 Yanis M. Macé
Yanis M. Macé Nicolas Bidère
Nicolas Bidère Tiphaine Douanne
Tiphaine Douanne
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Death, 09 April 2025
Sec. Cellular Stress and Survival: Crosslinks, Intersections, and Pathways
Volume 4 - 2025 | https://doi.org/10.3389/fceld.2025.1583221
This article is part of the Research TopicPosttranslational Modifications in Life and Death DecisionsView all articles
The deubiquitinating enzyme CYLD hydrolyzes Lys63-and Met1-linked ubiquitin chains, playing a crucial role in regulating various cellular processes such as immune cell development, innate and adaptive immunity, spermatogenesis, ciliogenesis, and cell survival. CYLD also functions as a tumor suppressor and is mutated in familial cylindromatosis. This pleiotropic function implies tight regulatory mechanisms. In this review, we summarize the current knowledge on CYLD’s molecular characteristics, subcellular location, and binding partners, with a focus on its involvement in life-and-death decisions. In addition, we discuss how post-translational modifications, including phosphorylation, ubiquitination, and proteolysis, shape CYLD’s function, unveiling the potential for therapeutic intervention. Finally, we highlight the remaining challenges that need to be overcome to deepen our understanding of this crucial enzyme.
Ubiquitination is a pivotal post-translational modification (PTM) regulating numerous cellular processes in eukaryotes. It consists of the covalent attachment of ubiquitin to substrate proteins, either as a single moiety or in linkage-specific polyubiquitin chains, modulating protein stability, activity, or localization to drive diverse cellular outcomes (Komander and Rape, 2012). Ubiquitination is dynamically reversible through the action of deubiquitinating enzymes (DUBs), a specialized group of proteases that selectively trim ubiquitin from target proteins. CYLD is a member of the DUB family known as ubiquitin-specific proteases (USP). Initially identified as a gene mutated in familial cylindromatosis, an autosomal dominant condition that predisposes individuals to skin appendage tumors (Bignell et al., 2000), CYLD has since been implicated in numerous human diseases, including infectious conditions, neurodegenerative disorders, and various cancers (Marín-Rubio et al., 2023). In most cases, CYLD truncations and mutations found in patients negatively affect its expression. Genetic mouse models have demonstrated the wide functions of CYLD in diverse biological processes such as immune cell development and function, innate immunity, spermatogenesis, hepatocellular homeostasis, osteoclastogenesis, and ciliogenesis (Lork et al., 2017).
CYLD is best known for its role as a negative regulator of NF-κB signaling, where it deubiquitinates key components such as NEMO, TRAF2, TRAF6, TAK1, RIPK1, and Bcl3 (Trompouki et al., 2003; Brummelkamp et al., 2003; Kovalenko et al., 2003; Massoumi et al., 2006; Reiley et al., 2007; Wright et al., 2007). By curbing NF-κB-mediated gene expression following stimulation of cytokine receptors, antigen receptors, and pattern recognition receptors, CYLD prevents excessive inflammation and maintains immune homeostasis. Beyond NF-κB, CYLD also regulates other pathways like MAPK (Liang et al., 2011), Wnt/βCat (Tauriello et al., 2010; Van Andel et al., 2017), TGF-β (Zhao et al., 2011; Tang et al., 2019), and JNK signaling (Reiley et al., 2004; Staal et al., 2011), underscoring its broad influence on cellular physiology. In addition, CYLD is involved in cell cycle progression (Stegmeier et al., 2007), p53 DNA damage response (Fernández-Majada et al., 2016), autophagy (Qi et al., 2020; Colombo et al., 2021; Zajicek et al., 2022), centriolar satellite homeostasis (Douanne et al., 2019; Renaud et al., 2023), and primary cilia formation (Douanne et al., 2019; Eguether et al., 2014; Yang et al., 2014).
How CYLD acts on such various processes continues to be investigated. One key factor underlying the DUB pleiotropy is its tight regulation through many PTMs. In this review, we focus on the role of CYLD in programmed cell death, examine its regulation by PTMs, and explore emerging questions in this field.
CYLD is a member of the DUB family known as USPs. Together with at least 56 identified members, the USP family represents the largest DUB class and plays critical roles in various cellular processes (Clague et al., 2019). Structurally, CYLD contains three N-terminal cytoskeleton-associated protein-glycine-rich (CAP-Gly) domains and a C-terminal USP catalytic domain (Figure 1A). The CYLD USP domain exhibits distinct structural features compared to other family members, characterized by several insertions and deletions. Notably, CYLD lacks a Fingers subdomain, enabling it to hydrolyze internal linkages within Lys63 and Met1-linked (also known as linear) polyubiquitin chains. This structural adaptation provides CYLD with a kinetic advantage for these chain types, unlike other USPs that exclusively cleave chains from the distal end. Additionally, differences in the active site, particularly the extended β12/β13 loop, contribute to CYLD’s affinity for Lys63 while other differences account for the lack of activity towards Lys48-linked chains. These unique features distinguish CYLD from other USPs, which typically exhibit broader linkage promiscuity (Komander et al., 2008; Komander et al., 2009).
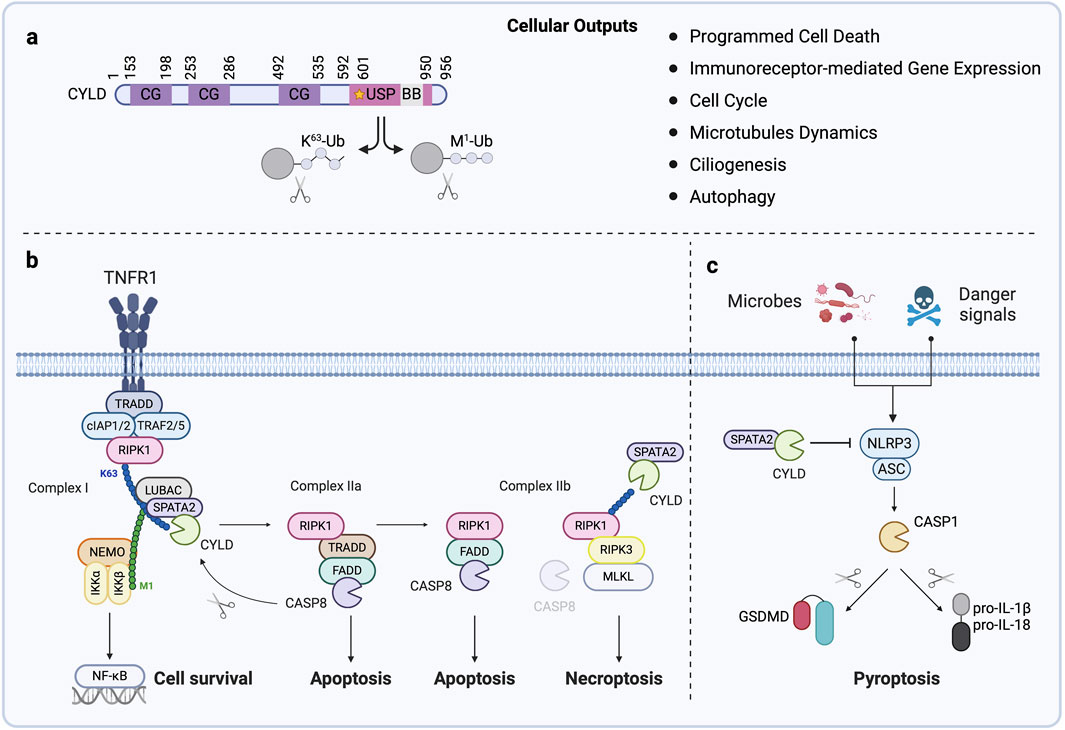
Figure 1. CYLD, a key Regulator of Programmed Cell Death (A) Domain structure of CYLD. CYLD contains three cytoskeleton-associated protein glycine-rich (CAP-Gly, CG) domains that mediate microtubule binding and the ubiquitin-specific protease domain (USP) harboring its catalytic activity and active cysteine (C601, illustrated by a star). The B box (BB) is embedded within the USP domain. CYLD hydrolyzes Lys63-and Met1-linked ubiquitin chains on substrates, leading to varied cellular outputs. (B) The engagement of TNF receptor 1 (TNFR1) triggers the formation of signaling complex I (RIPK1, cIAP1/2, TRADD, and TRAF2/5) at the cell membrane. This leads to the recruitment of the LUBAC together with CYLD and SPATA2, driving the ubiquitin-dependent activation of NF-κB and expression of anti-apoptotic genes. Deubiquitination of RIPK1 by CYLD inhibits NF-κB signaling and leads to the formation of complex IIa (RIPK1, TRADD, caspase-8 (CASP8), and FADD) in the cytosol. Activation of CASP8 within this complex results in apoptosis and also the CASP8-mediated cleavage of CYLD, restoring NF-κB signaling and establishing a finely tuned balance between cell survival and apoptosis. When NF-κB independent checkpoint is disrupted, RIPK1 engages another signaling complex called complex IIb, together with FADD and CASP8, to initiate apoptosis. Blockade of CASP8 drives binding of RIPK1 with RIPK3 and cell death by necroptosis. CYLD-mediated deubiquitination of RIPK1 in the necrosome facilitates necroptosis. (C) Various microbial motifs, endogenous and exogenous danger signals trigger the formation of the NLRP3 inflammasome, which serves as a cytosolic platform for the activation of CASP1. Active CASP1 matures the inflammatory cytokines IL-1β and IL-18 and the pore-forming protein Gasdermin D (GSDMD) into biologically active forms. Plasma membrane-assembled GSDMD pores allow the release of IL-1β and IL-18 and cause cell rupture and release of damage-associated molecular patterns leading to pyroptosis. CYLD restricts NLRP3 activity, limiting pyroptosis.
