 Christine A. King
Christine A. King Adam D. Wegman
Adam D. Wegman Timothy P. Endy
Timothy P. Endy- Department of Microbiology and Immunology, State University of New York (SUNY) Upstate Medical University, Syracuse, NY, United States
Dengue virus is an important human pathogen, infecting an estimated 400 million individuals per year and causing symptomatic disease in a subset of approximately 100 million. Much of the effort to date describing the host response to dengue has focused on the adaptive immune response, in part because of the well-established roles of antibody-dependent enhancement and T cell original sin as drivers of severe dengue upon heterotypic secondary infection. However, the innate immune system is a crucial factor in the host response to dengue, as it both governs the fate and vigor of the adaptive immune response, and mediates the acute inflammatory response in tissues. In this review, we discuss the innate inflammatory response to dengue infection, focusing on the role of evolutionarily conserved innate immune cells, their effector functions, and clinical course.
Introduction
Dengue Virus and Clinical Spectra
Dengue virus (DENV) is an arbovirus transmitted by the mosquito vectors Aedes aegypti and, to a lesser extent, Aedes albopictus (Scott and Morrison, 2010). Dengue virus belongs to the family Flaviviridae and is a single stranded, positive sense, enveloped, RNA virus. The genome is approximately 11 kb and encodes 10 proteins. Upon infection the viral genome is delivered to the cytoplasm and translated into one long polyprotein that is then cleaved by both host and viral specific proteases to yield 10 individual proteins. Three are structural proteins (envelope, core, and membrane) and seven are non-structural (NS) proteins (NS1, NS2a and NS2b, NS3, NS4a and NS4b, and NS5). Dengue is endemic in tropical and subtropical regions of the world where 2.5 billion people are at risk for infection. With approximately 400 million infections annually (WHO, 2009; Bhatt et al., 2013), dengue disease is a serious public health threat with no specific treatments currently available. There are currently four circulating serotypes (DENV-1 to 4) that exhibit up to 70% sequence homology (Blok, 1985; Green and Rothman, 2006). All four serotypes can cause a spectrum of disease with manifestations ranging from a subclinical infection to a mild febrile illness termed dengue fever (DF). In a subset of infections, severe hemorrhagic manifestations or shock syndrome known as dengue hemorrhagic fever (DHF) and dengue shock syndrome (DSS) (WHO, 2009) can develop. While the majority of patients develop only mild symptoms and recover after defervescence, approximately 5% develop life threating vascular dysfunction (Gubler, 1998; Halstead, 2007). The pathogenesis of severe dengue disease has been the focus of countless studies, and some progress in understanding disease associations and mechanisms has been made. What is known is that severe dengue disease most often occurs during a secondary DENV infection with a heterologous serotype (Halstead, 1994; Gubler, 1998; Halstead, 2007). This phenomenon is thought to involve antibody-dependent enhancement (ADE) which is characterized by the enhanced infection of target cells via Fcγ receptor bearing cell-mediated internalization of IgG coated virus. The hypothesis suggests that cross-reactive antibodies that bind virus are not neutralizing, or are at sub-neutralizing concentrations, (Halstead and O’Rourke, 1977a) thereby facilitating infection, rather than preventing. Several in vivo lines of evidence support this hypothesis (Halstead and O’Rourke, 1977a; Halstead and O’Rourke, 1977b; Zellweger et al., 2010). Both in vitro experiments in K562 cells and in vivo experiments with juvenile rhesus macaques demonstrated that ADE led to increased titers, with up to a 1000-fold increase in vitro and a 100-fold increase in vivo (Goncalvez et al., 2007). Higher levels of viremia are correlated with increased dengue disease severity in humans (Vaughn et al., 2000). There is also evidence that immature DENV virions are rendered highly infectious by anti-prM antibodies (Goncalvez et al., 2007; Rodenhuis-Zybert et al., 2010). Moreover, Fc receptor signaling during immune complex binding is not restricted to the internalization event; other signaling includes suppression of IFN-gamma transcription and translation, increased synthesis of IL-6, and downregulation of IRF-1 and STAT1 [reviewed in (Halstead et al., 2010)]. Fc receptor engagement also reportedly downregulates RIG-I/MDA5 signaling and decreases production of type I interferon (Chareonsirisuthigul et al., 2007).
The host-specific immune responses to DENV likely play a large role in the pathophysiology of disease and subsequent clinical manifestation of dengue infection. Dengue disease is a complex viral-host interaction with not only cross reactive antibody and T cell immunity as important determinants of severity (Mongkolsapaya et al., 2003; Friberg et al., 2011; Midgley et al., 2011), but also host genetics including polymorphisms in the TNF and lyphotoxin receptor (Fernandez-Mestre et al., 2004; Vejbaesya et al., 2009)and MHC class I alleles (Stephens et al., 2002; Zompi and Harris, 2012). These studies have found that several polymorphisms in these alleles are associated with more severe dengue disease, while others, particularly in the MHC alleles can be associated with less severe disease. For example, HLA A*0203 is associated with less severe dengue fever, while HLA*0207 is associated with more severe DHF and DSS in secondary infection. By contrast, HLA B44, B62, B76, and B77 are associated with protection against developing clinical disease after secondary dengue infection (Stephens et al., 2002). Virus virulence factors are also associated with severity of disease including the sequence of specific serotypes causing infection (Halstead et al., 1983; Morens and Halstead, 1987; Halstead, 1998). The wide range in clinical presentation is likely the result of the interaction of many variables, both virus and host. Clinical presentation can include severe headache; retro-orbital eye pain; muscle, joint, and bone pain; nausea; vomiting, macular or maculopapular rash; a positive tourniquet test; or other hemorrhagic manifestations such as petechia, ecchymosis, purpura, epistaxis, bleeding gums, and hematuria. Severity is associated with warning signs including abdominal pain or tenderness, persistent vomiting, clinical fluid accumulation, mucosal bleeding, lethargy, restlessness, and liver enlargement (Deen et al., 2006). The hallmark for DHF and DSS is plasma leakage characterized by endothelial damage and leakage of intravascular plasma to the extravascular space (Srikiatkhachorn, 2009; Rothman, 2010).
In this review, we focus on the innate inflammatory responses to DENV infection by innate immune cells, their effector functions and clinical course.
Models for Dengue Virus Immune Response
DENV, and accompanying clinical disease, are almost entirely restricted to humans and non-human primates, and the latter are largely asymptomatic. Few small animal models exist (Yauch and Shresta, 2008; Zellweger and Shresta, 2014), as rodent cells are generally not permissive to DENV infection, and each presents significant challenges for use and extrapolation to humans. To circumvent the issue of infectability, mouse models are genetically modified to be permissive to virus infection, most often by targeting the IFN system, and by adapting the virus. As such, immunocompromised mice are a common model for studying DENV pathogenesis and immunity. The most widely used model is the AG129 IFNα/β/γ receptor knockout mouse that when infected with a mouse-adapted DENV strain, recapitulates aspects of severe dengue disease (Yauch et al., 2009; Watanabe et al., 2012) including vascular leak and ADE (Shresta et al., 2006; Balsitis et al., 2010). The NOD/SCID/IL-2RγKO mice engrafted with human CD34+ stem cells develop symptoms of mild DF (Bente et al., 2005; Mota and Rico-Hesse, 2011), while the RAG-hu mouse model develops fever only (Kuruvilla et al., 2007). These models are not ideal in which to study the immunopathogenesis of a human-constrained virus. Dengue does not naturally infect rodents. Moreover, the use of these models requires adaptation of human dengue strains in order to establish any infection. This, coupled with the required immune knockouts to generate infection and disease, makes it difficult to apply knowledge gained in immune deficient murine systems to the events occurring in human hosts with intact immune systems. Much of the pathogenesis of dengue disease is thought to be due to activation of the immune system, and these models do not recapitulate a fully functioning immune system. However, immunocompetent C57BL/6j and BALB/c Mice infected IP with a mouse adapted passaged clinical isolate of DENV 3 exhibit severe disease and die by day 6–7 post-infection, recapitulating many of the observed clinical signs of severe dengue including thrombocytopenia, decrease in systolic blood pressure, increased liver enzymes, and viremia (Goncalves et al., 2012).
These animal models are not sufficient to test antivirals, understand mechanisms of clinical symptoms, or select vaccine candidates. Limited knowledge of the range and complexity of the immune response generated in humans makes it particularly challenging to design an effective vaccine. Illustrating this, the only available vaccine, licensed in 2019, was shown to sensitize some seronegative recipients to more severe dengue disease upon infection with wild-type virus (Biswal et al., 2019). These limitations arise, in part, from an incomplete understanding of the human immune response to DENV, which is essential to exploit when designing a vaccine. Importantly, the early innate immune response governs the fate and vigor of the subsequent adaptive immune response (Fearon and Locksley, 1996), which confers the long-term protection desired from a vaccine. Compounding this problem, epidemiological studies in dengue-endemic areas are limited in several ways: they cannot control for many important factors in their subjects (e.g. prior flavivirus exposure), and are limited to enrolling virologically-confirmed cases of dengue who present with symptoms. These limitations leave a substantial gap in our knowledge of the early innate immune response to DENV.
Controlled human infection models (CHIMs) have been used successfully and safely for a number of human pathogens including cholera, influenza, malaria, typhoid and other enteric pathogens. CHIMs for DENV challenge studies have been historically and currently used as a safe means to test vaccine product viability (Thomas, 2013; Endy, 2014). The Walter Reed Army Institute of Research (WRAIR) initiated the development of a DENV human infection model (DHIM) in 2001 using previous DENV vaccine viruses that were found to be too reactogenic for a vaccine, but safe in human trials and potential candidates for a challenge virus (Lyons, 2014). The first study was conducted in 2001 in 15 volunteers. Two subjects received DENV-1, three subjects received DENV-2, three subjects received DENV-3, four subjects received DENV-4, and three subjects received placebo. DENV-1 strain 45AZ5 was administered subcutaneously (SC) at a dose of 0.5 ml containing 1.6 x 104 PFU (Mammen et al., 2014). Challenge resulted in a mild dengue fever-like illness with fever, chills, myalgias, arthralgias, headache, eye pain, malaise, anorexia, backache, abdominal pain, pruritus, photosensitivity, lymphadenopathy, and loose stools. Also observed were a morbilliform truncal rash, lymphadenopathy, leukopenia, neutropenia, and small perihepatic effusion. A follow-up study was performed in 2008 using both previously vaccinated and non-vaccinated volunteers. In those who received DENV-1, all 5 subjects previously vaccinated with a tetravalent live-attenuated dengue vaccine were protected against DENV-1 virus challenge (Sun et al., 2013). In volunteers who did not receive the vaccine, dengue fever like symptoms and laboratory findings occurred. In both trials, volunteers had resolution of their symptoms and viremia without serious adverse events. We recently completed a phase I study of 12 healthy adult volunteers using a challenge virus, DENV-1-LVHC, strain 45AZ5 (Endy et al., 2020). All subjects developed neutralizing antibody to DENV-1, and 11 of the 12 developed viremia with peak viral loads similar to wild-type DENV infection. There were no serious consequences to infection and all recovered without problems. The DHIM offers a platform in which to test the viability of candidate vaccines and therapeutics. Equally important, it offers a reproducible model in which to study the viral-host interactions and the innate and adaptive immune response to DENV infection.
Innate Immune System
The ability to respond to and orchestrate effective defenses against invading pathogens is a key element of survival. The human immune system achieves this by effectively controlling dangerous pathogens and ignoring the rest. Composed of two arms, the innate arm and the adaptive arm, The innate arm is activated in response to both injury and infection; the adaptive arm is recruited and activated in response to innate immune activation and direction, is specific, and is tailored for individual pathogens [reviewed in (Dorothy and Lewis)].
Evolutionarily conserved, the innate system harnesses the power of molecular and danger patterns to activate a highly specific inflammatory response aimed at alerting and mobilizing immune cells, with the ultimate goal being to clear the invading pathogen and/or repair the damage that initiated the response [reviewed in (Janeway and Medzhitov, 2002; Barton, 2008)]. By contrast, the adaptive system is highly specific, with recognition mediated by and activation tailored to antigen encountered, and requires appropriate secondary signals. The effectors of the adaptive immune system include T cell and B cells. The innate system is more complex [reviewed in (Douglas and McDonald, 2019)]; it is composed of monocytes, macrophages, neutrophils, mast cells, basophils, eosinophils, NK cells and ILCs. Many of these are granulocytes that store preformed mediators, including specific inflammatory proteases and cytokines, for immediate release. These granulocytes also de novo synthesize a plethora of highly inflammatory cytokines, chemokines, and Cox-2- and 5-lipoxygenase-derived lipid mediators (Alvarez et al., 2010) that are necessary to induce and direct an appropriate adaptive immune response. Activation of tissue resident macrophages and mast cells at the area of wound or pathogen entry initiates a cascade of events aimed ultimately at healing and pathogen clearance, often with the help of the adaptive arm. The direct effect of these inflammatory mediators is to modify endothelial tight junctions and adhesion molecules to allow for influx of immune cells from the circulation, to recruit other innate immune effectors to the site, and to activate nearby tissue resident cells to mount a response against the insult. Upon activation of mast cells, dendritic cells are activated and directed to egress from the site, homing to the draining lymph node to activate the adaptive arm of the immune system (Jawdat et al., 2006; Suto et al., 2006; Dudeck et al., 2019). The evolutionary importance of these tissue resident cells is underscored by the finding that both phagocytes (macrophages) and granulocytes (mast cells) are highly conserved, found in a range of Kingdoms and species including invertebrates and primitive chordates (Rhodes et al., 1982; Crivellato and Ribatti, 2010; Wittamer et al., 2011). It is these tissue resident cells that initiate the acute inflammation required to alert and instruct both the innate and adaptive arms of the immune system to the danger/pathogen, allowing for effective mobilization of the appropriate cellular compartment to the site.
