaudhuri and Nussenzweig, 2017). PARP1 has a pivotal role in DNA repair and is involved in various repair pathways, such as single-strand break repair (SSBR) (Leppard et al., 2003; Fisher et al., 2007), base excision repair (BER) (Masson et al., 1998; Dantzer et al., 2000; Lavrik et al., 2001; El-Khamisy, 2003; Ronson et al., 2018), nucleotide excision repair (NER) (Pines et al., 2012; Robu et al., 2017), NHEJ (Wang et al., 2006; Mansour et al., 2010; Cheng et al., 2011; Spagnolo et al., 2012; Luijsterburg et al., 2016), and HR (Hochegger et al., 2006; Hu et al., 2014).
Interplay Between KSHV and DDR
Roles of the DDR in De Novo Infection and Latent Infection
During the latent phase, the KSHV genome persists in the host nucleus as a double-stranded circular DNA—i.e., as an extra-chromosomal viral genome (episome)—and a very limited set of viral genes such as LANA (ORF73), vFLIP (ORF71), v-cyclin (ORF72), Kaposin (ORF K12), vIRF3 (LANA2), and 12 miRNAs are expressed (Dittmer et al., 1998; Sun et al., 1999; Parravicini et al., 2000; Cai et al., 2005; Pfeffer et al., 2005; Samols et al., 2005; Lieberman, 2013; Qin et al., 2017).
Like the proteins of other viruses, the KSHV proteins interact with DDR components and activate or prevent the signaling response (Figure 1 and Table 1). De novo infection of KSHV in human PBMCs or primary endothelial cells upregulates the level of γH2AX, which is the phosphorylated form of H2AX (Jha et al., 2013; Singh et al., 2014). γH2AX interacts with LANA and contributes to LANA-mediated episome maintenance (Jha et al., 2013). It has been reported that H2AX knockdown reduces the expression of LANA and viral genome copies, suggesting that γH2AX has a role in latent gene expression and establishment of KSHV latency (Singh et al., 2014). In the same manner as phosphorylated H2AX, phosphorylated ATM was induced within 30 min post infection, and the inhibition of ATM activity caused a reduction of LANA expression, while knockdown of Chk1 and Chk2 did not affect LANA expression (Singh et al., 2014). These results suggest that selective activation of the DDR pathway is critical for the initial stages of KSHV infection and establishment of viral latency.
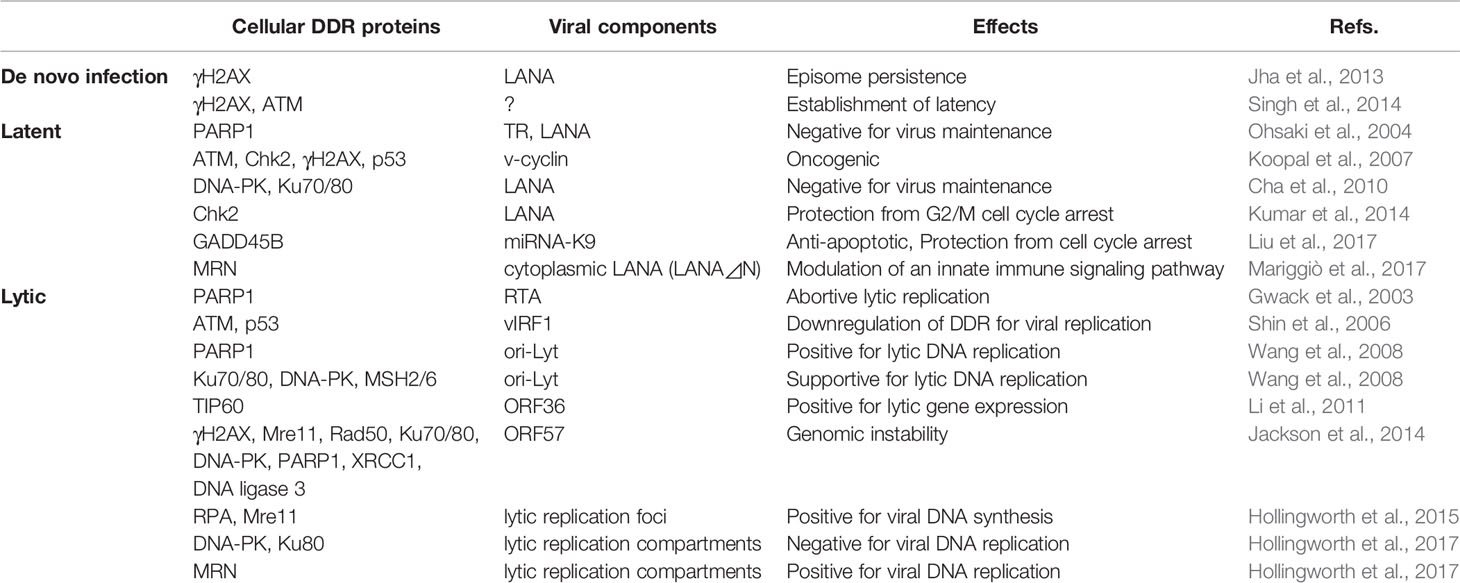
Table 1 List of DDR-KSHV interaction during de novo infection, latency, and lytic reactivation and its effects.
