Abstract
There are currently three oral medications approved for the treatment of multiple sclerosis (MS). Two of these medications, Fingolimod, and Teriflunomide, are considered to be anti-inflammatory agents, while dimethyl fumarate (DMF) is thought to trigger a robust antioxidant response, protecting vulnerable cells during an MS attack. We previously proposed that epsilon toxin from the gut bacterium, Clostridium perfringens, may initiate newly forming MS lesions due to its tropism for blood-brain barrier (BBB) vasculature and central nervous system myelin. Because gut microbiota will be exposed to these oral therapies prior to systemic absorption, we sought to determine if these compounds affect C. perfringens growth in vitro. Here we show that Fingolimod, Teriflunomide, and DMF indeed inhibit C. perfringens growth. Furthermore, several compounds similar to DMF in chemical structure, namely α, β unsaturated carbonyls, also known as Michael acceptors, inhibit C. perfringens. Sphingosine, a Fingolimod homolog with known antibacterial properties, proved to be a potent C. perfringens inhibitor with a Minimal Inhibitory Concentration similar to that of Fingolimod. These findings suggest that currently approved oral MS therapies and structurally related compounds possess antibacterial properties that may alter the gut microbiota. Moreover, inhibition of C. perfringens growth and resulting blockade of epsilon toxin production may contribute to the clinical efficacy of these disease-modifying drugs.
Introduction
Multiple sclerosis (MS) is the most common non-traumatic neurological disease of young adults in Western Europe and North America (Conway and Cohen, 2010). Although traditionally considered an autoimmune disease that specifically targets central nervous system myelin (Frohman et al., 2006), increasingly, investigators have been pursuing the idea that host-pathogen interactions may play a role in the MS disease process (Collins et al., 2012). Indeed, investigations into how the gut microbiota may trigger or modulate MS relapses are currently under way. With the advent of the first oral treatments for MS, Fingolimod, Teriflunomide, and dimethyl fumarate (DMF), a reasonable question arises. Do these oral medications possess antibacterial properties, and if so, could modulation of gut bacteria contribute to protection against MS relapse?
We have previously proposed that a bacterial neurotoxin, epsilon toxin, from the anaerobic gut bacterium, Clostridium perfringens, may play a pivotal role in triggering newly forming MS lesions (Rumah et al., 2013, 2015; Linden et al., 2015). Epsilon toxin (ETX) is a rational candidate MS trigger due to its tropism for the blood-brain barrier (BBB) and for the myelin sheath; both of which are specifically damaged during each MS relapse (Dorca-Arévalo et al., 2008; Rumah et al., 2013; Linden et al., 2015). Remarkably, newly forming MS lesions display evidence of BBB breakdown, oligodendrocyte cell death and early microglial activation in the absence of a peripheral inflammatory infiltrate (Barnett and Prineas, 2004). While the triggering agent of these early pathologic changes remains unknown, C. perfringens epsilon toxin serves as a provocative candidate due to its tissue specificity and resultant mechanistic plausibility (Rumah et al., 2013, 2015; Linden et al., 2015).
C. perfringens is an anaerobic, spore forming, gram-positive bacillus that is sub-classified into five distinct toxinotypes based on differential exotoxin production (Table 1). C. perfringens type A typically colonizes the human gut with a prevalence of 63% among healthy individuals (Carman et al., 2008), while C. perfringens types B and D, the producers of ETX, are commonly found in the intestines of ruminant animals such as sheep, goats, and cattle but not humans (Popoff, 2011). ETX toxin is a potent neurotoxin secreted as a 33 kDa inactive precursor during the logarithmic growth phase of C. perfringens in the mammalian intestine. This poorly active precursor is cleaved by gut trypsin, chymotrypsin and/or an additional clostridial exotoxin, lamda toxin. The 28.6 kDa enzymatic cleavage product permeablizes the gut epithelium, enters the blood stream and binds to receptors on the luminal surface of brain endothelial cells. Once bound to brain microvessels, ETX oligomerizes and forms a heptameric pore in the endothelial cell plasma membrane. Brain endothelial cell damage leads to breakdown of the BBB (Popoff, 2011). In addition to its known effects on BBB vasculature, ETX has been found to specifically bind to and damage myelin when incubated with mammalian brain slices (Dorca-Arévalo et al., 2008; Linden et al., 2015; Wioland et al., 2015). This unique ability to interact specifically with the tissues that are damaged in MS, the BBB, and CNS myelin, makes it a promising candidate as an environmental MS trigger.
Table 1
| A | α | Humans: Gangrene, toxic enteritis, food poisoning, sporadic diarrhea, some cases of SIDS. |
| Fowl: Necrotic enteritis. | ||
| Foals, pigs: Diarrhea. | ||
| B | α, β, ε | Newborn Lambs: Dysentery. |
| Newborn Calves and Foals: Hemorrhagic enteritis. | ||
| Sheep and Goats: Enterotoxemia. Focal symmetric encephalomalacia. | ||
| C | α, β | Humans: Necrotic enteritis (Pigbel). |
| Piglets, Lambs, Calves and Foals: Necrotic enteritis. | ||
| Sheep: Enterotoxemia. | ||
| D | α, ε | Lambs, Sheep, Calves and Goats: Enterotoxemia. Focal symmetric encephalomalacia. |
| E | α, ι | Calves: Enterotoxemia. |
Clostridium perfringens toxinotypes, genotypes, and associated diseases.
Fingolimod was the first oral therapy to be approved for the treatment of MS. It was rationally engineered from the antifungal molecule Myriocin, which was later shown to possess immunosuppressive properties. Fingolimod and Myriocin are both structurally homologous to sphingosine, a lipid that is a necessary component of cell membrane sphingolipids (Strader et al., 2011). Similar to Myriocin, sphingosine is also known to possess antimicrobial properties. However, while Myriocin is antifungal in nature, sphingosine is antibacterial (Fischer et al., 2012). Interestingly, Fingolimod has been shown to mimic sphingosine's antibacterial properties by protecting the cystic fibrosis transmembrane conductance regulator (CFTR) knockout mouse from luminal airway infection by Pseudomonas auerginosa (Pewzner-Jung et al., 2014).
In the context of MS, Fingolimod is phosphorylated in the bloodstream and subsequently binds to the lymphocyte sphingosine-1-phosphate receptor 1 (S1PR1), causing rapid internalization of S1PR1. In the absence of surface S1PR1, lymphocytes are unable to egress from lymphoid tissues and cannot traffic to target tissues such as the brain; thus the rationale that Fingolimod may reduce the risk of MS relapse and the severity of attacks through immune modulation (Strader et al., 2011).
Teriflunomide is the active metabolite of the immunosuppressant Lenflunomide, which is currently approved for the treatment of rheumatoid arthritis (Munier-Lehmann et al., 2013). Teriflunomide inhibits de novo pyrimidine synthesis in rapidly dividing cells such as clonally expanding lymphocytes, potentially mitigating an autoimmune attack against myelin. More specifically, Teriflunomide non-competitively inhibits dihydroorotate dehydrogenase, an enzyme involved in the first step of de novo pyrimidine synthesis. Memory B cells and T cells remain unaffected by Teriflunomide as they divide more slowly and can synthesize DNA by utilizing the pyrimidine salvage pathway (Bar-Or et al., 2014). Interestingly, dihydroorotate dehydrogenase inhibitors have been shown to arrest the growth of unicellular organisms such as plasmodium falciparum presumably by inhibiting de novo pyrimidine synthesis (Pavadai et al., 2016).
DMF is a fumaric acid ester, which was originally investigated for use as an antimicrobial preservative1. It was first used therapeutically to treat psoriasis based on a hypothesis that psoriasis is caused by a defect in fumarate mediated carbohydrate metabolism in the skin. In the early 2000s, a German neurologist noticed that MS patients taking DMF for concurrent psoriasis showed stabilization of their MS symptoms and a reduction in relapse rates (Phillips and Fox, 2013).
