
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Cell. Infect. Microbiol. , 14 August 2018
Sec. Clinical Microbiology
Volume 8 - 2018 | https://doi.org/10.3389/fcimb.2018.00275
This article is part of the Research Topic Towards Host-Directed Drug Therapies for Infectious and Non-Communicable Diseases View all 11 articles
 Lu Huang1
Lu Huang1 Nicole L. Kushner1
Nicole L. Kushner1 Monique E. Theriault1Davide Pisu1Shumin Tan2Case W. McNamara3
Monique E. Theriault1Davide Pisu1Shumin Tan2Case W. McNamara3 H. Mike Petrassi3
H. Mike Petrassi3 David G. Russell1*
David G. Russell1* Amanda C. Brown1
Amanda C. Brown1Mycobacterium tuberculosis (Mtb) continues to be a threat to Global Public Health, and its control will require an array of therapeutic strategies. It has been appreciated that high-throughput screens using cell-based assays to identify compounds targeting Mtb within macrophages represent a valuable tool for drug discovery. However, the host immune environment, in the form of lymphocytes and cytokines, is completely absent in a chemical screening platform based on infected macrophages alone. The absence of these players unnecessarily limits the breadth of novel host target pathways to be interrogated. In this study, we detail a new drug screening platform based on dissociated murine TB granulomas, named the Deconstructed Granuloma (DGr), that utilizes fluorescent Mtb reporter strains screened in the host immune environment of the infection site. The platform has been used to screen a collection of known drug candidates. Data from a representative 384-well plate containing known anti-bacterial compounds are shown, illustrating the robustness of the screening platform. The novel deconstructed granuloma platform represents an accessible, sensitive and robust high-throughput screen suitable for the inclusive interrogation of immune targets for Host-Directed Therapeutics.
Mycobacterium tuberculosis, the causative agent of human tuberculosis (TB), poses an ongoing threat to human health. The escalating incidence of drug resistant strains provides a new impetus to understand this complex pathogen and to identify novel therapeutics. Ginsberg and Spigelman (Ginsberg and Spigelman, 2007) list the goals and challenges for improved TB therapy, one of which is to “discover and develop new drugs that have novel mechanisms of action and are effective against persistent bacilli,” as well as to “develop new preclinical approaches to identify optimized drug combinations….” As a first step toward these aims, we have directed our efforts to developing improved drug screening tools. Traditional drug discovery programs for the identification of new anti-microbial compounds have focused on either target-based screening against enzymatic activities that have been shown genetically to be essential for bacterial survival (Mdluli and Spigelman, 2006; Lamichhane, 2011), or phenotypic screens exploring bacterial survival in rich broth (Collins and Franzblau, 1997; Pethe et al., 2010). Both approaches have had limited success in recent times, calling into question whether we should continue to devote our major efforts down these paths. Whilst complicated, the most frequently cited reasons for the failure of these approaches are limited drug permeability in the former approach, and the inappropriate nature of rich bacterial broth to mirror the physiology within the host in the latter approach (Koul et al., 2011; VanderVen et al., 2015; Zuniga et al., 2015).
There is also the sense that the “low-hanging fruit” have already been picked, and compounds with known modes of action continue to re-emerge from ongoing screens. For these reasons we feel that it is critical to develop new, innovative approaches to drug discovery that explore and exploit the host-derived pressures on Mycobacterium tuberculosis (Mtb) to identify compounds with enhanced efficacy within the host.
Host-direct therapy (HDT) is an emerging theme in the treatment of infectious diseases; small molecules are utilized to modulate or perturb host responses, to reduce replication or persistence of pathogens, to limit tissue damage, and to enhance the efficacy of current treatments (Kim and Yang, 2017; Kolloli and Subbian, 2017). Current HDT approaches for Mtb target an array of pathways, including: disintegration of granuloma structure, autophagy, anti-inflammatory response, and, more recently, checkpoint blockage to unleash the immune suppression induced by Mtb infection in the lung (Kolloli and Subbian, 2017; Kaufmann et al., 2018). However, our knowledge of immune protection against TB is heavily reliant on studies based on failed immunity in genetically-modified animals and catastrophic human genetic lesions, and this has likely limited the development of both vaccines and new host-dependent therapies (Huang and Russell, 2017). Recent work has indicated that, in addition to immune-mediated control, host macrophage ontogeny also plays a major role in expansion of bacterial numbers and disease progression (Huang et al., 2018). Furthermore, the efficacy of existing anti-tuberculosis drugs in vivo is markedly different from their in vitro activity, and is impacted by the immune status of the host tissue and cellular environment (Liu et al., 2016; Russell, 2017). These layers of host-dependent complexity represent an increased challenge to development of effective new drugs, but they also represent an un-mined opportunity if we can develop appropriate in vitro screens that incorporate this biology. Therefore, to accelerate anti-TB drugs and HDT discovery, we need a discovery platform with minimal assumptions that provides a more holistic representation of the complex biology of the infected host.
