 Wolfgang Eisenreich
Wolfgang Eisenreich Thomas Rudel
Thomas Rudel Jürgen Heesemann
Jürgen Heesemann Werner Goebel
Werner Goebel
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 13 July 2017
Sec. Bacteria and Host
Volume 7 - 2017 | https://doi.org/10.3389/fcimb.2017.00316
This article is part of the Research Topic Pathometabolism of Intracellular Bacteria View all 7 articles
Intracellular bacterial pathogens (IBPs) invade and replicate in different cell types including immune cells, in particular of the innate immune system (IIS) during infection in the acute phase. However, immune cells primarily function as essential players in the highly effective and integrated host defense systems comprising the IIS and the adaptive immune system (AIS), which cooperatively protect the host against invading microbes including IBPs. As countermeasures, the bacterial pathogens (and in particular the IBPs) have developed strategies to evade or reprogram the IIS at various steps. The intracellular replication capacity and the anti-immune defense responses of the IBP's as well as the specific antimicrobial responses of the immune cells of the innate and the AIS depend on specific metabolic programs of the IBPs and their host cells. The metabolic programs of the immune cells supporting or counteracting replication of the IBPs appear to be mutually exclusive. Indeed, recent studies show that upon interaction of naïve, metabolically quiescent immune cells with IBPs, different metabolic activation processes occur which may result in the provision of a survival and replication niche for the pathogen or its eradication. It is therefore likely that within a possible host cell population subsets exist that are metabolically programmed for pro- or anti-microbial conditions. These metabolic programs may be triggered by the interactions between different bacterial agonistic components and host cell receptors. In this review, we summarize the current status in the field and discuss metabolic adaptation processes within immune cells of the IIS and the IBPs that support or restrict the intracellular replication of the pathogens.
Metabolism governs virtually all dynamic processes of living cells. A functioning metabolism requires sufficient supply of basic nutrients, including carbon-, nitrogen-, and sulfur-sources, for (a) the generation of energy in form of ATP, (b) the production of low-molecular and high-molecular cellular building blocks, and (c) the maintenance of an intracellular redox status.
For achieving these three goals, prokaryotic microorganisms have evolved many different autotrophic and heterotrophic metabolic strategies including assimilation of carbon dioxide, specific transport systems and catabolic pathways for many different carbon sources, a large number of anabolic pathways, various aerobic and anaerobic ATP-generating reactions and the adaptation to a wide range of temperature, pH and pressure.
Mammalian cells, on the other hand, carry out a rather common type of heterotrophic metabolism which supports cell growth and proliferation through catabolism of a limited number of carbon sources, mainly glucose, fatty acids and amino acids (especially glutamine), thereby gaining (a) energy (ATP) preferentially by oxidative phosphorylation (OXPHOS, for a list of all abbreviations, see above and the Supplementary Table) via aerobic respiration and/or by substrate phosphorylation mainly via glycolysis and (b) intermediates for a limited number of anabolic processes (Ghesquiere et al., 2014). This metabolism functions optimally at mesophilic temperature, close to neutral pH and (micro)aerophilic conditions and is controlled (similar to prokaryotic cells) by a complex network of signaling pathways, transcription factors, microRNAs and post-transcriptional mechanisms (Storey and Storey, 2004; Storey, 2015) (for more details, see below). Most differentiated mammalian cells can reversibly switch from a quiescent metabolic state characterized by low catabolic and anabolic activity and OXPHOS (Palsson-Mcdermott and O'Neill, 2013) to a proliferating state with high catabolic and (often) high anabolic activities. This activated metabolism requires fast energy (mainly ATP) generation often driven by “aerobic glycolysis” (also known as “Warburg effect”).
Most bacterial pathogens that are able to colonize and grow in mammalian niches are optimally adapted to a mesophilic and heterotrophic metabolic life style. This is especially true for all human intracellular bacterial pathogens (IBPs) which are characterized by the ability to invade and proliferate in different mammalian cell types (Fuchs et al., 2012), including professional phagocytes of the innate immune system (IIS), in particular macrophages (MPs), dendritic cells (DCs) and (less frequently) neutrophilic polymorphonuclear leukocytes (PMNs), or neutrophils (NPs). Uptake of the IBPs by these cells occurs by receptor-triggered phagocytosis involving e.g., Fc-γ receptors, scavenger receptors, integrin receptors or C-type lectin receptors (Drummond and Brown, 2013; Freeman and Grinstein, 2014). Internalization of IBPs by non-professional phagocytes, e.g., epithelial and endothelial cells, requires the interaction between specific bacterial invasion factors and appropriate host cell receptors which triggers internalization (Pizarro-Cerdá and Cossart, 2006).
Once internalized, the IBPs replicate within the host cells in specialized vacuoles, in the cytosol or in both compartments (Knodler, 2015). Successful replication of the IBPs in these compartments requires adaptation of the bacteria to the metabolism of host cells which in turn may be reprogrammed by the infection with IBPs. Since the metabolic potentials of the IBPs significantly differ, the metabolic adaptation of IBPs to their host cells must require different, yet still poorly understood strategies on part of the IBPs and the host cells (Eisenreich et al., 2010; Fuchs et al., 2012; Abu Kwaik and Bumann, 2015).
For the protection against microbial pathogens, the mammalian hosts have evolved a highly efficient integrated immune system, consisting of the IIS and the adaptive immune system (AIS) which cooperatively interact. The professional phagocytes as well as the mucosal epithelial cells and the skin keratinocytes belonging to the IIS, and also the primed and activated lymphocytes of the AIS possess potent antimicrobial capacities against invading pathogens, including IBPs.
On the other hand, most of the immune cells, especially those of the IIS, may also serve as proficient host cells for IBPs. Compared to the hospitality of IIS immune cells, the capacity of the immune cells of the AIS, i.e., T and B lymphocytes, to function as host cells for IBPs appears to be limited (Menon et al., 2003; Geddes et al., 2007; Krocova et al., 2008; Konradt et al., 2011; Goenka et al., 2012; Nothelfer et al., 2014).
It is an apparent paradox that the same cell types of the IIS may represent convenient replication niches for the IBPs but also the most important tools for the destruction of IBPs. The immune (defense) responses triggered by IBPs have been extensively studied while the metabolic aspect has been rather neglected (Price and Vance, 2014; O'Neill and Pearce, 2016). In this review, we will therefore mainly focus on this latter aspect with emphasis on IBP-infected immune cells of the IIS.
There is growing evidence that both, the efficient expression of the antimicrobial activities of the immune cells and the successful replication of IBPs within these cells require specific metabolic programs in both interacting partners for which the terms “immunometabolism” (Rathmell, 2012; Pollizzi and Powell, 2014) and “pathometabolism” (Eisenreich et al., 2015), respectively, have been coined. These two metabolic entities seem to mutually influence each other when the immune cells meet IBPs.
In the first part of this review, we provide some relevant background information on the metabolism and the regulatory networks of IBPs and of mammalian host cells in general, however with a focus on the immune cells of the IIS. In the second part, we discuss the present, but still rather limited knowledge concerning the metabolic adaptation processes which occur in both partners during the interaction of IBPs with these immune cells.
Intracellular bacterial pathogens (IBPs) represent a group of disease-causing bacteria that are able to invade a variety of differentiated mammalian cells of various differentiation states, including immune cells (in particular those of the IIS), and to survive and proliferate in specialized membrane-surrounded vacuoles or in the cytosol (Figure 1) for extended periods of time before leaving the host cells by cell disruption or cell-to-cell spread (Cossart and Sansonetti, 2004; Kumar and Valdivia, 2009; Ray et al., 2009; Fredlund and Enninga, 2014). Most of the IBPs are facultative intracellular, i.e., they are able to replicate within host cells, but also extracellularly in tissues or mucosal excretions, in natural environments and in more or less complex cell-free (“axenic”) media (Omsland et al., 2009). The efficient replication of a minority of IBPs (so-called “obligate IBPs”) appears to be strictly dependent on host cells (Wood et al., 2014).
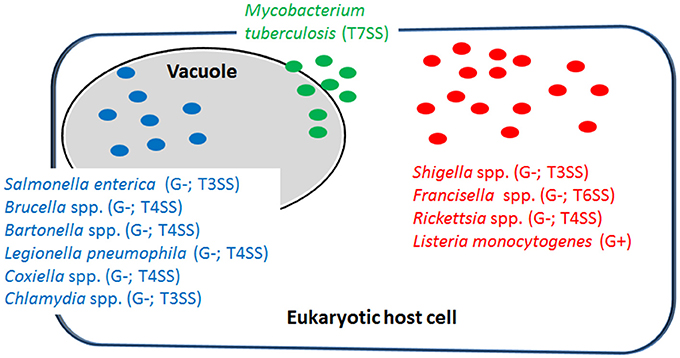
Figure 1. Representative examples of intracellular bacterial pathogens (IBPs; mentioned in this review) replicating in specialized vacuoles (in blue) or in the host cell's cytosol (in red). Some IBPs (e.g., M. tuberculosis) are able to replicate in both cellular compartments (in green). G−, Gram-negative; G+, Gram-positive; T3SS, type 3-secretion system; T4SS, type 4-secretion system; T6SS, type 6-secretion system; T7SS, type 7-secretion system.
Most IBPs have a typical Gram-negative cell envelope structure and inject (often many) effector proteins by specific protein secretion/injection systems (mainly type 3, and 6 secretion systems, T3SS, T4SS or T6SS; see Figure 1) into the host cells cytosol (Costa et al., 2015). Most of these effectors, whose functions are currently intensively investigated, are apparently involved in the uptake and/or the survival/proliferation processes of these IBPs by interfering mainly with different signaling pathways of host cells affecting cytoskeletal dynamics, survival, innate immune responses and possibly also metabolism of the contacted host cells (Alto and Orth, 2012; Eisenreich et al., 2013). The best-studied representative among the less frequently occurring Gram-positive IBPs is Listeria monocytogenes that replicates in the cytosol of infected host cells. The pathogenic Mycobacterium species have a unique cell envelope different from the typical cell envelopes of Gram-positive and Gram-negative bacteria. These pathogens possess type 7 protein secretion systems (ESX/T7SS) that secrete proteins some of which are clearly involved in pathogenicity and host cell interaction (Simeone et al., 2009; Houben et al., 2014).
Numerous virulence factors essential for invasion, intracellular survival and proliferation of the IBPs have been characterized (Cossart and Sansonetti, 2004). Although L. monocytogenes as Gram-positive pathogen lacks a protein injection apparatus, it produces a variety of secreted and cell-bound internalins that interact as ligands with different host cell receptors thereby performing in part similar trigger functions as the Gram-negative effector proteins (Bierne et al., 2007; Mcgann et al., 2007).
The expression of the major virulence genes of IBPs is often controlled by master transcription regulators, with links to the metabolism of the IBP and even of the host cell (Stoll et al., 2008; Poncet et al., 2009; De Las Heras et al., 2011; Gillmaier et al., 2012; Reniere et al., 2015). Not surprisingly, all IBPs are heterotrophic, aerobic or facultative anaerobic bacteria, able to survive and replicate under the normoxic and hypoxic conditions which they may encounter during their infection cycles. The metabolic potentials of most IBPs are reduced compared to the metabolic capacity of typical heterotrophic generalists (e.g., E. coli; see Figure 2). The extent of the metabolic reduction differs, however, strongly among these IBPs in view of the catabolic as well as the anabolic capabilities (Eisenreich et al., 2010; Fuchs et al., 2012), suggesting that the dependency on nutrient supply to be delivered by the host cell and the adaptation of the bacterial metabolism to that of the host cell must be specific for each IBP. Interestingly, there are, however some metabolic functions that are maintained by all IBPs and hence might be indispensable for intracellular bacterial survival (Figure 3).
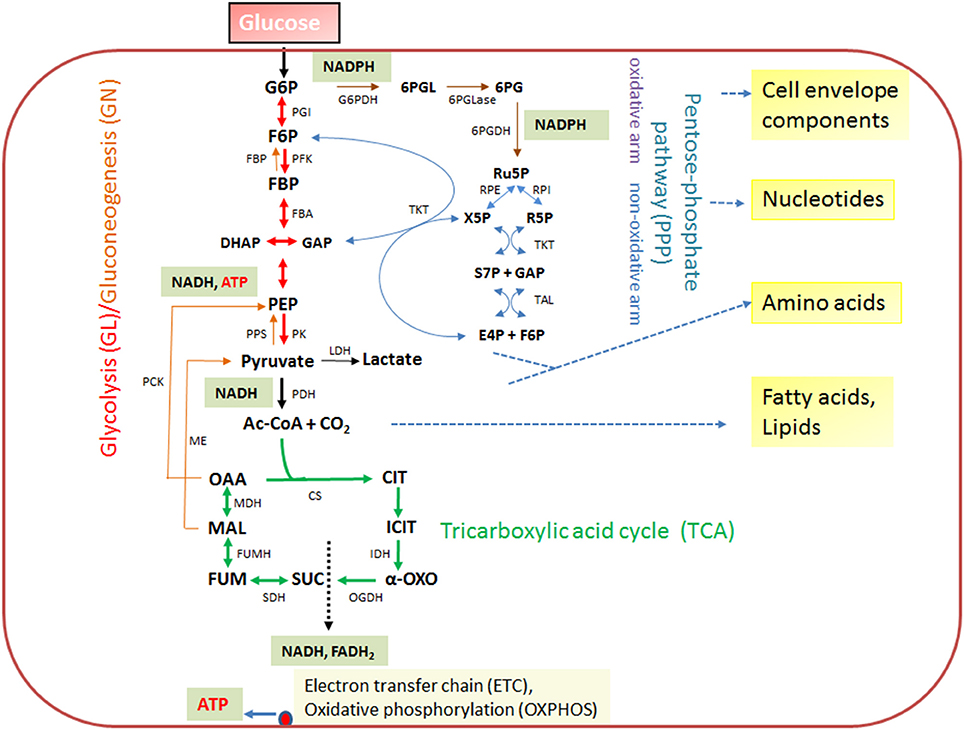
Figure 2. Schematic presentation of the basic catabolic and anabolic processes of a typical heterotrophic prokaryotic cell: glycolytic pathway (GL), pentose-phosphate pathway (PPP) with its oxidative and non-oxidative arms, and tricarboxylic acid cycle (TCA). The specific reactions essential for gluconeogenesis (GN) (in addition to the reversible GL reactions) are catalyzed by PEP carboxykinase (PCK), PEP synthetase (PPS) and fructose-1,6-diphosphatase (FBPase). The anaplerotic reactions are not shown with the exception of the reaction leading from malate to pyruvate, catalyzed by the malic enzyme (ME) which may play an important role the intracellular metabolism of IBPs (see Figure 3). The ATP production by oxidative phosphorylation (OXPHOS) via the electron transfer chain (ETC) is also indicated. Metabolites of GL: G6P, glucose-6P; F6P, fructose-6P; FBP, fructose-1,6 diphosphate; DHAP, dihydroxyacetone phosphate; GAP, glyceraldehyde-3P; PEP, phosphoenolpyruvate. Metabolites of the PPP: 6PGL, 6-phosphogluconolactone; 6PG, 6-phosphogluconate; Ru5P, ribulose-5P; X5P, xylulose-5P; R5P, ribose-5P; S7P, sedoheptulose-7P; E4P, erythrose-4P. Metabolites of the tricarboxylic acid cycle (TCA): OAA, oxaloacetate; CIT, citrate; ICIT, isocitrate; α-OXO, alpha-oxoglutarate; SUC, succinate, FUM, fumarate, MAL, malate. Enzymes of the GL: PGI, phosphoglucoisomerase; PFK, phosphofructokinase; FBA, fructobisphosphate aldolase; PK, pyruvatekinase. PDH, pyuvate dehydrogenase. LDH, lactate dehydrogenase. Enzymes of the PPP: G6PDH, glucose-6P dehydrogenase; 6PGLase, 6-phosphogluconolactonase; 6PGDH, 6-phosphogluconate dehydrogenase; RPE, Ru5P-epimerase; RPI, Ru5P-isomerase; TKT, transketolase; TAL, transaldolase. Enzymes of the TCA cycle: CS, citrate synthase; IDH, isocitrate dehydogenase; OGDH, oxoglutarate dehydrogenase; SDH, succinate dehydrogenase; FUMH, fumarate hydratase; MDH, malate dehydrogenase.
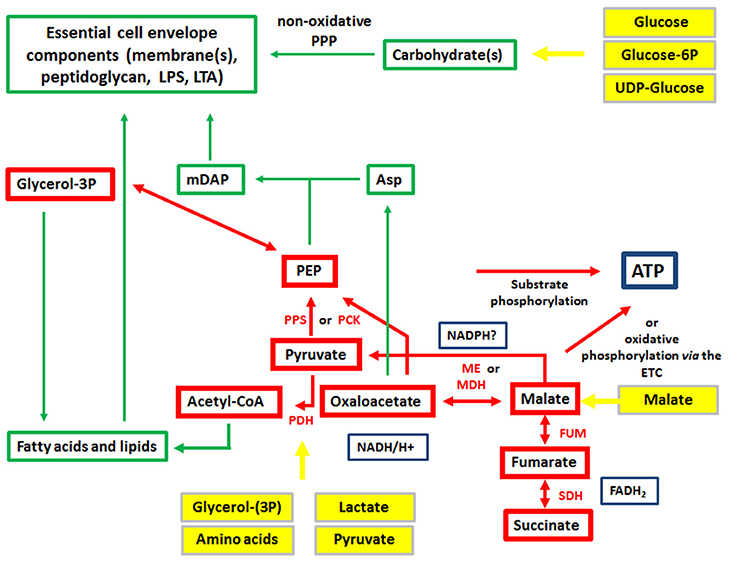
Figure 3. Metabolism of IBPs: metabolites and metabolic reactions common to all IBPs. Red boxes and arrows show the common catabolic products and reactions, respectively and green boxes and arrows the common anabolic products and reactions. Produced co-factors are in blue-framed boxes. Products in yellow boxes and arrows represent nutrients that can be delivered by the host cell.
The metabolic burden put on the host cell by these IBP infections has a strong impact on the host cells metabolism, causing activation of specific signaling pathways which may result in increased catabolic, energy and anabolic metabolism but also in induction of apoptosis/pyroptosis and autophagy (Eisenreich et al., 2013). These metabolic changes may in turn influence the metabolism of the intracellular bacteria. Thus, a complex network of metabolic interactions between the IBPs and host cells exists which determines the fate of IBPs within host cells, especially immune cells of the IIS (Eisenreich et al., 2015).
In contrast to the highly diverse metabolic potentials of the IBPs, the metabolic equipment of mammalian cells is quite similar (Figure 4). But in spite of this, the metabolic activities of host cells can change dramatically depending on the differentiation state, external and internal signals and (in case of cancer cells) genetic alterations.
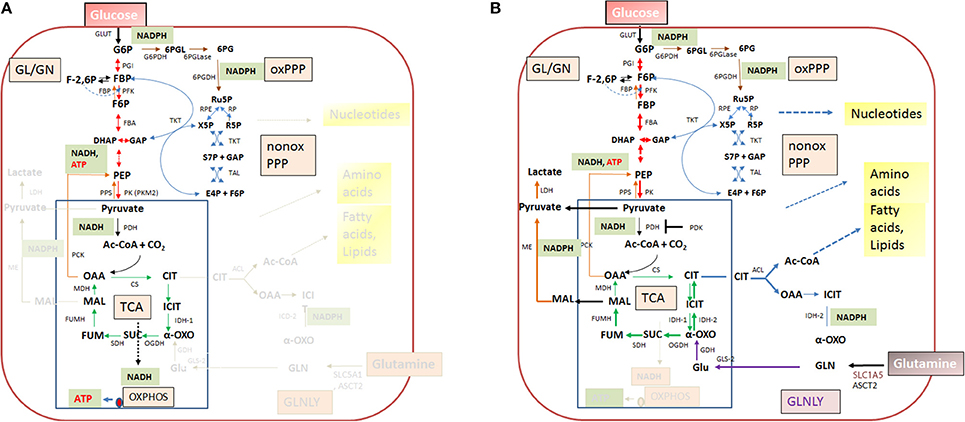
Figure 4. Quiescent (A) and activated (B) metabolism in differentiated mammalian cells. In differentiated mammalian cells, including mucosal epithelial cells (MEC) and non-activated immune cells, the metabolism is characterized by low catabolic and anabolic activities. Low carbon fluxes fed by glucose and/or fatty acids run through glycolysis, PPP, and the TCA cycle; oxidative phosphorylation (OXPHOS) is the predominant source of ATP. The metabolism of metabolically activated cells (B) is characterized by enhanced glucose uptake, highly activated (“aerobic”) glycolysis, reduced entrance of pyruvate into the TCA cycle and reduced OXPHOS. ATP is mainly produced by substrate phosphorylation in the glycolytic pathway. In addition these cells show often increased glutaminolysis which replenishes the TCA cycle, leading to citrate (eventually via reverse IDH) which is transported into the cytosol where it is converted by the ATP-dependent citrate lyase (ACL) into oxaloacetate and acetyl-CoA, two metabolites serving for biosynthesis of fatty acids/lipids and amino acids. ß-oxidation is inhibited. Metabolites of GL: G6P, glucose-6P; F6P, fructose-6P; FBP, fructose-1,6 biphosphate; DHAP, dihydroxyacetonephosphate; GAP, glyceraldehyde-3P; PEP, phosphoenolpyruvate. Metabolites of the PPP: 6PGL, 6-phosphogluconolactone; 6PG, 6-phosphogluconate; Ru5P, ribulose-5P; X5P, xylulose-5P; R5P, ribose-5P; S7P, sedoheptulose-7P; E4P, erythrose-4P. Metabolites of the tricarboxylic acid (TCA) cycle: OAA, oxaloacetate; CIT, citrate; ICIT, Isocitrate; α-OXO, alpha-oxoglutarate; SUC, succinate, FUM, fumarate, MAL, malate. Enzymes of the GL: GLUT, glucose transporter; PGI, phosphoglucoisomerase; PFK, phosphofructokinase; FBA, fructobisphosphate aldolase; PK, pyruvatekinase. PDH, pyuvate dehydrogenase. LDH, lactate dehydrogenase. Enzymes of the PPP: G6PDH, glucose-6P dehydrogenase; 6PGLase, 6-phosphogluconolactonase; 6PGDH, 6-phosphogluconate dehydrogenase; RPE, Ru5P-epimerase; RPI, Ru5P-isomerase; TKT, transketolase; TAL, transaldolase. Enzymes of the TCA cycle: CS, citrate synthase; ICD, isocitratedehydogenase; OGDH, oxoglutarate dehydrogenase; SDH, succinate dehydrogenase; FUMH, fumarate hydratase; MDH, malate dehydrogenase. Glutaminolysis (GLNLY): SLC1A5, ASCT2, glutamine transporters, GLS, glutaminase; GDH, glutamate dehydrogenase.
