 M. Fata Moradali
M. Fata Moradali Shirin Ghods
Shirin Ghods Bernd H. A. Rehm
Bernd H. A. Rehm- Institute of Fundamental Sciences, Massey University, Palmerston North, New Zealand
Pseudomonas aeruginosa is an opportunistic pathogen affecting immunocompromised patients. It is known as the leading cause of morbidity and mortality in cystic fibrosis (CF) patients and as one of the leading causes of nosocomial infections. Due to a range of mechanisms for adaptation, survival and resistance to multiple classes of antibiotics, infections by P. aeruginosa strains can be life-threatening and it is emerging worldwide as public health threat. This review highlights the diversity of mechanisms by which P. aeruginosa promotes its survival and persistence in various environments and particularly at different stages of pathogenesis. We will review the importance and complexity of regulatory networks and genotypic-phenotypic variations known as adaptive radiation by which P. aeruginosa adjusts physiological processes for adaptation and survival in response to environmental cues and stresses. Accordingly, we will review the central regulatory role of quorum sensing and signaling systems by nucleotide-based second messengers resulting in different lifestyles of P. aeruginosa. Furthermore, various regulatory proteins will be discussed which form a plethora of controlling systems acting at transcriptional level for timely expression of genes enabling rapid responses to external stimuli and unfavorable conditions. Antibiotic resistance is a natural trait for P. aeruginosa and multiple mechanisms underlying different forms of antibiotic resistance will be discussed here. The importance of each mechanism in conferring resistance to various antipseudomonal antibiotics and their prevalence in clinical strains will be described. The underlying principles for acquiring resistance leading pan-drug resistant strains will be summarized. A future outlook emphasizes the need for collaborative international multidisciplinary efforts to translate current knowledge into strategies to prevent and treat P. aeruginosa infections while reducing the rate of antibiotic resistance and avoiding the spreading of resistant strains.
Introduction
Pseudomonas aeruginosa is a Gram-negative and ubiquitous environmental bacterium. It is an opportunistic human pathogen capable of causing a wide array of life-threatening acute and chronic infections, particularly in patients with compromised immune defense. It has been of particular importance since it is the main cause of morbidity and mortality in cystic fibrosis (CF) patients and one of the leading nosocomial pathogens affecting hospitalized patients while being intrinsically resistant to a wide range of antibiotics.
P. aeruginosa strains possess large genomes (~5–7 Mbp). Their metabolic capacity is extensive as exemplified by their ability to produce multiple secondary metabolites and polymers as well as their ability to use various carbon sources and electron acceptors. The repertoire of P. aeruginosa genes which are substantially conserved suggest the highest proportion of regulatory genes and networks observed in known bacterial genomes and is foundational to respond and adapt to diverse environments (Stover et al., 2000; Mathee et al., 2008; Frimmersdorf et al., 2010). The ubiquitous presence of P. aeruginosa as well as its prevalence and persistence in clinical settings including intrinsic resistance to therapeutics are attributed to its extraordinary capability of survival by recruiting an arsenal of responsive mechanisms.
In the present review, we have attempted to summarize the diversity of these mechanisms causing the versatility of P. aeruginosa to adapt and thrive in unfavorable conditions particularly during pathogenesis. To this end, we will describe the clinical importance of P. aeruginosa followed by the well-characterized and most recent findings about key strategic adaptation mechanisms including quorum sensing (QS), motility-sessility switch, biofilm formation, antibiotic resistance mechanisms, adaptive radiation for persistence, stringent response and persisters, and the CRISPR-Cas systems. Recent findings on adaptive mechanisms will be set into context of the overall physiology of P. aeruginosa by also depicting on future research needs.
Clinical Importance
The CF patients suffer from a multisystem disease due to inheritable genetic defects in the CF transmembrane conductance regulator (CFTR) gene. However, the recurrence of bacterial infections in the abnormal mucus layers is the main cause of morbidity and mortality of CF patients (Khan et al., 1995; Rosenfeld et al., 2001). The CFTR regulator is responsible for regulating the transport of electrolytes and chloride across epithelial cell membranes to maintain normal mucus properties and homeostasis. Therefore, the loss of function of the CFTR protein results in abnormally thick, dehydrated and sticky mucus layers in the lung (Flume and Van Devanter, 2012). Hence, the CF patients are largely susceptible to respiratory infections by P. aeruginosa from infancy. When they are under a year old, almost 30% of CF infants can acquire initial P. aeruginosa strains from the environment leading to acute infections. This rate increases to about 50% by the age of 3 years while mucoid phenotypes causing chronic infections have been reported emerging at the age of 3 to16 years (median of 13 years) (Rehm et al., 1994; da Silva et al., 2013; Jones et al., 2016). P. aeruginosa will adapt to CF airways and persist as overwhelming, predominant and ineradicable infections to the end of patients' life in almost 70% of adults (Döring et al., 2000).
Furthermore, P. aeruginosa is also largely associated with hospital acquired infections including ventilator-associated pneumonia, central line-associated bloodstream infection, urinary catheter-related infection, and surgical/transplantation infections (Cardo et al., 2004; Nathwani et al., 2014; Trubiano and Padiglione, 2015). The International Nosocomial Infection Control Consortium reported that P. aeruginosa nosocomial infections have become a worldwide healthcare issue (Rosenthal et al., 2016). A cohort study reported that P. aeruginosa had the highest burden of healthcare-acquired infections in European intensive care units (Lambert et al., 2011). In the United States healthcare-associated P. aeruginosa infections were estimated to contribute to 51,000 cases each year (Eurosurveillance Editorial Team, 2013). P. aeruginosa is prevalent in healthcare settings because it is a common companion of patients under medical care and also it can survive on abiotic and biotic surfaces such as medical equipment resisting disinfection methods while being transmissible from patient-to-patient (Russotto et al., 2015).
P. aeruginosa infections are becoming more difficult to treat because this bacterium is naturally resistant to many antibiotics and the number of multidrug- and pan-drug-resistant strains is increasing worldwide. Strains have been reported which are resistant to almost all class of commonly used antibiotics including aminoglycosides, cephalosporins, fluoroquinolones, and carbapenems (Hancock and Speert, 2000; Poole, 2011; Eurosurveillance Editorial Team, 2013). In the United States about 13% of P. aeruginosa infections are caused by multidrug resistant strains (Eurosurveillance Editorial Team, 2013).
P. aeruginosa utilizes sophisticated genotypic events to support various phenotypes and molecular mechanisms required for survival during pathogenesis and antibiotic treatment.
Therefore, at initial stages of CF lung colonization, a large number of virulence and intrinsic antibiotic resistance mechanisms mediate survival. After infection, bacteria are exposed to inflammatory responses including oxidative stress followed by treatment with antibiotics (Furukawa et al., 2006; Turner et al., 2014). These environmental stress factors induce the expression of different sets of genes enabling P. aeruginosa to adapt and switch to persisting and resistant phenotypes, while becoming less virulent, such as upon formation of mucoid biofilms (MacDougall et al., 2005; Poole, 2012; Gellatly and Hancock, 2013). Due to the existence of an arsenal of molecular mechanisms conferring resistance to multiple classes of antibiotics, therapeutic options are increasingly limited for treatment of infections, while the number of infection incidences and multi-drug resistance strains are increasing.
Central Regulatory Role of Quorum Sensing (QS) for Virulence and Adaptation
Communication between individual cells using specific chemical signals is a well-known capability of bacteria and is called quorum sensing. Indeed, QS controls social behavior of bacteria by multiple interconnected signaling pathways (LaSarre and Federle, 2013). It allows bacterial communities to regulate a variety of biological processes important for bacterial adaptation and survival. Basically, this phenomenon relies on regulating the expression of specific sets of genes in response to a critical threshold of signaling molecules known as autoinducers (AIs). QS will mediate population density dependent collective responses and is therefore beneficial for community survival. A study showed that cells' responses to QS signals and the corresponding gene expression profile is heterogeneous within a given community leading to increasing fitness and chance of survival (Grote et al., 2015).
During pathogenesis P. aeruginosa QS plays a critical role for survival and colonization by coordinating phenotypic alterations at early stages of infection i.e., after attachment (González and Keshavan, 2006). The progress of acute to chronic infection is critically influenced by QS-dependent gene expression. More than 10% of P. aeruginosa genes are regulated by QS. These genes are mainly involved in virulence factor production, motility, motility-sessility switch and biofilm development, antibiotic resistance mechanisms and the adjustment of metabolic pathways for stress responses (Venturi, 2006; Williams and Camara, 2009; Barr et al., 2015). The role of QS in each physiological adaptation will be discussed below.
Molecular Mechanisms Underlying QS
As shown in Figure 1, four main pathways of QS dependent signaling exist in P. aeruginosa. These constitute a hierarchal network mediating integration of multiple signals via cross-talk between the QS signaling pathways. The most recently discovered IQS signaling pathway is less understood and its integration and impact on gene expression still needs to be unraveled. It was previously proposed that the IQS molecule (an aeruginaldehyde) is the product of enzymatic activity of proteins encoded by ambBCDE genes (Lee et al., 2013), while new findings showed the IQS molecule is a byproduct of the pyochelin biosynthesis pathway and AmbBCDE proteins are responsible for the biosynthesis of the toxin L-2-amino-4-methoxy-trans-3-butenoic acid (AMB) (Ye et al., 2014; Rojas Murcia et al., 2015; Sun et al., 2016).
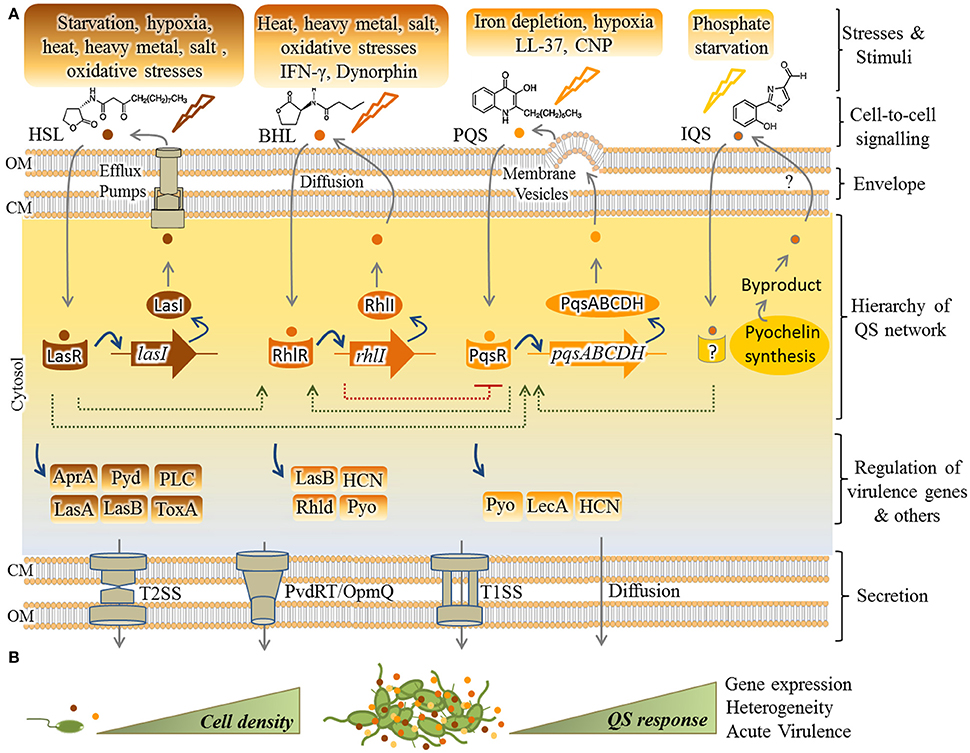
Figure 1. Hierarchical QS network in P. aeruginosa and regulation of virulence factors. (A) So far, four pathways including Las, Rhl, Pqs, and IQS have been understood as mediating QS responses in P. aeruginosa while LasR resides at the top of the cascade. In response to specific stimuli/stress, each pathway synthesizes cognate autoinducers (AIs) [HSL(3-oxo-C12-homoserine lactone)], BHL (N-butyrylhomoserine lactone or C4-HSL), PQS (2-heptyl-3-hydroxy-4-quinolone) and IQS [2-(2-hydroxyphenyl)-thiazole-4-carbaldehyde (aeruginaldehyde)]. Export and import of HSL, BHL, and PQS is mediated by the efflux pumps MexAB-OprM/MexEF-OprN, free diffusion and membrane vesicles, respectively. Question mark indicates unknown pathway of IQS transportation. As fine-tuned individual circuits, but interconnected (dashed lines), transcriptional factors (i.e., LasR, RhlR, and PqsR) are activated by AIs for upregulating expression of cognate AI synthases (respectively, LasI, RhlI, PqsABCDH) as well as others such as virulence factor genes. The IQS pathway remains unraveled and the IQS receptor is still unknown. Various secretion systems mainly type 1 and 2 secretion systems (T1SS/T2SS) and also PvdRT-OpmQ efflux pump mediate the secretion of virulence factors. (B) QS initiates upon cumulative production of AIs (small colorful circles) by increasing cell density and results in collective responses. AprA, alkaline protease; Pyd, pyoverdine; PLC, phospholipase C; Tox, toxin A; LasA, LasA elastase; LasB, LasB elastase; HCN, hydrogen cyanide; Pyo, pyocyanin; Rhld, rhamnolipids; Lec A, lectin A; CM, cytoplasmic membrane; OM, outer membrane.
In regard to the other three QS pathways, each system consists of at least two major functional elements; the first category of proteins (i.e., LasR, RhlR, or PqsR, respectively) is activated upon sensing specific autoinducers (AIs) and acts as transcriptional activator for genes encoding the second tier proteins, the cognate AI synthases (i.e., LasI, RhlI, and PqsABCDH, respectively). These activation steps constitute a fine-tuned circuit by which the synthesized AIs are exported outside the cells followed by being imported again (Figure 1). Transportation of these signals is not well understood, but it is proposed as being mediated by free diffusion, membrane transporters such as specific efflux systems or membrane vesicles (Mashburn and Whiteley, 2005; Martinez et al., 2009; Alcalde-Rico et al., 2016). Beyond this regulatory circuit the activated AI-sensing proteins act as transcriptional factors for activating the expression of other set of genes such as virulence genes in response to environmental stimuli (Figure 1). Transcriptional activation occurs via binding to conserved las/rhl boxes acting as operators residing upstream of these genes (González-Valdez et al., 2014; Lee and Zhang, 2015; Banerjee and Ray, 2016; Papenfort and Bassler, 2016). In the hierarchy of this network, LasR resides at the top of the cascade and along with RhlR mediates QS signaling at early stages of exponential growth phase while the PQS signaling pathway is active at late exponential growth phase (Choi et al., 2011). As abovementioned, cumulative and cell density-dependent production of AIs is required for reaching a specific threshold triggering collective responses by individual cells.
QS-Controlled Virulence Factors and Stress Responses
Production of virulence factors is a survival strategy for pathogens to evade the host immune defense resulting in progression of pathogenesis particularly at early stage of colonization and acute infection. A large number of virulence factors including cell-associated or secreted compounds, both low and high molecular weight compounds have been reported as important in colonization and establishment of infections by P. aeruginosa. Although they play a critical role in promoting bacterial growth and survival, they can cause devastating injuries to the host tissues and impair the immune responses. QS deficient mutants cause considerably less tissue damage and pathological changes during infections due to a significant decrease in the virulence and cytotoxicity (Nelson et al., 2009; Feng et al., 2016).
Production of many virulence factors is metabolically costly and requires community involvement. Hence, they are mainly under the regulatory control of the QS systems (Figure 1, Table 1; Whiteley et al., 1999; Diggle et al., 2002; Wagner et al., 2003; García-Contreras, 2016).
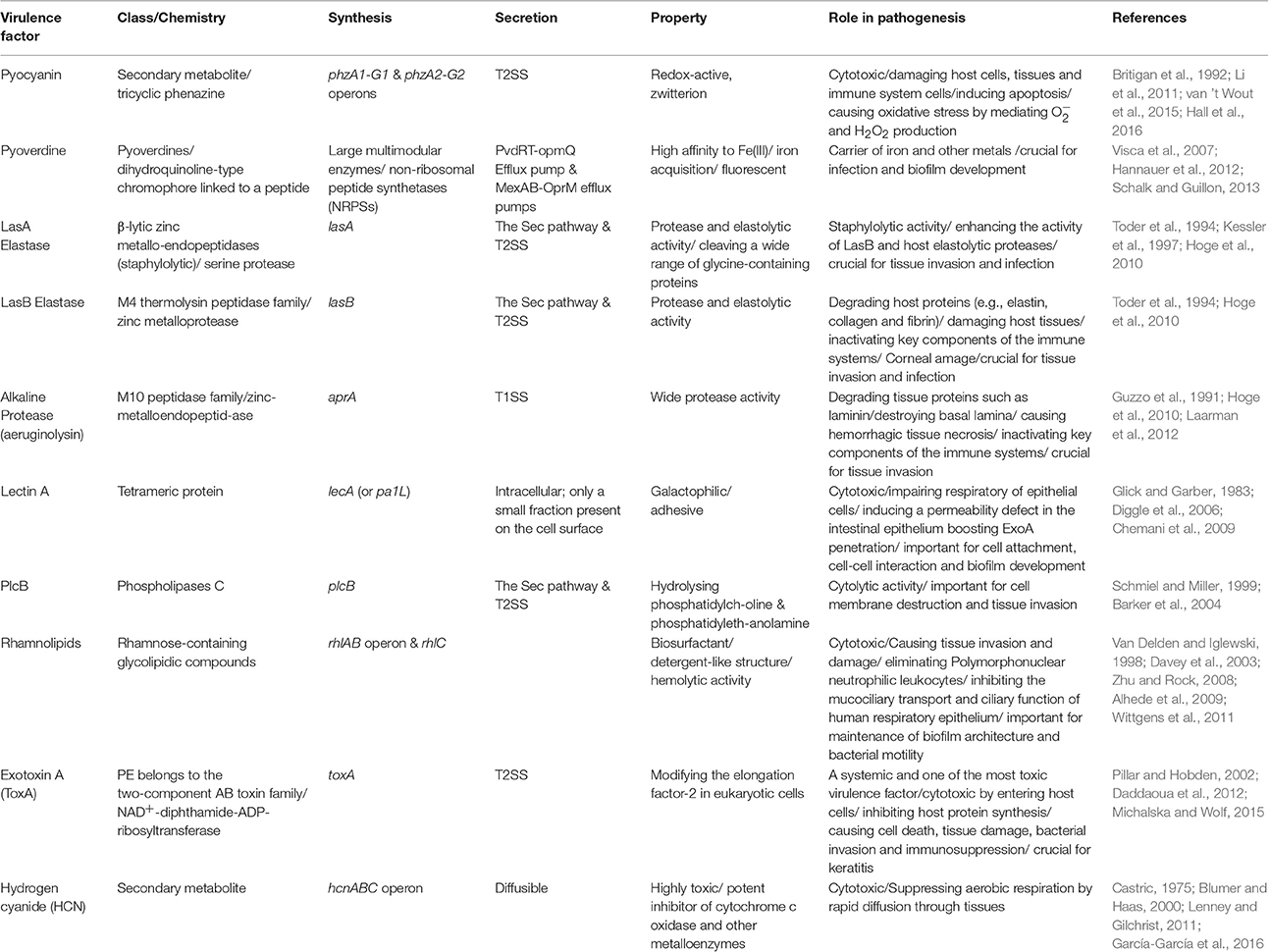
Table 1. Key QS-dependent virulence factors produced by P. aeruginosa.
Analysis of bronchial secretions of CF patients during different stages of pulmonary exacerbations showed that QS upregulates the expression of genes involved in the production of some destructive virulence factors such as proteases (elastase, alkaline protease), phenazines (pyocyanin), toxins (exotoxin A), rhamnolipids and hydrogen cyanide (Jaffar-Bandjee et al., 1995; Lee and Zhang, 2015). Production of these toxic compounds is destructive to the host cells/tissues by impairing permeability barrier and by inhibiting protein production promoting cell death.
Recent findings suggested a correlation between systemic concentrations of some QS signaling molecules with the clinical status of pulmonary exacerbation and at least some QS signaling molecules were elevated at the start of either pulmonary exacerbation or antibiotic treatment when assessing different biofluids (Barr et al., 2015). In conclusion, virulence factors assist bacteria in colonization and survival aligned with worsened clinical course of infections. Thus, QS can determine the degree of pathological damages and clinical stages of infections in response to environmental factors.
Pathogenesis encompasses various stresses such as host immune factors, bacterial interspecies competition, phosphate/iron depletion and starvation. QS systems and production of some virulence factors mediate appropriate responses to these stresses to promote survival and adaptation (García-Contreras et al., 2015). Here, we provide some examples of stress responses mediated by the QS systems.
Interferon-γ (IFN-γ) is a crucial cytokine of the human immune system during infection and it coordinates a wide array of immunological responses such as up-regulation of pathogen recognition and the activation of bactericidal effector functions (Schroder et al., 2004). IFN-γ produced by T-cells was shown to bind directly to P. aeruginosa OprF, an outer membrane protein. Upon formation of IFN-γ-OprF complexes the rhl QS system was activated and resulted in up-regulation of the expression of lecA (or PA-I lectin) and synthesis of pyocyanin. The lecA gene encodes the virulence determinant, galactophilic lectin (or LecA) (Wu et al., 2005) which is cytotoxic and acts as adhesion factor mediating initiation of host recognition by P. aeruginosa (Chemani et al., 2009). It induces an increased permeability of the intestinal and respiratory epithelial cells enabling cytotoxic exoproducts such as exotoxin A (Laughlin et al., 2000) to enter host cells (Bajolet-Laudinat et al., 1994). In addition it also contributes to biofilm development (Diggle et al., 2006). Furthermore, the QS system has been reported to mediate a response to the host antimicrobial factor LL-37 by increasing the production of pyocyanin, hydrogen cyanide, elastase and rhamnolipids (Strempel et al., 2013). The QS-dependent production of rhamnolipids has a crucial role in neutralizing the attack of neutrophils due to their necrotic property (Jensen et al., 2007; Van Gennip et al., 2009).
Recent findings indicated that the LasR and RhlR QS systems, but not the PQS system, play major roles in adaptation and response to environmental stresses such as oxidative, heat, heavy metal and salt stresses (García-Contreras et al., 2015).
The stress response of P. aeruginosa to the depletion of phosphate and iron was found to be linked (Slater et al., 2003). Different studies showed that acquisition of phosphate and iron are important for survival and pathogenesis of P. aeruginosa and the expression of cognate genes mediating acquisition of these elements are upregulated upon interaction with human respiratory epithelial cells (Frisk et al., 2004; Chugani and Greenberg, 2007). Various studies unraveled that phosphate- and iron-deficient conditions can trigger the activation of the QS system especially via the IQS- or PQS-dependent pathway leading to boosted activation of central QS and the production of virulence factors such as rhamnolipids, phenazines, cyanide, exotoxin A, LasA protease, elastase, and antimicrobials (Kim et al., 2003; Long et al., 2008; Zaborin et al., 2009; Bains et al., 2012; Lee et al., 2013; Nguyen et al., 2015). Production of such virulence factors can increase cytotoxic impact of bacteria on host tissue and promote pathogen survival.
The recently discovered IQS system which controls the expression of a large set of virulence factors was shown to be directly activated by phosphate limitation in P. aeruginosa (Lee et al., 2013; Lee and Zhang, 2015).
P. aeruginosa QS signaling molecules such as 2-heptyl-4-hydroxyquinoline (HHQ) and 2-heptyl-4-hydroxyquinoline-N-oxide (HQNO) can serve as antimicrobial agents against Staphylococcus aureus which is commonly present during early stages of pulmonary infections in CF patients. It is proposed that this antibacterial activity supports the dominance of P. aeruginosa during the course of infection. Interestingly, this inter-species competition is linked to the availability of iron as the depletion of iron potentiates the antistaphylococcal activity of these metabolites (Nguyen et al., 2016). Also, LasA is a staphylolytic protease produced by P. aeruginosa and it is under the regulation of the las QS system (Toder et al., 1991). Furthermore, when P. aeruginosa was grown together with the yeast Candida albicans in a mixed biofilm, the QS system upregulated the production of virulence factors such as pyoverdine, rhamnolipids and pyocyanin (Trejo-Hernández et al., 2014).