DMF has been shown to react with thiol-containing molecules such as the cellular antioxidant, glutathione, and the cysteine residues of proteins via a chemical reaction called the Michael addition (Brennan et al., 2015). Although DMF initially depletes mammalian cells of glutathione, its proposed protective action in MS stems from its ability to alkylate key cysteine residues in the redox sensitive protein Kelch-Like ECH-Associated Protein 1 (Keap1). Keap1 normally inhibits Nuclear factor (erythroid-derived 2)-like 2 (Nrf2) from translocating to the nucleus and activating antioxidant gene expression. When the cysteine residues of Keap1 are oxidized by reactive oxygen species (ROS) or organic electrophiles such as DMF, Keap1 dissociates from Nrf-2, allowing nuclear translocation to occur. This elicits a robust antioxidant cellular response. The initial decrease in cellular glutathione after DMF treatment is followed by a sharp glutathione increase via the Nrf-2 pathway, which may protect vulnerable cells in MS (Phillips and Fox, 2013).
Although Fingolimod, Teriflunomide, and DMF have proposed mechanisms for how they protect the central nervous system from MS mediated damage, one unexplored possibility is that these orally administered agents may inhibit the growth of neurotoxin-secreting gut bacteria. Because, during log-phase growth, C. perfringens secretes ETX, a toxin that specifically targets the BBB and the myelin sheath, we chose to investigate the effect of these oral MS therapies on C. perfringens growth in vitro.
Methods
Drugs and compounds
All drugs and compounds used in this study were purchased from Sigma Aldrich.
Bacterial strains and growth conditions
C. perfringens ATCC 13124 (type A), ATCC 3626 (type B), ATCC 51880 (type C), ATCC 3631 (type D), ATCC 27324 (type E), and two type A clinical isolates provided by New York Presbyterian Hospital were used for the initial screen while the “type strain,” ATCC 13124, was used for all subsequent experiments. Bacteria were cultured anaerobically at 37°C overnight using the GasPak 100 system (BD). Anaerobiosis was achieved by pre-reducing the culture media using an anaerobic jar containing a GasPak EZ Anaerobe System sachet for a minimum of 6 h before inoculation. After inoculation, the GasPak sachet was replaced for the overnight culture.
Experimental procedures
The compounds used for the initial growth inhibition screen were diluted to a final concentration of 500 μg/ml in Mueller Hinton broth (BD) and media was inoculated with 5 × 106 colony-forming units (CFUs) of different C. perfringens strains. Minimal Inhibitory Concentration values (≥95% growth inhibition, MIC95) were determined for inhibitory compounds using cation adjusted Mueller Hinton II broth (BD). Inhibitory compounds were diluted serially from 512 μg/ml down to 0.5 μg/ml, inoculated with 5 × 105 CFUs of C. perfringens and then anaerobically cultured at 37°C overnight as previously described. Culture conditions for each compound were performed in triplicate and bacterial growth was determined by measuring OD600-values from 1 ml of re-suspended bacteria.
Statistical analysis
Results are representative of data obtained from repeated independent experiments. Each value represents the mean ± SD for three replicates. Statistical analysis was performed using the two-tailed Student t-test (GraphPad Software, San Diego, CA, USA).
Results
With renewed interest in gut bacteria and their potential involvement in the pathogenesis of MS (Bhargava and Mowry, 2014), we wished to determine if oral disease-modifying drugs (DMDs) have the ability to modulate growth of C. perfringens since type B and D strains secrete ETX during log-phase growth. Therefore, we tested if oral DMDs affected the growth of C. perfringens toxinotypes A–E. We compared the growth inhibitory effects of Fingolimod, DMF, and Teriflunomide to that of oral symptom management drugs (SMDs) Baclofen, Bupropion, and Gabapentin; drugs not thought to alter the disease course of MS. We exposed C. perfringens cultures to 500 μg/ml of each compound, allowed for overnight anaerobic growth, and determined the optical density (OD600) the following day. We found that each oral DMD significantly inhibited all C. perfringens toxinotypes and strains tested, while the oral SMDs did not (Figure 1A). We then plotted the minimal inhibitory concentration (MIC) values for each inhibitory compound and found that Fingolimod was the most potent inhibitor at 4 μg/ml (Figure 1B).
Figure 1
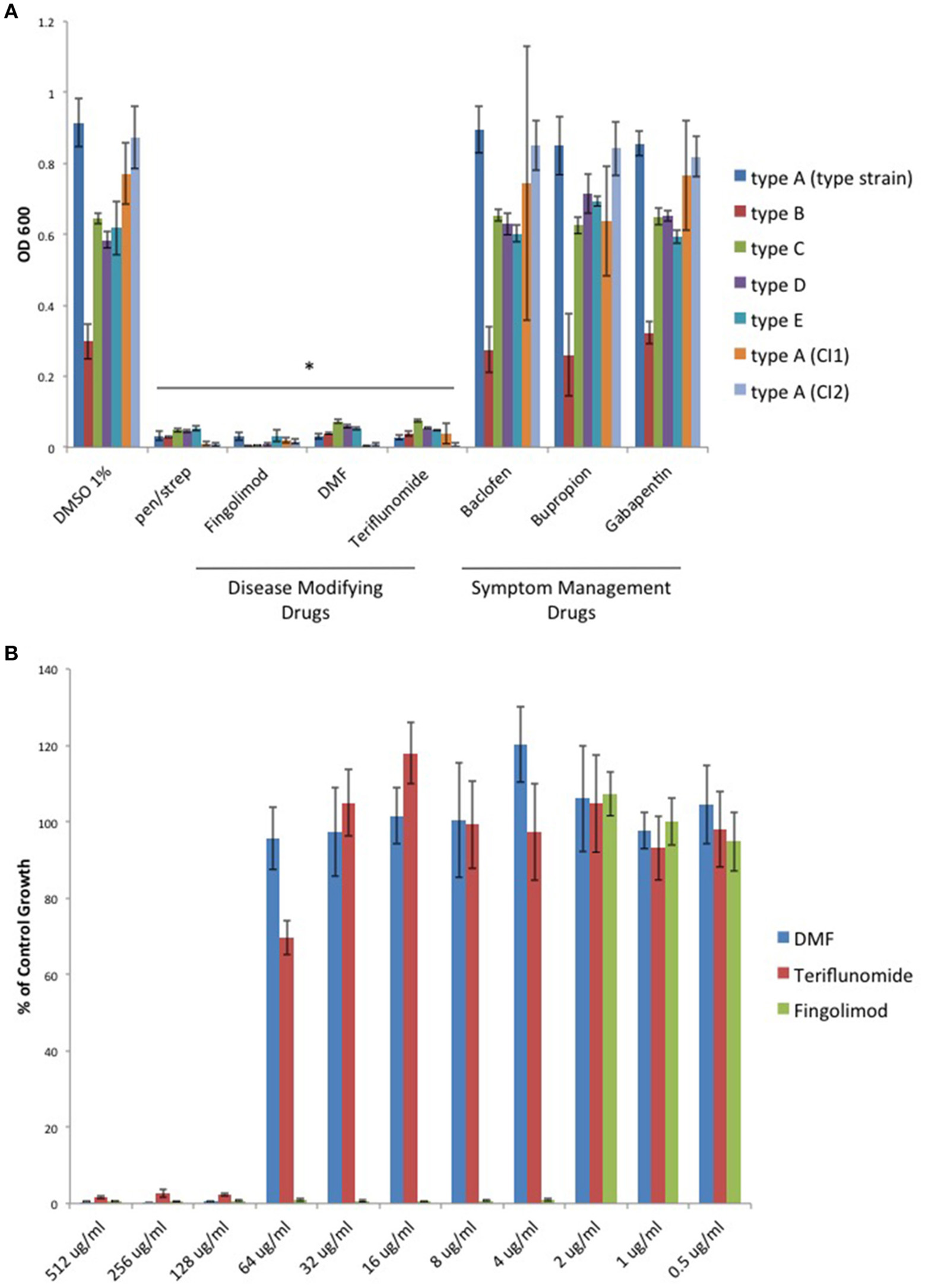
Oral disease modifying MS drugs inhibit C. perfringens growth, while oral drugs used for MS symptom management do not. (A) C. perfringens toxinotypes A, B, C, D, E, and two type A clinical isolates (CI1 and CI2) were anaerobically cultured in the presence of 500 μg/ml of oral DMDs, Fingolimod, DMF, and Teriflunomide, each of which significantly inhibited bacterial growth for all strains tested, similar to what was observed when bacteria were cultured in the presence of known antibiotic mixture penicillin/streptomycin (pen/strep, 100 U/ml). In contrast, oral SMDs failed to inhibit C. perfringens growth, yielding OD600-values similar to that of the DMSO vehicle control. Data are presented as means from three independent experiments. Error bars represent standard deviations, and asterisks indicate that results are statistically significant compared with the DMSO vehicle controls (Student's t-test, *P < 0.001). (B) Serial dilutions of inhibitory oral DMDs were performed and the type strain, C. perfringens ATCC 13124 (type A), was cultured in each condition. OD600-values for each concentration were divided by that of the corresponding dilution for the DMSO vehicle control. MIC-values were plotted for each oral DMD revealing that Fingolimod was the most potent compound with an MIC95 of 4 μg/ml, compared to 128 μg/ml for DMF and Teriflunomide.