Professional Phagocytes of the Skin: Macrophages, DCs, Langerhans Cells
There are several valid ways of subcategorizing phagocytic immune cell populations. The macrophage/DC/LC field is currently undergoing a shift away from defining these cell types and subtypes by their functional/phenotypic properties, and towards defining the populations by ontogeny (developmental lineage) and transcription factor expression/transcriptome profiles. In consequence, LCs/DCs and subsets and macrophages have not always been defined identically. Where appropriate, we have denoted specific subsets with their definitive CD antigens in order to facilitate comparisons between human and mouse data, as well as to aid in comparing studies where immune cell subtypes may not have been defined identically.
Dendritic cells are phagocytic cells derived from CD34+ hematopoietic cell precursors that give rise to both myeloid and lymphoid precursors. Dendritic cells are considered the most efficient antigen-presenting cell whose canonical function is to activate naïve T cells. In this capacity, dendritic cells capture, process and present antigens in MHC Class II to CD4 + T cells [reviewed in (Banchereau et al., 2000)]. They express high levels of MHC Class II and CD11c in addition to a range of other surface markers that identify distinct subtypes. As tissue-resident cells, they are relatively short-lived, and require replenishment from bone marrow-derived precursors in order to maintain their numbers in peripheral organs [reviewed in (Doebel et al., 2017; Collin and Bigley, 2018; Macri et al., 2018)]. Dendritic cells exist in two functional states, immature and mature [reviewed in (Worbs et al., 2017)]. In tissues they are immature, with a decreased ability to induce naïve lymphocyte effector responses but with a robust ability to capture antigen. Dendritic cells undergo a maturation program both as a result of macrophage and mast cell activation-derived signals and also by sensing of danger- or pathogen-associated molecular patterns. Maturation activates dendritic cells to migrate to secondary lymphoid organs where they function to present antigen to T cells and promote the initiation of adaptive immunity [reviewed in (Patente et al., 2019)]. Langerhans cells are epidermal innate immune cells of myeloid origin. They bear some functional similarities to dendritic cells, including the capacity to migrate to lymph nodes and stimulate T cells. However, classified according to developmental origin (ontogeny), they are considered a specialized subset of macrophages: they arise from embryonic precursors rather than bone marrow. Langerhans cells are long-lived in tissue and self-maintain their population without replenishment (Doebel et al., 2017).
The Infection Event: Anatomy and Mosquito Factors
DENV is introduced into the skin via saliva deposition as a mosquito takes a blood meal (Figure 1, Step 1). The mosquito proboscis must necessarily penetrate through the epidermis and into the dermis in order to access the capillary beds, though the specific distribution of virions introduced during the feeding event is unclear. Some sources suggest that DENV is directly injected into the dermis instead of the epidermis (Begum et al., 2019); others assert that the virus is introduced into both layers (Garcia et al., 2017; Rathore and St John, 2018); and still others claim that virions are introduced directly into the bloodstream, with “spillover” into both the dermis and epidermis (Martina et al., 2009). As discussed below, susceptible and permissive cells from both layers are infected soon after inoculation, supporting the notion that DENV is not restricted to a specific layer of the skin after the inoculation event.
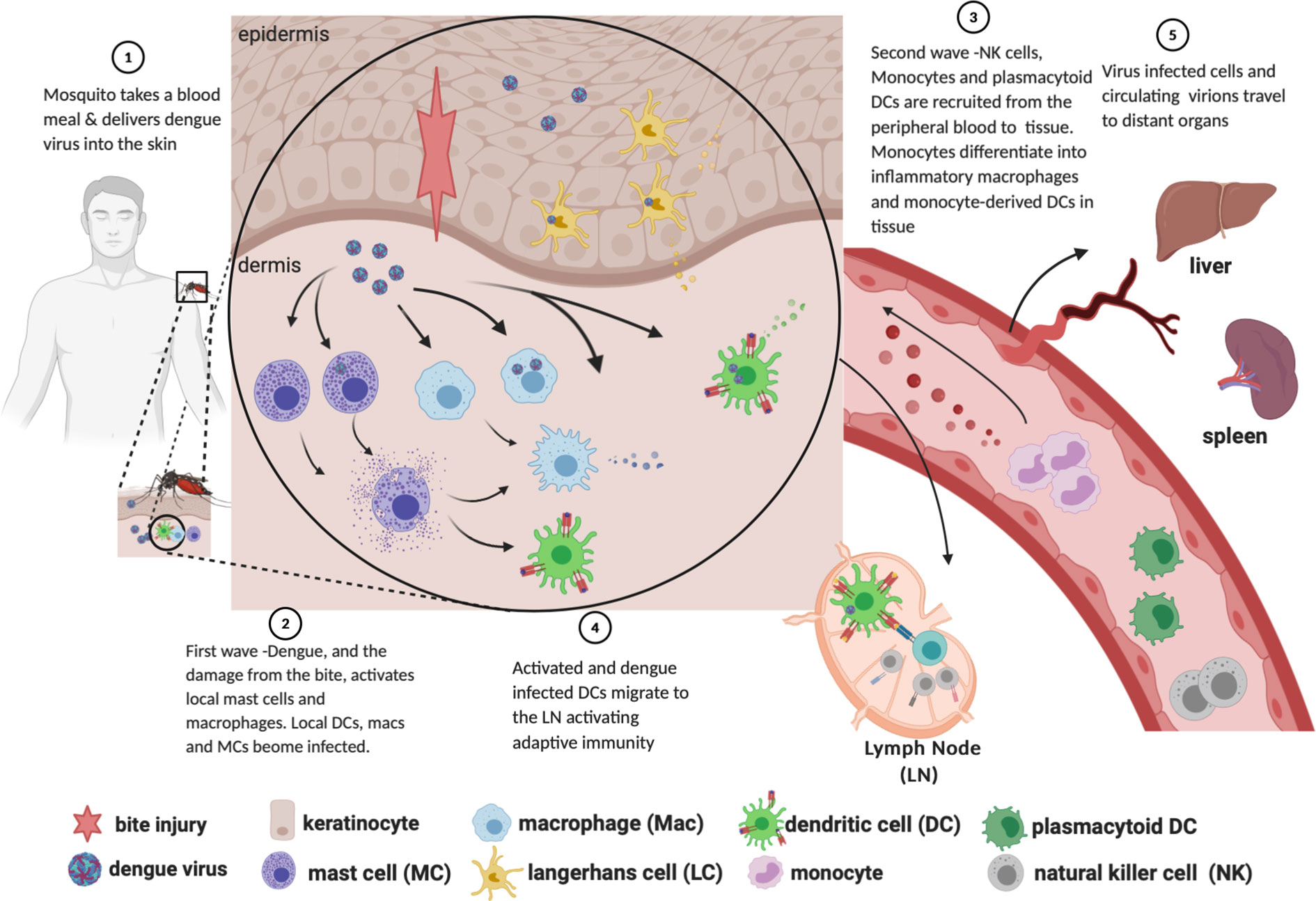
Figure 1 Path of dengue infection from mosquito bite to dissemination to organs. Created with Biorender.com.
In addition to viral particles, feeding mosquitos also inject several salivary proteins. These, in addition to local anticoagulant activity, have been shown to facilitate the establishment and virulence of several flaviviruses. Inoculation of C57BL/6 mice with West Nile Virus with infected Culex tarsalis mosquitoes results in increased viremia 24 and 48 h.p.i. compared to needle injection of a comparable inoculum (Styer et al., 2011). While the precise mechanism was not determined, possibilities include local immunomodulation at the bite site increasing WNV tropism, and/or leukocyte infiltration increasing the number of WNV-susceptible cells at the bite site. C57BL/6 mice passively immunized against the Aedes aegypti salivary factor NeSt1 showed reduced early ZIKV replication, reduced macrophage infiltration at the bite site, and increased 30-day survival; this is possibly due to reduced levels of pro-IL-1β and CXCL2 at the bite site (Hastings et al., 2019). Humanized (NSG) mice infected by A. aegypti mosquitoes exhibit prolonged DENV viremia; moreover, a mosquito bite containing no virus induced an innate immune cytokine response comprising TNF, IL-4, and IL-10, underscoring that mosquito saliva is itself immunomodulatory in the absence of virus (Cox et al., 2012). From a mechanistic standpoint, the A. aegypti salivary protein CLIPA3 has been implicated as a specific factor contributing to increased DENV infectivity, facilitating attachment of viral particles to cell surface receptors and cell migration via digestion of extracellular matrix (Conway et al., 2014). These experiments have not elaborated the range of mosquito salivary factors involved, but point to the initial inoculation conditions, and thus the innate immune response of the skin, as an important variable influencing the course of infection. It should be noted that much of the dengue field involving mouse or non-human primates, as well as the current human infection models, do not account for this variable when inoculating with DENV.
Once deposited, DENV is capable of infecting and replicating in keratinocytes and fibroblasts (Garcia et al., 2017) as well as the several populations of professional skin-resident phagocytic cells: Langerhans cells, dendritic cells, and macrophages (Wu et al., 2000; Cerny et al., 2014; Schmid and Harris, 2014; Garcia et al., 2017; Rathore and St John, 2018). A specific entry receptor or family of receptors for DENV, and thus the range of cellular tropism, has not yet been established. Aside from antibody-dependent enhancement, native cell-surface candidates for DENV internalization include L-SIGN, DC-SIGN, C-type lectins, the mannose receptor, glycosaminoglycans such as heparin sulfate, TIM-1, TAM, CD14, and CD300a (Crill and Roehrig, 2001; Cruz-Oliveira et al., 2015; Ngono and Shresta, 2018). The DENV E protein structural domain III is the most likely candidate for binding the cellular entry receptor (Crill and Roehrig, 2001; Cruz-Oliveira et al., 2015) but, owing in part to the range of putative entry receptors, the exact binding motif(s) remains unknown.
FIRST WAVE: Tissue-Resident Phagocytes and Keratinocytes Productively Infected
Work primarily done in C57BL/6 mice lacking the interferon alpha receptor (Ifnar -/-) (Schmid and Harris, 2014) and thoroughly reviewed in (Rathore and St John, 2018), demonstrated that there are two “waves” of cells infected with DENV following inoculation. In the first wave (infection by the initial inoculum), mast cells, macrophages, CD103+ DCs, and Ly6C-CD11b+ DCs—analogous to human CD141+ conventional (myeloid) dendritic cell subtypes 1 and 2, respectively (St John et al., 2011; Collin and Bigley, 2018)—are infected with DENV (Figure 1, Step 2). Other studies employing non-human primates and cadaveric human skin explants, in addition to the above studies in Ifnar -/- mice, have shown that Langerhans cells are among those initially infected (Taweechaisupapong et al., 1996a; Taweechaisupapong et al., 1996b; Wu et al., 2000).
While the infection of LCs, DCs, and macrophages is well known, there is some disagreement as to which layer of the skin carries the highest burden of infection. Studies done in intradermally inoculated Ifnar -/- mice show up to 100-fold more DENV-infected cells in the dermis as compared to the epidermis at 72 h post-inoculation (Schmid and Harris, 2014; Schmid et al., 2014). These flow cytometry data were gated on CD45+ cells taken from the skin of the mice, thereby excluding from consideration keratinocytes, which do not express CD45. By contrast, subsequent work by Duangkhae and colleagues demonstrated that keratinocytes alone accounted for up to 60% of DENV-infected cells in a human skin explant model, and that the epidermis contained approximately six-fold more DENV-infected cells than the dermis at 48 hpi (Duangkhae et al., 2018). While a consensus of infection burden between the skin layers has yet to be reached, the available evidence is generally clear that keratinocytes and skin-resident phagocytes are infected after introduction of virus and prior to development of viremia. These infected cells, while mounting an innate immune response against the virus, simultaneously create a milieu conducive to propagating the number of infected cells. For example, administration of neutralizing Ab to IL-1β into skin at 2 h.p.i. decreased the burden of infected dermal cells by 65% (Duangkhae et al., 2018). These data illustrate the feed-forward effect that leads to the second wave of infection.
General Cellular Host Response to DENV and Clinical Consequences
During viral replication, intracellular DENV ssRNA and dsRNA, intermediates of viral genome replication, are recognized as pathogen-associated molecular patterns (PAMPs) by host cell pattern recognition receptors (PRRs) including MDA5, RIG-I, TLR3, and TLR7 (Pichyangkul et al., 2003; Rothman et al., 2003; Morrison and García-Sastre, 2014; Cumberworth et al., 2017; Ngono and Shresta, 2018; Uno and Ross, 2018; Tremblay et al., 2019). Engagement of these receptors triggers an antiviral response, which is initially characterized by the induction of the type 1 interferon (IFN) response. Briefly, intracellular signaling pathways downstream of the above PAMP receptors culminate in activation of the master innate immune transcription factors IRF3, IRF7, and NF-κB, which direct the transcription and secretion of IFN-α and IFN-β. These proteins in turn act in both an autocrine and paracrine fashion to produce a general antiviral intracellular milieu hostile to virion production (Rothman et al., 2003; Morrison and García-Sastre, 2014; Cumberworth et al., 2017; Uno and Ross, 2018; Tremblay et al., 2019). Thus, the initial stages of DENV infection proceed in a positive-feedback cycle of an increasing burden of cells infected, as well as an increasing capacity to mount a type 1 interferon response.
