 Rebecca A. Drummond
Rebecca A. Drummond Michail S. Lionakis
Michail S. Lionakis
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell. Infect. Microbiol. , 05 April 2016
Sec. Fungal Pathogenesis
Volume 6 - 2016 | https://doi.org/10.3389/fcimb.2016.00039
Human CARD9 deficiency is an autosomal recessive primary immunodeficiency disorder caused by biallelic mutations in the gene CARD9, which encodes a signaling protein that is found downstream of many C-type lectin receptors (CLRs). CLRs encompass a large family of innate recognition receptors, expressed predominantly by myeloid and epithelial cells, which bind fungal carbohydrates and initiate antifungal immune responses. Accordingly, human CARD9 deficiency is associated with the spontaneous development of persistent and severe fungal infections that primarily localize to the skin and subcutaneous tissue, mucosal surfaces and/or central nervous system (CNS). In the last 3 years, more than 15 missense and nonsense CARD9 mutations have been reported which associate with the development of a wide spectrum of fungal infections caused by a variety of fungal organisms. The mechanisms by which CARD9 provides organ-specific protection against these fungal infections are now emerging. In this review, we summarize recent immunological and clinical advances that have provided significant mechanistic insights into the pathogenesis of human CARD9 deficiency. We also discuss how genetic mutations in CARD9-coupled receptors (Dectin-1, Dectin-2) and CARD9-binding partners (MALT1, BCL10) affect human antifungal immunity relative to CARD9 deficiency, and we highlight major understudied research questions which merit future investigation.
The last 3 years have seen an increasing number of reports of patients with overwhelming fungal disease attributed to a genetic mutation in the gene CARD9 (Caspase Recruitment Domain Family, member 9). Human CARD9 deficiency was first described in 2009 in a consanguineous family afflicted with mucosal and lethal systemic infections caused by Candida species, (Glocker et al., 2009), which are common human commensal yeasts (Lionakis and Netea, 2013). Since then, more than 15 other CARD9 mutations have been identified in 46 patients, that uniquely associate with mucosal and/or systemic infections caused by specific fungal phyla (Drewniak et al., 2013; Lanternier et al., 2013; Gavino et al., 2014; Lanternier et al., 2014; Wang et al., 2014; Drummond et al., 2015; Gavino et al., 2015; Grumach et al., 2015; Herbst et al., 2015; Jachiet et al., 2015; Lanternier et al., 2015; Liang et al., 2015; Celmeli et al., 2016; Yan et al., 2016). These reports unequivocally demonstrate the critical dependence on CARD9 in human antifungal immunity.
Fungi are recognized by the innate immune system via pattern recognition receptors (PRRs) belonging to several different classes, including the C-type lectin receptors (CLRs), Toll-like receptors (TLRs), and the intracellular nucleotide-binding oligomerization domain-like receptors (NLRs), which are predominantly expressed by myeloid cells including neutrophils, monocytes, macrophages, and dendritic cells (Hardison and Brown, 2012). Although all of these receptor families are involved in innate antifungal recognition, only mutations in the CLR pathway have thus far been associated with the spontaneous development of fungal infections in humans.
Many members of the CLR superfamily bind fungal carbohydrates and initiate complex intracellular signaling pathways to mediate cellular functions, such as fungal killing and production of pro-inflammatory cytokines (Lionakis and Netea, 2013). Several CLRs utilize a common signaling pathway involving the kinase Syk and the signaling adaptor, CARD9 (Gross et al., 2006; Drummond et al., 2011). Thus, CARD9 is centrally positioned downstream of many CLRs and is critical for their function.
In this review, we highlight recent novel immunological insights into the pathogenesis of human CARD9 deficiency and summarize the different human CARD9 mutations described to date along with their clinical and functional characteristics (Table 1). In addition, we discuss how genetic mutations in the CARD9-coupled receptors (e.g., Dectin-1, Dectin-2) and molecules that form signaling complexes with CARD9 (e.g., MALT1, BCL10) affect human antifungal immunity (Table 2), while highlighting major understudied areas that merit future investigation.
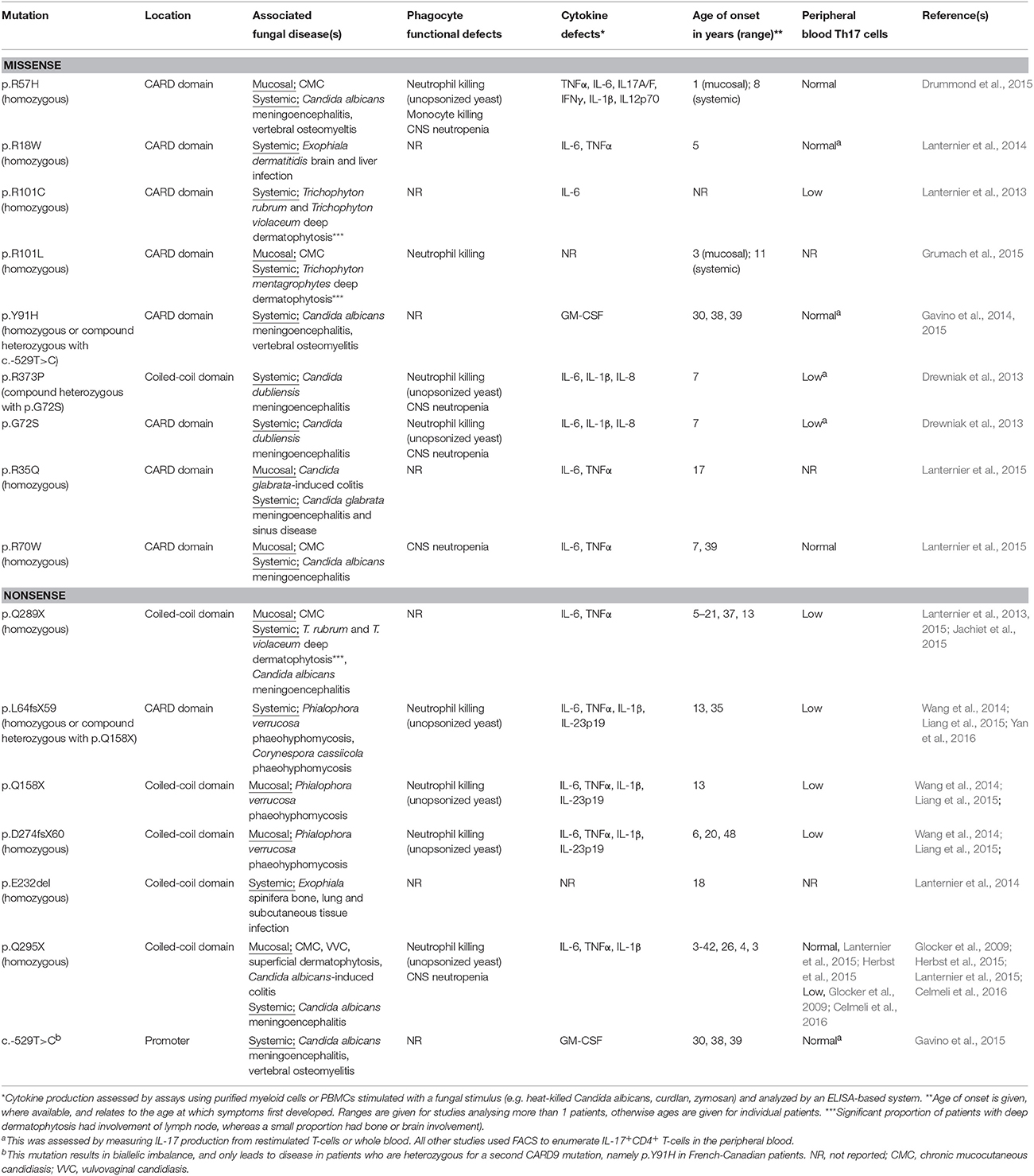
Table 1. Summary of human CARD9 mutations reported to date, with their associated clinical, immunological and functional characteristics.
