
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 25 October 2019
Sec. Microbiome in Health and Disease
Volume 9 - 2019 | https://doi.org/10.3389/fcimb.2019.00364
 Paul Metz1,2,3
Paul Metz1,2,3 Martijn J. H. Tjan2,3Shaoguang Wu3Mehrosh Pervaiz1
Martijn J. H. Tjan2,3Shaoguang Wu3Mehrosh Pervaiz1 Susanne Hermans1Aishwarya Shettigar3Cynthia L. Sears3Tina Ritschel1
Susanne Hermans1Aishwarya Shettigar3Cynthia L. Sears3Tina Ritschel1 Bas E. Dutilh1,4
Bas E. Dutilh1,4 Annemarie Boleij2*
Annemarie Boleij2*Objective: The human intestinal microbiome plays an important role in inflammatory bowel disease (IBD) and colorectal cancer (CRC) development. One of the first discovered bacterial mediators involves Bacteroides fragilis toxin (BFT, also named as fragilysin), a metalloprotease encoded by enterotoxigenic Bacteroides fragilis (ETBF) that causes barrier disruption and inflammation of the colon, leads to tumorigenesis in susceptible mice, and is enriched in the mucosa of IBD and CRC patients. Thus, targeted inhibition of BFT may benefit ETBF carrying patients.
Design: By applying two complementary in silico drug design techniques, drug repositioning and molecular docking, we predicted potential BFT inhibitory compounds. Top candidates were tested in vitro on the CRC epithelial cell line HT29/c1 for their potential to inhibit key aspects of BFT activity, being epithelial morphology changes, E-cadherin cleavage (a marker for barrier function) and increased IL-8 secretion.
Results: The primary bile acid and existing drug chenodeoxycholic acid (CDCA), currently used for treating gallstones, cerebrotendinous xanthomatosis, and constipation, was found to significantly inhibit all evaluated cell responses to BFT exposure. The inhibition of BFT resulted from a direct interaction between CDCA and BFT, as confirmed by an increase in the melting temperature of the BFT protein in the presence of CDCA.
Conclusion: Together, our results show the potential of in silico drug discovery to combat harmful human and microbiome-derived proteins and more specifically suggests a potential for retargeting CDCA to inhibit the pro-oncogenic toxin BFT.
Using in silico drug discovery and complementary in vitro analyses, chenodeoxycholic acid (CDCA) was found to inhibit the major gut pathogen-derived oncogenic Bacteroides fragilis toxin (BFT). Thus, CDCA may be a promising drug that limits ETBF-related inflammatory bowel disease (IBD) and colorectal cancer (CRC) development. CDCA is especially interesting as a BFT-inhibitor since it is one of several naturally occurring bile acids with gut concentrations well within the dose to effectively inhibit BFT. Hence, inter-individual variation in bile-acid concentrations could explain differences in the human colon response to ETBF and the risk for cancer. Moreover, CDCA has additional anti-inflammatory properties via activation of the Farnesoid X Receptor (FXR) and is an interesting compound to explore in IBD-patients. The newly discovered interaction between CDCA and BFT offers new opportunities in battling ETBF-related CRC and IBD, brings an additional option for experimental manipulation by “switching off” BFT and opens new avenues for understanding host-microbiome interactions.
In 2018, an estimated 861,663 people died from CRC globally, i.e., >9% of the total cancer deaths in that year (Bray et al., 2018). Over the last decade, the importance of microbial factors from our gut microbiome in CRC pathogenesis has emerged. An important CRC-driving bacterium identified in the mucosa of more than 80% of CRC-patients is enterotoxigenic Bacteroides fragilis (ETBF) carrying the Bacteroides fragilis toxin (BFT), also known as fragilysin (Boleij et al., 2015). ETBF has been specifically associated with diarrhea, inflammatory bowel disease (IBD) and anaerobic bacteremia (Wexler, 2007; Rabizadeh and Sears, 2008; Housseau and Sears, 2010; Zamani et al., 2017). ETBF is often found together with E. coli carrying the oncotoxin colibactin in mucosal biofilms of Familial Adenomatous Polyposis (FAP). These two oncogenic bacteria have been shown to synergize in vivo, creating a favorable environment for CRC development (Sears and Garrett, 2014; Dejea et al., 2018). Exposure to BFT is therefore proposed as an important microbiome-derived risk factor for CRC development in humans.
ETBF contains a 6 kilobase pair (kbp) pathogenicity island within a 40 kbp conjugative transposon that harbors the bft gene, encoding a 397 amino acid zinc-dependent metalloprotease (Moncrief et al., 1995; Wexler, 2007). BFT induces the degradation of the intercellular protein E-cadherin (Wu et al., 1998) of colonic epithelial cells (CECs), causing morphological changes in vitro such as rounding, swelling, and cluster dissolution (Rhee et al., 2009). Moreover, BFT activates the NF-B and β-catenin/Tcf signaling pathways, resulting in the expression of proinflammatory IL-8 and the c-myc oncogene, which have been associated with CRC (Wu et al., 2004; Ning et al., 2011).
BFT is the only recognized virulence factor of ETBF and considered to be fully responsible for its pathogenicity. Targeted BFT inhibition could therefore limit ETBF-related pathogenicity. Three different BFT isoforms have been identified, bft-1, bft-2, and bft-3 (Kato et al., 2000), whose encoded amino acid sequences are >93% identical (Chung et al., 1999; Kato et al., 2000).
BFT is encoded as a protoxin, where the active site responsible for the pro-oncogenic activity is completely blocked by a 9 amino acid pro-domain (Franco et al., 2005; Goulas et al., 2011). Below, we combine two in silico drug discovery techniques, drug repositioning and molecular docking, to predict potential inhibitors for BFT that interfere with its toxic activity by blocking the active site. The strongest in silico predicted inhibitors were tested in a CEC-model. We show for the first time that in silico drug prediction is particularly well suited to identify compounds effectively targeting microbial toxins.
