 Anna Maria Gorska
Anna Maria Gorska Eliseo A. Eugenin
Eliseo A. Eugenin
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 24 June 2020
Sec. Virus and Host
Volume 10 - 2020 | https://doi.org/10.3389/fcimb.2020.00261
This article is part of the Research Topic Direct and Indirect Interactions of HIV with Host Cells View all 10 articles
Glutamate (Glu) is the most abundant excitatory neurotransmitter in the central nervous system (CNS). HIV-1 and viral proteins compromise glutamate synaptic transmission, resulting in poor cell-to-cell signaling and bystander toxicity. In this study, we identified that myeloid HIV-1-brain reservoirs survive in Glu and glutamine (Gln) as a major source of energy. Thus, we found a link between synaptic compromise, metabolomics of viral reservoirs, and viral persistence. In the current manuscript we will discuss all these interactions and the potential to achieve eradication and cure using this unique metabolic profile.
Neurons are not able to perform de novo synthesis of the neurotransmitters glutamate (Glu) and gamma-aminobutyric acid (GABA) from glucose due to the lack of enzymes involved in their metabolism. Thus, a multicellular metabolic pathway known as the Glu/GABA-glutamine (Gln) cycle maintains the balance among these metabolites to support neuronal synaptic transmission (Bak et al., 2006).
Glu is the most abundant excitatory neurotransmitter in the central nervous system (CNS) (Zhou and Danbolt, 2014), and GABA is considered an inhibitory neurotransmitter in adulthood (McCormick, 1989; Harris-Warrick, 2005). A balance between these two neurotransmitters plays a critical role in several brain functions including learning and memory, development, pain, synaptogenesis, motor stimuli, and synaptic signaling (Petroff, 2002; Allen et al., 2004; Bak et al., 2006; Bonansco and Fuenzalida, 2016; Ford et al., 2017). Alterations in this balance have been associated with brain damage and several neurodegenerative diseases including Alzheimer's, Parkinson's, and NeuroHIV. In the particular case of NeuroHIV, the equilibrium of Glu/GABA/Gln is altered and contributes to neuronal and glial dysfunction as well as to cognitive impairment observed in at least half of the HIV-1-infected population, even in the current antiretroviral treatment (ART) era (Sailasuta et al., 2009; Cohen et al., 2010; Ernst et al., 2010; Young et al., 2014; Mohamed et al., 2018; Cysique et al., 2019). Briefly, the pathogenesis of NeuroHIV involves the early (7–10 days post-injection) transmigration of leukocytes carrying the virus across the blood-brain barrier (BBB) using chemokine gradients only sensed by HIV-1-infected monocytes due to their enhanced expression of key chemokine receptors such as CCR2 (Eugenin et al., 2006; Williams et al., 2013). Upon crossing the BBB, the few transmigrated HIV-1-infected leukocytes infect CNS resident cells such as microglia, macrophages, and a small population of astrocytes. If systemic HIV-1 replication is not controlled by ART, localized HIV-1-CNS replication and infection results in HIV-1 encephalitis and dementia (Gatanaga et al., 1999; Bingham et al., 2011; Gelman et al., 2013; Gelman, 2015; de Almeida et al., 2017; Mangus et al., 2018). However, in the current ART era, CNS damage is mild due to controlled peripheral and CNS replication as well as limited HIV-1 infection; despite this, 50% of the HIV-1-infected individuals still show significant signs of cognitive impairment, but the mechanism of CNS dysfunction is unknown (Eggers et al., 2017; Yoshimura, 2017; Bandera et al., 2019; Fernandes and Pulliam, 2019; Kim-Chang et al., 2019; Paul, 2019; Portilla et al., 2019; Swinton et al., 2019; Angelovich et al., 2020). Several groups have proposed that CNS damage in the current ART era corresponds to a combination of HIV-1 reservoirs within the brain, low level expression and secretion of viral proteins, as well as associated inflammation (Wong and Yukl, 2016; Veenstra et al., 2017, 2019). However, the nature and size of the viral reservoir within the CNS under effective ART is unknown (Churchill et al., 2006, 2009; Eugenin et al., 2011a; Russell et al., 2017; Al-Harti et al., 2018; Ko et al., 2019; Wallet et al., 2019). Our laboratory demonstrated that myeloid and glial cells within the CNS are viral reservoirs. These few viral reservoirs, despite the low to undetectable viral replication, are able to amplify toxicity and inflammation to neighboring uninfected cells by a gap junction, hemichannel, and tunneling nanotube mediated mechanism (Eugenin et al., 2009a,b; Berman et al., 2016; Malik and Eugenin, 2016, 2019; Ariazi et al., 2017; Okafo et al., 2017; Valdebenito et al., 2018).
Another potential mechanism of toxicity is mediated by the low-level production of viral proteins, not blocked by ART, and subsequent secretion into neighboring cells such as neurons and glia (Nath, 2002; Kovalevich and Langford, 2012; Sami Saribas et al., 2017). However, the extent and concentrations of viral proteins in the CNS and other tissues are unknown, but nanograms/ml of viral proteins (Nef, Tat) were detected in the serum or plasma of HIV-1-positive individuals (Westendorp et al., 1995; Goldstein, 1996; Xiao et al., 2000). Almost all HIV-1 proteins are neuro- or glial-toxic. For example, neurotoxicity has been described for several viral proteins including gp120 and the transactivator of transcription (Tat) (Table 1), as well as for host factors such as TNF-α, IL-1β, and IL-6 released from latently HIV-1-infected or -activated cells (Koller et al., 2001; Zhou et al., 2017). In addition, several ART drugs have been demonstrated to generate toxicity on their own (Brier et al., 2015; Underwood et al., 2015; Latronico et al., 2018). Thus, the combination of HIV-1-infection, low-level secretion of viral proteins, and ART toxicity probably contribute to CNS dysfunction.

Table 1. Effect of HIV-1 proteins on neurotoxicity induced by glutamatergic system dysregulation.
Another accepted mechanism of toxicity is Glu-mediated toxicity. Glu is a neurotransmitter dysregulated in HIV-1 infection that can contribute to HIV-associated neurocognitive disorder (HAND) (Huang et al., 2011; Potter et al., 2013; Vazquez-Santiago et al., 2014). In addition to synaptic dysregulation, a supplemental source of Glu in HIV-1 conditions is the persistently activated resident microglia and invading macrophages, which have been shown to increase Glu synthesis and to release into the extracellular space (Potter et al., 2013; Wu et al., 2015). Furthermore, in several cell types, HIV-1 infection leads to mitochondrial membrane destabilization and the release of phosphate-activated glutaminase (PAG) from the mitochondria matrix into the cytosol through the transition pore (Erdmann et al., 2007, 2009; Huang et al., 2011; Tian et al., 2012). Glutaminase (GLS) is an enzyme responsible for de novo synthesis of Glu from Gln resulting in the increased levels of intracellular and extracellular Glu due to mitochondrial dysfunction.
In individuals with NeuroHIV the extracellular level of Glu is elevated in cerebrospinal fluid (CSF) and plasma in correlation with severity of brain atrophy and dementia (Ferrarese et al., 2001; Cassol et al., 2014; Anderson et al., 2015). However, MRI/MRS imaging data are controversial. Some reports indicated that HIV-1-infected individuals with dementia have decreased levels of Glu in frontal gray and white matter as well as the basal ganglia in correlation with cognitive deficits (Sailasuta et al., 2009; Ernst et al., 2010; Mohamed et al., 2018). In contrast, other publications using MRS indicate that Glu is increased in the frontal white and gray matter and basal ganglia in HIV-1-infected individuals before and after initiation of ART as compared to uninfected individuals. However, treatment of HIV-1-infected individuals for extended periods of time with ART results in the reduction of extracellular levels of Glu, suggesting a positive effect of ART treatment (Young et al., 2014). Thus, most of these changes in Glu assessed by brain imaging are probably due to individual-to-individual variability, HIV-1 time course, HIV-1 replication, time of infection, ART, and several other potential CNS complications. Despite the mechanism of Glu dysregulation, we recently identified that glial and myeloid viral reservoirs within the brain generate energy from unusual sources of energy such as Glu and Gln, both of which are highly abundant in the brain, providing them with an almost endless source of energy. We recently demonstrated that blocking some of these metabolic pathways results in the effective killing of viral reservoirs, even in the absence of HIV-1 reactivation.
In this review, we will describe the overall Glu/GABA synaptic system, its dysregulation, and the role of HIV-1 in CNS dysfunction with a major focus on viral reservoirs.
Glu binding activates ionotropic and metabotropic receptors expressed on neuronal, glial, and immune cells (Zhou and Danbolt, 2014). Membrane-specific Glu receptors (GluRs) are expressed in neurons and glial cells, and they mediate most, but not all, excitatory and inhibitory effects (D'Antoni et al., 2008; Petralia, 2012; Skowronska et al., 2019). The activation of ionotropic Glu receptors mediates fast excitatory synaptic transmission, whereas the metabotropic receptors play a modulatory role (Lau and Tymianski, 2010).
Ionotropic Glu receptors (iGluRs) are proteins composed of different subunits that form ion channels. They are divided into three groups based on their structural similarities and named according to the type of agonist that activates them: amino-3- hydroxy-5-methylisoxazole-4-propionate (AMPAR), N-methyl- D-aspartate (NMDAR), and 2-carboxy-3-carboxymethyl-4-isopropenylpyrrolidine (kainate, KA) receptors (KAR) (Traynelis et al., 2010). The NMDAR family is composed of seven receptor families. They are divided in three main subunits, GluN1, GluN2, and GluN3, which present different subtypes: GluN2–N2A, N2B, N2C, N2D; GluN3–N3A, N3B (Kumar, 2015). For a functional channel, an NR1 and an NR2 subunit are required. Glu binds to the NR2 subunit while the co-agonist, glycine, binds to the NR1 subunit. The binding of the co-agonist enables the removal of the magnesium blockade (Guo et al., 2017; Gibb et al., 2018). Similar to NMDAR, the family of AMPA receptors is composed of four subunits, GluR1/2/3/4, and the GluR2 subunit plays a critical role in Ca2+ permeability (Lomeli et al., 1994). The KAR family is composed of two types of subunits: GluR5/6/7 and KA1 and KA2, which form homo- or heterotetramers. Depending on the receptor's subunit composition, the channel will have different electrophysiological properties (Contractor et al., 2011).
Metabotropic Glu receptors (mGluRs) have been subdivided in regard to sequence similarity and signaling into three groups (group I: mGluR 1 and 5; group II: mGluR 2 and 3; and group III: mGluR 4, 6, 7, and 8). The mGluRs are G protein-coupled receptors that are linked to various intracellular second messenger cascades. Group I mGluRs are coupled to Gq, and signaling through these receptors is carried out by the phospholipase C and stimulation of intracellular calcium (Ca2+) release. Receptors belonging to this group (mGluR1 and mGluR5) are located predominantly postsynaptically. However, the presynaptic receptors control Glu release. Thus, depending on the receptor location, they have different activities. The second (mGluR2 and mGluR3) and the third (mGluR4, 6, 7, and 8) group are coupled to Gi/o proteins, and they inhibit the activity of adenylate cyclase, resulting in a reduction of the intracellular level of cyclic AMP and the inhibition of protein kinase A (Pilc et al., 2008; Wieronska and Pilc, 2009; Niswender and Conn, 2010). In contrast to group I, receptors from the second and third groups are located predominantly presynaptically, and they inhibit the function of glutamatergic neurons, but the receptors from the third group also activate GABA release (Conn and Pin, 1997).
mGluRs receptors modulate neuronal excitability via regulation of voltage-sensitive calcium channels, G protein-coupled inwardly rectifying potassium (GIRK) channels, and GABA, AMPA, and NMDA receptors (Conn and Pin, 1997). Moreover, in several brain regions, iGluRs were identified on synaptic terminals (i.e., presynaptic receptors) as functional autoreceptors to modulate Glu and GABA release (Duguid and Smart, 2004; Fiszman et al., 2005). Interestingly, all these receptors are also expressed in immune cells, non-excitable, providing a unique neuroimmune cross talk that still is under examination (Eck et al., 1989; Pacheco et al., 2007; Moroni et al., 2012; Yang et al., 2013) and can further contribute to NeuroHIV development.
The release of intracellular Glu into the extracellular space can be achieved by four different mechanisms: (1) calcium-dependent exocytosis of neurotransmitter from vesicular storage; (2) reverse action of Glu transporters located pre- and postsynaptically on neurons and astrocytes plasma membrane; (3) reverse cystine/glutamate transporter activity on the plasma membrane; and (4) hemichannels (Allen et al., 2004; Santello et al., 2012; Li et al., 2014).
The first system corresponds to the synaptic regulation of the release and uptake of Glu. Glu in the CNS is synthetized and stored (millimolar concentration) in synaptic vesicles of glutamatergic neurons. The millimolar concentration of Glu in the synaptic cleft is kept in nanomolar range after synaptic stimulation by reverse excitatory amino acid transporters (EAATs). The second system corresponds to Glu transporters belonging to solute carrier family (SLC): Na+- dependent (XAG) excitatory amino acid transporter (EAAT-1), also known as Na+-dependent glutamate/aspartate transporter (GLAST) and EAAT-2 or glutamate transporter 1 (GLT-1). These transporters work based on the maintainence of the Na+ gradient between intracellular space and extracellular fluid by Na+/K+-ATPase. In the CNS, the Glu transporters are located on astrocytes (Rothstein et al., 1996), neurons (Kanai and Hediger, 2004), and the BBB (Kanai and Hediger, 2004), and their main function is to remove the excess of Glu from the synaptic cleft after signal transduction (Nedergaard et al., 2002; Grewer et al., 2008). The third system, Xc−, is an exchanger of intracellular Glu for extracellular cystine. Cystine is reduced to glutathione (GSH) in the cytoplasm (Lu, 2013). It was published that disruption in the Xc− the system may lead to an increase of extracellular Glu concentration, which is a source of oxidative stress and cell death (Bridges et al., 2012b; Lewerenz et al., 2013). The fourth system correspond to hemichannels composed of connexin or pannexin proteins. They are expressed on the surface of glial cells including astrocytes and microglia/macrophages, which allow for the rapid intercellular exchange of ions and metabolites. Opening of hemichannels occurs only under stress conditions to serve as a backup system for Glu buffering, calcium wave propagation, and synaptic plasticity (Figure 1). In astrocytes, unopposed hemichannels on astrocytes membrane are responsible for the release of gliotransmitters, including ATP, Glu, nicotinamide adenine dinucleotide (NAD), and D-serine to the extracellular space. Under physiological conditions, hemichannels are in a closed state. However, upon inflammation and damage, hemichannels become open and have been associated with several neurodegenerative disorders (Xing et al., 2019) including NeuroHIV (Eugenin et al., 2011a; Orellana et al., 2014; Berman et al., 2016; Malik and Eugenin, 2016, 2019). Only recently, we identified that circulating levels of ATP can be used to provide an early detection of cognitive impairment in HIV-infected individuals (Velasquez et al., 2020).
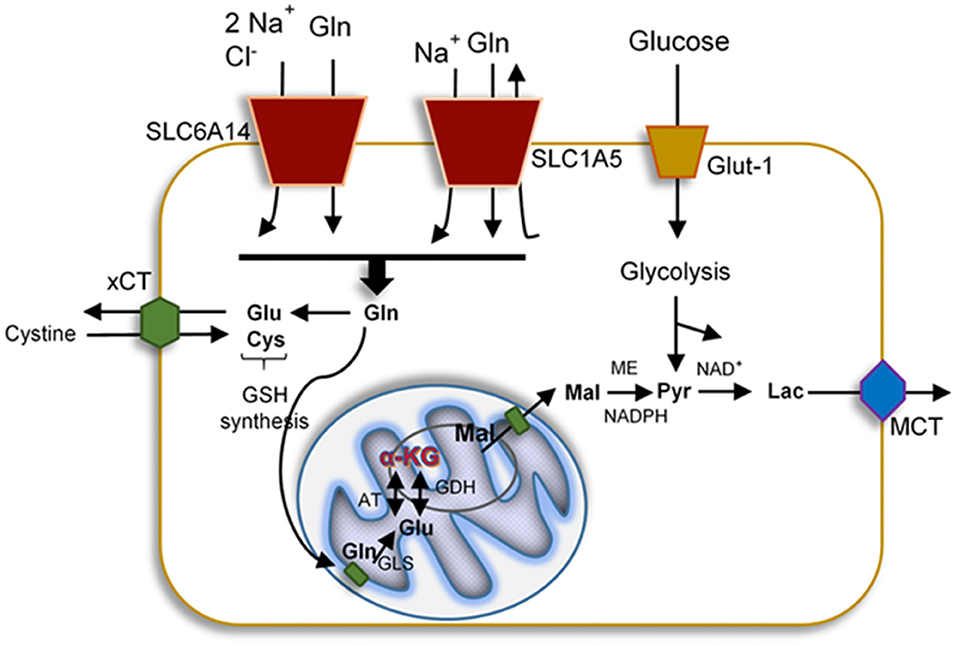
Figure 1. Representation of the uptake and mitochondrial delivery system of glucose, glutamate, and glutamine. Glucose enters the cells by glucose transporter-1 (Glut-1), cystine, Gln, and Glu also are uptake by specific transporters such as cystine/glutamate antiporter (xCT), Na+ and Cl−-dependent neurotransmitter transporter (SLC6A14), and neutral amino acid transporter (SLC1A5), respectively. All proteins affected by HIV-1 infection. Upon uptake, these molecules can become part of the glycolysis, GSH synthesis, or the TCA. In the brain, metabolism of these mediators generates lactate that exits the cells via the monocarboxylete transporter (MCT).
