
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 12 April 2019
Sec. Parasite and Host
Volume 9 - 2019 | https://doi.org/10.3389/fcimb.2019.00105
This article is part of the Research Topic Immunobiology of Leishmaniasis View all 25 articles
 Joao Luiz Mendes Wanderley1*
Joao Luiz Mendes Wanderley1* Poliana Deolindo2
Poliana Deolindo2 Eric Carlsen3
Eric Carlsen3 Arieli Bernardo Portugal1
Arieli Bernardo Portugal1 Renato Augusto DaMatta4
Renato Augusto DaMatta4 Marcello Andre Barcinski5†
Marcello Andre Barcinski5† Lynn Soong6*†
Lynn Soong6*†Leishmania amazonensis amastigotes can make use of surface-exposed phosphatidylserine (PS) molecules to promote infection and non-classical activation of macrophages (MΦ), leading to uncontrolled intracellular proliferation of the parasites. This mechanism was quoted as apoptotic mimicry. Moreover, the amount of PS molecules exposed on the surface of amastigotes correlates with the susceptibility of the host. In this study, we tested whether host cellular responses influence PS expression on intracellular amastigotes. We found that the level of PS exposure on intracellular amastigotes was modulated by CD4+ T cell and MΦ activation status in vitro and in vivo. L. amazonensis infection generated a Th1/Th2-mixed cytokine profile, providing the optimal MΦ stimulation that favored PS exposure on intracellular amastigotes. Maintenance of PS exposed on the parasite was dependent on low, but sustained, levels of nitric oxide and polyamine production. Amastigotes obtained from lymphopenic nude mice did not expose PS on their surface, and adoptive transfer of CD4+ T cells reversed this phenotype. In addition, histopathological analysis of mice treated with anti-PS antibodies showed increased inflammation and similarities to nude mouse lesions. Collectively, our data confirm the role of pathogenic CD4+ T cells for disease progression and point to PS as a critical parasite strategy to subvert host immune responses.
Leishmania amazonensis (L. amazonensis) is the causative agent of cutaneous Leishmaniasis in South America. This species is associated with most cases of diffuse/disseminated cutaneous Leishmaniasis (DCL), a very severe clinical manifestation (Leon et al., 1990). Experimentally, most inbred mouse strains develop progressive cutaneous lesions, although the disease severity varies among mice of different genetic backgrounds (Terabe et al., 2004). Both DCL patients and experimentally infected mice show deficient cellular immune responses to the pathogen, as judged by delayed-type hypersensitivity (DTH) responses or cytokine/chemokine profiles (Ji et al., 2003; Silveira et al., 2005). In fact, when compared to the classical L. major infection models, L. amazonensis-infected mice failed to elicit a polarized Th response because activated CD4+ T cells produced a mix of Th1/Th2/Th17 and modulatory cytokines (Ji et al., 2003; Ramer et al., 2006; Vargas-Inchaustegui et al., 2009). This phenotype is consistent with the poor activation presented by infected dendritic cells, which is not sufficient to turn these cells into efficient, functional antigen-presenting cells (Xin et al., 2008; Wanderley et al., 2013). Consequently, macrophages (MΦs), the preferential host cells for parasite growth, are not efficiently activated and not capable of controlling the infection. Competent MΦ activation is necessary for disease control, since those cells are the main effector cells for parasite killing, usually dependent on the expression of nitric oxide synthase (iNOS) (Xie et al., 1993) or the production of reactive oxygen species (Carneiro et al., 2018). On the other hand, alternative or non-classical MΦ activation leads to an increased activation of arginase I, an enzyme responsible for the first step of polyamine synthesis, which is mandatory for parasite growth (Franca-Costa et al., 2015) and restrains NO production by competing for the same substrate, L-arginine (Wanasen and Soong, 2008). Those intracellular pathways control the fate of the intracellular parasite.
Apoptotic cells are known to display several distinctive molecular patterns, which are recognized by phagocytic cells for efficient internalization (Poon et al., 2014). In addition, phagocytes stimulated by apoptotic cell recognition are prompted to produce modulatory cytokines such as TGF-β and IL-10 (Fadok et al., 1998). Phosphatidylserine (PS) is a structural phospholipid that is actively maintained in the cytoplasmic leaflet of the plasma membrane but is translocated to the surface at the early stages of apoptotic death (Fadok et al., 1992). Recognition of PS exposed at the surface of apoptotic cells is sufficient to induce apoptotic cell clearance and non-classical activation of phagocytic cells (Hoffmann et al., 2001). We had previously shown that the amastigote forms of L. amazonensis, when purified from mice lesions, exposed PS at their surface without additional signs of apoptotic death. Since PS-exposing amastigotes are fully viable and highly competent in infecting and maintaining a productive disease in mice, we termed this phenotype apoptotic mimicry (de Freitas Balanco et al., 2001). As in the case of apoptotic cell/phagocyte interactions, the host cell is induced to produce immunosuppressive cytokines, which, in turn, signal for MΦ non-classical activation and consequent parasite growth (de Freitas Balanco et al., 2001; Wanderley et al., 2006). PS exposure on L. amazonensis amastigotes correlates with the severity of the disease, since amastigotes purified from BALB/c mice, which are highly susceptible to the infection, exhibit a higher density of PS moieties than do those from parasites purified from semi-resistant C57BL/6 mice (Wanderley et al., 2006). In addition, in vivo treatment of infected mice with anti-PS monoclonal antibodies delays disease progression and up-regulates the efficiency of dendritic cells to present antigen and activate parasite-specific T cells (Wanderley et al., 2013). PS exposure on pathogens operates in several different models of infection, such as those using Trypanosoma cruzi (Damatta et al., 2007), Toxoplasma gondii (Seabra et al., 2004), enveloped and non-enveloped viruses in which they confirm PS as a strategy to silently invade host cells (Seabra et al., 2004; Damatta et al., 2007; Mercer and Helenius, 2008; Feng et al., 2013). Additionally, by inducing transient PS exposure on the surface of host cells, viral infections can spread signals derived from PS recognition, such as TGF-β and IL-10 production by neighbor phagocytes, to avoid full activation of the immune system (Soares et al., 2008).
In this study, we tested whether PS exposure is an adaptive response of L. amazonensis amastigotes to the hostile environment of the parasitophorous vacuole generated by MΦ immune activation. We observed that intracellular amastigotes infecting activated MΦs are able to increase PS exposure. This is dependent on iNOS and arginase I concomitant expression. We confirmed our findings by demonstrating that PS exposure on amastigotes purified from lesions of T cell-deficient nude mice was nearly absent, but the adoptive transfer of primed CD4+ T cells recovered this phenotype. We also demonstrated that lesions of anti-PS antibody-treated infected mice were similar to lesions of immunodeficient mice. Our data lead us to conclude that PS exposed by intracellular amastigotes of L. amazonensis is a phenotype acquired as a response to host immune activation, and thus an important adaptive strategy employed by those intracellular parasites.
Female nude BALB/c mice (C.Cg/AnNTac-Foxn1nu NE9), C57BL/6 mice deficient in iNOS (C57BL/6NTac-Nos2tm1N12), and their corresponding wide-type (WT) controls were purchased from Taconic Farms (Germantown, NY) or Harlan Sprague Dawley (Indianapolis, IN), respectively. All mice were maintained under specific pathogen-free conditions and used at 6–8 weeks of age, according to the protocols approved by the Animal Care and Use Committee of the University of Texas Medical Branch (#9803016A). Promastigotes of L. amazonensis (LV78) were cultured at 23°C in Schneider's Drosophila medium (Invitrogen, Carlsbad, CA), pH 7.0, supplemented with 20% FBS (Sigma, St. Louis, MO) and 50 μg/ml of gentamicin. Axenic amastigotes of L. amazonensis (LV78) were cultured at 33°C in complete Grace's insect cell culture medium (Invitrogen), pH 5.0, supplemented with 20% FBS. Parasite infectivity was maintained by in vivo passages in BALB/c mice, and cultures of <6 passages were used for infection.
Otherwise stated, all recombinant cytokines were purchased from Peprotech (Rocky Hill, NJ, USA). Superoxide scavenger MnTBAP (Mn3 tetrakis (4-benzoic acid) porphyrin chloride) was purchased from Enzo Life Sciences (Farmingdale, NY, USA), iNOS inhibitor L-NIL- [L-N6-(1iminoethyl) lysine], and (ODC) decarboxylase inhibitor DFMO (DL-α-Difluoromethylornithine, Hydrochloride) were purchased from Calbiochem (Darmstadt, Germany).
Infected tissues or infected MΦs were finely minced and homogenized with a tissue grinder (Thomas Scientific, NJ). The cell suspension was centrifuged at 50 g for 10 min at 4°C. The supernatant was carefully collected, and further centrifuged and washed for 3 more times at 1,450 g for 17 min at 4°C. After 2 h incubation under rotation at 34°C to liberate endocytic membranes (Saraiva et al., 1983), amastigotes were further centrifuged and incubated for 16 h at 34°C to complete release of endocytic membranes and to test for bacterial contamination. After this time, they were centrifuged and washed 3 times before use. Prior to amastigote purification from in vitro infected cells, MΦs were thoroughly washed with HBSS.
