
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell. Infect. Microbiol. , 04 August 2017
Sec. Bacteria and Host
Volume 7 - 2017 | https://doi.org/10.3389/fcimb.2017.00351
 Andrew J. Fleetwood1*
Andrew J. Fleetwood1* Man K.S. Lee2
Man K.S. Lee2 William Singleton3
William Singleton3 Adrian Achuthan1
Adrian Achuthan1 Ming-Chin Lee1
Ming-Chin Lee1 Neil M. O'Brien-Simpson3Andrew D. Cook1Andrew J. Murphy2
Neil M. O'Brien-Simpson3Andrew D. Cook1Andrew J. Murphy2 Stuart G. Dashper3
Stuart G. Dashper3 Eric C. Reynolds3John A. Hamilton1
Eric C. Reynolds3John A. Hamilton1Porphyromonas gingivalis is one of the bacterial species most closely associated with periodontitis and can shed large numbers of outer membrane vesicles (OMVs), which are increasingly thought to play a significant role in bacterial virulence and pathogenicity. Macrophages are amongst the first immune cells to respond to bacteria and their products, so we sought to directly compare the response of macrophages to P. gingivalis or its purified OMVs. Macrophages stimulated with OMVs produced large amounts of TNFα, IL-12p70, IL-6, IL-10, IFNβ, and nitric oxide compared to cells infected with P. gingivalis, which produced very low levels of these mediators. Both P. gingivalis and OMVs induced a shift in macrophage metabolism from oxidative phosphorylation (OXPHOS) to glycolysis, which was supported by enhanced lactate release, decreased mitochondrial oxygen consumption with reduced spare respiratory capacity, as well as increased mitochondrial reactive oxygen species (ROS) production. Corresponding to this metabolic shift, gene expression analysis of macrophages infected with P. gingivalis or stimulated with OMVs revealed a broad transcriptional upregulation of genes critical to glycolysis and a downregulation of genes associated with the TCA cycle. Upon examination of inflammasome signaling and pyroptosis it was found that P. gingivalis did not activate the inflammasome in macrophages as the mature forms of caspase-1, IL-1β, and IL-18 were not detected and there was no extracellular release of lactate dehydrogenase (LDH) or 7-AAD staining. In comparison, macrophages stimulated with OMVs potently activated caspase-1, produced large amounts of IL-1β, IL-18, released LDH, and were positive for 7-AAD indicative of pyroptotic cell death. These data directly quantitate the distinct effects of P. gingivalis and its OMVs on macrophage inflammatory phenotype, mitochondrial function, inflammasome activation, and pyroptotic cell death that may have potential implications for their roles in chronic periodontitis.
Porphyromonas gingivalis is recognized as a keystone pathogen (Hajishengallis et al., 2012) and is one of the bacterial biofilm species isolated from subgingival plaque most strongly associated with clinical indicators of periodontitis, including increased pocket depth and bleeding on probing (Socransky et al., 1998; Komiya et al., 2000). A common feature of Gram-negative bacteria, like P. gingivalis, is the biogenesis of outer membrane vesicles (OMVs), which are spherical membrane structures that are increasingly thought to play a significant role in microbial virulence and not simply by-products of bacterial cell wall damage or lysis (Kuehn and Kesty, 2005; Ellis and Kuehn, 2010). P. gingivalis OMVs are enriched for the pathogen's major virulence factors such as gingipains (Arg- and Lys-specific proteolytic enzymes) and lipopolysaccharide (LPS) (Veith et al., 2014). Due to the small size of OMVs (50–70 nm in diameter) they spread more readily in tissues than their larger parent cells (Kuehn and Kesty, 2005; Darveau, 2010). As a result, P. gingivalis OMVs are highly immunogenic and have been found to induce infiltration of neutrophils in connective tissue (Srisatjaluk et al., 1999) and promote macrophage foam cell formation (Qi et al., 2003).
Recently, metabolic reprogramming in host immune cells, particularly in macrophages and dendritic cells has been implicated in regulating their phenotype and function (O'Neill and Pearce, 2016). Macrophages activated with LPS and IFNγ (so called M1 macrophages) shift their glucose metabolism from oxidative phosphorylation (OXPHOS) to glycolysis and this metabolic shift is central to their production of mediators associated with an M1 phenotype (e.g., NO) (Tannahill et al., 2013). Likewise the commitment of IL-4 stimulated macrophages (so called M2 macrophages) to OXPHOS to generate ATP is critical to their adoption of a M2 phenotype (Vats et al., 2006; Huang et al., 2014). A detailed comparison of metabolism in M1 vs. M2 macrophages identified specific metabolic pathways in both cell types that were critical in governing their polarization (Jha et al., 2015). Many recent studies have examined the links between glycolysis and cell effector function. For example, LPS-induced glycolysis enables dendritic cell maturation (Everts et al., 2014) whilst glycolysis is involved in inflammasome activation (Masters et al., 2010; Tannahill et al., 2013; Moon et al., 2015) and promotion of antibacterial responses in macrophages (Cordes et al., 2016; Lampropoulou et al., 2016). Much of this important information has been generated with purified LPS (reviewed in O'Neill et al., 2016) with relatively few studies (Garaude et al., 2016; Gleeson et al., 2016) addressing the impact of viable bacteria on cellular metabolism.
P. gingivalis has been shown to survive within macrophages (Wang et al., 2007; Wang and Hajishengallis, 2008; Slocum et al., 2014) and myeloid dendritic cells where it reprograms them to induce an immunosuppressive T cell effector response (Zeituni et al., 2009). Indeed, myeloid dendritic cells have been suggested to disseminate P. gingivalis from the oral mucosa to atherosclerotic plaques (Carrion et al., 2012). The ability of P. gingivalis to persist intracellularly is intriguing given the link between periodontal disease and certain systemic inflammatory conditions (Hajishengallis, 2015). Pyroptosis is a programmed form of proinflammatory cell death that allows the elimination of intracellular pathogens (Franchi et al., 2012; Aachoui et al., 2013). Pyroptosis occurs following activation of the cytosolic inflammasome signaling complex, which generates active caspase-1 leading to pore formation and the release of cytosolic contents (e.g., LDH) and production of the inflammatory cytokines IL-1β and IL-18 (Shi et al., 2015). There is conflicting evidence as to whether P. gingivalis can activate the inflammasome in macrophages, which in large part seems to be due to differences in cell populations studied (Taxman et al., 2006, 2012; Slocum et al., 2014). Another complication has been the gingipain-mediated degradation of the major readouts used to determine inflammasome activation and pyroptosis (Jung et al., 2015). P. gingivalis evasion of inflammasome activation would provide an intracellular niche for the pathogen to survive and disseminate to distant sites.
P. gingivalis OMVs can also persist within the lysosomes of host cells (Gui et al., 2016), are potent activators of Toll-like receptors (TLRs) (Cecil et al., 2016) and can deliver virulence factors deep into host tissues (Mashburn-Warren and Whiteley, 2006; Darveau, 2010). It is currently unknown whether P. gingivalis OMVs activate the inflammasome or induce pyroptotic cell death. The host response to OMVs is likely to be different to the parent bacterium as their phospholipid, gingipain and LPS profiles are distinct (Veith et al., 2014; Ho et al., 2015; Xie, 2015; Gui et al., 2016). It has been speculated that P. gingivalis OMVs act as a virulence factor secretion system that contributes to pathogenicity partly by supporting evasion of host immune defense mechanisms (Gui et al., 2016). In agreement with this a recent study found that the ability of P. gingivalis to subvert a local immune response was due to OMVs rendering monocytes unresponsive to live P. gingivalis (Waller et al., 2016). To better understand the immunostimulatory capabilities of OMVs, we recently developed a high-sensitivity flow cytometry method to enumerate P. gingivalis-derived OMVs (Cecil et al., 2016). This method allows removal of non-OMV associated material (e.g., fimbriae and secreted proteins; Nakao et al., 2014) to yield highly purified single lipid-bilayer vesicles. We found that the immunogenicity of vesicles purified from three periodontal pathogens (as measured by TLR activation) could be better differentiated when standardized using vesicle number compared to protein concentration (Cecil et al., 2016). To our knowledge there have been no studies directly comparing the immune responses elicited by P. gingivalis or its purified OMVs. By utilizing our protocol for enumerating vesicles (Cecil et al., 2016) we were able to directly compare the effect of P. gingivalis and OMVs at identical infection ratios on macrophage biology. Such studies may provide a more accurate understanding of their immunogenicity.
In the current study, we show that P. gingivalis and its OMVs induce a metabolic shift in macrophages from OXPHOS to glycolysis whilst OMVs potently activate proinflammatory cytokine production, inflammasome signaling, and pyroptotic cell death in macrophages. P. gingivalis only weakly activated macrophage cytokine production and did not activate the inflammasome or induce pyroptosis. These findings highlight the distinct ways in which P. gingivalis and its OMVs modulate the host immune response and underscore the potent stimulatory nature of OMVs, which may help explain their unique functions in periodontal disease.
Murine BMM (from 8 to 12 week old C57BL/6 mice) and human MDM were prepared as before (Fleetwood et al., 2015). Briefly, bone marrow cells were isolated from femurs of mice and cultured in RPMI 1640 medium, supplemented with 10% heat-inactivated FCS, 2 mM GlutaMax-1, 100 U/ml penicillin, and 100 μg/ml streptomycin in the presence of M-CSF (2,000 U/ml). On day 4, non-adherent cells were collected and cultured for a further 3 days in M-CSF (2,000 U/ml) to derive BMM. Adherent BMM were harvested on day 7 at which stage they express the major macrophage surface markers (e.g., CSF-1R, F4/80, and Mac-1) (Lari et al., 2007). Human monocytes were purified from buffy coats (Red Cross Blood Bank, Melbourne, VIC, Australia), using RosetteSep Ab mixture (Stem Cell Technologies, Vancouver, BC, Canada), which negatively selects CD14+ monocytes, followed by Ficoll-Paque density gradient centrifugation (Way et al., 2009). Cells were then cultured in RPMI 1640 (supplemented as above) for 7 days in M-CSF (2,000 U/ml) to differentiate them into MDM (Lacey et al., 2012). All experiments were approved by the Royal Melbourne Hospital Research Foundation and Animal Ethics Committee and conducted in compliance with the guidelines of the National Health and Medical Research Council.
P. gingivalis strain W50 (ATCC 53978) was obtained from the culture collection of the Oral Health Cooperative Research Centre at the Melbourne Dental School. P. gingivalis W50 was grown and harvested as described (O'Brien-Simpson et al., 2000; Lam et al., 2016). Growth conditions of batch cultures were monitored at 650 nm using a spectrophotometer (model 295E, Perkin-Elmer, Germany). As before (Cecil et al., 2016), cells were harvested during late exponential growth by centrifugation (7,000 g, 20 min at 4°C) and enumerated (number/ml) by flow cytometry using a Cell Lab Quanta SC flow cytometer (Beckman Coulter, Australia) and a LIVE/DEAD BacLight™ Bacterial Viability Kit (Life Technologies, Australia). Bacteria were resuspended in 0.01 M phosphate buffered saline (PBS, Sigma-Aldrich), pH 7.4, before incubation with macrophages. Aliquots of P. gingivalis were heat-killed at 70°C for 1 h, as described (Palm et al., 2013).
