 Vivek Anantharaman1
Vivek Anantharaman1
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
HYPOTHESIS AND THEORY article
Front. Cell. Infect. Microbiol. , 29 June 2012
Sec. Molecular Bacterial Pathogenesis
Volume 2 - 2012 | https://doi.org/10.3389/fcimb.2012.00089
This article is part of the Research Topic Microbial genomics challenge Darwin View all 15 articles
The endosymbiotic origin of eukaryotes brought together two disparate genomes in the cell. Additionally, eukaryotic natural history has included other endosymbiotic events, phagotrophic consumption of organisms, and intimate interactions with viruses and endoparasites. These phenomena facilitated large-scale lateral gene transfer and biological conflicts. We synthesize information from nearly two decades of genomics to illustrate how the interplay between lateral gene transfer and biological conflicts has impacted the emergence of new adaptations in eukaryotes. Using apicomplexans as example, we illustrate how lateral transfer from animals has contributed to unique parasite-host interfaces comprised of adhesion- and O-linked glycosylation-related domains. Adaptations, emerging due to intense selection for diversity in the molecular participants in organismal and genomic conflicts, being dispersed by lateral transfer, were subsequently exapted for eukaryote-specific innovations. We illustrate this using examples relating to eukaryotic chromatin, RNAi and RNA-processing systems, signaling pathways, apoptosis and immunity. We highlight the major contributions from catalytic domains of bacterial toxin systems to the origin of signaling enzymes (e.g., ADP-ribosylation and small molecule messenger synthesis), mutagenic enzymes for immune receptor diversification and RNA-processing. Similarly, we discuss contributions of bacterial antibiotic/siderophore synthesis systems and intra-genomic and intra-cellular selfish elements (e.g., restriction-modification, mobile elements and lysogenic phages) in the emergence of chromatin remodeling/modifying enzymes and RNA-based regulation. We develop the concept that biological conflict systems served as evolutionary “nurseries” for innovations in the protein world, which were delivered to eukaryotes via lateral gene flow to spur key evolutionary innovations all the way from nucleogenesis to lineage-specific adaptations.
Ever since the emergence of the endosymbiotic hypothesis as the primary model for the origin of eukaryotes there has been considerable interest in two major issues which it brought forth, namely large-scale lateral gene flow and genetic conflicts. While the exact details of the nature of this endosymbiotic event are still debated, by its very nature the endosymbiotic hypothesis implies gene flow between the alphaproteobacterial mitochondrial progenitor and the nucleo-cytoplasmic progenitor of archaeal ancestry (Martin and Muller, 1998; Esser et al., 2004; Rivera and Lake, 2004; Aravind et al., 2006; Gabaldon and Huynen, 2007; Pisani et al., 2007; Sapp, 2007). This phenomenon is not just relevant to the origin of eukaryotes, but also several other symbiogenic events that shaped the subsequent evolution of eukaryotes, such as the origin of the primary photosynthetic eukaryotes, including the plants, and the numerous secondary or tertiary photosynthetic eukaryotes (Delwiche, 1999; Palmer, 2003; Bhattacharya et al., 2004; Keeling, 2004; Huang and Gogarten, 2007; Oborník et al., 2009). In the former event, not just the well-known gene flow from cyanobacteria, but also complementary contributions from a chlamydia-like endosymbiont have been postulated (Huang and Gogarten, 2007). Additionally, there are other inter-organismal interactions that have occurred throughout eukaryotic evolution, which have resulted in comparable gene flow, albeit in a more episodic fashion (Anantharaman et al., 2007). Eukaryotes are characterized by a wide-range of close organismal associations. Indeed, cytoplasmic symbiotic bacteria, comparable to the progenitors of the mitochondria and chloroplasts, and infection by several types of large DNA viruses are a common feature of many eukaryotes, including representatives of the metazoan and amoebozoan lineages (Batut et al., 2004; Collingro et al., 2005; Ogata et al., 2006; Iyer et al., 2006b; Nikoh et al., 2008; Bertelli et al., 2010; Raoult and Boyer, 2010; Schmitz-Esser et al., 2010; Georgiades et al., 2011). There are also examples of some rather dramatic inter-eukaryotic associations, like endoparasitism as exhibited by apicomplexans, karyoklepty, or “theft” of chlorophyte nuclei (along with the chloroplasts) observed among ciliates, or karyoparasitism, involving injection of parasitic nuclei into host cells, which is observed in certain rhodophytes (Fields and Rhodes, 1991; Goff and Coleman, 1995; Johnson et al., 2007). Further, it has been noted that the phagotrophic nutrition of many eukaryotes can also result in a more general form of genetic chimerism, facilitated by the constant engulfment of genetic material of particular types of bacteria and eukaryotes (Doolittle, 1998). Yet other eukaryotes, such as the rotifers, appear to even actively engage in uptake and incorporation of genetic material from their environments—in addition to the proposed role in compensating for the lack of sexual reproduction, this phenomenon also serves as a conduit for notable “alien” gene flow (Gladyshev et al., 2008). Thus, it has become increasingly clear in the past two decades that gene flow between distant lineages and the consequent genomic chimerism might have a notable role in the evolution of eukaryotes.
Inter-organismal and intra-organismal genetic conflicts are a quotidian feature across all organizational levels of life (Smith and Price, 1973; Maynard Smith and Szathmáry, 1995; Hurst et al., 1996; Burt and Trivers, 2006; Werren, 2011). In their simplest form they include various trophic interactions between organisms, such as predation. Such conflicts might also arise between different cells of the same species cooperatively aggregating to form a multicellular assembly or developing as a multicellular organism due the emergence of “cheaters,” whose genetic interests do not align with the remaining cooperating cells (Dao et al., 2000). At the level of a single cell, as the interests of different genomes residing within it are not necessarily aligned with each other, there is potential for yet another level of genetic conflicts (Burt and Trivers, 2006). Such conflicts have a long evolutionary history in the prokaryotic superkingdoms in the form of the interactions between plasmids and the cellular genome. However, the endosymbiotic origin of eukaryotes made it one of their quintessential features because it brought together multiple distinct genomes (i.e., the nuclear and mitochondrial) in a single cell (Maynard Smith and Szathmáry, 1995; Werren, 2011). Such inter-genomic conflicts within the cell further expanded in course of eukaryotic evolution due to additional associations introducing interactions with genomes from plastids, nucleomorphs, and endosymbiotic/parasitic and intra-cellular bacterial predators of mitochondria (Sassera et al., 2006; Werren, 2011). In several cases symbiotic bacteria are involved in multi-level cooperation-conflict relationships: For instance, the bacterial symbiont Photorhabdus enables predatory nematodes to feed on insects by killing them with toxins (Bowen et al., 1998), whereas the endosymbiotic bacterium Hamiltonella defensa protects aphids against parasitoid wasps by deploying toxins against them (Degnan et al., 2009). Conflicts between the cellular genomes and viruses that exploit them for their own reproduction add yet another dimension to conflicts occurring within cells (Iyer et al., 2006b; Raoult and Boyer, 2010). Finally, there might be genetic conflicts within a single genome itself, arising from a wide variety of selfish elements trying to maximize their own fitness at the expense of the remaining genes (Burt and Trivers, 2006; Werren, 2011). These selfish elements are often characterized by a degree of intra- and/or inter-genomic mobility and assume a bewildering array of forms, including numerous distinct types of transposable elements, restriction-modification, and toxin-antitoxin systems (Kobayashi, 2001; Anantharaman and Aravind, 2003; Burt and Trivers, 2006; Ishikawa et al., 2010; Leplae et al., 2011). The former elements catalyze or facilitate their own proliferation, while the latter elements enforce cellular genomes to retain them by killing cells in which they are disrupted. Despite being primarily selfish elements, they might on occasions confer a fitness advantage to genomes, as this indirectly augments their own fitness (Burt and Trivers, 2006; Werren, 2011).
These conflicts are often directly mediated by particular molecules, either proteins or small molecules which act as “chemical armaments”; although in multicellular forms it might be reflected as morphological features that serve as weaponry (Smith and Price, 1973; Anantharaman and Aravind, 2003; Degnan et al., 2009; Ishikawa et al., 2010; Leplae et al., 2011; Werren, 2011; Zhang et al., 2011; Iyer et al., 2011b). Not surprisingly, each of the many levels of organismal conflict have sparked off intense “arms races” between the interacting organisms (Dawkins and Krebs, 1979), whose signatures are often seen in the form of extensive diversification of the proteins directly participating in, or synthesizing molecules deployed in conflict (Cascales et al., 2007; Zhang et al., 2011). Concomitantly, there is also a similar rapid diversification of proteins directly involved in defending or serving as antidotes against the chemical armaments deployed in the conflict (Anantharaman and Aravind, 2003; Leplae et al., 2011; Zhang et al., 2011; Iyer et al., 2011b). Importantly, both the offensive and defensive molecular adaptations involved in these conflicts can be transmitted between genomes by way of lateral transfer and is an important factor both in the spread of antibiotic production and resistance among prokaryotes (Walsh, 2003; Aminov and Mackie, 2007; Skippington and Ragan, 2011).
The ever-expanding genomic data from both eukaryotes and prokaryotes, along with genome-scale analysis, has considerably elucidated the major trends in the genomic chimerism arising from the bacterial and archaeal progenitors of the eukaryotes (Martin and Muller, 1998; Esser et al., 2004; Rivera and Lake, 2004; Aravind et al., 2006; Gabaldon and Huynen, 2007; Pisani et al., 2007). These analyses have particularly helped differentiate the cellular systems which have a primarily archaeal provenance (e.g., core DNA replication, core RNA metabolism, and translation) as against those with a primarily bacterial provenance (various aspects of energy, anabolic, and catabolic metabolism). However, uncovering the origins of specific systems, which appear to be eukaryotic synapomorphies (or shared derived characters), have required a somewhat distinct computational approach relying on in-depth analysis of protein sequences and structures (Aravind et al., 2006, 2011; Burroughs et al., 2011). Such analyses revealed glimpses of a collusion between gene flow through lateral transfer and the selective forces acting on molecular players in organismal and intra-genomic conflict in shaping the evolution of key components of systems such as eukaryotic chromatin, RNA-based gene regulation, and certain signaling pathways. However, this aspect of eukaryotic evolution is considerably under-appreciated. Hence, in this article we present a synthetic overview of: (1) how large-scale lateral gene flow between interacting organisms has facilitated the emergence of new adaptations deployed in inter-organismal conflict. (2) How adaptations developed due to the intense selection for diversity in the molecular participants in organismal and genomic conflicts were dispersed by lateral transfer and subsequently exapted for various eukaryote-specific adaptations. Due to limitations of space, we do not provide a comprehensive survey of all known instances of the above processes. Instead, we attempt to highlight the importance of these processes in the emergence of key adaptations, not just in early eukaryotes, but also during their subsequent evolution, with diverse illustrations emerging from recent investigations. We must emphasize that in this article we mainly use published examples that have been reported in several individual studies on various biological systems or protein families. However, this is the first time they are being brought together to create a coherent picture. A detailed presentation of the methodological apparatus for sequence, structure and phylogenetic analysis of the presented examples is precluded due to limitations of space. However, we refer readers to the individual studies from which we draw our examples for details regarding the computational analysis of the proteins considered here. We use these to develop a conceptual framework for understanding the importance of the diversifying forces acting during biological conflicts in facilitating adaptations that played a role in the so-called “major transitions” of eukaryotic evolutions (Maynard Smith and Szathmáry, 1995).
Sequence profile searches to establish the relationships between protein domains were performed using the PSI-BLAST (Altschul et al., 1997) and JACKHMMER (Eddy, 2009) programs that run against the non-redundant (NR) protein database of National Center for Biotechnology Information (NCBI). For most searches which were used to report the relationships presented in this work a cut-off e-value of 0.01 was used to assess significance. This was further confirmed with other aids such as secondary structure prediction and superposition on known structures, if available. Protein sequences were clustered using the BLASTCLUST program (ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.html) to identify related sequences in gene neighborhoods. Multiple sequence alignments of all domains were built by the Kalign (Lassmann et al., 2009) and PCMA programs (Pei et al., 2003), followed by manual adjustments on the basis of profile-profile and structural alignments. Secondary structures were predicted using the JPred program (Cuff et al., 1998). A comprehensive database of profiles was then constructed using these multiple alignments and was used extensively in the annotation and analysis of protein domain architectures and gene neighborhoods. For other known domains, the Pfam database (Finn et al., 2010) was used as a guide, though the profiles were augmented in several cases by addition of newly detected divergent members that were not detected by the original Pfam models. Clustering with BLASTCLUST, followed by multiple sequence alignment, and further sequence profile searches were used to identify other domains that were not present in the Pfam database. Signal peptides and transmembrane segments were detected using the TMHMM and Phobius programs (Kall et al., 2007). The HHpred program was used for profile-profile comparisons to either unify poorly characterized families of proteins or find homologous structures in the PDB database (Soding et al., 2005). Structure similarity searches were performed using the DaliLite program (Holm et al., 2008). Preliminary phylogenetic analysis was conducted using a rapid but approximate-maximum-likelihood method implemented in the FastTree 2.1 program under default parameters (Price et al., 2010). In-house bench-marking suggested that these results are generally comparable to complete ML implemented in the Phylip (Proml) and Molphy packages (Felsenstein, 1989; Adachi and Hasegawa, 1992). Predicted lateral transfers to eukaryotes were further evaluated for false positives by ensuring they were embedded in contigs or complete chromosome sequences with other genes typical of eukaryotes, comparing exon-intron structure of the genes, studying their phyletic distribution within eukaryotes and comparing the protein distances of the predicted eukaryotic proteins (as measured by bit scores) with bacterial homologs. Structural visualization and manipulations were performed using the PyMol (http://www.pymol.org) program. Automatic aspects of large-scale analysis of sequences, structures and genome context were performed by using the in-house TASS package, which comprises a collection of Perl scripts.
Apicomplexa are a remarkable clade of alveolate eukaryotes entirely comprised of highly specialized metazoan parasites (Levine, 1988; Vivier and Desportes, 1990). With other alveolates, such as ciliates, colpodellids, perkinsids and dinoflagellates, they share organelles known as extrusomes, which allow delivery of a payload of proteins into target cells, such as their prey or hosts (Leander and Keeling, 2003). While basal apicomplexans, the archigregarines, are partial endoparasites that insert only the forepart of their cell into the host cells to suck nutrients, the derived apicomplexans are obligate endoparasites that reside entirely within the cells they invade (Leander et al., 2006). Basal apicomplexans typically have a single-host, but many of the derived apicomplexans like the malarial parasite Plasmodium and Theileria have evolved lifecycles with two distinct hosts (Levine, 1988; Vivier and Desportes, 1990). Genome analysis of multiple apicomplexans ranging from the relatively basal Cryptosporidium to the highly derived Plasmodium have shown that they have evolved a remarkable set of secreted or membrane-anchored (surface) proteins that interact with host molecules as a part of the invasion process or other cytoadherance events during their lifecycle (Kaslow et al., 1988; Kappe et al., 1998, 1999; Anantharaman et al., 2007; Arredondo et al., 2012). While surface proteins in each apicomplexan lineage show a wide-range of lineage-specific domains (e.g., the Rifins and Dbl domain proteins in P. falciparum), they also contain a striking array of domains that are also found in surface proteins of animals (Patthy, 1999; Anantharaman et al., 2007) (Figure 1). Case by case phylogenetic analysis revealed that at least 18 types of non-catalytic domains from apicomplexans are otherwise found only in the animal lineage, or alternatively are most closely related to versions found in the animal lineage (Anantharaman et al., 2007) (Figure 1). Functional studies in metazoans suggest that majority of these domains, such as the thrombospondin-1 (TSP1), sushi/CCP, MAM, fibronectin-type 2, scavenger receptor, kringle, and vWA domains are involved in adhesive interactions between proteins or proteins and carbohydrates on the cell-surface (Bork, 1993; Patthy, 1999). More recently structural analysis has revealed that the SRS and s48/45 domains, respectively, from coccidian and aconoidasidan apicomplexans, were probably derived through rapid sequence divergence from the ephrin-like domain found in metazoan signaling molecules (Arredondo et al., 2012). Genome analysis suggests that while some of these “animal-like” domains were acquired early in apicomplexan evolution, yet others were acquired only later by specific lineages (Figure 1) (Anantharaman et al., 2007). This suggests that the acquisition of a structurally diverse, but functionally comparable group of domains from their animal hosts has been a persistent feature of apicomplexan evolution. Although functional studies on apicomplexan surface proteins with animal domains are still in relatively early stages, two major themes are beginning to emerge: (1) Some of these proteins appear to have a parasite-specific function in relation to their sexual development, such as in gamete fusion (Pradel et al., 2004; Arredondo et al., 2012). (2) Most others have been adapted for a diverse set of interactions pertaining to invasion of host cells or localization to particular tissues and are often secreted via specialized extrusomes of apicomplexans known as rhoptries (Bradley and Sibley, 2007; Santos and Soldati-Favre, 2011). Particularly striking is the recruitment of the TSP1-domain-containing adhesins early in apicomplexan evolution as part of the conserved invasion apparatus that depends on a cytoskeletal gliding motor unique to apicomplexans (Soldati-Favre, 2008).