Because Fingolimod is a homolog of D-sphingosine and Myriocin, both of which have been shown to possess antimicrobial properties (Fischer et al., 2012), we compared the inhibitory activity of Fingolimod to these related sphingoid molecules. We exposed the type strain, C. perfringens ATCC 13124 (type A), to Fingolimod, D-sphingosine, and Myriocin and identified that, like Fingolimod, D-sphingosine also displayed inhibitory activity. Myriocin failed to inhibit C. perfringens, but instead, enhanced bacterial growth (Figure 2A). We then plotted and compared MICs for Fingolimod and D-sphingosine and determined that D-sphingosine displayed a similar inhibitory potency to that of Fingolimod with an MIC95 of 4 μg/ml (Figure 2B).
Figure 2
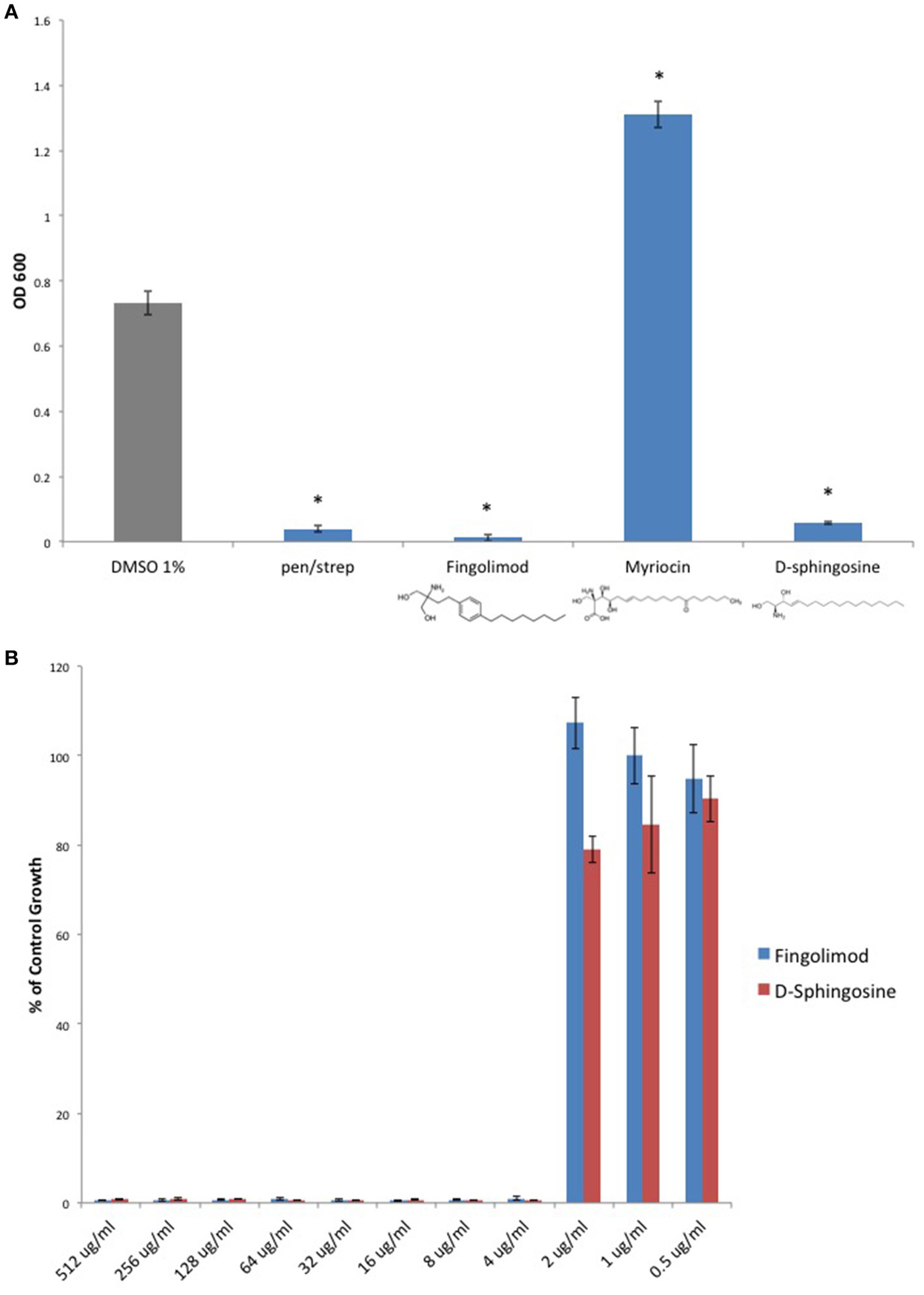
D-sphingosine, a Fingolimod related compound, inhibits C. perfringens growth. (A) Type strain, C. perfringens ATCC 13124, was anaerobically cultured in the presence of 500 μg/ml Fingolimod and other sphingoid compounds, D-sphingosine and Myriocin. Like Fingolimod, D-sphingosine also inhibited C. perfringens growth. However, Myriocin failed to inhibit the bacterium. Instead, Myriocin enhanced C. perfringens growth above that of the DMSO vehicle control. Data are presented as means from three independent experiments. Error bars represent standard deviations, and asterisks indicate that results are statistically significant compared with the DMSO vehicle control (gray); Student's t-test, *P < 0.0001. (B) Serial dilutions of inhibitory sphingoid molecules were performed and C. perfringens ATCC 13124 was cultured at each dilution. OD600-values for each dilution were divided by that of the corresponding DMSO vehicle control dilution. MIC-values were plotted for each of the inhibitory sphingoid compounds revealing that D-sphingosine mimics Fingolimod's antibacterial potency with an MIC95-value of 4 μg/ml.
Although an antioxidant mechanism has been proposed for how DMF protects cells against MS mediated damage, DMF was originally investigated for use as an antimicrobial compound1. Interestingly, DMF was also found to inhibit the growth of Clostridium botulinum (Dymicky et al., 1987), a bacterial species closely related to C. perfringens. DMF is known to be a Michael acceptor and its ability to affect the redox status of cells stems from its electrophilic nature. Michael acceptors accept electrons during the Michael reaction, while nucleophilic thiols (Michael donors) donate electrons. The Michael reaction results in covalent alkylation of the sulfhydryl group. This covalent linkage permanently inactivates thiol-containing molecules if the thiol is necessary for the molecule's function, as is the case for glutathione and its antioxidant properties (Brennan et al., 2015).
We sought to determine if DMF's antimicrobial activity extended to C. perfringens. Furthermore, we examined DMF's Michael acceptor activity as pertaining to its antimicrobial properties. We screened DMF and its metabolites, monomethyl fumarate (MMF), and fumarate and found that each compound inhibited the growth of type strain, C. perfringens ATCC 13124. However, their saturated succinate counterparts dimethyl succinate (DMS), monomethyl succinate (MMS), and succinate, molecules devoid of Michael acceptor activity due to reduction of the α, β carbon double bond, failed to inhibit C. perfringens (Figure 3A). We plotted MIC-values for DMF, MMF, and fumarate and found that DMF was four times more potent than either MMF or fumarate (Figure 3B).
