 Tatiane S. Lima
Tatiane S. Lima Melissa B. Lodoen
Melissa B. Lodoen
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell. Infect. Microbiol. , 16 April 2019
Sec. Parasite and Host
Volume 9 - 2019 | https://doi.org/10.3389/fcimb.2019.00103
This article is part of the Research Topic Toxoplasma gondii Host Interactions: A Story of Immune Attack and Parasite Counterattack View all 19 articles
Toxoplasma gondii is an intracellular protozoan parasite of global importance that can remarkably infect, survive, and replicate in nearly all mammalian cells. Notably, 110 years after its discovery, Toxoplasmosis is still a neglected parasitic infection. Although most human infections with T. gondii are mild or asymptomatic, T. gondii infection can result in life-threatening disease in immunocompromised individuals and in the developing fetus due to congenital infection, underscoring the role of the host immune system in controlling the parasite. Recent evidence indicates that T. gondii elicits a robust innate immune response during infection. Interestingly, however, T. gondii has evolved strategies to successfully bypass or manipulate the immune system and establish a life-long infection in infected hosts. In particular, T. gondii manipulates host immunity through the control of host gene transcription and dysregulation of signaling pathways that result in modulation of cell adhesion and migration, secretion of immunoregulatory cytokines, production of microbicidal molecules, and apoptosis. Many of these host-pathogen interactions are governed by parasite effector proteins secreted from the apical secretory organelles, including the rhoptries and dense granules. Here, we review recent findings on mechanisms by which T. gondii evades host innate immunity, with a focus on parasite evasion of the human innate immune system.
Toxoplasma gondii is an obligate intracellular protozoan parasite that infects an estimated one-third of the global human population. Although most infections are asymptomatic, T. gondii can cause life-threatening infections in immunocompromised individuals and the developing fetus (Montoya and Liesenfeld, 2004). During infection, T. gondii disseminates via the circulatory system and establishes chronic infection in several organs, including the heart and brain (Harker et al., 2015).
Although both humans and rodents are hosts for T. gondii, there are key differences in the innate immune responses between these species. In the mouse, innate immunity is mediated by TLR11 and TLR12 recognition of T. gondii profilin, which is the dominant mechanism driving IL-12 production (Yarovinsky et al., 2005; Koblansky et al., 2013). Notably, TLR11 is non-functional in humans, and TLR12 does not exist in the human genome, indicating alternative mechanisms of parasite sensing in human cells. Although these mechanisms have not been completely defined, it is known that IL-12 is produced by human neutrophils and monocytes in response to T. gondii (Bliss et al., 1999; Aldebert et al., 2007). In human monocytes, unlike in mouse macrophages (Robben et al., 2004), phagocytosis of the parasite drives this IL-12 response (Tosh et al., 2016). IL-12 induces the production of IFN-γ, a key mediator of immunity in humans and mice (Suzuki et al., 1988; Ceravolo et al., 1999) that initiates protective type 1 immunity (Gazzinelli et al., 1994; Däaubener et al., 1995).
In addition to activating T cell-mediated immunity, IFN-γ functions in a cell-autonomous manner to control intracellular parasites. IFN-γ increases tryptophan degradation in human fibroblasts, inhibiting parasite replication (Pfefferkorn, 1984). More recently, IFN-γ inducible proteins were found on the parasitophorous vacuole membrane (PVM). Interestingly, these immunity-related GTPases (IRGs) play an important role in cell-intrinsic antimicrobial defense in the mouse (Zhao et al., 2009), but this locus is considerably smaller in humans and does not appear to be involved in immune defense. A parallel IFN-γ-dependent mechanism of resistance in humans and mice consists of the guanylate binding proteins (GBPs), which are recruited to the parasitophorous vacuole membrane and cause vacuolar membrane disruption and parasite clearance (Yamamoto et al., 2012; Degrandi et al., 2013; Selleck et al., 2013). Human GBP1 restricts replication of type II T. gondii in epithelial cells without targeting the parasitophorous vacuole (Johnston et al., 2016), suggesting that GBPs can participate in host defense without causing classical vacuolar membrane disruption. In human cells, ubiquitination at the parasite vacuole has also emerged as a key mechanism of parasite control, leading to non-canonical autophagy and parasite growth stunting in HeLa cells (Selleck et al., 2015) or endolysosomal fusion and parasite clearance in umbilical vein endothelial cells (Clough et al., 2016).
As an obligate intracellular parasite, T. gondii has evolved strategies to successfully manipulate the host immune system to establish a productive infection and maintain an optimal replicative niche. Here we review recently described strategies by which T. gondii specifically evades human innate immunity, with brief mention of related studies in the mouse.
The manipulation of signaling pathways leading to cytokine production is an effective strategy to impair immune responses that compromise pathogen survival. Although T. gondii resides within a vacuole in infected cells, effector proteins released through the parasite's specialized secretory organelles, the rhoptries or dense granules, are instrumental in manipulating host cell signaling and transcriptional responses (Figure 1).