Early biochemical and bioinformatic analysis revealed the presence of a small Zn-binding module inserted within the CYLD catalytic domain bearing similarities to B box domains and RING fingers of E3 ligases (Komander et al., 2008). The presence of this B box in CYLD’s USP domain represents another singularity. In vitro deubiquitination assay using recombinant CYLD lacking this region demonstrated that this structure is dispensable for the catalytic activity and specificity of the enzyme. Furthermore, the isolated B box neither exhibited E3 ligase activity nor interacted with ubiquitin or ubiquitin chains, excluding it as a ubiquitin-binding domain (UBD) (Komander et al., 2008). Subsequent work identified a role for the B box in CYLD dimerization and its interaction with the canonical partner spermatogenesis-associated protein 2 (SPATA2) (Elliott et al., 2016). Interestingly, in HEK293 T cells, CYLD is predominantly localized in the cytosol, while mutants lacking the B box partially translocate to the nucleus. This suggests the B box is involved in retaining CYLD within the cytosol (Komander et al., 2008; Xie et al., 2017).
Upstream of its C-terminal catalytic domain, CYLD contains three CAP-Gly domains (Komander and Rape, 2012; Bignell et al., 2000). These approximately 70-amino acid SRC homology 3 (SH3)-fold domains mediate interactions with microtubules and microtubule-associated proteins (Yan et al., 2015). The first and second CAP-Gly domains bind directly to microtubules, with CAP-Gly1 displaying the highest binding affinity (Gao et al., 2008; Wickström et al., 2010), while CAP-Gly3 interacts with the non-catalytic subunit of the IKK complex NEMO (Saito et al., 2004). Pre-treatment of in vitro-assembled microtubules with subtilisin (also known as PCSK9), a serine protease that removes the C-terminal tails of ⍺-and β-tubulin, blocks the interaction of purified CYLD with microtubules, suggesting that CYLD binds to the C-terminal tails of tubulin (Yang et al., 2015). Of note, the three CYLD CAP-Gly domains possess divergent canonical GKNDG motifs: GFTDG, GNWDG, and GCTDG, respectively, suggesting relatively low microtubule binding affinity. This could explain why CYLD is predominantly localized to the cytoplasm, with only a small portion colocalizing with microtubules. Notably, both CAP-Gly2 and CAP-Gly3 function as UBDs, and CAP-Gly3 is essential for CYLD full catalytic activity in vitro and CYLD-mediated NOD2 signaling in cells (Elliott et al., 2021). Interestingly, the ubiquitin-binding properties observed in CYLD CAP-Gly domains do not seem to extend to other CAP-Gly domains. This difference may be attributed to several unique structural features within CYLD’s CAP-Gly domains compared to canonical CAP-Gly domains.
Historically, CYLD has been identified as a negative regulator of signal transduction leading to NF-κB (Trompouki et al., 2003; Brummelkamp et al., 2003; Kovalenko et al., 2003; Regamey et al., 2003) and JNK (Reiley et al., 2004; Nikolaou et al., 2012; Pannem et al., 2014) activation in response to TNF, thereby modulating the balance between cell survival and programmed cell death by apoptosis (Huyghe et al., 2023). Specifically, TNF binding to its receptor 1 (TNFR1) assembles a complex named TNF-RSC (TNF-receptor signaling complex, also called complex I), which comprises TRADD, RIPK1, TRAF2/5, and cIAP1/2 (Figure 1B). Within this complex, Lys63-ubiquitination of RIPK1 facilitates the recruitment of the linear ubiquitin chain assembly complex (LUBAC), a tripartite complex of the E3 ligases HOIP and HOIL-1, and the adaptor SHARPIN, which catalyzes Met1 ubiquitin chains, thereby enabling NF-κB activation (Tokunaga et al., 2009; Haas et al., 2009; Tokunaga et al., 2011; Gerlach et al., 2011). Notably, a subset of CYLD is bound to the LUBAC and consequently recruited to the TNFR1, where it counteracts the ubiquitination of several TNF-RSC components (Takiuchi et al., 2014; Draber et al., 2015). At a later point after following TNFR1 engagement, a secondary complex, termed complex IIa, is formed containing TRADD, FADD, cFLIP, and cysteine aspartyl protease caspase-8 (CASP8). When NF-κB is active, the transient formation of this complex is inhibited. However, if NF-κB is repressed, the inactive CASP8 homolog cFLIP levels are downregulated, driving CASP8 activation and resulting in apoptosis (Figure 1B) (Draber et al., 2015; Wang et al., 2008; Ting and Bertrand, 2016). If the early NF-κB independent checkpoint is disrupted, RIPK1 engages another signaling complex called complex IIb, together with CASP8, to initiate apoptosis (Ting and Bertrand, 2016). Inhibition or absence of CASP8 can cause RIPK1 to associate with RIPK3, activating the pore-forming protein MLKL, and ultimately triggering necroptosis, an inflammatory form of cell death (Figure 1B). In both forms of complex IIb-mediated death, RIPK1 kinase activity seems required. The balance between survival and different death modalities is tightly controlled by DUBs, which dismantle the ubiquitin network associated with these signaling complexes. By deubiquitinating RIPK1, CYLD promotes the formation of complex IIa favoring apoptosis (Figure 1B) (Draber et al., 2015; Wang et al., 2008). Moreover, CYLD’s deubiquitinating activity toward RIPK1 induces TNF-mediated necroptosis, while its depletion or cleavage limits cell death (O’Donnell et al., 2011; Hitomi et al., 2008; Vanlangenakker et al., 2011; Legarda et al., 2016; Ganjam et al., 2018; Moquin et al., 2013). Additionally, CYLD downregulation or deficiency has been linked to increased cell survival in gefitinib-treated non-small cell lung cancer (Yuan et al., 2020), enhanced proliferation in skin or cervical cancer (Massoumi et al., 2006; Huang et al., 2023), and attenuated germ cell apoptosis (Wright et al., 2007), as well as TNF-mediated apoptosis in a panel of cancer cell lines (Wang et al., 2008).
Recently, CYLD has been shown to limit pyroptosis, a lytic inflammatory form of cell death triggered by various infectious and sterile insults (Yu et al., 2021). For instance, CYLD deficiency accelerates LPS-induced pyroptosis in astrocytes (Li et al., 2019). Furthermore, CYLD restricts NLRP3 inflammasome activity by deubiquitinating PLK4 in macrophages (Yang et al., 2020) or WNK1 in an osteoporosis mouse model (Jiang et al., 2024) (Figure 1C). In estrogen receptor-positive and triple-negative breast cancer cell lines, hypoxia drives the RNF213-mediated ubiquitination and subsequent degradation of CYLD, leading to NF-κB-induced gene expression of NLRP3, which, in conjunction with hypoxia-induced endoplasmic reticulum stress, triggers pyroptosis (Bhardwaj et al., 2025). Finally, recent work proposed that CYLD could enhance the sensitivity of prostate cancer cells to ferroptosis via YAP deubiquitination, thereby inducing ferroptosis-related gene expression (Gu et al., 2024).
