 Tuodi Li
Tuodi Li Juan Wang
Juan Wang Qianqian Cao
Qianqian Cao Fei Li
Fei Li Jiangyuan Han
Jiangyuan Han Bingdong Zhu
Bingdong Zhu Ying Zhang2
Ying Zhang2 Hongxia Niu
Hongxia Niu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 30 September 2020
Sec. Molecular Bacterial Pathogenesis
Volume 10 - 2020 | https://doi.org/10.3389/fcimb.2020.581986
Persisters are metabolically quiescent phenotypic variants of the wild type that are tolerant to cidal antibiotics, and the mechanisms of persister formation and survival are complex and not completely understood. To identify genes involved in persistence to tosufloxacin, which has higher activity against persisters than most other quinolones, we screened the E. coli KEIO mutant library using a different condition from most persister mutant screens (6 h) with a longer exposure of 18 h with tosufloxacin. We identified 18 mutants (acrA, acrB, ddlB, dnaG, gltI, hlpA, lpcA, recG, recN, rfaH, ruvC, surA, tatC, tolQ, uvrD, xseA, and ydfI) that failed to form tosufloxacin tolerant persisters. Among them, gltI, hlpA, ruvC, ddlB, ydfI, and tatC are unique genes involved in E. coli persistence to tosufloxacin which have not been reported before. Furthermore, deletion mutants in genes coding periplasmic proteins (surA, lpcA, hlpA, and gltI) had more defect in persistence to tosufloxacin than the other identified mutants, with surA and lpcA mutants being the most prominent. The “deep” persister phenotype of surA and lpcA mutants was further confirmed both in vitro and in vivo. Compared with the wild type strain E. coli BW25113 in vitro, the persister phenotype of the surA and lpcA mutants was decreased more than 100–1,000-fold in persistence to various antibiotics, acidic, hyperosmotic and heat conditions. In addition, in both stationary phase bacteria and biofilm bacteria infection mouse models, the surA and lpcA mutants had lower survival and persistence than the parent uropathogenic strain UTI89, suggesting that the in vitro identified persister mechanisms (surA and lpcA) are operative and valid for in vivo persistence. Our findings provide new insight into the mechanisms of persister formation and maintenance under tosufloxacin and will likely provide novel therapeutic and vaccine targets for developing more effective treatment and prevention of persistent E. coli infections.
Persisters are a sub-population of genetically drug susceptible quiescent cells that are tolerant to cidal stresses (antibiotic, drug, heat, acidic pH and oxidative, etc.) and revive to metabolically active bacteria upon exiting stresses (Zhang, 2014). Persisters are implicated in many persistent bacterial infections, including tuberculosis, Lyme disease, urinary tract infections, and biofilm infections (Lewis, 2005; Stricker and Johnson, 2011; Zhang et al., 2012). As the quiescent persister bacteria have the capacity to survive antibiotics and revert to replicating forms, persisters are responsible for relapse after treatment.
Persisters have been regarded as phenotypic variants of the wild type that are not all the same. As expressed in a Yin-Yang model (Zhang, 2014), persisters have varying hierarchy and enormous heterogeneity, such as “shallow” persisters or “deep” persisters. The level and proportion of persister formation are affected by many factors, including bacterial age, environments that the bacteria reside in, and antibiotic exposure (Joers et al., 2010; Zhang, 2014). Due to its complexity, the mechanism of persister formation and survival are not completely understood. Mutagenesis screens have been traditionally used to isolate genes involved in persister formation. For example, screening the Escherichia coli KEIO mutant library identified several mutants (relA, phoU, sucB, and ubiF, etc.) with reduced antibiotic tolerance in different studies (Korch et al., 2003; Li and Zhang, 2007; Ma et al., 2010). It is observed that different persister genes can be identified at different exposure conditions of screening, including antibiotic type, concentration and exposure length.
We previously found that tosufloxacin (50 μM) compared with other quinolones had higher activity against both Escherichia coli (E. coli) and Staphylococcus aureus (S. aureus) persisters in vitro (Niu et al., 2015a,b), but it could not kill the biofilm persisters in vivo (unpublished observation). However, it is not clear what determines its unique activity against bacterial persisters. To identify genes involved in persistence to tosufloxacin, we screened the E. coli KEIO mutant library with tosufloxacin and identified several interesting candidate genes that encode periplasmic proteins SurA, LpcA, HlpA, and GltI whose mutations caused reduced persistence to tosufloxacin.
Uropathogenic E. coli strain UTI89, the KEIO collection of Escherichia coli K-12 single-gene knockout mutants and its parent strain BW25113 were used in this study. All the strains were cultured in Luria-Bertani (LB) medium. Tosufloxacin, levofloxacin, ofloxacin and gentamicin were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA) and were used at different concentrations (see below).
Human bladder epithelial cell strain (ATCC HTB-9) was purchased from Bio-feng company (Shanghai, China) and was cultured in RPMI 1640 with 10% newborn calf serum.
C57BL/6 or Bab/c female mice (6–8 weeks old) were obtained from Lanzhou Veterinary Research Institute and maintained in clean animal facility at Lanzhou University. Animals received free access to water and standard mouse chow throughout the study. The use of animals was in compliance with the guidelines set by the Institutional Animal Care and Use Committee of Lanzhou University.
The 3,985 single-gene knockout mutants in the E. coli KEIO mutant library were arrayed in 46 96-well plates. Then, the mutants were transferred to 200 μl fresh LB broth containing 30 μg/mL kanamycin using a 96-pin replicator and cultured overnight at 37°C without shaking. The overnight culture (~3 × 109 CFU/mL cells) was exposed to tosufloxacin (10 μM, 333 × MIC) for 18 h. Finally, bacteria were plated onto LB agar plates using 96-pin replicator and mutants with defective persistence, as indicated by the lack of growth, were identified for further analysis. The active hits were confirmed by rescreens using the same procedure for three times.
The identified mutants with defective persistence were cultured to stationary phase and dispensed into 1.5 mL Eppendorf tubes in 1 mL followed by addition of tosufloxacin (10 μM, 333 × MIC). Each day after exposure, 100 μl of the culture samples was taken out, washed with phosphate buffered saline (PBS), serially 10-fold diluted and plated for colony formation unit (CFU) count on LB agar plates.
To complement the gene knockout mutants of interest (ΔlpcA and ΔsurA), plasmids encoding lpcA and surA were generated by inserting the gene fragments into the multiple cloning sites of pTrc99a separately as follows. Initially, the DNA sequences of lpcA and surA were generated by PCR amplification from the DNA of wild type strain with primers lpcA A-F and lpcA A-R, surA A-F, and surA A-R (Table 1). Then the fragments were cloned into the unique sites of pTr99a plasmid to obtain the recombinant plasmids pTrc99a-lpcA and pTrc99a-surA. Finally, the recombinant plasmids or empty vector pTrc99a were transformed into the corresponding mutants to construct complementation strains.

Table 1. Primers used in this study.
E. coli wild type strain BW25113, mutants (ΔlpcA and ΔsurA) and complemented strains (ΔlpcA-pTrc99a-lpcA and ΔsurA-pTrc99a-surA) were all cultured overnight to stationary phase, and then treated with tosufloxacin (10 μM), levofloxacin (10 μM), ofloxacin (10 μM), and gentamicin (20 μM). Three days post exposure, the surviving bacteria were determined by CFU counting.
For acidic, hypertonic and heat stresses assays, stationary phase cultures were washed and diluted 100-fold, and then exposed to acidic condition (pH3.0 LB broth) for 2 days, hyperosmotic condition (3M NaCl LB broth) for 2 days, or heat condition (52°C water bath) for 3 h. Surviving bacteria were also determined by CFU counting.