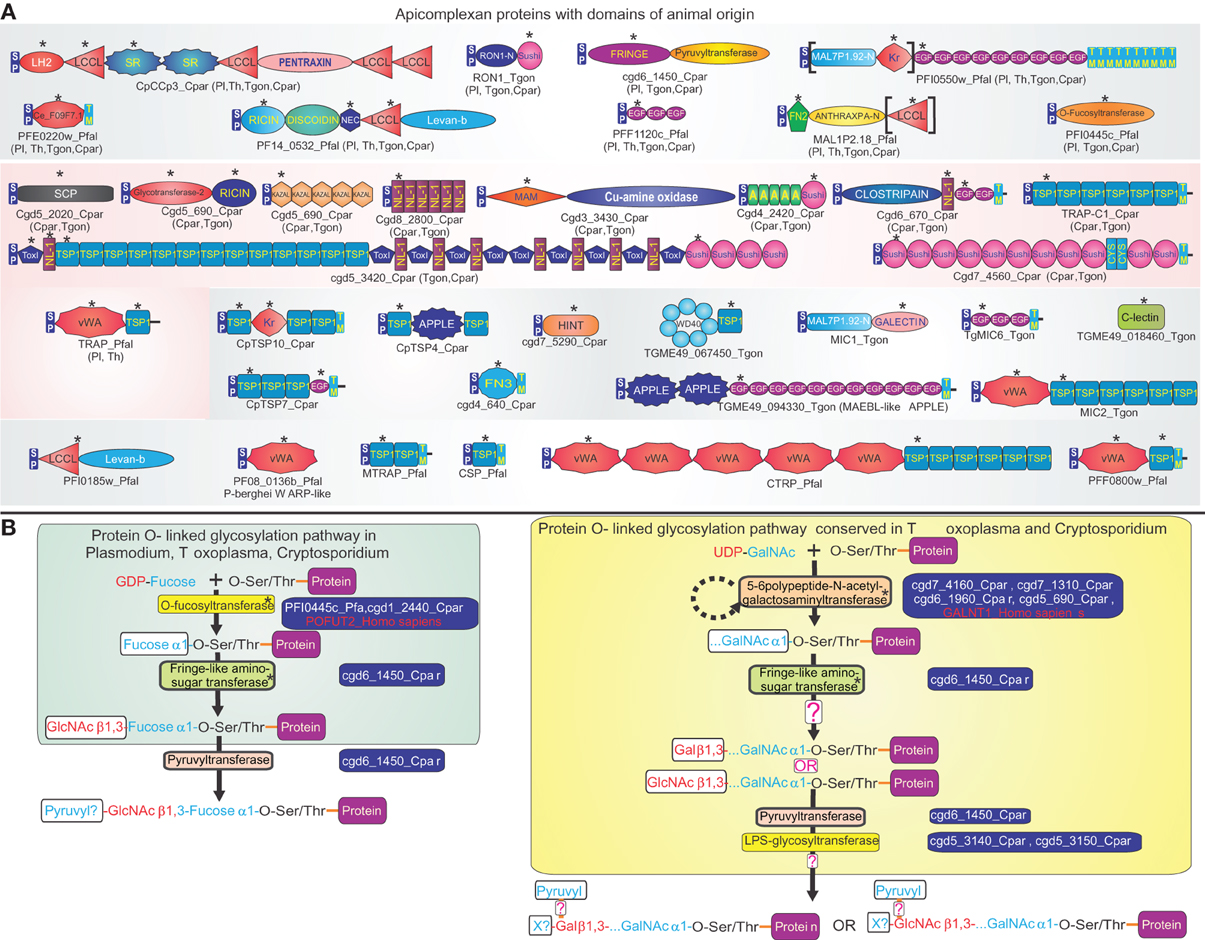
Figure 1. Animal domains and animal-type O-glycosylation systems in apicomplexa. (A) Domain architectures of apicomplexan proteins containing adhesion domains of animal origin. Proteins are labeled by their gene names/common names and species abbreviation separated by an underscore, and are grouped based on their conservation in apicomplexans. If a domain architecture is present in more than one distinct apicomplexan lineage, the additional lineages are shown in brackets. Domains of animal origin are marked with an asterisk above the domain. If a domain is present in multiple copies in a protein, only one (the first) instance of it is labeled with an asterisk. Domains not present in all orthologs of a protein are enclosed in square brackets. Standard abbreviations are used for domains. Species abbreviations are as follows: Cpar: Cryptosporidium parvum, Pl: Plasmodium, Pfal: Plasmodium falciparum, Th: Theileria, Tgon: Toxoplasma gondii. (B) Protein O-linked glycosylation pathways of animal provenance in apicomplexans. Gene names of enzymes involved in these pathways are shown to the right of the enzyme, along with examples of orthologous proteins from animals. The reconstructed oligosaccharide chain is represented using abbreviations for various sugars and functional groups. Speculative parts are marked with a “?”. GalNAc: N-acetylgalactosamine, GlcNAc: N-acetylglucosamine, X? indicates an uncharacterized sugar added by the LPS glycosyltransferase. Enzymes of animal origin are marked with an asterisk. Species abbreviations are as in (A).
Genome analysis has also revealed that apicomplexans possess an animal O-like glycosylation system with two separate arms performing the fucosylation and N-acetylgalactosaminylation of hydroxyl groups of serines or threonine on target proteins (Anantharaman et al., 2007) (Figure 1). The first of these has at its core two enzymes, the protein O-fucosyltransferase and a Drosophila fringe-like glycosyltransferase that elongates the initial fucose chain with N-acetylglucosamine (Varki et al., 1999; Luo et al., 2006). Also associated with this pathway is the fucose-GDP transporter that allows parasites to take up fucose (Luhn et al., 2001). Interestingly, this pathway modifies TSP1 and EGF domains, both of which appear to have been acquired by apicomplexans through lateral transfer from animals (Figure 1). The second pathway displays three distinct orthologous groups of proteins, which constitute the enzyme complex that transfers UDP-linked N-acetylgalactosamine to mucin-like target proteins typified by homopolymeric stretches of serines and threonines (Varki et al., 1999). Phyletic and phylogenetic analysis revealed that enzymes of both these arms of the O-linked glycosylation system and the fucose transporter are specifically related to their animal counterparts to the exclusion of homologs from any other lineage (Anantharaman et al., 2007). Furthermore, their phyletic patterns suggest that the glycosylation pathways were acquired in the common ancestor of endoparasitic apicomplexans, though they were either partially lost in haemosporidians or completely lost in piroplasms. Interestingly, in the more basal apicomplexans, like Cryptosporidium and the coccidians, there is a lineage-specific expansion of surface proteins with mucin-like S/T stretches, which are likely to be primary targets of the second arm of the glycosylation system (Stwora-Wojczyk et al., 2004; Anantharaman et al., 2007). Given the gut parasitism of these apicomplexans, it is possible that these glycosylated mucin-like proteins helped homotypic interactions with the gut mucosa, which is also enriched in surface mucins (McGuckin et al., 2011). However, emergence of vertebrate blood parasitism in haemosporidians and piroplasms probably rendered these useless, and perhaps even maladaptive due to the immune response directed against them, thereby favoring their loss.
Thus, apicomplexan genomics suggests that not just adhesion domains of surface proteins, but also entire modification pathways for them were acquired on account of lateral gene flow from their hosts. It appears likely that gene transfer from the host facilitated by the initial parasitic contact allowed the development of elaborate host interaction proteins that might have been central to the emergence of the intimate endoparasitism observed in apicomplexans.
In contrast to the above-discussed example, where a unique set of adaptations emerged due to lateral transfer in course of an evolving host-parasite conflict, several other molecular adaptations appear to be common across a wide-range of biological conflicts. These commonalities appear to be a consequence of two disparate forces: (1) Convergent evolution due to strong selection for particular types of molecular interactions in conflicts; (2) Rapid dispersion over wide phylogenetic distances of certain highly effective adaptations by lateral transfer. We briefly outline some of these adaptations below.
Proteinaceous toxins are the mainstay across all major levels of biological conflict. Such toxins are seen in competition between multicellular eukaryotes (e.g., castor bean ricin, Aspergillus sarcin and various snake venom proteins) and between them and their pathogens (e.g., anti-microbial peptide toxins and defensive RNases such as RNase A and RNase L)(Rochat and Martin-Eauclaire, 2000; Rosenberg, 2008; Wiesner and Vilcinskas, 2010). Conversely, such toxins are also used by pathogenic and symbiotic bacteria directed against their hosts (e.g., the cholera toxin and the shiga toxin) (Aepfelbacher et al., 2000; Alouf and Popoff, 2006). Similarly, the importance of protein toxins is becoming apparent in inter-bacterial conflicts (Schwarz et al., 2010; Russell et al., 2011; Iyer et al., 2011b; Zhang et al., 2011). In this regard, an exciting recent discovery has been made of a highly prevalent system of secreted multi-domain toxins, primarily involved in intra-specific conflict between related strains of prokaryotes (Aoki et al., 2011; Iyer et al., 2011b; Zhang et al., 2011). These proteins are typified by the tendency to vary their C-terminal toxin domains through a process of recombination that replaces an existing toxin domain by a distinct one encoded by standalone cassettes, while retaining the rest of the protein's architecture (i.e., N-terminal regions related to trafficking and presentation) intact (Zhang et al., 2011). Hence, these toxins are termed polymorphic toxins. They include contact-dependent versions, which have long N-terminal stalks comprised of RHS/YD or filamentous haemagglutinin repeats that present the C-terminal toxin domain at the tip, shorter diffusible versions, and versions injected or delivered via type VI and ESX/type VII secretory systems (Aoki et al., 2011; Iyer et al., 2011b; Zhang et al., 2011). Importantly, they share these delivery/presentation mechanisms with those toxins using conflicts with hosts (Schwarz et al., 2010). However, they are distinguished from them by the presence a specific immunity protein encoded by a gene downstream of the toxin gene (Aoki et al., 2011; Zhang et al., 2011). Given their role in intra-specific conflict, they are an important determinant of kin-recognition and thereby have an effect on the included fitness in prokaryotes. Inter-genomic conflicts between cellular genomes and selfish replicons residing in the same cell (e.g., classical bacteriocins and plasmid addiction toxins) and intra-genomic conflicts between selfish elements and the host genome (restriction-modification (R-M) systems and genomic toxin-antitoxin (TA) systems) also use protein toxins with related domains (Cascales et al., 2007; Zhang et al., 2011). The protein toxins of TA systems enable them to act as selfish elements that favor their own retention or “addiction” by killing cells where they are lost or disrupted. However, they might also enhance the fitness of their prokaryotic host. Indeed, expression of chromosomally embedded TA systems has been observed in diverse pathogens such as Mycobacterium tuberculosis and Brucella abortus when they are replicating within human cells. Here, the action of the toxin actually helps the bacteria to persist effectively in the hosts (Korch et al., 2009; Heaton et al., 2012).
There are some frequently recurrent themes in these toxins deployed across different levels of biological conflict: Most prominent are enzymatic toxins that disrupt the flow of biological information—nucleases targeting genomic DNA, tRNAs and rRNAs, nucleic acid base glycosylases, nucleic acid-modifying enzymes such as deaminases, peptidases that cleave key protein targets, and protein-modifying enzymes such as ADP-ribosyltransferases and AMP/UMPylating enzymes that alter the properties of proteins, such as components of the signaling and translation apparatus (Anantharaman and Aravind, 2003; Cascales et al., 2007; Leplae et al., 2011; Zhang et al., 2011). For example, toxins with the restriction endonuclease (REase) or HNH/ENDOVII folds are seen in intra-specific, inter-specific, inter-genomic (i.e., plasmid-encoded colicins) and intra-genomic conflicts (Stoddard, 2005; Cascales et al., 2007; Zhao et al., 2007; Zhang et al., 2011). Alternatively they disrupt cellular integrity by forming pores in cellular membranes (Gilbert, 2002). The enzymatic domains deployed in these conflicts are characterized by rapid sequence and structure divergence due to selection arising from immunity proteins and resistance against them.
Deployment of small molecule toxins or antibiotics, synthesized via dedicated secondary metabolism pathways, is another common strategy, primarily observed in inter-organismal conflicts (Walsh, 2003). They are particularly common in bacteria, and in certain eukaryotic clades, such as fungi and plants. Several distinct types of such molecules are synthesized, with aminoglycosidic, fatty-acid-based (polyketide) and peptide-based skeletons being prevalent (Walsh, 2003). These basic skeletons, which are often synthesized by large multi-domain or multi-protein complexes catalyzing one or more rounds of endoergic condensations of acyl moieties or amino acids, are typically subject to a wide variety of modifications enzymes such as 2-oxoglutarate-dependent hydroxylases, methylases and oxidoreductases (Walsh, 2003; Iyer et al., 2009, 2010). Related to antibiotics are siderophores that are secreted for chelation of essential environmental metals (Barry and Challis, 2009). While not being toxic, they are the center of inter-organismal conflict because several bacteria have evolved receptors for uptake of “non-self” siderophores that allow them to benefit from siderophores produced by other organisms in the environment (Lee et al., 2012). Organisms combat such siderophore-stealing by diversifying their siderophores through modifications similar to those of antibiotics (Samel et al., 2008). Similar pressures also apply to small molecule signals that are used, especially by bacteria, to communicate with each other, as they can also be potentially exploited by non-kin organisms (Brady et al., 2004). Thus, the related secondary metabolism pathways for antibiotic, signaling molecule and siderophore biosynthesis are under pressure for rapid diversification due to pressures from resistance and stealing. In most bacteria, components of these secondary metabolism pathways are encoded by multi-gene operons, which, as indicated by the large number of dioxygenases and oxidoreductases encoded by them, appear to have radiated concomitant with the first oxygenation event in Earth's history (Iyer et al., 2010). Subsequently, they appear to have undergone diversification through recruitment of multiple non-ribosomal peptide ligases and acyl condensation enzymes, sequence divergence of individual enzymatic components, and recombination between distinct biosynthetic operons to synthesize new products (Walsh, 2003; Samel et al., 2008; Iyer et al., 2009, 2010).
The fitness of intra-genomic and intra-cellular selfish elements depends on a variety of enzymes that allow their efficient propagation. One group of these enzymes is directly involved in the replication and transcription of the selfish DNA and provides autonomy from the host replication and transcription systems (Galun, 2003; Burt and Trivers, 2006). These enzymes include DNA polymerases, RNA polymerases, primases and reverse transcriptases, which in certain cases are distantly related to the cellular counterparts and in other cases, represent distinct, non-homologous enzymes with analogous activities. These enzymes often face selective pressures for diversification due to exploitation by defective or satellite element which lack their own replication or due to host defensive mechanisms (Galun, 2003; Burt and Trivers, 2006). Another widely used group of enzymes that do not directly catalyze nucleic acid synthesis are transposases/integrases, which often display nuclease domains related to the nuclease domain of toxins (see above) (Lilley and White, 2000; Galun, 2003; Stoddard, 2005; Burt and Trivers, 2006; Zhao et al., 2007; Mak et al., 2010; Zhang et al., 2011). One frequently encountered catalytic domain across a wide-range of transposons is a transposase/integrase domain of the RNAseH fold which is related to the nuclease domain found in the archaeal NurA and the argonaute nucleases (Aravind et al., 2000; Nowotny, 2009). This suggests that several of these mobile elements share an ultimate common ancestry in the form of an ancient RNAseH integrase. Additionally, these enzymes from selfish elements are characterized by a mélange of structurally distinct DNA-binding domains (DBDs), which diversify considerably due to pressures for specific recognition of sequences in the selfish elements (Babu et al., 2006).