Neurons cannot synthesize Glu de novo due to the lack of expression of glutamine synthetase (GS), which converts Glu to Gln, and pyruvate carboxylase (PC), which converts Gln into Glu (Pardo et al., 2011). In contrast, astrocytes express both enzymes (Bak et al., 2006). After the release of neurotransmitters to the synaptic cleft, the excess of Glu or GABA is removed by neighboring astrocytes using specific transporters and both neurotransmitters are metabolized into Gln via GS or transformed into Glu and then to α-KG to become part of the tricarboxylic acid cycle (TCA cycle, Krebs cycle) (Bak et al., 2006). In glutamatergic neurons, Glu is synthetized on three pathways: (1) from α-KG, a product from the TCA cycle, which is converted by the action of glutamate dehydrogenase (GDH) and aspartate aminotransferase (AAT) into Glu. GDH is a reversible enzyme that can transform a-KG, ammonia, and NADH or NADPH into Glu (Hara et al., 2003). (2) Gln from astrocytes is delivered into neurons by amino acids transporters (SLC6A14 and SLC1A5) and metabolized directly via enzyme glutaminase (GLS)/phosphate-activated GLS (PAG) into Glu. GLS is a main Gln-utilizing enzyme in the brain, converting the Gln to Glu, and it is located on the inner membrane of mitochondria (Holcomb et al., 2000). The two types of GLS are well-characterized as the “kidney type” (GLS1) and “liver type” (GLS2). In the human brain, GLS1 has two isoforms: kidney-type glutaminase (KGA) and glutaminase C, which are expressed in the various cell types including neurons, microglia, macrophages, and astrocytes (Cardona et al., 2015). GLS is known to promote cancer cell proliferation and differentiation (Erickson and Cerione, 2010). Moreover, GLS is an initial enzyme in the glutaminolysis pathway, providing the energy source for protein and lipid synthesis. During glutaminolysis, Gln is converted to Glu and subsequently into α-KG, which then enters the TCA cycle (Figure 1). Also, Glu can be converted into GSH that can protect the cells from oxidative stress and promote their survival. The third pathway (3) of Glu synthesis is from Glu reuptake from extracellular space and transformation into Gln to go back to neurons (Peng et al., 1993; Schousboe et al., 2013) (Figure 2). GABA, similarly to Glu, is metabolized on two pathways: (1) reuptake from extracellular space and incorporation into the TCA cycle and then conversion to α-KG via GABA-transaminase (GABA-T) and succinic semialdehyde dehydrogenase (SSADH), and (2) from Gln, which is converted to Glu, and next via decarboxylation (glutamate decarboxylase enzyme, GAD) to GABA (Schousboe et al., 2013) (Figure 2). The synaptic pool of Glu can be divided into two compartments: (1) intracellular/cytosolic, in which Glu is synthetized from Gln inside glutamatergic neurons via enzyme GLS; and (2) mitochondrial, in which Glu synthesis from Gln is followed by Glu transformation to α-KG, which enters into the TCA cycle. Inside the mitochondria, Gln, even if it is considered as a source of energy, is called the “Trojan horse” because it is not the preferred source of energy. During the metabolism of Glu from Gln, Gln-derived ammonia () is also generated and affects mitochondrial function by increasing the production of ROS and inducing mitochondrial permeability transition (MPT). This process introduces the collapse of the inner mitochondrial membrane and leads to mitochondrial dysfunction and free radicals production (Zoratti et al., 2005; Albrecht and Norenberg, 2006) (Figure 3). Later we will discuss why HIV-1-reservoirs prefer this metabolic pathway to survive for extended periods of time within the CNS. Also, glucose, the main brain energy substrate, is involved in the Glu/GABA synthesis cycle. In astrocytes, glucose is metabolized to lactate and pyruvate, which is a substrate for the TCA cycle, which produces α-KG and Gln, the fundamental substrate for Glu/GABA synthesis (Hertz and Chen, 2017). These tight connections between neurotransmission, neurotransmitter metabolism, and energetics are used in pathological conditions to promote the survival of glioblastoma stem cells as well as HIV-1-CNS reservoirs.
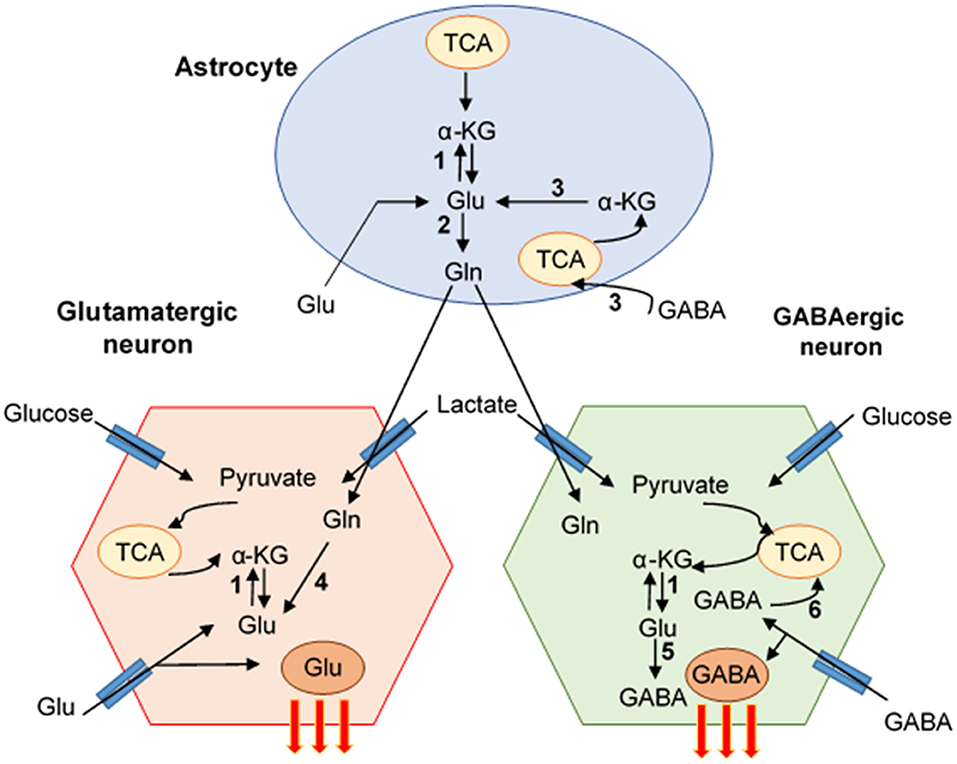
Figure 2. Pathways of Glutamate/GABA synthesis-neuro-glia interaction. The cartoon represents the mutual interaction of astrocyte and glutamate (Glu) and GABA neuron. Glu-GABA cycle enzymes: (1) aminotransferases (AT), (2) glutamine synthetase (GS), (3) glutamate dehydrogenase (GDH)/aspartate aminotransferase (AAT), (4) glutaminase (PAG), (5) glutamate decarboxylase (GAD), (6) GABA transaminase (GABA-T)/succinate-semialdehyde dehydrogenase (SSADH) (Schousboe et al., 2013).
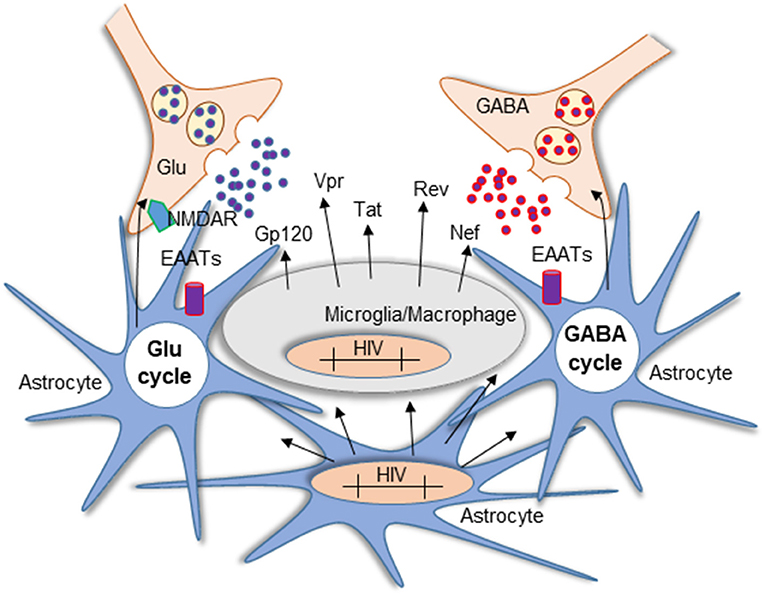
Figure 3. Mechanism of Glu neurotoxicity in NeuroHIV pathogenesis. Glu is a critical neurotransmitter dysregulated in HIV-1 infection and HAND. Imbalance in the Glu/GABA cycle impaired the excitatory/inhibitory state of the neurons resulting in persistent stimulation of NMDARs. The diagram represents the activated state of astrocytes and microglia/macrophages. In the CNS, microglia, macrophages, and a small population of astrocytes become infected and carry HIV-integrated DNA. Although astrocytes cannot produce the virus, they contribute to neurotoxicity mediated by the production of viral proteins that are not blocked by ART and the subsequent secretion into neighboring cells such as neurons and glia. Another source of toxicity is the persistent activation of resident glial cells that increase Glu synthesis and release into the extracellular space.
HIV-1 infection cannot be eradicated due to the early generation of viral reservoirs in several tissue compartments (Wong and Yukl, 2016). Despite common knowledge that the elimination of viral reservoirs is essential to cure HIV-1, the nature, characteristics, and mechanisms of latent and long-lasting HIV-1-reservoirs are unknown. Currently, the best-characterized viral reservoirs are different circulating subpopulations of CD4+ T lymphocytes; however, several groups propose that circulating viral reservoirs correspond to a poor representation of the real reservoir pool that is present in several tissues (Chomont et al., 2009; Soriano-Sarabia et al., 2014; Kandathil et al., 2016; Abreu et al., 2019).
In most cases, within the first few weeks of HIV-1 infection, the viral reservoirs are established in multiple tissues and anatomic compartments (Wong and Yukl, 2016). Compartmentalization of HIV-1 occurs as a result of differential selective pressures between tissues or restricted virus flow within organs (Borderia et al., 2007). Moreover, the different concentrations of antiretroviral drugs within compartments can result in divergent viral evolution (Zarate et al., 2007). It was shown that, in individuals diagnosed with HIV-1 infection, the pick level of HIV-1 replication was established in blood, oropharyngeal, and genital tract tissue, but not in the CSF (Pilcher et al., 2001). Furthermore, upon ART interruption, viral rebound was different between the blood and CSF, suggesting a differential compartmentalization (Garcia et al., 1999; Gianella et al., 2016; Mukerji et al., 2018). Early on in HIV-1-infection, the CNS becomes a unique compartment due to the presence of the BBB and the blood-CSF barrier, both of which restrict virus trafficking between the CNS and blood compartment, resulting in the rise of at least two separate HIV-1 compartments with different viral and cell evolutions (Smit et al., 2001; Wang et al., 2001; Ohagen et al., 2003; Abbate et al., 2005; Ritola et al., 2005; Yukl et al., 2013; Ganor et al., 2019). These differences in reactivation time and evolution can also be explained by the cell type infected, tissue microenvironment, access to ART, type of ART, pK distribution, proliferating cell properties, antigen dependency, or independent activation (Alexaki et al., 2008; Bingham et al., 2011; Pinkevych et al., 2015, 2016; Gianella et al., 2016).
Viral reservoirs are defined as cells with an integrated HIV-1 genome that have the capacity to silence viral replication, survive for extended periods of time, and become competent upon reactivation (Blankson et al., 2002). The best-described viral reservoirs are resting CD4+ T lymphocytes, including central memory T lymphocytes, transitional T lymphocytes, and effector memory T lymphocytes, which maintain the infection by homeostatic proliferation in central memory and transitional memory T lymphocytes (Lee and Lichterfeld, 2016). It was shown that activated T lymphocytes have a higher copy number of HIV-1-integrated DNA as compared to lymphocytes in the resting state of individuals with or without ART. Further characterization of the types of T lymphocytes indicate that HIV-1-DNA is present mostly in resting central memory and transitional memory T lymphocytes. These results indicate that both T lymphocyte populations are the circulating viral reservoirs (Chomont et al., 2009; Josefsson et al., 2013). Using the quantitative viral outgrowth assay (QVOA), it has been demonstrated that replication-competent viruses reside in transitional memory T lymphocytes even in samples obtained from HIV-1-infected individuals on long-lasting ART (Siliciano et al., 2003; Soriano-Sarabia et al., 2014). During early infection, CD4+ T lymphocytes are the major target cells (Haase, 1999). Next, within the first few weeks of primary HIV-1 infection, the virus population doubles every 6 to 10 h, resulting in infection of another 20 cells for each cell infected early on (Nowak et al., 1997; Little et al., 1999). During the symptomatic primary infection, the level of circulating virus in the blood (often >106 to 108/ml viral particles) and infected cell are high, and this initiates the compartmentalization early on in several tissues including the gut-associated lymphoid tissue (GALT). In 1995 studies by Wei et al. (1995) and Ho et al. (1995) showed viral evolution was a dynamic process because replacement of the wild-type virus can be achieved in 14 days, suggesting that viral replication and cell turnover can contribute to viral variability and the generation of reservoirs. In these studies, treatment with the HIV-1 reverse transcriptase and protease inhibitors (nevirapine and ritonavir) prevented infection of new cells but did not block virus replication in the cells already carrying integrated HIV-1 DNA. Both anti-viral drugs induced a decrease in plasma virus levels in the first 2 weeks of therapy. Further studies using mathematical models to calculate HIV-1 replication, half-life of replicating and reservoirs cells, as well as the potential contribution of tissue associated viral reservoirs indicates that ART is extremely effective in reducing systemic viral replication after 1–2 weeks after initiation by preventing new infections. However, a second phase of replication was detected that corresponded to the loss of long-lasting infected cells. However, a pool of tissue resident cells still maintained a slow decay in systemic replication. The authors estimate that 2.3 to 3.1 years are required to eliminate these HIV-1 cells from the tissue compartment, however, this cleaning does not mean eradication (Perelson et al., 1997). It was calculated that the resting T helper lymphocytes population in HIV-1 patients on ART may contain ~1 replication competent viral genome per 106 CD4+ lymphocytes. Considering that the half-life on the latently infected memory T lymphocytes is around 4 years, 88 years of treatment would be required to reach a level of cure using current ART (Finzi et al., 1999; Shan and Siliciano, 2013; Hill, 2018).
HIV-1 enters the CNS early after primary infection (7–10 days), which contributes to the establishment of viral reservoirs in the CNS (Fischer-Smith et al., 2001; Valcour et al., 2012). CD14+CD16+ monocytes enter the CNS using physiological gradients of CCL2 from the brain and an upregulated expression of CCR2 on HIV-1-infected cells despite ART (Eugenin et al., 2006). More specifically, the mature CD14+CD16+ monocytes are strongly associated with CNS inflammation and HAND development (Williams et al., 2012, 2013). It was shown that in HIV+ CD14+CD16+ monocytes preferentially transmigrate across the BBB, and also junctional proteins JAM-A and ALCAM support their transmigration. Blocking of JAM-A and ALCAM by specific antibodies and the dual inhibitor of chemokine receptors CCR2/CCR5 prevent preferential transmigration of HIV+ CD14+CD16+ monocytes. Moreover, the surface of CCR2 is increased on HIV-1-infected CD14+CD16+ monocytes from individuals who manifested HANDs, independently of ART status, viral load, and CD4+ T lymphocytes count (Williams et al., 2014). The increase of CCR2 on CD14+CD16+ monocytes correlate with a higher level of HIV-1 DNA and neuronal damage, suggesting that CD14+CD16+ monocytes could have a key role in HAND pathogenesis and neuronal damage observed in infected individuals. After entering into the brain, HIV-1-infected monocytes differentiate into macrophages resulting in the release of hosts and viral components inducing infection of host cells such as microglia and astrocytes (Churchill et al., 2006).
Pericytes have a crucial role in the formation and maintenance of BBB integrity. Reduction in pericyte coverage is a typical feature of BBB impairment, observed in Alzheimer's disease (AD), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), and also HAND (Sengillo et al., 2013; Winkler et al., 2013; Persidsky et al., 2016). The main cause of loss of pericyte integrity observed in these diseases is associated with loss of adhesion into the basal membrane and dedifferentiation of smooth muscle cells resulting in their detachment from endothelial cells (Persidsky et al., 2016). HIV-1 Tat also induce changes in pericytes, suggesting that the loss of pericyte coverage and their detachment from the endothelium contribute to loss of BBB integrity in HAND (Niu et al., 2014). Within the CNS, microglia, and macrophages are the main cells supporting HIV-1 replication, however, astrocytes also are infected but HIV-1 replication is minimal to undetectable. Even though only ~5% of the astrocytes become infected, their HIV-1-infection, but not replication, contributes to loss of BBB integrity (Churchill et al., 2006; Eugenin et al., 2011a). In pericytes, after HIV-1-infection, the viral replication decreased. Viral infection was demonstrated by Alu-PCR and infection was subjected to reactivation (Persidsky et al., 2016).
ART is a key factor in viral reservoir formation within the CNS. It has been demonstrated that initiation of ART less than 6 months after primary infection is associated with lower levels of T-cells activation and smaller circulating reservoir size during long-term therapy, supporting the hypothesis that reservoirs are formed early during infection (Jain et al., 2013; Abrahams et al., 2019). Moreover, by using mathematical models, it has been demonstrated that ART interruption with subsequent drug “washout” induces virus rebound (Hill et al., 2014, 2016; Pinkevych et al., 2015, 2016). Despite the efforts in re-using the success of ART to cure the disease, it is clear at this point that HIV-1-infected cells, including HIV-1 reservoirs, also have an alternative mechanism of survival, not only depending on silent or active replication. Some of these viral mechanisms are anti-apoptotic, metabolic, proliferating, and tissue sequestrating, and there is a variability between people and infected cells. All these mechanisms contribute to perpetuating the virus.
