 Lu Jia
Lu Jia Nannan Han1
Nannan Han1 Juan Du
Juan Du
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 18 July 2019
Sec. Microbes and Innate Immunity
Volume 9 - 2019 | https://doi.org/10.3389/fcimb.2019.00262
Periodontitis is a common intraoral infection and is inextricably linked to systemic diseases. Recently, the regulation between host immunologic response and periodontal pathogens has become a hotspot to explain the mechanism of periodontitis and related systemic diseases. Since Porphyromonas gingivalis (P. gingivalis) was proved as critical periodontal pathogen above all, researches focusing on the mechanism of its virulence factors have received extensive attention. Studies have shown that in the development of periodontitis, in addition to the direct release of virulent factors by periodontal pathogens to destroy periodontal tissues, over-low or over-high intrinsic immune and inflammatory response mediated by Toll-like receptors (TLRs) can lead to more lasting destruction of periodontal tissues. It is very necessary to sort out how various cytopathic factors of P. gingivalis mediate inflammation and immune responses between the host through TLRs so as to help precisely prevent, diagnose, and treat periodontitis in clinic. This review summarizes the role of three most widely studied pathogenic factors produced by P. gingivalis (lipopolysaccharide, gingipains, pili) and their interactions with TLRs at the cellular and molecular level in the progress of periodontitis.
Periodontitis refers to inflammatory pathological damage of the gums and periodontal support tissues, including the gums, alveolar bone, periodontal ligament, and cementum. Untreated periodontitis can cause the formation of deep periodontal pockets, which eventually lead to loosening of the teeth (Pihlstrom et al., 2005; Bostanci and Belibasakis, 2012). According to a survey by the World Health Organization, 10–15% of adults worldwide suffer from periodontitis (Petersen and Ogawa, 2012). The factor initiating periodontitis is biofilm plaques, in which Porphyromonas gingivalis (P. gingivalis), a Gram-negative anaerobe and component of the “red complex” (categorized together with Tannerella forsythia and Treponema denticola, highly relevant to periodontitis), has been irrefutably shown to be the key pathogen underlying the pathogenesis of chronic periodontitis (CP) (Parahitiyawa et al., 2010). Furthermore, host inflammatory and immune responses to microbial communities change the subgingival environment, causing low-abundant key opportunistic pathogens such as P. gingivalis to become the dominant bacteria in the biofilm, thus breaking the homeostasis between symbiotic microorganisms and the host, promoting the development of periodontitis, and even triggering systemic diseases (Abdi et al., 2017). For example, animal experiments have been used to demonstrate that P. gingivalis can also colonize some distant organs, such as the coronary artery, placenta, liver, and even the brain, causing specific infections associated with the activation of Toll-like receptors (TLRs) (Olsen and Yilmaz, 2016; Huck et al., 2018). Based on these data, an increasing number of scholars are investing in molecular biology to explore the pathogenic mechanisms of P. gingivalis (Olsen et al., 2018). However, the specific pathogenic effect of P. gingivalis virulence factors remain incompletely understood, and many topics remain controversial.
The so-called virulence factors are molecules that cause damage to the host at different stages of the organism's (bacteria, viruses, fungi, and protozoa) life cycle, increasing their effectiveness. They mainly involve the following functions: (1) colonization in the host; (2) immune escape; (3) immunosuppression; (4) cellular entry and exit; (5) extraction of nutrients from the host; and (6) release of virulence factors (How et al., 2016). Porphyromonas gingivalis can produce various virulence factors, such as lipopolysaccharide (LPS), gingipains, fimbriae/pili, collagenase, (erythrocyte) lectins, capsule, protease, and superoxide dismutase, to evade the host immune defense system and destroy host periodontal tissues. Recent studies have confirmed that LPS, gingipains, and fimbriae/pili are the most important pathogenic substances of P. gingivalis and the most widely studied in the field of periodontitis, and each of these factors play key roles in the progression of periodontitis (Mysak et al., 2014).
Although periodontal pathogens play very important roles in the initial stage of periodontitis, directly destructing host periodontal tissues by releasing toxic factors and metabolites, the progression of periodontitis is regulated by mainly the interaction between the host immune response and periodontal pathogens. Because the indirect damage caused by host congenital and adaptive immunological responses that are overactivated or blocked by periodontal pathogens is more traumatic and lasts longer, either low reactivity or hyperresponsiveness of the immune response results in persistent periodontal tissue damage (Meyle et al., 2017). As we know, an organism first relies on innate immune responses to resist pathogenic microbial invasion, which is also a prerequisite for adaptive immunity. Typical pathogenic molecules are identified by multiple cell surface receptors, which are called pattern recognition receptors (PRRs) and include TLRs, NOD-like receptors (NLRs), C-type lectin receptors (CLRs), and RIG-I-like receptors (RLRs). After numerous pathogen-associated molecular patterns (PAMPs) are distinguished by these receptors, the intracellular signal transmission pathways are initiated, thereby stimulating the expression of inducible costimulators and releasing inflammatory factors, chemotactic factors, and interferons (except those of the gamma type), among others (Akira et al., 2006). TLRs are the most characteristic PRRs that activate and are widely expressed in multiple cell types, including neutrophils, macrophages, keratinocytes, and fibroblasts (Akira and Takeda, 2004). The key point in the innate immune response during the pathogenesis of periodontitis lies in the recognition between pathogenic factors and PRRs. A new case-control study comparing the relationship between early periodontitis and single nucleotide polymorphisms (SNPs) of TLRs, NLRs, and RLRs showed that TLR polymorphisms are associated with the susceptibility of adolescents to periodontitis (Leite et al., 2018). Some scholars performing a meta-analysis of the susceptibility of TLR4 polymorphism to periodontitis revealed that TLR4 C>G (rs7873784) may be associated with CP in the Asian population and be transmitted to the next generation in a recessive form (Jin et al., 2016). However, controversy exists regarding this topic (Song et al., 2013; Chrzeszczyk et al., 2015).
This review focuses specifically on how three critical P. gingivalis pathogenic factors explicitly contribute to the pathogenesis of periodontitis and how they mediate innate immunoinflammatory host responses via different TLRs.
Porphyromonas gingivalis-LPS, located on the lateral lobule of bacterial adventitia, is a bacterial endotoxin composed of lipid A (a conserved inner region without species specificity), core oligosaccharide (the bridge lipid A and O-polysaccharide), and O-specific polysaccharide (has a highly variable outer region) that has numerous biological activities and plays a strong pathogenic role in periodontal tissues (Schromm et al., 2000). Lipid A is the core factor underlying the immunological activity of LPS and is structurally composed of a phosphorylated β (1-6) D-glucosamine disaccharide backbone and multiacyl chains acylated by fatty acids at specific positions on the backbone (Dixon and Darveau, 2005; the schematic diagram of LPS structure shown in Figure 1A). P. gingivalis-LPS is released after the lysis of bacteria or as free vesicles outward from the outer membranes of living bacteria. These LPS-containing microvesicles, which function like “microbullets” that land on the host, further perpetuate the invasiveness of P. gingivalis, giving it the ability to destroy periodontal tissues and trigger inflammation (Zhang et al., 2015; Singhrao and Olsen, 2018). The basic chemical composition of P. gingivalis-LPS is nearly identical to that of a typical bacterial endotoxin. The key difference is that the LPS lipid A structure produced by P. gingivalis can undergo isomeric acylation by two modes (tetra-acylation and penta-acylation) depending on the environmental factors, such as the hemin levels, phosphate availability, and incubation temperatures, thus initiating differential immunoinflammatory responses (Rangarajan et al., 2017). Accordingly, P. gingivalis-LPS with tetra-acylated lipid A has been designated LPS1435/1449 based on its molecular weights of 1435 and 1449 Da, while the other molecule is named P. gingivalis-LPS1690 based on its molecular weight of 1690 Da (Curtis et al., 2011). Thus, TLR4 and TLR2 can be simultaneously activated after recognition of P. gingivalis-LPS, while E. coli-LPS can activate only TLR4 (Liu et al., 2008; Coats et al., 2009; different acylation structures of P. gingivalis-LPS and E. coli-LPS shown in Figure 1B). Therefore, the heterogeneous form of P. gingivalis-LPS is identified as a PAMP, and its regulatory role associated with host cell-specific TLRs has been extensively studied (Nichols et al., 2012).
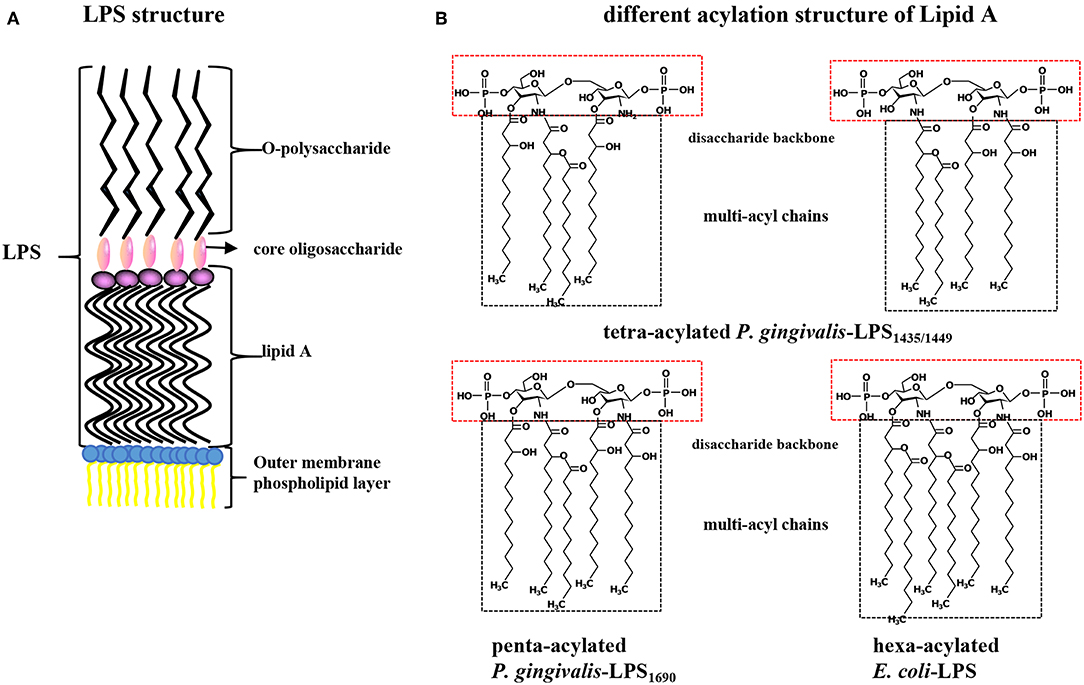
Figure 1. (A) Structure diagram of P. gingivalis-LPS. LPS is composed of lipid A (the conserved inner region without species specificity), core oligosaccharide (the bridge between lipid A and O-polysaccharide), and O-specific polysaccharide (a highly variable outer region) and located on bacterial outer membrane. (B) Contrast different acylation structure of P. gingivalis and E. coli LPS. Lipid A is composed of a phosphorylated β (1-6) D-glucosamine disaccharide backbone and multi-acyl chains acylated by fatty acids at specific positions. P. gingivalis-LPS with tetra-acylated chains is designated as LPS1435/1449 with its molecular weight of 1435 and 1449 Da, while the penta-acylated is named P. gingivalis-LPS1690 with a molecular weight of 1690 Da. The lipid A of E. coli-LPS is hexa-acylated.
