Abstract
Over the last decade, there has been significant advances in the understanding of the cross-talk between metabolism and immune responses. It is now evident that immune cell effector function strongly depends on the metabolic pathway in which cells are engaged in at a particular point in time, the activation conditions, and the cell microenvironment. It is also clear that some metabolic intermediates have signaling as well as effector properties and, hence, topics such as immunometabolism, metabolic reprograming, and metabolic symbiosis (among others) have emerged. Viruses completely rely on their host's cell energy and molecular machinery to enter, multiply, and exit for a new round of infection. This review explores how viruses mimic, exploit or interfere with host cell metabolic pathways and how, in doing so, they may evade immune responses. It offers a brief outline of key metabolic pathways, mitochondrial function and metabolism-related signaling pathways, followed by examples of the mechanisms by which several viral proteins regulate host cell metabolic activity.
Introduction
Several recent comprehensive reviews have highlighted the key role of eukaryotic cell metabolism in immunity (Ganeshan and Chawla, 2014; O'Neill and Pearce, 2016; O'Neill et al., 2016). Six main and interconnected metabolic pathways have a role in the immune response: glycolysis; the pentose phosphate pathway (PPP); the tricarboxylic acid cycle (TCA), also known as Krebs cycle; the fatty acid oxidation (FAO), also known as β-oxidation; as well as the fatty acid and amino acid synthesis pathways (Figure 1).
Figure 1
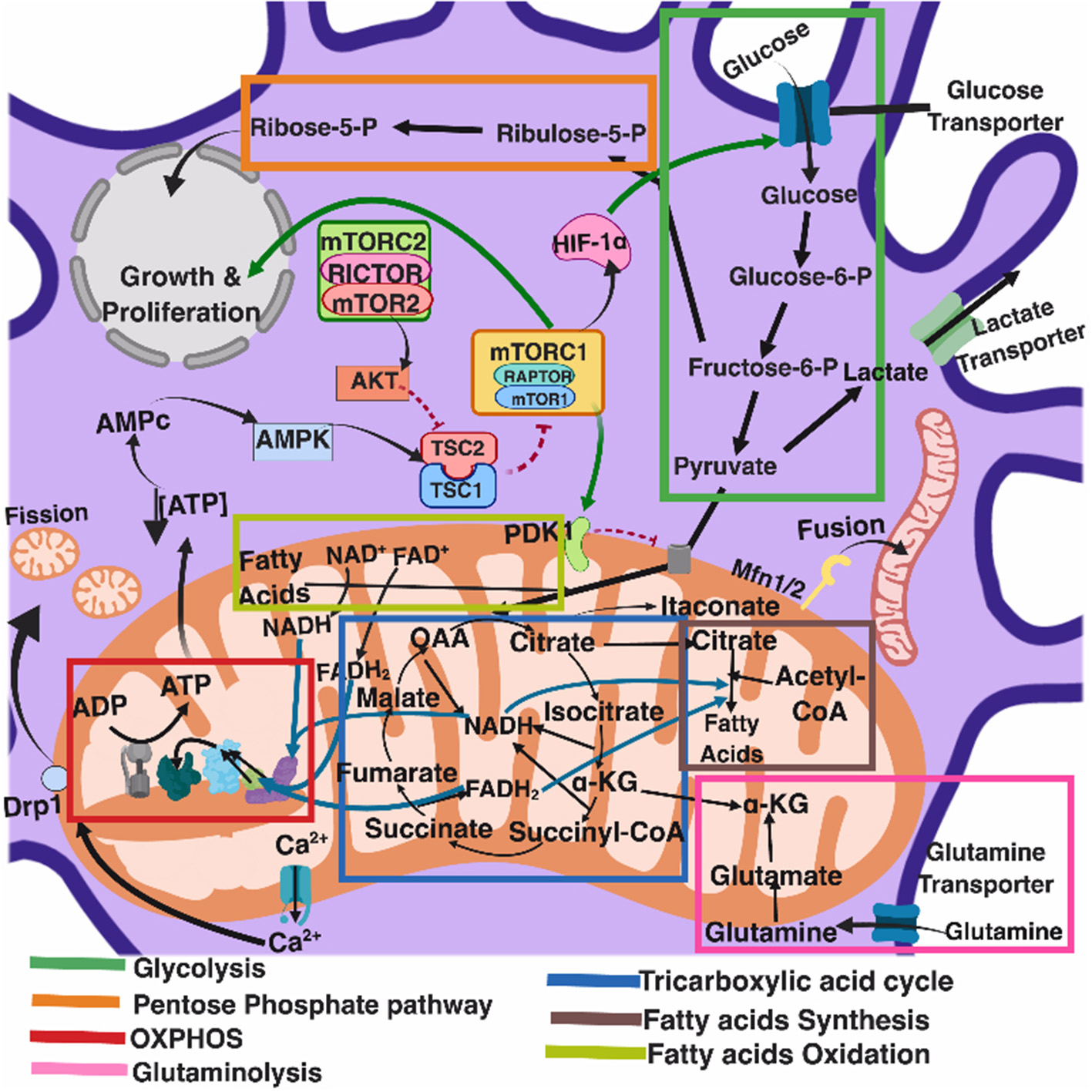
Eukaryotic cell metabolism. Bioenergetic and biosynthetic pathways interconnect glycolysis, glutaminolysis, PPP (pentose phospahate pathway), TCA (tricarboxylic acid cycle), FAO (fatty acid oxidation), fatty acid synthesis, aminoacid synthesis, metabolic sensors such as the AMPK, mTORC1, and mTORC2 pathways, and are also dependent on calcium homeostasis, mitochondrial membrane potential and mitochondrial dynamics. All together they influence cell function and may be the targets of several viruses.
Mitochondria take central stage in cellular metabolism since TCA, FAO, oxidative phosphorylation (OXPHOS), calcium buffering, and heme biosynthesis take place within this organelle (Mishra and Chan, 2016).
Energetic and biosynthetic metabolism is fueled by carbon sources, including glucose and glutamine (DeBerardinis and Cheng, 2010), which are taken up by the cells by glucose and glutamine transporters, respectively (Bhutia and Ganapathy, 2016; Navale and Paranjape, 2016).
Once in the cytosol, glucose is converted to pyruvate, via glycolysis, yielding two molecules of ATP and two molecules of NADH (which acts as a cofactor in several enzymatic reactions) per unit of glucose. The glycolysis pathway is also the source of biosynthetic intermediates that serve the purpose of ribose and nucleotides synthesis (glucose-6-phosphate into ribulose 5-phosphate), amino acids (3-phosphoglycerate enters the serine biosynthetic pathway), and fatty acids (by the sequential conversion of glycolysis-derived pyruvate into the TCA intermediate citrate that may be exported from the mitochondria to the cytosol, where it is converted into acetyl-coA).
Glycolysis-derived pyruvate is either converted to lactate, which is exported out of the cells, or converted into acetyl-CoA that enters the TCA cycle through the aldol condensation with oxaloacetate to form citrate (O'Neill et al., 2016). Citrate is then sequentially converted to isocitrate, α-ketoglutarate, succinyl CoA, succinate, fumarate, malate, and oxaloacetate, which starts a new round of the TCA cycle by its reaction with pyruvate-derived acetyl CoA. Fatty acids can also be converted into acetyl CoA through FAO, linking this metabolic pathway with the TCA cycle. Two major products of both the TCA cycle and FAO are NADH and FADH2, which can transfer electrons to the mitochondrial electron transport chain coupled with OXPHOS and the generation of ATP (O'Neill et al., 2016). In addition, succinate, an intermediate in the TCA cycle, is also an electron donor for the mitochondrial respiratory chain at complex II (succinate dehydrogenase) (Rich and Maréchal, 2010).
The pentose phosphate pathway involves a non-oxidative as well as an oxidative branch; the first allows for the diversion from glycolysis intermediates toward the synthesis of nucleotide and amino acid precursors, while the second generates reducing equivalents of nicotinamide adenine dinucleotide phosphate hydrogen (NADPH), which maintain a favorable cellular redox environment and allows fatty acid synthesis (O'Neill et al., 2016).
Fatty acid synthesis uses glycolysis, TCA cycle, and pentose phosphate pathway metabolic intermediates. TCA cycle-derived citrate may be exported from the mitochondria to the cytosol and then ATP citrate lyase converts citrate to acetyl-coA, which in turn may be carboxylated by acetyl-CoA carboxylase (ACC) producing malonyl-CoA. Furthermore, fatty acid synthase and NADPH elongate fatty acid chains (O'Neill et al., 2016).
Glutamine is also a primary source of energy as it is converted to glutamate and then to α-ketoglutarate, which enters the TCA cycle (DeBerardinis and Cheng, 2010).
Immune system cells preferentially follow one or other metabolic pathway, depending on cell type, differentiation status, activation conditions, and microenvironment. Resting T lymphocytes rely mostly on OXPHOS, whereas activated and proliferating T lymphocytes upregulate the expression of the glucose transporter glut-1 and key glycolytic enzymes, relying mostly on glycolysis (Frauwirth et al., 2002; Pearce and Pearce, 2013).
Memory T lymphocytes use OXPHOS (Pearce and Pearce, 2013), “classically activated” macrophages (stimulated with LPS plus IFN-γ)—also referred to as M1 macrophages—engage in glycolysis, whereas alternatively activated macrophages (stimulated with IL-4)—also referred to as M2 macrophages—use OXPHOS and FAO to generate energy (Rodríguez-Prados et al., 2010). Stimulated macrophages and dendritic cells engage in glycolysis after activation through pattern recognition receptors (PRRs) (O'Neill and Pearce, 2016).
Neutrophils rely mostly on glycolysis (Pearce and Pearce, 2013) and the release of neutrophil extracellular traps (NETs) is dependent on the increase in cell membrane glut-1, glucose uptake, and the glycolytic rate (Rodriguez-Espinosa et al., 2015).
Activated B lymphocytes undergo metabolic reprogramming in response to changing energetic and biosynthetic demands, and long-lived plasma cells uptake glucose and glutamine at a higher rate; glucose is used to generate pyruvate for spare respiratory capacity, and glutamine is used as a carbon source for mitochondrial anaplerotic reactions and respiration, promoting cell survival (Jellusova and Rickert, 2016; Lam et al., 2018).
Switching metabolic pathways (metabolic reprograming) leads to changes in cell function (Buck et al., 2017) and the metabolic microenvironment, i.e., tissue O2 tension, or the concentration of metabolites such as lactate determines cell immune responses (Romero-Garcia et al., 2016).
Interestingly, viral infections such as ocular infection with herpes simplex virus-1 (HSV-1) may change blood glucose levels in the course of infection (Varanasi et al., 2017). Moreover, if glucose utilization is pharmacologically limited in vivo in the inflammatory phase, lesions diminish but, if glucose utilization is limited in the acute phase of infection when the replicating virus is still present in the eye, infected mice become susceptible to the lethal effects of HSV-1 infection as the virus spreads to the brain, causing encephalitis (Varanasi et al., 2017). This highlights the fundamental relationship between cell metabolism, immune response, and viral pathogenesis.
Anti-Viral Immune Responses
Among the most effective antiviral immune responses is the production of several type I interferons (Figure 2); interferon-α (IFN-α) subtypes and interferon-β (IFN-β), which along with IFN-ε, IFN-τ, IFN-κ, IFN-ω, IFN-δ, and IFN-ζ, are collectively referred to as type I interferons; most cells can produce IFN-α and IFN-β following cell activation through the recognition of viral nucleic acids (McNab et al., 2015).