Uropathogenic E. coli UTI89 mutant strains ΔsurA and ΔlpcA were generated using λ red homologous recombination system (Datsenko and Wanner, 2000). Firstly, linear fragment of FRT (flippase recognition target)-flanked chloramphenicol resistance gene were generated by PCR from pKD3 using primers surA B-F and surA B-R, lpcA B-F and lpcA B-R (Table 1). Then, pKD46 carrying the λ red recombinase gene was transformed into E. coli UTI89, and the transformants were grown in 5-mL LB broth with ampicillin and L-arabinose at 30°C to an OD600 of 0.4 and then made electrocompetent cells by concentrating and washing three times with ice-cold 10% glycerol. Next, the amplified linear fragments were transformed into the electrocompetent cells using Bio-Rad Gene Pulser Xcell™. The shocked cells were added to 1-mL LB, incubated 2 h at 37°C, and then the culture was spread onto LB agar with chloramphenicol to select Cm-resistant tansformants. Finally, PCR verify the deletion with primers surA C-F and surA C-R, lpcA C-F, and lpcA C-R (Table 1).
The biofilm formation of uropathogenic E. coli UTI89 and its mutant strains ΔsurA and ΔlpcA was investigated in microtiter plates as described (O'Toole, 2011). Briefly, uropathogenic E. coli UTI89 and the mutant strains were cultured overnight in 10 mL LB broth at 37°C, and then the bacterial culture was diluted 1:100 into fresh LB broth for biofilm assay. The dilutions were added in a 96 well dish (100 μL/well) with 8 replicate wells for each strain, and the microtiter plate was incubated for 24 h at 37°C. After incubation, each well was washed twice with PBS, added crystal violet solution (0.1%) and incubated for 10 min. Next, the biofilm stain was dissolved with 30% acetic acid and quantified absorbance in a plate reader at 550 nm.
The persistence of uropathogenic E. coli UTI89 mutant strains ΔsurA and ΔlpcA in vivo were analyzed in UTI mouse models (Hung et al., 2009). Firstly, C57BL/6 or Balb/c mice were anesthetized with 4% chloral hydrate (10 μL/g) by intraperitoneal injection. Then, the mice were inoculated with 50 μL stationary phase bacteria or biofilm bacteria (107 CFU per mice) via transurethral route. For stationary phase inoculum, bacteria were cultured over 24 h, and then were washed and resuspended in PBS prior to injection into C57BL/6 mice. For biofilm inoculum (O'Toole, 2011; Yee et al., 2019b), biofilms were first grown in LB medium in 96-well plates. After 24 h of incubation, the planktonic cells were removed, and the wells were washed with PBS. The biofilms in the bottom or the side wall of the wells were mechanically dislodged by scraping the bottom of the microtiter well using a pipette and resuspended in PBS. Quantification of all inoculum was performed by serial dilution and plating. On the 1st, 3rd, 5th, or 6th day post infection, mice were sacrificed and the whole bladder or kidney were harvested and homogenized aseptically. Finally, the individual homogenate was serial diluted with sterile PBS and plated on LB agar in triplicates for CFU count.
Adhesion and invasion assays were performed as described (Elsinghorst, 1994; Martinez et al., 2000). Firstly, human epithelial cells (ATCC HTB-9) were seed in 90 mm plates in 500 μL of RPMI 1640 with 10% newborn calf serum, and single-cell suspension of the epithelial cells was prepared by trypsinization. The suspension was diluted in fresh, pre-warmed 1640 medium, and the diluted cell suspension was added to a 24-well plate at 2 × 106 cell in 1 mL per well. E. coli UTI89 and its mutant strains ΔsurA and ΔlpcA were cultured overnight and diluted l: 1000 into fresh LB broth and grown at 37° to mid-log phase (OD600 = ~0.5). The mid-log phase bacteria were then centrifuged, resuspended in PBS and adjusted to appropriate concentration according to the results of CFU count. Next, epithelial cells were infected with bacteria in a multiplicity of infection (m.o.i.) of 5~10 per cell in three sets of triplicate wells. After 2 h co-incubation at 37°C, one set of the infected cells was lysed with 0.1% Triton X-100, and the lysates were plated on LB-agar plates to calculate the total number of infected bacteria (represented as A).
For adherence assays, a second set of cells were firstly washed five times with PBS, lysed with 0.1% Triton X-100, and then the lysates were plated on LB-agar plates to calculate the number of bacteria recovered after PBS washes (represented as B). Adherence frequency was calculated as B/A. For invasion assays, after the initial 2 h co-incubation, a third set of cells were firstly washed twice with PBS and then continue to incubate for 2 h with gentamicin (100 μg/mL) in RPM-1640 to kill extracellular bacteria. Cells were then washed additional three times with PBS, lysed with 0.1% Triton X-100, and plated on LB-agar plates to calculate the number of bacteria surviving incubation with gentamicin (represented as C). Invasion frequency was calculated as C/B.
All the experiments in this study were done in triplicates and repeated for three times, and the results were expressed as means ± SD. The significance of experimental differences between the mutants and the wild type strains was evaluated by unpaired Student's t-test using SPSS13.0 software. Values of p < 0.05 were considered as statistically significant.
To better understand the genes involved in persistence to tosufloxacin, we performed a genetic screen using the E. coli KEIO collection which contained 3,985 single-gene knockout mutants. Screening procedure and analysis are shown in methods. The overnight culture was exposed to tosufloxacin (10 μM), which has the highest bactericidal activity among different quinolones for E. coli (Hongxia et al., 2018), for 18 h. Using a 96-pin replicator, bacteria were stamped onto LB agar plates and mutants with defective persistence, as indicated by the lack of growth, were identified for further examination.
Of the 3,985 mutants in the E. coli KEIO collection, we identified 18 mutants (acrA, acrB, ddlB, dnaG, gltI, hlpA, lpcA, recG, recN, rfaH, ruvC, surA, tatC, tolQ, uvrD, xseA, and ydfI), that failed to grow on LB plates after exposure with tosufloxacin (10 μM) (Table 2). The target mutants were selected and rescreened twice to confirm a stable phenotype with defective persistence to tosufloxacin (10 μM). Of the mutant genes in the 18 identified mutants, dnaG, recG, recN, rfaH, ruvC, xseA, and xseB play a role in genetic processes such as DNA replication and repair; ddlB, ydfI, and tatC are enzymes (e.g., protein translocase, ligase or oxidoreductase); gltI, lpcA, hlpA, and surA are periplasmic proteins; acrA and acrB are acriflavine efflux proteins; tolQ is an outer membrane protein involved in the integrity of the bacterial envelope and the import of both filamentous phage and group A colicins.
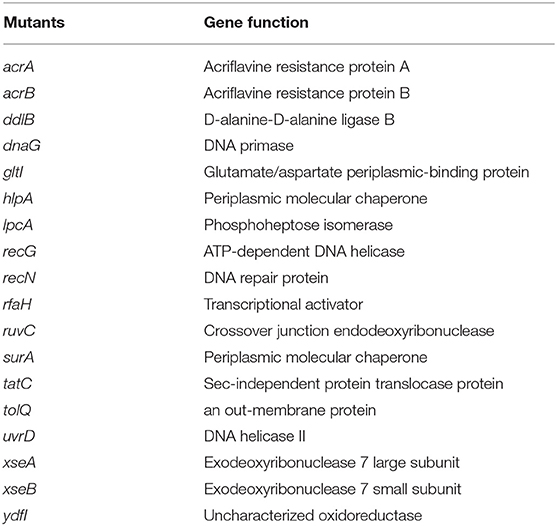
Table 2. Function of the identified 18 genes involved in tosufloxacin persistence.
To further validate and compare the persistence ability of the identified mutants above, we performed time-dependent killing studies as described in methods. Compared with the wild-type BW25113 strain, all the mutants exhibited increased susceptibility and defective persistence to tosufloxacin (10 μM) at various time points. However, after only 3 days of tosufloxacin (10 μM) exposure, there were no surviving cells in surA and lpcA mutants, whereas more than 102 CFU/mL bacteria still remained in the rest of the mutants and 105-106 CFU/mL bacteria in the wild-type BW25113 strain (Figures 1A–C). Next, gltI, hlpA, tatC, rfaH, ruvC mutants, died completely 4 days post tosufloxacin exposure (10 μM) (Figures 1B,C). The results showed that the surA and lpcA mutants were the most susceptible and had most defect in persistence to tosufloxacin.