The combination of LPS and the MD-2/TLR4 complex triggers the host innate immune response, induces inflammation and cytokine production, and activates effector cells and complementary systems. In the TLR family, TLR4 is unique because it must form a dimer complex with MD-2 to capture its ligand LPS. Because multiple acyl chains of LPS lipid A are key to the MD-2/TLR4 complex, the LPS poly-acyl chains must be protected until they are incorporated into the MD-2/TLR4 complex. In this process, LPS-binding protein (LBP) and CD14, key auxiliary molecules, improve the efficiency of LPS transport, and sensitivity of detection (Ryu et al., 2017). LBP, a 60 kDa serum glycoprotein, is mainly produced by hepatocytes, lung, and gastrointestinal epithelial cells. Compared with those of other acute-phase proteins, the serum concentration of LBP increases slowly, peaking at ~2–3 days after acute infection. Surprisingly, a high concentration of LBP inhibits inflammation induced by LPS to some extent (Zweigner et al., 2001). Low concentrations of LBP have a high affinity for lipid A and thus promote formation of the LBP-LPS complex, which is subsequently transported to membrane CD14 (mCD14) or soluble CD14 (sCD14) (Ding and Jin, 2014). Soluble CD14 in the serum makes CD14-deficient cells, including most epithelial and endothelial cells, respond to LPS. Once CD14 binds to the LBP-LPS complex, LPS is transiently transferred to CD14, which has a hydrophobic pocket at its N-terminus that serves as the LPS binding site (Kelley et al., 2013). Transport of LPS to surface MD-2/TLR4 in the form of the LBP-LPS-CD14 complex is a prerequisite for TLR4 internalization and subsequent immune response (Tsukamoto et al., 2018). Notably, LBP expression in gingival epithelial cells (GECs) of the gingival-dental junction is significantly lower in CP patients than in healthy people (Ding and Jin, 2014). Research has shown that the addition of exogenous sCD14 substantially promotes the ability of human periodontal ligament stem cells (PDLSCs) to produce the inflammatory factors activated by P. gingivalis-LPS, including IL-6, IL-8 and chemokine (C-C motif) ligand 2 (CCL2) (Andrukhov et al., 2016). In fact, one study showed that both LBP and exogenous sCD14 could promote the internalization of TLR4, but the concentration of sCD14 required was higher than that of LBP (Tsukamoto et al., 2018).
In terms of the TLR-mediated innate immune response induced by Gram-negative bacteria, such as P. gingivalis and C. burnetii (the pathogen that causes Q fever), TLR2 and TLR4 are the most widely studied (Ramstead et al., 2016; Song et al., 2017). In particular, TLR2 requires heterodimerization with TLR1 or TLR6 to function properly, wherein the TLR2/TLR1 binding region is triacylated lipopeptides, such as Pam3CSK4, and the TLR2/TLR6 ligand is diacylated lipid/lipopeptides, such as lipoteichoic acid (LTA) (Nguyen et al., 2017). Based on this information, different subspecies of C. burnetii function differently in innate immune recognition; for example, C. burnetii Nine Mile is mediated by only the TLR1/TLR2 heterodimer, while C. burnetii 3262 is recognized by both TLR1/TLR2 and TLR2/TLR6 (Ammerdorffer et al., 2015). TLRs, as type I transmembrane proteins, are composed of a transmembrane structure, an extracellular amino terminus domain (recognizing PAMPs) and an intracellular Toll/interleukin-1 receptor (TIR) homology domain. To date, ten TLR subtypes have been found in humans, among which TLR2/TLR1, TLR2/TLR6, TLR4, TLR5, and TLR10 are cell surface receptors mainly identifying proteins or lipids, and TLR3, TLR8, and TLR9 bind the endoplasmic reticulum membrane, mainly identifying nucleic acids (Akira and Takeda, 2004). In conjunction with the downstream TLRs, some adaptor protein molecules recognize the TIR domain, including myeloid differentiation factor-88 (MyD88), MyD88-adapter-like/TIR-domain-containing adaptor protein (Mal/TIRAP), TIR-domain-containing adaptor-inducing IFN-β (TRIF), TRIF-related adaptor molecule (TRAM) and selective androgen receptor modulators (SARM). According to the adaptor protein recruited, TLR active pathways are divided into MyD88-dependent and TRIF-dependent pathways. TLR2/TLR1, TLR2/TLR6, TLR5, TLR7/TLR8, and TLR9 are all related to the MyD88-dependent pathway. TLR3 is activated through a TRIF-dependent pathway. TLR4 can activate the MyD88 pathway on the plasma membrane and then trigger the TRIF pathway by activating the TRAM adaptor on the endosomes (Liu et al., 2014; Chen et al., 2016). When MyD88 is activated as the downstream binding protein, nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), activating protein (AP-1) and interferon response factors 1, 5, and 7 (IRF-1, IRF-5, IRF-7) became active, inducing the participation of inflammatory factor genes, while the TRIF pathway ultimately results in the sensitization of IRF-3 and activation of type I IFN-inducible genes (Roshan et al., 2016). The extracellular domain of TLR4 must initially bind to the MD-2 molecule to achieve the most powerful recognition and transmission of LPS. By forming a homodimer or a larger complex with the help of MD-2, TLR4 exhibits more binding sites for PAMPs on the cell membrane, significantly enhancing the activation of the NF-κB pathway (Visintin et al., 2001). The radioprotective 105 kDa protein (RP105, also termed CD180), originally discovered to protect B cells from radiation-induced apoptosis, is a specific inhibitor of TLR4. As the specific homolog of TLR4, CD180 is not the same Toll family receptor as other TLRs due to the loss of an intracellular signaling domain. Because it is physically associated with the MD-2-like molecule MD-1, substantial structural similarity but contrasting regulation exists between CD180 and TLR4 (Yang et al., 2018). Therefore, both MD-1/CD180 and MD-2/TLR4 are cell surface receptors that mediate LPS signaling pathways that have similar structures but opposite functions (Maeshima and Fernandez, 2013).
Previous research reported that the mediation of TLR2 is essential for the loss of alveolar bone caused by P. gingivalis in animal models, and treatment of E. coli-LPS-tolerant bone marrow-derived macrophages (BMDM) with P. gingivalis resulted in upregulation of TLR2 expression and excessive tumor necrosis factor (TNF) production in vitro (Papadopoulos et al., 2013). In fact, questions regarding the leading signaling pathway activated in P. gingivalis-LPS-triggered immunoinflammatory reactions are controversial, as studies found that TLR4 exerts a dominant function, while others demonstrated that TLR2 is the primary contributor (Wang and Ohura, 2002). Some studies have reported that downstream cytokine changes are mainly caused by the NF-κB pathway, while others believe that the mitogen-activated protein kinase (MAPK) pathway and others, such as Jun N-terminal kinases (JNKs) and phosphatidylinositol 3-kinase (PI3K), are active during this process (Bainbridge and Darveau, 2001; Liu et al., 2013). For example, researchers found that P. gingivalis-LPS reduced the osteogenic polarization potential of PDLSCs via the TLR4-NF-κB signaling pathway. Moreover, blocking either TLR4 or NF-κB signaling hindered LPS-induced alveolar bone loss (Li et al., 2014). Other results indicated that P. gingivalis-LPS activates M1 and M2 macrophages mainly via TLR2, accompanied by the phenomena that high concentrations of LPS stimulate M1 macrophages to produce nitric oxide (NO), while low concentrations primarily increase the expression of TNF-α and IL-6 (Holden et al., 2014).
Regarding the involvement of the TLR4 pathway, penta-acylated P. gingivalis-LPS1690 is similar to classical hexa-acylated E. coli-LPS to some extent (Bozkurt et al., 2017). For example, both P. gingivalis-LPS1690 and E. coli-LPS elevate the expression of the LBP protein through TLR4 in human oral keratinocytes, among which the former induction is achieved via NF-κB and p38/MPAK pathways, whereas the latter regulation is controlled by the NF-κB, p38/MPAK and JNK signaling pathways (Ding et al., 2013). Another study showed that P. gingivalis-LPS distinctly enhanced IL-6 mRNA expression but downregulated the cell surface molecules TLR2 and TLR4, but not at the transcriptional level, while E. coli-LPS induced a similar but more obvious alteration than P. gingivalis-LPS (Andrukhov et al., 2014). However, some differences do exist between P. gingivalis-LPS1690 and E. coli-LPS based on cell types and their exact molecular biological activities. For example, P. gingivalis-LPS1690 significantly enhanced the transcription of NF-κB in mouse cementoblasts via TLR2, while E. coli-LPS was regulated by TLR4 (Nemoto et al., 2006). In addition, a study revealed that P. gingivalis-LPS1690 not only upregulated TLR4 expression in human gingival fibroblasts (hGFs) with positive correlations of dose and time, but also induced the expression of TLR2 (Herath et al., 2013a). Another study further demonstrated that the 16 kDa lipoprotein from P. gingivalis-LPS extracted by the phenol-water method activated hGFs to induce IL-8 production via the NF-κB pathway by means of TLR2, while E. coli-LPS was assisted by only TLR4 (Hashimoto et al., 2004).
In fact, signaling pathways triggered by P. gingivalis-LPS are closely related to its acylated lipid A isomer and target cells. As mentioned earlier, P. gingivalis can be classified into two groups based on the different acylation configurations of lipid A, namely, penta-acylated LPS1690 and tetra-acylated LPS1435/1449, which differentially modulate innate host response and produce diverse substances, e.g., skin-antimicrobial peptide 1 (SAP1), inflammatory cytokines (IL-6, IL-8), and MMP-3 (Reife et al., 2006; Herath et al., 2011, 2013b). For example, P. gingivalis-LPS1690, but not LPS1435/1449, significantly increased TLR4, and MD-2 expression in hGFs at concentrations ≥0.1 μg/mL. Both of LPS1690 and LPS1435/1449 could significantly enhance the expression of TLR2 in hGFs, but the effect of LPS1435/1449 was stronger than that of LPS1690 (Herath et al., 2013a). Subsequent experiments demonstrated that P. gingivalis-LPS1690 significantly activated the NF-κB (mainly), p38/MAPK, and extracellular signal-regulated kinase (ERK)1/2 signaling pathways, while P. gingivalis-LPS1435/1449 primarily induced the p38/MAPK and ERK1/2 signaling pathways with little sensitization of the NF-κB pathway (Figure 2).
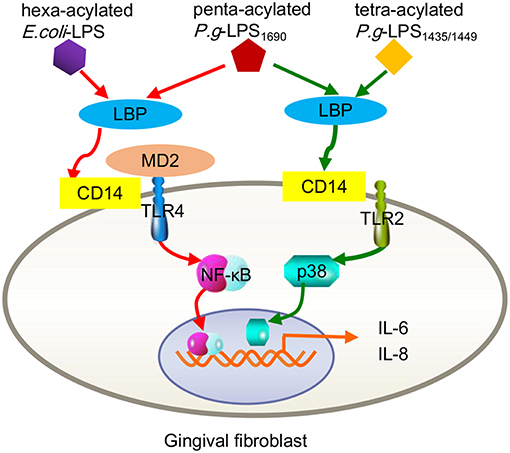
Figure 2. Contrast inflammatory signals in human gingival fibroblasts triggered by the different acylation structures of P. gingivalis LPS and E. coli LPS. P.g-LPS1690 upregulates the expression of IL-6 and IL-8, mainly via the MD-2/TLR4-NF-κB pathway. This process seemed to mimic hexa-acylated E. coli-LPS. Both P.g LPS1435/1449 and LPS1690 stimulate increased expression of TLR2, but E. coli LPS did not trigger TLR2. P.g-LPS1435/1449 primarily induced the p38/MAPK signaling pathway with little sensitization of the NF-κB pathway.