Figure 2
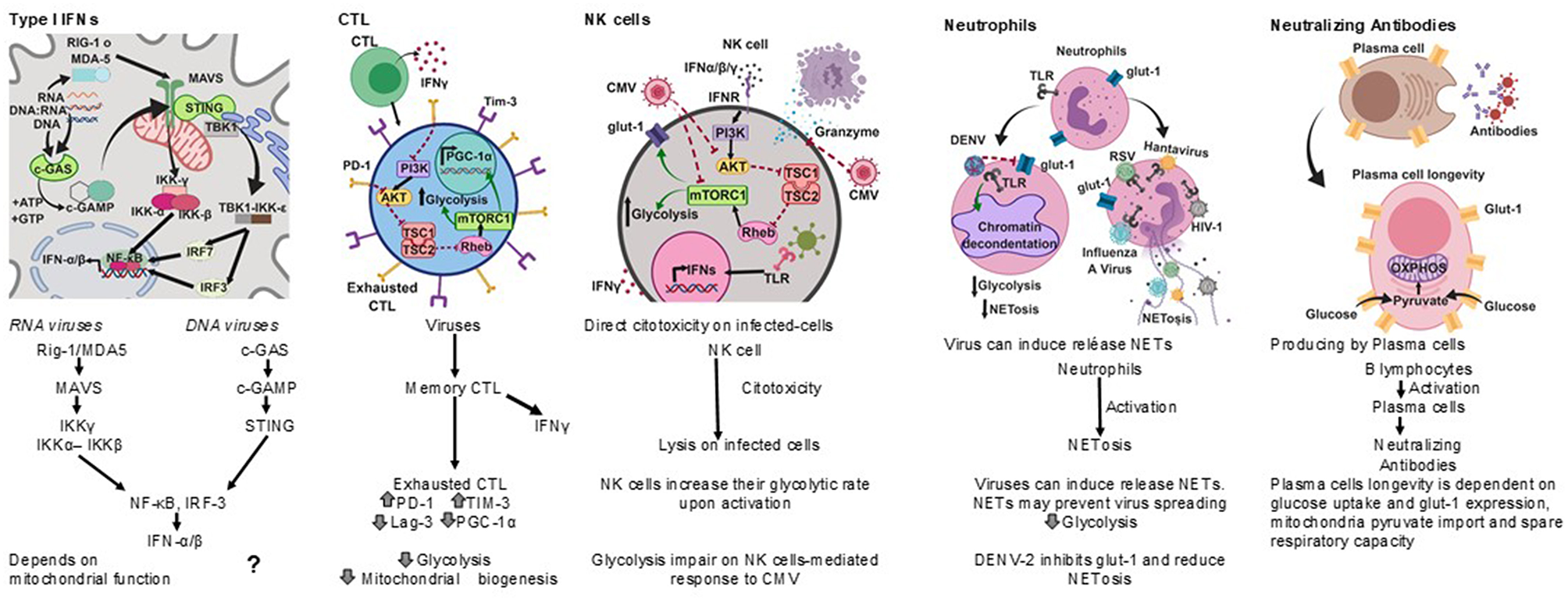
Antiviral immune responses. Type I interferons, cell cytotoxicity, neutrophil extracellular traps and neutralizing antibodies protect against viral infections, each type of response has a metabolic hallmark. Viruses may target specific metabolic pathways for immune evasion.?, not known.
The RIG-I-MDA5-mitochondrial antiviral-signaling protein (MAVS) axis is the major sensing pathway for RNA viruses, while the axis composed of the cyclic guanosine monophosphate (cGMP)-adenosine monophosphate (AMP) synthase (cGAS) and the stimulator of interferon genes (STING) is the major sensing pathway for DNA viruses (Wu and Chen, 2014). However, there is recent evidence that the cGAS-STING pathway may also restrict the infection by RNA viruses, thus suggesting a connection between the sensing of cytosolic DNA and RNA (Ni et al., 2018).
Both anti-viral pathways converge in the activation of two main transcription factors that regulate the expression of type-I interferons, nuclear factor kappa B (NFκB) and interferon regulatory factor 3 (IRF3). In the case of the RIG-I-MDA5-MAVS pathway, their activation depends on mitochondrial function (Seth et al., 2005; Koshiba, 2013).
Both IFN-α and IFN-β activate the expression of interferon-stimulated genes (ISGs) through the Janus kinase-signal transducer and activator of transcription (JAK-STAT) signaling pathway, leading to the inhibition of viral replication and assembly (Darnell et al., 1994; Seth et al., 2005).
Cytotoxic T lymphocytes (CTL) play an important role in the clearance of viral infections (Figure 2); memory CTL can be activated by low concentrations of antigen, readily producing cytokines and the lysis of infected cells, thus preventing dissemination (Veiga-Fernandes et al., 2000).
Upon acute viral infection, virus-specific memory CTL quickly produce IFN-γ. However, around 18 h after infection the number of IFN-γ producing CTL drops concomitantly with the upregulation of inhibitory receptors. It has been suggested that the decrease in the synthesis of IFN-γ by CTL is an active regulatory process (Hosking et al., 2013) reminiscent of T cell exhaustion, a process also known to take place during chronic viral infections (Yi et al., 2010; Wherry, 2011).
A hallmark of T cell exhaustion is the upregulation of inhibitory receptors such as programmed death-1 (PD-1), T cell immunoglobulin mucin-3 (Tim-3), and lymphocyte activation gene-3 (Lag-3) (Freeman et al., 2000; Barber et al., 2006). Interestingly, PD-1 negatively regulates glycolysis, represses the transcriptional co-activator peroxisome proliferator-activated receptor-gamma co-activator (PGC)-1alpha (PGC-1α), which plays an important role in the regulation of carbohydrate and lipid metabolism, and impairs CTL responses (Bengsch et al., 2016).
Other anti-viral cell-mediated immune responses include NK cell cytotoxicity (Hammer et al., 2018) and neutrophil extracellular traps (NETs) (Schönrich and Raftery, 2016) (Figure 2).
Natural killer (NK) cells have anti-viral activities as they exert direct cytotoxicity on virus infected-cells, and readily produce IFN-γ. NK cells increase their glycolytic rate upon activation (Gardiner and Finlay, 2017), and disruption of glycolysis impairs NK cell-mediated responses to Cytomegalovirus (CMV), for instance (Mah et al., 2017).
Neutrophils are considered a first line of defense against pathogens. However, their role in the control of viral infections is not as clear as for other pathogens (Galani and Andreakos, 2015). It has recently been recognized that viruses can induce the release of neutrophil extracellular traps (NETs), and the mechanisms by which NETs could contribute to anti-viral immunity are emerging (Hammer et al., 2018).
Several viruses, including Hantaan virus (HTNV), H1N1 Influenza A virus (IAV), human immunodeficiency virus (HIV-1), and Respiratory Syncytial virus (RSV), directly stimulate neutrophils to release NETs (Raftery et al., 2014; Delgado-Rizo et al., 2017), and both IFN-α and IFN-γ can prime mature neutrophils to release NETs upon further stimulation (Martinelli et al., 2004; Hammer et al., 2018).
HIV-1 may also prevent the release of NETs by inducing dendritic cells to produce IL-10, which in turn suppresses the reactive oxygen species (ROS)-dependent release of NETs (Saitoh et al., 2012; Hammer et al., 2018).
The Dengue virus serotype-2 (DENV-2) down-modulates the phorbol 12- myristate 13- acetate-(PMA) induced release of NETs, and it has been proposed that one of the mechanisms for this is the interference with the mobilization of the glucose transporter glut-1 to the cell membrane and consequently with the glucose uptake (Moreno-Altamirano et al., 2015).
NETs may prevent virus spreading by being trapped by electrostatic attraction or be inactivated by molecules associated with NETs, such as myeloperoxidase and α-defensins (Saitoh et al., 2012; Hammer et al., 2018).
Antibodies are also important anti-viral effectors (Figure 2), and whereas cytotoxic lymphocytes can eliminate infected cells, antibodies are capable of both eliminating infected cells and neutralizing viruses, thereby preventing cell infection. The production of protective antibodies over prolonged periods constitutes a first line of defense against reinfection and, therefore, survival of antibody-producing plasma cells is determinant (Dörner and Radbruch, 2007). It is now known that plasma cell longevity is dependent on enhanced glut-1 expression and glucose uptake, mitochondria pyruvate import and spare respiratory capacity, and that nutrient uptake and catabolism distinguish plasma cell subsets with different lifespans and rates of secreted antibodies (Lam et al., 2016, 2018).
Taken together, it emerges that the activity of immune system cells is dependent on cell metabolism and that viruses could target cell metabolism to evade anti-viral immune responses. The next sections explore some specific mechanisms by which viruses may interfere with cell metabolism.
Mitochondrial Anti-Viral Signaling (MAVS) and Virus Subversion of MAVS
Mitochondria constitute a metabolic hub, so if a virus is to take control of host metabolism, targeting mitochondria is perhaps the best way.
In 2005 Seth et al. reported the identification of a new protein essential for the activation of the transcription factors NFκB and IRF3 by RNA viruses. They named the protein MAVS and showed that this contains a C-terminal transmembrane domain that targets the mitochondrial outer membrane. Strikingly, they found that this transmembrane domain and the targeting to mitochondria are essential for MAVS signaling, opening a new avenue of research in which mitochondria took center stage in antiviral immunity (Seth et al., 2005).
In non-stimulated cells, NFκB is located in the cytoplasm, associated with its inhibitor IκBα. Upon stimulation with viruses, other pathogens or cytokines, the IκB kinase (IKK) is activated, leading to the phosphorylation of IκBα and its subsequent ubiquitination and proteasomal degradation. NFκB is then released and translocated to the nucleus, where it activates immune and inflammatory genes (Silverman and Maniatis, 2001; Seth et al., 2005).
IRF3 is located in the cytoplasm of non-stimulated cells, and following viral or other pathogen infection it becomes phosphorylated by TANK-binding kinase 1 (TBK1) and IKK kinases, allowing the formation of homodimers that can translocate into the nucleus and activate the synthesis of IFN-β, acting in synergy with NFκB (Yoneyama et al., 2002; Fitzgerald et al., 2003; Hiscott et al., 2003; Seth et al., 2005).
IRF7 can also be phosphorylated by TBK1 and IKK (tenOever et al., 2004), leading to the production of interferon-α (Honda et al., 2005; Seth et al., 2005). NFκB and IRFs are activated by RNA viruses as well as by other pathogens.
The entry of RNA viruses to the cells produces double-stranded RNA intermediates, which can be recognized by host cell pathogen recognition receptors (PRRs) including TLR -3, -7, -8, and -9 (Akira and Takeda, 2004; Seth et al., 2005).
The receptor Retinoic Acid-Induced Gene I (RIG-1) recognizes intracellular dsRNA and the interaction of viral RNA with RIG-1 leads to a change in its conformation, which then activates NFκB and IRF3 (Yoneyama et al., 2002; Sumpter et al., 2005).
The melanoma differentiation-associated gene 5 (MDA5) is a RIG-I-like protein involved in dsRNA signaling and apoptosis (Kovacsovics et al., 2002; Seth et al., 2005).
In 2011, Koshiba (Koshiba, 2013) demonstrated that mitochondrial fusion and mitochondrial membrane potential (Δψm) are required for MAVS-mediated signaling. They showed that the deletion-targeting of mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2), two molecules involved in mitochondrial fusion, prevented cells from producing interferons and pro-inflammatory cytokines in response to viral infection. This resulted in increased viral replication along with a reduced Δψm, correlating with a reduced antiviral response. Interestingly, the reduction in Δψm did not affect the activation of IRF3, which acts downstream of MAVS, suggesting that Δψm and MAVS are coupled at the same stage in the RIG-1-like Receptor (RLR) signaling pathway (Koshiba, 2013).
In addition to mitochondria, MAVS are also found in peroxisomes and mitochondrial-associated membranes (MAMs) (Seth et al., 2005; Vazquez and Horner, 2015).
A natural target for the subversion of IFN type I-mediated antiviral response is the MAVS protein (Table 1). As an example, the influenza A virus encodes a protein called PB1-F2, which inhibits the induction of type I interferon at the level of the MAVS (Varga et al., 2012). PB1-F2 is an 87–90-amino-acid-long protein with a serine at position 66 (66S), which accounted for the virulence of the Spanish and avian flu pandemic viruses (H1N1 and H5N1, respectively). Interestingly, PB1-F2 66S has a higher affinity for MAVS than PB1-F2 66N, and more efficiently affects the Δψm than the wild-type PB1-F2 (Conenello et al., 2007).
Table 1
| Virus | Viral proteins | Effect | References |
|---|---|---|---|
| Influenza A virus (IAV) | PB1-F2 | Inhibition of type I IFN at the level of MAVS | Conenello et al., 2007 |
| Influenza A virus H1N1(1918) and H5N1 | PB1-F2 66S, PB1-F2 66N | Disruption of mitochondrial membrane potential and type I IFN response | Conenello et al., 2007 |
| Hepatitis C virus (HCV) | NS3/4A | Inhibition of type I IFN response by cleaving of MAVS | Meylan et al., 2005 |
Viruses that subvert MAVS.