Without external stimuli (e.g., growth factors) most terminally differentiated mammalian cells, including the immune cells of the IIS are non-proliferating or only slowly proliferating, i.e., these cells are in a (more or less) metabolically quiescent state (Figure 4A). In this state they take up, at a low rate, glucose—dependent on the cell type—by one of several glucose transporters (GLUTs) (Chen et al., 2015). Glucose is then phosphorylated by hexokinase (HK) to glucose-6-phosphate (G6P) and further metabolized in the cytosol through the glycolytic pathway (GL) and/or the pentose phosphate pathway (PPP). G6P is oxidized in the GL to pyruvate which is shuttled into mitochondria where it is further oxidized to carbon dioxide by pyruvate dehydrogenase (PDH) and the tricarboxylic acid cycle (TCA), generating NADH/H+, FADH2, GTP, and intermediates, essential for anabolic pathways (especially fatty acids/lipids, amino acids). G6P can be also oxidized by G6P dehydrogenase (G6PDH) and 6-phosphogluconate dehydrogenase (6PGDH) to ribulose-5P (Ru5P) in the oxidative branch of the PPP. Both reactions generate NADPH, an important co-factor for reductive biosynthetic pathways (e.g., fatty acids/lipids, deoxyribonucleotides) and scavenging of reactive oxygen species (ROS). In the non-oxidative branch of the PPP, Ru5P is reversibly converted by ribose-5-phosphate isomerase (RPI) to ribose-5P (R5P) which is an essential precursor to many biomolecules, especially nucleotides, and to xylulose-5P (X5P) by ribulose-5-phosphate 3-epimerase (RPE). This non-oxidative PPP branch consists of additional reversible reactions catalyzed by transketolase (TKT) and transaldolase (TAL) and recruits the glycolytic intermediates fructose-6P (F6P) and glyceraldehyde-3P (G3P), which can thereby also be converted into pentose phosphates (and vice versa) thus linking GL and PPP (see Figure 4). Indeed, GL and PPP are coordinately regulated to support cell growth and survival as discussed below in more detail.
Under non-activated cell conditions, ATP production occurs mainly by OXPHOS in the electron transport chain (ETC) whereby the electrons of NADH/H+ (generated in GL and TCA cycle) are transferred to oxygen as final acceptor. Under these normal physiological conditions growth factors are available only in limited and strictly controlled amounts (Rathmell et al., 2000). Hence, most terminally differentiated cells perform a catabolic metabolism which allows optimal efficiency of ATP production from limited nutrient supply (DeBerardinis et al., 2006; Vander Heiden et al., 2009; Ward and Thompson, 2012).
When growth factor concentration is increased, nutrient uptake (most notably by glucose and glutamine transporters) is induced and the intracellular nutrient pools rise. These cells now adopt a more anabolic metabolism (i.e., enhanced biosynthesis of nucleotides, “non-essential” amino acids, fatty acids/lipids and the macromolecules), double the cellular biomass and initiate cell division (DeBerardinis et al., 2008; Vander Heiden et al., 2009). Compared to the non-activated differentiated cells, the growth factor-stimulated, proliferating cells do not optimize efficiency of ATP production but rather the production of larger amounts of intermediary metabolites and NADPH for glutathione production (essential for antioxidant defense) and other biosynthetic reactions (Figure 4B). ATP is now mainly produced through glycolysis by substrate phosphorylation. Excess pyruvate generated as final product in the GL is reduced to lactate which is secreted. The reaction also restores NAD necessary for the maintenance of the glycolytic flux. This kind of metabolism (“aerobic glycolysis”), which is energetically inefficient but leads to fast ATP production and favors anabolic processes, is characteristic for many fast proliferating cancer cells (DeBerardinis, 2008; Ngo et al., 2015). Although the cells of the mononuclear phagocyte system (MPS), comprising monocytes (MOs), MPs and DCs, do not proliferate in tissue after extravasation, they can switch their metabolism upon transition from the quiescent to the activated state. The metabolism in the activated state of MP cells shows similarities to that of cancer cells (O'Neill and Pearce, 2016).
Expression of the pathways involved in carbon and energy metabolism of mammalian cells is controlled by a complex net of nutrient sensors, growth hormone receptors, several downstream signaling pathways, canonical transcription factors including NF-κB, AP-1, NFAT, ChREBP, PPARα, SREBP as well as non-coding RNAs (Eberle et al., 2004; Postic et al., 2007; Fritz and Fajas, 2010; Cairns et al., 2011; Thompson, 2011; Harris et al., 2012; Iizuka et al., 2013; Ganeshan and Chawla, 2014; Jiang et al., 2014; Pollizzi and Powell, 2014; Yu et al., 2015). In the last decade, studies in particular on the dysregulated metabolism of cancer cells also highlighted the significance of (proto)oncogenes and tumor suppressors as key regulators of cellular metabolism. Most important players are here PI3K/AKT1, mTORC1, AMPK, RAS, HIF-1, cMYC, and p53 (Figure 5). Whereas, the impact of these factors on the regulation of growth cycle, differentiation, proliferation, immune responses, survival, and apoptosis of mammalian cells were already extensively investigated in the past, their involvement in the regulation of central metabolic pathways was recognized only recently. For recent reviews, see (Yin et al., 2012; Iurlaro et al., 2014). In the following, we focus mainly on the impact of the latter factors for the regulation of the central carbon metabolism, since they may be primary targets for the interaction with specific effector proteins of IBPs resulting in the reprogramming of the host cell metabolism during infection (Table 1).
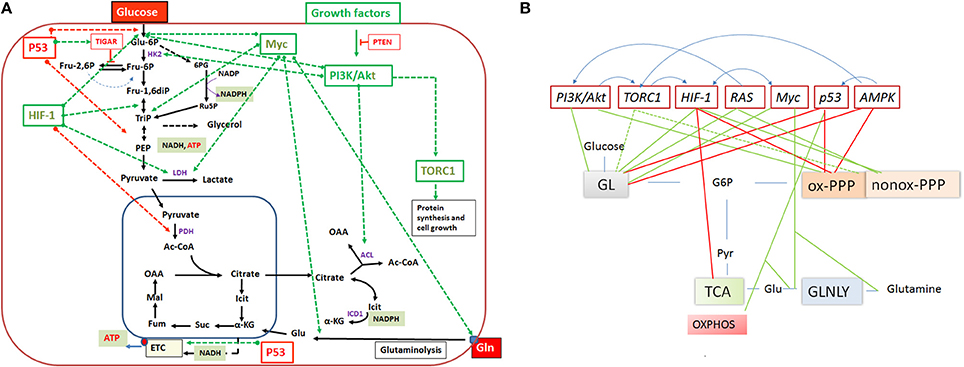
Figure 5. (A) Important regulators of the mammalian cell metabolism are (proto)-oncogenes and tumor suppressors and interacting regulatory factors (e.g., PI3K/AKT, RAS, Myc, TORC1, HIF-1 and P53, PTEN, TIGAR, respectively) that control key reactions of the central catabolic pathways (including glutaminolysis) either positively (green dashed arrows) or negatively (red dashed arrows). (B) A schematic overview on the catabolic pathways including glycolysis (GL), pentose phosphate pathway (PPP), tricarboxylic acid (TCA) cycle and glutaminolysis (GLNLY) controlled by these key regulators. Green lines indicate upregulation of catabolic pathways and red lines downregulation of the pathways. Anabolic pathways (i.e., those leading to synthesis of nucleotides, amino acids, glutathione, lipids) which depend on intermediates of the catabolic pathways are (indirectly) also influenced by these regulators. The blue arrows above the regulators indicate interactions between them. See text for further details.
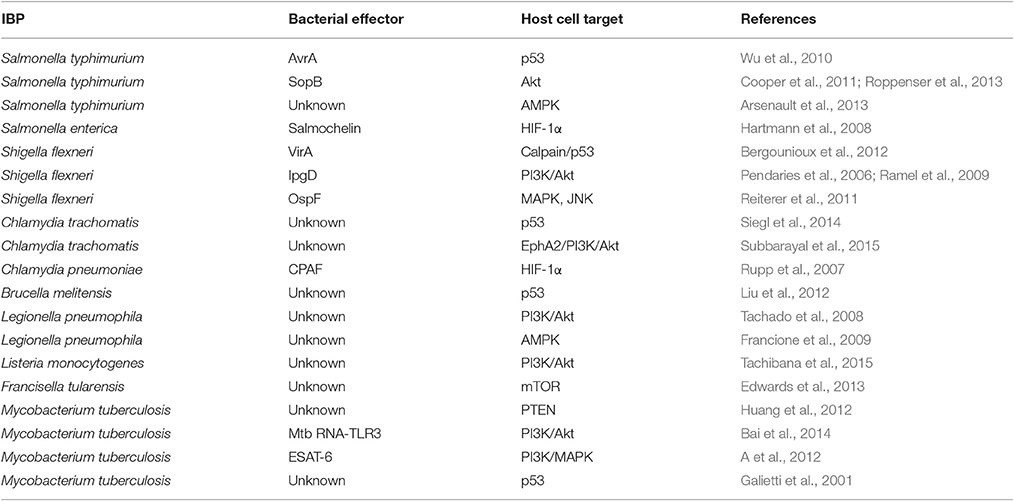
Table 1. Interactions of IBP effectors with their host cell targets.
The major signaling pathway downstream of growth factor receptors activating glucose flux is the phosphoinositide-3-kinase/Akt (PI3K/Akt) pathway (Hemmings and Restuccia, 2012). This pathway is also one of the most commonly altered signaling pathways in human cancer cells. PI3K generates phosphatidylinositol-3,4,5-trisphosphate (PIP3) from phosphatidylinositol-4,5-bisphosphate (PIP2) and PIP3 recruits Akt to the cell membrane permitting its activation by upstream kinases (e.g., 3-phosphoinositide dependent protein kinase-1 (PDPK1). The activated Akt then stimulates expression of several proteins (Figure 5) involved in glucose uptake and glycolysis (DeBerardinis et al., 2008), including the glucose transporter 1 (GLUT1) (through enhanced translation and translocation to the cell surface), hexokinase 2 (HK2), and phosphofructokinase 1 (PFK1). In addition to its regulatory function in glycolysis, the PI3K-Akt pathway may regulate G6PDH and thus the oxidative arm of PPP (Wagle et al., 1998). Furthermore, Akt activates ATP-dependent citrate lyase (ACL). This cytosolic enzyme cleaves citrate (exported from the mitochondria into the cytosol) into oxaloacetate and acetyl-CoA, thereby providing essential precursors for aspartate and fatty acid/lipid biosynthesis, respectively. Finally, it is noteworthy that chemokine-triggered migration of invasive cells involves the PI3K/Akt pathway.
The target of rapamycin (mTOR) complex 1 (mTORC1) is an important regulator of metabolism downstream of PI3K-Akt and is often constitutively activated during tumorigenesis (Guertin and Sabatini, 2007). Phosphorylated Akt activates mTORC1 by direct phosphorylation and by inactivation of the tuberous sclerosis protein 2 (TSC2), an inhibitor of mTORC1. A genomic approach (Duvel et al., 2010) suggests that mTORC1 induces transcription of several genes involved in glycolysis and both branches of PPP. The molecular nature of the activation of these genes by mTORC1 remains, however, elusive. The activity of this complex is also modulated by sensing essential amino acids (especially leucine) and hence represents a central regulator of amino acid metabolism. In response to these signals, mTORC1 positively regulates protein biosynthesis by activation of S6 kinase (S6K) which enhances expression of ribosomal proteins and by inhibiting the initiation factor 4E-binding protein (4E-BP). The mTOR complex 2 (mTORC2) lies upstream of Akt, is positively regulated by PI3K signaling and is also involved in the activation of mTORC1 (Liu et al., 2013).
The Ras proteins (RAS including K-Ras, H-Ras, and N-Ras) are low molecular weight G proteins whose activity is regulated by binding of guanine nucleotides. In the activated GTP-bound state, RAS modulates a number of signaling pathways. The two best-studied downstream pathways that are induced by activated RAS are the mitogen-activated protein kinases (MAPK) and the PI3K/Akt pathways (Gysin et al., 2011). Oncogenic RAS mutations, leading to loss of GTPase activity, tend to lock RAS in the active (GTP-bound) state which results in constitutive RAS signaling (Pratilas and Solit, 2010). As shown by Telang and colleagues (Telang et al., 2007), activated H-Ras increases in immortalized human bronchial epithelial cells, the glycolytic flux to lactate, TCA cycle activity and oxygen consumption. The same group showed that activation of H-Ras enhances cytochrome C oxidase (COX) activity and mitochondrial respiration in part via up-regulation of the COX Vb subunit (Telang et al., 2012; Chesney and Telang, 2013). Based on these data, it was postulated that H-Ras activation may cause simultaneous stimulation of COX activity and uncoupling of ATP synthase from the proton gradient in order to facilitate the continued oxidation of NADH to NAD necessary for glycolytic flux and TCA cycling.
In pancreatic tumors, oncogenic K-Ras caused stimulation of glucose uptake and channeling of glycolysis intermediates into the non-oxidative arm of the PPP, thereby promoting ribose-5P biogenesis and decoupling it from NADPH production (Ying et al., 2012). The oxidative branch of PPP and the TCA cycle were unaffected by the activated K-Ras. The authors further showed that the MAPK pathway and Myc-directed transcriptional control but not the PI3K/Akt pathway played the crucial role for the K-Ras-mediated metabolic reprogramming in these tumors.
The hypoxia-inducible transcription factor 1 (HIF-1), a heterodimer consisting of the HIF-1α and HIF-1β subunits (Iyer et al., 1998), plays also a central role in the regulation of cellular glucose catabolism. Whereas, HIF-1β is stable and constitutively expressed, expression of HIF-1α is induced and its active form is stabilized under low cellular oxygen (hypoxic) conditions. The stability of HIF-1α is regulated by prolyl hydroxylase (PHD)-mediated hydroxylation of proline residues of the HIF-1α subunit. This reaction targets HIF-1α for its recognition by the ubiquitin E3 ligase and its subsequent proteasomal degradation. Oxygen is required for the PHD activity. However, at low oxygen and in the presence of elevated levels of ROS or the two TCA intermediates, succinate and fumarate, PHD activity and hence HIF-1α degradation are inhibited. But even under normoxic conditions, HIF-1 can be positively regulated (on the transcriptional and the translational level) downstream of growth factor receptors by the PI3K/Akt/mTOR pathway (Dery et al., 2005; Lisy and Peet, 2008; Kuschel et al., 2012; Burroughs et al., 2013).
HIF-1 enhances transcription of genes encoding GLUTs and most enzymes of the glycolytic pathway. HIF-1 activation may also lead to a reduced flux through the oxidative arm of PPP while enhancing the non-oxidative arm, thereby providing R5P essential for nucleotide synthesis and cellular proliferation. In addition, HIF-1 inhibits pyruvate entry into the mitochondrial TCA cycle by promoting the expression of pyruvate dehydrogenase kinase (PDK), an enzyme which inhibits PDH activity, and activates expression of lactate dehydrogenase A (LDH-A) which converts pyruvate to lactate. The latter reaction regenerates NAD+ necessary for maintaining oxidation of glucose by glycolysis (Brahimi-Horn and Pouyssegur, 2009; Semenza, 2009; Zhao et al., 2010).
Although HIF-1 inhibits flux through the TCA cycle, citrate production can be maintained in the presence of active HIF-1 through a reverse TCA reaction which generates isocitrate/citrate by reductive carboxylation from glutamine-derived α-ketoglutarate (Figure 4) catalyzed by isocitrate dehydrogenase 2 (IDH2) (Wise et al., 2011).
The oncogene Myc is another important transcriptional regulator of carbon and nitrogen metabolism. Myc is activated downstream of growth factor signaling pathways, in particular of those involving RAS (Morrison, 2012; Chesney and Telang, 2013). Myc enhances glutamine uptake and glutaminolysis by positively regulating the expression of glutamine transporters (e.g., SLC1A5) and glutaminase (GLS), thereby supplementing the mitochondrial TCA cycle by the generated α-ketoglutarate (see Figure 4B). In addition, Myc promotes biosynthesis of nucleotides and amino acids through increased production of the necessary TCA intermediates via enhanced glutaminolysis, but also by enhanced transcription of genes which encode enzymes involved in the biosynthesis of these compounds (Tong et al., 2009). Myc in collaboration with HIF-1 may also stimulate the conversion of glucose to lactate by activating the expression of the genes encoding GLUT1, HK2, LDH-A, and pyruvate kinase M2 (PKM2) (Yeung et al., 2008; Dang et al., 2009).
Pyruvate kinase (PK), which catalyzes the last step of glycolysis converting phosphoenolpyruvate (PEP) to pyruvate with concomitant ATP production, represents an important metabolic switch point. This enzyme exists in two isoforms, PKM1 and PKM2. The constitutively active PKM1 is the major PK controlling the glycolytic flow into the TCA cycle in non-proliferating cells. PKM2, a less active isoform, has been shown to be involved in cancer metabolism and tumor growth (Christofk et al., 2008). Myc enhances the expression of PKM2. Activity of PKM2 is allosterically activated by the upstream glycolytic intermediate fructose-1,2-diphosphate (FBP). Growth factor signaling leads to the release of FBP from PKM2 resulting in its decreased pyruvate kinase activity. This causes the accumulation of upstream glycolytic intermediates and activation of the PPP which provides R5P and NADPH. Thus, the switch to PKM2 favors anabolic processes, especially biosynthesis of serine/glycine, nucleotides and the antioxidant glutathione (Soga, 2013). PKM2 can also act as a protein kinase using PEP as a phosphate donor, thereby converting PEP to pyruvate without ATP generation (Gao et al., 2012). The switch of PKM2 between the protein kinase activity (in the dimer form) and pyruvate kinase activity (in the tetramer form) may be an important factor in cell proliferation (Gao et al., 2013).
The p53 transcription factor, an extensively studied key tumor suppressor, is known to regulate in particular cell cycle arrest, apoptosis and DNA repair (Levine and Oren, 2009). But p53 is also an important regulator of carbon and energy metabolism. As a potent inhibitor of glycolysis (Vousden and Ryan, 2009), wild-type p53 downregulates glucose uptake and promotes expression of the tumor suppressor TP53-inducible glycolysis and apoptosis regulator (TIGAR). TIGAR is a phosphatase that lowers the level of fructose-2,6-diphosphate (F26BP), an allosteric activator of PFK1 (Figure 2). A reduced level of this metabolite therefore inhibits the glucose flux through glycolysis and may direct glucose-6P to PPP (Olovnikov et al., 2009). However, wild-type p53 binds directly to G6PDH thereby inhibiting its enzymatic activity which limits the glucose flux through PPP. Mitochondrial respiration is enhanced by p53-mediated induction of the cytochrome c oxidase assembly protein 2 (SCO2), an important regulator of cytochrome C oxidase. Another p53-activated metabolic target gene is GLS2 which encodes glutaminase 2. As described above, this enzyme converts glutamine to glutamate which—upon deamination to 2-oxoglutarate—replenishes the TCA cycle thereby enhancing the rate of the TCA pathway and OXPHOS.
Tumor-associated gain-of-function mutant p53 proteins have been identified (Brosh and Rotter, 2009) which may activate different metabolic pathways, e.g., the mevalonate pathway and hence lipid metabolism, glucose uptake with subsequent enhanced glycolysis or loss of the ability to block G6PDH activity, thereby activating the PPP (Jiang et al., 2011).
Metabolic effects mediated by p53 may also be due to its ability to inhibit the PI3K/Akt/mTOR pathway through the two tumor suppressors, phosphatase and tensin homolog (PTEN) and AMP-activated protein kinase (AMPK).
PTEN is a phosphatidylinositol-3,4,5-triphosphate (PIP3) 3-phosphatase which preferentially dephosphorylates phosphoinositides. Through this activity, PTEN reduces the intracellular level of PIP3, thereby negatively affecting the PI3K/Akt signaling pathway (Song et al., 2012).