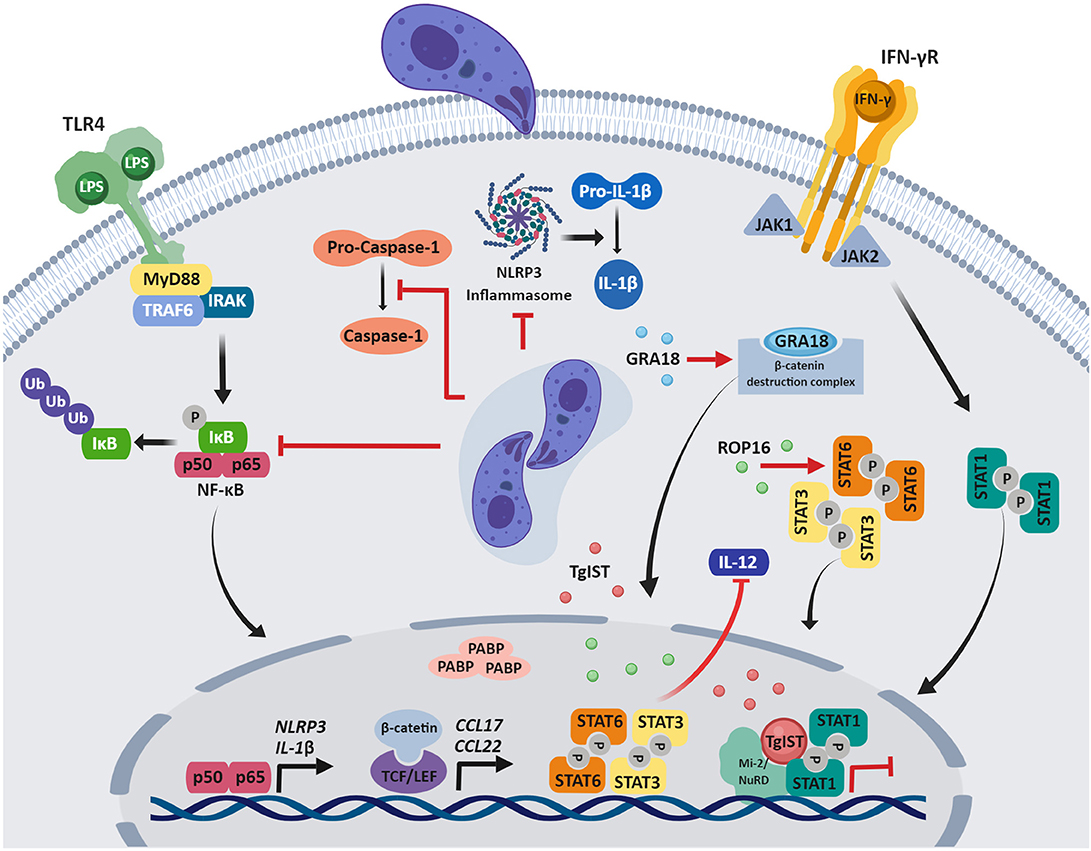
Figure 1. Modulation of host immune signaling by T. gondii. After invasion of the host cell, T gondii manipulation of host signaling pathways and gene expression impair innate immune responses. Parasite effector proteins that govern many of these host-pathogen interactions are secreted from the apical secretory organelles and are found in the host cytosol, associate with the PVM, or translocate to the host nucleus. Although a variety of mechanisms of immune evasion are shown, it should be noted that the function of specific effector proteins may depend on the T gondii strain and human cell type that is infected, as described in the review. Figures were created using BioRender.
The three dominant clonal lineages of T. gondii (types I, II, III) notably differ in their effects on host cells. Type I and III, but not type II strains activate the signal transducer and activator of transcription 3 and 6 (STAT3 and STAT6) in human and mouse cells, thereby down-regulating IL-12 (Saeij et al., 2007). Similarly, in mouse macrophages, constitutive activation of STAT3 by type I strains prevents LPS-induced IL-12p40 production (Butcher et al., 2005). The rhoptry kinase ROP16 is responsible for these effects, by phosphorylating and activating STAT3 and STAT6 in human and mouse cells (Saeij et al., 2007; Yamamoto et al., 2009; Ong et al., 2010).
T. gondii infection induces a robust IFN-γ-driven immune response that is critical to resolve acute infection and control chronic infection (Suzuki et al., 1988, 1989). IFN-γ stimulation induces a vast transcriptional program (Platanias, 2005), and genome-wide microarray analysis in human foreskin fibroblasts (HFFs) revealed that T. gondii infection blocks up-regulation of all 127 genes that were induced by IFN-γ treatment in this study (Kim et al., 2007). Subsequent research determined that type I, II and III strains inhibit STAT1 transcriptional activity through mechanisms independent of ROP16 or GRA15, a dense granule protein that activates sustained NF-κB signaling (Rosowski et al., 2011; Rosowski and Saeij, 2012). IFN-γ stimulation initiates JAK/STAT signaling, whereby STAT1 homodimers translocate to the nucleus and bind to gamma-activated sequences (GAS) in DNA to activate transcription (Sadzak et al., 2008). Notably, T. gondii inhibits the expression of IFN-γ responsive genes by preventing the dissociation of STAT1 from DNA, hampering its recycling and further cycles of STAT1-mediated transcription (Rosowski et al., 2014). Recent studies identified a parasite factor conserved among T. gondii strains that is required for blocking transcription of IFN-stimulated genes in HFFs: T. gondii inhibitor of STAT1-dependent transcription (TgIST) is a dense granule protein that binds to activated STAT1 dimers in the nucleus of IFN-γ-treated cells and also to the chromatin-modifying Mi2/NuRD complex, resulting in altered chromatin and blockade of IFN-γ-dependent transcription (Gay et al., 2016; Olias et al., 2016). Ectopic expression of TgIST in human cells demonstrated that it is sufficient to repress STAT1-dependent promoter activity (Gay et al., 2016). Moreover, in IFN-γ-treated mouse macrophages, TgIST blocks IRG-mediated clearance of type II T. gondii (Gay et al., 2016).
Another major signaling cascade dysregulated by T. gondii is the NF-κB pathway, which leads to the production of pro-inflammatory cytokines involved in host immunity. In infected HFFs, type I T. gondii limits NF-κB activation by reducing p65/RelA phosphorylation and translocation to the nucleus (Shapira et al., 2005). Type I T. gondii also inhibits LPS-induced IL-1β production in primary human neutrophils, and this effect is associated with inhibition of NF-κB signaling. In T. gondii-infected neutrophils, IκBα degradation and p65/RelA phosphorylation are reduced, as are transcripts for IL-1β and the inflammasome sensor NLRP3. T. gondii also inhibits caspase-1 cleavage and activation in infected neutrophils (Lima et al., 2018), but not in infected human monocytes (Gov et al., 2013, 2017), representing different human cell type-specific mechanisms of IL-1β regulation.
Recently, GRA18 was identified as a dense granule protein that reprograms inflammatory responses. GRA18 forms complexes with regulatory elements of the β-catenin destruction complex, which includes β-catenin, GSK3α/β, and the PP2A-B56 holoenzyme, promoting stabilization and nuclear translocation of β-catenin, and inducing β-catenin-dependent gene expression (He et al., 2018). β-catenin is the main effector of the Wnt pathway, functioning as a coactivator of the T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors (Cadigan and Waterman, 2012). In murine macrophages, GRA18 induces β-catenin-dependent genes associated with anti-inflammatory responses, including CCL17 and CCL22 (He et al., 2018), which may counterbalance type I inflammatory responses.
Although cell death caused by infection can be detrimental to the host, apoptosis is also an important means of eliminating intracellular pathogens (Williams, 1994). Perhaps unsurprisingly, viruses, bacteria, and parasites have evolved strategies to inhibit this programmed cell death (Friedrich et al., 2017). Indeed, T. gondii can arrest both cell-intrinsic (mitochondrial) and extrinsic (death receptor-mediated) pathways of apoptosis (Figure 2) in the cells it has invaded. This may help the parasite to preserve its intracellular niche, replicate, and avoid clearance by humoral immunity.