Oxygen depletion known as hypoxia is another stress factor for P. aeruginosa during pathogenesis. Hypoxia condition is influenced by various factors such as reduced ventilation through viscose layers, chronic inflammation, microbial population and biofilm formation (Hassett, 1996; Worlitzsch et al., 2002; Yoon et al., 2002; Alvarez-Ortega and Harwood, 2007; Hassett et al., 2009). However, P. aeruginosa can survive and grow under hypoxia to high cell densities. Under hypoxia stress P. aeruginosa retains the capability of microaerobic respiration, although occurrence of nitrate respiration was thought to be possible (Alvarez-Ortega and Harwood, 2007). The QS regulon expression occurs at low oxygen conditions. Hammond et al. (2015) unraveled that the 4-hydroxy-2-alkylquinolines (HAQ)-dependent QS pathway is active during hypoxia via the ANR protein as the master transcriptional regulator of anaerobic respiration while it is in the absence of LasR signaling (Hammond et al., 2015). Under low oxygen tension, the ANR protein positively regulated the production of the QS signaling molecule 4-hydroxy-2-alkylquinolines and in turn the regulation of virulence-related genes could continue via PQS system (Hammond et al., 2015). Furthermore, under hypoxia stress the ANR protein and the QS systems cooperatively regulate hydrogen cyanide biosynthesis (Castric, 1983, 1994; Pessi and Haas, 2000). This study provided further evidence that low oxygen-dependent QS inversely correlates with denitrification i.e., suppresses nitrate respiration (Hammond et al., 2015).
Overall, these examples have provided further insight into the versatility of P. aeruginosa to adapt to various environmental conditions by processing signals via integrated and cross-talking QS pathways resulting in enhanced survival i.e., in a medical context establishment of acute and chronic infections.
Persistence and Biofilm Formation
During acute infection the relationship between pathogen and host is reciprocally devastating as a variety of cytotoxic molecules produced by bacteria impair the host cellular processes while bacteria on the other hand encounter immune system responses such as production of antimicrobial compounds and reactive oxygen species, as well as enhanced phagocytosis. In this context, motile P. aeruginosa display a more virulent phenotype. Various modes of P. aeruginosa motility such as swimming and swarming involving flagella and twitching using type 4 pili are associated with virulent traits (Winstanley et al., 2016). A motile cell is readily detectable by the host immune system via flagellar and/or other motility components mediating recognition and induction of signaling pathways which trigger inflammatory responses and phagocytosis by murine or human macrophages (Amiel et al., 2010).
Switching to sessile lifestyle along with lower virulency is a survival advantage by which many pathogenic bacteria such as P. aeruginosa evade stresses and adverse conditions. They lose motility and attach to surfaces and form cellular aggregations or microcolonies which are embedded in extracellular polymeric substances (EPS) to protect bacteria from the surrounding environment. These structures are so called biofilms conferring an extreme capacity for persistence against phagocytosis, oxidative stresses, nutrient/oxygen restriction, metabolic waste accumulation, interspecies competitions, and conventional antimicrobial agents (Leid, 2009; Olsen, 2015).
Formation of mucoid biofilm by P. aeruginosa is the hallmark of chronic infections and indicative of disease progression and long-term persistence. As a consequence, P. aeruginosa dominates the microbial community of CF lungs in patients older than 24 years (McDaniel et al., 2015).
Other P. aeruginosa biofilm associated infections include chronic wound infection, chronic otitis media, chronic rhinosinusitis, catheter-associated urinary tract infection, and contact lens-related keratitis (Römling and Balsalobre, 2012).
Composition of the P. aeruginosa Biofilm
Formed on abiotic and biotic surfaces, the matrix of most biofilms embedding bacterial cells may account for over 90% of dry weight of whole biofilm mass. In fact, this matrix creates a niche rendering bacteria for intense cell-cell interaction and communication as well as a reservoir of metabolic substances, nutrients and energy for promoting growth while shielding cells from unfavorable conditions (Flemming and Wingender, 2010). The matrix is mainly formed by extracellular polymeric substances (EPS) which are mainly polysaccharides, proteins, extracellular DNA (eDNA) and lipids (Strempel et al., 2013). Major polymers and relevant characteristics are listed in Table 2.
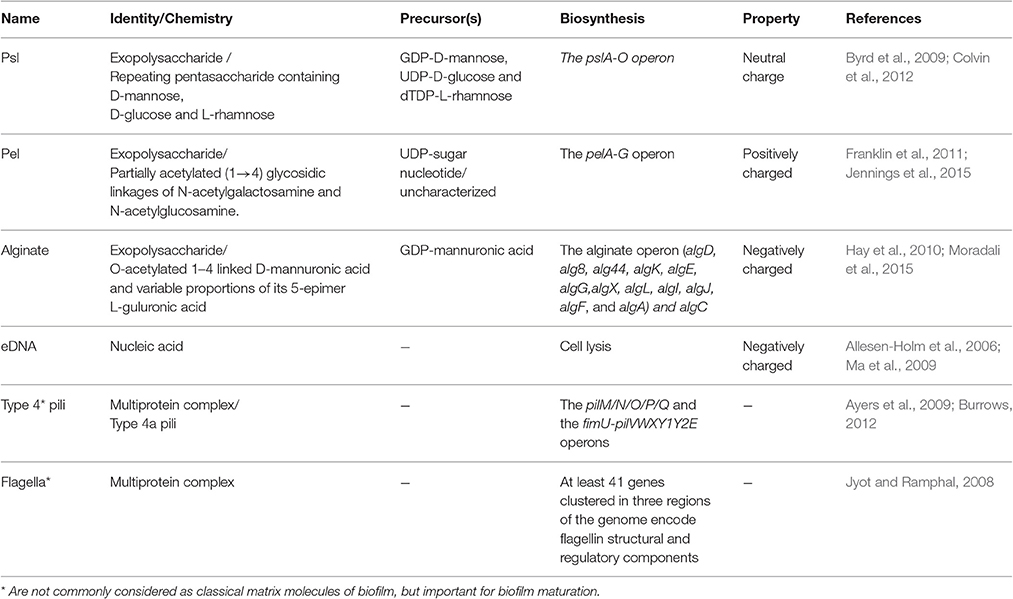
Table 2. Key polymeric substances in P. aeruginosa biofilm formation and development.
The exopolysaccharides Psl, Pel, and alginate are major constituents of the P. aeruginosa biofilm matrix involved in surface adhesion and together with eDNA determine the biofilm architecture. These EPS play an important role in resistance to immune responses and antibiotic treatments (Ghafoor et al., 2011; Gellatly and Hancock, 2013; Strempel et al., 2013). The differential role of each EPS has been analyzed at each stage of biofilm development. The various exopolysaccharides and eDNA were shown to interactively contribute to the biofilm architecture (Ghafoor et al., 2011). The presence of various EPS exhibiting different physiochemical properties confers a survival strategy for increasing the flexibility and stability of biofilms under various conditions (Jennings et al., 2015).
The Psl polysaccharide is a key element at early stage of biofilm formation when cells explore surfaces for adhesion (Overhage et al., 2005). It is anchored around cells in a helical arrangement initiating biofilm formation by enhancing cell migration, cell-cell interaction and cell-surface adhesion whereas in mature biofilms it is located to the periphery of mushroom shaped macrocolonies (Ma et al., 2009; Zhao et al., 2013). Psl can exist as a fiber-like matrix requiring type 4 pili-mediated migration of cells (Wang S. et al., 2013). It protects cells against phagocytosis and oxidative stress during infection (Mishra et al., 2012). Recent studies suggested that Psl can provide an instant protective role against anti-biofilm agents and a broad spectrum of antibiotics particularly at early stage of biofilm development (Zegans et al., 2012; Billings et al., 2013). Therefore, Psl provides a survival advantage during pathogenesis.
Similar to Psl, Pel is important for initiating and maintaining cell–cell interaction in biofilms (Colvin et al., 2011). Pel and/or Psl are the primary matrix structural polysaccharides in non-mucoid P. aeruginosa strains as a predominant environmental phenotype. However, contribution of Psl and Pel to the structure of mature biofilms is strain-dependent while both unique and functionally redundant roles have been reported for these exopolysaccharides (Colvin et al., 2012). Recent studies elucidating the chemical structure and biological function of Pel demonstrated that it is a major structural component of the biofilm stalk where it cross-links eDNA and structurally compensates for the absence of Psl in the periphery of mature biofilm (Jennings et al., 2015). Furthermore, Pel was shown to protect bacteria against certain aminoglycoside antibiotics (Colvin et al., 2011).
Overproduction of the exopolysaccharide, alginate, is characteristic for mucoid phenotype of most clinical isolates from CF patients. During adaptation to the CF lung environment, alginate is overproduced and predominantly constitutes the matrix of mature biofilms conferring a slimy or mucoid phenotype. Indeed, it is greatly important in biofilm maturation, structural stability and protection as well as persistence by shielding P. aeruginosa cells against opsonophagocytosis, free radicals released from immune cells, and antibiotics used for treatment (Hay et al., 2009a; Hay I. D. et al., 2013; Strempel et al., 2013). Some in vitro biofilm studies showed that the composition of the alginates can greatly influence biofilm characteristics such as viscoelastic property, bio-volume, cell density and architecture as well as cell-to-cell interaction, cell aggregation and surface attachment (Tielen et al., 2005; Moradali et al., 2015).
As abovementioned, eDNA is another important structural component for biofilm development and along with the Pel polysaccharide it can be detected within the stalks of mushroom-shaped macrocolonies. However, eDNA has multifaceted roles in biofilm formation such as contribution to forming cation gradients in the matrix via the chelating interaction of highly anionic DNA with cations such as Mg2+, Ca2+, Mn2+, and Zn2+, as a nutrient source during starvation, facilitating twitching motility and coordinating cell movements and conferring antibiotic resistance (Allesen-Holm et al., 2006; Mulcahy et al., 2008; Gloag et al., 2013).
Among the proteinaceous biofilm constituents, both flagella and the type 4 pili are important during maturation of the biofilm, however, these cell appendages are not commonly considered as classical matrix components of biofilms. Type 4 pili are important for adhesion and promote initial attachment of cells to surfaces at early stage of biofilm formation. Together with eDNA, flagella and the type 4 pili mediate migration required for the formation of the stalk and the cap in the mushroom-shaped microcolonies of the mature biofilm (Table 2; Barken et al., 2008; Mann and Wozniak, 2012).
Central Regulatory Network Governing the Motility-Sessility Switch
Transition from motility to sessility requires dynamic regulatory networks at transcriptional, post-transcriptional and post-translational levels resulting in coordinated timely expression of hundreds of genes. These events mainly arrest flagella based motility and the production of virulence factors such as exotoxins and proteases while positively regulating surface attachment, EPS production and biofilm maturation (Figure 2).
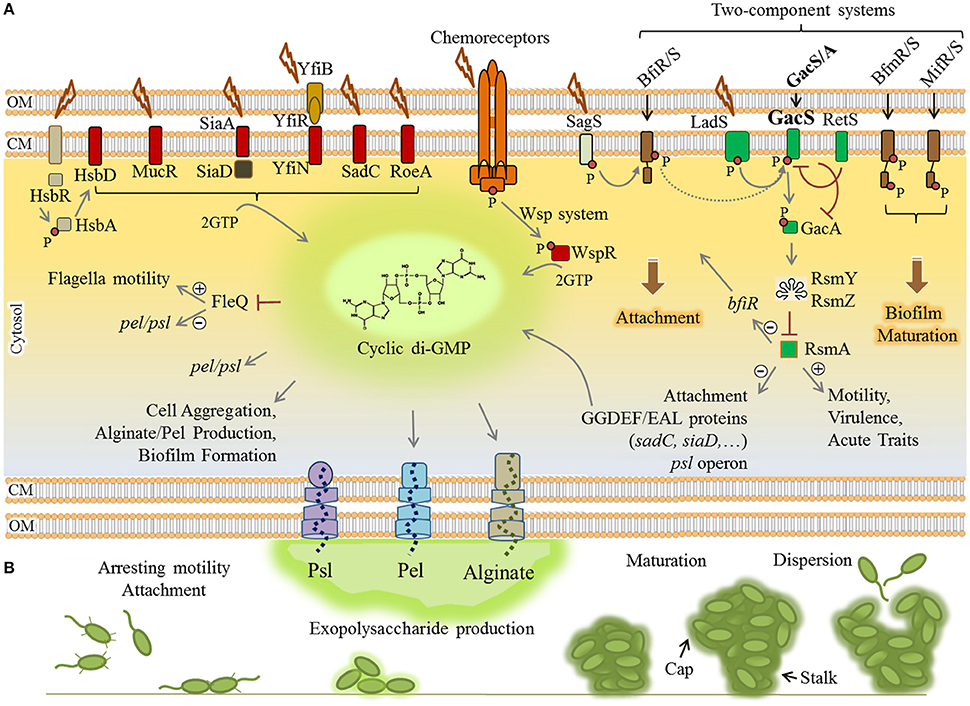
Figure 2. Regulatory networks underlying biofilm formation by P. aeruginosa. (A) Elevation of the cyclic di-GMP molecule is a key determinant for the motility-sessility switch. Environmental cues are sensed by various proteins localized in the envelope of the cells where these proteins contribute to two-component systems (brown/green rectangles), chemoreceptor-like system (orange complex) and other receptor mediated signaling pathways (arranged in the left side of figure). Either triggered as phosphorylation cascades (small red circle) or protein-protein interactions, the signals induce diguanylate cyclases (containing GGDEF motif) (red rectangles) to synthesize cyclic di-GMP from two molecules of GTP (guanosine-5′-triphosphate). Consequently, cyclic di-GMP sensing proteins act as receptor/effector for specific outputs such as induction of alginate and Pel polymerization, inhibition of motility and derepression of psl/pel expression via FleQ, induction of attachment and biofilm formation/maturation triggered by two component systems. The two-component systems are interconnected and the LadS/RetS/GacS/GacA/RsmA regulatory network (green rectangles) plays a key role in the phenotypic switch from motility to sessility and downregulation of QS and virulence factor production. (B) Various stages of biofilm formation and development were represented. Plus and minus signs represent positive and negative effect of transcriptional regulators, respectively. CM, cytoplasmic membrane; OM, outer membrane.
The small molecule cyclic-3′5′-diguanylic acid (cyclic di-GMP) is a key signal in post-transcriptional regulation of biofilm formation. It is an almost ubiquitous second messenger present in a wide range of bacteria that principally controls motility-sessility switch. The major determinant for this substantial phenotypic change is the cellular level of cyclic di-GMP, so that its elevation triggers biofilm formation while inhibiting motility. The cyclic di-GMP signaling system is very complex and two groups of proteins have been identified as main actors. The first group comprises cyclic di-GMP metabolizing enzymes including diguanylate cyclases (DGC) (containing GGDEF motif) and phosphodiesterases (containing EAL or HD-GYP motif) that respectively synthesize and degrade cyclic di-GMP in cells (Römling et al., 2013; Valentini and Filloux, 2016). At least 40 such proteins directly synthesize and/or degrade cyclic di-GMP in P. aeruginosa which controls cellular level of this molecule in response to perceived stimuli (Ryan et al., 2006). The second group is represented by cyclic di-GMP sensing proteins which also act directly as effectors or via protein-protein interactions to mediate the output response (Römling et al., 2013). For example, cyclic di-GMP is essential for the activation of alginate polysaccharide polymerization (Remminghorst and Rehm, 2006). Experimental evidence indicated that a pool of cyclic di-GMP is synthesized by MucR (a hybrid GGDEF/EAL domain-containing protein) in the proximity of the alginate biosynthesis/secretion multi-protein complex of P. aeruginosa (Hay et al., 2009b; Wang et al., 2015). Cyclic di-GMP binds to PilZ domain of Alg44 protein and allosterically activates alginate polymerization via interaction with Alg8 glycosyltransferase (Hay et al., 2009b; Moradali et al., 2015; Wang et al., 2015). Also, cyclic di-GMP binding to FleQ, a transcriptional master regulator represses flagella biosynthesis while it concomitantly derepresses the expression of pel and psl genes (Baraquet et al., 2012; Figure 2). Likewise, there are many other receptor/effector proteins which enhance required pathways for biofilm formation upon cyclic di-GMP binding while they inhibit motility and other virulence factor synthesis pathways.
In addition, the Wsp chemosensory system in P. aeruginosa is homologous to chemotaxis signaling pathways which regulate cyclic di-GMP synthesis via signal transduction (Figure 2). A cascade of phosphorylations is triggered upon surface attachment and possibly sensing mechanical stress or other environmental stimuli which then activate the cyclic di-GMP synthesizing protein WspR promoting biofilm formation (Hickman et al., 2005; Porter et al., 2011).
Furthermore, transduction of phosphorylation events via two-component regulatory systems controls biofilm formation in a stage-specific manner (Figure 2). This network consists of BfiRS, BfmRS, and MifRS and GacS/GacA regulatory components (Petrova and Sauer, 2009). The GacS/GacA two-component system is part of the global regulatory pathway comprising LadS/RetS/GacS/GacA/RsmA proteins (Figure 2). This pathway controls many physiological responses at post-transcriptional level and is involved in both motility-sessility and acute-chronic infection transitions. Of this regulatory pathway, the RNA-binding protein RsmA negatively controls biofilm formation pathways while it induces production of T3SS, type 4 pili and other virulence factors. RsmA binds to psl mRNA and inhibits the translation of required proteins for Psl polysaccharide biosynthesis (Irie et al., 2010; Jimenez et al., 2012). It also represses production of GGDEF/EAL encoding proteins; hence, it inhibits elevation of cyclic di-GMP levels. In P. aeruginosa, under stress conditions, this pathway generates non-coding RNAs (ncRNAs) known as RsmY and RsmZ which counteract RsmA translational repression activity, consequently derepressing biofilm formation mainly via cyclic di-GMP level increase resulting in exopolysaccharides production (Jimenez et al., 2012).
There are other regulatory pathways known to be involved in cyclic di-GMP turnover in response to external stimuli, but the further precise function still remains to be elucidated (Figure 2).
The Role of QS in Biofilm Development and Maturation
In addition to abovementioned regulatory networks, biofilm residents utilize QS systems for cell-to-cell communication and spatio-temporal regulation of expression of specific genes. During chronic infection, a major proportion of the colonizing population was thought to lose QS due to hypermutation events and phenotypic alterations. However, further investigations have now revealed that genes involved in the progress of biofilm maturation and persistence are positively regulated by QS in P. aeruginosa. Indeed QS-deficient mutants of P. aeruginosa (i.e., ΔlasRΔrhlR and ΔlasIΔrhlI) formed thin and much less developed biofilms which were more sensitive to antibiotic treatments and eradication (Shih and Huang, 2002; Nelson et al., 2009). Furthermore, Bjarnsholt et al. (2010) demonstrated that at least a part of QS pathways i.e., rhl encoded system and the production of C4-HSL signals was retained in predominantly mucoid population at the end of chronic stages coinciding with overproduction of alginate and rhamnolipids (Bjarnsholt et al., 2010). The biosurfactants, rhamnolipids, have been suggested to play an active role in maintenance of the biofilm architecture by contributing to the formation of internal cavities within the mature biofilm, allowing proper flow of water and nutrients (Davey et al., 2003; Boles et al., 2005; Dusane et al., 2010; Chrzanowski et al., 2012). Additionally, the production of pel polysaccharide, eDNA and QS-controlled production of pyocyanin are critical for biofilm maturation. Pel cross-links eDNA in the biofilm stalk via ionic interactions and it serves as important structural components of the biofilm matrix of P. aeruginosa (Jennings et al., 2015). Furthermore, pyocyanin molecules can promote eDNA release by inducing bacterial cell lysis. Pyocyanin binds to eDNA increasing its solution viscosity which influences the physicochemical interactions of the biofilm matrix with environment as well as facilitates cellular aggregations (Das et al., 2013, 2015). Collectively, such molecular and cellular interactions in combination with other polymeric substances lead to establishment of a robust and mature biofilm.
Antibiotic Resistance Mechanisms
Indeed, the emergence of antibiotic resistant bacteria is a global health issue. Among identified notoriously multi-drug resistant (MDR) bacteria, P. aeruginosa has been introduced as a major concern with a growing threat to global health resulting in dramatically increasing prevalence of nosocomial and chronic infections. This is due to the extraordinary capacity of these bacteria to develop resistance against a wide range of antimicrobials through various molecular mechanisms which are often simultaneously present in clinical isolates. Although each resistance mechanism is related to a specific class of antibiotics, multiple mechanisms mediate variably resistance to each class of antibiotics (Potron et al., 2015). Furthermore, the contribution of each mechanism varies from country to country. Loss or reduced copy numbers of OprD and overproduction of active efflux pumps, AmpC β-lactamase and extended-spectrum β-lactamases have been mainly reported as main contributors to multi-drug resistance phenotypes of P. aeruginosa isolates.
Recent reviews have described the prevalence and contribution of each resistance mechanism to each class of antibiotics in detail (Lister et al., 2009; Strateva and Yordanov, 2009; Sun et al., 2014; Hong et al., 2015; Potron et al., 2015). Here, we reviewed the most frequent and well-understood findings which are classified into intrinsic, acquired and adaptive mechanisms, and we provide an update on our understanding of how P. aeruginosa can survive antibiotic treatments.
Intrinsic Resistance Mechanisms
Like many Gram-negative bacteria, P. aeruginosa can be intrinsically resistant to particular antibiotics. Such intrinsic resistance mechanisms stem from the existence of genes in bacterial genome encoding inherent properties of cell structures and composition providing protection against toxic molecules and antimicrobials. It can also be contributed by the lack of susceptible sites which naturally exist in antibiotic sensitive species (e.g., resistance to triclosan) (Lambert et al., 2011; Blair et al., 2015; Figure 3).
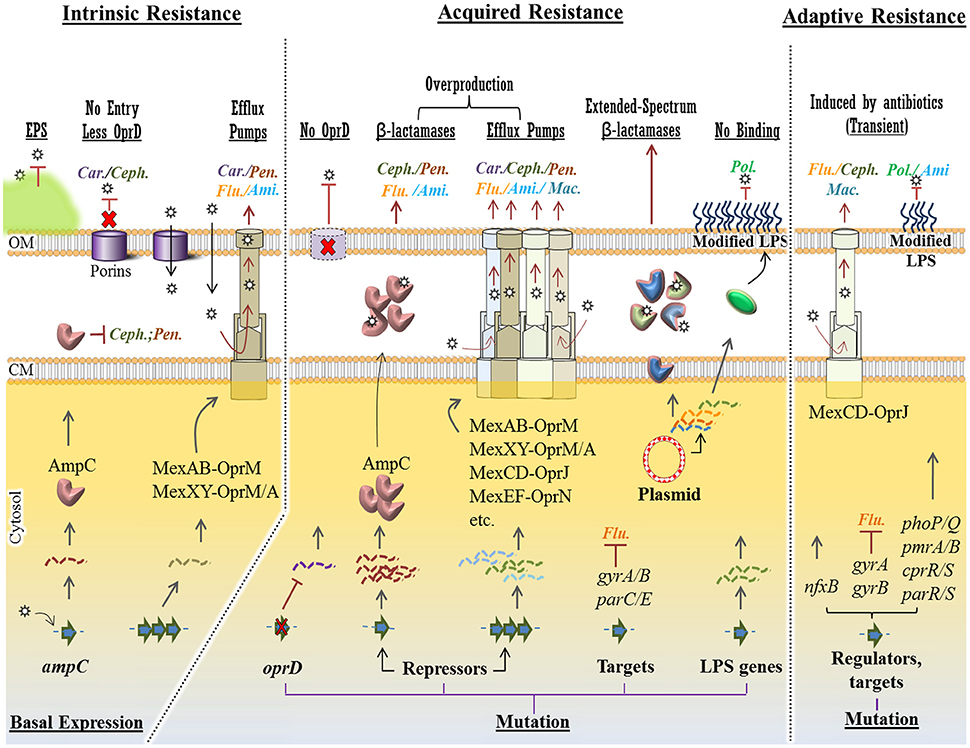
Figure 3. Intrinsic, acquired and adaptive mechanisms confer antibiotic resistance in P. aeruginosa. For each mechanism, various molecular strategies, which confer resistance to specific class of antipseudomonal antibiotics (Car., Carbapenems; Ceph., Cephalosporins; Pen., Penicillins; Ami., Aminoglycosides; Flu., Fluoroquinolones; Mac., Macrolides and Pol., Polymyxins), were presented at the top of the figure (underlined) Intrinsic mechanisms such as structural barriers [e.g., EPS (extracellular polymeric substances)], OprD reduction and basal production of AmpC β-lactamase and MexAB/XY efflux pumps confer a basal resistance to some group of antibiotics. However, in acquired resistance, mutational changes in the oprD gene, transcriptional repressors causingupregulation of resistance genes and efflux pumps conferring resistance against a wider spectrum of antibiotics. Plasmid-mediated resistance is very potent as a variety of resistance genes can be exchanged among bacteria. Either mediated by mutational changes in the genome or in plasmids, resistance to polymyxins occurs via modification of LPS (lipopolysaccharide) components hindering binding of the antibiotic to this layer. Adaptive resistance occurs in the presence of antibiotics mainly via mutation in regulatory genes. This is a transient and reversible resistance, which will reverse upon removal of antibiotics. Stars represent antibiotics and dashed/wavy lines represent transcriptional levels of each gene product. CM, cytoplasmic membrane; OM, outer membrane.