There are several viral and host proteins that have been described as mediating the over-activation of the Glu/GABA neurotransmission system (Eugenin et al., 2007; Ru and Tang, 2017). Some of these viral proteins are HIV-1 Tat and gp120. Gp120 is a viral envelope protein with neurotoxic activity. HIV-1 isolates from the CNS are mainly macrophage (M)-tropic (Gonzalez-Perez et al., 2012), but also the T-cell-tropic form was detected. The gp120 M-tropic and T-tropic glycoproteins can induce synaptodendritic degeneration by activation of chemokine co-receptors CXCR4 and CCR5, respectively (Kaul et al., 2007), localized on neurons and non-neuronal cells (Westmoreland et al., 2002). Gp120 induces a massive calcium release and changes in the morphology of neuronal mitochondria (Avdoshina et al., 2016; Rozzi et al., 2017). Also, gp120 is a glycoprotein with the ability to activate the NMDA receptors and increase the synaptic damage by overactivation. Direct mechanisms of neuronal injury induced by gp120/HIV via NMDAR involved NR2A and NR2B subunits based on enhanced NR2A- and NR2B-mediated EPSCs (Zhou et al., 2017). In the HIV/gp120-tg administration of the drug to mice, nitromemantine (a blocker of NMDAR), was protective against gp120 neurotoxic properties causing neuronal damage and synaptic loss. In the same studies, researchers demonstrated the crucial role of the NR3A subunit of NMDAR because the genetic overexpression of the NR3A subunit enhanced the synaptic injury in both gp120-tg and WT mice (Nakanishi et al., 2016). NMDAR phosphorylation in the cytoplasmic tail of this receptor has emerged as an important mechanism regulating its function and trafficking (Chen and Roche, 2007). Phosphorylation of Ser896 and Ser897 are crucial for channel activity but also for the promotion of trafficking from the Golgi to the plasma membrane (Scott et al., 2003). The T-cell-tropic gp120 promotes the trafficking and surface clustering of NMDAR, and this process was associated with the phosphorylation of the NMDA subunit NR1 at C terminal Ser896 and Ser897 (Xu et al., 2011). On the other hand, the M-tropic gp120 transiently decreased the level of pSer896 and pSer897 of the NR1 subunit, having the opposite effect on the T-tropic gp120 (Ru and Tang, 2016). Treatment with NMDAR and AMPAR antagonists prevents the effect of M-tropic gp120 on NR1 down-regulation, suggesting that phosphorylation of NR1 is dependent on synaptic activity and NMDAR activation (Ru and Tang, 2016). Moreover, both gp120 tropic forms present different neuropathological profiles—M-tropic gp120 has a weaker toxic property than T-tropic. Also, M-tropic (CCR5-preferring) is present in the early phase of HIV-1 infection, while T-tropic (CXCR4-preferring) in the late stage in patients with developed HAND (Bachis et al., 2010).
Tat is a potent excitotoxin and it stimulates NMDARs by direct cysteine–cysteine interaction with the extracellular domain of the receptor (Prendergast et al., 2002; Li et al., 2008). Moreover, Tat promotes the phosphorylation of the NMDA receptor and triggers the calcium efflux and receptor's stimulation (Haughey et al., 2001). Also, Tat can induce cell death in human neuroblastoma cells (SH-SY5Y) by stimulation of H2O2 release dependent on NMDAR activation (Capone et al., 2013). Amino acids 31–61 of Tat are necessary to cause neurotoxicity (Nath et al., 1996), and their cysteine residues are crucial for direct interaction with the NMDAR (Li et al., 2008). The role of Tat Δ31–61 was shown in studies with Tat Δ31–61 mutant protein where the modified protein was not able to bind to the NMDAR. Moreover, in the same studies, no interaction between Tat and NMDAR was observed (Li et al., 2008). The changes in the Tat protein structure prevented the interaction with the NMDAR, also the immune complex of the mutant Tat, nitrosylated Tat or anti-Tat antibody with NMDAR block neurotoxicity caused by NMDAR agonist (Rumbaugh et al., 2013). Tat as a transactivator of the HIV-1 long-terminal repeat (LTR) is necessary for viral replication (Moses et al., 1994; Taube et al., 1999). Tat causes neural injury by entering into the neurons by a low-density lipoprotein receptor-related protein-1 (LRP) (Liu et al., 2000; Eugenin et al., 2007). Tat binding to LRP on the neuron's membrane initiates the formation of a macro molecular complex among tat-LRP-PSD-95 (as an intracellular adaptor protein)-NMDAR, later nNOS is recruited to the complex with subsequent production of NO at the neuronal cell membrane (Eugenin et al., 2007). In this macro molecular complex, the NMDAR, especially the NR2A receptor subunit, play a crucial role in the process of Tat-induced apoptosis in human primary neurons (Eugenin et al., 2003; King et al., 2010). Application of the LRP antagonist, receptor-associated protein (RAP) completely blocked the Tat-dependent potentiation, which suggests that Tat potentiates NMDAR function via LRP (Krogh et al., 2014b). Application of Tat into human fetal neuron cell culture followed by calcium efflux mediated by excitatory amino acid receptors induced the calcium release via stimulation of IP3 receptor and activation of neuronal nitric oxide synthetase (nNOS). This process was abolished by inhibition of IP3 receptors, also had a neuroprotective effect from Tat toxicity (Haughey et al., 1999). Moreover, Tat potentiates the effect of NMDA on calcium release, by neuronal NMDAR sensitization so that the physiological level of Glu causes significant excitotoxicity and increases the intracellular calcium level. Our data indicates that blocking LRP, NMDAR, or nNOS activation reduces HIV-1 Tat internalization into neurons and prevents HIV-1 Tat-mediated apoptosis (Eugenin et al., 2007).
Also, HIV-1 protein gp120 acts synergistically with Tat and potentiates each protein's neurotoxic actions, promoting neuronal cell death (Nath et al., 2000). Most studies concerning the effect of Tat on NMDAR report that the changes in receptors function are acute (minutes to hours) (Haughey et al., 2001; Green and Thayer, 2016), while the neurotoxic properties of Tat occur after hours to days (Popescu, 2014). Tat-induced potentiation of the calcium release induced by NMDAR by 8 h then adapted, as later it was reported that it returned to the baseline by 24 h and dropped below control by 48 h (Krogh et al., 2014a).
Oligodendrocytes (OLs) are myelinating CNS cells. Their dysfunction leads to abnormal myelination, impairment of cell–cell signaling, and axon degeneration. OLs express iGluRs, including NMDA and AMPA receptors, and the expression of these receptors is highly heterogeneous (Salter and Fern, 2005). The NMDARs are mainly present on the myelin sheath, while AMPARs are equally distributed on the cell body (Micu et al., 2006). Also, the pattern of iGluRs distribution is different on immature and mature OLs; immature OLs represent a higher level of AMPARs and mature NMDARs, respectively. OL cells are also very sensitive to the toxic actions of Glu since the OL NMDARs are less susceptible to magnesium blockage; the level of Glu necessary to induce toxic effect in OLs may be lower than required for neurons (Karadottir et al., 2005). Activation of iGluR induced the accumulation of calcium in myelin cells in response to chemical ischemia. This effect was completely reversed by AMPA/KA antagonist (NBOX) in the oligodendroglial cell body but only moderately in myelin. In contrast, the broad spectrum of NMDAR antagonists (MK-801, 7-chloro-kynurenic acid, or d-AP5) decreased the level of calcium in myelin.
The massive release of Glu stimulated the NMDAR located on myelin and the accumulation of calcium inside myelin cells. The toxic action of Tat was also present in OL cells. In studies using transgenic mice expressing HIV-1 Tat, the primary corpus callosum and anterior commissure cell culture represent the increased level of OLs with aberrant morphology. Moreover, in the caudate-putamen, the myelin protein expression was abnormal. Tat protein caused the death of immature OLs and reduced myelin-like membrane production by mature OLs in a dose-dependent manner. Both toxic effects of Tat were reversed by the blocking of NMDARs by specific antagonist MK-801, while CNQX (AMPAR/KAR antagonist) only blocked the effect on immature OLs (Zou et al., 2015). This data confirms the results from Karadottir et al. (2005) that, depending on the stage of development (mature vs. immature), the pattern of iGluR is different.
In the current ART era, it still is a matter of debate whether viral proteins are produced within the brain. However, data from several laboratories including ours indicates that HIV-1 Tat, Nef, and gp120 are still produced and patients are developing antibodies to these proteins (Nath et al., 1987; Re et al., 1996; Rao, 2003; Eugenin et al., 2011b; Bachani et al., 2013; Nicoli et al., 2016). Besides, antiretroviral drugs do not affect the Tat protein level in HIV-1-infected patients, even when the viral blood load is low (Dickens et al., 2017). Thus, bystander damage by viral protein secretion is still a major concern mainly because ART blocks HIV-1 replication but is unable to reduce viral protein synthesis and secretion as well as associated inflammation and damage.
The major function of astrocytes is to support neurons by maintaining the balance of local ion concentration and pH homeostasis, regulating neurotransmitters level in extracellular space and clearing the metabolic waste (He and Sun, 2007). The dysfunction of the astrocyte network is a serious reason for the decline in neurons' survival. One of the main causes of neuron death is a massive release of Glu and NMDARs overactivation (Petralia, 2012; Wroge et al., 2012). Astrocytes prevent the neuronal loss by removing excess Glu from extracellular space by two Na+-dependent glutamate transporters (EAATs): GLAST/EAAT1 and GLT-1/EAAT2 (Rothstein et al., 1996). Loss of astrocytic Glu uptake and metabolism is one of the main factors responsible for neurotoxicity associated with neurodegenerative diseases, including HAND. It was shown that the most important factor in HIV-1-induced neurotoxicity is dysregulation of astrocytes Glu clearance (Potter et al., 2013). In monocyte-derived macrophages (MDM) HIV-1-infection up-regulate the EAAT-2 gene expression, but the transporter activity was decreased (Porcheray et al., 2006). Moreover, GS gene expression was decreased but the GS activity was increased (Porcheray et al., 2006). In HIV-1-positive people with HAND the astrocytic level of EAAT2 was reduced at the same time the extracellular level of Glu was increased (Xing et al., 2009; Borjabad et al., 2010). In the CNS a multiple factors cause the reduction of Glu transporters expression and in consequence astrocyte-dependent Glu clearance such as the pro-inflammatory mediators—tumor necrosis factor (TNF-α), interleukin (IL)-1β (Lindl et al., 2010) and HIV-1 viral protein—envelope glycoprotein (gp120), transactivator of transcription (Tat), and viral protein R (Vpr). The number of studies using cell cultures models has shown that gp120 significantly reduced the expression and function of Glu transporters, resulting in excessive accumulation of extracellular Glu in the synaptic cleft, overstimulation of Glu receptors and neurotoxicity (Nath et al., 2000; Wang et al., 2003; Visalli et al., 2007; Xu et al., 2011; Yang et al., 2013; Melendez et al., 2016; Zhou et al., 2017). HIV-1 protein, gp120 alters the neuronal function homeostasis and often cause synaptodendritic injury (Kaul et al., 2001; Visalli et al., 2007; Melendez et al., 2016; Zhou et al., 2017). In primary human astrocytes HIV-1 and gp120 by reducing the expression of EAAT2, impaired the clearance of Glu (Wang et al., 2003). In pharmacology studies of enzymes kinetics, the reduction in Vmax of both glutamate systems (XAG and Xc−) was observed in striatal glial and neuronal cells from gp120 mice, with no effect on Km for Glu (Melendez et al., 2016). These results suggest that gp120 reduces the density of Glu transporters in the mouse striatum without effect on Glu affinity to the transporters. Moreover, the total expression of GLT-1 protein was also altered in gp120 mice, which is involved in the development of HAND (Melendez et al., 2016). In Tat-transgenic mice the stimulus-evoked Glu release in the hippocampus and cortex was increased as compared to control animals, but GABA exocytosis was unaltered in the hippocampus and decreased in the cortex (Zucchini et al., 2013).
Protease inhibitors—Amprenavir (APV) and Lopinavir (LPV) caused the reduction of EAAT2 expression and diminished the intracellular concentration of Glu. LPV induced monocytes/microglia and astrocytes activation (Gupta et al., 2012), also in astrocytes LPV and APV increased Glu-dependent calcium efflux (Vivithanaporn et al., 2016). Moreover, the in vivo studies show a reduction in EAAT2 protein and mRNA level followed by the LPV treatment and suppression of the cortical level of L-glutamate and L-aspartate in conjunction with L-serine level reduction. The reduction of EAAT2 expression induced by LPV and APV on astrocytes reduces Glu extracellular clearance resulting in Glu overstimulation and calcium increase in glial cells (Vivithanaporn et al., 2016). Thus, a combination of HIV-1 infection and ART could also be additive negative effects in the glutamatergic system within the brain.
Tat specifically induced the macrophage/microglia activation and microglia-mediate neurotoxicity (Minghetti et al., 2004). Further it was shown that this process is caused by Tat-dependent activation of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Turchan-Cholewo et al., 2009). NADPH oxidase is a key component of ROS production for oxidative burst and cell signaling pathways. The activation of NADPH oxidase in microglia induce the release of Glu (Barger et al., 2007). It was shown that, Tat induced the dose-dependent Glu release from microglial which is associated with increased expression of the Xc− glutamate-cystine antiporter, and this effect was blocked by NADPH oxidase and glutamate-cystine inhibitors (Gupta et al., 2010). Furthermore, the extracellular cystine is transported inside of the cell by the counter-transporter cystine and then transformed to GSH. In HIV-1-infection GSH levels are decreased enhancing further oxidative damage induced by HIV-1 (Wanchu et al., 2009; Ferrucci et al., 2012). GSH function is not only antioxidant but is also very important for a conjunction of various drugs and xenobiotics elimination (Rieder et al., 1995; Singh et al., 2017). Also the proper function of Xc− system can be interrupted by the upregulation of activity of nuclear factor erythroid 2-releated factor 2 (Nrf2), the main activator of the antioxidant response by neutralization of ROS accumulation and GSH depletion (Zhang et al., 2013). In astrocytes, the upregulation of xCT – Xc− system subunit altered by Nrf2 could be a potential source of excitotoxicity and neuron death induced by massive Glu release (Fogal et al., 2007; Bridges et al., 2012a,b). In astrocytes Tat protein induced the Nrf2 activation and Xc− system up-regulation (Mastrantonio et al., 2019). Moreover, it reduced the neuron viability in a mechanism dependent on Nrf2 activation and Glu release from astrocytes. This effect was blocked by Xc− system inhibitor—sulfasalazine, thus blockade of the transporter confirms the mechanism of neurotoxicity dependent on astrocytes activation (Mastrantonio et al., 2019). During oxidative stress conditions, cells can up-regulate the activity of Nrf2 to neutralize the ROS accumulation and not lead to GSH depletion (Niture et al., 2014). Astrocyte more resistant to oxidative stress in comparison to neurons which can strongly up-regulate the Nrf2 expression (Baxter and Hardingham, 2016). In response for cellular redox stare, Nrf2 migrates to the nucleus and binds to promoter region—antioxidant response element (ARE) of many antioxidant genes such as system Xc− subunit xCT, superoxide dismutase (SOD), glutathione peroxidase (GPX) (Niture et al., 2014). Treatment of human U373 astroglial cells with recombinant Tat increased the mRNA expression of several antioxidant enzymes such as Xc−, Quinone Dehydrogenase 1 (NQO1), catalyze (CAT), Cu/Zn superoxide dismutase (SOD1 and SOD2), glutamate-cystine ligase (GCLC), and GPX (Mastrantonio et al., 2019). Also, U373 astroglial cells treated with Tat increased the nuclear Nrf2 level which was compatible with the transcriptional induction of antioxidant ARE genes, as well as decreased cell viability, suggesting that Tat-associated neurodegeneration could be caused by the Xc− system (Mastrantonio et al., 2019).
The peroxisome proliferator-activated receptors (PPARs) ligand-activated transcription factors belonging to the nuclear receptors for steroids, thyroid hormones, and retinoids. These receptors play major roles in cell differentiation, lipid homeostasis, and glucose regulation. Nowadays, the agonist of PPAR gamma (PPARγ) is promising treatment for neuroinflammation-related condition eg. Alzheimer's disease, Parkinson's disease, and stroke (Govindarajulu et al., 2018; Wen et al., 2018; Lee et al., 2019). It was demonstrated that PPAR attenuated the release of pro-inflammatory cytokines and oxidative stress promotors. Several laboratories demonstrated that PPAR agonist can be neuroprotective against HIV-1-associated neuroinflammation (Potula et al., 2008; Huang et al., 2014, 2015). In astrocytes and microglial PPARγ agonists, rosiglitazone and pioglitazone, prevented oxidative stress induced by cytokines and NO as well as GLT-1 expression induced by HIV-1 and gp120 exposure (Omeragic et al., 2017).
The astrocyte elevated gene-1 (AEG-1) is a novel protein involved in HAND development. AEG-1 is a multifunctional oncogene, identify as an HIV-1- and TNF-α-inducible transcript in astrocytes. Its higher expression has been demonstrated in HIV-1-infected astrocytes, as well as in astrocytes treated with gp120 and TNF-α (Kang et al., 2005). AEG-1 was studied in the cancer field, describe as oncogene implicated in the initiation of tumorigenesis, proliferation, metastasis, angiogenesis, and chemotherapy resistance of malignancies (Emdad et al., 2009, 2010; Lee et al., 2009; Chen et al., 2017). In malignant gliomas, expression of AEG-1 was high and associated with necrosis and neurodegeneration in correlation with reduced expression of EAAT2 (Lee et al., 2011). Moreover, the AEG-1 overexpression increased the expression of YY1, a transcriptional repressor of EAAT2 in astrocytes. The same effect was presented in the glioma cell line and is connected to glioma-associated necrosis (Lee et al., 2011; Vartak-Sharma et al., 2014). Present that inflammation-induced the expression of AEG-1 in astrocytes and loss of EAAT2 could contribute to neurodegeneration that has a neuroinflammatory manifestation, such as HAND. HIV-1-associated neuroinflammation is caused by cytokines and chemokines such as IL-1β, TNF-α which is followed by an imbalance in Glu homeostasis caused by EAAT2 and increased CCL2 production (Vartak-Sharma et al., 2014). Moreover, all the inflammation stimulus significantly increased AEG-1 expression in astrocytes and nuclear translocation induced by the nuclear factor (NF)-κB pathway which increases expression of NF- κB-responsive chemokine such as CCL2 (Vartak-Sharma et al., 2014).