AMPK is a key regulator of energy metabolism and glucose homeostasis. Liver kinase B1 (LKB1)-mediated phosphorylation activates AMPK by many stimuli, including cellular stress, hormones, cytokines and adipokines (Jones et al., 2005; Shackelford and Shaw, 2009; Hardie, 2011). AMPK inhibits mTORC1 and stimulates catabolic rather than anabolic pathways. Under low cellular energy conditions (high AMP/ATP ratio), AMPK triggers glycolysis if sufficient external glucose is provided. This results in decreased AMP and glucose levels. At low glucose level, sustained activity of AMPK activates p53 which—as described above—intervenes with glycolysis, PPP and OXPHOS thereby slowing down and balancing carbon flux through glycolysis and PPP (Jiang et al., 2014; Patra and Hay, 2014) and promoting OXPHOS. P53 further up-regulates transcription of the SESN gene and the produced sestrin proteins stimulate AMPK, thus eliciting a positive feedback loop.
The retinoblastoma protein (RB) is a tumor suppressor protein that is absent or inactivated in several human cancers. The best-characterized function of RB is the prevention of excessive cell growth by inhibiting premature cell division. When the cell is ready to divide, RB is phosphorylated, becomes inactive and allows cell cycle progression. RB is involved in metabolism through the transcriptional regulation of enzymes essential for nucleotide biosynthesis (Angus et al., 2002) and seems to be involved in switching an oxidative to a glycolytic metabolism (Nicolay and Dyson, 2013).
Lastly, it should be noted that many of the metabolic regulators converge or depend for activation directly or indirectly on mTORC1, the central check point of cell proliferation (Dunlop and Tee, 2009) and show mutual interactions (Figure 5B). Furthermore, it is worth mentioning that the interaction of members of the NF-κB family of pleiotropic transcriptional factors with p53, HIF-1 and other metabolic regulators also influences the energy metabolism in mammalian cells (reviewed in Johnson and Perkins, 2012).
In contrast to environmentally exposed interfaces, mammalian tissues are kept free of microorganisms by an efficient defense system including humoral and cellular components. Here, we will focus on the early phase of bacterial infections when the first line of defense, mainly comprising the IIS, is activated.
While commensal bacteria (as members of the healthy microbiota) support the host immune system by nutrient- and metabolite-dependent mechanisms (Brestoff and Artis, 2013), bacterial pathogens, especially IBPs, are able to break the cell and tissue sterility (Odegaard and Chawla, 2013) by using—under certain conditions—some of the immune cells, especially those of the IIS, as host cells where they may successfully survive, replicate and counteract the critical defense responses of these cells (Diacovich and Gorvel, 2010; Swart and Hensel, 2012).
The IIS consists of a collection of different subsystems (Medzhitov, 2007a). The most essential cellular players of the IIS are cells of the MPS, including MOs, MPs, DCs, and NPs as well as mucosal epithelial cells (MECs) and skin keratinocytes (SKs) (Guilliams et al., 2014). The specific molecules produced and in part secreted by these cell types upon activation act as signals for further immune responses or as direct antimicrobial agents. However, these cells may also serve as host cells for specific IBPs.
All cell types of the IIS and in particular MPs which show a high local and physiological flexibility (Murray and Wynn, 2011; Guilliams et al., 2014; Murray et al., 2014; Gross et al., 2015) express various sets of membrane-inserted, surface-exposed and cytosolic pattern recognition receptors (PRRs), which, as sensors for microbial pathogens, include the membrane-associated Toll-like receptors (TLRs) and C-type lectins, as well as the cytosolic nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs) and the retinoic acid-inducible gene I (RIG.I)-like receptors (RLRs) (Kawai and Akira, 2011). The most important PRRs for recognition of bacterial pathogens are TLRs and NLRs. The major pathogen-associated molecular patterns (PAMPs1) recognized by TLRs include lipopolysaccharide (LPS), lipoteichoic acid (LTA), peptidoglycan (PGN), and other cell envelope components of Gram-positive and Gram-negative bacteria, flagellin, CpG DNA, RNA (Medzhitov, 2007a,b; Kawai and Akira, 2011; Oldenburg et al., 2012; Krüger et al., 2015), as well as cyclic di-AMP (Woodward et al., 2010; Commichau et al., 2015; Whiteley et al., 2015) and also the host-derived danger-associated molecular patterns (DAMPs) (Escamilla-Tilch et al., 2013) representing endogenous molecules released from damaged host cells including mitochondrial DNA (Mills et al., 2017),. The cytosolic NLRs comprise more than 20 members (Franchi et al., 2008). Among the NLRs, NOD1 and NOD2 recognize specific fragments of the bacterial PGN, but for most NLRs the specific interacting ligands are yet unknown.
Exposure of MAMPs to PRRs differs between intracellular and extracellular bacterial pathogens. Extracellular bacterial pathogens may release MAMPs through various processes which then can directly bind to PRRs on immune cells, triggering host responses. In contrast, IBPs produce MAMPs which—due to their intracellular localization—may have more limited access to the immune system. The host responds to these MAMPs likely through different programs; it becomes increasingly clear that exosomes and other extracellular vesicles released from infected cells play an important role in this dissemination process (for a recent review, see Schorey et al., 2015). These vesicles not only include MAMPs, but also T- and B-cell antigens, as well as pathogen-derived virulence factors.
Interactions of MAMPs with TLRs and with NOD1/2 activate the MyD88-dependent signaling pathway leading (among others) to the activation of the transcription factors NF-kB, AP-1 and interferon regulatory factors (IRFs). These transcription factors are known to induce the expression of genes encoding the production of pro-inflammatory cytokines and chemokines, pro-IL-1ß, pro-IL18 and IL-8, TNF-α and of type I interferons (especially IFN-α and IFN-ß) essential for further immune responses (Ozinsky et al., 2000).
A subset of the NLRs (AIM2, IPAF, NLRP1, and NLRP3) initiates, e.g., by interaction with bacterial flagellin and DNA, the formation of inflammasomes, multiprotein complexes present in epithelial cells and immune cells of the myeloid lineage. The inflammasomes activate in particular caspase 1 and 11 which cleave pro-IL-1ß and pro-IL-18 to their active forms which are subsequently released, followed by pyroptosis. These cytokines in their mature form are key regulators of the host response against microbial pathogens (Schroder and Tschopp, 2010).
A hallmark of activated professional phagocytes (MOs, MPs, and NPs) and activated MECs is the production of microbicidal ROS and reactive nitrogen intermediates (RNIs) catalyzed by the NADPH oxidase and the inducible NO synthase (iNOS), respectively. In addition, the proinflammatory cytokines IL-1ß, IL-6, and TNF-α cause an inflammatory environment. Finally, restriction of accessible intracellular iron ions, essential for IBP replication, appears to be also an important defense mechanism of MPs and possibly other immune cells (Nairz et al., 2014, 2015).
PMNs represent the most abundant phagocytic cell population in human blood. These professional phagocytes are rapidly mobilized to sites of infection (Kennedy and Deleo, 2009) and, upon phagocytosis, kill bacterial pathogens and other microbes by a combination of ROS, cytotoxic granule components, hydrolytic enzymes and antimicrobial peptides which generate a highly lethal intraphagosomal environment (Nauseef, 2007; Kennedy and Deleo, 2009). NPs are short-lived and are pre-programmed to undergo under normal conditions spontaneous apoptosis, 18–24 h after release into circulation.
Besides these professional phagocytes, invading microorganisms encounter less aggressive IIS immune cells which form a dendritic network in lymphoid and non-lymphoid tissue (LT and NLT) and therefore are named DCs. The DCs comprise several subtypes with characteristic properties, such as sentinel functions and transport antigen and microorganisms to lymph nodes for the induction of an adaptive immune response (Mildner and Jung, 2014).
The past decade has experienced a focus on the convergence of metabolism and immune functions for which the term “immunometabolism” has been coined (for reviews, see Rathmell, 2012; Pearce and Pearce, 2013; Delgoffe and Powell, 2015; O'Neill and Pearce, 2016). Like most terminally differentiated mammalian cells, the non-activated immune cells of the IIS perform a quiescent metabolism which is characterized by a low glucose flow through glycolysis and the TCA cycle and the formation of ATP predominantly by OXPHOS often fueled by ß-oxidation of fatty acids/lipids in the mitochondria (DeBerardinis et al., 2006). Their biosynthetic activities are low under these conditions and even in presence of sufficient nutrient supply, trophic signals, such as cytokines must be present to avoid cell death (Frauwirth and Thompson, 2004). This quiescent metabolic state is activated in all immune cells of the IIS by various ligand/receptor interactions as shortly summarized in the following.
Resting NPs which contain few mitochondria and consume little oxygen (Van Raam et al., 2008) are mainly activated by TLR agonists, formylated peptides or by the local growth factor G-CSF. Activated NPs show increased oxygen consumption, glucose uptake, and now produce ATP exclusively by aerobic glycolysis (Gabig et al., 1979). PPP activity required especially for the generation of NADPH is also induced. NADPH is essential for the induction of NADPH oxidase responsible for the production of H2O2, the characteristic microbicidal product of NPs. Netosis, the special ability of NPs to release extracellular nets containing DNA and microbicidal proteins (Urban et al., 2009) is also dependent on NADPH oxidase (Kirchner et al., 2012).
DCs, a heterogeneous cell population, reside in tissues exposed to the external environment and represent a surveillance system sensing infections and the first line of defense against invading pathogens (Dominguez and Ardavin, 2010; Arnold-Schrauf et al., 2014; Mildner and Jung, 2014; Durai and Murphy, 2016). DCs also act at the transition from IIS and AIS by presenting microbial antigens to T cells and providing inflammatory signals that modulate T cell differentiation (Steinman, 2012).
Resident DCs undergo, upon interaction of microbial MAMPs and cellular DAMPs with the corresponding PRRs, a rapid switch from the resting metabolic state (characterized by fatty acid oxidation and OXPHOS) to an activated state. This is accompanied by the extensively studied induction of cytokines, chemokines and other factors functioning in IIS (and AIS). Most of the encoding genes are controlled by NF-κB, the expression of which is also highly induced under these conditions. The activated metabolic state of (MO-derived) DCs (triggered e.g., by E. coli or only by its LPS) is characterized by the induced expression of genes encoding proteins involved in glucose catabolism via glycolysis and PPP and transcription factors activating these pathways, in particular HIF-1 (Huang et al., 2001; Jantsch et al., 2008). An in-depth analysis (Krawczyk et al., 2010) showed that the interactions of TLR2, TLR4, and TLR9 with their corresponding ligands, i.e., bacterial PGN, LPS, and CpG, respectively, cause a switch from the OXPHOS-engaged resting state to the activated state characterized by aerobic glycolysis and lactate production similar to the “Warburg effect” in cancer cells (Warburg et al., 1927). These so-called Tip-DCs2 induce expression of iNOS resulting in increased NO production from arginine which seems to be responsible for the breakdown of mitochondrial respiration (Everts et al., 2012). This TLR-induced metabolic change is driven by the PI3K/Akt pathway (probably in a MyD 88-dependent manner (Gelman et al., 2006) and relies on HIF-1α (Jantsch et al., 2008). AMPK and the anti-inflammatory IL-10 antagonize the TLR-induced activation process (Krawczyk et al., 2010; Carroll et al., 2013). In contrast to cancer cells and activated T and B cells in which the metabolic switch to aerobic glycolysis causes enhanced cell division, the Warburg effect in DCs is mainly associated with increased biosynthetic activities (in particular production of immune mediators, like IL-12, IL-6, and TNF) rather than with cell proliferation. A more detailed overview on the present knowledge of the metabolic changes in the various activated DC subtypes is given in a recent review (Pearce and Everts, 2015).
MPs can be functionally grouped, in a simplified view, into pro-inflammatory bactericidal M1 and anti-inflammatory M2 subsets (Mantovani et al., 2005; Mosser and Edwards, 2008). In vivo studies show, however, that a broad spectrum of MP subtypes seems to exist with considerable plasticity rather than solely the M1- or M2-polarized subtypes (Murray and Wynn, 2011; Stewart et al., 2012; Vergadi et al., 2017).
Classical activation of resident, metabolically quiescent MPs, e.g., by LPS and IFN-γ, induces the M1 phenotype which is characterized (similar to the activated DC state) by HIF-1 activation, leading to increased glucose uptake and enhanced aerobic glycolysis, increased flux through the PPP, inhibition of mitochondrial OXPHOS and enhanced lipid biosynthesis (Rathmell, 2012). The LPS/TLR4-mediated activation also results in increased RNI and ROS production which mainly contributes to the antimicrobial activity of M1 macrophages. ROS is not only produced by the induced phagosomal NADPH oxidase, but also in the mitochondria by translocation of the tumor necrosis factor receptor-associated factor 6 (TRAF6) into these organelles (West et al., 2011). These MPs use significant amounts of the glycolytically generated ATP to maintain the mitochondrial membrane potential, thereby preventing apoptosis (Garedew et al., 2010). The M1 state is characterized by the production of pro-inflammatory cytokines TNF-α, IL-1ß, and IL-12.
Alternatively activated M2 macrophages (activated by IL-4 and IL-13) exhibit AMPK-controlled enhanced mitochondrial OXPHOS, fatty acid oxidation and a low rate of glycolysis (Vats et al., 2006). These MPs produce the anti-inflammatory cytokines IL-4 and IL-5. IL-4 induces the expression of arginase 1, a characteristic marker of M2 macrophages. This enzyme converts L-arginine to L-ornithine (and urea) which may lead via glutamic semialdehyde to L-proline. In contrast, in M1 macrophages imported L-arginine is used for the generation of NO catalyzed by iNOS (Gordon and Martinez, 2010; Odegaard and Chawla, 2011; Martinez and Gordon, 2014).
An interesting finding concerning the M1/M2 polarization has been reported by Haschemi et al. (2012). These authors showed that the sedoheptulose kinase SHPK (formerly carbohydrate kinase-like CARKL), which catalyzes an orphan reaction in the PPP, is an important modulator for M1/M2-polarization during LPS activation. SHPK downregulation (or loss) induced by LPS leads to induction of M1-like polarization while SHPK upregulation results in M2-like activation of MPs.
In summary, the metabolic hallmarks of activated NPs, DCs, and M1 macrophages are aerobic glycolysis, absence of OXPHOS and enhanced PPP leading to increased biosynthetic activity but not to cell proliferation. The high rate of glycolysis seems to be necessary to meet the increased demand of energy and precursors for the biosynthesis of antimicrobial metabolites and pro-inflammatory proteins. In contrast, M2 macrophages perform OXPHOS, fuelled mainly by fatty acid oxidation, while aerobic glycolysis does not occur.
Surface epithelia, such as the mucosal epithelia and the skin, are only one or a few cell layers thick but represent effective protective barriers against most microorganisms. Both, MECs, and SKs are non-professional phagocytes, but produce a diversity of antimicrobial proteins that directly kill or inhibit the growth of most of the frequently encountered microorganisms. Due to their antimicrobial potential (Gallo and Hooper, 2012), MECs and SKs are also considered as part of the IIS. Many IBPs are able to actively invade the epithelial cells and to replicate in the intracellular environment during infection (Pizarro-Cerdá and Cossart, 2006). The metabolic responses of MECs and SKs caused by IBP infections are poorly studied.
In response to disturbance of the tissue integrity, due to different events including infections by IBPs, MOs and NPs are recruited to the sites of infection where the activated phagocytes contribute to local inflammation. These inflammatory sites are characterized by low concentrations of oxygen and glucose, but high levels of lactate and ROS (Saadi et al., 2002; Campbell et al., 2014). The surrounding epithelial cells are affected by the oxygen depletion and ROS generation leading (among others) to HIF-1 stabilization (Campbell and Colgan, 2015) and enhanced HIF-regulated expression of antimicrobial factors (Peyssonnaux et al., 2008) and probably also to HIF-triggered shifts of metabolic pathways (Kominsky et al., 2010).
The responses of the effector immune cells to microbial pathogens, especially to IBPs, are aimed to eliminate (through the various induced defense responses discussed above) the respective pathogen and to return to cell sterility (Odegaard and Chawla, 2013). On the other hand, as repeatedly mentioned, effector immune cells, in particular those of the IIS, may also serve as niches for intracellular survival, replication and dissemination of many IBPs (Kumar and Valdivia, 2009; Rikihisa, 2010; Fabrik et al., 2013; Fredlund and Enninga, 2014). The fact that the same cell type is able to perform these two apparently contradictory functions and may allow intracellular replication of IBPs with highly diverse metabolic potentials suggests that different metabolic adaptation processes have to take place in both partners. Several possible solutions to this “paradox problem” are outlined in Figure 7. In the following, we will discuss investigations that shed some light on this intriguing problem.
Different IBPs prefer different carbon compounds for energy generation and production of necessary catabolic intermediates (Eisenreich et al., 2010; Abu Kwaik and Bumann, 2013; Barel et al., 2015). The few metabolic generalists among the IBPs, mainly the facultative intracellular Enterobacteriaceae (Salmonella, Shigella and the related enteroinvasive E. coli) and possibly also Brucella spp. seem to use glucose as preferred carbon source for their intracellular catabolic and anabolic activities (Bowden et al., 2009; Götz et al., 2010; Xavier et al., 2013; Waligora et al., 2014). However, glucose is also the primary carbon source for metabolically activated immune cells that serve as host cells for IBPs. Extensive glucose deprivation by the IBPs may therefore lead to cell death by apoptosis or pyroptosis (Fink and Cookson, 2007; Suzuki et al., 2007; Lembo-Fazio et al., 2011).
On the other hand, IBPs that have adapted to a mainly intracellular life, like Rickettsia, Chlamydia, Coxiella, Francisella, and Legionella are more or less auxotrophic and often lack pathways and reactions for efficient glucose uptake and/or catabolism. These IBPs, but even the above mentioned metabolic generalists may pursue an intracellular metabolic strategy to achieve optimal intracellular replication which we recently termed “bipartite metabolism” (Grubmüller et al., 2014; Abu Kwaik and Bumann, 2015; Eisenreich et al., 2015). In this scenario, a combination of several host cell-derived carbon compounds drives the intracellular microbial metabolism and thereby allows a fine-tuned adaptation to the host cell (Figure 6). One (or more) host cell-derived energy-rich carbon substrate(s), less critical for the host cell metabolism than glucose, may function as major energy source for ATP production by substrate phosphorylation or oxidative phosphorylation via the ETC. These substrates include glycolysis-derived compounds, like glycerol(-3P), pyruvate and lactate, but also TCA intermediates, like succinate and malate, or amino acids that can be converted to such substrates, such as serine, alanine, aspartate, glutamate, and fatty acids.
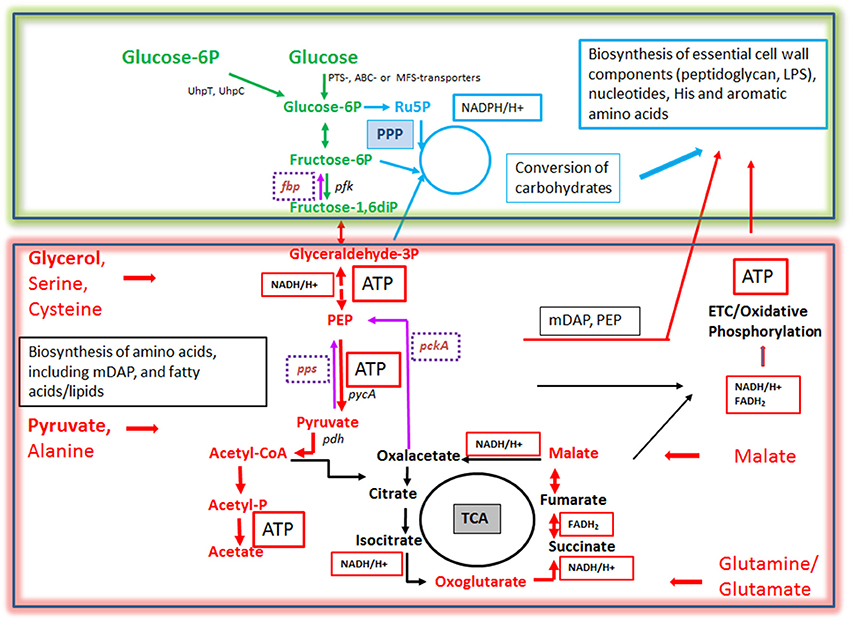
Figure 6. Bipartite metabolism of IBPs aiming to avoid the exclusive use of glucose (an indispensable carbon source of their mammalian host cells) by the IBPs for their intracellular metabolism. This strategy divides the bacterial carbon metabolism into two modules. Module 1 (green-framed), contains the upper (energy-dependent) part of the glycolytic pathway, as well as the oxidative and non-oxidative arms of PPP. These metabolic pathways are fed mainly by glucose, glucose-6P or polymers (e.g., glycogen) yielding these hexoses and deliver intermediates essential for the (energy-dependent) biosynthesis of cell envelope components, nucleotides, aromatic amino acids and NADPH, a co-factor essential for several biosynthetic pathways (especially fatty acids/lipids). The synthesis of certain cell envelope components and NADPH is indispensable for intracellular growth of the IBPs while all other components, including aromatic amino acids and nucleotides produced in module 1 can be also provided by the host cell (see Figure 3). Module 2 (red-framed) comprises the lower part of glycolysis, the TCA cycle and electron transport chain (ETC). Module 2 can be fed by C3-carbon sources, such as glycerol-(3P), serine, cysteine and pyruvate, by glutamine/glutamate and possibly by TCA intermediates, e.g., malate. It delivers mainly ATP by (a) substrate phosphorylation through NAD-dependent oxidation of these carbon sources and (b) by oxidative phosphorylation via ETC which converts NADH back to NAD necessary for maintaining the oxidation of the above carbon substrates. Module 2 delivers also intermediates for the biosynthesis of several amino acids, all of which can be imported from the host cell. Indispensable is, however the production of meso-diaminopimelic acid (mDAP) and PEP for the synthesis of peptidoglycan and of Ac-CoA for the biosynthesis for specific cell-envelope-associated fatty acids/lipids (see Figure 3).