With this aim in mind we have developed a high throughput screening (HTS) assay utilizing cells recovered from Mtb-infected mouse lungs (Deconstructed Granuloma, DGr), which we are using to screen compounds with efficacy against Mtb within the context of host-derived immune cells. These cells, which comprise mainly of resident alveolar macrophages, monocyte-derived macrophages, neutrophils and T- and B-lymphocytes, have been recruited and educated by the infection environment and therefore reflect the host immune pressures active within the TB granuloma. The DGr platform has been adapted to an HTS-compatible, 384-well format and generates a robust reproducible readout with an extensive dynamic range. We believe that this novel HTS platform will reveal new avenues toward the manipulation of the host immune environment to restrict bacterial growth and survival, and to act in concert with existing drug treatments and increase their in vivo potency.
C57BL/6 mice were purchased from The Jackson Laboratory. Mice were used at 6–8 week old. All mice were maintained in a specific pathogen-free biosafety level-3 facility at Cornell University. Animal care was in accordance with the guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care. All animal procedures were approved by the Institutional Animal Care and Use Committee of Cornell University.
Mtb strains were cultured in Middlebrook 7H9 liquid medium supplemented with 10% v/v OADC (oleic acid, bovine serum albumin, D-glucose, catalase, sodium chloride; Becton Dickinson) and 0.05% w/v Tyloxapol, or on solid Middlebrook 7H10 agar supplemented with 10% v/v OADC at 37°C, with 25 μg/ml kanamycin or 30 μg/ml hygromycin B where appropriate. Cultures for HTS were grown for 5 days in 100 ml roller cultures (7H9/OADC/Tyloxapol).
The Erdman background of Mtb was used to generate all reporter strains, this is a reference strain commonly used for studies of pathogenesis. We chose to use two different fluorescent reporter strains for the initial infection (mCherry) and the ex-vivo challenge (mKO) to allow us to identify cells infected from each of the separate infections. We found that after incubation, following the ex-vivo challenge, the intensity of mKO signal increased, however the mCherry readings remained constant. We favor mKO as a reporter for the final plate assay over mCherry because it has a greater range/sensitivity and a lower background. (1) mCherry strain: For mouse infection we used a smyc'::mCherry strain, which has been previously described (Tan et al., 2013); mice were inoculated intranasally with ~1,000 CFU of Erdman(smyc'::mCherry) in 30 μl PBS containing 0.05% Tween 80. The inoculum dosage was confirmed by plating different dilutions on 7H10 plates. Plates were incubated at 37°C and colonies enumerated after 3 weeks. (2) monomeric Kusabira Orange (mKO) strain: For the pilot HTS screen we created a tetracycline (tet) inducible mKO Mtb reporter strain, Erdman(P606'::mKO-tetON), by introduction of a replicating plasmid containing the tet-ON regulator upstream of P606 driven mKO, in the destination Gateway vector pDE43-MEK. The fluorescent protein Kusabira Orange was originally isolated from the stony coral Fungia concinna (Karasawa et al., 2004), and the mKO derivative used here was codon-optimized for Mtb by GenScript. (3) Dual mCherry-mKO strain: For imaging purposes we generated a dual reporter strain, Erdman(P606'::mKO-tetON, smyc'::mCherry), which constitutively expressed the mCherry fluorescent protein under the smyc promoter (Tan et al., 2013), and also inducibly expressed mKO upon the addition of exogenous tetracycline, on the replicating Gateway plasmid pDE43-MEK backbone.
BMDM were isolated from C57BL/6J mice and cultured in DMEM (Corning Cellgro) supplemented with 10% FBS (Thermo Scientific), 20% L929-conditioned media, 2 mM L-glutamine, 1 mM sodium pyruvate, and 1% penicillin/streptomycin (Corning Cellgro) at 37°C, 5% CO2, for 9 days prior to seeding for experiments. J774 cells were obtained from ATCC. Cells were maintained in DMEM +10% FBS + 2 mM L-glutamine + 1 mM sodium pyruvate (complete DMEM) + 1% penicillin/streptomycin at 37°C, 5% CO2, until 90% confluence, in T300 flasks. Cells were scraped from the flasks into cold, sterile PBS, centrifuged at 700 rpm for 10 mins, and re-suspended at 1 x 106/ml in complete DMEM. 30 μl of the cell suspension was then seeded into each well of 384-well optically clear, black plates (~3 × 104 cells/well) and incubated overnight at 37°C, 5% CO2.