However, hydrophilic antibiotics can enter cells by diffusing through membrane channels or porin proteins in a non-specific manner. As one of the intrinsic mechanisms, P. aeruginosa limits antibiotic entry by reducing the number of non-specific porin proteins and replacing them with specific or more-selective channels for taking up required nutrients resulting in lowered permeability to toxic chemicals (Tamber and Hancock, 2003; Figure 3). P. aeruginosa resistance to currently used broad-spectrum drugs such as carbapenems and cephalosporins is commonly caused by this adaptation (El Amin et al., 2005; Baumgart et al., 2010). Many of the clinical strains of P. aeruginosa displaying resistance to carbapenems such as imipenem are deficient in the OprD porin which specifically facilitates the diffusion of basic amino acids, small peptides as well as carbapenems into the cell (Trias and Nikaido, 1990; Strateva and Yordanov, 2009).
Active multidrug efflux pumps greatly contribute to antibiotic resistance observed in P. aeruginosa. The involved genes are ubiquitous in Gram-negative bacteria and they are located on the genome or plasmids. The multidrug efflux pumps are multi-protein complexes spanning the envelope of Gram-negative bacteria. They are responsible for expelling various toxic materials and a wide range of antimicrobials. Because of their broad substrate specificities they display resistance against different classes of antibiotics which are chemically unrelated (Blair et al., 2015; Venter et al., 2015).
P. aeruginosa possesses four well known active multidrug efflux pumps including MexAB-OprM, MexXY/OprM(OprA), MexCD-OprJ, and MexEF-OprN (Figure 3). The gene sets encoding these systems are under different regulatory factors; therefore, the expression levels of these systems significantly differ under various conditions. The MexAB-OprM and MexXY/OprM(OprA) are the most clinically important sets due to their large prevalence in clinical strains and significant contribution to a wide range of antibiotics (Avrain et al., 2013). The mexAB-oprM genes show a stable and constitutive expression in the cell guaranteeing a protective basal level production of the MexAB-OprM system to consistently expel a wide range of toxic molecules and antibiotics (Li et al., 2015). Hence it mainly contributes to natural resistance to antibiotics. The mexXY-(oprA) genes show lower basal expression levels and are mainly induced in response to protein synthesis inhibitors that target the ribosomal machinery (Matsuo et al., 2004; Hay T. et al., 2013). Both mexCD-oprJ and mexEF-oprN genes are not typically expressed in wild-type strains or their expression is very low and they have been proposed not to contribute significantly to natural antibiotic resistance (Llanes et al., 2011; Li et al., 2015).
There are other forms of multidrug efflux pumps such as MexJK, MexGHI-OpmD, MexVW, MexPQ-OpmE, MexMN, and TriABC. They are not expressed in wild-type strains but may contribute to adaptive resistance against antibiotic or biocide agents when expressed in resistant strains (Lister et al., 2009; Avrain et al., 2013).
On the other hand, they might play role in other physiological pathways as well. For example, The MexEF-OprN and MexGHI-OpmD sets can modulate QS systems by exporting the quinolone signaling molecule PQS reducing its cellular concentration resulting in the reduction of virulence factor production, which is presumably in favor of establishment of chronic infections (Köhler et al., 2001; Aendekerk et al., 2005; Lamarche and Déziel, 2011). However, many of these mechanisms remain still unclear with regard to their connection with other physiological pathways and their clinical relevance.
Another player of intrinsic resistance and basal lower level antibiotic susceptibility in P. aeruginosa is the gene encoding an inducible β-lactamase (AmpC) (Figure 3). Particularly, chromosomal expression and production of AmpC confers low level resistance to aminopenicillins and most cephalosporins because these antibiotics strongly induce the production of AmpC which consequently hydrolyzes these substrates (Oliver et al., 2015). However, through adaptive or acquired resistance mechanisms AmpC can be overproduced, consequently conferring resistance to a wider range of antibiotics such as aminoglycosides and fluoroquinolones (Umadevi et al., 2011). These mechanisms will be further discussed later.
Acquired Resistance Mechanisms
P. aeruginosa can acquire resistance to antibiotics through mutation of intrinsic genes or horizontal acquisition from other bacteria through transferring plasmids carrying genetic materials encoding for antibiotic resistance (Davies, 1997; Davies and Davies, 2010). Contrary to intrinsic mechanisms, acquired resistance is related to antibiotic selection and this selective advantage occurs in the presence of antibiotic compounds leading to irreversible resistant population (Lee et al., 2016). Therefore, similar to intrinsic resistance, acquired resistance is stable too and it can be transmitted to progeny.
However, due to over-expression of resistance genes and transmissibility by plasmids, acquired resistance is a potent mechanism which confers resistance to a wide spectrum of antibiotics as well as leads to increased prevalence among clinical and environmental strains.
Boosted Antibiotic Resistance via Mutations
Intrinsic resistance genes are negatively or positively regulated by one or more regulatory mechanisms which confer a basal lower susceptibility of P. aeruginosa to a narrow spectrum of antibiotics. However, mutation in regulatory pathway could increase promoter activities resulting in unleashing gene expression and overproduction of protein products such as AmpC and multi-drug efflux pumps systems. Consequently, it causes higher level of resistance to antibiotics (Blair et al., 2015; Figure 3).
As a common mutational feature of P. aeruginosa isolates, resistant clinical mutants display a constitutive high level of AmpC production even in the absence of antibiotic inducers. This is mainly due to mutational inactivation of ampD (repressor of ampC) and specific point mutations of ampR, both encoding two regulatory proteins important in induction of the ampC gene (Juan et al., 2005; Figure 3). Consequently, it turns into a major cause of greater resistance to a wide range of antibiotics such as most of the β-lactams (e.g., ticarcillin and piperacillin) as well as monobactams, third-generation and fourth-generation cephalosporins (Lister et al., 2009; Berrazeg et al., 2015). One study showed that 73% of tested clinical strains showed AmpC overproduction (Henrichfreise et al., 2007).
Several regulatory loci such as mexR, nalD, nalB, and nalC negatively control the expression of the mexAB-oprM operon in P. aeruginosa. On the other hand, various loss-of-function mutations in these loci derepress the expression of the mexAB-oprM operon leading to the overproduction of MexAB-OprM complex conferring a greater resistance to carbapenem antibiotics (Quale et al., 2006; Lister et al., 2009; Kao et al., 2016; Figure 3). Likewise, overproduction of other multidrug efflux pumps such as MexXY and MexCD-OprJ can occur via mutations in regulatory loci leading to unleashing gene expression and a greater resistance to a variety of antimicrobial agents (Lister et al., 2009; Figure 3).
Another clinically important and prevalent mutational alteration is attributed to OprD porin channel. This porin channel is localized in the outer membrane of P. aeruginosa and it is characterized as a carbapenem-specific porin (Figure 3). Therefore, loss or reduction of OprD can reduce permeability of the outer membrane to carbapenems (Epp et al., 2001; Gutiérrez et al., 2007; Kao et al., 2016). The emergence of resistance to imipenem and reduced susceptibility to meropenem has been reported upon the occurrence of oprD mutations. Genetic alteration in oprD can occur via nucleotide insertion or deletion and point mutations resulting in frameshift of the gene sequence, amino acid substitution, shortened putative loop L7 and premature stop codons (Kao et al., 2016). Furthermore, downregulation of oprD expression can be mediated by other regulatory factors such as MexT which itself concurrently upregulates mexEF-oprN expression (Köhler et al., 1997; Ochs et al., 1999).
Additionally, fluoroquinolone resistance among P. aeruginosa isolates can be mediated by either mutational changes within the fluoroquinolone targets i.e., DNA gyrase (gyrA and gyrB) and/or topoisomerase IV (parC and parE) or overproduction of active or inducible efflux pumps (Lee et al., 2005; Sun et al., 2014; Figure 3).
Plasmid-Mediated Resistance
Bacterial plasmids serve a central role as a potent vehicle for acquiring resistance genes and subsequent delivery to recipient host. This is so-called horizontal gene transfer whereby genetic elements can be transferred between bacterial cells particularly via conjugation. Some resistance plasmids are broad host range which can be transferred among various species via bacterial conjugation, while narrow host range plasmids are transferred among a small number of cells from similar bacterial species. For example, plasmid RP1 can transfer resistance genes to most Gram-negative bacteria (Kenward et al., 1978).
Plasmid-encoded antibiotic resistance confers resistance to different classes of antibiotics that are currently applied in frontline of clinical treatments such as β-lactams, fluoroquinolones and aminoglycosides (Bennett, 2008; Figure 3). So far, P. aeruginosa resistance via horizontal gene transfer has been reported for the genes encoding β-lactam-hydrolyzing enzymes known as the extended-spectrum β-lactamases and the carbapenemases, aminoglycoside-modifying enzymes, 16S rRNA methylases resulting in high-level pan-aminoglycoside resistance (Poole, 2011).
The genes encoding extended-spectrum β-lactamases and carbapenemase are clinically important not only due to their hydrolyzing activity on a wide range of β-lactams such as carbapenems and extended-spectrum cephalosporins, but also for their worldwide prevalence (Paterson and Bonomo, 2005; Blair et al., 2015; Sullivan et al., 2016). The global epidemiology of carbapenem-resistant P. aeruginosa was recently analyzed by Hong et al (Hong et al., 2015). They reported that the geographical prevalence of these genes differs from country to country, whereas the genes encoding carbapenemases such as IMP, VIM, and NDM type metallo-β-lactamases have been found in all continents (Johnson and Woodford, 2013; Meletis and Bagkeri, 2013; Hong et al., 2015). Almost all types of transferable carbapenemases, except SIM-1, have been detected in P. aeruginosa, and the prevalence of carbapenem-resistant isolates of P. aeruginosa is gradually increasing (Meletis and Bagkeri, 2013; Hong et al., 2015).
It is of concern that transferable plasmids carrying some of the resistance genes are mobile among a wide range of unrelated Gram-negative bacteria which increases the antimicrobial resistance transfer rate causing increasing treatment complications (Hong et al., 2015). Recent findings about antibiotic resistance have been even more concerning and warning. Liu et al. reported the first evidence of plasmid-mediated colistin resistance from China (Liu et al., 2016; Figure 3). Colistin (or polymyxin E) belongs to the family of polymyxins. The members of this class of antibiotics such as polymyxin B and colistin have been the last resort for antibiotic treatment of carbapenem-resistant bacteria such as P. aeruginosa isolates and Enterobacteriaceae (Falagas and Kasiakou, 2005). Resistance to polymyxins was previously reported to occur via chromosomal mutations (Moskowitz et al., 2012; Gutu et al., 2013), however, new evidence suggests plasmid-mediated resistance through the mobilization of the mcr-1 gene which consequently confers resistance to colistin (Figure 3). This gene was discovered in E. coli strain SHP45 collected from agricultural products. It is more concerning that the plasmid carrying mcr-1 was mobilized into K. pneumoniae and P. aeruginosa via conjugation (Liu et al., 2016). This finding has triggered serious concerns about the emergence of pan-drug-resistant Gram-negative bacteria leading to almost untreatable infections. Recent findings provided some evidence of the spreading high-risk of clone ST654 of P. aeruginosa containing the genomic blaNDM−1 resistance gene which also conferred resistance to colistin. It is likely that blaNDM−1 was acquired via genetic exchange between P. aeruginosa and K. pneumoniae isolate in the same patient (Mataseje et al., 2016).
Adaptive Resistance Mechanisms
Compared to other types of resistance mechanisms, adaptive mechanisms are not really well understood. Adaptive resistance is an unstable and transient form of resistance, which is induced in the presence of specific antibiotics and other environmental stresses. This type of resistance mainly relies on induced alterations in gene expression and protein production or alterations in antibiotic targets and it is reversal upon removal of external stimuli leading to re-gaining susceptibility (Barclay et al., 1992; Xiong et al., 1996; Fernández et al., 2011). This mechanism has been seen mediating the resistance of P. aeruginosa isolates to β-lactams, aminoglycosides, polymyxins and fluoroquinolones (Zhang et al., 2001; Poole, 2005; Fernández et al., 2010; Khaledi et al., 2016).
It has been seen that once strains encounters certain concentrations of antibiotics, they can tolerate higher concentrations in subsequent exposures, while cross-resistance to other antibiotics may occur as well (Mouneimné et al., 1999; Fujimura et al., 2009; Fernández et al., 2011; Pagedar et al., 2011). Furthermore, these alterations may link to other physiological events triggered by other stimuli and stresses as well as mutations in some specific genes (Xiong et al., 1996; Karlowsky et al., 1997; Fernández et al., 2011).
Using isolates from CF patients, it was shown that adaptive resistance of P. aeruginosa to fluoroquinolones such as ciprofloxacin is due to multiple mutations in the known-resistance genes including the gyrA, gyrB, nfxB, and orfN which were concomitant with mutations in the genes involved in cyclic di-GMP signaling (Figure 3). Mutations of nfxB were prevalent leading to loss of function of NfxB transcriptional repressor and consequently leading to the overproduction of MexCD-OprJ efflux pump (Wong et al., 2012; Figure 3). This efflux pump is an important determinant of resistance to fluoroquinolone antibiotics (Hirai et al., 1987). On the other hand, another study showed that expression of the mexCD-oprJ genes depends on the sigma factor AlgU and leads to resistance to the biocide chlorhexidine (Fraud et al., 2008). AlgU is well-known stress response sigma factor which positively regulates overproduction of alginate in mucoid isolates (Hershberger et al., 1995).
Another group showed that P. aeruginosa can acquire and lose resistance in the presence and absence of colistin, respectively. This occurred via adaptive multiple mutational mechanisms and genetic reversion (Lee et al., 2016). It was also demonstrated that resistance to certain polycationic antimicrobials such as aminoglycosides, polymyxins and cationic antimicrobial peptides can be mediated by altering the lipid A structure in LPS. This was caused by multiple mutations in cognate regulatory proteins such as the two-component systems PhoP-PhoQ, PmrA-PmrB, CprR-CprS, and ParR-ParS (Barrow and Kwon, 2009; Fernández et al., 2012; Figure 3). Other studies showed that further and complex genetic alterations affecting regulatory pathways including those causing amino acid substitutions in these cognate regulatory proteins such as PhoQ and PmrB are involved in polymyxin resistance. This is why the mechanism of resistance of P. aeruginosa to colistin was found to vary among isolates (Lee et al., 2016). Interestingly, this study showed that the acquisition of colistin resistance via many amino acid substitutions is reversible in colistin-susceptible revertants. However, even in the absence of colistin, resistance was preserved for some time and emergence of revertants may not occur so fast (Lee et al., 2016).
QS-Dependent Antibiotic Resistance
Some direct and indirect evidences have been found linking the QS systems with antibiotic resistance mechanisms in P. aeruginosa (Rasamiravaka and El Jaziri, 2016), but further exploration is needed for better understanding. Using clinical strains of P. aeruginosa, it was shown that the las system positively links to the expression of mexY gene encoding the inner-membrane drug/H+ antiporter protein MexY (Pourmand et al., 2015) which is a key subunit of the MexXY-oprM complex known as a major determinant of aminoglycoside resistance (Lau et al., 2014). On the other hand, some studies showed that CF-infecting strains with the common lasR loss-of-function mutations were more resistant to therapeutic antibiotics such as tobramycin, ciprofloxacin and ceftazidime. The reported antibiotic resistances in the lasR mutants were attributed to increased β-lactamase activity, bacterial metabolic adaptation or metabolic shifts (D'Argenio et al., 2007; Hoffman et al., 2010). However, the relationship of antibiotics susceptibility with the rhl encoded QS system and production of C4-HSL signals remains unclear (Bjarnsholt et al., 2010). Some supporting evidence was obtained by, treating P. aeruginosa biofilms with ciprofloxacin which upregulated the production and secretion of the virulent factor LasB, which is under the control of Rhl QS system (Oldak and Trafny, 2005; Figure 1).
Furthermore, two independent studies reported that the clinical strains of P. aeruginosa with QS-deficient phenotypes and negative for the production of QS-dependent virulence factors could cause infections and tend to be less susceptible to antimicrobial agents (Karatuna and Yagci, 2010; Wang H. et al., 2013). However, it was not shown how these mechanisms might link to each other while many of these clinical strains could also form biofilms with antibiotic resistance traits and many regulatory pathways for biofilm development are under the control of QS systems. Zhao et al. reported some supporting information showing the importance of QS systems in both biofilm formation and antimicrobials induced expression of ampC (Zhao et al., 2015). Earlier studies showed that by overexpressing the chromosomal type 1 β-lactamase, QS-dependent virulence factors were reduced and strains were less virulent (Ramisse et al., 2000). Also, Kong et al. analyzed the dual role of the AmpR transcriptional regulator where it positively regulated β-lactamases and negatively regulated the virulence factors through QS systems (Kong et al., 2005).
Balasubramanian et al. analyzed co-regulatory and transcriptional networks of three co-existing mechanisms involved in β-lactam resistance, alginate production and modulation of virulence factor expression. They showed that while AmpR positively and negatively regulates β-lactamases and QS-dependent proteases, respectively, there is an intimate crosstalk between the AmpR regulon and the master regulator AlgU which positively regulates alginate production (Balasubramanian et al., 2011). This gave more insight into the complexity of such co-existing networks. Recent findings also showed that high levels of cyclic di-GMP mediated by the SagS regulator contributes to elevated antibiotic resistance via BrlR regulon-dependent upregulation of cognate genes encoding MexAB-OprM and MexEF-OprN multidrug efflux pumps (Gupta et al., 2014).
The periplasmic TpbA tyrosine phosphatase was also reported as a regulatory candidate for linking QS signaling and biofilm formation. This protein was shown to be positively regulated by the las QS system at transcriptional level. Upon production of TpbA and its phosphatase activity in the periplasm, the cyclic di-GMP synthesizing protein TpbB is dephosphorylated at a tyrosine residue in periplasmic domain leading to inactivation of TpbB and a reduction in cyclic di-GMP levels and in turn Pel production, hence inhibiting biofilm development (Ueda and Wood, 2009). TpbA-dependent cyclic di-GMP reduction was also linked to increasing eDNA release by cell lysis (Ueda and Wood, 2010).
Overall, the reason for inconclusive information about the relation of the QS system and antibiotic resistance mechanisms is based on the fact that there are various layers of regulatory pathways associated with both QS systems and antibiotic resistance mechanisms in P. aeruginosa. Therefore, understanding the interplay between hierarchical QS systems and various antibiotic resistance mechanisms needs further exploration.
Adaptive Radiation for Persistence
Adaptation to the surrounding environments is an extraordinary capability of P. aeruginosa. It enables P. aeruginosa to inhabit diverse ecological niches such as colonization of various hosts as well as long term persisting infections. The adaptation process is designated as adaptive radiation by which initial clones would diversify into a variety of genotypes and phenotypes over time until the most favorable and adapted descendants are selected for long term persistence (Hogardt and Heesemann, 2010). A typical example is adaptation of P. aeruginosa isolates to the CF airways. Various studies have shown that initial colonization of CF lungs is caused by wild-type strains existing in the environment. For bacteria, the CF lungs encompass various stresses such as oxidative stresses and immune responses and inter-species competition followed by antibiotic treatment. Therefore, initial clones undergo substantial adaptation processes to survive such hostile environments.
Here, adaptive radiation is mainly due to intense genetic adaptations leading to the thousands of generations displaying diverse genotypes and phenotypes that emerge in vivo, while subjected to selection pressure imposed by the CF lung milieu (Figure 4). Therefore, selected variants display different genotypes when compared with initial wild-type colonizers and persist in the CF lungs leading to clonal expansion within patients and establishment of chronic infections (Mathee et al., 1999; Kong et al., 2005; Boles and Singh, 2008; Driffield et al., 2008; Workentine et al., 2013). By assessing a wide selection of phenotypes, Workentine et al. showed that the overall population structure in one chronically infected patient can be much more heterogeneous in phenotypes than what has been previously documented (Workentine et al., 2013). Furthermore, it has been reported that transmission of strains from patient to patient can result in the coexistence of highly divergent bacterial lineages (Winstanley et al., 2016).
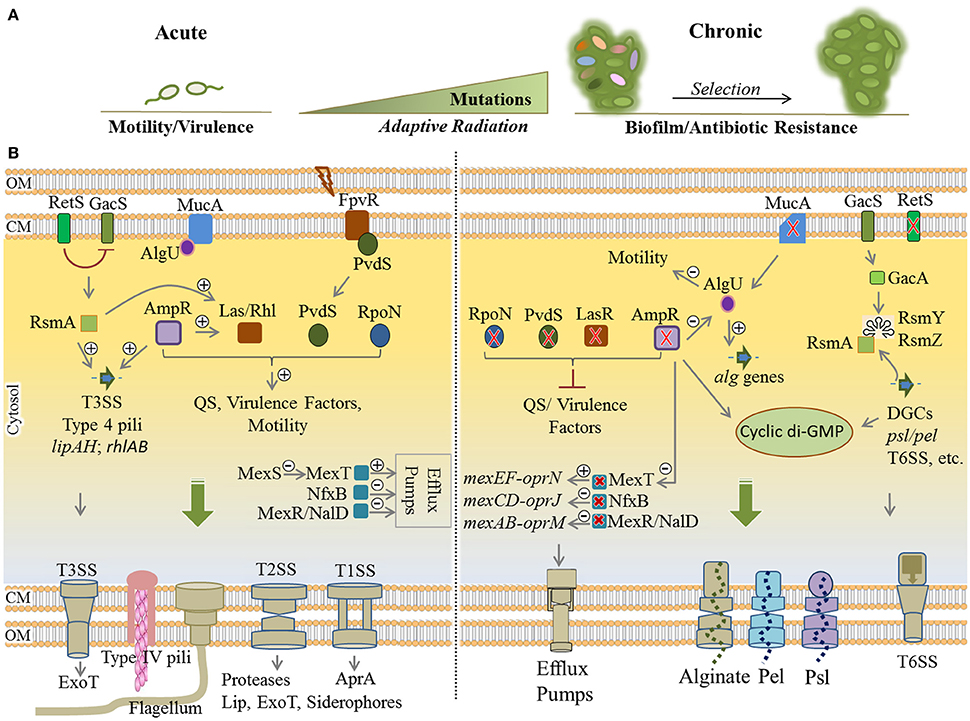
Figure 4. Remodeling of regulatory networks in P. aeruginosa during adaptive radiation and transition from acute to chronic infections. (A) During pathogenesis, adaptation to the CF lung environment occurs through “adaptive radiation” where intense genetic mutations lead to diverse genotypes and phenotypes (colorful ellipsoids) within bacterial populations followed by the selection of colonizers. Mutational adaptation and selection of generations drive bacterial transition from acute to chronic traits. (B) Remodeling of key regulatory networks between acute and chronic infections occurs mainly via mutational adaptation in cognate genes. Mutated lasR, ampR, and retS genes are key determinants in this process by which QS, virulence factor production and motility are downregulated, while synthesis of cyclic di-GMP, exopolysaccharides and various multidrug efflux pumps are upregulated. Mutation of mucA results in a defect in MucA (anti-sigma factor) releasing AlgU (positive regulator of alginate operon) that induces overproduction of alginate and the mucoid phenotype. Of important acute traits are flagellum, type 4 pili, T1SS, T2SS, T3SS (types 1 to 3 secretion systems), ExoT (exotoxins), Lip (lipases), AprA (alkaline proteases). The type 6 secretion system (biofilm-associated and toxin-delivering device to other bacteria) and efflux pumps and the production of EPS are part of chronic traits which confer antibiotic resistance and/or mediate biofilm formation. Plus and minus signs represent positive and negative effect of transcriptional regulators, respectively. Red cross indicates mutagenesis. CM, cytoplasmic membrane; OM, outer membrane.
Generally, phenotypic adaptation of these strains include slow growth, auxotrophy, virulence deficiency via downregulation of QS systems, loss of motility, biofilm formation, alginate overproduction and mucoid phenotype, antibiotic resistance, hypermutability, and lipopolysaccharide modifications. Downregulation of virulence factors such as flagella motility, T2SS/T3SS apparatus, and toxic components results in less inflammatory and phagocytic responses since the pathogen is less detectable for the immune system (Mahenthiralingam et al., 1994; Hogardt and Heesemann, 2010). Analysis of many clinical isolates showed these alterations represent convergent molecular evolution among many clinical isolates and mutation of 52 genes are mainly responsible for substantial phenotypic alterations associated with virulence traits and resistance (Diaz Caballero et al., 2015; Marvig et al., 2015). Of these genes, common adaptive mutations occur in regulatory genes including lasR, pvdS, rpoN, mucA, mexT, nfxB, mexR, nalD, retS, and ampR (Figure 4). Collectively, this leads to remodeling regulatory networks and developing a general adaptation pattern as explained below (Higgins et al., 2003; Hogardt and Heesemann, 2010; Rau et al., 2010; Winstanley et al., 2016).