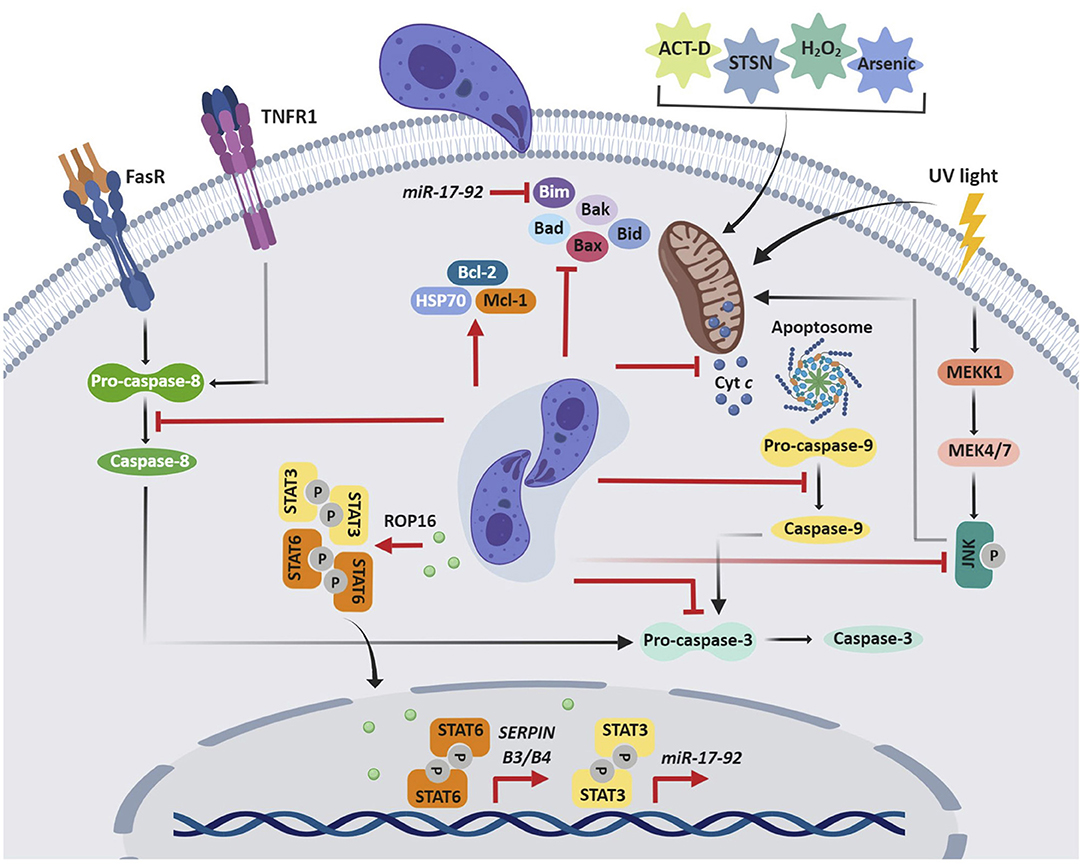
Figure 2. T. gondii inhibition of host cell apoptosis. T gondii impairs both cell-intrinsic (mitochondrial) and -extrinsic (death receptor-mediated) pathways of apoptosis, which allows the parasite to maintain its replicative niche. T gondii can interfere with the initiation, activation, or signaling of the apoptotic cascade, which may result from an indirect mechanism or the direct effect of secreted parasite effector proteins. ACT-D, actinomycin D; STSN, staurosporine; Cyt c, cytochrome c.
The initial observations that T. gondii inhibits host cell apoptosis were in mouse cell lines (Nash et al., 1998); however, over the last 20 years, multiple studies have revealed these effects in human cell lines and primary cells. Collectively, these data show that both type I and II T. gondii inhibit the extrinsic and intrinsic apoptotic pathways through similar mechanisms. The first study on human cells demonstrated that T. gondii-infected HL-60-derived macrophages are protected from actinomycin D-induced apoptosis (Goebel et al., 1999). This effect on the mitochondrial apoptotic pathway is associated with inhibition of cytochrome c release, which in turn reduces cleavage of apoptotic caspase-9 and caspase-3. In addition, Mcl-1, an anti-apoptotic factor from the Bcl-2 family is up-regulated by T. gondii infection (Goebel et al., 2001). T. gondii inhibition of UV-induced apoptosis of infected HeLa cells is also associated with decreased cytochrome c release and apoptotic caspase activity (Carmen et al., 2006). This pathway is known to rely on c-Jun NH2-terminal kinase (JNK) signaling (Tournier et al., 2000), and indeed, JNK activity was repressed in T. gondii-infected cells. Subsequent studies of staurosporine-treated HeLa cells and human Jurkat T cells provided evidence for how T. gondii impairs cytochrome c release. The oligomerization of the Bcl-2 pro-apoptotic proteins Bax and Bak permeabilizes the mitochondrial membrane, allowing the release of apoptogenic proteins, including cytochrome c (Jürgensmeier et al., 1998; Annis et al., 2005). Although T. gondii infection does not affect Bax or Bak expression, it inhibits conformational changes in these proteins, translocation of Bax from the cytosol to the mitochondria, and oligomerization of Bax, which contributes to decreased cytochrome c release (Hippe et al., 2009). Similarly, in arsenic trioxide-treated THP-1 macrophages, T. gondii increases expression of Bcl-2 and the anti-apoptotic chaperone heat-shock protein 70 (HSP70), which in turn reduces cytochrome c release and caspase-3 activation (Hwang et al., 2010). In staurosporine-treated cells, the mechanism is associated with induction of the serine protease inhibitors B3 and B4 (SERPIN B3/B4) via STAT6 activation (Song et al., 2012). In Jurkat cells, T. gondii inhibits apoptosis mediated by granzyme B, a death-inducing serine protease, by inhibiting granzyme B activity (Yamada et al., 2011).
The anti-apoptotic effects of T. gondii in diverse cell types appear to converge on inhibition of cytochrome c and apoptotic caspases. Interestingly, in a cell-free system with Jurkat cell extracts, the parasite can directly affect cytochrome c-induced caspase activation, independent of cytochrome c release from host cell mitochondria or upregulation of antiapoptotic molecules (Keller et al., 2006). Notably, parasite lysates mediated this effect, suggesting that a soluble parasite molecule specifically interferes with cytochrome c-induced caspase activation (Keller et al., 2006). Binding of cytochrome c and of dATP or ATP to the protease activating factor 1 (Apaf-1) allows the formation of a wheel-like heptameric complex, the apoptosome, which in turn activates caspase-9 (Reubold et al., 2009). Interestingly, T. gondii inhibits the binding of caspase-9 to Apaf-1, which prevents caspase-9 activity and subsequent caspase-7 and caspase-3 activation (Graumann et al., 2015).