The main source of Glu overproduction in HIV-1 infection, includes the release of Glu from dying cells, disturbance in neurotransmitter clearance via impaired enzyme activity, and activated macrophages/microglia that also can release significant amounts of Glu into the extracellular space. Glu neurotoxicity is mainly caused by the up-regulation of Glu-generated enzyme—GLS isoform C in HIV-1-infected microglia. Also, the level of GLS isoform C was elevated in HIV-1-positive post-mortem brains which may be a compensatory effect induced by chronic exacerbate levels of Glu (Huang et al., 2011). The GLS C overexpression is also correlated with several neurocognitive and neurodegenerative disorders (Wang et al., 2017). The overproduction of Glu during HIV-1 infection in monocyte-derived macrophage (MDM) is caused by the release of GLS from the mitochondria followed by the generation of oxidative stress (Erdmann et al., 2009; Tian et al., 2012). In HIV-1-infected human MDM the Gln-dependent production of Glu was blocked by a GLS inhibitor (6-diazo-5-oxo-l-norleucine) and by the antiretroviral drug (zidovudine) (Zhao et al., 2004). Moreover, the excessive release of Glu from HIV-1-infected MDMs was inhibited by using a drugs that target N-acetylaspartylglutamate (NAAG) to specifically inhibit GLS (Erdmann et al., 2007). It was shown that Glu antagonist 6-diazo-5-oxo-L-norleucin (DON), a compound with GLS inhibition activity significantly decreased the Glu overproduction induced by HIV-1 and Glu-dependent neurotoxicity (Conti and Minelli, 1994; Zhao et al., 2004; Thomas et al., 2014). The Pro-drug of DON that is metabolized by carboxylesterases increasing the levels of the active drug mainly in the brain showing a higher CSF/plasma ratio (Nedelcovych et al., 2017). Furthermore, the new DON prodrug, JHU083, in a mouse model of HAND, reversed the deficits in spatial learning and working memory induced by HIV-1 infection. In biochemical tests JHU083 reduced extracellular level of Glu in CNS of mice infected with Eco-HIV, and decreased overstimulation of GLS in microglial cells (Nedelcovych et al., 2019).
HIV-1 viral protein R (Vpr) is known to promote virus replication in monocytes and differentiated macrophages (Connor et al., 1995). It was shown that soluble Vpr is detected in the CSF of HIV-1-positive patients, and it is a source of neurotoxicity and antioxidant system dysfunction (Patel et al., 2000; Ferrucci et al., 2012). Vpr transduced human derived macrophages and subsequent SILAC analysis indicates that around 50% of the proteins identified are involved in glycolytic and citrate pathways (Barrero et al., 2013). Next, labeling of glucose with Carbon-13 (13C) and subsequent treatment of macrophages transduced with Vpr indicates that labeled glucose was uptaked faster than untransduced cells. Measure of release of Gln, Glu, and α-KG in Vpr transduced cells indicate a faster glycolytic and TCA metabolism (Datta et al., 2016). Imbalance in glutaminolysis (deamination of Gln by GLS to Glu with subsequent conversion of Glu to α-KG) affects the production of antioxidants such as GSH and NADPH. Moreover, α-KG an intermediate product in Glu metabolism is an ROS homeostasis stabilizer. Thus maintenance of ROS homeostasis in HIV-1-infected macrophages is sufficient for cell survival (Datta et al., 2016). Vpr also up-regulates the GLS isoform C expression, resulting in an increased level of Glu in a media of HIV-1-infected macrophages (Erdmann et al., 2009; Datta et al., 2016).
The analysis of circulating lipids of two HIV-1-infected populations, responding and non-responding to ART for 36 months, detected higher levels of HDL in the responding group and a high VLDL and low LDL in the non-responding group, suggesting that healthy lipids also are associated with better ART response (Palmer et al., 2014; Rodriguez-Gallego et al., 2018). Furthermore, examination of different groups of HIV-1-infected individuals separated by neurocognitive status (normal: stable neurocognitive impairments; worsening group: progression of neurocognitive impairments; and improving group: improving of their cognitive status) were analyzed by NMR. In the worsening group, the CSF levels of citrate and pyroglutamate were decreased, and the levels of glutamine and lactate were increased as compared to the normal cognition group. In contrast, stable and improved cognition showed decreased glutamine and glucose and increased citrate, pyroglutamate, and lactate as compared to the worsening group. This pattern suggests that the worsening group was associated with a higher glucose metabolism based on higher levels of creatine, suggesting higher energy demand; in contrast, the improving group was associated with a decrease in glycolysis and TCA cycle intermediates, suggesting a shift from aerobic to anaerobic metabolism as observed in the worsening group (Dickens et al., 2015). In HIV-1, it is known and well-described that the virus compromises most of the synaptic control of Glu as well as glial regulation of the glutamatergic system; however, the interpretation of these data was to increase synaptic compromise by overactivation of Glu receptors. Now, our data also indicate that high levels of extracellular Glu and Gln that are abound in HIV-1-conditons are a major source of energy to viral reservoirs (Castellano et al., 2019).
Overall, significant changes in brain volume have been observed, which may be related to synaptic compromise. In the pre-ART era, people with advanced disease had a significant reduction of cortical volume (Heindel et al., 1994; Peavy et al., 1994; Aylward et al., 1995) as compared to age-matched seronegative controls (Tate et al., 2010). The introduction of ART stopped the progression of brain volume loss in the thalamus, caudate, and cerebellum and caused cortical thinning in the frontal and temporal lobes and cingulate cortex (Sanford et al., 2018). Using MRS, the most common metabolic changes observed in HIV-1-positive patients is a decreased level of creatine (Cr); an increased level of choline (Cho) and myo-inositole (MI), which reflect neuroinflammation and microglial proliferation; and a decreased level of N-acetyl aspartate (NAA) and Glu as a markers of neuronal dysfunction and injury (Young et al., 2014). In patients before ART, the cortical level of inflammatory biomarkers Cho/Cr and MI/Cr was increased. In the same patients, the level of Glu/Cr in the basal ganglia was elevated and significantly declined after therapy initiation, suggesting a massive release of Glu from HIV-1-infected macrophages and microglia, as activated astrocytes function dysregulation. Introduction of ART within 5 years after infection attenuated the increase of these inflammatory markers (Young et al., 2014). Furthermore, in HIV-1-individuals under ART who manifest HAND, a lower ratio of NAA/Cr in frontal white matter, post cingulate, and precuneus was observed. Also, a decrease in the Glu/Cr ratio correlated with worse performance on verbal recall, psychomotor speed, and reaction time (Mohamed et al., 2018). In agreement with the previous data, a decreased level of NAA and Glu-Gln was also observed in the cortical gray matter during early HIV-1 infection, suggesting that HIV-1 causes neuronal and astroglial dysfunction soon after infection (Lentz et al., 2009).
In other studies, it has been demonstrated that in CSF of HIV-1-positive individuals who manifest HAND, the level of Glu was elevated, which suggests astrocyte activation—impairment of BBB integrity and Glu clearance. Moreover, in CSF, of all the examined individuals had an increased level of ketone bodies (BHBA and 1,2 propenodiol), which suggests the dysregulation of lipid metabolism (Cassol et al., 2014). This result is in line with our and other studies showing that HIV-1-infected cells can rapidly adapt and use an alternative sources of energy. In several pathologies, including cancer, glucose deficit results in the use of alternative sources of energy such as ketone bodies or amino acids (Laffel, 1999; Castellano et al., 2017).
CD4+ T lymphocytes are characterized as a circulating viral reservoir, which maintains survival by increased glucose transporter-Glut-1 expression and elevated glycolysis (Loisel-Meyer et al., 2012). It was shown that Glut-1 expression on CD4+ T lymphocytes is increased in HIV-1-infected individuals, and expression of Glut-1 could be a biomarker of CD4+ T lymphocyte activation in infected individuals (Palmer et al., 2014). Also, increased pyruvate delivery may be supported via the Glu-Gln cycle to cope with the high metabolic activity of infected cells. Delivery of a higher amount of ATP can enhance the proliferation of infected cells and induce viral replication (Loftus and Finlay, 2016).
Next to the CD4+ T lymphocytes, tissue macrophages can also establish HIV-1 reservoirs. First, HIV-1 replication occurs intracellularly in specific compartments described as a virus-containing compartments (VCCs), in which newly created virus particles sustain their infectious potential for extended periods of time (Gaudin et al., 2013), and, second, HIV-1-infected macrophages can survive the apoptotic response of the host cells against viral infection (Swingler et al., 2007; Castellano et al., 2017). It was shown that HIV-1-infected macrophages increase GSH synthesis to limit excitotoxicity and neutralize generated oxygen free radicals (Rimaniol et al., 2001). Furthermore, in latently infected macrophages, Bim, a highly pro-apoptotic negative regulator of Bcl-2, was upregulated and prevented Bcl-2-dependent sequencing of pro-apoptotic proteins in mitochondria. Furthermore, HIV-1 infection prevents the release of apoptosis-inducing factor (AIF) or cytochrome C from the mitochondria into the cytoplasm and apoptosome formation. Thus, HIV-1 infection and latency prevent apoptosis by compromising the formation of the transition pore in the mitochondria and by preventing the formation of the apoptosome (Castellano et al., 2017).
Previously we demonstrated that latent HIV-1-infected macrophages used unusual pathways to block apoptosis of infected cells by a Bim-mediated mechanism (Castellano et al., 2017). We characterized three different stages of HIV-1 infection in macrophages: an early stage (1–3 days post-infection) with increased HIV-1 replication; a middle stage (7–14 days post-infection) characterized by higher viral replication and high cell death, which affects mainly uninfected macrophages; and a late state (14–21 days post-infection) with minimal viral replication and cell death similar to the middle stage, resulting in the survival of a small population of infected macrophages. The stage of infection was associated with changes in mitochondrial metabolism where the basal oxygen consumption rate (OCR) was reduced in the early stage in contrast to middle stage where OCR was reduced and correlated with increased cell death. In last stage, where most surviving cells were infected, no future changes of OCR were observed. Moreover, blocking of mitochondrial complex V, which is responsible for ATP production, induced different cell responses in the early and late stages, but not in the middle stage where the level of apoptosis was the highest; this suggests that during minimal apoptosis respiration, ATP is reduced by the virus. Next, we established the metabolic changes in latently infected macrophages, which in contrast to CD4+ T lymphocytes, did not rely on glucose in the HIV-1 condition but used alternative sources of energy coming from the TCA cycle and accumulated lipids. Observed changes in macrophages metabolism suggest that compromised TCA is an additional source of carbon to accumulate lipids, which can be used as an alternative source of energy. The role of the TCA cycle was established by treatment of human macrophages with succinate, an intermediate in TCA that plays a crucial role in ATP production in mitochondria. Succinate induced lipid accumulation in HIV-1-infected macrophages in contrast to uninfected cells. Furthermore, we characterized that under HIV-1 conditions, latently infected macrophages are using Glu/Gln to provide α-KG and succinate for TCA cycle, whereas in uninfected macrophages, ATP is produced from fatty acids and glucose. Moreover, in the HIV-1 condition, blockade delivery of substrate for energy production (fatty acids, Gln, and glucose) induce a shift for use of alternative sources of energy. HIV-1-infected macrophages, in contrast to uninfected macrophages, cannot use alternative sources of energy, opening a unique opportunity to kill this type of reservoir. Furthermore, we demonstrated that latently HIV-1-infected macrophages used Gln, Glu, and α-KG to survive because the blocking of GLS or an amino acid transporter required for amino acid importation into the mitochondria induced a specific killing of the surviving HIV-1-infected macrophages (Castellano et al., 2019). Moreover, proinflammatory molecules, such as IFNγ and LPS, stimulate macrophage activation inducing glucose uptake with concomitant suppression of fatty acid uptake and oxidation (Vats et al., 2006). It was shown that released ROS and treatment with LPS increased the Glut-1 expression (Freemerman et al., 2014).
In this review, we presented the current knowledge about the role of Glu in NeuroHIV pathogenesis. Excessive release of Glu with concomitant blockade of reuptake and metabolism induce excitotoxicity and constant inflammation leads to HAND development. In a successful ART era, HIV-1 cannot yet be cured, due to viral reservoirs that are established in peripheral and CNS compartments. However, recent clinical observations have hypothesized that an early initiation of ART is crucial to a progressive contraction of the latent HIV-1 reservoir (“shrink”). This could possibly be accomplished with simultaneous strategies that activate (“kick” or “shock”) the latent reservoir and increase the clearance of virus-infected cells (“kill”), known as a “kick-kill” or “shock-kill” strategy (Chun et al., 1999; Archin et al., 2012; Van Lint et al., 2013; Pace and Frater, 2019; Edara et al., 2020). Although ART suppresses viremia in HIV-1 infected individuals, infected cells used Glu to maintain their survival and latent virus reservoirs. Moreover, Glu as a toxic molecule can induce viral rebound in latent infected cells. Thus, suppressing the overproduction of Glu shortly after infection may prevent neuroinflammation and successfully reduce the size of viral reservoirs that relay a significant amount of Glu to survive.
