 Yi-Lin Chan
Yi-Lin Chan Ching-Len Liao3,4
Ching-Len Liao3,4 Yi-Ling Lin
Yi-Ling Lin- 1Department of Life Science, Chinese Culture University, Taipei, Taiwan
- 2Institute of Biomedical Sciences, Academia Sinica, Taipei, Taiwan
- 3Department of Microbiology and Immunology, National Defense Medical Center, Taipei, Taiwan
- 4National Institute of Infectious Diseases and Vaccinology, National Health Research Institutes, Miaoli, Taiwan
- 5Genomics Research Center, Academia Sinica, Taipei, Taiwan
Japanese encephalitis virus (JEV), a mosquito-borne flavivirus, causes acute encephalitis in humans with high mortality. Not much is known about the interactions between viral and cellular factors that regulate JEV infection. By using a kinase/phosphatase-wide RNAi screening approach, we identified a cell cycle-regulating molecule, checkpoint kinase 2 (CHK2), that plays a role in regulating JEV replication. JEV infection induced G1 arrest and activated CHK2. Inactivation of CHK2 and its upstream ataxia-telangiectasia mutated kinase in JEV-infected cells by using inhibitors reduced virus replication. Likewise, JEV replication was significantly decreased by knockdown of CHK2 expression with shRNA-producing lentiviral transduction. We identified CHK2 as a cellular factor participating in JEV replication, for a new strategy in addressing JEV infection.
Introduction
Japanese encephalitis virus (JEV) is the most important cause of viral encephalitis in Southeast Asia, with 30,000–50,000 cases reported annually (Hegde and Gore, 2017). JEV is transmitted in a zoonotic cycle between mosquitoes and vertebrate-amplifying hosts, mainly swine and wading birds (Mansfield et al., 2017). As a member of the Flavivirus genus, the JEV virion is enveloped and has a positive-sense single-stranded RNA genome. The initial steps of JEV infection include virus attachment to cell-surface receptors and entry via receptor-mediated endocytosis. Translation of the viral genome produces a polyprotein that is processed to structural core (C), precursor of membrane (prM), and envelope (E) proteins and the nonstructural proteins NS1~NS5. Flaviviral genome replication occurs by the viral replicase complex via RNA-dependent RNA polymerization. The positive-sense genomic RNA is transcribed to a replication-intermediate negative-sense RNA, which is then used as a template to synthesize genomic RNA for subsequent translation and assembly of virion progeny (Tiroumourougane et al., 2002; Fields et al., 2007).
How a virus triggers DNA damage signaling is not fully understood, but previous reports have suggested that the cellular DNA repair machinery can recognize viral genetic materials, such as replicating nucleic acids and viral proteins, upon infection (Weitzman et al., 2004). Some viruses have been shown to interact with and/or affect components of the ATM DNA damage pathway (Lilley et al., 2007; Bagga and Bouchard, 2014). DNA viruses, such as human cytomegalovirus (CMV) activate the ATM checkpoint pathway during DNA replication and inhibit DNA damage responses by mislocalizing checkpoint proteins from the nucleus to cytoplasm (Gaspar and Shenk, 2006). Herpes simplex virus (HSV) induces an ATM-damage response that is essential for viral replication (Lilley et al., 2005; Shirata et al., 2005). Inhibition of CHK2 kinase activity by the CHK2 inhibitor II significantly reduces the CPE and genome replication of HSV-1 in corneal epithelium (Alekseev et al., 2015). Hepatitis C virus (HCV), an RNA virus belonging to Flaviviridae, induces G2/M phase arrest (Wu et al., 2008) and activates the dsDNA damage response pathway by causing DSBs and enhancing the mutation of cellular genes (Machida et al., 2004). Knockdown of ATM or CHK2 suppresses RNA replication of HCV (Ariumi et al., 2008), and HCV NS3-NS4A interacts with ATM, whereas HCV NS5B interacts with both ATM and CHK2 (Lai et al., 2008). JEV is able to manipulate the cell cycle, which reduces the proliferation of neural progenitor cells (Das and Basu, 2008) and allows for persistent infection (Kim et al., 2015). JEV is also able to manipulate the cell cycle, which reduces the proliferation of neural progenitor cells (Das and Basu, 2008) and allows for persistent infection (Kim et al., 2015).
Only a few host proteins have been found involved in JEV infection, with the roles in JEV replication remaining largely unknown. In this study, we identified CHK2, which participates in cellular regulation of JEV infection, by using a kinase/phosphatase-wide siRNA screening strategy. Both CHK2 activation and G1 phase arrest were noted in JEV infected cells and the activation of CHK2 appeared to be beneficial for JEV replication.
Materials and Methods
Cell Lines and Reagents
U87 cells (ATCC HTB-14), a human glioblastoma cell line, were cultured in minimal essential medium (Gibco) containing 10% fetal bovine serum (FBS), 1% sodium pyruvate, and 2 mM L-glutamine. A549 cells, a human lung carcinoma cell line, were cultured as previously described (Chang et al., 2006). BE(2)C (ATCC CRL68), a human neuroblastoma cell line were cultured in10% FBS and 2 mM L-glutamine containing RPMI 1640 (Gibco) medium. HEK293T, a human embryonic kidney cells, were cultured in 10% FBS and 2 mM L-glutamine containing Dulbecco's Modified Eagle's Medium (Gibco). Experiments were done using cells with passage 3–5. The ATM inhibitor (KU-55933) was from Calbiochem and the CHK2 inhibitor II was from Merck.