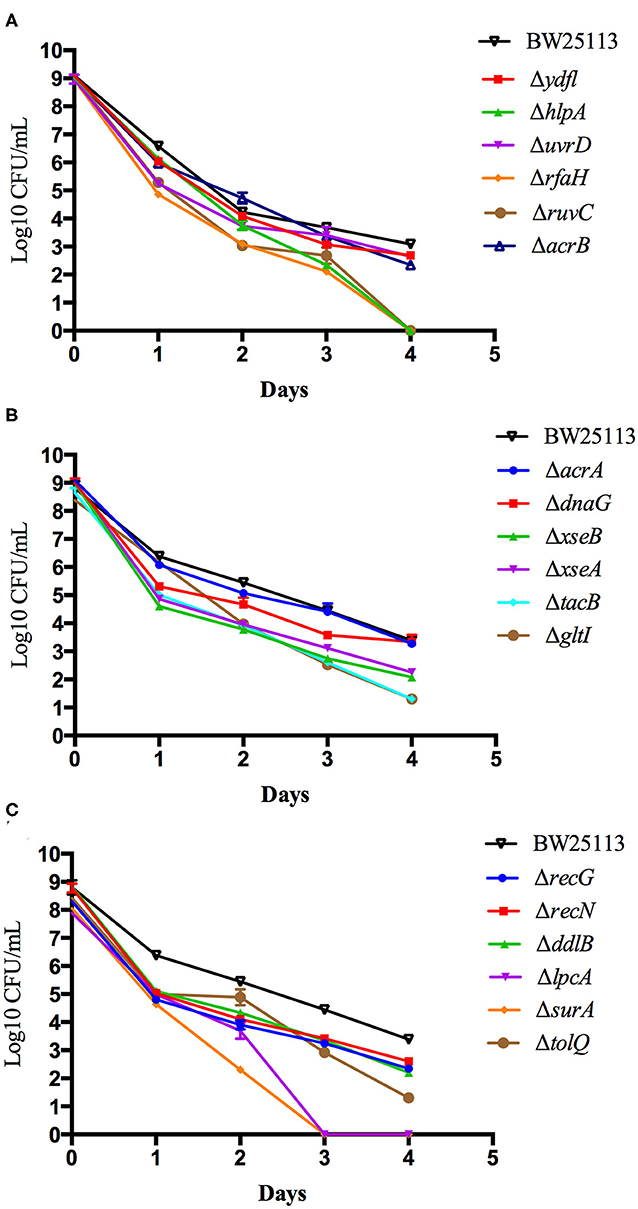
Figure 1. Compare the persistence ability of the identified mutants by CFU count. (A) Tosufloxacin killing curve of ydfl, hlpA, uvrD, rfaH, ruvC, acrB mutants. (B) Tosufloxacin killing curve of acrA, dnaG, xseB, xseA, tacB, gltI mutants. (C) Tosufloxacin killing curve of recG, recN, ddlB, lpcA, surA, tolQ mutants. Grow the identified mutants and their parent strain E. coli BW25113 in LB broth medium in tube. The cultures were grown to stationary phase (final concentration, >1 × 109 CFU/mL) and treated with tosufloxacin (10 μM) for the indicated days. Samples were diluted and spot plated on LB agar per day. The experiments were done in triplicates and repeated for three times. The results are expressed as means ± SD.
To further validate the role of surA and lpcA in persistence, the surA and lpcA mutants were complemented with their wild type gene using vector pTrc99a. Then we exposed all the mutants and their complemented strains and control strain to antibiotics including tosufloxacin, gentamicin, levofloxacin, ofloxacin and more stress conditions such as acidic, hypertonic and heat conditions.
Before the persistence assay, we first conducted a growth curve study by CFU counts to exclude the possibility that surA and lpcA mutants have any growth defects. The results showed that, under non-stress conditions, surA and lpcA mutants had the same growth ability in log phase and stationary phase compared with BW25113 strain (Figures 2, 3A). We then performed an MIC experiment for tosufloxacin, ciprofloxacin, ofloxacin, gentamicin and fosfomycin with these two mutants. Compared with control strain BW25113 strain, surA and lpcA mutants showed at least 4-folds decrease in MICs to tosufloxacin, while lpcA mutant had the same MICs to ciprofloxacin, ofloxacin, and surA mutant showed 2-fold decrease of MICs to ciprofloxacin, ofloxacin. And all the mutants had the same MICs to gentamicin and fosfomycin as BW25113 strain (Table 3).
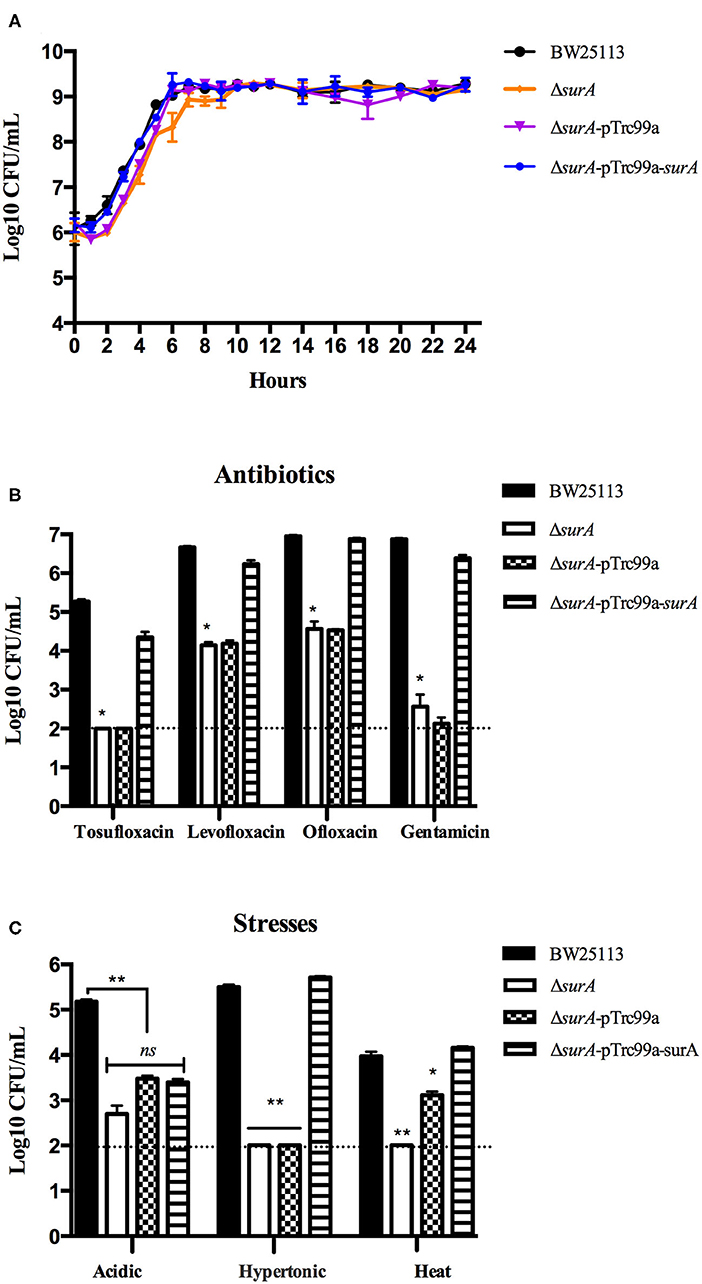
Figure 2. Survival of E. coli mutantΔsurA and its complemented strain exposed to different antibiotics and stress conditions. (A) Growth curve of the wild type strain E. coli BW25113, mutant strain ΔsurA, and complemented strain ΔsurA transformed with plasmid pTrc99a-surA. (B) Persistence ability of the stationary phase culture to antibiotics. All the strains were cultured overnight and then treated with tosufloxacin (10 μM), levofloxacin (10 μM), ofloxacin (10 μM), and gentamicin (20 μM) for 3 days. Survival of bacteria was determined by CFU counting. (C) Persistence ability of the stationary phase cultures to acidic, hypertonic and heat stresses. The stationary phase culture was diluted 100-fold and then exposed to acidic condition (pH3.0 LB broth) for 2 days, hyperosmotic condition (3M NaCl LB broth) for 2 days, or heat condition (52°C water bath) for 3 h. Survival of bacteria was determined by CFU counting. The experiments were done in triplicates and repeated for three times. The results are expressed as means ± SD. *p < 0.05, relative to BW25113 and ΔsurA-pTrc99a-surA groups. **p < 0.01, relative to BW25113 and ΔsurA-pTrc99a-surA groups. ns means no significance between the groups of ΔsurA and ΔsurA-pTrc99a-surA.
