

- 1Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, TX, USA
- 2Department of Pathology, University of Texas Medical Branch, Galveston, TX, USA
Alveolar type II pneumocytes (ATII) and alveolar macrophages (AM) play a crucial role in the lung's innate immune response. Burkholderia pseudomallei (BP) and Burkholderia mallei (BM) are facultative Gram-negative bacilli that cause melioidosis and glanders, respectively. The inhalation of these pathogens can cause lethal disease and death in humans. We sought to compare the pathogenesis of and host responses to BP and BM through contact with human primary ATII cells and monocytes-derived macrophages (MDM). We hypothesized that because BP and BM induce different disease outcomes, each pathogen would induce distinct, unique host immune responses from resident pulmonary cells. Our findings showed that BP adhered readily to ATII cells compared to BM. BP, but not BM, was rapidly internalized by macrophages where it replicated to high numbers. Further, BP-induced significantly higher levels of pro-inflammatory cytokine secretion from ATII cells (IL-6, IL-8) and macrophages (IL-6, TNFα) at 6 h post-infection compared to BM (p < 0.05). Interestingly, BM-induced the anti-inflammatory cytokine, IL-10, in ATII cells and macrophages at 6 h post-infection, with delayed induction of inflammatory cytokines at 24 h post-infection. Because BP is flagellated and produces LPS, we confirmed that it stimulated both Toll-like receptor (TLR) 4 and TLR5 via NF-κb activation while the non-flagellated BM stimulated only TLR4. These data show the differences in BP and BM pathogenicity in the lung when infecting human ATII cells and macrophages and demonstrate the ability of these pathogens to elicit distinct immune responses from resident lung cells which may open new targets for therapeutic intervention to fight against these pathogens.
Introduction
Burkholderia pseudomallei (BP) and Burkholderia mallei (BM) are facultative-intracellular Gram-negative bacilli that cause melioidosis and glanders, respectively (White, 2003; Gilad, 2007; Galyov et al., 2010). BP is a highly motile pathogen (Dance, 1991; Leelarasamee, 2004; Lazar Adler et al., 2009; Wikraiphat et al., 2009) that causes melioidosis in animals and humans (White, 2003; Gilad, 2007). The clinical symptoms of BP are difficult to diagnose because they present as flu-like symptoms, pneumonia, or septicemia (Dance, 1991; Leelarasamee, 2004; De Keulenaer and Cheng, 2006; Cheng et al., 2007). Melioidosis is endemic in subtropical and tropical areas, with most cases found in Southeast Asia and northern Australia where it is the most common cause of community-acquired pneumonia (White, 2003; Gilad, 2007; Galyov et al., 2010). A pulmonary BP infection can disseminate to distal organs with consequential development of bacteremia that can lead to death (White, 2003; Lazar Adler et al., 2009). In contrast, the closely related BM is a non- motile pathogen that is the etiological agent of glanders that persists in its equine host to survive (Redfearn et al., 1966; Gilad, 2007; Galyov et al., 2010). If transmitted to humans, the clinical signs of the disease are febrile pneumonia accompanied resulting in dissemination from the lungs to distal organs causing bacteremia and necrosis (Redfearn et al., 1966; Gilad, 2007; Galyov et al., 2010). Therefore, both of these pathogens are considered viable candidates for use as bioweapon agents via aerosolization effecting a large populated area.
BP and BM are phylogenetically very similar containing nearly identical 16S ribosomal DNA sequences with genome variations between the strains (Godoy et al., 2003; Holden et al., 2004). The BP genome contains two chromosomes, a large chromosome of 4.07 Mb that carries genes associated for metabolism and growth, and a smaller chromosome (3.17 Mb) encoding genes responsible for environmental adaptation and bacterial survival including the type III secretion system, capsule, and flagellum (Holden et al., 2004). BM also has two chromosomes but has a downsized genome. Chromosome 1 (3.5 Mb) encodes for genes involved in exopolysaccharide capsule, LPS, type IV pili, and type III and IV secretion systems (Holden et al., 2004) while chromosome 2 (2.3 Mb) carries genes encoding for LPS biosynthesis, capsule production and bacterial metabolism (Holden et al., 2004). Unlike BP, BM is non-flagellated and non-motile due to IS elements that disrupts the fliP gene, an essential gene for flagellum biogenesis, and a frameshift mutation in the flagellum motor gene that eliminates its function (Holden et al., 2004).
Although both pathogens share some antigenicity and virulence-associated similarities their disease outcomes are quite unique. While both BP and BM have been shown to readily adhere and invade transformed human and murine macrophage-like cell lines (Jones et al., 1996; Stevens and Galyov, 2004; Pudla et al., 2011) and ATII cell lines (Kespichayawattana et al., 2004; Utaisincharoen et al., 2004, 2005; Boddey et al., 2006; Sim et al., 2009; Whitlock et al., 2009; Balder et al., 2010), relatively few studies have examined the immunological outcomes-induced following infection of primary human cells (Nathan and Puthucheary, 2005; Puthucheary and Nathan, 2006) or performed direct comparisons of their differential infectivity process. This is likely due to the level 3 bio-containment facilities necessary to perform experiments with BM and BP. With the atmosphere of uncertainty following the tragic events of September 11th, 2001 the potential for these highly pathogenic organisms to be used as a biological weapon via aerosolization is a real possibility. Therefore, to gain a better understanding of BM and BP pathogenesis in the lung, we examined the induction of innate immune responses by primary human pulmonary cells in the early stages of BM or BP infection. The lung is constantly exposed to harmful inhaled environmental factors (i.e., bacteria, viruses, and chemicals). Pulmonary defenses have evolved such that the physiological positioning of resident alveolar cells are able to clear and/or fight against harmful particles. For example, alveolar macrophages (AM) are considered the first line of defense against inhaled pathogens. Successful pathogenic bacteria have developed mechanisms to subvert host defenses. Murine studies with BP showed macrophage invasion without activation of inducible nitric oxide synthase responsible for production of nitric oxide crucial to host attacks on intracellular bacteria (Utaisincharoen et al., 2001). The studies reported by Puthucheary and colleagues (Nathan and Puthucheary, 2005; Puthucheary and Nathan, 2006) indicated that BP was able to survive and replicate more successfully in macrophages collected from patients with melioidosis than normal macrophages from healthy volunteers. Additionally, Brett et al. (Balder et al., 2010) reported that BM LPS was a potent stimulator of cytokine secretion from human monocyte-derived macrophages (MDM) via TLR4, however, these studies did not examine the intact live pathogen. The interaction of BM with primary human macrophages is less studied and poorly understood.
