 Lucia Gonzales-Siles1,2,3*
Lucia Gonzales-Siles1,2,3* Roger Karlsson1,2,3
Roger Karlsson1,2,3 Patrik Schmidt1
Patrik Schmidt1 Francisco Salvà-Serra1,2,3,4,5
Francisco Salvà-Serra1,2,3,4,5 Daniel Jaén-Luchoro1,3Susann Skovbjerg1,2,3
Daniel Jaén-Luchoro1,3Susann Skovbjerg1,2,3 Edward R. B. Moore1,2,3,4*
Edward R. B. Moore1,2,3,4* Margarita Gomila5
Margarita Gomila5- 1Department of Infectious Diseases, Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
- 2Department of Clinical Microbiology, Sahlgrenska University Hospital, Gothenburg, Sweden
- 3Centre for Antibiotic Resistance Research (CARe), University of Gothenburg, Gothenburg, Sweden
- 4Culture Collection University of Gothenburg (CCUG), Sahlgrenska Academy, University of Gothenburg, Gothenburg, Sweden
- 5Department of Biology-Microbiology, Universitat de les Illes Balears, Palma, Spain
Correct identifications of isolates and strains of the Mitis-Group of the genus Streptococcus are particularly difficult, due to high genetic similarity, resulting from horizontal gene transfer and homologous recombination, and unreliable phenotypic and genotypic biomarkers for differentiating the species. Streptococcus pneumoniae and Streptococcus pseudopneumoniae are the most closely related species of the clade. In this study, publicly-available genome sequences for Streptococcus pneumoniae and S. pseudopneumoniae were analyzed, using a pangenomic approach, to find candidates for species-unique gene markers; ten species-unique genes for S. pneumoniae and nine for S. pseudopneumoniae were identified. These species-unique gene marker candidates were verified by PCR assays for identifying S. pneumoniae and S. pseudopneumoniae strains isolated from clinical samples. All determined species-level unique gene markers for S. pneumoniae were detected in all S. pneumoniae clinical isolates, whereas fewer of the unique S. pseudopneumoniae gene markers were present in more than 95% of the clinical isolates. In parallel, taxonomic identifications of the clinical isolates were confirmed, using conventional optochin sensitivity testing, targeted PCR-detection for the “Xisco” gene, as well as genomic ANIb similarity analyses for the genome sequences of selected strains. Using mass spectrometry-proteomics, species-specific peptide matches were observed for four of the S. pneumoniae gene markers and for three of the S. pseudopneumoniae gene markers. Application of multiple species-level unique biomarkers of S. pneumoniae and S. pseudopneumoniae, is proposed as a protocol for the routine clinical laboratory for improved, reliable differentiation, and identification of these pathogenic and commensal species.
Introduction
Streptococcus pneumoniae, also known as “pneumococcus,” is an important human pathogen, causing severe infections, such as meningitis, pneumonia, and bacteremia and responsible for considerable mortality worldwide (O'Brien et al., 2009). Among the pneumococci, a subset of atypical strains have been recognized that are optochin-resistant, bile insoluble, and lacking capsule, an essential virulence factor for disease (Sá-Leão et al., 2006; Rolo et al., 2013). Arbique et al. (2004) described some of these atypical pneumococci as a new species, Streptococcus pseudopneumoniae, which is characterized by being bile insoluble and optochin-resistant, when incubated in 5% CO2 atmosphere, but optochin–susceptible when incubated under ambient atmosphere.
Streptococcus pseudopneumoniae is a recognized member of the Viridans-Group streptococci (VGS) and is closely related, phylogenetically and taxonomically to S. pneumoniae and S. mitis, sharing nearly 100% 16S rRNA gene sequence similarity (Kilian et al., 2008). The high genotypic similarities between these species are associated with high rates of horizontal gene transfer and homologous recombination between the pathogenic S. pneumoniae and commensal Viridans-Group streptococci (Whatmore et al., 2000; Kilian et al., 2008). Because of the close relationships between these species, definitive identifications in the clinical laboratory can be problematic. Phenotypic biochemical and metabolic testing are often insufficient to distinguish S. pneumoniae, including atypical S. pneumoniae, from S. pseudopneumoniae or other closely related streptococci. Molecular-based techniques, such as Multi-Locus Sequence Analysis (MLSA) or Multi-Locus Sequence Typing (MLST), which use sequence analyses of multiple genes or alleles, enabling high resolution differentiation between species, have been shown to be reliable for differentiating S. pneumoniae from its closest related species, S. pseudopneumoniae (Bishop et al., 2009; Simões et al., 2010). In contrast, individual house-keeping gene PCR- and sequence-based assays have proven to be less reliable for differentiating the other closely related species of the Mitis-Group streptococci from each other (Gonzales-Siles et al., 2019). Several PCR-based methods, mostly targeting genes for specific pneumococcal virulence factors (lytA, ply, cpsA, pspA), specific intergenic DNA sequences, and specific regions of the 16S rRNA gene, have been proposed to be pneumococcal species-specific (El Aila et al., 2010; Varghese et al., 2017). Recently, a unique molecular marker, SPS0002, was described (Croxen et al., 2018). However, different studies have shown that these gene markers can be detected also in closely related species, and therefore, are not always reliable for S. pneumoniae species-level identifications (Whatmore et al., 2000; Kilian et al., 2008; Johnston et al., 2010; Simões et al., 2010; Rolo et al., 2013).
The availability of whole genome sequence data for a continuously-expanding number of strains of an extensive range of bacterial species makes it increasingly feasible to take advantage of comparative genomic analysis as a tool to recognize the global complexity of bacterial species and for identifying unique biomarkers for taxonomic, as well as functional characterization (Rouli et al., 2015). Pangenome analysis is a comparative whole-genome sequence-based method that enables construction of a framework for assessing the genomic diversity of entire repertoires of genes, as well as identifying core genomic elements. The pangenome includes the “core-genome,” which contains the genes present in all individuals of a lineage or taxon or other defined group, an “accessory or dispensable genome,” formed by the genes not present in all genomes (Medini et al., 2005; Tettelin et al., 2008; Vernikos et al., 2015). Additionally, the pangenome can be divided into four sub-sets, depending on the frequencies with which the genes appear in the different genomes considered: “core,” as defined above; “soft-core,” defined as genes present in at least 95% of the genomes; “shell,” defined as genes which are moderately common; and “cloud,” which represent the genes that are shared by a small fraction of the genomes studied (Koonin and Wolf, 2008; Kaas et al., 2012).
The aim of this study was to identify unique genomic markers, using pangenome analyses, for reliable differentiation and identification of both S. pneumoniae and S. pseudopneumoniae. For purposes of comparisons, strains of S. mitis, which is the next most-closely related species of the Mitis-Group of the genus Streptococcus, were also included in this study, to increase the specificities of the gene markers for S. pneumoniae and S. pseudopneumoniae. The species-dependent presence of these unique markers was confirmed by PCR of reference strains and clinical isolates and validated, using genomic and proteomic approaches.
Materials and Methods
Genome Selection
All complete, closed genome sequences for Streptococcus pneumoniae (n = 32) and all genome sequences available in GenBank (September 19, 2017) for S. pseudopneumoniae (n = 36, including one closed genome) and S. mitis (n = 65, including five closed genomes) were downloaded and included in the analysis for generating the species pangenomes (Supplementary Table 1). The taxonomic affiliations of each of the 134 bacterial genomes were assessed by calculating pair-wise genomic Average Nucleotide Identity (ANI) similarities between all genome sequences, based on the BLAST algorithm (ANIb) (Goris et al., 2007), using the JspeciesWS online service (Richter et al., 2016). A similarity matrix was generated and used to create a dendrogram of genomic relatedness, using the PermutMatrix software (Caraux and Pinloche, 2005), applying hierarchical clustering, the average linkage algorithm (UPGMA) and Pearson's distance correlation (Gomila et al., 2015). The dendrogram was visualized on-line with the Interactive Tree Of Life (iTOL) (Letunic and Bork, 2016). The workflow from genome selection to the identification of unique gene markers is summarized in Figure 1.
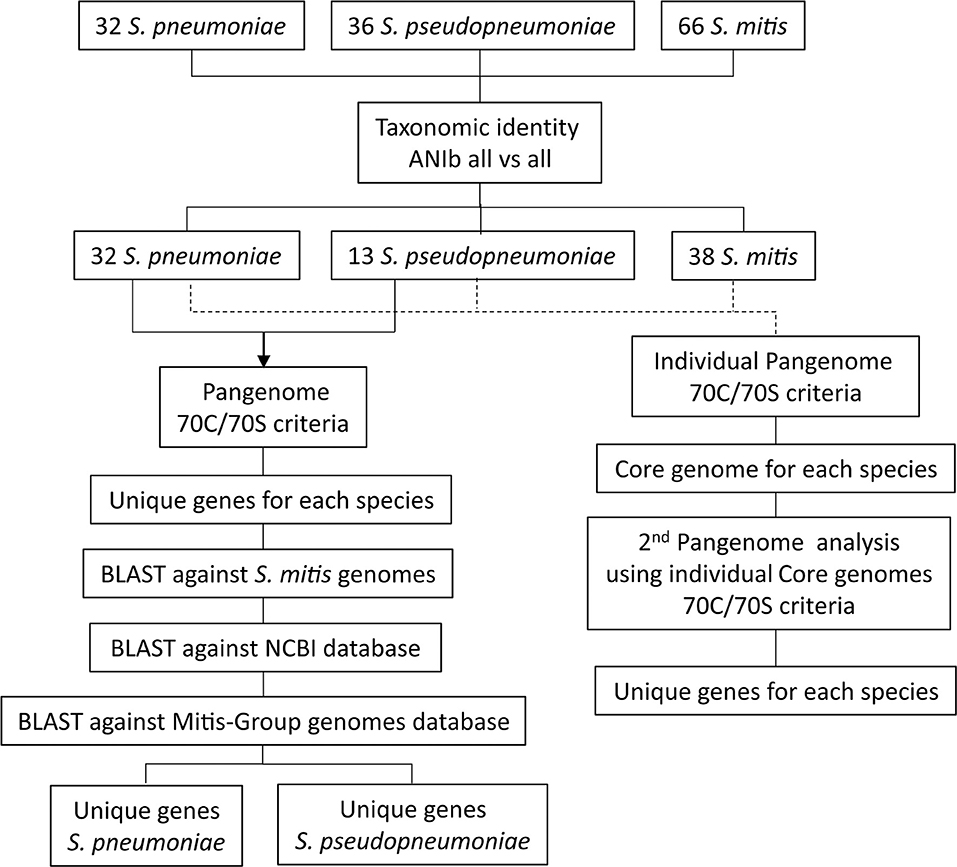
Figure 1. Schematic representation of the workflow used for identifications of unique genes for S. pneumoniae and S. pseudopneumoniae by applying a pangenome approach.