BMMΦs were generated from mice by cultivating fresh bone marrow cells in complete IMDM (Invitrogen) containing 10% FBS, supplemented with 20 ng/ml of recombinant M-CSF (eBioscience, San Diego, CA). To generate BMMΦs, we replaced the medium at 5–6 days of culture and harvested adhered cells after 10–12 days. To recover adhered MΦs, we washed the petri dishes twice with warm PBS (Invitrogen) and incubated the cells with 5 ml of cell dissociation solution (CellGro, Manassas, VA) for 20 min at room temperature. We detached the cells by pipetting up and down and washed the cell pellet twice with complete medium prior to use.
Parasites were quantified and 106 amastigotes were washed and suspended in annexin V binding buffer, which contains 10 mM HEPES, 150 mM NaCl, and 2.5 mM CaCl2, at pH 7.2. Cells were incubated at room temperature for 15 min with annexin V-FITC (Molecular Probes, Eugene, OR) at the concentration indicated by the manufacturer and diluted in the binding buffer. All incubation procedures were performed on ice. At the time of acquisition, 0.4 μg/ml of propidium iodide (PI, Sigma) was added to the control and Annexin V-FITC-labeled samples to determine parasite viability. Data were collected in a BD FACSCalibur® (20,000 gated events per sample) and analyzed by Cellquest Pro® (BD Biosciences, San Jose, CA) and FlowJo software (TreeStar, Ashland, OR).
Mice were infected in the footpad with 2 × 106 promastigote forms of L. amazonensis. After 5–7 weeks of infection, popliteal lymph nodes (LNs) were harvested, and a single-cell suspension was obtained. Total LN cells from infected or naïve mice were plated in U-bottomed, 96-well plates, 4 × 105 cells per well, in the presence of 40 μg/ml of soluble Leishmanial antigens (SLA). After 4 days of culture, supernatants from stimulated LN cells from naïve mice or infected mice were pooled, filtered, and stored in aliquots at −70°C. To generate SLA, promastigote forms were submitted to 5 cycles of freeze-and-thaw and centrifugation to dispose of insoluble materials.
BALB/c WT or nude mice were infected in the footpad with 1–2 × 106 promastigote forms of L. amazonensis. After 6–8 weeks of infection, popliteal LNs from WT mice were harvested and a single-cell suspension was obtained. CD4+ or CD8+ T cells were purified by selected cell isolation kits (Miltenyi Biotech, Alburn, CA), following the manufacturer's instructions. Infected BALB/c nude mice were i.v. injected with 1–3 × 106 purified CD4+ or CD8+ T cells. At 2–3 weeks post-transfer, mice were euthanized to obtain popliteal LNs and lesion-derived amastigotes to evaluate T cell activation and PS exposure on amastigotes, respectively.
Thioglycolate-elicited peritoneal MΦs or BMMΦs were placed on 24-well plates and allowed to attach overnight. Cells were incubated with axenic amastigotes or promastigotes in a 3:1 ratio. After 4 h at 33°C, free parasites were removed by washing and, if necessary, cells were activated and/or treated with SNs, cytokines or drugs. Cultures proceeded for an additional 24-h period. In some cases, MΦs were attached on 13-mm2 glass coverslips (Fisher Scientific, Pittsburgh, PA) and, after infection and activation/treatment, were stained with Giemsa (Sigma) to evaluate host cell morphology and infection efficiency.
Total RNA was extracted from 1 × 106 MΦs 24 h post-infection and/or activation by using the RNeasy system (QIAGEN, Valencia, CA). Immediately cDNA was generated by using up to 5 μg of total RNA and the Superscript III Synthesis System (Invitrogen) and following the manufacturer's instructions. Amplifications of specific cDNAs were performed by using the GoTaq® Green Master Mix system (Promega, San Luis Obispo, CA). Briefly, Arginase I cDNA was subsequently amplified by use of the following cDNA primers: sense, 5′-AGACATCGTGTACATTG-3′ and antisense, 5′-GAGTTCCGAAGCAAGCCAAG-3′. Amplification occurred over 30 cycles, with the first cycle for primary denaturing at 95°C for 2 min; the next 28 cycles each comprising three steps for denaturing (94°C, 35 s), primer annealing (59°C for 45 s) and primer extension (72°C, 45 s); and a final cycle of denaturing (95°C, 30 s), annealing (69°C, 30 s), and extension (72°C, 5 min). To amplify inducible nitric oxide synthase (iNOS) and β-actin cDNA, we used the following cDNA primers: iNOS sense, 5′-GTTTCTGGCAGCAGCGGCTC-3′; antisense, 5′-GCTCCTCGCTCAAGTTCAGC-3′. β-actin sense, 5′-CGTGGGCCGCCCTAGGCACCAGGG-3′; antisense, 5′-GGGAGGAAGAGGATGCCGCAGTGG-3′. Amplification occurred over 36 cycles, by using the following approaches:the first cycle for primary denaturing at 95°C for 2 min; 34 cycles each comprising three steps for denaturing (95°C, 30 s), annealing (69°C, 30 s), extension (72°C, 20 s); and a final cycle of denaturing (95°C, 30 s), annealing (69°C, 30 s), and extension (72°C, 5 min). All reactions were performed by using a GeneAmp PCR System 2700 (Applied Biosystems, Foster City, CA), and the PCR products were separated by electrophoresis on 1.2% agarose gels. Real-time RT-PCR assays were performed with TaqMan Universal PCR Master Mix (Applied Biosystems, Foster City, CA, USA), using the following primer-probe sets purchased from Applied Biosystems: inos (Mm00440502_m1), arginase I (Mm00475988_m1), and β-actin endogenous control. The reactions were performed using Bio-Rad CFX96 Real-Time PCR detection system. Data were normalized to the expression of β-actin.
Cytokine production was measured in the supernatant of LN cell or MΦ cultures by using the Bio-Plex Pro-Mouse Cytokine 23-plex Assay from Bio-Rad (Hercules, CA) and following the manufacturer's instructions. Total and biologically reactive TGF-β1 were measured by using the ELISA Ready-SET-Go system (eBioscience), and data for biological reactive TGF-β1 are presented, The level of nitric oxide was measured by using a Griess assay (Caymann Chemical, Ann Arbor, MI).
Parasite loads were quantified by measuring the gene of L. amazonensis cysteine proteinase isoform 1 (Llacys1), which is a single-copy gene per haploid genome and expressed in both developmental stages (Lasakosvitsch et al., 2003). Infected MΦs were collected for DNA extraction with a DNeasy kit (Qiagen, Valencia, CA). DNA (10 ng) was used for parasite detection by the UTMB Real-time PCR Core Facility (all reagents were purchased from Applied Biosystems, Foster City, CA). Each sample was run in duplicate and normalized by the amount of total DNA extracted. The number of parasites per sample was calculated based on a standard curve, as described in our previous studies (Xin et al., 2010).
Mice were infected i.d. in the right ear with 106 promastigotes. After 2 weeks of infection and every 3 days thereafter they were given i.p. injections of 100 μg of PGN635, a second-generation fully humanized anti-PS monoclonal antibody (Zhou et al., 2014). Other groups of mice received PBS or the isotype control C44 antibody that binds to colchicine (Edmond Rouan et al., 1989). Mice were treated for 6 weeks and the infected ears were collected, fixed in 4% paraformaldehyde, dehydrated, embedded in paraffin, and mounted slides were stained with hematoxylin and eosin.
The sizes parasitophorous vacuoles from lesions of mice treated with anti-PS, isotype antibodies or PBS were observed under an Axioplan (Zeiss) microscopy and images were captured using a MRc5 AxioCam digital camera and processed with the software ImageJ version 1.47t (Wayne Rasband–NIH). Values are shown as the area in μm2 for at least 200 PVs in each tested sample.
BMMΦs (1 × 106) were infected with axenic amastigotes at a 3:1 parasite-to-cell ratio. At 24 h post-infection and/or indicated treatments, cells were harvested, washed, and suspended in 10 μl of PBS and 10 μl of 2X lysis buffer (2% Triton X-100, 100 mM Tris-Cl, 600 mM NaCl, 10 mM EDTA, 2 mM PMSF, 250 mM sucrose) that contained an inhibitor cocktail (Roche, Indianapolis, IN). Protein concentrations were determined by using the BCA protein assay kit (Pierce Biotechnology). Equal amounts of proteins were loaded onto 10% SDS-polyacrylamide gels, and then transferred to polyvinylidene difluoride membranes (BioRad Laboratories, Hercules, CA). Rabbit anti-mouse iNOS and arginase I antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse anti-actin mAb (Sigma) was obtained from Dr. Jiaren Sun (Department of Microbiology and Immunology, UTMB, TX). Membranes were incubated with primary Abs (diluted 1:200 in TBS buffer containing 5% non-fat milk and 0.05% Tween-20) at 4°C overnight, washed, and incubated with an HRP-conjugated secondary Ab (1:2000) for 1 h. Blots were developed with the enzyme chemiluminescence kit ECL (Amersham Biosciences, Piscataway, NJ).
One- or two-way ANOVA was used for multiple group comparisons (GraphPad Software v5.0, San Diego, CA). Statistically significant values are referred to as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001.