Another residential pulmonary cell that will inevitably come into contact with inhaled Burkholderia is ATII cells, that represent approximately 4–5% of the cells in the alveolar space but comprises 15% of all the cells in the lung (Crapo et al., 1982, 2000; Novick et al., 1996; Mason, 2006). These non-professional immune cells are absolutely necessary for maintaining pulmonary homeostasis, specifically via the secretion of surfactants (Novick et al., 1996; Wright, 2005; Mason, 2006; Halliday, 2008; Pudla et al., 2011). ATII cells also are crucial, unconventional contributors to innate immunity via cytokine secretion, specifically chemokines and signaling molecules for professional immune cells including AM (O'Brien et al., 1999; Pechkovsky et al., 2000, 2005; Gentry et al., 2007). We have shown that the lethal pathogen, Francisella tularensis, stimulated polarized, primary human ATII cells to secrete chemotactic mediators that induced immune migration (Gentry et al., 2007). While there are various publications that describe BM (Whitlock et al., 2009) and BP (Stevens and Galyov, 2004; Utaisincharoen et al., 2005; West et al., 2008) interactions with non-polarized, transformed human A549 ATII cells, there are no reports using human primary ATII cells. Thus to remain physiologically, we utilized our ex vivo primary human ATII cell model as well as human MDM in the current studies to examine the pathogenic differences between BM and BP as revealed through their interactions with these residential pulmonary cells to induce different innate immune responses. These studies revealed that in the early stages of infection, BP, but not BM, readily adhered to ATII cells and macrophages and stimulated both TLR4 and TLR5 via its LPS and flagellin respectively. BP also induced high levels of cytokine secretion from ATII cells and MDM in the first 6 h after infection. Conversely, upon initial infection, BM-induced an inflammatory suppressor, IL-10, from ATII cells and macrophages with delayed secretion of inflammatory cytokines via TLR4 at 20 h post-infection.
Materials and Methods
Bacterial Strains
Burkholderia mallei (BM) China 7 (3873, MM-B, NBL7) and Burkholderia pseudomallei (BP) China 3 (MP-H, NBL 104) were obtained from BEI Resources (Manassas, VA) and propagated overnight at 37°C in Luria-Bertani (LB) broth (Difco Laboratories, Detroit, MI) from frozen stocks. Bacterial titers were determined using a Bel-Art—KLETT™ Colorimeter (Bel-Art Products, Pequannock, NJ) and confirmed by plating serial dilutions on LB agar plates. Both BM and BP are CDC classified category A select agent thus they were used in UTMB's certified BSL3 facility as per CDC and Federal regulations.
Human Cell Cultures
Primary human type II alveolar epithelial (ATII) cells (ScienCell, San Diego, CA) were seeded on BioCoat Growth Factor Reduced Matrigel®-coated transwell filters (BD; Franklin Lakes, NJ) and maintained using Bronchial Epithelial Medium enhanced with the Bullet kit supplements provided by the manufacturer (BEGM; Lonza, Basel, Switzerland) supplemented with 10% charcoal-stripped FBS (Hyclone; Logan, UT) and 10 ng/mL of keratinocyte growth factor (KGF; PeproTech, Rockhill, UT). ATII cells were grown to confluence (4–5 days) then used for experiments. Before each experiment, the A549 or ATII cells were washed 2X with appropriate medium containing no serum or antibiotics. The serum-free medium used during experimentation ensured no interference by serum proteins and did not adversely affect the health or metabolic activity of the cells.
Human monocytes were isolated from peripheral blood mononuclear cells obtained from healthy donors with full approval of the UTMB IRB. Monocytes were isolated using a Hypaque-Ficoll (GE Healthcare, Piscataway, NJ) density gradient (800 × g at 21°C) followed by selection using a human monocyte enrichment kit without cd16 depletion (Stemcell #19058) and Easysep magnet (Stemcell #18000) counterflow centrifugal elutriation (Beckman J2-21 M/E centrifuge with JE-B6 elutriator rotor, Beckman Instruments, Palo Alto, CA). The purified monocytes were cultured in RPMI 1640, 10% FBS supplemented with GM-CSF (100 ng/mL) for 7 days to ensure differentiation into macrophages as previously described (Eaves-Pyles et al., 2011). The macrophages were detached from the culture plate using a non-enzymatic cell dissociation solution (Sigma-Aldrich). MDM differentiation was confirmed by flow cytometry of cell surface markers expression (CD11b, CD80).
Infection Model and Cytokine Analyses
ATII cells or MDM (5 × 105/condition) were infected with BM or BP at an MOI of 100:1. Un-stimulated cells were used as negative controls. At 6 h post-infection, supernatants were collected from ATII cells and MDM then frozen at −80°C until analysis. At the same time point, parallel ATII cells or MDM cultures were washed with 2 mL of PBS then incubated for 30 min with serum-free medium containing 50 ug/mL of gentamicin (Sigma). ATII cells and MDM were washed then resuspended in serum-free medium containing 5 ug/mL of gentamicin and incubated an additional 14 h at 37°C/5% CO2. Supernatants were collected at 20 h post-infection then 6 and 20 h supernatants were analyzed via individual human ELISA kits (Pierce-Endogen) for the following cytokines: IL-6, IL-8, IL-10, and/or TNF-α.
Scanning Electron Microscopy (SEM)
ATII cells grown on transwell filters were exposed apically to BM or BP at an MOI of 100:1 for 4 h. Then filters were fixed in a mixture of 2.5% formaldehyde, 0.1% glutaraldehyde in 0.05 M cacodylate buffer pH 7.2 containing 0.03% trinitrophenol and 0.03% CaCl2, washed in 0.1 M cacodylate buffer pH 7.2 and post-fixed in 1% OsO4 in the same buffer. The filters were then dehydrated in ethanol and incubated with hexamethyldisalazane. After the filters were air-dried they were sputter coated for 40 s with iridium in an Emitech K575X turbo sputter coater (Emitech, Houston, TX). Samples were visualized using a Hitachi S4700 field emission scanning electron microscope (Hitachi High Technologies America, Electron Microscope Division, Pleasanton, CA) at 2 kV (UTMB Electron Microscopy Laboratory Facility).
Bacterial Adhesion and Invasion Assays
The ability of BM and BP to adhere to ATII cells was performed as previously described (Eaves-Pyles et al., 2008). Briefly, the cells were grown on 24-well plates to confluence at 37°C in 5% CO2 then infected with BM or BP at an MOI of 100:1 for 4 h at 37°C. To quantify adherence of the pathogens, the infected cells were washed twice with PBS then lysed with 200 μl of 0.1% Triton X-100 in PBS buffer. The adherent bacteria were plated, incubated and then colonies were counted the following day. To obtain accurate counts of the pathogen that adhered to ATII cells, bacterial invasion experiments were performed in parallel to the bacterial adhesion. Then the number of bacteria that invaded the cells was subtracted from the number of bacteria that adhered to the cells. Therefore, pathogen invasion was determined by infecting ATII cells with BM or BP as described above. Four hours post-infection, cells were washed twice with sterile PBS and then incubated with DMEM containing 50 ug/mL gentamicin for 1 h to kill extracellular bacteria. Cells were washed with PBS, lysed with 0.1% Triton X-100, plated and colonies were counted the following day as above.
