 Coralie Boulet
Coralie Boulet Christian D. Doerig
Christian D. Doerig Teresa G. Carvalho
Teresa G. Carvalho- 1Molecular Parasitology Laboratory, Department of Physiology, Anatomy and Microbiology, La Trobe University, Bundoora, VIC, Australia
- 2Infection and Immunity Program, Biomedicine Discovery Institute, Department of Microbiology, Monash University, Clayton, VIC, Australia
Malaria is a major global health burden, affecting over 200 million people worldwide. Resistance against all currently available antimalarial drugs is a growing threat, and represents a major and long-standing obstacle to malaria eradication. Like many intracellular pathogens, Plasmodium parasites manipulate host cell signaling pathways, in particular programmed cell death pathways. Interference with apoptotic pathways by malaria parasites is documented in the mosquito and human liver stages of infection, but little is known about this phenomenon in the erythrocytic stages. Although mature erythrocytes have lost all organelles, they display a form of programmed cell death termed eryptosis. Numerous features of eryptosis resemble those of nucleated cell apoptosis, including surface exposure of phosphatidylserine, cell shrinkage and membrane ruffling. Upon invasion, Plasmodium parasites induce significant stress to the host erythrocyte, while delaying the onset of eryptosis. Many eryptotic inducers appear to have a beneficial effect on the course of malaria infection in murine models, but major gaps remain in our understanding of the underlying molecular mechanisms. All currently available antimalarial drugs have parasite-encoded targets, which facilitates the emergence of resistance through selection of mutations that prevent drug-target binding. Identifying host cell factors that play a key role in parasite survival will provide new perspectives for host-directed anti-malarial chemotherapy. This review focuses on the interrelationship between Plasmodium falciparum and the eryptosis of its host erythrocyte. We summarize the current knowledge in this area, highlight the different schools of thoughts and existing gaps in knowledge, and discuss future perspectives for host-directed therapies in the context of antimalarial drug discovery.
Introduction
Malaria is a vector-borne parasitic disease that affects millions of people worldwide. It is estimated that half the world population is at risk of infection and in 2016, malaria was responsible for 200 million new cases and half a million deaths (WHO|World Malaria Report, 2017). An efficient malaria vaccine has yet to be developed, and resistance against all antimalarial drugs has been recorded, including to artemisinin (reviewed in Tilley et al., 2016), the front-line drug treatment recommended by the World Health Organization (WHO). The path to controlling and eliminating malaria is a difficult one, and the systematic emergence of drug resistant parasites against every anti-malarial drug introduced over the last century represents a major road block (Menard and Dondorp, 2017). The encouraging decrease in malaria morbidity and mortality observed in recent decades has now been reported to be slowing down, and be reversing in parts of the world (WHO|World Malaria Report, 2017). To reach the strategic objective set by the WHO to reduce global malaria incidence and mortality by at least 90% by 2030 (WHO|Global Technical Strategy for Malaria, 2016–2030), new antimalarial drugs with a novel mode of action are urgently needed.
Malaria is caused by the unicellular apicomplexan parasite Plasmodium. A human infection starts when an infected Anopheles mosquito injects parasites (in the form of sporozoites) during a blood meal. Sporozoites circulate in the blood stream and reach the liver, where they invade hepatocytes and establish an asymptomatic infection. Plasmodium hepatic stages replicate by schizogony, ultimately releasing tens of thousands progeny merozoites in the blood stream. Once in the blood stream, Plasmodium merozoites invade red blood cells, where they proliferate by schizogony in an asexual replication cycle, known as the erythrocytic cycle (Figure 1). The cycle begins when an extracellular merozoite invades an erythrocyte. Once intracellular, the parasite develops into a ring stage, grows into a metabolically active trophozoite, and, following DNA replication and asynchronous nuclear divisions matures into a multi-nucleated schizont. After cytokinesis, up to 32 new merozoites egress from each schizont, lysing the host red blood cell and allowing for a new cycle to begin. Alternatively, early ring stage parasites can mature into female or male gametocytes (immature sexual stage of the parasite), which, once taken up by an Anopheles mosquito, complete maturation and fertilization within the mosquito's gut. The resulting oocyst produces sporozoites that travel to the salivary gland of the mosquito, allowing for further transmission of the parasite. The erythrocytic stages of infection are responsible for malaria pathogenesis, whose clinical manifestations include severe anemia, organ failure and cerebral malaria (Autino et al., 2012). Among the five Plasmodium species that infect humans, P. falciparum is the most virulent. Here, we focus on host-parasite interaction mechanisms that allow the development of P. falciparum inside human erythrocytes (Figure 1).
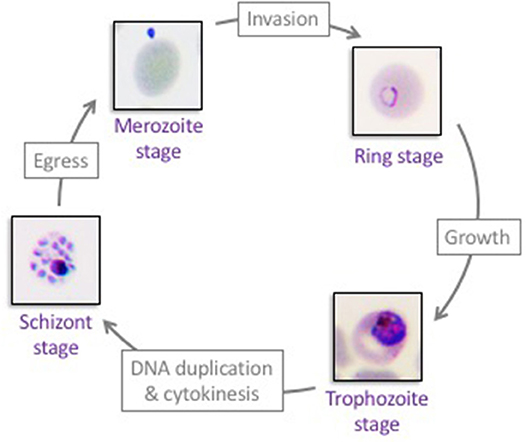
Figure 1. The asexual proliferation cycle of Plasmodium falciparum in human erythrocytes. Extracellular merozoites invade red blood cells to establish the erythrocytic asexual cycle. Each intracellular merozoite develops into an intra-erythrocytic ring stage, matures into a trophozoite stage, and subsequently forms a multi-nucleated schizont. Forty-eight hours post-merozoite infection, 8–32 new merozoites egress from each schizont-infected erythrocyte and a new erythrocytic cycle begins. Repeated cycles of erythrocyte invasion by Plasmodium falciparum parasites lead to all aspects of malaria pathogenesis.
Avoiding Antimalarial Drug Resistance by Targeting The Host Cell?
Curative antimalarials target the asexual proliferation of parasites in erythrocytes, and all antimalarial drugs developed to date directly target parasite factors. These include artemisinin (whose mechanism of action is yet to be fully understood), chloroquine (interferes with haemozoin formation, a process that detoxifies free haem released by hemoglobin digestion), atovaquone (inhibits mitochondrial respiration), proguanil/pyrimethamine (inhibits folate biosynthesis by targeting dihydrofolate reductase, PfDHFR), and various antibiotics which inhibit protein synthesis (Antony and Parija, 2016). Parasite resistance against anti-malarial drugs is a major long-standing issue, resulting in failure of many malaria eradication attempts. For instance, in 1955, the WHO launched a Global Malaria Eradication campaign, introducing Mass Drug Administration of chloroquine, the cheapest and most widely used antimalarial drug. However, in the 1960's, chloroquine resistance was reported in various South American and South-East Asian countries, and quickly spread, reaching African nations in the early 1970's (D'Alessandro and Buttiëns, 2001). Remarkably, all anti-malarial drugs saw resistance emerging within a couple of years of being commercialized (McClure and Day, 2014). Alarmingly, this also includes resistance against the current front-line drug, artemisinin, commonly used in combination therapies (Menard and Dondorp, 2017). Over relatively short periods of time, Plasmodium parasites have acquired genetic modifications, typically point mutations or copy number variations, resulting in resistance to antimalarial drugs. Such alterations have been shown to either affect the product directly targeted by the drug, or a membrane transporter, increasing the efflux of the drug to the outside environment. Such transporters are located on the plasma membrane of the parasite (e.g., multidrug resistance-associated protein, PfMRP) or on the food vacuole of the parasite (e.g., multidrug resistant protein 1, PfMDR1) (Antony and Parija, 2016).
Considering that Plasmodium is an obligate intracellular pathogen, and therefore relies on host cell factors to thrive, an alternative drug target strategy based on host cell factors can be envisaged. Host factors are not under the genetic control of the parasite, therefore host-directed therapy approaches bypass the most direct path to resistance, i.e., the selection of parasite genotypes encoding a mutated drug target.
For example, the host programmed cell death pathway could be targeted. Indeed, most intracellular pathogens avoid being eliminated by the immune system by “hiding” inside a host cell, while also gaining direct access to the host cell intracellular environment. Although this is highly beneficial to the pathogen, the stress induced by infection leads the host cell to trigger a cell death response or apoptosis. As a consequence, the pathogen is required to inhibit the host cell apoptotic response to ensure host cell survival until completion of its own replication cycle. In the case of Plasmodium, interference with host cell apoptosis has been established in the hepatic stage of infection (van de Sand et al., 2005; Kaushansky et al., 2013). However, the existence of a similar process in the erythrocytic stage of infection remains to be explored, likely because of the unique nature of the host erythrocyte cell.
Red Blood Cells—a Unique Cell Type
Maturation of erythrocyte progenitors in the bone marrow leads to the formation of enucleated reticulocytes that are released in the blood stream. Reticulocytes mature into erythrocytes, a process involving the loss of all intracellular organelles, including the nucleus, mitochondria, and endoplasmic reticulum (Lang and Föller, 2012). In the absence of de novo protein biosynthesis and mitochondria (which house the TCA cycle enzymes in nucleated cells), the survival and metabolism of mature erythrocytes rely exclusively on the existing pool of proteins and on glycolysis for the production of ATP (Lang and Föller, 2012). Further, a specialized function of oxygen transporter combined with a lack of organelles, have led RBCs to develop unique mechanisms of cell survival and cell death, which we outline below.
Erythrocytes Protect Themselves From Oxidative Stress
Red blood cells (RBCs) have a high haem iron content, essential for their role as oxygen and carbon dioxide transporters (Lang and Föller, 2012). For this reason, RBCs are constantly producing, and consequently exposed to, reactive oxygen species (ROS), including superoxide anion (O), hydrogen peroxide (H2O2) and hydroxyl radicals (OH−) (Baynes, 2005; Cimen, 2008; Schieber and Chandel, 2014). Reactive oxygen species impose oxidative stress on the cell, due to their inherent capability to damage lipids, proteins and DNA (although the latter is irrelevant in the context of RBCs). Erythrocytes possess a number of antioxidant strategies to counteract this stress, including ROS scavengers, such as glutathione and vitamins C and E, as well as various redox enzymes such as superoxide dismutase, catalase and glutathione peroxidase (Baynes, 2005). Glutathione is a Glu-Cys-Gly tri-peptide of crucial importance to maintain the intracellular environment in a reduced state (Wu et al., 2004). The ratio between reduced glutathione (GSH) and oxidized glutathione (GSSG) is a good indicator of the cellular redox state. Glutathione is naturally most required in tissues exposed to high ROS and oxidative stress levels, such as the liver and erythrocytes (Wu et al., 2004; Lu, 2009).
How Do RBCs Die? Senescence vs. Eryptosis
Mature RBCs have a life span of ~115 days (Franco, 2012), after which they undergo senescence, a gradual deterioration of the cell functional capacity, and clearance by the spleen. ROS accumulation has been proposed as a key life-span determinant (Hattangadi and Lodish, 2007). During this aging process, erythrocytes lose membranes through microvesiculation, become denser, intracellular enzymes display decreased activity, oxidative damages accumulate, and the membrane becomes more rigid (Lutz and Bogdanova, 2013). The Band 3 membrane protein (also called anion exchanger 1 or AE1) forms clusters (partly due to oxidation and hyperphosphorylation), that enhances deposition of complement C3 and subsequent binding of autoantibodies present in the serum, followed by clearance by macrophages (Lutz, 2004; Arese et al., 2005, p. 3; Lutz and Bogdanova, 2013). Additionally, the progressive exposure of the “eat-me” signal phosphatidylserine (PS), along with the reduced exposure of the “do-not eat-me” signal Cluster of Differentiation 47 (CD47, further discussed below), on the outside of a senescent erythrocyte further stimulate clearance by macrophages (Lutz and Bogdanova, 2013).
In addition to this type of “death by exhaustion,” RBCs can undergo a form of programmed cell death throughout their lifetime. This phenomenon was first described in 2001 (Bratosin et al., 2001) and the term eryptosis was proposed in 2005 (Lang K. et al., 2005). Eryptosis shares numerous similarities with apoptosis (Bratosin et al., 2001; Lang K. et al., 2005). Both apoptosis and eryptosis share the common purpose of destruction of damaged cells without inducing an inflammatory response (i.e., absence of cell lysis). This is particularly relevant in the case of RBC death, as the release of free hemoglobin in the blood induces renal impairment and thrombosis (Tolosano et al., 1999; Buehler et al., 2012; Schaer et al., 2013). Further, eryptotic and apoptotic cells share physiological characteristics including increased intracellular calcium concentrations, cell shrinkage, exposure of phosphatidylserine on the outer cell surface and membrane ruffling and blebbing (Bratosin et al., 2001; Lang K. et al., 2005). Like apoptosis, eryptosis can be triggered by a wide range of xenobiotics some of which have been listed in Table 1.
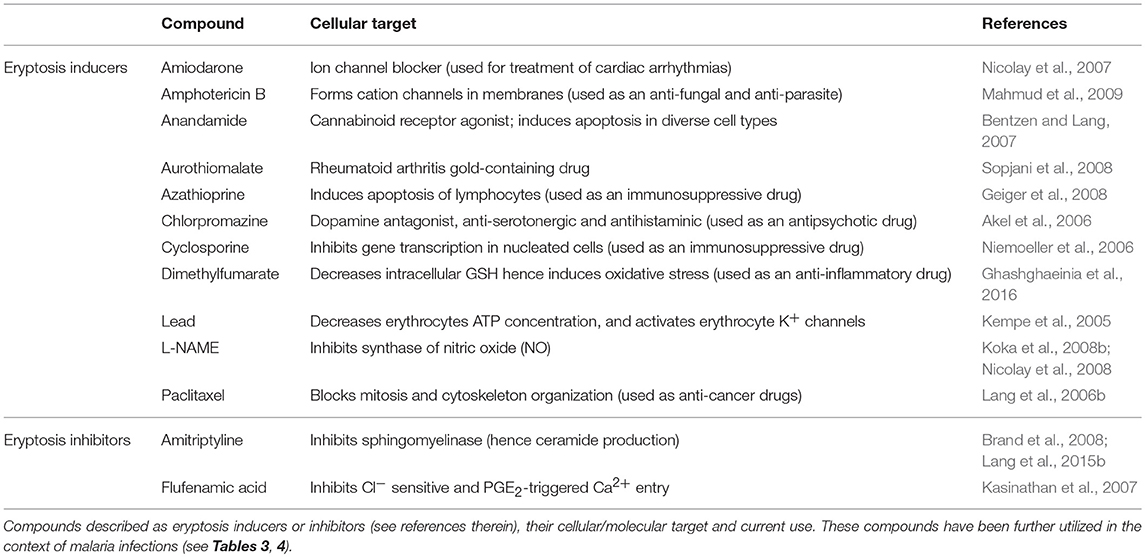
Table 1. Examples of relevant eryptosis inducers and inhibitors and their molecular targets.