Figure 3
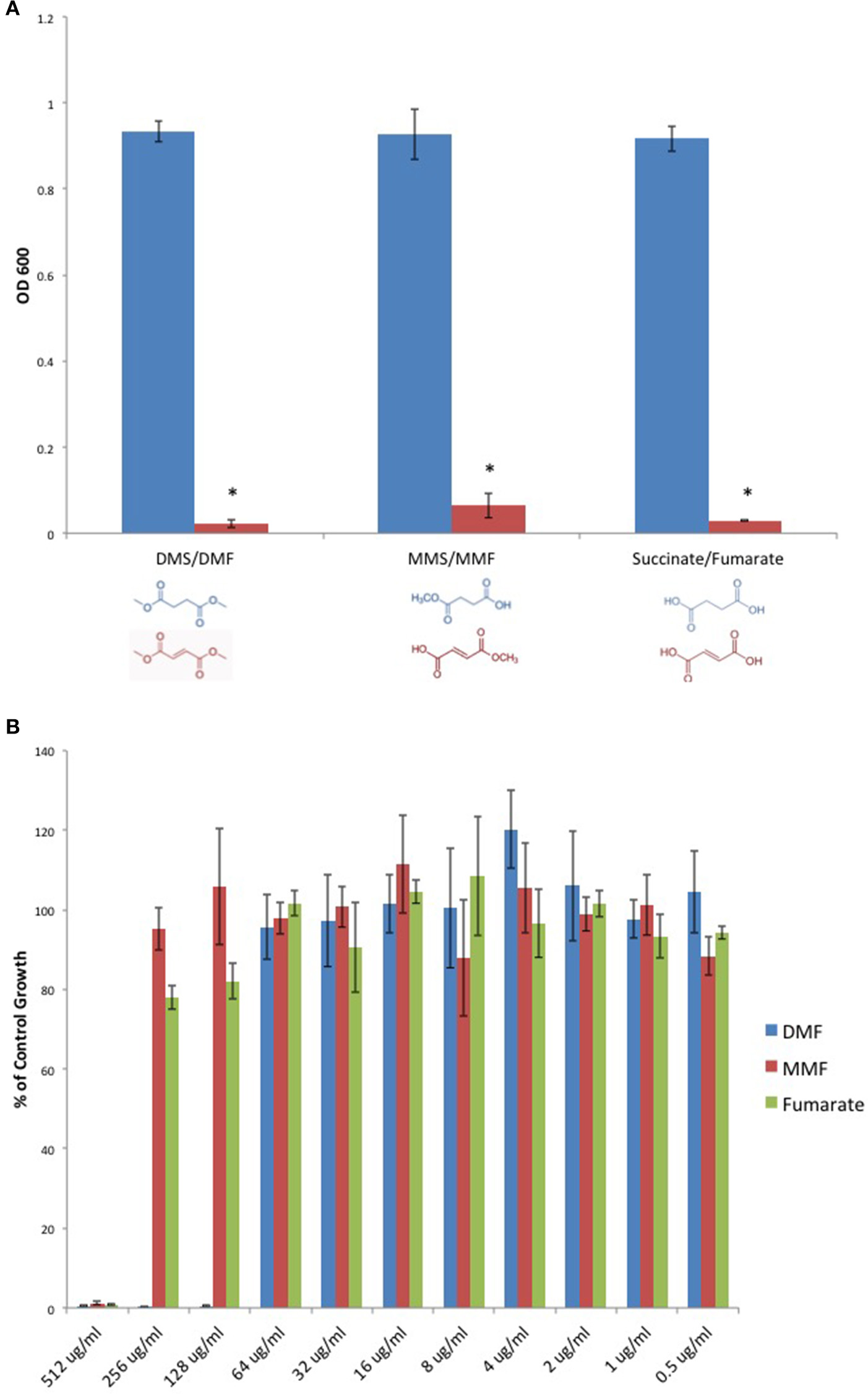
Fumarates inhibit C. perfringens growth while their related saturated succinates do not. (A) The anaerobic growth of C. perfringens ATCC 13124 was compared for DMF and its corresponding succinate (DMS), MMF and its corresponding succinate MMS, fumarate, and succinate. In each case, the unsaturated fumarate compounds (red) displayed inhibitory activity, while the saturated succinates (blue) did not. Data are presented as means from three independent experiments. Error bars represent standard deviations, and asterisks indicate that the inhibition of bacterial growth observed in the presence of unsaturated fumarates is statistically significant when compared to the bacterial growth observed in the presence of corresponding saturated succinates; Student's t-test, *P < 0.0001). (B) Serial dilutions of inhibitory fumarate compounds were performed and C. perfringens ATCC 13124 was cultured at each dilution. OD600-values for each dilution were divided by that of the corresponding DMSO vehicle control dilution. MIC-values were plotted for each of the inhibitory fumarates revealing that DMF is approximately four times more potent that MMF and fumarate.
Given that Michael acceptor activity was necessary for C. perfringens inhibition by DMF and its fumarate metabolites, we sought to determine if unrelated molecules that share the α, β unsaturated carbonyl structure could also inhibit C. perfringens. We screened Michael acceptors from a diverse group of chemical families and found that natural product Michael acceptors Gambogic acid (a xanthonoid), Parthenolide (a sesquiterpenoid), and Curcumin (a curcuminoid) each inhibited C. perfringens (Figure 4A). Interestingly, we found that Gambogic acid was particularly inhibitory with an MIC95 of 1 μg/ml (Figure 4B).
Figure 4
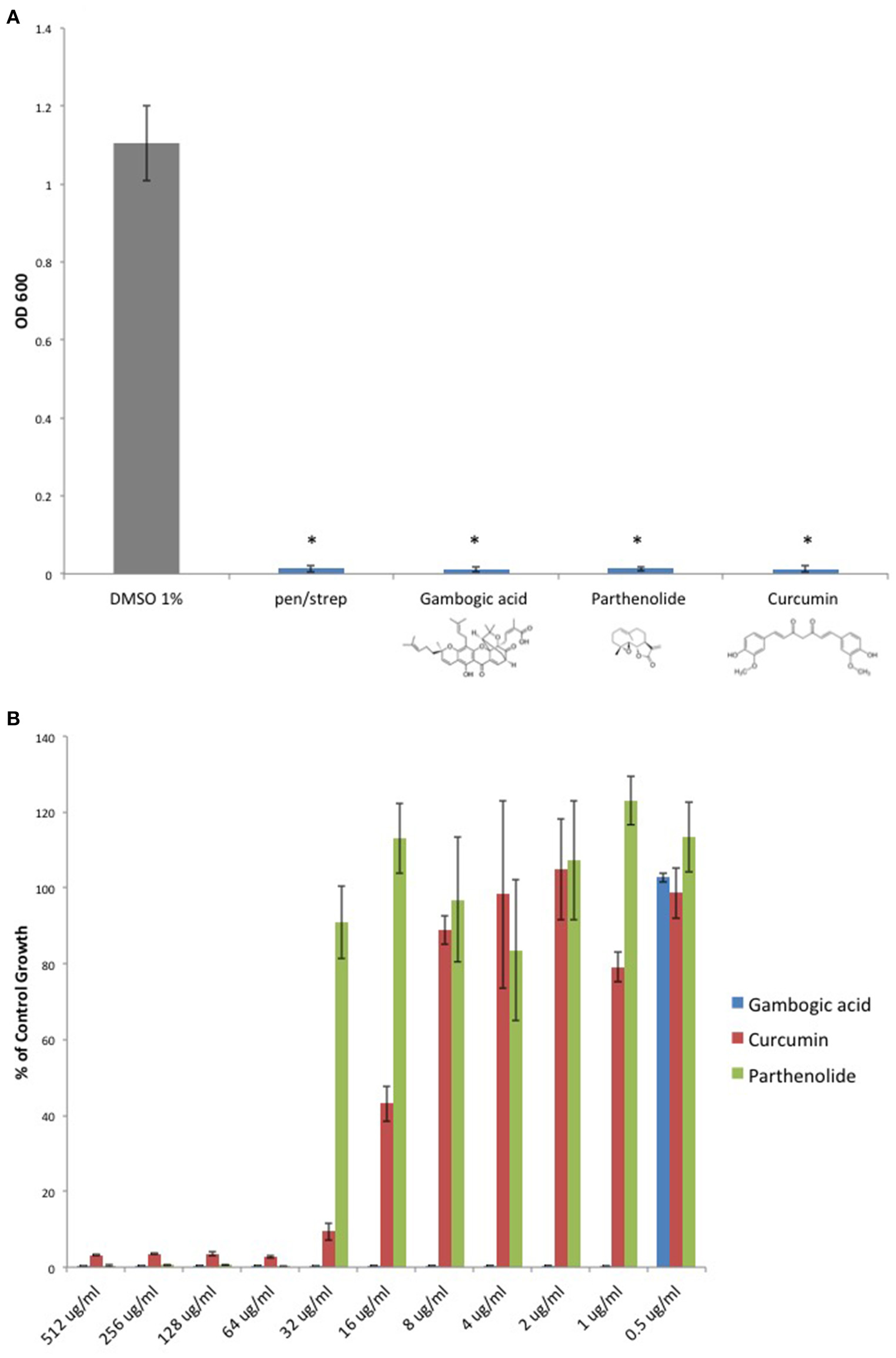
Natural product Michael acceptors inhibit C. perfringens growth. (A) Plant derived Michael acceptors were tested for inhibitory activity against C. perfringens ATCC 13124. Bacteria were grown anaerobically in the presence of 500 μg/ml Gambogic acid, Parthenolide, and Curcumin. Each natural product Michael acceptor successfully inhibited C. perfringens growth, similar to what was observed when bacteria were cultured in the presence of known antibiotic penicillin/streptomycin (pen/strep, 100 U/ml). Data are presented as means from three independent experiments. Error bars represent standard deviations, and asterisks indicate that results are statistically significant compared with the DMSO vehicle control (gray); Student's t-test, *P < 0.0001. (B) Serial dilutions of inhibitory natural product Michael acceptors were performed and C. perfringens ATCC 13124 was cultured at each dilution. OD600-values for each dilution were divided by that of the corresponding DMSO vehicle control dilution. MIC-values were plotted for each compound revealing Gambogic acid as the most potent with an MIC95 of 1 μg/ml when compared to Parthenolide and Curcumin, which each inhibit C. perfringens at 64 μg/ml.