More specifically, for this purpose L. monocytogenes seems to use glycerol (Eylert et al., 2008), Shigella pyruvate (Kentner et al., 2014), Francisella tularensis various amino acids, glycerol, glycerol-3P, or pyruvate (Brissac et al., 2015), Salmonella enterica glycerol, fatty acids and lactate (Steeb et al., 2013), Legionella pneumophila amino acids especially serine (Price et al., 2011; Schunder et al., 2014), Brucella abortus possibly glutamate (Zuniga-Ripa et al., 2014), Chlamydia trachomatis possibly malate (Iliffe-Lee and Mcclarty, 2000; Mehlitz et al., 2017), and Mycobacterium tuberculosis fatty acids, cholesterol (Ehrt et al., 2015; Lovewell et al., 2016) or glycolytic C3 compounds (Beste et al., 2013), respectively. These carbon substrates are not necessarily utilized for the generation of hexoses (and subsequently other carbohydrates needed for cell envelope synthesis) via gluconeogenesis. Indeed, several IBPs (Rickettsia, Chlamydia, Coxiella, and Legionella) lack the typical bacterial type I-III fructose-1,6-diphosphatases (FBPase, an essential enzyme for gluconeogenesis). It is, however, possible that these IBPs, some of which (e.g., L. pneumophila) are definitely able to perform gluconeogenesis, might compensate the missing conventional FBPase by alternative FBPase activities, e.g., by the reversible pyrophosphate-dependent phosphofructokinase or fructose-6-phosphate aldolase (Schürmann and Sprenger, 2001; Arimoto et al., 2002; Ganapathy et al., 2015). On the other hand, in Brucella abortus which possesses type I and type II FBPases, both enzymes are dispensible for intracellular replication (Zuniga-Ripa et al., 2014), whereas in Francisella tularensis inactivation of the glpX gene (encoding a type II FBPase) leads to reduced intracellular multiplication (Brissac et al., 2015), indicating the need of gluconeogenesis for intracellular replication of this IBP particularly in those stages of infection in which glucose is limited. However, when glucose is available most IBPs take up glucose in addition to the above mentioned carbon source(s) from the host cell, but use it for anabolic rather than for energy-producing catabolic pathways (Figure 6). In this context, it is interesting to note that most IBPs (exceptions are the facultative intracellular Enterobacteriaceae) lack the highly efficient glucose-specific phosphoenolpyruvate-dependent transport system (PTS), but may possess less efficient ABC or major facilitator superfamily GLUTs (Aguilar et al., 2012). Interestingly, L. monocytogenes possess a large number of PTS, but lack the most efficient glucose-specific PTS-G (Stoll and Goebel, 2010). Several IBPs are able to import phosphorylated hexoses (especially glucose-1P and/or glucose-6P, deriving eventually from host cell glycogen) by means of specific transporters: UhpT in case of L. monocytogenes (Chico-Calero et al., 2002) and UhpC in case of Chlamydia pneumoniae (Schwöppe et al., 2002) and Legionella pneumophila (Schwöppe et al., 2002; Chien et al., 2004). The expression of these transporters may be induced under the intracellular conditions (Joseph et al., 2006).
The imported glucose or glucose-P seems to be mainly channeled into the PPP. NADPH, produced in the oxidative branch of the PPP (present in IBPs with the exception of Francisella, Coxiella, and Rickettsia spp.), serves as cofactor for the reductive biosynthesis of nucleotides and fatty acids and for the generation of glutathione which provides protection against oxidative damage. Through the non-oxidative arm of the PPP (present in all IBPs except Rickettsia spp.) specific phosphorylated carbohydrates are generated which are required for the biosynthesis of aromatic amino acids, nucleotides and bacterial cell envelope components.
Most other basic anabolic compounds, especially amino acids (even those which in principle could be synthesized by the IBPs), are imported from the host cell (Grubmüller et al., 2014). An exception is meso-diaminopimelate (mDAP), an essential component of peptidoglycan synthesis which cannot be delivered by the host cell and has to be apparently produced by the IBPs (Figure 1). Among all IBPs for which genome sequences are known, only Francisella tularensis and other Francisella spp. lack most genes encoding the enzymes necessary for mDAP biosynthesis.
The withdrawal of amino acids from the host cells by the IBPs will cause amino acid starvation in the host cells (Tattoli et al., 2012; Tsalikis et al., 2013) which may lead, as discussed below, to induction of “prosurvival autophagy” thus prohibiting early host cell death by apoptosis/pyroptosis. Some IBPs possess anti-autophagic properties and in this case the induced autophagy of the host cell may even support intracellular replication of the IBPs (see below).
The concept of bipartite metabolism may also explain the frequently observed persistent state of IBPs, i.e., their intracellular survival without active replication. Interruption of the major glucose (or glucose-6P) supply to the IBP will stop bacterial cell wall synthesis and cell division, but as long as the catabolism of an alternative carbon substrate provides sufficient ATP (and eventually necessary amounts of phosphorylated carbohydrates generated by gluconeogenesis and PPP) to maintain the integrity of indispensable macromolecules (especially of DNA and cell envelope structures), the IBP will survive but not multiply.
As outlined above, the well-characterized defense mechanisms executed by the immune cells of the IIS include ROS and RNI production, synthesis of antibacterial peptides, induction of inflammasomes with the concomitant synthesis of inflammatory cytokines (IL1-ß and IL18) and subsequent pyroptosis as well as induction of autophagy (Sauer et al., 2010). These defense mechanisms are predominantly triggered by the interactions of different bacterial MAMPs with their corresponding TLRs and NLRs. Certain T3SS- and T4SS-effector proteins, e.g., SopE and SopB of S. Typhimurium secreted by T3SS-1, can also promote inflammatory host defense responses (Bruno et al., 2009).
The induced expression of these antimicrobial host responses requires activation of the metabolism of the immune cells involved (Lewis et al., 2011; Amiel et al., 2012; Everts et al., 2012). Interactions of LPS with TLR4 and of LTAs and lipoproteins with TLR2 and TLR6—presumably early events in the encounter of Gram-negative and Gram-positive IBPs, respectively with the IIS immune cells in vivo—trigger these defense reactions and activate the metabolism of the initially metabolically quiescent phagocytes (Krawczyk et al., 2010). This activation step leads to PI3K/Akt-mediated induction of aerobic glycolysis and NF-kB activation causing (among others) increased production of ROS and NO with subsequent loss of OXPHOS and inhibition of the TCA cycle (Everts et al., 2012). Induced expression of PKM2 seems to play an important role in this metabolic switch and is characteristic to inflammatory immune cells, such as M1 macrophages, Tip-DCs (M1-related cells that release TNF and NO, see above) (Schmid et al., 2012; Guilliams et al., 2014; Martinez and Gordon, 2014; Murray et al., 2014) and Th17 cells (Korn et al., 2009). This type of inflammatory cell metabolism represents, however, a rather hostile setting for the intracellular replication of invading IBPs (Price and Vance, 2014; Palsson-Mcdermott et al., 2015). Indeed, mice with impaired NO generation show dramatically enhanced bacterial proliferation in Kupffer cells upon infection with S. Typhimurium (Vazquez-Torres et al., 2004), suggesting that, without the antimicrobial NO production, intracellular replication of the S. Typhimurium in these metabolically activated MPs proceeds much more efficient.
To counteract the plethora of antimicrobial host cell reactions, IBPs have developed a variety of defense mechanisms: (a) inhibition of MAMP/PRR recognition through modification and shutting down the production of MAMPs, like LPS, LTA, and flagellin, (b) inactivation of ROS by induced production of superoxide dismutase and catalase, (c) reduction of the NO level through induction of arginase, (d) inhibition of inflammasome formation and pyroptosis, and (e) suppression of autophagy (Hedrick, 2004; Shen and Higgins, 2006; Bhavsar et al., 2007; Coats et al., 2007; Phalipon and Sansonetti, 2007; Ishii et al., 2008; Pieters, 2008; Taxman et al., 2010; Kullas et al., 2012; Fabrik et al., 2013; Smith and May, 2013; Al-Khodor et al., 2014; Okumura and Nizet, 2014; Wynosky-Dolfi et al., 2014). The realization of most of these bacterial countermeasures against the host cell defense reactions requires, however, a host cell metabolism that allows active intracellular replication of the IBPs and it remains to identify the appropriate metabolic host cell conditions that activate the IBP-specific countermeasures.
The paradox situation that the same immune cells may carry out antimicrobial (i.e., kill IBPs) but also promicrobial functions (i.e., serve as convenient host cells for the IBPs) was discussed by the example of MPs in a recent review (Price and Vance, 2014). An important message was that the anti-inflammatory M2 but not the inflammatory M1 phenotype of activated MPs represents a comfortable replication niche for IBPs. Indeed, recent studies (Eisele et al., 2013; Xavier et al., 2013) showed that alternatively activated M2 macrophages represent the predominant MP population inhabited by replicating Salmonella and Brucella strains, respectively, during chronic infection in the mouse model. This ability correlates with higher levels of unconsumed glucose in the M2 compared to M1 macrophages. Signal transduction through the nuclear peroxisome proliferator-activated receptors (PPAR-γ and PPAR-δ), respectively, was shown to be crucial for this process. Induction of these metabolic regulators enhance OXPHOS and fatty acid ß-oxidation in M2 macrophages thereby leading to increased levels of unconsumed glucose. M1 macrophages on the other hand catabolize glucose through the highly activated glycolytic pathway and thereby withdraw the bacteria this essential carbon source.
These findings led to the suggestion that the PPAR-induced increase in intracellular glucose availability may be a rather general mechanism promoting growth of persistent pathogens within M2 macrophages (Xavier et al., 2013). Indeed, M2 polarization of human macrophages was recently shown to favor also replication of Chlamydia pneumoniae (Buchacher et al., 2015).
However, the typical M2 metabolism does not seem to be the only metabolic condition which supports replication and proliferation of IBPs. Established MO- and MP-like cell lines, such as J774A.1, P388.D1, RAW264.7, THP-1, and U-973, which are routinely used as host cells for studying intracellular replication of IBPs, perform a permanently activated metabolism which is mainly triggered by the constitutive expression of oncogenes (e.g., Myc), or by the inactivation of tumor suppressors (e.g., p53). In these cell lines, most IBPs are able to replicate efficiently, indicating that this type of activated, glycolysis-driven host cell metabolism also supports successful intracellular replication of many IBPs (Fuchs et al., 2012; Eisenreich et al., 2013; Kentner et al., 2014). The dysregulated metabolism of these transformed cell lines is clearly different from the metabolism of M2 macrophages (characterized by ß-oxidation of fatty-acids driven OXPHOS), but apparently provides all nutrients necessary for the bipartite metabolism of most IBPs.
It should also be noted that in contrast to Salmonella and Brucella, the intracellular replication capacity of Listeria, Franciscella and Mycobacterium strains is not influenced by the modulation of the PPAR levels and hence the intracellular glucose concentration of the host cells (Eisele et al., 2013).
These data show that not only the strategy exemplified by M1/M2-polarized MPs (strategy A, outlined in Figure 7A), but also other (still rather hypothetical) strategies (exemplified e.g., by Figures 7B–D) may result in a host cell metabolism that supports IBP replication: (A) in an already activated immune cell population different subsets may exist (e.g., M1/M2 macrophages and their subgroups Martinez and Gordon, 2014) that are either metabolically favorable or adverse for intracellular replication of IBPs, (B) anti- or pro-replicative metabolic conditions may be induced in individual cells of a quiescent immune cell population depending on the interaction between different host cell receptors and different IBP ligands, (C) before or shortly after internalization the IBPs may translocate effector protein(s) that specifically counteract the antimicrobial activities (e.g., ROS or RNI production) without changing the metabolic conditions of the host cell that are otherwise favorable for IBP replication, or (D) phagocytosis of the IBPs and activation of pro-replicative metabolic conditions in the immune cells may be separately triggered by interaction of different host cell receptors with different IBP-specific ligands.
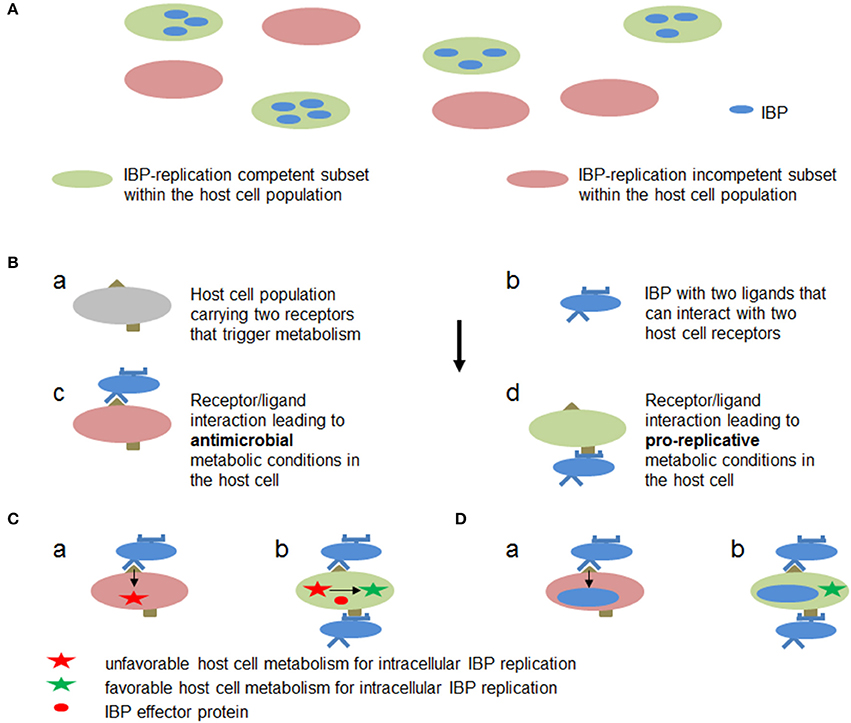
Figure 7. Hypothetic metabolic host cell conditions and interactions between host cell targets and IBP components leading to metabolic conditions in the host cell that are favorable or adverse for intracellular replication of IBPs. (A) Within a host cell population (e.g., MPs) metabolically different subsets (a and b) may already exist which allow (blue) or inhibit (yellow) active metabolism and hence replication of the IBP. (B) Cells within a host cell population may carry two receptors (a) which can interact with two different ligands of the IBP (b). One receptor/ligand combination (c) leads to an antibacterial metabolic host cell state (red), while the other (d) triggers metabolic host cell conditions that allow intracellular replication of IBP (green). (C) IBP (yellow circle) is internalized by the interaction of a host cell receptor (blue half circle) that triggers phagocytosis of the IBP but induces at the same time a host cell metabolism (a) that is unfavorable for intracellular IBP replication (red star). However, before or during internalization (b) the IBP may translocate an effector protein (red circle) via specific secretion (injection) systems that alters the host cell metabolism to become favorable for IBP replication (green star). (D) The IBP activates by a first interaction (a) a host cell metabolism with bacteriocidal activity. The IBP may inactivate, however, by a second bacterial ligand/host cell receptor interaction (b) the bacteriocidal activity thereby rendering the host cell metabolism favorable for IBP replication (green star).
The data discussed in Interactions between host cell targets and bacterial effectors leading to metabolic changes within host cells that may support intracellular replication of IBPs, suggest that the exclusive use of glucose is not necessarily the best strategy for fueling the intracellular metabolism of all IBPs. Rather, IBPs according to their different metabolic capacities may depend—for performing an optimally balanced intracellular metabolism and hence efficient proliferation—on IBP-specific combinations of catabolic and anabolic metabolites that are provided by the host cell. This in turn requires IBP-specific metabolic conditions within (non-cancer) host cells which (in vivo) may already exist in a subset of a potential host cell population or may be induced by invading pathogens in metabolically quiescent (primary) cells through the interaction of specific bacterial ligands (e.g., effector proteins or other virulence factors) and appropriate host cell receptors (Figure 7).
As discussed in Metabolic activation of quiescent host cells that may prohibit or favor intracellular growth of IBPs, the alternatively activated M2 macrophages seem to represent such a subpopulation performing a metabolism that supports intracellular replication of certain IBPs (Price and Vance, 2014). Alternatively, specific T3SS- or T4SS-effector proteins or other virulence factors of IBPs which directly or indirectly interact with the above described central regulators of host cell metabolism, including PI3K, Akt, mTOR, cMyc, HIF-1α, PKM2, P53, TIGAR, PTEN, and AMPK, might also be candidates for modulating the metabolism of (quiescent) primary host cells in such a way that it becomes supportive for intracellular IBP replication. Indeed, interactions of IBP effectors with several of these oncogenes and tumor suppressors have already been demonstrated (Table 1). However, the outcome of these interactions has been so far mainly studied with respect to the pathogen internalization and/or immune responses, while the likely impact on the host cell metabolism has hardly been addressed. To the best of our knowledge, only few reports (see below) show how IBPs may manipulate the host cell metabolism, in particular that of immune cells by such interactions.
Gillmaier et al. (2012) showed that primary murine bone marrow-derived MPs (BMDMs) which perform in the uninfected state the typical quiescent OXPHOS-based metabolism (see above), are metabolically highly activated upon internalization of wild-type L. monocytogenes. However, in contrast to LPS- and IFN-γ-mediated metabolic activation, the L. monocytogenes-induced BMDM metabolism does not only lead to induction of the glycolytic pathway but also to induction of the initial steps of the TCA cycle thus enabling synthesis of citrate, its subsequent export into the cytosol and the generation of acetyl-CoA and oxaloacetate by ACL. This may allow enhanced anabolic activities in the BMDMs (especially fatty acid/lipid and amino acid biosynthesis). This type of induced host cell metabolism is similar to that occurring in the murine macrophage cell line J774 (its metabolism is permanently activated due to constitutively expressed Myc) where L. monocytogenes replicates without significantly altering the metabolism of these host cells. The efficiency and the mode of intracellular metabolism of L. monocytogenes are similar in the activated BMDM and in J774 cells (Gillmaier et al., 2012).
Interestingly, only a small population of the BMDMs (<5%) contains a large number (>20 bacteria/host cell) of replicating listeriae, suggesting that this type of metabolic activation, obviously favorable for intracellular replication of L. monocytogenes, may be induced only in a subset of the BMDMs. Notably, this L. monocytogenes-triggered metabolism of the BMDMs mimics more the growth factor-induced metabolism governed by the PI3K/Akt/mTORC1 pathway which leads to the activation of glycolysis, PPP and TCA (Kaplan et al., 2000; Jones and Thompson, 2009; Salminen and Kaarniranta, 2010). It has been shown that internalin B (InlB) of L. monocytogenes has structural and functional similarities to the hepatocyte growth factor (HGF), recognizes the same receptor Met as HGF and InlB/Met interaction activates PI3K (Ireton et al., 1996; Shen et al., 2000; Bierne and Cossart, 2002; Li et al., 2005; Niemann, 2013; Gessain et al., 2015). It is therefore intriguing to speculate that InlB/Met interaction in a subset of the BMDM population (Galimi et al., 2001; Moransard et al., 2010) might be responsible for the induction of the above described metabolism that supports intracellular L. monocytogenes replication. Direct experimental evidence for this hypothesis is still missing, but it has been shown (Gillmaier et al., 2012) that a non-pathogenic prfA mutant (also unable to express inlB) and heat-inactivated L. monocytogenes wild-type bacteria are unable to induce this type of metabolism in BMDM.
As discussed above, the tumor suppressor p53 blocks glucose uptake, glycolysis and PPP (Vousden and Ryan, 2009) whereas inactivation or down-regulation of p53 leads to the activation of these metabolic pathways which occurs in many cancer cells. Caco-2 cells contain a deleted and mutated p53 gene and no detectable p53 protein (Djelloul et al., 1997). These metabolically activated cells are excellent hosts for many IBPs including L. monocytogenes. Indeed, down-regulation of p53 in RAW264.7 and HeLa cells and p53 knockout mice also results in significantly increased intracellular replication of L. monocytogenes, whereas intracellular replication is inhibited when p53 was overexpressed in the two cell lines (Wang et al., 2016).