Core- and Pangenome Analyses
After confirmation of genome taxonomic classifications, species core-genomes of S. pneumoniae (n = 32), S. pseudopneumoniae (n = 13), and S. mitis (n = 38) were determined. All genomes correctly classified were annotated, with the software Prokka, version 1.11 (Seemann, 2014). For each genome, the file containing all protein sequences was used for comparison against the rest of the strains by BLASTP (Altschul et al., 1990). Protein sequences were clustered into homologous groups, using the Get_Homologues software (Contreras-Moreira and Vinuesa, 2013). Two proteins were considered homologous if they fulfilled the “70C/70S” (i.e., 70% Contiguously aligned/70% sequence Similarity) criteria, referring to, at least, 70% similarity in, at least, 70% of the sequence (Tettelin et al., 2005). Using the core genomes of each of the different species, second core and pangenome analyses were calculated, applying the 70C/70S criteria, to identify the unique genes for each of the species. A Venn diagram, based on these results, was generated, using the web tool: http://bioinformatics.psb.ugent.be/webtools/Venn/.
Finally, another pangenome analysis, including only the S. pneumoniae and S. pseudopneumoniae genome sequences (n = 45), was performed, using the Get_Homologues software, employing the 70C/70S criteria. Based on this analysis, a list of unique genes for each species, i.e., present in one species and absent in the other one, was generated. In order to confirm the specificities of these genes, a search of the unique genes listed for each species was performed, using BLASTN version 2.6.0+, firstly, against all S. mitis genomes and, secondly, against the NCBI database for prokaryotes. Additionally, the genes shown to be unique for S. pneumoniae and S. pseudopneumoniae, were analyzed again with BLASTN against an internal database including 328 S. pneumoniae genome sequences, including the S. pneumoniae type strain, and 248 genome sequences assigned to 12 other species of the Mitis-Group of the genus Streptococcus, including the type strains of S. australis, S. cristatus, S. gordonii, S. infantis, S. massiliensis, S. mitis, S. oralis, S. parasanguinis, S. peroris, S. pseudopneumoniae, S. sanguinis, and S. sinensis. The nucleotide sequences of the genes that were confirmed to be unique for a given species were aligned, using Bionumerics 7.5 (Applied Maths, Sint-Martens-Latem, Belgium), to calculate the pair-wise sequence similarities. The nomenclature used to name each unique gene included the species and locus tag provided during annotation with Prokka.
Confirmation of Gene Markers
For validation of the suggested unique genes as species biomarkers, 29 strains of S. pseudopneumoniae, isolated from clinical samples, characterized, and archived at the Culture Collection University of Gothenburg (CCUG), Gothenburg, Sweden, were analyzed by genotypic- and phenotypic-based methods. The strains were screened by PCR for the presence of the unique gene markers, determined from the pangenome analysis. In parallel, the strains were tested by traditional clinical microbiology methods for identification of S. pseudopneumoniae, including optochin-sensitivity testing and absence of virulence genes. Whole-genome sequence analyses were performed for 14 of the 29 S. pseudopneumoniae strains.
Streptococcus pneumoniae unique genes were confirmed, using 20 strains, isolated from clinical samples, archived in the CCUG, for which genome sequences had been determined. The presence or absence of six of the S. pneumoniae unique genes was confirmed by PCR.
The presence or absence of the “Xisco” gene, which has been reported to be specific for S. pneumoniae strains (Salvà-Serra et al., 2018), was determined, by PCR, for all S. pneumoniae and S. pseudopneumoniae strains.
BLASTN analysis for each of the unique gene markers for S. pneumoniae and S. pseudopneumoniae was performed against 29 whole-genome sequences of S. pseudopneumoniae and 42 complete genome sequences of S. pneumoniae available in GenBank, November 22, 2019, that were not included in the pangenome analysis (Supplementary Table 2). Additionally, the genome sequences of 20 S. pneumoniae (Salvà-Serra et al., 2018) and 14 S. pseudopneumoniae clinical strains (Supplementary Table 3) were also included in the analysis. Previous confirmation of the taxonomic affiliations of the genomes was performed, as described above.
DNA Extraction, Whole-Genome Sequencing, and Assembly
Fourteen S. pseudopneumoniae strains that were negative for, at least, one of the proposed gene markers or that produced ambiguous results for the optochin-sensitivity testing were selected for whole genome sequence determination and analysis. Genomic DNA (gDNA) was isolated from bacterial strains of pure-culture, fresh biomass, using a Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA). The obtained DNA samples were purified, using the DNA Clean & ConcentratorTM-100 kit (Zymo Research, Irvine, CA, USA). Isolated and purified gDNA was sequenced, using the Illumina MiSeq platform (Eurofins Genomics, Konstanz, Germany). Sequences were trimmed, using Sickle version 1.33 (Joshi and Fass, 2011), with a Phred quality score threshold of Q30. Paired-end reads were assembled, using SPAdes, version 3.11.1 (Bankevich et al., 2012). Taxonomic affiliations were confirmed by ANIb (Richter and Rosselló-Móra, 2009), using JSpeciesWS (Richter et al., 2016), against the type strains S. pneumoniae NCTC 7465T and S. pseudopneumoniae CCUG 49455T. Deposits of sequences were made to GenBank at NCBI under the accession numbers listed in Supplementary Table 3.
PCR-Amplification
Specific PCR-amplification primers were designed for six unique S. pseudopneumoniae and six S. pneumoniae gene markers that had a gene size longer than 500 bp (Table 1). Amplification-primers were tested on strains of the respective species. A detail description of the PCR conditions is presented in the Supplementary Information. Strain CCUG 49455T, the type strain of S. pseudopneumoniae, was used as positive control for S. pseudopneumoniae and negative control for S. pneumoniae, whereas strain CCUG 28588T, the type strain of S. pneumoniae, was used as positive control for S. pneumoniae and negative control for S. pseudopneumoniae. The type strains of the closest phylogenetically-related species in the Mitis-Group of the genus Streptococcus (S. mitis CCUG 31611T and S. oralis CCUG 13229T) were also used as negative controls. The presence of the S. pneumoniae virulence genes, cpsA, lytA, and ply, which have been suggested to be present in other species of the Mitis-Group streptococci, were analyzed by PCR in all S. pseudopneumoniae strains, according to Nagai et al. (2001), using the primers listed in Table 1.
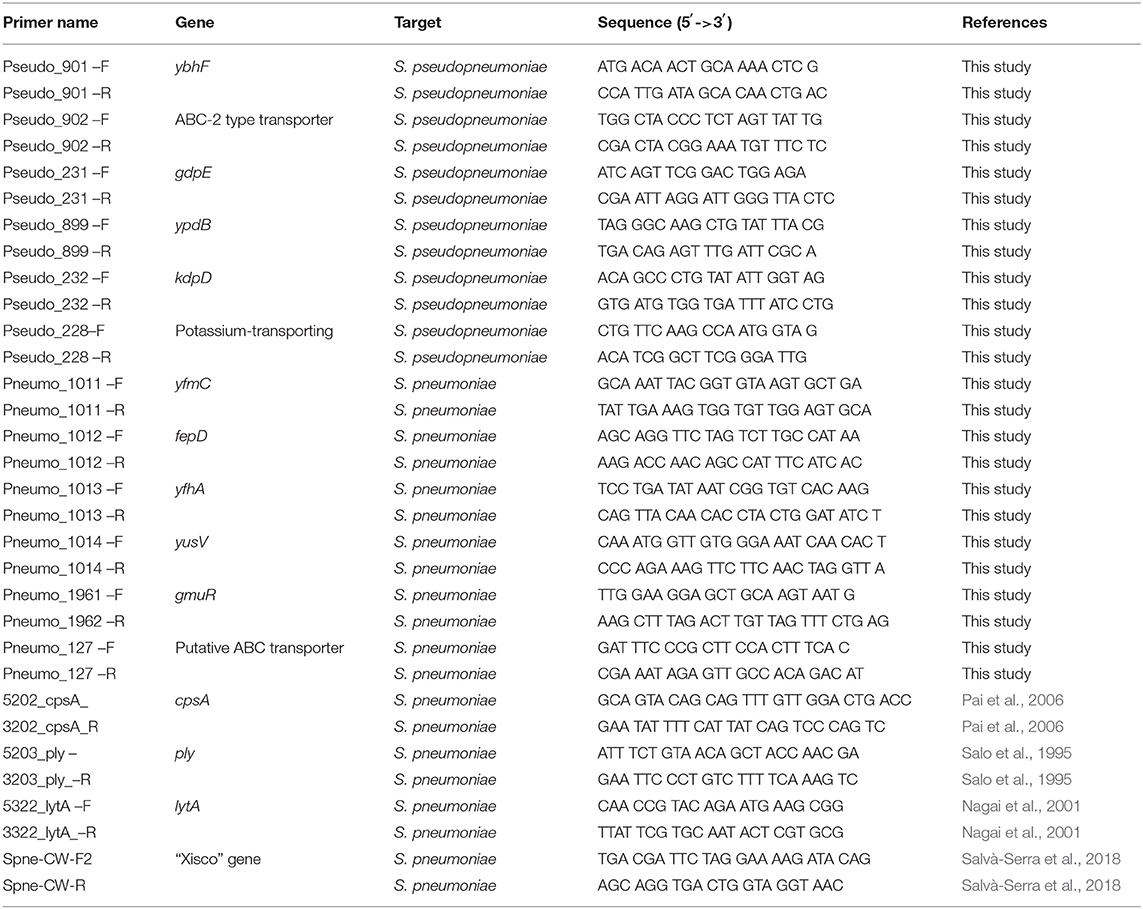
Table 1. List of PCR-primers used for amplification of unique gene markers of S. pneumoniae and S. pseudopneumoniae.