Mutation of lasR, pvdS, and rpoN impairs central QS system signal processing leading to the deficiency in virulence traits. In wild-type strains, the LasR and PvdS regulators control the expression of a large number of genes including key virulence factors (Table 1, Figure 4) and pyoverdine for iron acquisition, respectively (Table 1; Hoffman et al., 2009; Imperi et al., 2010; Jiricny et al., 2014; LaFayette et al., 2015). The alternative sigma factor RpoN has been also found to regulate many cell functions such as motility and virulence factors production via QS system (Cai et al., 2015; Figure 4). Additionally, knocking out mutation of mucA locus encoding anti-sigma factor MucA results in releasing the RNA polymerase sigma factor σ22 (AlgU) which itself positively regulates expression of the alginate biosynthesis operon and stress response mechanisms while it negatively regulates several virulence factors such as flagella, pili, T3SS, and Rhl quorum sensing signals (Folkesson et al., 2012). Development of the mucoid phenotype mediated by alginate overproduction as well as the formation of highly structured biofilms is the hallmark of chronic infections (Schurr et al., 1996; Hay I. D. et al., 2013; Figure 4).
Another common mutation has been reported in the mexT locus resulting in activation of MexT, the positive regulator of MexEF-OprN efflux pump, which in turn led to antibiotic resistance. In addition, the induction of MexEF-oprN production is linked to extruding QS signaling molecules and reduction of virulence factor production (Tian et al., 2009; Figure 4). The MexEF-OprN production is undetectable in wild-type strains due to the non-functionality of the mexT gene (Maseda et al., 2000, 2010). Furthermore, mutations of repressor genes nfxB and mexR/nalD in clinical strains upregulated the production of the MexCD-OprJ and MexAB-OprM efflux pumps, respectively, conferring resistance to a wider range of antibiotics (Higgins et al., 2003; Sobel et al., 2005; Jeannot et al., 2008; Rau et al., 2010; Figure 4). Also, mutation of other genes such as gyrA/gyrB (DNA gyrase), mexZ (transcriptional regulator of the mexXY) and mexS (transcriptional regulator of the mexEF) are commonly attributed to antibiotic resistance during mutational adaptations (Marvig et al., 2015; Figure 4).
As part of RetS/GacS/GacA/RsmA regulatory pathway, the retS gene is important for phenotypic shifting from acute to chronic infections (Lapouge et al., 2008; Moscoso et al., 2011). The retS mutation repressed the production of virulence factors such as T3SS and swarming motility while it upregulated production of the T6SS (type 6 secretion system) and exopolysaccharides Pel/Psl required for biofilm formation (Moscoso et al., 2011; Figure 4). The T6SS is a puncturing device for delivery of proteins and toxins into the competing bacteria and the host cells and an important survival advantage for P. aeruginosa. It is also required for biofilm formation while being considered as a virulence factor (Chen et al., 2015). This transitional impact was shown to be mediated by high levels of cyclic di-GMP (Boehm et al., 2010; Paul et al., 2010; Moscoso et al., 2011).
Mutation of the ampR gene is another common mutational adaptation with a large impact on remodeling of physiological traits (Figure 4). The AmpR global regulator in P. aeruginosa regulates not only resistance to different classes of clinically relevant antibiotics, but also expression of hundreds of genes involved in diverse physiological processes such as virulence, QS systems and stress responses (Balasubramanian et al., 2015). It is understood that the ampR mutation induces adaptations leading to chronic infection including the downregulation of stress responses and virulence factors via downregulating QS systems, and boosting biofilm formation and alginate overproduction by causing elevation of cyclic di-GMP levels. Additionally, it induced AlgU activity, and resistance to fluoroquinolone through activation of MexT upregulating the MexEF-OprN efflux pump as well as increasing twitching motility and T6SS production (Balasubramanian et al., 2011, 2012, 2014, 2015; Figure 4).
Other adaptive mutations of CF isolates have been commonly reported in anti-mutator genes including mutS, mutT, mutL, mutY, mutM, and uvrD conferring a “hypermutability phenotype” with elevated mutation rates due to the lack of DNA repair mechanisms. This phenotype has been described as being caused by later mutational events as they are understood to occur after mutation of the lasR and mucA genes known as earlier mutations. However, other reports postulated that mutation of anti-mutator loci may increase the rate of other adaptive mutations (Oliver and Mena, 2010).
Hypermutators are very prevalent in CF isolates and they are shown to have correlation with higher antibiotic resistance particularly in the late stage of chronic infections. However, hypermutators also display other phenotypes such as mucoidity, lack of motility and LPS production (Oliver et al., 2000; Ciofu et al., 2010; Varga et al., 2015).
Other distinct phenotypes correlated with adaptation to CF airways are the small-colony variants (SCVs). They are associated with prolonged persistence and chronic infections in CF lungs and obstructive pulmonary diseases (Malone, 2015). They have been characterized as variants forming rugose small colonies on solid media (1–3 mm in diameter) with slow growing, autoaggregative and enhanced biofilm formation characteristics combined with enhanced surface attachment and hyperpiliation for twitching (Häussler et al., 1999, 2003; Kirisits et al., 2005). In vitro analyses showed that the SCVs display increased resistance to a wide range of antibiotics (Wei et al., 2011). Different studies have demonstrated that the presence of SCVs in the CF lung is associated with poorer lung function and clinical outcomes (Häussler et al., 1999, 2003; Schneider et al., 2008).
It has been understood that SCVs show high levels of cyclic di-GMP production aligned with increased production of Pel and Psl exopolysaccharides (Starkey et al., 2009). So far investigations regarding the molecular mechanisms underlying SCVs formation confirmed loss-of-function mutations in regulatory genes such as wspF, yfiR and rsmA and some other genes which alter regulatory networks in favor of enhanced cyclic di-GMP production (Irie et al., 2010; Malone et al., 2012; Blanka et al., 2015; Malone, 2015). On the other hand, an upregulating cyclic di-GMP synthesis pathway is a key determinant of exopolysaccharide production leading to highly developed biofilms. However, it still remains unclear how two distinct phenotypes i.e., cell within mucoid biofilm and SCV differ in regard to the cyclic di-GMP mediated signaling pathways.
Survival by Stringent Response and Persister Formation
Stringent response to environmental stresses such as nutritional starvation and response to antibiotics and oxidative stresses share a similar outcome of adaptation i.e., all are leading to dormancy and persister formation. In both responses, bacteria slow down their metabolism through downregulating the expression of genes participating in the biosynthesis of proteins, ribosomes, cell wall, nucleic acid metabolism, and virulence factors. These dramatic metabolic alterations result in arresting cell growth and cell division in favor of bacterial survival (Eymann et al., 2002; Hesketh et al., 2007; Durfee et al., 2008).
Persisters are defined as subpopulations of cells, occurring at very low frequency, which stochastically emerge in the presence of stress. They show very slow growth enhancing survival under stress while viability of the majority of the population is severely impaired. Upon stress removal, persisters turn back to normal growth to propagate, which coincides with regained sensitivity to stress. Such persistence was suggested to be based on the heterogeneity of population by means of epigenetic mechanisms, not genetic mutations (Fasani and Savageau, 2015).
Various studies have provided evidences showing the link between stringent response and persistence, but mostly using E. coli as a model which can be informative for P. aeruginosa as it possesses homologous signaling pathways (Fung et al., 2010; Maisonneuve et al., 2013; Amato et al., 2014; Ramisetty et al., 2016). There are only a few studies aiming to explain such responses, but they provided inconclusive explanations. Therefore, we summarized the general findings in order to propose the underlying molecular mechanisms in P. aeruginosa.
Molecular Mechanisms Underlying Stringent Responses and Persisters
Notably, increased levels of (p)ppGpp (collectively designated for guanosine pentaphosphate and guanosine tetraphosphate) molecules in the cells is a central triggering alarmone for both persistence and stringent response (Potrykus and Cashel, 2008; Wu et al., 2010; Amato et al., 2014). The cellular levels of (p)ppGpp are mediated by the activity of the (p)ppGpp-synthesizing and degrading enzymes such as RelA and SpoT in response to external stimuli (Bremer and Dennis, 2008).
In stringent response when E. coli encounters amino acid deprivation, the ribosome-associated RelA synthesizes ppGpp molecules to an upper level. In association with the transcriptional regulator DksA (global regulator of metabolism), ppGpp interacts with RNA polymerase and inhibits the transcription of ribosomal RNA promoters. This inhibitory impact is concomitant with activation and upregulation of pathways for amino acid biosynthesis and the transcription of stress response genes (Potrykus and Cashel, 2008; Dalebroux and Swanson, 2012; Amato et al., 2014). Amato et al. (2013) found that stringent responses are linked to the emergence of persisters by involving ppGpp based regulatory events (Amato et al., 2013).
The persister state is typically based on the activity of genetically encoded toxin-antitoxin (TA) modules particularly in response to antibiotics, but proposed as being activated by the same acting elements i.e., RelA or SpoT and (p)ppGpp in E. coli (Maisonneuve et al., 2013). These TA system are widely distributed in genomes or plasmids of bacteria and archaea (Van Melderen, 2010). Basically, the toxin element is a stable protein while the cognate antitoxin element either is a protein or a small RNA molecule which are metabolically unstable under unfavorable conditions. A small RNA antitoxin directly inhibits the toxin translation by pairing with toxin mRNA or inactivate toxin by direct binding. A protein antitoxin on the other hand either degrades toxin mRNA or blocks the activity of cognate protein toxin via direct protein-protein interaction or protective interaction with toxin substrates. Impairment of antitoxin function under certain stresses such as antibiotics as well as nutritional stresses leads to the accumulation and activation of toxin proteins. Consequently, the toxin targets and interferes with key cellular processes such as DNA replication, and the synthesis of tRNA, membrane components, and ATP leading to the inhibition of cell growth and cell division to form a dormant or persister cell (Christensen-Dalsgaard et al., 2010; Wen et al., 2014). Presumably, bacteria switch off their active metabolism upon exposure to stress to evade starvation or antibiotic impact on their cellular targets. Christensen et al. demonstrated that E. coli encodes at least 10 TA loci and all of which could be induced by nutritional starvation and antibiotics (Christensen et al., 2001; Christensen-Dalsgaard et al., 2010).
So far, Maisonneuve et al. (2013) have proposed the best model presenting the hierarchical molecular mechanisms for E. coli persistence with the involvement of the key players including RelA/SpoT enzymes, (p)ppGpp signaling, Lon protease (ATP-dependent protease), inorganic polyphosphate (PolyP), and toxin-antitoxins systems (Maisonneuve et al., 2013; Figure 5). This model explains that (p)ppGpp synthesized by RelA/SpoT inhibits PolyP degradation, while PolyP accumulation stimulates Lon protease to degrade the antitoxin leading to the activation of toxin for arresting cellular processes and growth. Previously, it was shown in E. coli that in response to nutritional stress and antibiotics, transcriptional activation of TA loci depended on protease activity of Lon protease on antitoxin which itself repressed the TA promoter (Christensen-Dalsgaard et al., 2010). Furthermore, it was suggested that the complex of Lon protease with PolyP could promote ribosomal protein degradation for supplying required amino acids during starvation (Kuroda et al., 2001; Figure 5).
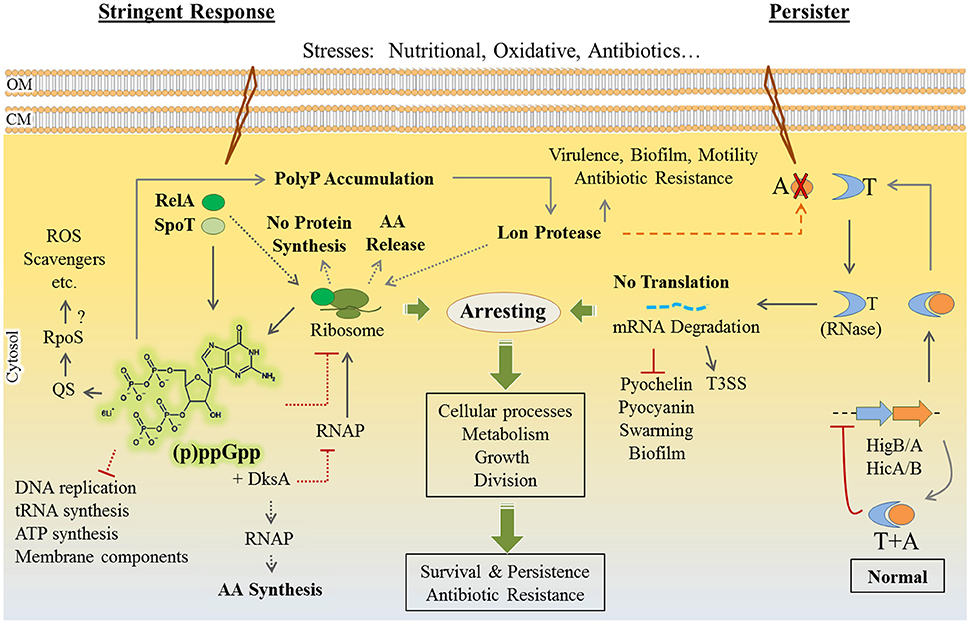
Figure 5. P. aeruginosa stringent response and persister formation. Stringent response is triggered by particular stresses such as amino acid and fatty acid starvation, iron/phosphor depletion and oxidative stress [e.g., reactive oxygen species (ROS)]. The (p)ppGpp alarmone is a key determinant for stringent response and it is elevated by RelA/SpoT enzymes. Generally, (p)ppGpp elevation and the PolyP (inorganic polyphosphate) and Lon protease complex interfere with normal biological processes in favor of bacterial survival via arresting metabolism, cell growth and cell division (dashed gray pathways are best understood for the E. coli model, but not or partially characterized in P. aeruginosa). In E.coli, (p)ppGpp signaling is linked to toxin (T)-antitoxin (A) system via activation of the Lon protease leading to the formation of persisters displaying dormant and antibiotic resistance phenotypes (dashed orange line). Generally, the TA complex is stable under normal conditions suppressing toxin activity and further expression of cognate genes. Upon antitoxin degradation, toxin becomes active to hinder biological processes. In the case of P. aeruginosa HigB/A, HicA/B, the toxin components perform endoribonuclease (RNase) activity on mRNA molecules. In P. aeruginosa, the (p)ppGpp alarmone is linked to the production of ROS scavengers probably via QS or RpoS regulators and Lon activity is required for biofilm formation, motility, virulence and antibiotic resistance. Furthermore, the TA system downregulates biofilm formation and virulence factor production while T3SS (type 3 secretion system) can be found upregulated. Although, the (p)ppGpp signaling, Lon protease activity and TA modules (i.e., HigB/A, HicA/B, and likely more complexes) are present in P. aeruginosa, their link to resistance to antibiotics and other stresses is poorly understood. AA, amino acids; QS, quorum sensing; RNAP, RNA polymerase. CM, cytoplasmic membrane; OM, outer membrane.
P. aeruginosa Stringent Response and Persisters
Required elements of stringent response and persistence including the ppGpp alarmone, SpoT, RelA, DksA, and the TA modules have been characterized in P. aeruginosa (Figure 5). Mutants deficient in relA and/or spoT genes showed increased sensitivity to heat shock, oxidative and osmotic stresses as well as antibiotics while becoming less virulent. Stringent response and RelA/SpoT activity for production of ppGpp were found to be crucial for regulation of virulence factor production (Erickson et al., 2004; Viducic et al., 2006; Boes et al., 2008; Nguyen et al., 2011; Vogt et al., 2011). Also, there is some experimental support that P. aeruginosa utilizes stringent response to protect cells from oxidative stresses generated by toxic reactive oxygen species (ROS) under aerobic conditions. The mutants deficient in relA and spoT genes were highly susceptible to multiple oxidants. This study showed that (p)ppGpp signaling is necessary for optimal expression of catalase and superoxide dismutase enzymes as major ROS scavengers, but it was assumed to be indirectly regulated through a complex regulatory network (Sampathkumar et al., 2016; Figure 5). Because, (p)ppGpp signaling is also required for full expression of other regulatory pathways controlling antioxidant response such as the stress response regulator RpoS as well as both Las and Rhl QS systems (van Delden et al., 2001; Kohanski et al., 2008; Schafhauser et al., 2014; Sampathkumar et al., 2016; Figure 5). The proposed E. coli model for the activation of the TA system via Lon protease has not been shown for P. aeruginosa, yet. However, different studies indicated that Lon protease activity was induced by aminoglycosides and lon mutants were highly susceptible to ciprofloxacin while lon was also required for biofilm formation, motility and virulence (Marr et al., 2007; Breidenstein et al., 2012). Molecular mechanism of these pathways have not been well characterized, but as proposed previously, interconnection of various regulatory and signaling pathways for appropriate responses leading to either as stringent/persistence, biofilm formation or virulence is anticipated (Kim et al., 1998).
To date five types (I-V) of TA systems have been described in bacteria based on the nature and mode of action of antitoxin (Wang et al., 2012). So far, HigB/HigA and HicA/HicB TA modules encoded by genomic loci have been experimentally demonstrated in P. aeruginosa, while other TA systems such as the relBE and parDE loci were predicted but they have not been characterized, yet (Pandey and Gerdes, 2005; Fernández-García et al., 2016; Figure 5). The HigB/HigA and HicA/HicB TA modules have been also widely reported for other bacteria (Pandey and Gerdes, 2005; Li G. et al., 2016; Wood and Wood, 2016).
These TA modules belong to the type II TA system where both toxin (i.e., HigB/HicA) and antitoxin (i.e., HigA/HicB) are proteins directly interacting with each other retaining the toxin inactivated, such as inhibiting the RNase activity of HigB or HicA (Christensen et al., 2001; Rocker and Meinhart, 2016; Figure 5). The HigB/HigA TA module was found to influence P. aeruginosa pathogenicity where toxin HigB was shown to reduce the production of the virulence factors pyochelin, pyocyanin, swarming, and impaired biofilm formation representing a novel function for a TA systems (Wood and Wood, 2016; Figure 5). Another study showed that the HigB/HigA TA module was necessary for the ciprofloxacin induced persister formation by P. aeruginosa. Concurrently, HigB overproduction upregulated the expression of T3SS genes leading to the accumulation of T3SS proteins in persisters as well as increasing bacterial cytotoxicity against host immune cells (Li M.et al., 2016; Figure 5).
Furthermore, these TA systems have been shown to be highly prevalent in the clinical strains (Pandey and Gerdes, 2005; Williams et al., 2011; Li G. et al., 2016). It is believed that persisters are one of the main reasons for recurring and chronic infections where persisters withstand antibiotic treatments and spawn new infecting population upon removal of antibiotic treatment (Lewis, 2007; Wang and Wood, 2011). They are abundant in P. aeruginosa biofilms which is the hallmark of long-term infections particularly in CF patients (Lewis, 2008; Mulcahy et al., 2010).
P. aeruginosa Resistance to Foreign DNA
Infection of bacteria with viruses or bacteriophages is a natural phenomenon which can lead to bacterial lysis. Bacteria harness various mechanisms to destroy such foreign DNAs leading to resistance. The CRISPR (clustered regularly interspaced short palindromic repeats)-Cas (CRISPR associated proteins) systems form the only adaptive immune system in prokaryotic cells which also mediates P. aeruginosa survival during viral invasions (Cady et al., 2012; Bondy-Denomy and Davidson, 2014). A CRISPR region is an array of multiple repeated sequences on the bacterial genome or a plasmid ranging from 21 to 48 bp in length and separated by 26 to 72 bp hypervariable spacers (Bhaya et al., 2011; Cady et al., 2012). The cas locus encoding Cas proteins is located in the vicinity of the CRISPR region (Bhaya et al., 2011). In principle, the molecular mechanism is based on acquisition and integration of small fragments of foreign DNAs such as derived from viruses into the spacer regions between two adjacent repeats within the CRISPR locus mediated by Cas proteins with nuclease activity. Subsequently, the CRISPR region is transcribed resulting in pre-CRISPR RNA (pre-crRNA) which undergoes hydrolysis by endoribonucleases forming small CRISPR RNAs (crRNAs). The mature crRNAs in association with a multiprotein complex known as CASCADE (CRISPR-associated complex for antiviral defense) recognizes invasive DNAs upon complementarity which results in the initiation of the cleavage of the crRNA–foreign DNA hybridization complex, mediating survival of bacteria after viral infections while protecting themselves from lysis (Brouns et al., 2008; Mojica et al., 2009; Deveau et al., 2010; Garneau et al., 2010; Bhaya et al., 2011). The CRISPR-Cas systems have been classified into three major types (I, II and III) and at least 11 subtypes (IA-F, IIA-C and IIIA-B) encoding distinct crRNA-guided surveillance complexes (Makarova et al., 2011).
A study showed that 36% of tested P. aeruginosa clinical isolates harbored CRISPR-Cas systems developing adaptive immunity against various mobile genetic elements such as temperate phages, prophages, pathogenicity island transposons which were integrated into the genome (Cady et al., 2011). Different studies demonstrated that the types I-F and I-E CRISPR-Cas systems are naturally active in P. aeruginosa isolates (Cady et al., 2012; Pawluk et al., 2014). A recent phylogenetic study revealed the existence of the type I-C CRISPR-Cas system in some isolates of P. aeruginosa (van Belkum et al., 2015).
On the other hand, the activity of CRISPR/Cas system can be inhibited by anti-CRISPR/Cas genes harbored by phages infecting P. aeruginosa which counteract the type I-F and I-E systems (Bondy-Denomy et al., 2015; Maxwell, 2016). Also it has been shown that phages producing anti-CRISPR activity are closely related to each other and with high sequence similarity to bacteriophage DMS3 (Bondy-Denomy et al., 2013). Bacteriophage DMS3 was isolated from clinical isolates of P. aeruginosa and it was shown to inhibit biofilm formation and swarming motility, and P. aeruginosa cannot develop immunity against it due to the lack of complementarity between crRNA and protospacers of DMS3 genome (Budzik et al., 2004; Zegans et al., 2009). Furthermore, the CRISPR-Cas systems show a strong correlation with antibiotic resistance/susceptibility (van Belkum et al., 2015). Additionally, the same study showed that the CRISPR-Cas systems play an important role in shaping the accessory genomes of globally distributed P. aeruginosa strains. Accessory genome is referred to highly variable regions of the genome versus a relatively invariable core genome. P. aeruginosa accessory genome varies from strain to strain, ranging from 6.9 to 18.0% of the total genome, and is mainly comprised of integrative and conjugative elements, replacement islands, prophages and phage-like elements, transposons, insertion sequences and integrons (Kung et al., 2010; Ozer et al., 2014). According to this finding the CRISPR typing with regard to the frequency of spacer integration and deletion between related strains can potentially be used for identifying the lineage of strains especially within outbreaks (van Belkum et al., 2015).
Overall, understanding of the CRISPR-Cas and anti-CRISPR-Cas systems is gradually becoming important in the context of pathogenesis and strain lineage identification. These links were highlighted by the discovery of the interaction of bacteriophage DMS3 and the type I-F CRISPR, and its impact on biofilms (Zegans et al., 2009; Palmer and Whiteley, 2011) as well as the role of different CRISPR/Cas systems on virulence and antibiotic resistance (Louwen et al., 2014). Overall, these findings suggest a more diverse function of CRISPR/Cas systems within the context of pathogenesis, requiring further in depth studies to elucidate the underlying molecular mechanisms.
Conclusions and Perspectives
For many years, P. aeruginosa has been a model organism and received much attention from scientific community to study the bacterial lifestyle and pathogenesis. It always has been of particular importance due to causing persistent infections in CF and immunocompromised patients. Nowadays, this ubiquitous bacterial pathogen is accepted worldwide as a public health risk due to its increasing prevalence in healthcare acquired infections combined with its ability to develop resistances to multiple classes of antibiotics. Over the past decade, extensive research studies have focused on these growing concerns aiming at deciphering the nature of P. aeruginosa capability and underlying molecular mechanisms applying different modes of persistence and antibiotic resistance.