We previously showed that lesion-derived amastigotes purified from BALB/c mice expose higher amounts of PS than do those parasites derived from C57BL/6 mice (Wanderley et al., 2006), a finding that may indicate that the host can modulate this phenotype of the parasite. To evaluate the role of host macrophages (MΦs) in modulating PS exposure on the parasite, we obtained thioglycollate-elicited peritoneal MΦs from BALB/c and C57BL/6 mice, infected them either with promastigotes or lesion-derived amastigotes and collected intracellular amastigotes every 24 h to evaluate PS exposure by flow cytometry. We found no major differences between parasites derived from BALB/c vs. C57BL/6 MΦs (Figure 1A), regardless of the form of parasite used for infection or the time post-infection. Consequently, we suspected that the interaction between MΦs and other types of immune cells was responsible for the differential levels of PS on lesion-derived parasites in vivo. To investigate this possibility, we obtained lymph node (LN) cells from infected BALB/c mice and incubated them with peritoneal MΦs infected with lesion-derived amastigotes. These interactions stimulated an about 30% higher PS exposure on intracellular amastigotes compared to that in parasites obtained from isolated MΦs, in a dose-dependent manner (Figure 1B).
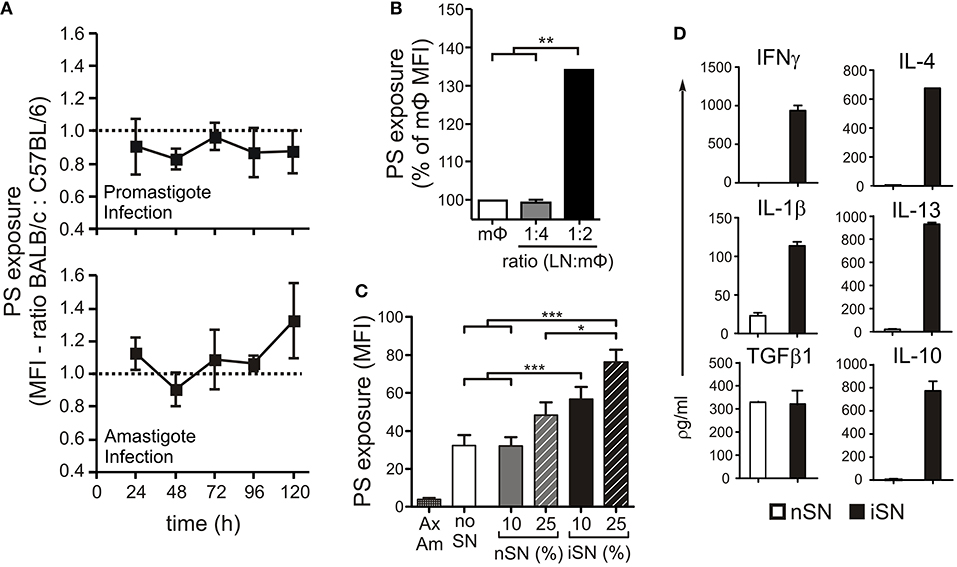
Figure 1. PS exposure on the surface of intracellular amastigotes is induced through MΦ activation by lymph node cells. (A) BALB/c- or C57BL/6-derived peritoneal MΦs were infected with stationary-phase promastigotes (upper panel) or amastigotes (lower panel). Every 24 h post-infection, intracellular parasites were purified, and PS exposure was analyzed by flow cytometry. Results are shown as the ratio between the MFIs of annexin V staining of amastigotes derived from BALB/c and C57BL/6 MΦs. BALB/c-derived BMMΦs were infected with amastigotes in the absence or presence of (B) LN cells from infected BALB/c mice (at indicated LN cell-to-MΦ ratios), or (C) supernatants of in vitro-stimulated LN cells from naïve BALB/c mice (nSN) or infected mice (iSN). After 24 h of treatment, amastigotes were purified for PS exposure analysis by flow cytometry. Results are pooled from 3 to 5 independent experiments. (D) Cytokine levels in supernatants of in vitro-stimulated LN cells from naïve BALB/c mice (nSN) or infected mice (iSN) were measured by bioplex assays. Graph represents data from 3 independent batches of SNs, produced from LNs of 2 mice per batch. *p < 0.05, **p < 0.01, ***p < 0.001.
To determine whether cytokine production by LN cells was a requirement to induce PS exposure on intracellular parasites, we generated supernatants from LN cells obtained from naïve (nSN) or infected (iSN) mice and stimulated with SLA for 4 days. We treated MΦs infected with axenic amastigotes with different concentrations of those supernatants (SNs) and evaluate PS exposure on intracellular amastigotes 24 h post-infection and stimulation. As shown in Figure 1C, axenic amastigotes exposed very low amounts of PS, which represented a technical advantage for minimizing background levels of PS exposure and indicated that parasite-host interactions should be necessary to stimulate PS exposure on amastigotes. Indeed, intracellular amastigotes from unstimulated MΦs (“no SN”) increased PS on their surface, 24 h post-infection. In addition, treatment of infected MΦs with iSNs induced PS exposure on intracellular amastigotes. This effect was dependent on the activation of those LN cells, since iSN was significantly more efficient than nSN and a clear positive correlation with the concentration of SN was observed: 10% of iSN added to the culture induced a 50% increase, while 25% of iSN induced about a 70% increase in PS exposure on intracellular amastigotes (Figure 1C).
As expected, the cytokine profile of those SNs corroborated the previous data regarding T cell activation during L. amazonensis infection in the mice (Ji et al., 2003). Moderate amounts of Th1, Th2, and modulatory cytokines such as IL-4, IL-13, IFNγ, IL-1β, TGF-β1, and IL-10 (Figure 1D), were present, especially in the SN generated from re-stimulated, in vivo-primed cells (iSN), which indicates that this response is antigen-specific. One of the hallmarks of the apoptotic mimicry, described to operate during amastigote infection, is the fact that amastigotes exposing PS are perfectly viable and infective and do not display any other sign of apoptotic death (de Freitas Balanco et al., 2001; Wanderley et al., 2006). To evaluate whether MΦ activation leads to PS exposure on intracellular amastigotes due to apoptotic mimicry, we measured parasite loads on infected MΦs at 24 and 72 h post-infection. As shown in Figure 2A, MΦ activation with SNs derived from stimulated LN cells from either infected or naïve mice promoted parasite proliferation 72 h post-infection, as opposed to LPS and IFNγ activation. In addition, the morphological characteristics of L. amazonensis infection are maintained, and include, for example, enlarged parasitophorous vacuoles with parasites attached to the vacuole internal membrane (Figure 2B). Our data suggest that MΦ activation through interactions with primed LN cells and their soluble products modulate active PS exposure by intracellular amastigotes in an apoptotic mimicry fashion.
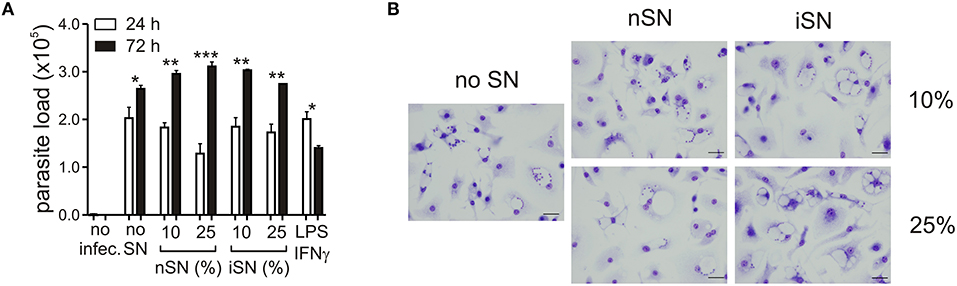
Figure 2. PS exposure on intracellular amastigotes, induced by MΦ stimulation is not due to apoptotic death. (A) Parasite loads in infected BALB/c-derived BMMΦs activated with different concentrations of nSN and iSN were measured by real-time PCR. Asterisks indicate comparison between black and white bars. LPS (100 ng/ml) plus IFN-γ (100 ng/ml) was used as positive control. *p < 0.05, **p < 0.01, ***p < 0.001. All comparisons were made among the same samples at 24 and 72 h post infection. (B) Photomicrography of infected BMMΦs activated with different concentrations of nSN and iSN. Bars indicate 10 μm. Graph represents data from 5 independent experiments.
It is known that the survival of Leishmania parasites inside macrophages (MΦs) is mostly mediated by the balance between iNOS and arginase I activation. These are induced enzymes and therefore their activity is directly related to their cellular expression. Arginase I is the first enzyme in the polyamine synthesis pathway, while iNOS is the enzyme responsible for all steps of NO synthesis. Those intracellular signaling pathways compete with each other, since they depend on the same substrate. In addition, some molecules, produced as secondary metabolites, act as cross-inhibitors (Wanasen and Soong, 2008). Interactions between infected MΦs and other immune cells determine which pathway is activated, since inflammatory cytokines stimulate iNOS expression and decrease mRNA levels of arginase I and vice-versa (Corraliza et al., 1995; Modolell et al., 1995). Since MΦ activation seems to be important for the cytokine-dependent PS exposure by intracellular parasites, we aimed to understand the role of those pathways in this mechanism. We observed that SN from re-stimulated, in vivo-primed LN cells induced the expression of both arginase I and iNOS at the mRNA and protein levels (Figures 3A,B and Figure S1). The expression of both enzymes was the highest when 25% of iSN was used to stimulate infected MΦs, the same concentration that induced an optimal increase of PS exposure on intracellular amastigotes. However, iSN stimulation was weak, when compared to that in the positive controls for arginase I and iNOS expression, TGFβ1+IL-4 or IFNγ+TNFα, respectively (Figures 3A,B). The expression of both enzymes by activated MΦs correlates with the profile of cytokines present on those SNs, since they are constituted by a mix of Th1/Th2/modulatory cytokines (Figure 1D).