OMVs were purified (from W50 ATCC 53978) and enumerated as described in detail here (Cecil et al., 2016). Briefly, bacteria were grown to late exponential phase and whole bacterial cells were removed by centrifugation at 8,000 × g for 30 min at 4°C (using a F10BCI-6x500 g rotor installed in an Avanti J-25I Centrifuge, Beckman Coulter). The collected supernatant was filtered through a 0.22 μm filter (Merck-Millipore, Australia) and then concentrated through a 100-kDa filter using a tangential flow filtration Minimate TFF System (PALL Life Sciences, Australia). The collected concentrate was centrifuged at 100,000 × g for 2 h at 4°C (using a JA-30.50 Ti fixed angle rotor installed in an Avanti J-30I Centrifuge, Beckman Coulter) to yield a crude OMV preparation. Highly purified OMVs were prepared using OptiPrep™ density gradient centrifugation. Gradient fractions containing purified OMVs were identified using a Qubit 2.0 Fluorometer (Life Technologies). Fractions containing the purified OMVs were pooled and washed with 0.01 M PBS (Sigma-Aldrich), pH 7.4 at 150,000 × g for 2 h at 4°C (using a SW40 Ti rotor installed in an Optima L-80XP Ultracentrifuge, Beckman-Coulter) and resuspended in 0.22 μm filtered 0.01 M PBS. Aliquots of washed OMVs were enumerated (number/ml) using an Apogee A50-Micro Flow Cytometer calibrated for flow and event rates with Apogee Flow Systems Calibration Beads. Enumerated OMVs were resuspended in 0.01 M PBS (Sigma-Aldrich), pH 7.4, and the protein concentration quantitated by Qubit Protein Assay Kit (Life Technologies). Aliquots of OMVs were heat-inactivated at 70°C for 1 h, as described (Palm et al., 2013).
Macrophages were infected with viable P. gingivalis, heat-killed P. gingivalis (HK-Pg), OMVs or heat-inactivated OMVs (HI-OMVs) at a multiplicity of infection (MOI) of 10:1, 25:1 or 100:1 bacilli or OMV/cell for 2 h. After 2 h the extracellular bacteria/OMVs were removed and the macrophages were washed with PBS and then treated with fresh media (containing 100 U/ml penicillin, 100 μg/ml streptomycin to kill any remaining extracellular bacteria), and M-CSF (2,000 U/ml) for 24 h. This stimulation protocol was used throughout the study and based on our initial dose-response experiments (see Supplementary Figures 2A,B) and other studies (Taxman et al., 2012; Cecil et al., 2016; Waller et al., 2016) an MOI of 25:1 was chosen as the optimal dose for this study. For reference, OMVs (at an MOI of 25:1) was equal to a protein concentration of 3.0 μg/ml. In certain experiments cells were stimulated with P. gingivalis-derived LPS (Invivogen; LPS-PG Ultrapure at 100 ng/ml) for 24 h as indicated. These infection protocols were used throughout the study. In specific experiments the competitive gingipain inhibitors, KYT-1 and KYT-36 (Peptide Institute, Osaka, Japan) (Kadowaki et al., 2004) were included in the media (at 10 μM).
Total RNA was purified from BMM infected with P. gingivalis or OMVs using RNeasy Plus Mini kit (Qiagen, Valencia, CA) and was reverse transcribed using the RT2 First Strand cDNA kit (Qiagen). A RT2 Profiler PCR Array for Mouse Glucose Metabolism (PAMM-006, Qiagen) was performed according to the manufacturer's instructions (Thermal profile: Stage 1 95°C for 10 min, Stage 2 95°C for 15 s followed by 60°C for 1 min with 40 cycles) using an ABI PRISM 7900HT sequence detection system (Applied Biosystems, Carlsbad, CA). Genes on this array are grouped according to function as follows: TCA cycle, Glycolysis, Pentose Phosphate Pathway, Gluconeogenesis and Glucose Regulation, and Glycogen Metabolism. Data analysis was performed using the RT2 Profiler PCR Array Data Analysis Template v3.3 from Qiagen. A fold-change of 3, p-value < 0.05 was used as a cutoff. Data were normalized to five housekeeping genes—Gusb, Hprt1, Hsp90ab1, Gapdh, and Actb. For individual quantitative PCR analysis TaqMan probe/primer combinations for murine Glut-1 (Mm00441480_m1), Pfkfb3 (Mm00504650_m1), Hk1 (Mm00439344_m1), and Irg-1 (Mm01224532_m1) (Applied Biosystems) as before (Fleetwood et al., 2007). Threshold cycle numbers were transformed to ΔCt values, and results expressed relative to reference gene, HPRT. Assays were performed in triplicate and results expressed relative to HPRT from four independent experiments.
IL-1β, IL-18, TNFα, IL-6, IL-12p70, IL-10 (Ready-Set-Go!; eBioscience), and IFNβ (PBL Assay Science) were quantified in the culture supernatants by ELISA according to manufactures instructions. NO production was measured by quantification in a Griess reaction (Sigma). Extracellular LDH was detected with a Cytotoxicity Detection kit (Roche Diagnostics) and results are presented as the percentage of total intracellular LDH. Lactate concentration in supernatant was measured by colorimetric assay (Sigma) and read at 570 nM on a BioRad 680 Plate Reader. Five millimolars of 2-deoxyglucose (2DG; Sigma) was used as indicated.
A Seahorse utility plate (Seahorse Bioscience) containing calibrant media (200 μl/well) together with Seahorse injector port and probe plate were incubated overnight in 37°C without CO2. The following day, media from Seahorse cell culture plate was replaced with Seahorse XF assay buffer (supplemented with 10 mM glucose and 2 mM glutamine) and incubated in CO2-free incubator at 37°C for at least half an hour. Macrophages that were infected (see stimulation protocol above) were then plated at 2 × 105 cells/well in a 96-well Seahorse cell culture plate (Agilent XFe96) with one well per corner of the plate containing supplemented-media without cells, as background control. The designated injector ports were filled with 25 μl of the following MitoStress Test inhibitors; 1 μM Oligomycin (an ATP synthase inhibitor that allows calculation of mitochondrial O2 consumption); 1 μM Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone [(FCCP), an ionophore that uncouples ATP synthesis from the electron transport chain to reveal maximal mitochondrial OXPHOS]; 1.5 μM Antimycin and 1.5 μM Rotenone (Complex I and III inhibitors that are used to reveal mitochondrial and non-mitochondrial O2 consumption) (Van den Bossche et al., 2015). The MitoStress Test Assay was run over 80 min on a Seahorse XF-e96 Bioanalyzer (Agilent) after calibration of utility plate with injector port plate as per manufacturer's instruction.
As per manufacturer's instructions, stock solutions of MitoTracker Red CMXRos (1 mM), MitoSox Red (5 mM), and MitoTracker Green FM (1 mM) were made in high-quality, anhydrous dimethyl sulfoxide (DMSO) (Sigma). MitoSox Red was further diluted to a working solution of 5 μM in pre-warmed Hanks balanced salt solution (HBSS) with calcium and magnesium. Macrophages that were infected (see stimulation protocol above) were collected in ice-cold PBS and stained for 15 min at 37°C in a CO2 incubator with pre-warmed PBS (1% BSA) containing MitoTracker Red CMXRos (25 nM) or MitoTracker Green FM (25 nM) or with HBSS/Ca/Mg buffer containing MitoSox Red (5 μM). Following staining the cells were washed twice with fresh medium (at 37°C) and analyzed immediately to determine mitochondrial mass, membrane potential, and ROS production. In parallel cells were stained with 7-amino actinomycin D (7-AAD; R&D Systems) for 30 min at 4°C. 7-AAD is a membrane impermeant dye that is excluded from viable cells. After staining, cells were washed twice in pre-warmed PBS (1% BSA) and then analyzed immediately using a CyAn ADP Flow Cytometer (Beckman Coulter). As a positive control for 7-AAD staining, macrophages (uninfected) were treated with H2O2 for 60 min (1 mM; Sigma) prior to viability analysis (Fink and Cookson, 2006). For flow cytometric analysis, a typical forward and side-scatter gate was set to exclude dead cells and aggregates and a total of 104 events in the gate were collected and analyses were performed using Kaluza v1.2 (Beckman Coulter) and data are expressed as mean fluorescence intensity (MFI). For 7-AAD staining the data are expressed as the percentage of positive cells for 7-AAD (± SD) compared to the untreated control.
Briefly, macrophages (3 × 106) were lysed with RIPA buffer (25 mM Tris-HCl pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, and complete protease inhibitors) at 4°C and whole cell extracts collected. Lysates were clarified by centrifugation at 13,000 × g for 10 min at 4°C and protein concentrations were determined with a Bio-Rad protein assay kit and equal amounts of protein were separated on a NuPAGE Novex 4–12% Bis-Tris gel (Thermo Fisher Scientific) and transferred onto PVDF membrane (Bio-Rad). Membranes were probed with antibodies against caspase-1 (clone 14F468) (Novus Biologicals CO, USA), NLRP3 (D4D8T), Asc (D2W8U), IL-1β (AF-401-NA) (R&D Systems, MN, USA), β-actin (clone AC-74, Sigma-Aldrich, St. Louis, MO). Binding was visualized by incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies and chemilluminescence (ECL). In particular experiments protein was precipitated from cell supernatants for detection of extracellular caspase-1, as described (Vanaja et al., 2016).
ANOVA was used to analyse the statistical significance of the data. The Bonferroni post-hoc test was employed for the comparisons between groups (GraphPad Prism7). p < 0.05 indicate significance.
To compare cytokine production induced by the pathogen and its components, murine bone-marrow-derived macrophages (BMM) were infected with viable P. gingivalis or its purified OMVs for 2 h, after which the culture medium was removed and replaced with fresh medium containing antibiotics. To account for possible cytokine degradation by heat-labile proteases (e.g., gingipains) present on OMVs or its parent cell (Abe et al., 1998; Calkins et al., 1998; Stathopoulou et al., 2009), we also challenged cells with heat-killed P. gingivalis (HK-Pg) and HI-OMVs.
BMM infected with P. gingivalis produced only modest amounts of TNFα, IL-12p70, IL-6, IL-10, and IFNβ compared to those stimulated with OMVs, which produced significantly (p < 0.05) higher levels of all of these mediators (Figure 1). BMM stimulated with OMVs produced large amounts of NO whilst cells infected with P. gingivalis failed to produce detectable NO (Figure 1). HK-Pg infection of BMM induced similar levels of TNFα, IL-12p70, IL-6, IL-10, and IFNβ compared to infection with viable P. gingivalis. Similar to P. gingivalis, HK-Pg did not induce detectable NO release from infected BMM. Of note, significantly (p < 0.05) higher levels of TNFα (Figure 1) were detected in the supernatants of BMM stimulated with HI-OMVs vs. OMVs, which is consistent with reports of gingipain-mediated degradation of this cytokine (Calkins et al., 1998). Interestingly, OMV-induced NO release from BMM was abolished when heat-inactivated (p < 0.05), suggesting a role for heat-labile protein(s) or enzyme(s) in promoting macrophage NO production. These data demonstrate the powerful immunostimulatory ability of OMVs by inducing large amounts of TNFα, IL-12p70, IL-6, IL-10, IFNβ, and NO from macrophages. In comparison, P. gingivalis-infected macrophages produce very low levels of these cytokines and failed to induce detectable NO. These findings are consistent with OMVs being enriched for virulence factors like LPS relative to their parent cells (Veith et al., 2014; Ho et al., 2015; Xie, 2015; Gui et al., 2016).