Antagonistic actions in biological conflicts are countered by a variety of dedicated immunity mechanisms, which act over and beyond the immunity gained via sequence divergence of targeted proteins. The polymorphic toxins, plasmid-borne bacteriocins, TA, and R-M systems are all characterized by the presence of an antidote or immunity protein that neutralizes the toxin produced by them (Kobayashi, 2001; Leplae et al., 2011; Russell et al., 2011; Zhang et al., 2011). Thus, they channelize their antagonistic effects primarily against non-self replicons lacking the protective immunity proteins. Conflicts between cellular and viral genomes have selected for the emergence of multiple dedicated immunity mechanisms. Both prokaryotes and eukaryotes have evolved their own dedicated RNA-based mechanisms, respectively, the CAS/CRISPR and the RNAi systems, which utilize the complementarity of processed RNA to target invasive replicons (Allis et al., 2006; Grewal, 2010; Makarova et al., 2011). Bacteria additionally have evolved less-understood DNA-based mechanisms such as the Abi and the Pgl systems to counter bacteriophages (Sumby and Smith, 2002; Chopin et al., 2005). In eukaryotes, lineage-specific expansions and concomitant sequence diversification of particular receptor molecules, commonly those with leucine-rich repeats (LRRs) are exploited to provide receptors for recognition of viral and bacterial pathogens (“antigen receptors”) (Pancer and Cooper, 2006). In some cases, LRR and other domains might be combined with the SCF-type ubiquitin E3-ligases to allow degradation of proteins encoded by invasive replicons or cells (Thomas, 2006). In the vertebrate lineage, on two independent occasions, elaborate mechanisms involving mutagenesis and recombination have evolved to enable diversification of pathogen receptors, which respectively, utilize the immunoglobulin domain and LRRs (Pancer and Cooper, 2006).
Endosymbiotic/parasitic bacteria utilize a multipronged approach by often simultaneously deploying several toxins or effectors, each with its own mode of action to manipulate the behavior of the eukaryotic hosts in which they reside. Yet genomics of these bacteria suggests that there is a relatively small set of strategies that are exploited by intra-cellular bacteria from across the bacterial tree, including representatives of alphaproteobacteria, gammaproteobacteria, chlamydiae, and bacteroidetes (Collingro et al., 2005; Ogata et al., 2006; Penz et al., 2010; Schmitz-Esser et al., 2010; Georgiades et al., 2011). The most commonly used approach is the deployment of proteins that alter action of the ubiquitin system, including E3-ligases with RING, U-Box and F-Box domains, deubiquitinating and desumoylating peptidases, especially of the OTU and SMT4/Ulp1-like families and ubiquitin-like (Ubl) proteins (Loureiro and Ploegh, 2006; Lomma et al., 2010; Penz et al., 2010; Schmitz-Esser et al., 2010). Such effectors are seen in several bacteria such as the chlamydiae, like Chlamydia, Protochlamydia and Waddlia, proteobacteria like Odysella, Wolbachia and Legionella, and the bacteroidetes Amoebophilus (Figure 2). Protein modification by the action of toxins/effectors with ADP-ribosyltransferase, DOC-type AMP/UMPylase, protein methylases and protein kinase domains is another widely used strategy common to several bacteria such as Yersinia, Xanthomonas, Legionella, Amoebophilus, and Waddlia (Yarbrough et al., 2009; Aravind et al., 2011; Feng et al., 2012). These modifying enzymes target proteins from various host systems such as chromatin and signaling proteins. Recent studies have indicated that deployment of diverse nucleic-acid-targeting effectors is also a common feature of numerous endoparasites/endo-symbionts. For example, effectors/toxins with nucleic deaminase domains are seen in Orientia, Wolbachia, and Amoebophilus (Zhang et al., 2011; Iyer et al., 2011b). Likewise, several of these bacteria also share effectors with different nuclease domains that might target both DNA and RNA. Interestingly, studies on eukaryotic viruses suggest that several of viruses also deploy a similar class of molecules. For example, numerous ubiquitin system components, including ubiquitin, SUMO and Apg8-like proteins, E3-ligases and deubiquitinating/desumoylating peptidases are encoded by nucleo-cytoplasmic large DNA viruses, baculoviruses and herpesviruses (Iyer et al., 2006b). Several Ubl proteins are also observed in polyproteins of eukaryotic RNA viruses (Burroughs et al., 2012). Similarly, protein kinases, ADP-ribosyltransferases and some other protein-modifying enzymes are also observed in several NCDLVs and baculoviruses such as the Agrotis segetum granulovirus (Iyer et al., 2006b; De Souza and Aravind, 2012).
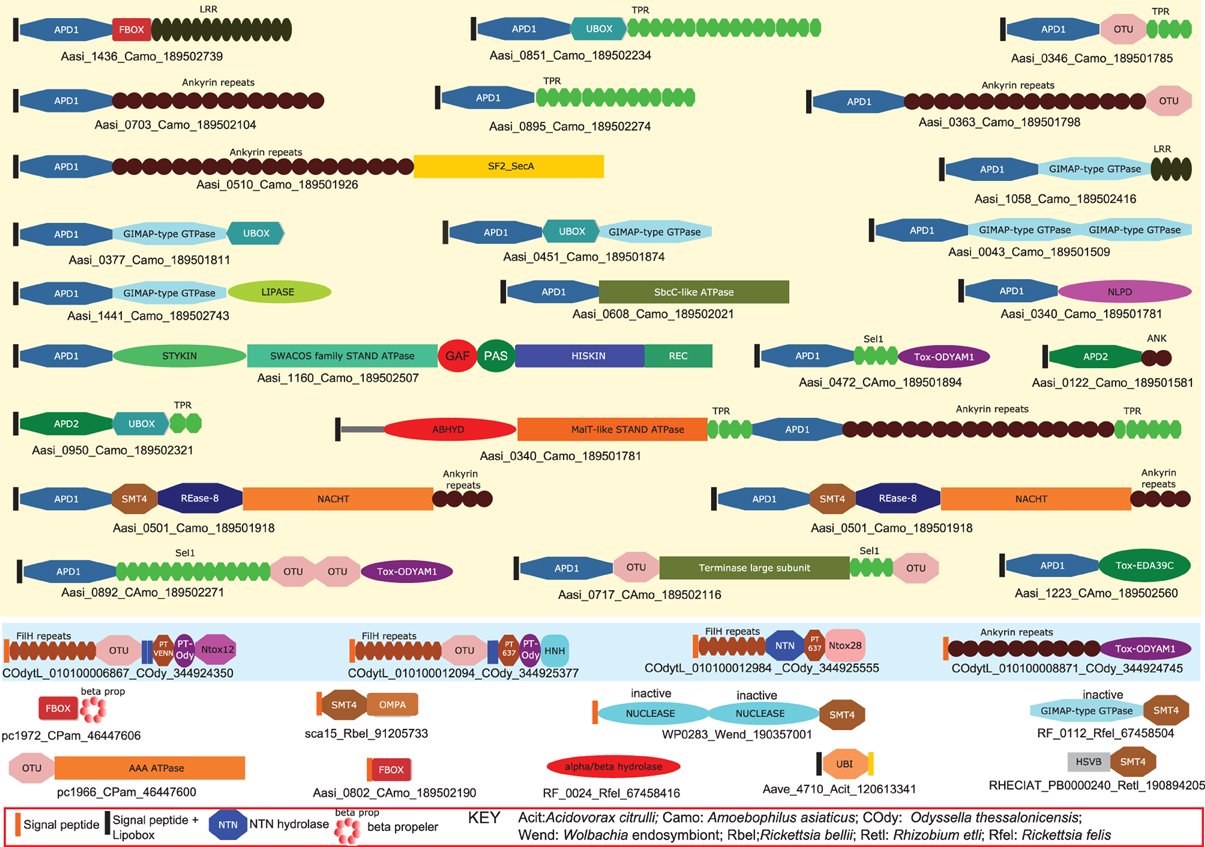
Figure 2. Domain architectures of effectors deployed by endosymbiotic/parasitic bacteria illustrating certain common functional strategies. Proteins are labeled by their gene names, species abbreviations and genbank index (GI) numbers separated by underscores. Non-standard domain names and expansion of species abbreviations are given in the key below the figure. Additionally, Amoebophilus prodomain 1 (APD1) and Amoebophilus prodomain 2 (APD2) are Amoebophilus-specific N-terminal domains that are present immediately downstream of a signal peptide and a lipobox. These domains are likely to help in the specific localization and/or clustering of effectors from this organism.
Among the endosymbiotic bacteria, Amoebophilus and Protochlamydia, which infect amoebozoan eukaryotes, are particularly striking in that a notable fraction of their proteomes is comprised of diverse effectors with different kinds of catalytic domains (Collingro et al., 2005; Schmitz-Esser et al., 2010). These include numerous ubiquitin system proteins, kinases and α/β hydrolases, which might function as lipases, RNases and REase-fold DNAses (Figure 2). Also notable are the Amoebophilus effectors with a GTPase domain related to the animal GIMAP GTPases and the AIG1-like GTPases of plants, which play a role in providing scaffolds on intra-cellular membranes (Schwefel et al., 2010). It is conceivable that bacterial effectors with these GTPase domains play a comparable role in remodeling the host membranes surrounding intra-cellular bacteria. Interestingly, such GIMAP GTPases are also encoded by certain animal RNA viruses (e.g., Duck hepatitis A virus) and herpesviruses (e.g., Anguillid herpesvirus 1). Together, the above observations suggest that there are relatively few ancient routes to achieve successful colonization of eukaryotic cells. These appear to have emerged, in part convergently, and in part via lateral transfer of certain effective catalytic toxin/effector domains between unrelated or distant intra-cellular residents of eukaryotes. Interestingly, the genomes of such endosymbiotic bacteria [e.g., Wolbachia (Nikoh et al., 2008)] and or DNA viruses [e.g., a Herpesvirus inserted into the genome of the amphioxus (De Souza et al., 2010)] can be integrated into host genomes. Thus, they serve as an effective conduit for transfer of symbiont/parasite adaptations to their hosts.
In this section of the article we discuss with examples as to how several of the above-discussed players deployed in biological conflicts have played a major role in the emergence and elaboration of various eukaryotic adaptations. In doing so we take examples both from early events close to eukaryogenesis and also systems that evolved in particular eukaryotic lineages, such as metazoans.
Eukaryotes are distinguished from the two prokaryotic superkingdoms by their dynamic chromatin organized by histones with low complexity tails, which provides a veritable “ecosystem” for several protein-modifying and ATP-dependent remodelers (Allis et al., 2006; Kouzarides, 2007; Aravind et al., 2011; Iyer et al., 2011a). The mysterious origins of several of the unique components of eukaryotic chromatin have begun to considerably clear up with recent genomic data. SWI2/SNF2 ATPases, which had at least six representatives by the time of the last eukaryotic common ancestor (LECA), had already diversified to perform several distinct chromatin remodeling activities, such as sliding/ejection of nucleosomes, exchange of canonical nucleosomes with those containing alternative histones, or altering nucleosomal spacing (Iyer et al., 2008b; Hauk and Bowman, 2011; Hota and Bartholomew, 2011). Phylogenetic, domain architecture, and gene neighborhood analysis revealed that SWI2/SNF2 ATPases are superfamily II DNA helicases, which had their most extensive diversification as part of R-M systems and related systems that are likely to function as a defensive mechanism against bacteriophages (related to the phage growth limitation or Pgl system) (Iyer et al., 2008b) (Figure 3). In phylogenetic trees, the eukaryotic versions are nested within the radiation of SWI2/SNF2 ATPases from prokaryotic selfish elements and were transferred on at least three independent occasions to eukaryotes (Figure 3A). The first of these transfers occurred prior to the LECA, and by the time of the LECA had proliferated to spawn at least six distinct lineages (Iyer et al., 2008b). The remaining two transfers occurred much later in eukaryotic evolution, and gave rise to the Strawberry Notch and HARP-like SWI2/SNF2 ATPases (Figure 3) (Iyer et al., 2008b). Bacterial R-M systems contributed a second ATP-dependent chromatin remodeling enzyme to eukaryotes, the MORC ATPase, which contains a composite module comprised of gyrase, histidine kinase, and MutL (GHKL) and S5 domains (Iyer et al., 2008a). Analysis of R-M bacterial systems showed that they display a vast radiation of several different types of GHKL-S5 module ATPases, of which the MORCs form one distinct clade (Figure 3B). Given that basal excavate lineages, such as parabasalids and diplomonads lack MORCs, they appear to have been acquired by eukaryotes post-LECA, prior to the radiation of the large eukaryotic clade uniting animals, fungi, amoebozoans, and plants (Iyer et al., 2008a) (Figure 4). Both the MORCs and the SWI2/SNF2 ATPases use ATP hydrolysis to catalyze DNA-unwinding or large-scale looping of DNA in aiding the restriction activity of the REases. This activity has been reused in a biochemically comparable, but functionally distinct, context to remodel protein-DNA contacts or facilitate higher-order looping in eukaryotic chromatin. In a similar vein, R-M systems might also account for the origin of the eukaryotic phosphoesterase enzyme TDP1, which hydrolyzes 3'-phosphotyrosyl bonds between DNA and the active tyrosine of topoisomerase Ib to release DNA from topoisomerase adducts (Gajewski et al., 2012). Sequence relationships of TDP1 suggest that it is likely to have been derived from HKD phosphoesterase domains found fused to SWI2/SNF2 ATPases in bacterial R-M systems (Iyer et al., 2006a).
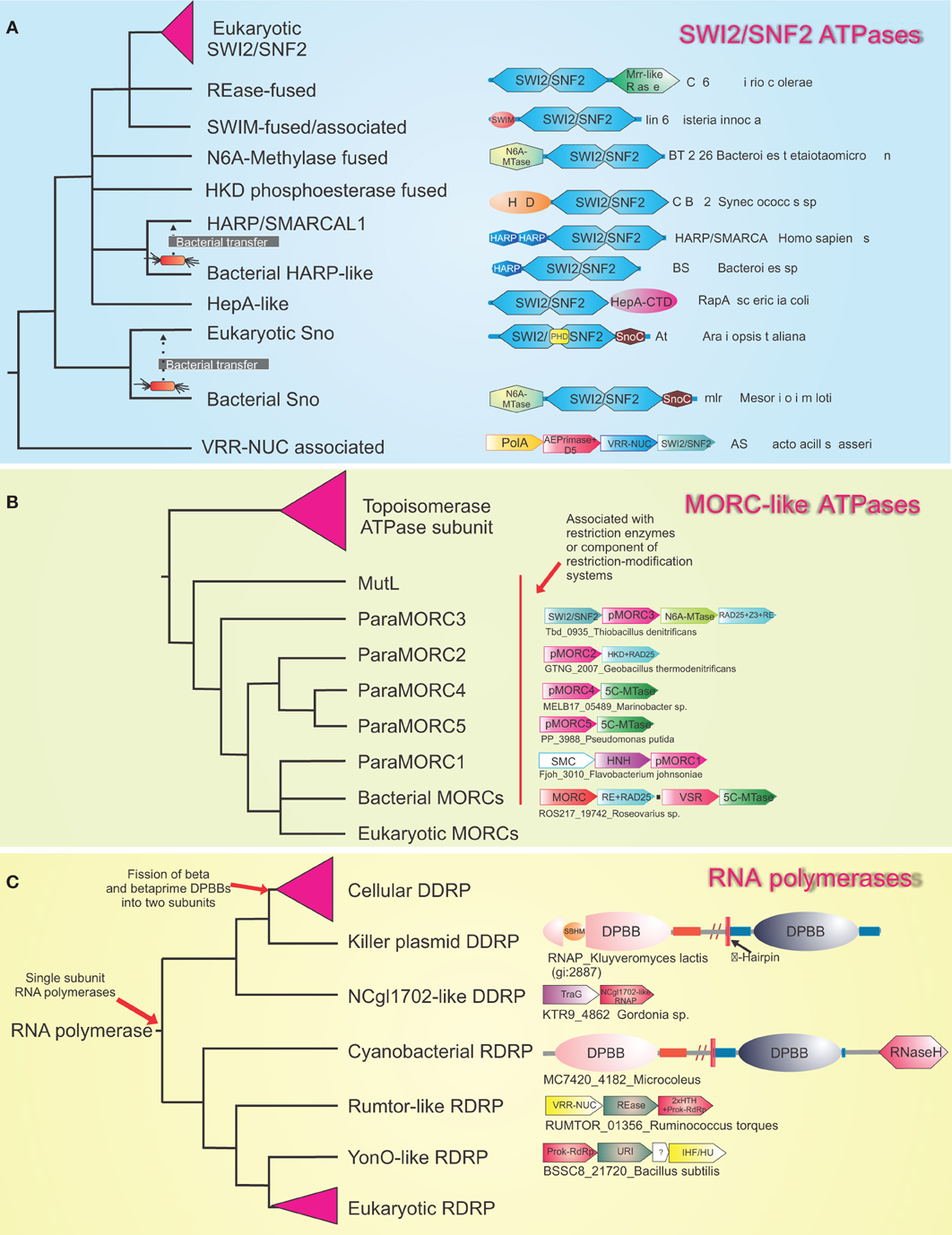
Figure 3. Evolutionary relationships of various families of enzymes illustrating the origin of eukaryotic versions within radiations of systems involved in inter- and intra-genomic conflicts. Reconstructed phylogenetic trees are shown for (A) The bacterial radiation of the SWI2/SNF2 ATPases. (B) MORC-like ATPases and (C) The Double-psi beta barrel containing RNA polymerases. Certain clades with multiple families such as the eukaryotic SWI2/SNF2 ATPases, the Topoisomerase ATPase subunits, the cellular DDRP and eukaryotic RdRPs are collapsed into triangles for clarity. Illustrative domain architectures or gene neighborhoods are shown next to the leaf. Genes in gene neighborhoods are shown in block arrows with the arrow head pointing from the 5′ to the 3′ gene. Proteins and gene neighborhoods are labeled by the gene name and species name separated by underscores. The trees represent only the overall topology because they were obtained by a combination of conventional phylogenetic tree construction and structure-based determination of higher-order relationships.