Based on the idea that similar binding sites can bind similar ligands, the in silico structure-based pharmacophore fingerprint comparison method KRIPO (Wood et al., 2012) was used to screen the Protein Data Bank (www.rcsb.org) (Berman et al., 2000) for proteins with a structurally similar binding site as BFT-3, whose crystal structure has been resolved (PDB ID: 3P24) (Goulas et al., 2011). The catalytic site of BFT-3 is shown in Figures 1A,B: a zinc ion is complexed by three histidine residues of the catalytic domain (HIS348, HIS352, HIS358) and one aspartate (ASP194) from the pro-domain.
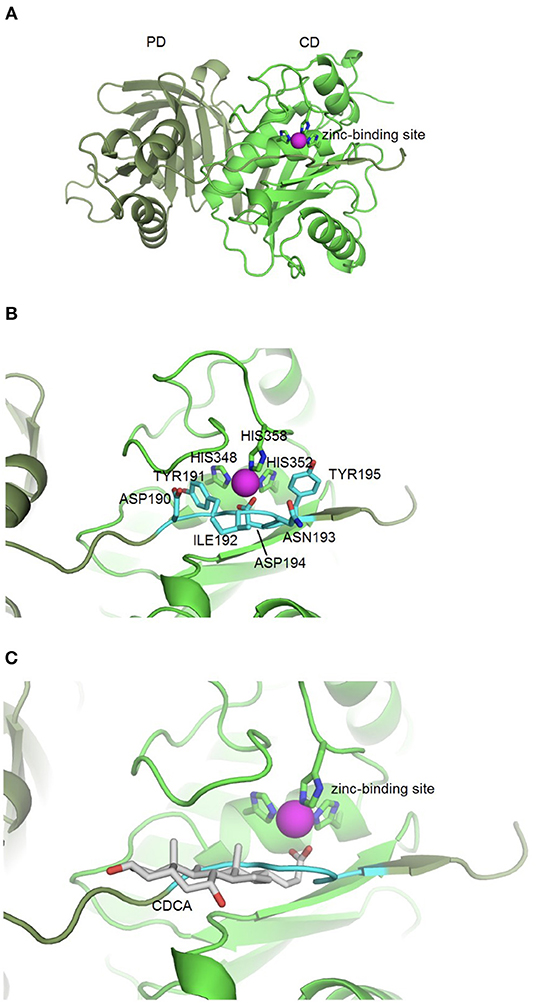
Figure 1. BFT-3 structure. (A) BFT structure and active site BFT-3 structure including pro-domain (PD, dark green) and catalytic domain (CD, light green) and zinc-binding site (zinc in purple). (B) Pro-domain residues (ASP190, TYR191, ILE192, ANS193, ASP194, and TYR195) used for defining the BFT-3 binding pocket (light blue) and zinc coordinating residues from the CD (HIS348, HIS352, and HIS358). (C) Chenodeoxycholic acid docked into the BFT-3 zinc-binding site using MOE.
After cleavage of the pro-domain, the active site of the protein is exposed. To define the binding site of BFT-3, atoms ≤6 Å from the active site blocking residues (ASP190, TYR191, ILE192, ANS193, ASP194, and TYR195) of the uncleaved BFT-3 pro-domain were selected and their pharmacophore features (aromatic interactions, hydrogen bond acceptors and donors, hydrophobic contacts, and positive/negative charges) described (Figure 1B). All pharmacophore features of this binding pocket ≤2.5 Å of any atom of the previously mentioned pro-domain residues were defined as the final protein structure-based pharmacophore of the BFT-3 binding site, which was then encoded into a fingerprint bit string representation (Wood et al., 2012). With a fingerprint bit string similarity of ≥0.45 to this structure-based pharmacophore of the BFT-3 binding site, all (sub)binding pockets and their associated ligands were selected from the Protein Data Bank.
From this preselection by KRIPO, ligands were extracted and subsequently docked into the BFT-3 binding site using MOE 2013.08021 (Figure 1C). The docking was guided by a pharmacophore model (Figure 2) of the BFT binding site to select compounds that could interact with the zinc atom. This additional docking step, using a pharmacophore description of the zinc interactions, was essential because KRIPO does not directly encode for metal interactions.
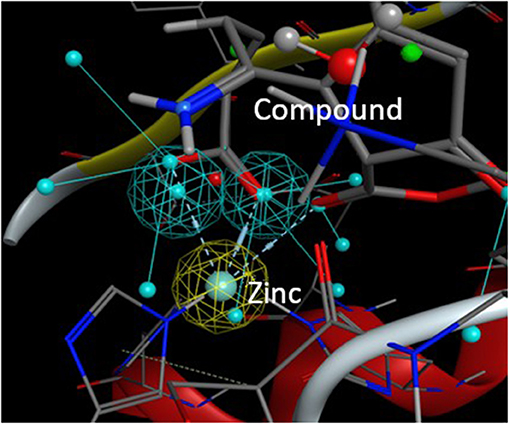
Figure 2. Pharmacophore model BFT-3 active site. Docking and interaction of a compound (sticks) with the BFT-3 active site (lines) guided by a pharmacophore model of the BFT-3 active site, including a pharmacophore description of the zinc atom (spheres).
HT29/c1 CECs were maintained in DMEM (Gibco/BRL 12800) supplemented with 10% Fetal Bovine Serum (FBS), 44 mM NaHCO3, 10 μg/mL Human apo-transferrin (Sigma T1147), 50 units/mL penicillin and 50 μg/mL streptomycin with pH 7.4 (Weikel et al., 1992; Sears, 2001). For morphology assays and subsequent ELISA's, HT29/c1 cells were grown in 96-well plates to a confluence of 70–80%.
Wild type ETBF strains VPI 13784 (BFT-1), 086–5443-2–2 (BFT-2) and k570 (BFT-3) were grown anaerobically in 3.7% BHI supplemented with 0.5% yeast extract, 0.05% cysteine HCL, 10 μL/mL hemin and 0.2 μL/L vitamin K, at 37°C for 24 h. After adjusting to get approximately equal optical densities of 0.8-1.0 (A600), pooled supernatants were sterilized by filtration through a 0.2 μm filter and concentrated 5–10-fold using a 10 kDa filter (Millipore). The concentration of these concentrated toxins was calibrated using HT29c1 morphological assays (Supplementary Figure 1). Concentrated toxins were stored at −20°C until further use.