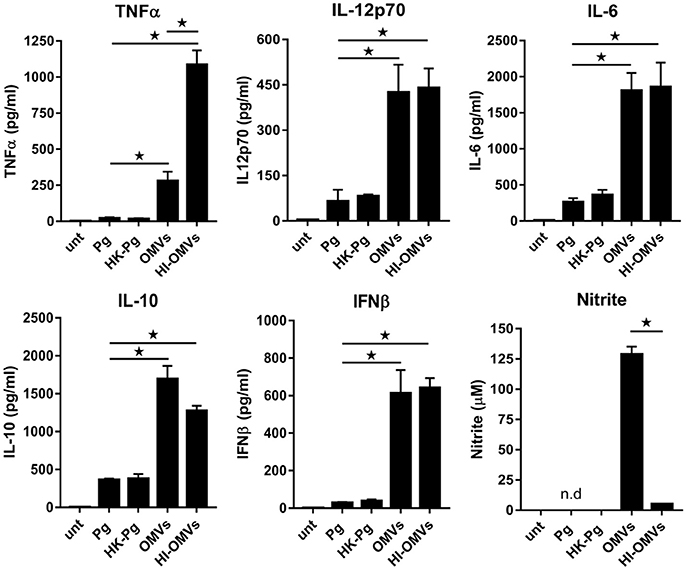
Figure 1. P. gingivalis and its OMVs differentially induce macrophage cytokine and nitric oxide release. BMM were infected (2 h at MOI of 25:1, see Materials and Methods) with viable P. gingivalis (Pg), heat-killed-Pg (HK-Pg), OMVs or heat-inactivated-OMVs (HI-OMVs), and release of TNFα, IL-12p70, IL-6, IL-10, IFNβ, and nitrite were measured at 24 h (6 h for TNFα). Cytokine levels (ELISA) and nitrite production (Griess reaction) were quantified and data are mean ± SD from four independent experiments (*p < 0.05; n.d, not detected).
The studies of macrophage remodeling of their metabolism have mostly been done in response to purified Escherichia coli LPS (O'Neill et al., 2016). Thus, we wanted to investigate the impact of P. gingivalis and its purified OMVs on macrophage glucose metabolism. To investigate this, BMM and human monocyte-derived macrophages (MDM) were infected with viable P. gingivalis or purified OMVs (as above) and lactate release was measured as the end product of the glycolytic pathway.
BMM (Figure 2A) and MDM (Figure 2D) demonstrated significantly (p < 0.05) increased lactate production at 24 h following infection with P. gingivalis or stimulation with OMVs relative to untreated cells. In the case of P. gingivalis-infected BMM (Figure 2B) and MDM (Figure 2E) the lactate levels increased over 72 h and could be blocked (p < 0.05) by pre-treatment of cells with the glycolytic inhibitor 2-deoxyglucose (2DG) (Figures 2C,F). HK-Pg and HI-OMVs also significantly (p < 0.05) increased lactate production from BMM (Supplementary Figure 1A) and MDM (Supplementary Figure 1D). We further found that P. gingivalis-derived LPS increased lactate production at 24 h in BMM and MDM relative to untreated cells (Supplementary Figures 1A,D). This shift toward glycolysis in BMM in response to P. gingivalis or OMVs was accompanied by an increased expression of Glut-1, Pfkfb3, and Hk1 (Figure 2G), which encode proteins critically involved in the glycolytic pathway. Interestingly, we also found that BMM infected with P. gingivalis or stimulated with OMVs increased expression of Irg-1 (Figure 2G), which has recently been postulated to impair OXPHOS in LPS-activated macrophages (Lampropoulou et al., 2016; Nemeth et al., 2016). Notably, BMM stimulated with OMVs had significantly (p < 0.05) higher expression of Glut-1, Pfkfb3, Hk1, and Irg-1 relative to P. gingivalis-infected BMM. These data show for the first time that P. gingivalis (and its major components, OMVs and LPS) induce glycolysis in murine and human macrophages.
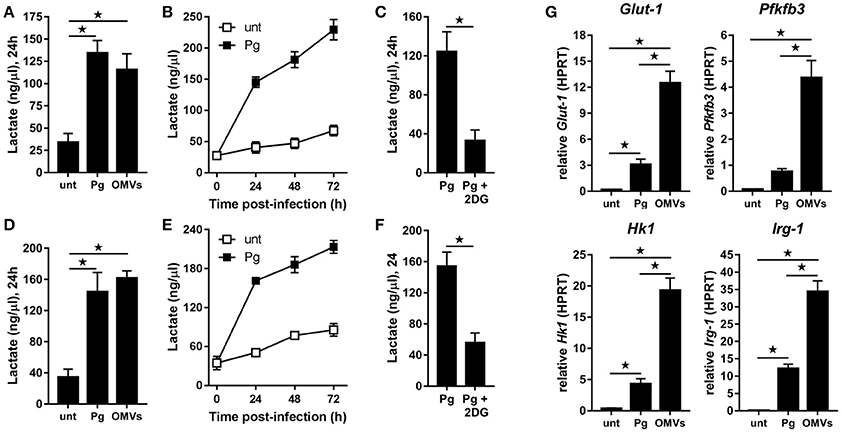
Figure 2. P. gingivalis and its OMVs induce glycolysis in macrophages. Secreted lactate concentrations from murine BMM (A–C) and human MDM (D–F) at indicated times (24–72 h) after infection with P. gingivalis (Pg) or its OMVs (25:1 MOI) (see Materials and Methods). Macrophages were treated with 2DG (5 mM) as indicated. (G) Gene expression by qPCR of Glut-1, Pfkfb3, Hk-1, and Irg-1 were quantitated in BMM at 24 h (treated as in A). Data are mean ± SD from four independent experiments (*p < 0.05).
To determine whether the observed increase in glycolysis was paralleled with changes to mitochondrial respiration we next performed extracellular flux analysis to measure the mitochondrial activity of untreated macrophages vs. those that were infected (as above) with P. gingivalis or stimulated with OMVs. As detailed in Figures 3A–C (for BMM) and Figures 3D–F (for MDM), changes in the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured in response to oligomycin (oligo), FCCP, and Antimycin + Rotenone (Ant + Rot) injection (Everts et al., 2012). These analyses revealed BMM and MDM infected with P. gingivalis or stimulated with OMVs had a markedly diminished spare respiratory capacity (SRC), as indicated by the difference between maximal OCR (after FCCP injection) and the basal OCR (Figures 3A,D), consistent with a reduced commitment to OXPHOS in these cells (Huang et al., 2016). Basal OCR was significantly (p < 0.05) reduced in MDM that were infected with P. gingivalis or stimulated with OMVs (Figure 3E) whilst basal OCR was significantly (p < 0.05) reduced in OMV-stimulated BMM (Figure 3B). P. gingivalis-infected BMM had a reduced OCR but this did not reach statistical significance (Figure 3B). HK-Pg, HI-OMVs, and P. gingivalis-derived LPS also reduced the SRC of BMM and MDM (Supplementary Figures 1B,E) whilst HI-OMVs and LPS also significantly reduced their basal OCR (Supplementary Figures 1C,F). Consistent with their increased lactate generation (Figures 2A,D), BMM and MDM infected with P. gingivalis or stimulated with OMVs has a significantly (p < 0.05) increased ECAR (Figures 3C,F). Upon further examination of their mitochondrial function, we observed that P. gingivalis-infected and OMV-stimulated BMM had decreased mitochondrial membrane potential, increased mitochondrial ROS release with no significant change in mitochondrial mass (Figure 3G). These data show for the first time that murine and human macrophages switch their glucose metabolism from OXPHOS to glycolysis and generate mitochondrial ROS in response to P. gingivalis (and its major components, OMVs and LPS).
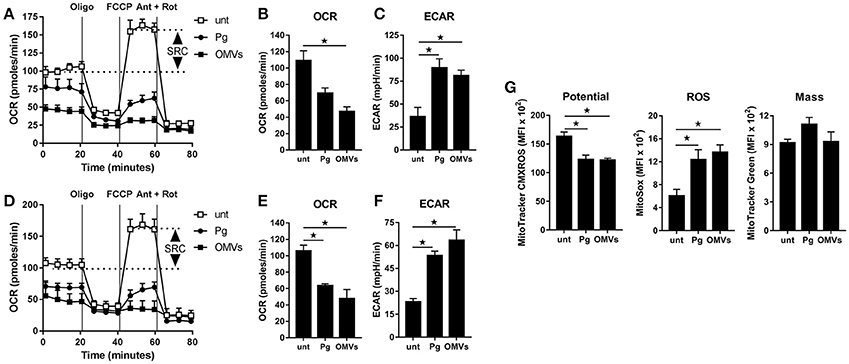
Figure 3. P. gingivalis and its OMVs disrupt mitochondrial respiration in macrophages. The mitochondrial respiration of BMM (A–C) and MDM (D–F) following infection with P. gingivalis (Pg) or OMVs (2 h at MOI of 25:1, see Materials and Methods) measured in a Seahorse XF-24 analyzer after 24 h. (A,D) Real-time oxygen consumption (OCR) was determined during sequential treatments with oligomycin (ATP-synthase inhibitor), FCCP, and antimycin-A + rotenone (ETC inhibitors) over 80 min. Spare respiratory capacity (SRC), the difference between maximal OCR, and the basal OCR, is depicted in the plots. Data represent means ± SD of triplicates. One of three experiments is shown. Basal OCR (B,E) and extracellular acidification rate (ECAR) (C,F) were analyzed (at 24 h) as readouts for oxidative phosphorylation (OXPHOS) and glycolysis, respectively. Data are mean ± SD from three independent experiments. (G) Mitochondrial membrane potential, ROS production and mass were determined at 24 h in BMM (treated as in A) by flow cytometry after staining with MitoTracker CMXROS, MitoSOX Red and MitoTracker Green FM, respectively. Data are mean fluorescence intensity (MFI) ± SD from three independent experiments (*p < 0.05).
Our earlier data (see Figure 2G) revealed that P. gingivalis and its OMVs induced the expression of several key genes involved in glucose metabolism. As such, a qPCR array focused on glucose metabolism was used to measure changes in gene expression in P. gingivalis or OMV stimulated macrophages. BMM were infected (as above) with P. gingivalis or OMVs and changes in expression (of a panel of 84 genes) were compared to untreated cells at 24 h. The gene expression data relative to untreated cells for P. gingivalis and OMV treated BMM are summarized in Volcano plots (Figure 4A) and are grouped according to gene function (Figures 4B–F). Genes with greater than three-fold difference from untreated cells were considered significant.