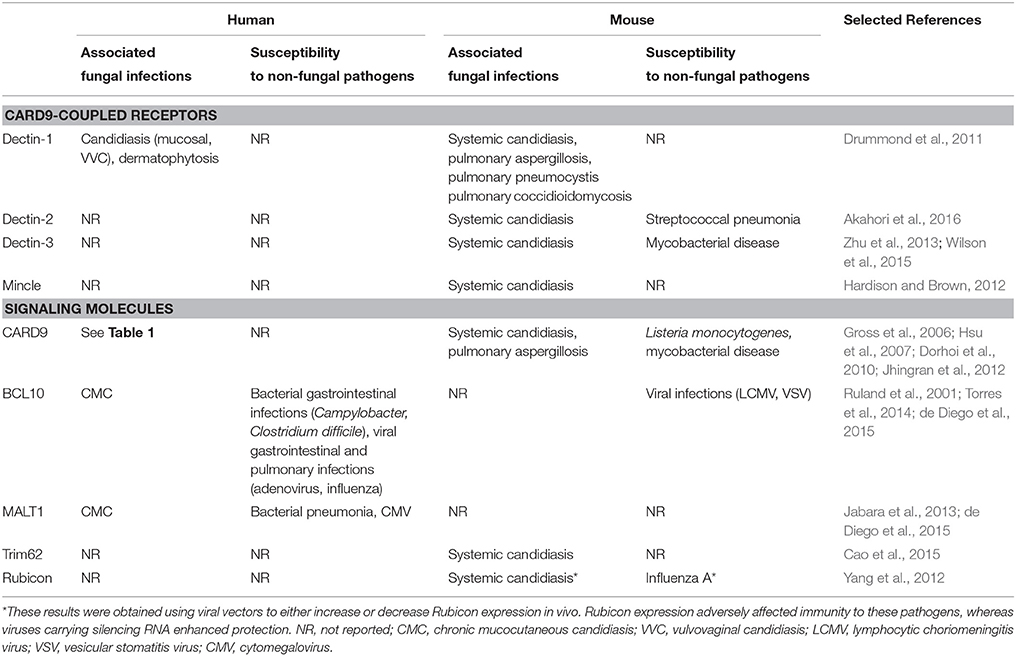
Table 2. Summary of infection susceptibilities to fungal and non-fungal pathogens in humans and mice deficient (or expressing non-functional variants) in the indicated CARD9-coupled receptors and associated signaling molecules.
The vast majority of inborn errors in immunity that predispose to fungal disease typically also lead to the development of non-fungal infections. Furthermore, these disorders usually segregate cleanly between mucosal or systemic fungal disease susceptibility, whereas CARD9 deficiency predisposes to both mucosal and systemic fungal disease. The only other inherited conditions that match the combined susceptibility to mucosal and systemic fungal infections seen in CARD9 deficiency are gain-of-function STAT1 mutations and autosomal-dominant STAT3 mutations; however, these mutations also result in bacterial, mycobacterial and/or viral infections (reviewed in detail here Lionakis et al., 2014). In contrast, CARD9 mutations in humans have thus far only been associated with fungal infections (Table 2), although Card9−∕− mice have been reported to be susceptible to intracellular bacterial infections (Hsu et al., 2007; Dorhoi et al., 2010) in addition to their susceptibility to fungal disease.
In mouse models of fungal challenge, Card9 genetic deletion abolishes the ability to control systemic Candida albicans infections, with 100% mortality observed as early as 72 h post-infection (Gross et al., 2006; Drummond et al., 2015). Similar results were recently obtained with Candida tropicalis, an emerging non-albicans Candida species, where fungal growth was found to be uncontrolled in the kidney, spleen, liver, and brain (Whibley et al., 2015). Card9−∕− animals were also found to be mildly susceptible to mucosal candidiasis; however, this was only evident during secondary infection suggesting that the dependence on Card9 lies within the adaptive immune compartment for this particular disease (Bishu et al., 2014). The difference between human and mouse susceptibility to mucosal fungal disease in the setting of CARD9-deficiency may be a reflection of the fact that, as opposed to humans, Candida is not a commensal organism in mice (Table 2). Susceptibility to pulmonary disease caused by the inhaled mold Aspergillus fumigatus is modestly enhanced in Card9−∕− animals, but only when these animals are challenged with an overwhelming number of conidia (8 × 107; Jhingran et al., 2012), whereas challenge with the still supra-physiologic inoculum, 3 × 107 conidia, does not confer heightened susceptibility in Card9−∕− mice (Jhingran et al., 2015). These mouse data are in line with the lack of an association between pulmonary mold infections and human CARD9-deficiency to date. More work will be required in mice to determine the role of Card9 in host defense against non-Candida, non-Aspergillus fungal pathogens that have been shown to cause disease in CARD9-deficient patients (Table 1).
Sixteen human CARD9 mutations have now been identified and characterized in patients originating from the USA, Canada, Europe, Middle East, South America, and China (Glocker et al., 2009; Drewniak et al., 2013; Lanternier et al., 2013; Gavino et al., 2014; Lanternier et al., 2014; Wang et al., 2014; Drummond et al., 2015; Grumach et al., 2015; Herbst et al., 2015; Jachiet et al., 2015; Lanternier et al., 2015; Yan et al., 2016). These mutations are found in the promoter region and the coiled-coil and CARD protein domains of CARD9, and both nonsense and missense mutations have been described (Table 1). Consistent with an autosomal recessive mode of inheritance, 80% of the reported patients were from consanguineous families; 89% were homozygous for the same allele, whereas 11% were compound heterozygous for different alleles. These mutations have variable penetrance, age of onset and effects, such that CARD9-deficiency manifests with multiple diseases caused by a variety of fungal organisms (Table 1). Based on the observed clinical variability thus far, the manifestations of human CARD9 deficiency can be broadly divided into three disease syndromes.