Viruses and Viral Infection
JEV strain RP-9 (Chen et al., 1996) was propagated in C6/36 cells. In Taiwan, JEV is classified as a BSL-2 pathogen according to the “Guidelines for research involving recombinant DNA molecules” issued by National Science Council, Taiwan (NSC., 2004). For viral infection, cells were adsorbed with viruses at the indicated multiplicity of infection (MOI) for 1 h at 37°C. Virus titers (PFU/ml) were determined by plaque-forming assays by using BHK-21 cells as described (Wu et al., 2002).
Human Kinase/Phosphatase-Wide RNAi Screening
U87 cells were transduced with VSV-G pseudotyped lentiviruses expressing shRNAs targeting 1,260 human kinases and phosphatases (obtained from the National RNAi Core Facility, Taiwan). After puromycin selection, cells were infected with JEV and surviving cell colonies were cultured. Genomic DNAs from surviving cells were extracted by using the QIAamp DNA Mini and Blood Mini Kit (Qiagen). The DNA regions containing shRNAs were amplified by PCR with primers (5′-TAATTTCTTGGGTAGTTTGCAGTT-3′ and 5′-CCCCAATCCCCCCTTTTC-3′) and cloned into pcDNA3.1/V5-His-TOPO (Invitrogen). The genes targeted by shRNA were identified by sequencing and BLAST search.
Lentivirus Preparation
Lentiviral vectors (pLKO.1-puro) carrying the shRNAs targeting CHK2 (5′-GAACAGATAAATACCGAACAT-3′) and control LacZ (5′-TGTTCGCATTATCCGAACCAT-3′) were co-transfected into 293T cells with pMD.G and pCMVΔR8.91 (obtained from the National RNAi Core Facility, Taiwan) by using lipofectamine 2000 reagent. The culture supernatants were harvested and further concentrated by centrifugation at 20,000 × g for 3 h at 4°C.
Cell Survival
Cells were collected for survival determination by a trypan blue exclusion method. The virus-induced cytopathic effect (CPE) was analyzed by measuring the release of a cytoplasmic enzyme, lactate dehydrogenase (LDH), by using a commercial kit (Cytotoxicity Detection Kit; Roche).
Immunoblotting
Cells were lysed and subjected to western blot analysis as described (Chan et al., 2008). Primary antibodies used in this study included anti-CHK2 (sc5278, Santa Cruz Biotechnology; H00011200-M01, Abnova), anti-CHK2 (Thr68) (#2661, Cell Signaling Technology), and anti-β actin (Chemicon). The mouse monoclonal antibody specific to JEV NS3 was described previously (Wu et al., 2002).
RNA Preparation and RT-PCR Analysis
RNA was extracted by using an RNeasy kit (Qiagen) according to the manufacturer's instructions. cDNA was reverse transcribed from 2 μg RNA with a random hexamer and a ThermoScript RT Kit (Invitrogen). PCR involved specific primer sets for CHK2 (5′-ATGTCTCGGGAGTCGGATGTT-3′ and 5′-ACTTTATTTCTGCTTAGTGACAGTGCA-3′) and β-actin (5′-TCCTGTGGCATCCACGAAACT-3′ and 5′-GAAGCATTTGCGGTGGACGAT-3′), respectively.
Cell Cycle Analysis by Flow Cytometry
The cell cycle status was analyzed by propidium iodide (PI) staining for nuclear DNA contents and by flow cytometry. At the indicated times after viral infection, cells were collected and fixed with 70% ethanol. Cells were stained with PI solution containing RNaseA at 4°C for 30 min, and then analyzed by flow cytometry (Attune NxT, Thermo Fisher).
Statistical Analysis
Data are presented as mean ± standard deviation (SD). Two categorical data were compared by independent Student t-test. The statistical tests were two-tailed and significance was set at P < 0.05, < 0.01, and < 0.005. For immunoblotting, the band density was quantified by use of ImageJ (US National Institutes of Health).
Results
Human Kinase/Phosphatase-Wide RNAi Screening Identified CHK2 as a Cellular Factor Involved in JEV Infection
We used a human kinase/phosphatase-wide RNAi screening strategy to search for potential kinases and phosphatases involved in JEV infection. U87, a human glioma cell line, was transduced by each of the seven VSV-G pseudotyped lentivirus pool (Human kinase and phosphatase set) provided by the National RNAi Core Facility. Each kinases/phosphatases pooled tube contains ~180 kinase/phosphatase genes; each gene is targeted by 5 shRNAs that bind to distinct target sequences. The VSV-G pseudotyped lentivirus set that carries these shRNAs knocked down 1260 genes encoding kinase/phosphatases, which accounts for ~90% of all kinase/phosphatase in accordance of the NCBI database. After selection with puromycin for lentivirus-transduced cells, cells were infected JEV at an MOI of 10 (Figure 1A). Surviving cell colonies were cultured to extract genomic DNA. DNAs encoding shRNA were amplified by PCR and sequenced to determine their targets by BLAST alignment with the NCBI database to further confirm the identities of these genes as kinase/phosphatase encoding genes. Seven host candidate genes (Figure 1B), were identified in cells survived from JEV challenge.