In this review, we summarized several of the well characterized molecular mechanisms which enable P. aeruginosa to survive various hostile conditions such as during pathogenesis and antibiotic treatment. These mechanisms form multiple layers of physiological adaptations correlating with social behavior and lifestyle of bacteria while responding environmental stimuli. Such extraordinary adaptive capability relies on extensive numbers of regulatory or controlling factors within integrated and complex signal processing pathways. These enable bacteria perceive and process environmental cues in order orchestrate physiological changes to promote adaptation to unfavorable conditions. Many of these regulatory pathways, their cognate player, their signals and how they are integrated with global regulatory networks still remain poorly understood.
Furthermore, we highlighted key molecular pathways driving P. aeruginosa survival and persistence at different stages of pathogenesis such as QS elements for virulence traits, cyclic di-GMP signaling in biofilm formation and development of chronic traits, (p)ppGpp signaling/TA system in persister formation and various strategic adaptations for developing resistance to divers classes of antibiotics.
Recent technological advances in genomic characterization of pathogens have provided invaluable information about the dynamics of P. aeruginosa populations and their heterogeneity at different stages of pathogenesis. These results which were explained under “adaptive radiation” term emphasized that this species shows a stunning capability to become resilient during pathogenesis to withstand antibacterial treatment.
Available information indicated that sole therapy which only relies on bacteriostatic/bactericidal compounds can readily be defeated by bacterial resiliency and a management program is still required to combat infections. This program should be able to predict and evaluate physiological adaptations at each stage of infection for exerting appropriate treatments which could interfere with adaptation rather than increasing the chance of bacterial survival. A good example is improper and frequent application of antibiotics which must be avoided. Instead a comprehensive hygiene program must be applied in healthcare settings and among personnel to stop the spread of nosocomial infections specifically caused by multidrug resistance strains. Also, further research on identification of new drugs and developing new alternative prevention and treatment strategies for interfering with key regulatory pathways is needed.
At the end we suggest all efforts should consider international coordinated multidisciplinary programs with results of laboratory outputs being deposited in centralized accessible databases to expedite advances in control of infections and its implementation into clinical settings. The steadily growing concern of emerging antibiotic resistance strains in the world, would justify the set-up of such databases which then allow developing world-wide guidelines for monitoring and recording antibiotic resistance cases around the world. This should provide healthcare experts with appropriate guidelines for well managing bacterial infections and preventing the rate and spread of resistance strains.
Author Contributions
MM and BR conceived and wrote the majority of the manuscript. SG contributed biofilm and alginate related aspects to the manuscript.
Funding
The work was funded via the Massey University Research fund.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are grateful to all past and present members of the BR Research group at the Institute of Fundamental Sciences (Massey University) for their contributions to the Pseudomonas aeruginosa research.
References
Aendekerk, S., Diggle, S. P., Song, Z., Høiby, N., Cornelis, P., Williams, P., et al. (2005). The MexGHI-OpmD multidrug efflux pump controls growth, antibiotic susceptibility and virulence in Pseudomonas aeruginosa via 4-quinolone-dependent cell-to-cell communication. Microbiology 151(Pt 4), 1113–1125. doi: 10.1099/mic.0.27631-0
Alcalde-Rico, M., Hernando-Amado, S., Blanco, P., and Martínez, J. L. (2016). Multidrug efflux pumps at the crossroad between antibiotic resistance and bacterial virulence. Front. Microbiol. 7:1483. doi: 10.3389/fmicb.2016.01483
Alhede, M., Bjarnsholt, T., Jensen, P. Ø., Phipps, R. K., Moser, C., Christophersen, L., et al. (2009). Pseudomonas aeruginosa recognizes and responds aggressively to the presence of polymorphonuclear leukocytes. Microbiology 155(Pt 11), 3500–3508. doi: 10.1099/mic.0.031443-0
Allesen-Holm, M., Barken, K. B., Yang, L., Klausen, M., Webb, J. S., Kjelleberg, S., et al. (2006). A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol. Microbiol. 59, 1114–1128. doi: 10.1111/j.1365-2958.2005.05008.x
Alvarez-Ortega, C., and Harwood, C. S. (2007). Responses of Pseudomonas aeruginosa to low oxygen indicate that growth in the cystic fibrosis lung is by aerobic respiration. Mol. Microbiol. 65, 153–165. doi: 10.1111/j.1365-2958.2007.05772.x
Amato, S. M., Fazen, C. H., Henry, T. C., Mok, W. W., Orman, M. A., Sandvik, E. L., et al. (2014). The role of metabolism in bacterial persistence. Front. Microbiol. 5:70. doi: 10.3389/fmicb.2014.00070
Amato, S. M., Orman, M. A., and Brynildsen, M. P. (2013). Metabolic control of persister formation in Escherichia coli. Mol. Cell 50, 475–487. doi: 10.1016/j.molcel.2013.04.002
Amiel, E., Lovewell, R. R., O'Toole, G. A., Hogan, D. A., and Berwin, B. (2010). Pseudomonas aeruginosa evasion of phagocytosis is mediated by loss of swimming motility and is independent of flagellum expression. Infect. Immun. 78, 2937–2945. doi: 10.1128/IAI.00144-10
Avrain, L., Mertens, P., and Van Bambeke, F. (2013). RND efflux pumps in P. aeruginosa: an underestimated resistance mechanism. Antibiot. Susceptibility 26321, 26–28. Available online at: http://www.uclouvain.be/cps/ucl/doc/ir-ldri/images/Avrain-2013-1.pdf
Ayers, M., Sampaleanu, L. M., Tammam, S., Koo, J., Harvey, H., Howell, P. L., et al. (2009). PilM/N/O/P proteins form an inner membrane complex that affects the stability of the pseudomonas aeruginosa type iv pilus secretin. J. Mol. Biol. 394, 128–142. doi: 10.1016/j.jmb.2009.09.034
Bains, M., Fernández, L., and Hancock, R. E. (2012). Phosphate starvation promotes swarming motility and cytotoxicity of Pseudomonas aeruginosa. Appl. Environ. Microbiol. 78, 6762–6768. doi: 10.1128/AEM.01015-12
Bajolet-Laudinat, O., Girod-de Bentzmann, S., Tournier, J. M., Madoulet, C., Plotkowski, M. C., Chippaux, C., et al. (1994). Cytotoxicity of Pseudomonas aeruginosa internal lectin PA-I to respiratory epithelial cells in primary culture. Infect. Immun. 62, 4481–4487.
Balasubramanian, D., Kong, K. F., Jayawardena, S. R., Leal, S. M., Sautter, R. T., and Mathee, K. (2011). Co-regulation of β-lactam resistance, alginate production and quorum sensing in Pseudomonas aeruginosa. J. Med. Microbiol. 60(Pt 2), 147–156. doi: 10.1099/jmm.0.021600-0
Balasubramanian, D., Kumari, H., Jaric, M., Fernandez, M., Turner, K. H., Dove, S. L., et al. (2014). Deep sequencing analyses expands the Pseudomonas aeruginosa AmpR regulon to include small RNA-mediated regulation of iron acquisition, heat shock and oxidative stress response. Nucleic Acids Res. 42, 979–998. doi: 10.1093/nar/gkt942
Balasubramanian, D., Kumari, H., and Mathee, K. (2015). Pseudomonas aeruginosa AmpR: an acute-chronic switch regulator. Pathog. Dis. 73, 1–14. doi: 10.1111/2049-632X.12208
Balasubramanian, D., Schneper, L., Merighi, M., Smith, R., Narasimhan, G., Lory, S., et al. (2012). The regulatory repertoire of Pseudomonas aeruginosa AmpC ss-lactamase regulator AmpR includes virulence genes. PLoS ONE 7:e34067. doi: 10.1371/journal.pone.0034067
Banerjee, G., and Ray, A. K. (2016). The talking language in some major Gram-negative bacteria. Arch. Microbiol. 198, 489–499. doi: 10.1007/s00203-016-1220-x
Baraquet, C., Murakami, K., Parsek, M. R., and Harwood, C. S. (2012). The FleQ protein from Pseudomonas aeruginosa functions as both a repressor and an activator to control gene expression from the pel operon promoter in response to c-di-GMP. Nucleic Acids Res. 40, 7207–7218. doi: 10.1093/nar/gks384
Barclay, M. L., Begg, E. J., and Chambers, S. T. (1992). Adaptive resistance following single doses of gentamicin in a dynamic in vitro model. Antimicrob. Agents Chemother. 36, 1951–1957. doi: 10.1128/AAC.36.9.1951
Barken, K. B., Pamp, S. J., Yang, L., Gjermansen, M., Bertrand, J. J., Klausen, M., et al. (2008). Roles of type IV pili, flagellum-mediated motility and extracellular DNA in the formation of mature multicellular structures in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 10, 2331–2343. doi: 10.1111/j.1462-2920.2008.01658.x
Barker, A. P., Vasil, A. I., Filloux, A., Ball, G., Wilderman, P. J., and Vasil, M. L. (2004). A novel extracellular phospholipase C of Pseudomonas aeruginosa is required for phospholipid chemotaxis. Mol. Microbiol. 53, 1089–1098. doi: 10.1111/j.1365-2958.2004.04189.x
Barr, H. L., Halliday, N., Cámara, M., Barrett, D. A., Williams, P., Forrester, D. L., et al. (2015). Pseudomonas aeruginosa quorum sensing molecules correlate with clinical status in cystic fibrosis. Eur. Respir. J. 46, 1046–1054. doi: 10.1183/09031936.00225214
Barrow, K., and Kwon, D. H. (2009). Alterations in two-component regulatory systems of phoPQ and pmrAB are associated with polymyxin B resistance in clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 53, 5150–5154. doi: 10.1128/AAC.00893-09
Baumgart, A. M., Molinari, M. A., and Silveira, A. C. (2010). Prevalence of carbapenem resistant Pseudomonas aeruginosa and Acinetobacter baumannii in high complexity hospital. Braz. J. Infect. Dis. 14, 433–436. doi: 10.1590/S1413-86702010000500002
Bennett, P. M. (2008). Plasmid encoded antibiotic resistance: acquisition and transfer of antibiotic resistance genes in bacteria. Br. J. Pharmacol. 153(Suppl. 1), S347–357. doi: 10.1038/sj.bjp.0707607
Berrazeg, M., Jeannot, K., Ntsogo Enguene, V. Y., Broutin, I., Loeffert, S., Fournier, D., et al. (2015). Mutations in β-lactamase AmpC increase resistance of Pseudomonas aeruginosa isolates to antipseudomonal cephalosporins. Antimicrob. Agents Chemother. 59, 6248–6255. doi: 10.1128/AAC.00825-15
Bhaya, D., Davison, M., and Barrangou, R. (2011). CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45, 273–297. doi: 10.1146/annurev-genet-110410-132430
Billings, N., Millan, M., Caldara, M., Rusconi, R., Tarasova, Y., Stocker, R., et al. (2013). The extracellular matrix component Psl provides fast-acting antibiotic defense in Pseudomonas aeruginosa biofilms. PLoS Pathog. 9:e1003526. doi: 10.1371/journal.ppat.1003526
Bjarnsholt, T., Jensen, P. Ø., Jakobsen, T. H., Phipps, R., Nielsen, A. K., Rybtke, M. T., et al. (2010). Quorum sensing and virulence of Pseudomonas aeruginosa during lung infection of cystic fibrosis patients. PLoS ONE 5:e10115. doi: 10.1371/journal.pone.0010115
Blair, J. M., Webber, M. A., Baylay, A. J., Ogbolu, D. O., and Piddock, L. J. (2015). Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42–51. doi: 10.1038/nrmicro3380
Blanka, A., Düvel, J., Dotsch, A., Klinkert, B., Abraham, W. R., Kaever, V., et al. (2015). Constitutive production of c-di-GMP is associated with mutations in a variant of Pseudomonas aeruginosa with altered membrane composition. Sci. Signal. 8:ra36. doi: 10.1126/scisignal.2005943
Blumer, C., and Haas, D. (2000). Mechanism, regulation, and ecological role of bacterial cyanide biosynthesis. Arch. Microbiol. 173, 170–177. doi: 10.1007/s002039900127
Boehm, A., Kaiser, M., Li, H., Spangler, C., Kasper, C. A., Ackermann, M., et al. (2010). Second messenger-mediated adjustment of bacterial swimming velocity. Cell 141, 107–116. doi: 10.1016/j.cell.2010.01.018
Boes, N., Schreiber, K., and Schobert, M. (2008). SpoT-triggered stringent response controls usp gene expression in Pseudomonas aeruginosa. J. Bacteriol. 190, 7189–7199. doi: 10.1128/JB.00600-08
Boles, B. R., and Singh, P. K. (2008). Endogenous oxidative stress produces diversity and adaptability in biofilm communities. Proc. Natl. Acad. Sci. U.S.A. 105, 12503–12508. doi: 10.1073/pnas.0801499105
Boles, B. R., Thoendel, M., and Singh, P. K. (2005). Rhamnolipids mediate detachment of Pseudomonas aeruginosa from biofilms. Mol. Microbiol. 57, 1210–1223. doi: 10.1111/j.1365-2958.2005.04743.x
Bondy-Denomy, J., and Davidson, A. R. (2014). To acquire or resist: the complex biological effects of CRISPR-Cas systems. Trends Microbiol. 22, 218–225. doi: 10.1016/j.tim.2014.01.007
Bondy-Denomy, J., Garcia, B., Strum, S., Du, M. J., Rollins, M. F., Hidalgo-Reyes, Y., et al. (2015). Multiple mechanisms for CRISPR-Cas inhibition by anti-CRISPR proteins. Nature 526, 136–139. doi: 10.1038/nature15254
Bondy-Denomy, J., Pawluk, A., Maxwell, K. L., and Davidson, A. R. (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432. doi: 10.1038/nature11723
Breidenstein, E. B. M., Janot, L., Strehmel, J., Fernandez, L., Taylor, P. K., Kukavica-Ibrulj, I., et al. (2012). The Lon protease is essential for full virulence in Pseudomonas aeruginosa. PLoS ONE 7:e49123. doi: 10.1371/journal.pone.0049123
Bremer, H., and Dennis, P. P. (2008). Modulation of chemical composition and other parameters of the cell at different exponential growth rates. EcoSal Plus 3, 1–49. doi: 10.1128/ecosal.5.2.3
Britigan, B. E., Roeder, T. L., Rasmussen, G. T., Shasby, D. M., McCormick, M. L., and Cox, C. D. (1992). Interaction of the Pseudomonas aeruginosa secretory products pyocyanin and pyochelin generates hydroxyl radical and causes synergistic damage to endothelial cells - Implications for Pseudomonas associated tissue injury. J. Clin. Invest. 90, 2187–2196. doi: 10.1172/JCI116104
Brouns, S. J., Jore, M. M., Lundgren, M., Westra, E. R., Slijkhuis, R. J., Snijders, A. P., et al. (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. doi: 10.1126/science.1159689
Budzik, J. M., Rosche, W. A., Rietsch, A., and O'Toole, G. A. (2004). Isolation and characterization of a generalized transducing phage for Pseudomonas aeruginosa strains PAO1 and PA14. J. Bacteriol. 186, 3270–3273. doi: 10.1128/JB.186.10.3270-3273.2004
Burrows, L. L. (2012). Pseudomonas aeruginosa twitching motility: type IV pili in action. Annu. Rev. Microbiol. 66, 493–520. doi: 10.1146/annurev-micro-092611-150055
Byrd, M. S., Sadovskaya, I., Vinogradov, E., Lu, H., Sprinkle, A. B., Richardson, S. H., et al. (2009). Genetic and biochemical analyses of the Pseudomonas aeruginosa Psl exopolysaccharide reveal overlapping roles for polysaccharide synthesis enzymes in Psl and LPS production. Mol. Microbiol. 73, 622–638. doi: 10.1111/j.1365-2958.2009.06795.x
Cady, K. C., Bondy-Denomy, J., Heussler, G. E., Davidson, A. R., and O'Toole, G. A. (2012). The CRISPR/Cas adaptive immune system of Pseudomonas aeruginosa mediates resistance to naturally occurring and engineered phages. J. Bacteriol. 194, 5728–5738. doi: 10.1128/JB.01184-12
Cady, K. C., White, A. S., Hammond, J. H., Abendroth, M. D., Karthikeyan, R. S., Lalitha, P., et al. (2011). Prevalence, conservation and functional analysis of Yersinia and Escherichia CRISPR regions in clinical Pseudomonas aeruginosa isolates. Microbiology 157(Pt 2), 430–437. doi: 10.1099/mic.0.045732-0
Cai, Z., Liu, Y., Chen, Y., Yam, J. K., Chew, S. C., Chua, S. L., et al. (2015). RpoN regulates virulence factors of Pseudomonas aeruginosa via modulating the PqsR quorum sensing regulator. Int. J. Mol. Sci. 16, 28311–28319. doi: 10.3390/ijms161226103
Cardo, D., Horan, T., Andrus, M., Dembinski, M., Edwards, J., Peavy, G., et al. (2004). National nosocomial infections surveillance (NNIS) system report, data summary from January 1992 through June 2004, issued October 2004. Am. J. Infect. Control 32, 470–485. doi: 10.1016/j.ajic.2004.10.001
Castric, P. (1994). Influence of oxygen on the Pseudomonas aeruginosa hydrogen cyanide synthase. Curr. Microbiol. 29, 19–21. doi: 10.1007/BF01570186
Castric, P. A. (1975). Hydrogen cyanide, a secondary metabolite of Pseudomonas aeruginosa. Can. J. Microbiol. 21, 613–618. doi: 10.1139/m75-088
Castric, P. A. (1983). Hydrogen cyanide production by Pseudomonas aeruginosa at reduced oxygen levels. Can. J. Microbiol. 29, 1344–1349. doi: 10.1139/m83-209
Chemani, C., Imberty, A., de Bentzmann, S., Pierre, M., Wimmerová, M., Guery, B. P., et al. (2009). Role of LecA and LecB lectins in Pseudomonas aeruginosa-induced lung injury and effect of carbohydrate ligands. Infect. Immun. 77, 2065–2075. doi: 10.1128/IAI.01204-08
Chen, L., Zou, Y., She, P., and Wu, Y. (2015). Composition, function, and regulation of T6SS in Pseudomonas aeruginosa. Microbiol. Res. 172, 19–25. doi: 10.1016/j.micres.2015.01.004
Choi, Y., Park, H. Y., Park, S. J., Park, S. J., Kim, S. K., Ha, C., et al. (2011). Growth phase-differential quorum sensing regulation of anthranilate metabolism in Pseudomonas aeruginosa. Mol. Cells 32, 57–65. doi: 10.1007/s10059-011-2322-6
Christensen, S. K., Mikkelsen, M., Pedersen, K., and Gerdes, K. (2001). RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. U.S.A. 98, 14328–14333. doi: 10.1073/pnas.251327898
Christensen-Dalsgaard, M., Jørgensen, M. G., and Gerdes, K. (2010). Three new RelE-homologous mRNA interferases of Escherichia coli differentially induced by environmental stresses. Mol. Microbiol. 75, 333–348. doi: 10.1111/j.1365-2958.2009.06969.x
Chrzanowski, L., Lawniczak, L., and Czaczyk, K. (2012). Why do microorganisms produce rhamnolipids? World J. Microb. Biot. 28, 401–419. doi: 10.1007/s11274-011-0854-8
Chugani, S., and Greenberg, E. P. (2007). The influence of human respiratory epithelia on Pseudomonas aeruginosa gene expression. Microb. Pathog. 42, 29–35. doi: 10.1016/j.micpath.2006.10.004
Ciofu, O., Mandsberg, L. F., Bjarnsholt, T., Wassermann, T., and Hoiby, N. (2010). Genetic adaptation of Pseudomonas aeruginosa during chronic lung infection of patients with cystic fibrosis: strong and weak mutators with heterogeneous genetic backgrounds emerge in mucA and/or lasR mutants. Microbiology 156, 1108–1119. doi: 10.1099/mic.0.033993-0
Colvin, K. M., Gordon, V. D., Murakami, K., Borlee, B. R., Wozniak, D. J., Wong, G. C. L., et al. (2011). The Pel polysaccharide can serve a structural and protective role in the biofilm matrix of Pseudomonas aeruginosa. PLoS Pathog. 7:e1001264. doi: 10.1371/journal.ppat.1001264
Colvin, K. M., Irie, Y., Tart, C. S., Urbano, R., Whitney, J. C., Ryder, C., et al. (2012). The Pel and Psl polysaccharides provide Pseudomonas aeruginosa structural redundancy within the biofilm matrix. Environ. Microbiol. 14, 1913–1928. doi: 10.1111/j.1462-2920.2011.02657.x
Daddaoua, A., Fillet, S., Fernández, M., Udaondo, Z., Krell, T., and Ramos, J. L. (2012). Genes for carbon metabolism and the ToxA virulence factor in Pseudomonas aeruginosa are regulated through molecular interactions of PtxR and PtxS. PLoS ONE 7:e39390. doi: 10.1371/journal.pone.0039390
Dalebroux, Z. D., and Swanson, M. S. (2012). ppGpp: magic beyond RNA polymerase. Nat. Rev. Microbiol. 10, 203–212. doi: 10.1038/nrmicro2720
D'Argenio, D. A., Wu, M., Hoffman, L. R., Kulasekara, H. D., Déziel, E., Smith, E. E., et al. (2007). Growth phenotypes of Pseudomonas aeruginosa lasR mutants adapted to the airways of cystic fibrosis patients. Mol. Microbiol. 64, 512–533. doi: 10.1111/j.1365-2958.2007.05678.x
da Silva, L. V. R. F., Ferreira, F. D., Reis, F. J. C., de Britto, M. C. A., Levy, C. E., Clark, O., et al. (2013). Pseudomonas aeruginosa infection in patients with cystic fibrosis: scientific evidence regarding clinical impact, diagnosis, and treatment. J. Bras. Pneumol. 39, 495–512. doi: 10.1590/S1806-37132013000400015
Das, T., Kutty, S. K., Kumar, N., and Manefield, M. (2013). Pyocyanin facilitates extracellular DNA binding to Pseudomonas aeruginosa influencing cell surface properties and aggregation. PLoS ONE 8:e58299. doi: 10.1371/journal.pone.0058299
Das, T., Kutty, S. K., Tavallaie, R., Ibugo, A. I., Panchompoo, J., Sehar, S., et al. (2015). Phenazine virulence factor binding to extracellular DNA is important for Pseudomonas aeruginosa biofilm formation. Sci. Rep. 5:8398. doi: 10.1038/srep08398
Davey, M. E., Caiazza, N. C., and O'Toole, G. A. (2003). Rhamnolipid surfactant production affects biofilm architecture in Pseudomonas aeruginosa PAO1. J. Bacteriol. 185, 1027–1036. doi: 10.1128/JB.185.3.1027-1036.2003
Davies, J., and Davies, D. (2010). Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 74, 417–433. doi: 10.1128/MMBR.00016-10
Davies, J. E. (1997). Origins, acquisition and dissemination of antibiotic resistance determinants. Ciba Found. Symp. 207, 15–27; discussion 27–35.