For initial examination of the putative BFT inhibitors, BFT-induced morphological changes were assessed with a previously described in vitro toxin activity assay (Weikel et al., 1992). HT29/c1 cells were exposed to BFT-3 at a final calibrated concentration of 100 pM and to each of the selected compounds, at 850 nM or 8.5 μM concentrations dissolved in DMEM (total volume was 200 μL) (Table 1 and Supplementary Table 1).

Table 1. The twelve in silico selected compounds predicted to inhibit BFT.
After this first in vitro screening, promising compounds were tested further at higher concentrations (85 and 175 μM). Compounds EBSHIPA, PPAA, CDCA, and DS-DHP were dissolved in 100% dimethyl sulfoxide (DMSO) and all other compounds in deionized water (dH2O). Positive controls contained BFT-3 (100 pM) without compounds, but with DMSO/H2O. Negative controls included either compound only, DMSO/H2O or neither. HT29/c1 cells were pre-incubated with the selected compounds in DMEM at 37°C for 30 min, before addition of BFT-3. Morphology changes were scored on a scale from 0 to 4 (Supplementary Figure 2), by 2 individual observers, every hour for a maximum of 4 h. Scores of all testing conditions were compared to positive and negative controls by using the independent unpaired t-test. Compounds that demonstrated BFT inhibition, were tested further in morphology assays with BFT-1 (200 pM) and BFT-2 (300 pM). The supernatants collected in these experiments were used to measure E-cadherin and IL-8 release by ELISA after 3 and 4 h respectively (Supplementary Method 1).
Protein Thermal Shift dye (Thermofisher) 2.5 μL was mixed with 5 μL of Protein Thermal Shift buffer, 1 μg of recombinant BFT-1 (Supplementary Method 1, Supplementary Table 2) in Buffer B (20 mM Tris-HCl, 150 mM NaCl and pH 7.4) and different concentrations of CDCA (0, 8.5, 85, 175, and 850 μM) (Goulas et al., 2011). Negative controls contained only CDCA [ligand only control (LOC)] or only buffer and dye [No-protein control (NPC)]. Reagents were added to a MicroAmp Optical Reaction plate spun at 1000 rpm for 1 min, and incubated in the 7500 Fast SDS Real-Time PCR system (Applied Biosystems), starting at 25°C, at a ramp rate of 1% until 99°C. Melting curves were generated by capturing fluorescent signal ROX after every cycle of temperature increase. First the raw fluorescence data were plotted in GraphPad Prism 6.00 and the post-peak fluorescence was truncated. Subsequently, non-linear fitting of the truncated data was performed to calculate the change in melting temperature (ΔTm) with a Boltzmann sigmoidal curve fitting (Sigmoidal dose-response) (Huynh and Partch, 2015). Changes in melting temperature were compared using a 1-way ANOVA.
To identify molecules that bind to the active site of BFT, two complementary in silico drug discovery techniques were used; KRIPO and MOE. The KRIPO screen resulted in 476 small molecules that were predicted to bind to the (sub)pockets of BFT-3. Subsequently, the MOE-screening resulted in a selection of 43 compounds that interact with the pharmacophore description of the zinc atom. Finally, a total of 12 compounds, available in regular vendor catalogs, was purchased and screened in vitro (Table 1 and Supplementary Table 3).
Sequence alignment of the three BFT isoforms (Accession numbers NCBI: BFT-1: BAA77276, BFT-2: AAB50410, BFT-3: AAD33214) showed a high protein sequence identity (≥ 93%) among all isoforms (Supplementary Figure 3). For BFT-1 and BFT-2, a homology model was built with the automated protocol of YASARA (http://www.yasara.org/) using the x-ray structure of BFT-3 as template (Goulas et al., 2011). Overlaying BFT isoform structures reveal that residue 357 is the only residue in the active site that differs between every isoform. For residue positions 277, 280, 316, 319, 369, and 370, that also help frame the active site (Goulas et al., 2011), BFT-1 contains different amino acids, while BFT-2 and BFT-3 are identical (Supplementary Figure 3). The high similarity between the 3 BFT isoforms increases the likelihood that the 12 selected compounds interact with all isoforms, although they may not be equally effective due to small differences in the active site and biologic activity (Sears, 2001). Therefore, inhibition of biological activity was tested on all isoforms.
To assess the efficacy of the 12 selected potential drug molecules, HT29/c1 cells were treated with BFT-3, in combination with each selected compound. Because the in silico drug design was based on the BFT-3 isoform, the initial screening focused on BFT-3 inhibition after 2- (Figure 3A) and 3-h (Figure 3B).
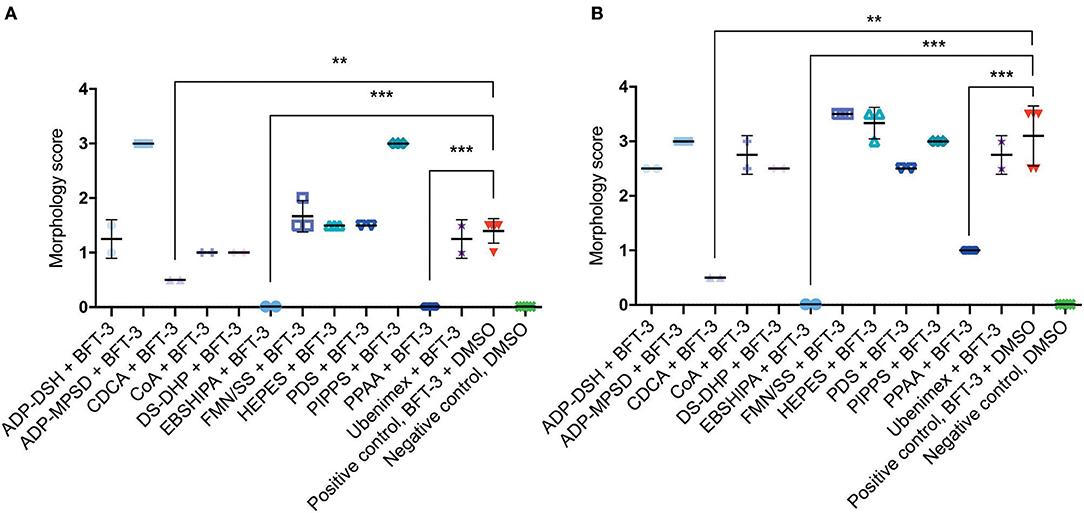
Figure 3. Morphological changes of in silico selected compounds. Drugs EBSHIPA, PPAA, and CDCA showed a 100%, 68–100%, and 64–84% reduction of BFT-3 related morphology changes, respectively, after 2 (A) and 3 (B) hours. N = 2–5 for each condition with (error) bars indicating means and standard deviations. (unpaired t-test, **P < 0.01; ***P < 0.001).