Mice were infected with Mtb, and sacrificed after 3 weeks. In the actual drug screen we process and pool cells from 5 infected mice to generate 6 × 384 well plates. The pooling of the granuloma cells minimizes variation and renders the screen much more reproducible from run to run by limiting mouse-to-mouse variation. Mouse lungs lungs were removed and placed into Miltenyi GentleMACS C-tubes, containing 5 ml lung dissociation buffer (5% FBS/PBS solution containing 250 U/ml collagenase IV and 20 U/ml DNase). Lungs were processed using the GentleMACS Lung 37C_m_LDK_1 program, with 1 set of lungs per tube. Cells were then pooled and re-suspended in 10 ml 5% FBS/PBS per set of lungs, passed through a 70 μm cell strainer, and then pelleted at 1,200 rpm for 10 min. 3 ml of ACK buffer (Lonza) was used to re-suspend the cell pellet and incubated at room temperature for 5 min to lyse red blood cells, following which 10 ml 5% FBS/PBS was added and the cell suspension passed through a 40 μm cell strainer. After a final spin at 1,200 rpm for 10 min the resulting pellet was re-suspended in 20 ml complete DMEM and cell counts performed. The cells were diluted to 3.33 × 106/ml in complete DMEM with Amphotericin B (final concentration 20 μg/ml). 30 μl of cells were seeded into each well of a 384 well optically clear, black plates (~105 cells/well). Lung cells were then followed by a secondary ex vivo infection with a desired fluorescent Mtb reporter strain at MOI 0.1 as detailed below.
Test drugs were arrayed from a 2 mM stock solution in DMSO via acoustic dispense (ECHO 550 liquid handler; Labcyte Inc.) into 384-well Axygen P-384-120SQ-C-S plates, sealed and stored frozen prior to use. Each well contained a final volume of 500 nl 1.5 mM compound. Pre-spotted assay plates were thawed and 24.5 μl of incomplete DMEM was added to each well to give a final drug concentration of 30 μM/well. These compound plates were centrifuged at 1,000 rpm and stored at 4°C prior to addition to assay plates.
Erdman(P606'::mKO-tetON) was grown for a week in 100 ml supplemented Middlebrook 7H9 broth with kanamycin. On the day of infection, 50 ml of culture was harvested by centrifugation at 3,000 rpm for 10 mins, re-suspended in 1 ml sterile cold basal uptake buffer (25 mM dextrose, 0.5% bovine serum albumin, 0.1% gelatin (Sigma), 1 mM CaCl2, 0.5 mM MgCl2 in PBS), passed through a tuberculin needle 6 times, and re-suspended in complete DMEM media at a final OD600 of 0.2. DGr cells were then infected with 10 μl/well of this bacterial suspension using a Perkin Elmer Janus liquid handling robot. Plates were returned to incubator (37°C, 5% CO2) for 1 hr to allow bacteria uptake by host cells. Following this incubation, the DGr cells received 10 μl /well complete DMEM with 60 μg/ml Amphotericin B (final concentration in assay 20 μg/ml) and 0.075 μg/ml isoniazid (INH), where appropriate (final concentration in assay 0.0125 μg/ml). Plates were returned to the incubator following this addition. Finally, test inhibitor compounds were transferred to the assay plates, with each well receiving 10 μl of 30 μM test compound (final compound concentration 5 μM). Plates were incubated at 37°C, 5% CO2. On day 4, 10 μl of 1.4 μg/ml anhydrotetracycline (ATc) was added per well (final concentration 200 ng/ml). Assay plates were returned to the incubator at 37°C, 5% CO2 for a further 3 days. On day 7, the fluorescence was read from the bottom on a Perkin Elmer Envision plate reader—excitation 530/8 nm, dichroic 555 nm, emission 579/25 nm.
The inclusion of Amphotericin B is a potential confounder because of its known immune-modulatory capacity (Hedges et al., 2015) but, because the lung is not a sterile environment, we needed to add the Amphotericin B as an antifungal. We have a series of counter screen assays to verify and validate hits from the primary screen therefore spurious positives will be removed from those compounds advanced in further analysis.
Lung cells were incubated with Fc block (eBioscience) for 15 min. Cells were then counted and incubated for 30 min in the dark with fluorophore-conjugated antibodies. Fluorophore-conjugated mAb specific to mouse CD45 (30-F11; BD Biosciences), CD11b (M1/70; BD Biosciences), CD64 (X54-5/7.1; Biolegend), CX3CR1 (SA011F11; BioLegend), Ly6G (1A8; BioLegend), MerTK (DS5MMER), SiglecF (E50-2440; BD Biosciences), CD4 (GK1.5), EpCAM (G8.8) were purchased from eBioscience unless otherwise indicated. Fixable viability dye was purchased from eBioscience. Cells were analyzed with a Becton Dickenson LSRII flow cytometer and data analyzed with FlowJo software (Tree Star).
Cells were imaged using a Leica SP5 confocal microscope and images were exported and analyzed using Volocity software (PerkinElmer).