DDR proteins are equally distributed in the nucleus, but recent works have suggested that cytoplasmic DDR proteins sensor the cytoplasmic exogenous DNA and activate the innate immune signaling (Roth et al., 2014). Mariggio et al. demonstrated that the cytoplasmic LANA recruits the MRN complex in the cytoplasm of KSHV-infected B cells to inhibit NF-κB activation and blocks the role of innate immune sensors of cytoplasmic DNA (Mariggiò et al., 2017).
The cell cycle profiles of KSHV-positive cells suggest that LANA inhibits nocodazole-induced G2/M arrest (Kumar et al., 2014). In the same study, Kumar et al. (2014) reported that LANA interacts with Chk2 through the serine rich N-terminal domain of Chk2, and that downregulation of Chk2 expression promotes G2/M arrest in KSHV-positive BC3 cells. These results suggested that LANA interacts with Chk2 to escape from the G2/M cell cycle arrest due to the ATM/ATR signaling pathway.
Some of the DDR proteins play negative roles in latent DNA replication (Koopal et al., 2007; Cha et al., 2010). Using a proteomics approach, Cha et al. (2010) identified DNA-PK, Ku70, and Ku80 as LANA-binding proteins. They further showed that LANA is phosphorylated by DNA-PK/Ku and reduces transient DNA replication. Finally, they reported that overexpression of Ku70 downregulates transient DNA replication, suggesting that the DNA-PK/Ku complex binds with LANA and negatively regulates latent replication (Cha et al., 2010).
KSHV v-cyclin induces replicative stress in EA.hy926—which is a HUVEC-epithelial A549 hybrid cell line and has been used as an endothelial cell model—and also induces DDR and senescence by activating γH2AX, ATM, Chk2, p53, and p21 (Koopal et al., 2007). This v-cyclin-induced DDR is dependent on CDK6, a catalytic subunit of the v-cyclin. From this study, the DDR response appears to be activated by host defense mechanisms.
In addition, a recent study suggested that KSHV miRNAs target GADD45B to protect infected cells from cell cycle arrest and apoptosis (Liu et al., 2017). KSHV infection in primary endothelial cells causes repression of growth arrest DNA damage-inducible gene 45 (GADD45B). This study also demonstrated that KSHV miRNA-K9 inhibits the expression of GADD45B induced by a p53 activator, Nutlin-3, and suggested that KSHV miRNAs play essential roles in protecting cells from the DDR-induced cell cycle arrest and apoptosis.
Roles of the DDR in Lytic Replication
Murine γ-herpesvirus 68 (MHV68) ORF36, which is a conserved serine/threonine protein kinase in Herpesviridae and is similar to the cellular kinase cdk2 (Romaker et al., 2006), phosphorylates H2AX (Tarakanova et al., 2007) during lytic replication. This ORF36-mediated H2AX phosphorylation is dependent on ATM activity, and is critical for viral replication, suggesting that the association between viral kinase and cellular DDR proteins synergistically supports viral replication. Another study demonstrated that ORF36 phosphorylates histone acetyltransferase TIP60, an upstream regulator of the DDR pathway, and promotes lytic gene expression (Li et al., 2011).
MDM2, an E3 ubiquitin ligase and a repressor of p53, has a negative role in viral reactivation (Balistreri et al., 2016). Lytic reactivation induces a p53 response in PEL cell lines and arrests the cells at G2 phase, which enables efficient lytic replication, as a positive effect of DDR on viral replication.
A proteomics analysis based on SILAC (stable isotope labelling by amino acids in cell culture) identified a large number of NHEJ proteins, including Rad50, Mre11, DNA-PK, Ku70, Ku80, PARP1, XRCC1 (X-ray repair cross-complementing protein 1), and DNA ligase3, that are enriched upon the expression of KSHV ORF57, a viral early protein and post-transcriptional regulator of gene expression (Jackson et al., 2014). In addition, as a consequence of the interaction between ORF57 and hTREX (human transcription and export complex), which is an mRNA export complex, the newly transcribed mRNA becomes unstable and forms R-loops, leading to genomic instability.
Affinity purification and mass spectrometry assays have identified MSH2, MSH3, MSH6, PARP1, DNA-PK, and Ku70/Ku80 as TR-binding proteins (Ohsaki et al., 2004), as ori-Lyt-binding proteins (Wang et al., 2008), and as LANA-binding proteins (Cha et al., 2010). Lytic reactivation in RTA-inducible BCBL1 and KSHV-infected endothelial cells causes DDR activation through the phosphorylation of H2AX, ATM, and DNA-PK and modulates cell cycle progression (Hollingworth et al., 2015). RPA (replication protein A), which is a single-stranded DNA-binding protein, and Mre11 accumulate at viral replication foci, suggesting that DDR proteins contribute to viral DNA replication. These studies suggested that NHEJ proteins such as the MSH2/6 heterodimer and DNA-PK/Ku70/Ku80 heterotrimer are recruited to latent/lytic replication origins and have some roles in the formation of a replication initiation complex.