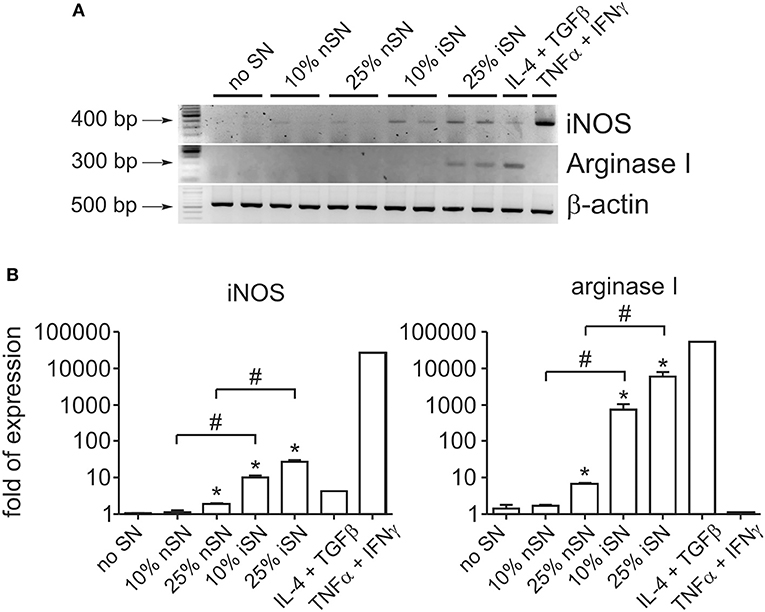
Figure 3. Macrophage stimulation leads to concomitant expression of iNOS and arginase I. (A) RT-PCR and (B) real-time RT-PCR analyses of iNOS and Arginase I expression by BALB/c-derived BMMΦs at 24 h post-infection and activation with SNs from SLA-stimulated, naïve LN cells (nSN) or re-stimulated, in vivo-primed LN cells (iSN). (A) Representative photographs of 2–3 experiments. TGF-β (10 ng/ml) plus IL-4 (1 ng/ml) stimulation was used as a positive control for arginase expression and IFN-γ (100 ng/ml) plus TNF-α (10 ng/ml) stimulation for iNOS expression. *p < 0.05 (compared with medium controls), #p < 0.05 (between the groups).
To understand the contribution of both pathways individually, we activated infected MΦs in the presence of L-NIL, a relatively selective inhibitor of iNOS, or, as a control, we used MnTBAP, a superoxide scavenger molecule. L-NIL has IC50 values of 0.4–3.3 μM for iNOS as opposed to 8–38 and 17–92 μM for eNOS and nNOS, respectively (Moore et al., 1994; Stenger et al., 1995). At 24 h of infection, we evaluated PS exposure on purified intracellular amastigotes. The iNOS inhibitor abrogated the cytokine-dependent induction of PS exposure, while scavenging superoxide molecules had no effect (Figure 4A). Treatment with both inhibitors brought the levels of PS exposure on amastigotes to the same levels of L-NIL alone, indicating that there was no synergistic effect between NO and superoxide to induce PS exposure on the parasite (Figure 4A). To further demonstrate the role of iNOS expression by MΦs in the induction of PS exposure on amastigotes, we generated BMMΦs from wild-type (WT) and iNOS−/− C57BL/6 mice, infected them, and purified intracellular amastigotes from activated MΦs to evaluate PS exposure on the parasite. C57BL/6-derived MΦs were more sensitive to activation, since 25% of nSN was able to induce the same levels of PS on the parasite when compared with iSN (Figure 4B). Activation of infected iNOS−/− MΦs did not increase PS exposure on amastigotes regardless of the source or the concentration of SN (Figure 4B). Similar to what happened in BALB/c-derived MΦs, the differences observed on amastigotes purified from WT or iNOS−/− C57BL/6 MΦs were not related to the death of the amastigotes, since parasite loads 72 h post-infection were not altered among the different SN treatments (Figure 4C). Despite the increased iNOS expression, MΦ activation with 10 or 25% of iSN or nSN did not induce detectable amounts of NO, evaluated by Griess reaction (Figure 4D). To test whether polyamines play a role in the induction of PS exposure on amastigotes, we purified amastigotes from infected- and activated-MΦs treated with different concentrations of difluoromethylornithine (DFMO), an specific and irreversible inhibitor of ornithine decarboxylase, the enzyme responsible for metabolizing L-arginine-derived ornithine into the polyamine putrescine (Canellakis et al., 1979). Independent on the concentration of DFMO used, this drug had no effect on the PS exposure on the amastigotes (Figure 4E). However, when we quantified parasite loads on infected-MΦs activated with 25% of iSN, we observed that DFMO treatment was detrimental for the proliferation of intracellular parasites at 72 h post-infection (Figure 4F). This finding may indicate that polyamine synthesis is a requirement for parasite proliferation and maintenance in these conditions, as described in other models (Majumder and Kierszenbaum, 1993; Vannier-Santos et al., 2008). Nevertheless, our data imply that PS exposure on intracellular amastigotes is modulated by cytokine-mediated, iNOS-dependent MΦ activation, rather than by arginase I expression. Arginase I, however, is necessary for parasite proliferation and survival in activated MΦs.
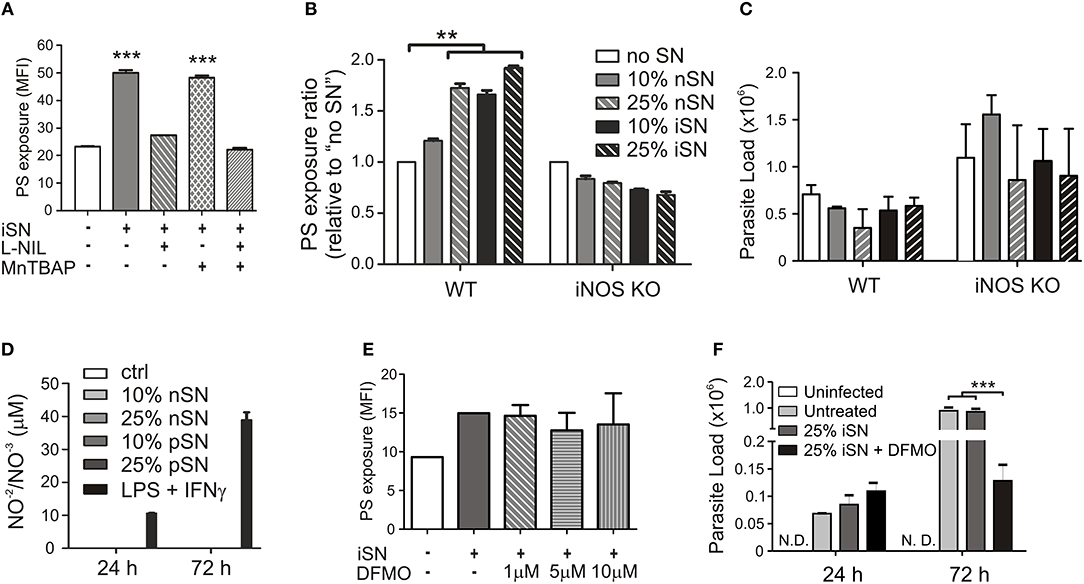
Figure 4. Cytokine-induced PS exposure on amastigotes depends on iNOS function. Infected BALB/c-derived BMMΦs were activated with SNs from re-stimulated, in vivo-primed LN cells (iSN) in the presence of (A) 50 μg/ml of L-NIL (a selective inhibitor of iNOS), 200 μM of MnTBAP (a superoxide scavenger molecule), or (E) different concentrations of DFMO (a selective ODC inhibitor). After 24 h of infection, amastigotes were purified for PS exposure analysis by flow cytometry. (B) BMMΦs were obtained from WT or iNOS KO mice, infected and activated with different concentrations of iSN and nSNs. After 24 h of infection, amastigotes were purified for PS exposure analysis by flow cytometry. Parasite loads in infected BMMΦs (F) treated with 10 μM of DFMO or (C) obtained from iNOS KO mice and activated with iSN were measured by real-time PCR. (D) NO production by BMMΦs stimulated with different concentrations of iSN, nSNs or LPS+IFN-γ (100 ng/ml for both), measured by Griess reaction. (A) Asterisks indicate comparison with untreated MΦs. Graphs represent data from 3 to 5 pooled experiments. **p < 0.01, ***p < 0.001.
To further understand the role of different cellular populations present in the lymph nodes (LNs) for the modulation of PS exposure on intracellular amastigotes, we infected WT or nude BALB/c mice in the footpad and purified lesion-derived amastigotes to evaluate PS exposure on a weekly basis. Confirming previous data from the literature, we observed a delay in the development of lesions in immunodeficient mice, which, eventually, reached the same size of lesions from WT mice (Soong et al., 1997; Figure 5A). PS exposure on lesion-derived amastigotes purified from WT mice was 2- to 6-times higher than on parasites obtained from nude mice, depending on the time post infection (Figure 5B), confirming previous results (Franca-Costa et al., 2012). Since CD4+ T cells are the major regulators of host immune response to Leishmania infection, we hypothesized that the modulation of PS exposure on intracellular parasites observed by incubating infected MΦs with LN cells or culture SNs was due to CD4+ T cell-dependent MΦ activation. To test this hypothesis, we purified CD4+ T cells from draining LNs of infected WT mice and adoptively transferred these cells to infected nude mice. After 2–3 weeks post-transfer, we purified lesion-derived amastigotes to evaluate PS exposure. The adoptive transfer of CD4+ T cells increased PS exposure on the amastigotes by 2- or 3-fold, whereas transfer of CD8+ T cells, used as a control, did not alter the parasite phenotype (Figures 5C,D).