Results were statistically analyzed using Student's t-test, or one-way ANOVA test with multiple comparisons where appropriate, with Prism 6.0 (GraphPad Software).
We isolated lung cells from C57BL/6J mice challenged with 1000 CFU of Erdman(smyc'::mCherry) for 3 weeks, to examine the distribution of mCherry Mtb in the infected mouse lung, and investigate whether the fluorescent bacterial signal in the isolated lung cells could be detected by a plate reader. Three weeks post infection was chosen because this is the transition period from innate immunity to the initiation of adaptive immunity in the lung during Mtb infection (Urdahl, 2014). We found that although Mtb predominately infects CD45+CD11b+ myeloid cells, only 2.12% of the CD45+CD11b+ cells carried Mtb (Figure 1A). We also developed a comprehensive multi-color flow cytometry analysis and further revealed different myeloid populations in the Mtb infected murine lung, including neutrophils, monocytes, alveolar and interstitial macrophages (Figure 1B). The tissue homogenization process releases these immune cells preferentially, and we observe only a relatively modest contribution from epithelial lung cells, fewer than 5% CD45- cells, in the resulting suspension (Figure 1B). Unfortunately, mCherry fluorescence was below the level of detection by the plate reader due to the low penetrance of bacteria in the lung myeloid cell population. To overcome this issue, we subjected the cells to an ex vivo challenge with reporter Mtb to further enhance the signal. Indeed, the percentage of mCherry Mtb infected CD45+CD11b+ cells was significantly increased 3 days post ex vivo challenge (Figure 1C), and this was further confirmed by confocal microscopy (Figures 1D,E). We conclude that the lung myeloid cells isolated from Mtb-infected mice are highly phagocytic, enabling an ex vivo challenge to markedly increase the number of infected cells and enhance fluorescent signal.
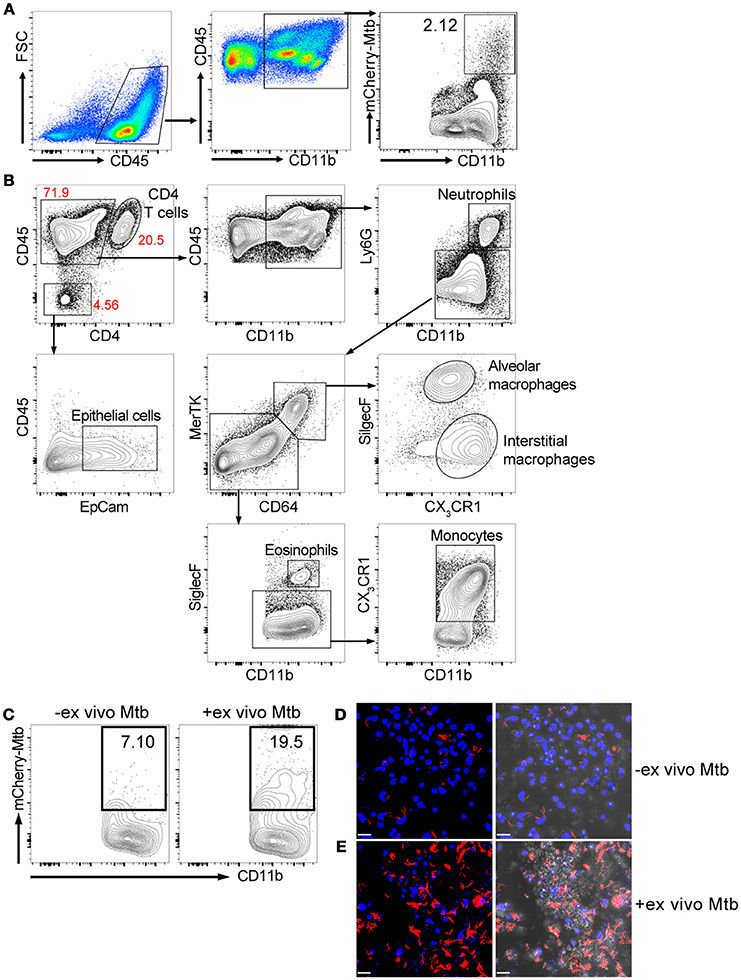
Figure 1. Ex vivo challenge with the fluorescent Mtb strain increases the numbers of infected myeloid cells. Mice were infected with ~1,000 CFU Erdman(smyc'::mCherry) for 3 weeks. Lung single cell suspensions were isolated for the analysis. (A) Gating strategy and flow cytometric analysis of percentage of Mtb-infected myeloid cells (CD45+CD11b+) in the mouse lung after 3 weeks infection. (B) Multi-color flow cytometry analysis of myeloid populations in Mtb infected mouse lung at 3 weeks post infection. (C) Flow cytometry analysis of percentage of Mtb-infected lung myeloid cells before and after ex vivo challenge with mCherry Mtb at MOI 0.1 for 24 h. (D,E) Confocal images of cultured lung cells isolated from mCherry Mtb-infected mice without (D) or with (E) ex vivo challenge. The left panels of (D,E) show the fluorescent images and the right panels show the same fluorescent images overlaid with the brightfield images. Scale bar = 25 μm in (D,E). The experiments were repeated at least two times.