Unbalanced levels of eryptosis have been proposed to play a key role in the pathology of numerous clinical disorders, including in metabolic syndrome (Zappulla, 2008), haemolytic uremic syndromes (Lang et al., 2006a), sickle-cell disease (Lang et al., 2002), thalassemia (Lang et al., 2002), and G6PD deficiency (Lang et al., 2002). The clinical relevance of red blood cell death in health and disease, has stimulated numerous studies over the last 20 years attempting to define the cellular and molecular mechanisms of eryptosis. Although much remains to be described, the section below summarizes our current knowledge of the regulatory mechanisms of eryptosis.
Eryptosis—What Do We Know About its Regulatory Mechanisms?
Eryptosis can be triggered by various signals, including osmotic shock (Huber et al., 2001; Lang et al., 2004), energy depletion (Klarl et al., 2006), oxidative stress (Lang K. et al., 2005; Lang et al., 2014), and xenobiotics (Lang and Lang, 2015; Pretorius et al., 2016). Regardless of the trigger, induction of an eryptotic state generally involves entry of extracellular calcium ions into the cell (Lang K. S. et al., 2003), which induces changes in membrane asymmetry/exposure of PS, cell shrinkage and membrane blebbing, detailed below and summarized in Figure 2.
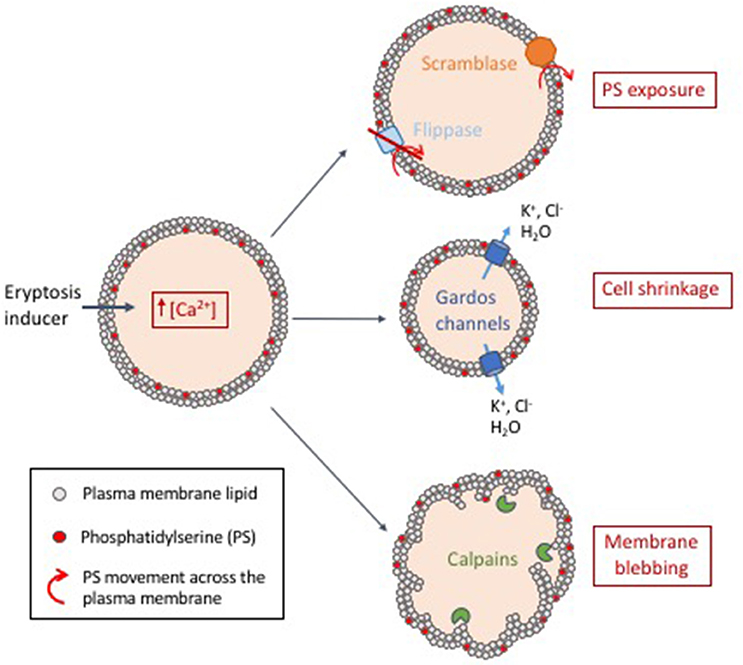
Figure 2. Proposed model of eryptosis mechanisms. Induction of eryptosis (energy depletion, oxidative stress or exposure to xenobiotics) leads to an increase of intracellular calcium concentration. (i) Intracellular calcium activates scramblases (membrane proteins that transport lipids non-specifically and bidirectionally) and inactivates flippases (membrane proteins that actively maintain phosphatidylserine (PS) in the membrane inner leaflet). This leads to exposure of PS at the surface of the cell, a signal that is recognized by macrophages, which then mediate clearance of eryptotic cells. (ii) Intracellular calcium activates calcium-sensitive potassium channels known as Gardos channels. This is followed by exit of potassium and chlorine, leading to loss of water by osmosis and subsequent cell shrinkage. (iii) Increased concentration of free intracellular calcium activates calpains (calcium-activated proteases). Calpains degrade cytoskeleton proteins which leads to membrane ruffling and blebbing.
Changes in Cell Membrane Asymmetry With Phosphatidylserine Exposure
In normal conditions, the RBC lipid bilayer is asymmetric, with specific lipids predominantly present on the inner or the outer leaflet. This is the case of the phospholipid phosphatidylserine, which is mainly present within the cytosolic monolayer (Leventis and Grinstein, 2010). The asymmetry is maintained by active membrane proteins termed flippases, that translocate phosphatidylserine and other phospholipids from the outer to the inner membrane leaflet (Sharom, 2011; Segawa and Nagata, 2015). In eryptotic cells, however, high intracellular calcium concentrations lead to the inactivation of flippases (Suzuki et al., 2010, 2013), leading to the rupture of membrane asymmetry and exposure of PS on the outer leaflet of the RBC membrane (Bratosin et al., 2001; Segawa and Nagata, 2015). High intracellular calcium levels also lead to the activation of another family of membrane proteins termed scramblases that translocate phospholipids non-specifically and bidirectionally in an ATP-independent manner (Segawa and Nagata, 2015). In addition, caspase 3 can also cleave and thus irreversibly activate scramblases and inactivate flippases (Berg et al., 2001; Schoenwaelder et al., 2009; Suzuki et al., 2010), committing the cell to PS exposure. The increased and abnormal exposure of PS on the outer RBC membrane is recognized by macrophages (McEvoy et al., 1986), which remove eryptotic cells from the circulation (Boas et al., 1998). “Flippase” and “scramblase” are generic terms used to describe lipid-transport enzymatic activities. The exact proteins responsible for PS exposure during eryptosis remain to be identified.
Cell Shrinkage and Gardos Channels Activation
Increased concentration of cytosolic calcium activates calcium-sensitive potassium channels, or Gardos channels (Bookchin et al., 1987; Brugnara et al., 1993), which allow the exit of K+ (Lang P. A. et al., 2003). The loss of intracellular K+ hyperpolarises the cell membrane, forcing the exit of Cl− (Lang P. A. et al., 2003). Consequently, the loss of water through osmosis leads to cell shrinkage (Lang P. A. et al., 2003). Cell shrinkage activates phospholipase A, which produces platelet-activating factor (PAF) (Lang P. A. et al., 2005). PAF in turn stimulates sphingomyelinase, an enzyme that breaks down sphingomyelin (Lang P. A. et al., 2005), the most prevalent sphingolipid in the cell membrane (Barenholz and Thompson, 1980). This leads to the formation of ceramide, a well-known inducer of eryptosis (Lang et al., 2004) and apoptosis (Dbaibo et al., 1997; Birbes et al., 2002). The role of ceramide in eryptosis has been review by Lang et al. (2015b).
Membrane Blebbing and Calpain Activation
Increased intracellular calcium concentration leads to the activation of calpains (Berg et al., 2001), a family of cytosolic calcium-dependent thiol proteases. Calpains I (or μ-calpains) are activated by micromolar amounts of calcium, whereas activation of calpains II (or M-calpains) requires millimolar amounts of calcium (Perrin and Huttenlocher, 2002). Activated calpains target RBC cytoskeleton proteins, such as spectrin, actin, and band 3/AE1 (Murachi et al., 1981; Schwarz-Ben Meir et al., 1991), whose degradation ultimately leads to ruffling and blebbing of the RBC membrane (Larsen et al., 2008).
Depending on the eryptotic trigger, different pathways can be activated. For instance, eryptosis due to oxidative stress seems to activate caspases (Matarrese et al., 2005), although there is some controversy about the activity of caspases in RBCs (Berg et al., 2001). Interestingly, it has been observed that not all erythrocytes have the same sensitivity to oxidative stress-induced eryptosis, differences being attributed to age and oxidative stress “history” of the cell (Ghashghaeinia et al., 2012). Eryptosis triggered by energy depletion has been shown to involve a number of kinases—further discussed below—as well as an impairment in antioxidant glutathione (GSH) replenishment, which requires energy (Tang et al., 2015).
Although some of the mechanisms leading to eryptosis have been described, the molecular players and signaling pathways underpinning eryptosis remain yet to be fully characterized. So far, a few key regulators of classical apoptotic pathways, protein kinases and “death” receptors have been suggested as putative molecular regulators of eryptosis and are summarized in the next section.
Molecular Regulators of Eryptosis
Death or apoptotic proteins, protein kinases and cell surface receptors are proposed to have a role in the execution or the inhibition of eryptotic pathways. The section below (and Table 2) summarizes available information on such molecular regulators of eryptosis, and highlights the many gaps in knowledge that remain to be filled.
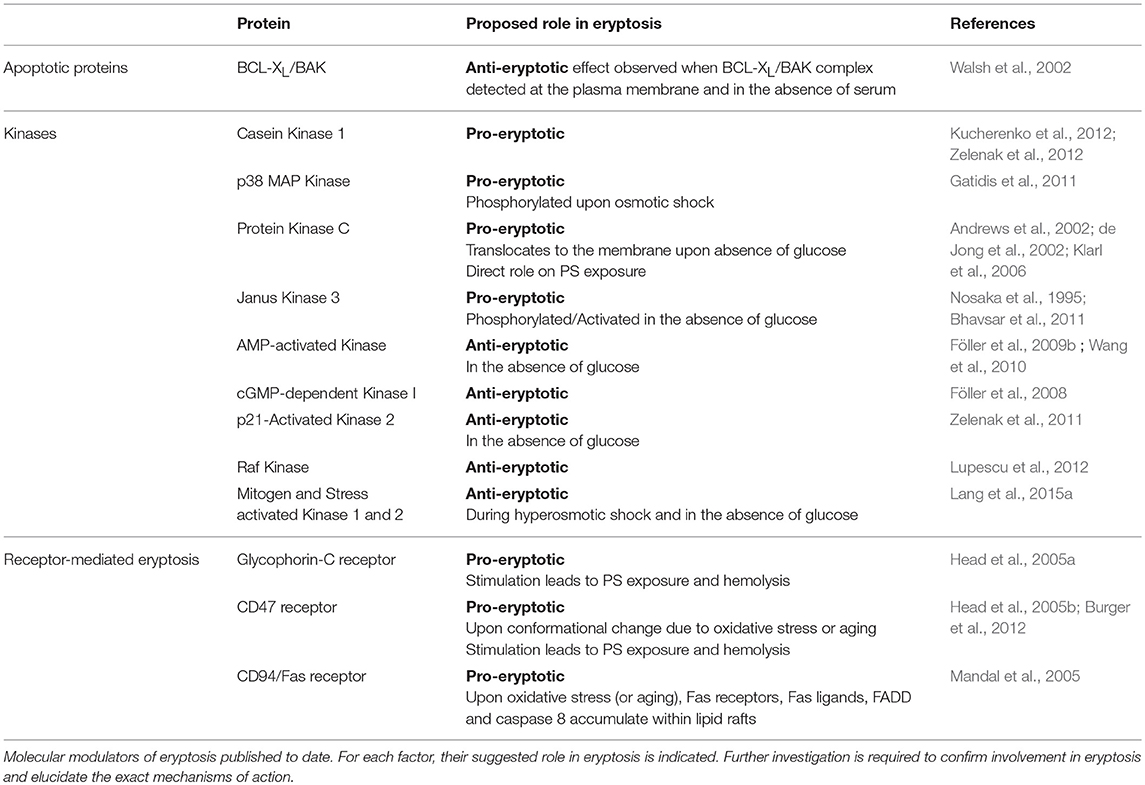
Table 2. Molecular regulators of eryptosis.
Apoptotic Proteins Involved in Erythrocyte Survival
Many cell death proteins are present in mature RBCs, including members of the BCL-2 family such as the pro-apoptotic proteins BAD, BID, BCL10, BAK, BAX, and the anti-apoptotic protein BCL-XL (Walsh et al., 2002; Roux-Dalvai et al., 2008; Lange et al., 2014). These proteins play crucial roles in mitochondria-dependent apoptosis, and are key gatekeepers of viability in nucleated cells.
In healthy nucleated cells, pro-survival BCL-XL and BCL-2 sequester BAX and BAK, while pro-apoptotic BAD is sequestered in a phosphorylation-dependent manner by protein 14-3-3 to repress apoptosis. When apoptotic signals overcome survival signals, the phosphorylation status of BAD changes, causing its release from 14-3-3 and binding to pro-survival BCL-XL and BCL-2, unleashing the apoptotic pore forming BAK and BAX. Perforation of the mitochondrial outer membrane commits the cell to apoptosis: cytochrome c is released, an apoptosome is formed, and effector caspases are activated, ultimately leading to DNA fragmentation, PS exposure and membrane blebbing.
BCL-XL is known to be crucial during haematopoiesis and is particularly strongly expressed during the late stages of erythroblasts maturation (Gregoli and Bondurant, 1997), while expression of BAX and BAK decreases during development of erythroid lineage cells (Gregoli and Bondurant, 1997). In reticulocytes (immature erythrocytes containing a mitochondrion), inhibition of BCL-XL leads to caspase-dependent apoptosis (Delbridge et al., 2017). Similarly, the interaction between BCL-XL and BAK in the plasma membrane is required for the survival of mature RBCs (which lack mitochondria) (Walsh et al., 2002). Indeed, rupture of this interaction using a BAK-mimetic small molecule (resulting in the release of BAK) increases intracellular calcium concentrations and induces PS exposure (Walsh et al., 2002). Interestingly, eryptosis induction through this pathway is inhibited by the addition of serum, suggestive of a key role for serum survival factors in erythrocyte viability. Nevertheless, the role of death proteins within mature erythrocytes, if any, remains to be investigated.
Protein Kinases Involved in Eryptosis
Phosphorylation is a key regulatory mechanism of all intracellular processes, including apoptosis in nucleated cells. Noteworthy, phosphorylation and other post-translational modifications are possibly even more important in mature erythrocytes, as these cells cannot synthesize de novo proteins. Accordingly, numerous protein kinases have been shown to regulate eryptosis. Some, including CK1α, p38 MAP kinase, PKC, and JAK3, stimulate eryptosis; others, such as AMPK, cGKI, PAK2, Raf kinase, and MSK1/2, inhibit this process. The section below summarizes the currently knowledge regarding the mechanisms by which these kinases stimulate or inhibit eryptosis in mature RBCs (also see Table 2).