Interestingly, P. gingivalis-LPS1435/1449 can block the expression of ELAM-1 induced by E. coli-LPS on human endothelial cells at the level of combination with the TLR4 receptor ectodomain, exhibiting a TLR4 antagonist ability (Coats et al., 2003). As a structure-determined function, researchers proposed that the shared MD-2 and MD-1 protein module mediates diverse biological functions through specific interactions with lipid structures (Nagai et al., 2005). As mentioned earlier, the MD-2/TLR4 compound-mediated immune response induced by bacterial LPS is closely associated with the combined MD-1/CD180 formation (Nagai et al., 2005). Due to the common features and functional interrelationship of MD-1/CD180 and TLR4/MD-2, the LPS-originating pathway can be restrained by MD-1/CD180 acting as an MD-2/TLR4 antagonist, which occurs in a wide ranges of cell types, including human embryonic kidney 293 (HEK293) cells, dendritic cells, and macrophages (Divanovic et al., 2005). In fact, this antagonistic activity results from the combination of the TLR4 SV1 splicing variant binding with MD-2, resulting in the absence of signaling molecules in the extracellular N-terminal domain (Coats et al., 2005). As another example, a recent study found that LBP alone can upregulate IL-6 expression in human oral keratinocytes, a process mediated by TLR2 and involving NF-κB, JNK/p38 and IRF. When P. gingivalis-LPS1435/1449 and LPS1690 interacted with LBP, both downregulated the expression of IL-6 in keratinocytes, and LPS1435/1449 was downregulated to a greater extent than LPS1690. Further studies verified that the expression levels of both CD180 and MD-1 were significantly increased after LPS1435/1449 bound LBP, while the combination of LPS1690 and LBP upregulated CD180 expression but decreased MD-1 expression. This result provided a precise explanation for the previously mentioned result that the downregulation of IL-6 caused by LPS1435/1449 binding with LBP was more obvious than that of LPS1690 (Figure 3). The above results lead to the conclusion that MD-1/CD180 acts as a fine-tuning mediator of the regulation of P. gingivalis-LPS heterogeneity induced signaling pathways (Ding et al., 2017).
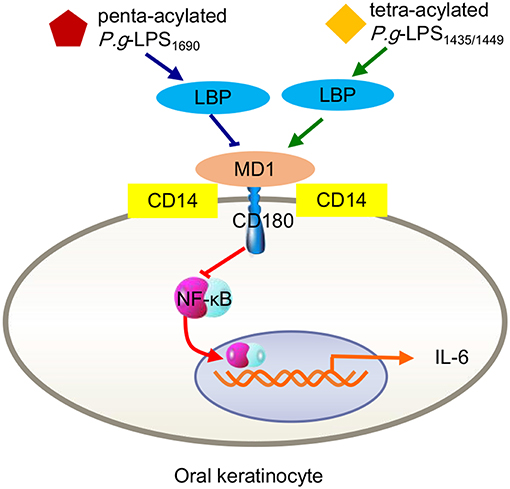
Figure 3. Inflammatory signals in human oral keratinocytes triggered by P. gingivalis-LPS heterogeneity. Either tetra-acylated P.g-LPS1435/1449 or penta-acylated LPS1690 downregulated the expression of IL-6 through the MD-2/TLR4-NF-κB pathway. Because MD-1/CD180 complexes negatively mediate the MD-2/TLR4 pathway, the degree of LPS1435/1449 downregulation via increasing MD-1 was more pronounced than that of LPS1690 via reducing MD-1.
In summary, we can draw the following conclusions: regarding the TLR2 pathway, either P. gingivalis LPS1690 or LPS1435/1449 serves as an excitomotor; in particular, LPS1690 is a strong TLR4 agonist, while LPS1435/1449 is only a weak stimulant, and more precisely, a potent antagonist of TLR4 (Herath et al., 2013a). In vitro experiments indicated that the response to P. gingivalis-LPS varied considerably depending on the cell types examined or the type of LPS produced (especially in some experiments, as LPS extracted from P. gingivalis was not completely pure; Darveau et al., 2004). Furthermore, both P. gingivalis LPS1690 and LPS1435/1449 increased the amount of IFN-γ in whole blood cell cultures (WBCCs) from CP patients, and the former showed higher secretion results. Moreover, P. gingivalis-LPS1690 could trigger the abnormal whole blood cell secretion of IL-10 in healthy individuals, while P. gingivalis-LPS1435/1449 could not, further proving that different types of P. gingivalis-LPS induce unequivocal changes in the blood systems of CP patients (Nogueira-Filho et al., 2014). On the other hand, considering the complex circumstances of local and systemic hosts in vivo, one study demonstrated that both LPS1690 and LPS1435/1449 were effective stimulators of inflammation, and LPS1435/1449 was more effective than LPS1690 but less harmful than E. coli-LPS at the site of inoculation (Liu et al., 2008). Currently, how host cells protect against different P. gingivalis phenotypes dependent on the microenvironmental condition is not fully understood (Ding et al., 2017). However, changing the heterogeneous lipid A structure to adapt to tissue-specific cellular receptor signaling pathways has clearly been demonstrated to be a mechanism by which P. gingivalis avoids the host's innate immune attack and accesses the periodontal tissue (Olsen and Singhrao, 2018).
Gingipains (named after P. gingivalis clostripain), belonging to the cysteine protease family and existing in the outer membranes, vesicles, and extracellular structures of P. gingivalis, are important virulence factors that exert the vital function of mediating the interaction between P. gingivalis bacteria and hosts (Yongqing et al., 2011). Gingipains can be divided into two categories: arginine-dependent gingipain R (Rgp) and lysine-dependent gingipain K (Kgp). Together, the two kinds of P. gingivalis gingipains complete 85% of the proteolysis outside the cell (de Diego et al., 2014). Rgp has been further subdivided into RgpA and RgpB on the basis of structure. The molecular weights of RgpA, RgpB, and Kgp, which are coded by the rgpA, rgpB, and kgp genes, are 95, 50, and 105 kDa, respectively. Collectively, these enzymes are primarily composed of the following components: a signal peptide, an N-terminal domain, a catalytic domain (CD), an immunoglobulin superfamily-like domain (IgSF), a hemagglutinin/adhesion (HA) domain and a C-terminal domain (Nakayama, 2015). Among these proteins, the structure of RgpB is the simplest, as it has no HA domain, while RgpA contains four HA domains (called RgpAA1-RgpAA4) located in the middle of the IgSF and C-terminal domains. Kgp also has 3 to 5 such domains (called KgpAA1-KgpAA5) in different bacterial strains (de Diego et al., 2013; structure of gingipains shown in Figure 4). The CDs of RgpB and RgpA are highly homologous in terms of their amino acid sequences, but no proteins with CD structures similar to those of Kgp have been reported. Based on analysis of the high-resolution crystal structure of Kgp competent fragments, the key catalytic mechanism of Kgp probably lies in the requirement of a triplet (C477-H444-D388) instead of a cysteine-histidine dimer (de Diego et al., 2014; Gorman et al., 2015). In addition, dimerization of the pro-domain also plays a substantial role in the specific latency mechanism of Kgp (Pomowski et al., 2017). In an animal experiment designed to test the efficacy of three gene vaccines (rgpA, rgpB, and kgp) in treating peri-implant inflammation, the kgp and rgpA DNA vaccines enhanced immune responses and significantly retarded alveolar bone loss in vivo, whereas the rgpB vaccine was ineffective (Guo et al., 2014). Given the different biological properties of RgpA, RgpB, and Kgp, the virulence rankings of the three gingipains explored using periodontitis model mice was Kgp > or = RgpB >> RgpA (Pathirana et al., 2007).

Figure 4. Structure of gingipains. Gingipains are divided into Rgp (arginine-dependent gingipain R) and Kgp (lysine-dependent gingipain K). Rgp has been further subdivided into RgpA and RgpB. The molecular weight of RgpA, RgpB and Kgp is 95, 50, and 105 kDa, respectively. Gingipains are primarily made up of the following components: the signal peptide, the N-terminal domain, the catalytic domain (CD), the immunoglobulin superfamily-like domain (IgSF), the hemagglutinin/adhesion (HA) domain, and the C-terminal domain. The structure of RgpB is the simplest without HA domains. RgpA has four HA domains (called RgpAA1 to RgpAA4) located in the middle of the IgSF and C-terminal domain. Kgp also has 3–5 such domains (called KgpAA1 to KgpAA5) in the light of different bacterial strains.
The biosynthesis of gingipains (secretion and activation) is a complicated process that has not yet been fully explored. The overall process is as follows: when gingipains have just been translated, they are present in the form of an inactive zymogen to block the unwanted proteolytic activity inside the cell. After posttranslational processing, gingipains are converted into catalytically active proteases and then transported to the extracellular environment. Although the process of converting from an inactive zymogen to a catalytic gingipain is not fully defined, acetylated-lysine residues were found in the structures of these three mature proteases, suggesting that acetylation is a potential mechanism underlying gingipain activation and maturation (Ren et al., 2017). Herein, Vim A must be mentioned as a necessary acetyltransferase in the maturation process of gingipains to modulate the virulence of P. gingivalis, and it is indispensable for the maturation of RgpA (Aruni et al., 2013). However, RgpB and Kgp can be synthesized and secreted via a Vim A-independent pathway, which accounts for the late-onset protease activity observed in Vim A-deficient mutants (Dou et al., 2015). A recent study showed that PG1842 can act as an acetyltransferase to replace Vim A in the process of RgpB acetylation. In any case, acetylation is definitively a requisite posttranslational protein modification during the process of gingipain maturation (Mishra et al., 2018).
Regarding the status of P. gingivalis during oral biofilm, gingipain protease activity affects the composition of multimicrobial biofilm quantitatively and qualitatively (Hocevar et al., 2018). Gingipains also function as ligands in the coaggregation of P. gingivalis with other oral bacteria, such as T. denticola, to promote the colonization of P. gingivalis in dental plaque (Ito et al., 2010). In a biofilm model consisting of ten subgingival bacteria, the most prominent outcome of replacing the normal strain with a P. gingivalis Rgp/Kgp mutant was the change in the T. denticola three-dimensional distribution throughout the biofilm. Among these proteins, Rgp enhanced the growth of T. denticola, and Kgp promoted the accumulation of T. denticola in the biofilm. This synergistic effect was shown to be beneficial to not only the survival and virulence of biofilm colonies but also to their ability to form the red bacterial complex (Bao et al., 2014). Kgp has been proven to play a very important role in the shedding and reduction of biofilms, improving the competitive advantage of P. gingivalis in plaque biofilms. In addition, both RgpA and Kgp participate in P. gingivalis adhesion to oral epithelial cells, aggregating with other bacterial species (Sakanaka et al., 2016).