To provide additional evidence that Michael acceptor activity is indeed critical to the antibacterial properties of α, β unsaturated carbonyls, we searched the literature to find compounds for which experimental values of Michael reaction potencies have been determined. Dinkova-Kostova et al. determined the potencies of several plant derived Michael acceptors for their ability to induce cellular quinone reductase activity; a cellular marker for a compound's reactivity with sulfhydryl containing molecules (Dinkova-Kostova et al., 2001). In our study, the antibacterial potencies of Cinnamic acid, trans-Chalcone, and Curcumin mirrored the Michael acceptor potencies described by Dinkova-Kostova and colleagues (Figure 5). Cinnamic acid was previously shown to be inactive as a Michael acceptor, and in our hands, this compound failed to inhibit C. perfringens. Furthermore, Curcumin was found to be approximately four times more potent than trans-Chalcone (ratio = 4.13; Dinkova-Kostova et al., 2001). Likewise, we found that Curcumin was four times more potent than trans-Chalcone as a C. perfringens inhibitor (ratio = 4).
Figure 5
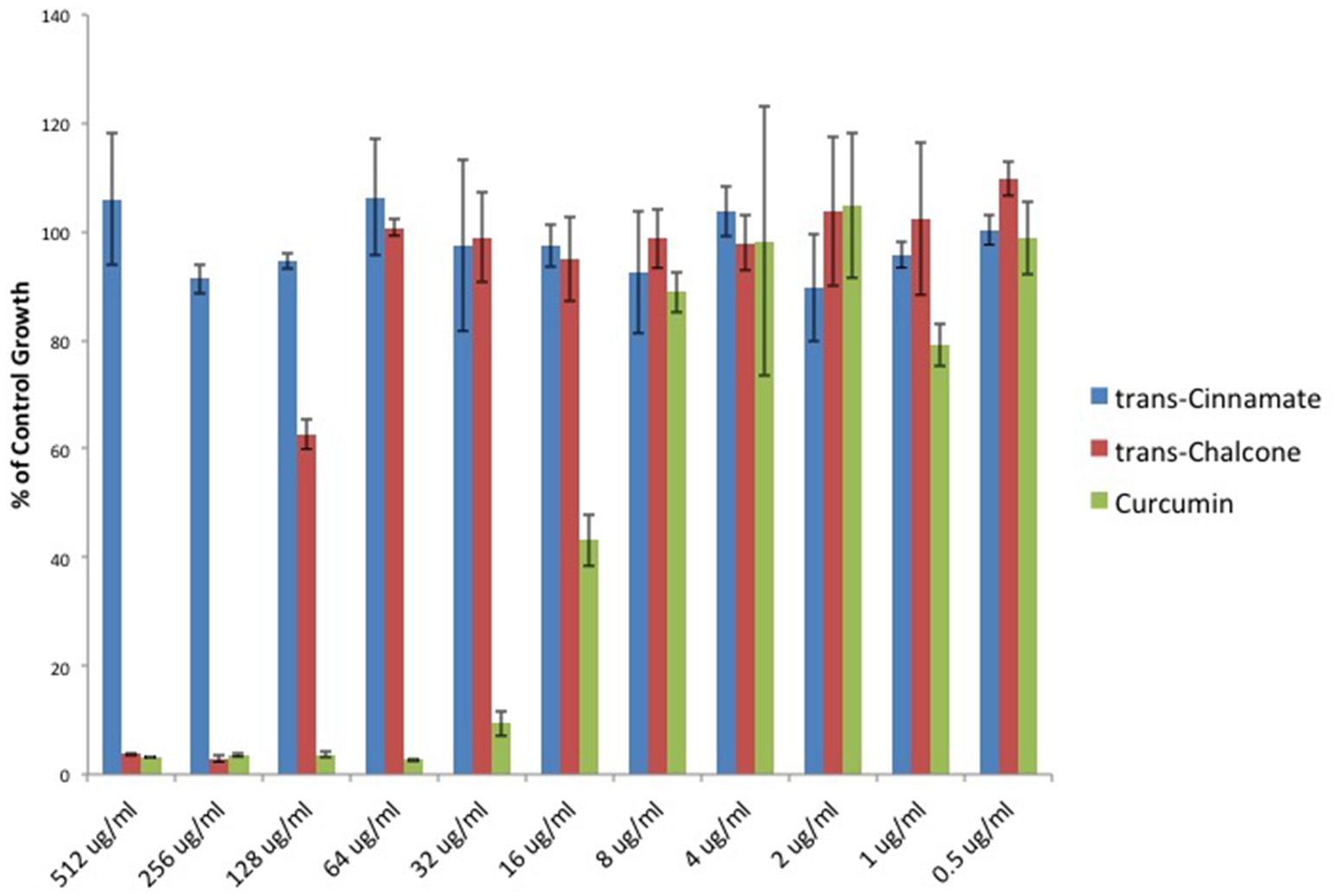
Michael acceptor C. perfringens growth inhibition reflects known reactivity potencies. Serial dilutions of α, β unsaturated carbonyls were performed and C. perfringens ATCC 13124 was cultured at each dilution. OD600-values for each dilution were divided by that of the corresponding DMSO vehicle control dilution. MIC-values were plotted for each compound revealing that Cinnamic acid displays no inhibitory activity, while Curcumin is four times more potent than trans-Chalcone. The relative inhibitory potencies of each compound corresponds almost exactly with the relative Michael acceptor potencies as demonstrated by Dinkova-Kostova et al. (2001).
Since Michael acceptors react with thiols and deplete cellular glutathione levels (Brennan et al., 2015), we surmised that Michael acceptor inhibition of C. perfringens might be abolished by the addition of exogenous glutathione. To test this, we compared C. perfringens growth in the presence of Michael acceptors with and without an equal quantity of exogenous glutathione. We also tested the effect of glutathione on the inhibitory activity of each of the oral MS DMDs. Glutathione completely abolished growth inhibition by the known Michael acceptors in our study, but failed to abolish the inhibitory effects of Fingolimod and Teriflunomide (Figure 6A). Because glutathione is an antioxidant, as are vitamins C and E, we sought to determine if the glutathione's abrogation of Michael acceptor antibacterial activity is based on its nucleophilic behavior or due, more generally, to its antioxidant properties. C. perfringens was challenged with DMF in the presence of vitamin C, vitamin E, or the Michael donor, glutathione. Of the antioxidant panel, only the Michael donor, glutathione, was able to neutralize DMF's inhibitory effect (Figure 6B).