Macrophage Phagocytosis and Intracellular Replication of BM and BP
MDM (5 × 105 cells) were stimulated with BP or BM (MOI 100:1). To examine phagocytosis and intracellular replication of BM and BP, at 1 h post-infection, macrophages were washed with 2 mL of PBS then incubated for 30 min with serum-free medium containing 50 ug/mL of gentamicin (Sigma) to kill extracellular bacteria (Eaves-Pyles et al., 2001a). Cells were lysed by adding 0.1% SDS and lysates were plated on BHI plates and incubated at 37°C in 5% CO2 overnight to allow quantification of bacterial phagocytosis. To quantify intracellular replication of the pathogens, parallel macrophage cultures were treated and infected as described above. However, following the 30 min gentamicin incubation, cells were washed then resuspended in serum-free medium containing 5 ug/mL of gentamicin. Cells were incubated an additional 3 h at 37°C/5% CO2 then lysed, plated, and single colonies were counted as described above.
TLR Stimulation by BM or BP
As described previously (Gentry et al., 2007), human embryonic kidney (HEK) 293 cells genetically manipulated to express human TLR4, or TLR5 (Invivogen, San Diego, CA) were cultured and maintained in DMEM (Cellgro Mediatech) supplemented with 10% FBS, 100 units/mL pencillin, 100 ug/mL streptomycin, 10 ug/mL basticidin, and 2 mM glutamine. To analyze activation of the transcription factor NF-κb, each of HEK293 cell lines were transfected with the pNF-κB-secreted alkaline phosphatase (pNF-κB-SEAP) reporter plasmid (Clontech, Mountain View, CA) using Lipofectamine Plus 188 (Invitrogen) per the manufacturer's instructions. The day after transfection, the existing medium was replaced with fresh DMEM containing no antibiotics followed by the addition of BM or BP at an MOI of 100:1. Commercial TLR agonists (Invivogen) were used as positive controls that included LPS (TLR4) and flagellin (TLR5). Medium alone and HEK293 cells with no agonist served as negative controls. The cells were incubated for 6 h then medium was aspirated and replaced with fresh DMEM with antibiotics and incubated overnight at 37°C and 5% CO2. Collected Supernatants were heat inactivated for 30 min at 65°C. SEAP activity in each sample was quantified (Mason, 2006) by the Phospha-Light Chemiluminescence Reporter Assay (Tropix, Bedford, MA) and a TR717 microplate 195 luminometer with WinGlow software (Tropix/PE Applied Biosystems, Bedford, MA196).
Western Immunoblot
Western blots were performed as we described previously (Eaves-Pyles et al., 2001a,b). Briefly, supernatants from BM and BP (as collected above) were analyzed for the amount of protein in each sample by the Bradford assay (Bio-Rad, Hercules, CA). The supernatants were boiled in loading buffer [4% SDS, 20% glycerol, 125 mM Tris-HCl (pH 6.8), and 10% 2-mercaptoethanol] then 40 μg of protein was loaded on an 8–16% Tris-glycine gradient gel (Novex, San Diego, CA). Electrophoresed proteins were transferred to a nitrocellulose membrane (Novex) and membranes were blocked with 10% nonfat dried milk for 30 min prior to incubation with rabbit polyclonal anti-(FliC) flagellin (BioLegend, San Diego, CA) at a dilution of 1:1000 overnight. Blots were washed, followed by the addition of peroxidase-conjugated anti-rabbit immunoglobulin G (Sigma Chemical Co., St. Louis, MO) at a dilution of 1:10,000 for 3 h. Blots were washed and then incubated for 1 min in enhanced chemiluminescence reagents (ECL kit; Amersham, Little Chalfont, Buckinghamshire, England). Processed blots were placed on X-ray film (Kodak®) for empirically optimized exposures.
Statistical Analysis
Numerical results are presented herein as mean ± SEM of two or three-independent experiments containing replicate experimental conditions per each experiment. Statistically analysis of numerical data was completed by Student's t-test or analysis of variance (ANOVA) using Prism software (Graph Pad v4.0, San Diego, CA). Differences were noted to be statistically significant when the p value was < 0.05.
Results
BP and BM Interact with the Apical Surface of ATII
We sought to visualize the interaction between BM or BP and ATII cells during early stages of a pulmonary infection. To remain biologically and physiologically relevant, ATII cells were grown on transwell filters to allow polarization before being infected apically with BM or BP. Four hours post-infection, scanning electron microscopy (SEM) was used to visualize interactions between the bacteria and the ATII cells. Figure 1 illustrates the distinct differences between BM and BP's contact with the ATII cells. BM contacted the apical surface of ATII cells but did not appear to readily adhere to the ATII cells at 4 h post-infection as such the pathogen could be seen floating free in the medium (Figures 1A,B). Conversely, BP consistently established intimate contact with the apical surface of ATII cells (Figures 1C–E) where ATII microvilli are wrapped around individual BP organisms (Figures 1D,E). In addition to these findings, quantitative analysis of pathogen adhesion to ATII cells showed that at 4 h post-infection BP adhered to the apical surface of ATII cells significantly better than BM (p < 0.05; Figure 1F). These data demonstrate distinct differences between BM and BP as the efficient adherence of BP to ATII cells is more pronounced in the early stages of infection than BM's ability to bind to host cells.
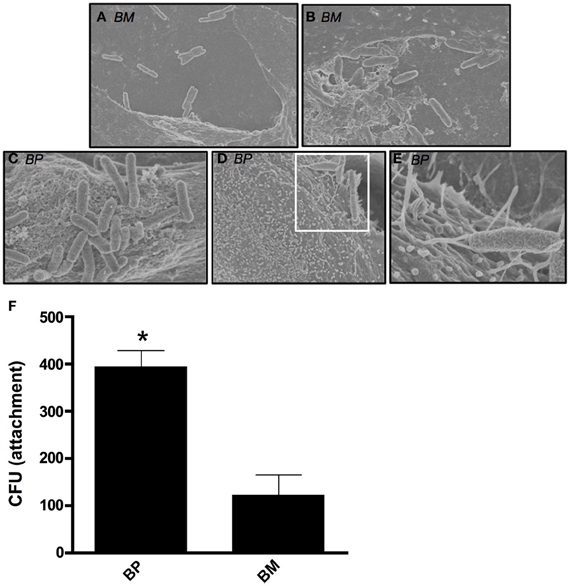
Figure 1. B. mallei (BM) and B. pseudomallei (BP) interaction with primary human ATII cells (ATII cells). ATII cells were grown on 3.0-um transwell filters to confluence prior to apical exposure of BM and BP at an MOI of approximately 100:1 for 4 h. Minimal contact of BM (A and B) with ATII cells was visualized via SEM. However, BP (C–E) adhesion to the apical surface of ATII cells was observed and the pathogen was entangled by the ATII microvilli [D (white boxed area) and E]. Further, quantitative analysis of attachment showed that BP significantly adhered better to ATII cells compared to BM (p < 0.05; F). Data is representative of triplicate samples of three experiments and is represented as mean ± SEM. *p < 0.05.