Overall, CYLD balances life and death decisions through its deubiquitinating activity. CYLD’s dual role in either promoting or restricting distinct types of cell death seems to also rely on its interactions, modifications, localization, and input signal.
In 2016, four independent groups used mass spectrometry to characterize either the TNF-RSC or CYLD interactome and concurrently identified SPATA2 as a partner for CYLD (Elliott et al., 2016; Schlicher et al., 2016; Wagner et al., 2016; Kupka et al., 2016). SPATA2 encompasses an N-terminal PUB domain that binds a non-PIM domain in the CYLD C-terminal USP domain. SPATA2 also connects CYLD to the LUBAC via a PIM (in SPATA2)-PUB (in HOIP) interaction (Elliott et al., 2016). This interaction facilitates the recruitment of CYLD to the TNF-RSC and NOD2-RSC and likely to other immune signaling complexes (Elliott et al., 2016; Draber et al., 2015; Kupka et al., 2016; Hrdinka et al., 2016). In vitro, SPATA2 binding enhances the enzyme activity of CYLD toward Met1-and Lys63-linked ubiquitin substrates, and the loss of SPATA2 results in an increased ubiquitination of CYLD substrates (Elliott et al., 2021; Schlicher et al., 2016; Griewahn et al., 2023). Besides bridging CYLD to the LUBAC, SPATA2 competes with the Met1 DUB OTULIN for binding to HOIP, thereby balancing the activity of the LUBAC (Griewahn et al., 2023). The combined absence of SPATA2 and CYLD causes the overactivation of the LUBAC and uncontrolled inflammation and results in penetrant perinatal lethality (Griewahn et al., 2023). Of note, Cyld−/−Spata2−/− MEFs exhibit reduced TNF-mediated caspase activation and apoptosis (Griewahn et al., 2023).
Whether SPATA2 contributes to all known functions controlled by CYLD is not fully understood. Interestingly, SPATA2 was initially identified through screening a human testis cDNA library expressed in the cytosol of Sertoli cells (Graziotto et al., 1999), echoing CYLD’s role during spermatogenesis (Wright et al., 2007). Furthermore, similar to CYLD defect, cells silenced or knocked out for SPATA2 resist RIPK1-dependent apoptosis and necroptosis (Hitomi et al., 2008; Schlicher et al., 2016; Wagner et al., 2016; Kupka et al., 2016; Wei et al., 2017). SPATA2 deficient cells display increased Met1-linked ubiquitination of RIPK1, causing inhibition of RIPK1 kinase activity (Wei et al., 2017). Consequently, SPATA2 deficiency sensitizes mice to systemic inflammatory response syndrome (SIRS) induced by TNF (Wei et al., 2017). The impact of SPATA2 on gene induction is debated, with some reports indicating a role (Schlicher et al., 2016; Wagner et al., 2016) and others not (Elliott et al., 2016; Kupka et al., 2016) in NF-κB signaling. Additionally, SPATA2 was shown to recruit CYLD to the centrosome, thereby limiting NLRP3-dependent inflammation and cell death (Yang et al., 2020).
Several groups have reported the phosphorylation of CYLD on multiple Ser/Thr residues within a “phospho-patch” between the CAP-Gly2 and CAP-Gly3 between amino acids 418 and 444 and have highlighted a role for the phospho-acceptor Serine 418. For example, phosphorylation at S418 has been observed in response to various stimuli, including mitogens, antigen receptor ligation, TNF, LPS, stimulation of the C-type lectin receptor DC-SIGN, MDP, and constitutively in some aggressive B-cell lymphoma and Adult T-cell leukemia/lymphoma (T-ALL) samples (Elliott et al., 2021; Reiley et al., 2005; Lork et al., 2018; Xu et al., 2021; Gringhuis et al., 2014; Xu et al., 2020; Ang et al., 2021). However, caution is advised when interpreting these results, as Lork et al. found that the commercially used phospho-CYLD antibody also recognizes an unknown protein of the size of CYLD (Lork et al., 2018). Recently, a proteomic analysis by mass spectrometry of TNF-stimulated U2OS cells identified six serine phosphorylation sites, out of which five are within the “phospho-patch” (S392, S418, S422, S439, S444) and one in a linker region between the CAP-Gly3 and the USP domain (S568) (Elliott et al., 2021).
The phosphorylation of CYLD is mediated by both the canonical IKKβ and noncanonical IKK TBK1/IKKε, depending on the nature of the input signal. For instance, IKKβ was associated with CYLD phosphorylation following stimulation with TNF and MDP and in T-ALL (Elliott et al., 2021; Reiley et al., 2005). TBK1 and IKKε phosphorylate CYLD in T lymphocytes upon antigen receptor ligation (Lork et al., 2018), in dendritic cells stimulated with the C-type lectin receptor DC-SIGN (Gringhuis et al., 2014), and in T-ALL (Xu et al., 2020). Moreover, TBK1 and IKKε bind and phosphorylate CYLD at S418 in vitro and in cellulo (Lork et al., 2018; Friedman et al., 2008; Hutti et al., 2009). Of note, CYLD was proposed to deubiquitinate TBK1 and IKKε (Friedman et al., 2008).
The impact of phosphorylation on CYLD’s DUB activity remains disputed, with reports of both downregulation and upregulation. For instance, chemical inhibition of IKKβ modulates TRAF2 ubiquitination, JNK activation, and cell death in response to TNF stimulation (Reiley et al., 2005; Ang et al., 2021). In T-ALL, CYLD phosphorylation by IKKβ and TBK1/IKKε results in its inactivation, promoting cell survival (Xu et al., 2020). A similar mechanism was proposed to explain IKKε-mediated cellular transformation (Hutti et al., 2009). Moreover, IKKε-mediated phosphorylation of CYLD at S418 also limits its DUB activity in dendritic cells after stimulation of the C-type lectin receptor DC-SIGN (Gringhuis et al., 2014). By contrast, one study shows that phosphorylation of CYLD at S418 by IKKβ increases its DUB activity (Thein et al., 2014a), while another reports that it is dispensable (Elliott et al., 2021). Interestingly, Elliott et al. discovered that IKKβ can phosphorylate a truncated (436–956) mutant of CYLD lacking S418 in vitro, increasing the Lys63 DUB activity of CYLD (Elliott et al., 2021). Conversely, a S568A mutant exhibited reduced Lys63 DUB activity. Nonetheless, the phosphorylation of both S418 and S568 is required for optimal signaling, and their combined mutation caused the accumulation of ubiquitinated species of RIPK2 in cells stimulated with NOD2. As a consequence, the production of cytokines was increased (Elliott et al., 2021) (Table 1).