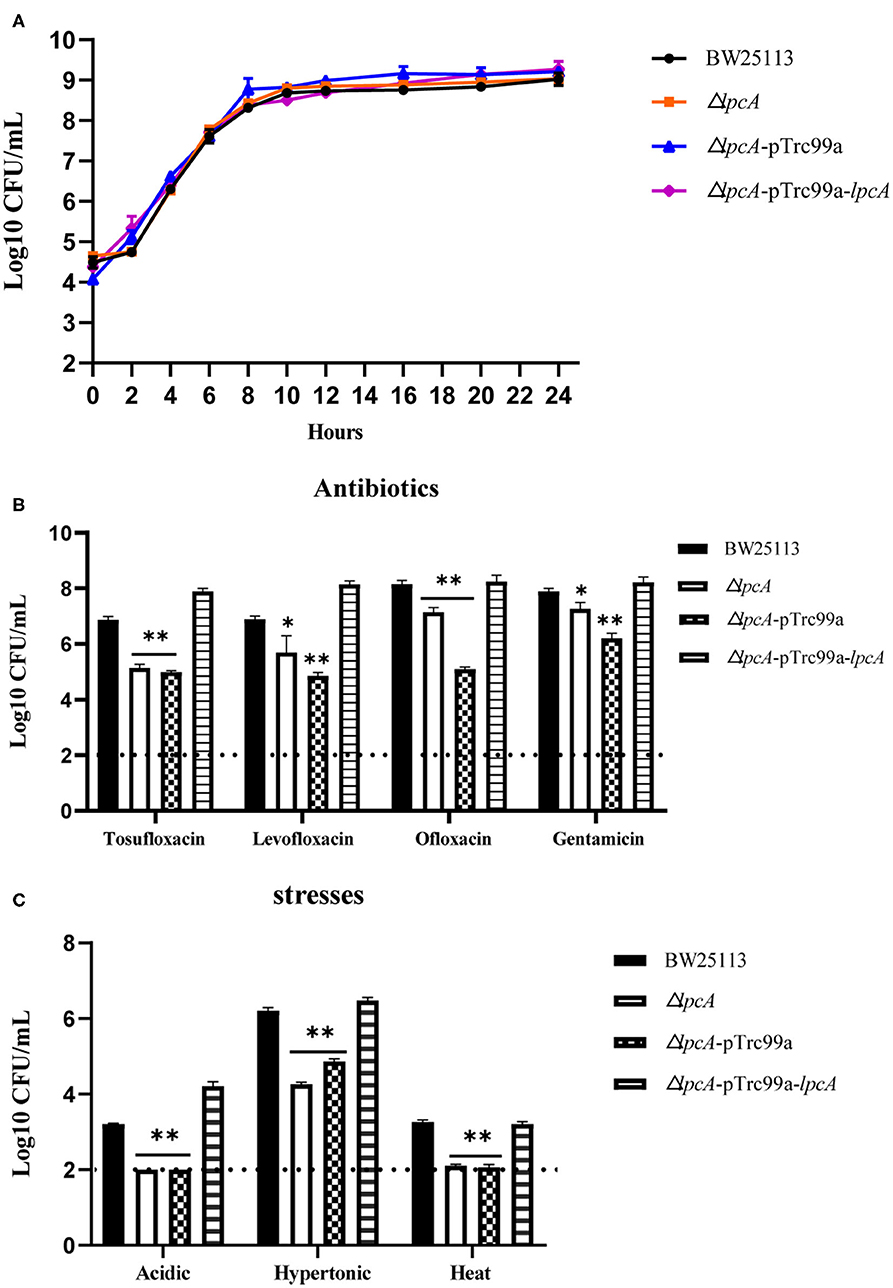
Figure 3. Survival of E. coli mutant ΔlpcA and its complemented strain exposed to different antibiotics and stress conditions. (A) Growth curve of the wild type strain E. coli BW25113, mutant strain ΔlpcA, and the complemented strain ΔlpcA transformed with plasmid pTrc99a-lpcA. (B) Persistence ability of the stationary phase culture to antibiotics. All the strains were cultured overnight and then treated with tosufloxacin (10 μM), levofloxacin (10 μM), ofloxacin (10 μM), and gentamicin (20 μM) for 3 days. Survival of bacteria was determined by CFU counting. (C) Persistent ability of the stationary phase cultures to acidic, hypertonic and heat stresses. The stationary phase culture was diluted 100-fold and then exposed to acidic condition (pH3.0 LB broth) for 2 days, hyperosmotic condition (3M NaCl LB broth) for 2 days, or heat condition (52 °C water bath) for 3 h. Survival of bacteria was determined by CFU counting. The experiments were done in triplicates and repeated for three times. The results are expressed as means ± SD. *p < 0.05, relative to BW25113 and ΔsurA-pTrc99a-surA groups. **p < 0.01, relative to BW25113 and ΔsurA-pTrc99a-surA groups.
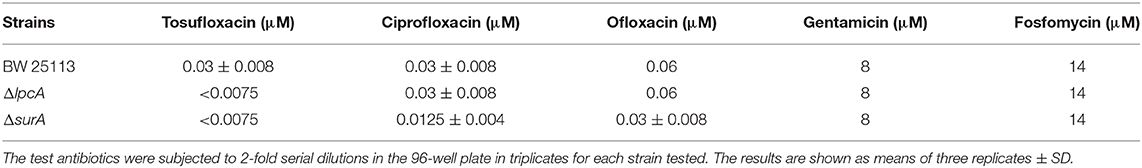
Table 3. The MICs of antibiotics (tosufloxacin, ciprofloxacin, oflofloxacin, gentamycin and fosfomycin) against E. coli BW25113 and its mutants ΔlpcA and ΔsurA.
Next, we performed the persistence assays, where stationary-phase bacteria were exposed to different stresses for different times separately, including treatment with tosufloxacin (10 μM), levofloxacin (10 μM), ofloxacin (10 μM) and gentamicin (20 μM) for 3 days, exposure to acidic condition (pH3.0 LB broth) for 2 days, hyperosmotic condition (3M NaCl LB broth) for 2 days, or heat condition (52°C water bath) for 3 h. surA mutant showed increased susceptibility and defective persistence to all the antibiotics (tosufloxacin, ciprofloxacin, ofloxacin and gentamicin), while its complemented strain ΔsurA-pTrc99a-surA restored the persistence level to that of the wild type strain (Figure 2B). When exposed to hyperosmotic and heat conditions, upon each time point, the surviving cells in the surA mutant was below the detection limit (10 CFU/mL), whereas the wild type strain BW25113 had more than 105 CFU/mL bacteria left in the hyperosmotic and 104 CFU/mL bacteria remained in heat treatment (Figure 2C). Under acidic condition, the remaining bacteria of the surA mutants was down to 102 CFU/mL, whereas wild type strain BW25113 had roughly 105 CFU/mL bacteria upon 2 days post exposure (Figure 2C). In addition, the persister levels of the complemented strain ΔsurA-pTrc99a-surA were restored to that of the wild type strain in hyperosmotic and heat conditions (Figures 2B,C).
Consistent with the surA mutant, lpcA mutant also displayed increased susceptibility and defective persistence to all the stresses (antibiotics, acidic, hypertonic and heat conditions) in different persistence assays, especially to acidic and heat conditions (Figures 3B,C). And, the persistence ability of its complemented strain ΔlpcA -pTrc99a-lpcA was restored to that of the wild type strain BW25113 (Figures 3B,C).
The results indicate that both surA and lpcA mutants had significantly less persistence ability to different antibiotics and stress conditions than the wild type strain BW25113 (p < 0.05) (Figures 2, 3).