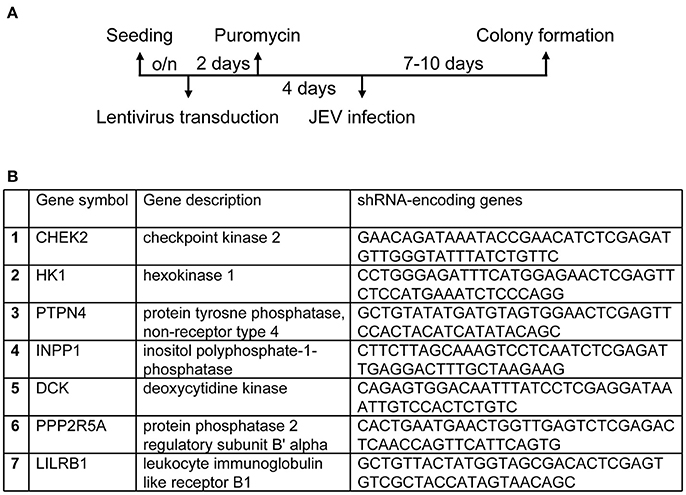
Figure 1. Establishing a human kinases/phophatases-wide RNAi screen system. (A) Overview of RNAi screening to genes involved in regulation of JEV infection. U87 cells transduced with lentiviruses expressing shRNAs targeting human kinases and phosphatase were selected with puromycin (10 μg/ml) for 4 days and infected JEV at an MOI of 10. (B) Cells survived from JEV infection were identified for candidate genes.
To verify whether knockdown of these candidate genes indeed rescued cells from JEV infection, we transduced U87 cells with the lentiviral vector targeting each candidate gene and infected the cells with JEV. Knockdown of one of these candidate genes, CHEK2, substantially rendered cell survival from JEV infection. U87 cells showed reduced expression of CHK2 by transduction with lentivirus expressing an shRNA targeting CHK2 (Figure 2A). Upon JEV infection, knockdown of CHK2 resulted in reduced CPE (Figure 2B), enhanced cell survival (Figure 2C) and reduced JEV progeny production (Figure 2D) as compared with control knockdown shLacZ cells. To ascertain the importance of CHK2 in JEV infection, we further checked the involvement of CHK2 in another human cell line, A549 cells. Similarly, JEV production was reduced in human A549 cells with knocked-down CHK2 expression (Figure 3).
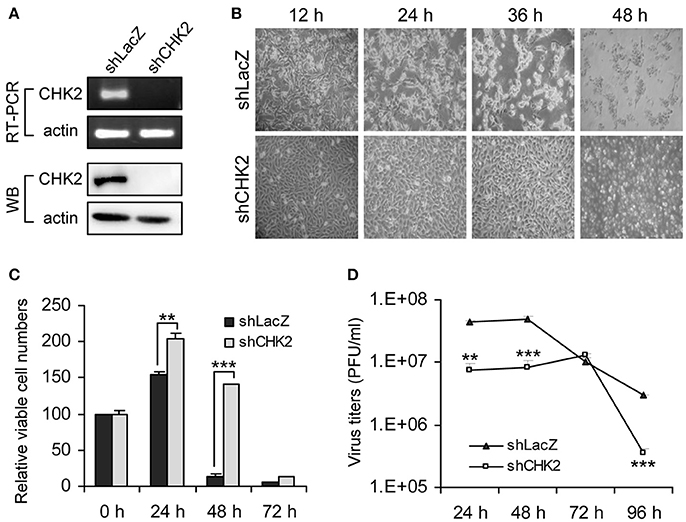
Figure 2. Human kinase/phosphatase-wide RNAi screening identified CHK2 as a cellular factor involved in JEV infection. (A) Human U87 cells transduced with lentivirus expressing shRNA targeting CHK2 (shCHK2) or LacZ control (shLacZ) were verified for CHK2 knockdown by RT-PCR for mRNA level and immunoblotting analysis for protein expression. The cells were infected with JEV (MOI 5) for the indicated times. Cell morphology (B), relative viable cell numbers (n = 3) were determined by trypan blue exclusion (C) and viral progeny production (n = 3) was determined by plaque-forming assays (D). Data are mean ± SD. **p < 0.01; ***p < 0.005 by two-tailed Student t-tests.
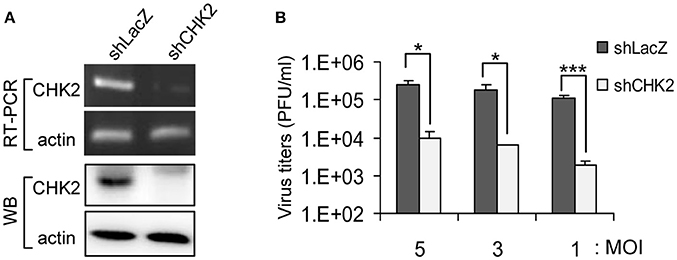
Figure 3. Reduced CHK2 expression decreases JEV replication. Human A549 cells were transduced with shCHK2-expressing lentivirus and selected by puromycin. ShRNA-targeting LacZ was the non-targeting sequence control. (A) The RNA and protein levels of CHK2 were verified by RT-PCR with CHK2-specific primers and immunoblotting with anti-CHK2 antibody, respectively. Cells were infected with JEV at the indicated MOIs (5, 3, and 1); (B) at 24 h post infection, culture supernatants were collected for virus titration by plaque-forming assays. Data are mean ± SD virus titers (n = 3). *p < 0.05; ***p < 0.005 by two-tailed Student t-test.
JEV-Activated CHK2 Is Beneficial for JEV Replication
CHK2 is a protein kinase activated by ATM in response to DNA damage (Bakkenist and Kastan, 2004). To understand how CHK2 participates in JEV replication, we determined whether the ATM/CHK2 pathway was activated by JEV infection. CHK2 activation, as manifested by phosphorylation at Thr-68, was evident in JEV-infected cells in an MOI dependent manner, and an ATM inhibitor KU-55933 (Lau et al., 2005) reduced the CHK2 activation (Figure 4A). Furthermore, the ATM inhibitor significantly reduced JEV production (Figure 4B).
