 David W. Reid†‡
David W. Reid†‡ Jonathan B. Muyskens†
Jonathan B. Muyskens† James T. Neal†‡ Gino W. Gaddini‡ Lucy Y. Cho‡
James T. Neal†‡ Gino W. Gaddini‡ Lucy Y. Cho‡ Anica M. Wandler Crystal M. Botham‡
Anica M. Wandler Crystal M. Botham‡ Karen Guillemin*
Karen Guillemin*- Institute of Molecular Biology, University of Oregon, Eugene, OR, USA
Helicobacter pylori strains containing the CagA protein are associated with high risk of gastric diseases including atrophic gastritis, peptic ulcers, and gastric cancer. CagA is injected into host cells via a Type IV secretion system where it activates growth factor-like signaling, disrupts cell-cell junctions, and perturbs host cell polarity. Using a transgenic Drosophila model, we have shown that CagA expression disrupts the morphogenesis of epithelial tissues such as the adult eye. Here we describe a genetic screen to identify modifiers of CagA-induced eye defects. We determined that reducing the copy number of genes encoding components of signaling pathways known to be targeted by CagA, such as the epidermal growth factor receptor (EGFR), modified the CagA-induced eye phenotypes. In our screen of just over half the Drosophila genome, we discovered 12 genes that either suppressed or enhanced CagA's disruption of the eye epithelium. Included in this list are genes involved in epithelial integrity, intracellular trafficking, and signal transduction. We investigated the mechanism of one suppressor, encoding the epithelial polarity determinant and junction protein Coracle, which is homologous to the mammalian Protein 4.1. We found that loss of a single copy of coracle improved the organization and integrity of larval retinal epithelia expressing CagA, but did not alter CagA's localization to cell junctions. Loss of a single copy of the coracle antagonist crumbs enhanced CagA-associated disruption of the larval retinal epithelium, whereas overexpression of crumbs suppressed this phenotype. Collectively, these results point to new cellular pathways whose disruption by CagA are likely to contribute to H. pylori-associated disease pathology.
Introduction
H. pylori infects approximately 50% of the world's population and is a leading cause of ulcers and gastric cancer (Amieva and El-Omar, 2008). Strains harboring the virulence factor, CagA, are up to three times more potent in contributing to cancer progression than strains lacking this factor (Blaser et al., 1995; Huang et al., 2003; Wu et al., 2003). In cell culture experiments, CagA has been shown to interact physically with at least 20 proteins, such as SHP-2 and Par1, and to modulate the activity of many other host proteins (Hatakeyama, 2008; Backert et al., 2010). However, progress in characterizing the in vivo significance of these putative host effectors of CagA has been hampered by a lack of experimental models to study CagA's effects on intact tissues. We have developed a transgenic Drosophila model to study the expression of CagA in epithelial tissues such as the larval and adult eye (Botham et al., 2008; Muyskens and Guillemin, 2011). In this system, CagA is expressed as a full-length protein that is tyrosine phosphorylated by host kinases and localizes to cell junctions, as in mammalian cells (Botham et al., 2008).
Using this system, we showed that CagA interacts genetically with proteins identified as its physical targets in tissue culture cells. Several of CagA's physical interaction partners include members of receptor tyrosine kinase (RTK) signaling pathways that are normally scaffolded together in the cell by the adaptor protein Grb2-associated binder (Gab) (Hatakeyama, 2003). We demonstrated that expression of CagA could rescue phenotypes associated with loss of the Drosophila Gab, Son of sevenless, indicating that CagA functions as a Gab mimic and restores the physical interactions required for efficient RTK signaling. In these studies we also discovered that ectopic expression of CagA in the developing Drosophila eye, unlike over-expression of Son of sevenless, profoundly disrupted the morphogenesis of the retinal epithelium, resulting in adult eyes with a “rough” phenotype in which the crystalline array of facets is perturbed. We went on to show that CagA's disruption of the larval retinal epithelium was due to over-activation of myosin light chain (Muyskens and Guillemin, 2011), which has been implicated in disruption of gastrointestinal epithelial barriers (Shen et al., 2009) and H. pylori pathogenesis (Wroblewski et al., 2009). In this study we describe a forward genetic screen to uncover additional host genes that influence CagA's activity in the retinal epithelium.
The Drosophila eye has been a fertile genetic system for discovering genes involved in cellular signaling pathways, including many RTK signaling pathway members (Voas and Rebay, 2004). Because of the Drosophila eye's repeating pattern of facets or ommatidia, even subtle perturbations in signaling pathways that regulate eye morphogenesis can be distinguished by the severity of the rough eye phenotype of the adults, making possible rapid, high throughput screens for dominant enhancer and suppressor mutations (St Johnston, 2002). These genetic screens have proven to be extremely fruitful because of the high degree of conservation in molecular signaling pathways in eukaryotic cells. For example, the important CagA interactor SHP-2 was originally identified in a genetic screen in the Drosophila eye (Simon et al., 1991), and subsequently identified in mammals (Freeman et al., 1992). The functional conservation between human and Drosophila SHP-2 is illustrated by the fact that expression of the human protein can rescue the eye defects of a csw mutant lacking the Drosophila SHP-2 (Oishi et al., 2006). The high degree of molecular conservation in cellular processes targeted by bacterial pathogens has allowed researchers to screen for host factors that interact genetically with bacterial effector proteins in genetically tractable systems such as fruit flies and yeast (Siggers and Lesser, 2008; Boyer et al., 2012).
Here we exploited the CagA-induced rough eye phenotype to identify host genes that are important for pathogenic mechanisms of CagA. We used molecularly defined chromosomal deficiencies to screen over half of the Drosophila genome for dominant suppressors or enhancers of CagA-induced epithelial disruption. Our deficiency screen identified 12 novel genetic interactors, capable of modulating the severity of CagA-induced disruption of the adult retinal epithelium. We refer to these genetic interactors collectively as the modifier of CagA (Moc) genes. Moc genes have been shown to function in numerous cellular pathways including those involved in maintenance of epithelial integrity, intracellular trafficking, and signal transduction. We further investigated CagA's genetic interactions with one Moc suppressor, the epithelial polarity determinant coracle that is the homolog of the mammalian 4.1 protein. In addition, we extended our genetic interaction network to show that other polarity determinants with antagonistic functions to coracle behave as dominant enhancers of CagA-associated epithelial phenotypes. The Moc genes provide new avenues of investigation toward understanding CagA's pathogenicity in humans.