The first syndrome associated with CARD9 deficiency affects the skin and subcutaneous tissues, and patients in this category present with dramatic persistent fungal infections of the skin, nails and/or scalp (Lanternier et al., 2013; Wang et al., 2014), with occasional contiguous dissemination to the subcutaneous layers, lymph nodes and bone (Lanternier et al., 2013). The etiological agents for these infections so far described include Trichophyton species (causing deep dermatophytosis; Lanternier et al., 2013), Phialophora verrucosa (an agent of phaeohyphomycosis; Wang et al., 2014), and one intriguing case of phaeohyphomycosis caused by the plant pathogen Corynespora cassiicola (Yan et al., 2016). The CARD9-dependent functions that provide organ-specific protection in the skin against dermatophytes and agents of phaehyphomycosis are currently not well defined. In mouse models of cutaneous C. albicans infection, it has been shown that IL-17A and IL-23 signaling are critical for antifungal host defense in the skin (Kagami et al., 2010), and that induction of Th17 immunity is dependent on the CARD9-coupled receptor Dectin-1 expressed by Langerhans cells (Kashem et al., 2015), a population of dendritic cells resident in the epidermal layers of the skin. Both of these studies used C. albicans to initiate infection and did not investigate the dependence on Card9 for these activities. It therefore remains to be explored whether IL-17-dependent mechanisms of clearance require CARD9 for antifungal defense in the skin, and how important these mechanisms are for clearance of dermatophytes and agents of phaeohyphomycosis, where innate killing mechanisms of neutrophils and macrophages appear important (de Sousa et al., 2015; Liang et al., 2015).
The second predominant manifestation of human CARD9-deficiency is systemic fungal disease, which primarily manifests as fungal meningoencephalitis caused by Candida species (Gavino et al., 2014; Drummond et al., 2015). Some of these patients have also developed accompanying bone infection of the vertebra (Gavino et al., 2014; Drummond et al., 2015). Less often, CARD9-deficient patients develop phaeohyphomycosis caused by Exophiala species that targets the central nervous system (CNS) and/or other deep tissues including the liver, lung, and bone (Lanternier et al., 2014). Strikingly, Candida infections involving the kidney, liver or spleen, the organs typically affected in patients with iatrogenic immunosuppression (Lionakis, 2014), have not been reported in CARD9-deficient patients. Similarly, pulmonary infections by ubiquitous inhaled molds, or infections by Cryptococcus neoformans, Cryptococcus gattii, Pneumocystis jirovecii, and the endemic dimorphic fungi (i.e., Histoplasma capsulatum, Coccidioides immitis, or Blastomyces dermatitidis) have also not been reported thus far in CARD9-deficient patients. Collectively, the tropism of organ-specific systemic fungal disease in CARD9 deficiency implies that CARD9 has fungal- and organ-specific innate immune effects, which we currently only partially understand.
In addition to cutaneous/subcutaneous and CNS fungal disease, many CARD9-deficient patients are also susceptible to persistent and recurrent infections of the mucosal surfaces by Candida species, collectively termed chronic mucocutaneous candidiasis (CMC). CMC can develop as a single entity or in association with the aforementioned systemic or cutaneous/subcutaneous fungal diseases, and the mouth, gut and vagina have all been reportedly affected in CARD9-deficient patients (Table 1). When CMC and systemic fungal disease occur in the same individual, it appears that CMC starts at an earlier age and often pre-dating the development of the more serious fungal diseases affecting the brain (Drummond et al., 2015; Grumach et al., 2015; Lanternier et al., 2015). CARD9 deficiency is the only reported human genetic disorder where mucosal and systemic Candida disease occur together in the absence of other non-fungal infections (Lionakis et al., 2014). This is a rare event for patients to develop both mucosal and systemic Candida infections, since each disease has differing requirements for immune protection. Specifically, mucosal candidiasis control requires IL-17 signaling via Th17 cells, γδ T-cells and innate lymphoid cell populations (Conti et al., 2009; Gladiator et al., 2013; Conti et al., 2014), whereas systemic defense depends heavily on the accumulation and effector function of neutrophils and mononuclear phagocytes (Brown, 2011; Lionakis and Netea, 2013; Lionakis, 2014). Therefore, CARD9 deficiency gives rise to detrimental effects in both lymphoid and myeloid cell accumulation and/or function.
As CARD9-deficiency is associated with a spectrum of fungal diseases, this could indicate at the varying requirements for CARD9-dependent functions at different anatomical sites, since antifungal immune responses vary drastically between organs (Lionakis et al., 2011). As mentioned above, Th17 responses are required for protection against mucosal candidiasis in both mice and humans, whereas there is far less dependence on these responses for systemic anti-Candida defense (Conti and Gaffen, 2015). The critical role of CARD9 in the induction of IL-17 responses (LeibundGut-Landmann et al., 2007; Bishu et al., 2014) may account for CMC susceptibility of affected patients. Indeed, the majority of CARD9 mutations are associated with a reduced number of circulating Th17 cells (Table 1). However, approximately half of the reported CARD9 missense mutations have no impact on the percent of peripheral blood Th17 cells following stimulation with PMA/ionomycin (Table 1). Thus, the role of CARD9 in human Th17 immunity and defense against CMC still remains unclear. Further work will be needed to decipher how human CARD9 deficiency affects the pathways governing Th17 differentiation and function, and how CARD9 mutations that do not affect peripheral blood Th17 cell numbers can still result in the development of CMC.
Our group's research recently uncovered novel insight into how human CARD9-deficiency may predispose to fungal infection of the CNS (Drummond et al., 2015). Prior to this work, the susceptibility to CNS fungal disease was thought to be the result of a selective defect in killing unopsonized C. albicans yeasts by CARD9-deficient neutrophils (Drewniak et al., 2013; Drummond et al., 2015). Since opsonization is naturally low in the CNS, this killing defect has been thought to be clinically relevant in this tissue thus potentially accounting for localized organ-specific disease. Neutrophils are critical for antifungal host defense in mice and humans (Lionakis and Netea, 2013), and an intrinsic functional defect in these cells is a plausible explanation for the susceptibility to fungal CNS disease in CARD9-deficient patients. However, C. albicans pseudohyphae, not yeast, is the predominant fungal morphology observed in infected CNS tissue of mice and humans (Lionakis et al., 2011; Drummond et al., 2015), and hyphal killing by CARD9-deficient human neutrophils appears intact (Drewniak et al., 2013; Drummond et al., 2015). Importantly, we found that neutrophils are not recruited to the fungal-infected CNS in a CARD9-deficient patient or infected Card9−∕− animals (Drummond et al., 2015). In line with our findings, other groups have also reported a lack of neutrophil accumulation in the Candida-infected CSF of CARD9-deficient patients; instead, large numbers of lymphocytes and eosinophils comprise the cellular inflammatory reaction in these patients (Drewniak et al., 2013; Lanternier et al., 2015; Celmeli et al., 2016). This is in stark contrast to patients with Candida meningoencephalitis who have intact CARD9 signaling, where cells recruited to the infected CSF are predominantly neutrophils (Drummond et al., 2015). Therefore, although the selective neutrophil killing defect against unopsonized Candida yeasts may contribute toward the development of CNS fungal disease in CARD9-deficient patients, the major contributor toward the CNS infection susceptibility of CARD9-deficient patients appears to be the lack of neutrophils in the infected CNS, thus creating a condition of “CNS-specific neutropenia.”