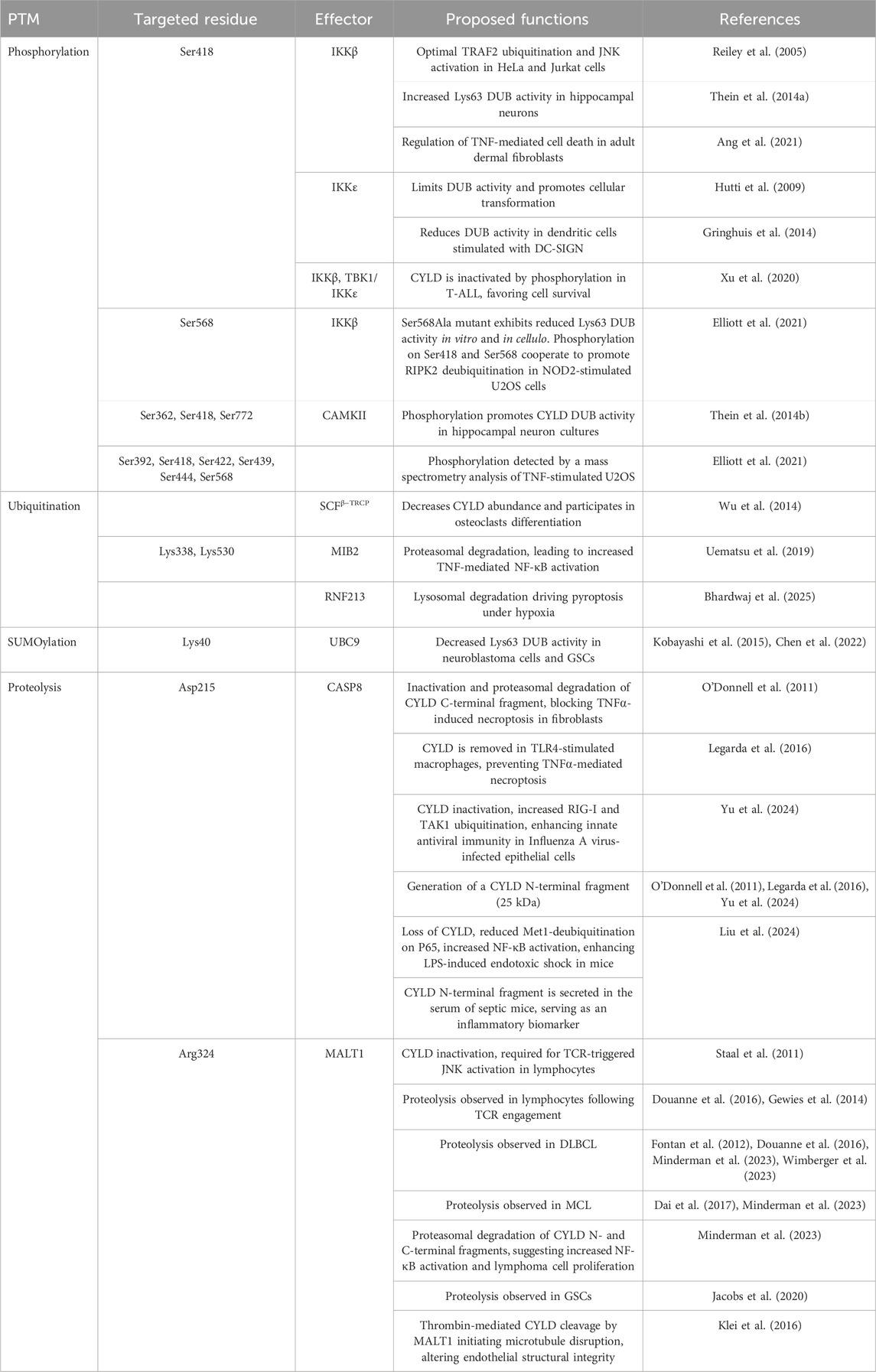
Table 1. CYLD regulations by PTMs shape Cell Fate.
Though specialized in removing ubiquitin chains, CYLD is itself subject to ubiquitination. While examining the interaction between CYLD and a panel of E3 ligases in an overexpression system in HEK293T, CYLD was found to interact with the beta-transducin repeat-containing protein Skp1-Cullin-F-box protein (SCFβ−TRCP). Further analysis in this system revealed that SCFβ−TRCP ubiquitinates CYLD to modulate its abundance, though the precise ubiquitination sites are not identified. Depletion of endogenous IKKα or IKKβ by shRNAs led to an accumulation of CYLD, while SCFβ−TRCP-mediated CYLD ubiquitination was lost on S432A/S436A mutants, suggesting that phosphorylation is a prerequisite to ubiquitination (Wu et al., 2014). This regulatory mechanism was further explored in the context of bone differentiation, where SCFβ−TRCP was found to regulate osteoclast differentiation by modulating CYLD levels, thereby influencing NF-κB activation in response to receptor activator NF-κB ligand (RANKL) stimulation (Wu et al., 2014).
CYLD stability is also controlled by the mind bomb homologue 2 (MIB2) E3 ligase (Uematsu et al., 2019). Overexpression experiments in HEK293T identified that the ankyrin repeat in MIB2 interacts with the third CAP-Gly domain of CYLD. Immunoprecipitation combined with the use of ubiquitin linkage-specific antibodies revealed that MIB2 decorated CYLD with Lys48-linked chains, driving its proteasomal degradation. Mutagenesis identified the acceptor lysines as Lys338 and Lys530. MIB2-dependent CYLD degradation led to an increase in NF-κB activation in response to TNF stimulation. In keeping with this, MIB2-knockout mice exhibited suppressed inflammatory responses in a K/BxN serum transfer arthritis model (Uematsu et al., 2019). A recent study identified the E3 ligase RNF213 as responsible for ubiquitinating CYLD and SPATA2 in hypoxic tumors (Bhardwaj et al., 2025). The use of a recombinant Lys63Arg mutant, which resists Lys63 ubiquitination, confirmed the involvement of this linkage, though the precise modification sites were not determined. This ubiquitination promoted the lysosomal degradation of CYLD and SPATA2, leading to decreased levels of both proteins, initiating pyroptotic cell death (Bhardwaj et al., 2025).
In addition to ubiquitination, CYLD undergoes SUMOylation. Small ubiquitin-like modifiers (SUMOs) are members of the ubiquitin-like protein family that, similar to ubiquitin, can be covalently conjugated to substrate proteins, altering their fate. In neuroblastoma cells, retinoic acid treatment induces CYLD SUMOylation (Kobayashi et al., 2015). Analysis using a SUMO predictor program combined with mutagenesis revealed that CYLD is modified on Lys40 outside of the USP domain. In vitro DUB assays and immunoprecipitation experiments revealed that CYLD SUMOylation impairs its catalytic activity against both free ubiquitin and substrate proteins, including TRAF6 and TRAF2. Interestingly, overexpression of a non-SUMOylatable CYLD mutant impaired retinoic acid-induced NF-κB activation and differentiation in neuroblastoma cells, instead promoting cell death (Kobayashi et al., 2015). More recently, CYLD was shown to be SUMOylated by UBC9 at Lys40 in a glioblastoma stem-like cells (GSCs) model. Consistent with previous findings, this modification impaired CYLD activity, resulting in excessive Lys63-linked polyubiquitination of NF-κB signaling intermediaries (TRAF2, TRAF6, RIPK1, and NEMO). This hyperactivation of NF-κB promoted the proneural-to-mesenchymal transition (PMT), a critical phenotypic shift in GSCs (Chen et al., 2022) (Table 1).
Proteolysis, unlike other PTMs, is irreversible, leading to significant effects on activity, interaction, and localization of the targeted protein. In 2011, the Ting group first reported CYLD cleavage by CASP8 at aspartic residue 215 (between CAP-Gly1 and 2) following TNF stimulation in fibroblasts. This proteolysis inactivated CYLD and promoted degradation of its C-terminal fragment, consequently blocking cell death by necroptosis. Mutation of the CYLD cleavage site promotes RIPK1 deubiquitination and interaction with the necrosome complex, leading to cell death (O’Donnell et al., 2011). A few years later, the same group extended this knowledge to macrophages, showing that CASP8-mediated CYLD cleavage following TLR4 stimulation prevents TNF-mediated necroptosis (Legarda et al., 2016). However, knock-in mice expressing non-cleavable CYLD D215A are viable, suggesting that CYLD cleavage by CASP8 is dispensable for necroptosis (Newton et al., 2019). More recently, CYLD processing by CASP8 was observed upon Influenza A virus infection in epithelial cells, likely enhancing innate antiviral immunity (Yu et al., 2024). A preprint highlighted a new role for CYLD processing by CASP8, showing that mice expressing the CASP8-resistant CYLD mutant present resistance to LPS-induced endotoxic shock by inhibiting NF-κB pathway activation and transcription of NLRP3 inflammasome. Interestingly, this work also suggests that CYLD N-terminal fragment following CASP8 cleavage is secreted outside the cells and may serve as a biomarker (Liu et al., 2024).