Other viruses, such as hepatitis C virus (HCV), induce the cleavage of MAVS from the outer membrane of mitochondria, reducing the interferon-producing response. In this case, the NS3/4A protein cleaves MAVS at cysteine 508 (Meylan et al., 2005; Bender et al., 2015; Vazquez and Horner, 2015).
Another family of pattern recognition receptors contain a nucleotide-binding and oligomerization domain (NOD) and is called the NLR (NOD-like receptor) family. NOD2 facilitates the activation of IRF3 and the synthesis of type I IFN in response to single-stranded RNA. Interestingly, the activation of NOD2 is dependent on MAVS (Sabbah et al., 2009; Moreira and Zamboni, 2012).
Recently, NLRX1 (also known as NOD5, NOD9, or NOD26), a member of the NLR family that localizes to the outer mitochondrial membrane, was shown to mediate MAVS degradation, allowing HCV to evade type I IFN-mediated antiviral response (Qin et al., 2017).
cGAS-STING Anti-Viral Pathway and Its Subversion by Viruses
The cyclic guanosine monophosphate (cGMP)-adenosine monophosphate (AMP) synthase (cGAS) recognizes viral as well as bacterial double-stranded DNA (dsDNA) (Wu and Chen, 2014; Ni et al., 2018). After binding to dsDNA, cGAS catalyzes the synthesis of the second messenger cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), which then binds to the stimulator of interferon genes (STING) adaptor protein on the endoplasmic reticulum (ER); STING, as a dimer, translocates from the endoplasmic reticulum to the Golgi complex, where it recruits TANK-binding kinase 1 (TBK1) which activates the transcription factors NFκB and IRF3, both of which translocate to the nucleus and induce the synthesis of type I interferons (Barber, 2015; Ni et al., 2018).
While the activation of the RIG-1-MDA5-MAVS antiviral signaling pathway clearly requires mitochondrial activity, in the form of mitochondrial dynamics and Δψm, a metabolism-related component in the cGAS-STING antiviral signaling pathway has not been explicitly identified. However, several lines of research suggest crosstalk between cGAS-STING and metabolism. Firstly, the ER has been regarded as a separate metabolic compartment on the basis that the ER luminal micro-environment is different from the cytosol, that it contains its own pool of pyridine nucleotides, and that several metabolic pathways related to carbohydrate and steroid metabolism, biotransformation, and protein processing take place in the ER (Csalaa et al., 2006); viral infections may lead to ER stress and to the unfolded protein response (UPR) (Zhang and Wang, 2012); and the mitochondrial function in cells undergoing ER stress is compromised, particularly at the level of mitochondrial membrane potential, oxygen consumption, and ATP production (Wang et al., 2011). The ER stress and UPR synergy with the cGAS-STING antiviral signaling pathway still needs to be fully elucidated (Smith, 2014).
Among the DNA viruses that activate the cGAS-STING pathway are herpes simplex virus 1 (HSV-1), vaccinia virus (VV), and murine gamma herpesvirus 68 (MHV68). Interestingly, RNA viruses such as HIV-1 generate RNA: DNA hybrids as well as dsDNA that may activate the cGAS-STING pathway (Ma and Damania, 2016; Ni et al., 2018).
Of note, dengue virus (DENV)-induced mitochondrial damage leads to mitochondrial DNA release to the cytosol, and the activation of the cGAS-STING pathway (Sun et al., 2007). Since other viruses may cause mitochondrial damage (see below) it is plausible that other RNA viruses may activate cGAS-STING through mitochondrial DNA release.
Several DNA virus-associated proteins are known to interfere with the cGAS-STING pathway, as reviewed by Ni et al. (2018), either by interfering with DNA binding to cGAS, as is the case of Kaposi's sarcoma-associated herpesvirus (KSHV), Epstein Barr virus (EBV), and murine gammaherpesvirus-68 (MHV68,γHV68) tegument protein open reading frame 52 (ORF52), and the KSHV latency-associated nuclear antigen (LANA) protein which interact with cGAS (Wu et al., 2015; Zhang et al., 2016), or by targeting STING, as is the case for the HSV-1-infected cell protein 27 (ICP27) and the UL46 protein, the KSHV viral interferon regulatory factor 1 (vIRF1), the human papillomavirus 18 (HPV18) E7 oncoprotein, the human adenovirus 5 (hAd5) E1A oncoprotein, and the Hepatitis B virus (HBV) polymerase (Lau et al., 2015; Liu et al., 2015; Ma et al., 2015; Christensen et al., 2016;Deschamps and Kalamvoki, 2017).
A more recent development in the field is the finding that some RNA viruses are also capable to interfere with the cGAS-STING pathway, subverting its anti-viral effect (Ni et al., 2018).
Finally, it has been shown that single- or double-stranded DNA may attenuate glucose metabolism, leading to ATP depletion and so constitute a metabolic barrier for viral replication. However, the mechanism seems to be dependent on the activation of adenosine monophosphate (AMP)-activated protein kinase (AMPK) and the activation of mechanistic target of rapamycin complex 1 (mTORC1) (see below), but independent of the cGAS-STING anti-viral pathway (Zheng et al., 2015).
Mitochondrial Proteins Other than MAVS as Targets of Viral Infection
Some viruses encode mitochondrial proteins, which allow them a direct functional intervention on host cells mitochondria (Table 2). In this regard, the Acanthamoeba polyphaga mimivirus (APMV), one of the largest known viruses (400 nm in its capside diameter) (La Scola et al., 2003; Monné et al., 2007), encodes a mitochondrial transport protein called VMC1 (viral mitochondrial carrier), whose function is to transport dATP and other nucleotide triphosphates (dTTP, TTP, UTP, and ADP). VMC1 can support the replication of the APMV genome by acquiring additional nucleotide triphosphates from the mitochondrial pool in exchange for cytosolic ADP (Monné et al., 2007). The APMV genome additionally encodes other five putative mitochondrial proteins (Monné et al., 2007).
Table 2
| Virus | Viral proteins | Effect | References |
|---|---|---|---|
| Acantthamoeba polyphaga mimivirus (APMV) | Virus mitochondrial carrier 1 (VMC1) | Increase of viral replication by transporting dATP from the motochondrial pool | Monné et al., 2007 |
| Epstein Barr virus (EBV) | BHRF1, BZLF1, BALF1, early Zta | Increase of viral replication, prevention of B cell apoptosis, blockage of mDNA replication | Cavallari et al., 2018 |
| Hepatitis C virus (HCV) | p7, NS3/4A, NS5A | Disruption of mitochondrial function, cleaveage of MAVS | Cavallari et al., 2018 |
| Hepatitis C virus (HCV) | Core | Mitochondria depolarization, increased production of mitochondrial ROS | Cavallari et al., 2018 |
| Influenza virus (IV) | PB1-F2, PB2, NS1 | Modulation of viral replication, viral mRNA synthesis | Cavallari et al., 2018 |
| Herpes simplex virus-1 (HSV-1) | UL 12.5 | Degradation of mitochondrial DNA early during infection | Cavallari et al., 2018 |
| Herpes simplex virus-1 (HSV-1) | UL 12 | Generation of mature viral genomes | Cavallari et al., 2018 |
Viruses that target other mitochondrial proteins.
The Human T-cell leukemia virus type 1 (HTLV-1) causes adult T-cell leukemia/lymphoma (ATLL) and encodes an 87-amino acid protein (p13) that helps this virus to establish a persistent infection. This protein primarily accumulates in the inner mitochondrial membrane of host cells and alters mitochondrial morphology toward a more rounded shape, fragments mitochondria (mitochondrial fission), and reduces mitochondrial Ca2+ uptake (Biasiotto et al., 2010; Cavallari et al., 2018).
Several proteins encoded by Epstein Barr virus (EBV) target mitochondria, such as BHRF1 (BamHI-H right reading frame), BZLF1 (also known as Zebra protein), BALF1 (BamHI-A left frame transcript), LMP2A (Latent membrane protein), and immediate early Zta protein. BHRF1 accumulates in the outer mitochondrial membrane (OMM) of B lymphocytes, preventing apoptosis and promoting survival of EBV-infected cells, viral persistence, and replication; BHRF presents homology with the transmembrane domains of some eukaryotic Bcl-2 family members (Kvansakul et al., 2017; Cavallari et al., 2018); and BZLF1 has the capacity to interact with mtSSB (mitochondrial single-stranded DNA-binding protein), which is required for the replication of the mitochondrial genome, and partially redirects mtSSB from mitochondria to the nucleus (LaJeunesse et al., 2005; Cavallari et al., 2018). BALF1 also shares homology with Bcl-2 family members and modulates apoptosis and promotes transformation (Hsu et al., 2012; Cavallari et al., 2018). LMP2A induces mitochondrial fission by a Drp1-dependent mechanism (Pal et al., 2014; Cavallari et al., 2018), and finally, the immediate early Zta protein can also bind mtSSB in the cytoplasm, inducing its re-location to the nuclei, blocking mitochondrial DNA replication and facilitating viral replication (Wiedmer et al., 2008).
Many other viruses encode mitochondrial proteins capable of regulating a broad spectrum of mitochondrial activities, as reviewed by Cavallari et al. (2018), including the control of intracellular Ca2+, apoptosis, mitochondrial dynamics, the levels of cytochrome c oxidase III (COXIII) and COX activity, as well as cellular ROS production, and the aggregation of mitochondria near the nucleus. Others promote mitophagy and interfere with the antiviral interferon response (Wu et al., 2007; Wang and Ryu, 2010). Proteins such as KS-Bcl-2 localize in mitochondria (Gallo et al., 2017), and others such as the KSHV-encoded K7 protein localize in mitochondria as well as in the ER and nucleus (Feng et al., 2002).
The non-structural proteins p7 of HCV can modify the mitochondrial function. The p7 protein is determinant for the assembly and later release of infectious virions, it is capable to form membrane-associated hexameric ion channels, induces mitochondrial membrane depolarization, and binds to the interferon inducible protein 6–16 (IFI6-16) (Nieva et al., 2012; Madan and Bartenschlager, 2015; Qi et al., 2017); HepG2 cells that express HCV core protein have increased levels of prohibitin, a protein that regulates mitochondrial function and apoptosis (Peng et al., 2015), by reducing the levels of COX subunits I and II. Therefore, the interaction between the HCV core protein and prohibitin may interfere with the assembly of the respiratory chain, which could lead to increased production of mitochondrial ROS and viral replication (Tsutsumi et al., 2009; Ren et al., 2016). Other molecular partners for viral-encoded mitochondrial proteins are voltage-dependent anion channel 3 (VDAC3) (Rahmani et al., 2000), and heat shock protein 60 (HSP60) (Tanaka et al., 2004).
Three influenza virus proteins are known to localize into mitochondria: PB1-F2, PB2, and NS1 (Chen et al., 2001; Yamada et al., 2004; Carr et al., 2006; Tsai et al., 2017). Although PB1-F2 is dispensable for viral replication, at least in some host cells, its expression accelerates influenza virus-induced apoptosis in human monocytes through mitochondrial ANT3 (adenine nucleotide translocator 3) and VDAC1 (voltage dependent anion channel 1) (Chen et al., 2001; Zamarin et al., 2005). The PB2 protein has a key role in viral mRNA synthesis and localizes in mitochondria, where it can regulate the viability of mitochondria during infection (Carr et al., 2006). The NS1 protein is highly expressed in Influenza A virus-infected cells, and predominantly localizes in the nucleus, although it may also be found in the cytoplasm at later stages of infection (Melén et al., 2007). Although NS1 does not harbor mitochondria-targeting sequences, it has also been found in mitochondria at early times (1.5 h) post-infection (Tsai et al., 2017).
The UL12 gene of herpes simplex virus type 1 (HSV-1) encodes two distinct but related alkaline DNases through two separately promoted 3' co-terminal mRNAs, producing full-length (UL12) and amino-terminal truncated (UL12.5) proteins. UL12 localizes to the nucleus while UL12.5 is predominantly located in mitochondria, where it degrades mitochondrial DNA early during infection. Whereas nuclear-targeted UL12 produces mature viral genomes from larger genome precursors (Draper et al., 1986; Saffran et al., 2007; Corcoran et al., 2009), the role of UL12.5 is not well-defined since mitochondrial DNA degradation is not required for HSV-1 replication (Duguay et al., 2014).