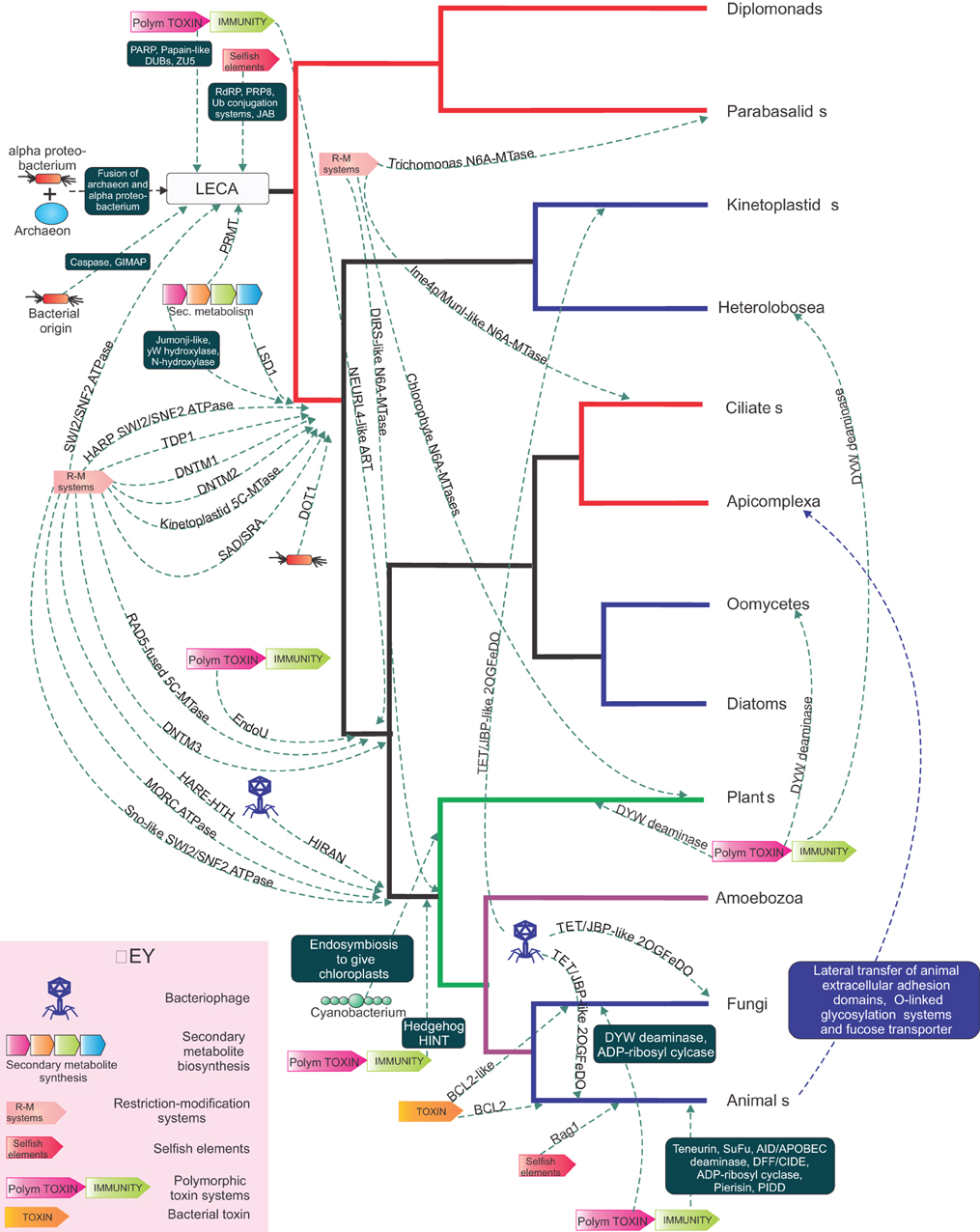
Figure 4. A tree of the eukaryotic relationships illustrating the points of recruitment in eukaryotes in different functional systems of various domains from different biological conflict systems. With a eukaryotic tree as reference, the source and reconstructed point of transfer of various domains recruited from different conflict systems and symbiogenic events are shown. The transfers are shown as dashed arrows with the arrow head pointing to the ancestor in which the transfer is proposed to have taken place. The dashed lines are labeled either with a single gene or a set of genes enclosed in a box. The conflict systems are shown in the key at the bottom left.
Similar studies have shown that the DNA methylases of eukaryotes, which play an important role as encoders of epigenetic information that goes over and beyond the basic genetic information, also largely owe their origin to R-M systems and related methylation systems that protect prokaryotic genomes against restriction attacks by selfish R-M systems (Bestor, 1990; Iyer et al., 2011a). Both DNA cytosine (C5) and adenine (N6) methylases of eukaryotes appear to have been derived from bacterial R-M system and dcm methylases on more than 10 independent occasions (Iyer et al., 2011a). As none of the conserved eukaryotic lineages of DNA methylases can be detected in the parabasalids and diplomonads, it appears that the classical epigenetic DNA modification of cytosine was absent in the LECA. The primary conserved cytosine DNA methylase of eukaryotes, DNMT1, appears to have emerged only just before the time the heterolobosean-kinetoplastid clade branched off from the remaining eukaryotes, and phylogenetic analysis strongly supports its origin from a bacterial R-M system methylase-related to M.NgoFVII (Iyer et al., 2011a). Most other DNA methylases of eukaryotes can be attributed to comparable later acquisitions, primarily from other types of R-M systems. Recent discoveries have indicated that the reversal of cytosine DNA methylation in several eukaryotic lineages occurs via the action of Tet-JBP family of 2-oxoglutarate and iron-dependent dioxygenases (2OGFeDOs), which remove the methyl group through oxidative conversion to hydroxymethylcytosine and further oxidized cytosine derivatives that are then cleared by base excision repair (He et al., 2011; Iyer et al., 2011a). Interestingly, related enzymes, JBP1/2, catalyze the hydroxylation of thymine in the synthesis of base J, an epigenetic modification observed in kinetoplastids (Vainio et al., 2009). Prior studies on the evolution of 2OGFeDOs revealed that the eukaryotic Tet-JBP enzymes were derived from precursors encoded by caudate bacteriophages (Iyer et al., 2011a). Bacteriophages have been known to display a rich variety of DNA modifications, including hydroxymethylated pyrimidines, which enable them to evade restriction by different R-M systems in the host genome (Gommers-Ampt and Borst, 1995). Thus, the bacteriophage Tet-JBP enzymes appear to have first emerged as part of their counter-restriction strategy, and subsequently recruited to generating and erasing epigenetic marks on DNA upon being transferred to eukaryotes. Multiple studies have also revealed that not just enzymatic domains, but also specific DBDs found in eukaryotic chromatin proteins might have been acquired from bacterial R-M systems and replication apparatus of caudate bacteriophages. The SAD/SRA domain, which is a key player in eukaryotic chromatin as an epigenetic “reader” of hemimethylated cytosine marks, has been derived from the DNA-binding domain of REases from R-M systems that discriminate between hemimethylated and fully methylated sites (Iyer et al., 2011a). Likewise, the recently described HARE-HTH domain, which might have an important role in discriminating the DNA modification generated by the cytosine methylases, and the Tet/JBP enzymes has also evolved from bacterial R-M systems, where it is combined with several distinct REase domains (Aravind and Iyer, 2012). On the other hand, another DNA-binding domain, the HIRAN domain, which among other proteins is associated with the eukaryotic chromatin remodeling RAD5-type SWI2/SNF2 ATPases appears to have emerged from the replication apparatus of caudate bacteriophages (Iyer et al., 2006a).
In stark contrast to chromatin remodeling and epigenetic DNA modifications, enzymes catalyzing epigenetic modifications of proteins in eukaryotic chromatin appear to have extensively drawn from very different types of prokaryotic systems involved in inter-organismal conflict. Two key epigenetic modifications are acetylation of lysines and methylation of both lysines and arginines in histones and other proteins in eukaryotic chromatin (Allis et al., 2006; Kouzarides, 2007). Sequence comparisons show that the eukaryotic arginine methylases (PRMT) have been derived from within a bacterial radiation of peptide methylases (Aravind et al., 2011). The closest bacterial sister groups of the eukaryotic PRMTs are encoded in antibiotic-like secondary metabolite biosynthesis operons that also contain genes for peptide dioxygenases, non-ribosomal peptide synthetases and other peptide-oxidizing enzymes such as LSD1-related amine oxidases (Aravind et al., 2011). Bacterial PRMT domains are also incorporated as domains of gigantic antibiotic biosynthesis enzymes, such as anabaenopeptilide synthetase that synthesizes a peptide toxin of the cyanobacterium Anabaena (Rouhiainen et al., 2000; Aravind et al., 2011). Interestingly, the LSD1-like amine oxidases observed in these and other peptide antibiotic/toxin biosynthesis operons are also the precursors of eukaryotic histone demethylases that catalyze oxidative removal of methyl groups from mono- and di-methylated histone H3K4 (Allis et al., 2006; Kouzarides, 2007). All the remaining histone demethylases in eukaryotes belong to one large superfamily of 2-oxoglutarate-dependent dioxygenases known as the Jumonji-related dioxygenases (Iyer et al., 2010). These, along with LSD1, are absent in the earliest-branching eukaryotes such as parabasalids and diplomonads, and first appear as multiple paralogous copies just prior to the divergence of the heterolobosean-kinetoplastid clade from the other eukaryotes (Iyer et al., 2010). However, each of these multiple eukaryotic paralogous lineages have their own bacterial counterparts suggesting that they had already diverged in bacteria before being acquired. In bacteria, like LSD1, they appear in one or more copies in peptide antibiotic/toxin and siderophore biosynthesis operons (Iyer et al., 2010), where they are likely to catalyze multiple oxidative modifications of peptides as previously observed in the biosynthesis of penicillin and its derivatives (Liras and Demain, 2009). Thus, it is plausible that eukaryotes acquired multiple paralogous jumonji-related dioxygenases via the transfer of a single secondary metabolism gene-cluster with multiple versions of these enzymes. In eukaryotes, other than histone demethylation, they also radiated to give rise to enzymes catalyzing the last step in the generation of the eukaryote-specific tRNAPhe modification, hydroxywybutosine, and protein asparagine hydroxylation (Iyer et al., 2010). In contrast to these, the histone H3K79 methylase Dot1 appears to have emerged from a methylase effector delivered by intra-cellular symbionts and is seen in diverse bacterial endo-symbionts/pathogens of amoeboid protozoans and metazoans, like Parachlamydia and Legionella (Aravind et al., 2011).
Thus, components from R-M and virus-restriction systems, viral replication apparatus, peptide antibiotic/siderophore biosynthesis systems and effectors of intra-cellular bacteria, which are exemplars of intra-genomic, inter-genomic and inter-organismal conflict systems, have been harnessed as progenitors of distinguishing components of eukaryotic chromatin.
Eukaryotes are characterized by the unique RNAi system, which is typified by small RNAs (usually 23–35 nt in length) that perform a number of roles ranging from post-transcription gene regulation to regulation of chromatin structure (Allis et al., 2006; Grewal, 2010). Of these small RNAs, the siRNA-type RNAs are particularly important in gene-silencing, and might be amplified by a distinctive enzyme of this system, the RNA-dependent RNA-polymerase (RdRP), which can be traced back to the LECA (Salgado et al., 2006; Ruprich-Robert and Thuriaux, 2010; Iyer and Aravind, 2011). Sequence-structure analysis of the RdRP revealed that its two catalytic double-ψ−β-barrel (DPBB) domains are related to the catalytic domain found in the two largest subunits of the cellular RNA polymerases from all life forms (Salgado et al., 2006; Ruprich-Robert and Thuriaux, 2010; Iyer and Aravind, 2011). The search for RdRP cognates outside eukaryotes showed that they are prevalent in certain bacteriophages of firmicutes and also a variety of recently identified novel selfish elements in bacterial genomes (Figure 3C) (Iyer and Aravind, 2011). In these potential selfish elements they are often encoded alongside genes for different DNase domains such as those belonging to the REase and URI endonuclease fold, which might aid in the mobility of the elements (Figure 3C). The RdRPs might also be combined with RNAse H domain in the cyanobacterial versions suggesting that might function in the context of RNA-DNA hybrids (Iyer and Aravind, 2011). Furthermore, structural analysis of the RNA-polymerases with DPBB catalytic domains showed that the RdRP-like enzymes belonged to a radiation of single-subunit RNA polymerases encoded by variety of selfish elements, from within which the cellular multi-subunit versions emerged via fission of the two catalytic domain-containing segments of the single-subunit enzyme (Figure 3C). It appears plausible that these RdRP-like enzymes of intra-genomic selfish elements and bacteriophages primarily arose as enzymes that aided their mobility by potentially acting as primases enabling their replication (Iyer and Aravind, 2011). Upon acquisition by the eukaryotic lineage, prior to the LECA, the enzyme appears to have been recruited as a part of the RNAi systems for amplification of small RNAs. Interestingly, the RdRP is not the only nucleic acid polymerase that has been recruited to RNA metabolism from a prokaryotic selfish element. Recent studies on the domain architectures and sequence relationships of the most conserved splicing factor of eukaryotes Prp8, which is part of the spliceosomal catalytic center, has revealed that it has been derived from the polyprotein of a retroelement replete with the reverse transcriptase, “thumb” and RNaseH domains (Dlakic and Mushegian, 2011). However, in Prp8 the active site of the reverse transcriptase domain is disrupted, suggesting that it merely functions in a nucleic acid-binding capacity rather than as an active enzyme (Dlakic and Mushegian, 2011). It is conceivable that this retroelement was associated with the ancestral group-II introns that invaded the genome in the pre-LUCA period to give rise to the spliceosomal introns of eukaryotes.
On several occasions, components of yet another prokaryotic inter-organismal conflict system, namely the recently characterized polymorphic toxin systems, appear to have contributed to eukaryotic RNA-processing and modification systems (Zhang et al., 2011). In eukaryotes, small nucleolar RNAs (snoRNAs) are required for modification and maturation of rRNA in the nucleolus. In several eukaryotes certain snoRNA, like U16 and U86, are directly released from the introns encoding them by the endonucleolytic action of the EndoU RNase (Laneve et al., 2003). Sequence and structure analysis revealed that the EndoU RNase of eukaryotes is nested within a vast radiation of RNase domains that function as toxins in bacterial polymorphic toxin and related secreted toxin systems (Zhang et al., 2011). Thus, acquisition of the EndoU domain appears to have enabled eukaryotes to bypass splicing to directly release snoRNAs from introns. RNA-editing via deamination of cytosine and adenine has considerably expanded in eukaryotes and is observed not just in tRNAs but also in mRNAs and as part of a counter-viral strategy (Iyer et al., 2011b). The origins of certain divergent metal-dependent nucleic acid deaminase domains, such as those of the AID-APOBEC clade and the DYW clade, which catalyzes massive RNA-editing in plant chloroplasts and mitochondria, were rather unclear until recently (Zehrmann et al., 2011). Analysis of the polymorphic toxins revealed that one of the widely used toxin domains was the nucleic acid deaminase that had greatly diversified in such and related secreted toxins (Iyer et al., 2011b). Importantly, the origin of the both the DYW and AID-APOBEC-like deaminases could be placed within specific prokaryotic toxin groups (see below for details).