BM and BP-Induced Distinct as Well as Time-Dependent Cytokine Secretion from ATII Cells and MDM
Both ATII cells and AM will be contacted by inhaled BM and BP over the course of pulmonary infections. As such, we sought to evaluate the responses of these residential professional immune cells and epithelial cells responded to the pathogens in different the stages of infection. MDM and ATII cells were infected with BM or BP at an MOI of 100:1 then cells supernatants were collected at 6 and 20 h post-infection. Results showed that BP stimulated significantly higher levels of cytokine secretion from MDM [Figures 2A (IL-6), B (TNFα)] and ATII cells [Figures 2D (IL-6) and E (IL-8)] at both tested time points compared to BM or uninfected controls (p < 0.05). At the later time point it was observed that BM provoked responses. Specifically, secretion of IL-6 and TNF-α from MDM and IL-6 and IL-8 from ATII cells increased at 20 h post-infection (Figures 2A,B,D,E). Interestingly, the anti-inflammatory cytokine, IL-10 was secreted by ATII cells and MDM to significantly higher levels at 6 h post-BM infection compared to BP, then IL-10 levels decreased by 20 h post-infection, but remained higher than uninfected controls and BP-infected cells (p < 0.05; Figures 2C,F).
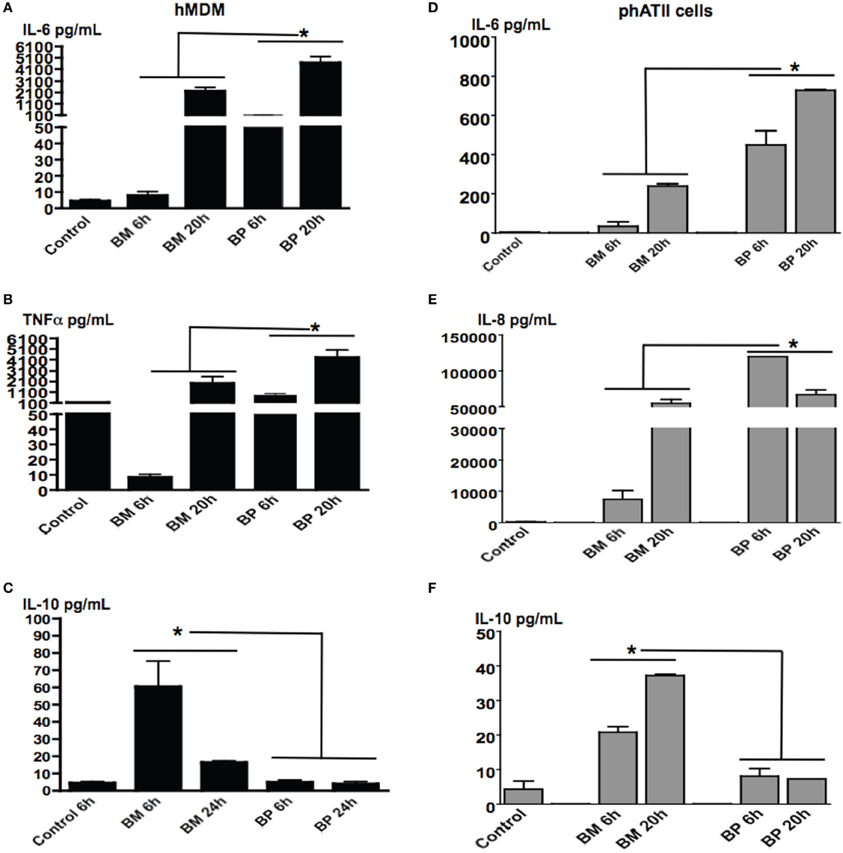
Figure 2. BM and BP-induced time-dependent secretion of cytokines from ATII cells and MDM. ATII cells and MDM were infected with BM or BP (MOI 100:1) for 6 or 20 h. Supernatant was collected and analyzed for cytokine secretion. Un-stimulated cells served as baseline controls. BP stimulated higher levels of IL-6 (A) and TNF-α (B) from MDM as well as IL-6 (D) and IL-8 (E) from ATII cells at 6 and 20 h post-infection compared to BM. However, BM-induced significantly higher IL-10 secretion from MDM compared to BP at 6 h post-infection (C). Additionally, IL-10 was significantly secreted from BM-ATII cells compared to BP at both 6 and 20 h post-infection (F). Data is representative of two experiments containing duplicate conditions/experiment and is represented as mean ± SEM. *denotes significance of p < 0.05.
These data demonstrate that BP and BM induce distinct cytokines that benefits their specific survival in ATII cells and macrophages. In the initial stages of infection, BP-induced vigorous immune responses in both cell types, while BM-induced an anti-inflammatory response (i.e., IL-10) in the early stages of infection then transitioned into a traditional immune response.
Phagocytosis and Intracellular Replication of BM and BP
Because BM and BP interacted with MDM and ATII cells to induce cytokines, we sought to determine the invasion capabilities and intracellular replication of BM and BP in these host cells. To this end, MDM and ATII cells were infected with BM or BP (MOI 100:1) for 1 h to determine phagocytosis of the pathogens and at 3 h post-infection to assess intracellular replication. Cells were lysed at each time point, plated then the bacterial CFU were quantified. Several observations were made from these data results. First, BP was more readily phagocytosed at 1 h post-infection by hMDM (Figure 3A) compared to phATII cells (p < 0.05; Figure 3B). However, BP replicated in both cell types to relatively high numbers at 3 h post-infection (Figures 3A,B). Second, the phagocytosis of BM by hMDM and ATII cells was minimal (Figures 3A,B) but the intracellular replication of BM in hMDM, but not in phATII cells, was considerably high at 3 h post-infection (Figure 3A).
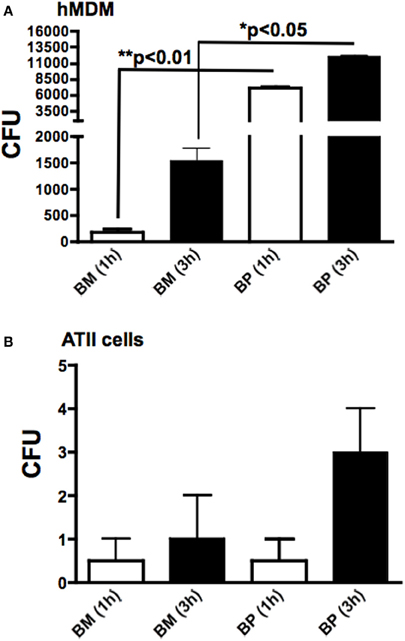
Figure 3. Phagocytosis and intracellular replication of BM and BP. MDM and ATII cells were infected with BM or BP (MOI of 100:1). As determined by CFUs, phagocytosis (accessed at 1 h) and intracellular replication (accessed at 3 h post-infection) of BP by MDM was significantly higher compared to BM (A). Alternatively, neither BM nor BP invaded or replicated to high numbers in ATII cells (B). Data is representative of two experiments containing duplicate conditions/experiment and is represented as mean ± SEM. *denotes significance of p < 0.05 and **p < 0.01.