We investigated the underlying mechanisms for the CNS-specific neutropenia observed in our CARD9-deficient patient and Card9−∕− animals. Impaired neutrophil accumulation in the Candida-infected CNS was not the result of defective neutrophil production in the bone marrow, neutrophil egress into the blood, neutrophil survival or neutrophil-intrinsic chemotaxis (Drummond et al., 2015). Instead, we found that CARD9 deficiency confers a lack of CXC-chemokine induction in the CNS during fungal infection, in both mice and humans (Drummond et al., 2015). Using mouse models, we found that CXC-chemokines were induced in a proportional manner to brain fungal burden, and this relationship was lost in the absence of Card9 (Drummond et al., 2015). We showed that this defect is specific to the CNS, since CXC-chemokine production and neutrophil recruitment were affected in a significantly lesser extent in the kidney of infected Card9−∕− mice, similar to recent findings using C. tropicalis (Whibley et al., 2015). Furthermore, we also showed that this defect was specific to fungal infection, as neutrophil recruitment was also unaffected following bacterial brain infection (Drummond et al., 2015), in agreement to the lack of susceptibility to bacterial meningoencephalitis in CARD9-deficient patients. Future work will be required to define the Card9-dependent cellular sources of neutrophil-targeted CXC-chemokines that are required for neutrophil recruitment to the CNS during fungal invasion, as well as the CARD9-coupled receptors critical for this effector cell accumulation. Of interest, Dectin-1 has been shown to promote neutrophil recruitment in the C. albicans-infected mouse peritoneum (Taylor et al., 2007), but its role in neutrophil recruitment in the fungal-infected CNS has not yet been explored.
Consistent with a role of CARD9 in neutrophil accumulation during fungal disease, the Hohl group recently characterized a neutrophil recruitment defect to the A. fumigatus-infected lung in Card9−∕− mice, which was also linked to poor CXC-chemokine production in the lung (Jhingran et al., 2015). However, in that model, Card9 was only partially required in the later stages of infection for neutrophil accumulation in the infected lung. Instead, neutrophil recruitment in the early phase was controlled by the TLR/IL-1R signaling adaptor, MyD88, which was required for chemokine induction by epithelial cells (Jhingran et al., 2015). Thus, the collective dependence on Card9 for neutrophil recruitment during pulmonary mold infections appears relatively mild compared to the universal reliance required for neutrophil accumulation and protection against Candida infection in the CNS (Gross et al., 2006; Drummond et al., 2015; Whibley et al., 2015). This dichotomy observed in mice between the Card9-dependence for protection against molds and yeasts may help explain why human CARD9 deficiency does not appear to associate with pulmonary fungal infections, despite the continuous environmental exposure to fungal spores. In order to clarify the fungal- and organ-specific functions of CARD9, future studies will be needed to examine the mechanistic role of CARD9 on other myeloid cells, besides neutrophils, such as monocytes, resident macrophages, dendritic cells, and myeloid-derived suppressor cells (Rieber et al., 2015), as well as resident epithelial and stromal cells in mediating systemic antifungal host defense.
Immune-based strategies have recently been reported to be an effective adjunct treatment for fungal meningoencephalitis in CARD9-deficient patients (Gavino et al., 2014, 2015; Celmeli et al., 2016), as long-term antifungal usage does not always lead to clinical remission, especially when treatment is not aggressive early on (Drummond et al., 2015). For instance, Gavino et al. showed that exogenous treatment with granulocyte-macrophage colony stimulating factor (GM-CSF) successfully led to clinical remission in a cohort of French-Canadian patients with Candida CNS infection, carrying the p.Y91H allele (Gavino et al., 2014, 2015). More recently, this work was mirrored by a second group using the related colony stimulating factor, G-CSF, administration of which resulted in clinical improvement in a Turkish patient with the p.Q295X mutation who had uncontrolled Candida CNS infection (Celmeli et al., 2016). Long-term GM-CSF treatment appeared necessary in the French-Canadian patients as its discontinuation led to clinical relapse, whereas short-term G-CSF treatment appeared adequate as its discontinuation did not result in infection relapse (Gavino et al., 2014, 2015; Celmeli et al., 2016).
At the mechanistic level, the Y91H mutation was shown to cause a defect in the formation of a CARD9-Ras protein-specific guanine nucleotide releasing factor 1 (Ras-GRF1) complex, necessary for downstream NF-κB and ERK signaling, which subsequently led to defective GM-CSF production in response to fungal ligands (Gavino et al., 2015). Impaired ERK activation via altered Card9 association with Ras-GRF1 and H-Ras was confirmed in Card9−∕− mice (Jia et al., 2014). Presumably, this GM-CSF specific defect may be amenable to correction by exogenous GM-CSF administration in these patients. In the study by the Grimbacher group, G-CSF treatment was shown to correct defective IL-17 production from CARD9-deficient cells, although not to the same levels as CARD9+ cells donated by a healthy volunteer (Celmeli et al., 2016). Both GM-CSF and G-CSF have pleiotropic effects on the immune system, and could therefore have potentially rescued CNS infection via boosting neutrophil recruitment to the site of infection and/or enhancing myeloid cell effector function, or other as yet unknown mechanisms may be involved. As these parameters were not examined in these studies, it is unclear whether GM-CSF/G-CSF could bypass multiple immune defects associated with human CARD9 deficiency, or if these success stories rely on correcting specific defects caused by the CARD9 mutation identified in these particular patients. Additional work testing the efficacy of GM-CSF/G-CSF in other CARD9-deficient patients with different mutations and other clinical manifestations beyond Candida CNS infection, corroborated by similar studies in Card9−∕− mouse models of fungal infections, will be required to determine the general applicability and the accompanying mechanisms of these treatments in human CARD9-deficiency.
After ligand binding, CLRs initiate intracellular signaling either directly via their intracellular tails (e.g., Dectin-1) or indirectly via association with a signaling adaptor such as FcRγ (e.g., Dectin-2, Mincle; Drummond et al., 2011). In the Syk/CARD9 pathway, tyrosine residues in the signaling motif of the CLR tail or adaptor become phosphorylated by SHP-2 leading to recruitment and activation of Syk kinase (Deng et al., 2015). Downstream of Syk, CARD9 forms a signaling complex with BCL10 and MALT1 which provides a scaffold for further signaling events such as NF-κB activation and the formation and activation of inflammasomes (Lionakis and Netea, 2013). The formation and regulation of the CARD9-BCL10-MALT1 (CBM) signaling complex is only partially understood, however recent reports have identified CARD9 binding partners that inhibit or activate CLR-CARD9 signaling. For example, TRIM62 is a ubiquitin ligase that activates CARD9 and thus positively regulates the CLR-CARD9 pathway (Cao et al., 2015). Accordingly, Trim62−∕− mice were recently found to be more susceptible to systemic candidiasis, with increased tissue fungal burdens and accelerated mortality (Cao et al., 2015), although this increase was modest relative to that seen in Card9−∕− animals that develop overwhelming rapidly-lethal disease (Gross et al., 2006; Drummond et al., 2015). In contrast, the protein Rubicon was shown to be a negative regulator of CARD9 signaling, acting to dissemble the CBM complex and interrupt downstream cytokine production (Yang et al., 2012). For both of these molecules, as well as other yet unknown CARD9 signaling partners, their role in human antifungal immunity remains to be determined (Table 2).