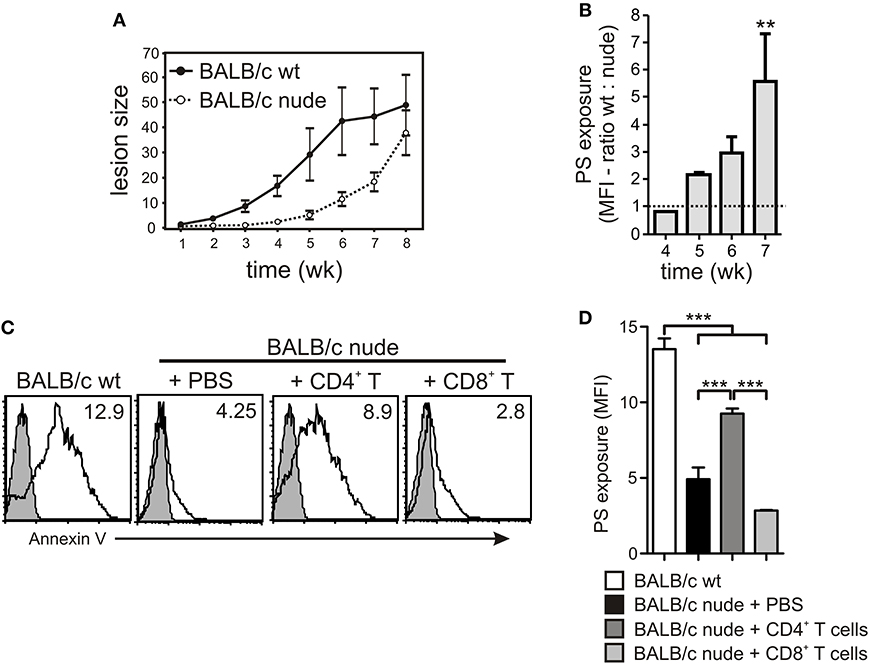
Figure 5. PS exposure on intracellular amastigotes is dependent on the presence of CD4+ T cells in vivo. (A) WT or nude BALB/c mice were infected with 2 × 106 stationary-phase promastigotes on the hind footpad. Lesion sizes were measured weekly by using a Vernon caliper. (B) PS exposure on lesion-derived amastigotes was assessed by flow cytometry. Results are shown as the ratio between the MFIs of annexin V staining of amastigotes derived from WT vs. nude mice. (C,D) After 7–8 weeks of infection, 4 × 106 CD4+ or CD8+ T cells, purified from draining LNs of infected WT BALB/c mice, were adoptively transferred to infected nude mice. At 2–3 weeks post-transfer, lesion-derived amastigotes were purified for PS exposure analysis by flow cytometry. (C) The shaded histograms represent amastigotes stained with annexin V in the absence of CaCl2. **p < 0.01, ***p < 0.001. (B,D) Graphs represent data from 2 to 3 pooled experiments.
In addition to the decreased PS exposure on amastigotes derived from nude mice (Franca-Costa et al., 2012; Figure 5B), histopathological analysis of BALB/c nude mice lesions showed a marked decrease in the size of the parasitophorous vacuoles of infected MΦs (Franca-Costa et al., 2012). This is also observed in lesions from MHC class II−/− mice (Soong et al., 1997). We observed that infected mice treated intraperitoneally with anti-PS mAb displayed decreased parasitophorous vacuole size (Figures 6D–F) when compared to untreated mice (Figures 6A–C). The difference in the vacuole size was not observed in infected mice treated with isotype control antibodies (Figure 6G and Supplementary Table 1). Our data demonstrate that PS exposure on intracellular amastigotes is a response of the parasite when it senses MΦ activation through primed CD4+ T cells.
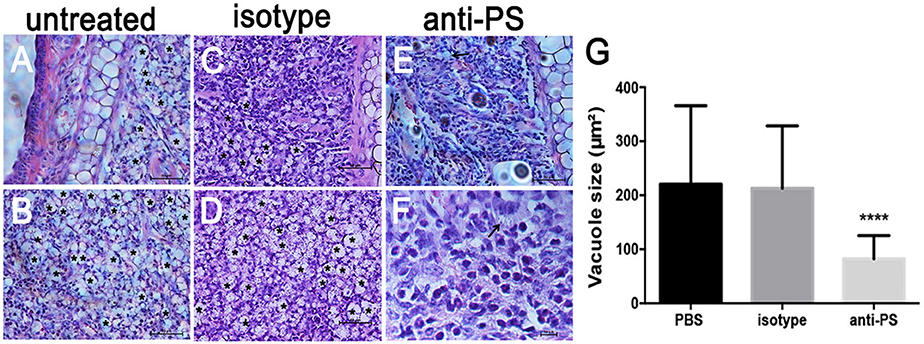
Figure 6. Histopathological analysis of mice lesions treated with anti-PS blocking monoclonal antibodies. (A–F) Mice were infected in the ear dermis with 106 stationary-phase promastigotes. After 2 weeks of infection and every 3 days thereafter, mice were given i.p. injections of 100 μg of anti-PS or isotype control antibody. After 6 weeks of treatment the lesions were processed for histopathological analysis. (A–D) Asterisks in the images indicate large vacuoles containing amastigotes, (E,F) arrows indicate vacuoles containing parasites in anti PS treated infected mice. (G) Quantification of parasitophorous vacuole size from 2 independent experiments. ****p < 0.0001 compared to isotype and PBS treated mice.
L. amazonensis, particularly its amastigotes, can infect host cells without triggering overt cellular activation. Actually, those parasites are able to down-modulate signaling pathways involved in dendritic cell activation, suppress cytokine production and expression of MHC class II molecules and, therefore, to inhibit antigen presentation (Prina et al., 2004; Xin et al., 2008). In addition, the inhibitory effects of L. amazonensis amastigotes on macrophages (MΦs) are also well known, including sequestration and degradation of MHC class II molecules, inhibition of endosomal proteases and blocking of NO production (Prina et al., 1990, 1993; Antoine et al., 1999). Consequently, L. amazonensis infection models are typically characterized by presenting a weak and non-polarized T cell response that is not sufficient to induce proper MΦ activation and control of parasite loads (Soong et al., 1997; Ji et al., 2002, 2003).
Our group has previously shown that the ability of amastigotes of L. amazonensis to silently infect host cells is mainly due to the exposure of PS molecules at their surface in a mechanism referred to as apoptotic mimicry (de Freitas Balanco et al., 2001; Wanderley et al., 2006). The recognition of this molecule and opsonizing ligands such as antibodies and complement factors mediate amastigote uptake by MΦs. In addition PS is highly effective in triggering anti-inflammatory cytokine production by MΦs, creating a permissive environment for parasite intracellular proliferation (de Freitas Balanco et al., 2001; Wanderley et al., 2006; Birge et al., 2016). Interestingly, PS exposure on amastigotes is correlated with disease severity, since amastigotes derived from BALB/c mice, which develop a more severe disease when exposed to higher amounts of PS than do those obtained from moderately susceptible C57BL/6 mice (Wanderley et al., 2006). In the context of parasite/host interactions, there are several reports showing a similar effect in the opposite direction: infection modulating host cell functions such as apoptosis, and expression of molecules involved with microbicidal effects or immune functions (Osorio y Fortea et al., 2007; Soong, 2008; Lecoeur et al., 2010; Muxel et al., 2017b). However, the presence of this phenotype suggests that, in the case of BALB/c mice infection, the host is able to modulate parasite virulence by inducing or selecting amastigotes with an increased capacity to expose PS. Definitely, this modulation depends on host cell types and activation status, since the internalization of axenically-cultured amastigotes by MΦs induces a basal level of PS exposure on these parasites, which is absent in vitro. However, MΦs with different genetic backgrounds are not able, by themselves, to differentially modulate PS exposure by the intracellular parasites. Differential modulation only occurs in the presence of lymph node (LN) cells or soluble molecules produced by antigen-stimulated LN cells.
PS recognition is clearly related to the internalization of amastigotes by MΦs (de Freitas Balanco et al., 2001; Wanderley et al., 2006) and to the modulation of dendritic cells and MΦ responses by both promastigotes and amastigotes (Wanderley et al., 2006, 2009, 2013; Franca-Costa et al., 2012). These events are dependent on PS recognition by surface receptors expressed by MΦs and other phagocytic cells. However, our results indicate that PS exposure on intracellular amastigotes is a counteraction of MΦ activation, suggesting that the parasite is able to modulate the host cells from within the parasitophorous vacuole. Therefore it should be determined whether there are endosomal PS receptors expressed by the MΦ, especially those ones involved in the regulation of host cell inflammatory activation such as TAM receptors (Axl, Tyro3, and Mer; Rothlin et al., 2007). Since signal transduction can be mediated by endosomal receptors it is likely to assume that PS receptors either coopted from the plasma membrane during parasite uptake or specifically trafficked to the parasitophorous vacuole.