In addition to blocking the intrinsic pathway, T. gondii also inhibits the extrinsic pathway of apoptosis. T. gondii prevents apoptosis in infected U937 monocytic cells treated with TNF-α and cycloheximide (Goebel et al., 2001). Fas/CD95-induced apoptosis is blocked in the human B cell line SKW6.4 by T. gondii interference with the initiator caspase-8, in the absence of a mitochondrial amplification loop (Vutova et al., 2007). Reduced levels of pro-caspase-8 decrease its association with the death-inducing signaling complex (DISC) and impair activation of effector caspases (Vutova et al., 2007). In HeLa cells, in which Fas/CD95-ligation is amplified via the mitochondrial amplification loop, T. gondii inhibits cleavage of the pro-apoptotic BH3-only protein Bid, the release of cytochrome c, and the activity of the initiator caspase-8 and caspase-9 and the effector caspase-3 and caspase-7 (Hippe et al., 2008).
All of the previously noted human studies characterize the anti-apoptotic effect of T. gondii in human cell lines; however, more recently, this effect has been demonstrated in primary human cells. T. gondii infection of human peripheral blood mononuclear cell (PBMCs)-derived macrophages blocks staurosporine-induced apoptosis via increased expression of the miR-17-92 gene cluster (Cai et al., 2014). The promoter of this cluster contains two putative STAT3 binding sites, and T. gondii TgCtwh3 with atypical genotype China 1, activates STAT3, similar to type I T. gondii. STAT3 activation leads to increased miR-17-92 expression and decreased expression of Bim, a BH3-only member of the Bcl-2 family that contributes to pore formation in the mitochondrial membrane and cytochrome c release (O'Connor et al., 1998; Cai et al., 2014). The miRNA miR-20a is a member of the miR-17-92 gene cluster and its expression is up-regulated in human macrophages infected with type I T. gondii. Inhibition of this miRNA reverses the effect, resulting in apoptosis of human macrophages (Rezaei et al., 2018).
Glycosylphosphatidylinositols (GPIs) are glycolipids that link proteins to eukaryotic cell membranes. GPI anchors are abundantly expressed on many protozoan parasite surfaces, including T. gondii (Lekutis et al., 2001). Since exposing macrophages to Trypanosoma cruzi GPIs enhances expression of the anti-apoptotic A1 and Bcl-2-like genes (Ropert et al., 2002), a similar mechanism for T. gondii GPIs was investigated; however, highly purified T. gondii GPIs do not affect apoptosis of HL-60, Jurkat, or SKW6.4 cells (Debierre-Grockiego et al., 2007). Despite the many studies describing the anti-apoptotic effect of T. gondii in human cells, the parasite factor(s) that trigger this response remain unknown.
In phagocytes, such as neutrophils and macrophages, ROS production is an important antimicrobial response for the elimination of pathogens. Interestingly, however, ROS is not induced in T. gondii-infected human macrophages (Wilson et al., 1980), potentially due to an immune evasion mechanism, as noted below. Non-phagocytic cells also generate low levels of ROS (Bedard and Krause, 2007), and in ARPE-19 cells, T. gondii targets the main NADPH oxidase by reducing Nox4 at the transcript and protein levels, resulting in decreased intracellular ROS. The effect on Nox4 expression was associated with activation of PI3K/AKT signaling in infected cells (Zhou et al., 2013). Proliferation of type I T. gondii in murine inflammatory macrophages was also associated with decreased ROS production (Shrestha et al., 2006). Recent studies in mouse macrophages showed that clearance of type III, but not type I, T. gondii relies on NADPH activity, increased ROS production, and induction of GBP5, suggesting that virulent strains may block ROS production, enabling parasite survival (Matta et al., 2018).
Microbicidal enzymes also contribute to destroying intracellular and extracellular pathogens. Neutrophil granule enzymes are secreted into the phagolysosome and released during NETosis. A Kazal family serine protease inhibitor, T. gondii protease inhibitor 1 (TgPI-1), is secreted by the dense granules and inhibits neutrophil elastase activity (Morris et al., 2002). It is known that both tachyzoites and bradyzoites are resistant to physiological concentrations of trypsin (Sharma and Dubey, 1981), which the parasite encounters in the intestine. TgPI-1 also inhibits trypsin and chymotrypsin, two proteolytic enzymes of the small intestine (Pszenny et al., 2000; Morris et al., 2002). Together, these data suggest a role for TgPI-1 in evading neutrophils and in protecting the parasite in the gut.
T. gondii also affects the cell cycle in infected human cells. In HFFs, the parasite induces progression from G0/G1 to S phase and an arrest toward G2/M (Molestina et al., 2008). This response is associated with sustained activation of extracellular signal-regulated kinase (ERK) signaling, which may act as a positive feedback to maintain HFFs in S phase (Molestina et al., 2008). Similar G2 arrest was observed in the human BeWo trophoblast cell line and in primary normal dermal human fibroblasts (NHDFs). T. gondii infection induces expression of the E3 ubiquitin-protein ligase UHRF1 and down-regulates the cyclin B1, which may cause the cell cycle arrest (Brunet et al., 2008). GRA16 is a dense granule protein that is exported from the PV into the cytoplasm and accumulates in the host nucleus. This protein binds to two host enzymes, the deubiquitinase HAUSP and the PP2A phosphatase, which regulate p53 and cell cycle, suggesting that GRA16 controls host cell arrest in G2/M phase (Bougdour et al., 2013). Modulation of the host cell cycle may influence how T. gondii controls its own replication and suggests a preference for proliferation in G2/M phase.
The first microarrays performed on T. gondii-infected cells revealed up-regulation of host genes involved in nutrient scavenging and metabolism, which the parasite requires for replication (Blader et al., 2001). Interestingly, the hypoxia-inducible factor-1 alpha (HIF-1α) transcription factor, which is required for parasite replication, is stabilized and activated in T. gondii-infected HFFs (Spear et al., 2006). HIF-1 stabilization occurs because PHD2, a prolyl hydroxylase that targets HIF-1 for proteasomal degradation, is down-regulated during infection via activin-like receptor kinase signaling (Wiley et al., 2010).
Poly-adenosine-binding proteins (PABPs) are RNA binding proteins that bind to polyadenylated RNA and are involved in metabolic pathways of the mRNA, and their sub-cellular distribution changes in response to cellular stress (Gray et al., 2015). Nuclear granulation of PABPs is induced in T. gondii-infected HFFs (Fischer et al., 2018), which may enable the parasite to influence the host cell transcriptome. Quantitative proteomic analysis of HFFs also indicates that T. gondii globally reprograms key metabolic pathways in the host cell, including glycolysis, lipid, and sterol metabolism, mitosis, apoptosis, and structural-protein expression (Nelson et al., 2008). Together, these processes may facilitate T. gondii establishment of its replicative niche.