Persister cells can be found to be heavily enriched inside biofilms due to the high cell density, and nutrient and oxygen limiting environment inside the biofilm matrix (Kavanaugh and Horswill, 2016). To evaluate whether mutations in surA and lpcA influence the biofilm formation of E. coli, we did a biofilm formation assay in a microtiter plate. We observed that uropathogenic E. coli UTI89 was capable of adhering to the walls and bottoms of the microtiter plate wells to form biofilm. However, the mutant strains UTI89 ΔsurA and ΔlpcA formed significant weaker biofilm than the wild type strain UTI89 (p < 0.001), with at least 50% reduction (Figure 4). Besides, mutant strain UTI89 ΔsurA showed much less biofilm formation than ΔlpcA (p < 0.05).
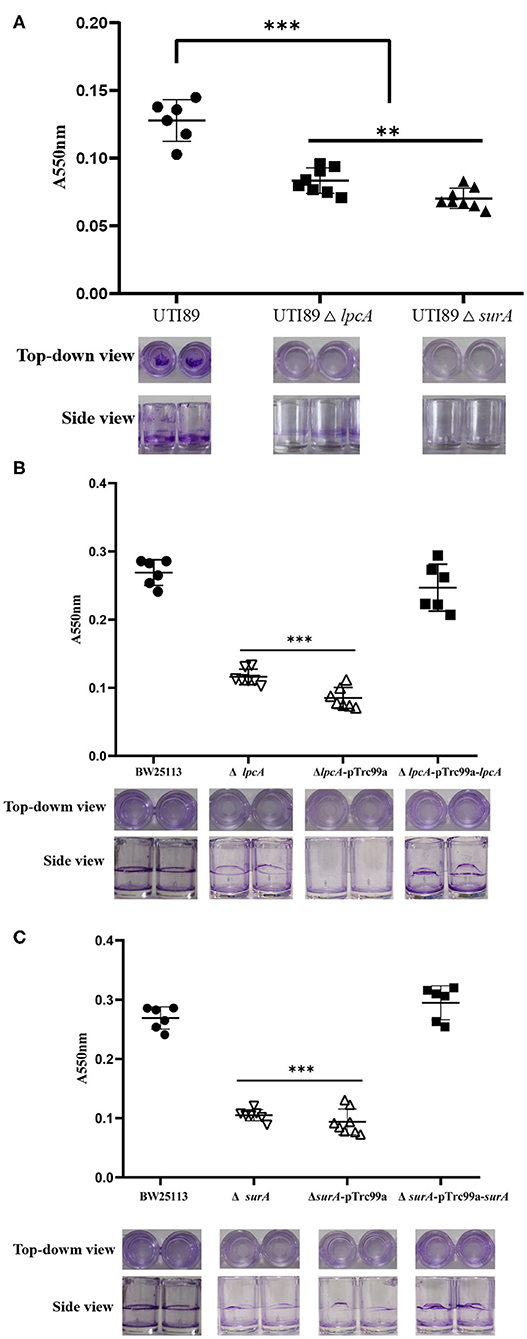
Figure 4. The biofilm formation level of E. coli ΔsurA and ΔlpcA mutants in microtiter dish. (A) Uropathogenic E. coli UTI89 ΔsurA and ΔlpcA mutants. (B) E. coli BW25113 ΔsurA and its complemented strain. (C) E. coli BW25113 ΔlpcA and its complemented strain. E. coli UTI89, E. coli BW25113 and their mutant strains ΔsurA and ΔlpcA were grown in a 96-well dish (100 μL/well) with 8 replicate wells for each strain, and the microtiter plate was incubated for 24 h at 37°C. Biofilm level was measured spectrophotometrically at 550 nm. Representative images of stained biofilms were shown at the bottom of the figure. The experiments were done with 8 replicates and repeated for three times. The results are expressed as means ± SD. **p < 0.01, compared with each other between UTI89 ΔsurA and ΔlpcA groups. ***p < 0.001, relative to UTI89 group.
Our in vitro persister assays revealed that both surA and lpcA mutants had defect in persistence to antibiotics and stresses. To further evaluate the ability of the surA and lpcA mutants to survive and persist in vivo, we could not use the non-pathogenic strain E. coli BW25113 and instead we constructed mutant strains of ΔsurA and ΔlpcA in uropathogenic E. coli UTI89 and evaluated their persistence ability in C57BL/6 and Bab/c mice. The C57BL/6 mice were infected with stationary phase persisters of E. coli UTI89 and its mutants ΔsurA and ΔlpcA, and on the 1st, 3rd, and 5th day post infection, the bacterial loads in bladder and kidney of the infected mice were analyzed. The Bab/c mice were infected with biofilm persisters of E. coli UTI89 and its mutants ΔsurA and ΔlpcA, and the bacterial loads in bladder, kidney and urine were analyzed 6 days after infection.
In C57BL/6 mice infected with stationary phase persisters, the bacterial loads in bladder and kidney were decreased gradually as time went on post infection in general for different strains, though the mutant strains ΔsurA and ΔlpcA had significant lower bacterial loads than their parental strain UTI89 (p < 0.01) (Figure 5). In bladder (Figure 5A), both the surA and lpcA mutants had ~103 CFU on the 5th day post infection, whereas the parental strain had 104 CFU. In kidneys (Figure 5B), on the 1st day post infection, no bacteria were detected in surA mutant infected mice, whereas 104 CFU and 105CFU bacteria were detected in lpcA mutant and UTI89 wild type strain infected mice separately; on the 3rd day post infection, the bacterial load in lpcA mutant decreased to the level below the detection limit, whereas UTI89 wild type strain still had 103 CFU bacteria remaining. The results suggest that surA and lpcA mutants have lower persistence than the wild type strain UTI89. Besides, stationary phase surA mutant was incapable of infecting kidney (Figure 5B).
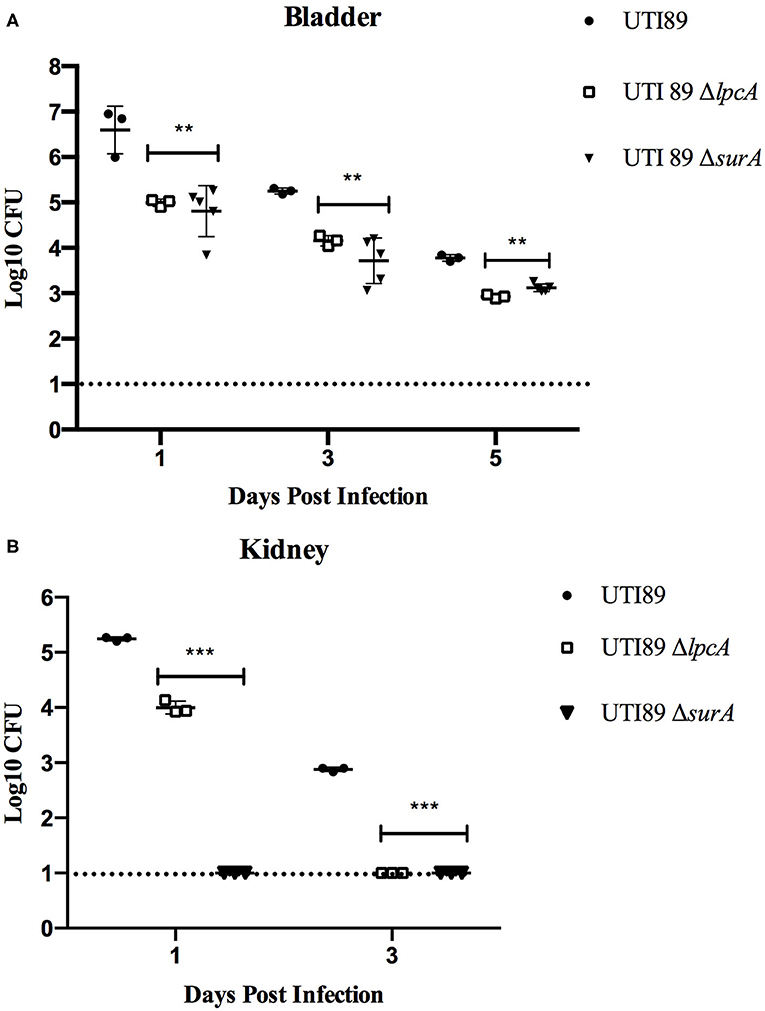
Figure 5. The persistence ability of stationary phase bacteria of uropathogenic E. coli UTI89 mutant strains ΔsurA and ΔlpcA in C57BL/6 mice. (A) Bacterial in bladder. (B) Bacterial in kidney. The mice were infected with E. coli UTI89, UTI89 ΔsurA, and UTI89 ΔlpcA (107CFU per mice) via transurethral route, respectively. On the 1st, 3rd, and 5th day post infection, mice were sacrificed and their whole bladder and kidney were homogenized. Then, the individual homogenization was serial diluted with sterile PBS and plated on LB agar in triplicates for CFU count (limit of detection is 10 bacteria/sample). The results (CFU/sample) are expressed as means of at least 3 mice ± SD. **p < 0.01, relative to UTI89 group. ***p < 0.001, relative to UTI89 group.