Despite sharing the CARD9 intracellular signaling pathway, many CLRs exhibit independent functions (reviewed extensively elsewhere Hardison and Brown, 2012) and form complex cross-talk relationships that can either be synergistic (Hadebe et al., 2015) or modulatory (Gringhuis et al., 2011). Although individual CLRs perform many critical functions, mutations that disrupt their expression do not lead to development of spontaneous invasive fungal disease, but are instead associated with mucosal infections (Table 2). For example, the best-studied human CLR mutation is p.Y238X in the CLEC7A gene, which encodes Dectin-1. Dectin-1 is the β-glucan receptor (Brown, 2006), predominantly expressed by myeloid cells, and is required for many antifungal activities including fungal killing, phagocytosis, cytokine production, inflammasome activation, and CD4+ T-cell survival and function (Drummond and Brown, 2011; Gringhuis et al., 2012; Drummond et al., 2016). The Y238X polymorphism causes poor expression of Dectin-1 and defective pro-inflammatory cytokine production following fungal restimulation (Ferwerda et al., 2009). A family of patients with this mutation was reported to suffer from frequent bouts of vulvovaginal candidiasis and onychomycosis (Ferwerda et al., 2009). In line with this, IL-17 responses were found to be defective in Y238X cells restimulated with C. albicans yeasts (van de Veerdonk et al., 2009), and thus disruption of this protective response could be responsible for the association between Y238X and mucosal fungal infections described in the above family. However, the precise role of the Y238X polymorphism in promoting mucosal fungal disease deserves further investigation, since this polymorphism has a high prevalence in several human populations (Ferwerda et al., 2009), yet it is unclear to what extent carriers of Y238X suffer from such infections. At the population level, patients carrying the Y238X polymorphism were shown to be more likely to have oral and gastrointestinal Candida colonization (Plantinga et al., 2009), and were at greater risk for developing invasive mold infections following hematopoietic stem cell transplantation (Cunha et al., 2010), at odds with the absence of pulmonary fungal infections in CARD9-deficient patients. At the mechanistic level, the Y238X polymorphism has no effect on phagocytosis and fungal killing by neutrophils in vitro, which may account for why these patients do not develop systemic disease. However, many CARD9 mutations also do not affect phagocytosis and have only minor effects on fungal killing (Drewniak et al., 2013; Drummond et al., 2015; Liang et al., 2015), thus there are likely other complex functions dependent on CARD9 and independent of Dectin-1 which we have yet to discover, that are critical for systemic defense.
Other CLR polymorphisms that have been associated with fungal disease in humans include the Dectin-1 polymorphism rs2078178 and the Dectin-2 polymorphism rs11045418. The Dectin-1 variant was found to associate with medically refractory ulcerative colitis, through an as yet unclear mechanism whereby the Dectin-1 variants aggravate existing inflammatory bowel disorders. The authors used mouse models to demonstrate that C. tropicalis was responsible for worsening colitis in Clec7a−∕− animals, indicative of the involvement of fungi and defective antifungal immunity in this model (Iliev et al., 2012). Dectin-2 is another CLR that binds α-mannans and has a similar expression profile to Dectin-1 (Kerscher et al., 2013), and has been shown to be particularly important for initiation of antifungal Th17 responses (Saijo et al., 2010; Gringhuis et al., 2011). Recently, a Dectin-2 polymorphism was found to associate with pulmonary cryptococcosis, but not with cryptococcal meningitis, in Chinese patients (Hu et al., 2015). The basis for this association remains unclear, since Dectin-2-deficient mice are not more susceptible to pulmonary cryptococcosis (Nakamura et al., 2015) and detailed studies into the functional consequences of human Dectin-2 polymorphisms are currently lacking. Identification of other human mutations in CLRs is still in its infancy (Table 2), as exemplified by the recent characterization of novel CLRs (e.g., Dectin-3, Mincle) and thus the full scope and spectrum of CLR-dependent antifungal immune functions is not yet well defined. More work is therefore required to comprehensively analyze the CLR-specific roles in immune responses to various fungal pathogens.
While human CARD9 deficiency is uniquely associated with fungal disease, human mutations in its partners BCL10 and MALT1 have wider consequences for the immune system (reviewed extensively here de Diego et al., 2015, and see Table 2).
MALT1 is required for NF-κB activation and is found downstream of many immune receptors, including antigen-specific T- and B-cell receptors, the CLRs, and some G-protein coupled receptors (de Diego et al., 2015). Recent reports of patients with MALT1 missense mutations demonstrated that disruption of MALT1 function has far-reaching consequences for innate and adaptive immunity resulting in combined immunodeficiency. These patients suffered from a variety of infectious and growth disorders, including bacterial pulmonary infections, CMC, persistent cytomegalovirus infection, and inflammatory defects in the gut (Jabara et al., 2013; McKinnon et al., 2014; Punwani et al., 2015). Functionally, MALT1-deficiency conferred an inability to degrade IκBα and defects in T-cell derived IL-2 production (Jabara et al., 2013; McKinnon et al., 2014; Punwani et al., 2015), in line with earlier work describing the immune defects of Malt1−∕− mice (de Diego et al., 2015). The effects of human MALT1 deficiency on antifungal immunity have not been studied in detail. However, Gringhuis and colleagues showed that MALT1 was required for production of Th17-polarizing cytokines from human dendritic cells, through a pathway involving Dectin-2 and the NF-κB subunit c-Rel (Gringhuis et al., 2011). Thus, the correlation between human MALT1 deficiency and CMC could be related to the critical role for Th17 cells in protecting mucous membranes from Candida infection, although this has yet to be fully explored.
BCL10, like MALT1, is involved in many immune signaling pathways and thus BCL10 deficiency also has broad effects on the hematopoietic and non-hematopoietic immune system. A young child with complete BCL10 deficiency was recently described, who died at 3 years of age following multiple viral infections, CMC, colitis, and gastrointestinal infections (Torres et al., 2014). Interestingly, macrophages and dendritic cells from this patient had normal TLR and Dectin-1 mediated responses, as measured by pro-inflammatory cytokine production following restimulation with specific ligands. Instead, most of the immune defects in this patient mapped to the adaptive immune system, where generation of memory B- and T-cells was severely compromised (Torres et al., 2014), in line with findings using Bcl10−∕− animals (Ruland et al., 2001). The study of more patients with MALT1 and BCL10 mutations will be required to define the full spectrum of susceptibility to mucosal and systemic fungal disease. In addition, work in mice will be critical to determine the mechanisms by which Malt1 and Bcl10 promote mucosal host defense and whether they mediate CNS-specific protective immunity, as does Card9.