Deveau, H., Garneau, J. E., and Moineau, S. (2010). CRISPR/Cas system and its role in phage-bacteria interactions. Annu. Rev. Microbiol. 64, 475–493. doi: 10.1146/annurev.micro.112408.134123
Diaz Caballero, J., Clark, S. T., Coburn, B., Zhang, Y., Wang, P. W., Donaldson, S. L., et al. (2015). Selective sweeps and parallel pathoadaptation drive Pseudomonas aeruginosa evolution in the cystic fibrosis lung. MBio 6, e00981–e00915. doi: 10.1128/mBio.00981-15
Diggle, S. P., Stacey, R. E., Dodd, C., Cámara, M., Williams, P., and Winzer, K. (2006). The galactophilic lectin, LecA, contributes to biofilm development in Pseudomonas aeruginosa. Environ. Microbiol. 8, 1095–1104. doi: 10.1111/j.1462-2920.2006.001001.x
Diggle, S. P., Winzer, K., Lazdunski, A., Williams, P., and Cámara, M. (2002). Advancing the quorum in Pseudomonas aeruginosa: mvat and the regulation of N-acylhomoserine lactone production and virulence gene expression. J. Bacteriol. 184, 2576–2586. doi: 10.1128/JB.184.10.2576-2586.2002
Döring, G., Conway, S. P., Heijerman, H. G., Hodson, M. E., Høiby, N., Smyth, A., et al. (2000). Antibiotic therapy against Pseudomonas aeruginosa in cystic fibrosis: a European consensus. Eur. Respir. J. 16, 749–767. doi: 10.1034/j.1399-3003.2000.16d30.x
Driffield, K., Miller, K., Bostock, J. M., O'Neill, A. J., and Chopra, I. (2008). Increased mutability of Pseudomonas aeruginosa in biofilms. J. Antimicrob. Chemother. 61, 1053–1056. doi: 10.1093/jac/dkn044
Durfee, T., Hansen, A. M., Zhi, H., Blattner, F. R., and Jin, D. J. (2008). Transcription profiling of the stringent response in Escherichia coli. J. Bacteriol. 190, 1084–1096. doi: 10.1128/JB.01092-07
Dusane, D. H., Zinjarde, S. S., Venugopalan, V. P., McLean, R. J., Weber, M. M., and Rahman, P. K. (2010). Quorum sensing: implications on rhamnolipid biosurfactant production. Biotechnol. Genet. Eng. Rev. 27, 159–184. doi: 10.1080/02648725.2010.10648149
El Amin, N., Giske, C. G., Jalal, S., Keijser, B., Kronvall, G., and Wretlind, B. (2005). Carbapenem resistance mechanisms in Pseudomonas aeruginosa: alterations of porin OprD and efflux proteins do not fully explain resistance patterns observed in clinical isolates. APMIS 113, 187–196. doi: 10.1111/j.1600-0463.2005.apm1130306.x
Epp, S. F., Köhler, T., Plésiat, P., Michéa-Hamzehpour, M., Frey, J., and Pechère, J. C. (2001). C-terminal region of Pseudomonas aeruginosa outer membrane porin OprD modulates susceptibility to meropenem. Antimicrob. Agents Chemother. 45, 1780–1787. doi: 10.1128/AAC.45.6.1780-1787.2001
Erickson, D. L., Lines, J. L., Pesci, E. C., Venturi, V., and Storey, D. G. (2004). Pseudomonas aeruginosa relA contributes to virulence in Drosophila melanogaster. Infect. Immun. 72, 5638–5645. doi: 10.1128/IAI.72.10.5638-5645.2004
Eurosurveillance Editorial Team (2013). CDC publishes report on antibiotic resistance threats in the United States for the first time. Eurosurveillance 18:5. Available online at: http://www.ecdc.europa.eu/en/activities/sciadvice/_layouts/forms/Review_DispForm.aspx?List=a3216f4c-f040-4f51-9f77-a96046dbfd72&ID=752&RootFolder=%2Fen%2Factivities%2Fsciadvice%2FLists%2FECDC%20Reviews&Web=0be238ef-4498-4962-8826-2e082a1f1639
Eymann, C., Homuth, G., Scharf, C., and Hecker, M. (2002). Bacillus subtilis functional genomics: global characterization of the stringent response by proteome and transcriptome analysis. J. Bacteriol. 184, 2500–2520. doi: 10.1128/JB.184.9.2500-2520.2002
Falagas, M. E., and Kasiakou, S. K. (2005). Colistin: the revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis. 40, 1333–1341. doi: 10.1086/429323
Fasani, R. A., and Savageau, M. A. (2015). Unrelated toxin-antitoxin systems cooperate to induce persistence. J. R. Soc. Interface 12:20150130. doi: 10.1098/rsif.2015.0130
Feng, L., Xiang, Q., Ai, Q., Wang, Z., Zhang, Y., and Lu, Q. (2016). Effects of quorum sensing systems on regulatory T cells in catheter-related Pseudomonas aeruginosa biofilm infection rat models. Mediators Inflamm. 2016:4012912. doi: 10.1155/2016/4012912
Fernández, L., Breidenstein, E. B., and Hancock, R. E. (2011). Creeping baselines and adaptive resistance to antibiotics. Drug Resist. Updat. 14, 1–21. doi: 10.1016/j.drup.2011.01.001
Fernández, L., Gooderham, W. J., Bains, M., McPhee, J. B., Wiegand, I., and Hancock, R. E. (2010). Adaptive resistance to the “last hope” antibiotics polymyxin B and colistin in Pseudomonas aeruginosa is mediated by the novel two-component regulatory system ParR-ParS. Antimicrob. Agents Chemother. 54, 3372–3382. doi: 10.1128/AAC.00242-10
Fernández, L., Jenssen, H., Bains, M., Wiegand, I., Gooderham, W. J., and Hancock, R. E. (2012). The two-component system CprRS senses cationic peptides and triggers adaptive resistance in Pseudomonas aeruginosa independently of ParRS. Antimicrob. Agents Chemother. 56, 6212–6222. doi: 10.1128/AAC.01530-12
Fernández-García, L., Blasco, L., Lopez, M., Bou, G., García-Contreras, R., Wood, T., et al. (2016). Toxin-antitoxin systems in clinical pathogens. Toxins (Basel). 8:227. doi: 10.3390/toxins8070227
Flemming, H. C., and Wingender, J. (2010). The biofilm matrix. Nat. Rev. Microbiol. 8, 623–633. doi: 10.1038/nrmicro2415
Flume, P. A., and Van Devanter, D. R. (2012). State of progress in treating cystic fibrosis respiratory disease. BMC Med. 10:88. doi: 10.1186/1741-7015-10-88
Folkesson, A., Jelsbak, L., Yang, L., Johansen, H. K., Ciofu, O., Høiby, N., et al. (2012). Adaptation of Pseudomonas aeruginosa to the cystic fibrosis airway: an evolutionary perspective. Nat. Rev. Microbiol. 10, 841–851. doi: 10.1038/nrmicro2907
Franklin, M. J., Nivens, D. E., Weadge, J. T., and Howell, P. L. (2011). Biosynthesis of the Pseudomonas aeruginosa extracellular polysaccharides, alginate, Pel, and Psl. Front. Microbiol. 2:167. doi: 10.3389/fmicb.2011.00167
Fraud, S., Campigotto, A. J., Chen, Z., and Poole, K. (2008). MexCD-OprJ multidrug efflux system of Pseudomonas aeruginosa: involvement in chlorhexidine resistance and induction by membrane-damaging agents dependent upon the AlgU stress response sigma factor. Antimicrob. Agents Chemother. 52, 4478–4482. doi: 10.1128/AAC.01072-08
Frimmersdorf, E., Horatzek, S., Pelnikevich, A., Wiehlmann, L., and Schomburg, D. (2010). How Pseudomonas aeruginosa adapts to various environments: a metabolomic approach. Environ. Microbiol. 12, 1734–1747. doi: 10.1111/j.1462-2920.2010.02253.x
Frisk, A., Schurr, J. R., Wang, G., Bertucci, D. C., Marrero, L., Hwang, S. H., et al. (2004). Transcriptome analysis of Pseudomonas aeruginosa after interaction with human airway epithelial cells. Infect. Immun. 72, 5433–5438. doi: 10.1128/IAI.72.9.5433-5438.2004
Fujimura, T., Anan, N., Sugimori, G., Watanabe, T., Jinushi, Y., Yoshida, I., et al. (2009). Susceptibility of Pseudomonas aeruginosa clinical isolates in Japan to doripenem and other antipseudomonal agents. Int. J. Antimicrob. Agents 34, 523–528. doi: 10.1016/j.ijantimicag.2009.07.008
Fung, D. K., Chan, E. W., Chin, M. L., and Chan, R. C. (2010). Delineation of a bacterial starvation stress response network which can mediate antibiotic tolerance development. Antimicrob. Agents Chemother. 54, 1082–1093. doi: 10.1128/AAC.01218-09
Furukawa, S., Kuchma, S. L., and O'Toole, G. A. (2006). Keeping their options open: acute versus persistent infections. J. Bacteriol. 188, 1211–1217. doi: 10.1128/JB.188.4.1211-1217.2006
García-Contreras, R. (2016). Is quorum sensing interference a viable alternative to treat Pseudomonas aeruginosa infections? Front. Microbiol. 7:1454. doi: 10.3389/fmicb.2016.01454
García-Contreras, R., Nuñez-López, L., Jasso-Chávez, R., Kwan, B. W., Belmont, J. A., Rangel-Vega, A., et al. (2015). Quorum sensing enhancement of the stress response promotes resistance to quorum quenching and prevents social cheating. ISME J. 9, 115–125. doi: 10.1038/ismej.2014.98
García-García, J. D., Sánchez-Thomas, R., and Moreno-Sánchez, R. (2016). Bio-recovery of non-essential heavy metals by intra- and extracellular mechanisms in free-living microorganisms. Biotechnol. Adv. 34, 859–873. doi: 10.1016/j.biotechadv.2016.05.003
Garneau, J. E., Dupuis, M. È., Villion, M., Romero, D. A., Barrangou, R., Boyaval, P., et al. (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. doi: 10.1038/nature09523
Gellatly, S. L., and Hancock, R. E. W. (2013). Pseudomonas aeruginosa: new insights into pathogenesis and host defenses. Pathog. Dis. 67, 159–173. doi: 10.1111/2049-632X.12033
Ghafoor, A., Hay, I. D., and Rehm, B. H. (2011). Role of exopolysaccharides in Pseudomonas aeruginosa biofilm formation and architecture. Appl. Environ. Microbiol. 77, 5238–5246. doi: 10.1128/AEM.00637-11
Glick, J., and Garber, N. (1983). The intracellular localization of Pseudomonas aeruginosa lectins. J. Gen. Microbiol. 129, 3085–3090. doi: 10.1099/00221287-129-10-3085
Gloag, E. S., Turnbull, L., Huang, A., Vallotton, P., Wang, H., Nolan, L. M., et al. (2013). Self-organization of bacterial biofilms is facilitated by extracellular DNA. Proc. Natl. Acad. Sci. U.S.A. 110, 11541–11546. doi: 10.1073/pnas.1218898110
González, J. E., and Keshavan, N. D. (2006). Messing with bacterial quorum sensing. Microbiol. Mol. Biol. Rev. 70, 859–875. doi: 10.1128/MMBR.00002-06
González-Valdez, A., Servín-González, L., Juarez, K., Hernandez-Aligio, A., and Soberón-Chávez, G. (2014). The effect of specific rhlA-las-box mutations on DNA binding and gene activation by Pseudomonas aeruginosa quorum-sensing transcriptional regulators RhlR and LasR. FEMS Microbiol. Lett. 356, 217–225. doi: 10.1111/1574-6968.12505
Grote, J., Krysciak, D., and Streit, W. R. (2015). Phenotypic heterogeneity, a phenomenon that may explain why quorum sensing does not always result in truly homogenous cell behavior. Appl. Environ. Microbiol. 81, 5280–5289. doi: 10.1128/AEM.00900-15
Gupta, K., Liao, J., Petrova, O. E., Cherny, K. E., and Sauer, K. (2014). Elevated levels of the second messenger c-di-GMP contribute to antimicrobial resistance of Pseudomonas aeruginosa. Mol. Microbiol. 92, 488–506. doi: 10.1111/mmi.12587
Gutiérrez, O., Juan, C., Cercenado, E., Navarro, F., Bouza, E., Coll, P., et al. (2007). Molecular epidemiology and mechanisms of carbapenem resistance in Pseudomonas aeruginosa isolates from Spanish hospitals. Antimicrob. Agents Chemother. 51, 4329–4335. doi: 10.1128/AAC.00810-07
Gutu, A. D., Sgambati, N., Strasbourger, P., Brannon, M. K., Jacobs, M. A., Haugen, E., et al. (2013). Polymyxin resistance of Pseudomonas aeruginosa phoQ mutants is dependent on additional two-component regulatory systems. Antimicrob. Agents Chemother. 57, 2204–2215. doi: 10.1128/AAC.02353-12
Guzzo, J., Pages, J. M., Duong, F., Lazdunski, A., and Murgier, M. (1991). Pseudomonas aeruginosa alkaline protease: evidence for secretion genes and study of secretion mechanism. J. Bacteriol. 173, 5290–5297. doi: 10.1128/jb.173.17.5290-5297.1991
Hall, S., McDermott, C., Anoopkumar-Dukie, S., McFarland, A. J., Forbes, A., Perkins, A. V, et al. (2016). Cellular effects of pyocyanin, a secreted virulence factor of Pseudomonas aeruginosa. Toxins (Basel). 8:236. doi: 10.3390/toxins8080236
Hammond, J. H., Dolben, E. F., Smith, T. J., Bhuju, S., and Hogan, D. A. (2015). Links between Anr and quorum sensing in Pseudomonas aeruginosa biofilms. J. Bacteriol. 197, 2810–2820. doi: 10.1128/JB.00182-15
Hancock, R. E. W., and Speert, D. P. (2000). Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and impact on treatment. Drug Resist Updat. 3, 247–255. doi: 10.1054/drup.2000.0152
Hannauer, M., Braud, A., Hoegy, F., Ronot, P., Boos, A., and Schalk, I. J. (2012). The PvdRT-OpmQ efflux pump controls the metal selectivity of the iron uptake pathway mediated by the siderophore pyoverdine in Pseudomonas aeruginosa. Environ. Microbiol. 14, 1696–1708. doi: 10.1111/j.1462-2920.2011.02674.x
Hassett, D. J. (1996). Anaerobic production of alginate by Pseudomonas aeruginosa: alginate restricts diffusion of oxygen. J. Bacteriol. 178, 7322–7325. doi: 10.1128/jb.178.24.7322-7325.1996
Hassett, D. J., Sutton, M. D., Schurr, M. J., Herr, A. B., Caldwell, C. C., and Matu, J. O. (2009). Pseudomonas aeruginosa hypoxic or anaerobic biofilm infections within cystic fibrosis airways. Trends Microbiol. 17, 130–138. doi: 10.1016/j.tim.2008.12.003
Häussler, S., Tümmler, B., Weissbrodt, H., Rohde, M., and Steinmetz, I. (1999). Small-colony variants of Pseudomonas aeruginosa in cystic fibrosis. Clin. Infect. Dis. 29, 621–625. doi: 10.1086/598644
Häussler, S., Ziegler, I., Löttel, A., von Götz, F., Rohde, M., Wehmhöhner, D., et al. (2003). Highly adherent small-colony variants of Pseudomonas aeruginosa in cystic fibrosis lung infection. J. Med. Microbiol. 52(Pt 4), 295–301. doi: 10.1099/jmm.0.05069-0
Hay, I. D., Gatland, K., Campisano, A., Jordens, J. Z., and Rehm, B. H. (2009a). Impact of alginate overproduction on attachment and biofilm architecture of a supermucoid Pseudomonas aeruginosa strain. Appl. Environ. Microbiol. 75, 6022–6025. doi: 10.1128/AEM.01078-09
Hay, I. D., Remminghorst, U., and Rehm, B. H. (2009b). MucR, a novel membrane-associated regulator of alginate biosynthesis in Pseudomonas aeruginosa. Appl. Environ. Microbiol. 75, 1110–1120. doi: 10.1128/AEM.02416-08
Hay, I. D., Ur Rehman, Z., Ghafoor, A., and Rehm, B. H. (2010). Bacterial biosynthesis of alginates. J. Chem. Techn. Biotechnol. 85, 752–759. doi: 10.1002/jctb.2372
Hay, I. D., Ur Rehman, Z., Moradali, M. F., Wang, Y., and Rehm, B. H. (2013). Microbial alginate production, modification and its applications. Microb. Biotechnol. 6, 637–650. doi: 10.1111/1751-7915.12076
Hay, T., Fraud, S., Lau, C. H., Gilmour, C., and Poole, K. (2013). Antibiotic inducibility of the MexXY multidrug efflux operon of Pseudomonas aeruginosa: involvement of the MexZ anti-repressor ArmZ. PLoS ONE 8:e56858. doi: 10.1371/journal.pone.0056858
Henrichfreise, B., Wiegand, I., Pfister, W., and Wiedemann, B. (2007). Resistance mechanisms of multiresistant Pseudomonas aeruginosa strains from Germany and correlation with hypermutation. Antimicrob. Agents Chemother. 51, 4062–4070. doi: 10.1128/AAC.00148-07
Hershberger, C. D., Ye, R. W., Parsek, M. R., Xie, Z. D., and Chakrabarty, A. M. (1995). The algT (algU) gene of Pseudomonas aeruginosa, a key regulator involved in alginate biosynthesis, encodes an alternative sigma factor (sigma E). Proc. Natl. Acad. Sci. U.S.A. 92, 7941–7945. doi: 10.1073/pnas.92.17.7941
Hesketh, A., Chen, W. J., Ryding, J., Chang, S., and Bibb, M. (2007). The global role of ppGpp synthesis in morphological differentiation and antibiotic production in Streptomyces coelicolor A3(2). Genome Biol. 8:R161. doi: 10.1186/gb-2007-8-8-r161
Hickman, J. W., Tifrea, D. F., and Harwood, C. S. (2005). A chemosensory system that regulates biofilm formation through modulation of cyclic diguanylate levels. Proc. Natl. Acad. Sci. U.S.A. 102, 14422–14427. doi: 10.1073/pnas.0507170102
Higgins, P. G., Fluit, A. C., Milatovic, D., Verhoef, J., and Schmitz, F. J. (2003). Mutations in GyrA, ParC, MexR and NfxB in clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 21, 409–413. doi: 10.1016/S0924-8579(03)00009-8
Hirai, K., Suzue, S., Irikura, T., Iyobe, S., and Mitsuhashi, S. (1987). Mutations producing resistance to norfloxacin in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 31, 582–586. doi: 10.1128/AAC.31.4.582
Hoffman, L. R., Kulasekara, H. D., Emerson, J., Houston, L. S., Burns, J. L., Ramsey, B. W., et al. (2009). Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J. Cyst. Fibros. 8, 66–70. doi: 10.1016/j.jcf.2008.09.006
Hoffman, L. R., Richardson, A. R., Houston, L. S., Kulasekara, H. D., Martens-Habbena, W., Klausen, M., et al. (2010). Nutrient availability as a mechanism for selection of antibiotic tolerant Pseudomonas aeruginosa within the CF airway. PLoS Pathog. 6:e1000712. doi: 10.1371/journal.ppat.1000712
Hogardt, M., and Heesemann, J. (2010). Adaptation of Pseudomonas aeruginosa during persistence in the cystic fibrosis lung. Int. J. Med. Microbiol. 300, 557–562. doi: 10.1016/j.ijmm.2010.08.008
Hoge, R., Pelzer, A., Rosenau, F., and Wilhelm, S. (2010). Weapons of a pathogen: proteases and their role in virulence of Pseudomonas aeruginosa. Current research, technology and education topics in applied microbiology and microbial biotechnology 2, 383–395.
Hong, D. J., Bae, I. K., Jang, I. H., Jeong, S. H., Kang, H. K., and Lee, K. (2015). Epidemiology and characteristics of metallo-β-lactamase-producing Pseudomonas aeruginosa. Infect Chemother. 47, 81–97. doi: 10.3947/ic.2015.47.2.81
Imperi, F., Tiburzi, F., Fimia, G. M., and Visca, P. (2010). Transcriptional control of the pvdS iron starvation sigma factor gene by the master regulator of sulfur metabolism CysB in Pseudomonas aeruginosa. Environ. Microbiol. 12, 1630–1642. doi: 10.1111/j.1462-2920.2010.02210.x
Irie, Y., Starkey, M., Edwards, A. N., Wozniak, D. J., Romeo, T., and Parsek, M. R. (2010). Pseudomonas aeruginosa biofilm matrix polysaccharide Psl is regulated transcriptionally by RpoS and post-transcriptionally by RsmA. Mol. Microbiol. 78, 158–172. doi: 10.1111/j.1365-2958.2010.07320.x
Jaffar-Bandjee, M. C., Lazdunski, A., Bally, M., Carrere, J., Chazalette, J. P., and Galabert, C. (1995). Production of elastase, exotoxin A, and alkaline protease in sputa during pulmonary exacerbation of cystic fibrosis in patients chronically infected by Pseudomonas aeruginosa. J. Clin. Microbiol. 33, 924–929.
Jeannot, K., Elsen, S., Kohler, T., Attree, I., van Delden, C., and Plésiat, P. (2008). Resistance and virulence of Pseudomonas aeruginosa clinical strains overproducing the MexCD-OprJ efflux pump. Antimicrob. Agents Chemother. 52, 2455–2462. doi: 10.1128/AAC.01107-07
Jennings, L. K., Storek, K. M., Ledvina, H. E., Coulon, C., Marmont, L. S., Sadovskaya, I., et al. (2015). Pel is a cationic exopolysaccharide that cross-links extracellular DNA in the Pseudomonas aeruginosa biofilm matrix. Proc. Natl. Acad. Sci. U.S.A. 112, 11353–11358. doi: 10.1073/pnas.1503058112
Jensen, P. Ø., Bjarnsholt, T., Phipps, R., Rasmussen, T. B., Calum, H., Christoffersen, L., et al. (2007). Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology 153(Pt 5), 1329–1338. doi: 10.1099/mic.0.2006/003863-0
Jimenez, P. N., Koch, G., Thompson, J. A., Xavier, K. B., Cool, R. H., and Quax, W. J. (2012). The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 76, 46–65. doi: 10.1128/MMBR.05007-11
Jiricny, N., Molin, S., Foster, K., Diggle, S. P., Scanlan, P. D., Ghoul, M., et al. (2014). Loss of social behaviours in populations of Pseudomonas aeruginosa infecting lungs of patients with cystic fibrosis. PLoS ONE 9:e83124. doi: 10.1371/journal.pone.0083124
Johnson, A. P., and Woodford, N. (2013). Global spread of antibiotic resistance: the example of New Delhi metallo-β-lactamase (NDM)-mediated carbapenem resistance. J Med. Microbiol. 62(Pt 4), 499–513. doi: 10.1099/jmm.0.052555-0
Jones, P., Palser, S. C., Pravle, A. P., Hurley, M. N., and Smyth, A. R. (2016). WS20.1 Secular trends in Pseudomonas aeruginosa acquisition in the United Kingdom: a registry study. J. Cyst. Fibros 15(Suppl. 1), S31. doi: 10.1016/S1569-1993(16)30169-2
Juan, C., Macia, M. D., Gutierrez, O., Vidal, C., Perez, J. L., and Oliver, A. (2005). Molecular mechanisms of β-lactam resistance mediated by AmpC hyperproduction in Pseudomonas aeruginosa clinical strains. Antimicrob. Agents Chemother. 49, 4733–4738. doi: 10.1128/AAC.49.11.4733-4738.2005
Jyot, J., and Ramphal, R. (2008). “Flagella and pili of Pseudomonas aeruginosa,” in Pseudomonas: Model Organism, Pathogen, Cell Factory, ed B. H. A. Rehm (Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA), 85–108.
Kao, C. Y., Chen, S. S., Hung, K. H., Wu, H. M., Hsueh, P. R., Yan, J. J., et al. (2016). Overproduction of active efflux pump and variations of OprD dominate in imipenem-resistant Pseudomonas aeruginosa isolated from patients with bloodstream infections in Taiwan. BMC Microbiol. 16:107. doi: 10.1186/s12866-016-0719-2
Karatuna, O., and Yagci, A. (2010). Analysis of quorum sensing-dependent virulence factor production and its relationship with antimicrobial susceptibility in Pseudomonas aeruginosa respiratory isolates. Clin. Microbiol. Infect. 16, 1770–1775. doi: 10.1111/j.1469-0691.2010.03177.x
Karlowsky, J. A., Hoban, D. J., Zelenitsky, S. A., and Zhanel, G. G. (1997). Altered denA and anr gene expression in aminoglycoside adaptive resistance in Pseudomonas aeruginosa. J. Antimicrob. Chemother. 40, 371–376. doi: 10.1093/jac/40.3.371
Kenward, M. A., Brown, M. R., Hesslewood, S. R., and Dillon, C. (1978). Influence of R-plasmid RP1 of Pseudomonas aeruginosa on cell wall composition, drug resistance, and sensitivity to cold shock. Antimicrob. Agents Chemother. 13, 446–453. doi: 10.1128/AAC.13.3.446
Kessler, E., Safrin, M., Abrams, W. R., Rosenbloom, J., and Ohman, D. E. (1997). Inhibitors and specificity of Pseudomonas aeruginosa LasA. J. Biol. Chem. 272, 9884–9889. doi: 10.1074/jbc.272.15.9884
Khaledi, A., Schniederjans, M., Pohl, S., Rainer, R., Bodenhofer, U., Xia, B., et al. (2016). Transcriptome profiling of antimicrobial resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 60, 4722–4733. doi: 10.1128/AAC.00075-16
Khan, T. Z., Wagener, J. S., Bost, T., Martinez, J., Accurso, F. J., and Riches, D. W. H. (1995). Early pulmonary inflammation in infants with cystic fibrosis. Am. J. Resp. Crit. Care 151, 1075–1082.