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
This work was funded by the National Institute of Mental Health grant, MH096625, the National Institute of Neurological Disorders and Stroke, NS105584, and UTMB funding (to EE).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbate, I., Cappiello, G., Longo, R., Ursitti, A., Spano, A., Calcaterra, S., et al. (2005). Cell membrane proteins and quasispecies compartmentalization of CSF and plasma HIV-1 from aids patients with neurological disorders. Infect. Genet. Evol. 5, 247–253. doi: 10.1016/j.meegid.2004.08.006
Abrahams, M. R., Joseph, S. B., Garrett, N., Tyers, L., Moeser, M., Archin, N., et al. (2019). The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci. Transl. Med. 11:aaw5589. doi: 10.1126/scitranslmed.aaw5589
Abreu, C. M., Veenhuis, R. T., Avalos, C. R., Graham, S., Parrilla, D. R., Ferreira, E. A., et al. (2019). Myeloid and CD4 T cells comprise the latent reservoir in antiretroviral therapy-suppressed SIVmac251-infected macaques. MBio 10:e01659-19. doi: 10.1128/mBio.01659-19
Albrecht, J., and Norenberg, M. D. (2006). Glutamine: a Trojan horse in ammonia neurotoxicity. Hepatology 44, 788–794. doi: 10.1002/hep.21357
Alexaki, A., Liu, Y., and Wigdahl, B. (2008). Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 6, 388–400. doi: 10.2174/157016208785861195
Al-Harti, L., Joseph, J., and Nath, A. (2018). Astrocytes as an HIV CNS reservoir: highlights and reflections of an NIMH-sponsored symposium. J. Neurovirol. 24, 665–669. doi: 10.1007/s13365-018-0691-8
Allen, N. J., Karadottir, R., and Attwell, D. (2004). Reversal or reduction of glutamate and GABA transport in CNS pathology and therapy. Pflugers Arch. 449, 132–142. doi: 10.1007/s00424-004-1318-x
Anderson, A. M., Harezlak, J., Bharti, A., Mi, D., Taylor, M. J., Daar, E. S., et al. (2015). Plasma and cerebrospinal fluid biomarkers predict cerebral injury in HIV-infected individuals on stable combination antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 69, 29–35. doi: 10.1097/QAI.0000000000000532
Angelovich, T. A., Churchill, M. J., Wright, E. J., and Brew, B. J. (2020). “New Potential Axes of HIV Neuropathogenesis with Relevance to Biomarkers and Treatment,” in Current Topics in Behavioral Neurosciences (Berlin; Heidelberg: Springer). doi: 10.1007/7854_2019_126
Archin, N. M., Liberty, A. L., Kashuba, A. D., Choudhary, S. K., Kuruc, J. D., Crooks, A. M., et al. (2012). Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 487, 482–485. doi: 10.1038/nature11286
Ariazi, J., Benowitz, A., De Biasi, V., Den Boer, M. L., Cherqui, S., Cui, H., et al. (2017). Tunneling nanotubes and gap junctions-their role in long-range intercellular communication during development, health, and disease conditions. Front. Mol. Neurosci. 10:333. doi: 10.3389/fnmol.2017.00333
Avdoshina, V., Fields, J. A., Castellano, P., Dedoni, S., Palchik, G., Trejo, M., et al. (2016). The HIV protein gp120 alters mitochondrial dynamics in neurons. Neurotox. Res. 29, 583–593. doi: 10.1007/s12640-016-9608-6
Aylward, E. H., Brettschneider, P. D., McArthur, J. C., Harris, G. J., Schlaepfer, T. E., Henderer, J. D., et al. (1995). Magnetic resonance imaging measurement of gray matter volume reductions in HIV dementia. Am. J. Psychiatry 152, 987–994. doi: 10.1176/ajp.152.7.987
Bachani, M., Sacktor, N., McArthur, J. C., Nath, A., and Rumbaugh, J. (2013). Detection of anti-tat antibodies in CSF of individuals with HIV-associated neurocognitive disorders. J. Neurovirol. 19, 82–88. doi: 10.1007/s13365-012-0144-8
Bachis, A., Cruz, M. I., and Mocchetti, I. (2010). M-tropic HIV envelope protein gp120 exhibits a different neuropathological profile than T-tropic gp120 in rat striatum. Eur. J. Neurosci. 32, 570–578. doi: 10.1111/j.1460-9568.2010.07325.x
Bak, L. K., Schousboe, A., and Waagepetersen, H. S. (2006). The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J. Neurochem. 98, 641–653. doi: 10.1111/j.1471-4159.2006.03913.x
Bandera, A., Taramasso, L., Bozzi, G., Muscatello, A., Robinson, J. A., Burdo, T. H., et al. (2019). HIV-associated neurocognitive impairment in the modern ART era: are we close to discovering reliable biomarkers in the setting of virological suppression? Front. Aging Neurosci. 11:187. doi: 10.3389/fnagi.2019.00187
Barger, S. W., Goodwin, M. E., Porter, M. M., and Beggs, M. L. (2007). Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J. Neurochem 101, 1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x
Barrero, C. A., Datta, P. K., Sen, S., Deshmane, S., Amini, S., Khalili, K., et al. (2013). HIV-1 Vpr modulates macrophage metabolic pathways: a SILAC-based quantitative analysis. PLoS ONE 8:e68376. doi: 10.1371/journal.pone.0068376
Baxter, P. S., and Hardingham, G. E. (2016). Adaptive regulation of the brain's antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 100, 147–152. doi: 10.1016/j.freeradbiomed.2016.06.027
Berman, J. W., Carvallo, L., Buckner, C. M., Luers, A., Prevedel, L., Bennett, M. V., et al. (2016). HIV-tat alters Connexin43 expression and trafficking in human astrocytes: role in NeuroAIDS. J. Neuroinflamm. 13:54. doi: 10.1186/s12974-016-0510-1
Bingham, R., Ahmed, N., Rangi, P., Johnson, M., Tyrer, M., and Green, J. (2011). HIV encephalitis despite suppressed viraemia: a case of compartmentalized viral escape. Int. J. STD AIDS 22, 608–609. doi: 10.1258/ijsa.2011.010507
Blankson, J. N., Persaud, D., and Siliciano, R. F. (2002). The challenge of viral reservoirs in HIV-1 infection. Annu. Rev. Med. 53, 557–593. doi: 10.1146/annurev.med.53.082901.104024
Bonansco, C., and Fuenzalida, M. (2016). Plasticity of hippocampal excitatory-inhibitory balance: missing the synaptic control in the epileptic brain. Neural Plast. 2016:8607038. doi: 10.1155/2016/8607038
Borderia, A. V., Codoner, F. M., and Sanjuan, R. (2007). Selection promotes organ compartmentalization in HIV-1: evidence from gag and pol genes. Evolution 61, 272–279. doi: 10.1111/j.1558-5646.2007.00025.x
Borjabad, A., Brooks, A. I., and Volsky, D. J. (2010). Gene expression profiles of HIV-1-infected glia and brain: toward better understanding of the role of astrocytes in HIV-1-associated neurocognitive disorders. J. Neuroimmune Pharmacol. 5, 44–62. doi: 10.1007/s11481-009-9167-1
Bridges, R., Lutgen, V., Lobner, D., and Baker, D. A. (2012a). Thinking outside the cleft to understand synaptic activity: contribution of the cystine-glutamate antiporter (System xc-) to normal and pathological glutamatergic signaling. Pharmacol. Rev. 64, 780–802. doi: 10.1124/pr.110.003889
Bridges, R. J., Natale, N. R., and Patel, S. A. (2012b). System xc(-) cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. Br. J. Pharmacol. 165, 20–34. doi: 10.1111/j.1476-5381.2011.01480.x
Brier, M. R., Wu, Q., Tanenbaum, A. B., Westerhaus, E. T., Kharasch, E. D., and Ances, B. M. (2015). Effect of HAART on brain organization and function in HIV-negative subjects. J. Neuroimmune Pharmacol. 10, 517–521. doi: 10.1007/s11481-015-9634-9
Capone, C., Cervelli, M., Angelucci, E., Colasanti, M., Macone, A., Mariottini, P., et al. (2013). A role for spermine oxidase as a mediator of reactive oxygen species production in HIV-Tat-induced neuronal toxicity. Free Radic. Biol. Med. 63, 99–107. doi: 10.1016/j.freeradbiomed.2013.05.007
Cardona, C., Sanchez-Mejias, E., Davila, J. C., Martin-Rufian, M., Campos-Sandoval, J. A., Vitorica, J., et al. (2015). Expression of Gls and Gls2 glutaminase isoforms in astrocytes. Glia 63, 365–382. doi: 10.1002/glia.22758
Cassol, E., Misra, V., Dutta, A., Morgello, S., and Gabuzda, D. (2014). Cerebrospinal fluid metabolomics reveals altered waste clearance and accelerated aging in HIV patients with neurocognitive impairment. AIDS 28, 1579–1591. doi: 10.1097/QAD.0000000000000303
Castellano, P., Prevedel, L., and Eugenin, E. A. (2017). HIV-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving Bim. Sci. Rep. 7:12866. doi: 10.1038/s41598-017-12758-w
Castellano, P., Prevedel, L., Valdebenito, S., and Eugenin, E. A. (2019). HIV infection and latency induce a unique metabolic signature in human macrophages. Sci. Rep. 9:3941. doi: 10.1038/s41598-019-39898-5
Chen, B. S., and Roche, K. W. (2007). Regulation of NMDA receptors by phosphorylation. Neuropharmacology 53, 362–368. doi: 10.1016/j.neuropharm.2007.05.018
Chen, C. Y., Chen, Y. Y., Chen, J. J. W., Chen, K. Y., Ho, C. C., Shih, J. Y., et al. (2017). Astrocyte-elevated gene-1 confers resistance to pemetrexed in non-small cell lung cancer by upregulating thymidylate synthase expression. Oncotarget 8, 61901–61916. doi: 10.18632/oncotarget.18717
Chomont, N., El-Far, M., Ancuta, P., Trautmann, L., Procopio, F. A., Yassine-Diab, B., et al. (2009). HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 15, 893–900. doi: 10.1038/nm.1972
Chun, T. W., Engel, D., Mizell, S. B., Hallahan, C. W., Fischette, M., Park, S., et al. (1999). Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat. Med. 5, 651–655. doi: 10.1038/9498
Churchill, M. J., Gorry, P. R., Cowley, D., Lal, L., Sonza, S., Purcell, D. F., et al. (2006). Use of laser capture microdissection to detect integrated HIV-1 DNA in macrophages and astrocytes from autopsy brain tissues. J. Neurovirol. 12, 146–152. doi: 10.1080/13550280600748946
Churchill, M. J., Wesselingh, S. L., Cowley, D., Pardo, C. A., McArthur, J. C., Brew, B. J., et al. (2009). Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 66, 253–258. doi: 10.1002/ana.21697
Cohen, R. A., Harezlak, J., Gongvatana, A., Buchthal, S., Schifitto, G., Clark, U., et al. (2010). Cerebral metabolite abnormalities in human immunodeficiency virus are associated with cortical and subcortical volumes. J. Neurovirol. 16, 435–444. doi: 10.1007/BF03210849
Conn, P. J., and Pin, J. P. (1997). Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 37, 205–237. doi: 10.1146/annurev.pharmtox.37.1.205
Connor, R. I., Chen, B. K., Choe, S., and Landau, N. R. (1995). Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 206, 935–944. doi: 10.1006/viro.1995.1016
Conti, F., and Minelli, A. (1994). Glutamate immunoreactivity in rat cerebral cortex is reversibly abolished by 6-diazo-5-oxo-L-norleucine (DON), an inhibitor of phosphate-activated glutaminase. J. Histochem. Cytochem. 42, 717–726. doi: 10.1177/42.6.7910617
Contractor, A., Mulle, C., and Swanson, G. T. (2011). Kainate receptors coming of age: milestones of two decades of research. Trends Neurosci 34, 154–163. doi: 10.1016/j.tins.2010.12.002
Cysique, L. A., Juge, L., Lennon, M. J., Gates, T. M., Jones, S. P., Lovelace, M. D., et al. (2019). HIV brain latency as measured by CSF BcL11b relates to disrupted brain cellular energy in virally suppressed HIV infection. AIDS 33, 433–441. doi: 10.1097/QAD.0000000000002076
D'Antoni, S., Berretta, A., Bonaccorso, C. M., Bruno, V., Aronica, E., Nicoletti, F., et al. (2008). Metabotropic glutamate receptors in glial cells. Neurochem. Res. 33, 2436–2443. doi: 10.1007/s11064-008-9694-9
Datta, P. K., Deshmane, S., Khalili, K., Merali, S., Gordon, J. C., Fecchio, C., et al. (2016). Glutamate metabolism in HIV-1 infected macrophages: role of HIV-1 Vpr. Cell Cycle 15, 2288–2298. doi: 10.1080/15384101.2016.1190054
de Almeida, S. M., Rotta, I., Ribeiro, C. E., Oliveira, M. F., Chaillon, A., de Pereira, A. P., et al. (2017). Dynamic of CSF and serum biomarkers in HIV-1 subtype C encephalitis with CNS genetic compartmentalization-case study. J. Neurovirol. 23, 460–473. doi: 10.1007/s13365-017-0518-z
Dickens, A. M., Anthony, D. C., Deutsch, R., Mielke, M. M., Claridge, T. D., Grant, I., et al. (2015). Cerebrospinal fluid metabolomics implicate bioenergetic adaptation as a neural mechanism regulating shifts in cognitive states of HIV-infected patients. AIDS 29, 559–569. doi: 10.1097/QAD.0000000000000580
Dickens, A. M., Yoo, S. W., Chin, A. C., Xu, J., Johnson, T. P., Trout, A. L., et al. (2017). Chronic low-level expression of HIV-1 Tat promotes a neurodegenerative phenotype with aging. Sci. Rep. 7:7748. doi: 10.1038/s41598-017-07570-5
Duguid, I. C., and Smart, T. G. (2004). Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat. Neurosci. 7, 525–533. doi: 10.1038/nn1227
Eck, H. P., Frey, H., and Droge, W. (1989). Elevated plasma glutamate concentrations in HIV-1-infected patients may contribute to loss of macrophage and lymphocyte functions. Int. Immunol. 1, 367–372. doi: 10.1093/intimm/1.4.367
Edara, V. V., Ghorpade, A., and Borgmann, K. (2020). Insights into the gene expression profiles of active and restricted red/green-HIV(+) human astrocytes: implications for shock or lock therapies in the brain. J. Virol. 94:e01563-19. doi: 10.1128/JVI.01563-19
Eggers, C., Arendt, G., Hahn, K., Husstedt, I. W., Maschke, M., Neuen-Jacob, E., et al. (2017). HIV-1-associated neurocognitive disorder: epidemiology, pathogenesis, diagnosis, and treatment. J. Neurol. 264, 1715–1727. doi: 10.1007/s00415-017-8503-2
Emdad, L., Lee, S. G., Su, Z. Z., Jeon, H. Y., Boukerche, H., Sarkar, D., et al. (2009). Astrocyte elevated gene-1 (AEG-1) functions as an oncogene and regulates angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 106, 21300–21305. doi: 10.1073/pnas.0910936106
Emdad, L., Sarkar, D., Lee, S. G., Su, Z. Z., Yoo, B. K., Dash, R., et al. (2010). Astrocyte elevated gene-1: a novel target for human glioma therapy. Mol. Cancer Ther. 9, 79–88. doi: 10.1158/1535-7163.MCT-09-0752
Erdmann, N., Tian, C., Huang, Y., Zhao, J., Herek, S., Curthoys, N., et al. (2009). In vitro glutaminase regulation and mechanisms of glutamate generation in HIV-1-infected macrophage. J. Neurochem. 109, 551–561. doi: 10.1111/j.1471-4159.2009.05989.x
Erdmann, N., Zhao, J., Lopez, A. L., Herek, S., Curthoys, N., Hexum, T. D., et al. (2007). Glutamate production by HIV-1 infected human macrophage is blocked by the inhibition of glutaminase. J. Neurochem. 102, 539–549. doi: 10.1111/j.1471-4159.2007.04594.x
Erickson, J. W., and Cerione, R. A. (2010). Glutaminase: a hot spot for regulation of cancer cell metabolism? Oncotarget 1, 734–740. doi: 10.18632/oncotarget.208
Ernst, T., Jiang, C. S., Nakama, H., Buchthal, S., and Chang, L. (2010). Lower brain glutamate is associated with cognitive deficits in HIV patients: a new mechanism for HIV-associated neurocognitive disorder. J. Magn. Reson. Imaging 32, 1045–1053. doi: 10.1002/jmri.22366
Eugenin, E. A., Clements, J. E., Zink, M. C., and Berman, J. W. (2011a). Human immunodeficiency virus infection of human astrocytes disrupts blood-brain barrier integrity by a gap junction-dependent mechanism. J. Neurosci. 31, 9456–9465. doi: 10.1523/JNEUROSCI.1460-11.2011
Eugenin, E. A., D'Aversa, T. G., Lopez, L., Calderon, T. M., and Berman, J. W. (2003). MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J. Neurochem. 85, 1299–1311. doi: 10.1046/j.1471-4159.2003.01775.x
Eugenin, E. A., Gaskill, P. J., and Berman, J. W. (2009a). Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: a potential mechanism for intercellular HIV trafficking. Cell. Immunol. 254, 142–148. doi: 10.1016/j.cellimm.2008.08.005
Eugenin, E. A., Gaskill, P. J., and Berman, J. W. (2009b). Tunneling nanotubes (TNT): a potential mechanism for intercellular HIV trafficking. Commun. Integr. Biol. 2, 243–244. doi: 10.4161/cib.2.3.8165
Eugenin, E. A., King, J. E., Hazleton, J. E., Major, E. O., Bennett, M. V., Zukin, R. S., et al. (2011b). Differences in NMDA receptor expression during human development determine the response of neurons to HIV-tat-mediated neurotoxicity. Neurotox. Res. 19, 138–148. doi: 10.1007/s12640-010-9150-x
Eugenin, E. A., King, J. E., Nath, A., Calderon, T. M., Zukin, R. S., Bennett, M. V., et al. (2007). HIV-tat induces formation of an LRP-PSD-95- NMDAR-nNOS complex that promotes apoptosis in neurons and astrocytes. Proc. Natl. Acad. Sci. U.S.A. 104, 3438–3443. doi: 10.1073/pnas.0611699104
Eugenin, E. A., Osiecki, K., Lopez, L., Goldstein, H., Calderon, T. M., and Berman, J. W. (2006). CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood-brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J. Neurosci. 26, 1098–1106. doi: 10.1523/JNEUROSCI.3863-05.2006