Eryptosis Stimulator: Casein Kinase 1
Casein kinase 1 (CK1) is a serine/threonine protein kinase with seven isoforms in humans, and plays a role in very diverse cellular processes, including cell cycle, gene expression, cytoskeleton modifications, cell adhesion, and modulation of receptor-mediated signaling (Schittek and Sinnberg, 2014). In the context of apoptosis in nucleated cells, CK1α phosphorylates BCL10, BID and Fas-associated death domain (FADD, further described below) (Schittek and Sinnberg, 2014). In RBCs, stimulation of CK1α enhances eryptosis following energy depletion, oxidative stress and osmotic shock (Kucherenko et al., 2012; Zelenak et al., 2012). Conversely, CK1α inhibition blunts eryptosis in these same conditions.
Eryptosis Stimulator: p38 MAP Kinase
p38 mitogen-activated protein kinase (p38 MAPK) is activated by diverse stimuli, such as UV light, heat, osmotic shock and cytokines (Zarubin and Han, 2005). In the context of apoptosis, p38 MAP kinase can have pro- and anti-apoptotic roles depending on the cell type and on the stimulus. In RBCs, p38 MAP kinase is phosphorylated upon osmotic shock, and participates in calcium uptake, PS exposure and cell shrinkage (as p38 MAP kinase inhibition blunts these effects) (Gatidis et al., 2011).
Eryptosis Stimulator: Protein Kinase C
Protein Kinase C (PKC) molecules constitute a family of eight serine/threonine protein kinase isoforms that are activated upon increase of Ca2+ and diacylglycerol (Mochly-Rosen et al., 2012). Upon activation, PKC is recruited to the membrane where it phosphorylates its substrates. PKC can regulate an important number of processes, including gene expression, protein secretion, cell division and inflammation (Mochly-Rosen et al., 2012). In RBCs, activation of PKC leads to increased calcium intake, cell shrinkage and PS exposure, whereas PKC inhibition prevents calcium entry and PS exposure (Andrews et al., 2002; de Jong et al., 2002). Furthermore, glucose depletion induces PKCα translocation to the membrane, significantly increasing PKC activity and phosphorylation of membrane proteins (Klarl et al., 2006). Interestingly, PS exposure following energy depletion was blunted with PKC-specific inhibitors (Klarl et al., 2006), consistent with a direct role of PKC on PS exposure.
Eryptosis Stimulator: Janus Kinase 3
Janus kinase 3 (JAK3) is a cytosolic tyrosine kinase, part of the larger JAK family, which includes JAK1, JAK2, and TYK2. All Janus kinases play a role in signaling triggered by extracellular cytokines and growth factors. Typically, upon binding of a cytokine to its receptors, the receptors multimerize, specific JAK kinases are recruited to the receptor cytosolic domain, and trans-phosphorylate (Rawlings et al., 2004). Janus kinases substrates include STATs (signal transduction and activators of transcription) which then enter the nucleus and regulate transcription. Of relevance, JAK/STAT signaling is crucial for haematopoiesis and JAK3 is predominantly expressed in haematopoietic cells (Nosaka et al., 1995, p. 3). In RBCs, following glucose depletion, JAK3 is phosphorylated on the activating Tyrosine 980 (Nosaka et al., 1995, p. 3). Further, inhibition of JAK3 (or absence of JAK3 in the case of jak3−/− mice) blunts the exposure of PS upon glucose depletion, but has no effect on cell size (Bhavsar et al., 2011). Overall, JAK3 is thought to be implicated in membrane scrambling upon energy depletion.
Eryptosis Inhibitor: AMP-Activated Kinase
AMP-activated kinase (AMPK) is a serine/threonine kinase that is activated by adenosine monophosphate (AMP) and plays a crucial role in energy homeostasis. Increased concentration of AMP (and a decrease in ATP concentration) is an indication of energy depletion. Upon activation, AMPK switches on catabolic processes that produce ATP, such as glycolysis, and switches off anabolic pathways that consume ATP, such as lipogenesis. Interestingly, mice lacking the AMPK subunit α exhibit erythrocytes with a shorter lifespan compared to their wild-type counterpart (Wang et al., 2010). Additionally, inhibition of AMPK exacerbates eryptosis phenotypes induced by glucose depletion: in an energy depletion context, inhibition of AMPK further increases PS exposure and intracellular calcium, suggesting a role of AMPK in protecting against eryptosis (Föller et al., 2009b).
Eryptosis Inhibitor: cGMP-Dependent Kinase I
cGMP-dependent protein kinase I (cGKI) is a serine/threonine kinase activated by cyclic guanosine monophosphate (cGMP), and is implicated in calcium regulation, platelet activation and many other processes (Butt et al., 1994). Noteworthy, mice lacking cGKI display increased levels of eryptosis, as detected by elevated PS exposure, faster RBC clearance and a higher rate of RBC turnover (Föller et al., 2008).
Eryptosis Inhibitor: p21-Activated Kinase 2
The p21-activated kinase (PAK) family of serine/threonine kinases comprises 6 members in humans. PAK1, 2 and 3 are effector kinases of the Rho-GTPase CDC42 and Rac, two key cytoskeletal regulatory proteins. PAK2 can also be activated by proteolytic cleavage by caspase 3 during apoptosis (Rane and Minden, 2014). In erythrocytes, PAK2 inhibition leads to PS exposure in the absence of glucose, an effect not exacerbated by the absence of AMPK (ampk knock-out mice), suggesting that both these enzymes are acting within the same pathway (Zelenak et al., 2011). Overall AMPK and PAK2 appear to have a pro-survival role during energy depletion.
Eryptosis Inhibitor: Raf Kinase
The Rapid accelerated fibrosarcoma (Raf) kinase family of serine/threonine kinases comprises 3 members that act upstream of mitogen-activated protein kinase (MAPK) pathways: A-Raf, B-Raf, and C-Raf. Typically, growth factor receptors activate the Ras GTPase, which activates Raf, which in turn phosphorylates MEK, leading to activation of ERK (Matallanas et al., 2011). Raf is implicated in apoptosis, and a specific inhibitor, Sorafenib, has been developed for cancer treatment (Wilhelm et al., 2004). In erythrocytes, inhibition of Raf with Sorafenib leads to eryptosis, suggesting a protective role of Raf against eryptosis (Lupescu et al., 2012).
Eryptosis Inhibitor: Mitogen and Stress Activated Kinase 1 and 2
MSK1 and MSK2 are serine kinases involved in the MAPK cascade. Once activated by ERK1 and 2, they phospho-activate transcription factors, which then promote cellular proliferation (Wiggin et al., 2002, p. 1). Mice lacking MSK1/2 present a higher turnover of erythrocytes (Lang et al., 2015a). Although in normal conditions, the levels of PS exposure were similar compared to their wild-type counterpart, when MSK1/2-deficient erythrocytes were stressed with hyperosmotic shock or energy depletion, eryptosis (as indicated by PS exposure and cell shrinkage) was significantly enhanced.
Although numerous protein kinases have been shown to be involved in eryptosis, the mechanisms leading to their activation, as well as the specific downstream effectors, are yet to be identified. This represents an important gap in knowledge in the field that requires further investigation.
Receptor-Mediated Eryptosis
The third category of molecular regulators of eryptosis encompasses cellular receptors that have the ability to transform extracellular signals into an intracellular cell death cascade. Indeed, similar to apoptosis of nucleated cells, eryptosis can be triggered upon the binding of specific extracellular ligands to various red blood cell surface receptors. RBC receptors that have been shown to trigger eryptosis include glycophorin-C, CD47, and CD95/Fas. We are yet to fully understand the links between activation of these surface receptors and the trigger of eryptosis; and this section summarizes the current knowledge of receptor-mediated eryptosis activation (also see Table 2).
Glycophorin-C Receptor
Glycophorin-C (GPC) is an important membrane glycoprotein that interacts with the membrane skeleton and plays a role in the maintenance of the shape of the erythrocyte (Tanner, 1993). Interestingly, it has been reported that binding of anti-GPC antibodies leads to PS exposure and hemolysis (Head et al., 2005a).
CD47 Receptor
Cluster of Differentiation 47 (CD47) is a transmembrane protein that has long been described as a “don't eat-me” signal directed toward the macrophage receptor SIRPα (Oldenborg et al., 2000, p. 47). However, recent studies indicate that CD47 can switch to an “eat-me” signal in aging and oxidative-stressed erythrocytes (Burger et al., 2012, p. 47). Indeed, oxidative stress induces a conformational change of CD47 which, despite retaining binding to the same macrophage SIRPα receptor, will under these conditions induce phagocytosis (Burger et al., 2012). Interestingly, induction of CD47 signaling through binding of its TSP-1 ligand, as well as anti-CD47 antibodies and a specific CD47-binding peptide, trigger PS exposure and hemolysis of RBCs (Head et al., 2005b).
CD94/Fas Receptor
Fas receptor (or CD94 or APO-1) is a well-studied death receptor in the context of apoptosis and other programmed cell death pathways (Schulze-Osthoff et al., 2001). Upon binding of the Fas ligand FasL (either as transmembrane proteins on cytotoxic T lymphocytes, or as soluble proteins) to the Fas receptors on the target cell, the latter form death-inducing signaling complexes (DISC). Aggregation of Fas receptors allows the recruitment of proteins that carry Fas-associated death domain (FADD), leading to the recruitment and activation of caspase 8. Caspase 8 in turn can cleave, and hence activate, BID (with subsequent mitochondria-dependent apoptosis activation) and/or directly activate effector caspase 3. Mandal et al. (2005) showed that Fas receptors, Fas ligands, FADD and caspase 8 accumulate within lipid rafts in old erythrocyte membranes, as well as in oxidative-stressed erythrocytes. Caspase 8 is activated in both old and oxidative-stressed erythrocytes, followed by activation of caspase 3 and reduced activity of flippases (and hence increased PS exposure).
Studying Eryptosis: the Tools of the Trade
As emphasized above, the molecular events leading to eryptosis remain largely unknown. A number of challenges, inherent to the specific cell type we are considering, explain this gap in knowledge. A key step toward further dissecting the molecular mechanisms of eryptosis relies on robust technical approaches. However, cell death detection techniques relying on nuclear DNA fragmentation or mitochondrial activity cannot be used in this context, for obvious reasons. Nevertheless, a key phenotypic hallmark of eryptosis (and apoptosis) is the cell surface exposure of phosphatidyl serine, which allows flow cytometry to be a key technique commonly used to detect and quantify eryptosis. The binding of fluorescent Annexin V to externalized PS and the cell volume (measured via forward scatter values) are the most common parameters used to assess eryptosis. Additionally, intracellular calcium concentrations can be measured with cell-permeable fluorescent indicators, such as Fluo-3 AM dye. Intracellular ceramide levels can be detected with ceramide-specific antibodies, and the redox state of the cells can be assessed through measurement of GSH/GSSG ratio (reviewed in Pretorius et al., 2016; Jemaà et al., 2017).
Another key hurdle is the study of the RBC proteome given the overwhelming relative amounts of the two most abundant proteins: hemoglobin represents 97% of RBCs' total proteins, and carbonic anhydrase I 1% (Barasa and Slijper, 2014). This implies that all other proteins are present in a much lower relative abundance, and therefore difficult to identify and quantify.
At the genetic level, as RBCs are enucleated, experiments requiring genetic manipulations aimed at investigating the role of specific genes in eryptosis cannot be conducted. However, recent advances in in vitro stem cell culture and haematopoietic lineage differentiation, as well as advances in genome editing, offer exciting perspectives to address a number of these issues in the near future (Hockemeyer and Jaenisch, 2016; Caulier et al., 2017). For example, with major advances in in vitro stem cell differentiation methods being made, in vitro production of large amounts of human mature erythrocytes for transfusion could become a reality (Lapillonne et al., 2010; Caulier et al., 2017). Further, genetic manipulation of stem cells, followed by maturation into erythrocytes, would allow the investigation of molecular events and signaling pathways essential for mature erythrocyte survival. This has indeed been recently explored in the context of indirect areas of RBC research, notably with respect to infection of RBCs with malaria parasites (Egan, 2017; Kanjee et al., 2017).
Finally, other challenges to the study of eryptosis (and erythrocytes in general) include RBC origin, availability, purification, storage conditions and the lack of well-characterized cell lines.
Plasmodium-Infected Erythrocytes: a Fight Between Life and Death
It is well documented that many pathogens manipulate (i.e., inhibit) the apoptotic pathway of their host cell in order to achieve intracellular survival (viruses: Hay and Kannourakis, 2002; parasites: James and Green, 2004; bacteria: Ashida et al., 2011). In the case of malaria parasites, this has been demonstrated during the infection of liver cells (van de Sand et al., 2005). However, little is known about host-parasite interactions of the erythrocytic stages of infection. Unraveling the molecular regulation of eryptosis is key to our understanding of the interrelationship between Plasmodium parasites and their host red cell. Plasmodium imposes oxidative stress and induces phosphatidylserine exposure on the host red blood cells, but the clinical outcome arising from PS-exposing Plasmodium-infected RBCs is debated. In the context of the tools currently available to study eryptosis, this section reviews published evidence and proposed models that attempt to characterize the interplay between Plasmodium and host RBC death mechanisms (also see Figure 3).