Of all the selected compounds, only chenodeoxycholic acid (CDCA) resulted in significant reduction of HT29/c1 morphological change due to BFT-3. The inhibition peaked 3 h after addition of BFT-3 with a mean reduction in morphology score of 84% (minimum 80%, maximum 86%) compared to the positive control (N = 2, unpaired t-test, P = 0.0014). While EBSHIPA and PPAA also seemed to reduce BFT-3 related morphology changes, they were excluded from further testing due to their effects on cell morphology change. Both EBSHIPA and PPAA resulted in CEC cell cluster shrinkage and detachment of cells from the wells. These effects interfered with morphology scoring and hence discriminating between the effects of the compounds and BFT. Next, the BFT inhibitory effect of CDCA was tested at different concentrations (8.5, 85, and 175 μM) with BFT-1, BFT-2, and BFT-3. Increasing concentrations of CDCA inhibited morphology changes for both BFT-1 and BFT-3 (Figures 4A–C). While BFT-1 activity was only fully inhibited (identical to the negative control experiment) by CDCA at 175 μM for the first 2 h (N = 8, unpaired t-test, P < 0.0001), BFT-3 activity was fully inhibited by CDCA at 85 and 175 μM for 4 h compared to the positive control (N = 8, unpaired t-test, P < 0.0001). Less but still significant inhibition of morphological change was observed with lower concentrations of CDCA. CDCA at a concentration of 8.5 μM only showed inhibition in morphological assays with BFT-2 after 1 h. Due to the high potency of the BFT-2 concentrate and the associated difficulties in titration to similar levels as BFT-1 and BFT-3, we decided not to test CDCA at higher concentrations in assays with BFT-2 and to focus on BFT-1 and BFT-3.
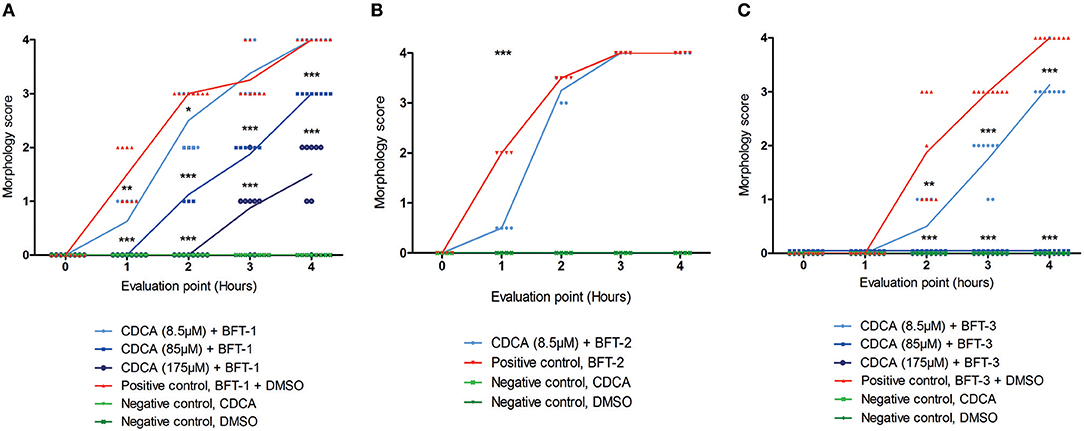
Figure 4. Morphological inhibition of BFT-1, BFT-2, and BFT-3 by CDCA. BFT-1, BFT-2, and BFT-3 related morphology changes were inhibited by CDCA, where an increase in CDCA concentration correlated with increased inhibition for BFT-1 and BFT-3. Depending on measurement times, CDCA at a concentration of 8.5, 85, and 175 μM resulted in 0–42%, 25–63%, and 73–100% inhibition of BFT-1 (A). BFT-2 related morphology changes, only tested at a CDCA-concentration of 8.5 μM, were reduced by 75% (B). BFT-3 was inhibited by CDCA at a concentration of 8.5, 85, and 175 μM with 25–73%, 100%, and 100% inhibition, respectively, depending on measurement times (C). Negative control experiments did not display any morphology changes (score = 0 for all 8 replicates). Lines indicate means and N = 8 for each condition. Significance is marked by asterisks (unpaired t-test, *P = 0.01–0.05; **P = 0.001–0.01; ***P < 0.001).
To better quantify the inhibitory action of CDCA on BFT biological activity, E-cadherin cleavage and IL-8 secretion were measured in cell supernatants. The amount of E-cadherin released in the supernatant is a measure for the activity of BFT, thus, effective inhibition of BFT by the selected inhibitors is predicted to reduce E-cadherin release. CDCA (8.5 μM) reduced E-cadherin release by 18% for BFT-1 (minimum 6%, maximum 30%, N = 2–3, unpaired t-test, P = 0.0389) and 15% for BFT-3 (minimum 8%, maximum 22%, N = 4, unpaired t-test, P = 0.005), when compared to the positive control (Figures 5A–C). This suggests that even at 8.5 μM CDCA, E-cadherin release by BFT-1 and BFT-3 is already inhibited. CDCA did not result in significantly reduced E-cadherin release by CECs exposed to BFT-2.