Mitochondrial Dynamics and Viruses
Mitochondria constantly undergo fusion and fission depending on the cell metabolic requirements, a process that has been dubbed as mitochondrial dynamics (Mishra and Chan, 2016).
Along with being the “powerhouse” of eukaryotic cells, mitochondria are also involved in cellular innate antiviral immunity (Seth et al., 2005). Mitochondrial fusion and fission processes depend on the activity of mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), and optic atrophy protein 1 (OPA1)—which promotes fusion—in addition to Dynamin-related protein 1 (Drp1)—which promotes mitochondrial fission (Mishra and Chan, 2016). There is evidence that antiviral immune responses can be regulated by mitochondrial dynamics (Arnoult et al., 2011; West et al., 2011). The close association between mitochondrial dynamics and several mitochondrial and cellular functions may suggest that mitochondrial dynamics could be a target for viruses to interfere with immune responses (Table 3). Likewise, the non-structural protein 4A (NS4A) from HCV, either alone or associated with the non-structural protein 3 (NS3), accumulates in mitochondria, altering the mitochondrial dynamics (Nomura-Takigawa et al., 2006). Infection with HIV-1 re-shapes mitochondrial distribution within the cells (Radovanović et al., 1999), while African swine fever virus (ASFV) induces the clustering of mitochondria around virus factories within infected cells, providing the local energy required for the release of virus (Rojo et al., 1998). The DENV NS2b3 protein partially cleaves Mfn1 and Mfn2, attenuating interferon responses (Yu et al., 2015), and induces mitochondrial fusion by inhibiting Drp1 activation and in turn the activation of the interferon response (Chatel-Chaix et al., 2016).
Table 3
| Virus | Viral proteins | Effect | References |
|---|---|---|---|
| Hepatitis C virus (HCV) | NS4A, NS3 | Change of mitochondria distribution | Nomura-Takigawa et al., 2006 |
| Human immunodeficiency virus-1 (HIV-1) | Clustering of mitochondria | Radovanović et al., 1999 | |
| African swine fever virus (ASFV) | Cluster of mitochondria around virus factories, providing ATP for virus release | Rojo et al., 1998 | |
| Dengue virus (DENV) | NS2b3 | Cleavege of Mfn1 and Mfn2, attenuation of IFN responses | Yu et al., 2015 |
| Mitochondrial fusion by inhibition of Drp1 | Chatel-Chaix et al., 2016 | ||
| Hepatitis B virus (HBV) | HBx | Mitochondrial fission, and mitochondrial injury | Kim et al., 2013 |
Viruses that disrupt mitochondrial dynamics.
Excessive mitochondrial fission may lead to mitochondrial damage, and this may have a role in hepatitis B virus (HBV)-induced liver disease (Kim et al., 2013).
Hepatitis B virus, through its HBx protein, triggers the translocation of Drp1 to the mitochondria by stimulating the phosphorylation of Drp1 at the Ser616 residue, and on the other hand, contributes to the degradation of Mfn2, favoring mitochondrial fission and mitophagy, attenuating the virus-induced apoptosis in the process (Kim et al., 2013).
Intracellular calcium concentrations also regulate mitochondrial dynamics since the calcium-dependent phosphatase calcineurin dephosphorylates Drp1 at S637, facilitating the recruitment of Drp1 to the mitochondria and the consequent mitochondrial fission (Cereghetti et al., 2008).
Intracellular Calcium Homeostasis and Viral Infections
Intracellular calcium participates in cell signaling, mitochondrial function, and cell death (Duchen, 2000; Contreras et al., 2010), and Ca2+ uptake by mitochondria activates Krebs cycle enzymes and oxidative phosphorylation, leading to higher ATP production (Nasr et al., 2003).
Several viruses regulate host cell calcium concentrations in the cytoplasm as well as in mitochondria, allowing viral gene expression, virus replication, and the control of host cell viability (Table 4). HSV1 downregulates the uptake of Ca2+ by mitochondria along its lytic cycle, modulating virus replication (Lund and Ziola, 1985). Other viruses such as HCV target mitochondria, increasing Ca2+ concentration (Li et al., 2007; Campbell et al., 2009). Among the HCV proteins known to interfere with Ca2+ homeostasis, are the core protein, the NS5A, and the p7 protein (Gong et al., 2001; Griffin et al., 2004; Kalamvoki and Mavromara, 2004; Dionisio et al., 2009).
Table 4
| Virus | Viral proteins | Effect | References |
|---|---|---|---|
| Human T leukemia virus (HTLV-1) | p13 | p13 accumulates in the inner mitochondrial membrane, reduces Dym and mCa2+ uptake | Biasiotto et al., 2010 |
| Herpes simplex virus 1 (HSV1) | ? | Modulation of viral replication by down-regulation of Ca2+ uptake by mitochondria | Lund and Ziola, 1985 |
| Hepatitis C virus (HCV) | NS5A, p7 | Increase of Ca2+ concentration | Gong et al., 2001; Griffin et al., 2004 |
| Hepatitis B virus (HBV) | HBx | Ca2+ release from mitochondria and ER | Bouchard et al., 2001 |
| Human immunodeficiency virus-1 (HIV-1) | Nef | Increase in viral replication by IP3R-dependent increase of cytosolic Ca2+ | Foti et al., 1999 |
| Rotavirus | NSP4 | virus release by decreasing Ca2+ concentration | Tian et al., 1995; Ruiz et al., 2007 |
| Poliovirus | 2BC | Increase in viral gene expression and apoptosis by increse in Ca2+ concentration | Aldabe et al., 1997 |
| Coxsackievirus B3 | 2B | Control of apoptosis and virus release by regulation of Ca2+ concentration | Campanella et al., 2004 |
| Human cytomegalovirus (HCMV) | pUL37x1 | Increased viral replication by mitochondria Ca2+ uptake and increased ATP | Sharon-Friling et al., 2006; Bozidis et al., 2010 |
Viruses that disrupt calcium homeostasis.
?, not known.
HBV induces the mobilization of Ca2+ from mitochondria and endoplasmic reticulum to the cytoplasm through the interaction of the HBV protein X with voltage-dependent anion channels (VDAC) (Bouchard et al., 2001; Choi et al., 2005). The HIV-1 protein Nef (nuclear elongation factor) interacts with the Inositol 1,4,5-trisphosphate receptor (IP3Rs), increasing cytosolic Ca2+ concentration, promoting the transcription of virus-encoded genes and viral replication (Kinoshita et al., 1997; Foti et al., 1999). Rotavirus, through its NSP4 protein, activates phospholipase C (PLC) and the release of Ca2+ from the endoplasmic reticulum to the cytosol. However, by the end of its life cycle there is a decrease in cellular Ca2+ concentrations enabling rotavirus release (Tian et al., 1995; Ruiz et al., 2007; Díaz et al., 2008).
Poliovirus increases intracellular Ca2+ concentrations shortly after infection, increasing viral gene expression (Irurzun et al., 1995; Aldabe et al., 1997). By the end of the virus life cycle Ca2+ accumulates within mitochondria at the expense of ER stores in a mitochondrial calcium uniporter (MCU) and voltage-dependent anion channel (VDAC)-dependent process, leading to mitochondrial dysfunction and apoptosis (Brisac et al., 2010).
Enteroviruses control apoptosis through Ca2+ regulation; in this way, low levels of cytosolic Ca2+ provide the conditions for viral replication while high concentrations of cytosolic Ca2+ lead to the formation of vesicles which allow virus release (Campanella et al., 2004; Van Kuppeveld et al., 2005).
Human cytomegalovirus (HCMV) protein pUL37 × 1, also known as viral mitochondrion-localized inhibitor of apoptosis (vMIA) localizes into mitochondria and induces the transfer of ER Ca2+ into mitochondria, increasing the production of ATP and virus replication (Sharon-Friling et al., 2006; Bozidis et al., 2010).
The maturation of viral glycoproteins is dependent on both pH and intracellular Ca2+ concentrations. Ca2+ acts as a cofactor for several enzymes including glycosyl- and sulfo-transferases (Vanoevelen et al., 2007). Measles virus (MV), Dengue virus (DENV), West Nile virus (WNV), Zika virus (ZIKV), and Chikungunya virus (CHIKV) use the host calcium pump secretory pathway calcium ATPase 1 (SPCA1) for Ca2+ loading into the trans Golgi network, which activates glycosyl transferases and proteases allowing viral maturation and spreading (Hoffmann et al., 2017).
mTOR and AMPK as Metabolic Hubs and Viral Targets for Evasion
The mechanistic target of rapamycin (mTOR) and the adenosine monophosphate-activated protein kinase (AMPK) constitute an integrated metabolic sensor. High levels of ATP (high ATP/AMP ratio) activate mTORC1, resulting in enhanced nutrient-dependent protein synthesis, cell growth and proliferation, whereas low levels of ATP (low ATP/AMP and ATP/ADP ratios), a hallmark of metabolic stress (starvation, hypoxia or viral infection), lead to AMPK-mediated inhibition of mTORC1 and activation of mTORC2, which restores energy homeostasis by switching the ATP-consuming biosynthetic pathways off and the ATP-producing catabolic pathways on (Hardie et al., 2012; Saxton and Sabatini, 2017).
MTOR acts as the catalytic subunit of either of two molecular complexes known as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2); mTORC1 is bound to the protein Raptor (Hara et al., 2002; Kim et al., 2002) and mTORC2 is bound to the protein Rictor (Hresko and Mueckler, 2005).
MTORC1 induces metabolic reprograming from OXPHOS to glycolysis by upregulating the transcription factor hypoxia-induced factor 1α (HIF1α) and, as a result, increases the expression of several glycolytic enzymes including phospho-fructo kinase (PFK). On the other hand, mTORC2 regulates cell proliferation and survival by activating the PI3K-Akt pathway (Düvel et al., 2010; Thomanetz et al., 2013; Saxton and Sabatini, 2017). The mTORC1 complex acts downstream of Akt and, as a way of regulation, the mTORC1 substrate p70S6K suppresses mTORC2, and the mTORC1 substrate Grb10 suppresses PI3K signaling (Hsu et al., 2011; Yu et al., 2011; Saxton and Sabatini, 2017), establishing a negative feedback that balances mTORC1 and mTORC2 activities (Meade et al., 2018).
Extracellular growth factors, the cell energy status, and different stressors such as viral infection are integrated into the mTOR pathway. Not surprisingly, viruses can modulate mTOR signaling to their advantage (Le Sage et al., 2016; Saxton and Sabatini, 2017) (Table 5). HSV-1 can enhance mTORC1 activity; whereas Poliovirus, HIV-1, Sindbis virus, and CHIKV can inhibit this same complex (Martin et al., 2012).
Table 5
| Virus | Viral proteins | Effect | References |
|---|---|---|---|
| Herpes simplex virus 1 (HSV1) | viral kinase Us3 | Enhancement of mTORC1 activity | Martin et al., 2012 |
| Poliovirus (PV) | Inhibition of mTORC1 activity | ||
| Human immunodeficiency virus-1 (HIV-1) | Env | Activation of mTORC1 activity | Le Sage et al., 2016 |
| Sindbis virus (SINV) | Activation of mTORC | Le Sage et al., 2016 | |
| Chikungunya virus (CHIKV) | ? | Controversial activation/Inhibition of mTOR | Le Sage et al., 2016 |
| Influenza A virus (IAV) | NS1 | Differential activation of mTORC1 and mTORC2, supports viral replication | Kuss-Duerkop et al., 2017 |
| Andes virus (ANDV) | glycoprotein Gn | Activation of mTOR, supports viral protein expression and replication | McNulty et al., 2013 |
| Hepatitis C virus (HCV) | NS5A | Activation of mTORC1 supports viral protein expression and replication | Stohr et al., 2016 |
| Poxviruses | F17 | Evasion of cytosolic sensing by disruption of the mTORC1-mTORC2 circuit | Meade et al., 2018 |
| Dengue virus (DENV) | ? | Viral replication by activation of AMPK and inhibition of mTORC1 | Jordan and Randall, 2017 |
| Zika virus (ZIKV) | ? | AMPK activation evokes antiviral innate responses and restricts virus replication | Kumar et al., 2018 |
Viruses that target mTOR or AMPK.