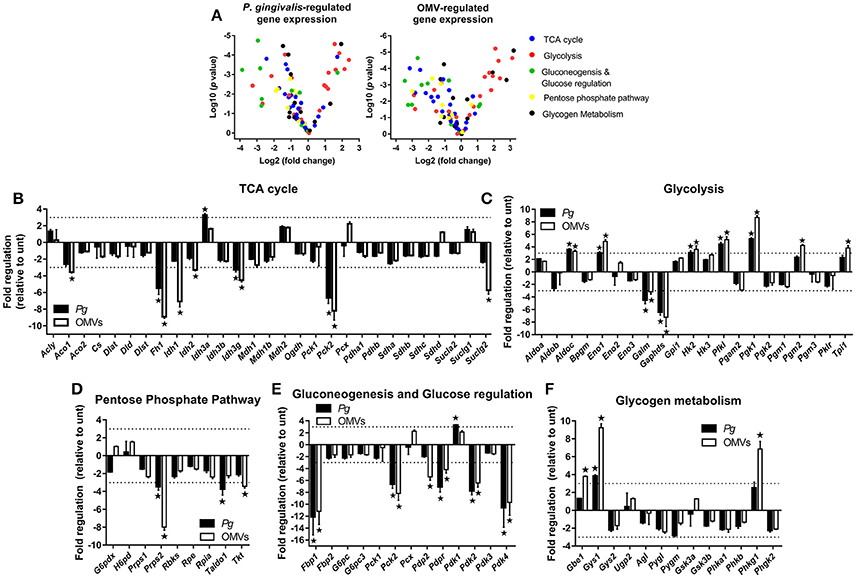
Figure 4. P. gingivalis and its OMVs regulate macrophage metabolic gene expression. (A) Volcano plots showing genes differentially expressed in BMM following infection with P. gingivalis or OMVs (25:1 MOI, see Materials and Methods) relative to untreated BMM after 24 h using a Glucose Metabolism PCR Array (PAMM-006Z). RNA was purified for cDNA synthesis and qPCR performed and (B–F) genes are grouped according to function and expression is shown relative to untreated BMM. Data are mean ± SD from three independent experiments. Genes with greater than three-fold difference from untreated cells (indicated with dotted line) were considered significant (*p < 0.05).
From the panel of 84 genes that were analyzed, 20 genes were significantly (p < 0.05) regulated in BMM infected with P. gingivalis relative to untreated cells whilst 27 genes were significantly (p < 0.05) regulated in OMV treated BMM relative to control cells. In broad terms both P. gingivalis and OMVs induced similar expression profiles in BMM compared to control cells, with both stimuli reducing the expression of several TCA cycle genes (Figure 4B) whilst enhancing the expression of many glycolytic genes (Figure 4C). Additionally, P. gingivalis and OMV stimulation of BMM led to reduced expression of transcripts involved in the pentose phosphate pathway (Figure 4D) and in gluconeogenesis and glucose regulation (Figure 4E). OMVs tended to be a more potent stimulus at least under the current experimental conditions than P. gingivalis. Specific alterations in gene expression are discussed in more detail below.
P. gingivalis and OMVs significantly (p < 0.05) downregulated BMM expression of many genes involved in the TCA cycle like fumarase-1 (Fh1), isocitrate dehydrogenase 3 gamma (Idh3g), and phosphoenolpyruvate carboxykinase 2 (Pck2) (Figure 4B). Three key enzymes of the TCA cycle involved in the reactions that convert citrate to succinate, aconitase 1 (Aco1), isocitrate dehydrogenase 1 and 2 (Idh1/2), and succinyl-CoA ligase (GDP-forming) subunit beta (Suclg2) were specifically downregulated (p < 0.05) in response to OMVs (Figure 4B). On the other hand, expression of genes related to glycolysis like aldolase C (Aldoc), enolase 1 (Eno1), hexokinase 2 (Hk2), phosphofructokinase (Pfk1), and phosphoglycerate kinase 1 (Pgk1) were all enhanced (p < 0.05) in BMM following P. gingivalis or OMV stimulation (Figure 4C). Not all glycolytic genes were upregulated with aldose 1-epimerase (Galm) and glyceraldehyde-3-phosphate dehydrogenase, spermatogenic (Gapdhs) both being downregulated (p < 0.05) in BMM by P. gingivalis and OMVs (Figure 4C).
Macrophages that have committed to glycolysis following activation (e.g., via LPS) have increased flux through the pentose phosphate pathway (Krawczyk et al., 2010; Freemerman et al., 2014). Interestingly, we found that BMM infected with P. gingivalis or stimulated with OMVs tended to downregulate genes in this pathway (Figure 4D). Expression of phosphoribosyl pyrophosphate synthetase 2 (Prps2), transaldolase 1 (Taldo1), and transketolase (Tkt) were all reduced in BMM infected with P. gingivalis (Prps2 and Taldo1) or stimulated with OMVs (Prps2 and Tkt) (Figure 4D, p < 0.05). Genes involved in gluconeogenesis, namely fructose bisphosphatase (Fbp1) and phosphoenolpyruvate carboxykinase 2 (Pck2) were significantly (p < 0.05) downregulated in BMM treated with P. gingivalis or OMVs (Figure 4E) as were pyruvate dehydrogenase phosphatase regulatory subunit (Pdpr) and pyruvate dehydrogenase kinase isoforms 2/4 (Pdk2/4), which are regulators of glucose metabolism (Figure 4E). Glycogen branching enzyme (Gbe1), glycogen synthase 1 (Gys1), and phosphorylase b kinase gamma catalytic chain (Phkg1) were all enhanced (p < 0.05) in OMV stimulated BMM (Figure 4F).
Overall these data reveal that P. gingivalis and its OMVs reprogram the metabolic gene expression profile of macrophages by enhancing expression of genes involved in the glycolytic pathway whilst simultaneously decreasing expression of genes involved in the TCA cycle, pentose phosphate pathway, and the gluconeogenesis and glucose regulation pathways.
Given that P. gingivalis and OMVs dramatically altered macrophage glucose metabolism and that cellular glucose metabolism is thought to regulate inflammasome activation (Everts and Pearce, 2014; O'Neill and Pearce, 2016), we next sought to address the effect of P. gingivalis and OMVs on macrophage inflammasome activation. In addition, we determined whether P. gingivalis and OMVs induced pyroptosis, which is a proinflammatory and lytic form of cell death (Schroder and Tschopp, 2010), that can occur following prolonged activation of the inflammasome (Miao et al., 2011). To investigate this BMM (Figure 5) and MDM (Figure 6) were infected with P. gingivalis, HK-Pg, OMVs, or HI-OMVs (as above) and inflammasome activation and pyroptosis measured at 24 h.
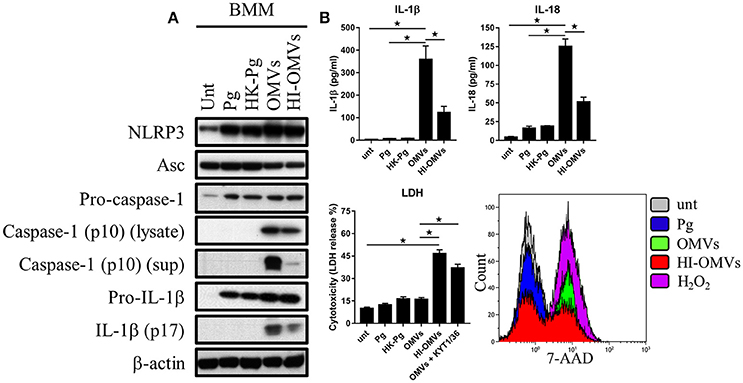
Figure 5. P. gingivalis and its OMVs differentially induce inflammasome signaling and pyroptosis in murine macrophages. BMM were infected as before (2 h at MOI of 25:1, see Materials and Methods) with viable P. gingivalis (Pg), heat-killed-Pg (HK-Pg), OMVs, or heat-inactivated-OMVs (HI-OMVs) and (A) the activation of inflammasome components in the lysates [or supernatants (sup) where indicated] measured after 24 h by Western blot; β-actin serves as a loading control throughout. Western blot data are representative of at least three independent experiments. (B) Production of IL-1β and IL-18 (by ELISA) from BMM was measured in the supernatant at 24 h. Data are mean ± SD from four independent experiments. Cell viability was determined by measuring extracellular LDH release and by 7-AAD exclusion (by flow-cytometry) at 24 h. 7-AAD is a membrane impermeant dye that is excluded from viable cells. The percent of 7-AAD positive (±SD) cells from three independent experiments was calculated relative to the untreated control. BMM treated with H2O2 (1 mM for 60 min) were included as positive control for 7-AAD. A representative histogram is shown. Cells were also treated where indicated with OMVs in the presence of KYT-1 and KYT-36 (10 μM of each). *p < 0.05.
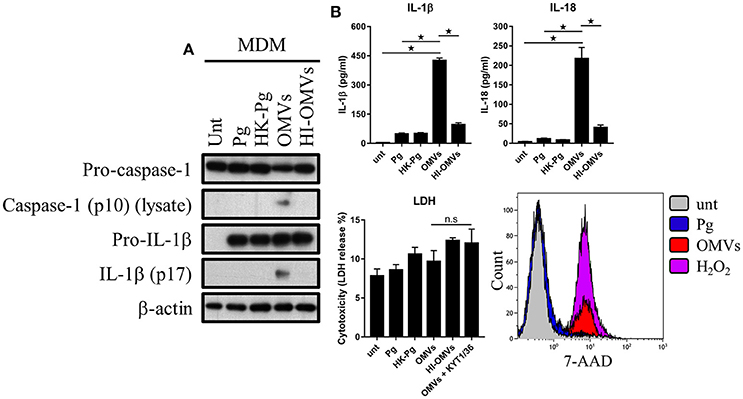
Figure 6. P. gingivalis and its OMVs differentially induce inflammasome signaling and pyroptosis in human macrophages. MDM were infected as before (2 h at MOI of 25:1, see Materials and Methods) with viable P. gingivalis (Pg), heat-killed-Pg (HK-Pg), OMVs, or heat-inactivated-OMVs (HI-OMVs) and (A) the activation of inflammasome components in the lysates was measured after 24 h by Western blot; β-actin serves as a loading control throughout. Western blot data are representative of at least three independent experiments. (B) Production of IL-1β and IL-18 (by ELISA) from MDM was measured in the supernatant at 24 h. Data are mean ± SD from four independent experiments. Cell viability was determined by measuring extracellular LDH release and by 7-AAD exclusion (by flow-cytometry) at 24 h. 7-AAD is a membrane impermeant dye that is excluded from viable cells. The percent of 7-AAD positive (±SD) cells from three independent experiments was calculated relative to the untreated control. MDM treated with H2O2 (1 mM for 60 min) were included as positive control for 7-AAD. A representative histogram is shown. Cells were also treated where indicated with OMVs in the presence of KYT-1 and KYT-36 (10 μM of each). *p < 0.05: n.s, not significant.
Treatment of BMM with P. gingivalis, HK-Pg, OMVs, or HI-OMVs induced the expression on the major inflammasome components NLRP3, pro-caspase-1, and pro-IL-1β. Asc was endogenously expressed and its levels were unchanged regardless of treatment (Figure 5A). Consistent with activation of the inflammasome, BMM treated with OMVs, and to a lesser extent HI-OMVs, triggered cleavage of pro-caspase-1 and pro-IL-1β into mature caspase-1 p10 (detected in the lysate and supernatant) and IL-1βp17 (Figure 5A), as well as inducing IL-1β and IL-18 secretion (Figure 5B). Moreover, OMV treatment of BMM led to the release of large amounts of IL-1β and IL-18 in an MOI-dependent manner (Supplementary Figure 2A) whereas infection with P. gingivalis only led to minimal amounts of these mediators being produced. A feature of pyroptosis is the formation of pores in the plasma membrane that allow the release of intracellular contents (Jorgensen and Miao, 2015). Consistent with the induction of pyroptosis, OMVs triggered significant release of the intracellular protein LDH from BMM (Figure 5B), which was only evident when BMM were treated with OMVs in the presence of the gingipain proteolytic inhibitors KYT-1 and KYT-36, or when stimulated with HI-OMVs (Figure 5B). These findings indicate that gingipains, which are enriched on OMVs (Veith et al., 2014), degrade extracellular LDH to mask this specific readout of pyroptosis. These findings concur with an earlier report of gingipain-mediated LDH proteolysis (Jung et al., 2015). As mentioned above, pyroptosis leads to the formation of pores in the plasma membrane that allow a membrane impermeant dye like 7-AAD to enter pyroptotic cells but not apoptotic or viable cells (Miao et al., 2011). We found that a significant (p < 0.05) percentage of BMM treated with OMVs (69% ± 8) and to a lesser extent HI-OMVs (46% ± 5) were positive for 7-AAD relative to untreated (7% ± 3) or P. gingivalis infected BMM (13% ± 5) (Figure 5B). BMM treated with OMVs at an MOI of 100:1 were nearly uniformly positive for 7-AAD (89% ± 7) (Supplementary Figure 2A). In summary, P. gingivalis did not trigger caspase-1 and IL-1β maturation, IL-1β, and IL-18 secretion or pyroptosis in BMM (Figure 5B). Thus, unlike their parent bacterium, OMVs trigger inflammasome activation and pyroptosis in murine macrophages.