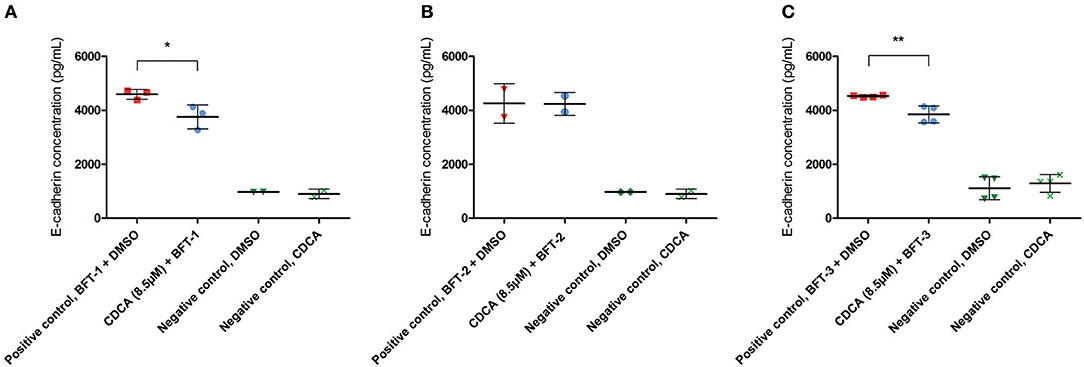
Figure 5. CDCA inhibits E-cadherin cleavage induced by BFT-1 and BFT-3. The release of E-cadherin into the cell medium supernatant for 3 h was measured with an E-cadherin ELISA. CDCA at 8.5 μM led to an average 18% (unpaired t-test, P = 0.0389) (A), 0% (B), and 15% (unpaired t-test, P = 0.005) (C) reduction of E-cadherin concentrations in morphology assays with BFT-1 (A), BFT-2 (B), and BFT-3 (C), respectively. Bars indicate means and standard deviations. BFT-1, N = 2–3, BFT-2, N = 2, and BFT-3, N = 4 (unpaired t-test, *P = 0.01–0.05; **P = 0.001–0.01).
Similar to E-cadherin release, effective inhibitors of BFT should lower the concentration of IL-8 in the cell culture supernatant after BFT-exposure. Indeed, CDCA significantly decreased IL-8 release of BFT-1 and BFT-3 treated HT29/c1 cells after 4 h. Furthermore, CDCA led to dose-dependent IL-8 reduction with CDCA at 175 μM yielding the highest rates, being 67% (minimum 43%, maximum 80%, N = 3–6, P = 0.0023) and 69% (minimum 32%, maximum 88%, N = 5–10, P < 0.0001) for BFT-1 and BFT-3 respectively (Figures 6A,B). At this peak reduction, IL-8 concentrations are almost equal to negative control values, indicating nearly full inhibition. The inhibition in E-cadherin release and IL-8 secretion correlates well with the observed morphological HT29/c1 cell changes induced by both BFT-1 and BFT-3 suggesting that inhibition of BFT by CDCA encompasses key aspects of BFT's effect on CECs.
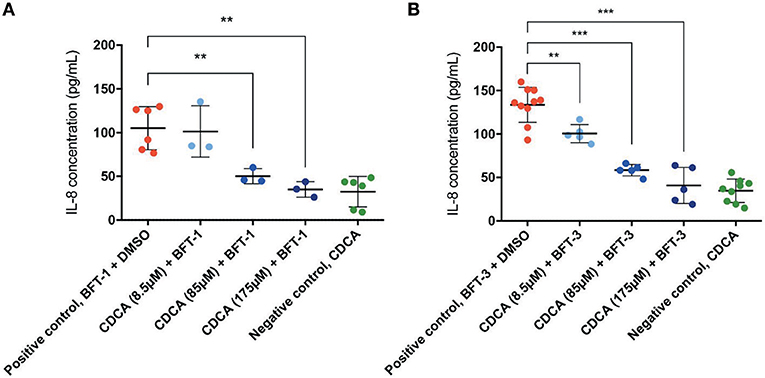
Figure 6. CDCA inhibits IL-8 secretion induced by BFT-1 and BFT-3. The amount of IL8 secreted by HT29/c1 cells for 4 h was measured by an IL-8 ELISA. CDCA in increasing concentrations shows significant inhibition of BFT-1 (A) and BFT-3 (B). In experiments with BFT-1, CDCA results in a significant mean 52% (unpaired t-test, P = 0.0080) and 67% (unpaired t-test, P = 0.0023) reduction of IL-8 at 85 and 175 μM, respectively (A). For BFT-3, CDCA significantly reduced IL-8 concentrations with a mean 24% (unpaired t-test, P = 0.0049), 56% (unpaired t-test, P < 0.0001) and 69% (unpaired t-test, P < 0.0001) at 8.5, 85, and 175 μM, respectively (B). BFT-1, N = 3–6 and BFT-3, N = 5–10. Bars indicate means and standard deviations. (unpaired t-test, **P = 0.001–0.01; ***P < 0.001).
While it was encouraging that CDCA inhibited the activity of BFT-1 and BFT-3 in morphology assays and ELISA, these observations might still be the result of indirect effects (e.g., action on CECs) rather than direct binding of CDCA to BFT. To test whether the BFT inhibition is due to direct binding of CDCA to BFT, we performed a thermal shift assay (TSA) with purified rBFT-1. A TSA can be used to detect protein-ligand interactions. Proteins tend to have a more hydrophobic core and hydrophilic surface (Lodish et al., 2000). When heated, proteins unfold and their hydrophobic core is exposed. In a TSA, fluorescent dye is used to bind to the hydrophobic core and emit a measurable signal. corresponding to the melting temperature (Tm) of the protein. Protein-binding ligands increase the thermal stability and an increased Tm. A significant shift in melting temperature of rBFT-1 (62°C) was observed with concentrations of 85 and 175 μM CDCA (ΔTm 1.10, and ΔTm 2.10, N = 3, P < 0.0001, 1-Way ANOVA; Figure 7).