Figure 6
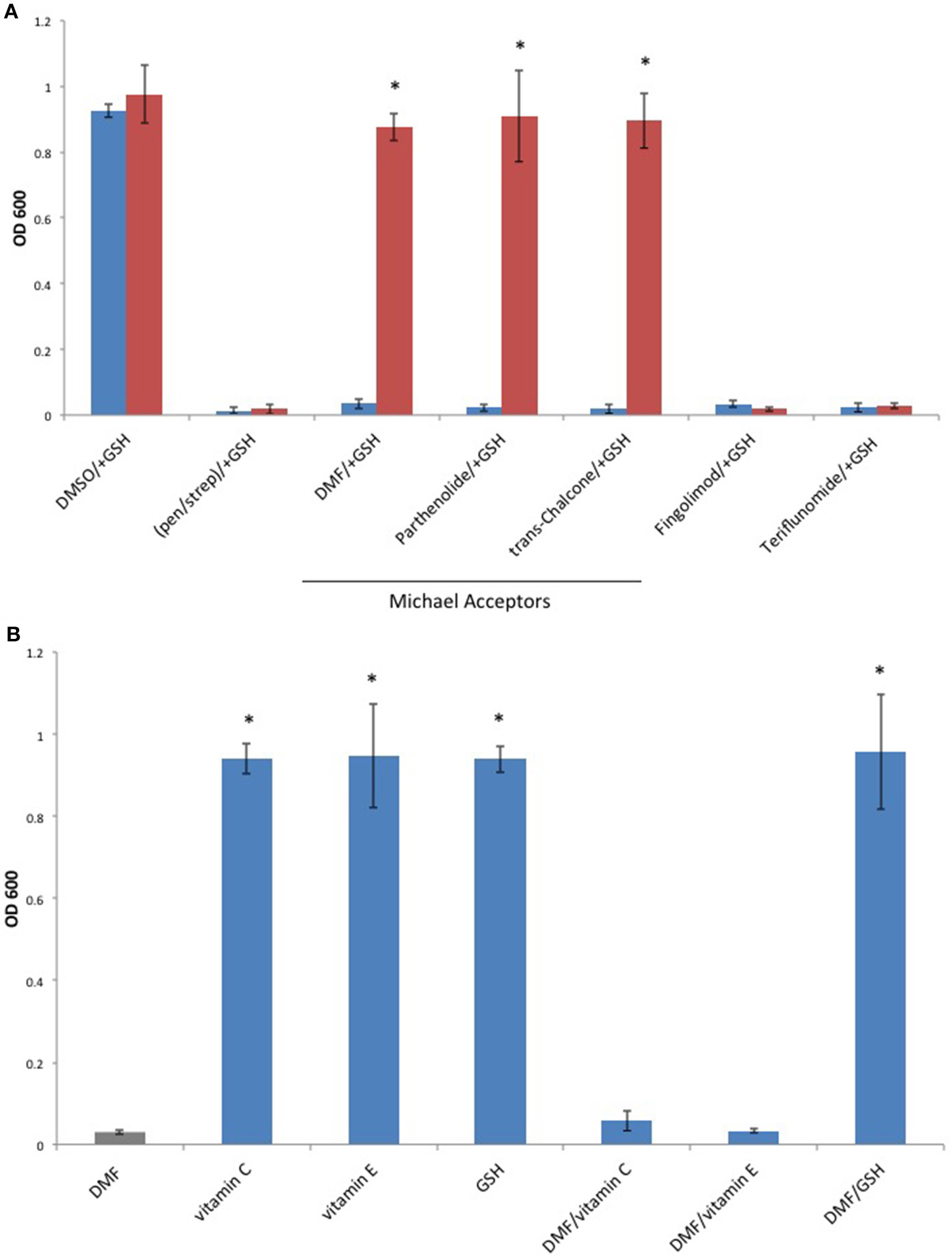
The Michael donor, glutathione (GSH), abolishes Michael acceptor inhibition of C. perfringens growth. (A) C. perfringens ATCC 13124 was cultured anaerobically at a Michael acceptor (DMF, Parthenolide, and trans-Chalcone), and non-Michael acceptor (Fingolimod and Teriflunomide) concentration of 500 μg/ml, with (red) or without (blue) the addition of an equal quantity of exogenous Michael donor, GSH. Only Michael acceptor mediated growth inhibition could be abolished by the addition of exogenous GSH. The inhibitory activity Fingolimod and Teriflunomide remained unaffected by the presence of GSH, similar to what was observed when bacteria were cultured in the presence of pen/strep and GSH. Data are presented as means from three independent experiments. Error bars represent standard deviations, and asterisks indicate that GSH aided growth recovery is statistically significant when compared to the lack of growth recovery in the absence of GSH; Student's t-test, *P < 0.001. (B)C. perfringens ATCC 13124 was cultured in the presence of DMF, vitamin C, vitamin E, and GSH each at concentration of 250 μg/ml. Only the Michael acceptor, DMF, inhibited bacterial growth. DMF was then paired with the antioxidants, vitamin C, vitamin E, and GSH at concentrations of 250 μg/ml for each compound. The Michael donor antioxidant, GSH, abolished DMF inhibition. However, the non-Michael donor antioxidants, vitamin C and vitamin E, were unable to abolish DMF's inhibitory effect on C. perfringens growth. Data are presented as means from three independent experiments. Error bars represent standard deviations, and asterisks indicate that results are statistically significant compared with the DMF control (gray); Student's t-test, *P < 0.001.
Discussion
In this study, we have shown that each of the oral DMDs approved for the treatment of MS, Fingolimod, Teriflunomide, and DMF, inhibits the in vitro growth of the epsilon toxin-secreting gut bacterium, C. perfringens. In contrast, oral therapies used specifically for symptomatic management fail to prevent C. perfringens growth. Of note, Fingolimod proved to be bactericidal, while Teriflunomide and DMF were bacteriostatic (Supplemental Figure 1). The antibacterial properties of oral DMDs raises the possibility that modulation of the intestinal microbiota may play a role in the clinical efficacy of these compounds. Preventing C. perfringens growth and toxin production may serve as a specific example of this. Furthermore, we have identified two distinct classes of molecules capable of inhibiting C. perfringens; namely sphingoid compounds such as Fingolimod and D-sphingosine, and Michael acceptors such as DMF, its fumarate metabolites, and various natural products that are α, β unsaturated carbonyls.
Important factors must be considered when attempting to extrapolate these in vitro findings to what may be occurring in the human gut. First, how do the in vitro inhibitory concentrations compare to concentrations found in the human gut? The resting volume of the human stomach is ~0.08 L (Johnson, 1994), which yields a calculated gut concentration of 6.3 μg/ml for Fingolimod (MIC95 = 4 μg/ml), 87–175 μg/ml for Teriflunomide (MIC95 = 128 μg/ml), and 1500–3000 μg/ml for DMF (MIC95 = 128 μg/ml). Therefore, each compound's MIC95 is within the calculated range of the therapeutic concentration that will enter the small intestine. Furthermore, DMF is a delayed released capsule that dissolves in the more basic pH of the small intestine (Gold et al., 2016). Local release of DMF may increase its concentration in the small intestine where C. perfringens resides.
Second, our experimental growth conditions are likely to be more favorable to C. perfringens growth than the intestinal milieu. Anexic, in vitro growth protects C. perfringens from competition with other bacteria for nutrients. In addition, C. perfringens will not be exposed to toxic molecules secreted by competing bacteria such as bacteriocins or host derived antibacterial molecules such as defensins. Therefore, the MIC95 for each of the oral DMDs may be considerably less in an environment such as the human intestine where C. perfringens must contend with a multitude of external factors.