The culmination of these data showed that the professional immune cell, macrophages, markedly surpassed the resident pulmonary epithelial cells, ATII cells, in phagocytosing BP and to a lesser extent BM. Both pathogens replicated rapidly to high numbers in macrophages suggesting bacterial manipulation and/or evasion of innate defenses.
Stimulation of TLRs by BM and BP
Because TLRs are at the forefront of recognizing invading pathogens resulting in the up-regulation of innate immune response via the NF-κb signal transduction pathway (Hatada et al., 2000; O'Neill and Bowie, 2007), we investigated the ability of BP and BM to stimulate TLR4 and TLR5. We chose TLR4 because it is the receptor for LPS and both pathogens are Gram-negative microbes. Additionally, because BP, but not BM, is flagellated, we hypothesized that BP flagellin would stimulate TLR5. Stability transfected HEK293 cells expressing human TLR4 or TLR5 were co-transfected with the NF-κb-SEAP reporter plasmid then infected with BP or BM at an MOI of 100:1. Following an overnight incubation, supernatants were collected and analyzed for SEAP secretion. Un-stimulated cells were used as negative controls while additional cultures of HEK293 TLR4 and TLR5 expressing cells were stimulated with their respective TLR agonist (purified LPS for TLR4 or flagellin for TLR5), which served as positive controls. Our results showed that both BP and BM-induced NF-κb activation via TLR4 stimulation that was equivalent to purified LPS and significantly higher than flagellin or un-stimulated controls (p < 0.05; Figure 4A). However, as predicted, only the flagellated BP-induced NF-κb via TLR5 stimulation compared to BM and LPS (p < 0.05; Figure 4B). To confirm the release of BP flagellin into the surrounding environment, immunoblot analysis detected the presence of flagellin in the BP supernatant compared to no detection of flagellin in the supernatant of BM (Figure 4C).
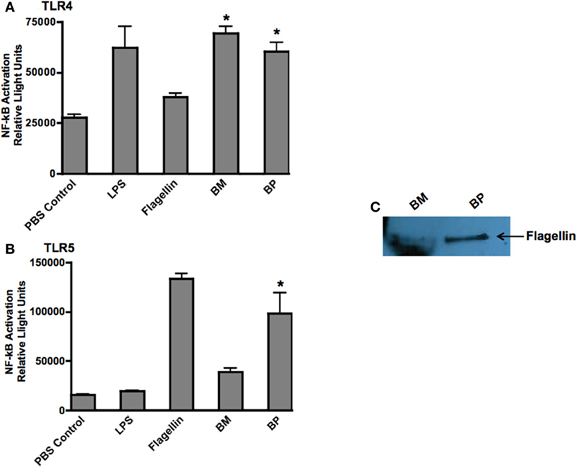
Figure 4. BM and BP stimulation of various Toll-Like Receptors. To determine if BM and/or BP stimulated various TLRs, HEK293 cells expressing TLRs 4 and 5 and transfected with the firefly luciferase plasmid pNiFty2-Luc were stimulated with BM or BP at an MOI of 100:1. Un-stimulated cells served as negative controls. The TLR agonist served as positive controls. The cells were incubated with live bacteria for 6 h. The medium was then aspirated and replaced with fresh DMEM without antibiotics and incubated overnight at 37°C and 5% CO2. The TLR agonists remained with the cells for an overnight incubation. The following day, samples were analyzed to determine luciferase activity. Data is expressed as relative luciferase activity. Both BM and BP (B) stimulated TLR4 as effectively as the LPS agonist (A). However, the flagellin from BP stimulated TLR5 (B) as we detected flagellin in the medium of B-infected cells but not BM (C). Three experiments were performed in using triplicate samples and data is represented as mean ± SEM and *denotes significant differences of p < 0.01.
These findings demonstrate that BP, but not BM, possesses various virulence factors that readily stimulate multiple TLRs thereby rapidly inducing innate immune responses.
Discussion
The importance of studying the pathogenesis of inhaled BM and BP has become more imminent as their potential for agents of bioterrorism has become known. Although BM and BP are in the same genus of bacteria, in this current study our findings showed that they demonstrate distinct pathogenicity characteristics upon the infection of resident lung cells, providing better understanding of their different disease outcomes.
Pathogen adhesion to host epithelial cells offers an opportunity for invasion and ultimately colonization of the host. It has been shown that both BM and BP possess certain genes that aid in host cell adherences such as boaA adhesion genes while others are exclusive to BP such as type IV pili, type III secretion system, flagella (Inglis et al., 2003; Kespichayawattana et al., 2004; Boddey et al., 2006; Balder et al., 2010). Flagella are recognized as important indirect contributors to bacterial virulence as the motility provided by the flagellum structure is frequently associated with the ability of a pathogen to move through its surrounding environment, as well as aid in attachment, adhesion, and invasion to host cells subsequently contributing to disease processes (Komoriya et al., 1999; Eaves-Pyles et al., 2001a; Chua et al., 2003; Ramos et al., 2004; Chuaygud et al., 2008). Our findings showed that the flagellated BP, but not the non-flagellated BM, readily adhered to ATII cells in the first 4 h post-infection. BP's motile and adhesion characteristics provided by flagella is likely allowing a more intimate interaction with host cells, facilitating the phagocytosis of the bacteria, while non-flagellated BM had minimal cellular adhesion in the early stages of infection. BP flagellum was shown to be necessary to penetrate the mucous lining the respiratory tract in order to gain access to host cells in intranasal and intraperitoneal infection mice models (Chua et al., 2003). Other studies have shown that an aflagellated BP mutant was internalized significantly less by macrophages and ATII cells as compared to wild type BP (Chuaygud et al., 2008). Likewise, flagellin negative variants have been observed to be less endocytosed than wild type flagellated BP, which signifies the involvement of the BP flagellum in cellular invasion (Inglis et al., 2003).