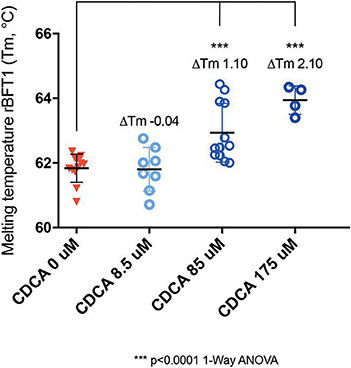
Figure 7. CDCA interacts directly with recombinant BFT-1. TSA with CDCA and rBFT-1 (N = 3), where CDCA resulted in a significant shift in melting temperature at concentrations of 85 and 175 μM (ΔTm 1.10 and ΔTm 2.10, respectively (P < 0.0001 for 1-Way ANOVA and Dunnett's multiple comparison test) when compared to the negative control condition (CDCA 0 μM). CDCA at a concentration of 8.5 μM did not result in a significant shift of the melting temperature of rBFT-1. Bars indicate means and standard deviations. N = 4–12 for each condition.
This indicates a direct interaction of CDCA with rBFT-1 that increases with the concentration of CDCA and corresponds to the inhibitory effect on the morphological assay for BFT-1 and CDCA. The raw fluorescent data used for calculation of ΔTm are presented in Supplementary Figure 4. These results strongly indicate that the two molecules form a physical bond, further supporting our in silico predictions. The predicted interaction of CDCA with the active site of BFT-3 is shown in Figure 1C.
The CRC-driving effect of ETBF is mediated by the toxin BFT, a metalloprotease that induces cleavage of E-cadherin resulting in marked changes in the biology of CECs. Here, we computationally predicted and screened a range of drug compounds for their ability to inhibit the proteolytic activity of BFT. Two compounds, EBSHIPA and PPAA, induced morphological effects on CECs resulting in cell cluster shrinkage and detachment. Because cell cluster shrinkage and detachment of cells interferes with morphology scoring and may be the reason of the observed BFT-inhibition they were excluded from further testing. We found that CDCA, a naturally-occurring primary bile acid, effectively inhibits BFT activity. Inhibition was observed in three complementary assays targeting different features of the cellular response to BFT-1 and BFT-3, including cell culture-based morphology change, E-cadherin cleavage, and IL-8 secretion. Additionally, direct interaction of CDCA to BFT was validated with a TSA.
The effect of CDCA on inhibition of the CEC response to BFT was different for the three BFT isoforms. CDCA most strongly inhibited BFT-3, than BFT-1, and only minimal inhibition was observed for BFT- 2. However, the inhibition of BFT-2 by CDCA was only tested at a concentration of 8.5 μM. This was due to the high potency of the BFT-2 concentrate and the associated difficulties in titration to similar levels as BFT-1 and BFT-3. Explanations for the difference in response to CDCA of the three BFT isoforms could be: (1) small differences in functional toxin concentration between isoforms, (2) the difference in amino acid sequence between isoforms may lead to altered protein folding or functionality of the binding pocket, and (3) the rate of activation of BFT by cleaving off the pro-domain and subsequent secretion may depend on the isoform or B. fragilis strain.
CDCA is a natural bile acid present in the human gut. Fast colonic transit is associated with higher concentrations of primary bile acids such as CDCA in the feces (Valdés Olmos et al., 1991). Delivery of CDCA via ileocolonic capsule accelerates colonic transit in healthy control subjects possibly by inducing pressure waves in the proximal colon (Bampton et al., 2002; Odunsi–Shiyanbade et al., 2010). Alternatively, loperamide, a drug used to reduce diarrhea, results in slower transit time and increases bile acid reabsorption, reducing primary bile acid concentrations in the feces. Natural CDCA concentrations range between 0.015 mM in healthy control subjects to 0.12 mM in patients with irritable bowel syndrome diarrhea. According to our results, the latter concentration is well within the range for BFT-inhibition by CDCA. Higher colonic transit due to diarrhea in these patients and hence higher fecal CDCA concentrations may thus target BFT activity (Peleman et al., 2017). Thus, natural CDCA concentrations could be a factor that modifies the outcome of ETBF-infection and colonization in individual patients, e.g., contributing to CRC or not.
An advantage of CDCA as a potential drug is that it is already being used as an oral treatment for gallstones, cerebrotendinous xanthomatosis, and constipation (Jiang et al., 2015; Fiorucci and Distrutti, 2019). This would simplify the transition from in vitro studies to in vivo and finally clinical studies, mainly because CDCA has already passed several toxicity tests (European Medicines Agency: Assessment report Chenodeoxycholic acid Leadiant) and has limited side effects at concentrations below 750 mg (Mok et al., 1974).
Aside from the well-known role of CDCA in lipid digestion in the bowel, CDCA is the strongest naturally occurring agonist of the farnesoid X receptor (FXR) (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999). FXR is expressed throughout the intestine and plays a pivotal role in the regulation of bile acid, lipid and glucose homeostasis, inflammatory responses, barrier function of the gut and prevention of bacterial translocation (Staels and Fonseca, 2009; Chai et al., 2015). Furthermore, FXR seems to play a thus far unexplained role in various diseases, including CRC, IBD and type 2 diabetes mellitus (Ding et al., 2015). This raises the question whether CDCA, while acting via FXR, could protect against CRC development, an idea that is partially supported by research suggesting that CDCA acts as an anti-inflammatory agent in the gut. Because CDCA directly binds to BFT (Wu et al., 2006), it may have a dual effect in altering ETBF colonic pathogenesis, both by binding to BFT directly as shown here, and by acting on FXR (Makishima et al., 1999; Parks et al., 1999; Wang et al., 1999).
This research was a first step to seek a compound that targets BFT to be used as putative preventive treatment for ETBF-related CRC and possibly other ETBF-related diseases. In vivo experiments are required to investigate the BFT-inhibitory effect of CDCA on tumorigenesis and inflammation in the colon and rectum. Eventually, clinical studies would be needed to determine which specific patient groups may benefit from treatment with CDCA.
In general, in silico drug screening serves as a very effective, time-saving and economical method applicable to research in which specific molecules are targeted. With an increasing performance of predicting drugs that interact in vitro and in vivo, in silico screening effectively complements laboratory screening methods.
The data that support the findings of this study are available from the corresponding author upon reasonable request.
PM performed experiments and wrote the manuscript. MT performed experiments. AB and BD designed the study, trained PM and MT, supervised the project, and wrote the manuscript. SW and CS trained and hosted PM and MT in their lab. AS created rBFT-1, MP, PM, and SH performed in silico modeling with KRIPO and MOE under the supervision of TR.