Conversely, each of the oral DMDs possesses a significant degree of hydrophobicity, and lipid-binding molecules within the gut lumen may sequester these compounds, preventing toxic interactions with gut bacteria. Specifically considering DMF, a Michael acceptor, extracellular nucleophiles present in the gut may react with its electrophilic β carbon before it can enter the bacterial cell, possibly diminishing its antibacterial activity within the gut.
DMF's Michael reaction-dependent inhibition of C. perfringens growth may be explained by its ability to deplete this bacterium of thiol containing compounds. It is striking that nucleophilic thiols not only play an important role in mammalian cell homeostasis, but are also necessary substrates for C. perfringens growth. This bacterium depends on an organic source of sulfur (thiols) and will not grow with strictly inorganic sources (, , S2O32−, and Si; Fuchs and Bonde, 1957). Therefore, depleting C. perfringens of thiols may contribute to Michael acceptor mediated growth inhibition.
Although Teriflunomide is an α, β unsaturated carbonyl, we have shown that glutathione has no effect on its ability to inhibit C. perfringens growth. It is tempting to speculate that Teriflunomide inhibits de novo pyrimidine synthesis in rapidly dividing bacterial cells, as it does in mammalian cells, via inhibition of dihydroorotate dehydrogenase; a gene that has been annotated for C. perfringens in the Uniprot Knowledgebase. However, we have not examined the inhibitory mechanism of Teriflunomide in the present study.
That Michael acceptors such as DMF and its fumarate metabolites inhibit C. perfringens may open the door to development of new oral MS therapies derived from the Michael acceptor functional class. Gambogic acid has been used in Eastern medicine for centuries to treat intestinal ailments and parasites (Wu et al., 2004), and in our hands, it displays an impressive antibacterial potency (MIC95 = 1 μg/ml).
We searched for Michael acceptors currently approved for human use that possess no known immunosuppressive properties. The naphthoquinone, Menadione (vitamin K3), is a synthetic precursor for vitamin K. It is commonly used as a dietary supplement for livestock and as a cost effective vitamin K replacement therapy in developing countries. Of note, Menadione has recently been shown to inhibit S. aureus and B. anthracis growth, and to suppress S. aureus secretion of toxic shock syndrome toxin 1 (TSST-1; Schlievert et al., 2013). Similarly, we find that Menadione inhibits C. perfringens growth, but related compounds with long aliphatic side chains, vitamin K1, vitamin K2, and ubiquinone do not (Supplemental Figure 2A). While Menadione's MIC95-value was found to be 64 μg/ml (Supplemental Figure 2B), the inactivity of the Menadione related compounds, all of which are electron carriers in the electron transport chain, might be due to the fact that they are sequestered in the cell membrane by their aliphatic side chains. Membrane sequestration may protect cytosolic nucleophiles from undergoing Michael addition and subsequent depletion. Additionally, unlike Menadione but similar to Teriflunomide, these molecules possess a third σ bond at the β carbon position. This may prevent nucleophilic attack due to steric hindrance and abolish Michael acceptor activity (Schwöbel et al., 2010).
In light of the serious side effects associated with current oral DMDs, this study may be of immediate clinical importance. Some of these adverse effects are due to immunosuppression of the CNS, as evidenced by increased risk of JC virus infection and progressive multifocal leukoencephalopathy (PML, FDA Drug Safety Communication, 2014; Brooks, 2015). Perhaps new antibacterial compounds based on these early oral DMDs, but lacking their immunosuppressive properties, may be of use in treating MS. For example, Fingolimod/D-sphingosine related compounds lacking hydroxyl head groups will not undergo phosphorylation and will not target lymphocyte S1PR1. Such compounds would not be immunosuppressive and may reduce the risk of JC virus infection and the development of PML. Along these lines, we have tabulated the MIC95-values for each inhibitory compound used in this study (Table 2).
Table 2
| Compound | MIC95 (μg/ml) |
|---|---|
| Gambogic Acid | 1 |
| Fingolimod | 4 |
| D-sphingosine | 4 |
| Curcumin | 64 |
| Parthenolide | 64 |
| Menadione (vitamin K3) | 64 |
| Teriflunomide | 128 |
| Dimethyl fumarate (DMF) | 128 |
| Trans-Chalcone | 256 |
| Monomethyl fumarate (MMF) | 512 |
| Fumarate | 512 |
Minimal Inhibitor Concentrations for all inhibitory compounds used in the study.
Funding
This work was generously supported by the Rockefeller University's Robertson Therapeutic Development Fund (RTDF), the Center for Disorders of the Digestive System (CDDS), and the Weill Cornell Tisch Family Research Fund.
Conflict of interest statement
All authors are named as inventors on a pending patent entitled, “Methods to protect against and treat multiple sclerosis,” (Publication number CA2899961 A1), which identifies Clostridium perfringens epsilon toxin as candidate trigger for multiple sclerosis.
Statements
Author contributions
KR conceived the study. KR, VF, and TV designed the study. KR, VF, and TV performed the literature search. KR collected the data and wrote the paper. All authors analyzed the data.
Acknowledgments
We wish to thank Mr. Nick Lewis of Downing LLP for his valuable insights.
Conflict of interest
All authors are named as inventors on a pending patent entitled, “Methods to protect against and treat multiple sclerosis,” (Publication number CA2899961 A1), which identifies Clostridium perfringens epsilon toxin as candidate trigger for multiple sclerosis.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00011/full#supplementary-material
Supplementary Figure 1Fingolimod is bactericidal, while DMF and Teriflunomide are bacteriostatic. C. perfringens ATCC 13124 was anaerobically cultured to stationary phase and exposed to Fingolimod (500 μg/ml), DMF (500 μg/ml), Teriflunomide (500 μg/ml), pen/strep (100 U/ml) or DMSO vehicle control for 4 h under anaerobic conditions. Treated cultures were diluted 1000 fold in fresh, pre-reduced Mueller Hinton broth and cultured anaerobically. Fingolimod inhibited C. perfringens growth in a bactericidal fashion, similar to what was observed with the known bactericidal antibiotic mixture pen/strep, as post-treatment dilution and repeat culture failed to restore bacterial growth. Conversely, DMF and Teriflunomide were shown to be bacteriostatic, as post-treatment dilution and culture successfully restored bacterial growth. Data are presented as means from three independent experiments. Error bars represent standard deviations, and asterisks indicate that results are statistically significant compared with the DMSO vehicle control (gray); Student's t-test, *P < 0.0001.
Supplementary Figure 2Synthetic vitamin K3, Menadione, inhibits C. perfringens growth. (A) C. perfringens ATCC 13124 was anaerobically cultured in the presence of vitamin K homologs, vitamins K1, K2, K3, and ubiquinone. Only the synthetic vitamin K3 (Menadione) inhibited bacterial growth, similar to what was observed when bacteria were cultured in the presence known antibiotic penicillin/streptomycin (pen/strep, 100 U/ml). Conversely, bacteria derived vitamin K2 enhanced C. perfringens growth, while plant derived vitamin K1 and mammalian ubiquinone yielded OD600-values similar to that of the DMSO vehicle control. Data are presented as means from three independent experiments. Error bars represent standard deviations, and asterisks indicate that results are statistically significant compared with the DMSO vehicle control (gray); Student's t-test, *P < 0.001. (B) Serial dilutions of Menadione were performed and C. perfringens ATCC 13124 was cultured at each dilution. OD600-values for each dilution were divided by that of the corresponding DMSO vehicle control dilution. MIC-values were plotted yielding an MIC95-value of 64 μg/ml.
Footnotes
1.^ Pest control. Patent Publication Number: US2218181 A.