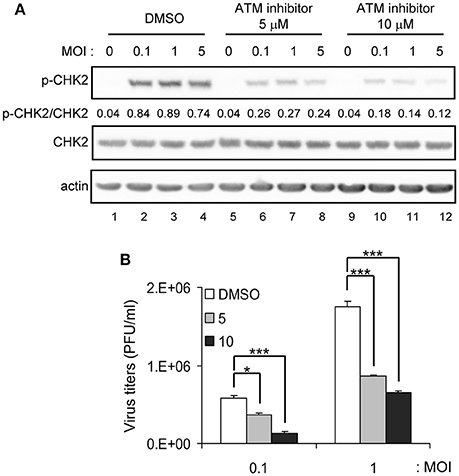
Figure 4. ATM inhibitor reduces JEV replication. A549 cells were infected with JEV at MOI 0.1, 1 or 5 in the absence (DMSO solvent control) or presence of 5 or 10 μM ATM inhibitor (KU-55933) as indicated. (A) Immunoblotting analysis of cell lysates at 24 h with antibodies against T68-phosphorylated CHK2, CHK2 and actin. (B) A549 cells were treated with ATM inhibitor (KU-55933) at the indicated dose, followed by JEV infection at MOI 0.1 or 1 for 24 h. Culture supernatants were collected for virus titration by plaque-forming assays. Data are mean ± SD virus titers (n = 3). *p < 0.05; ***p < 0.005 by two-tailed Student t-test.
The involvement of CHK2 in JEV replication was further assessed by using a cell-permeable CHK2 inhibitor, CHK2 inhibitor II (Arienti et al., 2005). U87 cells were infected with JEV (MOI 0.1 and 5) in the absence or presence of non-cytotoxic CHK2 inhibitor II (25 and 50 μM). Immunoblotting analysis with anti-CHK2 (Thr68) antibody showed that CHK2 inhibitor II effectively reduced the JEV induced phosphorylation of CHK2 protein (Figure 5A). CHK2 inhibitor II also reduced virus production. With low MOI, 25 μM CHK2 inhibitor II readily decreased JEV production by ~100-fold (Figure 5B). With high MOI, 25 μM CHK2 inhibitor II slightly suppressed JEV production, whereas 50 μM conferred 94% reduction in virus production. With the treatment of 50 μM CHK2 inhibitor II, virus progeny was inhibited in both high MOI (Figure 5C) and low MOI (Figure 5D) at various time points. The antiviral effect of the CHK2 inhibitor II was further verified in A549 cells and BE(2)C cells. CHK 2 inhibitor II reduced both the viral protein expression and CHK2 phosphorylation level (Figure 6A) Viral progeny production (Figure 6B) was suppressed by CHK2 inhibitor II at different time points (Figures 6C, D). Similar to U87, the CHK2 phosphorylation of BE(2)C neuroblastoma cells was induced by JEV and reduced by CHK2 inhibitor II (Figure 7A). The virus progeny in both high MOI (Figure 7B) and low MOI (Figure 7C) was inhibited by 50 μM CHK2 inhibitor II in various time points. Collectively, blocking the ATM/CHK2 pathway by an ATM inhibitor or a CHK2 inhibitor reduced JEV replication in human glioblastoma, lung cancer carcinoma, and neuroblastoma cell lines tested.
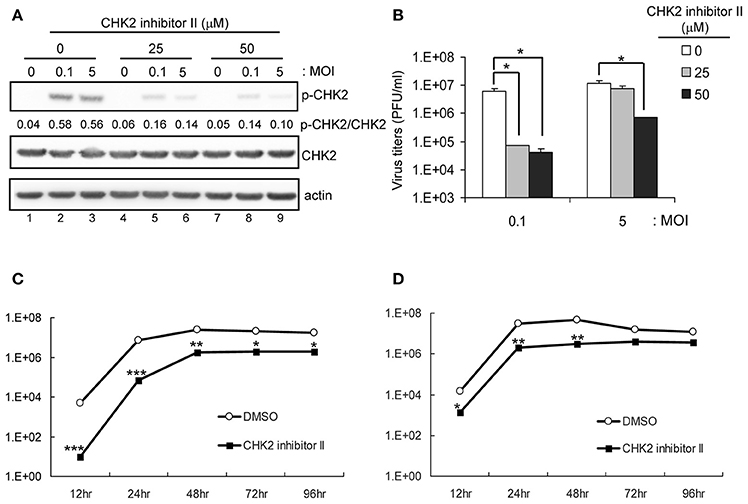
Figure 5. Inhibition of CHK2 activity reduces JEV replication in human U87 glioma cells. U87 cells were infected with JEV (MOI 0, 0.1 and 5) with or without of CHK2 inhibitor II (0, 25 and 50 μM). (A) At 24 h post treatment, immunoblotting analysis of cell lysates at 24 h with antibodies against, T68-phosphorylated CHK2, CHK2, and actin was performed; virus titration (B) in culture supernatant was determined by plaque-forming assays. Cells were treated with DMSO or CHK2 inhibitor II (50 μM), followed by JEV infection at MOIs of 0.1 (C) or 5 (D) for the indicated times. Culture supernatants were collected for virus titration by plaque-forming assays. Data are mean ± SD virus titers (n = 3). *p < 0.05; **p < 0.01; ***p < 0.005 by two-tailed Student t-test.