Materials and Methods
Drosophila Strains
All flies were raised on standard Drosophila media at 22°C unless otherwise noted. The P{w[UAS-CagA]} transgenic line was generated as described (Botham et al., 2008). Transgenes were expressed in the eye using P{w[+mC] = GAL4-ninaE.GMR}12 [GMR, Bloomington Stock Center (BSC # 1104)]. Deficiency lines used for the initial identification of genomic regions were generated by Exelixis (Parks et al., 2004). The genetic null allele of csw (cswC114) was obtained from Michael Simon (Stanford University). All other alleles used are described on FlyBase (Tweedie et al., 2009), including EGFRt1 (FBst0002079), par1k06323 (FBal0064446), rho172F (FBst0007326), and the Moc genes listed in Table 1.
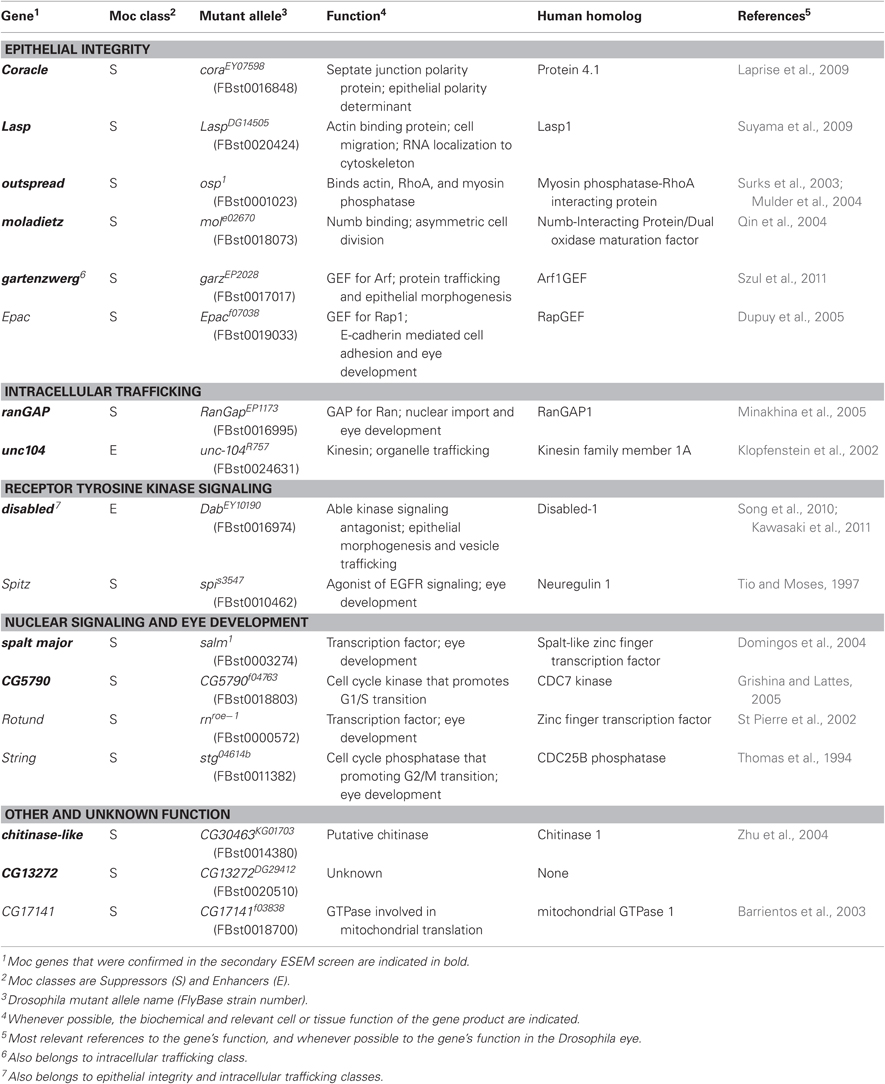
Table 1. Moc genes identified in Drosophila adult eyes.
Moc Genetic Screen
Males carrying a genetic deletion (generally spanning between 5 and 30 genes) on one chromosome and a visual marker such as CyO on the other were crossed to female virgins homozygous for the GMR-GAL4 driver and CagA. Moc mutants were identified by comparing the overall eye roughness of adult flies carrying a deficiency to siblings that carried the visual marker by light microscopy. We screened 237 deficiency stocks covering 7451 genes, or 53% of Drosophila genes. We found a surprisingly high proportion—49%—of the deficiencies resulted in suppression, while only a few enhancers were identified (Figure 1). Particularly severe Moc mutants were chosen for further investigation in an attempt to identify a single gene responsible for the modification. Additional deficiencies overlapping the genetic region of interest were used to narrow the number of potentially responsible genes. Assuming that a single gene were responsible for the modification of the rough-eyed phenotype, genes within an overlapping deficiency that did not act as a Moc could be eliminated as candidates, while genes not included within deficiencies that acted as a Moc could also be eliminated. Once the number of candidate genes was sufficiently low, males carrying null alleles for candidate genes were crossed to GMR-GAL4/GMR-GAL4; UAS-CagA/UAS-CagA female virgins and the eyes of adult progeny were screened for modification of the rough-eyed phenotype. This method allowed for identification of a single Moc gene in 17 of the 22 initial Moc deficiencies that were chosen for analysis.
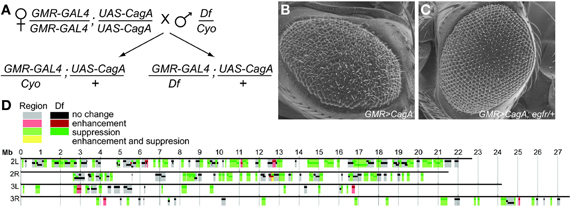
Figure 1. (A) Crossing scheme for the Moc deficiency screen. Flies containing the genetic deficiency were compared to those containing a visual marker such as CyO. Flies expressing CagA in a wild-type, (B) or egfr−/+ background, (C) were imaged by ESEM. (D) Chromosomal map of the genetic deficiency screen. The result from each deficiency (darker colors) is indicated along with the inferred functionality of each genetic region (lighter colors), where deficiencies that caused no change override those that cause enhancement or suppression.