By virtue of the proteolytic action of gingipains, P. gingivalis cleaves or degrades a variety of host proteins to escape immune defense, including immunomodulatory proteins, signaling pathway regulatory proteins, and adhesion molecules (Barth et al., 2013; Hocevar et al., 2018). P. gingivalis can also recognize NLRs in a gingipain-independent manner, activate NLR pyrin domain-containing 3 (NLRP3)-modified inflammatory corpuscle and release the inflammatory factors IL-1β and TNF-α, inducing an inflammatory response in the host. Interestingly, gingipains released by P. gingivalis itself can degrade mature IL-1β and TNF-α, weakening the inflammatory response of the host to some extent. Both the positive and negative immunomodulatory effects of P. gingivalis on the host mentioned above are actually beneficial to long-lasting colonization in periodontal tissues (Jung et al., 2015). For instance, gingipains inhibit the cellular PI3K/Akt signaling pathway by cleaving extracellular PI3Kp85α-associated membrane proteins, thereby achieving immune regulation of GECs, and this process is independent of virulence factor invasion (Nakayama et al., 2015). The purified gingipains RgpA and Kgp downregulate mCD14 expression in a time- and concentration-dependent manner, resulting in low responsiveness of macrophages to P. gingivalis infection (Wilensky et al., 2015). This reduced mCD14 expression relies on the presence of the HA domain, causing RgpA and Kgp to exhibit the shedding enzyme effect, while RgpB is not active because of its lack of the HA domain. The weakening of CD14 efficacy is not only beneficial for the reproduction of eosinophilic bacteria in bacterial biofilms but also leads to more severe chronic inflammation (Olsen et al., 2017).
For the most part, P. gingivalis withstands the bactericidal lytic activity in blood serum because gingipains are the main force in combating complement systems, which can be applicated in hosts via three mechanisms: classical, lectin, and alternative. The initial stages of these three pathways differ, but the final outcomes involve the insertion of membrane attack complexes into cell membranes, inducing the generation of chemicals and phagocytosis of targeted Gram-negative bacteria (Ricklin et al., 2010). P. gingivalis-specific gingipains decompose C5 into C5a and C5b in two ways, by directly exerting C5 convertase-like activity and activating thrombin to replace C5 convertase by activating prothrombin (Hajishengallis et al., 2013a). It is worth noting that gingipains induce the polarization of M1 macrophages as a regulatory factor, thereby facilitating P. gingivalis infection via a C5a-mediated pathway (Hou et al., 2017). C5a is an effective chemotactic agent and phagocyte activator that is unfavorable for P. gingivalis. However, P. gingivalis itself has some resistance to complement dissolution due to the anionic polysaccharide structure of lipid A anchored on the surface (Olsen et al., 2017). In addition, RgpA hijacks and adsorbs C4b-binding protein, a complement physiological regulator, on the surface of P. gingivalis, inhibiting the classical and lectin pathways of the complement system (Hertz et al., 2018). More importantly, recent studies have found an important mechanism of combined action between the C5a receptor (C5aR) and TLR2 in macrophages to promote the adaptability of P. gingivalis (Wang et al., 2010). While the pili or lipoproteins of P. gingivalis activate TLR2/TLR1 on macrophages and induce a small amount of cyclic adenosine monophosphate (cAMP), C5a-C5aR synergistically enhances the weak cAMP response activated by TLR2/TLR1 alone by stimulating calcium-dependent intracellular signaling, resulting in a large amount of cAMP. Moreover, the continuous increase in cAMP activates cAMP-dependent protein kinase A (PKA) in macrophages and destroys the bactericidal function of inducible NO synthase (iNOS), which is dependent on NF-κB (Wang et al., 2010). In fact, the cross-talk mechanism of C5aR-TLR2 resulting in maximal macrophage cAMP production stimulated by P. gingivalis is specifically attributable to the assistance of CXC-chemokine receptor 4 (CXCR4), although the coassociation of C5aR and TLR2 in lipid rafts can effectively increase cAMP production in a CXCR4-independent manner (Wang et al., 2010). The regulatory action of the cAMP/PKA pathway on cell activity depends on mainly the binding of phosphorylated cAMP response element binding protein (CREB) to nuclear coactivator CREB-binding protein (CBP) (Dyson and Wright, 2016). Because intracellular CBP is limited, CREB (Ser133) phosphorylated by PKA and NF-κB p65 (Ser276) competently binds to CBP. Because cAMP/PKA mediates CREB to capture the binding CBP sites, the NF-κB pathway is suppressed (Figure 5). Moreover, upon the addition of glycogen synthase kinase 3β (GSK3β) inhibitors, the iNOS and NO produced by the coactivation of P. gingivalis-excited C5a/TLR2 were reduced to some extent but not completely inhibited, demonstrating that GSK3β partially promotes the iNOS signaling pathway. On the other hand, the aggregation of PKA also inhibits IRF-1, which is the key to the IFN-γ-mediated synergistic promotion of iNOS transcription (Salim et al., 2016). Therefore, the cross-talk between C5aR and TLR2 induces a cAMP-dependent immune overthrow, which leads to the reduction of iNOS and weakening of the bactericidal efficacy of macrophages. At the same time, GSK3β (Ser9) is inactivated by PKA phosphorylation, which is key to regulating proinflammatory and anti-inflammatory factors, inhibiting the production of TLR-mediated proinflammatory mediators (e.g., IL-6, TNF-α, IL-12, and IFN-γ) in host cells (Martin et al., 2005). The C5a-C5aR signaling axis also inhibits TLR4-induced IL-12p35, IL-12/23p40, and IL-23p19 expression at the transcriptional level in macrophages and downregulates IL-12p70 and IL-23 at the translation level by the PI3K and ERK1/2 signaling pathways, thereby alleviating latent tissue damage regulated by effector Th1 and Th17 cells (Weaver et al., 2007).
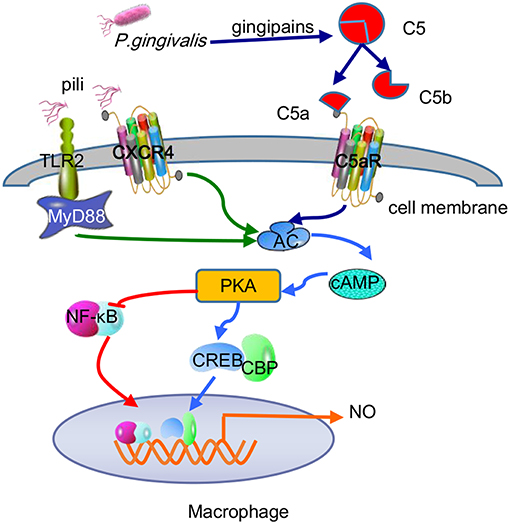
Figure 5. Antibactericidal mechanisms of C5a-TLR2-CXCR4 cross-talk induced by P. gingivalis in macrophages. P. gingivalis pili activated the TLR2-MyD88-dependent pathway and caused a small amount of cAMP production, while C5a-C5aR activation mediated by P. gingivalis gingipain-degradation of C5 synergistically enhances the production of cAMP. The combination of pili and CXCR4 helped maximize cAMP production via C5a-TLR2 cross-talk. The continuous increase in cAMP activated PKA to mediate CREB capturing the limited binding sites of CBP and inhibited NF-κB, thus reducing macrophage-forming NO and destroying the bactericidal function.
In addition to the powerful protein degradation function mentioned above, gingipains have recently been found to have an undeniable transpeptidation effect, which results from the transfer of several amino acids between peptide chains. Considering that heme must be ingested to provide iron and protoporphyrin IX and thus ensure viability and virulence (Ohya et al., 2016), P. gingivalis, an obligate anaerobic bacterium located in subgingival plaque, utilizes the transpeptidase activity of gingipains to extract the required nutrients from human hemoglobin (Smalley and Olczak, 2017). During this process, small peptides such as glycylglycine (GlyGly) not only enhance the proteolytic activity of gingipains but also act as receptor molecules participating in gingipain-catalyzed transpeptidation. In general, the trans-peptide reaction is much more efficient than the corresponding hydrolysis reaction, causing simple pathogenic proteins to become “transpeptidases” in a short time, which may induce the destruction of immune tolerance and trigger autoimmune diseases (Zhang et al., 2018). In addition, Kgp may not only act as a sensor of the hemoglobin concentration in the environment, thereby regulating the acetylation mode of lipid A in P. gingivalis-LPS, but also directly participate in the aggregation and degradation of hemoglobin, affecting its type, growth, and infection of P. gingivalis (Smalley and Olczak, 2017).
Pili are filamentous structures located on the P. gingivalis surface that enhance the bacterial adhesion to multiple types of surfaces, such as the extracellular matrix, host cells and other bacteria, and take part in the formation of biofilm (Nagano et al., 2013). P. gingivalis pili can be exhibited in at least two forms, namely, the major FimA and the minor Mfa, both of which regulate bacterial dependence on various molecules and oral substrates and are important for biofilm formation (Nagano et al., 2012). The major proteins, which protrude ~3 μm above the surface, are composed of the FimA protein subunit (the main P. gingivalis pilus subunit) and are encoded by the fimA gene, while the minor subunits, which are also called short fimbriae, are composed of mainly Mfa1 structural subunit proteins and are encoded by the mfa1 gene, and their lengths differ from 60 to 500 nm (Nakano and Amano, 2013). In addition to the primary Mfa1 protein, mature pili also have affiliated Mfa2-5 proteins. Mfa2 plays an anchor role, while Mfa3 can bind with Mfa1/2/4/5 in vitro, connecting with other pilus subunits as a binding protein (Ikai et al., 2015). Recent data indicate that the C-terminal domain of Mfa1, rather than Mfa3, affects the aggregation and maturation of downstream pilus proteins (Hall et al., 2018); however, the aggregation of Mfa1 does not depend on other pilus proteins but requires the proteolytic action produced by the gingipains RgpA and RgpB (Lee et al., 2018). Based on nucleotide sequence differences between the translational reading frame on the gene fimA, P. gingivalis is divided into six genotypes (type I to V and Ib). Among these genotypes, the fimA genotypes II and IV are widely distributed in periodontitis isolates (Nagano et al., 2018). The adhesive and invasive abilities of epithelial cells in patients with type II fimA clones were shown to be significantly enhanced compared with those of other fimA clones, suggesting that these clones are most closely associated with CP (Enersen et al., 2008). A recent study showed that type II fimA genotype is also detected at high rates in patients with both periodontitis and rheumatoid arthritis (RA) (Ayala-Herrera et al., 2018).
Adhesion is an indispensable process in the pathological development of periodontitis. Because the pili are the most prominent structure on the surface of bacteria, it is highly likely that their attachment is the first step in the reaction between microorganisms and the host (Mantri et al., 2015). It has been confirmed that pili can mediate the attachment of P. gingivalis to hydroxyapatite, hGFs and epithelia (Sojar et al., 2002; Baek et al., 2013). Porphyromonas gingivalis long fimbrial proteins were capable of activating human GECs through TLR2 with a complex of sCD14 and LBP and significantly upregulating IL-8 expression and NF-κB activation, which were involved in bone resorption (Kusumoto et al., 2004). Porphyromonas gingivalis minor pili not only enhanced the bone resorption of osteoclasts by producing IL-1β, TNF-α, and IL-6 but also promoted the differentiation of osteoclast precursor cells. The addition of anti-TLR2 antibody significantly inhibited the formation of osteoclasts caused by short fimbriae, while anti-TLR4 antibody did not obviously block pit formation on dentine blocks (Hiramine et al., 2003). Moreover, similar results demonstrated that the 67-kDa minor pili on the P. gingivalis surface stimulated the expression of TNF-α, IL-1α, IL-1β, and IL-6 cytokines in macrophages via the TLR2/complement receptor 3 (CR3) pathway, which showed the vital function of regulating immunity and mediating tissue damage during the development of periodontitis (Wang et al., 2007). Similar to CD14 regulating the sensitivity of TLR4-MD2 combination with LPS, P. gingivalis fimbriae upregulated IL-6 expression via the TLR2-p38/MAPK pathway in human monocytes, while LBP improved the reaction (Pollreisz et al., 2010). In addition, although sCD14 is a necessary molecule to recognize P. gingivalis by epithelial cells, mCD14 can cause a more violent reaction in response to activation of downstream pathways, producing IL-6, IL-8, granulocyte-macrophage colony-stimulating factor (GM-CSF), and TNF-α (Eskan et al., 2007).