In the last decade, significant progress has been made in characterizing mechanisms of immune evasion by T. gondii. Rodents are a natural host for T. gondii and a relevant model for studying many aspects of parasite immunity. However, the extension of these studies to human cells, which differ in key innate immune pathways, will be critical for understanding determinants of human disease. Future work on the contribution of parasite effector proteins to host-pathogens interactions in both hematopoietic and non-hematopoietic human cells will be of particular interest, as will studies investigating the synergistic effects of these proteins or their role in establishing chronic infection, potentially by altering pathways in brain cells (Schlüter et al., 2001; Xiao et al., 2011; Mammari et al., 2014). Ultimately, elucidation of the molecular mechanisms governing human immune evasion by T. gondii may provide new insights into potential therapeutic targets that contribute to reduced disease and improved outcomes for human health.
TSL wrote the first draft of the manuscript. TSL and MBL edited and revised the manuscript. Both authors read and approved the submitted version.
MBL is supported by NIH R01AI120846.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aldebert, D., Durand, F., Mercier, C., Brenier-Pinchart, M. P., Cesbron-Delauw, M. F., and Pelloux, H. (2007). Toxoplasma gondii triggers secretion of interleukin-12 but low level of interleukin-10 from the THP-1 human monocytic cell line. Cytokine 37, 206–211. doi: 10.1016/j.cyto.2007.03.012
Annis, M. G., Soucie, E. L., Dlugosz, P. J., Cruz-Aguado, J. A., Penn, L. Z., Leber, B., et al. (2005). Bax forms multispanning monomer s that oligomerize to permeabilize membranes during apoptosis. FMBO J. 24, 2096–2103. doi: 10.1038/sj.emboj.7600675
Bedard, K., and Krause, K. H. (2007). The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 87, 245–313. doi: 10.1152/physrev.00044.2005
Blader, I. J., Manger, I. D., and Boothroyd, J. C. (2001). Microarray analysis reveals previously unknown changes in Toxoplasma gondii-infected human cells. J. Biol. Chem. 276, 24223–24231. doi: 10.1074/jbc.M100951200
Bliss, S. K., Marshall, A. J., Zhang, Y., and Denkers, E. Y. (1999). Human polymorphonuclear leukocytes produce IL-12, TNF-alpha, and the chemokines macrophage-inflammatory protein-1 alpha and−1 beta in response to Toxoplasma gondii antigens. J. Immunol. 162, 7369–7375.
Bougdour, A., Durandau, E., Brenier-Pinchart, M. P., Ortet, P., Barakat, M., Kieffer, S., et al. (2013). Host cell subversion by Toxoplasma GRA16, an exported dense granule protein that targets the host cell nucleus and alters gene expression. Cell Host Microbe 13, 489–500. doi: 10.1016/j.chom.2013.03.002
Brunet, J., Pfaff, A. W., Abidi, A., Unoki, M., Nakamura, Y., Guinard, M., et al. (2008). Toxoplasma gondii exploits UHRF1 and induces host cell cycle arrest at G2 to enable its proliferation. Cell. Microbiol. 10, 908–920. doi: 10.1111/j.1462-5822.2007.01093.x
Butcher, B. A., Kim, L., Panopoulos, A. D., Watowich, S. S., Murray, P. J., and Denkers, E. Y. (2005). IL-10-independent STAT3 activation by Toxoplasma gondii mediates suppression of IL-12 and TNF-alpha in host macrophages. J. Immunol. 174, 3148–3152. doi: 10.4049/jimmunol.174.6.3148
Cadigan, K. M., and Waterman, M. L. (2012). TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb. Perspect. Biol. 4:a007906. doi: 10.1101/cshperspect.a007906
Cai, Y., Chen, H., Mo, X., Tang, Y., Xu, X., Zhang, A., et al. (2014). Toxoplasma gondii inhibits apoptosis via a novel STAT3-miR-17-92-Bim pathway in macrophages. Cell. Signal. 26, 1204–1212. doi: 10.1016/j.cellsig.2014.02.013
Carmen, J. C., Hardi, L., and Sinai, A. P. (2006). Toxoplasma gondii inhibits ultraviolet light-induced apoptosis through multiple interactions with the mitochondrion-dependent programmed cell death pathway. Cell. Microbiol. 8, 301–315. doi: 10.1111/j.1462-5822.2005.00622.x
Ceravolo, I. P., Chaves, A. C., Bonjardim, C. A., Sibley, D., Romanha, A. J., and Gazzinelli, R. T. (1999). Replication of Toxoplasma gondii, but not Trypanosoma cruzi, is regulated in human fibroblasts activated with gamma interferon: requirement of a functional JAK/STAT pathway. Infect. Immun. 67, 2233–2240.
Clough, B., Wright, J. D., Pereira, P. M., Hirst, E. M., Johnston, A. C., Henriques, R., et al. (2016). K63-linked ubiquitination targets Toxoplasma gondii for endo-lysossomal destruction in IFNγ-stimulated human cells. PLoS Pathog. 12:e1006027. doi: 10.1371/journal.ppat.1006027
Däaubener, W., Mackenzie, C., and Hadding, U. (1995). Establishment of T-helper type 1- and T-helper type 2-like human Toxoplasma antigen-specific T-cell clones. Immunology 86, 79–84.
Debierre-Grockiego, F., Hippe, D., Schwarz, R. T., and Lüder, C. G. (2007). Toxoplasma gondii glycosylphosphatidylinositols are not involved in T. gondii-induced host cell survival. Apoptosis 12, 781–790. doi: 10.1007/s10495-006-0038-4
Degrandi, D., Kravets, E., Konermann, C., Beuter-Gunia, C., Klümpers, V., Lahme, S., et al. (2013). Murine guanylate binding protein 2 (mGBP2) controls Toxoplasma gondii replication. Proc. Natl. Acad. Sci. U.S.A. 110, 294–299. doi: 10.1073/pnas.1205635110
Fischer, K., Roberts, M., Roscoe, S., Avci, Y., and Ananvoranich, S. (2018). Toxoplasma gondii infection induces the formation of host's nuclear granules containing poly(A)-binding proteins. Can. J. Microbiol. 64, 551–558. doi: 10.1139/cjm-2017-0755
Friedrich, A., Pechstein, J., Berens, C., and Lührmann, A. (2017). Modulation of host cell apoptotic pathways by intracellular pathogens. Curr. Opin. Microbiol. 35, 88–99. doi: 10.1016/j.mib.2017.03.001
Gay, G., Braun, L., Brenier-Pinchart, M. P., Vollaire, J., Josserand, V., Bertini, R. L., et al. (2016). Toxoplasma gondii TgIST co-opts host chromatin repressors dampening STAT1-dependent gene regulation and IFN-γ-mediated host defenses. J. Exp. Med. 213, 1779–1798. doi: 10.1084/jem.20160340
Gazzinelli, R. T., Wysocka, M., Hayashi, S., Denkers, E. Y., Hieny, S, Caspar, P, Trinchieri, G., et al. (1994). Parasite-induced IL-12 stimulates early IFN-gamma synthesis and resistance during acute infection with Toxoplasma gondii. J. Immunol. 153, 2533–2543.