Systemic fungal infections continue to represent a significant unmet medical problem associated with high mortality, making the development of vaccines and novel immune-based treatments highly desirable in order to improve patient outcomes. Deciphering the cellular and molecular immune functions that promote effective antifungal immunity will help us achieve these goals and improve current clinical approaches. Human primary immunodeficiency disorders that predispose to fungal infections give us crucial clues as to which molecules are important for antifungal host defense. CARD9 is a critical antifungal molecule yet there remain significant gaps in our understanding, including the organ-specific functions operating in the skin, mucosal surfaces, and deep tissues, why CARD9-deficiency doesn't predispose to more infections by other fungal organisms, and how some CARD9-deficient patients can live for 2 or 3 decades before developing any serious fungal infections while others with identical mutations develop severe fungal disease early in life. Further study into the pathogenesis of human CARD9-deficiency will yield exciting new insights into the mechanisms that protect humans from these devastating fungal diseases, which should lead to improved therapeutic strategies for affected patients in the future.
RAD and MSL co-wrote the manuscript and approved for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
This research was supported by the Intramural Research Program of the National Institutes of Health (National Institute of Allergy and Infectious Diseases).
Akahori, Y., Miyasaka, T., Toyama, M., Matsumoto, I., Miyahara, A., Zong, T., et al. (2016). Dectin-2-dependent host defense in mice infected with serotype 3 Streptococcus pneumoniae. BMC Immunol. 17:1. doi: 10.1186/s12865-015-0139-3
Bishu, S., Hernández-Santos, N., Simpson-Abelson, M. R., Huppler, A. R., Conti, H. R., Ghilardi, N., et al. (2014). The adaptor CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections. Infect. Immun. 82, 1173–1180. doi: 10.1128/IAI.01335-13
Brown, G. D. (2006). Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 6, 33–43. doi: 10.1038/nri1745
Brown, G. D. (2011). Innate antifungal immunity: the key role of phagocytes. Ann. Rev. Immunol. 29, 1–21. doi: 10.1146/annurev-immunol-030409-101229
Cao, Z., Conway, K. L., Heath, R. J., Rush, J. S., Leshchiner, E. S., Ramirez-Ortiz, Z. G., et al. (2015). Ubiquitin ligase TRIM62 regulates CARD9-mediated anti-fungal immunity and intestinal inflammation. Immunity 43, 715–726. doi: 10.1016/j.immuni.2015.10.005
Celmeli, F., Oztoprak, N., Turkkahraman, D., Seyman, D., Mutlu, E., Frede, N., et al. (2016). Successful granulocyte colony-stimulating factor treatment of relapsing Candida albicans meningoencephalitis caused by CARD9 deficiency. Pediatr. Infect. Dis. J. 35, 428–431. doi: 10.1097/INF.0000000000001028
Conti, H. R., and Gaffen, S. L. (2015). IL-17–Mediated immunity to the opportunistic fungal pathogen Candida albicans. J. Immunol. 195, 780–788. doi: 10.4049/jimmunol.1500909
Conti, H. R., Peterson, A. C., Brane, L., Huppler, A. R., Hernández-Santos, N., Whibley, N., et al. (2014). Oral-resident natural Th17 cells and γδ T cells control opportunistic Candida albicans infections. J. Exp. Med. 211, 2075–2084. doi: 10.1084/jem.20130877
Conti, H. R., Shen, F., Nayyar, N., Stocum, E., Sun, J. N., Lindemann, M. J., et al. (2009). Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 206, 299–311. doi: 10.1084/jem.20081463
Cunha, C., Di Ianni, M., Bozza, S., Giovannini, G., Zagarella, S., Zelante, T., et al. (2010). Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 116, 5394–5402. doi: 10.1182/blood-2010-04-279307
de Diego, R. P., Sanchez-Ramon, S., Lopez-Collazo, E., Martinez-Barricarte, R., Cubillos-Zapata, C., Cerdan, A. F., et al. (2015). Genetic errors of the human caspase recruitment domain-B-cell lymphoma 10-mucosa-associated lymphoid tissue lymphoma-translocation gene 1 (CBM) complex: molecular, immunologic, and clinical heterogeneity. J. Allergy Clin. Immunol. 136, 1139–1149. doi: 10.1016/j.jaci.2015.06.031
Deng, Z., Ma, S., Zhou, H., Zang, A., Fang, Y., Li, T., et al. (2015). Tyrosine phosphatase SHP-2 mediates C-type lectin receptor-induced activation of the kinase Syk and anti-fungal TH17 responses. Nat. Immunol. 16, 642–652. doi: 10.1038/ni.3155
de Sousa, M. D. T., Santana, G. B., Criado, P. R., and Benard, G. (2015). Chronic widespread dermatophytosis due to Trichophyton rubrum: a syndrome associated with a Trichophyton-specific functional defect of phagocytes. Front. Microbiol. 6:8. doi: 10.3389/fmicb.2015.00801
Dorhoi, A., Desel, C., Yeremeev, V., Pradl, L., Brinkmann, V., Mollenkopf, H. J., et al. (2010). The adaptor molecule CARD9 is essential for tuberculosis control. J. Exp. Med. 207, 777–792. doi: 10.1084/jem.20090067
Drewniak, A., Gazendam, R. P., Tool, A. T. J., van Houdt, M., Jansen, M. H., van Hamme, J. L., et al. (2013). Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 121, 2385–2392. doi: 10.1182/blood-2012-08-450551
Drummond, R. A., and Brown, G. D. (2011). The role of Dectin-1 in the host defence against fungal infections. Curr. Opin. Microbiol. 14, 392–399. doi: 10.1016/j.mib.2011.07.001
Drummond, R. A., Collar, A. L., Swamydas, M., Rodriguez, C. A., Lim, J. K., Mendez, L. M., et al. (2015). CARD9-dependent neutrophil recruitment protects against fungal invasion of the central nervous system. PLoS Pathog. 11:e1005293. doi: 10.1371/journal.ppat.1005293
Drummond, R. A., Dambuza, I. M., Vautier, S., Taylor, J. A., Reid, D. M., Bain, C. C., et al. (2016). CD4+ T-cell survival in the GI tract requires Dectin-1 during fungal infection. Mucosal Immunol. 9, 492–502. doi: 10.1038/mi.2015.79
Drummond, R. A., Saijo, S., Iwakura, Y., and Brown, G. D. (2011). The role of Syk/CARD9 coupled C-type lectins in antifungal immunity. Eur. J. Immunol. 41, 276–281. doi: 10.1002/eji.201041252
Ferwerda, B., Ferwerda, G., Plantinga, T. S., Willment, J. A., van Spriel, A. B., Venselaar, H., et al. (2009). Human Dectin-1 deficiency and mucocutaneous fungal infections. N. Eng. J. Med. 361, 1760–1767. doi: 10.1056/NEJMoa0901053
Gavino, C., Cotter, A., Lichtenstein, D., Lejtenyi, D., Fortin, C., Legault, C., et al. (2014). CARD9 deficiency and spontaneous central nervous system candidiasis: complete clinical remission with GM-CSF therapy. Clin. Infect. Dis. 59, 81–84. doi: 10.1093/cid/ciu215
Gavino, C., Hamel, N., Zeng, J. B., Legault, C., Guiot, M. C., Chankowsky, J., et al. (2015). Impaired RASGRF1/ERK–mediated GM-CSF response characterizes CARD9 deficiency in French-Canadians. J. Allergy Clin. Immunol. doi: 10.1016/j.jaci.2015.09.016. [Epub ahead of print].