CYLD is also processed at the evolutionary conserved site Arg324 (between CAP-Gly2 and CAP-Gly3) by the MALT1 paracaspase, a caspase-like protein that regulates the activation of immune cells (Jaworski and Thome, 2016; Thys et al., 2018). CYLD is cleaved by MALT1 in lymphocytes following antigen receptor activation, in a subset of aggressive B-cell diffuse large B-cell lymphoma (DLBCL), in mantle cell lymphoma (MCL) (Staal et al., 2011; Fontan et al., 2012; Douanne et al., 2016; Dai et al., 2017; Minderman et al., 2023; Wimberger et al., 2023), and in GSC patient-derived samples (Jacobs et al., 2020). However, the exact role of CYLD cleavage by MALT1 remains elusive. Jurkat T lymphocytes overexpressing a MALT1-resistant CYLD mutant (Arg324Ala) display reduced JNK signaling and interleukin-2 production upon T-cell receptor ligation (Staal et al., 2011). However, JNK is normally activated in cells from mice expressing catalytically dead MALT1 (Gewies et al., 2014). MALT1 proteolysis has also been proposed to favor the degradation of N-terminal and C-terminal fragments, inhibiting CYLD activity, and suggesting a role in lymphoma cell growth (Minderman et al., 2023). Besides this role in immune cells, thrombin-induced MALT1-dependent CYLD cleavage has been presented to initiate microtubule disruption, altering endothelial barrier integrity (Klei et al., 2016). Altogether, CYLD proteolysis appears to depend on cell type and input signal. Nevertheless, the fate of the cleavage fragments is still enigmatic in terms of localization, interaction, and activity (Table 1).
CYLD is a versatile DUB involved in numerous cellular processes. In this regard, CYLD is tightly regulated at multiple levels, including subcellular localization, interaction with various partners, and both translational and post-translational stages. However, several crucial questions remain unanswered. For instance, the complete landscape of CYLD’s PTMs has yet to be mapped. Specific cell death programs, in which the role of CYLD has not been assigned, are known to involve palmitoylation, oxidation, or caspase-independent proteolysis. Assessing the contribution of lysosomal proteases, calpains, or granzymes may yield interesting findings. Another important aspect to explore is the precise impact of PTMs on CYLD’s enzyme activity, proximitome, or location. In addition, understanding the potential interplay between PTMs and the cognate CYLD partner SPATA2 represents a compelling avenue for further exploration. Introducing an additional layer of complexity, PTMs may operate hierarchically, where one modification serves as a prerequisite for another. This seems to be the case for certain CYLD ubiquitination events that depend on prior phosphorylation. Investigating these cooperative interactions between PTMs will undoubtedly enhance our understanding of CYLD function and unveil novel layers of regulation.
YM: Writing – original draft, Writing – review and editing. NB: Funding acquisition, Writing – original draft, Writing – review and editing. TD: Writing – original draft, Writing – review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This work was funded by Fondation ARC contre le Cancer (NB), Institut National Du Cancer (INCa_18384), Ligue nationale contre le cancer (Equipe labellisée) et comités de Loire-Atlantique, Maine et Loire, Vendée, Ille-et-Vilaine, Mayenne, and Finistère (NB). YM received a fellowship from Ligue Nationale contre le Cancer. The team is part of the SIRIC ILIAD (INCA-DGOS-INSERM-ITMO Cancer_18011). TD received support from the Bettencourt Schueller Foundation as an awardee of the Young Researcher Prize.
We apologize to those whose important work could not be cited owing to space constraints.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ang, R. L., Chan, M., Legarda, D., Sundberg, J. P., Sun, S. C., Gillespie, V. L., et al. (2021). Immune dysregulation in SHARPIN-deficient mice is dependent on CYLD-mediated cell death. Proc. Natl. Acad. Sci. U. S. A. 118 (50), e2001602118. doi:10.1073/pnas.2001602118
Bhardwaj, A., Panepinto, M. C., Ueberheide, B., and Neel, B. G. (2025). A mechanism for hypoxia-induced inflammatory cell death in cancer. Nature 637 (8045), 470–477. doi:10.1038/s41586-024-08136-y
Bignell, G. R., Warren, W., Seal, S., Takahashi, M., Rapley, E., Barfoot, R., et al. (2000). Identification of the familial cylindromatosis tumour-suppressor gene. Nat. Genet. 25 (2), 160–165. doi:10.1038/76006
Brummelkamp, T. R., Nijman, S. M. B., Dirac, A. M. G., and Bernards, R. (2003). Loss of the cylindromatosis tumour suppressor inhibits apoptosis by activating NF-kappaB. Nature 424 (6950), 797–801. doi:10.1038/nature01811
Chen, Z., Wang, S., Li, H. L., Luo, H., Wu, X., Lu, J., et al. (2022). FOSL1 promotes proneural-to-mesenchymal transition of glioblastoma stem cells via UBC9/CYLD/NF-κB axis. Mol. Ther. 30 (7), 2568–2583. doi:10.1016/j.ymthe.2021.10.028
Clague, M. J., Urbé, S., and Komander, D. (2019). Breaking the chains: deubiquitinating enzyme specificity begets function. Nat. Rev. Mol. Cell. Biol. 20 (6), 338–352. doi:10.1038/s41580-019-0099-1
Colombo, E., Horta, G., Roesler, M. K., Ihbe, N., Chhabra, S., Radyushkin, K., et al. (2021). The K63 deubiquitinase CYLD modulates autism-like behaviors and hippocampal plasticity by regulating autophagy and mTOR signaling. Proc. Natl. Acad. Sci. 118 (47), e2110755118. doi:10.1073/pnas.2110755118
Dai, B., Grau, M., Aukema, S. M., Hoster, E., Vogt, N., Staiger, A. M., et al. (2017). B-cell receptor–driven MALT1 activity regulates MYC signaling in mantle cell lymphoma. Blood 129 (3), 333–346. doi:10.1182/blood-2016-05-718775
Douanne, T., André-Grégoire, G., Thys, A., Trillet, K., Gavard, J., and Bidère, N. (2019). CYLD regulates centriolar satellites proteostasis by counteracting the E3 ligase MIB1. Cell. Rep. 27 (6), 1657–1665.e4. doi:10.1016/j.celrep.2019.04.036
Douanne, T., Gavard, J., and Bidère, N. (2016). The paracaspase MALT1 cleaves the LUBAC subunit HOIL1 during antigen receptor signaling. J. Cell. Sci. 129 (9), 1775–1780. doi:10.1242/jcs.185025
Draber, P., Kupka, S., Reichert, M., Draberova, H., Lafont, E., de Miguel, D., et al. (2015). LUBAC-recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell. Rep. 13 (10), 2258–2272. doi:10.1016/j.celrep.2015.11.009
Eguether, T., Ermolaeva, M. A., Zhao, Y., Bonnet, M. C., Jain, A., Pasparakis, M., et al. (2014). The deubiquitinating enzyme CYLD controls apical docking of basal bodies in ciliated epithelial cells. Nat. Commun. 5 (1), 4585. doi:10.1038/ncomms5585
Elliott, P. R., Leske, D., Hrdinka, M., Bagola, K., Fiil, B. K., McLaughlin, S. H., et al. (2016). SPATA2 links CYLD to LUBAC, activates CYLD, and controls LUBAC signaling. Mol. Cell. 63 (6), 990–1005. doi:10.1016/j.molcel.2016.08.001