Kim, E. J., Sabra, W., and Zeng, A. P. (2003). Iron deficiency leads to inhibition of oxygen transfer and enhanced formation of virulence factors in cultures of Pseudomonas aeruginosa PAO1. Microbiology 149(Pt 9), 2627–2634. doi: 10.1099/mic.0.26276-0
Kim, H. Y., Schlictman, D., Shankar, S., Xie, Z., Chakrabarty, A. M., and Kornberg, A. (1998). Alginate, inorganic polyphosphate, GTP and ppGpp synthesis co-regulated in Pseudomonas aeruginosa: implications for stationary phase survival and synthesis of RNA/DNA precursors. Mol. Microbiol. 27, 717–725. doi: 10.1046/j.1365-2958.1998.00702.x
Kirisits, M. J., Prost, L., Starkey, M., and Parsek, M. R. (2005). Characterization of colony morphology variants isolated from Pseudomonas aeruginosa biofilms. Appl. Environ. Microbiol. 71, 4809–4821. doi: 10.1128/AEM.71.8.4809-4821.2005
Kohanski, M. A., Dwyer, D. J., Wierzbowski, J., Cottarel, G., and Collins, J. J. (2008). Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell 135, 679–690. doi: 10.1016/j.cell.2008.09.038
Köhler, T., Michéa-Hamzehpour, M., Henze, U., Gotoh, N., Curty, L. K., and Pechère, J. C. (1997). Characterization of MexE-MexF-OprN, a positively regulated multidrug efflux system of Pseudomonas aeruginosa. Mol. Microbiol. 23, 345–354. doi: 10.1046/j.1365-2958.1997.2281594.x
Köhler, T., van Delden, C., Curty, L. K., Hamzehpour, M. M., and Pechere, J. C. (2001). Overexpression of the MexEF-OprN multidrug efflux system affects cell-to-cell signaling in Pseudomonas aeruginosa. J. Bacteriol. 183, 5213–5222. doi: 10.1128/JB.183.18.5213-5222.2001
Kong, K.-F., Jayawardena, S. R., Indulkar, S. D., del Puerto, A., Koh, C.-L., Høiby, N., et al. (2005). Pseudomonas aeruginosa AmpR is a global transcriptional factor that regulates expression of AmpC and PoxB β-lactamases, proteases, quorum sensing, and other virulence factors. Antimicrob. Agents Chemother. 49, 4567–4575. doi: 10.1128/AAC.49.11.4567-4575.2005
Kung, V. L., Ozer, E. A., and Hauser, A. R. (2010). The accessory genome of Pseudomonas aeruginosa. Microbiol. Mol. Biol. Rev. 74, 621–641. doi: 10.1128/MMBR.00027-10
Kuroda, A., Nomura, K., Ohtomo, R., Kato, J., Ikeda, T., Takiguchi, N., et al. (2001). Role of inorganic polyphosphate in promoting ribosomal protein degradation by the Lon protease in E. coli. Science 293, 705–708. doi: 10.1126/science.1061315
Laarman, A. J., Bardoel, B. W., Ruyken, M., Fernie, J., Milder, F. J., van Strijp, J. A., et al. (2012). Pseudomonas aeruginosa alkaline protease blocks complement activation via the classical and lectin pathways. J. Immunol. 188, 386–393. doi: 10.4049/jimmunol.1102162
LaFayette, S. L., Houle, D., Beaudoin, T., Wojewodka, G., Radzioch, D., Hoffman, L. R., et al. (2015). Cystic fibrosis-adapted Pseudomonas aeruginosa quorum sensing lasR mutants cause hyperinflammatory responses. Sci. Adv. 1:e1500199. doi: 10.1126/sciadv.1500199
Lamarche, M. G., and Déziel, E. (2011). MexEF-OprN efflux pump exports the Pseudomonas quinolone signal (PQS) precursor HHQ (4-hydroxy-2-heptylquinoline). PLoS ONE 6:e24310. doi: 10.1371/journal.pone.0024310
Lambert, M. L., Suetens, C., Savey, A., Palomar, M., Hiesmayr, M., Morales, I., et al. (2011). Clinical outcomes of health-care-associated infections and antimicrobial resistance in patients admitted to European intensive-care units: a cohort study. Lancet Infect. Dis. 11, 30–38. doi: 10.1016/S1473-3099(10)70258-9
Lapouge, K., Schubert, M., Allain, F. H., and Haas, D. (2008). Gac/Rsm signal transduction pathway of gamma-proteobacteria: from RNA recognition to regulation of social behaviour. Mol. Microbiol. 67, 241–253. doi: 10.1111/j.1365-2958.2007.06042.x
LaSarre, B., and Federle, M. J. (2013). Exploiting quorum sensing to confuse bacterial pathogens. Microbiol. Mol. Biol. Rev. 77, 73–111. doi: 10.1128/MMBR.00046-12
Lau, C. H., Hughes, D., and Poole, K. (2014). MexY-promoted aminoglycoside resistance in Pseudomonas aeruginosa: involvement of a putative proximal binding pocket in aminoglycoside recognition. Mbio 5:e01068. doi: 10.1128/mBio.01068-14
Laughlin, R. S., Musch, M. W., Hollbrook, C. J., Rocha, F. M., Chang, E. B., and Alverdy, J. C. (2000). The key role of Pseudomonas aeruginosa PA-I lectin on experimental gut-derived sepsis. Ann. Surg. 232, 133–142. doi: 10.1097/00000658-200007000-00019
Lee, J. K., Lee, Y. S., Park, Y. K., and Kim, B. S. (2005). Alterations in the GyrA and GyrB subunits of topoisomerase II and the ParC and ParE subunits of topoisomerase IV in ciprofloxacin-resistant clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 25, 290–295. doi: 10.1016/j.ijantimicag.2004.11.012
Lee, J., Wu, J., Deng, Y., Wang, J., Wang, C., Wang, J., et al. (2013). A cell-cell communication signal integrates quorum sensing and stress response. Nat. Chem. Biol. 9, 339–343. doi: 10.1038/nchembio.1225
Lee, J. Y., Park, Y. K., Chung, E. S., Na, I. Y., and Ko, K. S. (2016). Evolved resistance to colistin and its loss due to genetic reversion in Pseudomonas aeruginosa. Sci. Rep. 6:25543. doi: 10.1038/srep25543
Lee, J., and Zhang, L. (2015). The hierarchy quorum sensing network in Pseudomonas aeruginosa. Protein Cell 6, 26–41. doi: 10.1007/s13238-014-0100-x
Lenney, W., and Gilchrist, F. J. (2011). Pseudomonas aeruginosa and cyanide production. Eur. Respir. J. 37, 482–483. doi: 10.1183/09031936.00122810
Lewis, K. (2007). Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 5, 48–56. doi: 10.1038/nrmicro1557
Lewis, K. (2008). Multidrug tolerance of biofilms and persister cells. Curr. Top. Microbiol. Immunol. 322, 107–131. doi: 10.1007/978-3-540-75418-3_6
Li, G., Shen, M., Lu, S., Le, S., Tan, Y., Wang, J., et al. (2016). Identification and characterization of the HicAB toxin-antitoxin system in the opportunistic pathogen Pseudomonas aeruginosa. Toxins (Basel). 8:113. doi: 10.3390/toxins8040113
Li, M., Long, Y., Liu, Y., Liu, Y., Chen, R., Shi, J., et al. (2016). HigB of Pseudomonas aeruginosa enhances killing of phagocytes by up-regulating the type III secretion system in ciprofloxacin induced persister cells. Front. Cell. Infect. Microbiol. 6:125. doi: 10.3389/fcimb.2016.00125
Li, X. Z., Plésiat, P., and Nikaido, H. (2015). The challenge of efflux-mediated antibiotic resistance in Gram-negative bacteria. Clin. Microbiol. Rev. 28, 337–418. doi: 10.1128/CMR.00117-14
Li, Y. Q., Du, X. L., Lu, Z. J., Wu, D. Q., Zhao, Y. L., Ren, B., et al. (2011). Regulatory feedback loop of two phz gene clusters through 5′-untranslated regions in Pseudomonas sp M18. PLoS ONE 6:e19413. doi: 10.1371/journal.pone.0019413
Lister, P. D., Wolter, D. J., and Hanson, N. D. (2009). Antibacterial-resistant Pseudomonas aeruginosa: clinical impact and complex regulation of chromosomally encoded resistance mechanisms. Clin. Microbiol. Rev. 22, 582–610. doi: 10.1128/CMR.00040-09
Liu, Y. Y., Wang, Y., Walsh, T. R., Yi, L. X., Zhang, R., Spencer, J., et al. (2016). Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect. Dis. 16, 161–168. doi: 10.1016/S1473-3099(15)00424-7
Llanes, C., Kohler, T., Patry, I., Dehecq, B., van Delden, C., and Plésiat, P. (2011). Role of the MexEF-OprN efflux system in low-level resistance of Pseudomonas aeruginosa to ciprofloxacin. Antimicrob. Agents Chemother. 55, 5676–5684. doi: 10.1128/AAC.00101-11
Long, J., Zaborina, O., Holbrook, C., Zaborin, A., and Alverdy, J. (2008). Depletion of intestinal phosphate after operative injury activates the virulence of P. aeruginosa causing lethal gut-derived sepsis. Surgery 144, 189–197. doi: 10.1016/j.surg.2008.03.045
Louwen, R., Staals, R. H. J., Endtz, H. P., van Baarlen, P., and van der Oost, J. (2014). The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol. Mol. Biol. Rev. 78, 74–88. doi: 10.1128/MMBR.00039-13
Ma, L., Conover, M., Lu, H., Parsek, M. R., Bayles, K., and Wozniak, D. J. (2009). Assembly and development of the Pseudomonas aeruginosa biofilm matrix. PLoS Pathog. 5:e1000354. doi: 10.1371/journal.ppat.1000354
MacDougall, C., Harpe, S. E., Powell, J. P., Johnson, C. K., Edmond, M. B., and Polk, R. E. (2005). Pseudomonas aeruginosa, Staphylococcus aureus, and fluoroquinolone use. Emerging Infect. Dis. 11, 1197–1204. doi: 10.3201/eid1108.050116
Mahenthiralingam, E., Campbell, M. E., and Speert, D. P. (1994). Nonmotility and phagocytic resistance of Pseudomonas aeruginosa isolates from chronically colonized patients with cystic fibrosis. Infect. Immun. 62, 596–605.
Maisonneuve, E., Castro-Camargo, M., and Gerdes, K. (2013). (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154, 1140–1150. doi: 10.1016/j.cell.2013.07.048
Makarova, K. S., Haft, D. H., Barrangou, R., Brouns, S. J. J., Charpentier, E., Horvath, P., et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol. 9, 467–477. doi: 10.1038/nrmicro2577
Malone, J. G. (2015). Role of small colony variants in persistence of Pseudomonas aeruginosa infections in cystic fibrosis lungs. Infect. Drug Resist. 8, 237–247. doi: 10.2147/IDR.S68214
Malone, J. G., Jaeger, T., Manfredi, P., Dötsch, A., Blanka, A., Bos, R., et al. (2012). The YfiBNR signal transduction mechanism reveals novel targets for the evolution of persistent Pseudomonas aeruginosa in cystic fibrosis airways. PLoS Pathog. 8:e1002760. doi: 10.1371/journal.ppat.1002760
Mann, E. E., and Wozniak, D. J. (2012). Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol. Rev. 36, 893–916. doi: 10.1111/j.1574-6976.2011.00322.x
Marr, A. K., Overhage, J., Bains, M., and Hancock, R. E. W. (2007). The Lon protease of Pseudomonas aeruginosa is induced by aminoglycosides and is involved in biofilm formation and motility. Microbiology 153(Pt 2), 474–482. doi: 10.1099/mic.0.2006/002519-0
Martinez, J. L., Sánchez, M. B., Martínez-Solano, L., Hernandez, A., Garmendia, L., Fajardo, A., et al. (2009). Functional role of bacterial multidrug efflux pumps in microbial natural ecosystems. FEMS Microbiol. Rev. 33, 430–449. doi: 10.1111/j.1574-6976.2008.00157.x
Marvig, R. L., Sommer, L. M., Molin, S., and Johansen, H. K. (2015). Convergent evolution and adaptation of Pseudomonas aeruginosa within patients with cystic fibrosis. Nat. Genet. 47, 57–64. doi: 10.1038/ng.3148
Maseda, H., Saito, K., Nakajima, A., and Nakae, T. (2000). Variation of the mexT gene, a regulator of the MexEF-oprN efflux pump expression in wild-type strains of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 192, 107–112. doi: 10.1111/j.1574-6968.2000.tb09367.x
Maseda, H., Uwate, M., and Nakae, T. (2010). Transcriptional regulation of the MexEF-oprN multidrug efflux pump operon by MexT and an unidentified repressor in nfxC-type mutant of Pseudomonas aeruginosa. FEMS Microbiol. Lett. 311, 36–43. doi: 10.1111/j.1574-6968.2010.02063.x
Mashburn, L. M., and Whiteley, M. (2005). Membrane vesicles traffic signals and facilitate group activities in a prokaryote. Nature 437, 422–425. doi: 10.1038/nature03925
Mataseje, L. F., Peirano, G., Church, D. L., Conly, J., Mulvey, M., and Pitout, J. D. (2016). Colistin-nonsusceptible Pseudomonas aeruginosa sequence type 654 with blaNDM−1 arrives in North America. Antimicrob. Agents Chemother. 60, 1794–1800. doi: 10.1128/AAC.02591-15
Mathee, K., Ciofu, O., Sternberg, C., Lindum, P. W., Campbell, J. I, Jensen, P., et al. (1999). Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology 145(Pt 6), 1349–1357. doi: 10.1099/13500872-145-6-1349
Mathee, K., Narasimhan, G., Valdes, C., Qiu, X., Matewish, J. M., Koehrsen, M., et al. (2008). Dynamics of Pseudomonas aeruginosa genome evolution. Proc. Natl. Acad. Sci. U.S.A. 105, 3100–3105. doi: 10.1073/pnas.0711982105
Matsuo, Y., Eda, S., Gotoh, N., Yoshihara, E., and Nakae, T. (2004). MexZ-mediated regulation of MexXY multidrug efflux pump expression in Pseudomonas aeruginosa by binding on the mexZ-mexX intergenic DNA. FEMS Microbiol. Lett. 238, 23–28. doi: 10.1016/j.femsle.2004.07.010
Maxwell, K. L. (2016). Phages fight back: inactivation of the CRISPR-Cas bacterial immune system by anti-CRISPR proteins. PLoS Pathog. 12:e1005282. doi: 10.1371/journal.ppat.1005282
McDaniel, C. T., Panmanee, W., and Hassett, D. J. (2015). “An overview of infections in cystic fibrosis airways and the role of environmental conditions on Pseudomonas aeruginosa biofilm formation and viability,” in Cystic Fibrosis in the Light of New Research, ed D. Wat (InTech), 171–198.
Meletis, G., and Bagkeri, M. (2013). “Pseudomonas aeruginosa: multi-drug-resistance development and treatment options,” in Infection Control, ed S. Basak (InTech), 33–56.
Michalska, M., and Wolf, P. (2015). Pseudomonas Exotoxin A: optimized by evolution for effective killing. Front. Microbiol. 6:963. doi: 10.3389/fmicb.2015.00963
Mishra, M., Byrd, M. S., Sergeant, S., Azad, A. K., Parsek, M. R., McPhail, L., et al. (2012). Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell. Microbiol. 14, 95–106. doi: 10.1111/j.1462-5822.2011.01704.x
Mojica, F. J., Díez-Villaseñor, C., García-Martínez, J., and Almendros, C. (2009). Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 155(Pt 3), 733–740. doi: 10.1099/mic.0.023960-0
Moradali, M. F., Donati, I., Sims, I. M., Ghods, S., and Rehm, B. H. (2015). Alginate polymerization and modification are linked in Pseudomonas aeruginosa. MBio 6, e00453–e00415. doi: 10.1128/mBio.00453-15
Moscoso, J. A., Mikkelsen, H., Heeb, S., Williams, P., and Filloux, A. (2011). The Pseudomonas aeruginosa sensor RetS switches type III and type VI secretion via c-di-GMP signalling. Environ. Microbiol. 13, 3128–3138. doi: 10.1111/j.1462-2920.2011.02595.x
Moskowitz, S. M., Brannon, M. K., Dasgupta, N., Pier, M., Sgambati, N., Miller, A. K., et al. (2012). PmrB mutations promote polymyxin resistance of Pseudomonas aeruginosa isolated from colistin-treated cystic fibrosis patients. Antimicrob. Agents Chemother. 56, 1019–1030. doi: 10.1128/AAC.05829-11
Mouneimné, H., Robert, J., Jarlier, V., and Cambau, E. (1999). Type II topoisomerase mutations in ciprofloxacin-resistant strains of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 43, 62–66.
Mulcahy, H., Charron-Mazenod, L., and Lewenza, S. (2008). Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog. 4:e1000213. doi: 10.1371/journal.ppat.1000213
Mulcahy, L. R., Burns, J. L., Lory, S., and Lewis, K. (2010). Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 192, 6191–6199. doi: 10.1128/JB.01651-09
Nathwani, D., Raman, G., Sulham, K., Gavaghan, M., and Menon, V. (2014). Clinical and economic consequences of hospital-acquired resistant and multidrug-resistant Pseudomonas aeruginosa infections: a systematic review and meta-analysis. Antimicrob. Resist. Infect. Control 3:32. doi: 10.1186/2047-2994-3-32
Nelson, L. K., D'Amours, G. H., Sproule-Willoughby, K. M., Morck, D. W., and Ceri, H. (2009). Pseudomonas aeruginosa las and rhl quorum-sensing systems are important for infection and inflammation in a rat prostatitis model. Microbiology 155(Pt 8), 2612–2619. doi: 10.1099/mic.0.028464-0
Nguyen, A. T., Jones, J. W., Camara, M., Williams, P., Kane, M. A., and Oglesby-Sherrouse, A. G. (2016). Cystic fibrosis isolates of Pseudomonas aeruginosa retain iron-regulated antimicrobial activity against Staphylococcus aureus through the action of multiple alkylquinolones. Front. Microbiol. 7:1171. doi: 10.3389/fmicb.2016.01171
Nguyen, A. T., Jones, J. W., Ruge, M. A., Kane, M. A., and Oglesby-Sherrouse, A. G. (2015). Iron depletion enhances production of antimicrobials by Pseudomonas aeruginosa. J. Bacteriol. 197, 2265–2275. doi: 10.1128/JB.00072-15
Nguyen, D., Joshi-Datar, A., Lepine, F., Bauerle, E., Olakanmi, O., Beer, K., et al. (2011). Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 334, 982–986. doi: 10.1126/science.1211037
Ochs, M. M., McCusker, M. P., Bains, M., and Hancock, R. E. (1999). Negative regulation of the Pseudomonas aeruginosa outer membrane porin OprD selective for imipenem and basic amino acids. Antimicrob. Agents Chemother. 43, 1085–1090.
Oldak, E., and Trafny, E. A. (2005). Secretion of proteases by Pseudomonas aeruginosa biofilms exposed to ciprofloxacin. Antimicrob. Agents Chemother. 49, 3281–3288. doi: 10.1128/AAC.49.8.3281-3288.2005
Oliver, A., Cantón, R., Campo, P., Baquero, F., and Blázquez, J. (2000). High frequency of hypermutable Pseudomonas aeruginosa in cystic fibrosis lung infection. Science 288, 1251–1253. doi: 10.1126/science.288.5469.1251
Oliver, A., and Mena, A. (2010). Bacterial hypermutation in cystic fibrosis, not only for antibiotic resistance. Clin. Microbiol. Infect. 16, 798–808. doi: 10.1111/j.1469-0691.2010.03250.x
Oliver, A., Mulet, X., López-Causapé, C., and Juan, C. (2015). The increasing threat of Pseudomonas aeruginosa high-risk clones. Drug Resist. Updat. 21–22, 41–59. doi: 10.1016/j.drup.2015.08.002
Olsen, I. (2015). Biofilm-specific antibiotic tolerance and resistance. Eur. J. Clin. Microbiol. Infect. Dis. 34, 877–886. doi: 10.1007/s10096-015-2323-z
Overhage, J., Schemionek, M., Webb, J. S., and Rehm, B. H. (2005). Expression of the psl operon in Pseudomonas aeruginosa PAO1 biofilms: PslA performs an essential function in biofilm formation. Appl. Environ. Microbiol. 71, 4407–4413. doi: 10.1128/AEM.71.8.4407-4413.2005
Ozer, E. A., Allen, J. P., and Hauser, A. R. (2014). Characterization of the core and accessory genomes of Pseudomonas aeruginosa using bioinformatic tools Spine and AGEnt. BMC Genomics 15:737. doi: 10.1186/1471-2164-15-737
Pagedar, A., Singh, J., and Batish, V. K. (2011). Efflux mediated adaptive and cross resistance to ciprofloxacin and benzalkonium chloride in Pseudomonas aeruginosa of dairy origin. J. Basic Microbiol. 51, 289–295. doi: 10.1002/jobm.201000292
Palmer, K. L., and Whiteley, M. (2011). DMS3-42: the secret to CRISPR-dependent biofilm inhibition in Pseudomonas aeruginosa. J. Bacteriol. 193, 3431–3432. doi: 10.1128/JB.05066-11
Pandey, D. P., and Gerdes, K. (2005). Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 33, 966–976. doi: 10.1093/nar/gki201
Papenfort, K., and Bassler, B. L. (2016). Quorum sensing signal-response systems in Gram-negative bacteria. Nat. Rev. Microbiol. 14, 576–588. doi: 10.1038/nrmicro.2016.89
Paterson, D. L., and Bonomo, R. A. (2005). Extended-spectrum β-lactamases: a clinical update. Clin. Microbiol. Rev. 18, 657–686. doi: 10.1128/CMR.18.4.657-686.2005
Paul, K., Nieto, V., Carlquist, W. C., Blair, D. F., and Harshey, R. M. (2010). The c-di-GMP binding protein YcgR controls flagellar motor direction and speed to affect chemotaxis by a “backstop brake” mechanism. Mol. Cell 38, 128–139. doi: 10.1016/j.molcel.2010.03.001
Pawluk, A., Bondy-Denomy, J., Cheung, V. H. W., Maxwell, K. L., and Davidson, A. R. (2014). A new group of phage Anti-CRISPR genes inhibits the type I-E CRISPR-Cas system of Pseudomonas aeruginosa. MBio 5:e00896-14. doi: 10.1128/mBio.00896–14
Pessi, G., and Haas, D. (2000). Transcriptional control of the hydrogen cyanide biosynthetic genes hcnABC by the anaerobic regulator ANR and the quorum-sensing regulators LasR and RhlR in Pseudomonas aeruginosa. J. Bacteriol. 182, 6940–6949. doi: 10.1128/JB.182.24.6940-6949.2000
Petrova, O. E., and Sauer, K. (2009). A novel signaling network essential for regulating Pseudomonas aeruginosa biofilm development. PLoS Pathog. 5:e1000668. doi: 10.1371/journal.ppat.1000668
Pillar, C. M., and Hobden, J. A. (2002). Pseudomonas aeruginosa exotoxin A and keratitis in mice. Invest. Ophthalmol. Vis. Sci. 43, 1437–1444. Available online at: http://iovs.arvojournals.org/article.aspx?articleid=2123690
Poole, K. (2005). Aminoglycoside resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49, 479–487. doi: 10.1128/AAC.49.2.479-487.2005
Poole, K. (2011). Pseudomonas aeruginosa: resistance to the max. Front. Microbiol. 2:65. doi: 10.3389/fmicb.2011.00065
Poole, K. (2012). Stress responses as determinants of antimicrobial resistance in Gram-negative bacteria. Trends Microbiol. 20, 227–234. doi: 10.1016/j.tim.2012.02.004
Porter, S. L., Wadhams, G. H., and Armitage, J. P. (2011). Signal processing in complex chemotaxis pathways. Nat. Rev. Microbiol. 9, 153–165. doi: 10.1038/nrmicro2505
Potron, A., Poirel, L., and Nordmann, P. (2015). Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int. J. Antimicrob. Agents 45, 568–585. doi: 10.1016/j.ijantimicag.2015.03.001
Potrykus, K., and Cashel, M. (2008). (p)ppGpp: still magical? Annu. Rev. Microbiol. 62, 35–51. doi: 10.1146/annurev.micro.62.081307.162903
Pourmand, M., Sadighian, H., and Naderi, M. (2015). Relation between expression of the las quorum-sensing system in clinical isolates of Pseudomonas aeruginosa and expression of efflux pump and ampC. J. Med. Bacteriol. 2, 32–40. Available online at: http://tums.ac.ir/1393/04/18/Sadighian.pdf-mpourmand-2014-07-09-06-33.pdf
Quale, J., Bratu, S., Gupta, J., and Landman, D. (2006). Interplay of efflux system, ampC, and oprD expression in carbapenem resistance of Pseudomonas aeruginosa clinical isolates. Antimicrob. Agents Chemother. 50, 1633–1641. doi: 10.1128/AAC.50.5.1633-1641.2006
Ramisetty, B. C. M., Ghosh, D., Chowdhury, M. R., and Santhosh, R. S. (2016). What is the link between stringent response, endoribonuclease encoding Type II Toxin-Antitoxin systems and persistence? bioRxiv. Front. Microbiol. 7:1882. doi: 10.1101/069831
Ramisse, F., van Delden, C., Gidenne, S., Cavallo, J., and Hernandez, E. (2000). Decreased virulence of a strain of Pseudomonas aeruginosa O12 overexpressing a chromosomal type 1 β-lactamase could be due to reduced expression of cell-to-cell signaling dependent virulence factors. FEMS Immunol. Med. Microbiol. 28, 241–245. doi: 10.1111/j.1574-695X.2000.tb01483.x
Rasamiravaka, T., and El Jaziri, M. (2016). Quorum-sensing mechanisms and bacterial response to antibiotics in P. aeruginosa. Curr Microbiol. doi: 10.1007/s00284-016-1101-1
Rau, M. H., Hansen, S. K., Johansen, H. K., Thomsen, L. E., Workman, C. T., Nielsen, K. F., et al. (2010). Early adaptive developments of Pseudomonas aeruginosa after the transition from life in the environment to persistent colonization in the airways of human cystic fibrosis hosts. Environ. Microbiol. 12, 1643–1658. doi: 10.1111/j.1462-2920.2010.02211.x
Rehm, B. H., Grabert, E., Hein, J., and Winkler, U. K. (1994). Antibody response of rabbits and cystic fibrosis patients to an alginate-specific outer membrane protein of a mucoid strain of Pseudomonas aeruginosa. Microb. Pathog. 16, 43–51. doi: 10.1006/mpat.1994.1004
Remminghorst, U., and Rehm, B. H. (2006). Alg44, a unique protein required for alginate biosynthesis in Pseudomonas aeruginosa. FEBS Lett. 580, 3883–3888. doi: 10.1016/j.febslet.2006.05.077
Rocker, A., and Meinhart, A. (2016). Type II toxin: antitoxin systems. More than small selfish entities? Curr. Genet. 62, 287–290. doi: 10.1007/s00294-015-0541-7
Rojas Murcia, N., Lee, X., Waridel, P., Maspoli, A., Imker, H. J., Chai, T., et al. (2015). The Pseudomonas aeruginosa antimetabolite L -2-amino-4-methoxy-trans-3-butenoic acid (AMB) is made from glutamate and two alanine residues via a thiotemplate-linked tripeptide precursor. Front. Microbiol. 6:170. doi: 10.3389/fmicb.2015.00170
Römling, U., and Balsalobre, C. (2012). Biofilm infections, their resilience to therapy and innovative treatment strategies. J. Intern. Med. 272, 541–561. doi: 10.1111/joim.12004
Römling, U., Galperin, M. Y., and Gomelsky, M. (2013). Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiol. Mol. Biol. Rev. 77, 1–52. doi: 10.1128/MMBR.00043-12
Rosenfeld, M., Gibson, R. L., McNamara, S., Emerson, J., Burns, J. L., Castile, R., et al. (2001). Early pulmonary infection, inflammation, and clinical outcomes in infants with cystic fibrosis. Pediatr. Pulmonol. 32, 356–366. doi: 10.1002/ppul.1144
Rosenthal, V. D., Al-Abdely, H. M., El-Kholy, A. A., AlKhawaja, S. A. A., Leblebicioglu, H., Mehta, Y., et al. (2016). International nosocomial infection control consortium report, data summary of 50 countries for 2010-2015: device-associated module. Am. J. Infect. Control. 44, 1495–1504. doi: 10.1016/j.ajic.2016.08.007
Russotto, V., Cortegiani, A., Raineri, S. M., and Giarratano, A. (2015). Bacterial contamination of inanimate surfaces and equipment in the intensive care unit. J. Intensive Care 3, 54. doi: 10.1186/s40560-015-0120-5
Ryan, R. P., Fouhy, Y., Lucey, J. F., and Dow, J. M. (2006). Cyclic di-GMP signaling in bacteria: recent advances and new puzzles. J. Bacteriol. 188, 8327–8334. doi: 10.1128/JB.01079-06
Sampathkumar, G., Khakimova, M., Chan, T., and Nguyen, D. (2016). “The stringent response and antioxidant defences in Pseudomonas aeruginosa,” in Stress and Environmental Regulation of Gene Expression and Adaptation in Bacteria, ed F. J. de Bruijn (Hoboken, NJ: John Wiley & Sons, Inc.), 500–506.