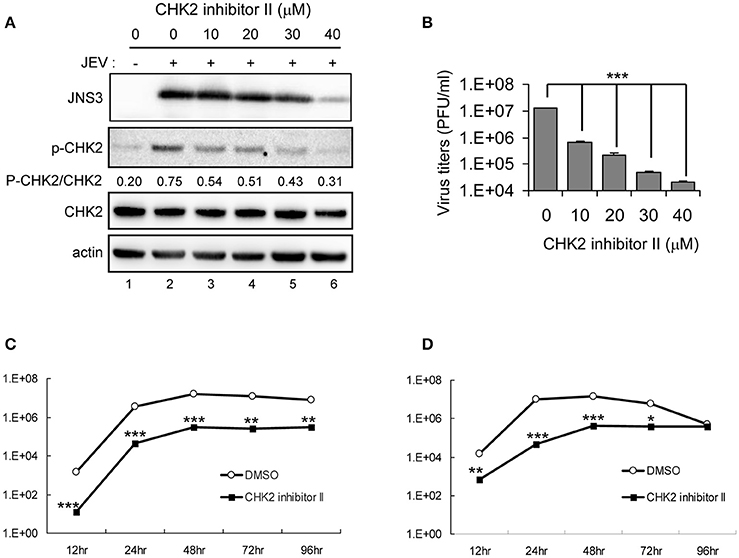
Figure 6. Inhibition of CHK2 activity reduces JEV replication in human A549 lung carcinoma cells. A549 cells were mock-infected or infected with JEV (MOI 5) with various doses of CHK2 inhibitor II. (A) At 24 h post treatment, immunoblotting analysis of cell lysates at 24 h with antibodies against JEV NS3, T68-phosphorylated CHK2, CHK2, and actin and was performed; (B) virus titers determination in culture supernatants were determined by plaque-forming assays. Cells were treated with DMSO or CHK2 inhibitor II (50 μM), followed by JEV infection at MOIs of 0.1 (C) or 5 (D) for the indicated times. Culture supernatants were collected for virus titration by plaque-forming assays. Data are mean ± SD virus titers (n = 3). *p < 0.05; **p < 0.01; ***p < 0.005 by two-tailed Student t-test.
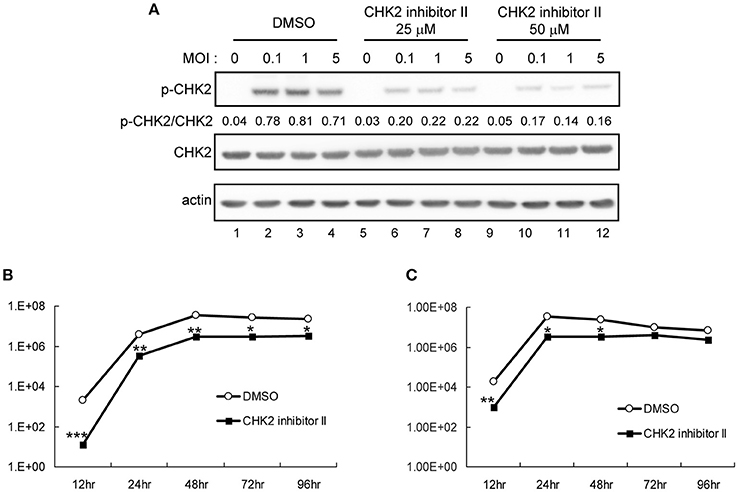
Figure 7. Inhibition of CHK2 activity reduces JEV replication in human BE(2)C neuroblastoma cells. BE(2)C cells were infected with JEV at MOI 0.1, 1 or 5 in the absence (DMSO solvent control) or presence of 25 or 50 μM CHK2 inhibitor II as indicated. (A) Immunoblotting analysis of cell lysates at 24 h with antibodies against T68-phosphorylated CHK2, CHK2 and actin. BE(2)C cells were treated with DMSO or CHK2 inhibitor II (50 μM), followed by JEV infection at MOIs of 0.1 (B) or 5 (C) for the indicated times. Culture supernatants were collected for virus titration by plaque-forming assays. Data are mean ± SD virus titers (n = 3). *p < 0.05; **p < 0.01 ***p < 0.005 by two-tailed Student t-test.
JEV Infection Resulted in G1 Arrest in U87, A549, BE(2)C Cells
Given that the ATM/CHK2 pathway is activated upon DNA damage in order to modulate cell cycle progression, we assessed whether JEV infection results in cell cycle arrest. We analyzed the cell cycle by measuring DNA content with PI staining for mock- and JEV-infected cells at 24 h post-infection. JEV infection increased the proportion of cells in the G1 phase from 55.46% to 78.87% in U87 cells (Figure 8A). Similar G1 phase arrest was also noted in A549 cells (Figure 8B) and BE(2)C cells (Figure 8C). Therefore, JEV infection induced cell cycle arrest in the G1 phase.
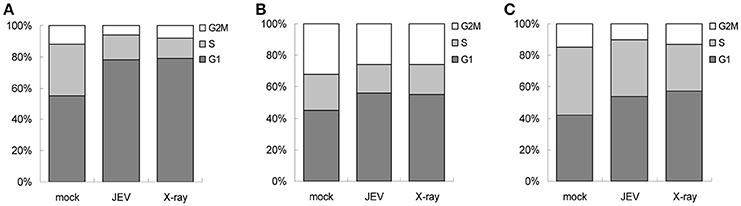
Figure 8. JEV infection induces cell cycle arrest at G1 phase. U87 (A), A549 (B), and BE(2)C (C) cells were mock-infected, JEV (MOI 5) infected, or X-ray (8 Gy) treated. At 24 h post-treatment, DNA content was determined by PI staining and analyzed by flow cytometry for proportion of cells in G1, S, and G2 phases.