?, not known.
Activation of mTORC1 supports viral protein expression and replication of Influenza A virus, Andes virus (ANDV), and HCV (McNulty et al., 2013; Stohr et al., 2016; Kuss-Duerkop et al., 2017). On the other hand, poxviruses are capable of evading their cytosolic sensing by means of a conserved structural protein that disrupts the mTORC1-mTORC2 regulatory circuit while maintaining the metabolic benefits of mTOR activity (Meade et al., 2018).
DENV activates AMPK, decreases the activity of mTORC1, and induces lipophagy, a process that is required for the robust DENV replication; the autophagic-mediated mobilization of lipids increases the β-oxidation in DENV-infected cells (Jordan and Randall, 2017) whereas AMPK activation evokes antiviral innate responses and restricts ZIKV replication (Kumar et al., 2018).
Can Viruses Replicate within Mitochondria?
In addition to the interaction of viral proteins with mitochondria, which modify mitochondrial function, the Alphanodavirus flock house virus (FHV) can infect yeast, insect, plant, and mammalian cells, and replicates its RNA in the mitochondrial outer membrane. Miller et al. showed that the FHV RNA-dependent RNA polymerase, required for FHV RNA replication, localizes to the outer mitochondrial membrane and by electron microscopy these authors identified 40–60 nm membrane-bound spherical structures, similar to other virus-induced membrane structures, within the mitochondrial intermembrane space of infected cells from Drosophila (Miller et al., 2001).
Concluding Remarks
This review explores how viruses may subvert immune responses by controlling host cell metabolism.
Viruses may target MAVS (RIG-I-MDA5-MAVS anti-viral pathway) interfering with RNA virus-induced type 1 interferon responses and target other mitochondrial-associated proteins, disrupting mitochondrial dynamics, mitochondrial membrane potential, and calcium handling—all of which may affect anti-viral immunity. They may also regulate the production of ATP to their advantage by interfering with mitochondrial calcium mobilization, mitochondrial enzymatic activities, and key metabolic sensors such as mTORC1, mTORC2, and AMPK. They may also induce cytotoxic T lymphocyte exhaustion, which implies metabolic reprogramming.
Viruses may also target the cGAS-STING anti-viral pathway, interfering with DNA virus-induced type I IFN responses. Since this anti-viral pathway is not directly connected with host cell metabolism (at least not in the way the RIG-I-MDA5-MAVS is), one key outstanding question is why anti-RNA viruses IFN responses are more “metabolically directed” compared to anti-DNA virus responses. Moreover, why do some RNA viruses induce the release of mitochondrial DNA and in this way recruit the RIG-I-MDA5-MAVS pathway?
In the context of HCV infection, there are at least two mechanisms accounting for the degradation of MAVS, direct cleaving by the HCV-encoded NS3/4A protein, and the NLRX1-induced proteosomal degradation. As both MAVS and NLRX1 localize in the outer mitochondrial membrane, and MAVS signaling is dependent on mitochondrial function, it remains to be determined whether NLRX1 activity is also dependent on mitochondrial function. However, it is currently known that NLRX1 regulates OXPHOS and cell integrity in a model of ischemia-reperfusion injury, and that loss of NLRX1 increases oxygen consumption and oxidative stress in epithelial cells (Stokman et al., 2017).
The role of glycolysis, β-oxidation, and oxidative phosphorylation on viral infections is continuing to emerge, but there are still outstanding questions on the role and mechanism that some metabolic intermediates may play in viral infection. For instance, dimethyl fumarate enhances the infection of cancer cell lines and human tumor biopsies with several oncolytic viruses (Selman et al., 2018), whereas ZIKV infection upregulates the enzyme cis-aconitate descarboxylase, which converts the TCA intermediate cis-aconitate to itaconate, an endogenous inhibitor of succinate dehydrogenase, inhibiting the conversion of succinate to fumarate and generating a metabolic state that restricts ZIKV replication in neurons (Daniels et al., 2019). These topics require further exploration.
On the other hand, the success of anti-viral antibody responses as well as of antibody-mediated anti-viral vaccine protection depends on plasma cell lifespan, which ultimately relies on plasma cell metabolism; something that differs from B lymphocyte metabolism (Lam et al., 2018). It would therefore be interesting to determine whether there are viruses that specifically target plasma cell metabolism, and in which case whether protecting plasma cell metabolism could be therapeutically useful in helping to support long-lasting anti-viral immune responses.
Statements
Author contributions
MM-A and FS-G conceived and designed the review, wrote the paper, edited, and approved the final draft. SK contributed to discussions on the paper, edited, and approved the final draft.
Acknowledgments
We thank Bruno Aguilar-Lopez for help with the figures. While we have tried to include all key references, we apologize to authors for not including all references due to space constraints. Current work in the Immunoregulation laboratory is being funded by Consejo Nacional de Ciencia y Tecnología (grant 284602). FS-G and MM-A are EDI, COFAA, and SNI fellows.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
- ACC
Acetyil-CoA carboxylase
- Akt
Akt/Protein kinase B
- AMP
Adenosine monophosphate
- AMPK
Adenosine monophosphate-activated protein kinase
- ATP
Adenosine three phosphate
- 2B
2B protein
- 2BC
2BC protein
- ANT3
Adenine nucleotide translocator 3
- ATLL
Adult T-cell leukemia/lymphoma
- BALF1
BamH1-A left frame transcript
- BHRF1
BamH1-Hright reading frame
- BZLF1
Zebra protein
- cGAS
cyclic guanosin monophosphate-adenosin monophosphate synthase
- cGMP
cyclic guanosine monophosphate
- CoA
Coenzyme A
- CTL
Cytotoxic T lymphocytes
- COXIII
Cytochrome c oxidase III
- Δψm
Mitochondrial membrane potential
- Drp1
Dynamin-related protein
- dTTP
Deoxythymidine triphosphate
- early Zta
early Zta protein
- Env
Envelope
- ER
Endoplasmic reticulum
- FADH2
reduced Flavin adenin dinucleotide
- FAO
Fatty acid oxidation
- FHV
Flock house virus
- F17
F17 protein
- Grb10
Growth factor receptor bound protein 10
- HBx
Hepatitis B virus x protein
- HIF1α
Hypoxia-induced factor 1α
- HPV 18
Human papillomavirus 18
- KSHV
Kaposi's sarcoma-associated herpesvirus
- HSP60
Heat shock protein 60
- IFI6-16
Interferon inducible protein 6-16
- IFNs
Interferons
- IκBα, nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor
alpha
- IKK
IκB kinase
- IL-4
Interleukin-4
- IP3Rs
Inositol 1,4,5-triphosphate receptors
- IRF3
Interferon regulatory factor 3
- ISGs
Interferon-stimulator genes
- JAK-STAT
Janus kinase-Signal transductor and activator of transcription
- Lag-3
Lymphocyte activation gene-3
- LANA
Latency-associated nuclear antigen
- LMP2A
Latent membrane protein 2A
- LPS
Lypopolysacharide
- M1
Macrophage type1
- M2
Macrophage type 2
- MAMs
Mitochondria-associated membranes
- MAVS
Mitochondrial antiviral-signaling protein
- MCU
Mitochondrial calcium uniporter
- MDA-5
Melanoma differentiation-associated gene 5
- Mfn1
Mitofusin 1
- MHV68γHV68
Murine gammaherpesvirus-68
- mTORC1
mechanistic target of rapamycin complex 1
- mTORC2
mechanistic target of rapamycin complex 2
- mtSSB
Mitochondrial single-stranded DNA binding protein
- NADPH
reduced Nicotinamide adenine dinucleotide phosphate
- Nef
Nuclear elongation factor
- NS
Non-structural Proteins
- NETs
Neutrophyl extracellular traps
- NFκB
Nuclear factor kappa B
- NK
Natural killer
- NLR
NOD-like receptor
- NOD
Nucleotide-binding and oligomerization domain
- NS1
Non-structural protein 1
- NS2b3
Non-structural protein 2b3
- OMM
outer mitochondrial membrane
- OPA1
Optic atrophy protein 1
- ORF52
Open reading frame 52
- OXPHOS
Oxidative phosphorylation
- PB1-F2
PB1-F2 protein
- PB1-F2 66S, PB1-F2 protein, serine 66 PB1-F2 66N, PB1-F2 protein
asparagine 66
- PB2
PB2 protein
- PD-1
Programmed death-1
- PFK
Phosphofructokinase
- PGC-1a
Peroxisome proliferator-activated receptor-gamma coactivator-1alpha
- PI3K
phosphatidylinositol 3-kinase
- PLC
Phospholipase C
- PMA
Phorbol 12-myristate 13-acetate
- PPP
pentose phosphate pathway
- PRRs
Pattern recognition receptors
- p7
protein 7
- p13
protein 13
- P70S6K
Ribosomal protein S6 kinase beta-1
- RIG-1
Retinoic acid-induced gene 1
- RLR
RIG-1-like Receptor
- ROS
Reactive Oxygen Species
- SPCA 1
Secretory pathway calcium ATPase 1
- STING
Stimulator of interferon genes
- TBK1
TANK binding kinase 1
- TCA
Tricarboxylic acid
- Tim-3
T cell immunoglobulin mucin-3
- TLRs
Toll like receptors
- TTP
thymidine triphosphate
- UL 12
full length UL 12 protein
- UL 12. 5
N-terminally truncated UL 12 protein
- UPR
Unfolded protein response
- UTP
Uridine triphosphate
- VV
Vaccinia virus
- VDAC3, Voltage dependent anion channel 3;VMC1
Viral mitochondrial carrier 1
- vIRF1
viral Interferon regulatory factor 1
- vMIA
viral mitochondrial-localized inhibitor of apoptosis. Note: Other viruses abbreviations are indicated in Tables 1–5.
Abbreviations
References
1
Akira S. Takeda K. (2004). Toll-like receptor signalling. Nat. Rev. Immunol.4,499–511. 10.1038/nri1391
2
Aldabe R. Irurzun A. Carrasco L. (1997). Poliovirus protein 2BC increase cytosolic free calcium concentrations. J. Virol.71, 6214–6217.