Fernandes, N., and Pulliam, L. (2019). “Inflammatory Mechanisms and Cascades Contributing to Neurocognitive Impairment in HIV/AIDS,” in Current Topics in Behavioral Neurosciences (Berlin; Heidelberg: Springer). doi: 10.1007/7854_2019_100
Ferrarese, C., Aliprandi, A., Tremolizzo, L., Stanzani, L., De Micheli, A., Dolara, A., et al. (2001). Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 57, 671–675. doi: 10.1212/WNL.57.4.671
Ferrucci, A., Nonnemacher, M. R., Cohen, E. A., and Wigdahl, B. (2012). Extracellular human immunodeficiency virus type 1 viral protein R causes reductions in astrocytic ATP and glutathione levels compromising the antioxidant reservoir. Virus Res. 167, 358–369. doi: 10.1016/j.virusres.2012.06.002
Finzi, D., Blankson, J., Siliciano, J. D., Margolick, J. B., Chadwick, K., Pierson, T., et al. (1999). Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 5, 512–517. doi: 10.1038/8394
Fischer-Smith, T., Croul, S., Sverstiuk, A. E., Capini, C., L'Heureux, D., Regulier, E. G., et al. (2001). CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: perivascular accumulation and reservoir of HIV infection. J. Neurovirol. 7, 528–541. doi: 10.1080/135502801753248114
Fiszman, M. L., Barberis, A., Lu, C., Fu, Z., Erdelyi, F., Szabo, G., et al. (2005). NMDA receptors increase the size of GABAergic terminals and enhance GABA release. J. Neurosci. 25, 2024–2031. doi: 10.1523/JNEUROSCI.4980-04.2005
Fogal, B., Li, J., Lobner, D., McCullough, L. D., and Hewett, S. J. (2007). System x(c)- activity and astrocytes are necessary for interleukin-1 beta-mediated hypoxic neuronal injury. J. Neurosci. 27, 10094–10105. doi: 10.1523/JNEUROSCI.2459-07.2007
Ford, T. C., Nibbs, R., and Crewther, D. P. (2017). Increased glutamate/GABA+ ratio in a shared autistic and schizotypal trait phenotype termed social disorganisation. Neuroimage Clin. 16, 125–131. doi: 10.1016/j.nicl.2017.07.009
Freemerman, A. J., Johnson, A. R., Sacks, G. N., Milner, J. J., Kirk, E. L., Troester, M. A., et al. (2014). Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 289, 7884–7896. doi: 10.1074/jbc.M113.522037
Ganor, Y., Real, F., Sennepin, A., Dutertre, C. A., Prevedel, L., Xu, L., et al. (2019). HIV-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat Microbiol. 4, 633–644. doi: 10.1038/s41564-018-0335-z
Garcia, F., Plana, M., Vidal, C., Cruceta, A., O'Brien, W. A., Pantaleo, G., et al. (1999). Dynamics of viral load rebound and immunological changes after stopping effective antiretroviral therapy. AIDS 13, F79–86. doi: 10.1097/00002030-199907300-00002
Gatanaga, H., Oka, S., Ida, S., Wakabayashi, T., Shioda, T., and Iwamoto, A. (1999). Active HIV-1 redistribution and replication in the brain with HIV encephalitis. Arch. Virol. 144, 29–43. doi: 10.1007/s007050050483
Gaudin, R., Berre, S., Cunha de Alencar, B., Decalf, J., Schindler, M., Gobert, F. X., et al. (2013). Dynamics of HIV-containing compartments in macrophages reveal sequestration of virions and transient surface connections. PLoS One 8:e69450. doi: 10.1371/journal.pone.0069450
Gelman, B. B. (2015). Neuropathology of HAND with suppressive antiretroviral therapy: encephalitis and neurodegeneration reconsidered. Curr. HIV/AIDS Rep. 12, 272–279. doi: 10.1007/s11904-015-0266-8
Gelman, B. B., Lisinicchia, J. G., Morgello, S., Masliah, E., Commins, D., Achim, C. L., et al. (2013). Neurovirological correlation with HIV-associated neurocognitive disorders and encephalitis in a HAART-era cohort. J. Acquir. Immune Defic Syndr. 62, 487–495. doi: 10.1097/QAI.0b013e31827f1bdb
Gianella, S., Kosakovsky Pond, S. L., Oliveira, M. F., Scheffler, K., Strain, M. C., De la Torre, A., et al. (2016). Compartmentalized HIV rebound in the central nervous system after interruption of antiretroviral therapy. Virus Evol. 2:vew020. doi: 10.1093/ve/vew020
Gibb, A. J., Ogden, K. K., McDaniel, M. J., Vance, K. M., Kell, S. A., Butch, C., et al. (2018). A structurally derived model of subunit-dependent NMDA receptor function. J. Physiol. 596, 4057–4089. doi: 10.1113/JP276093
Goldstein, G. (1996). HIV-1 Tat protein as a potential AIDS vaccine. Nat. Med. 2, 960–964. doi: 10.1038/nm0996-960
Gonzalez-Perez, M. P., O'Connell, O., Lin, R., Sullivan, W. M., Bell, J., Simmonds, P., et al. (2012). Independent evolution of macrophage-tropism and increased charge between HIV-1 R5 envelopes present in brain and immune tissue. Retrovirology. 9:20. doi: 10.1186/1742-4690-9-20
Govindarajulu, M., Pinky, P. D., Bloemer, J., Ghanei, N., Suppiramaniam, V., and Amin, R. (2018). Signaling mechanisms of selective PPARgamma modulators in alzheimer's disease. PPAR Res. 2018:2010675. doi: 10.1155/2018/2010675
Green, M. V., and Thayer, S. A. (2016). NMDARs adapt to neurotoxic HIV protein tat downstream of a GluN2A-ubiquitin ligase signaling pathway. J. Neurosci. 36, 12640–12649. doi: 10.1523/JNEUROSCI.2980-16.2016
Grewer, C., Gameiro, A., Zhang, Z., Tao, Z., Braams, S., and Rauen, T. (2008). Glutamate forward and reverse transport: from molecular mechanism to transporter-mediated release after ischemia. IUBMB Life 60, 609–619. doi: 10.1002/iub.98
Guo, H., Camargo, L. M., Yeboah, F., Digan, M. E., Niu, H., Pan, Y., et al. (2017). A NMDA-receptor calcium influx assay sensitive to stimulation by glutamate and glycine/D-serine. Sci. Rep. 7:11608. doi: 10.1038/s41598-017-11947-x
Gupta, S., Knight, A. G., Knapp, P. E., Hauser, K. F., Keller, J. N., and Bruce-Keller, A. J. (2010). HIV-Tat elicits microglial glutamate release: role of NAPDH oxidase and the cystine-glutamate antiporter. Neurosci. Lett 485, 233–236. doi: 10.1016/j.neulet.2010.09.019
Gupta, S., Knight, A. G., Losso, B. Y., Ingram, D. K., Keller, J. N., and Bruce-Keller, A. J. (2012). Brain injury caused by HIV protease inhibitors: role of lipodystrophy and insulin resistance. Antiviral Res. 95, 19–29. doi: 10.1016/j.antiviral.2012.04.010
Haase, A. T. (1999). Population biology of HIV-1 infection: viral and CD4+ T cell demographics and dynamics in lymphatic tissues. Annu. Rev. Immunol. 17, 625–656. doi: 10.1146/annurev.immunol.17.1.625
Hara, N., Yamada, K., Terashima, M., Osago, H., Shimoyama, M., and Tsuchiya, M. (2003). Molecular identification of human glutamine- and ammonia-dependent NAD synthetases. Carbon-nitrogen hydrolase domain confers glutamine dependency. J. Biol. Chem. 278, 10914–10921. doi: 10.1074/jbc.M209203200
Harris-Warrick, R. (2005). Synaptic chemistry in single neurons: GABA is identified as an inhibitory neurotransmitter. J. Neurophysiol. 93, 3029–3031. doi: 10.1152/classicessays.00026.2005
Haughey, N. J., Holden, C. P., Nath, A., and Geiger, J. D. (1999). Involvement of inositol 1,4,5-trisphosphate-regulated stores of intracellular calcium in calcium dysregulation and neuron cell death caused by HIV-1 protein tat. J. Neurochem. 73, 1363–1374. doi: 10.1046/j.1471-4159.1999.0731363.x
Haughey, N. J., Nath, A., Mattson, M. P., Slevin, J. T., and Geiger, J. D. (2001). HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J. Neurochem. 78, 457–467. doi: 10.1046/j.1471-4159.2001.00396.x
He, F., and Sun, Y. E. (2007). Glial cells more than support cells? Int. J. Biochem. Cell Biol. 39, 661–665. doi: 10.1016/j.biocel.2006.10.022
Heindel, W. C., Jernigan, T. L., Archibald, S. L., Achim, C. L., Masliah, E., and Wiley, C. A. (1994). The relationship of quantitative brain magnetic resonance imaging measures to neuropathologic indexes of human immunodeficiency virus infection. Arch. Neurol. 51, 1129–1135. doi: 10.1001/archneur.1994.00540230067015
Hertz, L., and Chen, Y. (2017). Integration between glycolysis and glutamate-glutamine cycle flux may explain preferential glycolytic increase during brain activation, requiring glutamate. Front. Integr. Neurosci. 11:18. doi: 10.3389/fnint.2017.00018
Hill, A. L. (2018). Mathematical models of HIV latency. Curr. Top. Microbiol. Immunol. 417, 131–156. doi: 10.1007/82_2017_77
Hill, A. L., Rosenbloom, D. I., Fu, F., Nowak, M. A., and Siliciano, R. F. (2014). Predicting the outcomes of treatment to eradicate the latent reservoir for HIV-1. Proc. Natl. Acad. Sci. U.S.A. 111, 13475–13480. doi: 10.1073/pnas.1406663111
Hill, A. L., Rosenbloom, D. I., Siliciano, J. D., and Siliciano, R. F. (2016). Insufficient evidence for rare activation of latent HIV in the absence of reservoir-reducing interventions. PLoS Pathog. 12:e1005679. doi: 10.1371/journal.ppat.1005679
Ho, D. D., Neumann, A. U., Perelson, A. S., Chen, W., Leonard, J. M., and Markowitz, M. (1995). Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature 373, 123–126. doi: 10.1038/373123a0
Holcomb, T., Taylor, L., Trohkimoinen, J., and Curthoys, N. P. (2000). Isolation, characterization and expression of a human brain mitochondrial glutaminase cDNA. Brain Res. Mol. Brain Res. 76, 56–63. doi: 10.1016/S0169-328X(99)00331-9
Huang, W., Chen, L., Zhang, B., Park, M., and Toborek, M. (2014). PPAR agonist-mediated protection against HIV Tat-induced cerebrovascular toxicity is enhanced in MMP-9-deficient mice. J. Cereb. Blood Flow Metab. 34, 646–653. doi: 10.1038/jcbfm.2013.240
Huang, W., Mo, X., Wu, X., Luo, W., and Chen, Y. (2015). Rosiglitazone suppresses HIV-1 Tat-induced vascular inflammation via Akt signaling. Mol. Cell. Biochem. 407, 173–179. doi: 10.1007/s11010-015-2467-2
Huang, Y., Zhao, L., Jia, B., Wu, L., Li, Y., Curthoys, N., et al. (2011). Glutaminase dysregulation in HIV-1-infected human microglia mediates neurotoxicity: relevant to HIV-1-associated neurocognitive disorders. J. Neurosci. 31, 15195–15204. doi: 10.1523/JNEUROSCI.2051-11.2011
Jain, V., Hartogensis, W., Bacchetti, P., Hunt, P. W., Hatano, H., Sinclair, E., et al. (2013). Antiretroviral therapy initiated within 6 months of HIV infection is associated with lower T-cell activation and smaller HIV reservoir size. J. Infect. Dis. 208, 1202–1211. doi: 10.1093/infdis/jit311
Josefsson, L., Palmer, S., Faria, N. R., Lemey, P., Casazza, J., Ambrozak, D., et al. (2013). Single cell analysis of lymph node tissue from HIV-1 infected patients reveals that the majority of CD4+ T-cells contain one HIV-1 DNA molecule. PLoS Pathog. 9:e1003432. doi: 10.1371/journal.ppat.1003432
Kanai, Y., and Hediger, M. A. (2004). The glutamate/neutral amino acid transporter family SLC1: molecular, physiological and pharmacological aspects. Pflugers Arch. 447, 469–479. doi: 10.1007/s00424-003-1146-4
Kandathil, A. J., Sugawara, S., and Balagopal, A. (2016). Are T cells the only HIV-1 reservoir? Retrovirology 13:86. doi: 10.1186/s12977-016-0323-4
Kang, D. C., Su, Z. Z., Sarkar, D., Emdad, L., Volsky, D. J., and Fisher, P. B. (2005). Cloning and characterization of HIV-1-inducible astrocyte elevated gene-1, AEG-1. Gene 353, 8–15. doi: 10.1016/j.gene.2005.04.006
Karadottir, R., Cavelier, P., Bergersen, L. H., and Attwell, D. (2005). NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature 438, 1162–1166. doi: 10.1038/nature04302
Kaul, M., Garden, G. A., and Lipton, S. A. (2001). Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature 410, 988–994. doi: 10.1038/35073667
Kaul, M., Ma, Q., Medders, K. E., Desai, M. K., and Lipton, S. A. (2007). HIV-1 coreceptors CCR5 and CXCR4 both mediate neuronal cell death but CCR5 paradoxically can also contribute to protection. Cell Death Differ. 14, 296–305. doi: 10.1038/sj.cdd.4402006
Kim-Chang, J. J., Donovan, K., Loop, M. S., Hong, S., Fischer, B., Venturi, G., et al. (2019). Higher soluble CD14 levels are associated with lower visuospatial memory performance in youth with HIV. AIDS 33, 2363–2374. doi: 10.1097/QAD.0000000000002371
King, J. E., Eugenin, E. A., Hazleton, J. E., Morgello, S., and Berman, J. W. (2010). Mechanisms of HIV-tat-induced phosphorylation of N-methyl-D-aspartate receptor subunit 2A in human primary neurons: implications for neuroAIDS pathogenesis. Am. J. Pathol. 176, 2819–2830. doi: 10.2353/ajpath.2010.090642
Ko, A., Kang, G., Hattler, J. B., Galadima, H. I., Zhang, J., Li, Q., et al. (2019). Macrophages but not astrocytes harbor HIV DNA in the brains of HIV-1-infected aviremic individuals on suppressive antiretroviral therapy. J. Neuroimmune Pharmacol. 14, 110–119. doi: 10.1007/s11481-018-9809-2
Koller, H., Schaal, H., Freund, M., Garrido, S. R., von Giesen, H. J., Ott, M., et al. (2001). HIV-1 protein Tat reduces the glutamate-induced intracellular Ca2+ increase in cultured cortical astrocytes. Eur. J. Neurosci. 14, 1793–1799. doi: 10.1046/j.0953-816x.2001.01808.x
Kovalevich, J., and Langford, D. (2012). Neuronal toxicity in HIV CNS disease. Future Virol. 7, 687–698. doi: 10.2217/fvl.12.57
Krogh, K. A., Green, M. V., and Thayer, S. A. (2014a). HIV-1 Tat-induced changes in synaptically-driven network activity adapt during prolonged exposure. Curr. HIV Res. 12, 406–414. doi: 10.2174/1570162X13666150121110402
Krogh, K. A., Wydeven, N., Wickman, K., and Thayer, S. A. (2014b). HIV-1 protein Tat produces biphasic changes in NMDA-evoked increases in intracellular Ca2+ concentration via activation of Src kinase and nitric oxide signaling pathways. J. Neurochem. 130, 642–656. doi: 10.1111/jnc.12724
Kumar, A. (2015). NMDA receptor function during senescence: implication on cognitive performance. Front. Neurosci. 9:473. doi: 10.3389/fnins.2015.00473
Laffel, L. (1999). Ketone bodies: a review of physiology, pathophysiology and application of monitoring to diabetes. Diabetes Metab. Res. Rev. 15, 412–426. doi: 10.1002/(SICI)1520-7560(199911/12)15:6<;412::AID-DMRR72>;3.0.CO;2-8
Latronico, T., Pati, I., Ciavarella, R., Fasano, A., Mengoni, F., Lichtner, M., et al. (2018). In vitro effect of antiretroviral drugs on cultured primary astrocytes: analysis of neurotoxicity and matrix metalloproteinase inhibition. J. Neurochem. 144, 271–284. doi: 10.1111/jnc.14269
Lau, A., and Tymianski, M. (2010). Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 460, 525–542. doi: 10.1007/s00424-010-0809-1
Lee, G. Q., and Lichterfeld, M. (2016). Diversity of HIV-1 reservoirs in CD4+ T-cell subpopulations. Curr. Opin. HIV AIDS 11, 383–387. doi: 10.1097/COH.0000000000000281
Lee, S. G., Jeon, H. Y., Su, Z. Z., Richards, J. E., Vozhilla, N., Sarkar, D., et al. (2009). Astrocyte elevated gene-1 contributes to the pathogenesis of neuroblastoma. Oncogene 28, 2476–2484. doi: 10.1038/onc.2009.93
Lee, S. G., Kim, K., Kegelman, T. P., Dash, R., Das, S. K., Choi, J. K., et al. (2011). Oncogene AEG-1 promotes glioma-induced neurodegeneration by increasing glutamate excitotoxicity. Cancer Res. 71, 6514–6523. doi: 10.1158/0008-5472.CAN-11-0782
Lee, Y., Cho, J. H., Lee, S., Lee, W., Chang, S. C., Chung, H. Y., et al. (2019). Neuroprotective effects of MHY908, a PPAR alpha/gamma dual agonist, in a MPTP-induced Parkinson's disease model. Brain Res. 1704, 47–58. doi: 10.1016/j.brainres.2018.09.036
Lentz, M. R., Kim, W. K., Lee, V., Bazner, S., Halpern, E. F., Venna, N., et al. (2009). Changes in MRS neuronal markers and T cell phenotypes observed during early HIV infection. Neurology 72, 1465–1472. doi: 10.1212/WNL.0b013e3181a2e90a
Lewerenz, J., Hewett, S. J., Huang, Y., Lambros, M., Gout, P. W., Kalivas, P. W., et al. (2013). The cystine/glutamate antiporter system x(c)(-) in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid. Redox Signal 18, 522–555. doi: 10.1089/ars.2011.4391
Li, T., Giaume, C., and Xiao, L. (2014). Connexins-mediated glia networking impacts myelination and remyelination in the central nervous system. Mol. Neurobiol. 49, 1460–1471. doi: 10.1007/s12035-013-8625-1
Li, W., Huang, Y., Reid, R., Steiner, J., Malpica-Llanos, T., Darden, T. A., et al. (2008). NMDA receptor activation by HIV-Tat protein is clade dependent. J. Neurosci. 28, 12190–12198. doi: 10.1523/JNEUROSCI.3019-08.2008
Lindl, K. A., Marks, D. R., Kolson, D. L., and Jordan-Sciutto, K. L. (2010). HIV-associated neurocognitive disorder: pathogenesis and therapeutic opportunities. J. Neuroimmune Pharmacol. 5, 294–309. doi: 10.1007/s11481-010-9205-z
Little, S. J., McLean, A. R., Spina, C. A., Richman, D. D., and Havlir, D. V. (1999). Viral dynamics of acute HIV-1 infection. J. Exp. Med. 190, 841–850. doi: 10.1084/jem.190.6.841