It was shown that the different combinations between the coreceptor and TLR2 directly affect the activation of microbial molecules and TLR2; for instance, CXCR4 acts as a pattern recognition receptor and is also a coreceptor for TLR2 (van Bergenhenegouwen et al., 2013). The combination of pili and CXCR4 blocks the production of TNF-α mediated by NF-κB activation and simultaneously promotes IL-10 production. Further studies indicated that cross-talk exists between the TLR2/CXCR4 signaling pathway and the inhibition of the NF-κB pathway following TLR2 recognition of the pili. The key factor of this mechanism is that cAMP-dependent PKA inhibits the MyD88-dependent antibacterial pathway induced by TLR2 activation, which is the signaling transducer downstream of CXCR4 as mentioned before, reducing macrophage-forming NO and enhancing P. gingivalis against host clearance (Hajishengallis et al., 2008). In addition, the recognition of CXCR4 by P. gingivalis pili can activate the β2 integrin CR3 induced by PI3K in a TLR2-dependent manner, representing another cellular adhesion signaling pathway widely distributed in neutrophils, natural killer (NK) cells and macrophages. Upon CR3 activation, the safe entry of P. gingivalis into macrophages is mediated, simultaneously inhibiting the production of IL-12p70 and enhancing its capacity to evade death (Hajishengallis et al., 2013b).
Many studies have concluded that TLR2-MyD88 is a classical inflammatory pathway that mediates the periodontal tissue destruction caused by P. gingivalis (Burns et al., 2010). Recently, the TLR2-MAL/TIRAP-PI3K pathway was shown to promote P. gingivalis infection-driven alveolar bone resorption in vivo, even in the absence of the typical TLR2 adapter protein MyD88. At the same time, in vitro experiments demonstrated that activation of the TLR2-PI3K signal caused neutrophils to produce the proinflammatory cytokine TNF-α, thus inhibiting the maturation of phagosomes and prolonging survival time in host cells (Makkawi et al., 2017). While MyD88 contributes to the defense of neutrophils against P. gingivalis, pathogens can eliminate this host anti-infection mechanism by inducing the degradation of MyD88 in both humans and mice. The ability of P. gingivalis to degenerate MyD88 in neutrophils has been proven to be achieved through the C5aR-TLR2 cross-talk mechanism, in which the ubiquitin-proteasome and smad ubiquitin regulatory factor 1 (Smurfl) are involved, rather than via cAMP-dependent PKA (Maekawa et al., 2014). Furthermore, the MyD88-independent signaling pathway was also shown to work together with C5aR-TLR2 to promote P. gingivalis infection, during which PI3K acts as an effector of C5aR-TLR2 induced by the adaptor TIRAP (Hajishengallis, 2015) and blocks Ras homolog gene family member A (RhoA) activation and actin polymerization, thereby inhibiting P. gingivalis phagocytosis (Figure 6). In this regard, this result differs from the C5aR-TLR2 cross-talk in macrophages mentioned earlier.
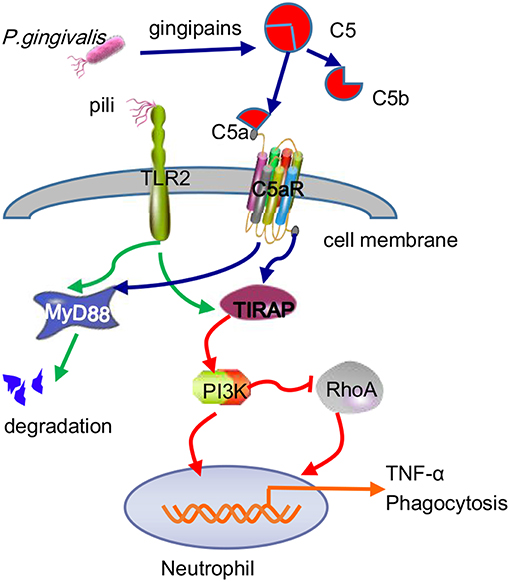
Figure 6. Antibactericidal mechanisms of C5a-TLR2 cross-talk induced by P. gingivalis in neutrophils. The C5aR-TLR2 cross-talk activated by P. gingivalis pili induced degradation of MyD88 in neutrophils. Without MyD88, the coassociation of C5aR-TLR2 promoted P. gingivalis infection through activation of the TIRAP-dependent PI3K signaling pathway, not only producing the inflammatory cytokine TNF-α but also blocking RhoA activation and actin polymerization to inhibit the maturation of phagosomes, thus blocking P. gingivalis phagocytosis.
The oral cavity is an open microecological environment with more than 700 species of microorganisms that normally maintain a dynamic balance with the host's immune system. When the balance between bacteria and the host is perturbed, opportunistic pathogens, including periodontal pathogens, will become dominant. Periodontitis caused by periodontal pathogens destroys the epithelial junction between the teeth and periodontal tissue, forming a periodontal pocket. Furthermore, the periodontal pocket provides an anaerobic and nutrient-rich growth microenvironment for periodontal pathogens to survive and spreading (Wolf and Lamster, 2011). As the most important pathogen of periodontal tissue infection, P. gingivalis can directly destroy periodontal tissues by secreting toxic factors such as LPS, gingipains and pili, and these important virulence factors can activate a wide range of host immune cells in periodontal tissues, triggering a local immune response, allowing the defense cells to release numerous inflammatory mediators and leading to secondary damage to the periodontal tissue. Regarding periodontitis pathogenesis, TLRs, as PAMP recognition receptors, can mediate the inherent immunological reactions of the host to P. gingivalis, which is the basis of adaptive immunity and plays a vital role during the occurrence and development of periodontitis (Nakayama and Ohara, 2017). This review therefore details how the most critical virulence mediators of P. gingivalis trigger host defense cells and regulate the microbial-host immune-inflammatory responses by interacting with TLRs at the molecular biology level, hopefully providing an opportunity to more clearly understand the pathogenesis of periodontitis.
Considering the different experimental methods, cell types, reagents, and other experimental details used by different scholars, contradictions and controversies in the experimental results remain. To date, we still do not fully understand how P. gingivalis can not only induce bodily inflammation but also escape the host immune surveillance and flourish under the microecologically imbalanced system by fully exploiting its key virulence molecules. In addition, other toxicity factors produced by P. gingivalis also play substantial roles in the pathogenesis of periodontitis, such as capsules activating the host complement system (Singh et al., 2011), the hemophore HmuY helping P. gingivalis capture and internalize heme from the host (Smalley et al., 2011), and outer membrane proteins adhering to the host outer membrane (Chen et al., 2011). Regarding other virulence factors, their specific pathogenic mechanisms and how they regulate host immunity through TLRs will be further summarized. Elucidating the unique pathogenic mechanism of P. gingivalis virulence factors is an arduous task, and a substantial amount of work remains and requires the joint efforts of scholars. We believe that with the rapid development of biomedicine, these problems will eventually be solved to provide more effective clinical therapy for patients with periodontitis and periodontal-related systemic diseases.
LJ analyzed literature and wrote the paper. NH, JD, LG, ZL, and YL edited and approved the manuscript.
This work was supported by grants from the National Nature Science Foundation of China (81470751 to YL, 81600891 to LG), the Beijing Municipal Administration of Hospitals Clinical Medicine Development of Special Funding Support (ZYLX201703), the Beijing Baiqianwan Talents Project (2017A17 to YL), Beijing Municipal Science & Technology Commission (Z16100000516203 to LG), Beijing Municipal Administration of Hospitals' Youth Programme (QML20181501 to LG), Beijing Dongcheng Excellent Talent (to LG).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abdi, K., Chen, T., Klein, B. A., Tai, A. K., Coursen, J., Liu, X., et al. (2017). Mechanisms by which Porphyromonas gingivalis evades innate immunity. PLoS ONE 12:e0182164. doi: 10.1371/journal.pone.0182164
Akira, S., and Takeda, K. (2004). Toll-like receptor signalling. Nat. Rev. Immunol. 4, 499–511. doi: 10.1038/nri1391
Akira, S., Uematsu, S., and Takeuchi, O. (2006). Pathogen recognition and innate immunity. Cell 124, 783–801. doi: 10.1016/j.cell.2006.02.015
Ammerdorffer, A., Schoffelen, T., Gresnigt, M. S., Oosting, M., den Brok, M. H., Abdollahi-Roodsaz, S., et al. (2015). Recognition of Coxiella burnetii by toll-like receptors and nucleotide-binding oligomerization domain-like receptors. J. Infect. Dis. 211, 978–987. doi: 10.1093/infdis/jiu526
Andrukhov, O., Andrukhova, O., Ozdemir, B., Haririan, H., Muller-Kern, M., Moritz, A., et al. (2016). Soluble CD14 enhances the response of periodontal ligament stem cells to P. gingivalis Lipopolysaccharide. PLoS ONE 11:e0160848. doi: 10.1371/journal.pone.0160848
Andrukhov, O., Ertlschweiger, S., Moritz, A., Bantleon, H. P., and Rausch-Fan, X. (2014). Different effects of P. gingivalis LPS and E. coli LPS on the expression of interleukin-6 in human gingival fibroblasts. Acta Odontol. Scand. 72, 337–345. doi: 10.3109/00016357.2013.834535
Aruni, A. W., Robles, A., and Fletcher, H. M. (2013). VimA mediates multiple functions that control virulence in Porphyromonas gingivalis. Mol. Oral. Microbiol. 28, 167–180. doi: 10.1111/omi.12017
Ayala-Herrera, J. L., Abud-Mendoza, C., Gonzalez-Amaro, R. F., Espinosa-Cristobal, L. F., and Martinez-Martinez, R. E. (2018). Distribution of Porphyromonas gingivalis fimA genotypes in patients affected by rheumatoid arthritis and periodontitis. Acta Odontol. Scand. 76, 520–524. doi: 10.1080/00016357.2018.1469788
Baek, K. J., Choi, Y., and Ji, S. (2013). Gingival fibroblasts from periodontitis patients exhibit inflammatory characteristics in vitro. Arch. Oral. Biol. 58, 1282–1292. doi: 10.1016/j.archoralbio.2013.07.007
Bainbridge, B. W., and Darveau, R. P. (2001). Porphyromonas gingivalis lipopolysaccharide: an unusual pattern recognition receptor ligand for the innate host defense system. Acta Odontol. Scand. 59, 131–138. doi: 10.1080/000163501750266710