The cellular immune response at large—not limited to type 1 IFN secretion—as well as an increasing burden of infected cells contributes to the subsequent clinical manifestations of dengue. In one human subject who received a tetravalent live DENV vaccine, infected dendritic cells and Langerhans cells were found in a cutaneous rash distant from the injection site (Wu et al., 2000). The investigators performed immunohistochemical staining for DENV glycoprotein from a skin biopsy and confirmed the presence of infected DCs and LCs, indicating an association between presence of DENV and clinical symptoms manifesting in the skin. Production of the pyrogen IL-1β begins with the first wave of infected cells; additionally, production of TNF, responsible for pyrexia, myalgia and appetite suppression, could begin at the stage of skin infection as well (Schaeffer et al., 2015). More broadly, type 1 interferon contributes to a gamut of clinical symptoms, the range of which is clearly delineated during administration of exogenous interferon in a therapeutic setting: fever, chills, headache, and fatigue (Todd and Goa, 1992; Sleijfer et al., 2005; Owen et al., 2013; Torkildsen et al., 2016).
DENV: Subversion of the Interferon Response
Though the IFN response is critical to dampen DENV replication (Diamond et al., 2000), it is insufficient to completely suppress production of infectious DENV. Indeed, historical studies in humans [reviewed in (Snow et al., 2014)] as well as human data from our lab (Endy et al., 2020) suggest that not every DENV inoculation results in detectable viremia. However, the typical finding of viremia in symptomatic cases, and the presence of DENV antigen in several cell types beyond the site of infection (Begum et al., 2019), indicates that infection is not routinely confined to the skin. This is likely due to subversion of the human interferon response by DENV. At a tissue level, data from human skin explants show transient, rather than sustained, production of IFN-α by skin cells after DENV infection, with peak levels occurring less than 12 h.p.i. (Duangkhae et al., 2018). At the cellular level, several DENV nonstructural (NS) proteins suppress both induction and downstream signaling of interferon. NS2a, NS4a, and NS4b block TBK1 phosphorylation, preventing the transcription of IFN-β (Dalrymple et al., 2015). NS2b targets cGAS for degradation, and NS2b/3 cleaves the intracellular DNA sensor STING, representing multiple points of disruption to the cGAS-STING pathway which would otherwise induce interferon (Lee et al., 2018). NS5 2’-O-methylates the 5’ cap of the DENV genome, which prevents detection by RIG-I (Lee et al., 2018). NS4a binds to the CARD-like and transmembrane domains of MAVS, preventing binding of RIG-I and therefore activation of IRF3 and consequent interferon induction (He et al., 2016).
Not all DENV antagonism of the IFN response occurs upstream of interferon production itself. This is important when considering that pDCs (discussed below in the Second Wave section) produce IFN without being infected. Thus, DENV also has the capability to disrupt the signaling downstream of the IFNAR: NS2a, NS4a, and NS4b inhibit STAT1 phosphorylation, and NS5 mediates proteasomal degradation of STAT2 (Figure 2) (Morrison and García-Sastre, 2014; Tremblay et al., 2019). DENV has genome size constraints and limited coding capacity expressing 10 proteins, seven of which are non-structural; five of those function to target an aspect of the IFN system. This intense focus of interfering with the IFN system underscores how important this system is in controlling DENV infection. Together, these mechanisms suppress IFN activation and downstream signaling, enabling DENV to replicate locally and to produce virions that infect cells beyond the bite site.
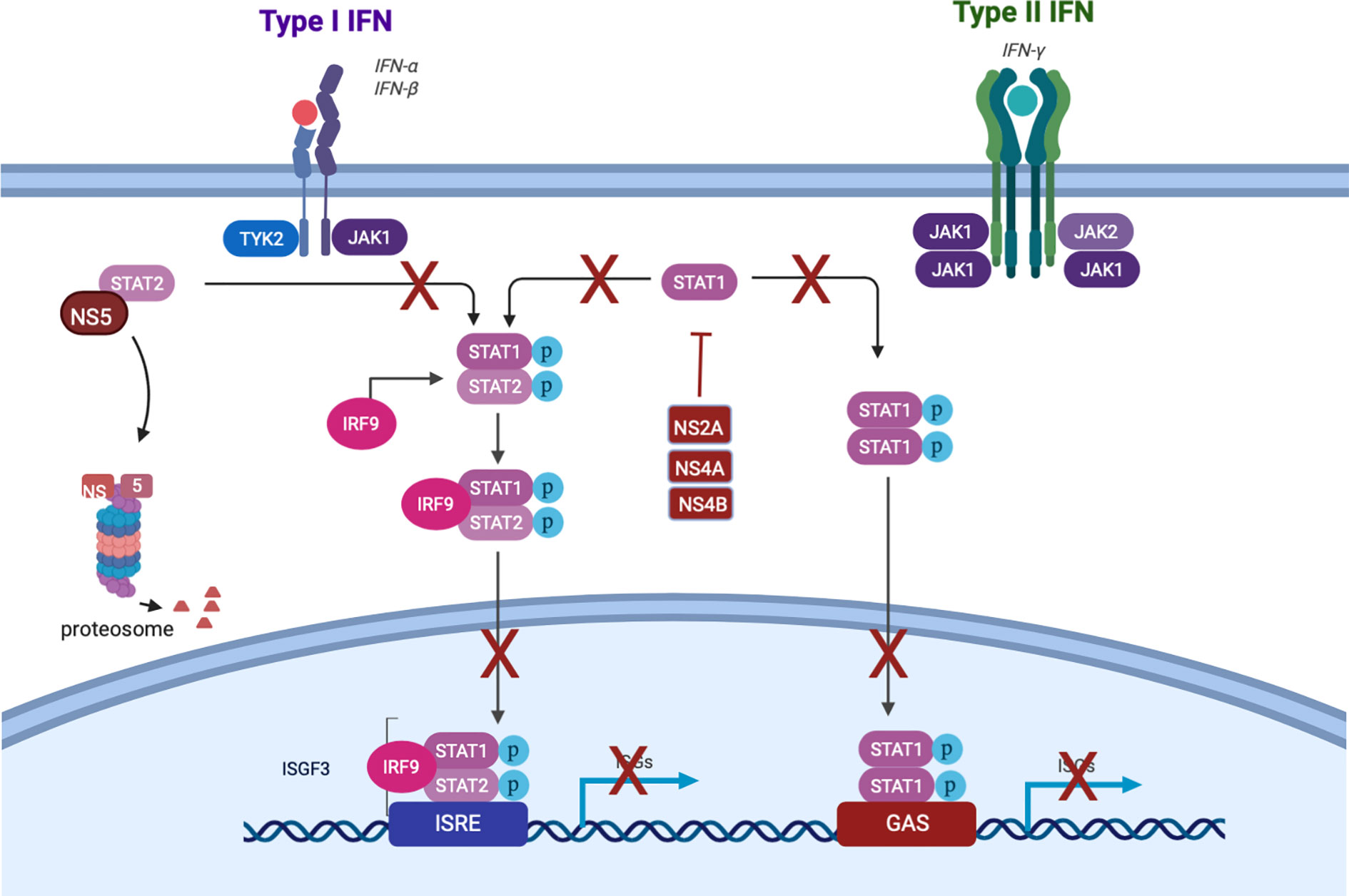
Figure 2 Dengue subversion of interferon signaling. Created with Biorender.com.
Tissue Resident Mast Cells Are Activated During Dengue Infection
Mast cells (MCs) are long-lived, CD34+ (Maaninka et al., 2013) tissue-resident innate immune inflammatory cells that reside in almost every tissue and organ, including in high levels in the skin (Galli et al., 2005a). In skin, mast cells are positioned in the dermis, adjacent to microvessels, where they serve as sentinels and sense blood borne and tissue localized pathogens to direct immune cell recruitment to the area. The perivascular location also facilitates MC-driven modulation of EC function (Klein et al., 1989; Brown et al., 2011; Kunder et al., 2011), and several mast cell specific -mediators have been shown to promote EC activation (Compton et al., 1998; Frossi et al., 2004), a response necessary for immune cell recruitment. MCs recognize and become activated by a wide range of stimuli, including foreign pathogens [reviewed in (Marshall and Bienenstock, 1994; Marshall and Jawdat, 2004; Abraham and St John, 2010)] to drive a rapid, pro-inflammatory response (Galli et al., 2005b; Metz et al., 2007; Metz and Maurer, 2007) involving protease release, eicosanoid synthesis and release, and de novo synthesis of cytokines, chemokines and growth factors, including Type 1 IFNs.
Mast cell activation in vivo in tissue and in vitro by DENV induces degranulation with release of granule contents (Figure 1, Step 2) (St John et al., 2011; Troupin et al., 2016) that stimulates activation of the endothelium (Brown et al., 2011; Kunder et al., 2011) and likely contributes to the clinical rash often seen during the clinical course of infection (Nakamura et al., 2009; Mishra et al., 2018). Activated mast cells synthesize and release CXCL2, a potent chemotactic factor for neutrophils, and have been shown to promote neutrophil activation and recruitment from the periphery (Zhang et al., 1992; Abraham and Malaviya, 1997). In vivo mouse studies and in vitro human models have demonstrated mast cells are permissive to dengue virus infection via ADE (King et al., 2000). One of the key responses is de novo synthesis of pro-inflammatory cytokines TNF (Brown et al., 2011) and IL-1β (King et al., 2000), and the chemokines CCL3, CCL4 and CCL5 (King et al., 2002), CXCL12, and CXCL1 (St John et al., 2011). Mast cells respond to DENV antibody-mediated infection with a robust IFN expression by 12 h post-infection that is maintained up to 72 h (Brown et al., 2011). Via degranulation, release of Type 1 IFNs, and chemotactic factors, MCs initiate local acute immune activation, providing the required signals for activation and chemotactic recruitment of circulating monocytes, local tissue macrophages, NK cells and neutrophils. Subsequent work with MC-deficient mice demonstrated the importance of mast cell-driven recruitment of natural killer and natural killer T cells into the infected skin. In this model, mast cells were critical for containing DENV in vivo, and without which there was increased viral burden within draining lymph nodes after subcutaneous infection compared to MC-sufficient mice (St John et al., 2011).
Human studies have demonstrated that mast cells exhibit an extensive activated phenotype with degranulation in the rash of infected subjects (Asher AN et al., 2002) and dengue disease severity associated with levels of mast cell granule stored mediators including chymase (Avirutnan and Matangkasombut, 2013; Tissera et al., 2017), histamine (Tuchinda et al., 1977), VEGF (Furuta et al., 2012), and tryptase (Rathore et al., 2019). Disease severity is also associated with IgE, a classical mast cell activating antibody (Koraka et al., 2003). Together, these data strongly suggest a role for dengue-driven mast cell mediator release in clinical symptoms of dengue disease. In addition, a large histological study of DHF patients in Thailand determined that mast cells in connective tissue showed evidence of activation including swelling, vacuolation of the cytoplasm, and loss of granule integrity (Bhamarapravati et al., 1967). Mast cell activation [reviewed in (Valent, 2013; Afrin et al., 2017)] is known to mediate several of the common clinical symptoms associated with dengue patients including rash, diarrhea, vomiting, headache, eye pain/inflammation and muscle pain. Given the potency of mast cell activation and the resultant inflammation and clinical symptoms, targeting vasoactive mast cell mediators such as histamine or tryptase may have a significant impact of clinical course. In a small study in Pakistan, treatment with anti-histamines and steroids dramatically reduced dengue symptoms, shortening duration to 3–5 days, as compared to the other treatment groups of 7–10 days (Siddique et al., 2008). An older study on 24 patients in the Armed Forces found even low dose targeting of histamine reduced the duration of clinical symptoms, suggesting that interfering with mast cell mediators or activation may have a role in treating dengue disease (Hoffman et al., 1954). More recently, a randomized clinical trial in 200 dengue patients demonstrated that a single daily dose of 10 mg montelukast, a cysteinyl leukotriene inhibitor that blocks leukotriene C4, D4, and E4 eicosanoids from binding their cognate cysteinyl leukotriene receptor 1, significantly reduced incidence of severe dengue shock syndrome by 71%, as compared to the control group (Ahmad et al., 2018). Montelukast is a standard cysteinyl leukotriene receptor inhibitor given to asthma patients to help control the mast cell-derived inflammatory leukotriene responses to reduce airway inflammation. Given that dengue virus induces degranulation of tissue resident mast cells, that rash exists at sites distant to inoculation, and that in our human infection model rash is present both before and after resolution of DENV viremia (Endy et al., 2020), we suggest that perhaps DENV is present at high levels in skin sites throughout disease. In this context, rash would then be a direct clinical sign of DENV virus replication, as has been observed in other severe viral infections, including measles and pox virus skin lesions.
SECOND WAVE—Monocyte-Derived Macrophages and DCs Recruited and Infected; Plasmacytoid DCs Recruited and Activated
Following the first wave of cells infected in the skin, local myeloid cells, monocyte-derived macrophages, monocyte-derived dendritic cells (moDCs, Ly6C+CD11b+), and circulating plasmacytoid dendritic cells (pDCs, CD123+) are recruited into the skin via chemokine signals (Figure 1, Step 4). These signals are secreted from tissue-resident macrophages, mast cells and other initially infected resident cells. Myeloid cells are recruited to the site of inflammation via CCL2, IL-1β and CCL20 (Dieu-Nosjean et al., 2000; Rider et al., 2011; Schmid and Harris, 2014; Schmid et al., 2014; Duangkhae et al., 2018); monocyte extravasation from peripheral blood is facilitated by CCL1 and CCL5 (Shi and Pamer, 2011); and pDC recruitment to inflamed skin is facilitated by CCL2, among others (Swiecki and Colonna, 2015).