Environmental Scanning Electron Microscopy
To evaluate the CagA-induced eye phenotypes at higher resolution, we used environmental scanning electron microscopy (ESEM). Flies were anesthetized with FlyNap (Carolina Biological Supply Company) and imaged using an FEI Quanta 200 environmental scanning electron microscope. Images of at least 10 flies of each genotype were recorded and scored in a blinded fashion by five investigators. Scoring classes were defined as follows: (0) Geometric organization intact. (1) Loss of geometric organization, fewer than 25% of ommatidia fused or malformed. (2) Loss of geometric organization, greater than 25% of ommatidia fused or malformed. (3) Loss of geometric organization, greater than 25% of ommatidia fused, malformed, and greater than 1% but less than 25% of the eye lacks a recognizable morphology. (4) Loss of geometric organization, greater than 25% of ommatidia fused or malformed, and greater than 25% of the eye lacks a recognizable morphology. (5) Loss of geometric organization, greater than 25% of ommatidia fused and malformed, greater than 25% of the eye lacks a recognizable morphology, and pronounced invaginations on the eye surface.
Immunohistochemistry
Eye discs were dissected from third instar larvae and fixed for 30 min (4% formaldehyde, 0.1 M PIPES (pH 6.9), 0.3% Triton X-100, 2 mM EGTA, 1 mM MgSO4), then washed (0.3% Triton X-100 in phosphate buffered saline, PBS) for 20 min and blocked for at least one hour in 1% bovine serum albumin and 0.3% Triton X-100 in PBS (PBSBT). Tissues were then incubated in primary antibody mouse anti-Dlg [4F3 (Developmental Studies Hybridoma Bank)], mouse anti-DCAD2 (DSHB), or mouse anti-HA (Covance) overnight at 1:100 in PBSBT. Tissues were rinsed for 1 h in PBSBT, then incubated with anti-mouse conjugated Cy3 (Jackson ImmunoResearch) at 1:200. Imaginal discs were mounted in VectaShield (Vector Laboratories) and visualized with a Nikon TE2000 U with C1 Digital Eclipse confocal microscope.
Evaluation of Larval Retinal Epithelial Morphology
Z-stacks of eye discs stained for the septate junction component Dlg were generated using a 0.2 μm step size and compiled in ImageJ. The areas chosen for Z-stacks were ∼1 mm2 in area and devoid of ectopic furrows. The intensity of fluorescence 4.8 microns below the peak intensity was taken to represent the relative integrity of the epithelium, with higher values representing a more disrupted tissue. Intensities were normalized to the maximum density in the Z-stack to generate the final metric.
Results
A Deficiency Screen for Modifiers of CagA-Induced Adult Eye Defects
In this study, we screened transgenic Drosophila expressing the H. pylori cagA gene for dominant modifiers of CagA-induced epithelial disruption. We had shown previously that CagA expression in the developing eye results in a rough eye phenotype that is easily detected using a dissecting microscope and that is sensitive to dosage, with expression of two copies of cagA resulting in a much more severe disruption of the adult structure than a single copy (Botham et al., 2008). We used the Gal4 transcription factor under the GMR promoter to drive expression of a UAS-CagA transgene in the developing eye beginning in the larval eye imaginal disc as photoreceptors are first being specified. GMR-Gal4 expression is maintained in the eye primordia throughout subsequent development and into adulthood.
For our genetic screen, we crossed homozygous GMR-GAL4; UAS-CagA females to males carrying molecularly defined chromosomal deletions maintained over a balancer chromosome with a dominant marker, such as the CyO balancer on the second chromosome (Figure 1A). Half of the resulting progeny would contain the deficiency and could be compared to the other half with the marker to look for enhancement or suppression of the CagA-induced rough eye phenotype.
To assess the feasibility of this genetic screening strategy, we tested whether deletion of single copies of genes encoding known genetic interactors of CagA would modify the CagA-associated phenotype. CagA is a potent activator of RTK pathway signaling in tissue culture cells (Backert et al., 2010). In the Drosophila eye, EGF receptor is a critical RTK required for multiple steps during development (Dominguez et al., 1998). We, therefore, asked whether reducing RTK signaling by removing a single copy of the egfr gene would reduce the severity of the CagA-induced rough eye phenotype. As predicted, the severity of eye disruption was significantly reduced in egfr−/+ flies expressing CagA as compared to CagA-expressing control flies (Figures 1B,C). This demonstrated that it is possible to genetically suppress CagA's disruption of the Drosophila adult eye, thus motivating us to use this system for an unbiased genetic screen for Moc genes.
We took advantage of a publicly available collection of Drosophila stocks containing deficiencies in defined genomic regions (Parks et al., 2004) to systematically search for chromosomal regions that modify CagA's disruption of the epithelium. Using this collection, we tested 237 stocks with genomic deletions for their ability to modify the CagA-induced rough eye phenotype. Combined, this collection covered 7451 genes, or approximately 53% of all Drosophila genes (Figure 1D).
From this initial panel of deficiency stocks, 22 chromosomal regions were identified that modified CagA's disruption of the eye epithelium with high expressivity and penetrance. To identify the individual genes responsible for the modification of CagA's activity, CagA-expressing flies were subsequently crossed to fly stocks containing smaller deficiencies located within the 22 chromosomal regions identified in the initial screen to narrow the number of candidate Moc genes. Once we whittled the number of candidates down to 5–10 genes, we obtained all available strains with mutations in the candidate genes within the interval to test for their ability to modify the rough-eyed phenotype. This method allowed us to identify a single Moc gene in 17 of the 22 initial Moc intervals. These 17 genes are listed in Table 1. Moc genes fit broadly into the functional classes of epithelial integrity, intracellular trafficking, signal transduction, and nuclear signaling, with three additional genes of miscellaneous or unknown function.
A Secondary Screen for Strong Modifiers of the CagA-Induced Phenotype
To assess the degree of modification caused by the identified modifiers, we used ESEM to obtain high-resolution images of adult eyes from multiple individuals expressing each of the Moc genes in the GMR>CagA background. At this high resolution, we saw that CagA expression induced mispolarized and supernumerary bristles, fusion of ommatidia, and in the most severe instances, loss of apparent ommatidial patterning and the development of large invaginations in the epithelium. From our large data set of images, we were able to discern a continuum of severity and develop a scoring system that enabled us to quantify the rough eye phenotype (Figures 2A–F). Eyes scoring 0 resembled wild-type flies and had no apparent sign of disruption. In contrast, eyes scoring 5 were the most severely disrupted. Eyes scoring 1–4 had intermediate levels of disruption. For a complete description of the scoring system, see Materials and Methods.