Optochin Testing
Optochin testing was performed, according to Arbique et al. (2004). The description of the method is presented in the Supplementary Information. S. pneumoniae CCUG 28588T and S. pseudopneumoniae CCUG 49455T were used as controls.
Proteotyping Analysis Using Bottom-Up Tandem Mass Spectrometry (LC-MS/MS)
Verification of expression of the selected candidate gene markers was performed, by Liquid Chromatography tandem Mass Spectrometry (LC-MS/MS) proteomics. The protein sequences for all unique genes determined for S. pseudopneumoniae and S. pneumoniae were extracted from the genomes included in the study. Three strains of S. pneumoniae (CCUG 28588T, CCUG 7206, and CCUG 35180) and three strains of S. pseudopneumoniae (CCUG 49455T, CCUG 62647, CCUG 63747) had been analyzed by proteotyping (Karlsson et al., 2015), in a previous study (Karlsson et al., 2018), for identifying species-unique peptide markers. A BLASTP search was done, using the protein sequences of the candidate gene marker products against the species-unique peptides found in that previous study.
Generation of Peptide Inclusion Lists for LC-MS/MS Analysis
Inclusion lists of species-unique peptides were generated for the three most prevalent, by PCR, unique genes of S. pseudopneumoniae and for the four most prevalent unique genes of S. pneumoniae. The amino acid sequences of the selected genes were digested, in-silico, with the enzyme trypsin, using the Peptide Cutter tool at Expasy website (https://web.expasy.org/peptide_cutter/). Peptides ranging from 6 to 25 amino acids in length were selected as these were of suitable length for detection by MS; the MS platform used was the Q Exactive (Thermo Fisher). Inclusion lists were generated, using the MacCoss Lab Software Skyline (https://skyline.ms/project/home/begin.view), and by selecting +2 and +3 as the possible charge states, followed by exporting an isolation list.
The S. pneumoniae (CCUG 28588T, CCUG 7206, and CCUG 35180) and S. pseudopneumoniae (CCUG 49455T, CCUG 62647, CCUG 63747) strains were cultivated and analyzed, according to Karlsson et al. (2018) for LC-MS/MS proteotyping. Inclusion lists containing the candidate peptide biomarkers were used during the MS analysis; by employing the inclusion lists, the m/z ratios corresponding to these peptide biomarkers were selected for fragmentation even if they were not among the most abundant peptides. Tandem MS data was evaluated, using TCUP (Typing and Characterization Using Proteomics), according to Boulund et al. (2017). In this study, the evaluation was performed at the species level and, thus, sets of species-unique peptides were identified from each strain analysis.
Results
Phylogenetic Assignation of Genomes
Taxonomic identifications of the 133 genomes included in the study, by ANIb similarity analyses, confirmed that all 32 S. pneumoniae genomes were correctly classified and were, therefore, included in the study. However, only 13 of the 36 S. pseudopneumoniae genomes in GenBank were confirmed, by ANIb similarity analyses, as S. pseudopneumoniae; 15 of the S. “pseudopneumoniae” genomes were identified as S. mitis and the remaining eight were identified as S. oralis. Among the 65 S. mitis genomes listed in GenBank, three genomes were identified as S. oralis, whereas the remaining 63 were confirmed to be S. mitis. After genome sequence-based taxonomic designations of all the selected genomes, 78 genomes were classified as S. mitis (Supplementary Table 1). In order to reduce the number of S. mitis genomes for the analysis and to attain approximate equality in the number of genomes analyzed for each species, two criteria were implemented. Firstly, genomes were selected, requiring a baseline of 40 or less contigs and, secondly, the selected genomes should cover the representative clades for S. mitis depicted in the dendrogram, based on ANIb analysis (Supplementary Figure 1). Based on these criteria, 38 strains of S. mitis were included in the analyses (Supplementary Table 1).
Pangenome Analysis
The pangenome analysis for each of the species showed that S. pneumoniae has the highest percentage of genes forming the core genome (35.0%), which are the genes shared by all genomes, compared to S. pseudopneumoniae (31.1%) and to S. mitis (19.5%). On the other hand, S. mitis was observed to have the highest percentage of genes forming the cloud group (55.4%), compared to S. pneumoniae (33.0%) and S. pseudopneumoniae (43.2%). The fact that S. mitis has the lowest relative number of core genes, but more than 50% of cloud genes, suggests that S. mitis is, genomically, a relatively heterogeneous species. In Table 2, the pangenome distribution of the genes for each species is indicated.
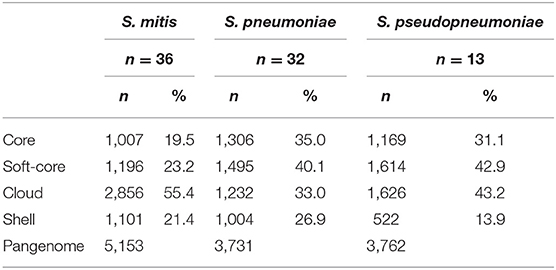
Table 2. Pangenome distribution of the genes for each species, using 70C/70S (70% Contiguously aligned/70% sequence Similarity) criteria.
The second core genome analysis, including only the genes belonging to species core genomes, obtained by individual analysis for each of the species, showed the number of unique genes for S. pneumoniae (n = 179) and S. pseudopneumoniae (n = 188) and a markedly lower number of unique genes for S. mitis (n = 52; Figure 2). These results are concordant with the high genomic intra-species variation among S. mitis genomes shown in previous studies (Kilian et al., 2008, 2014; Jensen et al., 2016).
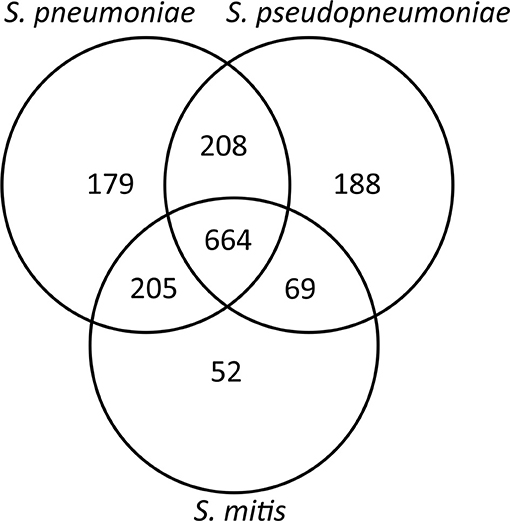
Figure 2. Venn diagram showing the number of shared and unique genes between S. pneumoniae. S. pseudopneumoniae and S. mitis.
Due to the low number of unique genes for S. mitis showed in the pangenome analysis, a second pangenome analysis was done, including only S. pneumoniae and S. pseudopneumoniae genome sequences. This analysis showed that 16.7% of the genes were present in the core and 46.4% were present in the cloud (Supplementary Table 4). Although S. pneumoniae and S. pseudopneumoniae are phylogenetically closely related, a high percentage of genes was observed to be strain-unique and only a small percentage of genes was present in all genomes of a species (Supplementary Table 4).
Based on the pangenome analyses, 94 genes of S. pseudopneumoniae and 77 genes of S. pneumoniae were observed to be species-unique genes, i.e., genes that are present in all the genomes of one species and absent in all genomes of other species. BLASTN analyses of these proposed unique genes, against the sequences of S. mitis genomes, demonstrated that 32 of the 94 genes of S. pseudopneumoniae and 39 of the 77 genes of S. pneumoniae were not present among any S. mitis genomes and were considered potential specific biomarkers for S. pseudopneumoniae and for S. pneumoniae. A second BLASTN analysis of these candidate genes against the NCBI prokaryote database revealed only 13 of the 32 genes were unique for S. pseudopneumoniae and 20 of the 39 genes were unique for S. pneumoniae (Figure 3). The discarded genes matched to other Streptococcus species and the Streptococcus phage IPP62. Furthermore, BLASTN analyses were performed against our internal database. The 13 S. pseudopneumoniae unique genes were analyzed, with respect to 567 non-S. pseudopneumoniae streptococcal genome sequences, whereas the 20 S. pneumoniae unique genes were analyzed with respect to 248 non-S. pneumoniae streptococcal genome sequences. From these analyses, nine genes were observed to be unique for S. pseudopneumoniae and ten genes unique for S. pneumoniae (Table 3) and could be considered potential gene biomarkers for these species. In general, the unique genes for both S. pseudopneumoniae and S. pneumoniae were observed to be highly conserved, as shown in Table 3.