Another study also demonstrated that the Ku70/Ku80 heterodimer and the MRN complex are recruited to viral replication compartments (RCs) during lytic replication, and the activation of ATM kinase promotes viral replication (Hollingworth et al., 2017). On the other hand, the other DDR proteins, such as γH2AX, MDC1, and 53BP1, localize on the periphery of viral RCs. In addition, knockdown or inhibition of NHEJ proteins such as Ku80 and DNA-PK enhances viral replication, suggesting a negative effect of the NHEJ pathway on viral replication (Hollingworth et al., 2017). Thus, it still remains to be clarified whether such DNA repair components have positive or negative roles in KSHV viral replication. Further work is necessary to elucidate how DNA repair components function in viral lytic replication.
PARP1 has essential roles in posttranslational modification of a large number of target proteins related to various kinds of cellular events and acts as a multifunctional enzyme. A previous study reported that PARP1 inhibits viral transcription through RTA ribosylation and leads to abortive lytic replication (Gwack et al., 2003). PARP1 directly binds to the TR and ribosylates LANA to modulate viral replication in latency (Ohsaki et al., 2004; Cha et al., 2010). From these studies, PARP1 has negative roles in latent viral DNA replication and in lytic gene expression. On the other hand, another study reported that PARP1 plays a positive role in the early stage of viral DNA replication in lytic reactivation (Wang et al., 2008).
Although the biological significance of DDR activation in KSHV lytic/latent replication is not clear, the activation of upstream signaling of DDR seems to benefit the viral replication. In contrast, some viral proteins directly interact with the downstream signaling components to prevent effectors from suppressing virus replication. Downstream of ATM pathway are inactivated in the late stage of lytic reactivation through the vIRF1 (viral interferon regulatory factor 1)-mediated pathway (Shin et al., 2006). vIRF1 interacts with ATM and downregulates ATM kinase activity and the p53 protein level (Shin et al., 2006).
Conclusions
Over the past decades, many studies have elucidated the relationships between KSHV and the DDR signaling pathway. The roles of the DDR in viral replication depend on the type of infection, the structure of the viral genome formed by replication, the cell type, and the cell cycle stage. Aberrant DNA structures and signaling in the course of viral genome replication lead to the DDR pathway as a host defense response. On the other hand, viruses have developed strategies to hijack the DDR signaling pathway for their survival (Figure 2). A number of studies introduced in this article suggest that activation of the upstream pathway of the DDR—which includes DNA damage sensors and transducers—contributes to the modulation of both cell cycle progression and viral replication, whereas the downstream signaling pathways, such as the apoptosis pathway, are unfavorable for viruses. Accordingly, KSHV probably has developed strategies to negate a part of the DDR pathway.
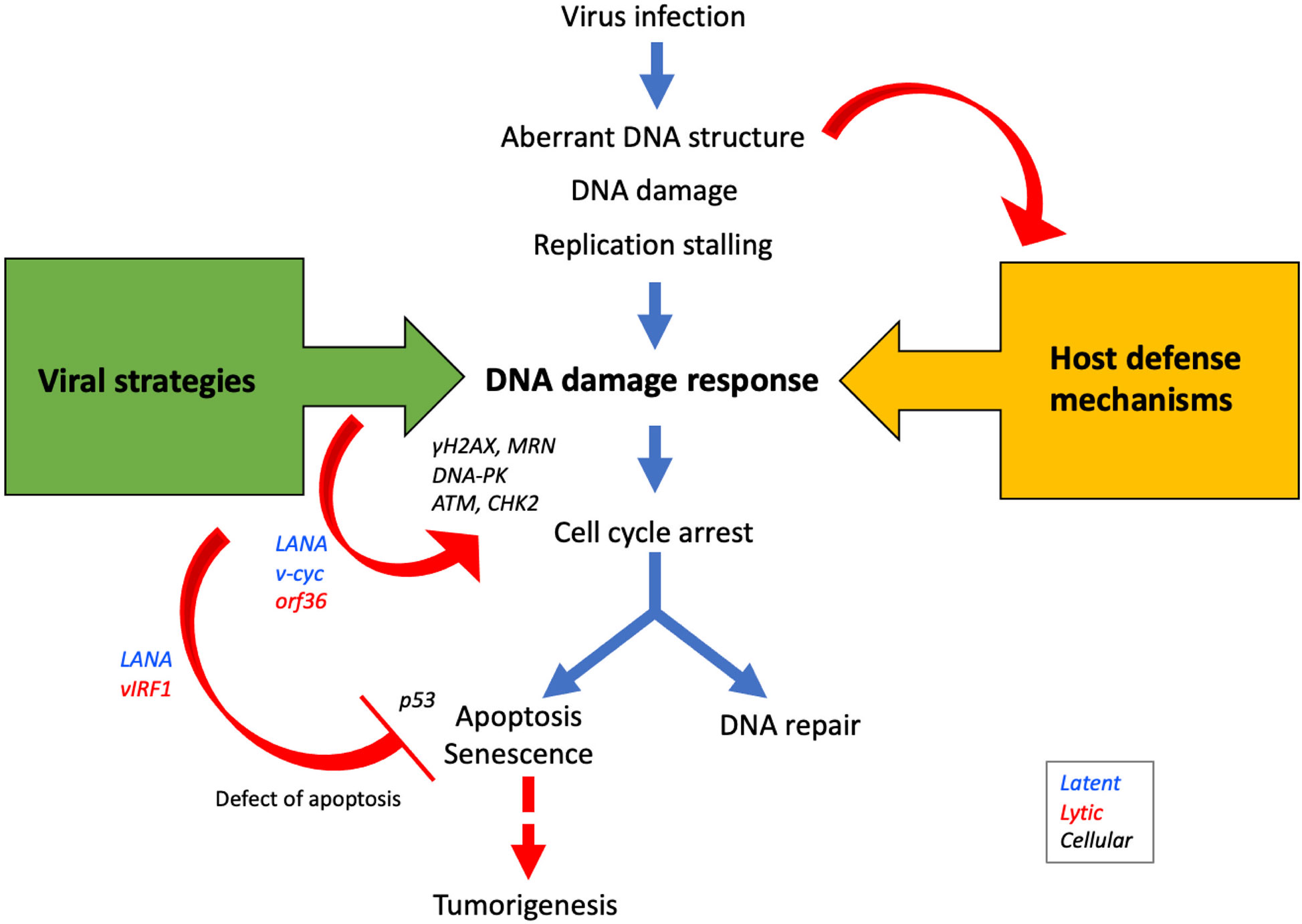
Figure 2 Viral proteins manipulate DDR signaling pathways to promote viral propagation. During infection, aberrant DNA structures or DNA damages caused by virus replication are recognized by DDR sensors/transducers as host defense mechanisms. On the other hand, viral proteins such as LANA, v-cyc, and ORF36 activate DDR proteins to modulate the cell cycle in the S-phase to promote viral replication. Since apoptosis decreases the opportunity for viral propagation, many viral proteins such as LANA, vIRF1, and vFLIP inhibit apoptosis and promote cell survival. The activation of the apoptotic pathway following the DDR occurs via p53, but because viral proteins deregulate this pathway, the risk of tumorigenesis is increased.