Recent studies on the diversity of catalytic toxin domains deployed in bacterial polymorphic and related secreted toxins systems are also throwing light on the emergence of what were previously considered uniquely eukaryotic signaling systems (Figure 4). One such is the polyADP-ribosylation system, which modifies aspartate, glutamate and lysine side chains in both cytoplasmic and nuclear proteins including histones, with profound effects on DNA repair, chromatin organization, telomere dynamics, centrosomal and mitotic spindle organization, and endosomal trafficking (Ame et al., 2004). The enzymes catalyzing this modification, polyADP-ribosyl polymerases (PARPs), can be traced back to the LECA, but their emergence in eukaryotes remained a mystery (Citarelli et al., 2010). The closest relatives of the PARPs are found among toxin domains of a toxin used in inter-bacterial conflicts delivered via a distinctive phage-derived, injecting secretory system known as the Photorhabdus virulence cassette (Hurst et al., 2004; Zhang et al., 2012). Related PARP domains are also found as effectors of intra-cellular symbionts/parasites of amoebae and metazoa such as Legionella drancourtii. Recently, a novel family of ADP-ribosyltransferases (ARTs), distinct from the PARPs, was identified, and typified by the Neurl4 protein of humans (De Souza and Aravind, 2012). These ARTs might have an important role in the organization of the eukaryotic centrosome among other processes. They also seem to have been derived from effectors delivered by endoparasitic bacteria, such as Waddlia (Hurst et al., 2004). The use of mono-ADP-ribosyltransferases by diverse bacteria as toxins in intra- and inter-specific conflicts (i.e., polymorphic toxins) and those directed at host proteins is well-known (Koch-Nolte et al., 2008; Laing et al., 2011; De Souza and Aravind, 2012). Indeed, other than the PARPs and Neurl4-like ARTs, the eukaryotes also possess several mono-ARTs which are nested within the radiation of bacterial toxin ARTs. Thus, on more than three occasions eukaryotes appear to have recruited the toxin ART/PARP domains as protein-modifying enzymes, with the event giving rise to the PARPs probably happening before the LECA (Figure 4). While in bacteria these enzymes appear to largely function as toxins, in eukaryotes they appear to have been utilized to post-translationally modify proteins and provide an additional level of coding information (Koch-Nolte et al., 2008; Laing et al., 2011). Beyond the events spawning pathways that are widespread in eukaryotes, polymorphic and related toxin systems also appear to have contributed to the origin of signaling systems unique to certain lineages, such as metazoans. In addition to ARTs, other bacterial toxin domains utilizing NAD as a substrate have also been recruited to metazoan signaling. The ADP-ribosyl cyclase domain was previously observed only in animals (in the CD38 and CD157 proteins) and generates two messenger molecules, namely cyclic ADP ribose (cADPr) and nicotinic acid adenine dinucleotide phosphate (NAADP), respectively, from NAD and NADP (Guse and Lee, 2008). The former two nucleotides function as messenger molecules that induce calcium signaling pathways via the ryanodine receptors (Guse and Lee, 2008). The discovery of the ADP-ribosyl cyclase as a toxin domain in bacterial polymorphic toxins provides a potential explanation for the sudden origin of this signaling enzyme in animals (Zhang et al., 2012). Additionally, fungi too appear to have independently acquired this domain from bacteria, suggesting that it might have been recruited on more than one occasion in eukaryotic evolution (Zhang et al., 2012).
The Teneurin/Odd Oz proteins found in metazoans and choanoflagellates function as developmental regulators with a potential role in cell-surface adhesion in diverse processes such as cell migration, neuronal path finding and fasciculation, gonad development, and basement membrane integrity (Minet et al., 1999; Silva et al., 2011). These proteins appear to have been derived from a complete bacterial polymorphic toxin, with both the N-terminal RHS/YD repeats, which form a stalk and the C-terminal toxin domain that is a derived version of the HNH/EndoVII fold (Zhang et al., 2012). While the C-terminal toxin domain has lost its active site residues in the animal lineages, it is cleaved and secreted as a potential neuromodulator (Qian et al., 2004). On the other hand the N-terminal RHS repeats appear to play a role in adhesion between different Teneurin/Odd molecules, which is a key aspect of their cell-cell signaling function (Silva et al., 2011). Other than the toxin domains, certain other domains in eukaryotic signaling pathways have also been acquired from bacterial polymorphic toxin systems. The hedgehog signaling pathway is a eukaryotic signaling pathway initiated by the hedgehog proteins, which undergo autoproteolytic cleavage to release signaling messengers (Ingham et al., 2011). The HINT domain, which catalyzes this autoproteolytic cleavage in the eukaryotic hedgehog proteins, is likely to have been derived from the HINT domains commonly found in bacterial polymorphic toxins, where they apparently facilitate the autoproteolytic release of the C-terminal toxin domain into target cells (Zhang et al., 2011). In metazoans, hedgehog activates a down-stream signaling cascade in target cells to activate the transcription factor Gli (Ingham et al., 2011). The Suppressor of Fused (SuFu) protein tethers the Gli in the cytoplasm in the absence of the hedgehog signal to prevent constitutive activation. This SuFu protein of the animal hedgehog pathway also has its origin in bacterial polymorphic toxin systems, where members of the SuFu superfamily function as immunity proteins that neutralize a structurally diverse range of toxin domains (Zhang et al., 2011).
One of the most remarkable features of eukaryotes is the ubiquitin system, which comprises of several parallel enzymatic cascades which ligate Ubiquitin or an Ubl protein to target proteins, typically on a lysine residue (Hochstrasser, 2009). These cascades are typified by an E1 enzyme, which activates the Ub/Ubl terminal COOH group by adenylation and trans-thiolation to transfer it to and E2 enzyme. The E2 enzyme may then either directly or via an E3 enzyme transfer the Ub/Ubl to the target protein. In eukaryotes, such modifications often target proteins for degradation via the proteasomal system, where the Ub/Ubl is first cleaved off and released by a JAB domain metallopeptidase (Kerscher et al., 2006). In addition to proteasomal degradation, Ub/Ubl modifications also alter the interactions, localization and biochemistry of the target proteins and are modulated by a series of peptidases (DUBs) that debiquitinate them (Burrows and Johnston, 2012). Until recently it was thought that the Ub-system was a purely eukaryotic innovation. However, multiple studies have shown that the antecedents of the Ub-system first emerged in prokaryotes as part of a dramatic radiation of Ubls and E1-like enzyme in operons for the biosynthesis of cofactors (e.g. thiamin and molybdopterin), cysteine, and peptide secondary metabolites such as siderophores, antibiotics/toxins and small molecule signals (Burroughs et al., 2011, 2012). A subset of these operons is highly mobile (i.e., widespread dispersal across distant lineages) and evolved features characteristic of the eukaryotic Ub-systems, namely the presence of E2 and sometimes E3 enzymes and the deubiquitinating JAB peptidase (Burroughs et al., 2011). The fact that these operons are mobile, and usually tend to couple the ubiquitinating enzymes with deubiquitinating JAB peptidases, presents parallels to the R-M systems (Iyer et al., 2006c). Like them these systems combine opposing actions in the modifying and de-modifying enzymes, and have no links to the metabolic enzymes that are typical of the operons with E1-like enzymes and Ubls that synthesize small molecule. Hence, we posit that these are potential selfish elements that act like the R-Ms, but at the protein level, by possibly destabilizing proteins through transfer of the Ubl and restoring the original protein by removal of the Ubl by the JAB peptidase. Since these Ub-like systems are closest to the eukaryotic versions, it is very likely that they were derived from them. On account of their mobility they are seen in several bacteria and certain archaea (e.g., the Caldiarchaeum) (Iyer et al., 2006c; Burroughs et al., 2011; Nunoura et al., 2011); hence, it is possible that eukaryotes might have acquired the precursor of their Ub-system either from their archaeal precursor or from endosymbiotic bacteria (Figure 4).
The only DUB that is consistently observed in prokaryotic Ub-like systems is the JAB peptidase domain (Iyer et al., 2006c; Burroughs et al., 2011). Eukaryotes, however, possess several other DUBs, most of which belong to the papain-like peptidase fold and a few to the Zincin-like metallopeptidase fold (Iyer et al., 2004). Interestingly, papain-like peptidases (e.g., Otu-like peptidase domain) and Zincin-like metallopeptidases are frequently found among the toxin domains of effectors delivered by a range of intra-cellular bacteria (Loureiro and Ploegh, 2006). These were previously thought to be lateral transfers from hosts to their endo-symbionts/parasites, which are used to interfere with the host Ub-system (Lomma et al., 2010; Schmitz-Esser et al., 2010). However, recent studies on polymorphic toxin systems suggest that such peptidase domains are far more widely distributed in bacterial toxins and often among toxins of free-living bacteria deployed in inter-bacterial conflicts (Zhang et al., 2012). Hence, it seems more likely that they first emerged in bacteria as part of the polymorphic and related secreted toxin systems and were acquired by eukaryotes and recruited as DUBs in course of the development of the mitochondrial endosymbiosis (Figure 4). Not surprisingly, these DUB-like peptidase domains are common among intra-cellular bacteria such as Wolbachia, Rickettsia and Odyssella, which are closely related to the mitochondrial precursors (Figure 2). Indeed, these DUBs probably originally emerged as part of the strategy utilized by these bacterial endo-symbionts/pathogens that countered the immunity mechanism based on ubiquitination of target proteins. Interestingly, several of these papain-like DUB domains are also related to polyprotein-processing peptidases of eukaryotic RNA viruses and retroviruses (Iyer et al., 2004). It is conceivable that the emergence of the Ub-system in eukaryotes also provided a means for RNA viruses to escape constraints placed by the eukaryotic mRNA cap on internal translation initiation, by simply enabling translation of polyproteins that are then processed by the DUB peptidases. In course of viral evolution many of the DUB domains were probably incorporated into their own polyproteins to allow autoproteolytic processing.
One of the simplest counter-pathogen strategies is regulated cell death or apoptosis, in which a cell might sacrifice its own fitness and prevent the pathogen from replicating within it. This typically works in situations where the inclusive fitness accrued from saving kin from infection might contribute to fixation of altruistic behaviors such as apoptosis (Aravind et al., 2009). Such mechanisms are likely to be further enhanced with the emergence of colonial or multicellular organization. Some of the simplest programmed cell death systems seen in bacteria are constituted by intra-genomic selfish elements. For example, in Escherichia coli a defective prophage produces a toxin known as Lit with a zincin-like metallopeptidase domain to cleave the elongation factor Tu and kill the cell when infected by the phage T4, thereby preventing the further spread of T4 to remaining cells in the colony (Snyder, 1995). Likewise, under conditions of starvation, when resources are limiting, chromosomally encoded toxin-antitoxin systems, such as the entericidin locus, mediate cell death in bacteria like E. coli and allow certain cells to survive and grow at the expense of kin that have undergone cell death (Bishop et al., 1998). Thus, the principle of the use of toxins as mediators of programmed cell death appears to be an ancient one (Jensen and Gerdes, 1995; Bishop et al., 1998). Although eukaryotes lack conventional toxin-antitoxin systems, the executioners of apoptosis resemble the prokaryotic toxins from these and other conflict systems in that they cleave or modify specific target proteins or permeabilize membranes in the cell committed to apoptosis. These effectors have been best studied in the animal lineage and include membrane-permeability regulators (the BCL2 superfamily), DNA-cleaving enzymes (e.g., the DNA fragmentation factor/CIDE), DNA-modifying enzymes (e.g., pierisin) and peptidases (e.g., the caspases) (Chou et al., 1999; Lugovskoy et al., 1999; Kanazawa et al., 2001; Riedl and Salvesen, 2007). Investigation into the provenance of these proteins has revealed multiple ancient connections to bacterial toxin systems. The core helical domain of the BCL2 superfamily (the first 6 helices) is specifically related to the translocation (T) domain of several host-directed toxins from distantly related bacteria such as the diphtheria, botulinum, tetanus and Vibrio toxins (Chou et al., 1999). The T-domain undergoes a pH induced conformational change to assume a BCL2-like structure, inserts into the endosomal membrane and transfers the catalytic domain of the toxin into host cytoplasm. Given its sudden emergence in metazoans, it is likely that it was derived from a bacterial toxin and recruited as regulator of the permeability of the mitochondrial membrane. In metazoans these domains diversified into anti-apoptotic versions, which prevent the release of cytochrome C from mitochondria and pro-apoptotic versions which foster its release (Chou et al., 1999; Riedl and Salvesen, 2007). From animals, the BCL2 superfamily was secondarily acquired by large DNA viruses that infect them, such as herpesviruses, poxviruses, iridoviruses and asfarviruses, and used as an anti-apoptotic effector to prevent hosts from using cell death as a defense against them (Iyer et al., 2006b). The T-domain of bacterial toxins appears to have been independently transferred to the fungus Metarhizium, where it appears to be utilized in multiple toxins directed against the insect host.
Among catalytic effectors of apoptosis, in metazoans the DFF/CIDE endonuclease catalyzes the genome fragmentation of DNA that is typical of apoptosis (Lugovskoy et al., 1999; Riedl and Salvesen, 2007). Structural studies had revealed that this domain contains an endonuclease domain of the HNH/EndoVII fold, but its origins remained unclear (Lugovskoy et al., 1999). Recent analysis of the bacterial polymorphic toxins revealed that a subset of them contains a toxin nuclease domain, which shares unique sequence signatures with the DFF/CIDE endonuclease domain to the exclusion of other representatives of HNH/EndoVII fold (Zhang et al., 2012). Here again, the relative abundance of the HNH/EndoVII fold among polymorphic and related toxin domains compared to its lone presence in DFF/CIDE, which is restricted to metazoans, points to an origin for the latter from a representative in the bacterial toxin systems. Pierisin-type ARTs are unusual enzymes that mediate apoptosis (thus far only known from lepidopterans) by ADP-ribosylating the N2 atom of guanine in DNA (Kanazawa et al., 2001). The lepidopteran pierisin-like ARTs are specifically related to the ART toxin domains found in certain bacterial polymorphic toxins and insecticidal toxins of insect pathogens, such as Bacillus sphaericus (Orth et al., 2011). This suggests that they were probably laterally transferred into lepidopterans from a bacterial symbiont or parasite, followed by their reuse as an apoptotic effector. In all the above examples the natural action of the bacterial toxins in disrupting or killing animal cells appears to have been harnessed as a mechanism to execute apoptosis.
Caspase-like peptidases are the central executers of apoptosis throughout eukaryotes and have been demonstrated to play a central role in cell death in animals, fungi, plants, and certain other eukaryotes (Aravind and Koonin, 2002; Riedl and Salvesen, 2007). Prior evolutionary analysis of the caspase-like superfamily revealed that they first diversified in bacteria into several clades such as the metacaspases, paracaspases and numerous other bacteria-specific lineages (Aravind and Koonin, 2002). Metacaspases were transferred to eukaryotes prior the LECA and are found in most eukaryotes (Figure 4). Subsequently, in the animal lineage and in dictyostelid slime molds metacaspases were displaced by a second acquisition from bacteria, the paracaspases, which then radiated in animal to give rise to the classical caspases (Aravind and Koonin, 2002). This phyletic pattern suggests that paracaspases are effectively functionally comparable to metacaspases, as they have displaced them on more than one occasion. Interestingly, several bacteria, particularly endosymbiotic/parasitic alphaproteobacteria (e.g., Agrobacterium, Labrenzia, Bradyrhizobium) encode metacaspases and paracaspases with N-terminal signal peptides that are likely to be secreted into their hosts (Aravind and Koonin, 2002). Hence, these peptidases were possibly first used in regulating endoparasite/symbiont-host conflicts to modulate the immune response and cell death in favor of the intra-cellular bacterium. Consistent with this, recent studies in humans have shown that the paracaspase modulates the T-cell-dependent immune response by cleaving A20, a deubiquitinating enzyme involved in the process, and is required for prevention of cell death in diffuse large B cell lymphoma (Coornaert et al., 2008; Ferch et al., 2009). This suggests that caspase-like peptidases might have been acquired on multiple occasions in eukaryotic evolution from endosymbiotic bacteria, which were probably utilizing them to regulate the survival of their host cells. On a similar note, the GIMAP/AIG1-like GTPases, which are deployed by certain endo-symbionts/parasites (Figure 2), could have given rise to the eukaryote representatives of this clade which are known to modulate both apoptosis and the immune response.
Thus, protein domains that originally diversified in prokaryotic conflict systems both as toxin and also as potential modulators of host defensive responses have had a notable effect on the evolution of apoptosis.