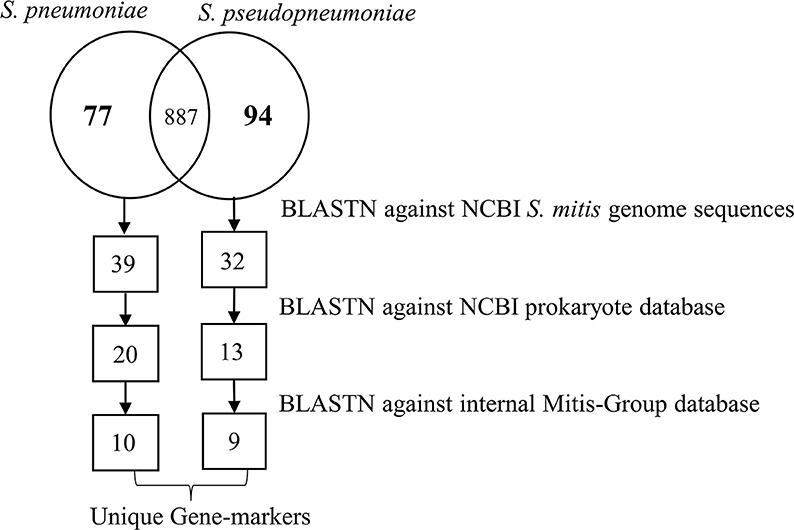
Figure 3. Schematic representation of the number of unique genes found at each step of the analysis for identifications of specific unique biomarkers for S. pneumoniae and S. pseudopneumoniae.
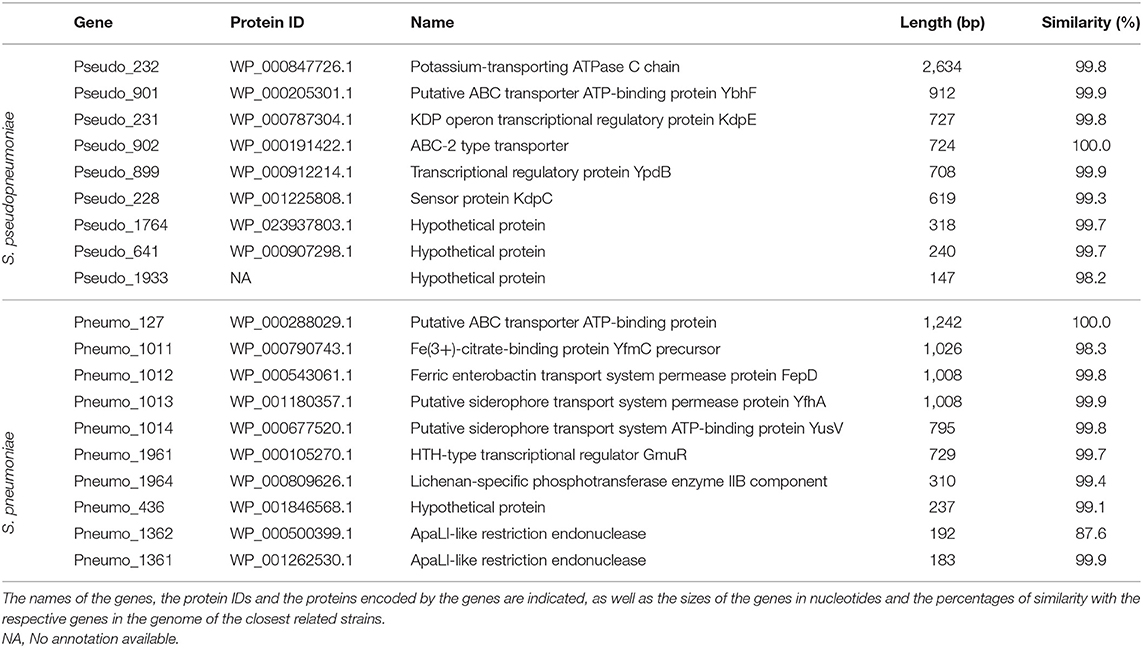
Table 3. List of unique genes of S. pseudopneumoniae and S. pneumoniae.
Confirmation of Gene Markers
PCR-amplification primers targeting the six S. pneumoniae unique genes longer than 500 bp (Table 1) were designed and tested on 20 strains isolated from clinical samples and classified as S. pneumoniae at the CCUG; all strains tested were positive for all six of the genes. Identifications of the strains were confirmed by gDNA ANIb similarities of each of the genomes, with respect to the genome sequence of S. pneumoniae type strain (CCUG 28588T). ANIb values were observed to be higher than 98.0% in all cases. In addition, all strains were positive for the presence of the “Xisco” gene (Table 4).
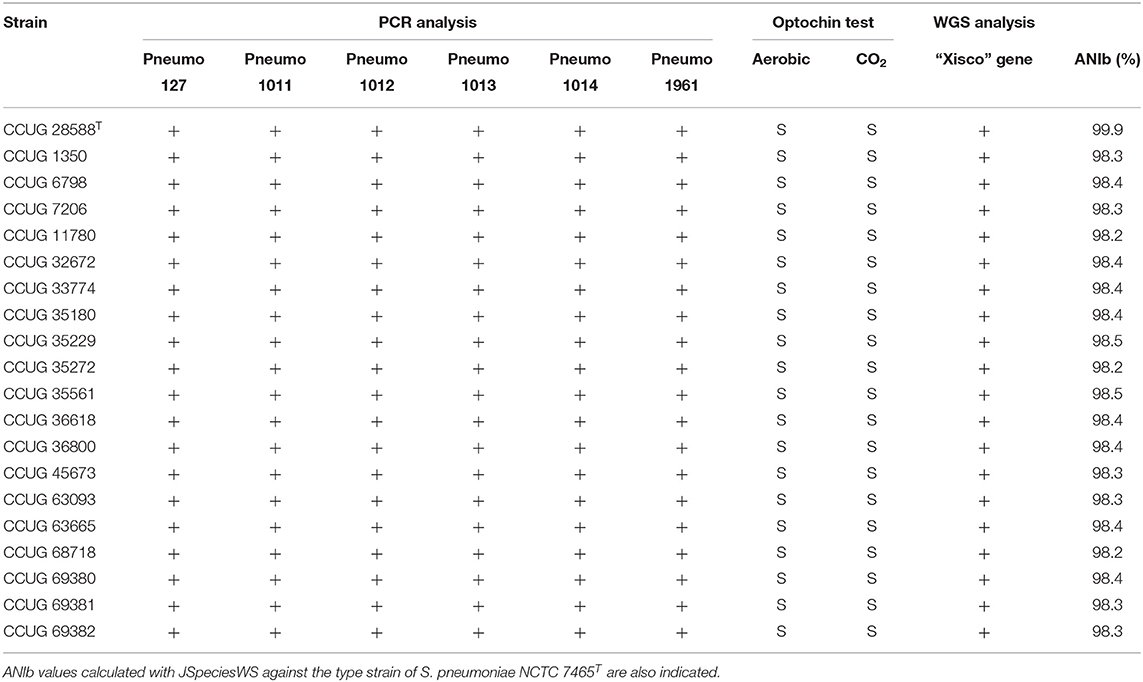
Table 4. Genotypic and phenotypic characterization of S. pneumoniae strains used for confirmation of gene markers.
Twenty-nine S. pseudopneumoniae CCUG strains isolated from clinical samples were analyzed by PCR for six S. pneumoniae unique genes longer than 500 bp (Table 1). All strains of S. pseudopneumoniae tested were positive for PCR-amplification of the Pseudo_899 gene, whereas 28 strains (97%) were positive for Pseudo_902 and Pseudo_228 genes, 26 strains (90%) were positive for Pseudo_231, 21 strains (73%) were positive for Pseudo_901, and 17 strains (60 %) were positive for Pseudo_232 genes (Supplementary Figure 2). Sixteen of the strains (57%) were positive for PCR-amplification of all six markers analyzed, whereas five strains (17%) were positive for five and four markers, respectively, two strains (7%) were positive for three markers, and one strain was positive for two markers (3%) (Table 5).
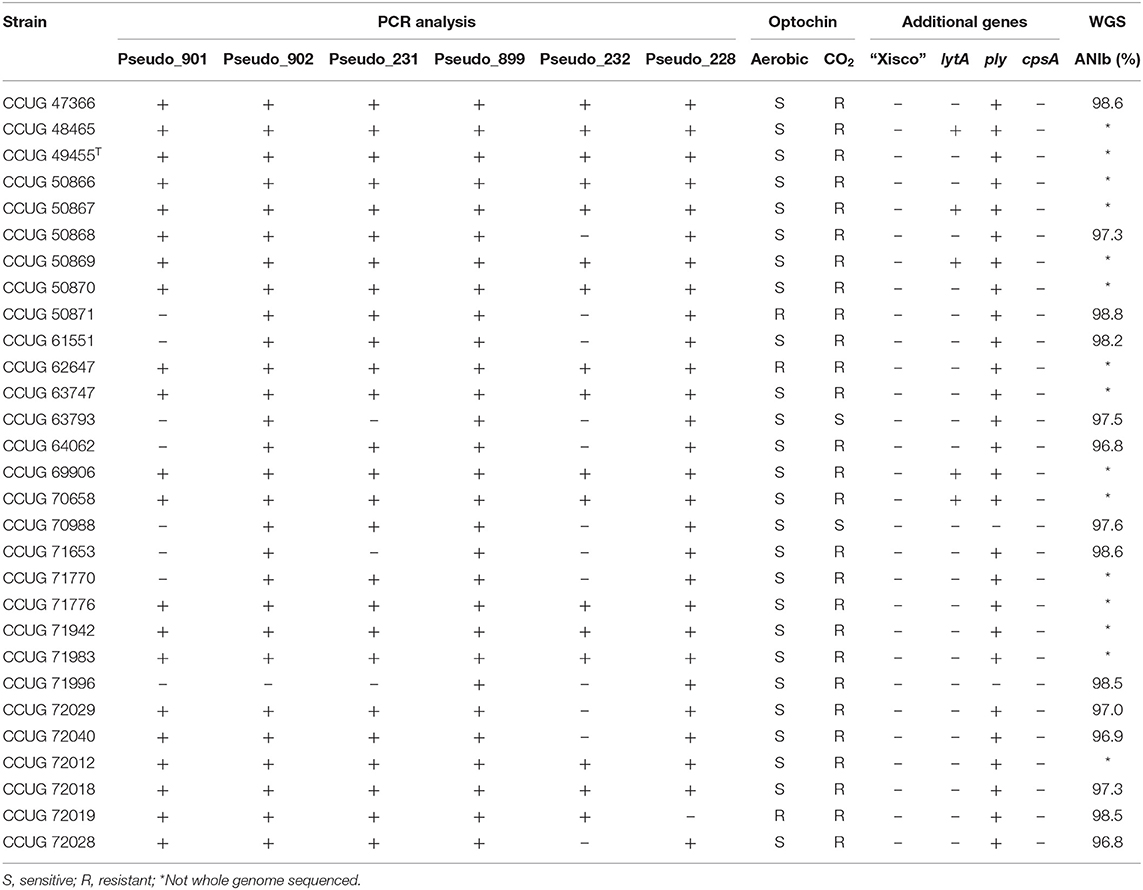
Table 5. Genotypic and phenotypic characterization of the 29 S. pseudopneumoniae strains used for confirmation of gene markers; ANIb similarity values calculated with JSpeciesWS against S. pseudopneumoniae type strain CCUG 49455T are indicated if the whole genome sequence was determined.