Elliott, P. R., Leske, D., Wagstaff, J., Schlicher, L., Berridge, G., Maslen, S., et al. (2021). Regulation of CYLD activity and specificity by phosphorylation and ubiquitin-binding CAP-Gly domains. Cell. Rep. 37 (1), 109777. doi:10.1016/j.celrep.2021.109777
Fernández-Majada, V., Welz, P. S., Ermolaeva, M. A., Schell, M., Adam, A., Dietlein, F., et al. (2016). The tumour suppressor CYLD regulates the p53 DNA damage response. Nat. Commun. 7 (1), 12508. doi:10.1038/ncomms12508
Fontan, L., Yang, C., Kabaleeswaran, V., Volpon, L., Osborne, M. J., Beltran, E., et al. (2012). MALT1 small molecule inhibitors specifically suppress ABC-DLBCL in vitro and in vivo. Cancer Cell. 22 (6), 812–824. doi:10.1016/j.ccr.2012.11.003
Friedman, C. S., O’Donnell, M. A., Legarda-Addison, D., Ng, A., Cárdenas, W. B., Yount, J. S., et al. (2008). The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 9 (9), 930–936. doi:10.1038/embor.2008.136
Ganjam, G. K., Terpolilli, N. A., Diemert, S., Eisenbach, I., Hoffmann, L., Reuther, C., et al. (2018). Cylindromatosis mediates neuronal cell death in vitro and in vivo. Cell. Death Differ. 25 (8), 1394–1407. doi:10.1038/s41418-017-0046-7
Gao, J., Huo, L., Sun, X., Liu, M., Li, D., Dong, J. T., et al. (2008). The tumor suppressor CYLD regulates microtubule dynamics and plays a role in cell migration. J. Biol. Chem. 283 (14), 8802–8809. doi:10.1074/jbc.M708470200
Gerlach, B., Cordier, S. M., Schmukle, A. C., Emmerich, C. H., Rieser, E., Haas, T. L., et al. (2011). Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471 (7340), 591–596. doi:10.1038/nature09816
Gewies, A., Gorka, O., Bergmann, H., Pechloff, K., Petermann, F., Jeltsch, K. M., et al. (2014). Uncoupling Malt1 threshold function from paracaspase activity results in destructive autoimmune inflammation. Cell. Rep. 9 (4), 1292–1305. doi:10.1016/j.celrep.2014.10.044
Graziotto, R., Foresta, C., Scannapieco, P., Zeilante, P., Russo, A., Negro, A., et al. (1999). cDNA cloning and characterization of PD1: a novel human testicular protein with different expressions in various testiculopathies. Exp. Cell. Res. 248 (2), 620–626. doi:10.1006/excr.1999.4449
Griewahn, L., Müller-Foxworth, M., Peintner, L., Wissler, M., Weiss, M., Brauns-Schubert, P., et al. (2023). SPATA2 restricts OTULIN-dependent LUBAC activity independently of CYLD. Cell. Rep. 42 (1), 111961. doi:10.1016/j.celrep.2022.111961
Gringhuis, S. I., Kaptein, T. M., Wevers, B. A., Mesman, A. W., and Geijtenbeek, T. B. H. (2014). Fucose-specific DC-SIGN signalling directs T helper cell type-2 responses via IKKε- and CYLD-dependent Bcl3 activation. Nat. Commun. 5, 3898. doi:10.1038/ncomms4898
Gu, Y., Wu, S., Fan, J., Meng, Z., Gao, G., Liu, T., et al. (2024). CYLD regulates cell ferroptosis through Hippo/YAP signaling in prostate cancer progression. Cell. Death Dis. 15 (1), 79. doi:10.1038/s41419-024-06464-5
Haas, T. L., Emmerich, C. H., Gerlach, B., Schmukle, A. C., Cordier, S. M., Rieser, E., et al. (2009). Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell. 36 (5), 831–844. doi:10.1016/j.molcel.2009.10.013
Hitomi, J., Christofferson, D. E., Ng, A., Yao, J., Degterev, A., Xavier, R. J., et al. (2008). Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 135 (7), 1311–1323. doi:10.1016/j.cell.2008.10.044
Hrdinka, M., Fiil, B. K., Zucca, M., Leske, D., Bagola, K., Yabal, M., et al. (2016). CYLD limits Lys63-and met1-linked ubiquitin at receptor complexes to regulate innate immune signaling. Cell. Rep. 14 (12), 2846–2858. doi:10.1016/j.celrep.2016.02.062
Huang, Y., Zhang, W., Yu, Z., Su, H., Zeng, B., Piao, J., et al. (2023). A tumor suppressive role of CYLD as a novel potential DUB of aurora B in cervical cancer. Clin. Med. Insights Oncol. 17, 11795549231180832. doi:10.1177/11795549231180832
Hutti, J. E., Shen, R. R., Abbott, D. W., Zhou, A. Y., Sprott, K. M., Asara, J. M., et al. (2009). Phosphorylation of the tumor suppressor CYLD by the breast cancer oncogene IKKepsilon promotes cell transformation. Mol. Cell. 34 (4), 461–472. doi:10.1016/j.molcel.2009.04.031
Huyghe, J., Priem, D., and Bertrand, M. J. M. (2023). Cell death checkpoints in the TNF pathway. Trends Immunol. 44 (8), 628–643. doi:10.1016/j.it.2023.05.007
Jacobs, K. A., André-Grégoire, G., Maghe, C., Thys, A., Li, Y., Harford-Wright, E., et al. (2020). Paracaspase MALT1 regulates glioma cell survival by controlling endo-lysosome homeostasis. EMBO J. 39 (1), e102030. doi:10.15252/embj.2019102030
Jaworski, M., and Thome, M. (2016). The paracaspase MALT1: biological function and potential for therapeutic inhibition. Cell. Mol. Life Sci. 73 (3), 459–473. doi:10.1007/s00018-015-2059-z
Jiang, G., Cai, Y., Cheng, D., Wang, H., Deng, G., and Xiang, D. (2024). CYLD alleviates NLRP3 inflammasome-mediated pyroptosis in osteoporosis by deubiquitinating WNK1. J. Orthop. Surg. 19 (1), 212. doi:10.1186/s13018-024-04675-2
Klei, L. R., Hu, D., Panek, R., Alfano, D. N., Bridwell, R. E., Bailey, K. M., et al. (2016). MALT1 protease activation triggers acute disruption of endothelial barrier integrity via CYLD cleavage. Cell. Rep. 17 (1), 221–232. doi:10.1016/j.celrep.2016.08.080
Kobayashi, T., Masoumi, K. C., and Massoumi, R. (2015). Deubiquitinating activity of CYLD is impaired by SUMOylation in neuroblastoma cells. Oncogene 34 (17), 2251–2260. doi:10.1038/onc.2014.159
Komander, D., Lord, C. J., Scheel, H., Swift, S., Hofmann, K., Ashworth, A., et al. (2008). The structure of the CYLD USP domain explains its specificity for lys63-linked polyubiquitin and reveals a B box module. Mol. Cell. 29 (4), 451–464. doi:10.1016/j.molcel.2007.12.018
Komander, D., and Rape, M. (2012). The ubiquitin code. Annu. Rev. Biochem. 81 (1), 203–229. doi:10.1146/annurev-biochem-060310-170328
Komander, D., Reyes-Turcu, F., Licchesi, J. D. F., Odenwaelder, P., Wilkinson, K. D., and Barford, D. (2009). Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 10 (5), 466–473. doi:10.1038/embor.2009.55
Kovalenko, A., Chable-Bessia, C., Cantarella, G., Israël, A., Wallach, D., and Courtois, G. (2003). The tumour suppressor CYLD negatively regulates NF-kappaB signalling by deubiquitination. Nature 424 (6950), 801–805. doi:10.1038/nature01802
Kupka, S., De Miguel, D., Draber, P., Martino, L., Surinova, S., Rittinger, K., et al. (2016). SPATA2-Mediated binding of CYLD to HOIP enables CYLD recruitment to signaling complexes. Cell. Rep. 16 (9), 2271–2280. doi:10.1016/j.celrep.2016.07.086
Legarda, D., Justus, S. J., Ang, R. L., Rikhi, N., Li, W., Moran, T. M., et al. (2016). CYLD proteolysis protects macrophages from TNF-mediated auto-necroptosis induced by LPS and licensed by type I IFN. Cell. Rep. 15 (11), 2449–2461. doi:10.1016/j.celrep.2016.05.032
Li, L., Shu, M. Q., and Chen, J. (2019). CYLD deficiency exacerbates lipopolysaccharide (LPS)-induced pyroptosis in astrocytes of mice with sepsis. Biochem. Biophys. Res. Commun. 514 (4), 1066–1073. doi:10.1016/j.bbrc.2019.05.033