Shigella flexneri (which similar to L. monocytogenes replicates in host cells cytosol) may invade and multiply within MPs but rapidly induce pyroptotic cell death of these host cells (Suzuki et al., 2007). Epithelial cells of the intestine are the preferred host cells which allow efficient replication of Shigella upon oral infection. In the infected cells, degradation of p53 has been reported (Bergounioux et al., 2012) which is mainly caused by calpain protease activation, promoted by the T3SS effector protein VirA. The p53 degradation could activate the host cell metabolism, but the calpain activation causes also cell death and thus elimination of this replication niche (Bergounioux et al., 2012). However, IpgD, another T3SS effector of S. flexneri, dephosphorylates PIP2 and the resulting phosphatidylinositol-5-phosphate leads to the activation of the PI3K pathway which promotes phosphorylation of Akt (Pendaries et al., 2006). This Akt activation could also activate the host cell metabolism.
Down-regulation of p53 has also been demonstrated in human cells upon infection by different Chlamydia species (González et al., 2014; Siegl et al., 2014) and this down-regulation appears to be crucial for the efficient intracellular replication of C. trachomatis which in this case mainly occurs in a membrane-bound compartment, the inclusion. However, it is still unknown which chlamydial factor(s) is responsible for the p53 down-regulation and what are the metabolic consequences.
Mycobacterium tuberculosis (Mbt) infection is initiated by inhalation into the alveolus. Although studies in lungs of aerosol-infected mice show extensive replication of Mbt in alveolar epithelial cells and other non-macrophage cells and dissemination to other organs early in infection (Ryndak et al., 2015), alveolar MPs are the typical host cells of Mbt where the bacteria primarily reside in phagosomes. Fatty acids and cholesterol are considered to be the most important carbon sources for optimal growth and persistence of Mbt during infection (Lovewell et al., 2016). These two nutrients are involved in the production of energy but also of intermediates for mycolic acids, the major and specific lipid components of the cell envelope of Mbt (Marrakchi et al., 2014). Mbt is also able to utilize several other substrates depending on the growth conditions (Marrero et al., 2013; Beste et al., 2013; Ehrt et al., 2015; Hampel et al., 2015; Rücker et al., 2015) Glucose has been reported to be used for growth in vivo and both, glycolysis and gluconeogenesis, are important pathways for the intracellular carbon metabolism (Ehrt et al., 2015).
Not surprisingly, Mbt significantly reprograms the metabolism of the infected host MPs. Metabolomic profiling in mice infected with Mbt showed increased levels of several metabolites in the lung (presumably within MPs) of infected mice which suggest elevated ß-oxidation of lipids and glyoxylate shunting, increased nucleotide and amino acids metabolism and increased anti-oxidative stress response in this tissue upon Mbtinfection (Shin et al., 2011). A hallmark of mycobacterial infection is the formation of foamy MPs characterized by the accumulation of large lipid bodies. The foamy phenotype appears to be associated with the temporary increase of the peroxisome proliferator-activated receptor gamma (PPAR-y) and dependent on TLR-2 (Almeida et al., 2012). It was further shown (Singh et al., 2012) that Mbt induces the foamy phenotype by diverting the glycolytic pathway of the host MPs toward ketone body synthesis. This metabolic dysregulation leads to the activation of the anti-lipolytic G protein-coupled receptor GPR109A which causes perturbations in lipid homeostasis and consequent accumulation of lipid bodies in the MP. ESAT-6, a secreted virulence factor of Mbt interacting with TLR2, seems to be responsible for this feedback loop. The increased MP glycolysis and the accompanied lipid droplet accumulation is in line with the observation that liver X transcriptional receptors (LXRA and LRXB) which control fatty acid, cholesterol and glucose homeostasis are critical regulators of lipid metabolism during intracellular Mbt replication (Han et al., 2014).
Autophagy is a catabolic process that sequesters and degrades dispensible cellular contents, like waste proteins and damaged organelles, but also microorganisms reaching the cytosol (reviewed by Gomes and Dikic, 2014; Randow and Youle, 2014). Induction of autophagy occurs in response to depletion of ATP, amino acids and glucose and is controlled mainly by AMPK and mTORC1. As outlined above, AMPK is activated by high AMP/ATP ratios while mTORC1 activity is modulated by amino acid levels (Bar-Peled et al., 2012; Inoki et al., 2012).
In the autophagic process, the targeted cytosolic components are engulfed in double membrane surrounded autophagosomes which then fuse with lysosomes leading to the degradation of the autosomal contents by digestive enzymes. This process results in the release of metabolites (in particular amino acids and carbohydrates) that can be reused for anabolic and catabolic processes, especially for protein biosynthesis and energy generation.
“Prosurvival autophagy” may thus promote cell survival under nutrient (especially amino acid) starvation conditions (Dibble and Manning, 2013; Jewell and Guan, 2013), but it may also support microbial replication. Manipulation of both, the energy-sensing AMPK and the nutrient-sensing mTORC1 kinase activities, has been shown to be crucial for the proliferation of several viral pathogens (Brunton et al., 2013); the resulting metabolic changes in the host cells which support viral replication have, however, hardly been analyzed.
The significance of these two key regulators of cell metabolism for the intracellular replication of bacterial pathogens has also been discussed and the importance of autophagy for intracellular growth (through the supply of additional nutrients) has been clearly demonstrated for Franciscella tularensis (Steele et al., 2013).
Other IBPs adapted to intracellular life in the host cells cytosol, like L. monocytogenes and Shigella flexneri, also induce autophagy in MPs (Mostowy et al., 2011). These IBPs have developed specific mechanisms which protect themselves from the autophagic attack. L. monocytogenes protects itself by ActA and InlK (Yoshikawa et al., 2009; Dortet et al., 2012) while S. flexneri escapes autophagy mainly by IcsB (Ogawa et al., 2005). Autophagy induced in the host cells infected by these IBPs may therefore provide additional nutrients, especially amino acids (Grubmüller et al., 2014) to these intracellular bacteria thus supporting their intracellular replication.
As discussed above, activated NPs infiltrating the intestinal epithelium infected by certain IBPs, e.g., S. Typhimurium, will affect the metabolism of the surrounding epithelial cells (e.g., by HIF-1 activation) and, hence, possibly also the replication of IBPs capable of invading these epithelial cells.
Activated NPs may, however, also positively affect the metabolism and proliferation of IBPs in the extracellular state (Winter et al., 2010a; Lopez et al., 2012). The best studied example is S. Typhimurium which induces acute inflammation in the intestine by T3SS-1 effectors translocated into epithelial cells and MPs. ROS produced by the attracted activated NPs convert luminal thiosulfate to tetrathionate which serves as Salmonella-specific electron acceptor in anaerobic respiration (Winter et al., 2010b). Specific S. Typhimurium strains lysogenized by a bacteriophage carrying the sopE gene are also able to enhance the production of host-derived nitrate which is an even more efficient electron acceptor than tetrathionate allowing anaerobic nitrate respiration by these bacteria (Lopez et al., 2012). Gut-derived ethanolamine has been suggested as electron donor for this anaerobic respiration chain (Thiennimitr et al., 2011). This metabolic capacity of S. Typhimurium (and possibly of other IBPs), supported by activated immune cells, enhances proliferation of these pathogens in the gastrointestinal tract at the expense of the other fermenting resident gut microbes.
The genes essential for this anaerobic metabolism of enteric S. Typhimurium appear to be part of a metabolic network which is decaying in the extraintestinal typhoid S. Typhi (Nuccio and Bäumler, 2014). S. Typhi tightly regulates the expression of T3SS-1 by the regulatory protein TviA, thereby suppressing the pro-inflammatory host responses which reduces intestinal inflammation (Winter et al., 2014) and even impairs activation and proliferation of naïve flagellin–specific CD4+ T cells in Peyer's patches and mesenteric lymph nodes (Atif et al., 2014). This allows increased bacterial dissemination finally leading to systemic infection.
Other metabolites produced by activated immune cells in excess, e.g., TCA intermediates like succinate, malate (due to enhanced glutaminolysis) and especially lactate (due to enhanced glycolysis) could be used by IBPs as additional catabolic carbon sources in bipartite metabolism during intracellular replication.
Immune cells, especially those belonging to the innate branch of the immune system, may function, on the one hand, as powerful defense weapons against microbial pathogens (including IBPs) and, on the other hand, as important host cells for IBPs. These two conflicting functions, executed by the same group of immune cells require specific metabolic programs to which both partners seem to contribute. There is substantial information on the mechanisms leading to the antimicrobial activities of these immune cells that are mainly triggered by the interaction of well-characterized bacterial components with their respective cell receptors. There is also growing knowledge on the various countermeasures which the IBPs have evolved to counteract these antimicrobial cell activities. However, rather little is known how the metabolism of the IBPs has to adapt to that of these host cells in order to achieve optimal intracellular bacterial growth. Even less is known how the metabolism of the immune cells is adjusted to serve either as an antimicrobial environment or as a comfortable replication niche for IBPs. The still quite limited number of studies (broadly discussed in this review) addressing these intriguing problems suggests that different, IBP-specific adaptation strategies are pursued when the pathogens encounter these target cells. This is not unexpected considering the different metabolic potentials of the IBPs and hence their varying nutritional dependency on the host cells, as well as the different arsenal of weapons developed by the IBPs (effector proteins and other virulence factors). A major problem also arises by the heterogeneity of the immune cells of the IIS, especially of MPs (Guilliams et al., 2014; Murray et al., 2014) (for more details, see Supplementary Material), which are often the initial host cells for IBPs during infection. In most reports describing the metabolic reprogramming induced in these cells (even when using primary MPs) it remains unclear to which subtype the actual IBP replication supporting host cell belongs. In vivo infection studies, combined with single cell analysis and sensitive techniques allowing the determination of the metabolism in the infected single cells might be necessary to unravel this unsolved problem. The answer to these open questions will not only widen our basic understanding of IBP infections, but may also reveal novel approaches to combat the often severe diseases caused by these pathogens (Bettencourt and Powell, 2017).
WG and WE designed the study. WG, WE, TR, and JH wrote to article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
WE thanks the Bundesministerium für Bildung und Forschung (BMBF) through ERA-NET Infect-ERA in the context of the EUGENPATH network and the Deutsche Forschungsgemeinschaft (EI 384/11, RU 631/9-2).
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00316/full#supplementary-material
4E-BP, factor 4E binding protein; 6PG, 6-phosphogluconate; 6PGDH, 6-phosphogluconate dehydrogenase; 6PGL, 6-phosphogluconolactone; 6PGLase, 6-phosphogluconolactonase; ACL, ATP-dependent citrate lyase; AIS, adaptive immune system; AMPK, AMP-activated protein kinase; α-OXO, α-oxoglutarate; BMDM, bone marrow-derived MP; ChREBP, carbohydrate-responsive element-binding protein; CIT, citrate; COX, cytochrome C oxidase; CS, citrate synthase; DAMP, danger-associated molecular patterns; DC, dentritic cell; DHAP, dihydroxyacetone phosphate; E4P, erythrose-4-phosphate; ESX, ESAT-6 secretion system; ETC, electron transport chain; F26BP, fructose-2,6-diphosphate; F6P, fructose-6-phosphate; FBA, fructobisphosphate aldolase; FBP, fructose-1,6-diphosphate; FBPase, fructose-1,6-diphosphatase; FUM, fumarate; FUMH, fumarate hydratase; G-, Gram-negative; G+, Gram-positive; G3P, glyceraldehyde-3-phosphate; G6P, glucose-6-phosphate; G6PDH, glucose-6-phosphate dehydrogenase; GAP, glyceraldehyde-3P; GDH, glutamate dehydrogenase; GLNLY, glutaminolysis; GL, glycolytic pathway; GLS, glutaminase; GLUT, glucose transporter; GLUT1, glucose transporter 1; GN, gluconeogenesis; HGF, hepatocyte growth factor; HIF-1, hypoxia-inducible transcription factor 1; HK2, hexokinase 2; IBP, intracellular bacterial pathogen; ICIT, isocitrate; IDH, isocitrate dehydrogenase; IFN, type-I interferon; IIS, innate immune system; IL, interleukin; InlB, internalin B; iNOS, inducible NO synthase; IRF, interferon regulatory factors; LDH, lactate dehydrogenase; LKB1, liver kinase B1; LPS, lipopolysaccharide; LT, lymphoid tissue; LTA, lipoteichoic acid; MAL, malate; MAMP, microbe-associated molecular pattern; MAPK, mitogen-activated protein kinase, Mbt, Mycobacterium tuberculosis; mDAP, meso-diaminopimelate; MDH, malate dehydrogenase; ME, malic enzyme; MEC, mucosal epithelial cell; MO, monocyte; MP, macrophage; MPS, mononuclear phagocyte system; mTOR, target of rapamycin; mTORC1, target of rapamycin complex 1; mTORC2, target of rapamycin complex 2; NLR, (NOD)-like receptor; NLT; non-lymphoid tissue; NOD, nucleotide-binding oligomerization domain; NP, neutrophile; OAA, oxaloacetate; OGDH, oxoglutarate dehydrogenase; OXPHOS, oxidative phosphorylation; PAMP, pathogen-associated molecular pattern; PCK, PEP carboxykinase; PDH, pyruvate dehydrogenase; PDK, pyruvate dehydrogenase kinase; PDPK1, 3-phosphoinositide dependent protein kinase-1; PEP, phosphoenolpyruvate; PFK, phosphofructokinase; PFK1, phosphofructokinase 1; PGI, phosphoglucoisomerase; PGN, peptidoglycan; PHD, prolyl hydroxylase; PI3K, phosphoinositide-3-kinase; PIP2, phosphatidylinositol-4,5-bisphosphate; PIP3, phosphatidylinositol-3,4,5-triphosphate; PK, pyruvate kinase; PKM1, pyruvate kinase M1; PKM2, pyruvate kinase M2; PMN, neutrophilic polymorphonuclear leukocytes; PPAR, peroxisome proliferator-activated receptor; PPP, pentose phosphate pathway; PPS, PEP synthetase; PRR, pattern recognition receptor; PTEN, phosphatase and tensin homolog; PTS, PEP-dependent transport system; R5P, ribose-5-phosphate; RAS, Ras protein; RB, retinoblastoma protein; RIG.I, retinoic acid-inducible gene I; RLR, (RIG.I)-like receptor; RNI, reactive nitrogen intermediate; ROS, reactive oxygen species; RPE, ribulose-5-phosphate 3-epimerase; RPI, ribose-5-phosphate isomerase; Ru5P, ribulose-5-phosphate; S6K, S6 kinase; S7P, sedoheptulose-7-phosphate; SCO2, cytochrome c oxidase assembly protein 2; SDH, succinate dehydrogenase; SHPK, sedoheptulose kinase; SK, skin keratinocyte; SREBP, sterol regulatory element-binding protein; SUC, succinate; T3SS, type 3-secretion system; T4SS, type 4-secretion system; T7SS, type 7-secretion system; TAL, transaldolase; TCA, tricarboxylic acid cycle; TIGAR, TP53-inducible glycolysis and apoptosis regulator; TKT, transketolase; TLR, Toll-like receptor; TNF, tumor necrosis factor; TRAF6, tumor necrosis factor receptor-associated factor 6; X5P, xylulose-5-phosphate.
1. ^More recently called “microbe-associated molecular pattern” (MAMP).
2. ^According to a new nomenclature based on ontology (for details, see also Supplementary Material), the assignment of this DC subtype has to be revised.
A, S. K., Bansal, K., Holla, S., Verma-Kumar, S., Sharma, P., and Balaji, K. N. (2012). ESAT-6 induced COX-2 expression involves coordinated interplay between PI3K and MAPK signaling. Mol. Immunol. 49, 655–663. doi: 10.1016/j.molimm.2011.11.011
Abu Kwaik, Y., and Bumann, D. (2013). Microbial quest for food in vivo: 'nutritional virulence' as an emerging paradigm. Cell. Microbiol. 15, 882–890. doi: 10.1111/cmi.12138
Abu Kwaik, Y., and Bumann, D. (2015). Host delivery of favorite meals for intracellular pathogens. PLoS Pathog. 11:e1004866. doi: 10.1371/journal.ppat.1004866
Aguilar, C., Escalante, A., Flores, N., De Anda, R., Riveros-Mckay, F., Gosset, G., et al. (2012). Genetic changes during a laboratory adaptive evolution process that allowed fast growth in glucose to an Escherichia coli strain lacking the major glucose transport system. BMC Genomics 13:385. doi: 10.1186/1471-2164-13-385
Al-Khodor, S., Marshall-Batty, K., Nair, V., Ding, L., Greenberg, D. E., and Fraser, I. D. (2014). Burkholderia cenocepacia J2315 escapes to the cytosol and actively subverts autophagy in human macrophages. Cell. Microbiol. 16, 378–395. doi: 10.1111/cmi.12223
Almeida, P. E., Carneiro, A. B., Silva, A. R., and Bozza, P. T. (2012). PPARγ expression and function in mycobacterial infection: roles in lipid metabolism, immunity, and bacterial killing. PPAR Res. 2012:383829. doi: 10.1155/2012/383829
Alto, N. M., and Orth, K. (2012). Subversion of cell signaling by pathogens. Cold Spring Harb. Perspect. Biol. 4:a006114. doi: 10.1101/cshperspect.a006114
Amiel, E., Everts, B., Freitas, T. C., King, I. L., Curtis, J. D., Pearce, E. L., et al. (2012). Inhibition of mechanistic target of rapamycin promotes dendritic cell activation and enhances therapeutic autologous vaccination in mice. J. Immunol. 189, 2151–2158. doi: 10.4049/jimmunol.1103741
Angus, S. P., Wheeler, L. J., Ranmal, S. A., Zhang, X., Markey, M. P., Mathews, C. K., et al. (2002). Retinoblastoma tumor suppressor targets dNTP metabolism to regulate DNA replication. J. Biol. Chem. 277, 44376–44384. doi: 10.1074/jbc.M205911200
Arimoto, T., Ansai, T., Yu, W., Turner, A. J., and Takehara, T. (2002). Kinetic analysis of PPi-dependent phosphofructokinase from Porphyromonas gingivalis. FEMS Microbiol. Lett. 207, 35–38. doi: 10.1111/j.1574-6968.2002.tb11024.x
Arnold-Schrauf, C., Dudek, M., Dielmann, A., Pace, L., Swallow, M., Kruse, F., et al. (2014). Dendritic cells coordinate innate immunity via MyD88 signaling to control Listeria monocytogenes infection. Cell Rep. 6, 698–708. doi: 10.1016/j.celrep.2014.01.023
Arsenault, R. J., Napper, S., and Kogut, M. H. (2013). Salmonella enterica Typhimurium infection causes metabolic changes in chicken muscle involving AMPK, fatty acid and insulin/mTOR signaling. Vet. Res. 44:35. doi: 10.1186/1297-9716-44-35
Atif, S. M., Winter, S. E., Winter, M. G., Mcsorley, S. J., and Bäumler, A. J. (2014). Salmonella enterica serovar Typhi impairs CD4 T cell responses by reducing antigen availability. Infect. Immun. 82, 2247–2254. doi: 10.1128/IAI.00020-14
Bai, W., Liu, H., Ji, Q., Zhou, Y., Liang, L., Zheng, R., et al. (2014). TLR3 regulates mycobacterial RNA-induced IL-10 production through the PI3K/AKT signaling pathway. Cell. Signal. 26, 942–950. doi: 10.1016/j.cellsig.2014.01.015
Barel, M., Ramond, E., Gesbert, G., and Charbit, A. (2015). The complex amino acid diet of Francisella in infected macrophages. Front. Cell. Infect. Microbiol. 5:9. doi: 10.3389/fcimb.2015.00009
Bar-Peled, L., Schweitzer, L. D., Zoncu, R., and Sabatini, D. M. (2012). Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell 150, 1196–1208. doi: 10.1016/j.cell.2012.07.032
Bergounioux, J., Elisee, R., Prunier, A. L., Donnadieu, F., Sperandio, B., Sansonetti, P., et al. (2012). Calpain activation by the Shigella flexneri effector VirA regulates key steps in the formation and life of the bacterium's epithelial niche. Cell Host Microbe 11, 240–252. doi: 10.1016/j.chom.2012.01.013
Beste, D. J., Nöh, K., Niedenfuhr, S., Mendum, T. A., Hawkins, N. D., Ward, J. L., et al. (2013). 13C-Flux spectral analysis of host-pathogen metabolism reveals a mixed diet for intracellular Mycobacterium tuberculosis. Chem. Biol. 20, 1012–1021. doi: 10.1016/j.chembiol.2013.06.012
Bettencourt, I. A., and Powell, J. D. (2017). Targeting metabolism as a novel therapeutic approach to autoimmunity, inflammation, and transplantation. J. Immunol. 198, 999–1005. doi: 10.4049/jimmunol.1601318
Bhavsar, A. P., Guttman, J. A., and Finlay, B. B. (2007). Manipulation of host-cell pathways by bacterial pathogens. Nature 449, 827–834. doi: 10.1038/nature06247
Bierne, H., and Cossart, P. (2002). InlB, a surface protein of Listeria monocytogenes that behaves as an invasin and a growth factor. J. Cell Sci. 115, 3357–3367.