The optochin-sensitivity testing for S. pseudopneumoniae strains observed 26/29 strains exhibiting optochin-resistance when cultivated with CO2 and optochin-sensitivity when cultivated in aerobic conditions. One strain was resistant and two were sensitive with both cultivation conditions (Table 5).
PCR assays for three of the most prevalent virulence genes found in S. pneumoniae strains, namely the genes cpsA, lytA, and the ply were performed on the S. pseudopneumoniae strains. None of the S. pseudopneumoniae strains tested carried the cpsA gene, whereas 5/29 (17%) samples were positive for lytA and 26/29 (93%) samples were positive for the ply gene. In only five strains both the lytA and ply genes were present. In contrast, the presence of the “Xisco” gene, which has been shown to be unique for S. pneumoniae, was absent in all S. pseudopneumoniae strains.
Fourteen strains of S. pseudopneumoniae that were negative for, at least, one of the proposed gene markers or produced ambiguous results for the optochin-sensitivity testing were selected for whole genome sequencing analysis. Genome sequencing yielded paired-end reads of 150 bp, distributed in a total output ranging from 1.7 to 2.7 Gb per strain. Draft genome sequences of total lengths between 2.1 and 2.2 Mb, distributed in a number of scaffolds ranging from 55 to 95, were obtained. N50 values ranged from 48 to 91 kb. The GC content of the genome sequences was between 39.7 and 40.0% (Supplementary Table 3). Genome-based identifications of the strains was done by ANIb, with respect to the genome sequence of S. pseudopneumoniae type strain (CCUG 49455T). ANIb values were observed to be higher than 96%, confirming that all 14 strains tested were S. pseudopneumoniae in all cases (Table 5).
The proposed gene markers were analyzed using BLASTN against additional genome sequences of S. pneumoniae and S. pseudopneumoniae available in GenBank databases in November 2019, but that had not been included in the pangenome analysis (S. pneumoniae, n = 42; S. pseudopneumoniae, n = 29), as well as the genome sequences of 20 S. pneumoniae and 14 S. pseudopneumoniae clinical strains. The analysis revealed that Pneumo_1011, Pneumo_1012, Pneumo_1013, and Pneumo_1014 were present in two S. pseudopneumoniae genome sequences, whereas Pneumo_1961 and Pneumo_1964 were present in one S. pseudopneumoniae genome sequence (Supplementary Table 5). On the other hand, S. pseudopneumoniae gene markers were present in all S. pseudopneumoniae genome sequences and absent in all S. pneumoniae genome sequences analyzed (Supplementary Table 5).
Identification of Peptide Biomarker Candidates
Expression of the proposed unique gene markers for S. pneumoniae and S. pseudopneumoniae was assessed by comparing the protein sequence for each of the unique markers to the list of species-specific peptides, for both species, that were generated by shotgun LC-MS/MS analysis (Karlsson et al., 2018). Among the species unique peptides for S. pneumoniae, four peptide matches were observed for the gene marker, Pneumo_1011, and one peptide match was found for each of the gene markers, Pneumo_1014, Pneumo_1361, Pneumo_1961, and Pneumo_1964. For S. pseudopneumoniae, two peptide matches were found for the gene marker, Pseudo_228, and one peptide match was found for Pseudo_232 and Pseudo_1933 (Supplementary Table 6). Identification of peptides of the candidate gene markers confirms their expression.
The peptide inclusion lists for S. pneumoniae targeted four of the unique gene markers that had a peptide match in the initial analysis. When employing these inclusion lists during LC-MS/MS analysis of three S. pneumoniae strains (CCUG 7206, CCUG 35180, and CCUG 28588T), 14 peptide matches were found for Pneumo_1011, seven for Pneumo_1014, four for Pneumo_1964 and one match for Pneumo_1961. Six of the matches from Pneumo_1011, three from Pneumo_1014, and one from Pneumo_1964 and Pneumo_1961 were present in all three analyzed strains (Table 6).
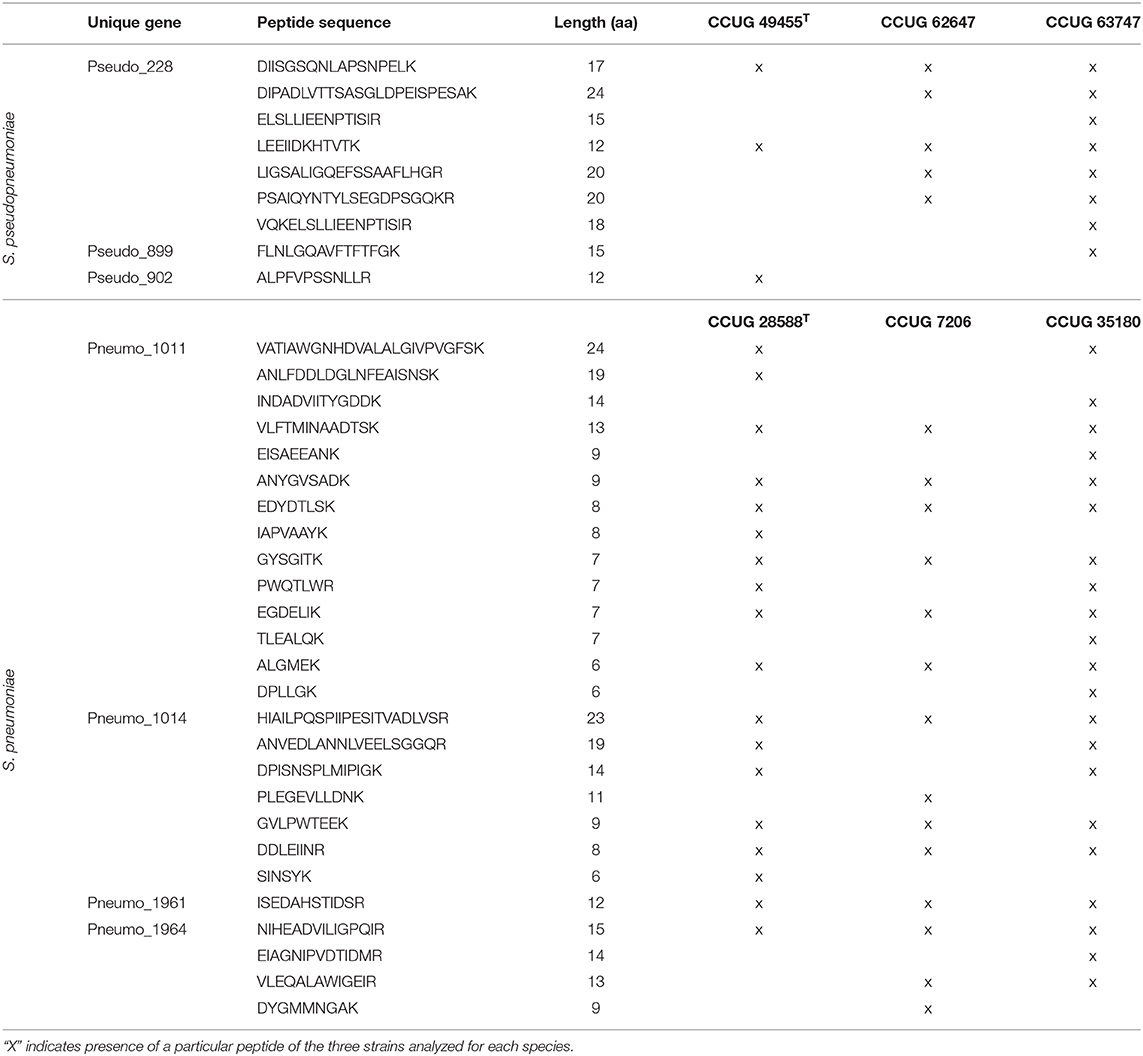
Table 6. List of the peptides detected from the unique genes of S. pneumoniae and S. pseudopneumoniae when analyzed using inclusion lists and targeted proteomics.
In-silico peptide inclusion lists were generated for the three most prevalent S. pseudopneumoniae gene markers. Species-specific peptide matches for the strains CCUG 62647, CCUG 63747, and the type strain CCUG 49455T were obtained by LC-MS/MS targeted proteomics and matched against the peptide inclusion list. Seven peptide matches were detected for Pseudo_228, from which two were present in the three analyzed strains (Table 6). Only one peptide match was detected for Pseudo_899 and Pseudo_902 and, in both cases, the matches were found in only one strain (Table 6).