Liang, G., Ahlqvist, K., Pannem, R., Posern, G., and Massoumi, R. (2011). “Serum response factor controls CYLD expression via MAPK signaling pathway,”. Editor S. Agarwal, 6. doi:10.1371/journal.pone.0019613PLoS ONE5e19613
Liu, J., Li, M., Xing, M., Liu, H., Wu, X., Wang, L., et al. (2024). Caspase-8-mediated CYLD cleavage boosts LPS-induced endotoxic shock. bioRxiv 2024. 04.03.587858. doi:10.1101/2024.04.03.587858
Lork, M., Kreike, M., Staal, J., and Beyaert, R. (2018). Importance of validating antibodies and small compound inhibitors using genetic knockout studies-T cell receptor-induced CYLD phosphorylation by ikkε/TBK1 as a case study. Front. Cell. Dev. Biol. 6, 40. doi:10.3389/fcell.2018.00040
Lork, M., Verhelst, K., and Beyaert, R. (2017). CYLD, A20 and OTULIN deubiquitinases in NF-κB signaling and cell death: so similar, yet so different. Cell. Death Differ. 24 (7), 1172–1183. doi:10.1038/cdd.2017.46
Marín-Rubio, J. L., Raote, I., Inns, J., Dobson-Stone, C., and Rajan, N. (2023). CYLD in health and disease. Dis. Model. Mech. 16 (6), dmm050093. doi:10.1242/dmm.050093
Massoumi, R., Chmielarska, K., Hennecke, K., Pfeifer, A., and Fässler, R. (2006). Cyld inhibits tumor cell proliferation by blocking Bcl-3-dependent NF-kappaB signaling. Cell. 125 (4), 665–677. doi:10.1016/j.cell.2006.03.041
Minderman, M., Lantermans, H. C., Grüneberg, L. J., Cillessen, SAGM, Bende, R. J., Van Noesel, C. J. M., et al. (2023). MALT1-dependent cleavage of CYLD promotes NF-κB signaling and growth of aggressive B-cell receptor-dependent lymphomas. Blood Cancer J. 13 (1), 37. doi:10.1038/s41408-023-00809-7
Moquin, D. M., McQuade, T., and Chan, F. K. M. (2013). CYLD deubiquitinates RIP1 in the TNFα-induced necrosome to facilitate kinase activation and programmed necrosis. PLoS ONE 8 (10), e76841. doi:10.1371/journal.pone.0076841
Newton, K., Wickliffe, K. E., Dugger, D. L., Maltzman, A., Roose-Girma, M., Dohse, M., et al. (2019). Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574 (7778), 428–431. doi:10.1038/s41586-019-1548-x
Nikolaou, K., Tsagaratou, A., Eftychi, C., Kollias, G., Mosialos, G., and Talianidis, I. (2012). Inactivation of the deubiquitinase CYLD in hepatocytes causes apoptosis, inflammation, fibrosis, and cancer. Cancer Cell. 21 (6), 738–750. doi:10.1016/j.ccr.2012.04.026
O’Donnell, M. A., Perez-Jimenez, E., Oberst, A., Ng, A., Massoumi, R., Xavier, R., et al. (2011). Caspase 8 inhibits programmed necrosis by processing CYLD. Nat. Cell. Biol. 13 (12), 1437–1442. doi:10.1038/ncb2362
Pannem, R. R., Dorn, C., Ahlqvist, K., Bosserhoff, A. K., Hellerbrand, C., and Massoumi, R. (2014). CYLD controls c-MYC expression through the JNK-dependent signaling pathway in hepatocellular carcinoma. Carcinogenesis 35 (2), 461–468. doi:10.1093/carcin/bgt335
Qi, L., Zang, H., Wu, W., Nagarkatti, P., Nagarkatti, M., Liu, Q., et al. (2020). CYLD exaggerates pressure overload-induced cardiomyopathy via suppressing autolysosome efflux in cardiomyocytes. J. Mol. Cell. Cardiol. 145, 59–73. doi:10.1016/j.yjmcc.2020.06.004
Regamey, A., Hohl, D., Liu, J. W., Roger, T., Kogerman, P., Toftgård, R., et al. (2003). The tumor suppressor CYLD interacts with TRIP and regulates negatively nuclear factor kappaB activation by tumor necrosis factor. J. Exp. Med. 198 (12), 1959–1964. doi:10.1084/jem.20031187
Reiley, W., Zhang, M., and Sun, S. C. (2004). Negative regulation of JNK signaling by the tumor suppressor CYLD. J. Biol. Chem. 279 (53), 55161–55167. doi:10.1074/jbc.M411049200
Reiley, W., Zhang, M., Wu, X., Granger, E., and Sun, S. C. (2005). Regulation of the deubiquitinating enzyme CYLD by IkappaB kinase gamma-dependent phosphorylation. Mol. Cell. Biol. 25 (10), 3886–3895. doi:10.1128/MCB.25.10.3886-3895.2005
Reiley, W. W., Jin, W., Lee, A. J., Wright, A., Wu, X., Tewalt, E. F., et al. (2007). Deubiquitinating enzyme CYLD negatively regulates the ubiquitin-dependent kinase Tak1 and prevents abnormal T cell responses. J. Exp. Med. 204 (6), 1475–1485. doi:10.1084/jem.20062694
Renaud, C. C. N., Trillet, K., Jardine, J., Merlet, L., Renoult, O., Laurent—Blond, M., et al. (2023). The centrosomal protein 131 participates in the regulation of mitochondrial apoptosis. Commun. Biol. 6 (1), 1271. doi:10.1038/s42003-023-05676-3
Saito, K., Kigawa, T., Koshiba, S., Sato, K., Matsuo, Y., Sakamoto, A., et al. (2004). The CAP-Gly domain of CYLD associates with the proline-rich sequence in NEMO/IKKgamma. Structure 12 (9), 1719–1728. doi:10.1016/j.str.2004.07.012
Schlicher, L., Wissler, M., Preiss, F., Brauns-Schubert, P., Jakob, C., Dumit, V., et al. (2016). SPATA2 promotes CYLD activity and regulates TNF-induced NF-κB signaling and cell death. EMBO Rep. 17 (10), 1485–1497. doi:10.15252/embr.201642592
Staal, J., Driege, Y., Bekaert, T., Demeyer, A., Muyllaert, D., Van Damme, P., et al. (2011). T-cell receptor-induced JNK activation requires proteolytic inactivation of CYLD by MALT1. EMBO J. 30 (9), 1742–1752. doi:10.1038/emboj.2011.85
Stegmeier, F., Sowa, M. E., Nalepa, G., Gygi, S. P., Harper, J. W., and Elledge, S. J. (2007). The tumor suppressor CYLD regulates entry into mitosis. Proc. Natl. Acad. Sci. 104 (21), 8869–8874. doi:10.1073/pnas.0703268104
Takiuchi, T., Nakagawa, T., Tamiya, H., Fujita, H., Sasaki, Y., Saeki, Y., et al. (2014). Suppression of LUBAC -mediated linear ubiquitination by a specific interaction between LUBAC and the deubiquitinases CYLD and OTULIN. Genes. cells. 19 (3), 254–272. doi:10.1111/gtc.12128
Tang, Y., Reissig, S., Glasmacher, E., Regen, T., Wanke, F., Nikolaev, A., et al. (2019). Alternative splice forms of CYLD mediate ubiquitination of SMAD7 to prevent TGFB signaling and promote colitis. Gastroenterology 156 (3), 692–707. doi:10.1053/j.gastro.2018.10.023
Tauriello, D. V. F., Haegebarth, A., Kuper, I., Edelmann, M. J., Henraat, M., Canninga-van Dijk, M. R., et al. (2010). Loss of the tumor suppressor CYLD enhances Wnt/beta-catenin signaling through K63-linked ubiquitination of Dvl. Mol. Cell. 37 (5), 607–619. doi:10.1016/j.molcel.2010.01.035
Thein, S., Pham, A., Bayer, K. U., Tao-Cheng, J. H., and Dosemeci, A. (2014a). IKK regulates the deubiquitinase CYLD at the postsynaptic density. Biochem. Biophys. Res. Commun. 450 (1), 550–554. doi:10.1016/j.bbrc.2014.06.019
Thein, S., Tao-Cheng, J. H., Li, Y., Bayer, K. U., Reese, T. S., and Dosemeci, A. (2014b). CaMKII mediates recruitment and activation of the deubiquitinase CYLD at the postsynaptic density. PloS One 9 (3), e91312. doi:10.1371/journal.pone.0091312
Thys, A., Douanne, T., and Bidère, N. (2018). Post-translational modifications of the CARMA1-BCL10-MALT1 complex in lymphocytes and activated B-cell like subtype of diffuse large B-cell lymphoma. Front. Oncol. 8, 498. doi:10.3389/fonc.2018.00498