This project was financially supported by the Radboud Honours Programme Medical Sciences and Johns Hopkins resources NIH R01CA179440 (CS, PI) and Bloomberg Philanthropies. PM received a student internship grant from the Koningin Wilhelmina Fonds (KWF) voor de Nederlandse Kankerbestrijding (Dutch Cancer Society). MT received a grant from Radboud University. CS was supported, in part, by a grant from Bristol Myer Squibb. BD was supported by Netherlands Organization for Scientific Research (NWO) Vidi grant 864.14.004. AB was supported by NWO Veni grant 016.166.089. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AG declared a past supervisory role with one of the authors CS to the handling editor.
We would like to thank researchers at the CMBI, Radboudumc (Aya Hefnawy, Gert Vriend, Fabio Baroni, and Joanna Lange), the Sears Laboratory at Johns Hopkins Medical Institutions (Christine Craig, Julia Drewes, Pauline Myles, Payam Fathi, and Xinqun Wu), and the Department of Pathology, Radboudumc (Blanca Scheijen and professor I. D. Nagtegaal) for their continued help and enthusiasm.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00364/full#supplementary-material
1. ^Molecular Operating Environment (MOE), 2013.08; Chemical Computing Group ULC, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2018.
Bampton, P. A., Dinning, P. G., Kennedy, M. L., Lubowski, D. Z., and Cook, I. J. (2002). The proximal colonic motor response to rectal mechanical and chemical stimulation. Am. J. Physiol. Liver Physiol. 282, G443–G449. doi: 10.1152/ajpgi.00194.2001
Berman, H. M., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T. N., Weissig, H., et al. (2000). The protein data bank. Nucleic Acids Res. 28, 235–224. doi: 10.1093/nar/28.1.235
Boleij, A., Hechenbleikner, E. M., Goodwin, A. C., Badani, R., Stein, E. M., Lazarev, M. G., et al. (2015). The Bacteroides fragilis toxin gene is prevalent in the colon mucosa of colorectal cancer patients. Clin. Infect. Dis. An Off. Publ. Infect. Dis. Soc. Am. 60, 208–215. doi: 10.1093/cid/ciu787
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A., and Jemal, A. (2018). Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 68, 394–424. doi: 10.3322/caac.21492
Chai, J., Zou, L., Li, X., Han, D., Wang, S., Hu, S., et al. (2015). Mechanism of bile acid-regulated glucose and lipid metabolism in duodenal-jejunal bypass. Int. J. Clin. Exp. Pathol. 8, 15778–15785.
Chung, G.-T., Franco, A. A., Wu, S., Rhie, G.-E., Cheng, R., Oh, H.-B., et al. (1999). Identification of a third metalloprotease toxin gene in extraintestinal isolates of Bacteroides fragilis. Infect. Immun. 67, 4945–4949.
Dejea, C. M., Fathi, P., Craig, J. M., Boleij, A., Taddese, R., Geis, A. L., et al. (2018). Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science 359, 592–597. doi: 10.1126/science.aah3648
Ding, L., Yang, L., Wang, Z., and Huang, W. (2015). Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 5, 135–144. doi: 10.1016/j.apsb.2015.01.004
Fiorucci, S., and Distrutti, E. (2019). Chenodeoxycholic acid: an update on its therapeutic applications. Handb. Exp. Pharmacol. 256, 265–282. doi: 10.1007/164_2019_226
Franco, A. A., Buckwold, S. L., Shin, J. W., Ascon, M., and Sears, C. L. (2005). Mutation of the zinc-binding metalloprotease motif affects Bacteroides fragilis toxin activity but does not affect propeptide processing. Infect. Immun. 73, 5273–5277. doi: 10.1128/IAI.73.8.5273-5277.2005
Goulas, T., Arolas, J. L., and Gomis-Rüth, F. X. (2011). Structure, function and latency regulation of a bacterial enterotoxin potentially derived from a mammalian adamalysin/ADAM xenolog. Proc. Natl. Acad. Sci. U.S.A. 108, 1856–1861. doi: 10.1073/pnas.1012173108
Housseau, F., and Sears, C. L. (2010). Enterotoxigenic Bacteroides fragilis (ETBF)-mediated colitis in Min (Apc+/-) mice: a human commensal-based murine model of colon carcinogenesis. Cell Cycle 9, 3–5. doi: 10.4161/cc.9.1.10352
Huynh, K., and Partch, C. L. (2015). Analysis of protein stability and ligand interactions by thermal shift assay. Curr. Protoc. Protein Sci. 79, 28.9.1–14. doi: 10.1002/0471140864.ps2809s79
Jiang, C., Xu, Q., Wen, X., and Sun, H. (2015). Current developments in pharmacological therapeutics for chronic constipation. Acta Pharm. Sin. B 5, 300–309. doi: 10.1016/j.apsb.2015.05.006
Kato, N., Liu, C.-X., Kato, H., Watanabe, K., Tanaka, Y., Yamamoto, T., et al. (2000). A new subtype of the metalloprotease toxin gene and the incidence of the three bft subtypes among Bacteroides fragilis isolates in Japan. FEMS Microbiol. Lett. 182, 171–176. doi: 10.1111/j.1574-6968.2000.tb08892.x
Lodish, H., Berk, A., and Zipursky, S. L. (2000). Molecular Cell Biology. 4th Edn. Section 3.1: Hierarchical Structure of Proteins. New York, NY: W.H. Freeman & Company.
Makishima, M., Okamoto, A. Y., Repa, J. J., Tu, H., Learned, R. M., Luk, A., et al. (1999). Identification of a nuclear receptor for bile acids. Science 284, 1362–1365. doi: 10.1126/science.284.5418.1362
Mok, H. Y. I., Bell, G. D., and Dowling, R. H. (1974). Effect of different doses of chenodeoxycholic acid on bile-lipid composition and on frequency of side-effects in patients with gallstones. Lancet 304, 253–257. doi: 10.1016/S0140-6736(74)91415-9
Moncrief, J. S., Obiso, R., Barroso, L. A., Kling, J. J., Wright, R. L., Van Tassell, R. L., et al. (1995). The enterotoxin of Bacteroides fragilis is a metalloprotease. Infect. Immun. 63, 175–181.