Discussion
Correct identifications and differentiation of the commensal S. pseudopneumoniae and S. mitis from the bacterial pathogen, S. pneumoniae, have been difficult due to conflicting results obtained with different types of analyses combined with the homogeneity and overlapping phenotypic traits of these species, which has led to frequent misidentifications of these species. Whole genome DNA sequence-based methods, such as core genome analysis, and in silico genomic calculations, such as ANIb similarities, are not always applicable to all laboratories. Importantly, in the present context, the traditional threshold of ANIb similarity values for species delineations does not apply in certain species of commensal Streptococcus species, thus other methodological approaches are needed (Jensen et al., 2016; Gonzales-Siles et al., 2019). In this study, we used the high-resolution capacity of genomics to identify specific molecular markers capable of reliably differentiating and identifying S. pneumoniae and S. pseudopneumoniae.
As a first criterion, only closed genomes sequences of S. pneumoniae were included for the analysis, given the high number of genome sequences available in the databases. Using closed genomes increases the quality of the analysis since the complete sets of genes have been determined for each genome. However, the number of genome sequences available for the commensal S. pseudopneumoniae and S. mitis species is more limited; therefore, all available genomes sequences, including draft genomes, as well as complete genomes, were included. Since a significant proportion of the genomes in the public databases were misclassified, only the 13 genomes that were confirmed to be S. pseudopneumoniae were included, whereas, for S. mitis, only genomes with <40 contigs and that represented all taxonomic clusters, based on ANIb analysis for the species, were included. The objective was to have similar numbers of genomes, as many as possible, for each species. Even though the correct taxonomic identifications for many of these genomes have been reported previously (Jensen et al., 2016; Gonzales-Siles et al., 2019), the correct taxonomic identities of these genomes have not been corrected in the public databases, to date. Thus, it was essential to confirm the taxonomic identity of each genome included in the study; this is a fundamental and necessary rule that should be followed in all studies relying upon genome sequence data from the public databases.
Pangenome analysis is used mainly to study the diversity and composition of the complete gene repertoire of a given species (Tettelin et al., 2008). In this study, individual pangenome analyses provided hints of the overall genomic variation within each of the species. S. pneumoniae and S. pseudopneumoniae possessed a similar percentage distribution of genes in each of the pangenome categories, in contrast to S. mitis, wherein the results correlated with the observed high genomic heterogeneity among S. mitis strains (Kilian et al., 2014; Jensen et al., 2016). The low percentage of genes belonging to the core (group of genes shared by all strains) and the high number of genes comprising the cloud (genes present in small fractions of strains) indicates high genetic intra-species variation among S. mitis strains. This intra-species variation is also corroborated by the low ANIb values of most of the S. mitis strains, with respect to the type strain of the species, as shown previously (Jensen et al., 2016; Gonzales-Siles et al., 2019). This feature makes finding species-unique genes difficult, given the probability that the size of the core genome will decrease when higher numbers of strains are analyzed. The low number of species-unique genes for S. mitis, compared to the two other species when the core genomes of the three strains were combined, clearly shows this intra-species variation.
The main benefit of using a pangenome approach for determination of unique gene markers is that the pangenome method compares the whole repertoire of genes in the genomes within a given species and between different species. An advantage of this method is that it is not based solely on the analysis of reference strains or previously well-characterized genes; instead, it considers any number of genome sequences, which currently are available in public databases. In addition, the 70C/70S criteria used for the pangenome analysis, in comparison with the traditional 50C/50S criteria used in most studies, increased the reliability and specificity for finding species-unique biomarkers, which is highly important when species that are difficult to differentiate are considered.
The validity of the identification of the 10 unique genes for S. pneumoniae found in this study is supported by a recent report based on the analysis of more than 7,500 genomes (Kilian and Tettelin, 2019). However, discrepancies in the results of identifications, using different techniques for isolates and strains of S. pneumoniae and S. pseudopneumoniae, have been observed when reference strains (from the CCUG) isolated from clinical samples were tested. All of the proposed species-unique markers for S. pneumoniae were present in all strains tested, in contrast to the S. pseudopneumoniae strains, in which the presence of the proposed species-unique biomarkers was variable between strains. The proposed S. pneumoniae biomarkers detected in the pangenome analysis were further analyzed in a larger set of genome sequences of our internal database, including 329 S. pneumoniae genome sequences, which increased the specificity of these biomarkers. Furthermore, since only limited numbers of S. pseudopneumoniae genomes are available, to date, and all of them were included in the pangenome analysis, the validation analysis was important for confirming the specificities of these markers. The observed variation in the presence of the proposed biomarkers among the different strains highlights the importance of using more than one biomarker for identifications of strains and isolates, particularly considering the high genetic exchange between strains of the Mitis-Group of the genus Streptococcus.
Among the species-unique genes found for S. pseudopneumoniae, three of the gene markers, Pseudo_228 (Potassium-transporting ATPase C), Pseudo_232 (kdpD), and Pseudo_231 (kdpE) are associated with the transport of potassium. The two-component system KdpD/KdpE governs K+ homeostasis by controlling synthesis of the high affinity K+ transporter KdpFABC. When sensing low environmental K+ concentrations, the dimeric kinase KdpD autophosphorylates in trans and transfers the phosphoryl-group to the response regulator KdpE, which subsequently activates kdpFABC transcription (Mörk-Mörkenstein et al., 2017). K+ is the major monovalent cation in bacteria and important for intracellular osmolarity, pH, cell turgor, enzyme activities, gene expression, and communication between cells (Epstein, 2003; Lee et al., 2010; Humphries et al., 2017). Why these genes are unique and conserved among S. pseudopneumoniae strains is not known but they may represent an advantage for the species to survive in the local environment of the human upper respiratory tract.
In contrast, four of the species-unique genes for S. pneumoniae gene markers, Pneumo_1011 (yfmC), Pneumo_1012 (fepD), Pneumo_1013 (yfhA), and Pneumo_1014 (yusV), are associated with the regulation of iron. Iron regulation is important for S. pneumoniae, as iron availability within the host environment can “make or break” a successful infection (Glanville et al., 2018). Compared to other bacterial species, S. pneumoniae possesses both high intracellular iron and peroxide levels, which is a perceived recipe for cell suicide (Weiser et al., 2001; Echlin et al., 2016). Thus, it is important for the bacteria to prevent its self-produced, extremely high levels of peroxide from reacting with intracellular iron. The way in which this regulation occurs is not well-known as S. pneumoniae lacks all typical redox-sensing factors known to alert the cell of such danger (Pericone et al., 2003). S. pneumoniae produces millimolar quantities of hydrogen peroxide in oxygen-rich environments as a metabolic by product of pyruvate oxidase, for regulating capsule formation, which is important for colonization of the upper respiratory tract (Echlin et al., 2016). Species-unique genes for both S. pneumoniae and S. pseudopneumoniae included ABC transporter proteins, that are vital for cell survival, since they function to counteract any undesirable change occurring in the cell; they could also be involved in the regulation of several physiological processes (Poolman et al., 2004). Finally, one of the genes of S. pneumoniae and three of S. pseudopneumoniae are hypothetical proteins. Since these genes are species unique, further studies to elucidate their functions are needed.
In order to determine whether the candidate biomarker genes were expressed or not, the presence of the genes was screened, using mass spectrometry “proteotyping” for discovery of species-unique peptide biomarkers (Karlsson et al., 2018). Proteotyping was able to differentiate the three most closely-related species of the Mitis-Group streptococci. More than 250 species-unique peptides were identified for each of the species S. pneumoniae, S. pseudopneumoniae, and S. mitis, enabling correct identifications, even when bacterial cells from these species were mixed, i.e., simulating a clinical sample (Karlsson et al., 2018).
In this study, the peptides detected by Karlsson et al. (2018) were used to experimentally verify expression of the candidate biomarker genes identified by the pan-genomics concept. In-silico digestion of the selected biomarker genes shown to be expressed, by the study of Karlsson et al. (2018), was performed. All possible peptides that, theoretically, could be generated by trypsin digestion and that was of suitable length for detection by the MS, were identified. Subsequently, “inclusion lists” of these peptides were generated containing which ions (mass-to-charge, m/z) to isolate and select for further fragmentation. In normal conditions, the MS normally operates in “Top10” mode, which means that the 10 most-abundant peptides in each scan are selected for fragmentation. By using “inclusion lists,” m/z ratios corresponding to the peptides in the inclusion list will be selected for fragmentation, even if they are not among the Top10 most-abundant peptides. As a result, fragmentation of a low-abundant peptide in the inclusion lists but not among the 10 most-abundant peptides in an MS-scan, is facilitated. The inclusion lists were used in the analysis of three strains each of S. pneumoniae, S. pseudopneumoniae, and S. mitis, described by Karlsson et al. (2018). The peptides found by this approach, could be considered to be experimentally verified to be expressed and, thus, suitable to be used as species-unique peptides for proteotyping- or other proteomic-based diagnostics of clinical samples, or suitable as the means for alternative detection methodologies.
This study proposes six S. pneumoniae unique gene markers (Pneumo_127, Pneumo_1011, Pneumo_1012, Pneumo_1013, Pneumo_1014, and Pneumo_1961), that have been found in all S pneumoniae strains analyzed, and three S. pseudopneumoniae unique gene markers (Pseudo_228, Pseudo_899, and Pseudo_902), that are present in more than 95% of analyzed S. pseudopneumoniae strains and have been experimentally verified to be expressed. However, additional BLASTN analyses performed against 29 additional genome sequences of S. pseudopneumoniae have revealed that four of the six gene markers of S. pneumoniae are present in two of these genome sequences (strains EL2652N1 and Spain939) and one present in another genome sequence (strain Spain3473). Therefore, identification of S. pneumoniae and S. pseudopneumoniae, including more than one unique marker, is proposed for reliable identification and separation of the two species.