Gladiator, A., Wangler, N., Trautwein-Weidner, K., and LeibundGut-Landmann, S. (2013). Cutting edge: IL-17-secreting innate lymphoid cells are essential for host defense against fungal infection. J. Immunol. 190, 521–525. doi: 10.4049/jimmunol.1202924
Glocker, E. O., Hennigs, A., Nabavi, M., Schaffer, A. A., Woellner, C., Salzer, U., et al. (2009). A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N. Engl. J. Med. 361, 1727–1735. doi: 10.1056/NEJMoa0810719
Gringhuis, S. I., Kaptein, T. M., Wevers, B. A., Theelen, B., van der Vlist, M., Boekhout, T., et al. (2012). Dectin-1 is an extracellular pathogen sensor for the induction and processing of IL-1b via a noncanonical caspase-8 inflammasome. Nat. Immunol. 13, 246–254. doi: 10.1038/ni.2222
Gringhuis, S. I., Wevers, B. A., Kaptein, T. M., van Capel, T. M. M., Theelen, B., Boekhout, T., et al. (2011). Selective C-Rel Activation via Malt1 Controls Anti-Fungal Th17 Immunity by Dectin-1 and Dectin-2. PLoS Pathog. 7:e1001259. doi: 10.1371/journal.ppat.1001259
Gross, O., Gewies, A., Finger, K., Schafer, M., Sparwasser, T., Peschel, C., et al. (2006). Card9 controls a non-TLR signalling pathway for innate anti-fungal immunity. Nature 442, 651–656. doi: 10.1038/nature04926
Grumach, A. S., de Queiroz-Telles, F., Migaud, M., Lanternier, F., Filho, N. R., Palma, S. M. U., et al. (2015). A homozygous CARD9 mutation in a brazilian patient with deep dermatophytosis. J. Clin. Immunol. 35, 486–490. doi: 10.1007/s10875-015-0170-4
Hadebe, S., Kirstein, F., Fierens, K., Chen, K., Drummond, R. A., Vautier, S., et al. (2015). Microbial ligand costimulation drives neutrophilic steroid-refractory asthma. PLoS ONE 10:e0134219. doi: 10.1371/journal.pone.0134219
Hardison, S. E., and Brown, G. D. (2012). C-type lectin receptors orchestrate antifungal immunity. Nat. Immunol. 13, 817–822. doi: 10.1038/ni.2369
Herbst, M., Gazendam, R., Reimnitz, D., Sawalle-Belohradsky, J., Groll, A., Schlegel, P. G., et al. (2015). Chronic Candida albicans meningitis in a 4-year-old girl with a homozygous mutation in the CARD9 gene (Q295X). Pediatr. Infect. Dis. J. 34, 999–1002. doi: 10.1097/INF.0000000000000736
Hsu, Y.-M. S., Zhang, Y., You, Y., Wang, D., Li, H., Duramad, O., et al. (2007). The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 8, 198–205. doi: 10.1038/ni1426
Hu, X.-P., Wang, R. Y., Wang, X., Cao, Y. H., Chen, Y. Q., Zhao, H. Z., et al. (2015). Dectin-2 polymorphism associated with pulmonary cryptococcosis in HIV-uninfected Chinese patients. Med. Mycol. 53, 810–816. doi: 10.1093/mmy/myv043
Iliev, I. D., Funari, V. A., Taylor, K. D., Quoclinh, N., Reyes, C. N., Strom, S. P., et al. (2012). Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336, 1314–1317. doi: 10.1126/science.1221789
Jabara, H. H., Ohsumi, T., Chou, J., Massaad, M. J., Benson, H., Megarbane, A., et al. (2013). A homozygous mucosa-associated lymphoid tissue 1 (MALT1) mutation in a family with combined immunodeficiency. J. Allergy Clin. Immunol. 132, 151–158. doi: 10.1016/j.jaci.2013.04.047
Jachiet, M., Lanternier, F., Rybojad, M., Bagot, M., Ibrahim, L., Casanova, J., et al. (2015). Posaconazole treatment of extensive skin and nail dermatophytosis due to autosomal recessive deficiency of CARD9. JAMA Dermatol. 151, 192–194. doi: 10.1001/jamadermatol.2014.2154
Jhingran, A., Kasahara, S., Shepardson, K. M., Junecko, B. A. F., Heung, L. J., Kumasaka, D. K., et al. (2015). Compartment-specific and sequential role of MyD88 and CARD9 in chemokine induction and innate defense during respiratory fungal infection. PLoS Pathog. 11:e1004589. doi: 10.1371/journal.ppat.1004589
Jhingran, A., Mar, K. B., Kumasaka, D. K., Knoblaugh, S. E., Ngo, L. Y., Segal, B. H., et al. (2012). Tracing conidial fate and measuring host cell antifungal activity using a reporter of microbial viability in the lung. Cell Rep. 2, 1762–1773. doi: 10.1016/j.celrep.2012.10.026
Jia, X.-M., Tang, B., Zhu, L. L., Liu, Y. H., Zhao, X. Q., Gorjestani, S., et al. (2014). CARD9 mediates Dectin-1–induced ERK activation by linking Ras-GRF1 to H-Ras for antifungal immunity. J. Exp. Med. 211, 2307–2321. doi: 10.1084/jem.20132349
Kagami, S., Rizzo, H. L., Kurtz, S. E., Miller, L. S., and Blauvelt, A. (2010). IL-23 and IL-17A, but Not IL-12 and IL-22, are required for optimal skin host defense against Candida albicans. J. Immunol. 185, 5453–5462. doi: 10.4049/jimmunol.1001153
Kashem, S. W., Igyártó, B. Z., Gerami-Nejad, M., Kumamoto, Y., Mohammed, J., Jarrett, E., et al. (2015). Candida albicans morphology and dendritic cell subsets determine T helper cell differentiation. Immunity 42, 356–366. doi: 10.1016/j.immuni.2015.01.008
Kerscher, B., Willment, J. A., and Brown, G. D. (2013). The Dectin-2 family of C-type lectin-like receptors: an update. Int. Immunol. 25, 271–277. doi: 10.1093/intimm/dxt006
Lanternier, F., Barbati, E., Meinzer, U., Liu, L., Pedergnana, V., Migaud, M., et al. (2014). Inherited CARD9 deficiency in 2 unrelated patients with invasive exophiala infection. J. Infect. Dis. 211, 1241–1250. doi: 10.1093/infdis/jiu412
Lanternier, F., Mahdaviani, S. A., Barbati, E., Chaussade, H., Koumar, Y., Levy, R., et al. (2015). Inherited CARD9 deficiency in otherwise healthy children and adults with Candida species–induced meningoencephalitis, colitis, or both. J. Allergy Clin. Immunol. 135, 1558–1568. doi: 10.1016/j.jaci.2014.12.1930
Lanternier, F., Pathan, S., Vincent, Q. B., Liu, L., Cypowyj, S., Prando, C., et al. (2013). Deep dermatophytosis and inherited CARD9 deficiency. N. Engl. J. Med. 369, 1704–1714. doi: 10.1056/NEJMoa1208487