It is known that the T cells response elicited by L. amazonensis infection is not sufficient to induce parasite killing, which was confirmed by our results. Cytokines produced by both in vivo primed (iSNs) or naïve LN cells (nSNs) stimulated with SLA were able to induce increased PS exposure, although this effect is prominent when in vivo primed cells were used, due to higher cytokine concentrations. In addition to this effect, we are demonstrating that MΦ stimulation with soluble factors produced by LN cells stimulated with L. amazonensis antigens generates a unique condition that triggers PS exposure on intracellular amastigotes, thereby increasing parasite virulence (Wanderley et al., 2006). We tried to activate infected MΦs with different combinations of cytokines, such as IFN-γ and TNF-α, in an attempt to create the exact conditions that drive PS exposure on the intracellular parasites. Although some induction of PS exposure was observed, it did not reach the same efficiency when compared to iSN (data not shown). We understand that PS exposure depends on a very unique combination of time-dependent and concentration-dependent cytokines, which favor parasite survival over parasite killing, but maintain stress-signals sufficient to induce PS exposure. This issue could be addressed by using SNs from LN cells from other infected mice strains, such as C57BL/6 or C3H.He mice (de Oliveira Cardoso et al., 2010). There are several reports demonstrating that CD4+ T cells are pathogenic for L. amazonensis infection; however, the mechanisms underlying this effect are not fully understood. It is well known that C57BL/6 mice deficient in the CIITA, MHC class II, and RAG2 genes or nude C57BL/6 mice exhibit a delay in lesion development and smaller parasite loads in infected tissues, indicating that CD4+ T cells play a role in lesion pathology and disease progression (Soong et al., 1997). However, those mice are persistently infected, developing lesions at later time points. The administration of competent CD4+CD25+ regulatory T cells is capable of transiently inhibiting those pathogenic effector cells, ameliorating the disease (Ji et al., 2005). Actually, amastigotes derived from BALB/c nude mice expose low amounts of PS, when compared to those in parasites obtained from their WT counterparts (Franca-Costa et al., 2012). The adoptive transfer of CD4+ T cells to infected nude mice stimulated PS exposure on lesion-derived amastigotes. These data reinforce the assertion that pathogenic CD4+ T cells affect Leishmania infection and suggest that these cells are necessary to generate amastigotes with high amounts of PS at their surface that, therefore, are highly capable of re-infecting new host cells, modulating MΦ functions and avoiding immune surveillance.
The outcome of Leishmania infection is determined by the efficiency of MΦ activation and by the enzyme metabolizing the aminoacid L-arginine. Classical MΦ activation through inflammatory cytokines leads to iNOS expression, NO production and parasite killing, whereas regulatory or anti-inflammatory cytokines lead to non-classical MΦ activation, arginase I expression, polyamine production and parasite survival and growth (Wanasen and Soong, 2008; Acuna et al., 2017; Muxel et al., 2017a). Both pathways use L-arginine as a primary substrate. Infected MΦs stimulated with supernatants from re-stimulated LN cells led to the simultaneous expression of iNOS and arginase I. This activation provides the necessary stimuli to increase PS exposure on intracellular amastigotes without interfering with the parasites' viability and proliferative capacity. The presence of very low concentrations of NO as a result of iNOS activation was sensed by intracellular amastigotes, triggering PS exposure, while arginase I expression and possible polyamine synthesis were necessary for maintenance of parasite viability and persistence in the host. NO is known to induce apoptosis in intracellular amastigotes (Murray and Nathan, 1999). However, it is possible that the low levels of NO produced, undetected by the Griess reaction, are sufficient to trigger PS exposure that does not lead to cell death due to the simultaneous presence of polyamines derived from arginase I/ODC activity. The latter can act as a protective factor, through DNA stabilization, protecting cells from DNA degradation or inducing autophagic processes, as shown in other models (Rowlatt and Smith, 1981; Ha et al., 1998; Madeo et al., 2010). However, our data do not exclude a possible participation polyamines derived from the parasite, since treatment with DFMO could block ODC expressed by the parasite. We are currently determining the optimal concentration of different NO-donor molecules, to induce PS exposure on axenic amastigotes. This information is necessary to study both the ability of NO to induce this phenotype and to better understand the role of polyamines for parasite survival. This mechanism configures a positive feedback cycle that is beneficial for the parasite. The poor activation of specific CD4+ T cell responses generates stimulatory conditions that induce non-classical MΦ activation, leading to concomitant and low expression of both iNOS and arginase I. Non-classical MΦ activation in turn, stimulates increased and sustained PS exposure on intracellular amastigotes, generating parasites more competent to infect new host cells and spread the anti-inflammatory signals derived from PS recognition. This mechanism seems to operate in BALB/c mice. The differential activation of CD4+ T cells in other mouse strains, such as C57BL/6, can explain the variations on PS exposure described in lesion-derived parasites from different mouse strains (Wanderley et al., 2006), and need to be further investigated. In addition, it is important to determine the impact of PS-dependent infection in human infections. Previous work showed that there is a positive correlation between PS exposure on parasites isolated from patients and the development of diffuse cutaneous Leishmaniasis (DCL). This correlation is also observed when comparing the level of PS on the surface of the isolated parasites and the number of lesions in the patient and the duration of the disease (Franca-Costa et al., 2012). DCL patients are characterized by low inflammatory and T cell response that leads to uncontrolled parasite dissemination and lesion development (Barroso et al., 2018). It is possible to suppose that in DCL patients, there is a unique combination of cytokines that induce augmented PS exposure on the parasite therefore leading to more severe disease.
One of the hallmarks of L. amazonensis infection is the peculiar parasitophorous vacuoles formed in infected MΦs. These vacuoles are large organelles shared by several parasites that continuously undergo fusion with lysosomes, exosomes and endosomes (Veras et al., 1996; Real et al., 2008). This feature is important for amastigotes to uptake nutrients (Borges et al., 1998), to dilute microbicidal molecules (Sacks and Sher, 2002; Wilson et al., 2008) and to evade the immune response (Antoine et al., 1999). Interestingly, different authors showed that enlarged vacuoles are less present in immunodeficient mice (Soong et al., 1997; Franca-Costa et al., 2012), suggesting that vacuole enlargement is also a counteractive response from amastigotes against a stressful environment. We observed that mice treated with anti-PS antibodies showed a marked and significant decrease in vacuole size when compared to untreated or isotype-treated mice. Surely, the antibodies are binding to released amastigotes since they do not have access to the parasitophorous vacuole. PS blockade could lead to a deviation in the endocytic pathway of parasite internalization since PS-dependent amastigote internalization occurs by macropinocytosis (Wanderley et al., 2006), which is characterized by the formation of enlarged endosomes (Basagiannis et al., 2016). The effect of anti-PS blocking antibodies on vacuole size provides a further explanation for the decreased parasite load of mice treated with anti-PS blocking antibodies (Wanderley et al., 2013). These results suggest that MΦ activation by T lymphocytes stimulate PS exposure and the consequences of this exposure are the alternative activation of MΦs, increase amastigote infectivity and enlargement of the parasitophorous vacuoles. The direct mechanism that link PS exposure and vacuole enlargement warrant further investigation.
In summary, this work describes that cytokine-dependent interactions between CD4+ T cells and infected MΦs are sensed by intracellular parasites, which counteract by exposing PS. Exposed PS, in turn, down-regulates the MΦ microbicidal capacity (Wanderley et al., 2006, 2013). Such cross-talk is obtained by a fine-tuned balance between iNOS activation, sufficient for stress-induced PS exposure, and arginase I activation, required for maintaining parasite survival and proliferation. We provide evidence that the increased PS exposure observed on amastigotes in vitro or from mouse lesions is due to MΦ stimulation by cytokines produced by CD4+ T cells. Therefore, the cellular immune response against the parasite can be exploited by the pathogen, generating amastigotes that are more competent to disseminate the disease and to escape from the host's immune system (Wanderley et al., 2006). In addition, we have provided further explanation for the pathogenic role of CD4+ T cells during L. amazonensis infection.
This study was carried out in accordance with the recommendations of University of Texas Medical Branch Animal Care and Use Committee. The protocol was approved by the University of Texas Medical Branch Animal Care and Use Committee under the number #9803016A.
JW and PD performed in vitro infections, cell cultures, and in vivo adoptive transfer experiments. EC performed qPCR and western blot analysis of iNOS and arginase I expression. AP performed the anti-PS in vivo treatment and histopathological analysis. JW, RD, MB, and LS designed experiments, wrote the manuscript, and made helpful critiques.
This study was supported by NIH grant AI043003 to LS, NIH training grant T32AI007526 to EDC, a Brazilian National Research Council Scholarship (CNPq) to JW, a scholarship to AP (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES)—Finance Code 001, a MCT/CNPq grant 471144/2008 and a CNPq senior investigator grant to MB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Lijun Xin and Dr. Yingwei Wang for technical assistance and helpful discussion, Eulógio Carlos Queiroz de Carvalho for assistance with histopathology sample processing, and Peregrine Pharmaceuticals for kindly donate anti-PS antibodies.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcimb.2019.00105/full#supplementary-material
Acuna, S. M., Aoki, J. I., Laranjeira-Silva, M. F., Zampieri, R. A., Fernandes, J. C. R., Muxel, S. M., et al. (2017). Arginase expression modulates nitric oxide production in Leishmania (Leishmania) amazonensis. PLoS ONE 12:e0187186. doi: 10.1371/journal.pone.0187186
Antoine, J. C., Lang, T., Prina, E., Courret, N., and Hellio, R. (1999). H-2M molecules, like MHC class II molecules, are targeted to parasitophorous vacuoles of Leishmania-infected macrophages and internalized by amastigotes of L. amazonensis and L. mexicana. J. Cell. Sci. 112(Pt 15), 2559–2570.