Ning, Y., Manegold, P. C., Hong, Y. K., Zhang, W., Pohl, A., Lurje, G., et al. (2011). Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int. J. Cancer 128, 2038–2049. doi: 10.1002/ijc.25562
Odunsi–Shiyanbade, S. T., Camilleri, M., McKinzie, S., Burton, D., Carlson, P., Busciglio, I. A., et al. (2010). Effects of Chenodeoxycholate and a Bile Acid Sequestrant, Colesevelam, on Intestinal Transit and Bowel Function. Clin. Gastroenterol. Hepatol. 8, 159–165.e5. doi: 10.1016/j.cgh.2009.10.020
Parks, D. J., Blanchard, S. G., Bledsoe, R. K., Chandra, G., Consler, T. G., Kliewer, S. A., et al. (1999). Bile acids: natural ligands for an orphan nuclear receptor. Science 284, 1365–1368. doi: 10.1126/science.284.5418.1365
Peleman, C., Camilleri, M., Busciglio, I., Burton, D., Donato, L., and Zinsmeister, A. R. (2017). Colonic transit and bile acid synthesis or excretion in patients with irritable bowel syndrome-diarrhea without bile acid malabsorption. Clin. Gastroenterol. Hepatol. 15, 720–727.e1. doi: 10.1016/j.cgh.2016.11.012
Rabizadeh, S., and Sears, C. (2008). New horizons for the infectious diseases specialist: how gut microflora promote health and disease. Curr. Infect. Dis. Rep. 10, 92–98. doi: 10.1007/s11908-008-0017-8
Rhee, K. J., Wu, S., Wu, X., Huso, D. L., Karim, B., Franco, A. A., et al. (2009). Induction of persistent colitis by a human commensal, enterotoxigenic Bacteroides fragilis, in wild-type C57BL/6 mice. Infect. Immun. 77, 1708–1718. doi: 10.1128/IAI.00814-08
Sears, C. L. (2001). The toxins of Bacteroides fragilis. Toxicon 39, 1737–1746. doi: 10.1016/S0041-0101(01)00160-X
Sears, C. L., and Garrett, W. S. (2014). Microbes, microbiota, and colon cancer. Cell Host Microbe 15, 317–328. doi: 10.1016/j.chom.2014.02.007
Staels, B., and Fonseca, V. A. (2009). Bile acids and metabolic regulation: mechanisms and clinical responses to bile acid sequestration. Diabetes Care 32(Suppl. 2), S237–S245. doi: 10.2337/dc09-S355
Valdés Olmos, R., den Hartog Jager, F., Hoefnagel, C., and Taal, B. (1991). Effect of loperamide and delay of bowel motility on bile acid malabsorption caused by late radiation damage and ileal resection. Eur. J. Nucl. Med. 18, 346–350. doi: 10.1007/BF02285463
Wang, H., Chen, J., Hollister, K., Sowers, L. C., and Forman, B. M. (1999). Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 3, 543–553. doi: 10.1016/S1097-2765(00)80348-2
Weikel, C. S., Grieco, F. D., Reuben, J., Myers, L. L., and Sack, R. B. (1992). Human colonic epithelial cells, HT29/C1, treated with crude Bacteroides fragilis enterotoxin dramatically alter their morphology. Infect. Immun. 60, 321–327.
Wexler, H. M. (2007). Bacteroides: the good, the bad, and the nitty-gritty. Clin. Microbiol. Rev. 20, 593–621. doi: 10.1128/CMR.00008-07
Wood, D. J., Vlieg, J., de Wagener, M., and Ritschel, T. (2012). Pharmacophore fingerprint-based approach to binding site subpocket similarity and its application to bioisostere replacement. J. Chem. Inf. Model. 52, 2031–2043. doi: 10.1021/ci3000776
Wu, S., Lim, K. C., Huang, J., Saidi, R. F., and Sears, C. L. (1998). Bacteroides fragilis enterotoxin cleaves the zonula adherens protein, E-cadherin. Proc. Natl. Acad. Sci. U.S.A. 95, 14979–14984. doi: 10.1073/pnas.95.25.14979
Wu, S., Powell, J., Mathioudakis, N., Kane, S., Fernandez, E., and Sears, C. L. (2004). Bacteroides fragilis enterotoxin induces intestinal epithelial cell secretion of interleukin-8 through mitogen-activated protein kinases and a tyrosine kinase-regulated nuclear factor-κB pathway. Infect. Immun. 72, 5832–5839. doi: 10.1128/IAI.72.10.5832-5839.2004
Wu, S., Shin, J., Zhang, G., Cohen, M., Franco, A., and Sears, C. L. (2006). The Bacteroides fragilis toxin binds to a specific intestinal epithelial cell receptor. Infect. Immun. 74, 5382–5390. doi: 10.1128/IAI.00060-06
Keywords: computational drug design and discovery, bacterial toxin, colorectal cancer, inflammatory bowel disease (IBD), Bacteroides fragilis enterotoxin, enterotoxigenic Bacteroides fragilis (ETBF), drug screening, chenodeoxycholic acid
Citation: Metz P, Tjan MJH, Wu S, Pervaiz M, Hermans S, Shettigar A, Sears CL, Ritschel T, Dutilh BE and Boleij A (2019) Drug Discovery and Repurposing Inhibits a Major Gut Pathogen-Derived Oncogenic Toxin. Front. Cell. Infect. Microbiol. 9:364. doi: 10.3389/fcimb.2019.00364
Received: 31 July 2019; Accepted: 08 October 2019;
Published: 25 October 2019.
Edited by:
Andrew T. Gewirtz, Georgia State University, United StatesReviewed by:
Ashu Sharma, University at Buffalo, United StatesCopyright © 2019 Metz, Tjan, Wu, Pervaiz, Hermans, Shettigar, Sears, Ritschel, Dutilh and Boleij. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Annemarie Boleij, YW5uZW1hcmllLmJvbGVpakByYWRib3VkdW1jLm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.