3
Arnoult D. Soares F. Tattoli S. Girardin S. E. (2011). Mitochondria in innate immunology. EMBO Rep.12, 901–910. 10.1038/embor.2011.157
4
Barber D. L. Wherry E. J. Masopust D. Zhu B. Allison J. P. Sharpe A. H. et al . (2006). Restoring function in exhausted CD8 T cells during chronic viral infection. Nature439, 682–687. 10.1038/nature04444
5
Barber G. N. (2015). STING: infection, inflammation and cancer. Nat. Rev. Immunol.15, 760–770. 10.1038/nri3921
6
Bender S. Reuter A. Eberle F. Einhorn E. Binder M. Bartenschlager R. (2015). Activation of type I and III interferon response by mitochondrial and peroxisomal MAVS and inhibition by hepatitis C virus. PLoS Pathog.11:e1005264. 10.1371/journal.ppat.1005264
7
Bengsch B. Johnson A. L. Kurachi M. Odorizzi P. M. Pauken K. E. Atanasio J. et al . (2016). Bioenergetic insufficiencies due to metabolic alterations regulated by PD-1 are an early driver of CD8+ T Cell Exhaustion. Immunity45,358–373. 10.1016/j.immuni.2016.07.008
8
Bhutia Y. D. Ganapathy V. (2016). Glutamine transporters in mammalian cells and their functions in physiology and cancer. Biochim. Biophys. Acta1863, 2531–2539. 10.1016/j.bbamcr.2015.12.017
9
Biasiotto R. Aguiari P. Rizzuto R. Pinton P. D'Agostino D. M. Ciminale V. (2010). The p13 protein of human T cell leukemia virus type 1 (HTLV-1) modulates mitochondrial membrane potential and calcium uptake. Biochim. Biophys. Acta1797, 945–951. 10.1016/j.bbabio.2010.02.023
10
Bouchard M. J. Wang L. H. Schneider R. J. (2001). Calcium signaling by HBx protein in hepatitis B virus DNA replication. Science294, 2376–2378. 10.1126/science.294.5550.2376
11
Bozidis P. Williamson C. D. Wong D. S. Colberg-Poley A. M. (2010). Trafficking of UL37 proteins into mitochondrion-associated membranes during permissive human cytomegalovirus infection. J. Virol.84, 7898–7903. 10.1128/JVI.00885-10
12
Brisac C. Téoul,é F Autret A. Pelletier I. Colbère-Garapin F. Brenner C. et al . (2010). Calcium flux between the endoplasmatic reticulum and mitochondrion contributes to poliovirus-induced apoptosis. J. Virol.84, 12226–12235. 10.1128/JVI.00994-10
13
Buck M. D. Sowell R. T. Kaech S. M. Pearce E. L. (2017). Metabolic instruction of immunity. Cell169, 570–586. 10.1016/j.cell.2017.04.004
14
Campanella M. de Jong A. S. Lanke K. W. Melchers W. J. Willems P. H. Pinton P. et al . (2004). The coxsackievirus 2B protein suppresses apoptotic host cell responses by manipulating intracellular Ca2+ homeostasis. J. Biol. Chem.279, 18440–18450. 10.1074/jbc.M309494200
15
Campbell R. V. Yang Y. Wang T. Rachamallu A. Li Y. Watowich S. J. et al (2009). Effects of hepatitis C core protein on mitochondrial electron transport and production of reactives oxygen species. Methods Enzymol.456, 363–380. 10.1016/S0076-6879(08)04420-0
16
Carr S. M. Carnero E. Garcia-Sastre A. Brownlee G. G. Fodor E. (2006). Characterization of a mitocondrial-targeting signal in the PB2 protein of influenza viruses. Virology344, 492–508. 10.1016/j.virol.2005.08.041
17
Cavallari I. Scattolin G. Silic-Benussi M. Raimondi V. D'Agostino D. M. Ciminale V. (2018). Mitochondrial proteins coded by human tumor viruses. Front Microbiol.9:81. 10.3389/fmicb.2018.00081
18
Cereghetti G. M. Stangherlin A. Martins de Brito O. Chang C. R. Blackstone C. Bernardi P. et al . (2008). Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. U.S.A.105, 15803–15808. 10.1073/pnas.0808249105
19
Chatel-Chaix L. Cortese M. Romero-Brey I. Bender S. Neufeldt C. J. Fischl W. et al . (2016). Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe14, 342–356. 10.1016/j.chom.2016.07.008
20
Chen W. Calvo P. A. Malide D. Gibbs J. Schubert U. Bacik I. et al . (2001). A novel influenza A virus mitochondrial protein that induces cell death. Nat. Med.7, 1306–1312. 10.1038/nm1201-1306
21
Choi Y. Park S. G. Yoo J. H. Jung G. (2005). Calcium ions affect the hepatitis B virus core assembly. Virology332, 454–463. 10.1016/j.virol.2004.11.019
22
Christensen M. H. Jensen S. B. Miettinen J. J Luecke S. Prabakaran T. Reinert L. S. et al . (2016). HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J.35, 1385–1399. 10.15252/embj.201593458
23
Conenello G. M. Zamarin D. Perrone L. A. Tumpey T. Palese P. (2007). A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog.4, 1414–1421. 10.1371/journal.ppat.0030141
24
Contreras L. Drago I. Zampese E. Pozzan T. (2010). Mitochondria: the calcium connection. Biochim. Biophys. Acta1797, 607–618. 10.1016/j.bbabio.2010.05.005
25
Corcoran J. A. Saffran H. A. Duguay B. A. Smiley J. R. (2009). Herpes simplex virus UL12.5 targets mitochondria through a mitochondrial localization sequence proximal to the N terminus. J. Virol.83, 2601–2610. 10.1128/JVI.02087-08
26
Csalaa M. Bánhegyib G. Benedettib A. (2006). Endoplasmic reticulum: a metabolic compartment. FEBS Lett.580, 2160–2165. 10.1016/j.febslet.2006.03.050
27
Daniels B. P. Kofman S. B. Smith J. R. Norris G. T. Snyder A. G. Kolb J. P. et al . (2019). The nucleotide sensor ZBP1 and kinase RIPK3 induce the enzyme IRG1 to promote an antiviral metabolic state in neurons. Immunity50, 64–76.e4. 10.1016/j.immuni.2018.11.017
28
Darnell J. E. Jr. Kerr I. M. Stark G. R. (1994). Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science264, 1415–1421. 10.1126/science.8197455
29
DeBerardinis R. J. Cheng T. (2010). Q's next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene29, 313–324. 10.1038/onc.2009.358
30
Delgado-Rizo V. Martínez-Guzmán M. A. I-iguez-Gutierrez L. García-Orozco A. Alvarado-Navarro A. Fafutis-Morris M. (2017). Neutrophil extracellular traps and its implications in inflammation: an overview. Front. Immunol8:81. 10.3389/fimmu.2017.00081
31
Deschamps T. Kalamvoki M. (2017). Evasion of the STING DNA-sensing pathway by VP11/12 of herpes simplex virus 1. J. Virol.91, e00535–e00517. 10.1128/JVI.00535-17
32
Díaz Y. Chemello M. E. Pe-a F. Aristimu-o O. C Zambrano J. L. Rojas H. et al (2008). Expression of nonstructural rotavirus protein NSP4 Mimics Ca2+ homeostasis changes induced by rotavirus infection in cultured cells. J. Virol.82, 11331–11343. 10.1128/JVI.00577-08
33
Dionisio N. Garcia-Mediavilla M. V. Sanchez-Campos S. Majano P. L. Benedicto I. Rosado J. A. et al (2009). Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signaling alterations in hepatocytes. J. Hepatol.50, 872–882. 10.1016/j.jhep.2008.12.026
34
Dörner T. Radbruch A. (2007). Antibodies and B cell memory in viral immunity. Immunity27, 384–392. 10.1016/j.immuni.2007.09.002
35
Draper K. G. Devi-Rao G. Costa R. H. Blair E. D. Thompson R. L. Wagner E. K. (1986). Characterization of the genes encoding herpes simplex virus type 1 and type 2 alkaline exonucleases and overlapping proteins. J. Virol.57, 1023–1036.
36
Duchen M. R. (2000). Mitochondria and calcium: from cell signalling to cell death. J. Physiol.529, 57–68. 10.1111/j.1469-7793.2000.00057.x
37
Duguay B. A. Saffran H. A. Ponomarev A. Duley S. A. Eaton H. E. Smiley J. R. (2014). Elimination of mitochondrial DNA is not required for herpes simplex virus 1 replication. J. Virol.88, 2967–2976. 10.1128/JVI.03129-13
38
Düvel K. Yecies J. L. Menon S. Raman P. Lipovsky A. I. Souza A. L. et al . (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell.39, 171–183. 10.1016/j.molcel.2010.06.022
39
Feng P. Park J. Lee B. S. Lee B. S. H. Bram R. J. Jung J. U. (2002). Kaposi's sarcoma-associated herpesvirus mitochondrial K7 protein targets a cellular calcium-modulating cyclophilin ligand to modulate intracellular calcium concentration and inhibit apoptosis. J. Virol.76, 11491–11504. 10.1128/JVI.76.22.11491-11504.2002
40
Fitzgerald K. A. McWhirter S. M. Faia K. L. Rowe D. C. Latz E. Golenbock D. T. et al . (2003). IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nat. Immunol.4, 491–496. 10.1038/ni921
41
Foti M. Cartier L. Piguet V. Lew D. P. Carpentier J. L. Trono D. et al . (1999). The HIV Nef protein alters Ca2+ signaling in myelomonocytic cells through SH3- mediated protein-protein interactions. J. Biol. Chem.274, 34765–34772. 10.1074/jbc.274.49.34765
42
Frauwirth K. A. Riley J. L. Harris M. H. Parry R. V. Rathmell J. C. Plas D. R. et al . (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity16, 769–777. 10.1016/S1074-7613(02)00323-0
43
Freeman G. J. Long A. J. Iwai Y. Bourque K. Chernova T. Nishimura H. et al . (2000). Engagement of the Pd-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med.192, 1027–1034. 10.1084/jem.192.7.1027
44
Galani I. E. Andreakos E. (2015). Neutrophils in viral infections: current concepts and caveats. J. Leukoc. Biol.98, 557–564. 10.1189/jlb.4VMR1114-555R
45
Gallo A. Lampe M. Günther T. Brune W. (2017). The viral Bcl-2 homologs of kaposi's sarcoma-associated herpesvirus and rhesus rhadinovirus share an essential Role for viral replication. J. Virol.91, e01875–e01816. 10.1128/JVI.01875-16
46
Ganeshan K. Chawla A. (2014). Metabolic regulation of immune responses. Annu Rev Immunol.32, 609–634. 10.1146/annurev-immunol-032713-120236
47
Gardiner C. M. Finlay D. K. (2017). What fuels natural killers? metabolism and NK cell responses. Front. Immunol.8:367. 10.3389/fimmu.2017.00367
48
Gong G. Waris G. Tanveer R. Siddiqui A. (2001). Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NF- B. Proc. Natl. Acad. Sci. U.S.A.98, 9599–9604. 10.1073/pnas.171311298
49
Griffin S. D. C. Harvey R. Clarke D. S. Barclay W. S. Harris M. Rowlands D. J. (2004). A conserved basic loop in hepatitis C virus p7 protein is required for amantadine-sensitive ion channel activity in mammalian cells but is dispensable for localization to mitochondria. J. Gen. Virol.85, 451–461. 10.1099/vir.0.19634-0
50
Hammer Q. Rückert T. Romagnani C. (2018). Natural killer cell specificity for viral infections. Nat. Immunol.19, 800–808. 10.1038/s41590-018-0163-6
51
Hara K. Maruki Y. Long X. Yoshino K. Oshiro N. Hidayat S. et al . (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell110, 177–189. 10.1016/S0092-8674(02)00833-4
52
Hardie D. G. Ross F. A. Hawley S. A. (2012). AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol.13, 251–262. 10.1038/nrm3311
53
Hiscott J. Grandvaux N. Sharma S. Tenoever B. R. Servant M. J. Lin R. (2003). Convergence of the NF-kappaB and interferon signaling pathways in the regulation of antiviral defense and apoptosis. Ann. N. Y. Acad. Sci.1010, 237–248. 10.1196/annals.1299.042
54
Hoffmann H. H. Schneider W. M. Blomen V. A. Scull M. A. Hovnanian A. Brummelkamp T. R. et al . (2017). Diverse viruses require the calcium transporter SPCA1 for maturation and spread. Cell Host Microbe22, 460–470. 10.1016/j.chom.2017.09.002
55
Honda K. Yanai H. Negishi H. Asagiri M. Sato M. Mizutani T. et al (2005). IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature434, 772–777. 10.1038/nature03464
56
Hosking M. P. Flynn C. T. Botten J. Whitton J. L. (2013). CD8+ memory T cells appear exhausted within hours of acute virus infection. J. Immunol.191, 4211–4222. 10.4049/jimmunol.1300920
57
Hresko R. C. Mueckler M. (2005). mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem.280, 40406–40416. 10.1074/jbc.M508361200
58
Hsu P. P. Kang S. A. Rameseder J. Zhang Y. Ottina K. A. Lim D. et al . (2011). The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1 mediated inhibition of growth factor signaling. Science332, 1317–1322. 10.1126/science.1199498
59
Hsu W. L. Chung P. J. Tsai M. H. Chang C. L. Liang C. L. (2012). A role for Epstein-Barr viral BALF1 in facilitating tumor formation and metastasis potential. Virus Res.163, 617–62710.1016/j.virusres.2011.12.017
60
Irurzun A. Arroyo J. Alvarez A. Carrasco L. (1995). Enhanced intracellular calcium concentration during poliovirus infection. J. Virol.69, 5142–5146.