Bao, K., Belibasakis, G. N., Thurnheer, T., Aduse-Opoku, J., Curtis, M. A., and Bostanci, N. (2014). Role of Porphyromonas gingivalis gingipains in multi-species biofilm formation. BMC Microbiol. 14, 258–258. doi: 10.1186/s12866-014-0258-7
Barth, K., Remick, D. G., and Genco, C. A. (2013). Disruption of immune regulation by microbial pathogens and resulting chronic inflammation. J. Cell. Physiol. 228, 1413–1422. doi: 10.1002/jcp.24299
Bostanci, N., and Belibasakis, G. N. (2012). Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol. Lett. 333, 1–9. doi: 10.1111/j.1574-6968.2012.02579.x
Bozkurt, S. B., Hakki, S. S., Hakki, E. E., Durak, Y., and Kantarci, A. (2017). Porphyromonas gingivalis lipopolysaccharide induces a pro-inflammatory human gingival fibroblast phenotype. Inflammation 40, 144–153. doi: 10.1007/s10753-016-0463-7
Burns, E., Eliyahu, T., Uematsu, S., Akira, S., and Nussbaum, G. (2010). TLR2-dependent inflammatory response to Porphyromonas gingivalis is MyD88 independent, whereas MyD88 is required to clear infection. J. Immunol. 184, 1455–1462. doi: 10.4049/jimmunol.0900378
Chen, J. Q., Szodoray, P., and Zeher, M. (2016). Toll-like receptor pathways in autoimmune diseases. Clin. Rev. Allergy Immunol. 50, 1–17. doi: 10.1007/s12016-015-8473-z
Chen, Y. Y., Peng, B., Yang, Q., Glew, M. D., Veith, P. D., Cross, K. J., et al. (2011). The outer membrane protein LptO is essential for the O-deacylation of LPS and the co-ordinated secretion and attachment of A-LPS and CTD proteins in Porphyromonas gingivalis. Mol. Microbiol. 79, 1380–1401. doi: 10.1111/j.1365-2958.2010.07530.x
Chrzeszczyk, D., Konopka, T., and Zietek, M. (2015). Polymorphisms of toll-like receptor 4 as a risk factor for periodontitis: meta-analysis. Adv. Clin. Exp. Med. 24, 1059–1070. doi: 10.17219/acem/47394
Coats, S. R., Jones, J. W., Do, C. T., Braham, P. H., Bainbridge, B. W., To, T. T., et al. (2009). Human Toll-like receptor 4 responses to P. gingivalis are regulated by lipid A 1- and 4'-phosphatase activities. Cell Microbiol 11, 1587–1599. doi: 10.1111/j.1462-5822.2009.01349.x
Coats, S. R., Pham, T. T., Bainbridge, B. W., Reife, R. A., and Darveau, R. P. (2005). MD-2 mediates the ability of tetra-acylated and penta-acylated lipopolysaccharides to antagonize Escherichia coli lipopolysaccharide at the TLR4 signaling complex. J. Immunol. 175, 4490–4498. doi: 10.4049/jimmunol.175.7.4490
Coats, S. R., Reife, R. A., Bainbridge, B. W., Pham, T.-T. T., and Darveau, R. P. (2003). Porphyromonas gingivalis lipopolysaccharide antagonizes Escherichia coli lipopolysaccharide at toll-like receptor 4 in human endothelial cells. Infect. Immun. 71, 6799–6807. doi: 10.1128/IAI.71.12.6799-6807.2003
Curtis, M. A., Percival, R. S., Devine, D., Darveau, R. P., Coats, S. R., Rangarajan, M., et al. (2011). Temperature-dependent modulation of Porphyromonas gingivalis lipid A structure and Interaction with the innate host defenses. Infect. Immun. 79, 1187–93. doi: 10.1128/IAI.00900-10
Darveau, R. P., Pham, T.-T. T., Lemley, K., Reife, R. A., Bainbridge, B. W., Coats, S. R., et al. (2004). Porphyromonas gingivalis lipopolysaccharide contains multiple lipid A species that functionally interact with both toll-like receptors 2 and 4. Infect. Immun. 72, 5041–5051. doi: 10.1128/IAI.72.9.5041-5051.2004
de Diego, I., Veillard, F., Sztukowska, M. N., Guevara, T., Potempa, B., Pomowski, A., et al. (2014). Structure and mechanism of cysteine peptidase gingipain K (Kgp), a major virulence factor of Porphyromonas gingivalis in periodontitis. J. Biol. Chem. 289, 32291–32302. doi: 10.1074/jbc.M114.602052
de Diego, I., Veillard, F. T., Guevara, T., Potempa, B., Sztukowska, M., Potempa, J., et al. (2013). Porphyromonas gingivalis virulence factor gingipain RgpB shows a unique zymogenic mechanism for cysteine peptidases. J. Biol. Chem. 288, 14287–14296. doi: 10.1074/jbc.M112.444927
Ding, P. H., Darveau, R. P., Wang, C. Y., and Jin, L. (2017). 3LPS-binding protein and its interactions with P. gingivalis LPS modulate pro-inflammatory response and Toll-like receptor signaling in human oral keratinocytes. PLoS ONE 12:e0173223. doi: 10.1371/journal.pone.0173223
Ding, P. H., and Jin, L. (2014). The role of lipopolysaccharide-binding protein in innate immunity: a revisit and its relevance to oral/periodontal health. J. Periodont. Res. 49, 1–9. doi: 10.1111/jre.12081
Ding, P. H., Wang, C. Y., Darveau, R. P., and Jin, L. J. (2013). Nuclear factor-kappaB and p38 mitogen-activated protein kinase signaling pathways are critically involved in Porphyromonas gingivalis lipopolysaccharide induction of lipopolysaccharide-binding protein expression in human oral keratinocytes. Mol. Oral. Microbiol. 28, 129–141. doi: 10.1111/omi.12010
Divanovic, S., Trompette, A., Atabani, S. F., Madan, R., Golenbock, D. T., Visintin, A., et al. (2005). Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat. Immunol. 6, 571–578. doi: 10.1038/ni1198
Dixon, D. R., and Darveau, R. P. (2005). Lipopolysaccharide heterogeneity: innate host responses to bacterial modification of lipid a structure. J. Dent. Res. 84, 584–595. doi: 10.1177/154405910508400702
Dou, Y., Robles, A., Roy, F., Aruni, A. W., Sandberg, L., Nothnagel, E., et al. (2015). The roles of RgpB and Kgp in late onset gingipain activity in the vimA-defective mutant of Porphyromonas gingivalis W83. Mol. Oral Microbiol. 30, 347–360. doi: 10.1111/omi.12098
Dyson, H. J., and Wright, P. E. (2016). Role of intrinsic protein disorder in the function and interactions of the transcriptional coactivators CREB-binding Protein (CBP) and p300. J. Biol. Chem. 291, 6714–6722. doi: 10.1074/jbc.R115.692020
Enersen, M., Olsen, I., Kvalheim, O., and Caugant, D. A. (2008). fimA genotypes and multilocus sequence types of Porphyromonas gingivalis from patients with periodontitis. J. Clin. Microbiol. 46, 31–42. doi: 10.1128/JCM.00986-07
Eskan, M. A., Hajishengallis, G., and Kinane, D. F. (2007). Differential activation of human gingival epithelial cells and monocytes by Porphyromonas gingivalis fimbriae. Infect. Immun. 75, 892–898. doi: 10.1128/IAI.01604-06
Gorman, M. A., Seers, C. A., Michell, B. J., Feil, S. C., Huq, N. L., Cross, K. J., et al. (2015). Structure of the lysine specific protease Kgp from Porphyromonas gingivalis, a target for improved oral health. Protein Sci. 24, 162–166. doi: 10.1002/pro.2589
Guo, M., Wang, Z., Fan, X., Bian, Y., Wang, T., Zhu, L., et al. (2014). kgp, rgpA, and rgpB DNA vaccines induce antibody responses in experimental peri-implantitis. J. Periodontol. 85, 1575–1581. doi: 10.1902/jop.2014.140240
Hajishengallis, G. (2015). Periodontitis: from microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 15, 30–44. doi: 10.1038/nri3785
Hajishengallis, G., Abe, T., Maekawa, T., Hajishengallis, E., and Lambris, J. D. (2013a). Role of complement in host-microbe homeostasis of the periodontium. Semin. Immunol. 25, 65–72. doi: 10.1016/j.smim.2013.04.004
Hajishengallis, G., McIntosh, M. L., Nishiyama, S. I., and Yoshimura, F. (2013b). Mechanism and implications of CXCR4-mediated integrin activation by Porphyromonas gingivalis. Mol. Oral Microbiol. 28, 239–249. doi: 10.1111/omi.12021
Hajishengallis, G., Wang, M., Liang, S., Triantafilou, M., and Triantafilou, K. (2008). Pathogen induction of CXCR4/TLR2 cross-talk impairs host defense function. Proc. Natl. Acad. Sci. U.S.A. 105, 13532–13537. doi: 10.1073/pnas.0803852105
Hall, M., Hasegawa, Y., Yoshimura, F., and Persson, K. (2018). Structural and functional characterization of shaft, anchor, and tip proteins of the Mfa1 fimbria from the periodontal pathogen Porphyromonas gingivalis. Sci. Rep. 8:1793. doi: 10.1038/s41598-018-20067-z
Hashimoto, M., Asai, Y., and Ogawa, T. (2004). Separation and structural analysis of lipoprotein in a lipopolysaccharide preparation from Porphyromonas gingivalis. Int. Immunol. 16, 1431–1437. doi: 10.1093/intimm/dxh146
Herath, T. D., Darveau, R. P., Seneviratne, C. J., Wang, C.-Y., Wang, Y., and Jin, L. (2013a). Tetra-and penta-acylated lipid A structures of Porphyromonas gingivalis LPS differentially activate TLR4-mediated NF-κB signal transduction cascade and immuno-inflammatory response in human gingival fibroblasts. PLoS ONE 8:e58496. doi: 10.1371/journal.pone.0058496
Herath, T. D., Wang, Y., Seneviratne, C. J., Lu, Q., Darveau, R. P., Wang, C. Y., et al. (2011). Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity differentially modulates the expression of IL-6 and IL-8 in human gingival fibroblasts. J. Clin. Periodontol. 38, 694–701. doi: 10.1111/j.1600-051X.2011.01741.x
Herath, T. D. K., Wang, Y., Seneviratne, C. J., Darveau, R. P., Wang, C.-Y., and Jin, L. (2013b). The expression and regulation of matrix metalloproteinase-3 is critically modulated by Porphyromonas gingivalis lipopolysaccharide with heterogeneous lipid A structures in human gingival fibroblasts. BMC Microbiol. 13:73. doi: 10.1186/1471-2180-13-73
Hertz, C. E., Bayarri-Olmos, R., Kirketerp-Møller, N., van Putten, S., Pilely, K., Skjoedt, M.-O., et al. (2018). Chimeric proteins containing MAP-1 and functional domains of C4b-binding protein reveal strong complement inhibitory capacities. Front. Immunol. 9, 1945–1945. doi: 10.3389/fimmu.2018.01945
Hiramine, H., Watanabe, K., Hamada, N., and Umemoto, T. (2003). Porphyromonas gingivalis67-kDa fimbriae induced cytokine production and osteoclast differentiation utilizing TLR2. FEMS Microbiol. Lett. 229, 49–55. doi: 10.1016/S0378-1097(03)00788-2
Hocevar, K., Potempa, J., and Turk, B. (2018). Host cell-surface proteins as substrates of gingipains, the main proteases of Porphyromonas gingivalis. Biol. Chem. 399, 1353–1361. doi: 10.1515/hsz-2018-0215