Upon arrival, moDCs and macrophages are productively infected with DENV (Schmid and Harris, 2014), with some in vitro evidence suggesting that moDCs are up to ten times more permissive to infection than either monocytes or macrophages (Navarro-Sanchez et al., 2003). Of interest, dengue virions shed by mosquito cells have a different tropism for human cells than those shed by human DCs, with the former able to bind both DC-SIGN and L-SIGN, and the latter only able to bind L-SIGN (Dejnirattisai et al., 2011). This factor, which is not controlled for in many experimental models, complicates the analysis of the kinetics and infected cell burden of the second wave. pDCs, on the other hand, are not productively infected themselves; they are instead activated by DENV-infected cells in a contact-dependent manner. Once activated, they are the predominant producers of type 1 interferons (Navarro-Sánchez et al., 2005; Webster et al., 2016; Webster et al., 2018; Assil et al., 2019). Though the roles of pDCs in viral immunopathology is complex (Swiecki and Colonna, 2015), contraction of the pDC compartment in peripheral blood was associated with a higher risk of severe dengue in children (Rothman et al., 2003) suggesting that pDCs are critical to the successful control of dengue virus.
NK Cells Are Activated and Recruited to Tissues During Infection
NK cells are bone marrow-derived innate immune lymphocytes that can kill virally infected cells, tumour cells, and antibody-opsonized cells/pathogens through a mechanism termed antibody-dependent cellular cytotoxicity (ADCC). Tissue resident cells including moDCs, macrophages [reviewed in (Michel et al., 2012)] and mast cells [reviewed in (Portales-Cervantes et al., 2019)] activated by initial infection (Figure 1, Step 2) and during the first “wave” rapidly recruit NK cells into infected organs and tissues in response to both DAMPS and viral infection, by secreting chemokines and cytokines (Figure 1, Step 3). Recruitment involves selective chemokine production in the microenviroment and corresponding receptor expression on NK cells. Several chemokines have been shown to mediate chemotaxis of NK cells to tissues including those typically known to direct chemotaxis and activation of monocytes [reviewed in (Shi and Pamer, 2011)] and mast cells [reviewed in (Halova et al., 2012)]. These include CCL2, CCL3, CCL4 (Morrison et al., 2003), CXCL2 (Burke et al., 2008), CXCL12 (Bernardini et al., 2008), and IFN (Paolini et al., 2015). Several chemokine receptors have been shown to mediate recruitment into infected tissues including CCR5, CXCR3, (Morrison et al., 2003; Huang et al., 2006), and sphingosine 1-phosphate receptor (Walzer et al., 2007), further expanding the chemokine ligands that can recruit NK cells.
Activation of NK-cell function is achieved by two distinct mechanisms: integration of signaling through a variety of inhibitory and activation receptors present on both the NK cells themselves and on host cells (Yokoyama et al., 2004; Jonsson and Yokoyama, 2009) and cytokine stimulation (Biron et al., 1999; Andrews et al., 2003; Cooper and Caligiuri, 2004). NK cell killing occurs by resting NK cells but is enhanced in response to cytokine stimulation. Typically, NK cells recognize “missing self” on infected cells when the host cell exhibits a down regulation in surface MHC class I molecules (Karre, 1995; Gasser and Raulet, 2006). NK cell killing activity is augmented by cytokine stimulation, including IL-12, IL-15, IL-18, and IL-21 (Cooper et al., 2001; Zwirner and Domaica, 2010; Brandstadter and Yang, 2011; Boieri et al., 2017) whereby cytokine-activated NK cells display enhanced cytotoxic activities (Henney et al., 1981; Boieri et al., 2017) and de novo cytokine synthesis that effectively amplifies the local immune response. Limiting infection spread early on is key to a rapid, effective, resolution and clearance of pathogen. NK are key effectors in controlling viral infections [reviewed in (Jost and Altfeld, 2013)] and are activated during dengue virus infection. In skin, St. John and colleagues, demonstrated early NK cell recruitment and activation at the site of DENV 2 inoculation in mouse footpads (St John et al., 2011). NK cell recruitment was dependent on mast cell activation and underscored how the innate immune system works in concert to mount an effective defense. More recently, human skin studies in DENV infected patients demonstrated CD69+CLA+ CXCR3+ CCR5+CD56 +NK cell recruitment to the skin during acute infection. These cells expressed proliferation markers and were increased during the febrile stage of illness, declining post-febrile, and in convalescence (Zimmer et al., 2019).
In several human studies circulating NK cells were found to express an activated phenotype with enhanced expression of CD69, a type II C‐lectin receptor and marker of lymphocyte activation. Homchampa et al. found evidence of NK cell cytotoxicity in children with acute dengue that correlated to disease severity (Homchampa et al., 1988) and later increased frequencies of circulating activated CD56+ CD69+ NK cells was observed in pediatric patients from Thailand with severe dengue disease as compared to patients with milder disease (Chen R. et al., 1999). In subsequent work, plasma from convalescent patients obtained after primary infection was found to mediate ADCC an in vitro NK cell killing assay (Laoprasopwattana et al., 2007) suggesting in the context of dengue, ADCC is likely also occurring.
An analysis of DENV patients from Brazil found an increase in circulating CD56+ NK cells during the acute phase of disease, defined as days 1–5 after onset of symptoms, with the majority of NK cells displaying early markers for activation including CD69, HLA-DR, and CD38, and increased expression of cytotoxic granule, TIA-1 (Azeredo et al., 2006) as compared to the late acute (days 6–10), or convalescence (days >11) patients. The group also showed that NK cell activating cytokine IL-15 was elevated in a significant number of patients during early acute infection (Azeredo et al., 2006).
Neutrophils
Neutrophils are phagocytic granulocytes that are primarily involved in control of bacteria. Neutrophils are activated by DAMPs, PAMPs and pro-inflammatory cytokines and complement split products, including CXCL2, TNF, C5a, and C3a [reviewed in (Silvestre-Roig et al., 2016)]. The roles of neutrophils during viral infections, and in relation to outcome, are not well understood. One of the most potent outcomes of neutrophil activation is the release of NETs. NET formation (NETosis) is characterized by nuclear decondensation and delobulation, rupture of the plasma membrane, and release of DNA fibers with antimicrobial peptides and histones (Brinkmann et al., 2004) NETosis is a potent anti-microbial mechanism, but excessive formation of NETs, or the inability to clear NETs from the circulation, contributes to pathogenesis of both autoimmune diseases (Kaplan and Radic, 2012; Knight et al., 2012) and exacerbation of several different viral infections. Excessive NET formation results in exacerbated allergic airway inflammation during rhinovirus (RV) infection (Toussaint et al., 2017) and airway obstruction during respiratory syncytial virus (RSV) infection (Cortjens et al., 2016). Influenza virus infection also induces NETs in lungs of infected mice, though inhibition of NET formation did not affect infection outcomes (Hemmers et al., 2011). Together, the emerging data suggest that a neutrophil response to a viral infection may be more detrimental than beneficial, though more research is needed.
Recent work has demonstrated that neutrophils are activated during DENV infection. Studies in Vietnamese children with severe dengue have demonstrated neutrophil activation at the transcriptomic level (Hoang et al., 2010). DENV-infected patients display an increased number of circulating neutrophils during infection, suggesting that they are being activated (Thein et al., 2014 #37)]. More recently, neutrophil elastase activity (a key component of neutrophil granules) was increased DENV-infected patients, as compared to healthy controls, and levels were associated with severity of disease (Kunder et al., 2018). These data suggest that neutrophils may play an unrecognized role in dengue disease. To fully understand any relationship more work is needed.
DENV: Escape From Skin and Replication in Lymph Nodes Leads to Development of Viremia
DENV escape is marked by migration of Langerhans cells and conventional dendritic cells stimulated by IL-1β and TNF (Stoitzner et al., 1999) out of the skin towards draining lymph nodes (Wu et al., 2000; Navarro-Sánchez et al., 2005; Cerny et al., 2014; Schmid and Harris, 2014; Duangkhae et al., 2018) (Figure 1, Step 4). Though some commentators presume that the mosquito feeding event results in viral particles being introduced directly into the bloodstream (Martina et al., 2009), this rarely, if ever, leads directly to viremia. Analysis of data from humans infected with DENV by mosquito bite has established a median intrinsic incubation period of 5.9 days, with 95% of subjects developing viremia between days 3 and 10 (Chan and Johansson, 2012). Therefore, it is much more likely that detection of DENV in the peripheral blood is a consequence of the egress of infected cells from the skin. This migration of infected phagocytes into regional lymph nodes 1) initiates the adaptive immune response, 2) precipitates further DENV replication in lymph node-resident and recruited mononuclear phagocytes, and 3) allows infectious DENV virions into the peripheral blood and the monocyte compartment (Castillo et al., 2018).
Peripheral Blood—Monocytes
There is some disagreement as to which circulating cell type comprises the majority of DENV-infected cells. Early work using flow cytometry and immunocytochemistry analysis of blood from acutely ill dengue patients claimed that B cells were the main mononuclear cell fraction containing DENV (King et al., 1999). Later reports directly contradicted that, claiming that monocytes were the predominant infected cell type (Durbin et al., 2008; Schmid et al., 2014). More recently still, virus-inclusive single-cell RNA seq of peripheral blood from a limited number of dengue-infected patients showed that the majority of cells containing DENV RNA were B lymphocytes (Zanini et al., 2018). Importantly, the authors noted that the gene expression profiles of the sequenced B cells suggested that the virus may not be actively replicating. Despite perhaps not being the most numerous DENV-infected circulating cell type, monocytes are critical cellular players in dengue pathogenesis, Monocytes are the main target for DENV replication in peripheral blood (Halstead et al., 1977; Chen Y.-C. et al., 1999; Durbin et al., 2008; Zanini et al., 2018), primarily entering via the mannose receptor [reviewed in (Reyes-del Valle et al., 2014)].
Infection of monocytes occurs at a higher frequency in secondary dengue infection as a result of ADE. Functionally, ADE results in an increased proportion of infected monocytes, at least a 14-fold increase in in vitro experiments with DENV-1; ADE in this model also resulted in an increase in TNF secretion (Halstead and O’Rourke, 1977b). Infection of peripheral monocytes activated endothelial cells in a TNF dependent manner (Anderson et al., 1997), suggesting increased monocyte infection may lead to enhanced levels of TNF, driving more severe clinical disease.
Distant Sites-Tissue Macrophages
Following dissemination of DENV from the skin to the peripheral blood (Figure 1, Step 5), and other organs in the body, other populations of specific tissue-resident macrophages are infected with DENV in the course of disease. These include Kupffer cells of the liver (Aye et al., 2014), and macrophages in the spleen (Balsitis et al., 2009). The infection of Kupffer cells and the resultant acute inflammation of the immediate area likely contributes to the observed elevation in liver enzymes ALT and AST (Fernando et al., 2016).
The Adaptive Immune Response
The adaptive immune response is composed of both a humoral and a cell mediated component and is absolutely essential for controlling viral pathogens. Dengue is known to activate B cells and results in the production of virus-specific IgM, IgG, and IgA antibodies, a portion of which bind the viral envelope protein and neutralize virions, thereby preventing entry into target cells. IgE is produced during dengue infection, and as noted in the mast cell section, would serve to activate innate immune cells through the high affinity Fc epsilon receptor one expressed at high levels on mast cells and upregulated on activated dendritic cells. Importantly, prior infection and antibody levels are a major risk factor for development of more severe disease as discussed in the Introduction: (dengue virus and clinical spectra). In the case of a heterotypic infection with a different dengue serotype, patients are at increased risk to develop dengue hemorrhagic fever and/or dengue shock syndrome. In these cases, sub-neutralizing levels of cross reactive antibodies facilitate entry into increased numbers of target cells via Fc receptor mediated uptake (Halstead, 1979; Halstead, 1988; Halstead, 1994). This is thought to increase the overall infection burden, leading to higher titers of circulating virus and greater inflammation. The cell-mediated arm of the adaptive system consists of CD4+ helper T cells and CD8+ cytotoxic T cells that function to promote B cell activation and killing of virally infected host cells. CD8+ and CD4+ T cells are known to be activated in large numbers during dengue virus infection (Tian et al., 2019). Several studies have demonstrated that T cell epitopes are present across the viral proteome and T cell activation can result in both protective and pathogenic outcomes. Significant evidence suggests that dengue can induce cross reactive T cell activation, termed T cell original sin (Mongkolsapaya et al., 2003; Rothman, 2011). In this setting cross-reactive T cells, specific for the primary infecting serotype, become predominant during a secondary heterologous infection. This expansion of preexisting cross-reactive and low-affinity memory T cells is thought to hamper effective viral control and contribute to severe disease through enhanced production of inflammatory cytokines. However several studies have also demonstrated a protective role for dengue specific T cells in controlling infection. In murine models both CD4+ T cells and CD8+ T cells play a protective role against DENV infection preventing severe disease and facilitating viral clearance (Yauch et al., 2009; Zellweger et al., 2010; Zellweger et al., 2014; Ngono and Shresta, 2018). The protective role of T cells during dengue infections is underscored by studies both in murine model and in humans, that identified protective HLA alleles that are associated with strong and multifunctional T cell responses (Stephens et al., 2002; Stephens, 2010).