Ting, A. T., and Bertrand, M. J. M. (2016). More to life than NF-κB in TNFR1 signaling. Trends Immunol. 37 (8), 535–545. doi:10.1016/j.it.2016.06.002
Tokunaga, F., Nakagawa, T., Nakahara, M., Saeki, Y., Taniguchi, M., Sakata, S. ichi, et al. (2011). SHARPIN is a component of the NF-κB-activating linear ubiquitin chain assembly complex. Nature 471 (7340), 633–636. doi:10.1038/nature09815
Tokunaga, F., Sakata, S. ichi, Saeki, Y., Satomi, Y., Kirisako, T., Kamei, K., et al. (2009). Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat. Cell. Biol. 11 (2), 123–132. doi:10.1038/ncb1821
Trompouki, E., Hatzivassiliou, E., Tsichritzis, T., Farmer, H., Ashworth, A., and Mosialos, G. (2003). CYLD is a deubiquitinating enzyme that negatively regulates NF-kappaB activation by TNFR family members. Nature 424 (6950), 793–796. doi:10.1038/nature01803
Uematsu, A., Kido, K., Takahashi, H., Takahashi, C., Yanagihara, Y., Saeki, N., et al. (2019). The E3 ubiquitin ligase MIB2 enhances inflammation by degrading the deubiquitinating enzyme CYLD. J. Biol. Chem. 294 (38), 14135–14148. doi:10.1074/jbc.RA119.010119
Van Andel, H., Kocemba, K. A., De Haan-Kramer, A., Mellink, C. H., Piwowar, M., Broijl, A., et al. (2017). Loss of CYLD expression unleashes Wnt signaling in multiple myeloma and is associated with aggressive disease. Oncogene 36 (15), 2105–2115. doi:10.1038/onc.2016.368
Vanlangenakker, N., Vanden Berghe, T., Bogaert, P., Laukens, B., Zobel, K., Deshayes, K., et al. (2011). cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell. Death Differ. 18 (4), 656–665. doi:10.1038/cdd.2010.138
Wagner, S. A., Satpathy, S., Beli, P., and Choudhary, C. (2016). SPATA2 links CYLD to the TNF-alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J. 35 (17), 1868–1884. doi:10.15252/embj.201694300
Wang, L., Du, F., and Wang, X. (2008). TNF-Α induces two distinct caspase-8 activation pathways. Cell. 133 (4), 693–703. doi:10.1016/j.cell.2008.03.036
Wei, R., Xu, L. W., Liu, J., Li, Y., Zhang, P., Shan, B., et al. (2017). SPATA2 regulates the activation of RIPK1 by modulating linear ubiquitination. Genes. Dev. 31 (11), 1162–1176. doi:10.1101/gad.299776.117
Wickström, S. A., Masoumi, K. C., Khochbin, S., Fässler, R., and Massoumi, R. (2010). CYLD negatively regulates cell-cycle progression by inactivating HDAC6 and increasing the levels of acetylated tubulin. EMBO J. 29 (1), 131–144. doi:10.1038/emboj.2009.317
Wimberger, N., Ober, F., Avar, G., Grau, M., Xu, W., Lenz, G., et al. (2023). Oncogene-induced MALT1 protease activity drives posttranscriptional gene expression in malignant lymphomas. Blood 142 (23), 1985–2001. doi:10.1182/blood.2023021299
Wright, A., Reiley, W. W., Chang, M., Jin, W., Lee, A. J., Zhang, M., et al. (2007). Regulation of early wave of germ cell apoptosis and spermatogenesis by deubiquitinating enzyme CYLD. Dev. Cell. 13 (5), 705–716. doi:10.1016/j.devcel.2007.09.007
Wu, X., Fukushima, H., North, B. J., Nagaoka, Y., Nagashima, K., Deng, F., et al. (2014). SCFβ-TRCP regulates osteoclastogenesis via promoting CYLD ubiquitination. Oncotarget 5 (12), 4211–4221. doi:10.18632/oncotarget.1971
Xie, S., Chen, M., Gao, S., Zhong, T., Zhou, P., Li, D., et al. (2017). The B-box module of CYLD is responsible for its intermolecular interaction and cytoplasmic localization. Oncotarget 8 (31), 50889–50895. doi:10.18632/oncotarget.15142
Xu, X., Kalac, M., Markson, M., Chan, M., Brody, J. D., Bhagat, G., et al. (2020). Reversal of CYLD phosphorylation as a novel therapeutic approach for adult T-cell leukemia/lymphoma (ATLL). Cell. Death Dis. 11 (2), 94. doi:10.1038/s41419-020-2294-6
Xu, X., Wei, T., Zhong, W., Ang, R., Lei, Y., Zhang, H., et al. (2021). Down-regulation of cylindromatosis protein phosphorylation by BTK inhibitor promotes apoptosis of non-GCB-diffuse large B-cell lymphoma. Cancer Cell. Int. 21 (1), 195. doi:10.1186/s12935-021-01891-2
Yan, S., Guo, C., Hou, G., Zhang, H., Lu, X., Williams, J. C., et al. (2015). Atomic-resolution structure of the CAP-Gly domain of dynactin on polymeric microtubules determined by magic angle spinning NMR spectroscopy. Proc. Natl. Acad. Sci. 112 (47), 14611–14616. doi:10.1073/pnas.1509852112
Yang, X., Li, W., Zhang, S., Wu, D., Jiang, X., Tan, R., et al. (2020). PLK 4 deubiquitination by Spata2-CYLD suppresses NEK7-mediated NLRP3 inflammasome activation at the centrosome. EMBO J. 39 (2), e102201. [Accessed 2022 Oct 18]. doi:10.15252/embj.2019102201
Yang, Y., Ran, J., Liu, M., Li, D., Li, Y., Shi, X., et al. (2014). CYLD mediates ciliogenesis in multiple organs by deubiquitinating Cep70 and inactivating HDAC6. Cell. Res. 24 (11), 1342–1353. doi:10.1038/cr.2014.136
Yang, Y., Ran, J., Sun, L., Sun, X., Luo, Y., Yan, B., et al. (2015). CYLD regulates noscapine activity in acute lymphoblastic leukemia via a microtubule-dependent mechanism. Theranostics 5 (7), 656–666. doi:10.7150/thno.10844
Yu, H., Sun, Y., Zhang, J., Zhang, W., Liu, W., Liu, P., et al. (2024). Influenza A virus infection activates caspase-8 to enhance innate antiviral immunity by cleaving CYLD and blocking TAK1 and RIG-I deubiquitination. Cell. Mol. Life Sci. 81 (1), 355. doi:10.1007/s00018-024-05392-z
Yu, P., Zhang, X., Liu, N., Tang, L., Peng, C., and Chen, X. (2021). Pyroptosis: mechanisms and diseases. Signal Transduct. Target Ther. 6 (1), 128. doi:10.1038/s41392-021-00507-5
Yuan, Y., Liu, L., Wang, Y., and Liu, S. (2020). Reduced expression of CYLD promotes cell survival and inflammation in gefitinib-treated NSCLC PC-9 cells: targeting CYLD may be beneficial for acquired resistance to gefitinib therapy. Cell. Biol. Int. 44 (9), 1911–1918. doi:10.1002/cbin.11397
Zajicek, A. S., Ruan, H., Dai, H., Skolfield, M. C., Phillips, H. L., Burnette, W. J., et al. (2022). Cylindromatosis drives synapse pruning and weakening by promoting macroautophagy through Akt-mTOR signaling. Mol. Psychiatry 27 (5), 2414–2424. doi:10.1038/s41380-022-01571-1
Zhao, Y., Thornton, A. M., Kinney, M. C., Ma, C. A., Spinner, J. J., Fuss, I. J., et al. (2011). The deubiquitinase CYLD targets Smad7 protein to regulate transforming growth factor β (TGF-β) signaling and the development of regulatory T cells. J. Biol. Chem. 286 (47), 40520–40530. doi:10.1074/jbc.M111.292961
Keywords: CYLD, DUB, cell death, post-translational modifications, phosphorylation, ubiquitin, proteolysis
Citation: Macé YM, Bidère N and Douanne T (2025) The good, the bad, and the modified: CYLD’s post-translational tale. Front. Cell Death 4:1583221. doi: 10.3389/fceld.2025.1583221
Received: 25 February 2025; Accepted: 27 March 2025;
Published: 09 April 2025.
Edited by:
Inna N. Lavrik, University Hospital Magdeburg, GermanyReviewed by:
Ulrich Maurer, University of Freiburg, GermanyCopyright © 2025 Macé, Bidère and Douanne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tiphaine Douanne, dGlwaGFpbmUuZG91YW5uZUBpbnNlcm0uZnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.