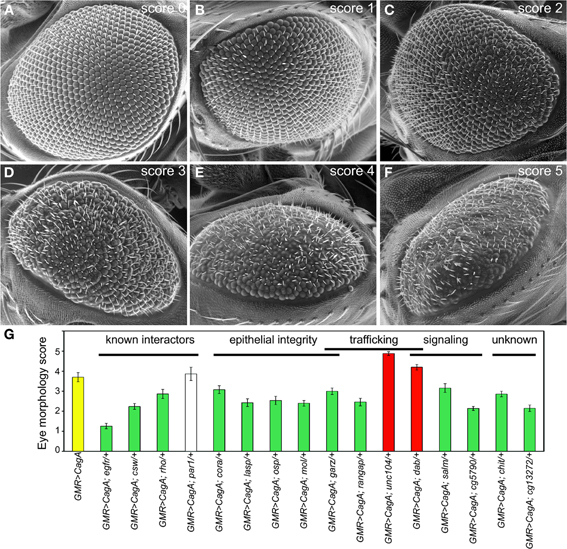
Figure 2. (A–F) Representative ESEM images for each class of disruption by CagA. The scoring rubric is described in Materials and Methods. (G) The mean ESEM-based eye disruption for CagA, known interactors and Moc genes of different functional classes. Error bars represent standard error.
For each of the Moc genes, at least 10 ESEM images of adult eyes from different flies were scored in a blinded fashion by five investigators, and the average score was tallied. From this analysis, we verified that 12 of the 17 genes were bona fide modifiers of CagA-induced eye disruption (Figure 2G and Table 1). Two of these behaved as enhancers and 10 were suppressors. The most abundant functional group among these 12 genes was the epithelial integrity class.
To calibrate the effects of the validated Moc genes, we quantified the ability of four known CagA signaling modulators to modify the CagA-induced eye phenotype: egfr (Keates et al., 2007; Bauer et al., 2009), csw (the homologue of SHP-2) (Higashi et al., 2002), rhoA (Muyskens and Guillemin, 2011), and par1 (Saadat et al., 2007). Of these, egfr proved to be the most potent suppressor of CagA-induced disruption. Loss of single copies of both csw and rhoA caused suppression, whereas loss of a single copy of par1 did not cause a significant modification of the CagA rough eye phenotype (Figure 2G). This analysis demonstrated that the 12 newly identified Moc genes had similar or stronger effects on the CagA-induced phenotype than known CagA signaling modulators.
Modification of CagA-Induced Disruption of the Larval Retinal Epithelium
Our Moc screen identified genetic modifiers of the CagA phenotype in the adult eye. We had previously reported that CagA expression with the GMR driver induces profound disruption of the morphogenesis of the larval retinal epithelium shortly after initiation of CagA expression (Muyskens and Guillemin, 2011). We showed that by overactivating Rho and non-muscle myosin in the larval epithelium, CagA causes ectopic furrowing of the epithelial sheet. Because so many of the Moc genes were implicated in epithelial integrity, we wished to determine whether any of them might modify CagA's effects at these early stages of epithelial disruption. We decided to focus on Coracle (Cora), because it had the best characterized function as an epithelial polarity determinant (Laprise et al., 2009, 2010).
To measure the integrity of the larval retinal epithelium, we stained the tissue with an antibody against Discs large (Dlg), a component of the septate junction, the invertebrate cell junction that is structurally homologous to the chordate tight junction. We used laser scanning confocal microscopy to image from the apical to basal poles a region of epithelium devoid of obvious ectopic furrows. We quantified the intensity of the Dlg signal as a function of depth from the apical surface. Maximal Dlg signal was just below the apical epithelial surface at the septate junction. When we compared the relative intensity of Dlg signal below this maximal point, we found that the CagA-expressing discs had significantly more Dlg signal at deeper positions relative to the GMR control discs (Figure 3A). For our further analysis, we quantified the relative Dlg intensity 4.8 microns below the point of peak Dlg intensity (arrow in Figure 3A), the point at which we observed the maximum difference between CagA-expressing and control larval retinal epithelia.
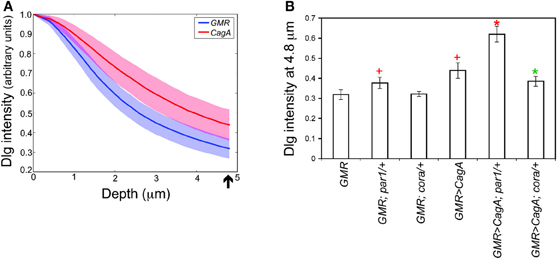
Figure 3. (A) Pattern of Dlg staining in Z-stacks of larval retinal imaginal discs. CagA-expressing eye discs are compared to those expressing the GMR-Gal4 driver alone. Shaded areas represent standard error. The arrow indicates the point at 4.8 microns below the peak intensity where the distribution was evaluated. (B) Quantification of larval retinal epithelial morphology for par1 and cora mutants, and their interactions with CagA.
We used this method to analyze the Dlg distribution in CagA-expressing larval eye discs lacking a single copy of a Moc gene. We first tested the consequence of depleting the CagA interactor and junctional protein Par1. Loss of a single copy of par1 caused a slight expansion of Dlg staining (Figure 3B). We also tested the consequence of depleting Cora, which is normally localized to the septate junctions. Loss of a single copy of cora had no effect on the distribution of Dlg at the septate junctions (Figure 3B). We then analyzed the Dlg distribution when these genes were deleted in the presence of CagA. In the larval epithelium, par1 behaved as a dominant enhancer of the CagA-associated disruption in Dlg protein distribution (Figure 3B), despite having no effect on the adult eye phenotype caused by CagA. In contrast, cora behaved as a dominant suppressor of the CagA phenotype in the larval epithelium, as it had done in the adult eye (Figures 2G,3B). The dominant enhancement of the CagA-induced epithelial disruption by par1 could be explained as the further impairment of a compromised tissue through the depletion of a junctional component. Less obvious was the mechanism by which cora depletion suppressed the CagA phenotype, which we sought to understand with further experiments.