Discussion
With the recent development of screening strategies, RNAi-based technology has been extensively used to identify genes involved in cellular processes, such as signal transduction, cell cycle, cancer biology and host–pathogen interactions (Houzet and Jeang, 2011). Viruses are obligate intracellular parasites that may utilize host cell machineries during every step of their life cycles (Goff, 2007). In this study, we used a human kinase/phosphatase-wide RNAi strategy for large scale screening to identify a gene(s) involved in regulating JEV infection in U87 human glioma cells. Among the 1260 human kinases/phosphatases found, we identified a cell cycle-regulating molecule, CHK2, that facilitated JEV infection in the cells.
For infectivity, viruses have been found to interact with CHK2 and its related responses in host cells. Human papillomaviruses activate the ATM DNA response (Lai et al., 2008; Moody and Laimins, 2009). HSV-1 and SV40 recruit members of the ATM DNA damage pathway to specific sites in the nucleus during replication (Taylor and Knipe, 2004; Wilkinson and Weller, 2004; Zhao et al., 2008). Furthermore, the NS5B of HCV was found to interact with both ATM and CHK2, and the replication of subgenomic replicon RNA of HCV was suppressed in cells with CHK2 knockdown (Ariumi et al., 2008). In this study, we also found that downregulation of CHK2 and inhibition of CHK activity enhanced cell resistance to JEV infection. Likewise, progeny virus production and expression of viral proteins were reduced in cells with knockdown of functional CHK2. Because viral replication induces cellular stresses and triggers an ATR response, viruses may hijack CHK2 to elongate the cell cycle arrest, thus creating a time window for viral replication. Consequently, downregulation of CHK2 becomes a suitable way to inhibit virus propagation.
Viruses are streamlined organisms featured in minimizing proteins required for genome replication and control of the host cell cycle. In analogy to some RNA viruses such as influenza A virus (He et al., 2010), mouse hepatitis virus (Chen and Makino, 2004), and severe acute respiratory syndrome coronavirus (Yuan et al., 2007), JEV infection is able to regulate cell-cycle arrest in the G1 phase, probably resulting from CHK2 activation (Lavin and Kozlov, 2007). Upon activation, CHK2 phosphorylates Cdc25a and Cdc25c (phosphatases), which results in G1/S or G2/M arrest via degradation or cytoplasmic sequestration, respectively (Antoni et al., 2007). Each phase of the cell cycle should represent a unique metabolic status. Indeed, the transcription activity of Pol II is much higher in the G1 phase than in the S or G2/M phase (Yonaha et al., 1995). As well, in non-dividing hepatocytes, the G0/G1 state has higher translation efficiency, which facilitates expression of the HCV genome (Fehr et al., 2012). Likewise, to proliferate the viral genome, JEV may arrest the cell cycle in the G1 phase to increase Pol II transcription activity and translation efficiency. Moreover, oncogenic viruses often possess mechanisms to induce G1/S cell cycle arrest via p53 inactivation, thus rescuing the infected cells from apoptosis (Yew and Berk, 1992; Scheffner et al., 1993; Härtl et al., 2008). JEV inducing G1 arrest may be advantageous to gain sufficient time and resources for viral replication and to avoid early apoptosis of infected cells.
Although the exact mechanism of how JEV induces the DNA damage response is not fully understood, our studies demonstrate that JEV executes its own replication by manipulating the host cell cycle via CHK2. The kinase/phosphatase-wide RNAi screening system can be an effective strategy to search for cellular factors involved in the regulation of JEV infection.
Author Contributions
Y-LC and Y-LL designed research, analyzed data and wrote the paper. Y-LC performed research. C-LL provided critical reagents.
Funding
This work was supported by grants awarded to Y-LL from Academia Sinica (106-2101-01-11-01) and Ministry of Science and Technology, Taiwan (MOST 106-0210-01-15-02).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the National RNAi Core Facility, Taiwan for shRNA and lentiviral constructs. We also thank the core facility of Institute of Biomedical Sciences for technical assistance.