Deregulation of the DDR pathway caused by such viral strategies increases the risk of tumorigenesis. More specifically, viruses affect cell cycle regulation to drive viral replication and manipulate the DDR pathway, and the resulting damage to the cellular repair system, increase in mutations, and resistance to apoptosis causes genomic instability and finally promotes tumorigenesis. An improved understanding of the battles between viruses and the DDR will lead to new therapeutic options for controlling viral replication and oncogenesis.
Author Contributions
EO contributed to the design of the manuscript. EO and KU contributed to the concept of the manuscript. KU proofread and modified the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Aneja K. K., Yuan Y. (2017). Reactivation and Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus: An Update. Front. Microbiol. 8:613:613. doi: 10.3389/fmicb.2017.00613
Balistreri G., Viiliäinen J., Turunen M., Diaz R., Lyly L., Pekkonen P., et al. (2016). Oncogenic Herpesvirus Utilizes Stress-Induced Cell Cycle Checkpoints for Efficient Lytic Replication. PloS Pathog. 12, e1005424. doi: 10.1371/journal.ppat.1005424
Barber G. N. (2014). STING-dependent cytosolic DNA sensing pathways. Trends Immunol. 35, 88–93. doi: 10.1016/j.it.2013.10.010
Bartek J., Lukas J. (2003). Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 3, 421–429. doi: 10.1016/S1535-6108(03)00110-7
Belgnaoui S. M., Fryrear K. A., Nyalwidhe J. O., Guo X., Semmes O. J. (2010). The Viral Oncoprotein Tax Sequesters DNA Damage Response Factors by Tethering MDC1 to Chromatin. J. Biol. Chem. 285, 32897–32905. doi: 10.1074/jbc.M110.146373
Blackford A. N., Jackson S. P. (2017). ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 66, 801–817. doi: 10.1016/j.molcel.2017.05.015
Burma S., Chen B. P., Murphy M., Kurimasa A., Chen D. J. (2001). ATM Phosphorylates Histone H2AX in Response to DNA Double-strand Breaks. J. Biol. Chem. 276, 42462–42467. doi: 10.1074/jbc.C100466200
Cai X., Lu S., Zhang Z., Gonzalez C. M., Damania B., Cullen B. R. (2005). Kaposi’s sarcoma-associated herpesvirus expresses an array of viral microRNAs in latently infected cells. Proc. Natl. Acad. Sci. 102, 5570–5575. doi: 10.1073/pnas.0408192102
Campbell M., Yang W.-S., Yeh W. W., Kao C.-H., Chang P.-C. (2020). Epigenetic Regulation of Kaposi’s Sarcoma-Associated Herpesvirus Latency. Front. Microbiol. 11:850:850. doi: 10.3389/fmicb.2020.00850
Carson C. T. (2003). The Mre11 complex is required for ATM activation and the G2/M checkpoint. EMBO J. 22, 6610–6620. doi: 10.1093/emboj/cdg630
Cesarman E., Knowles D. M. (1995). Kaposi’s Sarcoma–Associated Herpesvirus-Like DNA Sequences in AIDS-Related Body-Cavity–Based Lymphomas. THE New Engl. J. OF Med. 332, 6. doi: 10.1056/NEJM199505043321802
Cha S., Lim C., Lee J. Y., Song Y.-J., Park J., Choe J., et al. (2010). DNA-PK/Ku complex binds to latency-associated nuclear antigen and negatively regulates Kaposi’s sarcoma-associated herpesvirus latent replication. Biochem. Biophys. Res. Commun. 394, 934–939. doi: 10.1016/j.bbrc.2010.03.086
Chang H. H. Y., Pannunzio N. R., Adachi N., Lieber M. R. (2017). Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 18, 495–506. doi: 10.1038/nrm.2017.48
Cheng Q., Barboule N., Frit P., Gomez D., Bombarde O., Couderc B., et al. (2011). Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 39, 9605–9619. doi: 10.1093/nar/gkr656
Cimprich K. A., Cortez D. (2008). ATR: an essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 9, 616–627. doi: 10.1038/nrm2450
Daikoku T., Kudoh A., Sugaya Y., Iwahori S., Shirata N., Isomura H., et al. (2006). Postreplicative Mismatch Repair Factors Are Recruited to Epstein-Barr Virus Replication Compartments. J. Biol. Chem. 281, 11422–11430. doi: 10.1074/jbc.M510314200
Dantzer F., de la Rubia G., Ménissier-de Murcia J., Hostomsky Z., de Murcia G., Schreiber V. (2000). Base Excision Repair Is Impaired in Mammalian Cells Lacking Poly(ADP-ribose) Polymerase-1 †. Biochemistry 39, 7559–7569. doi: 10.1021/bi0003442