Barroso, D. H., Falcao, S. A. C., da Motta, J. O. C., Sevilha Dos Santos, L., Takano, G. H. S., Gomes, C. M., et al. (2018). PD-L1 may mediate T-cell exhaustion in a case of early diffuse leishmaniasis caused by Leishmania (L.) amazonensis. Front. Immunol. 9:1021. doi: 10.3389/fimmu.2018.01021
Basagiannis, D., Zografou, S., Murphy, C., Fotsis, T., Morbidelli, L., Ziche, M., et al. (2016). VEGF induces signalling and angiogenesis by directing VEGFR2 internalisation through macropinocytosis. J. Cell. Sci. 129, 4091–4104. doi: 10.1242/jcs.188219
Birge, R. B., Boeltz, S., Kumar, S., Carlson, J., Wanderley, J., Calianese, D., et al. (2016). Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell. Death Differ. 23, 962–978. doi: 10.1038/cdd.2016.11
Borges, V. M., Vannier-Santos, M. A., and de Souza, W. (1998). Subverted transferrin trafficking in Leishmania-infected macrophages. Parasitol. Res. 84, 811–822. doi: 10.1007/s004360050493
Canellakis, E. S., Viceps-Madore, D., Kyriakidis, D. A., and Heller, J. S. (1979). The regulation and function of ornithine decarboxylase and of the polyamines. Curr. Top. Cell. Regul. 15, 155–202. doi: 10.1016/B978-0-12-152815-7.50009-0
Carneiro, M. B. H., Roma, E. H., Ranson, A. J., Doria, N. A., Debrabant, A., Sacks, D. L., et al. (2018). NOX2-derived reactive oxygen species control inflammation during Leishmania amazonensis infection by mediating infection-induced neutrophil apoptosis. J. Immunol. 200, 196–208. doi: 10.4049/jimmunol.1700899
Corraliza, I. M., Soler, G., Eichmann, K., and Modolell, M. (1995). Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem. Biophys. Res. Commun. 206, 667–673. doi: 10.1006/bbrc.1995.1094
Damatta, R. A., Seabra, S. H., Deolindo, P., Arnholdt, A. C., Manhaes, L., Goldenberg, S., et al. (2007). Trypanosoma cruzi exposes phosphatidylserine as an evasion mechanism. FEMS Microbiol. Lett. 266, 29–33. doi: 10.1111/j.1574-6968.2006.00495.x
de Freitas Balanco, J. M., Moreira, M. E., Bonomo, A., Bozza, P. T., Amarante-Mendes, G., Pirmez, C., et al. (2001). Apoptotic mimicry by an obligate intracellular parasite downregulates macrophage microbicidal activity. Curr. Biol. 11, 1870–1873. doi: 10.1016/S0960-9822(01)00563-2
de Oliveira Cardoso, F., de Souza Cda, S., Mendes, V. G., Abreu-Silva, A. L., Goncalves da Costa, S. C., and Calabrese, K. S. (2010). Immunopathological studies of Leishmania amazonensis infection in resistant and in susceptible mice. J. Infect. Dis. 201, 1933–1940. doi: 10.1086/652870
Edmond Rouan, S. K., Otterness, I. G., Cunningham, A. C., and Rhodes, C. T. (1989). Specific, high affinity colchicine binding monoclonal antibodies: development and characterization of the antibodies. Hybridoma 8, 435–448. doi: 10.1089/hyb.1989.8.435
Fadok, V. A., Bratton, D. L., Konowal, A., Freed, P. W., Westcott, J. Y., and Henson, P. M. (1998). Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101, 890–898. doi: 10.1172/JCI1112
Fadok, V. A., Voelker, D. R., Campbell, P. A., Cohen, J. J., Bratton, D. L., and Henson, P. M. (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 148, 2207–2216.
Feng, Z., Hensley, L., McKnight, K. L., Hu, F., Madden, V., Ping, L., et al. (2013). A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature 496, 367–371. doi: 10.1038/nature12029
Franca-Costa, J., Van Weyenbergh, J., Boaventura, V. S., Luz, N. F., Malta-Santos, H., Oliveira, M. C., et al. (2015). Arginase I, polyamine, and prostaglandin E2 pathways suppress the inflammatory response and contribute to diffuse cutaneous Leishmaniasis. J. Infect. Dis. 211, 426–435. doi: 10.1093/infdis/jiu455
Franca-Costa, J., Wanderley, J. L., Deolindo, P., Zarattini, J. B., Costa, J., Soong, L., et al. (2012). Exposure of phosphatidylserine on Leishmania amazonensis isolates is associated with diffuse cutaneous Leishmaniasis and parasite infectivity. PLoS ONE 7:e36595. doi: 10.1371/journal.pone.0036595
Ha, H. C., Yager, J. D., Woster, P. A., and Casero, R. A. Jr. (1998). Structural specificity of polyamines and polyamine analogues in the protection of DNA from strand breaks induced by reactive oxygen species. Biochem. Biophys. Res. Commun. 244, 298–303. doi: 10.1006/bbrc.1998.8258
Hoffmann, P. R., deCathelineau, A. M., Ogden, C. A., Leverrier, Y., Bratton, D. L., Daleke, D. L., et al. (2001). Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell. Biol. 155, 649–659. doi: 10.1083/jcb.200108080
Ji, J., Masterson, J., Sun, J., and Soong, L. (2005). CD4+CD25+ regulatory T cells restrain pathogenic responses during Leishmania amazonensis infection. J. Immunol. 174, 7147–7153. doi: 10.4049/jimmunol.174.11.7147
Ji, J., Sun, J., Qi, H., and Soong, L. (2002). Analysis of T helper cell responses during infection with Leishmania amazonensis. Am. J. Trop. Med. Hyg. 66, 338–345. doi: 10.4269/ajtmh.2002.66.338
Ji, J., Sun, J., and Soong, L. (2003). Impaired expression of inflammatory cytokines and chemokines at early stages of infection with Leishmania amazonensis. Infect. Immun. 71, 4278–4288. doi: 10.1128/IAI.71.8.4278-4288.2003
Lasakosvitsch, F., Gentil, L. G., dos Santos, M. R., da Silveira, J. F., and Barbieri, C. L. (2003). Cloning and characterisation of a cysteine proteinase gene expressed in amastigotes of Leishmania (L.) amazonensis. Int. J. Parasitol. 33, 445–454. doi: 10.1016/S0020-7519(03)00010-9
Lecoeur, H., de La Llave, E., Osorio, Y. F. J., Goyard, S., Kiefer-Biasizzo, H., Balazuc, A. M., et al. (2010). Sorting of Leishmania-bearing dendritic cells reveals subtle parasite-induced modulation of host-cell gene expression. Microbes Infect. 12, 46–54. doi: 10.1016/j.micinf.2009.09.014
Leon, L. L., Machado, G. M., Paes, L. E., and Grimaldi Junior, G. (1990). Antigenic differences of Leishmania amazonensis isolates causing diffuse cutaneous Leishmaniasis. Trans. R. Soc. Trop. Med. Hyg. 84, 678–680. doi: 10.1016/0035-9203(90)90144-4
Madeo, F., Tavernarakis, N., and Kroemer, G. (2010). Can autophagy promote longevity? Nat. Cell. Biol. 12, 842–846. doi: 10.1038/ncb0910-842
Majumder, S., and Kierszenbaum, F. (1993). Inhibition of host cell invasion and intracellular replication of Trypanosoma cruzi by N,N'-bis(benzyl)-substituted polyamine analogs. Antimicrob. Agents Chemother. 37, 2235–2238. doi: 10.1128/AAC.37.10.2235
Mercer, J., and Helenius, A. (2008). Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science 320, 531–535. doi: 10.1126/science.1155164
Modolell, M., Corraliza, I. M., Link, F., Soler, G., and Eichmann, K. (1995). Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by TH1 and TH2 cytokines. Eur. J. Immunol. 25, 1101–1104. doi: 10.1002/eji.1830250436
Moore, W. M., Webber, R. K., Jerome, G. M., Tjoeng, F. S., Misko, T. P., and Currie, M. G. (1994). L-N6-(1-iminoethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J. Med. Chem. 37, 3886–3888. doi: 10.1021/jm00049a007
Murray, H. W., and Nathan, C. F. (1999). Macrophage microbicidal mechanisms in vivo: reactive nitrogen versus oxygen intermediates in the killing of intracellular visceral Leishmania donovani. J. Exp. Med. 189, 741–746. doi: 10.1084/jem.189.4.741
Muxel, S. M., Aoki, J. I., Fernandes, J. C. R., Laranjeira-Silva, M. F., Zampieri, R. A., Acuna, S. M., et al. (2017a). Arginine and Polyamines Fate in Leishmania Infection. Front. Microbiol. 8:2682. doi: 10.3389/fmicb.2017.02682
Muxel, S. M., Laranjeira-Silva, M. F., Zampieri, R. A., and Floeter-Winter, L. M. (2017b). Leishmania (Leishmania) amazonensis induces macrophage miR-294 and miR-721 expression and modulates infection by targeting NOS2 and L-arginine metabolism. Sci. Rep. 7:44141. doi: 10.1038/srep44141
Osorio y Fortea, J., Prina, E., de La Llave, E., Lecoeur, H., Lang, T., and Milon, G. (2007). Unveiling pathways used by Leishmania amazonensis amastigotes to subvert macrophage function. Immunol. Rev. 219, 66–74. doi: 10.1111/j.1600-065X.2007.00559.x
Poon, I. K., Lucas, C. D., Rossi, A. G., and Ravichandran, K. S. (2014). Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol. 14, 166–180. doi: 10.1038/nri3607
Prina, E., Abdi, S. Z., Lebastard, M., Perret, E., Winter, N., and Antoine, J. C. (2004). Dendritic cells as host cells for the promastigote and amastigote stages of Leishmania amazonensis: the role of opsonins in parasite uptake and dendritic cell maturation. J. Cell. Sci. 117(Pt 2), 315–325. doi: 10.1242/jcs.00860
Prina, E., Antoine, J. C., Wiederanders, B., and Kirschke, H. (1990). Localization and activity of various lysosomal proteases in Leishmania amazonensis-infected macrophages. Infect. Immun. 58, 1730–1737.