Holden, J. A., Attard, T. J., Laughton, K. M., Mansell, A., O'Brien-Simpson, N. M., and Reynolds, E. C. (2014). Porphyromonas gingivalis lipopolysaccharide weakly activates M1 and M2 polarized mouse macrophages but induces inflammatory cytokines. Infect. Immun. 82, 4190–4203. doi: 10.1128/IAI.02325-14
Hou, Y., Yu, H., Liu, X., Li, G., Pan, J., Zheng, C., et al. (2017). Gingipain of Porphyromonas gingivalis manipulates M1 macrophage polarization through C5a pathway. In Vitro Cell. Dev. Biol. Anim. 53, 593–603. doi: 10.1007/s11626-017-0164-z
How, K. Y., Song, K. P., and Chan, K. G. (2016). Porphyromonas gingivalis: an overview of periodontopathic pathogen below the gum line. Front. Microbiol. 7:53. doi: 10.3389/fmicb.2016.00053
Huck, O., You, J., Han, X., Cai, B., Panek, J., and Amar, S. (2018). Reduction of articular and systemic inflammation by Kava-241 in a Porphyromonas gingivalis-induced arthritis murine model. Infect. Immun. 86:e00356–18. doi: 10.1128/IAI.00356-18
Ikai, R., Hasegawa, Y., Izumigawa, M., Nagano, K., Yoshida, Y., Kitai, N., et al. (2015). Mfa4, an accessory protein of mfa1 fimbriae, modulates fimbrial biogenesis, cell auto-aggregation, and biofilm formation in Porphyromonas gingivalis. PLoS ONE 10:e0139454. doi: 10.1371/journal.pone.0139454
Ito, R., Ishihara, K., Shoji, M., Nakayama, K., and Okuda, K. (2010). Hemagglutinin/adhesin domains of Porphyromonas gingivalis play key roles in coaggregation with Treponema denticola. FEMS Immunol. Med. Microbiol. 60, 251–260. doi: 10.1111/j.1574-695X.2010.00737.x
Jin, S. H., Guan, X. Y., Liang, W. H., Bai, G. H., and Liu, J. G. (2016). TLR4 polymorphism and periodontitis susceptibility: a meta-analysis. Medicine. 95:e4845. doi: 10.1097/MD.0000000000004845
Jung, Y. J., Jun, H. K., and Choi, B. K. (2015). Contradictory roles of Porphyromonas gingivalis gingipains in caspase-1 activation. Cell. Microbiol. 17, 1304–1319. doi: 10.1111/cmi.12435
Kelley, S. L., Lukk, T., Nair, S. K., and Tapping, R. I. (2013). The crystal structure of human soluble CD14 reveals a bent solenoid with a hydrophobic amino-terminal pocket. J. Immunol. 190, 1304–1311. doi: 10.4049/jimmunol.1202446
Kusumoto, Y., Hirano, H., Saitoh, K., Yamada, S., Takedachi, M., Nozaki, T., et al. (2004). Human gingival epithelial cells produce chemotactic factors interleukin-8 and monocyte chemoattractant protein-1 after stimulation with Porphyromonas gingivalis via toll-like receptor 2. J. Periodontol. 75, 370–379. doi: 10.1902/jop.2004.75.3.370
Lee, J. Y., Miller, D. P., Wu, L., Casella, C. R., Hasegawa, Y., and Lamont, R. J. (2018). Maturation of the Mfa1 fimbriae in the oral pathogen Porphyromonas gingivalis. Front. Cell. Infect. Microbiol. 8, 137–137. doi: 10.3389/fcimb.2018.00137
Leite, F. R. M., Enevold, C., Bendtzen, K., Baelum, V., and Lopez, R. (2018). Pattern recognition receptor polymorphisms in early periodontitis. J Periodontol. 24:E1476. doi: 10.1002/JPER.18-0547
Li, C., Li, B., Dong, Z., Gao, L., He, X., Liao, L., et al. (2014). Lipopolysaccharide differentially affects the osteogenic differentiation of periodontal ligament stem cells and bone marrow mesenchymal stem cells through Toll-like receptor 4 mediated nuclear factor κB pathway. Stem Cell Res. Ther. 5:67. doi: 10.1186/scrt456
Liu, B., Wang, J., Cheng, L., and Liang, J. (2013). Role of JNK and NF-kappaB pathways in Porphyromonas gingivalis LPS-induced vascular cell adhesion molecule-1 expression in human aortic endothelial cells. Mol. Med. Rep. 8, 1594–1600. doi: 10.3892/mmr.2013.1685
Liu, R., Desta, T., Raptis, M., Darveau, R. P., and Graves, D. T. (2008). P. gingivalis and E. coli lipopolysaccharides exhibit different systemic but similar local induction of inflammatory markers. J. Periodontol. 79, 1241–1247. doi: 10.1902/jop.2008.070575
Liu, Y., Yin, H., Zhao, M., and Lu, Q. (2014). TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin. Rev. Allergy Immunol. 47, 136–147. doi: 10.1007/s12016-013-8402-y
Maekawa, T., Krauss, J. L., Abe, T., Jotwani, R., Triantafilou, M., Triantafilou, K., et al. (2014). Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 15, 768–778. doi: 10.1016/j.chom.2014.05.012
Maeshima, N., and Fernandez, R. C. (2013). Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front. Cell. Infect. Microbiol. 3:3. doi: 10.3389/fcimb.2013.00003
Makkawi, H., Hoch, S., Burns, E., Hosur, K., Hajishengallis, G., Kirschning, C. J., et al. (2017). Porphyromonas gingivalis stimulates TLR2-PI3K signaling to escape immune clearance and induce bone resorption independently of MyD88. Front. Cell. Infect. Microbiol. 7:359. doi: 10.3389/fcimb.2017.00359
Mantri, C. K., Chen, C.-H., Dong, X., Goodwin, J. S., Pratap, S., Paromov, V., et al. (2015). Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiologyopen 4, 53–65. doi: 10.1002/mbo3.221
Martin, M., Rehani, K., Jope, R. S., and Michalek, S. M. (2005). Toll-like receptor–mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat. Immunol. 6,777–784. doi: 10.1038/ni1221
Meyle, J., Dommisch, H., Groeger, S., Giacaman, R. A., Costalonga, M., and Herzberg, M. (2017). The innate host response in caries and periodontitis. J. Clin. Periodontol. 44, 1215–1225. doi: 10.1111/jcpe.12781
Mishra, A., Roy, F., Dou, Y., Zhang, K., Tang, H., and Fletcher, H. M. (2018). Role of acetyltransferase PG1842 in gingipain biogenesis in Porphyromonas gingivalis. J. Bacteriol. 200, e00385–e00318. doi: 10.1128/JB.00385-18
Mysak, J., Podzimek, S., Sommerova, P., Lyuya-Mi, Y., Bartova, J., Janatova, T., et al. (2014). Porphyromonas gingivalis: major periodontopathic pathogen overview. J. Immunol. Res. 2014:476068. doi: 10.1155/2014/476068
Nagai, Y., Kobayashi, T., Motoi, Y., Ishiguro, K., Akashi, S., Saitoh, S., et al. (2005). The radioprotective 105/MD-1 complex links TLR2 and TLR4/MD-2 in antibody response to microbial membranes. J. Immunol. 174, 7043–7049. doi: 10.4049/jimmunol.174.11.7043
Nagano, K., Abiko, Y., Yoshida, Y., and Yoshimura, F. (2013). Genetic and antigenic analyses of Porphyromonas gingivalis FimA fimbriae. Mol. Oral. Microbiol. 28, 392–403. doi: 10.1111/omi.12032
Nagano, K., Hasegawa, Y., Abiko, Y., Yoshida, Y., Murakami, Y., and Yoshimura, F. (2012). Porphyromonas gingivalis FimA fimbriae: fimbrial assembly by fimA alone in the fim gene cluster and differential antigenicity among fimA genotypes. PLoS ONE 7:e43722. doi: 10.1371/journal.pone.0043722
Nagano, K., Hasegawa, Y., Iijima, Y., Kikuchi, T., and Mitani, A. (2018). Distribution of Porphyromonas gingivalis fimA and mfa1 fimbrial genotypes in subgingival plaques. PeerJ. 6:e5581. doi: 10.7717/peerj.5581
Nakano, K., and Amano, A. (2013). Porphyromonas gingivalis fimbriae AU - Enersen, morten. J. Oral Microbiol. 5:20265. doi: 10.3402/jom.v5i0.20265
Nakayama, K. (2015). Porphyromonas gingivalis and related bacteria: from colonial pigmentation to the type IX secretion system and gliding motility. J. Periodontal Res. 50, 1–8. doi: 10.1111/jre.12255
Nakayama, M., Inoue, T., Naito, M., Nakayama, K., and Ohara, N. (2015). Attenuation of the phosphatidylinositol 3-kinase/Akt signaling pathway by Porphyromonas gingivalis gingipains RgpA, RgpB, and Kgp. J. Biol. Chem. 290, 5190–5202. doi: 10.1074/jbc.M114.591610
Nakayama, M., and Ohara, N. (2017). Molecular mechanisms of Porphyromonas gingivalis-host cell interaction on periodontal diseases. Jpn. Dent. Sci. Rev. 53, 134–140. doi: 10.1016/j.jdsr.2017.06.001
Nemoto, E., Darveau, R., Foster, B., Nogueira-Filho, G., and Somerman, M. (2006). Regulation of cementoblast function by P. gingivalis lipopolysaccharide via TLR2. J. Dental Res. 85, 733–738. doi: 10.1177/154405910608500809
Nguyen, M. T., Uebele, J., Kumari, N., Nakayama, H., Peter, L., Ticha, O., et al. (2017). Lipid moieties on lipoproteins of commensal and non-commensal staphylococci induce differential immune responses. Nat. Commun. 8:2246. doi: 10.1038/s41467-017-02234-4
Nichols, F. C., Bajrami, B., Clark, R. B., Housley, W., and Yao, X. (2012). Free lipid A isolated from Porphyromonas gingivalis lipopolysaccharide is contaminated with phosphorylated dihydroceramide lipids: recovery in diseased dental samples. Infect. Immun. 80, 860–874. doi: 10.1128/IAI.06180-11
Nogueira-Filho, G., Rosa, B. T., Santos, P. F., Tunes, U. R., Freire, S. M., Meyer, R., et al. (2014). Whole-blood cultures from patients with chronic periodontitis respond differently to Porphyromonas gingivalis but not Escherichia coli lipopolysaccharide. J. Periodontol. 85, e18–23. doi: 10.1902/jop.2013.120735
Ohya, M., Cueno, M. E., Tamura, M., and Ochiai, K. (2016). Varying hemin concentrations affect Porphyromonas gingivalis strains differently. Microb. Pathog. 94, 54–59. doi: 10.1016/j.micpath.2015.10.016
Olsen, I., Chen, T., and Tribble, G. D. (2018). Genetic exchange and reassignment in Porphyromonas gingivalis. J. Oral Microbiol. 10:1457373. doi: 10.1080/20002297.2018.1457373
Olsen, I., Lambris, J. D., and Hajishengallis, G. (2017). Porphyromonas gingivalis disturbs host-commensal homeostasis by changing complement function. J. Oral Microbiol. 9:1340085. doi: 10.1080/20002297.2017.1340085
Olsen, I., and Singhrao, S. K. (2018). Importance of heterogeneity in Porhyromonas gingivalis lipopolysaccharide lipid A in tissue specific inflammatory signalling. J. Oral Microbiol. 10:1440128. doi: 10.1080/20002297.2018.1440128
Olsen, I., and Yilmaz, Ö. (2016). Modulation of inflammasome activity by Porphyromonas gingivalis in periodontitis and associated systemic diseases. J. Oral Microbiol. 8:30385. doi: 10.3402/jom.v8.30385
Papadopoulos, G., Weinberg, E. O., Massari, P., Gibson, F. C. III., Wetzler, L. M., Morgan, E. F., et al. (2013). Macrophage-specific TLR2 signaling mediates pathogen-induced TNF-dependent inflammatory oral bone loss. J. Immunol. 190, 1148–1157. doi: 10.4049/jimmunol.1202511