Interestingly, other, previously reported gene markers for S. pneumoniae were not necessarily detected by our pangenomic approach. For instance, Tavares et al. (2019) indicated that “lytA, piaB, and SP2020 were found in non-pneumococcal strains.” Also, Carvalho et al. (2007) reported that a psaA-based real-time PCR-assay was positive in S. pseudopneumoniae strains. Salvà-Serra et al. (2018) proposed a “Xisco” gene-based PCR-assay for detection and identification of S. pneumoniae isolates, after analyzing hundreds of pneumococcal and non-pneumococcal genome sequences. Despite the robustness of the assay, in that same publication, they already reported the presence of a fragment (63%) of the gene in one genome of S. pseudopneumoniae and one genome of S. mitis. These previously-described marker genes did not pass the strict filters applied in this study.
Despite introduction of conjugate vaccines, S. pneumoniae remains a major cause of morbidity and mortality worldwide. S. pseudopneumoniae is less virulent, although, recent reports suggest the bacterium to be a potential pathogen in individuals having underlying conditions. It is, therefore, crucial for the clinical laboratory to correctly diagnose and differentiate S. pneumoniae and S. pseudopneumoniae in clinical samples. This is not least important in samples containing abundant commensal flora, such as sputum samples, in which high numbers of close-related species are present. Correct identification is also important for evaluation of introduced pneumococcal vaccines, including assessments of pneumococcal carriage in the healthy child population. Since pneumococci often tend to lyse and die, due to the activation of autolysin enzymes, molecular detection methods are sometimes superior to culture-dependent methods, and can also be applied in low-income settings. The same is true for patient samples collected after antibiotics have been given, where bacterial detection by culture is often negative, also in cases with severe infection.
The occurrence of horizontal gene transfer and homologous recombination between S. pneumoniae and commensal Mitis-Group streptococci (Whatmore et al., 2000; Kilian et al., 2008) makes it difficult to rely on the use of single biomarkers for identifications of these species. The proposition of using more than one marker is based on the findings that not all of the proposed species unique gene markers for S. pseudopneumoniae were present in all the strains tested. This is also supported by previous findings that markers reported to be unique for S. pneumoniae are also found in clinical isolates of other species of the Mitis-Group of the genus Streptococcus (Tavares et al., 2019). These facts further demonstrate the importance of using multi-locus approaches for identifications and differentiation of S. pneumoniae and S. pseudopneumoniae.
Data Availability Statement
The genome sequences of the 14 S. pseudopneumoniae clinical strains determined in this study have been deposited in DDBJ/ENA/GenBank under the following accession numbers: CCUG 47366 (RXXW01), CCUG 50868 (RXXV01), CCUG 50871 (RXXU01), CCUG 61551 (RXXT01), CCUG 63793 (RXXS01), CCUG 64062 (RXXR01), CCUG 70988 (RXXQ01), CCUG 71653 (RXXP01), CCUG 71996 (RXXN01), CCUG 72018 (RXXM01), CCUG 72019 (RXXL01), CCUG 72028 (RXXK01), CCUG 72029 (RXXJ01), and CCUG 72040 (RXXI01).
Author Contributions
LG-S, EM, and MG conceived the study. LG-S, RK, FS-S, SS, and MG designed the experiments. LG-S, PS, FS-S, DJ-L, RK, and MG performed the experiments and analyzed the data. LG-S, RK, EM, and MG drafted the manuscript. SS, FS-S, and DJ-L provided critical inputs. EM and SS acquired the project funding. MG was responsible for the overall direction of the project. All authors read and approved the final manuscript.
Funding
This work was supported by the European Commission 7th Framework Programme: Tailored-Treatment, Project No. 602860; the Swedish Västra Götaland regional funding: Project Nos. ALFGBG-437221, ALFGBG-720761, and ALF-grants for junior researchers ALFGBG-775231, 2018-2021. FS-S, DJ-L, and EM acknowledge support from the Swedish Västra Götaland Region, Lab Medicine Project No. 51060-6268. LG-S acknowledges the Kunglig. Vetenskaps- och Vitterhets-Samhället (KVVS) for the research stipendium support. FS-S and DJ-L were supported by a stipend for Basic and Advanced Research from the CCUG, through the Institute of Biomedicine, Sahlgrenska Academy, University of Gothenburg. The Culture Collection University of Gothenburg (CCUG) was supported by the Department of Clinical Microbiology, Sahlgrenska University Hospital, and the Sahlgrenska Academy of the University of Gothenburg.
Conflict of Interest
RK is affiliated with a company, Nanoxis Consulting AB.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The technical expertise and assistance with strain typing and identifications by the staff of the CCUG Typing Lab was acknowledged and appreciated. Beatriz Piñeiro-Iglesias was acknowledged for technical assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2020.00222/full#supplementary-material
Abbreviations
ANI, average nucleotide identity; ANIb, average nucleotide identity based on BLAST; WGS, whole genome sequencing; LC-MS/MS, Liquid chromatography tandem-mass spectrometry.
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Arbique, J. C., Poyart, C., Trieu-Cuot, P., Quesne, G., Carvalho Mda, G., Steigerwalt, A. G., et al. (2004). Accuracy of phenotypic and genotypic testing for identification of Streptococcus pneumoniae and description of Streptococcus pseudopneumoniae sp. nov. J. Clin. Microbiol. 42, 4686–4696. doi: 10.1128/JCM.42.10.4686-4696.2004
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bishop, C. J., Aanensen, D. M., Jordan, G. E., Kilian, M., Hanage, W. P., and Spratt, B. G. (2009). Assigning strains to bacterial species via the internet. BMC Biol. 7:3. doi: 10.1186/1741-7007-7-3
Boulund, F., Karlsson, R., Gonzales-Siles, L., Johnning, A., Karami, N., Al-Bayati, O., et al. (2017). Typing and characterization of bacteria using bottom-up tandem mass spectrometry proteomics. Mol. Cell. Proteomics 16, 1052–1063. doi: 10.1074/mcp.M116.061721
Caraux, G., and Pinloche, S. (2005). PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatics 21, 1280–1281. doi: 10.1093/bioinformatics/bti141
Carvalho Mda, G., Tondella, M. L., McCaustland, K., Weidlich, L., McGee, L., Mayer, L. W., Steigerwalt, A, et al. (2007) Evaluation improvement of real-time PCR assays targeting lytA, ply, psaA genes for detection of pneumococcal. DNA. J Clin Microbiol. doi: 10.1128/JCM.02498-06.
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Croxen, M. A., Lee, T. D., Azana, R., and Hoang, L. M. (2018). Use of genomics to design a diagnostic assay to discriminate between Streptococcus pneumoniae and Streptococcus pseudopneumoniae. Microb. Genom. 4:e000175. doi: 10.1099/mgen.0.000175
Echlin, H., Frank, M. W., Iverson, A., Chang, T. C., Johnson, M. D., Rock, C. O., et al. (2016). Pyruvate oxidase as a critical link between metabolism and capsule biosynthesis in Streptococcus pneumoniae. PLoS Pathog. 12:e1005951. doi: 10.1371/journal.ppat.1005951
El Aila, N. A., Emler, S., Kaijalainen, T., De Baere, T., Saerens, B., Alkan, E., et al. (2010). The development of a 16S rRNA gene based PCR for the identification of Streptococcus pneumoniae and comparison with four other species specific PCR assays. BMC Infect. Dis. 10:104. doi: 10.1186/1471-2334-10-104
Epstein, W. (2003). The roles and regulation of potassium in bacteria. Prog. Nucleic Acid Res. Mol. Biol. 75, 293–320. doi: 10.1016/S0079-6603(03)75008-9
Glanville, D. G., Han, L., Maule, A. F., Woodacre, A., Thanki, D., Abdullah, I. T., et al. (2018). RitR is an archetype for a novel family of redox sensors in the streptococci that has evolved from two-component response regulators and is required for pneumococcal colonization. PLoS Pathog. 14:e1007052. doi: 10.1371/journal.ppat.1007052
Gomila, M., Peña, A., Mulet, M., Lalucat, J., and García-Valdés, E. (2015). Phylogenomics and systematics in Pseudomonas. Front. Microbiol. 6:214. doi: 10.3389/fmicb.2015.00214
Gonzales-Siles, L., Salvà-Serra, F., Degerman, A., Norden, R., Lindh, M., Skovbjerg, S., et al. (2019). Identification and capsular serotype sequetyping of Streptococcus pneumoniae strains. J. Med. Microbiol. 68, 1173–1188. doi: 10.1099/jmm.0.001022
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Humphries, J., Xiong, L., Liu, J., Prindle, A., Yuan, F., Arjes, H. A., et al. (2017). Species-independent attraction to biofilms through electrical signaling. Cell 168, 200–209.e12. doi: 10.1016/j.cell.2016.12.014