Prina, E., Jouanne, C., de Souza Lao, S., Szabo, A., Guillet, J. G., and Antoine, J. C. (1993). Antigen presentation capacity of murine macrophages infected with Leishmania amazonensis amastigotes. J. Immunol. 151, 2050–2061.
Ramer, A. E., Vanloubbeeck, Y. F., and Jones, D. E. (2006). Antigen-responsive CD4+ T cells from C3H mice chronically infected with Leishmania amazonensis are impaired in the transition to an effector phenotype. Infect. Immun. 74, 1547–1554. doi: 10.1128/IAI.74.3.1547-1554.2006
Real, F., Pouchelet, M., and Rabinovitch, M. (2008). Leishmania (L.) amazonensis: fusion between parasitophorous vacuoles in infected bone-marrow derived mouse macrophages. Exp. Parasitol. 119, 15–23. doi: 10.1016/j.exppara.2007.12.013
Rothlin, C. V., Ghosh, S., Zuniga, E. I., Oldstone, M. B., and Lemke, G. (2007). TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131, 1124–1136. doi: 10.1016/j.cell.2007.10.034
Rowlatt, C., and Smith, G. J. (1981). Ultrastructural studies on chromatin digestion by micrococcal nuclease in the presence of polyamines. J. Cell. Sci. 48, 171–179.
Sacks, D., and Sher, A. (2002). Evasion of innate immunity by parasitic protozoa. Nat. Immunol. 3, 1041–1047. doi: 10.1038/ni1102-1041
Saraiva, E. M., Pimenta, P. F., Pereira, M. E., and de Souza, W. (1983). Isolation and purification of amastigotes of Leishmania mexicana amazonensis by a gradient of Metrizamide. J. Parasitol. 69, 627–629. doi: 10.2307/3281388
Seabra, S. H., de Souza, W., and Damatta, R. A. (2004). Toxoplasma gondii exposes phosphatidylserine inducing a TGF-beta1 autocrine effect orchestrating macrophage evasion. Biochem. Biophys. Res. Commun. 324, 744–752. doi: 10.1016/j.bbrc.2004.09.114
Silveira, F. T., Lainson, R., and Corbett, C. E. (2005). Further observations on clinical, histopathological, and immunological features of borderline disseminated cutaneous Leishmaniasis caused by Leishmania (Leishmania) amazonensis. Mem. Inst. Oswaldo Cruz. 100, 525–534. doi: 10.1590/S0074-02762005000500013
Soares, M. M., King, S. W., and Thorpe, P. E. (2008). Targeting inside-out phosphatidylserine as a therapeutic strategy for viral diseases. Nat. Med. 14, 1357–1362. doi: 10.1038/nm.1885
Soong, L. (2008). Modulation of dendritic cell function by Leishmania parasites. J. Immunol. 180, 4355–4360. doi: 10.4049/jimmunol.180.7.4355
Soong, L., Chang, C. H., Sun, J., Longley, B. J. Jr., Ruddle, N. H., Flavell, R. A., et al. (1997). Role of CD4+ T cells in pathogenesis associated with Leishmania amazonensis infection. J. Immunol. 158, 5374–5383.
Stenger, S., Thuring, H., Rollinghoff, M., Manning, P., and Bogdan, C. (1995). L-N6-(1-iminoethyl)-lysine potently inhibits inducible nitric oxide synthase and is superior to NG-monomethyl-arginine in vitro and in vivo. Eur. J. Pharmacol. 294, 703–712. doi: 10.1016/0014-2999(95)00618-4
Terabe, M., Wakana, S., Katakura, K., Onodera, T., Matsumoto, Y., and Ito, M. (2004). Influence of H2 complex and non-H2 genes on progression of cutaneous lesions in mice infected with Leishmania amazonensis. Parasitol. Int. 53, 217–221. doi: 10.1016/j.parint.2003.12.002
Vannier-Santos, M. A., Menezes, D., Oliveira, M. F., and de Mello, F. G. (2008). The putrescine analogue 1,4-diamino-2-butanone affects polyamine synthesis, transport, ultrastructure and intracellular survival in Leishmania amazonensis. Microbiology 154(Pt 10), 3104–3111. doi: 10.1099/mic.0.2007/013896-0
Vargas-Inchaustegui, D. A., Tai, W., Xin, L., Hogg, A. E., Corry, D. B., and Soong, L. (2009). Distinct roles for MyD88 and Toll-like receptor 2 during Leishmania braziliensis infection in mice. Infect. Immun. 77, 2948–2956. doi: 10.1128/IAI.00154-09
Veras, P. S., Topilko, A., Gouhier, N., Moreau, M. F., Rabinovitch, M., and Pouchelet, M. (1996). Fusion of Leishmania amazonensis parasitophorous vacuoles with phagosomes containing zymosan particles: cinemicrographic and ultrastructural observations. Braz. J. Med. Biol. Res. 29, 1009–1018.
Wanasen, N., and Soong, L. (2008). L-arginine metabolism and its impact on host immunity against Leishmania infection. Immunol. Res. 41, 15–25. doi: 10.1007/s12026-007-8012-y
Wanderley, J. L., Moreira, M. E., Benjamin, A., Bonomo, A. C., and Barcinski, M. A. (2006). Mimicry of apoptotic cells by exposing phosphatidylserine participates in the establishment of amastigotes of Leishmania (L) amazonensis in mammalian hosts. J. Immunol. 176, 1834–1839. doi: 10.4049/jimmunol.176.3.1834
Wanderley, J. L., Pinto da Silva, L. H., Deolindo, P., Soong, L., Borges, V. M., Prates, D. B., et al. (2009). Cooperation between apoptotic and viable metacyclics enhances the pathogenesis of Leishmaniasis. PLoS ONE 4:e5733. doi: 10.1371/journal.pone.0005733
Wanderley, J. L. M., Thorpe, P. E., Barcinski, M. A., and Soong, L. (2013). Phosphatidylserine exposure on the surface of Leishmania amazonensis amastigotes modulates in vivo infection and dendritic cell function. Parasite Immunol. 35, 109–119. doi: 10.1111/pim.12019
Wilson, J., Huynh, C., Kennedy, K. A., Ward, D. M., Kaplan, J., Aderem, A., et al. (2008). Control of parasitophorous vacuole expansion by LYST/Beige restricts the intracellular growth of Leishmania amazonensis. PLoS Pathog. 4:e1000179. doi: 10.1371/journal.ppat.1000179
Xie, Q. W., Whisnant, R., and Nathan, C. (1993). Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J. Exp. Med. 177, 1779–1784. doi: 10.1084/jem.177.6.1779
Xin, L., Li, K., and Soong, L. (2008). Down-regulation of dendritic cell signaling pathways by Leishmania amazonensis amastigotes. Mol. Immunol. 45, 3371–3382. doi: 10.1016/j.molimm.2008.04.018
Xin, L., Vargas-Inchaustegui, D. A., Raimer, S. S., Kelly, B. C., Hu, J., Zhu, L., et al. (2010). Type I IFN receptor regulates neutrophil functions and innate immunity to Leishmania parasites. J. Immunol. 184, 7047–7056. doi: 10.4049/jimmunol.0903273
Keywords: phosphatidylserine, amastigote, T cell, parasitophorous vacuole, macrophage, immune evasion
Citation: Wanderley JLM, Deolindo P, Carlsen E, Portugal AB, DaMatta RA, Barcinski MA and Soong L (2019) CD4+ T Cell-Dependent Macrophage Activation Modulates Sustained PS Exposure on Intracellular Amastigotes of Leishmania amazonensis. Front. Cell. Infect. Microbiol. 9:105. doi: 10.3389/fcimb.2019.00105
Received: 16 December 2018; Accepted: 26 March 2019;
Published: 12 April 2019.
Edited by:
Claudia Ida Brodskyn, Gonçalo Moniz Institute (IGM), BrazilReviewed by:
Veronica Jimenez, California State University, Fullerton, United StatesCopyright © 2019 Wanderley, Deolindo, Carlsen, Portugal, DaMatta, Barcinski and Soong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joao Luiz Mendes Wanderley, bG13am9hb0BtYWNhZS51ZnJqLmJy
Lynn Soong, bHlzb29uZ0B1dG1iLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.