Fluorescent reporter-based assays have been used extensively to test and screen antimicrobial compounds against mycobacteria (Stanley et al., 2012; VanderVen et al., 2015; Gupta et al., 2017). To achieve a higher sensitivity and an extended dynamic range, we constructed a dual inducible/constitutive fluorescent Mtb strain that constitutively expresses mCherry, and expresses mKO upon induction by tetracycline (or its derivative ATc), to enable the specific detection of viable bacteria. The rationale behind the construction of this strain is consistent with previous reports on the use of inducible fitness reporters in mycobacterium (Martin et al., 2012; Mouton et al., 2016). The performance of the new Mtb reporter strain was tested in murine bone marrow-derived macrophages (BMDM) and in J774 cells, a murine macrophage-like cell line. Both mCherry and mKO signal were readily detected by confocal microscopy upon ATc induction of infected cells. In contrast, mKO signal was completely absent in the presence of the anti-tuberculosis drug rifampicin (Rif) in both BMDM and J774 cells (Figure 2A). Analysis of the mKO fluorescence signal intensity with a plate reader demonstrates the robust dynamic range of the reporter strain in J774 cells, in the presence or absence of Rif (Figure 2B). This new inducible Mtb reporter strain therefore represents a highly sensitive tool suitable for HTS.
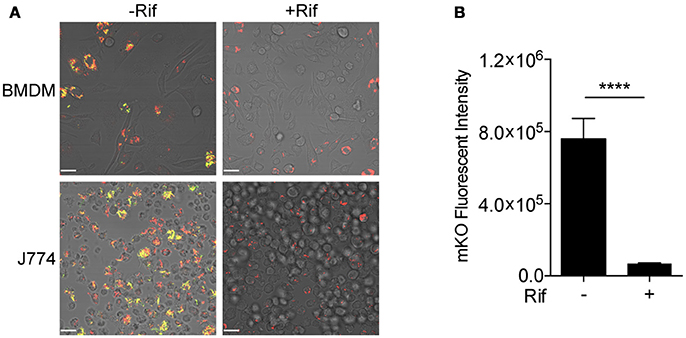
Figure 2. Validation of tet-ON inducible Mtb strain. (A) Confocal images of tet-ON inducible mKO/mCherry infected BMDM (MOI 3) and J774 (MOI 4) cells with or without Rif treatment. Rif (5 μM) was added to Mtb infected cells 3 h post infection. ATc (200 ng/ml) was added to cultures on day 4, and confocal images were taken on day 7. (B) The fluorescence signal of mKO was obtained with a plate reader. Data are shown as mean ± standard deviation. The experiments were repeated at least two times. P-value was calculated using Student's t-test. ****P < 0.0001.
We next combined the ex vivo challenge with a inducible tet-ON mKO reporter to validate the assay in 96- and 384-well plates and test its performance in a HTS format. We created single cell suspensions of the lungs removed from mice infected for 3 weeks with mCherry Mtb. These cells include the large numbers of innate immune cells that infiltrate into the lung, along with the developing lymphocyte response. The mKO Mtb strain was added ex vivo to lung cell cultures established in 96- or 384-well plates. Both Rif and INH were included in the test plates in control wells. Upon ATc induction, mKO signal was detected by the plate reader in the control wells but not in the control wells containing anti-tuberculosis drugs (Figure 3). The magnitude of the response likely reflects both bacteria numbers and bacterial fitness (ability to detect and respond to ATc).
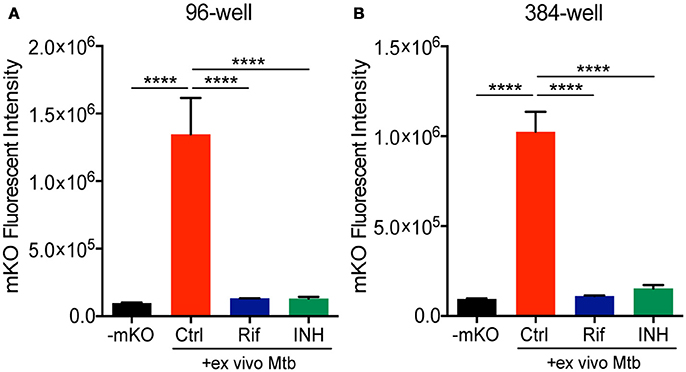
Figure 3. Deconstructed granuloma assay in 96- and 384-well platforms. Lung cells isolated from infected mice were cultured in (A) 96-well or (B) 384-well plates. Tet-ON inducible mKO/mCherry Mtb were added to the cultures at MOI 0.1 with or without addition of Rif and INH. The fluorescence signal of mKO was acquired with a plate reader. Data are shown as mean ± standard deviation. The experiments were repeated at least two times. P-value was calculated using one-way ANOVA test with Tukey's multiple comparisons. ****P < 0.0001.