While the intact flagella structure contributes to the adherence and invasion of a pathogen to host cells, it is the primary protein component of the flagellum structure, known as flagellin, that is now recognized as a major virulence factor of Gram-negative organisms (Eaves-Pyles et al., 2001a,b, 2011; Chua et al., 2003; Ramos et al., 2004; Chuaygud et al., 2008). The release of flagellin monomers into the surrounding environment occurs when the flagella are shed from the bacteria and the structure disassembles due to environmental and chemical factors or deliberate ejection from the bacteria (Ramos et al., 2004). Only after flagellin monomers are free from the flagella filament do they bind to and stimulate signaling pathways via their receptor, TLR5 (Smith et al., 2003), resulting in the secretion of inflammatory cytokines (Eaves-Pyles et al., 2001a,b, 2011). As such, we detected flagellin in the supernatant of BP, but not BM, of infected cultures, leading to NF-κb activation via TLR5 stimulation. Additionally, BM, as well as BP, stimulated TLR4 as both pathogens possess LPS as a major outer membrane component. Our findings correlate with other studies demonstrating the ability of BP to stimulate TLR4 and TLR5 (Hii et al., 2008; West et al., 2008; Wiersinga et al., 2008) and BM's stimulation of TLR4 only (Brett et al., 2007). Thus it is reasonable to conclude that as multiple TLRs are subject to stimulation by BP, but not BM, this likely contributed to the vigorous innate immune responses (e.g., cytokine secretion) induced by macrophages and ATII cells in the early stages of infection. Sim et al. (2009) showed that primary murine epithelial lung cells both in vitro and in vivo secrete significant levels of pro-inflammatory cytokines in response to BP. Our findings, as well as others (Stevens and Galyov, 2004; Sim et al., 2009), resemble the clinical situation of acute melioidosis in that this disease can lead to induction of pro-inflammatory cytokines with high levels of cytokine secretion, leading to poorer prognosis for infected patients (Simpson et al., 2000; Cheng et al., 2007; Wiersinga et al., 2007). Alternatively, and very interestingly, BM stimulated minimal cytokine secretion from macrophages and ATII cells in the initial infection stage but stimulated the secretion of pro-inflammatory suppressor, IL-10. However, cytokine secretion from ATII cells and macrophages increased by 20 h post-BM infection. A study by Brett et al. (2008) found that BM was a weak inducer of cytokines, including IL-10, from a macrophage cell line. Although both studies demonstrate a lack of cytokine secretion by BM-infected macrophages, the differences (i.e., IL-10 secretion) between these findings may be explained by the fact that our studies used primary human monocytes-derived macrophages in suspension while Brett et al. (Cheng et al., 2007) tested monolayers of a murine macrophage-like cell line infected with BM. As a result of our findings, we may hypothesize that the lack of inflammatory cytokine secretion but increased secretion of IL-10 by macrophages and ATII cells is a mechanism evolved by BM to avoid detection by the host. This evasion by BM would allow the pathogen time to adapt to the intracellular environment and replicate before the host detects it and is able to mount an effective, timely immune response.
Because alveolar macrophages are at the forefront of the host's immune defenses, it was not surprising that macrophages secreted higher levels of cytokines overall in the response to BM and BP compared to ATII cells. Additionally, although BP readily adhered to ATII cells, there was minimal invasion of and replication in ATII cells by both pathogens. Other studies have shown that BP adhere, invade, and replicate to adequate numbers in transformed ATII cell lines (Whitlock et al., 2009; Balder et al., 2010). However, our studies herein are the first to examine Burkholderia pathogenesis using primary human alveolar type II cells in a physiological relevant ex vivo model. While ATII cells are not professional immune cells, they contribute significantly to the pulmonary immune defenses against inhaled invaders via cytokine secretion and surfactant A and D (Crapo et al., 2000; Wright, 2005; Lhert et al., 2007; Chroneos et al., 2010). Our results suggest that ATII cells are an important cytokine secreting cells resulting from interactions with BM and BP, however, they are not a reservoir of invasion and intracellular replication for these pathogens, which may open new avenues of the host innate immune response for therapeutic treatment of melioidosis and glanders.
In conclusion, we have shown various distinct differences between BM and BP pathogenesis regarding their interaction with primary human ATII cells and macrophages. BM and BP are similar in various aspects of their pathogenesis they produce different disease outcomes. Our study demonstrates that each of these pathogens has several unique strain specific characteristics is revealed in their interactions with host cells to induce varied host immune responses. By delineating host immune responses to BM and BP we can better understand the pathogenesis of these pathogens and develop new directions for therapeutic intervention and vaccines.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The work was funded by NIH NIAID R03 AI078442-01A1 (Tonyia Eaves-Pyles).
References
Balder, R., Lipski, S., Lazarus, J., Grose, W., Wooten, R., Hogan, R., et al. (2010). Identification of Burkholderia mallei and Burkholderia pseudomallei adhesins for human respiratory epithelial cells. BMC Microbiol. 10:250. doi: 10.1186/1471-2180-10-250
Boddey, J. A., Fleg, C. P., Day, C. J., Beacham, I. R., and Peak, I. R. (2006). Temperature-regulated microcolony formation by Burkholderia pseudomallei requires pilA and enhances association with cultured human cells. Infect. Immun. 74, 5374–5381.
Brett, P. F., Burtnick, M. N., Snyder, D. S., Shannon, J. G., Azadi, P., and Gherardini, F. C. (2007). Burkholderia mallei expresses a unique lipopolysaccharide mixture that is a potent activator of human Toll-like receptor 4 complexes. Mol. Microbiol. 63, 379–390.
Brett, P. J., Burtnick, M. N., Su, H., Nair, V., and Gherardini, F. C. (2008). iNOS activity is critical for the clearance of Burkholderia mallei from infected RAW 264.7 murine macrophages. Cell. Microbiol. 10, 487–498.
Cheng, A. C., Limmathurotsakul, D., Chierakul, W., Getchalarat, N., Wuthiekanun, V., Stephens, D. P., et al. (2007). A randomized controlled trial of granulocyte colony-stimulating factor for the treatment of severe sepsis due to melioidosis in Thailand. Clin. Infect. Dis. 45, 308–314.
Chroneos, Z. C., Sever-Chroneos, Z., and Shephard, V. L. (2010). Pulmonary surfactant: an immunological perspective. Cell. Physiol. Biochem. 25, 13–26.
Chua, K., Chan, Y., and Gan, Y. (2003). Flagella are virulence determinants of Burkholderia pseudomallei. Infect. Immun. 71, 1622–1629.
Chuaygud, T., Tungpradabkul, S., Sirisinha, S., Chua, K. L., and Utaisincharoen, P. (2008). A role of Burkholderia pseudomallei flagella as a virulent factor. Trans. R. Soc. Trop. Med. Hyg. 102(Suppl. 1), S47–S54.
Crapo, J. D., Barry, B. E., Gehr, P., Bachofen, M., and Weibel, E. R. (1982). Cell number and cell characteristics of the normal human lung. Am. Rev. Respir. Dis. 125, 740–745.
Crapo, J. D., Harmsen, A. G., Sherman, M. P., and Musson, R. A. (2000). Pulmonary immunobiology and inflammation in pulmonary diseases. Am. J. Respir. Crit. Care Med. 162, 1983–1986.