We next addressed whether these observations were seen in MDM and found that OMVs, but interestingly not HI-OMVs, triggered caspase-1 and IL-1β maturation (Figure 6A) and IL-1β and IL-18 secretion (Figure 6B). Similar to BMM, OMVs potently induced IL-1β and IL-18 release from MDM in a MOI-dependent manner compared to P. gingivalis (Supplementary Figure 2B). Interestingly, despite clear inflammasome activation, there was no observable increase in LDH release indicative of pyroptosis from MDM in response to OMVs or OMVs + KYT-1/KYT-36 (Figure 6B). MDM are known to have diminished cell death responses, as measured by LDH release, relative to murine BMM (Bezbradica et al., 2017). A significant (p < 0.05) percentage of MDM treated with OMVs (23% ± 3) were positive for 7-AAD compared to untreated (4% ± 3) and P. gingivalis (6% ± 2) infected cells (Figure 6B). MDM treated with OMVs at an MOI of 100:1 were almost all positive for 7-AAD (84% ± 11) (Supplementary Figure 2B). Similar to BMM, MDM infected with P. gingivalis did not trigger caspase-1 and IL-1β maturation, IL-1β, and IL-18 secretion or pyroptosis. Combined these data show that P. gingivalis failed to activate the inflammasome in murine and human macrophages whereas purified OMVs provide the necessary priming and triggering signals to activate the inflammasome complex in both these populations. The ability of OMVs to activate the inflammasome (i.e., induce the mature forms of caspase-1 and IL-1β) appeared to be partly dependent on their expression of heat-labile protein(s) or enzyme(s), as these responses were impaired in BMM treated with HI-OMVs. In MDM, OMV-induced inflammasome activation was completely abolished when they were heat-inactivated. OMV enrichment for the gingipains relative to their parent cell (Veith et al., 2014) may partly explain the disparate macrophage inflammasome and pyroptotic responses to P. gingivalis compared to OMVs.
In this study we demonstrate that P. gingivalis, its OMVs and LPS, disrupt macrophage mitochondrial respiration and function and shift cellular metabolism toward glycolysis. We also provide important insights into the distinct responses of macrophages to OMVs compared to the parent bacterium. Macrophages stimulated with OMVs produce large amounts of inflammatory mediators and activate the inflammasome and pyroptotic cell death pathways. On the other hand, macrophages infected with P. gingivalis produce low levels of inflammatory cytokine and fail to activate the inflammasome or induce pyroptosis. These data help to further define the unique role of OMVs as the pathogen's major virulence factor and activator of host inflammation.
P. gingivalis OMVs are relatively small and stable and can penetrate host tissues where they activate an inflammatory host response (O'Brien-Simpson et al., 2009). However, quantitating their number in periodontitis is extremely difficult. In a previous study (Cecil et al., 2016), we found that for P. gingivalis cultures (at late exponential growth phase) the ratio of cells to OMVs was ~1:2,000. The levels of P. gingivalis in subgingival plaque are associated with disease severity and a threshold of around 106P. gingivalis cells per site is required for disease progression (Gmur et al., 1989; Jervoe-Storm et al., 2007; Byrne et al., 2009). If we extrapolate (using the 1:2,000 ratio of parent cells to OMVs) this equates to ~2 × 109 OMVs/site, which is well above the highest levels we use in the current study. Our data here do strongly support the notion that OMVs are potent activators of the host response (Ellis and Kuehn, 2010). We found macrophages produced large amounts of TNFα, IL-12p70, IL-6, IL-10, NO, and IFNβ in response to OMVs. Interestingly, IFNβ can act in an autocrine manner to promote IL-12p70, IL-10, and NO production in macrophages (Fleetwood et al., 2009), suggesting that IFNβ may be a central regulator of the macrophage cytokine response to OMVs. Consistent with this notion, macrophages infected with P. gingivalis produced comparatively low levels of IFNβ and were capable of only low level formation of IL-12p70 and IL-10 and failed to induce detectable NO. A recent study demonstrated a key role for IFNβ in P. gingivalis-induced periodontal disease (Mizraji et al., 2017). These data fit with the concept that a major role of OMVs is to activate host immune and inflammatory pathways to provide the nutrients, in the form of tissue breakdown products, that are necessary for the survival of the parent P. gingivalis cell (Hajishengallis, 2011, 2014).
There is a growing appreciation of the interdependency of metabolism and macrophage cell function (O'Neill et al., 2016). Much of this information has been generated via analysis of the metabolic reprogramming that occurs in LPS-stimulated macrophages with only a few studies using viable bacteria (Garaude et al., 2016; Gleeson et al., 2016). We demonstrate here that viable P. gingivalis induces a metabolic shift toward glycolysis in macrophages with attendant reduction in OXPHOS and mitochondrial function, as well as reduction in the expression of many key genes involved in the TCA cycle. A similar switch was observed in human and mouse macrophages stimulated with P. gingivalis OMVs and P. gingivalis-derived LPS. It might be expected then from these data that macrophages interacting locally with viable P. gingivalis or, for example, in the tissue with secreted OMVs (Mashburn-Warren and Whiteley, 2006; Darveau, 2010), will undergo metabolic reprogramming toward glycolysis. Interestingly, the disruption of OXPHOS and switch to glycolysis drives macrophage inflammatory responses (Cramer et al., 2003; Tannahill et al., 2013; Van den Bossche et al., 2015), and once the switch to glycolysis has been made these activated macrophages cannot be “repolarized” to adopt an anti-inflammatory phenotype (Van den Bossche et al., 2016). Activated macrophages are typically thought to utilize glycolysis for rapid ATP generation and clearance of intracellular pathogens by NO and ROS (O'Neill and Pearce, 2016; Van den Bossche et al., 2016). Here we found that P. gingivalis promoted glycolysis in macrophages without the attendant generation of NO, which in dendritic cells is required for the switch to glycolysis to occur (Everts et al., 2012; Everts and Pearce, 2014). In contrast, we found OMV induction of glycolysis was associated with high levels of NO production. Our data are consistent with reports of P. gingivalis inhibition (Wang et al., 2010) and OMV promotion (Imayoshi et al., 2011) of macrophage iNOS/NO generation. Unlike in dendritic cells, the switch to glycolysis in macrophages is not thought to be due to NO inhibition of OXPHOS (Everts et al., 2012; Everts and Pearce, 2014), but likely due to itaconate generation (Cordes et al., 2016; Lampropoulou et al., 2016). Itaconate is the product of an enzyme encoded by the immune responsive gene-1 (Irg-1) that converts cis-aconitate (derived from citrate) to itaconic acid (O'Neill and Pearce, 2016). Itaconate production is hugely increased in glycolytic macrophages where it functions as a central regulator of TCA cycle remodeling, via inhibition of succinate dehydrogenase (SDH) activity (Lampropoulou et al., 2016). In agreement with these studies the macrophage switch to glycolysis following P. gingivalis or OMV treatment was associated with increased expression of Irg-1. We are currently addressing the impact of itaconate on the mitochondrial remodeling observed in response to P. gingivalis. Besides its involvement in regulating macrophage metabolism, itaconate has antibacterial properties (Lampropoulou et al., 2016) and we are also addressing its impact on P. gingivalis survival.
The switch to glycolysis from OXPHOS in macrophages treated with P. gingivalis and OMVs was associated with significant reprogramming of their metabolic gene expression profile. These changes were broadly consistent to changes apparent in LPS-activated or M1 macrophages (McGettrick and O'Neill, 2013; Jha et al., 2015) and were exemplified by increased expression of key glycolytic genes (e.g., Glut-1, Hk1/2, Pfkfb3, and Pkfl) and decreased expression of TCA cycle genes (e.g., Fh1, Pck2, and Suclg2) following P. gingivalis or OMV treatment. Notably, one of the most highly regulated genes Idh1, which encodes a key enzyme of the TCA cycle and is downregulated in M1 cells leading to mitochondrial dysfunction (Jha et al., 2015), was decreased by OMVs. Indeed, decreased Idh1 and increased Irg-1 expression (discussed above) are transcriptional hallmarks of macrophages that have converted to glycolysis (Jha et al., 2015). Not all genes involved in either the glycolytic or TCA pathway were regulated as expected, for example, Gapdh, Galm, and Idh3a. As a gene involved in the glycolytic pathway Gapdh expression was significantly decreased by P. gingivalis and its OMVs. Intriguingly, GAPDH functions as a moonlighting protein with diverse functions (O'Neill et al., 2016), which include acting as a surface receptor on macrophages involved in iron acquisition (Raje et al., 2007). It is intriguing to speculate that following infection, P. gingivalis downregulates macrophage Gapdh expression, in an attempt to impair the removal of extracellular iron, which is an essential nutrient for the growth of P. gingivalis and the wider oral biofilm (Lewis, 2010). Flux through the pentose phosphate pathway is typically enhanced in glycolytic macrophages (Haschemi et al., 2012; Jha et al., 2015). Contrastingly, we found that expression of genes in this pathway tended to be decreased with several genes (i.e., Prps2, Taldo1, and Tkt) being significantly downregulated by P. gingivalis or OMVs. The pentose phosphate pathway generates nucleotides and is also a potential source of NADPH for generation of NO and ROS (Haschemi et al., 2012). Whether P. gingivalis targets this pathway for protection against macrophage bactericidal activity to contribute to its persistence is unknown. Collectively the metabolic gene expression changes observed in macrophages in response to P. gingivalis and OMVs fit with their commitment to ATP generation via glycolysis and the disruption of mitochondrial OXPHOS.