Goebel, S., Gross, U., and Lüder, C. G. (2001). Inhibition of host cell apoptosis by Toxoplasma gondii is accompanied by reduced activation of the caspase cascade and alterations of poly(ADP-ribose) polymerase expression. J. Cell. Sci. 114, 3495–3505.
Goebel, S., Lüder, C. G., and Gross, U. (1999). Invasion by Toxoplasma gondii protects human-derived HL-60 cells from actinomycin D-induced apoptosis. Med. Microbiol. Immunol. 187, 221–226. doi: 10.1007/s004300050096
Gov, L., Karimzadeh, A., Ueno, N., and Lodoen, M. B. (2013). Human innate immunity to Toxoplasma gondii is mediated by host caspase-1 and ASC and parasite GRA15. MBio. 4, e00255–e00213. doi: 10.1128/mBio.00255-13
Gov, L., Schneider, C. A., Lima, T. S., Pandori, W., and Lodoen, M. B. (2017). NLRP3 and potassium efflux drive rapid IL-1β release from primary human monocytes during Toxoplasma gondii infection. J. Immunol. 199, 2855–2864. doi: 10.4049/jimmunol.1700245
Graumann, K., Schaumburg, F., Reubold, T. F., Hippe, D., Eschenburg, S., and Lüder, C. G. (2015). Toxoplasma gondii inhibits cytochrome c-induced caspase activation in its host cell by interference with holo-apoptosomeassembly. Microb. Cell. 2, 150–162. doi: 10.15698/mic2015.05.201
Gray, N. K., Hrabálková, L., Scanlon, J. P., and Smith, R. W. (2015). Poly(A)-binding proteins and mRNA localization: who rules the roost? Biochem. Soc. Trans. 43, 1277–1284. doi: 10.1042/BST20150171
Harker, K. S., Ueno, N., and Lodoen, M. B. (2015). Toxoplasma gondii dissemination: a parasite's journey through the infected host. Parasite Immunol. 37, 141–149. doi: 10.1111/pim.12163
He, H., Brenier-Pinchart, M. P., Braun, L., Kraut, A., Touquet, B., Couté, Y., et al. (2018). Characterization of a Toxoplasma effector uncovers an alternative GSK3/β-catenin-regulatory pathway of inflammation. Elife 7:e39887. doi: 10.7554/eLife.39887
Hippe, D., Lytovchenko, O., Schmitz, I., and Lüder, C. G. (2008). Fas/CD95-mediated apoptosis of type II cells is blocked by Toxoplasma gondii primarily via interference with the mitochondrial amplification loop. Infect. Immun. 76, 2905–2912. doi: 10.1128/IAI.01546-07
Hippe, D., Weber, A., Zhou, L., Chang, D. C., Häcker, G., and Lüder, C. G. (2009). Toxoplasma gondii infection confers resistance against Bims-induced apoptosis by preventing the activation and mitochondrial targeting of pro-apoptotic Bax. J. Cell. Sci. 122, 3511–3521. doi: 10.1242/jcs.050963
Hwang, I. Y., Quan, J. H., Ahn, M. H., Ahmed, H. A., Cha, G. H., Shin, D. W., et al. (2010). Toxoplasma gondii infection inhibits the mitochondrial apoptosis through induction of Bcl-2 and HSP70. Parasitol. Res. 107, 1313–1321. doi: 10.1007/s00436-010-1999-3
Johnston, A. C., Piro, A., Clough, B., Siew, M., Virreira Winter, S., Coers, J., et al. (2016). Human GBP1 does not localize to pathogen vacuoles but restricts Toxoplasma gondii. Cell. Microbiol. 18, 1056–1064. doi: 10.1111/cmi.12579
Jürgensmeier, J. M., Xie, Z., Deveraux, Q., Ellerby, L., Bredesen, D., and Reed, J. C. (1998). Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl. Acad. Sci. U.S.A. 95, 4997–5002. doi: 10.1073/pnas.95.9.4997
Keller, P., Schaumburg, F., Fischer, S. F., Häcker, G., Gross, U., and Lüder, C. G. (2006). Direct inhibition of cytochrome c-induced caspase activation in vitro by Toxoplasma gondii reveals novel mechanisms of interference with host cell apoptosis. FEMS Microbiol. Lett. 258, 312–319. doi: 10.1111/j.1574-6968.2006.00241.x
Kim, S. K., Foutus, A. E., and Boothroyd, J. C. (2007). Toxoplasma gondii dysregulates IFN-γ-inducible gene expression in human fibroblasts: insights from a genome-wide transcriptional profiling. J. Immunol. 178, 5154–5165. doi: 10.4049/jimmunol.178.8.5154
Koblansky, A. A., Jankovic, D., Oh, H., Hieny, S., Sungnak, W., Mathur, R., et al. (2013). Recognition of profiling by Toll-like receptor 12 is critical for host resistance to Toxoplasma gondii. Immunity 38, 119–130. doi: 10.1016/j.immuni.2012.09.016
Lekutis, C., Ferguson, D. J., Grigg, M. E., Camps, M., and Boothroyd, J. C. (2001). Surface antigens of Toxoplasma gondii: variations on a theme. Int. J. Parasitol. 31, 1285–1292. doi: 10.1016/S0020-7519(01)00261-2
Lima, T. S., Gov, L., and Lodoen, M. B. (2018). Evasion of human neutrophil-mediated host defense during Toxoplasma gondii infection. mBio 9, e02027–e02017. doi: 10.1128/mBio.02027-17
Mammari, N., Vignoles, P., Halabi, M. A., Darde, M. L., and Courtioux, B. (2014). In vitro infection of human nervous cells by two strains of Toxoplasma gondii: a kinetic analysis of immune mediators and parasite multiplication. PLoS ONE 9:e98491. doi: 10.1371/journal.pone.0098491
Matta, S. K., Patten, K., Wang, Q., Kim, B. H., MacMicking, J. D., and Sibley, L. D. (2018). NADPH oxidase and guanylate binding protein 5 restrict survival of avirulent type III strains of Toxoplasma gondii in naive macrophages. MBio 9, e01393–e01318. doi: 10.1128/mBio.01393-18
Molestina, R. E., El-Guendy, N., and Sinai, A. P. (2008). Infection with Toxoplasma gondii results in the dysregulation of the host cell cycle. Cell. Microbiol. 10, 1153–1165. doi: 10.1111/j.1462-5822.2008.01117.x
Montoya, J. G., and Liesenfeld, O. (2004). Toxoplasmosis. Lancet 363, 1965–1976. doi: 10.1016/S0140-6736(04)16412-X
Morris, M. T., Coppin, A., Tomavo, S., and Carruthers, V. B. (2002). Functional analysis of Toxoplasma gondii protease inhibitor 1. J. Biol. Chem. 277, 45259–45266. doi: 10.1074/jbc.M205517200
Nash, P. B., Purner, M. B., Leon, R. P., Clarke, P., Duke, R. C., and Curiel, T. J. (1998). Toxoplasma gondii-infected cells are resistant to multiple inducers of apoptosis. J. Immunol. 160, 1824–1830.