Discussion
It is thought that the DENVs evolved into four distinct serotypes approximately 1,000 years ago and each of these four serotypes emerged into a cycle of transmission between humans and its mosquito vector approximately 125 to 320 years ago (Holmes and Twiddy, 2003; Twiddy et al., 2003a). Phylogenetic analysis suggests that the DENVs are rapidly evolving with major clade replacements and genetic shifts occurring in populations endemic for DENV (Holmes, 2003; Twiddy et al., 2003b; Zhang et al., 2005; Holmes, 2006). Asia in particular in this century has been pivotal in the evolution of DENV as the location of the first cases of the more severe form of DENV infection, dengue hemorrhagic fever (DHF), which made its first appearance in the 1950’s first in the Philippines then in Thailand (Halstead et al., 1967). This event was a hallmark denoting a change in the severity and pathogenesis of DENV viral-host interactions. The current Asian genotypes of each serotype are considered more severe and result in more severe dengue illness than the American genotypes (Kochel et al., 2002). Evidence suggests that DENV circulation in Asia due to its population growth and urbanization, high vector burden, and high level of pre-existing flavivirus seroprevalence, has contributed to the increase in genetic diversity of DENV which is estimated as increasing at a factor between 14 and 20 in the last 30 years (Twiddy et al., 2003a). The overall picture of DENV evolution in Asia and now the Americas is the active transmission of viruses in individuals who are highly flavivirus antibody experienced causing evolutionary pressure on the virus to evolve to escape and utilize pre-existing flavivirus immunity. By its nature the current evolving DENVs are adept at escaping heterologous neutralizing antibody and using it as a means to attain high viral load levels and more severe disease through antibody-dependent enhancement. Furthermore, in addition to the need to escape pre-existing adaptive immunity from antibody, the DENVs have evolved unique means to escape the host innate immune response as discussed above. Much needs to be understood about dengue pathogenesis and the viral-host interactions that result in severe disease. Utilization of the human infection model, improved techniques to understand the host genetic and immune response, and additional prospective human studies will further our understanding with application towards developing better drugs and vaccines to treat and prevent DENV infection.
Author Contributions
All authors listed have made a substantial, direct. and intellectual contribution to the manuscript, and approved it for publication.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abraham S. N., Malaviya R. (1997). Mast cells in infection and immunity. Infect. Immun. 65 (9), 3501–3508. doi: 10.1128/IAI.65.9.3501-3508.1997
Abraham S. N., St John A. L. (2010). Mast cell-orchestrated immunity to pathogens. Nat. Rev. Immunol. 10 (6), 440–452. doi: 10.1038/nri2782
Afrin L. B., Self S., Menk J., Lazarchick J. (2017). Characterization of Mast Cell Activation Syndrome. Am. J. Med. Sci. 353 (3), 207–215. doi: 10.1016/j.amjms.2016.12.013
Ahmad A., Waseem T., Butt N. F., Randhawa F. A., Malik U., Shakoori T. A. (2018). Montelukast reduces the risk of dengue shock syndrome in dengue patienys. Trop. Biomed. 35 (4), 1115–1122.
Alvarez Y., Valera I., Municio C., Hugo E., Padron F., Blanco L., et al. (2010). Eicosanoids in the innate immune response: TLR and non-TLR routes. Mediators Inflamm. 2010, 1–14. doi: 10.1155/2010/201929
Anderson R., Wang S., Osiowy C., Issekutz A. C. (1997). Activation of endothelial cells via antibody-enhanced dengue virus infection of peripheral blood monocytes. J. Virol. 71 (6), 4226–4232. doi: 10.1128/JVI.71.6.4226-4232.1997
Andrews D. M., Scalzo A. A., Yokoyama W. M., Smyth M. J., Degli-Esposti M. A. (2003). Functional interactions between dendritic cells and NK cells during viral infection. Nat. Immunol. 4 (2), 175–181. doi: 10.1038/ni880
Asher AN L. V. S., Krivda S., Wong H. K., Mammen M. P., Lyons A. G., Thomas S., et al. (2002). and DW Vaughn Degranulation of Mast Cells in Dengue Patients. Microscopy Microanal. 8 (Supplement S02), 920–921. doi: 10.1017/S1431927602107288
Assil S., Verin Colé S., Dong C., Dé Cembre E., Sherry L., Allatif O., et al. (2019). Plasmacytoid Dendritic Cells and Infected Cells Form an Interferogenic Synapse Required for Antiviral Responses. Cell Host Microbe 25 (5), 730–45.e6. doi: 10.1016/j.chom.2019.03.005
Avirutnan P., Matangkasombut P. (2013). Unmasking the role of mast cells in dengue. Elife 2, e00767. doi: 10.7554/eLife.00767
Aye K. S., Charngkaew K., Win N., Wai K. Z., Moe K., Punyadee N., et al. (2014). Pathologic highlights of dengue hemorrhagic fever in 13 autopsy cases from Myanmar. Hum. Pathol. 45 (6), 1221–1233. doi: 10.1016/j.humpath.2014.01.022
Azeredo E. L., De Oliveira-Pinto L. M., Zagne S. M., Cerqueira D. I., Nogueira R. M., Kubelka C. F. (2006). NK cells, displaying early activation, cytotoxicity and adhesion molecules, are associated with mild dengue disease. Clin. Exp. Immunol. 143 (2), 345–356. doi: 10.1111/j.1365-2249.2006.02996.x
Balsitis S. J., Coloma J., Castro G., Alava A., Flores D., McKerrow J. H., et al. (2009). Tropism of dengue virus in mice and humans defined by viral nonstructural protein 3-specific immunostaining. Am. J. Trop. Med. Hygiene 80 (3), 416–424. doi: 10.4269/ajtmh.2009.80.416
Balsitis S. J., Williams K. L., Lachica R., Flores D., Kyle J. L., Mehlhop E., et al. (2010). Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PloS Pathogens 6 (2), e1000790. doi: 10.1371/journal.ppat.1000790
Banchereau J., Briere F., Caux C., Davoust J., Lebecque S., Liu Y. J., et al. (2000). Immunobiology of dendritic cells. Annu. Rev. Immunol. 18, 767–811. doi: 10.1146/annurev.immunol.18.1.767
Barton G. M. (2008). A calculated response: control of inflammation by the innate immune system. J. Clin. Invest. 118 (2), 413–420. doi: 10.1172/JCI34431
Begum F., Das S., Mukherjee D., Mal S., Ray U. (2019). Insight into the Tropism of Dengue Virus in Humans. Viruses 11, 1–19. doi: 10.3390/v11121136
Bente D. A., Melkus M. W., Garcia J. V., Rico-Hesse R. (2005). Dengue fever in humanized NOD/SCID mice. J. Virol. 79 (21), 13797–13799. doi: 10.1128/JVI.79.21.13797-13799.2005
Bernardini G., Sciume G., Bosisio D., Morrone S., Sozzani S., Santoni A. (2008). CCL3 and CXCL12 regulate trafficking of mouse bone marrow NK cell subsets. Blood 111 (7), 3626–3634. doi: 10.1182/blood-2007-08-106203
Bhamarapravati N., Tuchinda P., Boonyapaknavik V. (1967). Pathology of Thailand haemorrhagic fever: a study of 100 autopsy cases. Ann. Trop. Med. Parasitol. 61, 500–510. doi: 10.1080/00034983.1967.11686519
Bhatt S., Gething P. W., Brady O. J., Messina J. P., Farlow A. W., Moyes C. L., et al. (2013). The global distribution and burden of dengue. Nature 496 (7446), 504–507. doi: 10.1038/nature12060
Biron C. A., Nguyen K. B., Pien G. C., Cousens L. P., Salazar-Mather T. P. (1999). Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu. Rev. Immunol. 17, 189–220. doi: 10.1146/annurev.immunol.17.1.189
Biswal S., Reynales H., Saez-Llorens X., Lopez P., Borja-Tabora C., Kosalaraksa P., et al. (2019). Efficacy of a Tetravalent Dengue Vaccine in Healthy Children and Adolescents. N Engl. J. Med. 381 (21), 2009–2019. doi: 10.1056/NEJMoa1903869
Blok J. (1985). Genetic relationships of the dengue virus serotypes. J. Gen. Virol. 66 ( Pt 6), 1323–1325. doi: 10.1099/0022-1317-66-6-1323
Boieri M., Ulvmoen A., Sudworth A., Lendrem C., Collin M., Dickinson A. M., et al. (2017). IL-12, IL-15, and IL-18 pre-activated NK cells target resistant T cell acute lymphoblastic leukemia and delay leukemia development in vivo. Oncoimmunology 6 (3), e1274478. doi: 10.1080/2162402X.2016.1274478
Brandstadter J. D., Yang Y. (2011). Natural killer cell responses to viral infection. J. Innate Immun. 3 (3), 274–279. doi: 10.1159/000324176
Brinkmann V., Reichard U., Goosmann C., Fauler B., Uhlemann Y., Weiss D. S., et al. (2004). Neutrophil extracellular traps kill bacteria. Science 303 (5663), 1532–1535. doi: 10.1126/science.1092385
Brown M. G., Hermann L. L., Issekutz A. C., Marshall J. S., Rowter D., Al-Afif A., et al. (2011). Dengue virus infection of mast cells triggers endothelial cell activation. J. Virol. 85 (2), 1145–1150. doi: 10.1128/JVI.01630-10
Burke S. M., Issekutz T. B., Mohan K., Lee P. W., Shmulevitz M., Marshall J. S. (2008). Human mast cell activation with virus-associated stimuli leads to the selective chemotaxis of natural killer cells by a CXCL8-dependent mechanism. Blood 111 (12), 5467–5476. doi: 10.1182/blood-2007-10-118547
Castillo J. A., Naranjo J. S., Rojas M., Castaño D., Velilla P. A. (2018). Role of Monocytes in the Pathogenesis of Dengue. Archivum. Immunol. Ther. Experiment. 67, 27–40. doi: 10.1007/s00005-018-0525-7
Cerny D., Haniffa M., Shin A., Bigliardi P., Tan B. K., Lee B., et al. (2014). Selective Susceptibility of Human Skin Antigen Presenting Cells to Productive Dengue Virus Infection. PloS Pathogens 10 (12), 1–15. doi: 10.1371/journal.ppat.1004548
Chan M., Johansson M. A. (2012). The Incubation Periods of Dengue Viruses. PloS One 7 (11), 1–7. doi: 10.1371/journal.pone.0050972
Chareonsirisuthigul T., Kalayanarooj S., Ubol S. (2007). Dengue virus (DENV) antibody-dependent enhancement of infection upregulates the production of anti-inflammatory cytokines, but suppresses anti-DENV free radical and pro-inflammatory cytokine production, in THP-1 cells. J. Gen. Virol. 88 (Pt 2), 365–375. doi: 10.1099/vir.0.82537-0
Chen R., Greene E. L., Collinsworth G., Grewal J. S., Houghton O., Zeng H., et al. (1999). Enrichment of transiently transfected mesangial cells by cell sorting after cotransfection with GFP. Am. J. Physiol. 276 (5 Pt 2), F777–F785. doi: 10.1152/ajprenal.1999.276.5.F777
Chen Y.-C., Wang S.-Y., King C.-C. (1999). Bacterial Lipopolysaccharide Inhibits Dengue Virus Infection of Primary Human Monocytes/Macrophages by Blockade of Virus Entry via a CD14-Dependent Mechanism. J. Virol. 73 (4), 2650–2657. doi: 10.1128/JVI.73.4.2650-2657.1999
Collin M., Bigley V. (2018). Human dendritic cell subsets: an update. Immunology 154, 3–20. doi: 10.1111/imm.12888
Compton S. J., Cairns J. A., Holgate S. T., Walls A. F. (1998). The role of mast cell tryptase in regulating endothelial cell proliferation, cytokine release, and adhesion molecule expression: tryptase induces expression of mRNA for IL-1 beta and IL-8 and stimulates the selective release of IL-8 from human umbilical vein endothelial cells. J. Immunol. 161 (4), 1939–1946.
Conway M. J., Watson A. M., Colpitts T. M., Dragovic S. M., Li Z., Wang P., et al. (2014). Mosquito Saliva Serine Protease Enhances Dissemination of Dengue Virus into the Mammalian Host. J. Virol. 88 (1), 164–175. doi: 10.1128/JVI.02235-13
Cooper M. A., Caligiuri M. A. (2004). Isolation and characterization of human natural killer cell subsets. Curr. Protoc. Immunol. 60, 7.34.1–7.34.12. doi: 10.1002/0471142735.im0734s60
Cooper M. A., Fehniger T. A., Caligiuri M. A. (2001). The biology of human natural killer-cell subsets. Trends Immunol. 22 (11), 633–640. doi: 10.1016/S1471-4906(01)02060-9
Cortjens B., de Boer O. J., de Jong R., Antonis A. F., Sabogal Pineros Y. S., Lutter R., et al. (2016). Neutrophil extracellular traps cause airway obstruction during respiratory syncytial virus disease. J. Pathol. 238 (3), 401–411. doi: 10.1002/path.4660
Cox J., Mota J., Sukupolvi-Petty S., Diamond M. S., Rico-Hesse R. (2012). Mosquito Bite Delivery of Dengue Virus Enhances Immunogenicity and Pathogenesis in Humanized Mice. J. Virol. 86 (14), 7637–7649. doi: 10.1128/JVI.00534-12
Crill W. D., Roehrig J. T. (2001). Monoclonal antibodies that bind to domain III of dengue virus E glycoprotein are the most efficient blockers of virus adsorption to Vero cells. J. Virol. 75 (16), 7769–7773. doi: 10.1128/JVI.75.16.7769-7773.2001
Crivellato E., Ribatti D. (2010). The mast cell: an evolutionary perspective. Biol. Rev. Camb. Philos. Soc 85 (2), 347–360. doi: 10.1111/j.1469-185X.2009.00105.x
Cruz-Oliveira C., Ao J., Freire M., Conceiç T. M., Conceição C., Higa L. M., et al. (2015). Receptors and routes of dengue virus entry into the host cells. FEMS Microbiol. Rev. 39, 155–170. doi: 10.1093/femsre/fuu004
Cumberworth S. L., Clark J. J., Kohl A., Donald C. L. (2017). Inhibition of type I interferon induction and signalling by mosquito-borne flaviviruses. Cell Microbiol. 19 (5), e12737. doi: 10.1111/cmi.12737
Dalrymple N. A., Cimica V., Mackow E. R., Buchmeier M. J. (2015). Dengue Virus NS Proteins Inhibit RIG-I/MAVS Signaling by Blocking TBK1/IRF3 Phosphorylation: Dengue Virus Serotype 1 NS4A Is a Unique Interferon-Regulating Virulence Determinant. mBio. 6 (3), e00553-15. doi: 10.1128/mBio.00553-15
Deen J. L., Harris E., Wills B., Balmaseda A., Hammond S. N., Rocha C., et al. (2006). The WHO dengue classification and case definitions: time for a reassessment. Lancet 368 (9530), 170–173. doi: 10.1016/S0140-6736(06)69006-5
Dejnirattisai W., Webb A. I., Chan V., Jumnainsong A., Davidson A., Mongkolsapaya J., et al. (2011). Lectin switching during dengue virus infection. J. Infect. Dis. 203 (12), 1775–1783. doi: 10.1093/infdis/jir173
Diamond M. S., Roberts T. G., Edgil D., Lu B., Ernst J., Harris E. (2000). Modulation of Dengue virus infection in human cells by alpha, beta, and gamma interferons. J. Virol. 74 (11), 4957–4966. doi: 10.1128/JVI.74.11.4957-4966.2000
Dieu-Nosjean M. C., Massacrier C., Homey B., Vanbervliet B., Pin J. J., Vicari A., et al. (2000). Macrophage inflammatory protein 3α is expressed at inflamed epithelial surfaces and is the most potent chemokine known in attracting langerhans cell precursors. J. Exp. Med. 192 (5), 705–717. doi: 10.1084/jem.192.5.705
Doebel T., Voisin B., Nagao K. (2017). Langerhans Cells-The Macrophage in Dendritic Cell Clothing. Trends Immunol. 38 (11), 817–828. doi: 10.1016/j.it.2017.06.008
Dorothy E., Lewis S. E. B. “Organization of the Immune System,” in Clinical Immunology, 5th ed. Eds. Rich TAF R. R., Shearer W. T., Schroeder H. W., Frew A. J., Weyand C. M. (Elsevier).