Despite the enormous disparities in the immune systems of different eukaryotes, there are a few common strategies that are observed across most of them. These include the use of a relatively small number of families of protein domains as antigen receptors. Diversification of antigen receptors in most eukaryotes is a passive process of sequence divergence, probably under positive selection, within families of lineage-specifically expanded proteins (e.g., LRR proteins). However, in both jawed and jawless vertebrates two distinct and directed mechanisms for their diversification have been observed, namely recombination and active mutagenesis, which result in different populations of lymphocytes expressing different types of antigen receptors (Pancer and Cooper, 2006; Schatz and Swanson, 2011). In both jawed and jawless vertebrates the process of directed mutagenesis by DNA cytosine deaminases of the AID-APOBEC superfamily is utilized (Rogozin et al., 2007). Such mutagenesis is used either as a trigger for antigen gene-conversion, or for hypermutation or for antibody class-switching. Additionally, certain representatives of the AID-APOBEC family of cytosine deaminases are also major line of defense against retroviruses by mutagenizing their genomes by cytosine deamination (Chiu and Greene, 2006). Although AID-APOBEC-like deaminases were, until recently, thought to be restricted to vertebrates, sensitive sequence analysis showed that more divergent members exist in nematodes, cnidarians and several distantly related algal groups. Identification of these sequences helped establish that the fast-evolving AID-APOBEC deaminases have their ultimate origin in the toxin domains of polymorphic and related secreted bacterial toxins (Iyer et al., 2011b). Indeed, effectors with toxin domains most closely related to the AID-APOBEC deaminases are observed in the Wolbachia endosymbiont of the moth Cadre cautella and the plant pathogen Pseudomonas brassicacearum (Iyer et al., 2011b). Thus, these mutagenic deaminase domains, which were originally part of toxins deployed by bacteria, appear to have provided the basis for the unique mechanism for antigen receptor diversification in vertebrates. However, their role in anti-retroviral response suggests that they were probably initially recruited merely as mutagenic enzymes that targeted viruses (i.e., similar to the original toxin role but merely directed at viruses). Interestingly, several filamentous fungi show a lineage-specific expansion of related nucleic acid deaminases that also appear to have been derived from toxin domains of bacterial provenance (Iyer et al., 2011b). It is conceivable that these play a similar role as the counter-retroviral deaminases in potentially mutating cytoplasmic parasitic elements or preventing anastomosis by unrelated hyphae.
In jawed vertebrates, antibody and T-cell receptor diversity is generated by the action (V-D-J and V-J recombination) of a dedicated recombination apparatus comprised of two proteins Rag1 and Rag2, of which Rag1 is the catalytic subunit of the recombinase (Schatz and Swanson, 2011). The origin of the Rag1 recombinase in animals had remained mysterious until it was shown that their recombinase domain is related to the transposase domain of a distinct class of eukaryotic transposons known as the Transib elements (Kapitonov and Jurka, 2005; Panchin and Moroz, 2008). This transposase domain contains a distinctive version of the RNAseH fold and cleaves sites associated with the termini of these transposons, which show sequence relationships to V-D-J and V-J recombination sites. Thus, the Rag1 recombinase appears to have evolved from a “domesticated” selfish element whose recombinase domain and terminal recognition sites were reused as a mechanism to generate diversity. Indeed, domestication of selfish elements for generation of diversity in host-pathogen interfaces is a general phenomenon, which is not restricted to the animal immune system: In certain caudate bacteriophages, the mutagenic reverse transcriptase of an integrated retroelement has been shown to play a role in creating sequence diversity in a tail-fiber-associated protein (Medhekar and Miller, 2007). This allows the bacteriophages to recognize a changing landscape of cell-surface proteins on their hosts.
As the endosymbiotic model for eukaryogenesis involves juxtaposition of two distinct genomes in the same cell, it implies an increased scope for genetic conflicts between the genomes and the intra-genomic selfish elements contained by them. Indeed, different scenarios exploiting such conflicts have been proposed. One of these argues that the mobile self-splicing group-II introns from the alphaproteobacterial mitochondrial progenitor invaded and proliferated in the progenitor of the nuclear genome (Koonin, 2006). As a consequence there was selection for the nuclear membrane as a physical barrier to protect unspliced intron-containing transcripts from the translation apparatus. This hypothesis posits that the pre-LECA eukaryotes were enormously enriched in introns (Koonin, 2006) as a consequence of reduced selection due to decreased effective population sizes (Lynch, 2007). However, direct evidence for highly intron-rich pre-LECA genomes is lacking based on available genomes and with the current data it is not possible to distinguish between: (1) the early proliferation of introns in eukaryotes being a consequence of the emergence of a protective barrier of the nucleus and (2) the nucleus being a consequence of the selective pressure imposed by intron proliferation. Moreover, there is little evidence for extensive proliferation of group-II introns in any prokaryotic lineage. In an alternative hypothesis, greater alignment of the genetic interests of the genomes of the pro-mitochondrion and the nucleus is likely to have happened with the transfer of genes, including those encoding ribosomal proteins, from the former genome to the latter (Jekely, 2008). This is likely to have resulted in chimeric ribosomes in the cytoplasm with potentially deleterious effects for both genomes. This hypothesis presents the nucleus as a physical barrier to prevent such chimerism and might also effectively explain the origin of the nucleolus, another defining feature of eukaryotes. It should be noted that nucleus-like structures have convergently evolved in certain representatives of the clade of bacteria uniting the planctomycetes, chlamydiae and verrucomicrobia (McInerney et al., 2011). In these cases there is no evidence for deleterious effects arising from intra-genomic selfish elements like group-II introns or ribosomal chimerism. Indeed alternative selective pressures could have facilitated nucleogenesis.
One key feature of bacterial endo-symbionts/parasites is their deployment of toxin/effector systems that contain nuclease and nucleic acid deaminase domains, both from polymorphic and host-directed toxin systems (Iyer et al., 2011b; Zhang et al., 2011, 2012). These are observed in a variety of extant endo-symbionts/parasites such as Wolbachia, Rickettsia, Orientia, Odyssella, Legionella, Amoebophilus, and Protochlamydia (Figure 2). Indeed, such genome-targeting toxins are likely to play a role in the chromosomal disruptions produced by Wolbachia in the process of regulating sex-specific survival and killing of incompatible hosts (Duron, 2008). Interestingly, a key nuclear pore component, Nup96/98, has an autoproteolytic ZU5 domain (Mans et al., 2004). ZU5 domains appear to have originated in bacterial cell-surface proteins, such as polymorphic toxins, and play a role in the autoproteolytic processing of toxins on the cell-surface [ZU5 domains were also secondarily acquired again from bacterial sources to give rise to the animal apoptosis regulator PIDD (Riedl and Salvesen, 2007; Zhang et al., 2012)]. It is possible that this key nuclear pore component was derived from a toxin system of the ancestral endosymbiont. Thus, it is likely that nucleic-acid-targeting toxins were deployed by the mitochondrial progenitor, which could have threatened the integrity of the DNA of the nuclear genome precursor. Hence, the nucleus was probably selected for, as a physical barrier to minimize this threat. In this scenario, once the initial endosymbiotic association between the mitochondrial precursor and the archaeon was underway, the selective pressure from the DNA-targeting toxins of the mitochondrial precursor favored the emergence of the nucleus very early in the development of the association. The early presence of the nucleus then favored the development of several characteristics of eukaryotes, including those that have been noted in the other hypotheses: (1) it would have allowed transfer of alphaproteobacterial ribosomal genes to the nuclear genome, as chimerism could be avoided due to presence of an additional compartment (Jekely, 2008), eventually leading the origin of the nucleolus. (2) It allowed retroelements associated with group-II introns to proliferate in nucleus (Koonin, 2006). This not only gave rise to introns but also the telomerase (Aravind et al., 2006). (3) The telomerase in turn facilitated the origin of multiple linear chromosomes, whose expression could now be coordinated as they were contained within the nuclear compartment. (4) Linear chromosomes, together with the nucleus, probably selected against the prokaryotic pumping mechanisms for chromosome segregation based on HerA-FtsK-like ATPases, and instead favored a cytoskeleton-based mechanism, which allowed for fixation of the microtubular apparatus. (5) The stabilization of multiple linear chromosomes contained with a nucleus also probably allowed for increased genome sizes in eukaryotes, as it removed the constraints coming from containing the entire genome on a large circular chromosome segregated by the ATPase pumps.
In conclusion a number of mechanistically distinct scenarios support a role for organismal and genomic conflict systems in eukaryotic nucleogenesis. Further investigations of alternative scenario presented here might provide a new handle to understand key events in eukaryogenesis.
In the above discussion, we provide a series of examples from across the eukaryotic phyletic spread for how the interplay between lateral gene flow inter-organismal, inter-genomic, and intra-genomic conflicts has shaped the evolution of numerous functional systems (Figure 4). These examples are by no means meant to be exhaustive—rather, they were chosen in order to provide a glimpse of the sheer variety of biological systems that are affected by the evolutionary contributions from such systems. One key theme that emerges from the above discussion is that domain families gained through lateral transfer in course of intimate inter-organismal associations, such as symbiosis and parasitism, can notably determine the very nature of these interactions. This is strikingly illustrated by the case of apicomplexan adhesion molecules implicated in host interaction: here manifold domains were acquired by the parasites via lateral transfer from their hosts, spawning unique “animal-like” interfaces for interacting with the host (Figure 1). The other recurrent theme, which transcends various biological systems, is how proteins/protein domains originally emerging in the context of various biological conflicts were recycled as regulatory molecules (Figure 4). Of these host-directed toxins, and the toxins, immunity proteins, structural modules and secretory components from bacterial polymorphic toxin systems have a distinct life beyond their locus of provenance in eukaryotic regulatory and defense systems (Iyer et al., 2011b; Zhang et al., 2011, 2012). We outline numerous occasions where these components were incorporated into regulatory systems of eukaryotes, and sometimes might have played a major role in the very origin of these systems. This process appears to be constantly on-going, all the way from the origin of eukaryotes to the terminal tips of the eukaryotic tree (Figure 4). The reason why proteins derived from biological conflict systems appear to be recruited for other functions might be attributed to the consequences of natural selection. Not surprisingly, toxin-immunity systems used in inter-organismal conflict have a large effect on the fitness of both the organisms producing toxins and those defending against them, thereby escalating an arms race situation. Many of the conflict systems deployed by bacteria might even function at the interface of symbiotic and parasitic interactions of bacteria and eukaryotes, thereby developing adaptations to effectively target components of eukaryotic systems. Toxins and immunity proteins of intra-genomic selfish elements are also under multiple levels of selection that foster their diversification. At one level they are under selection to evade host resistance to function effectively as addictive agents. At another level many of them might also be under selection to function as effective stress response mechanisms that allow their host genomes to survive adverse conditions. Consequently, there are strong selective pressures for constant diversification of toxins and the corresponding immunity proteins in various conflict systems. Hence, these biological conflicts could have functioned as evolutionary “nurseries” for innovations in both prokaryotic and eukaryotic proteins. Hence, lateral gene flow from symbionts, parasites and other modes of DNA uptake (Gladyshev et al., 2008; Nikoh et al., 2008) has enabled eukaryotes to have access to and import a “readymade” set of molecular innovations from such biological conflict systems. When recruited in non-conflict biological contexts, they can in turn spur the emergence of new interactions in eukaryotic systems. Thus, number of key eukaryotic innovations can be traced back to the above-described players in biological conflict systems, such as secondary metabolism operons, R-M, polymorphic and host-directed toxins systems, anti-phage systems, phage counter-restriction strategies, and mobile elements. These systems appear to have particularly expanded in bacteria on account of the presence of operons, extensive lateral transfer with several modes of DNA uptake and recombination, perhaps combined with high effective population sizes (Lynch, 2007). Thus, organismal and genomic conflicts as the basis for major molecular innovations, which in turn might facilitate major evolutionary transitions, can be considered a general evolutionary principle.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors' research is supported by the intramural funds of the US Department of Health and Human Services (National Library of Medicine, NIH).
Adachi, J., and Hasegawa, M. (1992). MOLPHY: Programs for Molecular Phylogenetics. Tokyo: Institute of Statistical Mathematics.
Aepfelbacher, M., Aktories, K., and Just, I. (2000). Bacterial Protein Toxins. Berlin, New York: Springer.
Allis, C. D., Jenuwein, T., Reinberg, D., and Caparros, M. (2006). Epigenetics. New York, NY: Cold Spring Harbor Laboratory Press.
Alouf, J. E., and Popoff, M. R. (2006). The Comprehensive Sourcebook of Bacterial Protein Toxins. Amsterdam, Boston: Elsevier Academic Press.
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402.
Aminov, R. I., and Mackie, R. I. (2007). Evolution and ecology of antibiotic resistance genes. FEMS Microbiol. Lett. 271, 147–161.
Anantharaman, V., and Aravind, L. (2003). New connections in the prokaryotic toxin-antitoxin network: relationship with the eukaryotic nonsense-mediated RNA decay system. Genome Biol. 4, R81.
Anantharaman, V., Iyer, L. M., Balaji, S., and Aravind, L. (2007). Adhesion molecules and other secreted host-interaction determinants in Apicomplexa: insights from comparative genomics. Int. Rev. Cytol. 262, 1–74.
Aoki, S. K., Poole, S. J., Hayes, C. S., and Low, D. A. (2011). Toxin on a stick: modular CDI toxin delivery systems play roles in bacterial competition. Virulence 2, 356–359.
Aravind, L., Abhiman, S., and Iyer, L. M. (2011). Natural history of the eukaryotic chromatin protein methylation system. Prog. Mol. Biol. Transl. Sci. 101, 105–176.
Aravind, L., Anantharaman, V., and Venancio, T. M. (2009). Apprehending multicellularity: regulatory networks, genomics, and evolution. Birth Defects Res. C Embryo Today 87, 143–164.
Aravind, L., and Iyer, L. M. (2012). The HARE-HTH and associated domains: novel modules in the coordination of epigenetic DNA and protein modifications. Cell Cycle 11, 119–131.
Aravind, L., Iyer, L. M., and Koonin, E. V. (2006). Comparative genomics and structural biology of the molecular innovations of eukaryotes. Curr. Opin. Struct. Biol. 16, 409–419.
Aravind, L., and Koonin, E. V. (2002). Classification of the caspase-hemoglobinase fold: detection of new families and implications for the origin of the eukaryotic separins. Proteins 46, 355–367.
Aravind, L., Makarova, K. S., and Koonin, E. V. (2000). Holliday junction resolvases and related nucleases: identification of new families, phyletic distribution and evolutionary trajectories. Nucleic Acids Res. 28, 3417–3432.
Arredondo, S. A., Cai, M., Takayama, Y., Macdonald, N. J., Anderson, D. E., Aravind, L., Clore, G. M., and Miller, L. H. (2012). Structure of the Plasmodium 6-cysteine s48/45 domain. Proc. Natl. Acad. Sci. U.S.A. 109, 6692–6697.
Babu, M. M., Iyer, L. M., Balaji, S., and Aravind, L. (2006). The natural history of the WRKY-GCM1 zinc fingers and the relationship between transcription factors and transposons. Nucleic Acids Res. 34, 6505–6520.
Barry, S. M., and Challis, G. L. (2009). Recent advances in siderophore biosynthesis. Curr. Opin. Chem. Biol. 13, 205–215.
Batut, J., Andersson, S. G., and O'callaghan, D. (2004). The evolution of chronic infection strategies in the alpha-proteobacteria. Nat. Rev. Microbiol. 2, 933–945.
Bertelli, C., Collyn, F., Croxatto, A., Ruckert, C., Polkinghorne, A. Kebbi-Beghdadi, C., Goesmann, A., Vaughan, L., and Greub, G. (2010). The waddlia genome: a window into chlamydial biology. PLoS ONE 5:e10890. doi: 10.1371/journal.pone.0010890
Bestor, T. H. (1990). DNA methylation: evolution of a bacterial immune function into a regulator of gene expression and genome structure in higher eukaryotes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 326, 179–187.
Bhattacharya, D., Yoon, H. S., and Hackett, J. D. (2004). Photosynthetic eukaryotes unite: endosymbiosis connects the dots. Bioessays 26, 50–60.
Bishop, R. E., Leskiw, B. K., Hodges, R. S., Kay, C. M., and Weiner, J. H. (1998). The entericidin locus of Escherichia coli and its implications for programmed bacterial cell death. J. Mol. Biol. 280, 583–596.
Bork, P. (1993). The modular architecture of a new family of growth regulators related to connective tissue growth factor. FEBS Lett. 327, 125–130.
Bowen, D., Rocheleau, T. A., Blackburn, M., Andreev, O., Golubeva, E., Bhartia, R., and Ffrench-Constant, R. H. (1998). Insecticidal toxins from the bacterium Photorhabdus luminescens. Science 280, 2129–2132.