Inflammasomes are cytosolic signaling complexes that specialize in the recognition of a multitude of microbial signals resulting in the generation of the active forms of caspase-1 and IL-1β, which may trigger pyroptotic cell death (Franchi et al., 2012). Pyroptosis allows the elimination of the intracellular niche of pathogens that infect macrophages (e.g., Shigella, Salmonella) and exposes them to antimicrobial effector functions (Schaale et al., 2016). Inflammasome activation is distinct in human monocytes compared to macrophages (Netea et al., 2009). In this important study, human monocytes (and PMA-treated THP-1 human monocytic cells) released mature IL-1β after a single stimulation with TLR2 or TLR4 ligands, due to endogenous release of ATP and activation of P2X7R. Macrophages on the other hand, are unable to process and secrete IL-1β solely in response to TLR ligands and do not release ATP. Thus, macrophages require a second signal (e.g., exogenous ATP) whereas monocytes (and THP-1 cells) only require one signal for inflammasome activation (Netea et al., 2009). This distinction may neatly explain why P. gingivalis activation of the inflammasome and IL-1β production has been demonstrated in THP-1 (Taxman et al., 2006; Park et al., 2014; Jung et al., 2015) and Mono-Mac-6 cell lines (Bostanci et al., 2009; Hamedi et al., 2009), as well as in human monocytes (Huang et al., 2009; Jung et al., 2015). In comparison, studies in mature macrophage populations found that P. gingivalis fails to activate the inflammasome (Taxman et al., 2012; Slocum et al., 2014) unless stimulated with a secondary signal (Morandini et al., 2014; Ramos-Junior et al., 2015). These findings are consistent with our own in human and mouse macrophages where we demonstrate that P. gingivalis provides the “priming” signal (i.e., upregulation of NLRP3 and pro-IL-1β) but not the secondary triggering signal that leads to maturation of caspase-1 and IL-1β. Thus the reported differences for P. gingivalis inflammasome activation are largely due to the nature of the cell type under investigation (i.e., immature monocyte or monocyte cell line vs. mature primary macrophage). Similar to macrophages, gingival epithelial cells do not activate the inflammasome in response to P. gingivalis infection and require a second stimulus in the form of exogenous ATP (Yilmaz et al., 2010). Subsequent studies in these cells revealed that P. gingivalis possesses a nucleoside-diphosphate kinase (NDK) that inhibits ATP (Johnson et al., 2015) and ROS (Choi et al., 2013; Hung et al., 2013) mediated inflammasome activation (Yilmaz and Lee, 2015). Interestingly, NDK can be secreted from infected cells (Choi et al., 2013) and utilizes the PNX1 membrane hemichannel to be translocated outside of the host cells (Atanasova et al., 2016). Whether similar mechanism(s) operate in macrophages is unknown but together these studies reveal the evasive strategies that enable P. gingivalis to circumvent inflammasome activation and host cell death in order to provide an intracellular niche for its survival. It should be noted that other periodontal pathogens, most notably Aggregatibacter actinomycetemcomitans, which possesses a potent leukotoxin, can also induce considerable inflammasome activation and pyroptosis. Uniquely, this pathogen's leukotoxin provides both the priming and triggering signals necessary to induce pyroptosis in macrophages (Kelk et al., 2011).
P. gingivalis OMVs were also able to provide the necessary priming and triggering signals to potently activate the inflammasome and induce pyroptosis in macrophages. A potential explanation for this finding is that gingipain levels are ~three to five-fold higher on OMVs compared to their parent cells (Mantri et al., 2015), and their proteolytic activity has been found to promote inflammasome activation and cell death responses in different cell types (Sheets et al., 2006; Jung et al., 2015). An additional explanation may be that OMVs do not contain NDK (Veith et al., 2014), so unlike their parent cells, OMVs cannot rely on the NDK inhibition of ATP/ROS-mediated inflammasome activation (Yilmaz and Lee, 2015). Consistent with a role for gingipain activity, we observed that when OMVs were heat-inactivated (which may deactivate gingipain proteolytic function; Stathopoulou et al., 2009) inflammasome and pyroptotic cell death responses in macrophages were diminished. Interestingly, P. gingivalis downregulates the production of the gingipains once it resides within the intracellular niche (Xia et al., 2007), which is a strategy not available to OMVs. Such a strategy would presumably lessen the damage to host proteins and reduce inflammasome activation contributing to the pathogen's survival within the host cell. P. gingivalis is a highly invasive intracellular oral pathogen that can persist in macrophages (Wang et al., 2007) and endothelial cells (Belanger et al., 2006), and can reside in gingival epithelial cells for extended periods without causing host cell death (Lamont et al., 1995; Lamont and Jenkinson, 1998). P. gingivalis is thought to be confined to the autophagosome in macrophages (Wang and Hajishengallis, 2008) whereas bacterial OMVs deliver LPS to the cytosol resulting in inflammasome activation and pyroptosis in macrophages (Vanaja et al., 2016). Thus, it is likely that OMV enrichment for gingipains, coupled to their ability to reach the cytosolic inflammasome complex, explain their marked ability to activate pyroptotic cell death in macrophages. Pyroptotic cell death induced by P. gingivalis OMVs (and by other pathogens e.g., A. actinomycetemcomitans) may partly explain the increased levels of LDH in the saliva of patients with periodontitis (De La Pena et al., 2007).
This study demonstrates that the major periodontal pathogen P. gingivalis promotes a metabolic shift toward glycolysis and disrupts mitochondrial function and the TCA cycle in macrophages. These changes were coupled to remodeling at the transcriptional level with a distinct downregulation of TCA cycle genes and increased glycolytic gene expression consistent with a commitment to ATP generation via aerobic glycolysis. These profound changes were also observed in macrophages in response to P. gingivalis OMVs. Uniquely, OMVs triggered considerable inflammatory cytokine release, inflammasome activation and pyroptotic cell death in macrophages. As mentioned (O'Brien-Simpson et al., 2009; Gui et al., 2016), OMVs can penetrate gingival tissue causing tissue damage and inflammation. Our study highlights that this may include activation of inflammatory cytokine release and promotion of glycolysis in gingival tissue macrophage populations leading to their programmed cell death via pyroptosis. The resulting inflammation and release of cytoplasmic compounds [e.g., damage-associated molecular pattern (DAMPs)] into the extracellular milieu would perpetuate local inflammation and, via the gingival exudate flow, return these micronutrients to P. gingivalis and other plaque bacteria (Gui et al., 2016). Further investigation of the mechanism(s) regulating this metabolic shift and its contribution to macrophage effector function in response to P. gingivalis is warranted.
AF, ML, WS, AA, and MCL performed the experiments. AF, ML, AC, AM, SD, ER, and JH analyzed and interpreted the data. ML, WS, AC, AM, SD, NO, and ER contributed reagents, materials, or analysis tools. AF and JH drafted the manuscript with all the authors provided the opportunity to comment, conceived and designed the study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This work was supported by the National Health and Medical Research Council (NHMRC; Project grant, APP1107001).
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00351/full#supplementary-material
Aachoui, Y., Sagulenko, V., Miao, E. A., and Stacey, K. J. (2013). Inflammasome-mediated pyroptotic and apoptotic cell death, and defense against infection. Curr. Opin. Microbiol. 16, 319–326. doi: 10.1016/j.mib.2013.04.004
Abe, N., Kadowaki, T., Okamoto, K., Nakayama, K., Ohishi, M., and Yamamoto, K. (1998). Biochemical and functional properties of lysine-specific cysteine proteinase (Lys-gingipain) as a virulence factor of Porphyromonas gingivalis in periodontal disease. J. Biochem. 123, 305–312. doi: 10.1093/oxfordjournals.jbchem.a021937
Atanasova, K., Lee, J., Roberts, J., Lee, K., Ojcius, D. M., and Yilmaz, O. (2016). Nucleoside-diphosphate-kinase of P. gingivalis is secreted from epithelial cells in the absence of a leader sequence through a pannexin-1 interactome. Sci. Rep. 6:37643. doi: 10.1038/srep37643
Belanger, M., Rodrigues, P. H., Dunn, W. A. Jr., and Progulske-Fox, A. (2006). Autophagy: a highway for Porphyromonas gingivalis in endothelial cells. Autophagy 2, 165–170. doi: 10.4161/auto.2828
Bezbradica, J. S., Coll, R. C., and Schroder, K. (2017). Sterile signals generate weaker and delayed macrophage NLRP3 inflammasome responses relative to microbial signals. Cell. Mol. Immunol. 14, 118–126. doi: 10.1038/cmi.2016.11
Bostanci, N., Emingil, G., Saygan, B., Turkoglu, O., Atilla, G., Curtis, M. A., et al. (2009). Expression and regulation of the NALP3 inflammasome complex in periodontal diseases. Clin. Exp. Immunol. 157, 415–422. doi: 10.1111/j.1365-2249.2009.03972.x
Byrne, S. J., Dashper, S. G., Darby, I. B., Adams, G. G., Hoffmann, B., and Reynolds, E. C. (2009). Progression of chronic periodontitis can be predicted by the levels of Porphyromonas gingivalis and Treponema denticola in subgingival plaque. Oral Microbiol. Immunol. 24, 469–477. doi: 10.1111/j.1399-302X.2009.00544.x
Calkins, C. C., Platt, K., Potempa, J., and Travis, J. (1998). Inactivation of tumor necrosis factor-alpha by proteinases (gingipains) from the periodontal pathogen, Porphyromonas gingivalis. Implications of immune evasion. J. Biol. Chem. 273, 6611–6614. doi: 10.1074/jbc.273.12.6611