LeibundGut-Landmann, S., Gross, O., Robinson, M. J., Osorio, F., Slack, E. C., Tsoni, S. V., et al. (2007). Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat. Immunol. 8, 630–638. doi: 10.1038/ni1460
Liang, P., Wang, X., Wang, R., Wan, Z., Han, W., and Li, R. (2015). CARD9 deficiencies linked to impaired neutrophil functions against Phialophora verrucosa. Mycopathologia 179, 347–357. doi: 10.1007/s11046-015-9877-2
Lionakis, M. S. (2014). New insights into innate immune control of systemic candidiasis. Med. Mycol. 52, 555–564. doi: 10.1093/mmy/myu029
Lionakis, M. S., Lim, J. K., Lee, C. C. R., and Murphy, P. M. (2011). Organ-specific innate immune responses in a mouse model of invasive Candidiasis. J. Innate Immun. 3, 180–199. doi: 10.1159/000321157
Lionakis, M. S., and Netea, M. G. (2013). Candida and host determinants of susceptibility to invasive Candidiasis. PLoS Pathog. 9:e1003079. doi: 10.1371/journal.ppat.1003079
Lionakis, M. S., Netea, M. G., and Holland, S. M. (2014). Mendelian genetics of human susceptibility to fungal infection. Cold Spring Harb. Perspect. Med. 4, 555–564. doi: 10.1101/cshperspect.a019638
McKinnon, M. L., Rozmus, J., Fung, S. Y., Hirschfeld, A. F., Del Bel, K. L., Thomas, L., et al. (2014). Combined immunodeficiency associated with homozygous MALT1 mutations. J. Allergy Clin. Immunol. 133, 1458–1462. doi: 10.1016/j.jaci.2013.10.045
Nakamura, Y., Sato, K., Yamamoto, H., Matsumura, K., Matsumoto, I., Nomura, T., et al. (2015). Dectin-2 deficiency promotes Th2 response and mucin production in the lungs after pulmonary infection with Cryptococcus neoformans. Infect. Immun. 83, 671–681. doi: 10.1128/IAI.02835-14
Plantinga, T., van der Velden, W., Ferwerda, B., van Spriel, A. B., Adema, G., Feuth, T., et al. (2009). Early stop polymorphism in human DECTIN1 is associated with increased candida colonization in hematopoietic stem cell transplant recipients. Clin. Infect. Dis. 49, 724–732. doi: 10.1086/604714
Punwani, D., Wang, H. P., Chan, A. Y., Cowan, M. J., Mallott, J., Sunderam, U., et al. (2015). Combined immunodeficiency due to MALT1 mutations, treated by hematopoietic cell transplantation. J. Clin. Immunol. 35, 135–146. doi: 10.1007/s10875-014-0125-1
Rieber, N., Singh, A., Öz, H., Carevic, M., Bouzani, M., Amich, J., et al. (2015). Pathogenic fungi regulate immunity by inducing neutrophilic myeloid-derived suppressor cells. Cell Host Microbe 17, 507–514. doi: 10.1016/j.chom.2015.02.007
Ruland, J., Duncan, G. S., Elia, A., del Barco Barrantes, I., Nguyen, L., Plyte, S., et al. (2001). Bcl10 is a positive regulator of antigen receptor–induced activation of NF-κB and neural tube closure. Cell 104, 33–42. doi: 10.1016/S0092-8674(01)00189-1
Saijo, S., Ikeda, S., Yamabe, K., Kakuta, S., Ishigame, H., Akitsu, A., et al. (2010). Dectin-2 recognition of a-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32, 681–691. doi: 10.1016/j.immuni.2010.05.001
Taylor, P. R., Tsoni, S. V., Willment, J. A., Dennehy, K. M., Rosas, M., Findon, H., et al. (2007). Dectin-1 is required for β-glucan recognition and control of fungal infection. Nat. Immunol. 8, 31–38. doi: 10.1038/ni1408
Torres, J. M., Martinez-Barricarte, R., García-Gómez, S., Mazariegos, M. S., Itan, Y., Boisson, B., et al. (2014). Inherited BCL10 deficiency impairs hematopoietic and nonhematopoietic immunity. J. Clin. Invest. 124, 5239–5248. doi: 10.1172/JCI77493
van de Veerdonk, F. L., Marijnissen, R. J., Kullberg, B. J., Koenen, H. J. P. M., Cheng, S. C., Joosten, I., et al. (2009). The macrophage mannose receptor induces IL-17 in response to Candida albicans. Cell Host Microbe 5, 329–340. doi: 10.1016/j.chom.2009.02.006
Wang, X., Wang, W., Lin, Z., Wang, X., Li, T., Yu, J., et al. (2014). CARD9 mutations linked to subcutaneous phaeohyphomycosis and TH17 cell deficiencies. J. Allergy Clin. Immunol. 133, 905–908.e3. doi: 10.1016/j.jaci.2013.09.033
Whibley, N., Jaycox, J. R., Reid, D., Garg, A. V., Taylor, J. A., Clancy, C. J., et al. (2015). Delinking CARD9 and IL-17: CARD9 Protects against Candida tropicalis Infection through a TNF-α–Dependent, IL-17–independent mechanism. J. Immunol. 195, 3781–3792. doi: 10.4049/jimmunol.1500870
Wilson, G. J., Marakalala, M. J., Hoving, J. C., van Laarhoven, A., Drummond, R. A., Kerscher, B., et al. (2015). The C-Type lectin receptor CLECSF8/CLEC4D is a key component of anti-mycobacterial immunity. Cell Host Microbe 17, 252–259. doi: 10.1016/j.chom.2015.01.004
Yan, X. X., Yu, C. P., Fu, X. A., Bao, F. F., Du, D. H., Wang, C., et al. (2016). CARD9 mutation linked to Corynespora cassiicola infection in a Chinese patient. Br. J. Dermatol. 174, 176–179. doi: 10.1111/bjd.14082
Yang, C.-S., Rodgers, M., Min, C. K., Lee, J. S., Kingeter, L., Lee, J. Y., et al. (2012). The autophagy regulator rubicon is a feedback inhibitor of CARD9-mediated host innate immunity. Cell Host Microbe 11, 277–289. doi: 10.1016/j.chom.2012.01.019
Keywords: CARD9, lectins, fungi, Candida, neutrophils, innate immunity
Citation: Drummond RA and Lionakis MS (2016) Mechanistic Insights into the Role of C-Type Lectin Receptor/CARD9 Signaling in Human Antifungal Immunity. Front. Cell. Infect. Microbiol. 6:39. doi: 10.3389/fcimb.2016.00039
Received: 08 February 2016; Accepted: 21 March 2016;
Published: 05 April 2016.
Edited by:
David Dockrell, University of Sheffield, UKCopyright © 2016 Drummond and Lionakis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michail S. Lionakis, bGlvbmFraXNtQG1haWwubmloLmdvdg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.