Parahitiyawa, N. B., Scully, C., Leung, W. K., Yam, W. C., Jin, L. J., and Samaranayake, L. P. (2010). Exploring the oral bacterial flora: current status and future directions. Oral Dis. 16, 136–145. doi: 10.1111/j.1601-0825.2009.01607.x
Pathirana, R. D., O'Brien-Simpson, N. M., Brammar, G. C., Slakeski, N., and Reynolds, E. C. (2007). Kgp and RgpB, but not RgpA, are important for Porphyromonas gingivalis virulence in the murine periodontitis model. Infect. Immun. 75, 1436–1442. doi: 10.1128/IAI.01627-06
Petersen, P. E., and Ogawa, H. (2012). The global burden of periodontal disease: towards integration with chronic disease prevention and control. Periodontol. 2000 60, 15–39. doi: 10.1111/j.1600-0757.2011.00425.x
Pihlstrom, B. L., Michalowicz, B. S., and Johnson, N. W. (2005). Periodontal diseases. Lancet 366, 1809–1820. doi: 10.1016/S0140-6736(05)67728-8
Pollreisz, A., Huang, Y., Roth, G. A., Cheng, B., Kebschull, M., Papapanou, P. N., et al. (2010). Enhanced monocyte migration and pro-inflammatory cytokine production by Porphyromonas gingivalis infection. J. Periodont. Res. 45, 239–245. doi: 10.1111/j.1600-0765.2009.01225.x
Pomowski, A., Uson, I., Nowakowska, Z., Veillard, F., Sztukowska, M. N., Guevara, T., et al. (2017). Structural insights unravel the zymogenic mechanism of the virulence factor gingipain K from Porphyromonas gingivalis, a causative agent of gum disease from the human oral microbiome. J. Biol. Chem. 292, 5724–5735. doi: 10.1074/jbc.M117.776724
Ramstead, A. G., Robison, A., Blackwell, A., Jerome, M., Freedman, B., Lubick, K. J., et al. (2016). Roles of toll-like receptor 2 (TLR2), TLR4, and MyD88 during pulmonary coxiella burnetii infection. Infect. Immun. 84, 940–949. doi: 10.1128/IAI.00898-15
Rangarajan, M., Aduse-Opoku, J., Paramonov, N., Hashim, A., and Curtis, M. (2017). Hemin binding by Porphyromonas gingivalis strains is dependent on the presence of A-LPS. Mol. Oral Microbiol. 32, 365–374. doi: 10.1111/omi.12178
Reife, R. A., Coats, S. R., Al-Qutub, M., Dixon, D. M., Braham, P. A., Billharz, R. J., et al. (2006). Porphyromonas gingivalis lipopolysaccharide lipid A heterogeneity: differential activities of tetra-and penta-acylated lipid A structures on E-selectin expression and TLR4 recognition. Cell. Microbiol. 8, 857–868. doi: 10.1111/j.1462-5822.2005.00672.x
Ren, J., Sang, Y., Lu, J., and Yao, Y.-F. (2017). Protein acetylation and its role in bacterial virulence. Trends Microbiol. 25, 768–779. doi: 10.1016/j.tim.2017.04.001
Ricklin, D., Hajishengallis, G., Yang, K., and Lambris, J. D. (2010). Complement: a key system for immune surveillance and homeostasis. Nat. Immunol. 11, 785–797. doi: 10.1038/ni.1923
Roshan, M. H., Tambo, A., and Pace, N. P. (2016). The Role of TLR2, TLR4, and TLR9 in the pathogenesis of atherosclerosis. Int. J. Inflam. 2016:1532832. doi: 10.1155/2016/1532832
Ryu, J. K., Kim, S. J., Rah, S. H., Kang, J. I., Jung, H. E., Lee, D., et al. (2017). Reconstruction of LPS transfer cascade reveals structural determinants within LBP, CD14, and TLR4-MD2 for efficient LPS recognition and transfer. Immunity 46, 38–50. doi: 10.1016/j.immuni.2016.11.007
Sakanaka, A., Takeuchi, H., Kuboniwa, M., and Amano, A. (2016). Dual lifestyle of Porphyromonas gingivalis in biofilm and gingival cells. Microb. Pathog. 94, 42–47. doi: 10.1016/j.micpath.2015.10.003
Salim, T., Sershen, C. L., and May, E. E. (2016). Investigating the role of TNF-α and IFN-γ activation on the dynamics of iNOS gene expression in LPS stimulated macrophages. PLoS ONE 11:e0153289. doi: 10.1371/journal.pone.0153289
Schromm, A. B., Brandenburg, K., Loppnow, H., Moran, A. P., Koch, M. H., Rietschel, E. T., et al. (2000). Biological activities of lipopolysaccharides are determined by the shape of their lipid A portion. Eur. J. Biochem. 267, 2008–2013. doi: 10.1046/j.1432-1327.2000.01204.x
Singh, A., Wyant, T., Anaya-Bergman, C., Aduse-Opoku, J., Brunner, J., Laine, M. L., et al. (2011). The capsule of Porphyromonas gingivalis leads to a reduction in the host inflammatory response, evasion of phagocytosis, and increase in virulence. Infect. Immun. 79, 4533–4542. doi: 10.1128/IAI.05016-11
Singhrao, S. K., and Olsen, I. (2018). Are Porphyromonas gingivalis outer membrane vesicles microbullets for sporadic alzheimer's disease manifestation? J. Alzheimers Dis. Rep. 2, 219–228. doi: 10.3233/ADR-180080
Smalley, J. W., Byrne, D. P., Birss, A. J., Wojtowicz, H., Sroka, A., Potempa, J., et al. (2011). HmuY haemophore and gingipain proteases constitute a unique syntrophic system of haem acquisition by Porphyromonas gingivalis. PLoS ONE 6:e17182. doi: 10.1371/annotation/8658727f-cc10-47ba-9c7e-8726f1f94b91
Smalley, J. W., and Olczak, T. (2017). Heme acquisition mechanisms of Porphyromonas gingivalis – strategies used in a polymicrobial community in a heme-limited host environment. Mol. Oral. Microbiol. 32, 1–23. doi: 10.1111/omi.12149
Sojar, H. T., Sharma, A., and Genco, R. J. (2002). Porphyromonas gingivalis fimbriae bind to cytokeratin of epithelial cells. Infect. Immun. 70, 96–101. doi: 10.1128/IAI.70.1.96-101.2002
Song, B., Zhang, Y. L., Chen, L. J., Zhou, T., Huang, W. K., Zhou, X., et al. (2017). The role of Toll-like receptors in periodontitis. Oral Dis. 23, 168–180. doi: 10.1111/odi.12468
Song, G. G., Kim, J. H., and Lee, Y. H. (2013). Toll-like receptor (TLR) and matrix metalloproteinase (MMP) polymorphisms and periodontitis susceptibility: a meta-analysis. Mol. Biol. Rep. 40, 5129–5141. doi: 10.1007/s11033-013-2616-1
Tsukamoto, H., Takeuchi, S., Kubota, K., Kobayashi, Y., Kozakai, S., Ukai, I., et al. (2018). Lipopolysaccharide (LPS)-binding protein stimulates CD14-dependent Toll-like receptor 4 internalization and LPS-induced TBK1-IKK-IRF3 axis activation. J. Biol. Chem. 293, 10186–10201. doi: 10.1074/jbc.M117.796631
van Bergenhenegouwen, J., Plantinga, T. S., Joosten, L. A., Netea, M. G., Folkerts, G., Kraneveld, A. D., et al. (2013). TLR2 & Co: a critical analysis of the complex interactions between TLR2 and coreceptors. J. Leukoc. Biol. 94, 885–902. doi: 10.1189/jlb.0113003
Visintin, A., Mazzoni, A., Spitzer, J. A., and Segal, D. M. (2001). Secreted MD-2 is a large polymeric protein that efficiently confers lipopolysaccharide sensitivity to Toll-like receptor 4. Proc. Natl. Acad. Sci. U.S.A. 98, 12156–12161. doi: 10.1073/pnas.211445098
Wang, M., Krauss, J. L., Domon, H., Hosur, K. B., Liang, S., Magotti, P., et al. (2010). Microbial Hijacking of complement–toll-like receptor crosstalk. Sci. Signal. 3:ra11. doi: 10.1126/scisignal.2000697
Wang, M., Shakhatreh, M. A., James, D., Liang, S., Nishiyama, S., Yoshimura, F., et al. (2007). Fimbrial proteins of porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J. Immunol. 179, 2349–2358. doi: 10.4049/jimmunol.179.4.2349
Wang, P. L., and Ohura, K. (2002). Porphyromonas gingivalis lipopolysaccharide signaling in gingival fibroblasts-CD14 and Toll-like receptors. Crit. Rev. Oral Biol. Med. 13, 132–142. doi: 10.1177/154411130201300204
Weaver, C. T., Hatton, R. D., Mangan, P. R., and Harrington, L. E. (2007). IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 25, 821–852. doi: 10.1146/annurev.immunol.25.022106.141557
Wilensky, A., Tzach-Nahman, R., Potempa, J., Shapira, L., and Nussbaum, G. (2015). Porphyromonas gingivalis gingipains selectively reduce CD14 expression, leading to macrophage hyporesponsiveness to bacterial infection. J. Innate Immun. 7, 127–135. doi: 10.1159/000365970
Wolf, D. L., and Lamster, I. B. (2011). Contemporary concepts in the diagnosis of periodontal disease. Dental Clinics 55, 47–61. doi: 10.1016/j.cden.2010.08.009
Yang, J., Yang, C., Yang, J., Ding, J., Li, X., Yu, Q., et al. (2018). RP105 alleviates myocardial ischemia reperfusion injury via inhibiting TLR4/TRIF signaling pathways. Int. J. Mol. Med. 41, 3287–3295. doi: 10.3892/ijmm.2018.3538
Yongqing, T., Potempa, J., Pike, R. N., and Wijeyewickrema, L. C. (2011). The lysine-specific gingipain of Porphyromonas gingivalis: importance to pathogenicity and potential strategies for inhibition. Adv. Exp. Med. Biol. 712, 15–29. doi: 10.1007/978-1-4419-8414-2_2
Zhang, D., Li, S., Hu, L., Sheng, L., and Chen, L. (2015). Modulation of protease-activated receptor expression by Porphyromonas gingivalis in human gingival epithelial cells. BMC Oral Health 15:128. doi: 10.1186/s12903-015-0105-8
Zhang, L., Veith, P. D., Huq, N. L., Chen, Y. Y., Seers, C. A., Cross, K. J., et al. (2018). Porphyromonas gingivalis gingipains display transpeptidation activity. J. Proteome Res. 17, 2803–2818. doi: 10.1021/acs.jproteome.8b00286
Zweigner, J., Gramm, H. J., Singer, O. C., Wegscheider, K., and Schumann, R. R. (2001). High concentrations of lipopolysaccharide-binding protein in serum of patients with severe sepsis or septic shock inhibit the lipopolysaccharide response in human monocytes. Blood 98, 3800–3808. doi: 10.1182/blood.V98.13.3800
Keywords: Porphyromonas gingivalis, Toll-like receptors, lipopolysaccharide, gingipains, fimbriae, virulence factor, periodontitis
Citation: Jia L, Han N, Du J, Guo L, Luo Z and Liu Y (2019) Pathogenesis of Important Virulence Factors of Porphyromonas gingivalis via Toll-Like Receptors. Front. Cell. Infect. Microbiol. 9:262. doi: 10.3389/fcimb.2019.00262
Received: 16 April 2019; Accepted: 04 July 2019;
Published: 18 July 2019.
Edited by:
Fulvio Reggiori, University of Groningen, NetherlandsReviewed by:
Maribasappa Karched, Kuwait University, KuwaitCopyright © 2019 Jia, Han, Du, Guo, Luo and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi Liu, bGlsaWxpdXlpQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.