De Keulenaer, B. L., and Cheng, A. C. (2006). Severe sepsis due to melioidosis. Chest 130:1282. doi: 10.1378/chest.130.4.1282
DeShazer, D., Brett, P. J., Carlyon, R., and Woods, D. E. (1997). Mutagenesis of Burkholderia pseudomallei with Tn5-OT182: isolation of motility mutants and molecular characterization of the flagellin structural gene. J. Bacteriol. 179, 2116–2125.
Eaves-Pyles, T., Allen, C. A., Taormina, J., Swidsinski, A., Tutt, C. B., Jezek, G. E., et al. (2008). Escherichia coli isolated from a Crohn's disease patient adheres, invades and induces inflammatory responses in polarized intestinal epithelial cells. IJMM 298, 397–409.
Eaves-Pyles, T., Bu, H., Tan, X., Cong, Y., Patel, J., Davey, R. A., et al. (2011). Luminal-Applied flagelin is internalized by polarized intestinal epithelial cells and elicits immune responses via the TLR5 dependent mechanism. PLoS ONE 6:e24869. doi: 10.1371/journal.pone.0024869
Eaves-Pyles, T., Murthy, K., Liaudet, L., Virág, L., Ross, G., Soriano, F. G., et al. (2001a). Flagellin, a novel mediator of Salmonella-induced epithelial activation and systemic inflammation: IêBá degradation, induction of nitric oxide synthase, induction of proinflammatory mediators, and cardiovascular dysfunction. J. Immunol. 166, 1248–1260.
Eaves-Pyles, T. D., Wong, H. R., Odoms, K., and Pyles, R. B. (2001b). Salmonella flagellin-dependent proinflammatory responses are localized to the conserved amino and carboxyl regions of the protein. J. Immunol. 167, 7009–7016.
Galyov, E. E., Brett, P. J., and DeShazer, D. (2010). Molecular Insights into Burkholderia pseudomallei and Burkholderia mallei Pathogenesis. Ann. Rev. Microbiol. 64, 495–517.
Gentry, M., Taormina, J., Pyles, R., Yeager, L., Kirtley, M., Popov, V., et al. (2007). Role of Primary human alveolar epithelial cells in host defense against Francisella tularensis Infection. Infect. Immun. 75, 3969–3978.
Gilad, J. (2007). Burkholderia mallei and Burkholderia pseudomallei: the causative micro-organisms of glanders and melioidosis. Recent Pat. Antiinfect. Drug Discov. 2, 233–241.
Godoy, D., Randle, G., Simpson, A. J., Aanensen, D. M., Pitt, T. L., Kinoshita, R., et al. (2003). Multilocus sequence typing and evolutionary relationships among the causative agents of melioidosis and glanders, Burkholderia pseudomallei and Burkholderia mallei. J. Clin. Microbiol. 41, 2068–2079.
Hatada, E. N., Krappmann, D., and Scheidereit, C. (2000). NF-kappaB and the innate immune response. Curr. Opin. Immunol. 12, 52–58.
Hii, C. S., Sun, G. W., Gow, J. W., Lu, J., Stevens, M. P., and Gan, Y. H. (2008). Interleukin-8 induction by Burkholderia pseudomallei can occur without toll-like receptor signaling but requires a functional type III secretion system. J. Infect. Dis. 197, 1537–1547.
Holden, M. T., Titball, R. W., Peacock, S. J., Cerdeño-Tárraga, A. M., Atkins, T., Crossman, L. C., et al. (2004). Genomic plasticity of the causative agent of melioidosis, Burkholderia pseudomallei. Proc. Natl. Acad. Sci. U.S.A. 101, 14240–14245.
Inglis, T. J., Robertson, T., Woods, D. E., Dutton, N., and Chang, B. J. (2003). Flagellum-Mediated adhesion by Burkholderia pseudomallei precedes invasion of Acanthamoeba astronyxis. Infect. Immun. 71, 2280–2282.
Jones, A. L., Beveridge, T. J., and Woods, D. E. (1996). Intracellular survival of Burkholderia pseudomallei. Infect. Immun. 64, 782–790.
Kespichayawattana, W., Intachote, P., Utaisincharoen, P., and Sirisinha, S. (2004). Virulent Burkholderia pseudomallei is more efficient than avirulent Burkholderia thailandensis in invasion and adherence to cultured human epithelial cells. Microb. Pathog. 36, 287–292.
Komoriya, K., Shibano, N., Higano, T., Azuma, N., Yamaguchi, S., and Aizawa, S. I. (1999). Flagellar proteins and type III-exported virulence factors are the predominant proteins secreted into the culture media of Salmonella typhimurium. Mol. Microbiol. 34, 767–779.
Larsen, J. C., and Johnson, N. H. (2009). Pathogenesis of Burkholderia pseudomallei and Burkholderia mallei. Mil. Med. 174, 647–651.
Lazar Adler, N. R., Govan, B., Cullinane, M., Harper, M., Adler, B., and Boyce, J. (2009). The molecular and cellular basis of pathogenesis in melioidosis: how does Burkholderia pseudomallei cause disease. FEMS Microbiol. Rev. 33, 1070–1099.
Lhert, F., Yan, W., Biswas, S. C., and Hall, S. B. (2007). Effects of hydrophobic surfactant proteins on collapse of pulmonary surfactant monolayers. Biophys. J. 93, 4237–4243.
McGowin, C. L., Ma, L., Martin, D. H., and Pyles, R. B. (2009). Mycoplasma genitalium-encoded MG309 activates NF-kappaB via Toll-like receptors 2 and 6 to elicit proinflammatory cytokine secretion from human genital epithelial cells. Infect. Immun. 77, 1175–1181.
Namba, K., Yamashita, I., and Vonderviszt, F. (1989). Structure of the core and central channel of bacterial flagella. Nature 342, 648–654.
Nathan, S. A., and Puthucheary, S. D. (2005). An electronmicroscopic study of the interaction of Burkholderia pseudomallei and human macrophages. Malays. J. Pathol. 27, 3–7.
Novick, R. J., Gehman, K. E., Ali, I. S., and Lee, J. (1996). Lung preservation: the importance of endothelial and alveolar type II cell integrity. Ann. Thorac. Surg. 62, 302–314.
O'Brien, A. D., Standiford, T. J., Bucknell, K. A., Wilcoxen, S. E., and Paine, R. (1999). Role of alveolar epithelial cell intercellular adhesion molecule-1 in host defense against Klebsiella pneumoniae. Am. J. Physiol. 276, 961–970.
O'Brien, A. D., Standiford, T. J., Christensen, P. J., Wilcoxen, S. E., and Paine, R. (1998). Chemotaxis of alveolar macrophages in response to signals derived from alveolar epithelial cells. J. Lab. Clin. Med. 131, 417–424.