Davis A. J., Chen B. P. C., Chen D. J. (2014). DNA-PK: A dynamic enzyme in a versatile DSB repair pathway. DNA Repair 17, 21–29. doi: 10.1016/j.dnarep.2014.02.020
Dittmer D., Lagunoff M., Renne R., Staskus K., Haase A., Ganem D. (1998). A Cluster of Latently Expressed Genes in Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 72, 8309–8315. doi: 10.1128/JVI.72.10.8309-8315.1998
Donzelli M., Draetta G. F. (2003). Regulating mammalian checkpoints through Cdc25 inactivation. EMBO Rep. 4, 671–677. doi: 10.1038/sj.embor.embor887
El-Khamisy S. F. (2003). A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 31, 5526–5533. doi: 10.1093/nar/gkg761
Everett R. D. (2006). Interactions between DNA viruses, ND10 and the DNA damage response. Cell Microbiol. 8, 365–374. doi: 10.1111/j.1462-5822.2005.00677.x
Fakhari F. D., Dittmer D. P. (2002). Charting Latency Transcripts in Kaposi’s Sarcoma-Associated Herpesvirus by Whole-Genome Real-Time Quantitative PCR. JVI 76, 6213–6223. doi: 10.1128/JVI.76.12.6213-6223.2002
Fisher A. E. O., Hochegger H., Takeda S., Caldecott K. W. (2007). Poly(ADP-Ribose) Polymerase 1 Accelerates Single-Strand Break Repair in Concert with Poly(ADP-Ribose) Glycohydrolase. MCB 27, 5597–5605. doi: 10.1128/MCB.02248-06
Fortunato E. A., Spector D. H. (2003). Viral induction of site-specific chromosome damage. Rev. Med. Virol. 13, 21–37. doi: 10.1002/rmv.368
Gillespie K. A., Mehta K. P., Laimins L. A., Moody C. A. (2012). Human Papillomaviruses Recruit Cellular DNA Repair and Homologous Recombination Factors to Viral Replication Centers. J. Virol. 86, 9520–9526. doi: 10.1128/JVI.00247-12
Gire V., Dulić V. (2015). Senescence from G2 arrest, revisited. Cell Cycle 14, 297–304. doi: 10.1080/15384101.2014.1000134
Gupta D. (2019). The mismatch repair-dependent DNA damage response_ Mechanisms and implications. DNA Repair 10, 60–69. doi: 10.1016/j.dnarep.2019.03.009
Gwack Y., Nakamura H., Lee S. H., Souvlis J., Yustein J. T., Gygi S., et al. (2003). Poly(ADP-Ribose) Polymerase 1 and Ste20-Like Kinase hKFC Act as Transcriptional Repressors for Gamma-2 Herpesvirus Lytic Replication. MCB 23, 8282–8294. doi: 10.1128/MCB.23.22.8282-8294.2003
Hau P., Tsao S. (2017). Epstein–Barr Virus Hijacks DNA Damage Response Transducers to Orchestrate Its Life Cycle. Viruses 9:341. doi: 10.3390/v9110341
Hochegger H., Dejsuphong D., Fukushima T., Morrison C., Sonoda E., Schreiber V., et al. (2006). Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 25, 1305–1314. doi: 10.1038/sj.emboj.7601015
Hollingworth R., Skalka G., Stewart G., Hislop A., Blackbourn D., Grand R. (2015). Activation of DNA Damage Response Pathways during Lytic Replication of KSHV. Viruses 7, 2908–2927. doi: 10.3390/v7062752
Hollingworth R., Horniblow R. D., Forrest C., Stewart G. S., Grand R. J. (2017). Localization of Double-Strand Break Repair Proteins to Viral Replication Compartments following Lytic Reactivation of Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 91, e00930–e00917. doi: 10.1128/JVI.00930-17
Hu Y., Petit S. A., Ficarro S. B., Toomire K. J., Xie A., Lim E., et al. (2014). PARP1-Driven Poly-ADP-Ribosylation Regulates BRCA1 Function in Homologous Recombination-Mediated DNA Repair. Cancer Discovery 4, 1430–1447. doi: 10.1158/2159-8290.CD-13-0891
Jackson B. R., Noerenberg M., Whitehouse A. (2014). A Novel Mechanism Inducing Genome Instability in Kaposi’s Sarcoma-Associated Herpesvirus Infected Cells. PloS Pathog. 10, e1004098. doi: 10.1371/journal.ppat.1004098
Jette N., Lees-Miller S. P. (2015). The DNA-dependent protein kinase: A multifunctional protein kinase with roles in DNA double strand break repair and mitosis. Prog. Biophys. Mol. Biol. 117, 194–205. doi: 10.1016/j.pbiomolbio.2014.12.003