References
1
Barnett M. H. Prineas J. W. (2004). Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol.55, 458–468. 10.1002/ana.20016
2
Bar-Or A. Pachner A. Menguy-Vacheron F. Kaplan J. Wiendl H. (2014). Teriflunomide and its mechanism of action in multiple sclerosis. Drugs74, 659–674. 10.1007/s40265-014-0212-x
3
Bhargava P. Mowry E. M. (2014). Gut microbiome and multiple sclerosis. Curr. Neurol. Neurosci. Rep.14:492. 10.1007/s11910-014-0492-2
4
Brennan M. S. Matos M. F. Li B. Hronowski X. Gao B. Juhasz P. et al . (2015). Dimethyl fumarate and monoethyl fumarate exhibit differential effects on KEAP1, NRF2 activation, and glutathione depletion in vitro. PLoS ONE10:e0120254. 10.1371/journal.pone.0120254
5
Brooks M. (2015). Third case of PML with fingolimod (Gilenya) in MS. Medscape. Available online at: http://www.medscape.com/viewarticle/849677
6
Carman R. J. Sayeed S. Li J. Genheimer C. W. Hiltonsmith M. F. Wilkins T. D. et al . (2008). Clostridium perfringens toxin genotypes in the feces of healthy North Americans. Anaerobe14, 102–108. 10.1016/j.anaerobe.2008.01.003
7
Collins S. M. Surette M. Bercik P. (2012). The interplay between the intestinal microbiota and the brain. Nat. Rev. Microbiol.10, 735–742. 10.1038/nrmicro2876
8
Conway D. Cohen J. A. (2010). Combination therapy in multiple sclerosis. Lancet Neurol.9, 299–308. 10.1016/S1474-4422(10)70007-7
9
Dinkova-Kostova A. T. Massiah M. A. Bozak R. E. Hicks R. J. Talalay P. (2001). Potency of Michael reaction acceptors as inducers of enzymes that protect against carcinogenesis depends on their reactivity with sulfhydryl groups. Proc. Natl. Acad. Sci. U.S.A.98, 3404–3409. 10.1073/pnas.051632198
10
Dorca-Arévalo J. Soler-Jover A. Gibert M. Popoff M. R. Martín-Satué M. Blasi J. (2008). Binding of epsilon-toxin from Clostridium perfringens in the nervous system. Vet. Microbiol.131, 14–25. 10.1016/j.vetmic.2008.02.015
11
Dymicky M. Bencivengo M. Buchanan R. L. Smith J. L. (1987). Inhibition of Clostridium botulinum 62A by fumarates and maleates and a relationship of activity to Inhibition of Clostridium botulinum 62A by fumarates and maleates and relationship of activity to some physicochemical constants. Appl. Environ. Microbiol.53, 110–113.
12
FDA Drug Safety Communication (2014). FDA Warns about Case of Rare Brain Infection PML with MS Drug TECFIDERA (Dimethyl Fumarate). Available online at: http://www.fda.gov/Drugs/DrugSafety/ucm424625.htm, Retrieved 2 December 2014.
13
Fischer C. L. Drake D. R. Dawson D. V. Blanchette D. R. Brogden K. A. Wertz P. W. (2012). Antibacterial activity of sphingoid bases and fatty acids against Gram-positive and Gram-negative bacteria. Antimicrob Agents Chemother.56, 1157–1161. 10.1128/AAC.05151-11
14
Frohman E. M. Racke M. K. Raine C. S. (2006). Multiple sclerosis–the plaque and its pathogenesis. N. Engl. J. Med.354, 942–955. 10.1056/NEJMra052130
15
Fuchs A. R. Bonde G. J. (1957). The availability of sulphur for Clostridium perfringens and an examination of hydrogen sulphide production. J. Gen. Microbiol.16, 330–340. 10.1099/00221287-16-2-330
16
Gold R. Arnold D. L. Bar-Or A. Hutchinson M. Kappos L. Havrdova E. et al . (2016). Long-term effects of delayed-release dimethyl fumarate in multiple sclerosis: interim analysis of ENDORSE, a randomized extension study. Mult. Scler. [Epub ahead of print]. 10.1177/1352458516649037
17
Johnson G. B. (1994). Holt Biology: Visualizing Life.Orlando, FL: Holt, Rinehart and Winston, 769.
18
Linden J. R. Ma Y. Zhao B. Harris J. M. Rumah K. R. Schaeren-Wiemers N. et al . (2015). Clostridium perfringens epsilon toxin causes selective death of mature oligodendrocytes and central nervous system demyelination. MBio6:e02513. 10.1128/mBio.02513-14
19
Munier-Lehmann H. Vidalain P. O. Tangy F. Janin Y. L. (2013). On dihydroorotate dehydrogenases and their inhibitors and uses. J. Med. Chem.56, 3148–3167. 10.1021/jm301848w
20
Pavadai E. El Mazouni F. Wittlin S. de Kock C. Phillips M. A. Chibale K. (2016). Identification of new human malaria parasite Plasmodium falciparum dihydroorotate dehydrogenase inhibitors by pharmacophore and structure-based virtual screening. J. Chem. Inf. Model.56, 548–562. 10.1021/acs.jcim.5b00680
21
Pewzner-Jung Y. Tavakoli Tabazavareh S. Grassmé H. Becker K. A. Japtok L. Steinmann J. et al . (2014). Sphingoid long chain bases prevent lung infection by Pseudomonas aeruginosa. EMBO Mol. Med.6, 1205–1214. 10.15252/emmm.201404075
22
Phillips J. T. Fox R. J. (2013). BG-12 in multiple sclerosis. Semin. Neurol.33, 56–65. 10.1055/s-0033-1343796
23
Popoff M. R. (2011). Epsilon toxin: a fascinating pore-forming toxin. FEBS J.278, 4602–4615. 10.1111/j.1742-4658.2011.08145.x
24
Rumah K. R. Linden J. Fischetti V. A. Vartanian T. (2013). Isolation of Clostridium perfringens type B in an individual at first clinical presentation of multiple sclerosis provides clues for environmental triggers of the disease. PLoS ONE8:e76359. 10.1371/journal.pone.0076359
25
Rumah K. R. Ma Y. Linden J. R. Oo M. L. Anrather J. Schaeren-Wiemers N. et al . (2015). The myelin and lymphocyte protein MAL is required for binding and activity of Clostridium perfringens ε-toxin. PLoS Pathog.11:e1004896. 10.1371/journal.ppat.1004896
26
Schlievert P. M. Merriman J. A. Salgado-Pabón W. Mueller E. A. Spaulding A. R. Vu B. G. et al . (2013). Menaquinone analogs inhibit growth of bacterial pathogens. Antimicrob Agents Chemother.57, 5432–5437. 10.1128/AAC.01279-13
27
Schwöbel J. A. Wondrousch D. Koleva Y. K. Madden J. C. Cronin M. T. Schüürmann G. (2010). Prediction of michael-type acceptor reactivity toward glutathione. Chem. Res. Toxicol.23, 1576–1585. 10.1021/tx100172x
28
Strader C. R. Pearce C. J. Oberlies N. H. (2011). Fingolimod (FTY720): a recently approved multiple sclerosis drug based on a fungal secondary metabolite. J. Nat. Prod.74, 900–907. 10.1021/np2000528
29
Wioland L. Dupont J. L. Doussau F. Gaillard S. Heid F. Isope P. et al . (2015). Epsilon toxin from Clostridium perfringens acts on oligodendrocytes without forming pores, and causes demyelination. Cell. Microbiol.17, 369–388. 10.1111/cmi.12373
30
Wu Z. Q. Guo Q. L. You Q. D. Zhao L. Gu H. Y. (2004). Gambogic acid inhibits proliferation of human lung carcinoma SPC-A1 cells in vivo and in vitro and represses telomerase activity and telomerase reverse transcriptase mRNA expression in the cells. Biol. Pharm. Bull.27, 1769–1774. 10.1248/bpb.27.1769
Summary
Keywords
multiple sclerosis, oral therapies, anti-bacterial agents, Clostridium perfringens , microbiome
Citation
Rumah KR, Vartanian TK and Fischetti VA (2017) Oral Multiple Sclerosis Drugs Inhibit the In vitro Growth of Epsilon Toxin Producing Gut Bacterium, Clostridium perfringens. Front. Cell. Infect. Microbiol. 7:11. doi: 10.3389/fcimb.2017.00011
Received
09 September 2016
Accepted
06 January 2017
Published
25 January 2017
Volume
7 - 2017
Edited by
Venkatakrishna Rao Jala, University of Louisville, USA
Reviewed by
Michael L. Vasil, University of Colorado Denver School of Medicine, USA; Caitlin S. L. Parello, Biomodels, LLC, USA
Updates
Copyright
© 2017 Rumah, Vartanian and Fischetti.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kareem R. Rumah rrumah@rockefeller.edu
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.