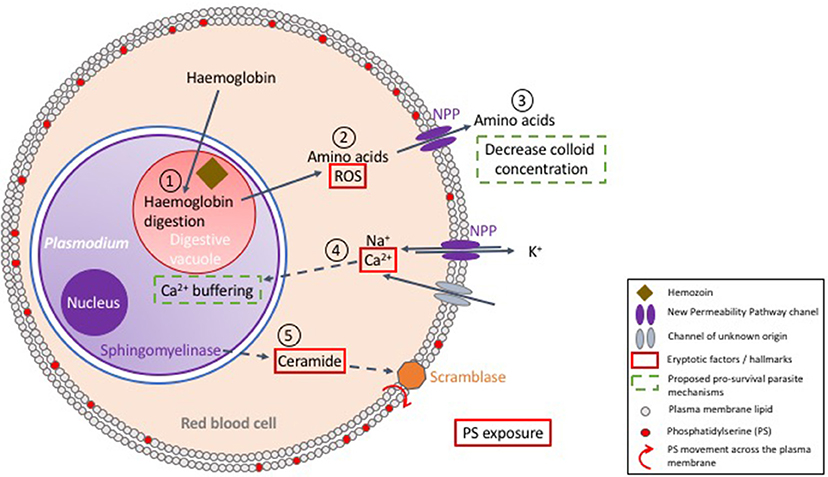
Figure 3. Proposed model of the interactions between Plasmodium and its host red blood cell. Upon erythrocyte invasion, Plasmodium digests host cell hemoglobin in its digestive vacuole (1). In this process, heme is detoxified by polymerisation into hemozoin and amino acids are utilized for parasite development. The surplus of amino acids is exported to the RBC cytosol (2), and further secreted to the extracellular milieu to decrease colloid concentration (3), which is proposed to enhance survival of the host cell. Digestion of hemoglobin also produces reactive oxygen species (ROS) (2), which enhances eryptosis. New Permeability Pathways (NPPs) and/or other transporters allow Ca2+ entry into the red cell cytosol, a key player in triggering eryptosis (4). However, it has been suggested that Plasmodium uptakes most of the intracellular calcium in its own cytosol, thus achieving delay of host cell death (4). Nevertheless, active parasite sphingomyelinase has been proposed to break down sphingomyelin, producing ceramide (5), which enhances exposure of phosphatidylserine (PS), and hence recognition and clearance by macrophages.
Plasmodium Imposes Oxidative Stress on Its Host Cell
During the 48h of its erythrocytic asexual cycle, Plasmodium falciparum increases its body mass by up to 32-fold. This requires a considerable supply of nutrients, which the parasite obtains from the digestion of hemoglobin (Zarchin et al., 1986). The ensuing generation of H2O2 and OH radicals imposes oxidative stress on the host cell (Atamna and Ginsburg, 1993; Ginsburg and Atamna, 1994), which, as discussed above, can lead to erythrocyte's death, which would represent a non-favorable outcome for the parasite (Figure 3).
Extensive Digestion of Hemoglobin Prevents Bursting of the Host Cell
In addition to the nutrients provided by hemoglobin degradation, parasite survival further relies on the import of nutrients and metabolites from the extracellular milieu, and on a mechanism of waste disposal to the extracellular environment (Kirk, 2001). For this purpose, Plasmodium exports parasite encoded transporters to the red cell membrane known as the New Permeability Pathways (NPP) transporters (Kirk, 2001; Huber et al., 2005). Consequently, during trophozoite development, extensive changes take place on the red cell membrane, leading to a dramatic decrease in K+, and increase in Na+ concentrations in the host cell cytosol to levels resembling those of the extracellular space (Kirk, 2001). The increased cytosolic Na+ concentration poses a risk of cell swelling and hemolysis. To prevent this and ensure osmotic stability, the parasite decreases the intracellular colloid osmotic pressure by digesting more hemoglobin than metabolically needed, and exports the excess of amino acids through the NPP transporters, consequently reducing cell swelling and decreasing risks of hemolysis (Lew et al., 2003) (Figure 3).
Plasmodium Might Sequester Intracellular Calcium to Delay Host Cell Death
In normal conditions, erythrocytes maintain low intracellular Ca2+ levels, corresponding to a concentration that is ~ 40,000-fold lower than that of free-Ca2+ in the blood plasma (Bogdanova et al., 2013). However, upon P. falciparum infection, the erythrocyte's calcium content increases by 10- to 20-fold (Kirk, 2001). Intracellular calcium is crucial for parasite invasion and development (Wasserman et al., 1982) and is thought to be key for many parasite signaling pathways (Enomoto et al., 2012). Interestingly, an increase of free Ca2+ in the RBC cytosol is not observed during infection; it is thought that Plasmodium accumulates Ca2+ in its own cytosol to maintain a low level of free Ca2+ in the host cell (Adovelande et al., 1993; Tiffert et al., 2000) (Figure 3). Ca2+ sequestration inside the parasite is thought to delay eryptosis, however direct measurements of in vivo calcium concentrations in the parasite are required to formally determine whether this is the case. Interestingly, during the liver stage of infection Plasmodium sequesters intracellular calcium from hepatocytes, therefore preventing/delaying PS exposure (Sturm et al., 2006). Noteworthy, calcium sequestration mechanisms of erythrocytic stages, if demonstrated, would likely be considered one of the basic foundations for parasite-manipulated eryptosis.
Increased Phosphatidylserine Exposure During Infection
Despite the fact that Plasmodium is proposed to control the levels of free Ca2+ in the RBC cytosol, an increase in PS exposure is observed in Plasmodium-infected RBCs (iRBCs) when compared to uninfected erythrocytes (uRBCs) (Schwartz et al., 1987). This has been observed not only in vitro with P. falciparum infection of human RBCs (Schwartz et al., 1987), but also in vivo in animal models of malaria infection, such as P. yoelii infection of mice (Totino et al., 2010) and P. knowlesi infection of monkeys (Joshi et al., 1987). Overall, PS-exposure tends to increase in the mature stages of parasite development (Joshi et al., 1987; Schwartz et al., 1987). A possible explanation for this phenomenon involves the formation of ceramides, a component of sphingomyelin, one of the major structural lipids of cell membranes. Hydrolysis of sphingomyelin, catalyzed by sphingomyelinase, generates ceramide, which, in erythrocytes, leads to PS exposure (see above) even at low cytosolic Ca2+ concentrations (Lang et al., 2004) (see Figure 3). Interestingly, erythrocytic forms of P. falciparum express a sphingomyelinase that is active during blood stages (Hanada et al., 2000). In line with this observation, a marked decrease of the sphingomyelin content is observed in RBC membranes upon infection by P. falciparum (Maguire and Sherman, 1990). Besides the role of ceramide in PS-exposure during infection, it is also interesting that GPC (an erythrocyte receptor able to mediate eryptosis) is used by P. falciparum to invade RBCs (Maier et al., 2003), and it has been suggested that this could be the trigger for PS exposure during early stages of parasite development (Head et al., 2005a).
Overall, Plasmodium induces eryptosis in the infected cell, by imposing oxidative stress, possibly by producing ceramide, activating GPC-pathways and modulating the erythrocyte intracellular ionic composition. At the same time, Plasmodium might also prevent / delay eryptosis and hemolysis by sequestering calcium, and exporting amino acids to the extracellular milieu, as summarized in Figure 3. A balance between induction and prevention (or delay) of eryptosis of the host cell by Plasmodium is likely to translate into a successful vs. unsuccessful infection. Although this appears key to our understanding of the clinical outcomes of malaria infections, much is yet to be uncovered in this area.
Plasmodium Induces Eryptosis of Bystander Erythrocytes
Further to the eryptosis that Plasmodium induces on its own infected cell, it has been observed that malaria infection induces eryptosis of bystander uninfected erythrocytes. In a study using a rodent malaria model, increased numbers of eryptotic uRBCs have been observed in mice 6–7 days post-infection with P. yoelii (Totino et al., 2013). Similarly, incubation of human RBCs with serum from patients infected with P. falciparum leads to increased PS exposure and decreased cell size of uRBCs (Totino et al., 2014), suggesting that P. falciparum is able to induce cell death of non-infected erythrocytes. Interestingly, this has not been observed in the case of P. vivax infections, so one cannot exclude that the proinflammatory response common in P. falciparum infections, but not in P. vivax infections, could be at least partially responsible for this observation (Totino et al., 2014). Further, another in vitro study where the role of proinflammatory responses cannot be taken into account, suggests the increase of PS exposure on bystander uRBCs is partially attributed to the presence of methaemoglobin in the extracellular medium. Methaemoglobin is a form of hemoglobin, in which the iron in the heme group is in the Fe3+ (ferric) state, not the Fe2+ (ferrous) of normal hemoglobin. Upon RBC lysis, hemoglobin is released and oxidized by oxygen to methaemoglobin, which in turn leads to PS exposure of bystander RBCs through oxidative stress (Balaji and Trivedi, 2013). Overall, increased eryptosis of uRBC is thought to be a major contributor of anemia observed during severe malaria infections (Jakeman et al., 1999; Totino et al., 2016), alongside the loss of iRBCs and decreased erythropoiesis (Pathak and Ghosh, 2016). Consequently, a full understanding of the molecular events leading to eryptosis of bystander uRBCs during a malaria infection is crucial to comprehensively address the clinical symptoms of severe anemia in malaria patients.
A Controversial Role of Eryptosis During Malaria Infection—is it Good or Bad?
While it has been established that Plasmodium infection contributes to eryptosis of the host cell, the clinical benefit of the phenomenon is debated. On one hand it is argued that PS-mediated clearance of infected cells is favorable to the host. On the other hand it is argued that PS exposure contributes to parasite immune evasion and malaria pathogenesis. This section summarizes the studies that have led to such observations.
Phosphatidylserine Exposure of Plasmodium-Infected Erythrocytes Implicated in Severe Disease
It has been argued that PS exposure of iRBCs may play a role in the cytoadherence of P. falciparum and enhance tissue sequestration of P. vivax (Eda and Sherman, 2002; Totino and Lopes, 2017). Adherence of PS-exposing RBCs to endothelial cells has been demonstrated to occur in various medical conditions and in in vitro experiments (Wali et al., 1988; Closse et al., 1999; Bonomini et al., 2002; Setty et al., 2002; Wautier et al., 2011). Therefore, it is plausible that exposure of PS in Plasmodium-infected RBCs contributes to adherence and sequestration, as well as to cell aggregation and rosetting (Ho et al., 1991). Consequently, increased PS exposure of iRBCs has two major detrimental effects for malaria patients: sequestered and adherent Plasmodium-iRBCs avoid immune clearance; and adherence to the endothelium and other cells induces thrombo-occlusion, which leads to severe disease. Both these events are well characterized in the case of P. falciparum infections, and have been to date mainly attributed to the cell surface exposure of PfEMP1, a parasite-derived molecule. Interestingly, in the case of P. vivax (which do not express PfEMP1), PS exposure might lead to a more severe course of infection, and there is some evidence that P. vivax-iRBCs are able to adhere to endothelial walls and to rosette, due at least in part to phosphatidylserine exposure (Costa et al., 2011; Totino and Lopes, 2017).
Does Phosphatidylserine Exposure of Plasmodium-Infected Erythrocytes Benefit Disease Outcome?
Despite the data discussed above suggesting that PS exposure in iRBCs is beneficial to parasite survival, two studies propose that eryptosis of Plasmodium-infected RBCs is beneficial for the outcome of malaria infection. One study has shown that P. falciparum preferentially invades non-eryptotic RBCs (Totino et al., 2010), therefore eryptosis of uninfected RBCs has been interpreted as “a host mechanism to fight malaria” (Totino et al., 2010). The decreased susceptibility of infection of eryptotic cells may in part be attributed to cytoskeleton changes in the host cell (Totino et al., 2010), since actin, spectrin and band 3 proteins, which are crucial for invasion by the parasite (Koch and Baum, 2016), are degraded during eryptosis (Murachi et al., 1981; Schwarz-Ben Meir et al., 1991). In this case, enhanced eryptosis is suggested to correlate with lower parasite burden. Another study proposed that PS-based clearance of ring stage-iRBCs prevents the formation of late-stage parasites, and hence sequestration and associated clinical complications (Föller et al., 2009a). The authors suggest that this may be part of the mechanisms leading to the relative protection against severe malaria in sickle-cell trait, homozygous hemoglobin-C and G6PD-deficiency (Cappadoro et al., 1998; Ayi et al., 2004).
Inducing Eryptosis to Treat Malaria?
Following from the idea that increased PS exposure of Plasmodium-infected RBCs benefits clearance and therefore disease outcome, numerous studies have assessed the effect of eryptosis modulators in the context of malaria. In the section below (and in Tables 2, 3), we summarize the studies that have attempted to date to induce or inhibit eryptosis with the aim of modulating malaria infection, and we further discuss the limitations of such approaches.
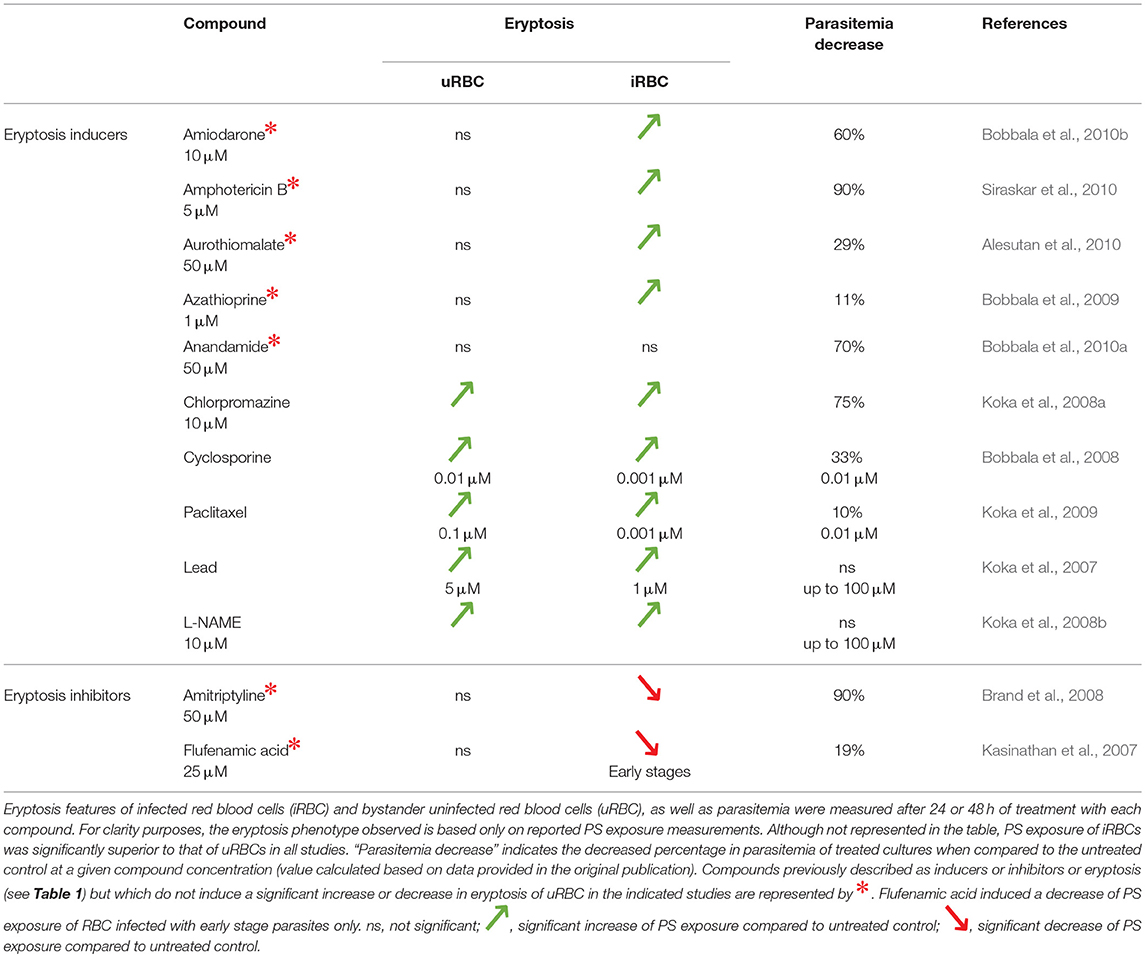
Table 3. Effect of eryptosis inducers and inhibitors on P. falciparum in vitro development.