Jha H. C., Upadhyay S. K., Aj M. P., Lu J., Cai Q., Saha A., et al. (2013). H2AX Phosphorylation Is Important for LANA-Mediated Kaposi’s Sarcoma-Associated Herpesvirus Episome Persistence. J. Virol. 87, 5255–5269. doi: 10.1128/JVI.03575-12
Jiricny J. (2006). The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 7, 335–346. doi: 10.1038/nrm1907
Kerur N., Veettil M. V., Sharma-Walia N., Bottero V., Sadagopan S., Otageri P., et al. (2011). IFI16 Acts as a Nuclear Pathogen Sensor to Induce the Inflammasome in Response to Kaposi Sarcoma-Associated Herpesvirus Infection. Cell Host. Microbe 9, 363–375. doi: 10.1016/j.chom.2011.04.008
Kilic S., Lezaja A., Gatti M., Bianco E., Michelena J., Imhof R., et al. (2019). Phase separation of 53 BP 1 determines liquid-like behavior of DNA repair compartments. EMBO J. 38, e101379. doi: 10.15252/embj.2018101379
Kleinberger T. (2020). En Guard! The Interactions between Adenoviruses and the DNA Damage Response. Viruses 12:996. doi: 10.3390/v12090996
Koopal S., Furuhjelm J. H., Järviluoma A., Jäämaa S., Pyakurel P., Pussinen C., et al. (2007). Viral Oncogene–Induced DNA Damage Response Is Activated in Kaposi Sarcoma Tumorigenesis. PloS Pathog. 3, e140. doi: 10.1371/journal.ppat.0030140
Kudoh A., Iwahori S., Sato Y., Nakayama S., Isomura H., Murata T., et al. (2009). Homologous Recombinational Repair Factors Are Recruited and Loaded onto the Viral DNA Genome in Epstein-Barr Virus Replication Compartments. JVI 83, 6641–6651. doi: 10.1128/JVI.00049-09
Kumar A., Sahu S. K., Mohanty S., Chakrabarti S., Maji S., Reddy R. R., et al. (2014). Kaposi Sarcoma Herpes Virus Latency Associated Nuclear Antigen Protein Release the G2/M Cell Cycle Blocks by Modulating ATM/ATR Mediated Checkpoint Pathway. PloS One 9, e100228. doi: 10.1371/journal.pone.0100228
Lavin M., Kozlov S., Gatei M., Kijas A. (2015). ATM-Dependent Phosphorylation of All Three Members of the MRN Complex: From Sensor to Adaptor. Biomolecules 5, 2877–2902. doi: 10.3390/biom5042877
Lavrik O. II, Prasad R., Sobol R. W., Horton J. K., Ackerman E. J., Wilson S. H. (2001). Photoaffinity Labeling of Mouse Fibroblast Enzymes by a Base Excision Repair Intermediate: EVIDENCE FOR THE ROLE OF POLY(ADP-RIBOSE) POLYMERASE-1 IN DNA REPAIR. J. Biol. Chem. 276, 25541–25548. doi: 10.1074/jbc.M102125200
Leppard J. B., Dong Z., Mackey Z. B., Tomkinson A. E. (2003). Physical and Functional Interaction between DNA Ligase III␣ and Poly(ADP-Ribose) Polymerase 1 in DNA Single-Strand Break Repair. Mol. Cell. Biol. 23, 9. doi: 10.1128/MCB.23.16.5919-5927.2003
Li R., Zhu J., Xie Z., Liao G., Liu J., Chen M.-R., et al. (2011). Conserved Herpesvirus Kinases Target the DNA Damage Response Pathway and TIP60 Histone Acetyltransferase to Promote Virus Replication. Cell Host. Microbe 10, 390–400. doi: 10.1016/j.chom.2011.08.013
Li G.-M. (2008). Mechanisms and functions of DNA mismatch repair. Cell Res. 18, 14. doi: 10.1038/cr.2007.115
Lieberman P. M. (2013). Keeping it quiet: chromatin control of gammaherpesvirus latency. Nat. Rev. Microbiol. 11, 863–875. doi: 10.1038/nrmicro3135
Lilley C. E., Chaurushiya M. S., Weitzman M. D. (2010). Chromatin at the intersection of viral infection and DNA damage. Biochim. Biophys. Acta (BBA) - Gene Regul. Mech. 1799, 319–327. doi: 10.1016/j.bbagrm.2009.06.007
Lilley C. E., Chaurushiya M. S., Boutell C., Everett R. D., Weitzman M. D. (2011). The Intrinsic Antiviral Defense to Incoming HSV-1 Genomes Includes Specific DNA Repair Proteins and Is Counteracted by the Viral Protein ICP0. PloS Pathog. 7, e1002084. doi: 10.1371/journal.ppat.1002084
Liu X., Happel C., Ziegelbauer J. M. (2017). Kaposi’s Sarcoma-Associated Herpesvirus MicroRNAs Target GADD45B To Protect Infected Cells from Cell Cycle Arrest and Apoptosis. J. Virol. 91, e02045–16, e02045-16. doi: 10.1128/JVI.02045-16
Liu K., Zheng M., Lu R., Du J., Zhao Q., Li Z., et al. (2020). The role of CDC25C in cell cycle regulation and clinical cancer therapy: a systematic review. Cancer Cell Int. 20, 213. doi: 10.1186/s12935-020-01304-w