The DGr has been utilized in a single dose drug screen of 10,000 bioactive compounds, where each compound was tested at 5 μM against the tet-ON mkO strain. The raw fluorescent data from a representative 384-well plate is shown in Figure 4A. These data were transformed into z-scores, calculated as z = x – m/s where m = median of sample plate, and s = sample standard deviation; a z-score of −2 or lower was considered to indicate a “hit” (Figure 4B). Known anti-bacterial compounds present in this particular plate e.g., solithromycin and sparfloxacin, (Figure 4C) were included to validate the platform. Both of these compound classes have been reported to have activity, albeit less than frontline drugs, against Mtb (Rastogi et al., 2000; Pranger et al., 2011). These data demonstrated that the DGr platform represents a viable, sensitive, and robust method for screening compounds for anti-tuberculosis activities. The inclusion of all the immune cell subsets from the infection site in the assay suggests that this platform has the capacity to identify anti-tuberculosis activities that are dependent on a diverse array of host pathways.
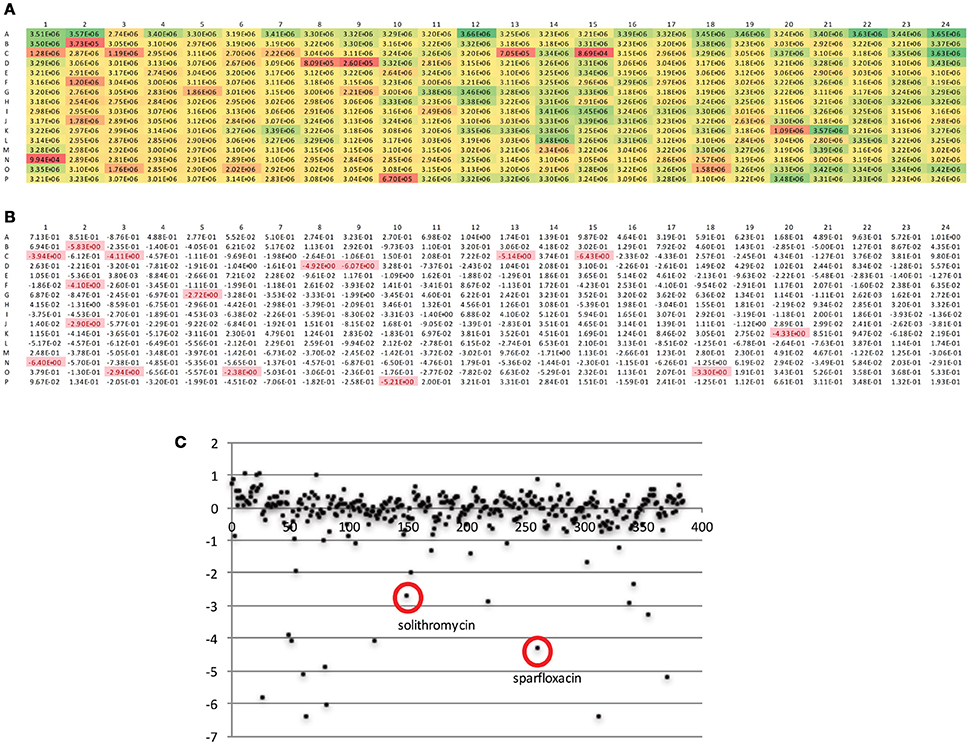
Figure 4. Analysis of fluorescent readout data from a representative 384-well plate. (A) Raw fluorescent data as obtained from plate read. (B) Data from (A) transformed into z-scores, calculated as z = x – m/s where m = median of sample plate, and s = sample standard deviation; a z-score of −2 or lower was considered to indicate a “hit.” (C) Scatter plot of z scores, x-axis is compound number (1-384), known anti-bacterials within this compound plate are circled.
Hits from the screen that we have completed have to be validated, and modes of action have to be determined, nonetheless we see considerable value in documenting the DGr platform in depth because this approach has broad application for the interrogation of chemical, genetic and immunological perturbation of the host components from the Mtb site of infection.