The Effect of Eryptosis Modulators on the Course of a Malaria Infection
Given the interrelationship between malaria and eryptosis, numerous studies have assessed the effect of eryptosis modulators in the context of malaria (Tables 2, 3 and references therein). Interestingly, various compounds described to induce or inhibit eryptosis in naïve RBCs (Table 1) fail to modulate eryptosis in bystander uRBCs of a Plasmodium culture (Table 3, compounds indicated by *). This discrepancy can be attributed to differences between naïve RBCs and Plasmodium in vitro culture conditions. Indeed, eryptosis of naïve RBCs is assessed in Ringer solution at 0.4% haematocrit (see references of Table 1), whereas eryptosis of Plasmodium cultures is assessed in complete RPMI medium at 4% haematocrit (see references of Table 3). Noteworthy, all but one of the eryptosis inducers tested on malaria parasites (regardless of their ability to induce eryptosis on bystander uRBCs), induce eryptosis of Plasmodium-infected RBCs, often at a lower concentration than the one required to induce eryptosis of uRBCs. Further, most of the compounds presented in Table 3 somehow affect parasite viability albeit at variable (and in some cases high) concentrations. Together, these observations provide the main argument in favor of the use of eryptosis inducers as anti-malarial treatments, particularly for those compounds where uRBCs remain unaffected at the concentrations required to affect infected cells. Similar observations were made when using in vivo mouse malaria models of infection (Table 4). Five eryptosis inducers have been reported to not modulate eryptosis in bystander uRBCs, while inducing eryptosis of iRBCs. Overall, these in vivo studies indicate that inducing eryptosis has a beneficial effect for the host. Indeed, a decrease of parasitemia and increase in mice survival are observed (Table 4). Importantly, few studies measured anemia levels of treated mice, although this is a crucial side effect to consider when testing the use of eryptosis manipulators in vivo (Table 4). With respect to the above studies, the possibility has not been excluded that some of the compounds may have off-targets in the parasite. Finally, it is interesting to note that some antimalarial drugs such as artesunate, mefloquine and quinine, have been shown to induce eryptosis (Alzoubi et al., 2014; Bissinger et al., 2015; Mischitelli et al., 2016), raising the question of a possible interference with the eryptotic process to their antimalarial effect.
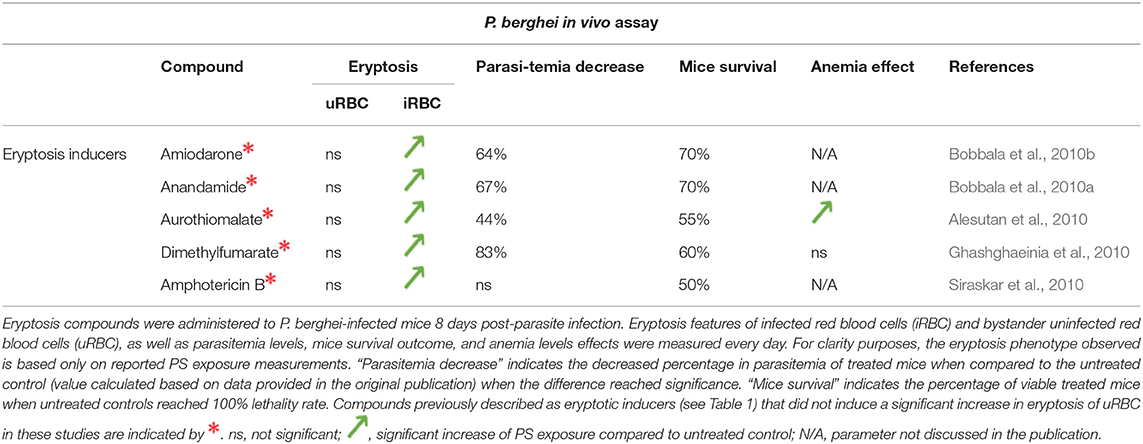
Table 4. Effect of eryptosis inducers on P. berghei in vivo development.
Inducing Eryptosis to Treat Malaria—Is It Realistic?
The majority of studies discussed in this review (and summarized in Tables 3, 4) support the notion that enhancing eryptosis of parasitised RBCs represents an attractive anti-malaria strategy. This is based on the hypothesis that exposing RBCs to eryptotic inducers could lead to early clearance of ring stages, preventing trophozoite development and sequestration, and therefore leading to a less severe course of infection. However, it is worth emphasizing that eryptosis inducers that do not specifically target iRBCs, will also increase PS exposure of non-infected RBCs and are therefore likely to lead to anemia. Perhaps the only exception to this, consists in the argument that iRBCs are stressed by the presence of the parasite, therefore the threshold to induce eryptosis may be lower for iRBCs than for uRBCs. Hence the idea of inducing eryptosis selectively (or predominantly) in Plasmodium-iRBC may be considered achievable by some authors. It is our view, however, that further work is required to assess the overall potential clinical benefits of eryptosis inducers in the context of a malaria infection. On the other hand, assuming that Plasmodium actively inhibits eryptosis of its host cell, an anti-malarial strategy aiming to prevent this inhibition appears more attractive and is discussed in the following section.
Preventing Plasmodium From Inhibiting Eryptosis—a Novel Approach to Treat Malaria?
Plasmodium interferes with apoptosis of its host cells in the liver of the vertebrate host (Heussler et al., 2006; Kakani et al., 2016) and in the mosquito midgut (Ramphul et al., 2015; Kakani et al., 2016). The parasite thus has a proven track record of manipulating host cell death pathways, and, as alluded to above, also appears to do so during infection of RBCs. Upon RBC invasion, buffering of host cell intracellular calcium and secretion of unused amino acids (Figure 3) have been proposed as the first signs of eryptosis manipulation by the parasite (Adovelande et al., 1993; Tiffert et al., 2000; Kirk, 2001). Consistent with this idea, inhibition of a P. falciparum cation-transporting ATPase was shown to induce eryptosis of the iRBC and its rapid clearance in vivo (Jiménez-Díaz et al., 2014), as if this inhibition reversed eryptosis silencing. Further, P. falciparum activates and relies on human kinases such as mitogen-activated protein kinase kinase (MEK) and p21 activated kinase (PAK), for its development inside RBCs (Sicard et al., 2011). Interestingly, PAK kinases have the ability to inhibit eryptosis in energy depletion conditions, as discussed above (Zelenak et al., 2011) and are often subverted by pathogens for different cellular functions, including manipulation of apoptosis (John von Freyend et al., 2017). Overall, even though there is currently no direct evidence that PAK activation in iRBCs is linked to eryptosis manipulation, it is an exciting hypothesis that opens great perspectives for anti-malaria intervention strategies. Indeed, inhibition of host kinases required by the parasite for survival would ensure disease treatment with smaller risks of emergence of parasite resistance. In addition, host-directed approaches offer the possibility of repurposing human kinase inhibitors already available in the drug-market (Doerig, 2004).
Conclusions and Future Directions
Although the complexity of eryptosis regulation in healthy individuals remains to be fully understood, numerous molecular mechanisms have been described to date. Understanding eryptosis regulation is particularly relevant to further our knowledge of Plasmodium-erythrocyte interactions. Indeed, manipulation of host cell pathways is a widely used strategy by a multitude of pathogens and offers novel and attractive host-direct therapy opportunities. Manipulation of eryptosis by Plasmodium parasites is an emergent area of research that has attracted attention in recent years, and where much is yet to be done. Importantly, host-directed antimalarial therapy offers the considerable advantage of limiting the major pathways toward drug resistance, namely the selection of mutated parasite-encoded targets. However, before this is realized, major gaps in knowledge prevail and need to be addressed. Below, we outline some of the key questions that need to be addressed.
Potential Manipulation of Eryptosis by Plasmodium
Although infection of erythrocytes by Plasmodium induces cell death hallmarks, it has been proposed that the parasite inhibits host cell death by buffering intracellular calcium levels (Adovelande et al., 1993), exporting amino acids (Lew et al., 2003), and manipulating host cell kinases (reviewed in Carvalho et al., 2016). Specific molecular mechanisms of calcium sequestration and host ceramide production for example are yet to be elucidated. However, host molecular mechanisms that are shown to be manipulated by the parasite represent exciting opportunities for host-directed therapy. In this context, modulators of eryptosis appear to have a protective effect toward the host in murine models, although possible off-target effects on parasite-encoded factors cannot be excluded, and the influence of these treatments on anemia levels are yet to be determined in most cases.
Role of PS Exposure During Plasmodium Infection
The clinical outcome arising from PS-exposing Plasmodium-infected RBCs is controversial. On the one hand, PS-exposure of erythrocytes infected by young parasites is thought to lead to early parasite clearance, hence being favorable to the host (Föller et al., 2009a). On the other hand it is argued that PS exposure enhances the cytoadherence of infected red blood cells, and therefore contributes to parasite immune evasion and malaria pathogenesis (Totino and Lopes, 2017). Although it is clear that Plasmodium infection enhances PS exposure of the host erythrocyte, the physiological role of this phenomenon remains to be elucidated and further in vivo studies are required to address this question.
Eryptosis of Bystander Erythrocytes
Another notable finding in malaria-related eryptosis is the increased eryptosis of uninfected bystander erythrocytes during a malaria infection (Totino et al., 2010, 2013). This is believed to be a key mechanism leading to malaria-induced anemia and the severe outcome of disease (Jakeman et al., 1999). However, the molecular mechanisms underpinning this phenomenon, including a possible role of the immune response, are not understood. There is little doubt that unraveling the mechanism underlying eryptosis of bystander erythrocytes can lead to critical clinical applications in malaria patients suffering from severe anemia.
Author Contributions
CB and TC drafted the manuscript. CD edited and commented on the manuscript.
Funding
The current work was conducted with funding from La Trobe University and Monash University.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Adovelande, J., Bastide, B., Délèze, J., and Schrével, J. (1993). Cytosolic free calcium in Plasmodium falciparum-infected erythrocytes and the effect of verapamil: a cytofluorimetric study. Exp. Parasitol. 76, 247–258.
Akel, A., Hermle, T., Niemoeller, O. M., Kempe, D. S., Lang, P. A., Attanasio, P., et al. (2006). Stimulation of erythrocyte phosphatidylserine exposure by chlorpromazine. Eur. J. Pharmacol. 532, 11–17. doi: 10.1016/j.ejphar.2005.12.037
Alesutan, I., Bobbala, D., Qadri, S. M., Estremera, A., Föller, M., and Lang, F. (2010). Beneficial effect of aurothiomalate on murine malaria. Malar. J. 9, 118. doi: 10.1186/1475-2875-9-118
Alzoubi, K., Calabrò, S., Bissinger, R., Abed, M., Faggio, C., and Lang, F. (2014). Stimulation of suicidal erythrocyte death by artesunate. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 34, 2232–2244. doi: 10.1159/000369666
Andrews, D. A., Yang, L., and Low, P. S. (2002). Phorbol ester stimulates a protein kinase C-mediated agatoxin-TK-sensitive calcium permeability pathway in human red blood cells. Blood 100, 3392–3399. doi: 10.1182/blood.V100.9.3392
Antony, H. A., and Parija, S. C. (2016). Antimalarial drug resistance: an overview. Trop Parasitol 6, 30–41. doi: 10.4103/2229-5070.175081
Arese, P., Turrini, F., and Schwarzer, E. (2005). Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes. Cell. Physiol. Biochem. 16, 133–146. doi: 10.1159/000089839
Ashida, H., Mimuro, H., Ogawa, M., Kobayashi, T., Sanada, T., Kim, M., et al. (2011). Cell death and infection: a double-edged sword for host and pathogen survival. J. Cell Biol. 195, 931–942. doi: 10.1083/jcb.201108081
Atamna, H., and Ginsburg, H. (1993). Origin of reactive oxygen species in erythrocytes infected with Plasmodium falciparum. Mol. Biochem. Parasitol. 61, 231–241.
Autino, B., Corbett, Y., Castelli, F., and Taramelli, D. (2012). Pathogenesis of malaria in tissues and blood. Mediterr. J. Hematol. Infect. Dis. 4:e2012061. doi: 10.4084/MJHID.2012.061
Ayi, K., Turrini, F., Piga, A., and Arese, P. (2004). Enhanced phagocytosis of ring-parasitized mutant erythrocytes: a common mechanism that may explain protection against falciparum malaria in sickle trait and beta-thalassemia trait. Blood 104, 3364–3371. doi: 10.1182/blood-2003-11-3820
Balaji, S. N., and Trivedi, V. (2013). Extracellular methemoglobin primes red blood cell aggregation in malaria: an in vitro mechanistic study. FEBS Lett. 587, 350–357. doi: 10.1016/j.febslet.2012.12.015
Barasa, B., and Slijper, M. (2014). Challenges for red blood cell biomarker discovery through proteomics. Biochim. Biophys. Acta 1844, 1003–1010. doi: 10.1016/j.bbapap.2013.10.002
Barenholz, Y., and Thompson, T. E. (1980). Sphingomyelins in bilayers and biological membranes. Biochim. Biophys. Acta Biomembr. 604, 129–158. doi: 10.1016/0005-2736(80)90572-6
Baynes, J. W. (2005). “Oxygen and life,” in Medical Biochemistry, eds J. W. Baynes, and M. H. Dominiczak (Philadelphia, PA: Elsevier Health Sciences), 497–506.