Cora Reduction Suppresses CagA-Induced Epithelial Disorganization but not CagA Protein Localization to Septate Junctions
Our finding that Dlg protein extended deeper from the apical surface in the CagA-expressing epithelium as compared to wild-type tissue could arise from multiple mechanisms. Two possible mechanisms are illustrated in Figures 4A–C. In the first model, CagA-expression could cause a loss of junctional integrity and expansion of Dlg protein toward the basal end of the cell (Figure 4B). Alternatively, CagA could cause disorganization of the epithelial sheet, resulting in a broader zone of Dlg expression when averaged across multiple cells (Figure 4C). To distinguish these possibilities, we examined the organization of the larval epithelium and cell junctions at high resolution. We co-stained larval retinal epithelia for both the septate junction marker Dlg and the adherens junction marker E-cadherin (E-cad). In Drosophila epithelia, the adherens junction is apical to the septate junction, and in the larval retinal epithelium these junctions are located at the apices of photoreceptors and supporting cells that surround the ommatidia (Figure 4D). In CagA-expressing eye imaginal disc epithelia, the zone of Dlg expression was frequently found to extend deeper from the apical surface. In some instances this appeared to be due to more disorganized junctions (arrowhead in Figure 4E), but frequently the integrity of the junctions looked normal and the Dlg staining was displaced deeper into the tissue due to irregularities in the epithelial sheet (arrow in Figure 4E). Because the integrity of Dlg and E-cad staining looked mostly normal in the CagA-expressing larval retinal epithelia, we concluded that loss of Cora suppresses CagA-associated phenotypes in this tissue by reducing the overall disorganization of the epithelial sheet.
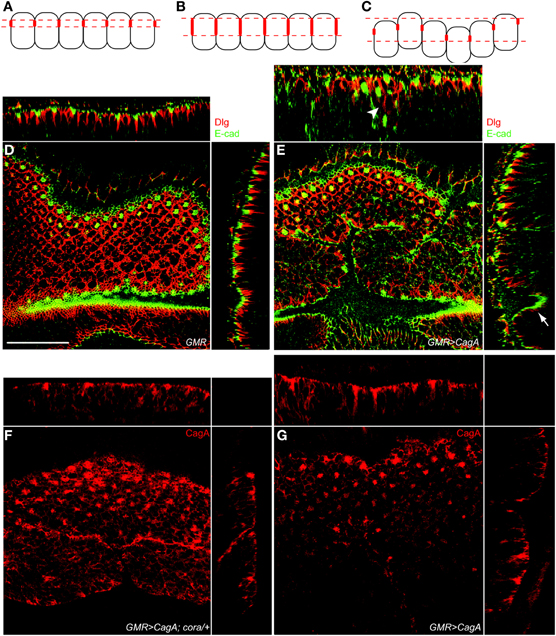
Figure 4. cora reduction suppresses CagA-induced epithelial disorganization but not CagA protein localization to septate junctions. (A–C) Model for the basal displacement of Dlg. Panel A represents the wild-type distribution of Dlg (represented as red structures on the lateral membranes of the epithelial cells). Panel B represents basally expanded Dlg expression due to expansion within individual cells. Panel C shows how epithelial disruption can cause basal mispositioning of Dlg expression by positioning cells deeper within the epithelium. (D) Control larval retinal epithelium (GMR-Gal4) stained with Dlg (red) and E-cad (green). YZ and XZ orthogonal planes are shown on the side and top, respectively, in D and E. Scale bar is 30 microns for all panels. (E) CagA-expressing larval retinal epithelium (GMR-Gal4; UAS-CagA) also stained with Dlg (red) and E-cad (green). Arrowhead in the upper orthogonal section shows basally mispositioned Dlg staining. Arrow indicates Dlg staining that is deep within the epithelium due to irregularities in the epithelial sheet. (F)cora+/− larval retinal epithelium expressing CagA (GMR-Gal4; UAS-CagA) showing CagA localization as labeled with anti-HA. Apical HA puncta are present. (G) A larval retinal disc expressing CagA (GMR-Gal4; UAS-CagA) labeled with HA antibody.
We had previously shown that CagA is localized to the apical junctional structures in the larval retinal epithelium and that a CagA mutant that fails to localize in this manner is a less potent disruptor of epithelial integrity (Muyskens and Guillemin, 2011). We wondered if loss of a single copy of cora could disrupt the localization of CagA to the apical cell junctions. We visualized CagA distribution using an HA epitope tag we had engineered into the protein. As we previously reported, we found that CagA was enriched in apical foci of CagA-expressing eye discs (Figure 4F). We found that this expression pattern was not different in eye discs lacking a single copy of cora, (Figure 4G). Therefore, cora's ability to suppress CagA-induced eye morphology does not appear to be due to failure of CagA protein to localize to apical cell junctions in the absence of one copy of cora.
Epithelial Polarity Determinants Modify CagA-Induced Disruption of the Larval Retinal Epithelium
To further explore the basis for cora suppression of CagA-induced larval retinal epithelial disorganization, we tested other epithelial polarity determinants for their ability to modify the CagA-induced larval retinal epithelium phenotype. Polarity in many epithelial tissues of both Drosophila and mammals is established and maintained by four conserved groups of polarity determinants: the apically localized Crumbs (Crb) group and three functionally distinct basolaterally distributed groups with defining members Cora, Scribble (Scrib), and Par1 (Laprise and Tepass, 2011). In contrast to cora, and similar to par1, crb, and scrib behaved as dominant enhancers of the CagA-induced larval epithelial disruption (Figure 5A), whereas loss of a single copy of these genes caused no epithelial disruption on their own (data not shown). To ask whether all Cora group members behaved as suppressors of CagA, we tested another Cora group member, Na, K-ATPase (encoded by the atpα gene). Unlike cora, atpα behaved as a dominant enhancer of CagA in the retinal epithelium (Figure 5A), and had no effect when depleted in the absence of CagA (data not shown). Cora and Crb mutually inhibit each other's activities in many epithelial structures (Laprise et al., 2009, 2010; Laprise and Tepass, 2011). We therefore asked whether over-expression of Crb would have the same effect as loss of Cora. Using a UAS-crb construct, we were able to achieve a significant suppression of CagA-induced epithelial disorganization, as measured by the basal distribution of Dlg (Figure 5A).
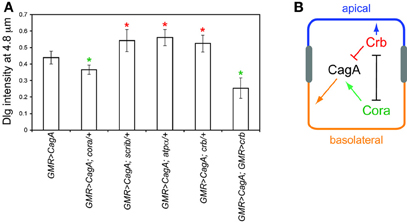
Figure 5. (A) Interactions of several epithelial polarity determinants with CagA, using the Dlg distribution assay described in Figure 3. Error bars represent standard error. Green asterisks represent mutants that significantly suppress CagA-induced epithelial disruption; red asterisks represent mutants that significantly enhance epithelial disruption (p < 0.05). (B) Model for the interactions of CagA with epithelial polarity determinants Cora and Crb.