On 3rd or 6th days post infection, Bab/c mice infected with biofilm persisters harbored elevated bacterial loads compared to the mice infected with stationary phase persisters or log-phase bacteria (data not published), indicating that biofilm persisters are the most persistent form. In this biofilm persister infection mouse model, at 6 days after infection, the mice infected with mutant strains UTI89 ΔsurA and ΔlpcA harbored 103~104 CFU in bladder, kidney or urine, whereas the mice infected with wild type strain UTI89 had at least 1,000-10,000-folds higher bacteria (p < 0.01) (Figure 6). These in-vivo findings suggest that the mutation in surA or lpcA caused defect in persistence not only in vitro but also in vivo in the mouse persister inocula model.
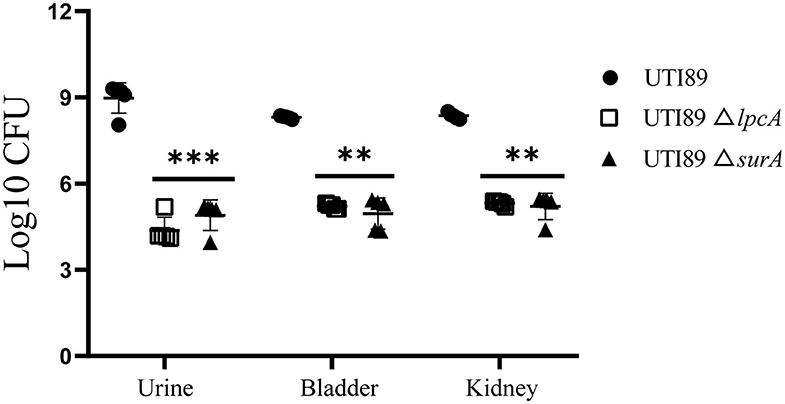
Figure 6. Defective persistence of the ΔsurA and ΔlpcA mutant strains compared with uropathogenic E. coli UTI89 parent strain in Bab/c mice. The mice were infected with biofilm persisters of E. coli UTI89 and its mutants ΔsurA and ΔlpcA (107 CFU per mice) via transurethral route, respectively. On 6th day post infection, mice were sacrificed and their whole bladder or kidneys were homogenized, and the homogenization was serial diluted and plated on LB agar for CFU count. The urine was serial diluted and plated on LB agar for CFU count directly, and the result is shown as CFU/mL. The results (CFU/sample or CFU/mL for urine) are expressed as means of 5 mice ± SD. **p < 0.01, relative to UTI89 group. ***p < 0.001, relative to UTI89 group.
To determine whether surA and lpcA participate in mediating bacterial adhesion and invasion of host cells, we further did the adhesion and invasion assays in human bladder epithelial cells. We infected the epithelial cells with E. coli UTI89 and its mutants ΔsurA and ΔlpcA for 2 h separately, and found that both the adhesion and invasion frequencies of the wild-type strain UTI89 were obviously higher than the mutant strains ΔsurA and ΔlpcA (p < 0.05) (Figure 7), suggesting that the expression of adhesin and invasin in E. coli might be regulated by surA and lpcA.
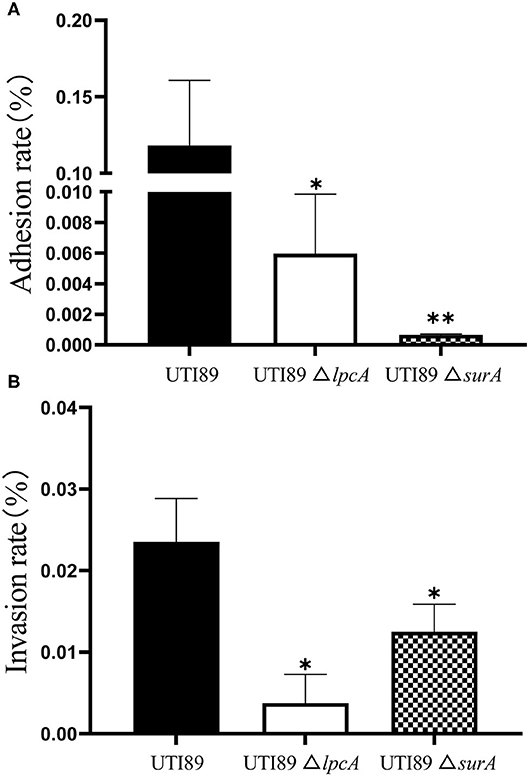
Figure 7. Defective adhesion and invasion of the ΔsurA and ΔlpcA mutants compared with uropathogenic E. coli UTI89 in bladder epithelial cells. (A) The adhesion ability of mutant strains. (B) The invasive capability of mutant strains. The human epithelial cells (ATCC HTB-9) (2 × 106 cell/well) were infected with log-phase E. coli UTI89 and its mutants ΔsurA and ΔlpcA in a multiplicity of infection (m.o.i.) of 5~10 per cell in 24-well plate. At 2 h post infection, the adhesion and invasion frequencies were analyzed as described in Methods. The experiments were done in triplicates and repeated for three times. The results are expressed as means ± SD. *p < 0.05, relative to UTI89 group. **p < 0.01, relative to UTI89 group.
We previously found that tosufloxacin (50 μM) had high activity against both E. coli and S. aureus persisters in vitro (Niu et al., 2015a,b), but it could not kill the biofilm persisters in vivo (unpublished observation). This means that persisters tolerant to tosufloxacin still exist, despite it is the most active quinolone against persisters in our previous study (Hongxia et al., 2018). In this study, using long exposure of 18 h with tosufloxacin (10 μM), we screened the E. coli KEIO mutant library to identify genes involved in persistence under tosufloxacin exposure. Though the KEIO collection had been used to identify persister related genes before, this is the first time it is used for identifying tosufloxacin tolerant persisters. We identified 18 mutants (acrA, acrB, ddlB, dnaG, gltI, hlpA, lpcA, recG, recN, rfaH, ruvC, surA, tatC, tolQ, uvrD, xseA, and ydfI) that failed to grow on LB plates after exposure with tosufloxacin (Table 2).
In contrast to using long exposure of 18 h with tosufloxacin as in this study, a previous study was done using short exposure of 6 h with ofloxacin to screen the same mutant library at stationary phase culture (Hansen et al., 2008). Within the identified mutants, apart from genes involved in DNA repair and recombination (recG, xseA, xseB) which were excluded from the previous study owing to increased sensitivity to ofloxacin, periplasmic molecular chaperone surA were identified in both screens. GltI, hlpA, ruvC, ddlB, ydfI, and tatC have not previously been described as persister genes, and we found that their mutations caused defect in persistence exposure to tosufloxacin, indicating that these are unique genes involved in E. coli persistence to tosufloxacin. It is of interest to note that recG, recN, lpcA, acrA, and acrB were identified as genes related to rifampin induced antagonism or persister formation to ofloxacin by our group previously (Cui et al., 2018). The results also further confirm the phenomenon that different persister genes will be identified using different antibiotics at different antibiotic exposure times.