Bradley, P. J., and Sibley, L. D. (2007). Rhoptries: an arsenal of secreted virulence factors. Curr. Opin. Microbiol. 10, 582–587.
Brady, S. F., Chao, C. J., and Clardy, J. (2004). Long-chain N-acyltyrosine synthases from environmental DNA. Appl. Environ. Microbiol. 70, 6865–6870.
Burroughs, A. M., Iyer, L. M., and Aravind, L. (2011). Functional diversification of the RING finger and other binuclear treble clef domains in prokaryotes and the early evolution of the ubiquitin system. Mol. Biosyst. 7, 2261–2277.
Burroughs, A. M., Iyer, L. M., and Aravind, L. (2012). The natural history of ubiquitin and ubiquitin-related domains. Front. Biosci. 17, 1433–1460.
Burrows, J. F., and Johnston, J. A. (2012). Regulation of cellular responses by deubiquitinating enzymes: an update. Front. Biosci. 17, 1184–1200.
Burt, A., and Trivers, R. (2006). Genes in Conflict: the Biology of Selfish Genetic Elements. Cambridge, MA: The Belknap Press of Harvard University Press.
Cascales, E., Buchanan, S. K., Duche, D., Kleanthous, C., Lloubes, R., Postle, K., Riley, M., Slatin, S., and Cavard, D. (2007). Colicin biology. Microbiol. Mol. Biol. Rev. 71, 158–229.
Chiu, Y. L., and Greene, W. C. (2006). APOBEC3 cytidine deaminases: distinct antiviral actions along the retroviral life cycle. J. Biol. Chem. 281, 8309–8312.
Chopin, M. C., Chopin, A., and Bidnenko, E. (2005). Phage abortive infection in lactococci: variations on a theme. Curr. Opin. Microbiol. 8, 473–479.
Chou, J. J., Li, H., Salvesen, G. S., Yuan, J., and Wagner, G. (1999). Solution structure of BID, an intracellular amplifier of apoptotic signaling. Cell 96, 615–624.
Citarelli, M., Teotia, S., and Lamb, R. S. (2010). Evolutionary history of the poly(ADP-ribose) polymerase gene family in eukaryotes. BMC Evol. Biol. 10, 308.
Collingro, A., Toenshoff, E. R., Taylor, M. W., Fritsche, T. R., Wagner, M., and Horn, M. (2005). ‘Candidatus Protochlamydia amoebophila’, an endosymbiont of Acanthamoeba spp. Int. J. Syst. Evol. Microbiol. 55, 1863–1866.
Coornaert, B., Baens, M., Heyninck, K., Bekaert, T., Haegman, M., Staal, J., Sun, L., Chen, Z. J., Marynen, P., and Beyaert, R. (2008). T cell antigen receptor stimulation induces MALT1 paracaspase-mediated cleavage of the NF-kappaB inhibitor A20. Nat. Immunol. 9, 263–271.
Cuff, J. A., Clamp, M. E., Siddiqui, A. S., Finlay, M., and Barton, G. J. (1998). JPred: a consensus secondary structure prediction server. Bioinformatics 14, 892–893.
Dao, D. N., Kessin, R. H., and Ennis, H. L. (2000). Developmental cheating and the evolutionary biology of Dictyostelium and Myxococcus. Microbiology 146(Pt 7), 1505–1512.
Dawkins, R., and Krebs, J. R. (1979). Arms races between and within species. Proc. R. Soc. Lond. B Biol. Sci. 205, 489–511.
De Souza, R. F., and Aravind, L. (2012). Identification of novel components of NAD-utilizing metabolic pathways and prediction of their biochemical functions. Mol. Biosyst. 8, 1661–1677.
De Souza, R. F., Iyer, L. M., and Aravind, L. (2010). Diversity and evolution of chromatin proteins encoded by DNA viruses. Biochim. Biophys. Acta 1799, 302–318.
Degnan, P. H., Yu, Y., Sisneros, N., Wing, R. A., and Moran, N. A. (2009). Hamiltonella defensa, genome evolution of protective bacterial endosymbiont from pathogenic ancestors. Proc. Natl. Acad. Sci. U.S.A. 106, 9063–9068.
Delwiche, C. F. (1999). Tracing the thread of plastid diversity through the tapestry of life. Am. Nat. 154, S164–S177.
Dlakic, M., and Mushegian, A. (2011). Prp8, the pivotal protein of the spliceosomal catalytic center, evolved from a retroelement-encoded reverse transcriptase. RNA 17, 799–808.
Doolittle, W. F. (1998). You are what you eat: a gene transfer ratchet could account for bacterial genes in eukaryotic nuclear genomes. Trends Genet. 14, 307–311.
Duron, O. (2008). Insights beyond Wolbachia-Drosophila interactions: never completely trust a model: insights from cytoplasmic incompatibility beyond Wolbachia-Drosophila interactions. Heredity (Edinb) 101, 473–474.
Eddy, S. R. (2009). A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211.
Esser, C., Ahmadinejad, N., Wiegand, C., Rotte, C., Sebastiani, F., Gelius-Dietrich, G., Henze, K., Kretschmann, E., Richly, E., Leister, D., Bryant, D., Steel, M. A., Lockhart, P. J., Penny, D., and Martin, W. (2004). A genome phylogeny for mitochondria among alpha-proteobacteria and a predominantly eubacterial ancestry of yeast nuclear genes. Mol. Biol. Evol. 21, 1643–1660.
Feng, F., Yang, F., Rong, W., Wu, X., Zhang, J., Chen, S., He, C., and Zhou, J. M. (2012). A Xanthomonas uridine 5'-monophosphate transferase inhibits plant immune kinases. Nature 485, 114–118.
Ferch, U., Kloo, B., Gewies, A., Pfander, V., Duwel, M., Peschel, C., Krappmann, D., and Ruland, J. (2009). Inhibition of MALT1 protease activity is selectively toxic for activated B cell-like diffuse large B cell lymphoma cells. J. Exp. Med. 206, 2313–2320.
Fields, S. D., and Rhodes, R. G. (1991). Ingestion and retention of Chroomonas spp. (cryptophyceae) by Gymnodinium acidotum (dinophyceae). J. Phycol. 27, 525–529.
Finn, R. D., Mistry, J., Tate, J., Coggill, P., Heger, A., Pollington, J. E., Gavin, O. L., Gunasekaran, P., Ceric, G., Forslund, K., Holm, L., Sonnhammer, E. L., Eddy, S. R., and Bateman, A. (2010). The Pfam protein families database. Nucleic Acids Res. 38, D211–D222.
Gabaldon, T., and Huynen, M. A. (2007). From endosymbiont to host-controlled organelle: the hijacking of mitochondrial protein synthesis and metabolism. PLoS Comput. Biol. 3:e219. doi: 10.1371/journal.pcbi.0030219
Gajewski, S., Comeaux, E. Q., Jafari, N., Bharatham, N., Bashford, D., White, S. W., and Van Waardenburg, R. C. (2012). Analysis of the active-site mechanism of tyrosyl-DNA phosphodiesterase I: a member of the phospholipase D superfamily. J. Mol. Biol. 415, 741–758.
Galun, E. (2003). Transposable elements: A Guide to the Perplexed and The Novice: With Appendices on RNAi, Chromatin Remodeling and Gene Tagging. Dordrecht, Boston: Kluwer Academic.
Georgiades, K., Madoui, M. A., Le, P., Robert, C., and Raoult, D. (2011). Phylogenomic analysis of Odyssella thessalonicensis fortifies the common origin of Rickettsiales, Pelagibacter ubique and Reclimonas americana mitochondrion. PLoS ONE 6:e24857. doi: 10.1371/journal.pone.0024857
Gladyshev, E. A., Meselson, M., and Arkhipova, I. R. (2008). Massive horizontal gene transfer in bdelloid rotifers. Science 320, 1210–1213.
Goff, L. J., and Coleman, A. W. (1995). Fate of parasite and host organelle dna during cellular transformation of red algae by their parasites. Plant Cell 7, 1899–1911.
Grewal, S. I. (2010). RNAi-dependent formation of heterochromatin and its diverse functions. Curr. Opin. Genet. Dev. 20, 134–141.
Hauk, G., and Bowman, G. D. (2011). Structural insights into regulation and action of SWI2/SNF2 ATPases. Curr. Opin. Struct. Biol. 21, 719–727.
He, Y. F., Li, B. Z., Li, Z., Liu, P., Wang, Y., Tang, Q., Ding, J., Jia, Y., Chen, Z., Li, L., Sun, Y., Li, X., Dai, Q., Song, C. X., Zhang, K., He, C., and Xu, G. L. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333, 1303–1307.
Heaton, B. E., Herrou, J., Blackwell, A. E., Wysocki, V. H., and Crosson, S. (2012). Molecular structure and function of the novel BrnT/BrnA toxin-antitoxin system of Brucella abortus. J. Biol. Chem. 287, 12098–12110.
Holm, L., Kaariainen, S., Rosenstrom, P., and Schenkel, A. (2008). Searching protein structure databases with DaliLite v.3. Bioinformatics 24, 2780–2781.
Hota, S. K., and Bartholomew, B. (2011). Diversity of operation in ATP-dependent chromatin remodelers. Biochim. Biophys. Acta 1809, 476–487.
Huang, J., and Gogarten, J. P. (2007). Did an ancient chlamydial endosymbiosis facilitate the establishment of primary plastids? Genome Biol. 8, R99.
Hurst, M. R., Glare, T. R., and Jackson, T. A. (2004). Cloning Serratia entomophila antifeeding genes–a putative defective prophage active against the grass grub Costelytra zealandica. J. Bacteriol. 186, 5116–5128.
Ingham, P. W., Nakano, Y., and Seger, C. (2011). Mechanisms and functions of Hedgehog signalling across the metazoa. Nat. Rev. Genet. 12, 393–406.
Ishikawa, K., Fukuda, E., and Kobayashi, I. (2010). Conflicts targeting epigenetic systems and their resolution by cell death: novel concepts for methyl-specific and other restriction systems. DNA Res. 17, 325–342.
Iyer, L. M., Abhiman, S., and Aravind, L. (2008a). MutL homologs in restriction-modification systems and the origin of eukaryotic MORC ATPases. Biol. Dir. 3, 8.
Iyer, L. M., Anantharaman, V., Wolf, M. Y., and Aravind, L. (2008b). Comparative genomics of transcription factors and chromatin proteins in parasitic protists and other eukaryotes. Int. J. Parasitol. 38, 1–31.
Iyer, L. M., Abhiman, S., and Aravind, L. (2011a). Natural history of eukaryotic DNA methylation systems. Prog. Mol. Biol. Transl. Sci. 101, 25–104.
Iyer, L. M., Zhang, D., Rogozin, I. B., and Aravind, L. (2011b). Evolution of the deaminase fold and multiple origins of eukaryotic editing and mutagenic nucleic acid deaminases from bacterial toxin systems. Nucleic Acids Res. 39, 9473–9497.
Iyer, L. M., Abhiman, S., De Souza, R. F., and Aravind, L. (2010). Origin and evolution of peptide-modifying dioxygenases and identification of the wybutosine hydroxylase/hydroperoxidase. Nucleic Acids Res. 38, 5261–5279.
Iyer, L. M., Abhiman, S. Maxwell Burroughs, A., and Aravind, L. (2009). Amidoligases with ATP-grasp, glutamine synthetase-like and acetyltransferase-like domains: synthesis of novel metabolites and peptide modifications of proteins. Mol. Biosyst. 5, 1636–1660.
Iyer, L. M., and Aravind, L. (2011). Insights from the architecture of the bacterial transcription apparatus. J. Struct. Biol. doi: 10.1016/j.jsb.2011.12.013. [Epub ahead of print].
Iyer, L. M., Babu, M. M., and Aravind, L. (2006a). The HIRAN domain and recruitment of chromatin remodeling and repair activities to damaged DNA. Cell Cycle 5, 775–782.
Iyer, L. M., Balaji, S., Koonin, E. V., and Aravind, L. (2006b). Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 117, 156–184.
Iyer, L. M., Burroughs, A. M., and Aravind, L. (2006c). The prokaryotic antecedents of the ubiquitin-signaling system and the early evolution of ubiquitin-like beta-grasp domains. Genome Biol. 7, R60.
Iyer, L. M., Koonin, E. V., and Aravind, L. (2004). Novel predicted peptidases with a potential role in the ubiquitin signaling pathway. Cell Cycle 3, 1440–1450.
Jekely, G. (2008). Origin of the nucleus and Ran-dependent transport to safeguard ribosome biogenesis in a chimeric cell. Biol. Dir. 3, 31.
Jensen, R. B., and Gerdes, K. (1995). Programmed cell death in bacteria: proteic plasmid stabilization systems. Mol. Microbiol. 17, 205–210.
Johnson, M. D., Oldach, D., Delwiche, C. F., and Stoecker, D. K. (2007). Retention of transcriptionally active cryptophyte nuclei by the ciliate Myrionecta rubra. Nature 445, 426–428.
Kall, L., Krogh, A., and Sonnhammer, E. L. (2007). Advantages of combined transmembrane topology and signal peptide prediction–the Phobius web server. Nucleic Acids Res. 35, W429–W432.
Kanazawa, T., Watanabe, M., Matsushima-Hibiya, Y., Kono, T., Tanaka, N., Koyama, K., Sugimura, T., and Wakabayashi, K. (2001). Distinct roles for the N- and C-terminal regions in the cytotoxicity of pierisin-1, a putative ADP-ribosylating toxin from cabbage butterfly, against mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 98, 2226–2231.
Kapitonov, V. V., and Jurka, J. (2005). RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 3:e181. doi: 10.1371/journal.pbio.0030181
Kappe, S., Bruderer, T., Gantt, S., Fujioka, H., Nussenzweig, V., and Menard, R. (1999). Conservation of a gliding motility and cell invasion machinery in Apicomplexan parasites. J. Cell Biol. 147, 937–944.
Kappe, S. H., Noe, A. R., Fraser, T. S., Blair, P. L., and Adams, J. H. (1998). A family of chimeric erythrocyte binding proteins of malaria parasites. Proc. Natl. Acad. Sci. U.S.A. 95, 1230–1235.
Kaslow, D. C., Quakyi, I. A., Syin, C., Raum, M. G., Keister, D. B., Coligan, J. E., Mccutchan, T. F., and Miller, L. H. (1988). A vaccine candidate from the sexual stage of human malaria that contains EGF-like domains. Nature 333, 74–76.
Keeling, P. J. (2004). Diversity and evolutionary history of plastids and their hosts. Am. J. Bot. 91, 1481–1493.
Kerscher, O., Felberbaum, R., and Hochstrasser, M. (2006). Modification of proteins by ubiquitin and ubiquitin-like proteins. Annu. Rev. Cell Dev. Biol. 22, 159–180.
Kobayashi, I. (2001). Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res. 29, 3742–3756.
Koch-Nolte, F., Kernstock, S., Mueller-Dieckmann, C., Weiss, M. S., and Haag, F. (2008). Mammalian ADP-ribosyltransferases and ADP-ribosylhydrolases. Front. Biosci. 13, 6716–6729.
Koonin, E. V. (2006). The origin of introns and their role in eukaryogenesis: a compromise solution to the introns-early versus introns-late debate? Biol. Dir. 1, 22.
Korch, S. B., Contreras, H., and Clark-Curtiss, J. E. (2009). Three Mycobacterium tuberculosis Rel toxin-antitoxin modules inhibit mycobacterial growth and are expressed in infected human macrophages. J. Bacteriol. 191, 1618–1630.
Laing, S., Unger, M., Koch-Nolte, F., and Haag, F. (2011). ADP-ribosylation of arginine. Amino Acids 41, 257–269.
Laneve, P., Altieri, F., Fiori, M. E., Scaloni, A., Bozzoni, I., and Caffarelli, E. (2003). Purification, cloning, and characterization of XendoU, a novel endoribonuclease involved in processing of intron-encoded small nucleolar RNAs in Xenopus laevis. J. Biol. Chem. 278, 13026–13032.
Lassmann, T., Frings, O., and Sonnhammer, E. L. (2009). Kalign2, high-performance multiple alignment of protein and nucleotide sequences allowing external features. Nucleic Acids Res. 37, 858–865.
Leander, B. S., and Keeling, P. J. (2003). Morphostasis in alveolate evolution. Trends Ecol. Evol. 18, 395–402.