Luijsterburg M. S., de Krijger I., Wiegant W. W., Shah R. G., Smeenk G., de Groot A. J. L., et al. (2016). PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol. Cell 61, 547–562. doi: 10.1016/j.molcel.2016.01.019
Mansour W. Y., Rhein T., Dahm-Daphi J. (2010). The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 38, 6065–6077. doi: 10.1093/nar/gkq387
Mariggiò G., Koch S., Zhang G., Weidner-Glunde M., Rückert J., Kati S., et al. (2017). Kaposi Sarcoma Herpesvirus (KSHV) Latency-Associated Nuclear Antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency. PloS Pathog. 13, e1006335. doi: 10.1371/journal.ppat.1006335
Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G. (1998). XRCC1 Is Specifically Associated with Poly(ADP-Ribose) Polymerase and Negatively Regulates Its Activity following DNA Damage. Mol. Cell. Biol. 18, 3563–3571. doi: 10.1128/MCB.18.6.3563
Menolfi D., Zha S. (2020). ATM, ATR and DNA-PKcs kinases—the lessons from the mouse models: inhibition ≠ deletion. Cell Biosci. 10:8. doi: 10.1186/s13578-020-0376-x
Ohsaki E., Ueda K., Sakakibara S., Do E., Yada K., Yamanishi K. (2004). Poly(ADP-Ribose) Polymerase 1 Binds to Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Terminal Repeat Sequence and Modulates KSHV Replication in Latency. J. Virol. 78, 9936–9946. doi: 10.1128/JVI.78.18.9936-9946.2004
Park J. W., Nickel K. P., Torres A. D., Lee D., Lambert P. F., Kimple R. J. (2014). Human papillomavirus type 16 E7 oncoprotein causes a delay in repair of DNA damage. Radiother. Oncol. 113, 337–344. doi: 10.1016/j.radonc.2014.08.026
Parravicini C., Chandran B., Corbellino M., Berti E., Paulli M., Moore P. S., et al. (2000). Differential Viral Protein Expression in Kaposi’s Sarcoma-Associated Herpesvirus-Infected Diseases. Am. J. Pathol. 156, 743–749. doi: 10.1016/S0002-9440(10)64940-1
Pfeffer S., Sewer A., Lagos-Quintana M., Sheridan R., Sander C., Grässer F. A., et al. (2005). Identification of microRNAs of the herpesvirus family. Nat. Methods 2, 269–276. doi: 10.1038/nmeth746
Pines A., Vrouwe M. G., Marteijn J. A., Typas D., Luijsterburg M. S., Cansoy M., et al. (2012). PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J. Cell Biol. 199, 235–249. doi: 10.1083/jcb.201112132
Planas-Silva M. D., Weinberg R. A. (1997). Estrogen-dependent cyclin E-cdk2 activation through p21 redistribution. Mol. Cell. Biol. 17, 4059–4069. doi: 10.1128/MCB.17.7.4059
Purushothaman P., Uppal T., Verma S. (2015). Molecular Biology of KSHV Lytic Reactivation. Viruses 7, 116–153. doi: 10.3390/v7010116
Qin J., Li W., Gao S.-J., Lu C. (2017). KSHV microRNAs: Tricks of the Devil. Trends Microbiol. 25, 648–661. doi: 10.1016/j.tim.2017.02.002
Ray Chaudhuri A., Nussenzweig A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621. doi: 10.1038/nrm.2017.53
Robu M., Shah R. G., Purohit N. K., Zhou P., Naegeli H., Shah G. M. (2017). Poly(ADP-ribose) polymerase 1 escorts XPC to UV-induced DNA lesions during nucleotide excision repair. Proc. Natl. Acad. Sci. U.S.A. 114, E6847–E6856. doi: 10.1073/pnas.1706981114
Romaker D., Schregel V., Maurer K., Auerochs S., Marzi A., Sticht H., et al. (2006). Analysis of the Structure–Activity Relationship of Four Herpesviral UL97 Subfamily Protein Kinases Reveals Partial but not Full Functional Conservation†. J. Med. Chem. 49, 7044–7053. doi: 10.1021/jm060696s
Ronson G. E., Piberger A. L., Higgs M. R., Olsen A. L., Stewart G. S., McHugh P. J., et al. (2018). PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 9, 746. doi: 10.1038/s41467-018-03159-2
Roth S., Rottach A., Lotz-Havla A. S., Laux V., Muschaweckh A., Gersting S. W., et al. (2014). Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1β production. Nat. Immunol. 15, 538–545. doi: 10.1038/ni.2888
Samols M. A., Hu J., Skalsky R. L., Renne R. (2005). Cloning and Identification of a MicroRNA Cluster within the Latency-Associated Region of Kaposi’s Sarcoma-Associated Herpesvirus. JVI 79, 9301–9305. doi: 10.1128/JVI.79.14.9301-9305.2005