References
Alekseev, O., Limonnik, V., Donovan, K., and Azizkhan-Clifford, J. (2015). Activation of checkpoint kinase 2 is critical for herpes simplex virus type 1 replication in corneal epithelium. Ophthalmic Res. 53, 55–64. doi: 10.1159/000366228
Antoni, L., Sodha, N., Collins, I., and Garrett, M. D. (2007). CHK2 kinase: cancer susceptibility and cancer therapy - two sides of the same coin? Nat. Rev. Cancer 7, 925–936. doi: 10.1038/nrc2251
Arienti, K. L., Brunmark, A., Axe, F. U., McClure, K., Lee, A., Blevitt, J., et al. (2005). Checkpoint kinase inhibitors: SAR and radioprotective properties of a series of 2-arylbenzimidazoles. J. Med. Chem. 48, 1873–1885. doi: 10.1021/jm0495935
Ariumi, Y., Kuroki, M., Dansako, H., Abe, K., Ikeda, M., Wakita, T., et al. (2008). The DNA damage sensors ataxia-telangiectasia mutated kinase and checkpoint kinase 2 are required for hepatitis C virus RNA replication. J. Virol. 82, 9639–9646. doi: 10.1128/JVI.00351-08
Bagga, S., and Bouchard, M. J. (2014). Cell cycle regulation during viral infection. Methods Mol. Biol. 1170, 165–227. doi: 10.1007/978-1-4939-0888-2_10
Bakkenist, C. J., and Kastan, M. B. (2004). Initiating cellular stress responses. Cell 118, 9–17. doi: 10.1016/j.cell.2004.06.023
Chan, Y. L., Chang, T. H., Liao, C. L., and Lin, Y. L. (2008). The cellular antiviral protein viperin is attenuated by proteasome-mediated protein degradation in Japanese encephalitis virus-infected cells. J. Virol. 82, 10455–10464. doi: 10.1128/JVI.00438-08
Chang, T. H., Liao, C. L., and Lin, Y. L. (2006). Flavivirus induces interferon-beta gene expression through a pathway involving RIG-I-dependent IRF-3 and PI3K-dependent NF-kappaB activation. Microbes Infect. 8, 157–171. doi: 10.1016/j.micinf.2005.06.014
Chen, C. J., and Makino, S. (2004). Murine coronavirus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 78, 5658–5669. doi: 10.1128/JVI.78.11.5658-5669.2004
Chen, L. K., Lin, Y. L., Liao, C. L., Lin, C. G., Huang, Y. L., Yeh, C. T., et al. (1996). Generation and characterization of organ-tropism mutants of Japanese encephalitis virus in vivo and in vitro. Virology 223, 79–88. doi: 10.1006/viro.1996.0457
Das, S., and Basu, A. (2008). Japanese encephalitis virus infects neural progenitor cells and decreases their proliferation. J. Neurochem. 106, 1624–1636. doi: 10.1111/j.1471-4159.2008.05511.x
Fehr, C., Conrad, K. D., and Niepmann, M. (2012). Differential stimulation of hepatitis C virus RNA translation by microRNA-122 in different cell cycle phases. Cell Cycle 11, 277–285. doi: 10.4161/cc.11.2.18699
Fields, B. N., Knipe, D. M., and Howley, P. M. (2007). Fields Virology. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins.
Gaspar, M., and Shenk, T. (2006). Human cytomegalovirus inhibits a DNA damage response by mislocalizing checkpoint proteins. Proc. Natl. Acad. Sci. U.S.A. 103, 2821–2826. doi: 10.1073/pnas.0511148103
Goff, S. P. (2007). Host factors exploited by retroviruses. Nat. Rev. Microbiol. 5, 253–263. doi: 10.1038/nrmicro1541
Härtl, B., Zeller, T., Blanchette, P., Kremmer, E., and Dobner, T. (2008). Adenovirus type 5 early region 1B 55-kDa oncoprotein can promote cell transformation by a mechanism independent from blocking p53-activated transcription. Oncogene 27, 3673–3684. doi: 10.1038/sj.onc.1211039
He, Y., Xu, K., Keiner, B., Zhou, J., Czudai, V., Li, T., et al. (2010). Influenza A virus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 84, 12832–12840. doi: 10.1128/JVI.01216-10
Hegde, N. R., and Gore, M. M. (2017). Japanese encephalitis vaccines: Immunogenicity, protective efficacy, effectiveness, and impact on the burden of disease. Hum. Vaccin. Immunother. 13, 1–18. doi: 10.1080/21645515.2017.1285472
Houzet, L., and Jeang, K. T. (2011). Genome-wide screening using RNA interference to study host factors in viral replication and pathogenesis. Exp. Biol. Med. 236, 962–967. doi: 10.1258/ebm.2010.010272
Kim, J. Y., Park, S. Y., Lyoo, H. R., Koo, E. S., Kim, M. S., and Jeong, Y. S. (2015). Extended stability of cyclin D1 contributes to limited cell cycle arrest at G1-phase in BHK-21 cells with Japanese encephalitis virus persistent infection. J. Microbiol. 53, 77–83. doi: 10.1007/s12275-015-4661-z
Lai, C. K., Jeng, K. S., Machida, K., Cheng, Y. S., and Lai, M. M. (2008). Hepatitis C virus NS3/4A protein interacts with ATM, impairs DNA repair and enhances sensitivity to ionizing radiation. Virology 370, 295–309. doi: 10.1016/j.virol.2007.08.037
Lau, A., Swinbank, K. M., Ahmed, P. S., Taylor, D. L., Jackson, S. P., Smith, G. C., et al. (2005). Suppression of HIV-1 infection by a small molecule inhibitor of the ATM kinase. Nat. Cell Biol. 7, 493–500. doi: 10.1038/ncb1250
Lavin, M. F., and Kozlov, S. (2007). ATM activation and DNA damage response. Cell Cycle 6, 931–942. doi: 10.4161/cc.6.8.4180
Lilley, C. E., Carson, C. T., Muotri, A. R., Gage, F. H., and Weitzman, M. D. (2005). DNA repair proteins affect the lifecycle of herpes simplex virus 1. Proc. Natl. Acad. Sci. U.S.A. 102, 5844–5849. doi: 10.1073/pnas.0501916102
Lilley, C. E., Schwartz, R. A., and Weitzman, M. D. (2007). Using or abusing: viruses and the cellular DNA damage response. Trends Microbiol. 15, 119–126. doi: 10.1016/j.tim.2007.01.003
Machida, K., Cheng, K. T., Sung, V. M., Lee, K. J., Levine, A. M., and Lai, M. M. (2004). Hepatitis C virus infection activates the immunologic (type II) isoform of nitric oxide synthase and thereby enhances DNA damage and mutations of cellular genes. J. Virol. 78, 8835–8843. doi: 10.1128/JVI.78.16.8835-8843.2004
Mansfield, K. L., Hernández-Triana, L. M., Banyard, A. C., Fooks, A. R., and Johnson, N. (2017). Japanese encephalitis virus infection, diagnosis and control in domestic animals. Vet. Microbiol. 201, 85–92. doi: 10.1016/j.vetmic.2017.01.014
Moody, C. A., and Laimins, L. A. (2009). Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLoS Pathog. 5:e1000605. doi: 10.1371/journal.ppat.1000605
NSC. (2004). 基 因 重 組 實 驗 守 則 “Guidelines of for research involving recombinant DNA molecules”, Appendix, I. I., Risk Factor Group 2:51.