Bentzen, P. J., and Lang, F. (2007). Effect of anandamide on erythrocyte survival. Cell. Physiol. Biochem. 20, 1033–1042. doi: 10.1159/000110714
Berg, C. P., Engels, I. H., Rothbart, A., Lauber, K., Renz, A., Schlosser, S. F., et al. (2001). Human mature red blood cells express caspase-3 and caspase-8, but are devoid of mitochondrial regulators of apoptosis. Cell Death Differ. 8, 1197–1206. doi: 10.1038/sj.cdd.4400905
Bhavsar, S. K., Gu, S., Bobbala, D., and Lang, F. (2011). Janus kinase 3 is expressed in erythrocytes, phosphorylated upon energy depletion and involved in the regulation of suicidal erythrocyte death. Cell. Physiol. Biochem. 27, 547–556. doi: 10.1159/000329956
Birbes, H., Bawab, S. E., Obeid, L. M., and Hannun, Y. A. (2002). Mitochondria and ceramide: intertwined roles in regulation of apoptosis. Adv. Enzy. Regul. 42, 113–129. doi: 10.1016/S0065-2571(01)00026-7
Bissinger, R., Barking, S., Alzoubi, K., Liu, G., Liu, G., and Lang, F. (2015). Stimulation of suicidal erythrocyte death by the antimalarial drug mefloquine. Cell. Physiol. Biochem. 36, 1395–1405. doi: 10.1159/000430305
Boas, F. E., Forman, L., and Beutler, E. (1998). Phosphatidylserine exposure and red cell viability in red cell aging and in hemolytic anemia. Proc. Natl. Acad. Sci. U.S.A. 95, 3077–3081.
Bobbala, D., Alesutan, I., Föller, M., Huber, S. M., and Lang, F. (2010a). Effect of anandamide in Plasmodium berghei-infected mice. Cell. Physiol. Biochem. 26, 355–362. doi: 10.1159/000320559
Bobbala, D., Alesutan, I., Föller, M., Tschan, S., Huber, S. M., and Lang, F. (2010b). Protective effect of amiodarone in malaria. Acta Trop. 116, 39–44. doi: 10.1016/j.actatropica.2010.05.005
Bobbala, D., Koka, S., Geiger, C., Föller, M., Huber, S. M., and Lang, F. (2009). Azathioprine favourably influences the course of malaria. Malar. J. 8:102. doi: 10.1186/1475-2875-8-102
Bobbala, D., Koka, S., Lang, C., Boini, K. M., Huber, S. M., and Lang, F. (2008). Effect of cyclosporine on parasitemia and survival of Plasmodium berghei infected mice. Biochem. Biophys. Res. Commun. 376, 494–498. doi: 10.1016/j.bbrc.2008.09.005
Bogdanova, A., Makhro, A., Wang, J., Lipp, P., and Kaestner, L. (2013). Calcium in red blood cells-a perilous balance. Int. J. Mol. Sci. 14, 9848–9872. doi: 10.3390/ijms14059848
Bonomini, M., Sirolli, V., Gizzi, F., Di Stante, S., Grilli, A., and Felaco, M. (2002). Enhanced adherence of human uremic erythrocytes to vascular endothelium: role of phosphatidylserine exposure. Kidney Int. 62, 1358–1363. doi: 10.1111/j.1523-1755.2002.kid560.x
Bookchin, R. M., Ortiz, O. E., and Lew, V. L. (1987). Activation of calcium-dependent potassium channels in deoxygenated sickled red cells. Prog. Clin. Biol. Res. 240, 193–200.
Brand, V., Koka, S., Lang, C., Jendrossek, V., Huber, S. M., Gulbins, E., et al. (2008). Influence of amitriptyline on eryptosis, parasitemia and survival of Plasmodium berghei-infected mice. Cell. Physiol. Biochem. 22, 405–412. doi: 10.1159/000185482
Bratosin, D., Estaquier, J., Petit, F., Arnoult, D., Quatannens, B., Tissier, J.-P., et al. (2001). Programmed cell death in mature erythrocytes: a model for investigating death effector pathways operating in the absence of mitochondria. Cell Death Differ. 8, 1143–1156. doi: 10.1038/sj.cdd.4400946
Brugnara, C., de Franceschi, L., and Alper, S. L. (1993). Inhibition of Ca(2+)-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J. Clin. Invest. 92, 520–526. doi: 10.1172/JCI116597
Buehler, P. W., Baek, J. H., Lisk, C., Connor, I., Sullivan, T., Kominsky, D., et al. (2012). Free hemoglobin induction of pulmonary vascular disease: evidence for an inflammatory mechanism. Am. J. Physiol. Lung. Cell. Mol. Physiol. 303, L312–L326. doi: 10.1152/ajplung.00074.2012
Burger, P., Hilarius-Stokman, P., de Korte, D., van den Berg, T. K., and van Bruggen, R. (2012). CD47 functions as a molecular switch for erythrocyte phagocytosis. Blood 119, 5512–5521. doi: 10.1182/blood-2011-10-386805
Butt, E., Abel, K., Krieger, M., Palm, D., Hoppe, V., Hoppe, J., et al. (1994). cAMP- and cGMP-dependent protein kinase phosphorylation sites of the focal adhesion vasodilator-stimulated phosphoprotein (VASP) in vitro and in intact human platelets. J. Biol. Chem. 269, 14509–14517.
Cappadoro, M., Giribaldi, G., O'Brien, E., Turrini, F., Mannu, F., Ulliers, D., et al. (1998). Early phagocytosis of glucose-6-phosphate dehydrogenase (G6PD)-deficient erythrocytes parasitized by Plasmodium falciparum may explain malaria protection in G6PD deficiency. Blood 92, 2527–2534.
Carvalho, T. G., Morahan, B., John von Freyend, S., Boeuf, P., Grau, G., Garcia-Bustos, J., et al. (2016). The ins and outs of phosphosignalling in Plasmodium: parasite regulation and host cell manipulation. Mol. Biochem. Parasitol. 208, 2–15. doi: 10.1016/j.molbiopara.2016.05.006
Caulier, A., Guyonneau Harmand, L., and Garçon, L. (2017). Immortalisation érythrocytaire pour production de globules rouges in vitro. Transfus. Clin. Biol. 24, 263–267. doi: 10.1016/j.tracli.2017.06.030
Cimen, M. Y. B. (2008). Free radical metabolism in human erythrocytes. Clin. Chim. Acta 390, 1–11. doi: 10.1016/j.cca.2007.12.025
Closse, C., Dachary-Prigent, J., and Boisseau, M. R. (1999). Phosphatidylserine-related adhesion of human erythrocytes to vascular endothelium. Br. J. Haematol. 107, 300–302.
Costa, F. T. M., Lopes, S. C. P., Ferrer, M., Leite, J. A., Martin-Jaular, L., Bernabeu, M., et al. (2011). On cytoadhesion of Plasmodium vivax: raison d'être? Mem. Inst. Oswaldo Cruz 106(Suppl. 1), 79–84. doi: 10.1590/S0074-02762011000900010
D'Alessandro, U., and Buttiëns, H. (2001). History and importance of antimalarial drug resistance. Trop. Med. Int. Health 6, 845–848. doi: 10.1046/j.1365-3156.2001.00819.x
Dbaibo, G. S., Perry, D. K., Gamard, C. J., Platt, R., Poirier, G. G., Obeid, L. M., et al. (1997). Cytokine Response Modifier A (CrmA) inhibits ceramide formation in response to Tumor Necrosis Factor (TNF)-α: CrmA and Bcl-2 target distinct components in the apoptotic pathway. J. Exp. Med. 185, 481–490.
de Jong, K., Rettig, M. P., Low, P. S., and Kuypers, F. A. (2002). Protein kinase C activation induces phosphatidylserine exposure on red blood cells. Biochemistry 41, 12562–12567. doi: 10.1021/bi025882o
Delbridge, A. R., Aubrey, B. J., Hyland, C., Bernardini, J. P., Rago, L. D., Garnier, J.-M., et al. (2017). The BH3-only proteins BIM and PUMA are not critical for the reticulocyte apoptosis caused by loss of the pro-survival protein BCL-XL. Cell Death Dis. 8:e2914. doi: 10.1038/cddis.2017.304
Doerig, C. (2004). Protein kinases as targets for anti-parasitic chemotherapy. Biochim. Biophys. Acta ProteinsProteomics 1697, 155–168. doi: 10.1016/j.bbapap.2003.11.021.
Eda, S., and Sherman, I. W. (2002). Cytoadherence of malaria-infected red blood cells involves exposure of phosphatidylserine. Cell. Physiol. Biochem. 12, 373–384. doi: 10.1159/000067908
Egan, E. S. (2017). Beyond hemoglobin: screening for malaria host factors. Trends Genet. 34, 133–141. doi: 10.1016/j.tig.2017.11.004
Enomoto, M., Kawazu, S., Kawai, S., Furuyama, W., Ikegami, T., Watanabe, J., et al. (2012). Blockage of spontaneous Ca2+ oscillation causes cell death in intraerythrocitic Plasmodium falciparum. PLoS ONE 7:e39499. doi: 10.1371/journal.pone.0039499
Föller, M., Bobbala, D., Koka, S., Huber, S. M., Gulbins, E., and Lang, F. (2009a). Suicide for survival–death of infected erythrocytes as a host mechanism to survive malaria. Cell. Physiol. Biochem. 24, 133–140. doi: 10.1159/000233238
Föller, M., Feil, S., Ghoreschi, K., Koka, S., Gerling, A., Thunemann, M., et al. (2008). Anemia and splenomegaly in cGKI-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 105, 6771–6776. doi: 10.1073/pnas.0708940105
Föller, M., Sopjani, M., Koka, S., Gu, S., Mahmud, H., Wang, K., et al. (2009b). Regulation of erythrocyte survival by AMP-activated protein kinase. FASEB J. 23, 1072–1080. doi: 10.1096/fj.08-121772
Franco, R. S. (2012). Measurement of red cell lifespan and aging. Transfus Med. Hemother. 39, 302–307. doi: 10.1159/000342232
Gatidis, S., Zelenak, C., Fajol, A., Lang, E., Jilani, K., Michael, D., et al. (2011). p38 MAPK activation and function following osmotic shock of erythrocytes. Cell. Physiol. Biochem. 28, 1279–1286. doi: 10.1159/000335859.
Geiger, C., Föller, M., Herrlinger, K. R., and Lang, F. (2008). Azathioprine-induced suicidal erythrocyte death. Inflamm. Bowel Dis. 14, 1027–1032. doi: 10.1002/ibd.20433
Ghashghaeinia, M., Bobbala, D., Wieder, T., Koka, S., Brück, J., Fehrenbacher, B., et al. (2010). Targeting glutathione by dimethylfumarate protects against experimental malaria by enhancing erythrocyte cell membrane scrambling. Am. J. Physiol. Cell Physiol. 299, C791–C804. doi: 10.1152/ajpcell.00014.2010
Ghashghaeinia, M., Cluitmans, J. C. A., Akel, A., Dreischer, P., Toulany, M., Köberle, M., et al. (2012). The impact of erythrocyte age on eryptosis. Br. J. Haematol. 157, 606–614. doi: 10.1111/j.1365-2141.2012.09100.x
Ghashghaeinia, M., Giustarini, D., Koralkova, P., Köberle, M., Alzoubi, K., Bissinger, R., et al. (2016). Pharmacological targeting of glucose-6-phosphate dehydrogenase in human erythrocytes by Bay 11-7082, parthenolide and dimethyl fumarate. Sci. Rep. 6:28754. doi: 10.1038/srep28754
Ginsburg, H., and Atamna, H. (1994). The redox status of malaria-infected erythrocytes: an overview with an emphasis on unresolved problems. Parasite 1, 5–13. doi: 10.1051/parasite/1994011005
Gregoli, P. A., and Bondurant, M. C. (1997). The roles of Bcl-XL and apopain in the control of erythropoiesis by erythropoietin. Blood 90, 630–640.
Hanada, K., Mitamura, T., Fukasawa, M., Magistrado, P. A., Horii, T., and Nishijima, M. (2000). Neutral sphingomyelinase activity dependent on Mg2+ and anionic phospholipids in the intraerythrocytic malaria parasite Plasmodium falciparum. Biochem. J. 346(Pt 3), 671–677. doi: 10.1042/bj3460671
Hattangadi, S. M., and Lodish, H. F. (2007). Regulation of erythrocyte lifespan: do reactive oxygen species set the clock? J. Clin. Invest. 117, 2075–2077. doi: 10.1172/JCI32559
Hay, S., and Kannourakis, G. (2002). A time to kill: viral manipulation of the cell death program. J. Gen. Virol. 83, 1547–1564. doi: 10.1099/0022-1317-83-7-1547
Head, D. J., Lee, Z. E., Poole, J., and Avent, N. D. (2005a). Expression of phosphatidylserine (PS) on wild-type and Gerbich variant erythrocytes following glycophorin-C (GPC) ligation. Br. J. Haematol. 129, 130–137. doi: 10.1111/j.1365-2141.2005.05407.x
Head, D. J., Lee, Z. E., Swallah, M. M., and Avent, N. D. (2005b). Ligation of CD47 mediates phosphatidylserine expression on erythrocytes and a concomitant loss of viability in vitro. Br. J. Haematol. 130, 788–790. doi: 10.1111/j.1365-2141.2005.05668.x
Heussler, V., Sturm, A., and Langsley, G. (2006). Regulation of host cell survival by intracellular Plasmodium and Theileria parasites. Parasitology 132, S49–S60. doi: 10.1017/S0031182006000850
Ho, M., Davis, T. M., Silamut, K., Bunnag, D., and White, N. J. (1991). Rosette formation of Plasmodium falciparum-infected erythrocytes from patients with acute malaria. Infect. Immun. 59, 2135–2139.