Furthermore, the deletion mutants in genes coding periplasmic proteins (surA, lpcA, hlpA, and gltI) had more defect in persistence to tosufloxacin than the other identified mutants, with surA and lpcA mutants being the most prominent. Although surA and lpcA were identified as possible persister genes before (Hansen et al., 2008; Cui et al., 2018), they have not been confirmed in vivo or in vitro. Thus, we chose surA and lpcA to further confirm their roles in E. coli persistence in vivo or in vitro separately. Our results showed that the persister phenotype showed more than 100–1,000-fold decrease in persistence against several stresses (antibiotics including tosufloxacin, ciprofloxacin, ofloxacin and gentamicin; acidic, hyperosmotic and heat conditions) for the surA and lpcA mutants than the wild type strain E. coli BW25113 in vitro. The “deep” persiser phenotype was further confirmed by complementation studies, and indeed the persistence phenotype of the complemented strains was restored to that of the wild type strain. Besides, both in the stationary phase bacteria and biofilm bacterial infection models, the surA and lpcA mutants had lower survival than the parent strain UTI89 in vivo in mice, and the stationary phase mutants had no or limited ability to infect kidney. Thus, our findings indicate that the in vitro identified persister mechanisms (surA and lpcA) are operative and valid for in vivo persisters, suggesting that proteins SurA, HlpA, GltI and LpcA are indeed important in the formation and survival of E. coli persisters not only in vitro but also in vivo. It is worth noting the unique genes identified in this study are mainly involved in periplasmic function of the cell, which may be related to the unique high activity of tosufloxacin against persisters. This is because the cell membrane is a known target of persister drugs (Hurdle et al., 2011). Future studies are needed to confirm this.
SurA was described as a parvulin-like peptidyl-prolyl isomerase (PPIase) and a periplasmic holdase chaperone, which facilitates the folding, assembling, delivery and maturation of β-barrel outer membrane proteins (OMPs) such as OmpA, OmpF, and LamB in Gram-negative bacteria (Thoma et al., 2015). In this study, we found that the persister phenotype of the surA mutant was decreased more than 100–1,000-fold in persistence to various antibiotics, acidic, hyperosmotic and heat conditions. The possible mechanism is that deletion of surA gene in E. coli may lead to a defect in outer membrane and thus decreased persistence of bacteria against antibiotics and other stresses in vitro. In addition, surA mutant had limited ability to form biofilm and decreased ability to attach and invade urothelial cells. Possible explanation for these phenomenons may be: (1) SurA functions as a chaperone for the outer membrane fimbrial usher FimD which is essential for type 1- and p-fimbriae (Justice et al., 2005). Accordingly, mutation in surA could influence the biogenesis of type 1-and p-fimbriae, which will impact the adherence and invasion of host cells. (2) Lack of surA in E. coli might induce the RcsCDB phosphorelay signaling pathway (Castanie-Cornet et al., 2006), which is required for biofilm development. Moreover, surA was still required for full virulence of E. coli, including the ability of the bacteria to compete with their host for iron (Vertommen et al., 2009), and insertion of LPS into the outer leaflet of the outer membrane (Bos et al., 2004). Thus, deletion of surA gene in E. coli UTI89, which decreased the pathogenicity and persistence of UTI89 bacteria in vivo, is consistent with its important role in maintaining fimbriae function. HlpA encodes periplasmic holdase chaperone Skp, which cooperates with surA in the biogenesis of β-barrel OMPs.In this study, we identified hlpA may be an important “deep” persister gene as surA.
Glutamate is important in the process of bacterial metabolism. E. coli contains several transporters for the uptake of glutamate, such as GltIJKL, GltP, GltS, and GadC. Among them, GltI is a known dual glutamate and aspartate periplasmic-binding protein which is a component of the sodium-dependent ABC transporter system (Willis and Furlong, 1975); GltP is a cytoplasmic membrane protein which is a carrier of an Na+-independent, binding protein-independent glutamate/aspartate transport system, but the GltP transport system is deficient in E. coli k12 strain (Deguchi et al., 1989); GltS is a carrier protein in cytoplasmic membrane which is a Na+-dependent, binding protein-independent glutamate transport system (Kahane et al., 1975); GadC is a glutamate antiporter in cytoplasmic membrane which regulates E. coli acid resistance (Richard and Foster, 2007). In this study, we found that gltI is involved in tosufloxacin-tolerant persister formation. In addition, we also found that gltS is involved in E. coli persister formation and survival both in vitro and in vivo (data not published). These findings reveal the pivotal role of glutamate metabolism in E. coli persistence. This finding is consistent with the previous observation in S. aureus where glutamate metabolism and transport have been found to be involved in persister survival (Yee et al., 2015, 2019a). The mechanism involved could be that glutamate metabolites such as NH3 allow bacteria to adapt to stress environments or work through regulating reactive oxygen species (ROS) generation by glutathione.
LpcA (also known as GmhA) is a soluble protein present in the cytoplasmic fraction, which functions as a phosphoheptose isomerase used in the biosynthesis of the core component of lipopolysaccharide (LPS) precursor glyceromannoheptose (Brooke and Valvano, 1996). The LPS of lpcA mutant lacks heptose (Tamaki et al., 1971) and the lpcA mutant had decreased persistence in vitro and in vivo, suggesting that the structural integrity of LPS is important for bacterial persistence and survival under stresses. We also found that lpcA mutant had limited ability to form biofilm and also had decreased ability to attach and invade urothelial cells (Figures 4, 7), suggesting the important role of bacterial LPS in biofilm formation and pathogenicity. Possible explanations for these phenomena may be: (1) mutant of lpcA may change the permeability barrier of outer membrane which prevents entry of small hydrophobic antibiotics (Leive, 1974; Nikaido, 2003), such as tosufloxacin, and allows Gram-negative bacteria to survive in stress environments, resulting in decreased persistence and survival under antibiotics or stresses. (2) LPS has the ability to induce expression of adhesion molecule and chemokines (Tanaka et al., 1997). Mutant of lpcA may change the structure of LPS, which will impact adherence and invasion and survival in the host cells.
In summary, we identified 18 mutants (acrA, acrB, ddlB, dnaG, gltI, hlpA, lpcA, recG, recN, rfaH, ruvC,surA, tatC, tolQ, uvrD, xseA, and ydfI) that failed to grow after exposure with tosufloxacin. Among these, GltI, hlpA, ruvC, ddlB, ydfI, and tatC have not previously been described as persister genes. The surA and lpcA mutants are the most prominent in persistence defect as shown by in vitro and in vivo tests followed by hlpA and gltI mutants. The genes identified by this study provide new insight into the mechanisms of persister formation and maintenance under tosufloxacin. Our findings will likely provide novel therapeutic and vaccine targets for developing more effective treatment and prevention of persistent E. coli infections.
All datasets generated for this study are included in the article/supplementary material.
The animal study was reviewed and approved by The Institutional Animal Care and Use Committee of Lanzhou University.