Leander, B. S., Lloyd, S. A., Marshall, W., and Landers, S. C. (2006). Phylogeny of marine Gregarines (Apicomplexa)–Pterospora, Lithocystis and Lankesteria–and the origin(s) of coelomic parasitism. Protist 157, 45–60.
Lee, W., Van Baalen, M., and Jansen, V. A. (2012). An evolutionary mechanism for diversity in siderophore-producing bacteria. Ecol. Lett. 15, 119–125.
Leplae, R., Geeraerts, D., Hallez, R., Guglielmini, J., Dreze, P., and Van Melderen, L. (2011). Diversity of bacterial type II toxin-antitoxin systems: a comprehensive search and functional analysis of novel families. Nucleic Acids Res. 39, 5513–5525.
Lilley, D. M., and White, M. F. (2000). Resolving the relationships of resolving enzymes. Proc. Natl. Acad. Sci. U.S.A. 97, 9351–9353.
Liras, P., and Demain, A. L. (2009). Chapter 16. Enzymology of beta-lactam compounds with cephem structure produced by actinomycete. Meth. Enzymol. 458, 401–429.
Lomma, M., Dervins-Ravault, D., Rolando, M., Nora, T., Newton, H. J., Sansom, F. M., Sahr, T., Gomez-Valero, L., Jules, M., Hartland, E. L., and Buchrieser, C. (2010). The Legionella pneumophila F-box protein Lpp2082 (AnkB) modulates ubiquitination of the host protein parvin B and promotes intracellular replication. Cell. Microbiol. 12, 1272–1291.
Loureiro, J., and Ploegh, H. L. (2006). Antigen presentation and the ubiquitin-proteasome system in host-pathogen interactions. Adv. Immunol. 92, 225–305.
Lugovskoy, A. A., Zhou, P., Chou, J. J., Mccarty, J. S., Li, P., and Wagner, G. (1999). Solution structure of the CIDE-N domain of CIDE-B and a model for CIDE-N/CIDE-N interactions in the DNA fragmentation pathway of apoptosis. Cell 99, 747–755.
Luhn, K., Wild, M. K., Eckhardt, M., Gerardy-Schahn, R., and Vestweber, D. (2001). The gene defective in leukocyte adhesion deficiency II encodes a putative GDP-fucose transporter. Nat. Genet. 28, 69–72.
Luo, Y., Nita-Lazar, A., and Haltiwanger, R. S. (2006). Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J. Biol. Chem. 281, 9385–9392.
Mak, A. N., Lambert, A. R., and Stoddard, B. L. (2010). Folding, DNA recognition, and function of GIY-YIG endonucleases: crystal structures of R.Eco29kI. Structure 18, 1321–1331.
Makarova, K. S., Aravind, L., Wolf, Y. I., and Koonin, E. V. (2011). Unification of Cas protein families and a simple scenario for the origin and evolution of CRISPR-Cas systems. Biol. Dir. 6, 38.
Mans, B. J., Anantharaman, V., Aravind, L., and Koonin, E. V. (2004). Comparative genomics, evolution and origins of the nuclear envelope and nuclear pore complex. Cell Cycle 3, 1612–1637.
Martin, W., and Muller, M. (1998). The hydrogen hypothesis for the first eukaryote. Nature 392, 37–41.
Maynard Smith, J., and Szathmáry, E. (1995). The Major Transitions in Evolution. Oxford, New York: W. H. Freeman Spektrum.
McGuckin, M. A., Linden, S. K., Sutton, P., and Florin, T. H. (2011). Mucin dynamics and enteric pathogens. Nat. Rev. Microbiol. 9, 265–278.
McInerney, J. O., Martin, W. F., Koonin, E. V., Allen, J. F., Galperin, M. Y., Lane, N., Archibald, J. M., and Embley, T. M. (2011). Planctomycetes and eukaryotes: a case of analogy not homology. Bioessays 33, 810–817.
Medhekar, B., and Miller, J. F. (2007). Diversity-generating retroelements. Curr. Opin. Microbiol. 10, 388–395.
Minet, A. D., Rubin, B. P., Tucker, R. P., Baumgartner, S., and Chiquet-Ehrismann, R. (1999). Teneurin-1, a vertebrate homologue of the Drosophila pair-rule gene ten-m, is a neuronal protein with a novel type of heparin-binding domain. J. Cell Sci. 112(Pt 12), 2019–2032.
Nikoh, N., Tanaka, K., Shibata, F., Kondo, N., Hizume, M., Shimada, M., and Fukatsu, T. (2008). Wolbachia genome integrated in an insect chromosome: evolution and fate of laterally transferred endosymbiont genes. Genome Res. 18, 272–280.
Nowotny, M. (2009). Retroviral integrase superfamily: the structural perspective. EMBO Rep. 10, 144–151.
Nunoura, T., Takaki, Y., Kakuta, J., Nishi, S., Sugahara, J., Kazama, H., Chee, G. J., Hattori, M., Kanai, A., Atomi, H., Takai, K., and Takami, H. (2011). Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Res. 39, 3204–3223.
Oborník, M., Janouškovec, J., Chrudimský, T., and Lukeš, J. (2009). Evolution of the apicoplast and its hosts: from heterotrophy to autotrophy and back again. Int. J. Parasitol. 39, 1–12.
Ogata, H., La Scola, B., Audic, S., Renesto, P., Blanc, G., Robert, C., Fournier, P. E., Claverie, J. M., and Raoult, D. (2006). Genome sequence of Rickettsia bellii illuminates the role of amoebae in gene exchanges between intracellular pathogens. PLoS Genet. 2:e76. doi: 10.1371/journal.pgen.0020076
Orth, J. H., Schorch, B., Boundy, S., Ffrench-Constant, R., Kubick, S., and Aktories, K. (2011). Cell-free synthesis and characterization of a novel cytotoxic pierisin-like protein from the cabbage butterfly Pieris rapae. Toxicon 57, 199–207.
Palmer, J. D. (2003). The symbiotic birth and spread of plastids: how many times and whodunit? J. Phycol. 39, 4–12.
Pancer, Z., and Cooper, M. D. (2006). The evolution of adaptive immunity. Annu. Rev. Immunol. 24, 497–518.
Panchin, Y., and Moroz, L. L. (2008). Molluscan mobile elements similar to the vertebrate Recombination-Activating Genes. Biochem. Biophys. Res. Commun. 369, 818–823.
Pei, J., Sadreyev, R., and Grishin, N. V. (2003). PCMA: fast and accurate multiple sequence alignment based on profile consistency. Bioinformatics 19, 427–428.
Penz, T., Horn, M., and Schmitz-Esser, S. (2010). The genome of the amoeba symbiont “Candidatus Amoebophilus asiaticus” encodes an afp-like prophage possibly used for protein secretion. Virulence 1, 541–545.
Pisani, D., Cotton, J. A., and McInerney, J. O. (2007). Supertrees disentangle the chimerical origin of eukaryotic genomes. Mol. Biol. Evol. 24, 1752–1760.
Pradel, G., Hayton, K., Aravind, L., Iyer, L. M., Abrahamsen, M. S., Bonawitz, A., Mejia, C., and Templeton, T. J. (2004). A multidomain adhesion protein family expressed in Plasmodium falciparum is essential for transmission to the mosquito. J. Exp. Med. 199, 1533–1544.
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE 5:e9490. doi: 10.1371/journal.pone.0009490
Qian, X., Barsyte-Lovejoy, D., Wang, L., Chewpoy, B., Gautam, N., Al Chawaf, A., and Lovejoy, D. A. (2004). Cloning and characterization of teneurin C-terminus associated peptide (TCAP)-3 from the hypothalamus of an adult rainbow trout (Oncorhynchus mykiss). Gen. Comp. Endocrinol. 137, 205–216.
Raoult, D., and Boyer, M. (2010). Amoebae as genitors and reservoirs of giant viruses. Intervirology 53, 321–329.
Riedl, S. J., and Salvesen, G. S. (2007). The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 8, 405–413.
Rivera, M. C., and Lake, J. A. (2004). The ring of life provides evidence for a genome fusion origin of eukaryotes. Nature 431, 152–155.
Rochat, H., and Martin-Eauclaire, M.-F. (2000). Animal Toxins: Facts and Protocols. Basel Boston, MA: Birkhauser Verlag.
Rogozin, I. B., Iyer, L. M., Liang, L., Glazko, G. V., Liston, V. G., Pavlov, Y. I., Aravind, L., and Pancer, Z. (2007). Evolution and diversification of lamprey antigen receptors: evidence for involvement of an AID-APOBEC family cytosine deaminase. Nat. Immunol. 8, 647–656.
Rosenberg, H. F. (2008). RNase A ribonucleases and host defense: an evolving story. J. Leukoc. Biol. 83, 1079–1087.
Rouhiainen, L., Paulin, L., Suomalainen, S., Hyytiainen, H., Buikema, W., Haselkorn, R., and Sivonen, K. (2000). Genes encoding synthetases of cyclic depsipeptides, anabaenopeptilides, in Anabaena strain 90. Mol. Microbiol. 37, 156–167.
Ruprich-Robert, G., and Thuriaux, P. (2010). Non-canonical DNA transcription enzymes and the conservation of two-barrel RNA polymerases. Nucleic Acids Res. 38, 4559–4569.
Russell, A. B., Hood, R. D., Bui, N. K., Leroux, M., Vollmer, W., and Mougous, J. D. (2011). Type VI secretion delivers bacteriolytic effectors to target cells. Nature 475, 343–347.
Salgado, P. S., Koivunen, M. R., Makeyev, E. V., Bamford, D. H., Stuart, D. I., and Grimes, J. M. (2006). The structure of an RNAi polymerase links RNA silencing and transcription. PLoS Biol. 4:e434. doi: 10.1371/journal.pbio.0040434
Samel, S. A., Marahiel, M. A., and Essen, L. O. (2008). How to tailor non-ribosomal peptide products–new clues about the structures and mechanisms of modifying enzymes. Mol. Biosyst. 4, 387–393.
Santos, J. M., and Soldati-Favre, D. (2011). Invasion factors are coupled to key signalling events leading to the establishment of infection in apicomplexan parasites. Cell. Microbiol. 13, 787–796.
Sapp, J. (2007). “Mitochondria and their host: morphology to molecular phylogeny,” in Origin of Mitochondria and Hydrogenosomes, eds W. F. Martin and M. Müller (Berlin, Heidelberg: Springer), 57–83.
Sassera, D., Beninati, T., Bandi, C., Bouman, E. A., Sacchi, L., Fabbi, M., and Lo, N. (2006). ‘Candidatus Midichloria mitochondrii’, an endosymbiont of the tick Ixodes ricinus with a unique intramitochondrial lifestyle. Int. J. Syst. Evol. Microbiol. 56, 2535–2540.
Schatz, D. G., and Swanson, P. C. (2011). V(D)J recombination: mechanisms of initiation. Annu. Rev. Genet. 45, 167–202.
Schmitz-Esser, S., Tischler, P., Arnold, R., Montanaro, J., Wagner, M., Rattei, T., and Horn, M. (2010). The genome of the amoeba symbiont “Candidatus Amoebophilus asiaticus” reveals common mechanisms for host cell interaction among amoeba-associated bacteria. J. Bacteriol. 192, 1045–1057.
Schwarz, S., West, T. E., Boyer, F., Chiang, W. C., Carl, M. A., Hood, R. D., Rohmer, L., Tolker-Nielsen, T., Skerrett, S. J., and Mougous, J. D. (2010). Burkholderia type VI secretion systems have distinct roles in eukaryotic and bacterial cell interactions. PLoS Pathog. 6:e1001068. doi: 10.1371/journal.ppat.1001068
Schwefel, D., Frohlich, C., Eichhorst, J., Wiesner, B., Behlke, J., Aravind, L., and Daumke, O. (2010). Structural basis of oligomerization in septin-like GTPase of immunity-associated protein 2 (GIMAP2). Proc. Natl. Acad. Sci. U.S.A. 107, 20299–20304.
Silva, J. P., Lelianova, V. G., Ermolyuk, Y. S., Vysokov, N., Hitchen, P. G., Berninghausen, O., Rahman, M. A., Zangrandi, A., Fidalgo, S., Tonevitsky, A. G., Dell, A., Volynski, K. E., and Ushkaryov, Y. A. (2011). Latrophilin 1 and its endogenous ligand Lasso/teneurin-2 form a high-affinity transsynaptic receptor pair with signaling capabilities. Proc. Natl. Acad. Sci. U.S.A. 108, 12113–12118.
Skippington, E., and Ragan, M. A. (2011). Lateral genetic transfer and the construction of genetic exchange communities. FEMS Microbiol. Rev. 35, 707–735.
Snyder, L. (1995). Phage-exclusion enzymes: a bonanza of biochemical and cell biology reagents? Mol. Microbiol. 15, 415–420.
Soding, J., Biegert, A., and Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248.
Soldati-Favre, D. (2008). Molecular dissection of host cell invasion by the apicomplexans: the glideosome. Parasite 15, 197–205.
Stwora-Wojczyk, M. M., Kissinger, J. C., Spitalnik, S. L., and Wojczyk, B. S. (2004). O-glycosylation in Toxoplasma gondii: identification and analysis of a family of UDP-GalNAc:polypeptide N-acetylgalactosaminyltransferases. Int. J. Parasitol. 34, 309–322.
Sumby, P., and Smith, M. C. (2002). Genetics of the phage growth limitation (Pgl) system of Streptomyces coelicolor A3. Mol. Microbiol. 44, 489–500.
Thomas, J. H. (2006). Adaptive evolution in two large families of ubiquitin-ligase adapters in nematodes and plants. Genome Res. 16, 1017–1030.
Vainio, S., Genest, P. A., Ter Riet, B., Van Luenen, H., and Borst, P. (2009). Evidence that J-binding protein 2 is a thymidine hydroxylase catalyzing the first step in the biosynthesis of DNA base J. Mol. Biochem. Parasitol. 164, 157–161.
Varki, A., Cummings, R., Esko, J., Freeze, H., Hart, G., and Marth, J. (1999). Essentials of Glycobiology. New York, NY: Cold Spring Harbor Laboratory Press.
Vivier, E., and Desportes, I. (1990). “Phylum Apicomplexa,” in Handbook of Protoctista, eds L. Margulis, J. O. Corliss, M. Melkonian, and D. J. Chapman (Boston, MA: Jones and Bartlett Publishers), 549–573.
Werren, J. H. (2011). Selfish genetic elements, genetic conflict, and evolutionary innovation. Proc. Natl. Acad. Sci. U.S.A. 108(Suppl. 2), 10863–10870.
Wiesner, J., and Vilcinskas, A. (2010). Antimicrobial peptides: the ancient arm of the human immune system. Virulence 1, 440–464.
Yarbrough, M. L., Li, Y., Kinch, L. N., Grishin, N. V., Ball, H. L., and Orth, K. (2009). AMPylation of Rho GTPases by Vibrio VopS disrupts effector binding and downstream signaling. Science 323, 269–272.
Zehrmann, A., Verbitskiy, D., Hartel, B., Brennicke, A., and Takenaka, M. (2011). PPR proteins network as site-specific RNA editing factors in plant organelles. RNA Biol. 8, 67–70.
Zhang, D., De Souza, R. F., Anantharaman, V., Iyer, L. M., and Aravind, L. (2012). Polymorphic toxin systems: comprehensive characterization of trafficking modes, processing, mechanisms of action, immunity and ecology using comparative genomics. Biol. Dir. doi: 10.1186/1745-6150-7-18. [Epub ahead of print].
Zhang, D., Iyer, L. M., and Aravind, L. (2011). A novel immunity system for bacterial nucleic acid degrading toxins and its recruitment in various eukaryotic and DNA viral systems. Nucleic Acids Res. 39, 4532–4552.
Keywords: antibiotics, biological conflict, endosymbiosis, immunity proteins, restriction-modfication, RNAi, selfish elements, toxins
Citation: Aravind L, Anantharaman V, Zhang D, de Souza RF and Iyer LM (2012) Gene flow and biological conflict systems in the origin and evolution of eukaryotes. Front. Cell. Inf. Microbio. 2:89. doi: 10.3389/fcimb.2012.00089
Received: 18 May 2012; Accepted: 13 June 2012;
Published online: 29 June 2012.
Edited by:
Didier Raoult, Université de la Méditerranée, FranceReviewed by:
Didier Raoult, Université de la Méditerranée, FranceCopyright: © 2012 Aravind, Anantharaman, Zhang, de Souza and Iyer. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: L. Aravind, National Center for Biotechnology Information, National Library of Medicine, National Institutes of Health, Bethesda, MD, USA. e-mail:YXJhdmluZEBuY2JpLm5sbS5uaWguZ292
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.