Bierne, H., Sabet, C., Personnic, N., and Cossart, P. (2007). Internalins: a complex family of leucine-rich repeat-containing proteins in Listeria monocytogenes. Microbes Infect. 9, 1156–1166. doi: 10.1016/j.micinf.2007.05.003
Bowden, S. D., Rowley, G., Hinton, J. C., and Thompson, A. (2009). Glucose and glycolysis are required for the successful infection of macrophages and mice by Salmonella enterica serovar Typhimurium. Infect. Immun. 77, 3117–3126. doi: 10.1128/IAI.00093-09
Brahimi-Horn, M. C., and Pouyssegur, J. (2009). HIF at a glance. J. Cell Sci. 122, 1055–1057. doi: 10.1242/jcs.035022
Brestoff, J. R., and Artis, D. (2013). Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 14, 676–684. doi: 10.1038/ni.2640
Brissac, T., Ziveri, J., Ramond, E., Tros, F., Kock, S., Dupuis, M., et al. (2015). Gluconeogenesis, an essential metabolic pathway for pathogenic Francisella. Mol. Microbiol. 98, 518–534. doi: 10.1111/mmi.13139
Brosh, R., and Rotter, V. (2009). When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer 9, 701–713. doi: 10.1038/nrc2693
Bruno, V. M., Hannemann, S., Lara-Tejero, M., Flavell, R. A., Kleinstein, S. H., and Galan, J. E. (2009). Salmonella Typhimurium type III secretion effectors stimulate innate immune responses in cultured epithelial cells. PLoS Pathog. 5:e1000538. doi: 10.1371/journal.ppat.1000538
Brunton, J., Steele, S., Ziehr, B., Moorman, N., and Kawula, T. (2013). Feeding uninvited guests: mTOR and AMPK set the table for intracellular pathogens. PLoS Pathog. 9:e1003552. doi: 10.1371/journal.ppat.1003552
Buchacher, T., Ohradanova-Repic, A., Stockinger, H., Fischer, M. B., and Weber, V. (2015). M2 Polarization of human macrophages favors survival of the intracellular pathogen Chlamydia pneumoniae. PLoS ONE 10:e0143593. doi: 10.1371/journal.pone.0143593
Burroughs, S. K., Kaluz, S., Wang, D., Wang, K., Van Meir, E. G., and Wang, B. (2013). Hypoxia inducible factor pathway inhibitors as anticancer therapeutics. Future Med. Chem. 5, 553–572. doi: 10.4155/fmc.13.17
Cairns, R. A., Harris, I. S., and Mak, T. W. (2011). Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95. doi: 10.1038/nrc2981
Campbell, E. L., Bruyninckx, W. J., Kelly, C. J., Glover, L. E., Mcnamee, E. N., Bowers, B. E., et al. (2014). Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity 40, 66–77. doi: 10.1016/j.immuni.2013.11.020
Campbell, E. L., and Colgan, S. P. (2015). Neutrophils and inflammatory metabolism in antimicrobial functions of the mucosa. J. Leukoc. Biol. 98, 517–522. doi: 10.1189/jlb.3MR1114-556R
Carroll, K. C., Viollet, B., and Suttles, J. (2013). AMPKα1 deficiency amplifies proinflammatory myeloid APC activity and CD40 signaling. J. Leukoc. Biol. 94, 1113–1121. doi: 10.1189/jlb.0313157
Chen, L. Q., Cheung, L. S., Feng, L., Tanner, W., and Frommer, W. B. (2015). Transport of sugars. Annu. Rev. Biochem. 84, 865–894. doi: 10.1146/annurev-biochem-060614-033904
Chesney, J., and Telang, S. (2013). Regulation of glycolytic and mitochondrial metabolism by ras. Curr. Pharm. Biotechnol. 14, 251–260. doi: 10.2174/1389201011314030002
Chico-Calero, I., Suarez, M., Gonzalez-Zorn, B., Scortti, M., Slaghuis, J., Goebel, W., et al. (2002). Hpt, a bacterial homolog of the microsomal glucose-6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc. Natl. Acad. Sci. U.S.A. 99, 431–436. doi: 10.1073/pnas.012363899
Chien, M., Morozova, I., Shi, S., Sheng, H., Chen, J., Gomez, S. M., et al. (2004). The genomic sequence of the accidental pathogen Legionella pneumophila. Science 305, 1966–1968. doi: 10.1126/science.1099776
Christofk, H. R., Vander Heiden, M. G., Harris, M. H., Ramanathan, A., Gerszten, R. E., Wei, R., et al. (2008). The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452, 230–233. doi: 10.1038/nature06734
Coats, S. R., Do, C. T., Karimi-Naser, L. M., Braham, P. H., and Darveau, R. P. (2007). Antagonistic lipopolysaccharides block E. coli lipopolysaccharide function at human TLR4 via interaction with the human MD-2 lipopolysaccharide binding site. Cell. Microbiol. 9, 1191–1202. doi: 10.1111/j.1462-5822.2006.00859.x
Commichau, F. M., Dickmanns, A., Gundlach, J., Ficner, R., and Stülke, J. (2015). A jack of all trades: the multiple roles of the unique essential second messenger cyclic di-AMP. Mol. Microbiol. 97, 189–204. doi: 10.1111/mmi.13026
Cooper, K. G., Winfree, S., Malik-Kale, P., Jolly, C., Ireland, R., Knodler, L. A., et al. (2011). Activation of Akt by the bacterial inositol phosphatase, SopB, is wortmannin insensitive. PLoS ONE 6:e22260. doi: 10.1371/journal.pone.0022260
Cossart, P., and Sansonetti, P. J. (2004). Bacterial invasion: the paradigms of enteroinvasive pathogens. Science 304, 242–248. doi: 10.1126/science.1090124
Costa, T. R., Felisberto-Rodrigues, C., Meir, A., Prevost, M. S., Redzej, A., Trokter, M., et al. (2015). Secretion systems in Gram-negative bacteria: structural and mechanistic insights. Nat. Rev. Microbiol. 13, 343–359. doi: 10.1038/nrmicro3456
Dang, C. V., Le, A., and Gao, P. (2009). MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin. Cancer Res. 15, 6479–6483. doi: 10.1158/1078-0432.CCR-09-0889
DeBerardinis, R. J. (2008). Is cancer a disease of abnormal cellular metabolism? New angles on an old idea. Genet. Med. 10, 767–777. doi: 10.1097/GIM.0b013e31818b0d9b
DeBerardinis, R. J., Lum, J. J., Hatzivassiliou, G., and Thompson, C. B. (2008). The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 7, 11–20. doi: 10.1016/j.cmet.2007.10.002
DeBerardinis, R. J., Lum, J. J., and Thompson, C. B. (2006). Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J. Biol. Chem. 281, 37372–37380. doi: 10.1074/jbc.M608372200
De Las Heras, A., Cain, R. J., Bielecka, M. K., and Vazquez-Boland, J. A. (2011). Regulation of Listeria virulence: PrfA master and commander. Curr. Opin. Microbiol. 14, 118–127. doi: 10.1016/j.mib.2011.01.005
Delgoffe, G. M., and Powell, J. D. (2015). Sugar, fat, and protein: new insights into what T cells crave. Curr. Opin. Immunol. 33, 49–54. doi: 10.1016/j.coi.2015.01.015
Dery, M. A., Michaud, M. D., and Richard, D. E. (2005). Hypoxia-inducible factor 1: regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 37, 535–540. doi: 10.1016/j.biocel.2004.08.012
Diacovich, L., and Gorvel, J. P. (2010). Bacterial manipulation of innate immunity to promote infection. Nat. Rev. Microbiol. 8, 117–128. doi: 10.1038/nrmicro2295
Dibble, C. C., and Manning, B. D. (2013). Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 15, 555–564. doi: 10.1038/ncb2763
Djelloul, S., Forgue-Lafitte, M. E., Hermelin, B., Mareel, M., Bruyneel, E., Baldi, A., et al. (1997). Enterocyte differentiation is compatible with SV40 large T expression and loss of p53 function in human colonic Caco-2 cells. Status of the pRb1 and pRb2 tumor suppressor gene products. FEBS Lett 406, 234–242. doi: 10.1016/S0014-5793(97)00208-1
Dominguez, P. M., and Ardavin, C. (2010). Differentiation and function of mouse monocyte-derived dendritic cells in steady state and inflammation. Immunol. Rev. 234, 90–104. doi: 10.1111/j.0105-2896.2009.00876.x
Dortet, L., Mostowy, S., and Cossart, P. (2012). Listeria and autophagy escape: involvement of InlK, an internalin-like protein. Autophagy 8, 132–134. doi: 10.4161/auto.8.1.18218
Drummond, R. A., and Brown, G. D. (2013). Signalling C-type lectins in antimicrobial immunity. PLoS Pathog. 9:e1003417. doi: 10.1371/journal.ppat.1003417
Dunlop, E. A., and Tee, A. R. (2009). Mammalian target of rapamycin complex 1: signalling inputs, substrates and feedback mechanisms. Cell. Signal. 21, 827–835. doi: 10.1016/j.cellsig.2009.01.012
Durai, V., and Murphy, K. M. (2016). Functions of murine dendritic cells. Immunity 45, 719–736. doi: 10.1016/j.immuni.2016.10.010
Duvel, K., Yecies, J. L., Menon, S., Raman, P., Lipovsky, A. I., Souza, A. L., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183. doi: 10.1016/j.molcel.2010.06.022
Eberle, D., Hegarty, B., Bossard, P., Ferre, P., and Foufelle, F. (2004). SREBP transcription factors: master regulators of lipid homeostasis. Biochimie 86, 839–848. doi: 10.1016/j.biochi.2004.09.018
Edwards, M. W., Aultman, J. A., Harber, G., Bhatt, J. M., Sztul, E., Xu, Q., et al. (2013). Role of mTOR downstream effector signaling molecules in Francisella tularensis internalization by murine macrophages. PLoS ONE 8:e83226. doi: 10.1371/journal.pone.0083226
Ehrt, S., Rhee, K., and Schnappinger, D. (2015). Mycobacterial genes essential for the pathogen's survival in the host. Immunol. Rev. 264, 319–326. doi: 10.1111/imr.12256
Eisele, N. A., Ruby, T., Jacobson, A., Manzanillo, P. S., Cox, J. S., Lam, L., et al. (2013). Salmonella require the fatty acid regulator PPARdelta for the establishment of a metabolic environment essential for long-term persistence. Cell Host Microbe 14, 171–182. doi: 10.1016/j.chom.2013.07.010
Eisenreich, W., Dandekar, T., Heesemann, J., and Goebel, W. (2010). Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nat. Rev. Microbiol. 8, 401–412. doi: 10.1038/nrmicro2351
Eisenreich, W., Heesemann, J., Rudel, T., and Goebel, W. (2013). Metabolic host responses to infection by intracellular bacterial pathogens. Front. Cell. Infect. Microbiol. 3:24. doi: 10.3389/fcimb.2013.00024
Eisenreich, W., Heesemann, J., Rudel, T., and Goebel, W. (2015). “Metabolic adaptations of intracellullar bacterial pathogens and their mammalian host cells during infection (“pathometabolism”),” in Metabolism and Bacterial Pathogenesis, eds T. Conway and P. Cohen (Washington, DC: ASM Press), 27–58.
Escamilla-Tilch, M., Filio-Rodriguez, G., Garcia-Rocha, R., Mancilla-Herrera, I., Mitchison, N. A., Ruiz-Pacheco, J. A., et al. (2013). The interplay between pathogen-associated and danger-associated molecular patterns: an inflammatory code in cancer? Immunol. Cell Biol. 91, 601–610. doi: 10.1038/icb.2013.58
Everts, B., Amiel, E., Van Der Windt, G. J., Freitas, T. C., Chott, R., Yarasheski, K. E., et al. (2012). Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 120, 1422–1431. doi: 10.1182/blood-2012-03-419747
Eylert, E., Schär, J., Mertins, S., Stoll, R., Bacher, A., Goebel, W., et al. (2008). Carbon metabolism of Listeria monocytogenes growing inside macrophages. Mol. Microbiol. 69, 1008–1017. doi: 10.1111/j.1365-2958.2008.06337.x
Fabrik, I., Hartlova, A., Rehulka, P., and Stulik, J. (2013). Serving the new masters-dendritic cells as hosts for stealth intracellular bacteria. Cell. Microbiol. 15, 1473–1483. doi: 10.1111/cmi.12160
Fink, S. L., and Cookson, B. T. (2007). Pyroptosis and host cell death responses during Salmonella infection. Cell. Microbiol. 9, 2562–2570. doi: 10.1111/j.1462-5822.2007.01036.x
Franchi, L., Park, J. H., Shaw, M. H., Marina-Garcia, N., Chen, G., Kim, Y. G., et al. (2008). Intracellular NOD-like receptors in innate immunity, infection and disease. Cell. Microbiol. 10, 1–8.
Francione, L., Smith, P. K., Accari, S. L., Taylor, P. E., Bokko, P. B., Bozzaro, S., et al. (2009). Legionella pneumophila multiplication is enhanced by chronic AMPK signalling in mitochondrially diseased Dictyostelium cells. Dis. Model. Mech. 2, 479–489. doi: 10.1242/dmm.003319
Frauwirth, K. A., and Thompson, C. B. (2004). Regulation of T lymphocyte metabolism. J. Immunol. 172, 4661–4665. doi: 10.4049/jimmunol.172.8.4661
Fredlund, J., and Enninga, J. (2014). Cytoplasmic access by intracellular bacterial pathogens. Trends Microbiol. 22, 128–137. doi: 10.1016/j.tim.2014.01.003
Freeman, S. A., and Grinstein, S. (2014). Phagocytosis: receptors, signal integration, and the cytoskeleton. Immunol. Rev. 262, 193–215. doi: 10.1111/imr.12212
Fritz, V., and Fajas, L. (2010). Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene 29, 4369–4377. doi: 10.1038/onc.2010.182
Fuchs, T. M., Eisenreich, W., Heesemann, J., and Goebel, W. (2012). Metabolic adaptation of human pathogenic and related non-pathogenic bacteria to extra- and intracellular habitats. FEMS Microbiol. Rev. 36, 435–462. doi: 10.1111/j.1574-6976.2011.00301.x
Gabig, T. G., Bearman, S. I., and Babior, B. M. (1979). Effects of oxygen tension and pH on the respiratory burst of human neutrophils. Blood 53, 1133–1139.
Galietti, F., Bollo, E., Cappia, S., Dondo, A., Pregel, P., Nicali, R., et al. (2001). p53 expression in cultured blood human monocytes infected with mycobacterial strains. Panminerva Med. 43, 249–255.
Galimi, F., Cottone, E., Vigna, E., Arena, N., Boccaccio, C., Giordano, S., et al. (2001). Hepatocyte growth factor is a regulator of monocyte-macrophage function. J. Immunol. 166, 1241–1247. doi: 10.4049/jimmunol.166.2.1241
Gallo, R. L., and Hooper, L. V. (2012). Epithelial antimicrobial defence of the skin and intestine. Nat. Rev. Immunol. 12, 503–516. doi: 10.1038/nri3228
Ganapathy, U., Marrero, J., Calhoun, S., Eoh, H., De Carvalho, L. P., Rhee, K., et al. (2015). Two enzymes with redundant fructose bisphosphatase activity sustain gluconeogenesis and virulence in Mycobacterium tuberculosis. Nat. Commun. 6, 7912. doi: 10.1038/ncomms8912
Ganeshan, K., and Chawla, A. (2014). Metabolic regulation of immune responses. Annu. Rev. Immunol. 32, 609–634. doi: 10.1146/annurev-immunol-032713-120236
Gao, X., Wang, H., Yang, J. J., Chen, J., Jie, J., Li, L., et al. (2013). Reciprocal regulation of protein kinase and pyruvate kinase activities of pyruvate kinase M2 by growth signals. J. Biol. Chem. 288, 15971–15979. doi: 10.1074/jbc.M112.448753
Gao, X., Wang, H., Yang, J. J., Liu, X., and Liu, Z. R. (2012). Pyruvate kinase M2 regulates gene transcription by acting as a protein kinase. Mol. Cell 45, 598–609. doi: 10.1016/j.molcel.2012.01.001
Garedew, A., Henderson, S. O., and Moncada, S. (2010). Activated macrophages utilize glycolytic ATP to maintain mitochondrial membrane potential and prevent apoptotic cell death. Cell Death Differ. 17, 1540–1550. doi: 10.1038/cdd.2010.27
Geddes, K., Cruz, F., and Heffron, F. (2007). Analysis of cells targeted by Salmonella type III secretion in vivo. PLoS Pathog. 3:e196. doi: 10.1371/journal.ppat.0030196
Gelman, A. E., Larosa, D. F., Zhang, J., Walsh, P. T., Choi, Y., Sunyer, J. O., et al. (2006). The adaptor molecule MyD88 activates PI-3 kinase signaling in CD4+ T cells and enables CpG oligodeoxynucleotide-mediated costimulation. Immunity 25, 783–793. doi: 10.1016/j.immuni.2006.08.023
Gessain, G., Tsai, Y. H., Travier, L., Bonazzi, M., Grayo, S., Cossart, P., et al. (2015). PI3-kinase activation is critical for host barrier permissiveness to Listeria monocytogenes. J. Exp. Med. 212, 165–183. doi: 10.1084/jem.20141406
Ghesquiere, B., Wong, B. W., Kuchnio, A., and Carmeliet, P. (2014). Metabolism of stromal and immune cells in health and disease. Nature 511, 167–176. doi: 10.1038/nature13312
Gillmaier, N., Götz, A., Schulz, A., Eisenreich, W., and Goebel, W. (2012). Metabolic responses of primary and transformed cells to intracellular Listeria monocytogenes. PLoS ONE 7:e52378. doi: 10.1371/journal.pone.0052378
Goenka, R., Guirnalda, P. D., Black, S. J., and Baldwin, C. L. (2012). B Lymphocytes provide an infection niche for intracellular bacterium Brucella abortus. J. Infect. Dis. 206, 91–98. doi: 10.1093/infdis/jis310
Gomes, L. C., and Dikic, I. (2014). Autophagy in antimicrobial immunity. Mol. Cell 54, 224–233. doi: 10.1016/j.molcel.2014.03.009
González, E., Rother, M., Kerr, M. C., Al-Zeer, M. A., Abu-Lubad, M., Kessler, M., et al. (2014). Chlamydia infection depends on a functional MDM2-p53 axis. Nat. Commun. 5:5201. doi: 10.1038/ncomms6201
Gordon, S., and Martinez, F. O. (2010). Alternative activation of macrophages: mechanism and functions. Immunity 32, 593–604. doi: 10.1016/j.immuni.2010.05.007
Götz, A., Eylert, E., Eisenreich, W., and Goebel, W. (2010). Carbon metabolism of enterobacterial human pathogens growing in epithelial colorectal adenocarcinoma (Caco-2) cells. PLoS ONE 5:e10586. doi: 10.1371/journal.pone.0010586
Gross, M., Salame, T. M., and Jung, S. (2015). Guardians of the gut-murine intestinal macrophages and dendritic cells. Front. Immunol. 6:254. doi: 10.3389/fimmu.2015.00254
Grubmüller, S., Schauer, K., Goebel, W., Fuchs, T. M., and Eisenreich, W. (2014). Analysis of carbon substrates used by Listeria monocytogenes during growth in J774A.1 macrophages suggests a bipartite intracellular metabolism. Front. Cell. Infect. Microbiol. 4:156. doi: 10.3389/fcimb.2014.00156
Guertin, D. A., and Sabatini, D. M. (2007). Defining the role of mTOR in cancer. Cancer Cell 12, 9–22. doi: 10.1016/j.ccr.2007.05.008
Guilliams, M., Ginhoux, F., Jakubzick, C., Naik, S. H., Onai, N., Schraml, B. U., et al. (2014). Dendritic cells, monocytes and macrophages: a unified nomenclature based on ontogeny. Nat. Rev. Immunol. 14, 571–578. doi: 10.1038/nri3712
Gysin, S., Salt, M., Young, A., and Mccormick, F. (2011). Therapeutic strategies for targeting ras proteins. Genes Cancer 2, 359–372. doi: 10.1177/1947601911412376
Hampel, A., Huber, C., Geffers, R., Spona-Friedl, M., Eisenreich, W., and Bange, F. C. (2015). Mycobacterium tuberculosis Is a natural ornithine aminotransferase (rocD) mutant and depends on Rv2323c for growth on arginine. PLoS ONE 10:e0136914. doi: 10.1371/journal.pone.0136914
Han, M., Liang, L., Liu, L. R., Yue, J., Zhao, Y. L., and Xiao, H. P. (2014). Liver X receptor gene polymorphisms in Tuberculosis: effect on susceptibility. PLoS ONE 9:e95954. doi: 10.1371/journal.pone.0095954
Hardie, D. G. (2011). AMP-activated protein kinase: a cellular energy sensor with a key role in metabolic disorders and in cancer. Biochem. Soc. Trans. 39, 1–13. doi: 10.1042/BST0390001
Harris, I., Mccracken, S., and Mak, T. W. (2012). PKM2: a gatekeeper between growth and survival. Cell Res. 22, 447–449. doi: 10.1038/cr.2011.203
Hartmann, H., Eltzschig, H. K., Wurz, H., Hantke, K., Rakin, A., Yazdi, A. S., et al. (2008). Hypoxia-independent activation of HIF-1 by enterobacteriaceae and their siderophores. Gastroenterology 134, 756–767. doi: 10.1053/j.gastro.2007.12.008
Haschemi, A., Kosma, P., Gille, L., Evans, C. R., Burant, C. F., Starkl, P., et al. (2012). The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 15, 813–826. doi: 10.1016/j.cmet.2012.04.023