HN designed the study, performed part of the experiments, analyzed the data, and wrote the manuscript. TL mainly performed the experiments and analyzed the data. YZ conceived the work and revised the manuscript. JW, QC, FL, and JH performed part of the experiments. All authors agree to be accountable for the content of the work. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Youth Fund of China [81701969] and the Fundamental Research Funds for the Central Universities of the Lanzhou University [lzujbky-2018-85].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Bos, M. P., Tefsen, B., Geurtsen, J., and Tommassen, J. (2004). Identification of an outer membrane protein required for the transport of lipopolysaccharide to the bacterial cell surface. Proc. Natl. Acad. Sci. U.S.A. 101, 9417–9422. doi: 10.1073/pnas.0402340101
Brooke, J. S., and Valvano, M. A. (1996). Biosynthesis of inner core lipopolysaccharide in enteric bacteria identification and characterization of a conserved phosphoheptose isomerase. J Biol. Chem. 271, 3608–3614. doi: 10.1074/jbc.271.7.3608
Castanie-Cornet, M. P., Cam, K., and Jacq, A. (2006). RcsF is an outer membrane lipoprotein involved in the RcsCDB phosphjorelay signaling pathway in Escherichia coli. J. Bacteriol. 188, 4264–4270. doi: 10.1128/JB.00004-06
Cui, P., Niu, H., Shi, W., Zhang, S., Zhang, W., and Zhang, Y. (2018). Identification of genesinvolved in bacteriostatic antibiotic-induced persister formation. Front. Microbiol. 9:413. doi: 10.3389/fmicb.2018.00413
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A. 97, 6640–6645. doi: 10.1073/pnas.120163297
Deguchi, Y., Yamato, I., and Anraku, Y. (1989). Molecular cloning of gitS and gltP, which encode glutamate carriers of Escherichia coli B. J. Bacteriol. 171, 1314–1319. doi: 10.1128/JB.171.3.1314-1319.1989
Elsinghorst, E. A. (1994). Measurement of invasion by gentamicin resistance. Methods Enzymol. 236, 405–420. doi: 10.1016/0076-6879(94)36030-8
Hansen, S., Lewis, K., and Vulic, M. (2008). Role of global regulators and nucleotide metabolism in antibiotic tolerance in Escherichia coli. Antimicrob. Agents Chemother. 52, 2718–2726. doi: 10.1128/AAC.00144-08
Hongxia, N., Yee, R., Cui, P., Zhang, S., Tian, L., Shi, W., et al. (2018). Identification and ranking of clinical compounds with activity against log-phase growing uropathogenic Escherichia coli. Curr. Drug Discov. Technol. 17, 191–196. doi: 10.2174/1570163815666180808115501
Hung, C. S., Dodson, K. W., and Hultgren, S. J. (2009). A murine model of urinary tract infection. Nat. Protoc. 4, 1230–1243. doi: 10.1038/nprot.2009.116
Hurdle, J. G., O'Neill, A. J., Chopra, I., and Lee, R. E. (2011). Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat. Rev. Microbiol. 9, 62–75. doi: 10.1038/nrmicro2474
Joers, A., Kaldalu, N., and Tenson, T. (2010). The frequency of persisters in Escherichia coli reflects the kinetics of awakening from dormancy. J. Bacteriol. 192, 3379–3384. doi: 10.1128/JB.00056-10
Justice, S. S., Hunstad, D. A., Harper, J. R., Duguay, A. R., Pinkner, J. S., Bann, J., et al. (2005). Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J. Bacteriol. 187, 7680–7686. doi: 10.1128/JB.187.22.7680-7686.2005
Kahane, S., Marcus, M., Barash, H., and Halpern, Y. S. (1975). Sodium-dependent glutamate transport in membrane vesicles of Escherichia coli K-12. FEBS Lett. 56, 235–239. doi: 10.1016/0014-5793(75)81099-4
Kavanaugh, J. S., and Horswill, A. R. (2016). Impact of environmental cues on staphylococcal quorum sensing and biofilm development. J. Biol. Chem. 291, 12556–12564. doi: 10.1074/jbc.R116.722710
Korch, S. B., Henderson, T. A., and Hill, T. M. (2003). Characterization of the hipA7 allele of Escherichia coli and evidence that high persistence is governed by (p)ppGpp synthesis. Mol. Microbiol. 50, 1199–1213. doi: 10.1046/j.1365-2958.2003.03779.x
Leive, L. (1974). The barrier function of the gram-negative envelope. Ann. N. Y. Acad. Sci. 235, 109–129. doi: 10.1111/j.1749-6632.1974.tb43261.x
Lewis, K. (2005). Persister cells and the riddle of biofilm survival. Biochemistry. 70, 267–274. doi: 10.1007/s10541-005-0111-6
Li, Y. F., and Zhang, Y. (2007). PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob. Agents Chemother. 51, 2092–2099. doi: 10.1128/AAC.00052-07
Ma, C., Sim, S., Shi, W., Du, L., Xing, D., and Zhang, Y. (2010). Energy production genes sucB and ubiF are involved in persister survival and tolerance to multiple antibiotics and stresses in Escherichia coli. FEMS Microbiol. Lett. 303, 33–40. doi: 10.1111/j.1574-6968.2009.01857.x
Martinez, J. J., Mulvey, M. A., Schilling, J. D., Pinkner, J. S., and Hultgren, S. J. (2000). Type 1 pilus-mediated bacterial invasion of bladder epithelial cells. EMBO J. 19, 2803–2812. doi: 10.1093/emboj/19.12.2803
Nikaido, H. (2003). Molecular basis of bacterial outer membrane permeability revisited. Microbiol. Mol. Biol. Rev. 67, 593–656. doi: 10.1128/MMBR.67.4.593-656.2003
Niu, H., Cui, P., Shi, W., Zhang, S., Feng, J., Wang, Y., et al. (2015a). Identification of anti-persister activity against Uropathogenic Escherichia coli from a clinical drug library. Antibiotics 4, 179–187. doi: 10.3390/antibiotics4020179
Niu, H., Cui, P., Yee, R., Shi, W., Zhang, S., Feng, J., et al. (2015b). A clinical drug library screen identifies tosufloxacin as being highly active against Staphylococcus aureus persisters. Antibiotics 4, 329–336. doi: 10.3390/antibiotics4030329
O'Toole, G. A. (2011). Microtiter dish biofilm formation assay. J. Vis. Exp. 30:2437. doi: 10.3791/2437
Richard, H., and Foster, J. W. (2007). Sodium regulates Escherichia coli acid resistance, and influences GadX- and GadW-dependent activation of gadE. Microbiology. 153, 3154–3161. doi: 10.1099/mic.0.2007/007575-0
Stricker, R. B., and Johnson, L. (2011). The pain of chronic Lyme disease: moving the discourse backward? FASEB J. 25, 4085–4087. doi: 10.1096/fj.11-1203LTR
Tamaki, S., Sato, T., and Matsuhashi, M. (1971). Role of lipopolysaccharides in antibiotic resistance and bacteriophage adsorption of Escherichia coli K-12. J. Bacteriol. 105, 968–975. doi: 10.1128/JB.105.3.968-975.1971
Tanaka, M., Aze, Y., and Fujita, T. (1997). Adhesion molecule LFA-1/ICAM-1 influences on LPS-induced megakaryocytic emperipolesis in the rat bone marrow. Vet. Pathol. 34, 463–466. doi: 10.1177/030098589703400511
Thoma, J., Burmann, B. M., Hiller, S., and Muller, D. J. (2015). Impact of holdase chaperones Skp and SurA on the folding of beta-barrel outer-membrane proteins. Nat. Struct. Mol. Biol. 22, 795–802. doi: 10.1038/nsmb.3087
Vertommen, D., Ruiz, N., Leverrier, P., Silhavy, T. J., and Collet, J. F. (2009). Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9, 2432–2443. doi: 10.1002/pmic.200800794
Willis, R. C., and Furlong, C. E. (1975). Purification and properties of a periplasmic glutamate-aspartate binding protein from Escherichia coli K12 strain W3092. J. Biol. Chem. 250, 2574–2580.
Yee, R., Cui, P., Shi, W., Feng, J., and Zhang, Y. (2015). Genetic screen reveals the role of purine metabolism in Staphylococcus aureus persistence to Rifampicin. Antibiotics 4, 627–642. doi: 10.3390/antibiotics4040627
Yee, R., Feng, J., Wang, J., Chen, J., and Zhang, Y. (2019a). Identification of genes regulating cell death in Staphylococcus aureus. Front. Microbiol. 10:2199. doi: 10.3389/fmicb.2019.02199
Yee, R., Yuan, Y., Shi, W., Brayton, C., Tarff, A., Feng, J., et al. (2019b). Infection with persister forms of Staphylococcus aureus causes a persistent skin infection with more severe lesions in mice: failure to clear the infection by the current standard of care treatment. Discov. Med. 28, 7–16.
Zhang, Y. (2014). Persisters, persistent infections and the Yin-Yang model. Emerg. Microbes Infect. 3:e3. doi: 10.1038/emi.2014.3
Keywords: Escherichia coli, persistence, tosufloxacin, persister, molecular mechanism
Citation: Li T, Wang J, Cao Q, Li F, Han J, Zhu B, Zhang Y and Niu H (2020) Identification of Novel Genes Involved in Escherichia coli Persistence to Tosufloxacin. Front. Cell. Infect. Microbiol. 10:581986. doi: 10.3389/fcimb.2020.581986
Received: 10 July 2020; Accepted: 01 September 2020;
Published: 30 September 2020.
Edited by:
Teresa Thurston, Imperial College London, United KingdomReviewed by:
Emily Mallick, Fluid-Screen, United StatesCopyright © 2020 Li, Wang, Cao, Li, Han, Zhu, Zhang and Niu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongxia Niu, bml1aHhAbHp1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.