Hockemeyer, D., and Jaenisch, R. (2016). Induced pluripotent stem cells meet genome editing. Cell Stem Cell 18, 573–586. doi: 10.1016/j.stem.2016.04.013
Huber, S. M., Duranton, C., and Lang, F. (2005). Patch-clamp analysis of the “new permeability pathways” in malaria-infected erythrocytes. Int. Rev. Cytol. 246, 59–134. doi: 10.1016/S0074-7696(05)46003-9
Huber, S. M., Gamper, N., and Lang, F. (2001). Chloride conductance and volume-regulatory nonselective cation conductance in human red blood cell ghosts. Pflügers. Arch. Eur. J. Physiol. 441, 551–558. doi: 10.1007/s004240000456
Jakeman, G. N., Saul, A., Hogarth, W. L., and Collins, W. E. (1999). Anaemia of acute malaria infections in non-immune patients primarily results from destruction of uninfected erythrocytes. Parasitology 119 (Pt 2), 127–133.
James, E. R., and Green, D. R. (2004). Manipulation of apoptosis in the host–parasite interaction. Trends Parasitol. 20, 280–287. doi: 10.1016/j.pt.2004.04.004
Jemaà, M., Fezai, M., Bissinger, R., and Lang, F. (2017). Methods employed in cytofluorometric assessment of eryptosis, the suicidal erythrocyte death. Cell. Physiol. Biochem. 43, 431–444. doi: 10.1159/000480469
Jiménez-Díaz, M. B., Ebert, D., Salinas, Y., Pradhan, A., Lehane, A. M., Myrand-Lapierre, M.-E., et al. (2014). (+)-SJ733, a clinical candidate for malaria that acts through ATP4 to induce rapid host-mediated clearance of Plasmodium. Proc. Natl. Acad. Sci. U.S.A. 111, E5455–E5462. doi: 10.1073/pnas.1414221111
John von Freyend, S., Kwok-Schuelein, T., Netter, H. J., Haqshenas, G., Semblat, J.-P., and Doerig, C. (2017). Subverting host cell P21-activated kinase: a case of convergent evolution across pathogens. Pathogens 6:E17. doi: 10.3390/pathogens6020017
Joshi, P., Dutta, G. P., and Gupta, C. M. (1987). An intracellular simian malarial parasite (Plasmodium knowlesi) induces stage-dependent alterations in membrane phospholipid organization of its host erythrocyte. Biochem. J. 246, 103–108.
Kakani, P., Suman, S., Gupta, L., and Kumar, S. (2016). Ambivalent outcomes of cell apoptosis: a barrier or blessing in malaria progression. Front. Microbiol. 7:302. doi: 10.3389/fmicb.2016.00302
Kanjee, U., Grüring, C., Chaand, M., Lin, K.-M., Egan, E., Manzo, J., et al. (2017). CRISPR/Cas9 knockouts reveal genetic interaction between strain-transcendent erythrocyte determinants of Plasmodium falciparum invasion. Proc. Natl. Acad. Sci. U.S.A. 114, E9356–E9365. doi: 10.1073/pnas.1711310114.
Kasinathan, R. S., Föller, M., Koka, S., Huber, S. M., and Lang, F. (2007). Inhibition of eryptosis and intraerythrocytic growth of Plasmodium falciparum by flufenamic acid. Naunyn. Schmiedebergs Arch. Pharmacol. 374, 255–264. doi: 10.1007/s00210-006-0122-x
Kaushansky, A., Metzger, P. G., Douglass, A. N., Mikolajczak, S. A., Lakshmanan, V., Kain, H. S., et al. (2013). Malaria parasite liver stages render host hepatocytes susceptible to mitochondria-initiated apoptosis. Cell Death Dis. 4:e762. doi: 10.1038/cddis.2013.286
Kempe, D. S., Lang, P. A., Eisele, K., Klarl, B. A., Wieder, T., Huber, S. M., et al. (2005). Stimulation of erythrocyte phosphatidylserine exposure by lead ions. Am. J. Physiol. Cell Physiol. 288, C396–C402. doi: 10.1152/ajpcell.00115.2004
Kirk, K. (2001). Membrane transport in the malaria-infected erythrocyte. Physiol. Rev. 81, 495–537. doi: 10.1152/physrev.2001.81.2.495
Klarl, B. A., Lang, P. A., Kempe, D. S., Niemoeller, O. M., Akel, A., Sobiesiak, M., et al. (2006). Protein kinase C mediates erythrocyte “programmed cell death” following glucose depletion. Am. J. Phys. Cell Physiol. 290, C244–C253. doi: 10.1152/ajpcell.00283.2005
Koch, M., and Baum, J. (2016). The mechanics of malaria parasite invasion of the human erythrocyte – towards a reassessment of the host cell contribution. Cell Microbiol. 18, 319–329. doi: 10.1111/cmi.12557
Koka, S., Bobbala, D., Lang, C., Boini, K. M., Huber, S. M., and Lang, F. (2009). Influence of paclitaxel on parasitemia and survival of Plasmodium berghei infected mice. Cell. Physiol. Biochem. 23, 191–198. doi: 10.1159/000204107
Koka, S., Huber, S. M., Boini, K. M., Lang, C., Föller, M., and Lang, F. (2007). Lead decreases parasitemia and enhances survival of Plasmodium berghei-infected mice. Biochem. Biophys. Res. Commun. 363, 484–489. doi: 10.1016/j.bbrc.2007.08.173
Koka, S., Lang, C., Boini, K. M., Bobbala, D., Huber, S. M., and Lang, F. (2008a). Influence of chlorpromazine on eryptosis, parasitemia and survival of Plasmodium berghe infected mice. Cell. Physiol. Biochem. 22, 261–268. doi: 10.1159/000149804
Koka, S., Lang, C., Niemoeller, O. M., Boini, K. M., Nicolay, J. P., Huber, S. M., et al. (2008b). Influence of NO synthase inhibitor L-NAME on parasitemia and survival of Plasmodium berghei infected mice. Cell. Physiol. Biochem. 21, 481–488. doi: 10.1159/000129641
Kucherenko, Y., Zelenak, C., Eberhard, M., Qadri, S. M., and Lang, F. (2012). Effect of casein kinase 1α activator pyrvinium pamoate on erythrocyte ion channels. Cell. Physiol. Biochem. 30, 407–417. doi: 10.1159/000339034
Lang, E., Bissinger, R., Fajol, A., Salker, M. S., Singh, Y., Zelenak, C., et al. (2015a). Accelerated apoptotic death and in vivo turnover of erythrocytes in mice lacking functional mitogen- and stress-activated kinase MSK1/2. Sci. Rep. 5:17316. doi: 10.1038/srep17316
Lang, E., Bissinger, R., Gulbins, E., and Lang, F. (2015b). Ceramide in the regulation of eryptosis, the suicidal erythrocyte death. Apoptosis 20, 758–767. doi: 10.1007/s10495-015-1094-4
Lang, E., and Lang, F. (2015). Triggers, inhibitors, mechanisms, and significance of eryptosis: the suicidal erythrocyte death. Bio Med. Res. Int. 2015:e513518. doi: 10.1155/2015/513518
Lang, F., Abed, M., Lang, E., and Föller, M. (2014). Oxidative stress and suicidal erythrocyte death. Antioxid. Redox Signal. 21, 138–153. doi: 10.1089/ars.2013.5747
Lang, F., and Föller, M. (2012). Erythrocytes: Physiology and Pathophysiology. Basel: World Scientific Publishing Company.
Lang, K., Lang, P., Bauer, C., Duranton, C., Wieder, T., Huber, S., et al. (2005). Mechanisms of suicidal erythrocyte death. CPB 15, 195–202. doi: 10.1159/000086406
Lang, K. S., Duranton, C., Poehlmann, H., Myssina, S., Bauer, C., Lang, F., et al. (2003). Cation channels trigger apoptotic death of erythrocytes. Cell Death Differ. 10, 249–256. doi: 10.1038/sj.cdd.4401144
Lang, K. S., Myssina, S., Brand, V., Sandu, C., Lang, P. A., Berchtold, S., et al. (2004). Involvement of ceramide in hyperosmotic shock-induced death of erythrocytes. Cell Death Differ. 11, 231–243. doi: 10.1038/sj.cdd.4401311
Lang, K. S., Roll, B., Myssina, S., Schittenhelm, M., Scheel-Walter, H.-G., Kanz, L., et al. (2002). Enhanced erythrocyte apoptosis in sickle cell anemia, thalassemia and glucose-6-phosphate dehydrogenase deficiency. Cell. Physiol. Biochem. 12, 365–372. doi: 10.1159/000067907
Lang, P. A., Beringer, O., Nicolay, J. P., Amon, O., Kempe, D. S., Hermle, T., et al. (2006a). Suicidal death of erythrocytes in recurrent hemolytic uremic syndrome. J. Mol. Med. 84, 378–388. doi: 10.1007/s00109-006-0058-0
Lang, P. A., Huober, J., Bachmann, C., Kempe, D. S., Sobiesiak, M., Akel, A., et al. (2006b). Stimulation of erythrocyte phosphatidylserine exposure by paclitaxel. Cell. Physiol. Biochem. 18, 151–164. doi: 10.1159/000095190
Lang, P. A., Kaiser, S., Myssina, S., Wieder, T., Lang, F., and Huber, S. M. (2003). Role of Ca2+-activated K+ channels in human erythrocyte apoptosis. Am. J. Physiol., Cell Physiol. 285, C1553–C1560. doi: 10.1152/ajpcell.00186.2003
Lang, P. A., Kempe, D. S., Tanneur, V., Eisele, K., Klarl, B. A., Myssina, S., et al. (2005). Stimulation of erythrocyte ceramide formation by platelet-activating factor. J. Cell Sci. 118, 1233–1243. doi: 10.1242/jcs.01730
Lange, P. F., Huesgen, P. F., Nguyen, K., and Overall, C. M. (2014). Annotating N termini for the human proteome project: N termini and Nα-acetylation status differentiate stable cleaved protein species from degradation remnants in the human erythrocyte proteome. J. Proteome Res. 13, 2028–2044. doi: 10.1021/pr401191w
Lapillonne, H., Kobari, L., Mazurier, C., Tropel, P., Giarratana, M.-C., Zanella-Cleon, I., et al. (2010). Red blood cell generation from human induced pluripotent stem cells: perspectives for transfusion medicine. Haematologica 95, 1651–1659. doi: 10.3324/haematol.2010.023556
Larsen, A. K., Lametsch, R., Elce, J. S., Larsen, J. K., Thomsen, B., Larsen, M. R., et al. (2008). Genetic disruption of calpain correlates with loss of membrane blebbing and differential expression of RhoGDI-1, cofilin and tropomyosin. Biochem. J. 411, 657–666. doi: 10.1042/BJ20070522
Leventis, P. A., and Grinstein, S. (2010). The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 39, 407–427. doi: 10.1146/annurev.biophys.093008.131234.
Lew, V. L., Tiffert, T., and Ginsburg, H. (2003). Excess hemoglobin digestion and the osmotic stability of Plasmodium falciparum-infected red blood cells. Blood 101, 4189–4194. doi: 10.1182/blood-2002-08-2654
Lu, S. C. (2009). Regulation of glutathione synthesis. Mol. Aspects Med. 30, 42–59. doi: 10.1016/j.mam.2008.05.005
Lupescu, A., Shaik, N., Jilani, K., Zelenak, C., Lang, E., Pasham, V., et al. (2012). Enhanced erythrocyte membrane exposure of phosphatidylserine following sorafenib treatment: an in vivo and in vitro study. CPB 30, 876–888. doi: 10.1159/000341465
Lutz, H. U. (2004). Innate immune and non-immune mediators of erythrocyte clearance. Cell. Mol. Biol. 50, 107–116.
Lutz, H. U., and Bogdanova, A. (2013). Mechanisms tagging senescent red blood cells for clearance in healthy humans. Front. Physiol. 4:387. doi: 10.3389/fphys.2013.00387
Maguire, P. A., and Sherman, I. W. (1990). Phospholipid composition, cholesterol content and cholesterol exchange in Plasmodium falciparum-infected red cells. Mol. Biochem. Parasitol. 38, 105–112.
Mahmud, H., Mauro, D., Qadri, S. M., Föller, M., and Lang, F. (2009). Triggering of suicidal erythrocyte death by amphotericin B. Cell. Physiol. Biochem. 24, 263–270. doi: 10.1159/000233251.
Maier, A. G., Duraisingh, M. T., Reeder, J. C., Patel, S. S., Kazura, J. W., Zimmerman, P. A., et al. (2003). Plasmodium falciparum erythrocyte invasion through glycophorin C and selection for Gerbich negativity in human populations. Nat. Med. 9, 87–92. doi: 10.1038/nm807
Mandal, D., Mazumder, A., Das, P., Kundu, M., and Basu, J. (2005). Fas-, caspase 8-, and caspase 3-dependent signaling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J. Biol. Chem. 280, 39460–39467. doi: 10.1074/jbc.M506928200
Matallanas, D., Birtwistle, M., Romano, D., Zebisch, A., Rauch, J., von Kriegsheim, A., et al. (2011). Raf family kinases. Genes Cancer 2, 232–260. doi: 10.1177/1947601911407323
Matarrese, P., Straface, E., Pietraforte, D., Gambardella, L., Vona, R., Maccaglia, A., et al. (2005). Peroxynitrite induces senescence and apoptosis of red blood cells through the activation of aspartyl and cysteinyl proteases. FASEB J. 19, 416–418. doi: 10.1096/fj.04-2450fje
McClure, N. S., and Day, T. (2014). A theoretical examination of the relative importance of evolution management and drug development for managing resistance. Proc. R. Soc. Lond. B Biol. Sci. 281:20141861. doi: 10.1098/rspb.2014.1861
McEvoy, L., Williamson, P., and Schlegel, R. A. (1986). Membrane phospholipid asymmetry as a determinant of erythrocyte recognition by macrophages. Proc. Natl. Acad. Sci. U.S.A. 83, 3311–3315.