Discussion
We have demonstrated that a transgenic Drosophila model can be used to identify conserved genes that modulate the effects of a virulence factor from a human pathogen. We show that CagA-induced perturbation of the Drosophila adult eye is a sensitive read-out for identification of genes that can alter CagA's ability to disrupt this tissue. Our approach is reductionist in that it characterizes the bacterial effector in isolation from other aspects of the infection process, such as immune responses to the bacteria and cellular interaction with the type IV secretion system that normally delivers CagA. The potential utility of the screen is limited by the extent of conservation between Drosophila and human genes and by the functional similarity between retinal and gastric epithelia. Nonetheless, we found that when depleted by one copy, genes encoding three known effectors of CagA, EGFR, Csw (SHP-2), and RhoA, significantly suppressed the eye morphological defects caused by CagA expression. This validated our approach to screen for dominant modifiers of CagA in this tissue. In our F1 screen we surveyed over half the Drosophila genome and identified 17 Moc genes, 12 of which we confirmed by high resolution ESEM.
Across the list of Moc genes, several themes of cellular and biochemical functions emerge. Eight of the 17 genes have known roles in epithelial integrity, including interactions with polarity determinants (coracle and moladietz) and the actin cytoskeleton (lasp and outspread). Three function in protein or organelle trafficking (gartenzwert, ranGAP, and unc104) with known or suspected roles in epithelial organization. Indeed, the ArfGEF, gartenzwerz, which is required for normal protein trafficking and morphogenesis of the Drosophila salivary gland epithelium (Szul et al., 2011), exemplified a growing appreciation of the connection between epithelial polarity and intracellular trafficking (Shivas et al., 2010). A frequent biochemical function among the Moc genes is interaction with GTPases or GTPase activity (outspread, gartenzwerz, epac, ranGAP, CG17141), which is interesting in light of the fact that we have shown that CagA's disruption of the larval retinal epithelium is due in part to excessive RhoA signaling (Muyskens and Guillemin, 2011). Another theme among the Moc genes is signal transduction and nuclear signaling, including two zinc finger transcription factors (spalt major and rotund) and two cell cycle regulators (homologs of CDC25B and CDC7). An additional signaling Moc, disabled, is an antagonist of Abl kinase (Song et al., 2010). In gastric epithelial cells, CagA has been shown to activate Abl and subsequently be phosphorylated by this kinase, resulting in enhanced CagA-mediated signaling, including RTK-dependent cell elongation (Tammer et al., 2007). Consistent with its molecular function as an inhibitor of Abl, loss of one copy of disabled results in enhancement of the CagA-mediated rough eye phenotype, the opposite effect of reduction of egfr, or the EGFR ligand, spitz. disabled has also been shown to be required for normal epithelial morphogenesis in Drosophila (Song et al., 2010) and to function in vesicle trafficking (Kawasaki et al., 2011), thereby linking the functions of RTK signaling, epithelial morphogenesis, and intracellular trafficking that run throughout the Moc list.
Within this group of modifiers, we focused our attention on cora because of its previously characterized role in epithelial polarity and septate junction regulation. The sepatate junction and its mammalian equivalent, the tight junction, regulate paracellular flux across epithelia. In the gastrointestinal tract, tight junctions are often targeted by enteric pathogens for invasion of deeper tissues or access to nutrients (Vogelmann et al., 2004). CagA has been shown to alter the distribution of the tight junction component ZO-1 and, over extended periods of time, impair tight junction integrity in H. pylori-infected cultured epithelial cells (Amieva et al., 2003). However, even under conditions when tight junctions remain intact, CagA confers on H. pylori the ability to replicate in the nutrient-poor environment of the epithelial apical surface (Tan et al., 2009). This CagA-mediated adaptation involves disruption of apical-basal polarity and expansion of basolateral markers to the apical surface (Tan et al., 2011).
We found that depletion of cora suppressed CagA-induced disruption of the larval retinal epithelium, but not by a perceptible change to the organization of the cell junctions or the localization of CagA to these structures. Intriguingly we found that over-expression of crb resulted in the same phenotypic suppression achieved by depletion of cora. Cora and Crb have mutually antagonistic activities, and in the absence of Cora, Crb will promote expansion of apical cell surfaces within the epithelium (Laprise et al., 2006, 2009). We hypothesize that this activity of Crb counteracts CagA's ability to promote more basolateral cell surface identities (Figure 5B). Thus, over-expressing Crb, or depleting its inhibitor, Cora, achieves a more balanced pull between apical promoting forces from Crb and basolateral promoting forces from CagA that is manifest as more normal epithelial organization in the CagA-expressing retinal epithelium in these genetic backgrounds.
In summary, our genetic screen has identified a number of host signaling pathways that modulate CagA's potency in disrupting host tissue. Further analysis of these Moc genes should lead new insights into CagA's mechanism of action in host tissue and may yield new strategies for pharmaceutical modulation of these pathways to treat H. pylori-associated pathologies.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Chris Doe, Tory Herman, Michael Simon, the Developmental Studies Hybridoma Bank, and the Bloomington Drosophila Stock Center for reagents and Kevin Bourzac for his contributions at the beginning of this project. This work was supported by Public Health Service grant R01 DK075667 to Karen Guillemin from the National Institutes of Health and by American Recovery and Reinvestment Act (ARRA) funds through the above grant.
Abbreviations
Moc, Modifier of CagA; ESEM, Environmental scanning electron microscopy.
References
Amieva, M. R., and El-Omar, E. M. (2008). Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 134, 306–323.
Amieva, M. R., Vogelmann, R., Covacci, A., Tompkins, L. S., Nelson, W. J., and Falkow, S. (2003). Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300, 1430–1434.
Backert, S., Tegtmeyer, N., and Selbach, M. (2010). The versatility of Helicobacter pylori CagA effector protein functions: the master key hypothesis. Helicobacter 15, 163–176.
Barrientos, A., Korr, D., Barwell, K. J., Sjulsen, C., Gajewski, C. D., Manfredi, G., Ackerman, S., and Tzagoloff, A. (2003). MTG1 codes for a conserved protein required for mitochondrial translation. Mol. Biol. Cell 14, 2292–2302.
Bauer, B., Bartfeld, S., and Meyer, T. F. (2009). H. pylori selectively blocks EGFR endocytosis via the non-receptor kinase c-Abl and CagA. Cell. Microbiol. 11, 156–169.
Blaser, M. J., Perez-Perez, G. I., Kleanthous, H., Cover, T. L., Peek, R. M., Chyou, P. H., Stemmermann, G. N., and Nomura, A. (1995). Infection with Helicobacter pylori strains possessing cagA is associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Res. 55, 2111–2115.