We have established an in vitro drug screening platform that incorporates the diverse cell populations that are recruited and educated within the murine TB granuloma. In contrast to existing screens on Mtb-infected macrophage mono-cultures, which have already proven useful (Christophe et al., 2010; Stanley et al., 2012; VanderVen et al., 2015), this new platform encompasses the broad range of immune pressures active in in vivo infection and therefore provides an increased number of potential targets for host-directed therapeutics. Although previous assays on heterologous cell types challenged in vitro have been detailed (Silva-Miranda et al., 2015), this current platform utilizes cells selected and programed by in vivo infection, thus making fewer assumptions regarding cell type and immune status. We have demonstrated that the platform is amenable to a HTS in a 384 well plate format and have called it the DGr (Deconstructed Granuloma) platform. Through the development process we found that an ex vivo challenge was required to amplify the bacterial signal for detection by plate reader. During optimization of the platform we used two different fluorescent reporter Mtb strains: a mCherry strain was used for the initial mouse infection, to generate the immune response in the animal, followed by ex vivo infection with a tet-ON mKO strain, employed for detection purposes in the HTS assay. We have performed a HTS screen with this DGr platform and provide data from a representative plate to demonstrate the robustness of the assay. Final assessment of the platform and its value as an HTS to discover novel HDTs is dependent on the analysis of the “hits” from the screening of this compound collection, which is ongoing.
In summary, we have developed a robust new, biologically-rich platform for screening for compounds with anti-tuberculosis activities. We believe that this complex platform encompasses much of the immune environment of the infection site, and is of particular value in the pursuit of Host-Directed Therapeutics. We also suggest that similar approaches using cells dissociated from the infected tissue harvested from experimental in vivo infections with other microbial pathogens might have comparable value to other drug discovery efforts.
LH and DR conceived the study, conducted the initial experiments and interpreted data; LH, NK, MT, and AB were responsible for conducting the experiments, data acquisition and interpretation. ST conceived of the use of mKO in Mtb, and generated and carried out initial tests on the inducible mKO strain. DP provided critical reagents. CM and HP provided the compound collection and assisted in the design and interpretation of the screen data. LH, AB and DR wrote the manuscript. All authors critically read and edited the manuscript.
This work was supported by National Institutes of Health Grant AI118582 and by Bill and Melinda Gates Foundation award OPP1108452 to DR. ST was supported in part by National Institutes of Health grant AI114952.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Dirk Schnappinger at Weill Cornell Medical College for providing Gateway cloning vectors and the synthetic promoters used in the construction of these reporter strains.
Christophe, T., Ewann, F., Jeon, H. K., Cechetto, J., and Brodin, P. (2010). High-content imaging of Mycobacterium tuberculosis-infected macrophages: an in vitro model for tuberculosis drug discovery. Future Med. Chem. 2, 1283–1293. doi: 10.4155/fmc.10.223
Collins, L., and Franzblau, S. G. (1997). Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 41, 1004–1009.
Ginsberg, A. M., and Spigelman, M. (2007). Challenges in tuberculosis drug research and development. Nat. Med. 13, 290–294. doi: 10.1038/nm0307-290
Gupta, R., Netherton, M., Byrd, T. F., and Rohde, K. H. (2017). reporter-based assays for high-throughput drug screening against Mycobacterium abscessus. Front. Microbiol. 8:2204. doi: 10.3389/fmicb.2017.02204
Hedges, J. F., Mitchell, A. M., Jones, K., Kimmel, E., Ramstead, A. G., Snyder, D. T., et al. (2015). Amphotericin B stimulates gammadelta T and NK cells, and enhances protection from Salmonella infection. Innate Immun. 21, 598–608. doi: 10.1177/1753425914567692
Huang, L., Nazarova, E. V., Tan, S., Liu, Y., and Russell, D. G. (2018). Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J. Exp. Med. 215, 1135–1152. doi: 10.1084/jem.20172020
Huang, L., and Russell, D. G. (2017). Protective immunity against tuberculosis: what does it look like and how do we find it? Curr. Opin. Immunol. 48, 44–50. doi: 10.1016/j.coi.2017.08.001
Karasawa, S., Araki, T., Nagai, T., Mizuno, H., and Miyawaki, A. (2004). Cyan-emitting and orange-emitting fluorescent proteins as a donor/acceptor pair for fluorescence resonance energy transfer. Biochem. J. 381, 307–312. doi: 10.1042/BJ20040321