Jensen, A., Scholz, C. F., and Kilian, M. (2016). Re-evaluation of the taxonomy of the mitis group of the genus Streptococcus based on whole genome phylogenetic analyses, and proposed reclassification of Streptococcus dentisani as Streptococcus oralis subsp. dentisani comb. nov., Streptococcus tigurinus as Streptococcus oralis subsp. tigurinus comb. nov., and Streptococcus oligofermentans as a later synonym of Streptococcus cristatus. Int. J. Syst. Evol. Microbiol. 66, 4803–4820. doi: 10.1099/ijsem.0.001433
Johnston, C., Hinds, J., Smith, A., Van Der Linden, M., Van Eldere, J., and Mitchell, T. J. (2010). Detection of large numbers of pneumococcal virulence genes in streptococci of the mitis group. J. Clin. Microbiol. 48, 2762–2769. doi: 10.1128/JCM.01746-09
Joshi, N. A., and Fass, J. N. (2011). Sickle: A Sliding-Window, Adaptive, Quality-Based Trimming Tool for FastQ Files (Version 1.33). Available online at: https://github.com/najoshi/sickle
Kaas, R. S., Friis, C., Ussery, D. W., and Aarestrup, F. M. (2012). Estimating variation within the genes and inferring the phylogeny of 186 sequenced diverse Escherichia coli genomes. BMC Genomics 13:577. doi: 10.1186/1471-2164-13-577
Karlsson, R., Gonzales-Siles, L., Boulund, F., Svensson-Stadler, L., Skovbjerg, S., Karlsson, A., et al. (2015). Proteotyping: proteomic characterization, classification and identification of microorganisms – a prospectus. Syst. Appl. Microbiol. 38, 246–257. doi: 10.1016/j.syapm.2015.03.006
Karlsson, R., Gonzales-Siles, L., Gomila, M., Busquets, A., Salvà-Serra, F., Jaén-Luchoro, D., et al. (2018). Proteotyping bacteria: characterization, differentiation and identification of pneumococcus and other species within the mitis group of the genus Streptococcus by tandem mass spectrometry proteomics. PLoS ONE 13:e0208804. doi: 10.1371/journal.pone.0208804
Kilian, M., Poulsen, K., Blomqvist, T., Havarstein, L. S., Bek-Thomsen, M., Tettelin, H., et al. (2008). Evolution of Streptococcus pneumoniae and its close commensal relatives. PLoS ONE 3:e2683. doi: 10.1371/journal.pone.0002683
Kilian, M., Riley, D. R., Jensen, A., Bruggemann, H., and Tettelin, H. (2014). Parallel evolution of Streptococcus pneumoniae and Streptococcus mitis to pathogenic and mutualistic lifestyles. MBio 5:e01490–14. doi: 10.1128/mBio.01490-14
Kilian, M., and Tettelin, H. (2019). Identification of virulence-associated properties by comparative genome analysis of Streptococcus pneumoniae, S. pseudopneumoniae, S. mitis, three S. oralis subspecies, and S. infantis. mBio 10: e01985–19. doi: 10.1128/mBio.02520-19
Koonin, E. V., and Wolf, Y. I. (2008). Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 36, 6688–6719. doi: 10.1093/nar/gkn668
Lee, C. R., Cho, S. H., Kim, H. J., Kim, M., Peterkofsky, A., and Seok, Y. J. (2010). Potassium mediates Escherichia coli enzyme IIANtr-dependent regulation of sigma factor selectivity. Mol. Microbiol. 78, 1468–1483. doi: 10.1111/j.1365-2958.2010.07419.x
Letunic, I., and Bork, P. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. doi: 10.1093/nar/gkw290
Medini, D., Donati, C., Tettelin, H., Masignani, V., and Rappuoli, R. (2005). The microbial pan-genome. Curr. Opin. Genet. Dev. 15, 589–594. doi: 10.1016/j.gde.2005.09.006
Mörk-Mörkenstein, M., Heermann, R., Gopel, Y., Jung, K., and Gorke, B. (2017). Non-canonical activation of histidine kinase KdpD by phosphotransferase protein PtsN through interaction with the transmitter domain. Mol. Microbiol. 106, 54–73. doi: 10.1111/mmi.13751
Nagai, K., Shibasaki, Y., Hasegawa, K., Davies, T. A., Jacobs, M. R., Ubukata, K., et al. (2001). Evaluation of PCR primers to screen for Streptococcus pneumoniae isolates and beta-lactam resistance, and to detect common macrolide resistance determinants. J. Antimicrob. Chemother. 48, 915–918. doi: 10.1093/jac/48.6.915
O'Brien, K. L., Wolfson, L. J., Watt, J. P., Henkle, E., Deloria-Knoll, M., Mccall, N., et al. (2009). Burden of disease caused by Streptococcus pneumoniae in children younger than 5 years: global estimates. Lancet 374, 893–902. doi: 10.1016/S0140-6736(09)61204-6
Pai, R., Gertz, R. E., and Beall, B. (2006). Sequential multiplex PCR approach for determining capsular serotypes of Streptococcus pneumoniae isolates. J. Clin. Microbiol. 44, 124–131. doi: 10.1128/JCM.44.1.124-131.2006
Pericone, C. D., Park, S., Imlay, J. A., and Weiser, J. N. (2003). Factors contributing to hydrogen peroxide resistance in Streptococcus pneumoniae include pyruvate oxidase (SpxB) and avoidance of the toxic effects of the fenton reaction. J. Bacteriol. 185, 6815–6825. doi: 10.1128/JB.185.23.6815-6825.2003
Poolman, B., Spitzer, J. J., and Wood, J. M. (2004). Bacterial osmosensing: roles of membrane structure and electrostatics in lipid-protein and protein-protein interactions. Biochim. Biophys. Acta 1666, 88–104. doi: 10.1016/j.bbamem.2004.06.013
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Richter, M., Rosselló-Móra, R., Oliver Glöckner, F., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Rolo, D., Simões, A. S., Domenech, A., Fenoll, A., Liñares, J., de Lencastre, H., et al. (2013). Disease isolates of Streptococcus pseudopneumoniae and non-typeable S. pneumoniae presumptively identified as atypical S. pneumoniae in Spain. PLoS ONE 8:e57047. doi: 10.1371/journal.pone.0057047
Rouli, L., Merhej, V., Fournier, P. E., and Raoult, D. (2015). The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microb. New Infect. 7, 72–85. doi: 10.1016/j.nmni.2015.06.005
Sá-Leão, R., Simões, A. S., Nunes, S., Sousa, N. G., Frazão, N., and De Lencastre, H. (2006). Identification, prevalence and population structure of non-typable Streptococcus pneumoniae in carriage samples isolated from preschoolers attending day-care centres. Microbiology 152, 367–376. doi: 10.1099/mic.0.28596-0
Salo, P., Ortqvist, A., and Leinonen, M. (1995). Diagnosis of bacteremic pneumococcal pneumonia by amplification of pneumolysin gene fragment in serum. J. Infect. Dis. 171, 479–482. doi: 10.1093/infdis/171.2.479
Salvà-Serra, F., Connolly, G., Moore, E. R. B., and Gonzales-Siles, L. (2018). Detection of “Xisco” gene for identification of Streptococcus pneumoniae isolates. Diagn. Microbiol. Infect. Dis. 90, 248–250. doi: 10.1016/j.diagmicrobio.2017.12.003
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Simões, A. S., Sá-Leão, R., Eleveld, M. J., Tavares, D. A., Carrico, J. A., Bootsma, H. J., et al. (2010). Highly penicillin-resistant multidrug-resistant pneumococcus-like strains colonizing children in Oeiras, Portugal: genomic characteristics and implications for surveillance. J. Clin. Microbiol. 48, 238–246. doi: 10.1128/JCM.01313-09
Tavares, D. A., Handem, S., Carvalho, R. J., Paulo, A. C., de Lencastre, H., Hinds, J., et al. (2019). Identification of Streptococcus pneumoniae by a real-time PCR assay targeting SP2020. Sci. Rep. 9:3285. doi: 10.1038/s41598-019-39791-1
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Varghese, R., Jayaraman, R., and Veeraraghavan, B. (2017). Current challenges in the accurate identification of Streptococcus pneumoniae and its serogroups/serotypes in the vaccine era. J. Microbiol. Methods 141, 48–54. doi: 10.1016/j.mimet.2017.07.015
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Weiser, J. N., Bae, D., Epino, H., Gordon, S. B., Kapoor, M., Zenewicz, L. A., et al. (2001). Changes in availability of oxygen accentuate differences in capsular polysaccharide expression by phenotypic variants and clinical isolates of Streptococcus pneumoniae. Infect. Immun. 69, 5430–5439. doi: 10.1128/IAI.69.9.5430-5439.2001
Whatmore, A. M., Efstratiou, A., Pickerill, A. P., Broughton, K., Woodard, G., Sturgeon, D., et al. (2000). Genetic relationships between clinical isolates of Streptococcus pneumoniae, Streptococcus oralis, and Streptococcus mitis: characterization of “Atypical” pneumococci and organisms allied to S. mitis harboring S. pneumoniae virulence factor-encoding genes. Infect. Immun. 68, 1374–1382. doi: 10.1128/IAI.68.3.1374-1382.2000
Keywords: Streptococcus, pneumococcus, S. pseudopneumoniae, pangenome, proteotyping, gene markers, identification
Citation: Gonzales-Siles L, Karlsson R, Schmidt P, Salvà-Serra F, Jaén-Luchoro D, Skovbjerg S, Moore ERB and Gomila M (2020) A Pangenome Approach for Discerning Species-Unique Gene Markers for Identifications of Streptococcus pneumoniae and Streptococcus pseudopneumoniae. Front. Cell. Infect. Microbiol. 10:222. doi: 10.3389/fcimb.2020.00222
Received: 05 February 2020; Accepted: 22 April 2020;
Published: 19 May 2020.
Edited by:
Jorge Eugenio Vidal, The University of Mississippi Medical Center, United StatesReviewed by:
Mogens Kilian, Aarhus University, DenmarkSrinivasan Velusamy, Centers for Disease Control and Prevention (CDC), United States
Copyright © 2020 Gonzales-Siles, Karlsson, Schmidt, Salvà-Serra, Jaén-Luchoro, Skovbjerg, Moore and Gomila. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lucia Gonzales-Siles, bHVjaWEuZ29uemFsZXNAZ3Uuc2U=; Edward R. B. Moore, ZWR3YXJkLm1vb3JlQGd1LnNl