Liu, Y., Jones, M., Hingtgen, C. M., Bu, G., Laribee, N., Tanzi, R. E., et al. (2000). Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat. Med. 6, 1380–1387. doi: 10.1038/82199
Loftus, R. M., and Finlay, D. K. (2016). Immunometabolism: cellular metabolism turns immune regulator. J. Biol. Chem. 291, 1–10. doi: 10.1074/jbc.R115.693903
Loisel-Meyer, S., Swainson, L., Craveiro, M., Oburoglu, L., Mongellaz, C., Costa, C., et al. (2012). Glut1-mediated glucose transport regulates HIV infection. Proc. Natl. Acad. Sci. U.S.A. 109, 2549–2554. doi: 10.1073/pnas.1121427109
Lomeli, H., Mosbacher, J., Melcher, T., Hoger, T., Geiger, J. R., Kuner, T., et al. (1994). Control of kinetic properties of AMPA receptor channels by nuclear RNA editing. Science 266, 1709–1713. doi: 10.1126/science.7992055
Lu, S. C. (2013). Glutathione synthesis. Biochim. Biophys. Acta 1830, 3143–3153. doi: 10.1016/j.bbagen.2012.09.008
Malik, S., and Eugenin, E. A. (2016). Mechanisms of HIV neuropathogenesis: role of cellular communication systems. Curr. HIV Res. 14, 400–411. doi: 10.2174/1570162X14666160324124558
Malik, S., and Eugenin, E. A. (2019). Role of connexin and pannexin containing channels in HIV infection and NeuroAIDS. Neurosci. Lett. 695, 86–90. doi: 10.1016/j.neulet.2017.09.005
Mangus, L. M., Beck, S. E., Queen, S. E., Brill, S. A., Shirk, E. N., Metcalf Pate, K. A., et al. (2018). Lymphocyte-dominant encephalitis and meningitis in simian immunodeficiency virus-infected macaques receiving antiretroviral therapy. Am. J. Pathol. 188, 125–134. doi: 10.1016/j.ajpath.2017.08.035
Mastrantonio, R., D'Ezio, V., Colasanti, M., and Persichini, T. (2019). Nrf2-mediated system xc(-) activation in astroglial cells is involved in HIV-1 tat-induced neurotoxicity. Mol. Neurobiol. 56, 3796–3806. doi: 10.1007/s12035-018-1343-y
McCormick, D. A. (1989). GABA as an inhibitory neurotransmitter in human cerebral cortex. J. Neurophysiol. 62, 1018–1027. doi: 10.1152/jn.1989.62.5.1018
Melendez, R. I., Roman, C., Capo-Velez, C. M., and Lasalde-Dominicci, J. A. (2016). Decreased glial and synaptic glutamate uptake in the striatum of HIV-1 gp120 transgenic mice. J. Neurovirol. 22, 358–365. doi: 10.1007/s13365-015-0403-6
Micu, I., Jiang, Q., Coderre, E., Ridsdale, A., Zhang, L., Woulfe, J., et al. (2006). NMDA receptors mediate calcium accumulation in myelin during chemical ischaemia. Nature 439, 988–992. doi: 10.1038/nature04474
Minghetti, L., Visentin, S., Patrizio, M., Franchini, L., Ajmone-Cat, M. A., and Levi, G. (2004). Multiple actions of the human immunodeficiency virus type-1 Tat protein on microglial cell functions. Neurochem. Res. 29, 965–978. doi: 10.1023/B:NERE.0000021241.90133.89
Mohamed, M., Barker, P. B., Skolasky, R. L., and Sacktor, N. (2018). 7T brain MRS in HIV infection: correlation with cognitive impairment and performance on neuropsychological tests. AJNR Am. J. Neuroradiol. 39, 704–712. doi: 10.3174/ajnr.A5547
Moroni, F., Cozzi, A., Sili, M., and Mannaioni, G. (2012). Kynurenic acid: a metabolite with multiple actions and multiple targets in brain and periphery. J. Neural. Transm. 119, 133–139. doi: 10.1007/s00702-011-0763-x
Moses, A. V., Ibanez, C., Gaynor, R., Ghazal, P., and Nelson, J. A. (1994). Differential role of long terminal repeat control elements for the regulation of basal and Tat-mediated transcription of the human immunodeficiency virus in stimulated and unstimulated primary human macrophages. J. Virol. 68, 298–307 doi: 10.1128/JVI.68.1.298-307.1994
Mukerji, S. S., Misra, V., Lorenz, D. R., Uno, H., Morgello, S., Franklin, D., et al. (2018). Impact of antiretroviral regimens on cerebrospinal fluid viral escape in a prospective multicohort study of antiretroviral therapy-experienced human immunodeficiency virus-1-infected adults in the United States. Clin. Infect. Dis. 67, 1182–1190. doi: 10.1093/cid/ciy267
Nakanishi, N., Kang, Y. J., Tu, S., McKercher, S. R., Masliah, E., and Lipton, S. A. (2016). Differential effects of pharmacologic and genetic modulation of NMDA receptor activity on HIV/gp120-induced neuronal damage in an in vivo mouse model. J. Mol. Neurosci. 58, 59–65. doi: 10.1007/s12031-015-0651-1
Nath, A. (2002). Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J. Infect. Dis. 186(Suppl. 2), S193–198. doi: 10.1086/344528
Nath, A., Haughey, N. J., Jones, M., Anderson, C., Bell, J. E., and Geiger, J. D. (2000). Synergistic neurotoxicity by human immunodeficiency virus proteins Tat and gp120: protection by memantine. Ann. Neurol. 47, 186–1943. doi: 10.1002/1531-8249(200002)47:2<;186::AID-ANA8>;3.0.CO;2-3
Nath, A., Psooy, K., Martin, C., Knudsen, B., Magnuson, D. S., Haughey, N., et al. (1996). Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J. Virol. 70, 1475–1480 doi: 10.1128/JVI.70.3.1475-1480.1996
Nath, N., Wunderlich, C., Darr, F. W. 2nd, Douglas, D. K., and Dodd, R. Y. (1987). Immunoglobulin level in donor blood reactive for antibodies to human immunodeficiency virus. J. Clin. Microbiol. 25, 364–369 doi: 10.1128/JCM.25.2.364-369.1987
Nedelcovych, M. T., Kim, B. H., Zhu, X., Lovell, L. E., Manning, A. A., Kelschenbach, J., et al. (2019). Glutamine antagonist JHU083 normalizes aberrant glutamate production and cognitive deficits in the EcoHIV murine model of HIV-associated neurocognitive disorders. J. Neuroimmune Pharmacol. 14, 391–400. doi: 10.1007/s11481-019-09859-w
Nedelcovych, M. T., Tenora, L., Kim, B. H., Kelschenbach, J., Chao, W., Hadas, E., et al. (2017). N-(Pivaloyloxy)alkoxy-carbonyl prodrugs of the glutamine antagonist 6-Diazo-5-oxo-l-norleucine (DON) as a potential treatment for HIV associated neurocognitive disorders. J. Med. Chem. 60, 7186–7198. doi: 10.1021/acs.jmedchem.7b00966
Nedergaard, M., Takano, T., and Hansen, A. J. (2002). Beyond the role of glutamate as a neurotransmitter. Nat. Rev. Neurosci. 3, 748–755. doi: 10.1038/nrn916
Nicoli, F., Chachage, M., Clowes, P., Bauer, A., Kowour, D., Ensoli, B., et al. (2016). Association between different anti-Tat antibody isotypes and HIV disease progression: data from an African cohort. BMC Infect. Dis. 16:344. doi: 10.1186/s12879-016-1647-3
Niswender, C. M., and Conn, P. J. (2010). Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu. Rev. Pharmacol. Toxicol. 50, 295–322. doi: 10.1146/annurev.pharmtox.011008.145533
Niture, S. K., Khatri, R., and Jaiswal, A. K. (2014). Regulation of Nrf2-an update. Free Radic. Biol. Med. 66, 36–44. doi: 10.1016/j.freeradbiomed.2013.02.008
Niu, F., Yao, H., Zhang, W., Sutliff, R. L., and Buch, S. (2014). Tat 101-mediated enhancement of brain pericyte migration involves platelet-derived growth factor subunit B homodimer: implications for human immunodeficiency virus-associated neurocognitive disorders. J. Neurosci. 34, 11812–11825. doi: 10.1523/JNEUROSCI.1139-14.2014
Nowak, M. A., Lloyd, A. L., Vasquez, G. M., Wiltrout, T. A., Wahl, L. M., Bischofberger, N., et al. (1997). Viral dynamics of primary viremia and antiretroviral therapy in simian immunodeficiency virus infection. J. Virol. 71, 7518–7525 doi: 10.1128/JVI.71.10.7518-7525.1997
Ohagen, A., Devitt, A., Kunstman, K. J., Gorry, P. R., Rose, P. P., Korber, B., et al. (2003). Genetic and functional analysis of full-length human immunodeficiency virus type 1 env genes derived from brain and blood of patients with AIDS. J. Virol. 77, 12336–12345. doi: 10.1128/JVI.77.22.12336-12345.2003
Okafo, G., Prevedel, L., and Eugenin, E. (2017). Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci. Rep. 7:16660. doi: 10.1038/s41598-017-16600-1
Omeragic, A., Hoque, M. T., Choi, U. Y., and Bendayan, R. (2017). Peroxisome proliferator-activated receptor-gamma: potential molecular therapeutic target for HIV-1-associated brain inflammation. J. Neuroinflammation 14:183. doi: 10.1186/s12974-017-0957-8
Orellana, J. A., Saez, J. C., Bennett, M. V., Berman, J. W., Morgello, S., and Eugenin, E. A. (2014). HIV increases the release of dickkopf-1 protein from human astrocytes by a Cx43 hemichannel-dependent mechanism. J. Neurochem. 128, 752–763. doi: 10.1111/jnc.12492
Pace, M., and Frater, J. (2019). Curing HIV by 'kick and kill': from theory to practice? Expert Rev. Anti Infect. Ther. 17, 383–386. doi: 10.1080/14787210.2019.1617697
Pacheco, R., Gallart, T., Lluis, C., and Franco, R. (2007). Role of glutamate on T-cell mediated immunity. J. Neuroimmunol. 185, 9–19. doi: 10.1016/j.jneuroim.2007.01.003
Palmer, C. S., Ostrowski, M., Gouillou, M., Tsai, L., Yu, D., Zhou, J., et al. (2014). Increased glucose metabolic activity is associated with CD4+ T-cell activation and depletion during chronic HIV infection. AIDS 28, 297–309. doi: 10.1097/QAD.0000000000000128
Pardo, B., Rodrigues, T. B., Contreras, L., Garzon, M., Llorente-Folch, I., Kobayashi, K., et al. (2011). Brain glutamine synthesis requires neuronal-born aspartate as amino donor for glial glutamate formation. J. Cereb. Blood Flow Metab. 31, 90–101. doi: 10.1038/jcbfm.2010.146
Patel, C. A., Mukhtar, M., and Pomerantz, R. J. (2000). Human immunodeficiency virus type 1 Vpr induces apoptosis in human neuronal cells. J. Virol. 74, 9717–9726. doi: 10.1128/JVI.74.20.9717-9726.2000
Paul, R. (2019). Neurocognitive phenotyping of HIV in the era of antiretroviral therapy. Curr. HIV/AIDS Rep. 16, 230–235. doi: 10.1007/s11904-019-00426-9
Peavy, G., Jacobs, D., Salmon, D. P., Butters, N., Delis, D. C., Taylor, M., et al. (1994). Verbal memory performance of patients with human immunodeficiency virus infection: evidence of subcortical dysfunction. The HNRC Group. J. Clin. Exp. Neuropsychol. 16, 508–523. doi: 10.1080/01688639408402662
Peng, L., Hertz, L., Huang, R., Sonnewald, U., Petersen, S. B., Westergaard, N., et al. (1993). Utilization of glutamine and of TCA cycle constituents as precursors for transmitter glutamate and GABA. Dev. Neurosci. 15, 367–377. doi: 10.1159/000111357
Perelson, A. S., Essunger, P., Cao, Y., Vesanen, M., Hurley, A., Saksela, K., et al. (1997). Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 387, 188–191. doi: 10.1038/387188a0
Persidsky, Y., Hill, J., Zhang, M., Dykstra, H., Winfield, M., Reichenbach, N. L., et al. (2016). Dysfunction of brain pericytes in chronic neuroinflammation. J. Cereb. Blood Flow Metab. 36, 794–807. doi: 10.1177/0271678X15606149
Petralia, R. S. (2012). Distribution of extrasynaptic NMDA receptors on neurons. Sci. World J. 2012:267120. doi: 10.1100/2012/267120
Petroff, O. A. (2002). GABA and glutamate in the human brain. Neuroscientist 8, 562–573. doi: 10.1177/1073858402238515
Pilc, A., Chaki, S., Nowak, G., and Witkin, J. M. (2008). Mood disorders: regulation by metabotropic glutamate receptors. Biochem. Pharmacol. 75, 997–1006. doi: 10.1016/j.bcp.2007.09.021
Pilcher, C. D., Shugars, D. C., Fiscus, S. A., Miller, W. C., Menezes, P., Giner, J., et al. (2001). HIV in body fluids during primary HIV infection: implications for pathogenesis, treatment and public health. AIDS 15, 837–845. doi: 10.1097/00002030-200105040-00004
Pinkevych, M., Cromer, D., Tolstrup, M., Grimm, A. J., Cooper, D. A., Lewin, S. R., et al. (2015). HIV reactivation from latency after treatment interruption occurs on average every 5-8 days–implications for HIV remission. PLoS Pathog. 11:e1005000. doi: 10.1371/journal.ppat.1005000
Pinkevych, M., Cromer, D., Tolstrup, M., Grimm, A. J., Cooper, D. A., Lewin, S. R., et al. (2016). Correction: HIV reactivation from latency after treatment interruption occurs on average every 5-8 days-implications for HIV remission. PLoS Pathog. 12:e1005745. doi: 10.1371/journal.ppat.1005745
Popescu, G. K. (2014). Dynamic plasticity of NMDA receptor-mediated calcium entry in neurons exposed to HIV-tat. J. Neurochem. 130, 609–611. doi: 10.1111/jnc.12737
Porcheray, F., Leone, C., Samah, B., Rimaniol, A. C., Dereuddre-Bosquet, N., and Gras, G. (2006). Glutamate metabolism in HIV-infected macrophages: implications for the CNS. Am. J. Physiol. Cell Physiol. 291, C618–626. doi: 10.1152/ajpcell.00021.2006
Portilla, I., Reus, S., Leon, R., van-der Hofstadt, C., Sanchez, J., Lopez, N., et al. (2019). Neurocognitive impairment in well-controlled HIV-infected patients: a cross-sectional study. AIDS Res. Hum. Retroviruses 35, 634–641. doi: 10.1089/aid.2018.0279
Potter, M. C., Figuera-Losada, M., Rojas, C., and Slusher, B. S. (2013). Targeting the glutamatergic system for the treatment of HIV-associated neurocognitive disorders. J. Neuroimmune Pharmacol. 8, 594–607. doi: 10.1007/s11481-013-9442-z
Potula, R., Ramirez, S. H., Knipe, B., Leibhart, J., Schall, K., Heilman, D., et al. (2008). Peroxisome proliferator-activated receptor-gamma activation suppresses HIV-1 replication in an animal model of encephalitis. AIDS 22, 1539–1549. doi: 10.1097/QAD.0b013e3283081e08
Prendergast, M. A., Rogers, D. T., Mulholland, P. J., Littleton, J. M., Wilkins, L. H. Jr., Self, R. L., et al. (2002). Neurotoxic effects of the human immunodeficiency virus type-1 transcription factor Tat require function of a polyamine sensitive-site on the N-methyl-D-aspartate receptor. Brain Res. 954, 300–307. doi: 10.1016/S0006-8993(02)03360-7
Rao, J. S. (2003). Molecular mechanisms of glioma invasiveness: the role of proteases. Nat. Rev. Cancer 3, 489–501. doi: 10.1038/nrc1121
Re, M. C., Furlini, G., Vignoli, M., Ramazzotti, E., Zauli, G., and La Placa, M. (1996). Antibody against human immunodeficiency virus type 1 (HIV-1) Tat protein may have influenced the progression of AIDS in HIV-1-infected hemophiliac patients. Clin. Diagn. Lab. Immunol. 3, 230–232 doi: 10.1128/CDLI.3.2.230-232.1996
Rieder, M. J., Krause, R., Bird, I. A., and Dekaban, G. A. (1995). Toxicity of sulfonamide-reactive metabolites in HIV-infected, HTLV-infected, and noninfected cells. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 8, 134–140 doi: 10.1097/00042560-199502000-00004
Rimaniol, A. C., Mialocq, P., Clayette, P., Dormont, D., and Gras, G. (2001). Role of glutamate transporters in the regulation of glutathione levels in human macrophages. Am. J. Physiol. Cell Physiol. 281, C1964–1970. doi: 10.1152/ajpcell.2001.281.6.C1964
Ritola, K., Robertson, K., Fiscus, S. A., Hall, C., and Swanstrom, R. (2005). Increased human immunodeficiency virus type 1 (HIV-1) env compartmentalization in the presence of HIV-1-associated dementia. J. Virol. 79, 10830–10834. doi: 10.1128/JVI.79.16.10830-10834.2005
Rodriguez-Gallego, E., Gomez, J., Pacheco, Y. M., Peraire, J., Vilades, C., Beltran-Debon, R., et al. (2018). A baseline metabolomic signature is associated with immunological CD4+ T-cell recovery after 36 months of antiretroviral therapy in HIV-infected patients. AIDS 32, 565–573. doi: 10.1097/QAD.0000000000001730
Rothstein, J. D., Dykes-Hoberg, M., Pardo, C. A., Bristol, L. A., Jin, L., Kuncl, R. W., et al. (1996). Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 16, 675–686. doi: 10.1016/S0896-6273(00)80086-0
Rozzi, S. J., Avdoshina, V., Fields, J. A., Trejo, M., Ton, H. T., Ahern, G. P., et al. (2017). Human immunodeficiency virus promotes mitochondrial toxicity. Neurotox. Res. 32, 723–733. doi: 10.1007/s12640-017-9776-z
Ru, W., and Tang, S. J. (2016). HIV-1 gp120Bal down-regulates phosphorylated NMDA receptor subunit 1 in cortical neurons via activation of glutamate and chemokine receptors. J. Neuroimmune Pharmacol. 11, 182–191. doi: 10.1007/s11481-015-9644-7
Ru, W., and Tang, S. J. (2017). HIV-associated synaptic degeneration. Mol. Brain 10:40. doi: 10.1186/s13041-017-0321-z
Rumbaugh, J. A., Bachani, M., Li, W., Butler, T. R., Smith, K. J., Bianchet, M. A., et al. (2013). HIV immune complexes prevent excitotoxicity by interaction with NMDA receptors. Neurobiol. Dis. 49, 169–176. doi: 10.1016/j.nbd.2012.08.013
Russell, R. A., Chojnacki, J., Jones, D. M., Johnson, E., Do, T., Eggeling, C., et al. (2017). Astrocytes resist HIV-1 fusion but engulf infected macrophage material. Cell Rep. 18, 1473–1483. doi: 10.1016/j.celrep.2017.01.027
Sailasuta, N., Shriner, K., and Ross, B. (2009). Evidence of reduced glutamate in the frontal lobe of HIV-seropositive patients. NMR Biomed. 22, 326–331. doi: 10.1002/nbm.1329
Salter, M. G., and Fern, R. (2005). NMDA receptors are expressed in developing oligodendrocyte processes and mediate injury. Nature 438, 1167–1171. doi: 10.1038/nature04301