Scheffner, M., Huibregtse, J. M., Vierstra, R. D., and Howley, P. M. (1993). The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 75, 495–505. doi: 10.1016/0092-8674(93)90384-3
Shirata, N., Kudoh, A., Daikoku, T., Tatsumi, Y., Fujita, M., Kiyono, T., et al. (2005). Activation of ataxia telangiectasia-mutated DNA damage checkpoint signal transduction elicited by herpes simplex virus infection. J. Biol. Chem. 280, 30336–30341. doi: 10.1074/jbc.M500976200
Taylor, T. J., and Knipe, D. M. (2004). Proteomics of herpes simplex virus replication compartments: association of cellular DNA replication, repair, recombination, and chromatin remodeling proteins with ICP8. J. Virol. 78, 5856–5866. doi: 10.1128/JVI.78.11.5856-5866.2004
Tiroumourougane, S. V., Raghava, P., and Srinivasan, S. (2002). Japanese viral encephalitis. Postgrad. Med. J. 78, 205–215. doi: 10.1136/pmj.78.918.205
Weitzman, M. D., Carson, C. T., Schwartz, R. A., and Lilley, C. E. (2004). Interactions of viruses with the cellular DNA repair machinery. DNA Repair. 3, 1165–1173. doi: 10.1016/j.dnarep.2004.03.018
Wilkinson, D. E., and Weller, S. K. (2004). Recruitment of cellular recombination and repair proteins to sites of herpes simplex virus type 1 DNA replication is dependent on the composition of viral proteins within prereplicative sites and correlates with the induction of the DNA damage response. J. Virol. 78, 4783–4796. doi: 10.1128/JVI.78.9.4783-4796.2004
Wu, S. C., Chang, S. C., Wu, H. Y., Liao, P. J., and Chang, M. F. (2008). Hepatitis C virus NS5A protein down-regulates the expression of spindle gene Aspm through PKR-p38 signaling pathway. J. Biol. Chem. 283, 29396–29404. doi: 10.1074/jbc.M802821200
Wu, S. F., Lee, C. J., Liao, C. L., Dwek, R. A., Zitzmann, N., and Lin, Y. L. (2002). Antiviral effects of an iminosugar derivative on flavivirus infections. J. Virol. 76, 3596–3604. doi: 10.1128/JVI.76.8.3596-3604.2002
Yew, P. R., and Berk, A. J. (1992). Inhibition of p53 transactivation required for transformation by adenovirus early 1B protein. Nature 357, 82–85.
Yonaha, M., Chibazakura, T., Kitajima, S., and Yasukochi, Y. (1995). Cell cycle-dependent regulation of RNA polymerase II basal transcription activity. Nucleic Acids Res. 23, 4050–4054. doi: 10.1093/nar/23.20.4050
Yuan, X., Yao, Z., Wu, J., Zhou, Y., Shan, Y., Dong, B., et al. (2007). G1 phase cell cycle arrest induced by SARS-CoV 3a protein via the cyclin D3/pRb pathway. Am. J. Respir. Cell Mol. Biol. 37, 9–19. doi: 10.1165/rcmb.2005-0345RC
Zhao, X., Madden-Fuentes, R. J., Lou, B. X., Pipas, J. M., Gerhardt, J., Rigell, C. J., et al. (2008). Ataxia telangiectasia-mutated damage-signaling kinase- and proteasome-dependent destruction of Mre11-Rad50-Nbs1 subunits in Simian virus 40-infected primate cells. J. Virol. 82, 5316–5328. doi: 10.1128/JVI.02677-07
Keywords: Japanese encephalitis virus (JEV), kinase/phosphatase-wide RNAi screening, checkpoint kinase 2 (CHK2), ataxia-telangiectasia mutated kinase (ATM), cell cycle G1 arrest
Citation: Chan Y-L, Liao C-L and Lin Y-L (2018) Human Kinase/Phosphatase-Wide RNAi Screening Identified Checkpoint Kinase 2 as a Cellular Factor Facilitating Japanese Encephalitis Virus Infection. Front. Cell. Infect. Microbiol. 8:142. doi: 10.3389/fcimb.2018.00142
Received: 13 December 2017; Accepted: 20 April 2018;
Published: 17 May 2018.
Edited by:
Alan G. Goodman, Washington State University, United StatesReviewed by:
Shenngbo Cao, Huazhong Agricultural University, ChinaMaureen T. Long, University of Florida College of Veterinary Medicine, United States
Copyright © 2018 Chan, Liao and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yi-Lin Chan, cGhkLmVsYWluZWNoYW5AZ21haWwuY29t
Yi-Ling Lin, eWxsQGlibXMuc2luaWNhLmVkdS50dw==