61
Jellusova J. Rickert R. C. (2016). The PI3K pathway in B cell metabolism. Crit. Rev. Biochem. Mol. Biol.51, 359–378. 10.1080/10409238.2016.1215288
62
Jordan T. X. Randall G. (2017). Dengue virus activates the AMP kinase-mTOR axis to stimulate a proviral lipophagy. J. Virol.91, e02020–e02016. 10.1128/JVI.02020-16
63
Kalamvoki M. Mavromara P. (2004). Calcium-dependent calpain proteases are implicated in processing of the hepatitis C virus NS5A protein. J. Virol.78, 11865– 11878. 10.1128/JVI.78.21.11865-11878.2004
64
Kim D. H. Sarbassov D. D. Ali S. M. King J. E. Latek R. R. Erdjument-Bromage H. et al . (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell110, 163–175. 10.1016/S0092-8674(02)00808-5
65
Kim S. J. Khan M. Quan J. Till A. Subramani S. Siddiqui A. (2013). Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. PLoS Pathog.9:e1003722. 10.1371/journal.ppat.1003722
66
Kinoshita S. Su L. Amano M. Timmerman L. A. Kaneshima H. Nolan G. P. (1997). The T cCell activation factor NFATc positively regulates HIV-1 replication and gene expression in T cells. Immunity6, 235–244. 10.1016/S1074-7613(00)80326-X
67
Koshiba T. (2013). Mitochondrial-mediated antiviral immunity. Biochim. Biophys. Acta1833, 225–232. 10.1016/j.bbamcr.2012.03.005
68
Kovacsovics M. Martinon F. Micheau O. Bodmer J. L. Hofmann K. Tschopp J. et al . (2002). Overexpression of Helicard, a CARD-containing helicase cleaved during apoptosis, accelerates DNA degradation. Curr Biol.12, 838–843. 10.1016/S0960-9822(02)00842-4
69
Kumar A. Kumar Singh P. Giri S. (2018). AMP-activated kinase (AMPK) promotes innate immunity and antiviral defense against Zika virus induced ocular infection. J. Immunol.200(Suppl.), 50.14.
70
Kuss-Duerkop S. K. Wang J. Mena I. White K. Metreveli G. Sakthivel R. et al . (2017). Influenza virus differentially activates mTORC1 and mTORC2 signaling to maximize late stage replication. PLoS Pathog.13:e1006635. 10.1371/journal.ppat.1006635
71
Kvansakul M. Caria S. Hinds M. G. (2017). The Bcl-2 family in host-virus interactions. Viruses9:E290. 10.3390/v9100290
72
La Scola B. Audic S. Robert C. Jungang L. de Lamballerie X. Drancourt M. et al . (2003). A giant virus in amoebae. Science299:2033. 10.1126/science.1081867
73
LaJeunesse D. R. Brooks K. Adamson A. L. (2005). Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 alter mitochondrial morphology during lytic replication. Biochem. Biophys. Res. Commun.333, 438–442. 10.1016/j.bbrc.2005.05.120
74
Lam W. Y. Becker A. M. Kennerly K. M. et al . (2016). Mitochondrial pyruvate import promotes long-term survival of antibody-secreting plasma cells. Immunity45, 60–73. 10.1016/j.immuni.2016.06.011
75
Lam W. Y. Jash A. Yao C. H. D'Souza L. Wong R. Nunley R. M. et al . (2018). Metabolic and transcriptional modules independently diversify plasma cell lifespan and function. Cell Rep.24, 2479–2492.e6. 10.1016/j.celrep.2018.07.084
76
Lau L. Gray E. E. Brunette R. L. Stetson D. B. (2015). DNA tumor virus oncogenes antagonize the cGAS-STING DNA-sensing pathway. Science350, 568–571. 10.1126/science.aab3291
77
Le Sage V. Cinti A. Amorim R. Mouland A. J. (2016). Adapting the stress response: viral subversion of the mTOR signaling pathway. Viruses8:152. 10.3390/v8060152
78
Li Y. Boehning D. F. Qian T. Popov V. L. Weinman S. A. (2007). Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J.21, 2474–2485. 10.1096/fj.06-7345com
79
Liu Y. Li J. Chen J. Li Y. Wang W. Du X. et al . (2015). Hepatitis B virus polymerase disrupts K63-linked ubiquitination of STING to block innate cytosolic DNA-sensing pathways. J. Virol.89, 2287–2300. 10.1128/JVI.02760-14
80
Lund K. Ziola B. (1985). Cell sonicates used in the analysis of how measles and herpes simplex type 1 virus infections influence Vero cell mitochondrial calcium uptake. Can. J. Biochem. Cell Biol.63, 1194–1197. 10.1139/o85-149
81
Ma Z. Damania B. (2016). The cGAS-STING defense pathway and its counteraction by viruses. Cell Host Microbe19,150–158. 10.1016/j.chom.2016.01.010
82
Ma Z. Jacobs S. R. West J. A. Stopford C. Zhang Z. Davis Z. et al . (2015). Modulation of the cGAS-STING DNA sensing pathway by gammaherpesviruses. Proc. Natl. Acad. Sci. U.S.A.112, E4306–E4315. 10.1073/pnas.1503831112
83
Madan V. Bartenschlager R. (2015). Structural and functional properties of the hepatitis C virus p7 viroporin. Viruses7, 4461–4481. 10.3390/v7082826
84
Mah A. Y. Rashidi A. Keppel M. P. Saucier N. Moore E. K. Alinger J. B. et al . (2017). Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight2:e95128. 10.1172/jci.insight.95128
85
Martin S. Saha B. Riley J. L. (2012). The battle over mTOR: an emerging theatre in host–pathogen immunity. PLoS Pathog.8, e1002894–e1002895. 10.1371/journal.ppat.1002894
86
Martinelli S. Urosevic M. Daryadel A. Oberholzer P. A. Baumann C. Fey M. F. et al . (2004). Induction of genes mediating interferon-dependent extracellular trap formation during neutrophil differentiation. J. Biol. Chem.279, 44123–44132. 10.1074/jbc.M405883200
87
McNab F. Mayer-Barber K. Sher A. Wack A. O'Garra A. (2015). Type I interferons in infectious disease. Nat. Rev. Immunol.15, 87–103. 10.1038/nri3787
88
McNulty S. Flint M. Nichol S. T. Spiropoulou C. F. (2013). Host mTORC1 signaling regulates andes virus replication. J. Virol.87, 912–922. 10.1128/JVI.02415-12
89
Meade N. Furey C. Li H. Verma R. Chai Q. Rollins M. G. et al . (2018). Poxviruses evade cytosolic sensing through disruption of an mTORC1-mTORC2. Cell174, 1143–1157.e17. 10.1016/j.cell.2018.06.053
90
Melén K. Kinnunen L. Fagerlund R. Ikonen N. Twu K. Y. Krug R. M. et al . (2007). Nuclear and nucleolar targeting of influenza A virus NS1 protein: striking differences between different virus subtypes. J. Virol.81, 5995–6006. 10.1128/JVI.01714-06
91
Meylan E. Curran J. Hofmann K. Moradpour D. Binder M. Bartenschlager R. et al . (2005). Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature437, 1167–1172. 10.1038/nature04193
92
Miller D. J. Schwartz M. D. Ahlquist P. (2001). Flock house virus RNA replicates on outer mitochondrial membranes in drosophila cells. J. Virol.75, 11664–11676. 10.1128/JVI.75.23.11664-11676.2001
93
Mishra P. Chan D. C. (2016). Metabolic regulation of mitochondrial dynamics. J. Cell. Biol.212, 379–387. 10.1083/jcb.201511036
94
Monné M. Robinson A. J. Boes C. Harbour M. E. Fearnley I. M. Kunji E. R. S. (2007). The mimivirus genome encodes a mitochondrial carrier that ransports dATP and dTTP. J. Virol.81, 3181–3186. 10.1128/JVI.02386-06
95
Moreira L. O. Zamboni D. S. (2012). NOD1 and NOD2 signaling in infection and inflammation. Front. Immunol.3:328. 10.3389/fimmu.2012.00328
96
Moreno-Altamirano M. M. Rodriguez-Espinosa O. Rojas-Espinosa O. Pliego-Rivero B. Sanchez-Garcia F. J. (2015). Dengue virus serotype-2 Interferes with the formation of neutrophil extracellular traps. Intervirology58, 250–259. 10.1159/000440723
97
Nasr P. Gursahani H. I. Pang Z. Bondada V. Lee J. Hadley R. W. et al . (2003). Influence of cytosolic and mitochondrial Ca2+, ATP, mitochondrial membrane potential, and calpain activity on the mechanism of neuron death induced by 3-nitropropionic acid. Neurochem. Int.43, 89–99. 10.1016/S0197-0186(02)00229-2
98
Navale A. M. Paranjape A. N. (2016). Glucose transporters: physiological and pathological roles. Biophys. Rev.8, 5–9. 10.1007/s12551-015-0186-2
99
Ni G. Ma Z. Damania B. (2018). cGAS and STING: at the intersection of DNA and RNA virus- sensing networks. PLoS Pathog.14:e1007148. 10.1371/journal.ppat.1007148
100
Nieva J. L. Madan V. Carrasco L. (2012). Viroporins: structure and biological functions. Nat. Rev. Microbiol.10, 563–574. 10.1038/nrmicro2820
101
Nomura-Takigawa Y. Nagano-Fujii M. Deng L. Kitazawa S. Ishido S. Sada K. et al . (2006). Non-structural protein 4A of Hepatitis C virus accumulates on mitochondria and renders the cells prone to undergoing mitochondria-mediated apoptosis. J. Gen. Virol.87,1935–1945. 10.1099/vir.0.81701-0
102
O'Neill L. A. Pearce E. J. (2016). Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med.213,15–23. 10.1084/jem.20151570
103
O'Neill L. A. J. Rigel J. K. Rathmell J. (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol.16, 553–565. 10.1038/nri.2016.70
104
Pal A. D. Basak N. P. Banerjee A. S. Banerjee S. (2014). Epstein-Barr virus latent membrane protein-2A alters mitochondrial dynamics promoting cellular migration mediated by Notch signaling pathway. Carcinogenesis35, 1592–1601. 10.1093/carcin/bgu069
105
Pearce E. L. Pearce E. J. (2013). Metabolic pathways in immune cell activation and quiescence. Immunity38, 633–643. 10.1016/j.immuni.2013.04.005
106
Peng Y. T. Chen P. Ouyang R. Y. Song L. (2015). Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis20, 1135–49. 10.1007/s10495-015-1143-z
107
Qi H. Chu V. Wu N. C. Chen Z. Truong S. Brar G. et al . (2017). Systematic identification of anti-interferon function on hepatitis C virus genome reveals p7 as an immune evasion protein. Proc. Natl. Acad. Sci. U.S.A.114, 2018–2023. 10.1073/pnas.1614623114
108
Qin Y. Xue B. Liu C. Wang X. Tian R. Xie Q. et al . (2017). NLRX1 mediates MAVS degradation to attenuate the hepatitis C virus-induced innate immune response through PCBP2. J. Virol.91, e01264–e01217. 10.1128/JVI.01264-17
109
Radovanović,ć J. S. Todorovi,ć V. Borici,ć I. M Janković-Hladni M. Korać A. (1999). Comparative ultrastructural studies on mitochondrial pathology in the liver of AIDS patients: clusters of mitochondria, protuberances, “minimitochondria,” vacuoles, and virus-like particles. Ultrastruct. Pathol.23, 19–24. 10.1080/019131299281798
110
Raftery M. J. Lalwani P. Krautkrämer E. Peters T. Scharffetter-Kochanek K. Kruger R. et al . (2014). β2 integrin mediates hantavirus-induced release of neutrophil extracellular traps. J. Exp. Med.211, 1485–1497. 10.1084/jem.20131092
111
Rahmani Z. Huh K. W. Lasher R. Siddiqui A. (2000). Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol.74, 2840–2846. 10.1128/JVI.74.6.2840-2846.2000
112
Ren J. H. Chen X. Zhou L. Tao N. N. Zhou H. Z. Liu B. et al . (2016). Protective role of sirtuin3 (SIRT3) in oxidative stress mediated by hepatitis B virus X protein expression. PLoS ONE.11:e0150961. 10.1371/journal.pone.0150961
113
Rich P. R. Maréchal A. (2010). The mitochondrial respiratory chain. Essays Biochem.47, 1–23. 10.1042/bse0470001
114
Rodriguez-Espinosa O. Rojas-Espinosa O. Moreno-Altamirano M. M. López-Villegas E. O. Sánchez-García F. J. (2015). Metabolic requirements for neutrophil extracellular traps formation. Immunology145, 213–224. 10.1111/imm.12437
115
Rodríguez-Prados J. C. Través P. G. Cuenca J. Rico D. Aragonés J. Martín-Sanz P. et al . (2010). Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J. Immunol.185, 605–614. 10.4049/jimmunol.0901698
116
Rojo G. Chamorro M. Salas M. L. Vi-uela E. Cuezva J. M. Salas J. (1998). Migration of mitochondria to viral assembly sites in African swine fever virus-infected cells. J. Virol.72, 7583–7588.