Sami Saribas, A., Cicalese, S., Ahooyi, T. M., Khalili, K., Amini, S., and Sariyer, I. K. (2017). HIV-1 Nef is released in extracellular vesicles derived from astrocytes: evidence for Nef-mediated neurotoxicity. Cell Death Dis. 8:e2542. doi: 10.1038/cddis.2016.467
Sanford, R., Ances, B. M., Meyerhoff, D. J., Price, R. W., Fuchs, D., Zetterberg, H., et al. (2018). Longitudinal trajectories of brain volume and cortical thickness in treated and untreated primary human immunodeficiency virus infection. Clin. Infect. Dis. 67, 1697–1704. doi: 10.1093/cid/ciy362
Santello, M., Cali, C., and Bezzi, P. (2012). Gliotransmission and the tripartite synapse. Adv. Exp. Med. Biol. 970, 307–331. doi: 10.1007/978-3-7091-0932-8_14
Schousboe, A., Bak, L. K., and Waagepetersen, H. S. (2013). Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front. Endocrinol. 4:102. doi: 10.3389/fendo.2013.00102
Scott, D. B., Blanpied, T. A., and Ehlers, M. D. (2003). Coordinated PKA and PKC phosphorylation suppresses RXR-mediated ER retention and regulates the surface delivery of NMDA receptors. Neuropharmacology 45, 755–767. doi: 10.1016/S0028-3908(03)00250-8
Sengillo, J. D., Winkler, E. A., Walker, C. T., Sullivan, J. S., Johnson, M., and Zlokovic, B. V. (2013). Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer's disease. Brain Pathol. 23, 303–310. doi: 10.1111/bpa.12004
Shan, L., and Siliciano, R. F. (2013). From reactivation of latent HIV-1 to elimination of the latent reservoir: the presence of multiple barriers to viral eradication. Bioessays 35, 544–552. doi: 10.1002/bies.201200170
Siliciano, J. D., Kajdas, J., Finzi, D., Quinn, T. C., Chadwick, K., Margolick, J. B., et al. (2003). Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 9, 727–728. doi: 10.1038/nm880
Singh, H. O., Lata, S., Angadi, M., Bapat, S., Pawar, J., Nema, V., et al. (2017). Impact of GSTM1, GSTT1 and GSTP1 gene polymorphism and risk of ARV-associated hepatotoxicity in HIV-infected individuals and its modulation. Pharmacogenomics J. 17, 53–60. doi: 10.1038/tpj.2015.88
Skowronska, K., Obara-Michlewska, M., Zielinska, M., and Albrecht, J. (2019). NMDA receptors in astrocytes: in search for roles in neurotransmission and astrocytic homeostasis. Int. J. Mol. Sci. 20:309. doi: 10.3390/ijms20020309
Smit, T. K., Wang, B., Ng, T., Osborne, R., Brew, B., and Saksena, N. K. (2001). Varied tropism of HIV-1 isolates derived from different regions of adult brain cortex discriminate between patients with and without AIDS dementia complex (ADC): evidence for neurotropic HIV variants. Virology 279, 509–526. doi: 10.1006/viro.2000.0681
Soriano-Sarabia, N., Bateson, R. E., Dahl, N. P., Crooks, A. M., Kuruc, J. D., Margolis, D. M., et al. (2014). Quantitation of replication-competent HIV-1 in populations of resting CD4+ T cells. J. Virol. 88, 14070–14077. doi: 10.1128/JVI.01900-14
Swingler, S., Mann, A. M., Zhou, J., Swingler, C., and Stevenson, M. (2007). Apoptotic killing of HIV-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 3, 1281–1290. doi: 10.1371/journal.ppat.0030134
Swinton, M. K., Carson, A., Telese, F., Sanchez, A. B., Soontornniyomkij, B., Rad, L., et al. (2019). Mitochondrial biogenesis is altered in HIV+ brains exposed to ART: implications for therapeutic targeting of astroglia. Neurobiol. Dis. 130:104502. doi: 10.1016/j.nbd.2019.104502
Tate, D. F., Conley, J., Paul, R. H., Coop, K., Zhang, S., Zhou, W., et al. (2010). Quantitative diffusion tensor imaging tractography metrics are associated with cognitive performance among HIV-infected patients. Brain Imaging Behav. 4, 68–79. doi: 10.1007/s11682-009-9086-z
Taube, R., Fujinaga, K., Wimmer, J., Barboric, M., and Peterlin, B. M. (1999). Tat transactivation: a model for the regulation of eukaryotic transcriptional elongation. Virology 264, 245–253. doi: 10.1006/viro.1999.9944
Thomas, A. G., O'Driscoll, C. M., Bressler, J., Kaufmann, W., Rojas, C. J., and Slusher, B. S. (2014). Small molecule glutaminase inhibitors block glutamate release from stimulated microglia. Biochem. Biophys. Res. Commun. 443, 32–36. doi: 10.1016/j.bbrc.2013.11.043
Tian, C., Sun, L., Jia, B., Ma, K., Curthoys, N., Ding, J., et al. (2012). Mitochondrial glutaminase release contributes to glutamate-mediated neurotoxicity during human immunodeficiency virus-1 infection. J. Neuroimmune Pharmacol. 7, 619–628. doi: 10.1007/s11481-012-9364-1
Traynelis, S. F., Wollmuth, L. P., McBain, C. J., Menniti, F. S., Vance, K. M., Ogden, K. K., et al. (2010). Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496. doi: 10.1124/pr.109.002451
Turchan-Cholewo, J., Dimayuga, V. M., Gupta, S., Gorospe, R. M., Keller, J. N., and Bruce-Keller, A. J. (2009). NADPH oxidase drives cytokine and neurotoxin release from microglia and macrophages in response to HIV-Tat. Antioxid. Redox Signal 11, 193–204. doi: 10.1089/ars.2008.2097
Underwood, J., Robertson, K. R., and Winston, A. (2015). Could antiretroviral neurotoxicity play a role in the pathogenesis of cognitive impairment in treated HIV disease? AIDS 29, 253–261. doi: 10.1097/QAD.0000000000000538
Valcour, V., Chalermchai, T., Sailasuta, N., Marovich, M., Lerdlum, S., Suttichom, D., et al. (2012). Central nervous system viral invasion and inflammation during acute HIV infection. J. Infect. Dis. 206, 275–282. doi: 10.1093/infdis/jis326
Valdebenito, S., Lou, E., Baldoni, J., Okafo, G., and Eugenin, E. (2018). The novel roles of connexin channels and tunneling nanotubes in cancer pathogenesis. Int. J. Mol. Sci. 19:1270. doi: 10.3390/ijms19051270
Van Lint, C., Bouchat, S., and Marcello, A. (2013). HIV-1 transcription and latency: an update. Retrovirology 10:67. doi: 10.1186/1742-4690-10-67
Vartak-Sharma, N., Gelman, B. B., Joshi, C., Borgamann, K., and Ghorpade, A. (2014). Astrocyte elevated gene-1 is a novel modulator of HIV-1-associated neuroinflammation via regulation of nuclear factor-kappaB signaling and excitatory amino acid transporter-2 repression. J. Biol. Chem. 289, 19599–19612. doi: 10.1074/jbc.M114.567644
Vats, D., Mukundan, L., Odegaard, J. I., Zhang, L., Smith, K. L., Morel, C. R., et al. (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. doi: 10.1016/j.cmet.2006.05.011
Vazquez-Santiago, F. J., Noel, R. J. Jr., Porter, J. T., and Rivera-Amill, V. (2014). Glutamate metabolism and HIV-associated neurocognitive disorders. J. Neurovirol. 20, 315–331. doi: 10.1007/s13365-014-0258-2
Veenstra, M., Byrd, D. A., Inglese, M., Buyukturkoglu, K., Williams, D. W., Fleysher, L., et al. (2019). CCR2 on peripheral blood CD14(+)CD16(+) monocytes correlates with neuronal damage, hiv-associated neurocognitive disorders, and peripheral HIV DNA: reseeding of CNS reservoirs? J. Neuroimmune Pharmacol. 14, 120–133. doi: 10.1007/s11481-018-9792-7
Veenstra, M., Leon-Rivera, R., Li, M., Gama, L., Clements, J. E., and Berman, J. W. (2017). Mechanisms of CNS viral seeding by HIV(+) CD14(+) CD16(+) monocytes: establishment and reseeding of viral reservoirs contributing to HIV-associated neurocognitive disorders. MBio 8:01280-17. doi: 10.1128/mBio.01280-17
Velasquez, S., Prevedel, L., Valdebenito, S., Gorska, A. M., Golovko, M., Khan, N., et al. (2020). Circulating levels of ATP is a biomarker of HIV cognitive impairment. EBiomedicine 51:102503. doi: 10.1016/j.ebiom.2019.10.029
Visalli, V., Muscoli, C., Sacco, I., Sculco, F., Palma, E., Costa, N., et al. (2007). N-acetylcysteine prevents HIV gp 120-related damage of human cultured astrocytes: correlation with glutamine synthase dysfunction. BMC Neurosci. 8:106. doi: 10.1186/1471-2202-8-106
Vivithanaporn, P., Asahchop, E. L., Acharjee, S., Baker, G. B., and Power, C. (2016). HIV protease inhibitors disrupt astrocytic glutamate transporter function and neurobehavioral performance. AIDS 30, 543–552. doi: 10.1097/QAD.0000000000000955
Wallet, C., De Rovere, M., Van Assche, J., Daouad, F., De Wit, S., Gautier, V., et al. (2019). Microglial cells: the main HIV-1 reservoir in the brain. Front. Cell. Infect. Microbiol. 9:362. doi: 10.3389/fcimb.2019.00362
Wanchu, A., Rana, S. V., Pallikkuth, S., and Sachdeva, R. K. (2009). Short communication: oxidative stress in HIV-infected individuals: a cross-sectional study. AIDS Res. Hum. Retroviruses 25, 1307–1311. doi: 10.1089/aid.2009.0062
Wang, T. H., Donaldson, Y. K., Brettle, R. P., Bell, J. E., and Simmonds, P. (2001). Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J. Virol. 75, 11686–11699. doi: 10.1128/JVI.75.23.11686-11699.2001
Wang, Y., Li, Y., Zhao, R., Wu, B., Lanoha, B., Tong, Z., et al. (2017). Glutaminase C overexpression in the brain induces learning deficits, synaptic dysfunctions, and neuroinflammation in mice. Brain Behav. Immun. 66, 135–145. doi: 10.1016/j.bbi.2017.06.007
Wang, Z., Pekarskaya, O., Bencheikh, M., Chao, W., Gelbard, H. A., Ghorpade, A., et al. (2003). Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology 312, 60–73. doi: 10.1016/S0042-6822(03)00181-8
Wei, X., Ghosh, S. K., Taylor, M. E., Johnson, V. A., Emini, E. A., Deutsch, P., et al. (1995). Viral dynamics in human immunodeficiency virus type 1 infection. Nature 373, 117–122. doi: 10.1038/373117a0
Wen, L., You, W., Wang, H., Meng, Y., Feng, J., and Yang, X. (2018). Polarization of microglia to the M2 phenotype in a peroxisome proliferator-activated receptor gamma-dependent manner attenuates axonal injury induced by traumatic brain injury in Mice. J. Neurotrauma 35, 2330–2340. doi: 10.1089/neu.2017.5540
Westendorp, M. O., Shatrov, V. A., Schulze-Osthoff, K., Frank, R., Kraft, M., Los, M., et al. (1995). HIV-1 Tat potentiates TNF-induced NF-kappa B activation and cytotoxicity by altering the cellular redox state. EMBO J. 14, 546–554 doi: 10.1002/j.1460-2075.1995.tb07030.x
Westmoreland, S. V., Alvarez, X., deBakker, C., Aye, P., Wilson, M. L., Williams, K. C., et al. (2002). Developmental expression patterns of CCR5 and CXCR4 in the rhesus macaque brain. J. Neuroimmunol. 122, 146–158. doi: 10.1016/S0165-5728(01)00457-X
Wieronska, J. M., and Pilc, A. (2009). Metabotropic glutamate receptors in the tripartite synapse as a target for new psychotropic drugs. Neurochem. Int. 55, 85–97. doi: 10.1016/j.neuint.2009.02.019
Williams, D. W., Byrd, D., Rubin, L. H., Anastos, K., Morgello, S., and Berman, J. W. (2014). CCR2 on CD14(+)CD16(+) monocytes is a biomarker of HIV-associated neurocognitive disorders. Neurol. Neuroimmunol. Neuroinflamm. 1:e36. doi: 10.1212/NXI.0000000000000036
Williams, D. W., Calderon, T. M., Lopez, L., Carvallo-Torres, L., Gaskill, P. J., Eugenin, E. A., et al. (2013). Mechanisms of HIV entry into the CNS: increased sensitivity of HIV infected CD14+CD16+ monocytes to CCL2 and key roles of CCR2, JAM-A, and ALCAM in diapedesis. PLoS ONE 8:e69270. doi: 10.1371/journal.pone.0069270
Williams, D. W., Eugenin, E. A., Calderon, T. M., and Berman, J. W. (2012). Monocyte maturation, HIV susceptibility, and transmigration across the blood brain barrier are critical in HIV neuropathogenesis. J. Leukoc. Biol 91, 401–415. doi: 10.1189/jlb.0811394
Winkler, E. A., Sengillo, J. D., Sullivan, J. S., Henkel, J. S., Appel, S. H., and Zlokovic, B. V. (2013). Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol. 125, 111–120. doi: 10.1007/s00401-012-1039-8
Wong, J. K., and Yukl, S. A. (2016). Tissue reservoirs of HIV. Curr. Opin. HIV AIDS 11, 362–370. doi: 10.1097/COH.0000000000000293
Wroge, C. M., Hogins, J., Eisenman, L., and Mennerick, S. (2012). Synaptic NMDA receptors mediate hypoxic excitotoxic death. J. Neurosci. 32, 6732–6742. doi: 10.1523/JNEUROSCI.6371-11.2012
Wu, B., Huang, Y., Braun, A. L., Tong, Z., Zhao, R., Li, Y., et al. (2015). Glutaminase-containing microvesicles from HIV-1-infected macrophages and immune-activated microglia induce neurotoxicity. Mol. Neurodegener. 10:61. doi: 10.1186/s13024-015-0058-z
Xiao, H., Neuveut, C., Tiffany, H. L., Benkirane, M., Rich, E. A., Murphy, P. M., et al. (2000). Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc. Natl. Acad. Sci. U.S.A. 97, 11466–11471. doi: 10.1073/pnas.97.21.11466
Xing, H. Q., Hayakawa, H., Gelpi, E., Kubota, R., Budka, H., and Izumo, S. (2009). Reduced expression of excitatory amino acid transporter 2 and diffuse microglial activation in the cerebral cortex in AIDS cases with or without HIV encephalitis. J. Neuropathol. Exp. Neurol. 68, 199–209. doi: 10.1097/NEN.0b013e31819715df
Xing, L., Yang, T., Cui, S., and Chen, G. (2019). Connexin hemichannels in astrocytes: role in CNS disorders. Front. Mol. Neurosci. 12:23. doi: 10.3389/fnmol.2019.00023
Xu, H., Bae, M., Tovar-y-Romo, L. B., Patel, N., Bandaru, V. V., Pomerantz, D., et al. (2011). The human immunodeficiency virus coat protein gp120 promotes forward trafficking and surface clustering of NMDA receptors in membrane microdomains. J. Neurosci. 31, 17074–17090. doi: 10.1523/JNEUROSCI.4072-11.2011
Yang, J., Hu, D., Xia, J., Liu, J., Zhang, G., Gendelman, H. E., et al. (2013). Enhancement of NMDA receptor-mediated excitatory postsynaptic currents by gp120-treated macrophages: implications for HIV-1-associated neuropathology. J. Neuroimmune Pharmacol. 8, 921–933. doi: 10.1007/s11481-013-9468-2
Yoshimura, K. (2017). Current status of HIV/AIDS in the ART era. J. Infect. Chemother. 23, 12–16. doi: 10.1016/j.jiac.2016.10.002
Young, A. C., Yiannoutsos, C. T., Hegde, M., Lee, E., Peterson, J., Walter, R., et al. (2014). Cerebral metabolite changes prior to and after antiretroviral therapy in primary HIV infection. Neurology 83, 1592–1600. doi: 10.1212/WNL.0000000000000932
Yukl, S. A., Shergill, A. K., Ho, T., Killian, M., Girling, V., Epling, L., et al. (2013). The distribution of HIV DNA and RNA in cell subsets differs in gut and blood of HIV-positive patients on ART: implications for viral persistence. J. Infect. Dis. 208, 1212–1220. doi: 10.1093/infdis/jit308
Zarate, S., Pond, S. L., Shapshak, P., and Frost, S. D. (2007). Comparative study of methods for detecting sequence compartmentalization in human immunodeficiency virus type 1. J. Virol. 81, 6643–6651. doi: 10.1128/JVI.02268-06
Zhang, M., An, C., Gao, Y., Leak, R. K., Chen, J., and Zhang, F. (2013). Emerging roles of Nrf2 and phase II antioxidant enzymes in neuroprotection. Prog. Neurobiol. 100, 30–47. doi: 10.1016/j.pneurobio.2012.09.003
Zhao, J., Lopez, A. L., Erichsen, D., Herek, S., Cotter, R. L., Curthoys, N. P., et al. (2004). Mitochondrial glutaminase enhances extracellular glutamate production in HIV-1-infected macrophages: linkage to HIV-1 associated dementia. J. Neurochem. 88, 169–180. doi: 10.1046/j.1471-4159.2003.02146.x
Zhou, Y., and Danbolt, N. C. (2014). Glutamate as a neurotransmitter in the healthy brain. J Neural Transm. 121, 799–817. doi: 10.1007/s00702-014-1180-8
Zhou, Y., Liu, J., and Xiong, H. (2017). HIV-1 Glycoprotein 120 enhancement of N-methyl-D-aspartate NMDA receptor-mediated excitatory postsynaptic currents: implications for HIV-1-associated neural injury. J. Neuroimmune Pharmacol. 12, 314–326. doi: 10.1007/s11481-016-9719-0
Zoratti, M., Szabo, I., and De Marchi, U. (2005). Mitochondrial permeability transitions: how many doors to the house? Biochim. Biophys. Acta 1706, 40–52. doi: 10.1016/j.bbabio.2004.10.006
Zou, S., Fuss, B., Fitting, S., Hahn, Y. K., Hauser, K. F., and Knapp, P. E. (2015). Oligodendrocytes are targets of HIV-1 Tat: NMDA and AMPA receptor-mediated effects on survival and development. J. Neurosci. 35, 11384–11398. doi: 10.1523/JNEUROSCI.4740-14.2015
Keywords: reservoirs, cure, NeuroHIV, dementia, glutamate, glutamine, HIV
Citation: Gorska AM and Eugenin EA (2020) The Glutamate System as a Crucial Regulator of CNS Toxicity and Survival of HIV Reservoirs. Front. Cell. Infect. Microbiol. 10:261. doi: 10.3389/fcimb.2020.00261
Received: 30 December 2019; Accepted: 04 May 2020;
Published: 24 June 2020.
Edited by:
Tetsuo Tsukamoto, Kindai University, JapanReviewed by:
Maria Teresa Sanchez-Aparicio, Icahn School of Medicine at Mount Sinai, United StatesCopyright © 2020 Gorska and Eugenin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eliseo A. Eugenin, ZWxldWdlbmlAdXRtYi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.