Nelson, M. M., Jones, A. R., Carmen, J. C., Sinai, A. P., Burchmore, R., and Wastling, J. M. (2008). Modulation of the host cell proteome by the intracellular apicomplexan parasite Toxoplasma gondii. Infect. Immun. 76, 828–844. doi: 10.1128/IAI.01115-07
O'Connor, L., Strasser, A., O'Reilly, L. A., Hausmann, G., Adams, J. M., Cory, S., et al. (1998). Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J. 15, 384–395.
Olias, P., Etheridge, R. D., Zhang, Y., Holtzman, M. J., and Sibley, L. D. (2016). Toxoplasma effector recruits the Mi-2/NuRD complex to repress STAT1 transcription and block IFN-γ-dependent gene expression. Cell Host Microbe 20, 72–82. doi: 10.1016/j.chom.2016.06.006
Ong, Y. C., Reese, M. L., and Boothroyd, J. C. (2010). Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J. Biol. Chem. 285, 28731–28740. doi: 10.1074/jbc.M110.112359
Pfefferkorn, E. R. (1984). Interferon gamma blocks the growth of Toxoplasma gondii in human fibroblasts by inducing the host cells to degrade tryptophan. Proc. Natl. Acad. Sci. U.S.A. 81, 908–912. doi: 10.1073/pnas.81.3.908
Platanias, L. C. (2005). Mechanisms of type-I and type-II-interferon-mediated signaling. Nat. Rev. Immunol. 5, 375–386. doi: 10.1038/nri1604
Pszenny, V., Angel, S. O., Duschak, V. G., Paulino, M., Ledesma, B., Yabo, M. I., et al. (2000). Molecular cloning, sequencing and expression of a serine protease inhibitor gene from Toxoplasma gondii. Mol. Biochem. Parasitol. 107, 241–249. doi: 10.1016/S0166-6851(00)00202-4
Reubold, T. F., Wohlgemuth, S., and Eschenburg, S. (2009). A new model for the transition of APAF-1 from inactive monomer to caspase-activating apoptosome. J. Biol. Chem. 284, 32717–32724. doi: 10.1074/jbc.M109.014027
Rezaei, F., Daryani, A., Sharifi, M., Sarvi, S., Jafari, N., Pagheh, A. S., et al. (2018). miR-20a inhibition using locked nucleic acid (LNA) technology and its effects on apoptosis of human macrophages infected by Toxoplasma gondii RH strain. Microb. Pathg. 121, 269–276. doi: 10.1016/j.micpath.2018.05.030
Robben, P. M., Mordue, D. G., Truscott, S. M., Takeda, K., Akira, S., and Sibley, L. D. (2004). Production of IL-12 by macrophages infected with Toxoplasma gondii depends on the parasite genotype. J. Immunol. 172, 3683–3694. doi: 10.4049/jimmunol.172.6.3686
Ropert, C., Ferreira, L. R., Campos, M. A., Procópio, D. O., Travassos, L. R., Ferguson, M. A., et al. (2002). Macrophage signaling by glycosylphosphatidylinositol-anchored mucin-like glycoproteins derived from Trypanosoma cruzi trypomastigotes. Microbes Infect. 4, 1015–1025. doi: 10.1016/S1286-4579(02)01609-X
Rosowski, E. E., Lu, D., Julien, L., Rodda, L., Gaiser, R. A., Jensen, K. D., et al. (2011). Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J. Exp. Med. 208, 195–212. doi: 10.1084/jem.20100717
Rosowski, E. E., Nguyen, Q. P., Camejo, A., Spooner, E., and Saeij, J. P. (2014). Toxoplasma gondii inhibits gamma interferon (IFN-γ)- and IFN-β induced host cell STAT1 transcriptional activity by increasing the association of STAT1 with DNA. Infect. Immun. 82, 706–719. doi: 10.1128/IAI.01291-13
Rosowski, E. E., and Saeij, J. P. (2012). Toxoplasma gondii clonal strains all inhibit STAT1 transcriptional activity but polymorphic effectors differentially modulate IFNγ induced gene expression and STAT1 phosphorylation. PLoS ONE 7:e51448. doi: 10.1371/journal.pone.0051448
Sadzak, I., Schiff, M., Gattermeier, I., Glinitzer, R., Sauer, I., Saalmüller, A., et al. (2008). Recruitment of Stat1 to chromatin is required for interferon-induced serine phosphorylation of Stat1 transactivation domain. Proc. Natl. Acad. Sci. U.S.A. 105, 8944–8949. doi: 10.1073/pnas.0801794105
Saeij, J. P., Coller, S., Boyle, J. P., Jerome, M. E., White, M. W., and Boothroyd, J. C. (2007). Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature 445, 324–327. doi: 10.1038/nature05395
Schlüter, D., Deckert, M., Hof, H., and Frei, K. (2001). Toxoplasma gondii infection of neurons induces neuronal cytoline and chemokine production, but gamma interferon- and tumor necrosis factor-stimulated neurons fail to inhibit the invasion and growth of T. gondii. Infect. Immun. 69, 7889–7893. doi: 10.1128/IAI.69.12.7889-7893.2001
Selleck, E. M., Fentress, S. J., Beatty, W. L., Degrandi, D., Pfeffer, K., Virgin, H. W., et al. (2013). Guanylate-binding protein 1 (Gbp1) contributes to cell-autonomous immunity against Toxoplasma gondii. PLoS Pathog. 9:e1003320. doi: 10.1371/journal.ppat.1003320
Selleck, E. M., Orchard, R. C., Lassen, K. G., Beatty, W. L., Xavier, R. J., Levine, B., et al. (2015). A noncanonical autophagy pathway restricts Toxoplasma gondii growth in a strain-specific manner in IFN-γ-activated human cells. MBio 6, e01157–e01115. doi: 10.1128/mBio.01157-15
Shapira, S., Harb, O. S., Margarit, J., Matrajt, M., Han, J., Hoffmann, A., et al. (2005). Initiation and termination of NF-κB signaling by the intracellular protozoan parasite Toxoplasma gondii. J. Cell. Sci. 118, 3501–3508. doi: 10.1242/jcs.02428
Sharma, S. P., and Dubey, J. P. (1981). Quantitative survival of Toxoplasma gondii tachyzoites and bradyzoites in pepsin and in trypsin solutions. Am. J. Vet. Res. 42, 128–130.