Schafhauser, J., Lepine, F., McKay, G., Ahlgren, H. G., Khakimova, M., and Nguyen, D. (2014). The stringent response modulates 4-hydroxy-2-alkylquinoline biosynthesis and quorum-sensing hierarchy in Pseudomonas aeruginosa. J. Bacteriol. 196, 1641–1650. doi: 10.1128/JB.01086-13
Schalk, I. J., and Guillon, L. (2013). Pyoverdine biosynthesis and secretion in Pseudomonas aeruginosa: implications for metal homeostasis. Environ. Microbiol. 15, 1661–1673. doi: 10.1111/1462-2920.12013
Schmiel, D. H., and Miller, V. L. (1999). Bacterial phospholipases and pathogenesis. Microbes Infect 1, 1103–1112. doi: 10.1016/S1286-4579(99)00205-1
Schneider, M., Mühlemann, K., Droz, S., Couzinet, S., Casaulta, C., and Zimmerli, S. (2008). Clinical characteristics associated with isolation of small-colony variants of Staphylococcus aureus and Pseudomonas aeruginosa from respiratory secretions of patients with cystic fibrosis. J. Clin. Microbiol. 46, 1832–1834. doi: 10.1128/JCM.00361-08
Schroder, K., Hertzog, P. J., Ravasi, T., and Hume, D. A. (2004). Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol. 75, 163–189. doi: 10.1189/jlb.0603252
Schurr, M. J., Yu, H., Martinez-Salazar, J. M., Boucher, J. C., and Deretic, V. (1996). Control of AlgU, a member of the sigma E-like family of stress sigma factors, by the negative regulators MucA and MucB and Pseudomonas aeruginosa conversion to mucoidy in cystic fibrosis. J. Bacteriol. 178, 4997–5004. doi: 10.1128/jb.178.16.4997-5004.1996
Shih, P. C., and Huang, C. T. (2002). Effects of quorum-sensing deficiency on Pseudomonas aeruginosa biofilm formation and antibiotic resistance. J. Antimicrob. Chemother. 49, 309–314. doi: 10.1093/jac/49.2.309
Slater, H., Crow, M., Everson, L., and Salmond, G. P. (2003). Phosphate availability regulates biosynthesis of two antibiotics, prodigiosin and carbapenem, in Serratia via both quorum-sensing-dependent and -independent pathways. Mol. Microbiol. 47, 303–320. doi: 10.1046/j.1365-2958.2003.03295.x
Sobel, M. L., Hocquet, D., Cao, L., Plesiat, P., and Poole, K. (2005). Mutations in PA3574 (nalD) lead to increased MexAB-OprM expression and multidrug resistance in laboratory and clinical isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 49, 1782–1786. doi: 10.1128/AAC.49.5.1782-1786.2005
Starkey, M., Hickman, J. H., Ma, L. Y., Zhang, N., De Long, S., Hinz, A., et al. (2009). Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J. Bacteriol. 191, 3492–3503. doi: 10.1128/JB.00119-09
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J., et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964. doi: 10.1038/35023079
Strateva, T., and Yordanov, D. (2009). Pseudomonas aeruginosa - a phenomenon of bacterial resistance. J. Med. Microbiol. 58, 1133–1148. doi: 10.1099/jmm.0.009142-0
Strempel, N., Neidig, A., Nusser, M., Geffers, R., Vieillard, J., Lesouhaitier, O., et al. (2013). Human host defense peptide LL-37 stimulates virulence factor production and adaptive resistance in Pseudomonas aeruginosa. PLoS ONE 8:e82240. doi: 10.1371/journal.pone.0082240
Sullivan, R., Schaus, D., John, M., and Delport, J. A. (2016). Extended spectrum beta-lactamases: a minireview of clinical relevant groups. J. Med. Microb. Diagn. 4:203. doi: 10.4172/2161-0703.1000203
Sun, J., Deng, Z., and Yan, A. (2014). Bacterial multidrug efflux pumps: mechanisms, physiology and pharmacological exploitations. Biochem. Biophys. Res. Commun. 453, 254–267. doi: 10.1016/j.bbrc.2014.05.090
Sun, S., Zhou, L., Jin, K., Jiang, H., and He, Y. W. (2016). Quorum sensing systems differentially regulate the production of phenazine-1-carboxylic acid in the rhizobacterium Pseudomonas aeruginosa PA1201. Sci. Rep. 6:30352. doi: 10.1038/srep30352
Tamber, S., and Hancock, R. E. (2003). On the mechanism of solute uptake in Pseudomonas. Front. Biosci. 8:s472–s483. doi: 10.2741/1075
Tian, Z. X., Mac Aogain, M., O'Connor, H. F., Fargier, E., Mooij, M. J., Adams, C., et al. (2009). MexT modulates virulence determinants in Pseudomonas aeruginosa independent of the MexEF-OprN efflux pump. Microb. Pathog. 47, 237–241. doi: 10.1016/j.micpath.2009.08.003
Tielen, P., Strathmann, M., Jaeger, K. E., Flemming, H. C., and Wingender, J. (2005). Alginate acetylation influences initial surface colonization by mucoid Pseudomonas aeruginosa. Microbiol. Res. 160, 165–176. doi: 10.1016/j.micres.2004.11.003
Toder, D. S., Ferrell, S. J., Nezezon, J. L., Rust, L., and Iglewski, B. H. (1994). lasA and lasB genes of Pseudomonas aeruginosa - Analysis of transcription and gene product activity. Infect. Immun. 62, 1320–1327.
Toder, D. S., Gambello, M. J., and Iglewski, B. H. (1991). Pseudomonas aeruginosa LasA: a second elastase under the transcriptional control of lasR. Mol. Microbiol. 5, 2003–2010. doi: 10.1111/j.1365-2958.1991.tb00822.x
Trejo-Hernández, A., Andrade-Domínguez, A., Herníndez, M., and Encarnación, S. (2014). Interspecies competition triggers virulence and mutability in Candida albicans-Pseudomonas aeruginosa mixed biofilms. ISME J. 8, 1974–1988. doi: 10.1038/ismej.2014.53
Trias, J., and Nikaido, H. (1990). Outer membrane protein D2 catalyzes facilitated diffusion of carbapenems and penems through the outer membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 34, 52–57. doi: 10.1128/AAC.34.1.52
Trubiano, J. A., and Padiglione, A. A. (2015). Nosocomial infections in the intensive care unit. Anaesth. Intensive Care 16, 598–602. doi: 10.1016/j.mpaic.2015.09.010
Turner, K. H., Everett, J., Trivedi, U., Rumbaugh, K. P., and Whiteley, M. (2014). Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet. 10:e1004518. doi: 10.1371/journal.pgen.1004518
Ueda, A., and Wood, T. K. (2009). Connecting quorum sensing, c-di-GMP, pel polysaccharide, and biofilm formation in Pseudomonas aeruginosa through tyrosine phosphatase TpbA (PA3885). PLoS Pathog. 5:e1000483. doi: 10.1371/journal.ppat.1000483
Ueda, A., and Wood, T. K. (2010). Tyrosine phosphatase TpbA of Pseudomonas aeruginosa controls extracellular DNA via cyclic diguanylic acid concentrations. Environ. Microbiol. 2, 449–455. doi: 10.1111/j.1758-2229.2010.00171.x
Umadevi, S., Joseph, N. M., Kumari, K., Easow, J. M., Kumar, S., Stephen, S., et al. (2011). Detection of extended spectrum beta lactamases, ampc beta-lactamases and metallobetalactamases in clinical isolates of ceftazidime resistant Pseudomonas aeruginosa. Braz. J. Microbiol. 42, 1284–1288. doi: 10.1590/S1517-83822011000400006
Valentini, M., and Filloux, A. (2016). Biofilms and cyclic di-GMP (c-di-GMP) signaling: lessons from Pseudomonas aeruginosa and other bacteria. J. Biol. Chem. 291, 12547–12555. doi: 10.1074/jbc.R115.711507
van Belkum, A., Soriaga, L. B., LaFave, M. C., Akella, S., Veyrieras, J. B., Barbu, E. M., et al. (2015). Phylogenetic distribution of CRISPR-Cas systems in antibiotic-resistant Pseudomonas aeruginosa. MBio 6:e01796–15. doi: 10.1128/mBio.01796-15
van Delden, C., Comte, R., and Bally, A. M. (2001). Stringent response activates quorum sensing and modulates cell density-dependent gene expression in Pseudomonas aeruginosa. J. Bacteriol. 183, 5376–5384. doi: 10.1128/JB.183.18.5376-5384.2001
Van Delden, C., and Iglewski, B. H. (1998). Cell-to-cell signaling and Pseudomonas aeruginosa infections. Emerg. Infect. Dis. 4, 551–560. doi: 10.3201/eid0404.980405
Van Gennip, M., Christensen, L. D., Alhede, M., Phipps, R., Jensen, P. Ø., Christophersen, L., et al. (2009). Inactivation of the rhlA gene in Pseudomonas aeruginosa prevents rhamnolipid production, disabling the protection against polymorphonuclear leukocytes. APMIS 117, 537–546. doi: 10.1111/j.1600-0463.2009.02466.x
Van Melderen, L. (2010). Toxin-antitoxin systems: why so many, what for? Curr. Opin. Microbiol. 13, 781–785. doi: 10.1016/j.mib.2010.10.006
Varga, J. J., Barbier, M., Mulet, X., Bielecki, P., Bartell, J. A., Owings, J. P., et al. (2015). Genotypic and phenotypic analyses of a Pseudomonas aeruginosa chronic bronchiectasis isolate reveal differences from cystic fibrosis and laboratory strains. BMC Genomics 16:883. doi: 10.1186/s12864-015-2069-0
Venter, H., Mowla, R., Ohene-Agyei, T., and Ma, S. (2015). RND-type drug e fflux pumps from Gram-negative bacteria: molecular mechanism and inhibition. Front. Microbiol. 6:377. doi: 10.3389/fmicb.2015.00377
Venturi, V. (2006). Regulation of quorum sensing in Pseudomonas. FEMS Microbiol. Rev. 30, 274–291. doi: 10.1111/j.1574-6976.2005.00012.x
Viducic, D., Ono, T., Murakami, K., Susilowati, H., Kayama, S., Hirota, K., et al. (2006). Functional analysis of spoT, relA and dksA genes on quinolone tolerance in Pseudomonas aeruginosa under nongrowing condition. Microbiol. Immunol. 50, 349–357. doi: 10.1111/j.1348-0421.2006.tb03793.x
Visca, P., Imperi, F., and Lamont, I. L. (2007). Pyoverdine siderophores: from biogenesis to biosignificance. Trends Microbiol. 15, 22–30. doi: 10.1016/j.tim.2006.11.004
Vogt, S. L., Green, C., Stevens, K. M., Day, B., Erickson, D. L., Woods, D. E., et al. (2011). The stringent response is essential for Pseudomonas aeruginosa virulence in the rat lung agar bead and Drosophila melanogaster feeding models of infection. Infect. Immun. 79, 4094–4104. doi: 10.1128/IAI.00193-11
Wagner, V. E., Bushnell, D., Passador, L., Brooks, A. I, and Iglewski, B. H. (2003). Microarray analysis of Pseudomonas aeruginosa quorum-sensing regulons: effects of growth phase and environment. J. Bacteriol. 185, 2080–2095. doi: 10.1128/JB.185.7.2080-2095.2003
Wang, H., Tu, F., Gui, Z., Lu, X., and Chu, W. (2013). Antibiotic resistance profiles and quorum sensing-dependent virulence factors in clinical isolates of Pseudomonas aeruginosa. Indian J. Microbiol. 53, 163–167. doi: 10.1007/s12088-013-0370-7
Wang, S., Parsek, M. R., Wozniak, D. J., and Ma, L. Z. (2013). A spider web strategy of type IV pili-mediated migration to build a fibre-like Psl polysaccharide matrix in Pseudomonas aeruginosa biofilms. Environ. Microbiol. 15, 2238–2253. doi: 10.1111/1462-2920.12095
Wang, X., and Wood, T. K. (2011). Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl. Environ. Microbiol. 77, 5577–5583. doi: 10.1128/AEM.05068-11
Wang, X. X., Lord, D. M., Cheng, H. Y., Osbourne, D. O., Hong, S. H., Sanchez-Torres, V., et al. (2012). A new type V toxin-antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat. Chem. Biol. 8, 855–861. doi: 10.1038/nchembio.1062
Wang, Y., Hay, I. D., Rehman, Z. U., and Rehm, B. H. (2015). Membrane-anchored MucR mediates nitrate-dependent regulation of alginate production in Pseudomonas aeruginosa. Appl. Microbiol. Biotechnol. 99, 7253–7265. doi: 10.1007/s00253-015-6591-4
Wei, Q., Tarighi, S., Dotsch, A., Haussler, S., Müsken, M., Wright, V. J., et al. (2011). Phenotypic and genome-wide analysis of an antibiotic-resistant small colony variant (SCV) of Pseudomonas aeruginosa. PLoS ONE 6:e29276. doi: 10.1371/journal.pone.0029276
Wen, Y. R., Behiels, E., and Devreese, B. (2014). Toxin- Antitoxin systems: their role in persistence, biofilm formation, and pathogenicity. Pathog. Dis. 70, 240–249. doi: 10.1111/2049-632X.12145
Whiteley, M., Lee, K. M., and Greenberg, E. P. (1999). Identification of genes controlled by quorum sensing in Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. U.S.A. 96, 13904–13909. doi: 10.1073/pnas.96.24.13904
Williams, J. J., Halvorsen, E. M., Dwyer, E. M., DiFazio, R. M., and Hergenrother, P. J. (2011). Toxin-antitoxin (TA) systems are prevalent and transcribed in clinical isolates of Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus. FEMS Microbiol. Lett. 322, 41–50. doi: 10.1111/j.1574-6968.2011.02330.x
Williams, P., and Camara, M. (2009). Quorum sensing and environmental adaptation in Pseudomonas aeruginosa: a tale of regulatory networks and multifunctional signal molecules. Curr. Opin. Microbiol. 12, 182–191. doi: 10.1016/j.mib.2009.01.005
Winstanley, C., O'Brien, S., and Brockhurst, M. A. (2016). Pseudomonas aeruginosa evolutionary adaptation and diversification in cystic fibrosis chronic lung infections. Trends Microbiol. 24, 327–337. doi: 10.1016/j.tim.2016.01.008
Wittgens, A., Tiso, T., Arndt, T. T., Wenk, P., Hemmerich, J., Müller, C., et al. (2011). Growth independent rhamnolipid production from glucose using the non-pathogenic Pseudomonas putida KT2440. Microb. Cell Fact. 10:80. doi: 10.1186/1475-2859-10-80
Wong, A., Rodrigue, N., and Kassen, R. (2012). Genomics of adaptation during experimental evolution of the opportunistic pathogen Pseudomonas aeruginosa. PLoS Genet. 8:e1002928. doi: 10.1371/journal.pgen.1002928
Wood, T. L., and Wood, T. K. (2016). The HigB/HigA toxin/antitoxin system of Pseudomonas aeruginosa influences the virulence factors pyochelin, pyocyanin, and biofilm formation. Microbiologyopen 5, 499–511. doi: 10.1002/mbo3.346
Workentine, M. L., Sibley, C. D., Glezerson, B., Purighalla, S., Norgaard-Gron, J. C., Parkins, M. D., et al. (2013). Phenotypic heterogeneity of Pseudomonas aeruginosa populations in a cystic fibrosis patient. PLoS ONE 8:e60225. doi: 10.1371/journal.pone.0060225
Worlitzsch, D., Tarran, R., Ulrich, M., Schwab, U., Cekici, A., Meyer, K. C., et al. (2002). Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J. Clin. Invest. 109, 317–325. doi: 10.1172/JCI0213870
van 't Wout, E. F. A., van Schadewijk, A., van Boxtel, R., Dalton, L. E., Clarke, H. J., Tommassen, J., et al. (2015). Virulence factors of Pseudomonas aeruginosa induce both the unfolded protein and integrated stress responses in airway epithelial cells. PLoS Pathog. 11:e1004946. doi: 10.1371/journal.ppat.1004946
Wu, J., Long, Q., and Xie, J. (2010). (p)ppGpp and drug resistance. J. Cell. Physiol. 224, 300–304. doi: 10.1002/jcp.22158
Wu, L., Estrada, O., Zaborina, O., Bains, M., Shen, L., Kohler, J. E., et al. (2005). Recognition of host immune activation by Pseudomonas aeruginosa. Science 309, 774–777. doi: 10.1126/science.1112422
Xiong, Y. Q., Caillon, J., Drugeon, H., Potel, G., and Baron, D. (1996). Influence of pH on adaptive resistance of Pseudomonas aeruginosa to aminoglycosides and their postantibiotic effects. Antimicrob. Agents Chemother. 40, 35–39.
Ye, L., Cornelis, P., Guillemyn, K., Ballet, S., and Hammerich, O. (2014). Structure revision of N-mercapto-4-formylcarbostyril produced by Pseudomonas fluorescens G308 to 2-(2-hydroxyphenyl)thiazole-4-carbaldehyde [aeruginaldehyde]. Nat. Prod. Commun. 9, 789–794.
Yoon, S. S., Hennigan, R. F., Hilliard, G. M., Ochsner, U. A., Parvatiyar, K., Kamani, M. C., et al. (2002). Pseudomonas aeruginosa anaerobic respiration in biofilms: relationships to cystic fibrosis pathogenesis. Dev. Cell 3, 593–603. doi: 10.1016/S1534-5807(02)00295-2
Zaborin, A., Romanowski, K., Gerdes, S., Holbrook, C., Lepine, F., Long, J., et al. (2009). Red death in Caenorhabditis elegans caused by Pseudomonas aeruginosa PAO1. Proc. Natl. Acad. Sci. U.S.A. 106, 6327–6332. doi: 10.1073/pnas.0813199106
Zegans, M. E., Wagner, J. C., Cady, K. C., Murphy, D. M., Hammond, J. H., and O'Toole, G. A. (2009). Interaction between bacteriophage DMS3 and host CRISPR region inhibits group behaviors of Pseudomonas aeruginosa. J. Bacteriol. 191, 210–219. doi: 10.1128/JB.00797-08
Zegans, M. E., Wozniak, D., Griffin, E., Toutain-Kidd, C. M., Hammond, J. H., Garfoot, A., et al. (2012). Pseudomonas aeruginosa exopolysaccharide Psl promotes resistance to the biofilm inhibitor polysorbate 80. Antimicrob. Agents Chemother. 56, 4112–4122. doi: 10.1128/AAC.00373-12
Zhang, L., Li, X. Z., and Poole, K. (2001). Fluoroquinolone susceptibilities of efflux-mediated multidrug-resistant Pseudomonas aeruginosa, Stenotrophomonas maltophilia and Burkholderia cepacia. J. Antimicrob. Chemother. 48, 549–552. doi: 10.1093/jac/48.4.549
Zhao, J., Jiang, H., Cheng, W., Wu, J., Wang, J., and Dong, L. (2015). The role of quorum sensing system in antimicrobial induced ampC expression in Pseudomonas aeruginosa biofilm. J. Basic Microbiol. 55, 671–678. doi: 10.1002/jobm.201300987
Zhao, K., Tseng, B. S., Beckerman, B., Jin, F., Gibiansky, M. L., Harrison, J. J., et al. (2013). Psl trails guide exploration and microcolony formation in Pseudomonas aeruginosa biofilms. Nature 497, 388–391. doi: 10.1038/nature12155
Keywords: Pseudomonas aeruginosa, virulence, biofilm, antibiotic resistance, persistence
Citation: Moradali MF, Ghods S and Rehm BHA (2017) Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell. Infect. Microbiol. 7:39. doi: 10.3389/fcimb.2017.00039
Received: 20 December 2016; Accepted: 02 February 2017;
Published: 15 February 2017.
Edited by:
Ghassan M. Matar, American University of Beirut, LebanonReviewed by:
Eduard Torrents, Institute for Bioengineering of Catalonia, SpainXingmin Sun, University of South Florida, USA
Copyright © 2017 Moradali, Ghods and Rehm. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bernd H. A. Rehm, Yi5yZWhtQG1hc3NleS5hYy5ueg==
†Present Address: M. Fata Moradali, Department of Oral Biology, College of Dentistry, University of Florida, Gainesville, FL, USA