Kaufmann, S. H. E., Dorhoi, A., Hotchkiss, R. S., and Bartenschlager, R. (2018). Host-directed therapies for bacterial and viral infections. Nat. Rev. Drug Discov. 17, 35–56. doi: 10.1038/nrd.2017.162
Kim, Y. R., and Yang, C. S. (2017). Host-directed therapeutics as a novel approach for tuberculosis treatment. J. Microbiol. Biotechnol. 27, 1549–1558. doi: 10.4014/jmb.1705.05032
Kolloli, A., and Subbian, S. (2017). Host-directed therapeutic strategies for tuberculosis. Front Med. 4:171. doi: 10.3389/fmed.2017.00171
Koul, A., Arnoult, E., Lounis, N., Guillemont, J., and Andries, K. (2011). The challenge of new drug discovery for tuberculosis. Nature 469, 483–490. doi: 10.1038/nature09657
Lamichhane, G. (2011). Mycobacterium tuberculosis response to stress from reactive oxygen and nitrogen species. Front. Microbiol. 2:176. doi: 10.3389/fmicb.2011.00176
Liu, Y., Tan, S., Huang, L., Abramovitch, R. B., Rohde, K. H., Zimmerman, M. D., et al. (2016). Immune activation of the host cell induces drug tolerance in Mycobacterium tuberculosis both in vitro and in vivo. J. Exp. Med. 213, 809–825. doi: 10.1084/jem.20151248
Martin, C. J., Booty, M. G., Rosebrock, T. R., Nunes-Alves, C., Desjardins, D. M., Keren, I., et al. (2012). Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe 12, 289–300. doi: 10.1016/j.chom.2012.06.010
Mdluli, K., and Spigelman, M. (2006). Novel targets for tuberculosis drug discovery. Curr. Opin. Pharmacol. 6, 459–467. doi: 10.1016/j.coph.2006.06.004
Mouton, J. M., Helaine, S., Holden, D. W., and Sampson, S. L. (2016). Elucidating population-wide mycobacterial replication dynamics at the single-cell level. Microbiology 162, 966–978. doi: 10.1099/mic.0.000288
Pethe, K., Sequeira, P. C., Agarwalla, S., Rhee, K., Kuhen, K., Phong, W. Y., et al. (2010). A chemical genetic screen in Mycobacterium tuberculosis identifies carbon-source-dependent growth inhibitors devoid of in vivo efficacy. Nat. Commun. 1:57. doi: 10.1038/ncomms1060
Pranger, A. D., Alffenaar, J. W., and Aarnoutse, R. E. (2011). Fluoroquinolones, the cornerstone of treatment of drug-resistant tuberculosis: a pharmacokinetic and pharmacodynamic approach. Curr. Pharm. Des. 17, 2900–2930. doi: 10.2174/138161211797470200
Rastogi, N., Goh, K. S., Berchel, M., and Bryskier, A. (2000). In vitro activities of the ketolides telithromycin (HMR 3647) and HMR 3004 compared to those of clarithromycin against slowly growing mycobacteria at pHs 6.8 and 7.4. Antimicrob. Agents Chemother. 44, 2848–2852. doi: 10.1128/AAC.44.10.2848-2852.2000
Russell, D. G. (2017). Microbiology: diversity breeds tolerance. Nature 546, 44–45. doi: 10.1038/546044a
Silva-Miranda, M., Ekaza, E., Breiman, A., Asehnoune, K., Barros-Aguirre, D., Pethe, K., et al. (2015). High-content screening technology combined with a human granuloma model as a new approach to evaluate the activities of drugs against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 693–697. doi: 10.1128/AAC.03705-14
Stanley, S. A., Grant, S. S., Kawate, T., Iwase, N., Shimizu, M., Wivagg, C., et al. (2012). Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem. Biol. 7, 1377–1384. doi: 10.1021/cb300151m
Tan, S., Sukumar, N., Abramovitch, R. B., Parish, T., and Russell, D. G. (2013). Mycobacterium tuberculosis responds to chloride and pH as synergistic cues to the immune status of its host cell. PLoS Pathog. 9:e1003282. doi: 10.1371/journal.ppat.1003282
Urdahl, K. B. (2014). Understanding and overcoming the barriers to T cell-mediated immunity against tuberculosis. Semin. Immunol. 26, 578–587. doi: 10.1016/j.smim.2014.10.003
VanderVen, B. C., Fahey, R. J., Lee, W., Liu, Y., Abramovitch, R. B., Memmott, C., et al. (2015). Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium's metabolism is constrained by the intracellular environment. PLoS Pathog. 11:e1004679. doi: 10.1371/journal.ppat.1004679
Keywords: tuberculosis, pulmonary, Mycobacterium tuberculosis, high-throughput screening assays, host-directed therapeutics, macrophages
Citation: Huang L, Kushner NL, Theriault ME, Pisu D, Tan S, McNamara CW, Petrassi HM, Russell DG and Brown AC (2018) The Deconstructed Granuloma: A Complex High-Throughput Drug Screening Platform for the Discovery of Host-Directed Therapeutics Against Tuberculosis. Front. Cell. Infect. Microbiol. 8:275. doi: 10.3389/fcimb.2018.00275
Received: 02 May 2018; Accepted: 23 July 2018;
Published: 14 August 2018.
Edited by:
Frank Brombacher, International Centre for Genetic Engineering and Biotechnology (ICGEB), South AfricaReviewed by:
Adam Cunningham, University of Birmingham, United KingdomCopyright © 2018 Huang, Kushner, Theriault, Pisu, Tan, McNamara, Petrassi, Russell and Brown. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David G. Russell, ZGdyOEBjb3JuZWxsLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.