Hedrick, S. M. (2004). The acquired immune system: a vantage from beneath. Immunity 21, 607–615. doi: 10.1016/j.immuni.2004.08.020
Hemmings, B. A., and Restuccia, D. F. (2012). PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 4:a011189. doi: 10.1101/cshperspect.a011189
Houben, E. N., Korotkov, K. V., and Bitter, W. (2014). Take five-Type VII secretion systems of Mycobacteria. Biochim. Biophys. Acta 1843, 1707–1716. doi: 10.1016/j.bbamcr.2013.11.003
Huang, G., Redelman-Sidi, G., Rosen, N., Glickman, M. S., and Jiang, X. (2012). Inhibition of mycobacterial infection by the tumor suppressor PTEN. J. Biol. Chem. 287, 23196–23202. doi: 10.1074/jbc.M112.351940
Huang, Q., Liu, D., Majewski, P., Schulte, L. C., Korn, J. M., Young, R. A., et al. (2001). The plasticity of dendritic cell responses to pathogens and their components. Science 294, 870–875. doi: 10.1126/science.294.5543.870
Iizuka, K., Wu, W., Horikawa, Y., Saito, M., and Takeda, J. (2013). Feedback looping between ChREBP and PPARα in the regulation of lipid metabolism in brown adipose tissues. Endocr. J. 60, 1145–1153. doi: 10.1507/endocrj.EJ13-0079
Iliffe-Lee, E. R., and Mcclarty, G. (2000). Regulation of carbon metabolism in Chlamydia trachomatis. Mol. Microbiol. 38, 20–30. doi: 10.1046/j.1365-2958.2000.02102.x
Inoki, K., Kim, J., and Guan, K. L. (2012). AMPK and mTOR in cellular energy homeostasis and drug targets. Annu. Rev. Pharmacol. Toxicol. 52, 381–400. doi: 10.1146/annurev-pharmtox-010611-134537
Ireton, K., Payrastre, B., Chap, H., Ogawa, W., Sakaue, H., Kasuga, M., et al. (1996). A role for phosphoinositide 3-kinase in bacterial invasion. Science 274, 780–782. doi: 10.1126/science.274.5288.780
Ishii, K. J., Koyama, S., Nakagawa, A., Coban, C., and Akira, S. (2008). Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe 3, 352–363. doi: 10.1016/j.chom.2008.05.003
Iurlaro, R., Leon-Annicchiarico, C. L., and Munoz-Pinedo, C. (2014). Regulation of cancer metabolism by oncogenes and tumor suppressors. Meth. Enzymol. 542, 59–80. doi: 10.1016/B978-0-12-416618-9.00003-0
Iyer, N. V., Kotch, L. E., Agani, F., Leung, S. W., Laughner, E., Wenger, R. H., et al. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 12, 149–162. doi: 10.1101/gad.12.2.149
Jantsch, J., Chakravortty, D., Turza, N., Prechtel, A. T., Buchholz, B., Gerlach, R. G., et al. (2008). Hypoxia and hypoxia-inducible factor-1α modulate lipopolysaccharide-induced dendritic cell activation and function. J. Immunol. 180, 4697–4705. doi: 10.4049/jimmunol.180.7.4697
Jewell, J. L., and Guan, K. L. (2013). Nutrient signaling to mTOR and cell growth. Trends Biochem. Sci. 38, 233–242. doi: 10.1016/j.tibs.2013.01.004
Jiang, P., Du, W., Wang, X., Mancuso, A., Gao, X., Wu, M., et al. (2011). p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 13, 310–316. doi: 10.1038/ncb2172
Jiang, P., Du, W., and Wu, M. (2014). Regulation of the pentose phosphate pathway in cancer. Protein Cell 5, 592–602. doi: 10.1007/s13238-014-0082-8
Johnson, R. F., and Perkins, N. D. (2012). Nuclear factor-kappaB, p53, and mitochondria: regulation of cellular metabolism and the Warburg effect. Trends Biochem. Sci. 37, 317–324. doi: 10.1016/j.tibs.2012.04.002
Jones, R. G., Plas, D. R., Kubek, S., Buzzai, M., Mu, J., Xu, Y., et al. (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 18, 283–293. doi: 10.1016/j.molcel.2005.03.027
Jones, R. G., and Thompson, C. B. (2009). Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev. 23, 537–548. doi: 10.1101/gad.1756509
Joseph, B., Przybilla, K., Stühler, C., Schauer, K., Slaghuis, J., Fuchs, T. M., et al. (2006). Identification of Listeria monocytogenes genes contributing to intracellular replication by expression profiling and mutant screening. J. Bacteriol. 188, 556–568. doi: 10.1128/JB.188.2.556-568.2006
Kaplan, O., Firon, M., Vivi, A., Navon, G., and Tsarfaty, I. (2000). HGF/SF activates glycolysis and oxidative phosphorylation in DA3 murine mammary cancer cells. Neoplasia 2, 365–377. doi: 10.1038/sj.neo.7900103
Kawai, T., and Akira, S. (2011). Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650. doi: 10.1016/j.immuni.2011.05.006
Kennedy, A. D., and Deleo, F. R. (2009). Neutrophil apoptosis and the resolution of infection. Immunol. Res. 43, 25–61. doi: 10.1007/s12026-008-8049-6
Kentner, D., Martano, G., Callon, M., Chiquet, P., Brodmann, M., Burton, O., et al. (2014). Shigella reroutes host cell central metabolism to obtain high-flux nutrient supply for vigorous intracellular growth. Proc. Natl. Acad. Sci. U.S.A. 111, 9929–9934. doi: 10.1073/pnas.1406694111
Kirchner, T., Möller, S., Klinger, M., Solbach, W., Laskay, T., and Behnen, M. (2012). The impact of various reactive oxygen species on the formation of neutrophil extracellular traps. Mediators Inflamm. 2012:849136. doi: 10.1155/2012/849136
Knodler, L. A. (2015). Salmonella enterica: living a double life in epithelial cells. Curr. Opin. Microbiol. 23, 23–31. doi: 10.1016/j.mib.2014.10.010
Kominsky, D. J., Campbell, E. L., and Colgan, S. P. (2010). Metabolic shifts in immunity and inflammation. J. Immunol. 184, 4062–4068. doi: 10.4049/jimmunol.0903002
Konradt, C., Frigimelica, E., Nothelfer, K., Puhar, A., Salgado-Pabon, W., Di Bartolo, V., et al. (2011). The Shigella flexneri type three secretion system effector IpgD inhibits T cell migration by manipulating host phosphoinositide metabolism. Cell Host Microbe 9, 263–272. doi: 10.1016/j.chom.2011.03.010
Korn, T., Bettelli, E., Oukka, M., and Kuchroo, V. K. (2009). IL-17 and Th17 cells. Annu. Rev. Immunol. 27, 485–517. doi: 10.1146/annurev.immunol.021908.132710
Krawczyk, C. M., Holowka, T., Sun, J., Blagih, J., Amiel, E., Deberardinis, R. J., et al. (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115, 4742–4749. doi: 10.1182/blood-2009-10-249540
Krocova, Z., Hartlova, A., Souckova, D., Zivna, L., Kroca, M., Rudolf, E., et al. (2008). Interaction of B cells with intracellular pathogen Francisella tularensis. Microb. Pathog. 45, 79–85. doi: 10.1016/j.micpath.2008.01.010
Krüger, A., Oldenburg, M., Chebrolu, C., Beisser, D., Kolter, J., Sigmund, A. M., et al. (2015). Human TLR8 senses UR/URR motifs in bacterial and mitochondrial RNA. EMBO Rep. 16, 1656–1663. doi: 10.15252/embr.201540861
Kullas, A. L., Mcclelland, M., Yang, H. J., Tam, J. W., Torres, A., Porwollik, S., et al. (2012). L-asparaginase II produced by Salmonella typhimurium inhibits T cell responses and mediates virulence. Cell Host Microbe 12, 791–798. doi: 10.1016/j.chom.2012.10.018
Kumar, Y., and Valdivia, R. H. (2009). Leading a sheltered life: intracellular pathogens and maintenance of vacuolar compartments. Cell Host Microbe 5, 593–601. doi: 10.1016/j.chom.2009.05.014
Kuschel, A., Simon, P., and Tug, S. (2012). Functional regulation of HIF-1α under normoxia—is there more than post-translational regulation? J. Cell. Physiol. 227, 514–524. doi: 10.1002/jcp.22798
Lembo-Fazio, L., Nigro, G., Noel, G., Rossi, G., Chiara, F., Tsilingiri, K., et al. (2011). Gadd45α activity is the principal effector of Shigella mitochondria-dependent epithelial cell death in vitro and ex vivo. Cell Death Dis. 2:e122. doi: 10.1038/cddis.2011.4
Levine, A. J., and Oren, M. (2009). The first 30 years of p53: growing ever more complex. Nat. Rev. Cancer 9, 749–758. doi: 10.1038/nrc2723
Lewis, R. S., Kolesnik, T. B., Kuang, Z., D'cruz, A. A., Blewitt, M. E., Masters, S. L., et al. (2011). TLR regulation of SPSB1 controls inducible nitric oxide synthase induction. J. Immunol. 187, 3798–3805. doi: 10.4049/jimmunol.1002993
Li, N., Xiang, G. S., Dokainish, H., Ireton, K., and Elferink, L. A. (2005). The Listeria protein internalin B mimics hepatocyte growth factor-induced receptor trafficking. Traffic 6, 459–473. doi: 10.1111/j.1600-0854.2005.00290.x
Lisy, K., and Peet, D. J. (2008). Turn me on: regulating HIF transcriptional activity. Cell Death Differ. 15, 642–649. doi: 10.1038/sj.cdd.4402315
Liu, P., Gan, W., Inuzuka, H., Lazorchak, A. S., Gao, D., Arojo, O., et al. (2013). Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 15, 1340–1350. doi: 10.1038/ncb2860
Liu, Q., Han, W., Sun, C., Zhou, L., Ma, L., Lei, L., et al. (2012). Deep sequencing-based expression transcriptional profiling changes during Brucella infection. Microb. Pathog. 52, 267–277. doi: 10.1016/j.micpath.2012.02.001
Lopez, C. A., Winter, S. E., Rivera-Chavez, F., Xavier, M. N., Poon, V., Nuccio, S. P., et al. (2012). Phage-mediated acquisition of a type III secreted effector protein boosts growth of Salmonella by nitrate respiration. MBio 3:e00143-12. doi: 10.1128/mBio.00143-12
Lovewell, R. R., Sassetti, C. M., and Vanderven, B. C. (2016). Chewing the fat: lipid metabolism and homeostasis during M. tuberculosis infection. Curr. Opin. Microbiol. 29, 30–36. doi: 10.1016/j.mib.2015.10.002
Mantovani, A., Sica, A., and Locati, M. (2005). Macrophage polarization comes of age. Immunity 23, 344–346. doi: 10.1016/j.immuni.2005.10.001
Marrakchi, H., Laneelle, M. A., and Daffe, M. (2014). Mycolic acids: structures, biosynthesis, and beyond. Chem. Biol. 21, 67–85. doi: 10.1016/j.chembiol.2013.11.011
Marrero, J., Trujillo, C., Rhee, K. Y., and Ehrt, S. (2013). Glucose phosphorylation is required for Mycobacterium tuberculosis persistence in mice. PLoS Pathog. 9:e1003116. doi: 10.1371/journal.ppat.1003116
Martinez, F. O., and Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 6:13. doi: 10.12703/P6-13
Mcgann, P., Ivanek, R., Wiedmann, M., and Boor, K. J. (2007). Temperature-dependent expression of Listeria monocytogenes internalin and internalin-like genes suggests functional diversity of these proteins among the listeriae. Appl. Environ. Microbiol. 73, 2806–2814. doi: 10.1128/AEM.02923-06
Medzhitov, R. (2007a). Recognition of microorganisms and activation of the immune response. Nature 449, 819–826. doi: 10.1038/nature06246
Medzhitov, R. (2007b). TLR-mediated innate immune recognition. Semin. Immunol. 19, 1–2. doi: 10.1016/j.smim.2007.02.001
Mehlitz, A., Eylert, E., Huber, C., Lindner, B., Vollmuth, N., Karunakaran, K., et al. (2017). Metabolic adaptation of Chlamydia trachomatis to mammalian host cells. Mol. Microbiol. 103, 1004–1019. doi: 10.1111/mmi.13603
Menon, A., Shroyer, M. L., Wampler, J. L., Chawan, C. B., and Bhunia, A. K. (2003). In vitro study of Listeria monocytogenes infection to murine primary and human transformed B cells. Comp. Immunol. Microbiol. Infect. Dis. 26, 157–174. doi: 10.1016/S0147-9571(02)00039-5
Mildner, A., and Jung, S. (2014). Development and function of dendritic cell subsets. Immunity 40, 642–656. doi: 10.1016/j.immuni.2014.04.016
Mills, E. L., Kelly, B., and O'Neill, L. A. J. (2017). Mitochondria are the powerhouses of immunity. Nat. Immunol. 18, 488–498. doi: 10.1038/ni.3704
Moransard, M., Sawitzky, M., Fontana, A., and Suter, T. (2010). Expression of the HGF receptor c-met by macrophages in experimental autoimmune encephalomyelitis. Glia 58, 559–571. doi: 10.1002/glia.20945
Morrison, D. K. (2012). MAP kinase pathways. Cold Spring Harb Perspect. Biol. 4:a011254. doi: 10.1101/cshperspect.a011254
Mosser, D. M., and Edwards, J. P. (2008). Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 8, 958–969. doi: 10.1038/nri2448
Mostowy, S., Sancho-Shimizu, V., Hamon, M. A., Simeone, R., Brosch, R., Johansen, T., et al. (2011). p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J. Biol. Chem. 286, 26987–26995. doi: 10.1074/jbc.M111.223610
Murray, P. J., Allen, J. E., Biswas, S. K., Fisher, E. A., Gilroy, D. W., Goerdt, S., et al. (2014). Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 41, 14–20. doi: 10.1016/j.immuni.2014.06.008
Murray, P. J., and Wynn, T. A. (2011). Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 11, 723–737. doi: 10.1038/nri3073
Nairz, M., Ferring-Appel, D., Casarrubea, D., Sonnweber, T., Viatte, L., Schroll, A., et al. (2015). Iron regulatory proteins mediate host resistance to Salmonella infection. Cell Host Microbe 18, 254–261. doi: 10.1016/j.chom.2015.06.017
Nairz, M., Haschka, D., Demetz, E., and Weiss, G. (2014). Iron at the interface of immunity and infection. Front. Pharmacol. 5:152. doi: 10.3389/fphar.2014.00152
Nauseef, W. M. (2007). How human neutrophils kill and degrade microbes: an integrated view. Immunol. Rev. 219, 88–102. doi: 10.1111/j.1600-065X.2007.00550.x
Ngo, D. C., Ververis, K., Tortorella, S. M., and Karagiannis, T. C. (2015). Introduction to the molecular basis of cancer metabolism and the Warburg effect. Mol. Biol. Rep. 42, 819–823. doi: 10.1007/s11033-015-3857-y
Nicolay, B. N., and Dyson, N. J. (2013). The multiple connections between pRB and cell metabolism. Curr. Opin. Cell Biol. 25, 735–740. doi: 10.1016/j.ceb.2013.07.012
Niemann, H. H. (2013). Structural basis of MET receptor dimerization by the bacterial invasion protein InlB and the HGF/SF splice variant NK1. Biochim. Biophys. Acta 1834, 2195–2204. doi: 10.1016/j.bbapap.2012.10.012
Nothelfer, K., Arena, E. T., Pinaud, L., Neunlist, M., Mozeleski, B., Belotserkovsky, I., et al. (2014). B lymphocytes undergo TLR2-dependent apoptosis upon Shigella infection. J. Exp. Med. 211, 1215–1229. doi: 10.1084/jem.20130914
Nuccio, S. P., and Bäumler, A. J. (2014). Comparative analysis of Salmonella genomes identifies a metabolic network for escalating growth in the inflamed gut. MBio 5, e00929–e00914. doi: 10.1128/mBio.00929-14
Odegaard, J. I., and Chawla, A. (2011). Alternative macrophage activation and metabolism. Annu. Rev. Pathol. 6, 275–297. doi: 10.1146/annurev-pathol-011110-130138
Odegaard, J. I., and Chawla, A. (2013). The immune system as a sensor of the metabolic state. Immunity 38, 644–654. doi: 10.1016/j.immuni.2013.04.001
Ogawa, M., Yoshimori, T., Suzuki, T., Sagara, H., Mizushima, N., and Sasakawa, C. (2005). Escape of intracellular Shigella from autophagy. Science 307, 727–731. doi: 10.1126/science.1106036
Okumura, C. Y., and Nizet, V. (2014). Subterfuge and sabotage: evasion of host innate defenses by invasive gram-positive bacterial pathogens. Annu. Rev. Microbiol. 68, 439–458. doi: 10.1146/annurev-micro-092412-155711
Oldenburg, M., Krüger, A., Ferstl, R., Kaufmann, A., Nees, G., Sigmund, A., et al. (2012). TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 337, 1111–1115. doi: 10.1126/science.1220363
Olovnikov, I. A., Kravchenko, J. E., and Chumakov, P. M. (2009). Homeostatic functions of the p53 tumor suppressor: regulation of energy metabolism and antioxidant defense. Semin. Cancer Biol. 19, 32–41. doi: 10.1016/j.semcancer.2008.11.005
Omsland, A., Cockrell, D. C., Howe, D., Fischer, E. R., Virtaneva, K., Sturdevant, D. E., et al. (2009). Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 106, 4430–4434. doi: 10.1073/pnas.0812074106
O'Neill, L. A., and Pearce, E. J. (2016). Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 213, 15–23. doi: 10.1084/jem.20151570
Ozinsky, A., Underhill, D. M., Fontenot, J. D., Hajjar, A. M., Smith, K. D., Wilson, C. B., et al. (2000). The repertoire for pattern recognition of pathogens by the innate immune system is defined by cooperation between toll-like receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 13766–13771. doi: 10.1073/pnas.250476497
Palsson-Mcdermott, E. M., Curtis, A. M., Goel, G., Lauterbach, M. A., Sheedy, F. J., Gleeson, L. E., et al. (2015). Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 21, 65–80. doi: 10.1016/j.cmet.2014.12.005
Palsson-Mcdermott, E. M., and O'Neill, L. A. (2013). The Warburg effect then and now: from cancer to inflammatory diseases. Bioessays 35, 965–973. doi: 10.1002/bies.201300084
Patra, K. C., and Hay, N. (2014). The pentose phosphate pathway and cancer. Trends Biochem. Sci. 39, 347–354. doi: 10.1016/j.tibs.2014.06.005
Pearce, E. J., and Everts, B. (2015). Dendritic cell metabolism. Nat. Rev. Immunol. 15, 18–29. doi: 10.1038/nri3771
Pearce, E. L., and Pearce, E. J. (2013). Metabolic pathways in immune cell activation and quiescence. Immunity 38, 633–643. doi: 10.1016/j.immuni.2013.04.005
Pendaries, C., Tronchere, H., Arbibe, L., Mounier, J., Gozani, O., Cantley, L., et al. (2006). PtdIns5P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 25, 1024–1034. doi: 10.1038/sj.emboj.7601001
Peyssonnaux, C., Boutin, A. T., Zinkernagel, A. S., Datta, V., Nizet, V., and Johnson, R. S. (2008). Critical role of HIF-1α in keratinocyte defense against bacterial infection. J. Invest. Dermatol. 128, 1964–1968. doi: 10.1038/jid.2008.27
Phalipon, A., and Sansonetti, P. J. (2007). Shigella's ways of manipulating the host intestinal innate and adaptive immune system: a tool box for survival? Immunol. Cell Biol. 85, 119–129. doi: 10.1038/sj.icb7100025
Pieters, J. (2008). Mycobacterium tuberculosis and the macrophage: maintaining a balance. Cell Host Microbe 3, 399–407. doi: 10.1016/j.chom.2008.05.006
Pizarro-Cerdá, J., and Cossart, P. (2006). Bacterial adhesion and entry into host cells. Cell 124, 715–727. doi: 10.1016/j.cell.2006.02.012
Pollizzi, K. N., and Powell, J. D. (2014). Integrating canonical and metabolic signalling programmes in the regulation of T cell responses. Nat. Rev. Immunol. 14, 435–446. doi: 10.1038/nri3701
Poncet, S., Milohanic, E., Maze, A., Nait Abdallah, J., Ake, F., Larribe, M., et al. (2009). Correlations between carbon metabolism and virulence in bacteria. Contrib. Microbiol. 16, 88–102. doi: 10.1159/000219374
Postic, C., Dentin, R., Denechaud, P. D., and Girard, J. (2007). ChREBP, a transcriptional regulator of glucose and lipid metabolism. Annu. Rev. Nutr. 27, 179–192. doi: 10.1146/annurev.nutr.27.061406.093618
Pratilas, C. A., and Solit, D. B. (2010). Targeting the mitogen-activated protein kinase pathway: physiological feedback and drug response. Clin. Cancer Res. 16, 3329–3334. doi: 10.1158/1078-0432.CCR-09-3064
Price, C. T., Al-Quadan, T., Santic, M., Rosenshine, I., and Abu Kwaik, Y. (2011). Host proteasomal degradation generates amino acids essential for intracellular bacterial growth. Science 334, 1553–1557. doi: 10.1126/science.1212868
Price, J. V., and Vance, R. E. (2014). The macrophage paradox. Immunity 41, 685–693. doi: 10.1016/j.immuni.2014.10.015