117
Romero-Garcia S. Moreno-Altamirano M. M. Prado-Garcia H. Sánchez-García F. J. (2016). Lactate contribution to the tumor microenvironment: mechanisms, effects on immune cells and therapeutic relevance. Front. Immunol.7:52. 10.3389/fimmu.2016.00052
118
Ruiz M. C. Aristimu-o O. C. Díaz Y. Pe-a F. Chemello M. E. Rojas H. et al . (2007). Intracellular disassembly of infectious rotavirus particles by depletion of Ca2+ sequestered in the endoplasmic reticulum at the end of virus cycle. Virus Res.130, 140–150. 10.1016/j.virusres.2007.06.005
119
Sabbah A. Chang T. H. Harnack R. Frohlich V. Tominaga K. Dube P. H. et al . (2009). Activation of innate immune antiviral responses by Nod2. Nat. Immunol.10, 1073–1080. 10.1038/ni.1782
120
Saffran H. A. Pare J. M. Corcoran J. A. Weller S. K. Smiley J. R. (2007). Herpes simplex virus eliminates host mitochondrial DNA. EMBO Rep.8, 188–193. 10.1038/sj.embor.7400878
121
Saitoh T. Komano J. Saitoh Y. Misawa T. Takahama M. Kozaki T et al . (2012). Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe12, 109–116. 10.1016/j.chom.2012.05.015
122
Saxton R. A. Sabatini D. M. (2017). mTOR signaling in growth, metabolism and disease. Cell168, 960–976. 10.1016/j.cell.2017.02.004
123
Schönrich G. Raftery M. J. (2016). Neutrophil extracellular traps go viral. Front. Immunol.7:366. 10.3389/fimmu.2016.00366
124
Selman M. Ou P. Rousso C. Bergeron A. Krishnan R. Pikor L. et al . (2018). Dimethyl fumarate potentiates oncolytic virotherapy through NF-κB inhibition. Sci. Transl. Med.10:425. 10.1126/scitranslmed.aao1613
125
Seth R. B. Sun L. Ea C. K. Chen Z. J. (2005). Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell122, 669–682. 10.1016/j.cell.2005.08.012
126
Sharon-Friling R. Goodhouse J. Colberg-Poley A. M. Anamaris M. Shenk T. (2006). Human cytomegalovirus pUL37x1 induces the release of endoplasmic reticulum calcium stores. Proc Natl Acad Sci U. S. A.103, 19117–19122. 10.1073/pnas.0609353103
127
Silverman N. Maniatis T. (2001). NF-kappaB signaling pathways in mammalian and insect innate immunity. Genes Dev.15, 2321–2342. 10.1101/gad.909001
128
Smith J. A. (2014). A new paradigm: innate immune sensing of viruses via the unfolded protein response. Front. Microbiol.5:222. 10.3389/fmicb.2014.00222
129
Stohr S. Costa R. Sandmann L. Westhaus S. Pfaender S. Anggakusuma et al . (2016). Host cell mTORC1 is required for HCV RNA replication. Gut65, 2017–2028. 10.1136/gutjnl-2014-308971
130
Stokman G. Kors L. Bakker P. J. Rampanelli E. Claessen N. Teske G. J. D. et al . (2017). NLRX1 dampens oxidative stress and apoptosis in tissue injury via control of mitochondrial activity. J. Exp. Med.214, 2405–2420. 10.1084/jem.20161031
131
Sumpter R. Jr. Loo Y. M. Foy E. Li K. Yoneyama M. Fujita T. et al . (2005). Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol.79, 2689–2699. 10.1128/JVI.79.5.2689-2699.2005
132
Sun B. Sundstrom K. B. Chew J. J. Bist P. Gan E. S. Tan H. C. et al . (2007). Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep.7:3594. 10.1038/s41598-017-03932-1
133
Tanaka Y. Kanai F. Kawakami T. Tateishi K. Ijichi H. Kawabe T. et al . (2004). Interaction of the hepatitis B virus X protein (HBx) with heat shock protein 60 enhances HBx-mediated apoptosis. Biochem. Biophys. Res. Commun.318, 461–9. 10.1016/j.bbrc.2004.04.046
134
tenOever B. R. Sharma S. Zou W. Sun Q. Grandvaux N. Julkunen I. et al . (2004). Activation of TBK1 and IKKvarepsilon kinases by vesicular stomatitis virus infection and the role of viral ribonucleoprotein in the development of interferon antiviral immunity. J. Virol.78, 10636–10649. 10.1128/JVI.78.19.10636-10649.2004
135
Thomanetz V. Angliker N. Cloetta D. Regula M. Lustenberger R. M. Schweighauser M. et al . (2013). Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol.201, 293–308. 10.1083/jcb.201205030
136
Tian P. Estes M. K. Hu Y. Ball J. M. Zeng C. Q. Schilling W. P. (1995). The rotavirus nonstructural glycoprotein NSP4 mobilizes Ca2+ from the endoplasmic reticulum. J Virol.69, 5763–5772.
137
Tsai C.-F. Lin H.-Y. Hsu W.-L. Tsai C.-H. (2017). The novel mitochondria localization of influenza A virus NS1 visualized by FlAsH labeling. FEBS Open Bio.7, 1960–1971. 10.1002/2211-5463.12336
138
Tsutsumi T. Matsuda M. Aizaki H. Moriya K. Miyoshi H. Fujie H. et al . (2009). Proteomics analysis of mitochondrial proteins reveals overexpression of a mitochondrial protein chaperon, prohibitin, in cells expressing hepatitis C virus core protein. Hepatology50, 378–86. 10.1002/hep.22998
139
Van Kuppeveld F. J. de Jong A. S. Melchers W. J. Willems P. H (2005). Enterovirus protein 2B po(u)res out the calcium: a viral strategy to survive?Trends Microbiol.13, 41–44. 10.1016/j.tim.2004.12.005
140
Vanoevelen J. Dode L. Raeymaekers L. Wuytack F. Missiaen L. (2007). Diseases involving the Golgi calcium pump. Subcell Biochem.45, 385–404. 10.1007/978-1-4020-6191-2_14
141
Varanasi S. K. Donohoe D. Jaggi U. Rouse B. T. (2017). Manipulating glucose metabolism during different stages of viral pathogenesis can have either detrimental or beneficial effects. J. Immunol.199, 1748–1761. 10.4049/jimmunol.1700472
142
Varga S. T. Grant A. Manicassamy B. Palesea P. (2012). Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol.86, 8359–8366. 10.1128/JVI.01122-12
143
Vazquez C. Horner S. M. (2015). MAVS coordination of antiviral innate immunity. J Virol.89, 6974 −6977. 10.1128/JVI.01918-14
144
Veiga-Fernandes H. Walter U. Bourgeois C. McLean A. Rocha B. (2000). Response of naïve and memory CD8+ T cells to antigen stimulation in vivo. Nat Immunol.1, 47–53. 10.1038/76907
145
Wang H. Ryu W. S . (2010). Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog.6:e1000986. 10.1371/journal.ppat.1000986
146
Wang X. Eno C. O. Altman B. J. Zhu Y. Zhao G. Olberding K. E. et al . (2011). ER stress modulates cellular metabolism. Biochem. J.435, 285–296. 10.1042/BJ20101864
147
West A. P. Shadel G. S. Ghosh S. (2011). Mitochondria in innate immune responses. Nat. Rev Immunol.11, 389–402. 10.1038/nri2975
148
Wherry E. J. (2011). T cell exhaustion. Nat. Immunol.12, 492–499. 10.1038/ni.2035
149
Wiedmer A. Wang P. Zhou J. Rennekamp A. J. Tiranti V. Zeviani M. et al . (2008). Epstein-barr virus immediate-early protein Zta Co-Opts mitochondrial single-stranded DNA binding protein to promote viral and inhibit mitochondrial DNA replication. J. Virol.82, 4647–4655. 10.1128/JVI.02198-07
150
Wu J. Chen Z. J. (2014). Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol.32, 461–488. 10.1146/annurev-immunol-032713-120156
151
Wu J. J. Li W. Shao Y. Avey D. Fu B. Gillen J. et al . (2015). Inhibition of cGAS DNA sensing by a herpesvirus virion protein. Cell Host Microbe18, 333–344. 10.1016/j.chom.2015.07.015
152
Wu M. Xu Y. Lin S. Zhang X. Xiang L. Yuan Z. (2007). Hepatitis B virus polymerase inhibits the interferon-inducible MyD88 promoter by blocking nuclear translocation of Stat1. J. Gen. Virol.88, 3260–3269. 10.1099/vir.0.82959-0
153
Yamada H. Chounan R. Higashi Y. Kurihara N. Kido H. (2004). Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett.578, 331–336. 10.1016/j.febslet.2004.11.017
154
Yi J. S. Cox M. A. Zajac A. J. (2010). T-cell exhaustion: characteristics, causes and conversion. Immunology129, 474–481. 10.1111/j.1365-2567.2010.03255.x
155
Yoneyama M. Suhara W. Fujita T. (2002). Control of IRF-3 activation by phosphorylation. J. Interferon Cytokine Res.22, 73–76. 10.1089/107999002753452674
156
Yu C. Y. Liang J. J. Li J. K. Lee Y. L. Chang B. L. Su C. I. et al . (2015). Dengue virus impairs mitochondrial fusion by cleaving mitofusins. PLoS Pathog.11:e1005350. 10.1371/journal.ppat.1005350
157
Yu Y. Yoon S. O. Poulogiannis G. Yang Q. Ma X. M. Ville'n J. et al . (2011). Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science332, 1322–1326. 10.1126/science.1199484
158
Zamarin D. Garcia-Sastre A. Xiao X. Wang R. Palese P. (2005). Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog.1:e4. 10.1371/journal.ppat.0010004
159
Zhang G. Chan B. Samarina N. Abere B. Weidner-Glunde M. Buch A. et al . (2016). Cytoplasmic isoforms of Kaposi sarcoma herpesvirus LANA recruit and antagonize the innate immune DNA sensor cGAS. Proc. Natl. Acad. Sci. USA.113, E1034–E1043. 10.1073/pnas.1516812113
160
Zhang L. Wang A. (2012). Virus-induced ER stress and the unfolded protein response. Front. Plant Sci.3:293. 10.3389/fpls.2012.00293
161
Zheng M. Xie L. Liang Y. Wu S. Xu H. Zhang Y. et al . (2015). Recognition of cytosolic DNA attenuates glucose metabolism and induces AMPK mediated energy stress response. Int. J. Biol. Sci.11, 587–594. 10.7150/ijbs.10945
Summary
Keywords
viruses, cell metabolism, mitochondria, immune response, viral evasion
Citation
Moreno-Altamirano MMB, Kolstoe SE and Sánchez-García FJ (2019) Virus Control of Cell Metabolism for Replication and Evasion of Host Immune Responses. Front. Cell. Infect. Microbiol. 9:95. doi: 10.3389/fcimb.2019.00095
Received
30 January 2019
Accepted
22 March 2019
Published
18 April 2019
Volume
9 - 2019
Edited by
Jonatas Abrahao, Federal University of Minas Gerais, Brazil
Reviewed by
Rafael Elias Marques, Centro Nacional de Pesquisa em Energia e Materiais (CNPEM), Brazil; Mauricio Teixeira Lima, Federal University of Minas Gerais, Brazil
Updates
Copyright
© 2019 Moreno-Altamirano, Kolstoe and Sánchez-García.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: María Maximina B. Moreno-Altamirano bertha.moreno.altamirano@gmail.comFrancisco Javier Sánchez-García fsanchez_1@yahoo.co.uk
This article was submitted to Virus and Host, a section of the journal Frontiers in Cellular and Infection Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.