Douglas R., McDonald O. L. (2018). “Innate Immunity,” in Clinical Immunology, 5th ed. Eds. Rich TAF R. R., Shearer W. T., Schroeder H. W., Frew A. J., Weyand C. M. (Elsevier), 39–53.e1.
Duangkhae P., Erdos G., Ryman K. D., Watkins S. C., Falo L. D., Marques E. T. A., et al. (2018). Interplay between Keratinocytes and Myeloid Cells Drives Dengue Virus Spread in Human Skin. J. Invest. Dermatol. 138, 618–626. doi: 10.1016/j.jid.2017.10.018
Dudeck J., Froebel J., Kotrba J., Lehmann C. H. K., Dudziak D., Speier S., et al. (2019). Engulfment of mast cell secretory granules on skin inflammation boosts dendritic cell migration and priming efficiency. J. Allergy Clin. Immunol. 143 (5), 1849–64 e4. doi: 10.1016/j.jaci.2018.08.052
Durbin A. P., Vargas M. J., Wanionek K., Hammond S. N., Gordon A., Rocha C., et al. (2008). Phenotyping of peripheral blood mononuclear cells during acute dengue illness demonstrates infection and increased activation of monocytes in severe cases compared to classic dengue fever. Virology 376 (2), 429–435. doi: 10.1016/j.virol.2008.03.028
Endy T. P., Wang D., Polhemus M. E., Jarman R. G., Jasper L. E., Gromowski G., et al. (2020). A Phase 1, Open Label- Assessment of a Dengue Virus-1 Live Virus Human Challenge - (DENV-1-LVHC) Strain. J. Infect. Dis. 10.1093/infdis/jiaa351
Endy T. P. (2014). Dengue human infection model performance parameters. J. Infect. Dis. 209 Suppl 2, S56–S60. doi: 10.1093/infdis/jiu112
Fearon D. T., Locksley R. M. (1996). The instructive role of innate immunity in the acquired immune response. Science 272 (5258), 50–53. doi: 10.1126/science.272.5258.50
Fernandez-Mestre M. T., Gendzekhadze K., Rivas-Vetencourt P., Layrisse Z. (2004). TNF-alpha-308A allele, a possible severity risk factor of hemorrhagic manifestation in dengue fever patients. Tissue Antigens 64 (4), 469–472. doi: 10.1111/j.1399-0039.2004.00304.x
Fernando S., Wijewickrama A., Gomes L., Punchihewa C. T., Madusanka S. D., Dissanayake H., et al. (2016). Patterns and causes of liver involvement in acute dengue infection. BMC Infect. Dis. 16, 319. doi: 10.1186/s12879-016-1656-2
Friberg H., Bashyam H., Toyosaki-Maeda T., Potts J. A., Greenough T., Kalayanarooj S., et al. (2011). Cross-reactivity and expansion of dengue-specific T cells during acute primary and secondary infections in humans. Sci. Rep. 1, 51. doi: 10.1038/srep00051
Frossi B., De Carli M., Pucillo C. (2004). The mast cell: an antenna of the microenvironment that directs the immune response. J. Leukocyte Biol. 75 (4), 579–585. doi: 10.1189/jlb.0603275
Furuta T., Murao L. A., Lan N. T., Huy N. T., Huong V. T., Thuy T. T., et al. (2012). Association of mast cell-derived VEGF and proteases in Dengue shock syndrome. PloS Negl. Trop. Dis. 6 (2), e1505. doi: 10.1371/journal.pntd.0001505
Galli S. J., Nakae S., Tsai M. (2005a). Mast cells in the development of adaptive immune responses. Nat. Immunol. 6 (2), 135–142. doi: 10.1038/ni1158
Galli S. J., Kalesnikoff J., Grimbaldeston M. A., Piliponsky A. M., Williams C. M., Tsai M. (2005b). Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu. Rev. Immunol. 23, 749–786. doi: 10.1146/annurev.immunol.21.120601.141025
Garcia M., Wehbe M., Lévêque N., Bodet C. (2017). Skin innate immune response to flaviviral infection. Cytokine Netw. 28 (2), 41–51. doi: 10.1684/ecn.2017.0394
Gasser S., Raulet D. H. (2006). Activation and self-tolerance of natural killer cells. Immunol. Rev. 214, 130–142. doi: 10.1111/j.1600-065X.2006.00460.x
Goncalves D., de Queiroz Prado R., Almeida Xavier E., Cristina de Oliveira N., da Matta Guedes P. M., da Silva J. S., et al. (2012). Immunocompetent mice model for dengue virus infection [corrected]. Sci. World J. 2012, 525947. doi: 10.1100/2012/525947
Goncalvez A. P., Engle R. E., St. Claire M., Purcell R. H., Lai C. J. (2007). Monoclonal antibody-mediated enhancement of dengue virus infection in vitro and in vivo and strategies for prevention. Proc. Natl. Acad. Sci. U. States A. 104 (22), 9422–9427. doi: 10.1073/pnas.0703498104
Green S., Rothman A. (2006). Immunopathological mechanisms in dengue and dengue hemorrhagic fever. Curr. Opin. Infect. Dis. 19 (5), 429–436. doi: 10.1097/01.qco.0000244047.31135.fa
Gubler D. J. (1998). Dengue and dengue hemorrhagic fever. Clin. Microbiol. Rev. 11 (3), 480–496. doi: 10.1128/CMR.11.3.480
Halova I., Draberova L., Draber P. (2012). Mast cell chemotaxis - chemoattractants and signaling pathways. Front. Immunol. 3, 119. doi: 10.3389/fimmu.2012.00119
Halstead S. B., O’Rourke E. J. (1977a). Antibody-enhanced dengue virus infection in primate leukocytes. Nature 265 (5596), 739–741. doi: 10.1038/265739a0
Halstead S. B., O’Rourke E. J. (1977b). Dengue viruses and mononuclear phagocytes. I. Infection enhancement by non-neutralizing antibody. J. Exp. Med. 146 (1), 201–217. doi: 10.1084/jem.146.1.201
Halstead S. B., Nimmannitya S., Yamarat C., Russell P. K. (1967). Hemorrhagic fever in Thailand; recent knowledge regarding etiology. Jpn. J. Med. Sci. Biol. 20, 96–103.
Halstead S. B., O’Rourke E. J., Allison A. C. (1977). Dengue viruses and mononuclear phagocytes. II. Identity of blood and tissue leukocytes supporting in vitro infection. J. Exp. Med. 146 (1), 218–229. doi: 10.1084/jem.146.1.218
Halstead S. B., Rojanasuphot S., Sangkawibha N. (1983). Original antigenic sin in dengue. Am. J. Trop. Med. Hyg. 32 (1), 154–156. doi: 10.4269/ajtmh.1983.32.154
Halstead S. B., Mahalingam S., Marovich M. A., Ubol S., Mosser D. M. (2010). Intrinsic antibody-dependent enhancement of microbial infection in macrophages: disease regulation by immune complexes. Lancet Infect. Dis. 10 (10), 712–722. doi: 10.1016/S1473-3099(10)70166-3
Halstead S. B. (1979). In vivo enhancement of dengue virus infection in Rhesus monkeys by passively transferred antibody. J. Infect. Dis. 140 (4), 527–533. doi: 10.1093/infdis/140.4.527
Halstead S. B. (1988). Pathogenesis of dengue: challenges to molecular biology. Science 239, 476–481. doi: 10.1126/science.239.4839.476
Halstead S. B. (1994). Antibody-dependent Enhancement of Infection: A Mechanism for Indirect Virus Entry into cells. Cellular Receptors for Animal Viruses (Long Island, New York: Cold Spring Harbor Laboratory Press), 493–515.
Halstead S. B. (1998). “Dengue viruses,” in Infectious Diseases, 2nd ed. Eds. Gorbach S. L., Bartlett J. G., Blacklow N. R. (Philadelphia: W.B. Saunders).
Hastings A. K., Uraki R., Gaitsch H., Dhaliwal K., Stanley S., Sproch H., et al. (2019). Aedes aegypti NeSt1 Protein Enhances Zika Virus Pathogenesis by Activating Neutrophils. J. Virol. 93 (13), 1–16. doi: 10.1128/JVI.00395-19
He Z., Zhu X., Wen W., Yuan J., Hu Y., Chen J., et al. (2016). Dengue Virus Subverts Host Innate Immunity by Targeting Adaptor Protein MAVS. J. Virol. 90, 7219–7230. doi: 10.1128/JVI.00221-16
Hemmers S., Teijaro J. R., Arandjelovic S., Mowen K. A. (2011). PAD4-mediated neutrophil extracellular trap formation is not required for immunity against influenza infection. PloS One 6 (7), e22043. doi: 10.1371/journal.pone.0022043
Henney C. S., Kuribayashi K., Kern D. E., Gillis S. (1981). Interleukin-2 augments natural killer cell activity. Nature 291 (5813), 335–338. doi: 10.1038/291335a0
Hoang L. T., Lynn D. J., Henn M., Birren B. W., Lennon N. J., Le P. T., et al. (2010). The early whole-blood transcriptional signature of dengue virus and features associated with progression to dengue shock syndrome in Vietnamese children and young adults. J. Virol. 84 (24), 12982–12994. doi: 10.1128/JVI.01224-10
Hoffman I., Monroe R. C., Abernathy R. S., Hall R. J., Picchi J., Speers R. W., et al. (1954). The possible role of histamine in epidemic hemorrhagic fever: an evaluation of antihistamine therapy. U. S. Armed Forces Med. J. 5 (5), 680–687.
Holmes E. C., Twiddy S. S. (2003). The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol. 3 (1), 19–28. doi: 10.1016/S1567-1348(03)00004-2
Holmes E. C. (2003). Patterns of intra- and interhost nonsynonymous variation reveal strong purifying selection in dengue virus. J. Virol. 77 (20), 11296–11298. doi: 10.1128/JVI.77.20.11296-11298.2003
Holmes E. C. (2006). The evolutionary biology of dengue virus. Novartis Foundation symposium, Vol. 277. 177–87; discussion 87-92, 251-3 (John Wiley & Sons Ltd.).
Homchampa P., Sarasombath S., Suvatte V., Vongskul M. (1988). Natural killer cells in dengue hemorrhagic fever/dengue shock syndrome. Asian Pac. J. Allergy Immunol. 6 (2), 95–102.
Huang D., Shi F. D., Jung S., Pien G. C., Wang J., Salazar-Mather T. P., et al. (2006). The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. 20 (7), 896–905. doi: 10.1096/fj.05-5465com
Janeway C. A. Jr., Medzhitov R. (2002). Innate immune recognition. Annu. Rev. Immunol. 20, 197–216. doi: 10.1146/annurev.immunol.20.083001.084359
Jawdat D. M., Rowden G., Marshall J. S. (2006). Mast cells have a pivotal role in TNF-independent lymph node hypertrophy and the mobilization of Langerhans cells in response to bacterial peptidoglycan. J. Immunol. 177 (3), 1755–1762. doi: 10.4049/jimmunol.177.3.1755
Jonsson A. H., Yokoyama W. M. (2009). Natural killer cell tolerance licensing and other mechanisms. Adv. Immunol. 101, 27–79. doi: 10.1016/S0065-2776(08)01002-X
Jost S., Altfeld M. (2013). Control of human viral infections by natural killer cells. Annu. Rev. Immunol. 31, 163–194. doi: 10.1146/annurev-immunol-032712-100001
Kaplan M. J., Radic M. (2012). Neutrophil extracellular traps: double-edged swords of innate immunity. J. Immunol. 189 (6), 2689–2695. doi: 10.4049/jimmunol.1201719
Karre K. (1995). Express yourself or die: peptides, MHC molecules, and NK cells. Science 267 (5200), 978–979. doi: 10.1126/science.7863341
King A. D., Nisalak A., Kalayanrooj S., Myint K. S., Pattanapanyasat K., Nimmannitya S., et al. (1999). B Cells are the Principal Circulating Mononuclear Cells Infected by Dengue Virus. Southeast Asian J. Trop. Med. Public Health 30 (4), 718–728.