Botham, C. M., Wandler, A. M., and Guillemin, K. (2008). A transgenic Drosophila model demonstrates that the Helicobacter pylori CagA protein functions as a eukaryotic Gab adaptor. PLoS Pathog. 4: e1000064. doi: 10.1371/journal.ppat.1000064
Boyer, L., Paquette, N., Silverman, N., and Stuart, L. M. (2012). Bacterial effectors: learning on the fly. Adv. Exp. Med. Biol. 710, 29–36.
Domingos, P. M., Mlodzik, M., Mendes, C. S., Brown, S., Steller, H., and Mollereau, B. (2004). Spalt transcription factors are required for R3/R4 specification and establishment of planar cell polarity in the Drosophila eye. Development 131, 5695–5702.
Dominguez, M., Wasserman, J. D., and Freeman, M. (1998). Multiple functions of the EGF receptor in Drosophila eye development. Curr. Biol. 8, 1039–1048.
Dupuy, A. G., L'Hoste, S., Cherfils, J., Camonis, J., Gaudriault, G., and De Gunzburg, J. (2005). Novel Rap1 dominant-negative mutants interfere selectively with C3G and Epac. Oncogene 24, 4509–4520.
Freeman, R. M. Jr., Plutzky, J., and Neel, B. G. (1992). Identification of a human src homology 2-containing protein-tyrosine-phosphatase: a putative homolog of Drosophila corkscrew. Proc. Natl. Acad. Sci. U.S.A. 89, 11239–11243.
Grishina, I., and Lattes, B. (2005). A novel Cdk2 interactor is phosphorylated by Cdc7 and associates with components of the replication complexes. Cell Cycle 4, 1120–1126.
Hatakeyama, M. (2003). Helicobacter pylori CagA–a potential bacterial oncoprotein that functionally mimics the mammalian Gab family of adaptor proteins. Microbes Infect. 5, 143–150.
Hatakeyama, M. (2008). SagA of CagA in Helicobacter pylori pathogenesis. Curr. Opin. Microbiol. 11, 30–37.
Higashi, H., Tsutsumi, R., Muto, S., Sugiyama, T., Azuma, T., Asaka, M., and Hatakeyama, M. (2002). SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 295, 683–686.
Huang, J. Q., Zheng, G. F., Sumanac, K., Irvine, E. J., and Hunt, R. H. (2003). Meta-analysis of the relationship between cagA seropositivity and gastric cancer. Gastroenterology 125, 1636–1644.
Kawasaki, F., Iyer, J., Posey, L. L., Sun, C. E., Mammen, S. E., Yan, H., and Ordway, R. W. (2011). The DISABLED protein functions in CLATHRIN-mediated synaptic vesicle endocytosis and exoendocytic coupling at the active zone. Proc. Natl. Acad. Sci. U.S.A. 108, E222–E229.
Keates, S., Keates, A. C., Katchar, K., Peek, R. M. Jr., and Kelly, C. P. (2007). Helicobacter pylori induces up-regulation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Infect. Dis. 196, 95–103.
Klopfenstein, D. R., Tomishige, M., Stuurman, N., and Vale, R. D. (2002). Role of phosphatidylinositol(4,5)bisphosphate organization in membrane transport by the Unc104 kinesin motor. Cell 109, 347–358.
Laprise, P., Beronja, S., Silva-Gagliardi, N. F., Pellikka, M., Jensen, A. M., Mcglade, C. J., and Tepass, U. (2006). The FERM protein Yurt is a negative regulatory component of the Crumbs complex that controls epithelial polarity and apical membrane size. Dev. Cell 11, 363–374.
Laprise, P., Lau, K. M., Harris, K. P., Silva-Gagliardi, N. F., Paul, S. M., Beronja, S., Beitel, G. J., Mcglade, C. J., and Tepass, U. (2009). Yurt, Coracle, Neurexin IV and the Na(+),K(+)-ATPase form a novel group of epithelial polarity proteins. Nature 459, 1141–1145.
Laprise, P., Paul, S. M., Boulanger, J., Robbins, R. M., Beitel, G. J., and Tepass, U. (2010). Epithelial polarity proteins regulate Drosophila tracheal tube size in parallel to the luminal matrix pathway. Curr. Biol. 20, 55–61.
Laprise, P., and Tepass, U. (2011). Novel insights into epithelial polarity proteins in Drosophila. Trends Cell Biol. 21, 401–408.
Minakhina, S., Myers, R., Druzhinina, M., and Steward, R. (2005). Crosstalk between the actin cytoskeleton and Ran-mediated nuclear transport. BMC Cell Biol. 6, 32.
Mulder, J., Ariaens, A., Van Den Boomen, D., and Moolenaar, W. H. (2004). p116Rip targets myosin phosphatase to the actin cytoskeleton and is essential for RhoA/ROCK-regulated neuritogenesis. Mol. Biol. Cell 15, 5516–5527.
Muyskens, J. B., and Guillemin, K. (2011). Helicobacter pylori CagA disrupts epithelial patterning by activating myosin light chain. PLoS One 6:e17856. doi: 10.1371/journal.pone.0017856
Oishi, K., Gaengel, K., Krishnamoorthy, S., Kamiya, K., Kim, I. K., Ying, H., Weber, U., Perkins, L. A., Tartaglia, M., Mlodzik, M., Pick, L., and Gelb, B. D. (2006). Transgenic Drosophila models of Noonan syndrome causing PTPN11 gain-of-function mutations. Hum. Mol. Genet. 15, 543–553.
Parks, A. L., Cook, K. R., Belvin, M., Dompe, N. A., Fawcett, R., Huppert, K., Tan, L. R., Winter, C. G., Bogart, K. P., Deal, J. E., Deal-Herr, M. E., Grant, D., Marcinko, M., Miyazaki, W. Y., Robertson, S., Shaw, K. J., Tabios, M., Vysotskaia, V., Zhao, L., Andrade, R. S., Edgar, K. A., Howie, E., Killpack, K., Milash, B., Norton, A., Thao, D., Whittaker, K., Winner, M. A., Friedman, L., Margolis, J., Singer, M. A., Kopczynski, C., Curtis, D., Kaufman, T. C., Plowman, G. D., Duyk, G., and Francis-Lang, H. L. (2004). Systematic generation of high-resolution deletion coverage of the Drosophila melanogaster genome. Nat. Genet. 36, 288–292.
Qin, H., Percival-Smith, A., Li, C., Jia, C. Y., Gloor, G., and Li, S. S. (2004). A novel transmembrane protein recruits numb to the plasma membrane during asymmetric cell division. J. Biol. Chem. 279, 11304–11312.