Carrion, J., Scisci, E., Miles, B., Sabino, G. J., Zeituni, A. E., Gu, Y., et al. (2012). Microbial carriage state of peripheral blood dendritic cells (DCs) in chronic periodontitis influences DC differentiation, atherogenic potential. J. Immunol. 189, 3178–3187. doi: 10.4049/jimmunol.1201053
Cecil, J. D., O'Brien-Simpson, N. M., Lenzo, J. C., Holden, J. A., Chen, Y. Y., Singleton, W., et al. (2016). Differential responses of pattern recognition receptors to outer membrane vesicles of three periodontal pathogens. PLoS ONE 11:e0151967. doi: 10.1371/journal.pone.0151967
Choi, C. H., Spooner, R., Deguzman, J., Koutouzis, T., Ojcius, D. M., and Yilmaz, O. (2013). Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell. Microbiol. 15, 961–976. doi: 10.1111/cmi.12089
Cordes, T., Wallace, M., Michelucci, A., Divakaruni, A. S., Sapcariu, S. C., Sousa, C., et al. (2016). Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J. Biol. Chem. 291, 14274–14284. doi: 10.1074/jbc.M115.685792
Cramer, T., Yamanishi, Y., Clausen, B. E., Forster, I., Pawlinski, R., Mackman, N., et al. (2003). HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell 112, 645–657. doi: 10.1016/S0092-8674(03)00154-5
Darveau, R. P. (2010). Periodontitis: a polymicrobial disruption of host homeostasis. Nat. Rev. Microbiol. 8, 481–490. doi: 10.1038/nrmicro2337
De La Pena, V. A., Diz Dios, P., and Tojo Sierra, R. (2007). Relationship between lactate dehydrogenase activity in saliva and oral health status. Arch. Oral Biol. 52, 911–915. doi: 10.1016/j.archoralbio.2007.04.008
Ellis, T. N., and Kuehn, M. J. (2010). Virulence and immunomodulatory roles of bacterial outer membrane vesicles. Microbiol. Mol. Biol. Rev. 74, 81–94. doi: 10.1128/MMBR.00031-09
Everts, B., Amiel, E., Huang, S. C., Smith, A. M., Chang, C. H., Lam, W. Y., et al. (2014). TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKvarepsilon supports the anabolic demands of dendritic cell activation. Nat. Immunol. 15, 323–332. doi: 10.1038/ni.2833
Everts, B., Amiel, E., Van Der Windt, G. J., Freitas, T. C., Chott, R., Yarasheski, K. E., et al. (2012). Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120, 1422–1431. doi: 10.1182/blood-2012-03-419747
Everts, B., and Pearce, E. J. (2014). Metabolic control of dendritic cell activation and function: recent advances and clinical implications. Front. Immunol. 5:203. doi: 10.3389/fimmu.2014.00203
Fink, S. L., and Cookson, B. T. (2006). Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 8, 1812–1825. doi: 10.1111/j.1462-5822.2006.00751.x
Fleetwood, A. J., Dinh, H., Cook, A. D., Hertzog, P. J., and Hamilton, J. A. (2009). GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J. Leukoc. Biol. 86, 411–421. doi: 10.1189/jlb.1108702
Fleetwood, A. J., Lawrence, T., Hamilton, J. A., and Cook, A. D. (2007). Granulocyte-macrophage colony-stimulating factor (CSF) and macrophage CSF-dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J. Immunol. 178, 5245–5252. doi: 10.4049/jimmunol.178.8.5245
Fleetwood, A. J., O'Brien-Simpson, N. M., Veith, P. D., Lam, R. S., Achuthan, A., Cook, A. D., et al. (2015). Porphyromonas gingivalis-derived RgpA-Kgp complex activates the macrophage urokinase plasminogen activator system: implications for periodontitis. J. Biol. Chem. 290, 16031–16042. doi: 10.1074/jbc.M115.645572
Franchi, L., Munoz-Planillo, R., and Nunez, G. (2012). Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332. doi: 10.1038/ni.2231
Freemerman, A. J., Johnson, A. R., Sacks, G. N., Milner, J. J., Kirk, E. L., Troester, M. A., et al. (2014). Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 289, 7884–7896. doi: 10.1074/jbc.M113.522037
Garaude, J., Acin-Perez, R., Martinez-Cano, S., Enamorado, M., Ugolini, M., Nistal-Villan, E., et al. (2016). Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 17, 1037–1045. doi: 10.1038/ni.3509
Gleeson, L. E., Sheedy, F. J., Palsson-Mcdermott, E. M., Triglia, D., O'leary, S. M., O'sullivan, M. P., et al. (2016). Cutting edge: Mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J. Immunol. 196, 2444–2449. doi: 10.4049/jimmunol.1501612
Gmur, R., Strub, J. R., and Guggenheim, B. (1989). Prevalence of Bacteroides forsythus and Bacteroides gingivalis in subgingival plaque of prosthodontically treated patients on short recall. J. Periodont. Res. 24, 113–120. doi: 10.1111/j.1600-0765.1989.tb00865.x
Gui, M. J., Dashper, S. G., Slakeski, N., Chen, Y. Y., and Reynolds, E. C. (2016). Spheres of influence: Porphyromonas gingivalis outer membrane vesicles. Mol. Oral Microbiol. 31, 365–378. doi: 10.1111/omi.12134
Hajishengallis, G. (2011). Immune evasion strategies of Porphyromonas gingivalis. J. Oral Biosci. 53, 233–240. doi: 10.1016/S1349-0079(11)80006-X
Hajishengallis, G. (2014). The inflammophilic character of the periodontitis-associated microbiota. Mol. Oral Microbiol. 29, 248–257. doi: 10.1111/omi.12065
Hajishengallis, G. (2015). Periodontitis: from microbial immune subversion to systemic inflammation. Nat. Rev. Immunol. 15, 30–44. doi: 10.1038/nri3785
Hajishengallis, G., Darveau, R. P., and Curtis, M. A. (2012). The keystone-pathogen hypothesis. Nat. Rev. Microbiol. 10, 717–725. doi: 10.1038/nrmicro2873
Hamedi, M., Belibasakis, G. N., Cruchley, A. T., Rangarajan, M., Curtis, M. A., and Bostanci, N. (2009). Porphyromonas gingivalis culture supernatants differentially regulate interleukin-1beta and interleukin-18 in human monocytic cells. Cytokine 45, 99–104. doi: 10.1016/j.cyto.2008.11.005
Haschemi, A., Kosma, P., Gille, L., Evans, C. R., Burant, C. F., Starkl, P., et al. (2012). The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 15, 813–826. doi: 10.1016/j.cmet.2012.04.023
Ho, M. H., Chen, C. H., Goodwin, J. S., Wang, B. Y., and Xie, H. (2015). Functional advantages of Porphyromonas gingivalis vesicles. PLoS ONE 10:e0123448. doi: 10.1371/journal.pone.0123448
Huang, M. T., Taxman, D. J., Holley-Guthrie, E. A., Moore, C. B., Willingham, S. B., Madden, V., et al. (2009). Critical role of apoptotic speck protein containing a caspase recruitment domain (ASC) and NLRP3 in causing necrosis and ASC speck formation induced by Porphyromonas gingivalis in human cells. J. Immunol. 182, 2395–2404. doi: 10.4049/jimmunol.0800909
Huang, S. C., Everts, B., Ivanova, Y., O'sullivan, D., Nascimento, M., Smith, A. M., et al. (2014). Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 15, 846–855. doi: 10.1038/ni.2956
Huang, S. C., Smith, A. M., Everts, B., Colonna, M., Pearce, E. L., Schilling, J. D., et al. (2016). Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity 45, 817–830. doi: 10.1016/j.immuni.2016.09.016
Hung, S. C., Choi, C. H., Said-Sadier, N., Johnson, L., Atanasova, K. R., Sellami, H., et al. (2013). P2X4 assembles with P2X7 and pannexin-1 in gingival epithelial cells and modulates ATP-induced reactive oxygen species production and inflammasome activation. PLoS ONE 8:e70210. doi: 10.1371/journal.pone.0070210
Imayoshi, R., Cho, T., and Kaminishi, H. (2011). NO production in RAW264 cells stimulated with Porphyromonas gingivalis extracellular vesicles. Oral Dis. 17, 83–89. doi: 10.1111/j.1601-0825.2010.01708.x
Jervoe-Storm, P. M., Alahdab, H., Koltzscher, M., Fimmers, R., and Jepsen, S. (2007). Comparison of curet and paper point sampling of subgingival bacteria as analyzed by real-time polymerase chain reaction. J. Periodontol. 78, 909–917. doi: 10.1902/jop.2007.060218
Jha, A. K., Huang, S. C., Sergushichev, A., Lampropoulou, V., Ivanova, Y., Loginicheva, E., et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430. doi: 10.1016/j.immuni.2015.02.005
Johnson, L., Atanasova, K. R., Bui, P. Q., Lee, J., Hung, S. C., Yilmaz, O., et al. (2015). Porphyromonas gingivalis attenuates ATP-mediated inflammasome activation and HMGB1 release through expression of a nucleoside-diphosphate kinase. Microbes Infect. 17, 369–377. doi: 10.1016/j.micinf.2015.03.010
Jorgensen, I., and Miao, E. A. (2015). Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 265, 130–142. doi: 10.1111/imr.12287
Jung, Y. J., Jun, H. K., and Choi, B. K. (2015). Contradictory roles of Porphyromonas gingivalis gingipains in caspase-1 activation. Cell. Microbiol. 17, 1304–1319. doi: 10.1111/cmi.12435
Kadowaki, T., Baba, A., Abe, N., Takii, R., Hashimoto, M., Tsukuba, T., et al. (2004). Suppression of pathogenicity of Porphyromonas gingivalis by newly developed gingipain inhibitors. Mol. Pharmacol. 66, 1599–1606. doi: 10.1124/mol.104.004366
Kelk, P., Abd, H., Claesson, R., Sandstrom, G., Sjostedt, A., and Johansson, A. (2011). Cellular and molecular response of human macrophages exposed to Aggregatibacter actinomycetemcomitans leukotoxin. Cell Death Dis. 2:e126. doi: 10.1038/cddis.2011.6
Komiya, A., Kato, T., Nakagawa, T., Saito, A., Takahashi, J., Yamada, S., et al. (2000). A rapid DNA probe method for detection of Porphyromonas gingivalis and Actinobacillus actinomycetemcomitans. J. Periodontol. 71, 760–767. doi: 10.1902/jop.2000.71.5.760
Krawczyk, C. M., Holowka, T., Sun, J., Blagih, J., Amiel, E., Deberardinis, R. J., et al. (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. doi: 10.1182/blood-2009-10-249540
Kuehn, M. J., and Kesty, N. C. (2005). Bacterial outer membrane vesicles and the host-pathogen interaction. Genes Dev. 19, 2645–2655. doi: 10.1101/gad.1299905
Lacey, D. C., Achuthan, A., Fleetwood, A. J., Dinh, H., Roiniotis, J., Scholz, G. M., et al. (2012). Defining GM-CSF–and macrophage-CSF–dependent macrophage responses by in vitro models. J. Immunol. 188, 5752–5765. doi: 10.4049/jimmunol.1103426
Lam, R. S., O'Brien-Simpson, N. M., Holden, J. A., Lenzo, J. C., Fong, S. B., and Reynolds, E. C. (2016). Unprimed, M1 and M2 macrophages differentially interact with Porphyromonas gingivalis. PLoS ONE 11:e0158629. doi: 10.1371/journal.pone.0158629
Lamont, R. J., and Jenkinson, H. F. (1998). Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol. Mol. Biol. Rev. 62, 1244–1263.
Lamont, R. J., Chan, A., Belton, C. M., Izutsu, K. T., Vasel, D., and Weinberg, A. (1995). Porphyromonas gingivalis invasion of gingival epithelial cells. Infect. Immun. 63, 3878–3885.