King C. A., Marshall J. S., Alshurafa H., Anderson R. (2000). Release of vasoactive cytokines by antibody-enhanced dengue virus infection of a human mast cell/basophil line. J. Virol. 74 (15), 7146–7150. doi: 10.1128/JVI.74.15.7146-7150.2000
King C. A., Anderson R., Marshall J. S. (2002). Dengue virus selectively induces human mast cell chemokine production. J. Virol. 76 (16), 8408–8419. doi: 10.1128/JVI.76.16.8408-8419.2002
Klein L. M., Lavker R. M., Matis W. L., Murphy G. F. (1989). Degranulation of human mast cells induces an endothelial antigen central to leukocyte adhesion. Proc. Natl. Acad. Sci. U. S. A. 86 (22), 8972–8976. doi: 10.1073/pnas.86.22.8972
Knight J. S., Carmona-Rivera C., Kaplan M. J. (2012). Proteins derived from neutrophil extracellular traps may serve as self-antigens and mediate organ damage in autoimmune diseases. Front. Immunol. 3, 380. doi: 10.3389/fimmu.2012.00380
Kochel T. J., Watts D. M., Halstead S. B., Hayes C. G., Espinoza A., Felices V., et al. (2002). Effect of dengue-1 antibodies on American dengue-2 viral infection and dengue haemorrhagic fever. Lancet 360 (9329), 310–312. doi: 10.1016/S0140-6736(02)09522-3
Koraka P., Murgue B., Deparis X., Setiati T. E., Suharti C., Van Gorp E. C. M., et al. (2003). Elevated Levels of Total and Dengue Virus-Specific Immunoglobulin E in Patients With Varying Disease Severity. J. Med. Virol. 70, 91–98. doi: 10.1002/jmv.10358
Kunder C. A., St John A. L., Abraham S. N. (2011). Mast cell modulation of the vascular and lymphatic endothelium. Blood 118 (20), 5383–5393. doi: 10.1182/blood-2011-07-358432
Kunder M., Lakshmaiah V., Moideen Kutty A. V. (2018). Plasma Neutrophil Elastase, alpha1-Antitrypsin, alpha2-Macroglobulin and Neutrophil Elastase-alpha1-Antitrypsin Complex Levels in patients with Dengue Fever. Indian J. Clin. Biochem. 33 (2), 218–221. doi: 10.1007/s12291-017-0658-1
Kuruvilla J. G., Troyer R. M., Devi S., Akkina R. (2007). Dengue virus infection and immune response in humanized RAG2(-/-)gamma(c)(-/-) (RAG-hu) mice. Virology 369 (1), 143–152. doi: 10.1016/j.virol.2007.06.005
Laoprasopwattana K., Libraty D. H., Endy T. P., Nisalak A., Chunsuttiwat S., Ennis F. A., et al. (2007). Antibody-dependent cellular cytotoxicity mediated by plasma obtained before secondary dengue virus infections: potential involvement in early control of viral replication. J. Infect. Dis. 195 (8), 1108–1116. doi: 10.1086/512860
Lee C.-K., Hirsch A. J., Xing J., Yu C.-Y., Kao Y.-T., Lai M. M. C. (2018). How Dengue Virus Circumvents Innate Immunity. Front. Immunol. 9, 2860–. doi: 10.3389/fimmu.2018.02860
Lyons A. G. (2014). The human dengue challenge experience at the Walter Reed Army Institute of Research. J. Infect. Dis. 209 Suppl 2, S49–S55. doi: 10.1093/infdis/jiu174
Maaninka K., Lappalainen J., Kovanen P. T. (2013). Human mast cells arise from a common circulating progenitor. J. Allergy Clin. Immunol. 132 (2), 463–9 e3. doi: 10.1016/j.jaci.2013.02.011
Macri C., Pang S., Patton T., O’Keeffe M. (2018). Dendritic cell subsets. Semin. Cell Dev. Biol. 84, 11–21. doi: 10.1016/j.semcdb.2017.12.009
Mammen M. P., Lyons A., Innis B. L., Sun W., McKinney D., Chung R. C., et al. (2014). Evaluation of dengue virus strains for human challenge studies. Vaccine 32 (13), 1488–1494. doi: 10.1016/j.vaccine.2013.12.040
Marshall J. S., Bienenstock J. (1994). The role of mast cells in inflammatory reactions of the airways, skin and intestine. Curr. Opin. Immunol. 6 (6), 853–859. doi: 10.1016/0952-7915(94)90004-3
Marshall J. S., Jawdat D. M. (2004). Mast cells in innate immunity. J. Allergy Clin. Immunol. 114 (1), 21–27. doi: 10.1016/j.jaci.2004.04.045
Martina B. E. E., Koraka P., Osterhaus A. D. M. E. (2009). Dengue Virus Pathogenesis: an Integrated View. Clin. Microbiol. Rev. 22 (4), 564–581. doi: 10.1128/CMR.00035-09
Metz M., Maurer M. (2007). Mast cells–key effector cells in immune responses. Trends Immunol. 28 (5), 234–241. doi: 10.1016/j.it.2007.03.003
Metz M., Grimbaldeston M. A., Nakae S., Piliponsky A. M., Tsai M., Galli S. J. (2007). Mast cells in the promotion and limitation of chronic inflammation. Immunol. Rev. 217, 304–328. doi: 10.1111/j.1600-065X.2007.00520.x
Michel T., Hentges F., Zimmer J. (2012). Consequences of the crosstalk between monocytes/macrophages and natural killer cells. Front. Immunol. 3, 403. doi: 10.3389/fimmu.2012.00403
Midgley C. M., Bajwa-Joseph M., Vasanawathana S., Limpitikul W., Wills B., Flanagan A., et al. (2011). An in-depth analysis of original antigenic sin in dengue virus infection. J. Virol. 85 (1), 410–421. doi: 10.1128/JVI.01826-10
Mishra A. K., George A. A., Abhilash K. P. P. (2018). The relationship between skin rash and outcome in dengue. J. Vector Borne Dis. 55 (4), 310–314. doi: 10.4103/0972-9062.256567
Mongkolsapaya J., Dejnirattisai W., Xu X. N., Vasanawathana S., Tangthawornchaikul N., Chairunsri A., et al. (2003). Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat. Med. 9 (7), 921–927. doi: 10.1038/nm887
Morens D. M., Halstead S. B. (1987). Disease severity-related antigenic differences in dengue 2 strains detected by dengue 4 monoclonal antibodies. J. Med. Virol. 22, 169–174. doi: 10.1002/jmv.1890220208
Morrison J., García-Sastre A. (2014). STAT2 signaling and dengue virus infection. JAK-STAT 3 (1), e27715–e2771e. doi: 10.4161/jkst.27715
Morrison B. E., Park S. J., Mooney J. M., Mehrad B. (2003). Chemokine-mediated recruitment of NK cells is a critical host defense mechanism in invasive aspergillosis. J. Clin. Invest. 112 (12), 1862–1870. doi: 10.1172/JCI18125
Mota J., Rico-Hesse R. (2011). Dengue virus tropism in humanized mice recapitulates human dengue fever. PloS One 6 (6), e20762. doi: 10.1371/journal.pone.0020762
Nakamura Y., Kambe N., Saito M., Nishikomori R., Kim Y. G., Murakami M., et al. (2009). Mast cells mediate neutrophil recruitment and vascular leakage through the NLRP3 inflammasome in histamine-independent urticaria. J. Exp. Med. 206 (5), 1037–1046. doi: 10.1084/jem.20082179
Navarro-Sanchez E., Altmeyer R., Amara A., Schwartz O., Fieschi F., Virelizier J. L., et al. (2003). Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 4 (7), 723–728. doi: 10.1038/sj.embor.embor866
Navarro-Sánchez E., Desprè P., Cedillo-Barrón L. (2005). Innate Immune Responses to Dengue Virus. Arch. Med. Res. 36, 425–435. doi: 10.1016/j.arcmed.2005.04.007
Ngono A. E., Shresta S. (2018). Immune Response to Dengue and Zika. Annu. Rev. Immunol. 36 (10), 1–30. doi: 10.1146/annurev-immunol-042617-053142
Owen J. A., Punt J., Stranford S. A., Jones P. (2013). Kuby Immunology. 7th ed (New York City: MacMillan), 120–1 p.
Paolini R., Bernardini G., Molfetta R., Santoni A. (2015). NK cells and interferons. Cytokine Growth Factor Rev. 26 (2), 113–120. doi: 10.1016/j.cytogfr.2014.11.003
Patente T. A., Pelgrom L. R., Everts B. (2019). Dendritic cells are what they eat: how their metabolism shapes T helper cell polarization. Curr. Opin. Immunol. 58, 16–23. doi: 10.1016/j.coi.2019.02.003
Pichyangkul S., Endy T. P., Kalayanarooj S., Nisalak A., Yongvanitchit K., Green S., et al. (2003). A blunted blood plasmacytoid dendritic cell response to an acute systemic viral infection is associated with increased disease severity. J. Immunol. 171 (10), 5571–5578. doi: 10.4049/jimmunol.171.10.5571
Portales-Cervantes L., Dawod B., Marshall J. S. (2019). Mast Cells and Natural Killer Cells-A Potentially Critical Interaction. Viruses, 11 (6), 514. doi: 10.3390/v11060514
Rathore A. P. S., St John A. L. (2018). Immune responses to dengue virus in the skin. Open Biol. 8, 1–9. doi: 10.1098/rsob.180087
Rathore A. P. S., Mantri C. K., Aman S. A. B., Syenina A., Ooi J., Jagaraj C. J., et al. (2019). Dengue virus-elicited tryptase induces endothelial permeability and shock. J. Clin. Invest. 129 (10), 4180–4193. doi: 10.1172/JCI128426
Reyes-del Valle J., Salas-Benito J., Soto-Acosta R., del Angel R. M. (2014). Dengue Virus Cellular Receptors and Tropism. Curr. Trop. Med. Rep. 1, 36–43. doi: 10.1007/s40475-013-0002-7
Rhodes C. P., Ratcliffe N. A., Rowley A. F. (1982). Presence of coelomocytes in the primitive chordate amphioxus (Branchiostoma lanceolatum). Science 217 (4556), 263–265. doi: 10.1126/science.7089565
Rider P., Carmi Y., Guttman O., Braiman A., Cohen I., Voronov E., et al. (2011). IL-1α and IL-1β Recruit Different Myeloid Cells and Promote Different Stages of Sterile Inflammation. J. Immunol. 187 (9), 4835–4843. doi: 10.4049/jimmunol.1102048
Rodenhuis-Zybert I. A., van der Schaar H. M., da Silva Voorham J. M., van der Ende-Metselaar H., Lei H.-Y., Wilschut J., et al. (2010). Immature Dengue Virus: A Veiled Pathogen? PloS Pathogens 6 (1), e1000718–e. doi: 10.1371/journal.ppat.1000718
Rothman L., Ennis F. A., Libraty D. H., Nisalak A., Yongvanitchit K., Green S., et al. (2003). A Blunted Blood Plasmacytoid Dendritic Cell Response to an Acute Systemic Viral Infection Is Associated with Increased Disease Severity. J. Immunol. References 171, 5571–5578. doi: 10.4049/jimmunol.171.10.5571
Rothman A. L. (2010). Cellular immunology of sequential dengue virus infection and its role in disease pathogenesis. Curr. topics Microbiol. Immunol. 338, 83–98. doi: 10.1007/978-3-642-02215-9_7
Rothman A. L. (2011). Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat. Rev. Immunol. 11 (8), 532–543. doi: 10.1038/nri3014
Schaeffer E., Flacher V., Papageorgiou V., Decossas M., Fauny J. D., Krämer M., et al. (2015). Dermal CD14 + dendritic cell and macrophage infection by dengue virus is stimulated by interleukin-4. J. Invest. Dermatol. 135 (7), 1743–1751. doi: 10.1038/jid.2014.525
Schmid M. A., Harris E. (2014). Monocyte Recruitment to the Dermis and Differentiation to Dendritic Cells Increases the Targets for Dengue Virus Replication. PloS Pathogens 10 (12), 1–18. doi: 10.1371/journal.ppat.1004541
Schmid M. A., Diamond M. S., Harris E. (2014). Dendritic cells in dengue virus infection: Targets of virus replication and mediators of immunity. Front. Immunol. 5 (647), 1–10. doi: 10.3389/fimmu.2014.00647
Scott T. W., Morrison A. C. (2010). Vector dynamics and transmission of dengue virus: implications for dengue surveillance and prevention strategies: vector dynamics and dengue prevention. Curr. Topics Microbiol. Immunol. 338, 115–128. doi: 10.1007/978-3-642-02215-9_9
Shi C., Pamer E. G. (2011). Monocyte recruitment during infection and inflammation. Nat. Rev. Immunol. 11 (11), 762–774. doi: 10.1038/nri3070
Shresta S., Sharar K. L., Prigozhin D. M., Beatty P. R., Harris E. (2006). Murine model for dengue virus-induced lethal disease with increased vascular permeability. J. Virol. 80 (20), 10208–10217. doi: 10.1128/JVI.00062-06
Siddique T., Ali M., Qadri A., Saeed K. (2008). Dengue fever, Presenation, and different treatment options in 30 patients. Pakastan J. Med. Helath Sci. 2 (3), 112–113.