O'Neill, L. A., and Bowie, A. G. (2007). The family of five: TIR-domain-containing adaptors in Toll-like receptor signaling. Nat. Rev. Immunol. 7, 353–364.
Pechkovsky, D. V., Goldmann, T., Ludwig, C., Prasse, A., Vollmer, E., Muller-Quernheim, J., et al. (2005). CCR2 and CXCR3 agonistic chemokines are differently expressed and regulated in human alveolar epithelial cells type II. Respir. Res. 6, 75–92.
Pechkovsky, D. V., Zissel, G., Ziegenhagen, M. W., Einhaus, M., Taube, C., Rabe, K. F., et al. (2000). Effect of proinflammatory cytokines on interleukin-8 mRNA expression and protein production by isolated human alveolar epithelial cells type II in primary culture. Eur. Cytokine Netw. 11, 618–625.
Pudla, M., Kananurak, A., Limposuwan, K., Sirisinha, S., and Utaisincharoen, P. (2011). Nucleotide-binding oligomerization domain-containing protein 2 regulates suppressor of cytokine signaling 3 expression in Burkholderia pseudomallei-infected mouse macrophage cell line RAW 264.7. Innate Immun. 17, 532–540.
Pudla, M., Limposuwan, K., and Utaisincharoen, P. (2011). Burkholderia pseudomallei-induced expression of a negative regulator, sterile-alpha and Armadillo motif-containing protein, in mouse macrophages: a possible mechanism for suppression of the MyD88-independent pathway. Infect. Immun. 79, 2921–2927.
Puthucheary, S. D., and Nathan, S. A. (2006). Comparison by electron microscopy of intracellular events and survival of Burkholderia pseudomallei in monocytes from normal subjects and patients with melioidosis. Singapore Med. J. 47, 697–703.
Ramos, H. C., Rumbo, M., and Sirard, J. C. (2004). Bacterial flagellins: mediators of pathogenicity and host immune responses in mucosa. Trends Microbiol. 12, 509–517.
Redfearn, M. S., Palleroni, N. J., and Stanier, R. Y. (1966). A comparative study of Pseudomonas pseudomallei and Bacillus mallei. J. Gen. Microbiol. 43, 293–313.
Sim, S. H., Liu, Y., Wang, D., Novem, V., Sivalingam, S. P., Thong, T. W., et al. (2009). Innate Immune Responses of Pulmonary Epithelial Cells to Burkholderia pseudomallei infection. PLoS ONE 4:e7308. doi: 10.1371/journal.pone.0007308
Simpson, A. J., Smit, M. D., Weverlind, G. J., Suputtamongkil, Y., Angus, B. J., Chaowgul, W., et al. (2000). Prognostic value of cytokine concentrations (tumor necrosis factor-α, interleukin-6, and interleukin 10) and clinical parameters in severe melioidosis. J. Infect. Dis. 181, 621–625.
Smith, K. D., Andersen-Nissen, E., Hayashi, F., Strobe, K., Bergman, M. A., Barrett, S. L., et al. (2003). Toll-like receptor 5 recognizes a conserved site on flagellin required for protofilament formation and bacterial motility. Nat. Immunol. 4, 1247–1253.
Stevens, M. P., and Galyov, E. E. (2004). Exploitation of host cells by Burkholderia pseudomallei. Int. J. Med. Microbiol. 293, 549–555.
Utaisincharoen, P., Anuntagool, N., Arjcharoen, S., Lengwehasatit, I., and Limposuwan, K. (2004). Burkholderia pseudomallei stimulates low interleukin-8 production in the human lung epithelial cell line A549. Clin. Exp. Immunol. 138, 61–65.
Utaisincharoen, P., Arjcharoen, S., Lengwehasatit, I., Limposuwan, K., and Sirisinha, S. (2005). Burkholderia pseudomallei invasion and activation of epithelial cells requires activation of p38 mitogen-activated protein kinase. Microb. Pathog. 38, 107–112.
Utaisincharoen, P., Tangthawornchaikul, N., Kespichayawattana, W., Chaisuriya, P., and Sirisinha, S. (2001). Burkholderia pseudomallei interferes with inducible nitric oxide synthase (iNOS) production: a possible mechanism of evading macrophage killing. Microbiol. Immunol. 45, 307–313.
West, T. E., Ernst, R. K., Jansson-Hutson, M. J., and Skerrett, S. J. (2008). Activation of Toll-like receptors by Burkholderia pseudomallei. BMC Immunol. 9:46. doi: 10.1186/1471-2172-9-46
Whitlock, G. C., Valbuena, G. A., Popov, V. L., Judy, B. M., Estes, D. M., and Torres, A. G. (2009). Burkholderia mallei cellular interactions in a respiratory cell model. J. Med. Microbiol. 58, 554–562.
Wiersinga, W. J., and Van der Poll, T. (2009). Immunity to Burkholderia pseudomallei. Curr. Opin. Infect. Dis. 22, 102–108.
Wiersinga, W. J., Wieland, C. W., Dessing, M. C., Chantratita, N., Cheng, A. C., Limmathurotsakul, D., et al. (2007). Toll-like receptor 2 impairs host defense in gram-negative sepsis caused by Burkholderia pseudomallei (melioidosis). PLoS MED. 4:e248. doi: 10.1371/journal.pmed.0040248
Wiersinga, W. J., Wieland, C. W., Roelofs, J. J., and Poll, T. (2008). MyD88 dependent signaling contributes to protective host defense against Burkholderia pseudomallei. PLoS ONE 3:e3494. doi: 10.1371/journal.pone.0003494
Wikraiphat, C., Charoensap, J., Utaisincharoen, P., Wongratanacheewin, S., Taweechaisupapong, S., Woods, D. E., et al. (2009). Comparative in vivo and in vitro analyses of putative virulence factors of Burkholderia pseudomallei using lipopolysaccharide, capsule and flagellin mutants. FEMS Immunol. Med. Microbiol. 56, 253–259.
Keywords: Burkholderia, alveoli, epithelial cells, macrophages, cytokines, innate immune response, Toll-like receptor
Citation: Lu R, Popov V, Patel J and Eaves-Pyles T (2012) Burkholderia mallei and Burkholderia pseudomallei stimulate differential inflammatory responses from human alveolar type II cells (ATII) and macrophages. Front. Cell. Inf. Microbio. 2:165. doi: 10.3389/fcimb.2012.00165
Received: 03 August 2012; Accepted: 07 December 2012;
Published online: 28 December 2012.
Edited by:
Nelson Gekara, Umea University, SwedenReviewed by:
Xin Li, The Ohio State University, USASung O. Kim, University of Western Ontario, Canada
Copyright © 2012 Lu, Popov, Patel and Eaves-Pyles. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Tonyia Eaves-Pyles, Department of Microbiology and Immunology, University of Texas Medical Branch, 301 University Blvd., Galveston, TX 77555-1070, USA. e-mail:dGRlYXZlc3BAdXRtYi5lZHU=