Shrestha, S. P., Tomita, T., Weiss, L. M., and Orlofsky, A. (2006). Proliferation of Toxoplasma gondii in inflammatory macrophages in vivo is associated with diminished oxygen radical production in the host cell. Int. J. Parasitol. 36, 433–441. doi: 10.1016/j.ijpara.2006.01.006
Song, K. J., Ahn, H. J., and Nam, H. W. (2012). Anti-apoptotic effects of SERPIN B3 and B4 via STAT6 activation in macrophages after infection with Toxoplasma gondii. Korean J. Parasitol. 50, 1–6. doi: 10.3347/kjp.2012.50.1.1
Spear, W., Chan, D., Coppens, I., Johnson, R. S., Giaccia, A., and Blader, I. J. (2006). The host cell transcription factor hypoxia-inducible factor 1 is required for Toxoplasma gondii growth and survival at physiological oxygen levels. Cell. Microbiol. 8, 339–352. doi: 10.1111/j.1462-5822.2005.00628.x
Suzuki, Y., Conley, F. K., and Remington, J. S. (1989). Importance of endogenous IFN-gamma for prevention of toxoplasmic encephalitis in mice. J. Immunol. 143, 2045–2050.
Suzuki, Y., Orellana, M. A., Schreiber, R. D., and Remington, J. S. (1988). Interferon-gamma: the major mediator of resistance against Toxoplasma gondii. Science 240, 516–518. doi: 10.1126/science.3128869
Tosh, K. W., Mittereder, L., Bonne-Annee, S., Hieny, S., Nutman, T. B., Singer, S. M., et al. (2016). The IL-12 response of primary human dentritic cells and monocytes to Toxoplasma gondii is stimulated by phagocytosis of live parasites rather than host cell invasion. J. Immunol. 196, 345–356. doi: 10.4049/jimmunol.1501558
Tournier, C., Hess, P., Yang, D. D., Xu, J., Turner, T. K., Nimnual, A., et al. (2000). Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288, 870–874. doi: 10.1126/science.288.5467.870
Vutova, P., Wirth, M., Hippe, D., Gross, U., Schulze-Osthoff, K., Schmitz, I., et al. (2007). Toxoplasma gondii inhibits Fas/CD95-triggered cell death by inducing aberrant processing and degradation of caspase-8. Cell. Microbiol. 9, 1556–1570. doi: 10.1111/j.1462-5822.2007.00893.x
Wiley, M., Sweeney, K. R., Chan, D. A., Brown, K. M., McMurtrey, C., Howard, E. W., et al. (2010). Toxoplasma gondii activates hypoxia-inducible factor (HIF) by stabilizing the HIF-1alpha subunit via type I activin-like receptor kinase receptor signaling. J. Biol. Chem. 285, 26852–26860. doi: 10.1074/jbc.M110.147041
Williams, G. T. (1994). Programmed cell death: a fundamental protective response to pathogens. Trends Microbiol. 2, 463–464. doi: 10.1016/0966-842X(94)90648-3
Wilson, C. B., Tsai, V., and Remington, J. S. (1980). Failure to trigger the oxidative metabolic burst by normal macrophages: possible mechanism of intracellular pathogens. J. Exp. Med. 151, 328–346. doi: 10.1084/jem.151.2.328
Xiao, J., Jones-Brando, L., Talbot, C. C. Jr., and Yolke, R. H. (2011). Differential effects of three canonical Toxoplasma strains on gene expression in human neuroepithelial cells. Infect. Immun. 79, 1363–1373. doi: 10.1128/IAI.00947-10
Yamada, T., Tomita, T., Weiss, L. M., and Orlofsky, A. (2011). Toxoplasma gondii inhibits granzyme B-mediated apoptosis by the inhibition of granzyme B function in host cells. Int. J. Parasitol. 41, 595–607. doi: 10.1016/j.ijpara.2010.11.012
Yamamoto, M., Okuyama, M., Ma, J. S., Kimura, T., Kamiyama, N., Saiga, H., et al. (2012). A cluster of interferon-γ-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37, 302–313. doi: 10.1016/j.immuni.2012.06.009
Yamamoto, M., Standley, D. M., Takashima, S., Saiga, H., Okuyama, M., Kayama, H., et al. (2009). A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J. Exp. Med. 206, 2747–2760. doi: 10.1084/jem.20091703
Yarovinsky, F., Zhang, D., Andersen, J. F., Bannenberg, G. L., Serhan, C. N., Hayden, M. S., et al. (2005). TLR11 activation of dendritic cells by a protozoan profiling-like protein. Science 308, 1626–1629. doi: 10.1126/science.1109893
Zhao, Y. O., Khaminets, A., Hunn, J. P., and Howard, J. C. (2009). Disruption of the Toxoplasma gondii parasitophorus vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog. 5:e1000288. doi: 10.1371/journal.ppat.1000288
Keywords: Toxoplasma gondii, immune evasion, innate immunity, IFN-γ, pro-inflammatory cytokines, apoptosis
Citation: Lima TS and Lodoen MB (2019) Mechanisms of Human Innate Immune Evasion by Toxoplasma gondii. Front. Cell. Infect. Microbiol. 9:103. doi: 10.3389/fcimb.2019.00103
Received: 01 February 2019; Accepted: 25 March 2019;
Published: 16 April 2019.
Edited by:
Nicolas Blanchard, INSERM U1043 Centre de Physiopathologie de Toulouse Purpan, FranceReviewed by:
Masahiro Yamamoto, Osaka University, JapanCopyright © 2019 Lima and Lodoen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Melissa B. Lodoen, bWxvZG9lbkB1Y2kuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.