Menard, D., and Dondorp, A. (2017). Antimalarial drug resistance: a threat to malaria elimination. Cold. Spring Harb. Perspect Med. 7:a025619. doi: 10.1101/cshperspect.a025619
Mischitelli, M., Jema,à, M., Almasry, M., Faggio, C., and Lang, F. (2016). Stimulation of erythrocyte cell membrane scrambling by Quinine. Cell. Physiol. Biochem. 40, 657–667. doi: 10.1159/000452578
Mochly-Rosen, D., Das, K., and Grimes, K. V. (2012). Protein kinase C, an elusive therapeutic target? Nat. Rev. Drug Dis. 11, 937–957. doi: 10.1038/nrd3871
Murachi, T., Tanaka, K., Hatanaka, M., and Murakami, T. (1981). Intracellular Ca2+-dependent protease (CALPAIN) and its high-molecular-weight endogenous inhibitor (CALPASTATIN). Adv. Enzy. Regul. 19, 407–424. doi: 10.1016/0065-2571(81)90026-1
Nicolay, J. P., Bentzen, P. J., Ghashghaeinia, M., Wieder, T., and Lang, F. (2007). Stimulation of erythrocyte cell membrane scrambling by amiodarone. Cell. Physiol. Biochem. 20, 1043–1050. doi: 10.1159/000110713
Nicolay, J. P., Liebig, G., Niemoeller, O. M., Koka, S., Ghashghaeinia, M., Wieder, T., et al. (2008). Inhibition of suicidal erythrocyte death by nitric oxide. Pflüg. Arch. Eur. J. Physiol. 456, 293–305. doi: 10.1007/s00424-007-0393-1
Niemoeller, O. M., Akel, A., Lang, P. A., Attanasio, P., Kempe, D. S., Hermle, T., et al. (2006). Induction of eryptosis by cyclosporine. Naunyn. Schmiedebergs Arch. Pharmacol. 374, 41–49. doi: 10.1007/s00210-006-0099-5
Nosaka, T., van Deursen, J. M., Tripp, R. A., Thierfelder, W. E., Witthuhn, B. A., McMickle, A. P., et al. (1995). Defective lymphoid development in mice lacking Jak3. Science 270, 800–802.
Oldenborg, P.-A., Zheleznyak, A., Fang, Y.-F., Lagenaur, C. F., Gresham, H. D., and Lindberg, F. P. (2000). Role of CD47 as a marker of self on red blood cells. Science 288, 2051–2054. doi: 10.1126/science.288.5473.2051
Pathak, V. A., and Ghosh, K. (2016). Erythropoiesis in malaria infections and factors modifying the erythropoietic response. Anemia 2016:e9310905. doi: 10.1155/2016/9310905
Perrin, B. J., and Huttenlocher, A. (2002). Calpain. Int. J. Biochem. Cell Biol. 34, 722–725. doi: 10.1016/S1357-2725(02)00009-2.
Pretorius, E., Plooy, J. N., and du, Bester J. (2016). A comprehensive review on eryptosis. CPB 39, 1977–2000. doi: 10.1159/000447895
Ramphul, U. N., Garver, L. S., Molina-Cruz, A., Canepa, G. E., and Barillas-Mury, C. (2015). Plasmodium falciparum evades mosquito immunity by disrupting JNK-mediated apoptosis of invaded midgut cells. Proc. Natl. Acad. Sci. U.S.A. 112, 1273–1280. doi: 10.1073/pnas.1423586112
Rane, C. K., and Minden, A. (2014). P21 activated kinases. Small GTPases 5:e28003. doi: 10.4161/sgtp.28003
Rawlings, J. S., Rosler, K. M., and Harrison, D. A. (2004). The JAK/STAT signaling pathway. J. Cell Sci. 117, 1281–1283. doi: 10.1242/jcs.00963
Roux-Dalvai, F., Gonzalez de Peredo, A., Sim,ó, C., Guerrier, L., Bouyssié, D., Zanella, A., et al. (2008). Extensive analysis of the cytoplasmic proteome of human erythrocytes using the peptide ligand library technology and advanced mass spectrometry. Mol. Cell Proteomics 7, 2254–2269. doi: 10.1074/mcp.M800037-MCP200
Schaer, D. J., Buehler, P. W., Alayash, A. I., Belcher, J. D., and Vercellotti, G. M. (2013). Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121, 1276–1284. doi: 10.1182/blood-2012-11-451229
Schieber, M., and Chandel, N. S. (2014). ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462. doi: 10.1016/j.cub.2014.03.034
Schittek, B., and Sinnberg, T. (2014). Biological functions of casein kinase 1 isoforms and putative roles in tumorigenesis. Mol Cancer 13:231. doi: 10.1186/1476-4598-13-231
Schoenwaelder, S. M., Yuan, Y., Josefsson, E. C., White, M. J., Yao, Y., Mason, K. D., et al. (2009). Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood 114, 663–666. doi: 10.1182/blood-2009-01-200345
Schulze-Osthoff, K., Ferrari, D., Los, M., Wesselborg, S., and Peter, M. E. (2001). Apoptosis signaling by death receptors. Eur. J. Biochem. 254, 439–459. doi: 10.1046/j.1432-1327.1998.2540439.x
Schwartz, R. S., Olson, J. A., Raventos-Suarez, C., Yee, M., Heath, R. H., Lubin, B., et al. (1987). Altered plasma membrane phospholipid organization in Plasmodium falciparum-infected human erythrocytes. Blood 69, 401–407.
Schwarz-Ben Meir, N., Glaser, T., and Kosower, N. S. (1991). Band 3 protein degradation by calpain is enhanced in erythrocytes of old people. Biochem. J. 275 (Pt 1), 47–52.
Segawa, K., and Nagata, S. (2015). An apoptotic ‘Eat Me' signal: phosphatidylserine exposure. Trends Cell Biol. 25, 639–650. doi: 10.1016/j.tcb.2015.08.003.
Setty, B. N. Y., Kulkarni, S., and Stuart, M. J. (2002). Role of erythrocyte phosphatidylserine in sickle red cell-endothelial adhesion. Blood 99, 1564–1571. doi: 10.1182/blood.V99.5.1564
Sharom, F. J. (2011). Flipping and flopping–lipids on the move. IUBMB Life 63, 736–746. doi: 10.1002/iub.515
Sicard, A., Semblat, J.-P., Doerig, C., Hamelin, R., Moniatte, M., Dorin-Semblat, D., et al. (2011). Activation of a PAK-MEK signalling pathway in malaria parasite-infected erythrocytes. Cell Microbiol. 13, 836–845. doi: 10.1111/j.1462-5822.2011.01582.x
Siraskar, B., Ballal, A., Bobbala, D., Föller, M., and Lang, F. (2010). Effect of amphotericin B on parasitemia and survival of plasmodium berghei-infected mice. Cell. Physiol. Biochem. 26, 347–354. doi: 10.1159/000320558
Sopjani, M., Föller, M., and Lang, F. (2008). Gold stimulates Ca2+ entry into and subsequent suicidal death of erythrocytes. Toxicology 244, 271–279. doi: 10.1016/j.tox.2007.12.001
Sturm, A., Amino, R., van de Sand, C., Regen, T., Retzlaff, S., Rennenberg, A., et al. (2006). Manipulation of host hepatocytes by the malaria parasite for delivery into liver sinusoids. Science 313, 1287–1290. doi: 10.1126/science.1129720
Suzuki, J., Fujii, T., Imao, T., Ishihara, K., Kuba, H., and Nagata, S. (2013). Calcium-dependent phospholipid scramblase activity of TMEM16 protein family members. J. Biol. Chem. 288, 13305–13316. doi: 10.1074/jbc.M113.457937
Suzuki, J., Umeda, M., Sims, P. J., and Nagata, S. (2010). Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838. doi: 10.1038/nature09583
Tang, H., Ho, H., Wu, P., Chen, S., Kuypers, F. A., Cheng, M., et al. (2015). Inability to maintain GSH pool in G6PD-deficient red cells causes futile AMPK activation and irreversible metabolic disturbance. Antioxid. Redox. Signal. 22, 744–759. doi: 10.1089/ars.2014.6142
Tanner, M. J. (1993). The major integral proteins of the human red cell. Baillieres Clin. Haematol. 6, 333–356.
Tiffert, T., Staines, H. M., Ellory, J. C., and Lew, V. L. (2000). Functional state of the plasma membrane Ca2+ pump in Plasmodium falciparum-infected human red blood cells. J. Physiol. 525, 125–134. doi: 10.1111/j.1469-7793.2000.00125.x
Tilley, L., Straimer, J., Gnädig, N. F., Ralph, S. A., and Fidock, D. A. (2016). Artemisinin action and resistance in Plasmodium falciparum. Trends Parasitol. 32, 682–696. doi: 10.1016/j.pt.2016.05.010
Tolosano, E., Hirsch, E., Patrucco, E., Camaschella, C., Navone, R., Silengo, L., et al. (1999). Defective recovery and severe renal damage after acute hemolysis in hemopexin-deficient mice. Blood 94, 3906–3914.
Totino, P. R., and Lopes, S. C. (2017). Insights into the cytoadherence phenomenon of Plasmodium vivax: the putative role of phosphatidylserine. Front. Immunol. 8:1148. doi: 10.3389/fimmu.2017.01148
Totino, P. R., Magalhaes, A. D., Silva, L. A., Banic, D. M., Daniel-Ribeiro, C. T., and Ferreira-da-Cruz Mde, F (2010). Apoptosis of non-parasitized red blood cells in malaria: a putative mechanism involved in the pathogenesis of anaemia. Malar. J. 9:350. doi: 10.1186/1475-2875-9-350
Totino, P. R. R., Daniel-Ribeiro, C. T., Ferreira-da-Cruz, M., and de, F (2016). Evidencing the role of erythrocytic apoptosis in malarial anemia. Front. Cell Infect. Microbiol. 6:176. doi: 10.3389/fcimb.2016.00176
Totino, P. R. R., Magalhães, A. D., Alves, E. B., Costa, M. R. F., de Lacerda, M. V. G., Daniel-Ribeiro, C. T., et al. (2014). Plasmodium falciparum, but not P. vivax, can induce erythrocytic apoptosis. Parasit. Vectors 7:484. doi: 10.1186/s13071-014-0484-8
Totino, P. R. R., Pinna, R. A., De-Oliveira, A. C. A. X., Banic, D. M., Daniel-Ribeiro, C. T., and Ferreira-da-Cruz Mde, F. (2013). Apoptosis of non-parasitised red blood cells in Plasmodium yoelii malaria. Mem. Inst. Oswaldo Cruz 108, 686–690. doi: 10.1590/0074-0276108062013003
van de Sand, C., Horstmann, S., Schmidt, A., Sturm, A., Bolte, S., Krueger, A., et al. (2005). The liver stage of Plasmodium berghei inhibits host cell apoptosis. Mol. Microbiol. 58, 731–742. doi: 10.1111/j.1365-2958.2005.04888.x
Wali, R. K., Jaffe, S., Kumar, D., and Kalra, V. K. (1988). Alterations in organization of phospholipids in erythrocytes as factor in adherence to endothelial cells in diabetes mellitus. Diabetes 37, 104–111.
Walsh, M., Lutz, R. J., Cotter, T. G., and O'Connor, R. (2002). Erythrocyte survival is promoted by plasma and suppressed by a Bak-derived BH3 peptide that interacts with membrane-associated Bcl-XL. Blood 99, 3439–3448. doi: 10.1182/blood.V99.9.3439.
Wang, S., Dale, G. L., Song, P., Viollet, B., and Zou, M. (2010). AMPKα1 deletion shortens erythrocyte life span in mice. J. Biol. Chem. 285, 19976–19985. doi: 10.1074/jbc.M110.102467
Wasserman, M., Alarcón, C., and Mendoza, P. M. (1982). Effects of Ca++ depletion on the asexual cell cycle of Plasmodium falciparum. Am. J. Trop. Med. Hyg. 31, 711–717.
Wautier, M.-P., Héron, E., Picot, J., Colin, Y., Hermine, O., and Wautier, J.-L. (2011). Red blood cell phosphatidylserine exposure is responsible for increased erythrocyte adhesion to endothelium in central retinal vein occlusion. J. Thromb. Haemost. 9, 1049–1055. doi: 10.1111/j.1538-7836.2011.04251.x
WHO|Global Technical Strategy for Malaria (2016–2030). WHO. Available online at: http://www.who.int/malaria/areas/global_technical_strategy/en/ (Accessed May 1, 2018).
WHO|World Malaria Report (2017). WHO. Available online at: http://www.who.int/malaria/publications/world-malaria-report-2017/report/en/ (Accessed February 12, 2018).
Wiggin, G. R., Soloaga, A., Foster, J. M., Murray-Tait, V., Cohen, P., and Arthur, J. S. C. (2002). MSK1 and MSK2 are required for the mitogen- and stress-induced phosphorylation of CREB and ATF1 in fibroblasts. Mol. Cell. Biol. 22, 2871–2881. doi: 10.1128/MCB.22.8.2871-2881.2002
Wilhelm, S. M., Carter, C., Tang, L., Wilkie, D., McNabola, A., Rong, H., et al. (2004). BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 64, 7099–7109. doi: 10.1158/0008-5472.CAN-04-1443
Wu, G., Fang, Y.-Z., Yang, S., Lupton, J. R., and Turner, N. D. (2004). Glutathione metabolism and its implications for health. J. Nutr. 134, 489–492. doi: 10.1093/jn/134.3.489
Zappulla, D. (2008). Environmental stress, erythrocyte dysfunctions, inflammation, and the metabolic syndrome: adaptations to CO2 increases? J. Cardiometab Syndr. 3, 30–34. doi: 10.1111/j.1559-4572.2008.07263.x
Zarchin, S., Krugliak, M., and Ginsburg, H. (1986). Digestion of the host erythrocyte by malaria parasites is the primary target for quinoline-containing antimalarials. Biochem. Pharmacol. 35, 2435–2442.
Zarubin, T., and Han, J. (2005). Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18. doi: 10.1038/sj.cr.7290257
Zelenak, C., Eberhard, M., Jilani, K., Qadri, S. M., Macek, B., and Lang, F. (2012). Protein kinase CK1α regulates erythrocyte survival. Cell. Physiol. Biochem. 29, 171–180. doi: 10.1159/000337598
Keywords: malaria, eryptosis, Plasmodium, apoptosis, programmed cell death, host-pathogen interaction, host-directed therapy
Citation: Boulet C, Doerig CD and Carvalho TG (2018) Manipulating Eryptosis of Human Red Blood Cells: A Novel Antimalarial Strategy? Front. Cell. Infect. Microbiol. 8:419. doi: 10.3389/fcimb.2018.00419
Received: 04 September 2018; Accepted: 14 November 2018;
Published: 30 November 2018.
Edited by:
Alexis Kaushansky, Seattle Children's Research Institute, United StatesReviewed by:
Marisa Ponpuak, Mahidol University, ThailandAnat Florentin, University of Georgia, United States
Copyright © 2018 Boulet, Doerig and Carvalho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa G. Carvalho, dC5jYXJ2YWxob0BsYXRyb2JlLmVkdS5hdQ==