Lampropoulou, V., Sergushichev, A., Bambouskova, M., Nair, S., Vincent, E. E., Loginicheva, E., et al. (2016). Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 24, 158–166. doi: 10.1016/j.cmet.2016.06.004
Lari, R., Fleetwood, A. J., Kitchener, P. D., Cook, A. D., Pavasovic, D., Hertzog, P. J., et al. (2007). Macrophage lineage phenotypes and osteoclastogenesis–complexity in the control by GM-CSF and TGF-beta. Bone 40, 323–336. doi: 10.1016/j.bone.2006.09.003
Lewis, J. P. (2010). Metal uptake in host-pathogen interactions: role of iron in Porphyromonas gingivalis interactions with host organisms. Periodontol. 2000 52, 94–116. doi: 10.1111/j.1600-0757.2009.00329.x
Mantri, C. K., Chen, C. H., Dong, X., Goodwin, J. S., Pratap, S., Paromov, V., et al. (2015). Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiologyopen 4, 53–65. doi: 10.1002/mbo3.221
Mashburn-Warren, L. M., and Whiteley, M. (2006). Special delivery: vesicle trafficking in prokaryotes. Mol. Microbiol. 61, 839–846. doi: 10.1111/j.1365-2958.2006.05272.x
Masters, S. L., Dunne, A., Subramanian, S. L., Hull, R. L., Tannahill, G. M., Sharp, F. A., et al. (2010). Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat. Immunol. 11, 897–904. doi: 10.1038/ni.1935
McGettrick, A. F., and O'Neill, L. A. (2013). How metabolism generates signals during innate immunity and inflammation. J. Biol. Chem. 288, 22893–22898. doi: 10.1074/jbc.R113.486464
Miao, E. A., Rajan, J. V., and Aderem, A. (2011). Caspase-1-induced pyroptotic cell death. Immunol. Rev. 243, 206–214. doi: 10.1111/j.1600-065X.2011.01044.x
Mizraji, G., Nassar, M., Segev, H., Sharawi, H., Eli-Berchoer, L., Capucha, T., et al. (2017). Porphyromonas gingivalis promotes unrestrained type I interferon production by dysregulating TAM signaling via MYD88 degradation. Cell Rep. 18, 419–431. doi: 10.1016/j.celrep.2016.12.047
Moon, J. S., Hisata, S., Park, M. A., Denicola, G. M., Ryter, S. W., Nakahira, K., et al. (2015). mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep. 12, 102–115. doi: 10.1016/j.celrep.2015.05.046
Morandini, A. C., Ramos-Junior, E. S., Potempa, J., Nguyen, K. A., Oliveira, A. C., Bellio, M., et al. (2014). Porphyromonas gingivalis fimbriae dampen P2X7-dependent interleukin-1beta secretion. J. Innate Immun. 6, 831–845. doi: 10.1159/000363338
Nakao, R., Kikushima, K., Higuchi, H., Obana, N., Nomura, N., Bai, D., et al. (2014). A novel approach for purification and selective capture of membrane vesicles of the periodontopathic bacterium, Porphyromonas gingivalis: membrane vesicles bind to magnetic beads coated with epoxy groups in a noncovalent, species-specific manner. PLoS ONE 9:e95137. doi: 10.1371/journal.pone.0095137
Nemeth, B., Doczi, J., Csete, D., Kacso, G., Ravasz, D., Adams, D., et al. (2016). Abolition of mitochondrial substrate-level phosphorylation by itaconic acid produced by LPS-induced Irg1 expression in cells of murine macrophage lineage. FASEB J. 30, 286–300. doi: 10.1096/fj.15-279398
Netea, M. G., Nold-Petry, C. A., Nold, M. F., Joosten, L. A., Opitz, B., Van Der Meer, J. H., et al. (2009). Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 113, 2324–2335. doi: 10.1182/blood-2008-03-146720
O'Brien-Simpson, N. M., Black, C. L., Bhogal, P. S., Cleal, S. M., Slakeski, N., Higgins, T. J., et al. (2000). Serum immunoglobulin G (IgG) and IgG subclass responses to the RgpA-Kgp proteinase-adhesin complex of Porphyromonas gingivalis in adult periodontitis. Infect. Immun. 68, 2704–2712. doi: 10.1128/IAI.68.5.2704-2712.2000
O'Brien-Simpson, N. M., Pathirana, R. D., Walker, G. D., and Reynolds, E. C. (2009). Porphyromonas gingivalis RgpA-Kgp proteinase-adhesin complexes penetrate gingival tissue and induce proinflammatory cytokines or apoptosis in a concentration-dependent manner. Infect. Immun. 77, 1246–1261. doi: 10.1128/IAI.01038-08
O'Neill, L. A., and Pearce, E. J. (2016). Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 213, 15–23. doi: 10.1084/jem.20151570
O'Neill, L. A., Kishton, R. J., and Rathmell, J. (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16, 553–565. doi: 10.1038/nri.2016.70
Palm, E., Khalaf, H., and Bengtsson, T. (2013). Porphyromonas gingivalis downregulates the immune response of fibroblasts. BMC Microbiol. 13:155. doi: 10.1186/1471-2180-13-155
Park, E., Na, H. S., Song, Y. R., Shin, S. Y., Kim, Y. M., and Chung, J. (2014). Activation of NLRP3 and AIM2 inflammasomes by Porphyromonas gingivalis infection. Infect. Immun. 82, 112–123. doi: 10.1128/IAI.00862-13
Qi, M., Miyakawa, H., and Kuramitsu, H. K. (2003). Porphyromonas gingivalis induces murine macrophage foam cell formation. Microb. Pathog. 35, 259–267. doi: 10.1016/j.micpath.2003.07.002
Raje, C. I., Kumar, S., Harle, A., Nanda, J. S., and Raje, M. (2007). The macrophage cell surface glyceraldehyde-3-phosphate dehydrogenase is a novel transferrin receptor. J. Biol. Chem. 282, 3252–3261. doi: 10.1074/jbc.M608328200
Ramos-Junior, E. S., Morandini, A. C., Almeida-Da-Silva, C. L., Franco, E. J., Potempa, J., Nguyen, K. A., et al. (2015). A dual role for P2X7 receptor during Porphyromonas gingivalis infection. J. Dent. Res. 94, 1233–1242. doi: 10.1177/0022034515593465
Schaale, K., Peters, K. M., Murthy, A. M., Fritzsche, A. K., Phan, M. D., Totsika, M., et al. (2016). Strain- and host species-specific inflammasome activation, IL-1beta release, and cell death in macrophages infected with uropathogenic Escherichia coli. Mucosal Immunol. 9, 124–136. doi: 10.1038/mi.2015.44
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Sheets, S. M., Potempa, J., Travis, J., Fletcher, H. M., and Casiano, C. A. (2006). Gingipains from Porphyromonas gingivalis W83 synergistically disrupt endothelial cell adhesion and can induce caspase-independent apoptosis. Infect. Immun. 74, 5667–5678. doi: 10.1128/IAI.01140-05
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526, 660–665. doi: 10.1038/nature15514
Slocum, C., Coats, S. R., Hua, N., Kramer, C., Papadopoulos, G., Weinberg, E. O., et al. (2014). Distinct lipid a moieties contribute to pathogen-induced site-specific vascular inflammation. PLoS Pathog. 10:e1004215. doi: 10.1371/journal.ppat.1004215
Socransky, S. S., Haffajee, A. D., Cugini, M. A., Smith, C., and Kent, R. L. Jr. (1998). Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25, 134–144. doi: 10.1111/j.1600-051X.1998.tb02419.x
Srisatjaluk, R., Doyle, R. J., and Justus, D. E. (1999). Outer membrane vesicles of Porphyromonas gingivalis inhibit IFN-gamma-mediated MHC class II expression by human vascular endothelial cells. Microb. Pathog. 27, 81–91. doi: 10.1006/mpat.1999.0287
Stathopoulou, P. G., Benakanakere, M. R., Galicia, J. C., and Kinane, D. F. (2009). The host cytokine response to Porphyromonas gingivalis is modified by gingipains. Oral Microbiol. Immunol. 24, 11–17. doi: 10.1111/j.1399-302X.2008.00467.x
Tannahill, G. M., Curtis, A. M., Adamik, J., Palsson-Mcdermott, E. M., Mcgettrick, A. F., Goel, G., et al. (2013). Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242. doi: 10.1038/nature11986
Taxman, D. J., Swanson, K. V., Broglie, P. M., Wen, H., Holley-Guthrie, E., Huang, M. T., et al. (2012). Porphyromonas gingivalis mediates inflammasome repression in polymicrobial cultures through a novel mechanism involving reduced endocytosis. J. Biol. Chem. 287, 32791–32799. doi: 10.1074/jbc.M112.401737
Taxman, D. J., Zhang, J., Champagne, C., Bergstralh, D. T., Iocca, H. A., Lich, J. D., et al. (2006). Cutting edge: ASC mediates the induction of multiple cytokines by Porphyromonas gingivalis via caspase-1-dependent and -independent pathways. J. Immunol. 177, 4252–4256. doi: 10.4049/jimmunol.177.7.4252
Van den Bossche, J., Baardman, J., and de Winther, M. P. (2015). Metabolic characterization of polarized M1 and M2 bone marrow-derived macrophages using real-time extracellular flux analysis. J. Vis. Exp. e53424. doi: 10.3791/53424
Van den Bossche, J., Baardman, J., Otto, N. A., Van Der Velden, S., Neele, A. E., Van Den Berg, S. M., et al. (2016). Mitochondrial dysfunction prevents repolarization of inflammatory macrophages. Cell Rep. 17, 684–696. doi: 10.1016/j.celrep.2016.09.008
Vanaja, S. K., Russo, A. J., Behl, B., Banerjee, I., Yankova, M., Deshmukh, S. D., et al. (2016). Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell 165, 1106–1119. doi: 10.1016/j.cell.2016.04.015
Vats, D., Mukundan, L., Odegaard, J. I., Zhang, L., Smith, K. L., Morel, C. R., et al. (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. doi: 10.1016/j.cmet.2006.05.011
Veith, P. D., Chen, Y. Y., Gorasia, D. G., Chen, D., Glew, M. D., O'Brien-Simpson, N. M., et al. (2014). Porphyromonas gingivalis outer membrane vesicles exclusively contain outer membrane and periplasmic proteins and carry a cargo enriched with virulence factors. J. Proteome Res. 13, 2420–2432. doi: 10.1021/pr401227e
Waller, T., Kesper, L., Hirschfeld, J., Dommisch, H., Kolpin, J., Oldenburg, J., et al. (2016). Porphyromonas gingivalis outer membrane vesicles induce selective tumor necrosis factor tolerance in a toll-like receptor 4- and mTOR-dependent manner. Infect. Immun. 84, 1194–1204. doi: 10.1128/IAI.01390-15
Wang, M., and Hajishengallis, G. (2008). Lipid raft-dependent uptake, signalling and intracellular fate of Porphyromonas gingivalis in mouse macrophages. Cell. Microbiol. 10, 2029–2042. doi: 10.1111/j.1462-5822.2008.01185.x
Wang, M., Krauss, J. L., Domon, H., Hosur, K. B., Liang, S., Magotti, P., et al. (2010). Microbial hijacking of complement-toll-like receptor crosstalk. Sci. Signal. 3:ra11. doi: 10.1126/scisignal.2000697
Wang, M., Shakhatreh, M. A., James, D., Liang, S., Nishiyama, S., Yoshimura, F., et al. (2007). Fimbrial proteins of Porphyromonas gingivalis mediate in vivo virulence and exploit TLR2 and complement receptor 3 to persist in macrophages. J. Immunol. 179, 2349–2358. doi: 10.4049/jimmunol.179.4.2349
Way, K. J., Dinh, H., Keene, M. R., White, K. E., Clanchy, F. I., Lusby, P., et al. (2009). The generation and properties of human macrophage populations from hemopoietic stem cells. J. Leukoc. Biol. 85, 766–778. doi: 10.1189/jlb.1108689
Xia, Q., Wang, T., Taub, F., Park, Y., Capestany, C. A., Lamont, R. J., et al. (2007). Quantitative proteomics of intracellular Porphyromonas gingivalis. Proteomics 7, 4323–4337. doi: 10.1002/pmic.200700543
Xie, H. (2015). Biogenesis and function of Porphyromonas gingivalis outer membrane vesicles. Future Microbiol. 10, 1517–1527. doi: 10.2217/fmb.15.63
Yilmaz, O., and Lee, K. L. (2015). The inflammasome and danger molecule signaling: at the crossroads of inflammation and pathogen persistence in the oral cavity. Periodontol. 2000 69, 83–95. doi: 10.1111/prd.12084
Yilmaz, O., Sater, A. A., Yao, L., Koutouzis, T., Pettengill, M., and Ojcius, D. M. (2010). ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell. Microbiol. 12, 188–198. doi: 10.1111/j.1462-5822.2009.01390.x
Keywords: macrophages, metabolism, inflammasome, P. gingivalis, vesicles, pyroptosis, cytokines and inflammation, glycolysis and oxidative phosphorylation
Citation: Fleetwood AJ, Lee MKS, Singleton W, Achuthan A, Lee M-C, O'Brien-Simpson NM, Cook AD, Murphy AJ, Dashper SG, Reynolds EC and Hamilton JA (2017) Metabolic Remodeling, Inflammasome Activation, and Pyroptosis in Macrophages Stimulated by Porphyromonas gingivalis and Its Outer Membrane Vesicles. Front. Cell. Infect. Microbiol. 7:351. doi: 10.3389/fcimb.2017.00351
Received: 03 May 2017; Accepted: 21 July 2017;
Published: 04 August 2017.
Edited by:
Georgios N. Belibasakis, Karolinska Institute (KI), SwedenReviewed by:
Özlem Yilmaz, Medical University of South Carolina, United StatesCopyright © 2017 Fleetwood, Lee, Singleton, Achuthan, Lee, O'Brien-Simpson, Cook, Murphy, Dashper, Reynolds and Hamilton. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrew J. Fleetwood, YW5kcmV3LmZsZWV0d29vZEB1bmltZWxiLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.