Saadat, I., Higashi, H., Obuse, C., Umeda, M., Murata-Kamiya, N., Saito, Y., Lu, H., Ohnishi, N., Azuma, T., Suzuki, A., Ohno, S., and Hatakeyama, M. (2007). Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature 447, 330–333.
Shen, L., Su, L., and Turner, J. R. (2009). Mechanisms and functional implications of intestinal barrier defects. Dig. Dis. 27, 443–449.
Shivas, J. M., Morrison, H. A., Bilder, D., and Skop, A. R. (2010). Polarity and endocytosis: reciprocal regulation. Trends Cell Biol. 20, 445–452.
Siggers, K. A., and Lesser, C. F. (2008). The Yeast Saccharomyces cerevisiae: a versatile model system for the identification and characterization of bacterial virulence proteins. Cell Host Microbe 4, 8–15.
Simon, M. A., Bowtell, D. D., Dodson, G. S., Laverty, T. R., and Rubin, G. M. (1991). Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell 67, 701–716.
Song, J. K., Kannan, R., Merdes, G., Singh, J., Mlodzik, M., and Giniger, E. (2010). Disabled is a bona fide component of the Abl signaling network. Development 137, 3719–3727.
St Johnston, D. (2002). The art and design of genetic screens: Drosophila melanogaster. Nat. Rev. Genet. 3, 176–188.
St Pierre, S. E., Galindo, M. I., Couso, J. P., and Thor, S. (2002). Control of Drosophila imaginal disc development by rotund and roughened eye: differentially expressed transcripts of the same gene encoding functionally distinct zinc finger proteins. Development 129, 1273–1281.
Surks, H. K., Richards, C. T., and Mendelsohn, M. E. (2003). Myosin phosphatase-Rho interacting protein. A new member of the myosin phosphatase complex that directly binds RhoA. J. Biol. Chem. 278, 51484–51493.
Suyama, R., Jenny, A., Curado, S., Pellis-Van Berkel, W., and Ephrussi, A. (2009). The actin-binding protein Lasp promotes Oskar accumulation at the posterior pole of the Drosophila embryo. Development 136, 95–105.
Szul, T., Burgess, J., Jeon, M., Zinn, K., Marques, G., Brill, J. A., and Sztul, E. (2011). The Garz Sec7 domain guanine nucleotide exchange factor for Arf regulates salivary gland development in Drosophila. Cell. Logist. 1, 69–76.
Tammer, I., Brandt, S., Hartig, R., Konig, W., and Backert, S. (2007). Activation of Abl by Helicobacter pylori: a novel kinase for CagA and crucial mediator of host cell scattering. Gastroenterology 132, 1309–1319.
Tan, S., Noto, J. M., Romero-Gallo, J., Peek, R. M. Jr., and Amieva, M. R. (2011). Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathog. 7:e1002050. doi: 10.1371/journal.ppat.1002050
Tan, S., Tompkins, L. S., and Amieva, M. R. (2009). Helicobacter pylori usurps cell polarity to turn the cell surface into a replicative niche. PLoS Pathog. 5:e1000407. doi: 10.1371/journal.ppat.1000407
Thomas, B. J., Gunning, D. A., Cho, J., and Zipursky, L. (1994). Cell cycle progression in the developing Drosophila eye: roughex encodes a novel protein required for the establishment of G1. Cell 77, 1003–1014.
Tio, M., and Moses, K. (1997). The Drosophila TGF alpha homolog Spitz acts in photoreceptor recruitment in the developing retina. Development 124, 343–351.
Tweedie, S., Ashburner, M., Falls, K., Leyland, P., Mcquilton, P., Marygold, S., Millburn, G., Osumi-Sutherland, D., Schroeder, A., Seal, R., and Zhang, H. (2009). FlyBase: enhancing Drosophila Gene Ontology annotations. Nucleic Acids Res. 37, D555–559.
Voas, M. G., and Rebay, I. (2004). Signal integration during development: insights from the Drosophila eye. Dev. Dyn. 229, 162–175.
Vogelmann, R., Amieva, M. R., Falkow, S., and Nelson, W. J. (2004). Breaking into the epithelial apical-junctional complex–news from pathogen hackers. Curr. Opin. Cell Biol. 16, 86–93.
Wroblewski, L. E., Shen, L., Ogden, S., Romero-Gallo, J., Lapierre, L. A., Israel, D. A., Turner, J. R., and Peek, R. M. Jr. (2009). Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology 136, 236–246.
Wu, A. H., Crabtree, J. E., Bernstein, L., Hawtin, P., Cockburn, M., Tseng, C. C., and Forman, D. (2003). Role of Helicobacter pylori CagA+ strains and risk of adenocarcinoma of the stomach and esophagus. Int. J. Cancer 103, 815–821.
Keywords: CagA, Helicobacter pylori, Drosophila, genetic modifier, epithelia, coracle, crumbs
Citation: Reid DW, Muyskens JB, Neal JT, Gaddini GW, Cho LY, Wandler AM, Botham CM and Guillemin K (2012) Identification of genetic modifiers of CagA-induced epithelial disruption in Drosophila. Front. Cell. Inf. Microbio. 2:24. doi: 10.3389/fcimb.2012.00024
Received: 16 December 2011; Accepted: 16 February 2012;
Published online: 13 March 2012.
Edited by:
D. Scott Merrell, Uniformed Services University, USAReviewed by:
Richard Peek, Vanderbilt University Medical Center, USASteffen Backert, Universtiy College Dublin, Ireland
Copyright: © 2012 Reid, Muyskens, Neal, Gaddini, Cho, Wandler, Botham and Guillemin. This is an open-access article distributed under the terms of the Creative Commons Attribution Non Commercial License, which permits non-commercial use, distribution, and reproduction in other forums, provided the original authors and source are credited.
*Correspondence: Karen Guillemin, Institute of Molecular Biology, University of Oregon, Eugene, OR 97403, USA. e-mail:Z3VpbGxlbWluQG1vbGJpby51b3JlZ29uLmVkdQ==
† These authors contributed equally to this work.
‡Current address: David W. Reid, Department of Biochemistry, Duke University School of Medicine, Durham, NC, USA
James T. Neal and Crystal M. Botham, Department of Medicine, Stanford University School of Medicine, Stanford, CA, USA
Gino W. Gaddini, College of Public Health and Human Sciences, Oregon State University, Corvallis, OR, USA
Lucy Y. Cho, Saint Louis University School of Medicine, St. Louis, Missouri, MO, USA.