Sarid R., Flore O., Bohenzky R. A., Chang Y., Moore P. S. (1998). Transcription Mapping of the Kaposi’s Sarcoma-Associated Herpesvirus (Human Herpesvirus 8) Genome in a Body Cavity-Based Lymphoma Cell Line (BC-1). J. Virol. 72, 1005–1012. doi: 10.1128/JVI.72.2.1005-1012.1998
Shin Y. C., Nakamura H., Liang X., Feng P., Chang H., Kowalik T. F., et al. (2006). Inhibition of the ATM/p53 Signal Transduction Pathway by Kaposi’s Sarcoma-Associated Herpesvirus Interferon Regulatory Factor 1. JVI 80, 2257–2266. doi: 10.1128/JVI.80.5.2257-2266.2006
Singh V. V., Dutta D., Ansari M. A., Dutta S., Chandran B., Longnecker R. (2014). Kaposi’s Sarcoma-Associated Herpesvirus Induces the ATM and H2AX DNA Damage Response Early during De Novo Infection of Primary Endothelial Cells, Which Play Roles in Latency Establishment. J. Virol. 88, 2821–2834. doi: 10.1128/JVI.03126-13
Smith H. L., Southgate H., Tweddle D. A., Curtin N. J. (2020). DNA damage checkpoint kinases in cancer. Expert Rev. Mol. Med. 22, e2. doi: 10.1017/erm.2020.3
Soulier J., Grollet L., Oksenhendler E., Cacoub P., Cazals-Hatem D., Babinet P. (1995). Kaposi’s Sarcoma-Associated Herpesvirus-Like DNA Sequences in Multicentric Castleman’s Disease. Blood 86 (4), 1276–1280. doi: 10.1182/blood.V86.4.1276.bloodjournal8641276
Spagnolo L., Barbeau J., Curtin N. J., Morris E. P., Pearl L. H. (2012). Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 40, 4168–4177. doi: 10.1093/nar/gkr1231
Stewart G. S., Wang B., Bignell C. R., Taylor A. M. R., Elledge S. J. (2003). MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature 421, 961–966. doi: 10.1038/nature01446
Sun R., Lin S.-F., Gradoville L., Yuan Y., Zhu F., Miller G. (1998). A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. 95, 10866–10871. doi: 10.1073/pnas.95.18.10866
Sun R., Lin S.-F., Staskus K., Gradoville L., Grogan E., Haase A., et al. (1999). Kinetics of Kaposi’s Sarcoma-Associated Herpesvirus Gene Expression. J. Virol. 73, 2232–2242. doi: 10.1128/JVI.73.3.2232-2242.1999
Tarakanova V. L., Leung-Pineda V., Hwang S., Yang C.-W., Matatall K., Basson M., et al. (2007). γ-Herpesvirus Kinase Actively Initiates a DNA Damage Response by Inducing Phosphorylation of H2AX to Foster Viral Replication. Cell Host. Microbe 1, 275–286. doi: 10.1016/j.chom.2007.05.008
Waldman T., Kinzler K. W., Vogelstein B. (1995). p21 Is Necessary for the p53-mediated G1 Arrest in Human Cancer Cells. Cancer Res. 55, 5187–1590.
Wang M., Wu W., Wu W., Rosidi B., Zhang L., Wang H., et al. (2006). PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 34, 6170–6182. doi: 10.1093/nar/gkl840
Wang Y., Li H., Tang Q., Maul G. G., Yuan Y. (2008). Kaposi’s Sarcoma-Associated Herpesvirus ori-Lyt-Dependent DNA Replication: Involvement of Host Cellular Factors. JVI 82, 2867–2882. doi: 10.1128/JVI.01319-07
Weitzman M. D., Fradet-Turcotte A. (2018). Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 5, 141–164. doi: 10.1146/annurev-virology-092917-043534
Williams A. B., Schumacher B. (2016). p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 6:a026070. doi: 10.1101/cshperspect.a026070
Keywords: Kaposi’s sarcoma-associated herpesvirus, DNA damage response, DNA repair, cell cycle, latency, lytic replication, KSHV, DDR
Citation: Ohsaki E and Ueda K (2020) Interplay Between KSHV and the Host DNA Damage Response. Front. Cell. Infect. Microbiol. 10:604351. doi: 10.3389/fcimb.2020.604351
Received: 09 September 2020; Accepted: 09 November 2020;
Published: 09 December 2020.
Edited by:
Subhash C. Verma, University of Nevada, Reno, United StatesReviewed by:
Paul Lesbats, UMR5234 Microbiologie Fondamentale et Pathogénicité (MFP), FranceAsuka Nanbo, Nagasaki University, Japan
Copyright © 2020 Ohsaki and Ueda. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eriko Ohsaki, ZW9oc2FraUB2aXJ1cy5tZWQub3Nha2EtdS5hYy5qcA==