Ramel, D., Lagarrigue, F., Dupuis-Coronas, S., Chicanne, G., Leslie, N., Gaits-Iacovoni, F., et al. (2009). PtdIns5P protects Akt from dephosphorylation through PP2A inhibition. Biochem. Biophys. Res. Commun. 387, 127–131. doi: 10.1016/j.bbrc.2009.06.139
Randow, F., and Youle, R. J. (2014). Self and nonself: how autophagy targets mitochondria and bacteria. Cell Host Microbe 15, 403–411. doi: 10.1016/j.chom.2014.03.012
Rathmell, J. C. (2012). Metabolism and autophagy in the immune system: immunometabolism comes of age. Immunol. Rev. 249, 5–13. doi: 10.1111/j.1600-065X.2012.01158.x
Rathmell, J. C., Vander Heiden, M. G., Harris, M. H., Frauwirth, K. A., and Thompson, C. B. (2000). In the absence of extrinsic signals, nutrient utilization by lymphocytes is insufficient to maintain either cell size or viability. Mol. Cell 6, 683–692. doi: 10.1016/S1097-2765(00)00066-6
Ray, K., Marteyn, B., Sansonetti, P. J., and Tang, C. M. (2009). Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat. Rev. Microbiol. 7, 333–340. doi: 10.1038/nrmicro2112
Reiterer, V., Grossniklaus, L., Tschon, T., Kasper, C. A., Sorg, I., and Arrieumerlou, C. (2011). Shigella flexneri type III secreted effector OspF reveals new crosstalks of proinflammatory signaling pathways during bacterial infection. Cell. Signal. 23, 1188–1196. doi: 10.1016/j.cellsig.2011.03.006
Reniere, M. L., Whiteley, A. T., Hamilton, K. L., John, S. M., Lauer, P., Brennan, R. G., et al. (2015). Glutathione activates virulence gene expression of an intracellular pathogen. Nature 517, 170–173. doi: 10.1038/nature14029
Rikihisa, Y. (2010). Anaplasma phagocytophilum and Ehrlichia chaffeensis: subversive manipulators of host cells. Nat. Rev. Microbiol. 8, 328–339. doi: 10.1038/nrmicro2318
Roppenser, B., Kwon, H., Canadien, V., Xu, R., Devreotes, P. N., Grinstein, S., et al. (2013). Multiple host kinases contribute to Akt activation during Salmonella infection. PLoS ONE 8:e71015. doi: 10.1371/journal.pone.0071015
Rücker, N., Billig, S., Bucker, R., Jahn, D., Wittmann, C., and Bange, F. C. (2015). Acetate dissimilation and assimilation in Mycobacterium tuberculosis depend on carbon availability. J. Bacteriol. 197, 3182–3190. doi: 10.1128/JB.00259-15
Rupp, J., Gieffers, J., Klinger, M., Van Zandbergen, G., Wrase, R., Maass, M., et al. (2007). Chlamydia pneumoniae directly interferes with HIF-1α stabilization in human host cells. Cell. Microbiol. 9, 2181–2191. doi: 10.1111/j.1462-5822.2007.00948.x
Ryndak, M. B., Singh, K. K., Peng, Z., and Laal, S. (2015). Transcriptional profile of Mycobacterium tuberculosis replicating in type II alveolar epithelial cells. PLoS ONE 10:e0123745. doi: 10.1371/journal.pone.0123745
Saadi, S., Wrenshall, L. E., and Platt, J. L. (2002). Regional manifestations and control of the immune system. FASEB J. 16, 849–856. doi: 10.1096/fj.01-0690hyp
Salminen, A., and Kaarniranta, K. (2010). Glycolysis links p53 function with NF-kappaB signaling: impact on cancer and aging process. J. Cell. Physiol. 224, 1–6. doi: 10.1002/jcp.22119
Sauer, J. D., Witte, C. E., Zemansky, J., Hanson, B., Lauer, P., and Portnoy, D. A. (2010). Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 7, 412–419. doi: 10.1016/j.chom.2010.04.004
Schmid, M., Wege, A. K., and Ritter, U. (2012). Characteristics of “Tip-DCs and MDSCs” and their potential role in Leishmaniasis. Front. Microbiol. 3:74. doi: 10.3389/fmicb.2012.00074
Schorey, J. S., Cheng, Y., Singh, P. P., and Smith, V. L. (2015). Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 16, 24–43. doi: 10.15252/embr.201439363
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140, 821–832. doi: 10.1016/j.cell.2010.01.040
Schunder, E., Gillmaier, N., Kutzner, E., Eisenreich, W., Herrmann, V., Lautner, M., et al. (2014). Amino acid uptake and metabolism of Legionella pneumophila hosted by Acanthamoeba castellanii. J. Biol. Chem. 289, 21040–21054. doi: 10.1074/jbc.M114.570085
Schürmann, M., and Sprenger, G. A. (2001). Fructose-6-phosphate aldolase is a novel class I aldolase from Escherichia coli and is related to a novel group of bacterial transaldolases. J. Biol. Chem. 276, 11055–11061. doi: 10.1074/jbc.M008061200
Schwöppe, C., Winkler, H. H., and Neuhaus, H. E. (2002). Properties of the glucose-6-phosphate transporter from Chlamydia pneumoniae (HPTcp) and the glucose-6-phosphate sensor from Escherichia coli (UhpC). J. Bacteriol. 184, 2108–2115. doi: 10.1128/JB.184.8.2108-2115.2002
Semenza, G. L. (2009). Regulation of cancer cell metabolism by hypoxia-inducible factor 1. Semin. Cancer Biol. 19, 12–16. doi: 10.1016/j.semcancer.2008.11.009
Shackelford, D. B., and Shaw, R. J. (2009). The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9, 563–575. doi: 10.1038/nrc2676
Shen, A., and Higgins, D. E. (2006). The MogR transcriptional repressor regulates nonhierarchal expression of flagellar motility genes and virulence in Listeria monocytogenes. PLoS Pathog. 2:e30. doi: 10.1371/journal.ppat.0020030
Shen, Y., Naujokas, M., Park, M., and Ireton, K. (2000). InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 103, 501–510. doi: 10.1016/S0092-8674(00)00141-0
Shin, J. H., Yang, J. Y., Jeon, B. Y., Yoon, Y. J., Cho, S. N., Kang, Y. H., et al. (2011). 1H NMR-based metabolomic profiling in mice infected with Mycobacterium tuberculosis. J. Proteome Res. 10, 2238–2247. doi: 10.1021/pr101054m
Siegl, C., Prusty, B. K., Karunakaran, K., Wischhusen, J., and Rudel, T. (2014). Tumor suppressor p53 alters host cell metabolism to limit Chlamydia trachomatis infection. Cell Rep. 9, 918–929. doi: 10.1016/j.celrep.2014.10.004
Simeone, R., Bottai, D., and Brosch, R. (2009). ESX/type VII secretion systems and their role in host-pathogen interaction. Curr. Opin. Microbiol. 12, 4–10. doi: 10.1016/j.mib.2008.11.003
Singh, V., Jamwal, S., Jain, R., Verma, P., Gokhale, R., and Rao, K. V. (2012). Mycobacterium tuberculosis-driven targeted recalibration of macrophage lipid homeostasis promotes the foamy phenotype. Cell Host Microbe 12, 669–681. doi: 10.1016/j.chom.2012.09.012
Smith, L. M., and May, R. C. (2013). Mechanisms of microbial escape from phagocyte killing. Biochem. Soc. Trans. 41, 475–490. doi: 10.1042/BST20130014
Soga, T. (2013). Cancer metabolism: key players in metabolic reprogramming. Cancer Sci. 104, 275–281. doi: 10.1111/cas.12085
Song, M. S., Salmena, L., and Pandolfi, P. P. (2012). The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296. doi: 10.1038/nrm3330
Steeb, B., Claudi, B., Burton, N. A., Tienz, P., Schmidt, A., Farhan, H., et al. (2013). Parallel exploitation of diverse host nutrients enhances Salmonella virulence. PLoS Pathog. 9:e1003301. doi: 10.1371/journal.ppat.1003301
Steele, S., Brunton, J., Ziehr, B., Taft-Benz, S., Moorman, N., and Kawula, T. (2013). Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PLoS Pathog. 9:e1003562. doi: 10.1371/journal.ppat.1003562
Steinman, R. M. (2012). Decisions about dendritic cells: past, present, and future. Annu. Rev. Immunol. 30, 1–22. doi: 10.1146/annurev-immunol-100311-102839
Stewart, D. A., Yang, Y., Makowski, L., and Troester, M. A. (2012). Basal-like breast cancer cells induce phenotypic and genomic changes in macrophages. Mol. Cancer Res. 10, 727–738. doi: 10.1158/1541-7786.MCR-11-0604
Stoll, R., and Goebel, W. (2010). The major PEP-phosphotransferase systems (PTSs) for glucose, mannose and cellobiose of Listeria monocytogenes, and their significance for extra-and intracellular growth. Microbiology 156, 1069–1083. doi: 10.1099/mic.0.034934-0
Stoll, R., Mertins, S., Joseph, B., Müller-Altrock, S., and Goebel, W. (2008). Modulation of PrfA activity in Listeria monocytogenes upon growth in different culture media. Microbiology 154, 3856–3876. doi: 10.1099/mic.0.2008/018283-0
Storey, K. B. (2015). Regulation of hypometabolism: insights into epigenetic controls. J. Exp. Biol. 218, 150–159. doi: 10.1242/jeb.106369
Storey, K. B., and Storey, J. M. (2004). Metabolic rate depression in animals: transcriptional and translational controls. Biol. Rev. Camb. Philos. Soc. 79, 207–233. doi: 10.1017/S1464793103006195
Subbarayal, P., Karunakaran, K., Winkler, A. C., Rother, M., Gonzalez, E., Meyer, T. F., et al. (2015). EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 11:e1004846. doi: 10.1371/journal.ppat.1004846
Suzuki, T., Franchi, L., Toma, C., Ashida, H., Ogawa, M., Yoshikawa, Y., et al. (2007). Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3:e111. doi: 10.1371/journal.ppat.0030111
Swart, A. L., and Hensel, M. (2012). Interactions of Salmonella enterica with dendritic cells. Virulence 3, 660–667. doi: 10.4161/viru.22761
Tachado, S. D., Li, X., Swan, K., Patel, N., and Koziel, H. (2008). Constitutive activation of phosphatidylinositol 3-kinase signaling pathway down-regulates TLR4-mediated tumor necrosis factor-α release in alveolar macrophages from asymptomatic HIV-positive persons in vitro. J. Biol. Chem. 283, 33191–33198. doi: 10.1074/jbc.M805067200
Tachibana, M., Hashino, M., Watanabe, K., Shimizu, T., and Watarai, M. (2015). Interferon γ-induced GTPase promotes invasion of Listeria monocytogenes into trophoblast giant cells. Sci. Rep. 5:8195. doi: 10.1038/srep08195
Tattoli, I., Sorbara, M. T., Vuckovic, D., Ling, A., Soares, F., Carneiro, L. A., et al. (2012). Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe 11, 563–575. doi: 10.1016/j.chom.2012.04.012
Taxman, D. J., Huang, M. T., and Ting, J. P. (2010). Inflammasome inhibition as a pathogenic stealth mechanism. Cell Host Microbe 8, 7–11. doi: 10.1016/j.chom.2010.06.005
Telang, S., Lane, A. N., Nelson, K. K., Arumugam, S., and Chesney, J. (2007). The oncoprotein H-RasV12 increases mitochondrial metabolism. Mol. Cancer 6:77. doi: 10.1186/1476-4598-6-77
Telang, S., Nelson, K. K., Siow, D. L., Yalcin, A., Thornburg, J. M., Imbert-Fernandez, Y., et al. (2012). Cytochrome c oxidase is activated by the oncoprotein Ras and is required for A549 lung adenocarcinoma growth. Mol. Cancer 11:60. doi: 10.1186/1476-4598-11-60
Thiennimitr, P., Winter, S. E., Winter, M. G., Xavier, M. N., Tolstikov, V., Huseby, D. L., et al. (2011). Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. U.S.A. 108, 17480–17485. doi: 10.1073/pnas.1107857108
Thompson, C. B. (2011). Rethinking the regulation of cellular metabolism. Cold Spring Harb. Symp. Quant. Biol. 76, 23–29. doi: 10.1101/sqb.2012.76.010496
Tong, X., Zhao, F., Mancuso, A., Gruber, J. J., and Thompson, C. B. (2009). The glucose-responsive transcription factor ChREBP contributes to glucose-dependent anabolic synthesis and cell proliferation. Proc. Natl. Acad. Sci. U.S.A. 106, 21660–21665. doi: 10.1073/pnas.0911316106
Tsalikis, J., Croitoru, D. O., Philpott, D. J., and Girardin, S. E. (2013). Nutrient sensing and metabolic stress pathways in innate immunity. Cell. Microbiol. 15, 1632–1641. doi: 10.1111/cmi.12165
Urban, C. F., Ermert, D., Schmid, M., Abu-Abed, U., Goosmann, C., Nacken, W., et al. (2009). Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 5:e1000639. doi: 10.1371/journal.ppat.1000639
Vander Heiden, M. G., Cantley, L. C., and Thompson, C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. doi: 10.1126/science.1160809
Van Raam, B. J., Sluiter, W., De Wit, E., Roos, D., Verhoeven, A. J., and Kuijpers, T. W. (2008). Mitochondrial membrane potential in human neutrophils is maintained by complex III activity in the absence of supercomplex organisation. PLoS ONE 3:e2013. doi: 10.1371/journal.pone.0002013
Vats, D., Mukundan, L., Odegaard, J. I., Zhang, L., Smith, K. L., Morel, C. R., et al. (2006). Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 4, 13–24. doi: 10.1016/j.cmet.2006.05.011
Vazquez-Torres, A., Vallance, B. A., Bergman, M. A., Finlay, B. B., Cookson, B. T., Jones-Carson, J., et al. (2004). Toll-like receptor 4 dependence of innate and adaptive immunity to Salmonella: importance of the Kupffer cell network. J. Immunol. 172, 6202–6208. doi: 10.4049/jimmunol.172.10.6202
Vergadi, E., Ieronymaki, E., Lyroni, K., Vaporidi, K., and Tsatsanis, C. (2017). Akt signaling pathway in macrophage activation and M1/M2 Polarization. J. Immunol. 198, 1006–1014. doi: 10.4049/jimmunol.1601515
Vousden, K. H., and Ryan, K. M. (2009). p53 and metabolism. Nat. Rev. Cancer 9, 691–700. doi: 10.1038/nrc2715
Wagle, A., Jivraj, S., Garlock, G. L., and Stapleton, S. R. (1998). Insulin regulation of glucose-6-phosphate dehydrogenase gene expression is rapamycin-sensitive and requires phosphatidylinositol 3-kinase. J. Biol. Chem. 273, 14968–14974. doi: 10.1074/jbc.273.24.14968
Waligora, E. A., Fisher, C. R., Hanovice, N. J., Rodou, A., Wyckoff, E. E., and Payne, S. M. (2014). Role of intracellular carbon metabolism pathways in Shigella flexneri virulence. Infect. Immun. 82, 2746–2755. doi: 10.1128/IAI.01575-13
Wang, S., Liu, P., Wei, J., Zhu, Z., Shi, Z., Shao, D., et al. (2016). Tumor suppressor p53 protects mice against Listeria monocytogenes infection. Sci. Rep. 6:33815. doi: 10.1038/srep33815
Warburg, O., Wind, F., and Negelein, E. (1927). The metabolism of tumors in the body. J. Gen. Physiol. 8, 519–530. doi: 10.1085/jgp.8.6.519
Ward, P. S., and Thompson, C. B. (2012). Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308. doi: 10.1016/j.ccr.2012.02.014
West, A. P., Brodsky, I. E., Rahner, C., Woo, D. K., Erdjument-Bromage, H., Tempst, P., et al. (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480. doi: 10.1038/nature09973
Whiteley, A. T., Pollock, A. J., and Portnoy, D. A. (2015). The PAMP c-di-AMP is essential for Listeria monocytogenes growth in rich but not minimal media due to a toxic increase in (p)ppGpp. [corrected]. Cell Host Microbe 17, 788–798. doi: 10.1016/j.chom.2015.05.006
Winter, S. E., Keestra, A. M., Tsolis, R. M., and Bäumler, A. J. (2010a). The blessings and curses of intestinal inflammation. Cell Host Microbe 8, 36–43. doi: 10.1016/j.chom.2010.06.003
Winter, S. E., Thiennimitr, P., Winter, M. G., Butler, B. P., Huseby, D. L., Crawford, R. W., et al. (2010b). Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429. doi: 10.1038/nature09415
Winter, S. E., Winter, M. G., Poon, V., Keestra, A. M., Sterzenbach, T., Faber, F., et al. (2014). Salmonella enterica Serovar Typhi conceals the invasion-associated type three secretion system from the innate immune system by gene regulation. PLoS Pathog. 10:e1004207. doi: 10.1371/journal.ppat.1004207
Wise, D. R., Ward, P. S., Shay, J. E., Cross, J. R., Gruber, J. J., Sachdeva, U. M., et al. (2011). Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. U.S.A. 108, 19611–19616. doi: 10.1073/pnas.1117773108
Wood, D. O., Wood, R. R., and Tucker, A. M. (2014). Genetic systems for studying obligate intracellular pathogens: an update. Curr. Opin. Microbiol. 17, 11–16. doi: 10.1016/j.mib.2013.10.006
Woodward, J. J., Iavarone, A. T., and Portnoy, D. A. (2010). c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705. doi: 10.1126/science.1189801
Wu, S., Ye, Z., Liu, X., Zhao, Y., Xia, Y., Steiner, A., et al. (2010). Salmonella typhimurium infection increases p53 acetylation in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G784–G794. doi: 10.1152/ajpgi.00526.2009
Wynosky-Dolfi, M. A., Snyder, A. G., Philip, N. H., Doonan, P. J., Poffenberger, M. C., Avizonis, D., et al. (2014). Oxidative metabolism enables Salmonella evasion of the NLRP3 inflammasome. J. Exp. Med. 211, 653–668. doi: 10.1084/jem.20130627
Xavier, M. N., Winter, M. G., Spees, A. M., Den Hartigh, A. B., Nguyen, K., Roux, C. M., et al. (2013). PPARγ-mediated increase in glucose availability sustains chronic Brucella abortus infection in alternatively activated macrophages. Cell Host Microbe 14, 159–170. doi: 10.1016/j.chom.2013.07.009
Yeung, S. J., Pan, J., and Lee, M. H. (2008). Roles of p53, MYC and HIF-1 in regulating glycolysis-the seventh hallmark of cancer. Cell. Mol. Life Sci. 65, 3981–3999. doi: 10.1007/s00018-008-8224-x
Yin, C., Qie, S., and Sang, N. (2012). Carbon source metabolism and its regulation in cancer cells. Crit. Rev. Eukaryot. Gene Expr. 22, 17–35. doi: 10.1615/CritRevEukarGeneExpr.v22.i1.20
Ying, H., Kimmelman, A. C., Lyssiotis, C. A., Hua, S., Chu, G. C., Fletcher-Sananikone, E., et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. doi: 10.1016/j.cell.2012.01.058
Yoshikawa, Y., Ogawa, M., Hain, T., Yoshida, M., Fukumatsu, M., Kim, M., et al. (2009). Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 11, 1233–1240. doi: 10.1038/ncb1967
Yu, C., Xue, J., Zhu, W., Jiao, Y., Zhang, S., and Cao, J. (2015). Warburg meets non-coding RNAs: the emerging role of ncRNA in regulating the glucose metabolism of cancer cells. Tumour Biol. 36, 81–94. doi: 10.1007/s13277-014-2875-z
Zhao, F., Mancuso, A., Bui, T. V., Tong, X., Gruber, J. J., Swider, C. R., et al. (2010). Imatinib resistance associated with BCR-ABL upregulation is dependent on HIF-1α-induced metabolic reprograming. Oncogene 29, 2962–2972. doi: 10.1038/onc.2010.67
Zuniga-Ripa, A., Barbier, T., Conde-Alvarez, R., Martinez-Gomez, E., Palacios-Chaves, L., Gil-Ramirez, Y., et al. (2014). Brucella abortus depends on pyruvate phosphate dikinase and malic enzyme but not on Fbp and GlpX fructose-1,6-bisphosphatases for full virulence in laboratory models. J. Bacteriol. 196, 3045–3057. doi: 10.1128/JB.01663-14
Keywords: pathometabolism, immunometabolism, innate immune system, metabolic adaptation, intracellular pathogens
Citation: Eisenreich W, Rudel T, Heesemann J and Goebel W (2017) To Eat and to Be Eaten: Mutual Metabolic Adaptations of Immune Cells and Intracellular Bacterial Pathogens upon Infection. Front. Cell. Infect. Microbiol. 7:316. doi: 10.3389/fcimb.2017.00316
Received: 31 March 2017; Accepted: 26 June 2017;
Published: 13 July 2017.
Edited by:
Eric Ghigo, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Jörg Stülke, University of Göttingen, GermanyCopyright © 2017 Eisenreich, Rudel, Heesemann and Goebel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wolfgang Eisenreich, d29sZmdhbmcuZWlzZW5yZWljaEBjaC50dW0uZGU=
Werner Goebel, Z29lYmVsQGJpb3plbnRydW0udW5pLXd1ZXJ6YnVyZy5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.