Silvestre-Roig C., Hidalgo A., Soehnlein O. (2016). Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood 127 (18), 2173–2181. doi: 10.1182/blood-2016-01-688887
Sleijfer S., Bannink M., Van Gool A. R., Kruit W. H. J., Stoter G. (2005). Side effects of interferon-α therapy. Pharm. World Sci. 27, 423–431. doi: 10.1007/s11096-005-1319-7
Snow G. E., Haaland B., Ooi E. E., Gubler D. J. (2014). Research on Dengue during World War II Revisited. Am. J. Trop. Med. Hyg. 91, 1203–1217. doi: 10.4269//ajtmh.14-0132
Srikiatkhachorn A. (2009). Plasma leakage in dengue haemorrhagic fever. Thromb. Haemostasis. 102 (6), 1042–1049. doi: 10.1160/TH09-03-0208
St John A. L., Rathore A. P., Yap H., Ng M. L., Metcalfe D. D., Vasudevan S. G., et al. (2011). Immune surveillance by mast cells during dengue infection promotes natural killer (NK) and NKT-cell recruitment and viral clearance. Proc. Natl. Acad. Sci. U. S. A. 108 (22), 9190–9195. doi: 10.1073/pnas.1105079108
Stephens H. A. F., Klaythong R., Sirikong M., Vaughn D. W., Green S., Kalayanarooj S., et al. (2002). HLA-A and -B allele associations with secondary dengue virus infections correlate with disease severity and the infecting viral serotype in ethnic Thais. Tissue Antigens 60 (4), 309–318. doi: 10.1034/j.1399-0039.2002.600405.x
Stephens H. A. (2010). HLA and other gene associations with dengue disease severity. Curr. Top. Microbiol. Immunol. 338, 99–114. doi: 10.1007/978-3-642-02215-9_8
Stoitzner P., Zanella M., Ortner U., Lukas M., Tagwerker A., Janke K., et al. (1999). Migration of Langerhans cells and dermal dendritic cells in skin organ cultures: augmentation by TNF-α and IL-1 β. J. Leukocyte Biol. 66 (3), 462–470. doi: 10.1002/jlb.66.3.462
Styer L. M., Lim P. Y., Louie K. L., Albright R. G., Kramer L. D., Bernard K. A. (2011). Mosquito Saliva Causes Enhancement of West Nile Virus Infection in Mice. J. Virol. 85 (4), 1517–1527. doi: 10.1128/JVI.01112-10
Sun W., Eckels K. H., Putnak J. R., Lyons A. G., Thomas S. J., Vaughn D. W., et al. (2013). Experimental dengue virus challenge of human subjects previously vaccinated with live attenuated tetravalent dengue vaccines. J. Infect. Dis. 207 (5), 700–708. doi: 10.1093/infdis/jis744
Suto H., Nakae S., Kakurai M., Sedgwick J. D., Tsai M., Galli S. J. (2006). Mast cell-associated TNF promotes dendritic cell migration. J. Immunol. 176 (7), 4102–4112. doi: 10.4049/jimmunol.176.7.4102
Swiecki M., Colonna M. (2015). The multifaceted biology of plasmacytoid dendritic cells. Nat. Rev. Immunol. 15, 471–485. doi: 10.1038/nri3865
Taweechaisupapong S., Sriurairatana S., Angsubhakorn S., Yoksan S., Bhamarapravati N. (1996a). In vivo and in vitro studies on the morphological change in the monkey epidermal Langerhans cells following exposure to dengue 2 (16681) virus. Southeast Asian J. Trop. Med. Public Health 27 (4), 664–672.
Taweechaisupapong S., Sriurairatana S., Angsubhakorn S., Yoksan S., Khin M. M., Sahaphong S., et al. (1996b). Langerhans cell density and serological changes following intradermal immunisation of mice with dengue 2 virus. J. Med. Microbiol. 45 (2), 138–145. doi: 10.1099/00222615-45-2-138
Thein T. L., Lye D. C., Leo Y. S., Wong J. G., Hao Y., Wilder-Smith Y., et al. (2014). Severe neutropenia in dengue patients: prevalence and significance. Am. J. Trop. Med. Hyg. 90 (6), 984–987. doi: 10.4269/ajtmh.14-0004
Thomas S. J. (2013). Dengue human infection model: Re-establishing a tool for understanding dengue immunology and advancing vaccine development. Hum. vaccines Immunother. 9 (7), 1587–1590. doi: 10.4161/hv.24188
Tian Y., Grifoni A., Sette A., Weiskopf D. (2019). Human T Cell Response to Dengue Virus Infection. Front. Immunol. 10, 2125. doi: 10.3389/fimmu.2019.02125
Tissera H., Rathore A. P. S., Leong W. Y., Pike B. L., Warkentien T. E., Farouk F. S., et al. (2017). Chymase Level Is a Predictive Biomarker of Dengue Hemorrhagic Fever in Pediatric and Adult Patients. J. Infect. Dis. 216 (9), 1112–1121. doi: 10.1093/infdis/jix447
Todd P. A., Goa K. L. (1992). Interferon Gamma-1b: A Review of its Pharmacology and Therapeutic Potential in Chronic Granulomatous Disease. Drugs 43 (1), 111–122. doi: 10.2165/00003495-199243010-00008
Torkildsen O., Myhr K. M., Bø L. (2016). Disease-modifying treatments for multiple sclerosis - a review of approved medications. Eur. J. Neurol. 23 (Suppl 1), 18–27. doi: 10.1111/ene.12883
Toussaint M., Jackson D. J., Swieboda D., Guedan A., Tsourouktsoglou T. D., Ching Y. M., et al. (2017). Host DNA released by NETosis promotes rhinovirus-induced type-2 allergic asthma exacerbation. Nat. Med. 23 (6), 681–691. doi: 10.1038/nm.4332
Tremblay N., Freppel W., Sow A. A., Chatel-Chaix L. (2019). The Interplay between Dengue Virus and the Human Innate Immune System: A Game of Hide and Seek. Vaccines 7 (4), 145–. doi: 10.3390/vaccines7040145
Troupin A., Shirley D., Londono-Renteria B., Watson A. M., McHale C., Hall A., et al. (2016). A Role for Human Skin Mast Cells in Dengue Virus Infection and Systemic Spread. J. Immunol. 197 (11), 4382–4391. doi: 10.4049/jimmunol.1600846
Tuchinda M., Dhorranintra B., Tuchinda P. (1977). Histamine content in 24-hour urine in patients with dengue haemorrhagic fever. Southeast Asian. J. Trop. Med. Public Health 8 (1), 80–83.
Twiddy S. S., Holmes E. C., Rambaut A. (2003a). Inferring the rate and time-scale of dengue virus evolution. Mol. Biol. Evol. 20 (1), 122–129. doi: 10.1093/molbev/msg010
Twiddy S. S., Pybus O. G., Holmes E. C. (2003b). Comparative population dynamics of mosquito-borne flaviviruses. Infect. Genet. Evol. 3 (2), 87–95. doi: 10.1016/S1567-1348(02)00153-3
Uno N., Ross T. M. (2018). Dengue virus and the host innate immune response. Emerg. Microbes. Infect. 7, 1–11. doi: 10.1038/s41426-018-0168-0
Valent P. (2013). Mast cell activation syndromes: definition and classification. Allergy 68 (4), 417–424. doi: 10.1111/all.12126
Vaughn D. W., Green S., Kalayanarooj S., Innis B. L., Nimmannitya S., Suntayakorn S., et al. (2000). Dengue viremia titer, antibody response pattern, and virus serotype correlate with disease severity. J. Infect. Dis. 181 (1), 2–9. doi: 10.1086/315215
Vejbaesya S., Luangtrakool P., Luangtrakool K., Kalayanarooj S., Vaughn D. W., Endy T. P., et al. (2009). TNF and LTA gene, allele, and extended HLA haplotype associations with severe dengue virus infection in ethnic Thais. J. Infect. Dis. 199 (10), 1442–1448. doi: 10.1086/597422
Walzer T., Chiossone L., Chaix J., Calver A., Carozzo C., Garrigue-Antar L., et al. (2007). Natural killer cell trafficking in vivo requires a dedicated sphingosine 1-phosphate receptor. Nat. Immunol. 8 (12), 1337–1344. doi: 10.1038/ni1523
Watanabe S., Tan K. H., Rathore A. P., Rozen-Gagnon K., Shuai W., Ruedl C., et al. (2012). The magnitude of dengue virus NS1 protein secretion is strain dependent and does not correlate with severe pathologies in the mouse infection model. J. Virol. 86 (10), 5508–5514. doi: 10.1128/JVI.07081-11
Webster B., Assil S., Dreux M. (2016). Cell-Cell Sensing of Viral Infection by Plasmacytoid Dendritic Cells. J. Virol. 90 (22), 10050–10053. doi: 10.1128/JVI.01692-16
Webster B., Werneke S. W., Zafirova B., This S., Coléon S., Décembre E., et al. (2018). Plasmacytoid dendritic cells control dengue and chikungunya virus infections via IRF7-regulated interferon responses. eLife 7, 1–23. doi: 10.7554/eLife.34273
WHO (2009). Dengue: Guidelines for diagnosis, treatment, prevention and control (Geneva, Switzerland: World Health Organization).
Wittamer V., Bertrand J. Y., Gutschow P. W., Traver D. (2011). Characterization of the mononuclear phagocyte system in zebrafish. Blood 117 (26), 7126–7135. doi: 10.1182/blood-2010-11-321448
Worbs T., Hammerschmidt S. I., Forster R. (2017). Dendritic cell migration in health and disease. Nat. Rev. Immunol. 17 (1), 30–48. doi: 10.1038/nri.2016.116
Wu S.-J. L., Grouard-Vogel G., Sun W., Mascola J. R., Brachtel E., Putvatana R., et al. (2000). Human skin Langerhans cells are targets of dengue virus infection. Nat. Med. 6 (7), 816–820. doi: 10.1038/77553
Yauch L. E., Shresta S. (2008). Mouse models of dengue virus infection and disease. Antiviral Res. 80 (2), 87–93. doi: 10.1016/j.antiviral.2008.06.010
Yauch L. E., Zellweger R. M., Kotturi M. F., Qutubuddin A., Sidney J., Peters B., et al. (2009). A protective role for dengue virus-specific CD8+ T cells. J. Immunol. 182 (8), 4865–4873. doi: 10.4049/jimmunol.0801974
Yokoyama W. M., Kim S., French A. R. (2004). The dynamic life of natural killer cells. Annu. Rev. Immunol. 22, 405–429. doi: 10.1146/annurev.immunol.22.012703.104711
Zanini F., Robinson M. L., Croote D., Sahoo M. K., Sanz A. M., Ortiz-Lasso E., et al. (2018). Virus-inclusive single-cell RNA sequencing reveals the molecular signature of progression to severe dengue. Proc. Nat. Acad. Sci. U. S. A. 115 (52), E12363–E123E9. doi: 10.1073/pnas.1813819115
Zellweger R. M., Shresta S. (2014). Mouse models to study dengue virus immunology and pathogenesis. Front. Immunol. 5, 151. doi: 10.3389/fimmu.2014.00151
Zellweger R. M., Prestwood T. R., Shresta S. (2010). Enhanced infection of liver sinusoidal endothelial cells in a mouse model of antibody-induced severe dengue disease. Cell Host Microbe 7 (2), 128–139. doi: 10.1016/j.chom.2010.01.004
Zellweger R. M., Eddy W. E., Tang W. W., Miller R., Shresta S. (2014). CD8+ T cells prevent antigen-induced antibody-dependent enhancement of dengue disease in mice. J. Immunol. 193 (8), 4117–4124. doi: 10.4049/jimmunol.1401597
Zhang Y., Ramos B. F., Jakschik B. A. (1992). Neutrophil recruitment by tumor necrosis factor from mast cells in immune complex peritonitis. Science 258 (5090), 1957–1959. doi: 10.1126/science.1470922
Zhang C., Mammen M. P. Jr., Chinnawirotpisan P., Klungthong C., Rodpradit P., Monkongdee P., et al. (2005). Clade replacements in dengue virus serotypes 1 and 3 are associated with changing serotype prevalence. J. Virol. 79 (24), 15123–15130. doi: 10.1128/JVI.79.24.15123-15130.2005
Zimmer C. L., Cornillet M., Sola-Riera C., Cheung K. W., Ivarsson M. A., Lim M. Q., et al. (2019). NK cells are activated and primed for skin-homing during acute dengue virus infection in humans. Nat. Commun. 10 (1), 3897. doi: 10.1038/s41467-019-11878-3
Zompi S., Harris E. (2012). Animal Models of Dengue Virus Infection. Viruses 4 (1), 62–82. doi: 10.3390/v4010062
Keywords: dengue, innate immunity, macrophages, mast cells, interferon, inflammation, pathogenesis, clinical symptoms
Citation: King CA, Wegman AD and Endy TP (2020) Mobilization and Activation of the Innate Immune Response to Dengue Virus. Front. Cell. Infect. Microbiol. 10:574417. doi: 10.3389/fcimb.2020.574417
Received: 19 June 2020; Accepted: 28 September 2020;
Published: 03 November 2020.
Edited by:
Yean Kong Yong, Xiamen University, Malaysia, MalaysiaReviewed by:
Rebecca Tweedell, St. Jude Children’s Research Hospital, United StatesEda K. Holl, Duke University, United States
Copyright © 2020 King, Wegman and Endy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Christine A. King, a2luZ2NoQHVwc3RhdGUuZWR1