 Ling Yang
Ling Yang Yan Zhang
Yan Zhang Zhuodong Chai
Zhuodong Chai Yuqi Zhou
Yuqi Zhou Zhenyu Li
Zhenyu Li Yinan Wei
Yinan Wei- Department of Pharmaceutical Sciences, Irma Lerma Rangel School of Pharmacy, Texas A&M University, College Station, TX, United States
Pyroptosis is a form of proinflammatory cell death characterized by inflammasome activation, pore formation, and the release of pro-inflammatory cytokines such as interleukin-1β (IL-1β) and IL-18 upon cell rupture. Nuclear factor-κB (NF-κB), a prototypical pro-inflammatory transcription factor, plays a critical role in immune system regulation. Recent research highlights the multifaceted roles of NF-κB signaling in pyroptosis. Various immunologically relevant ligands and their receptors can activate the NF-κB pathway to promote pyroptosis, with Toll-like receptors (TLRs), IL-1 receptors (IL-1Rs), and TNF receptors (TNFRs) being the most prominent. NF-κB regulates the transcription of key components of inflammasomes involved in pyroptosis, particularly the NLRP3 inflammasome. Recent studies also indicate that NF-κB modulates the activation of NLRC4 and AIM2 inflammasomes through distinct pathways in diverse inflammatory conditions, such as acute lung injury and neuroinflammation. Additionally, the NF-κB pathway mediates the production of inflammatory cytokines, including IL-1β, IL-33, and TNF-α, which further regulate pyroptosis. This review examines recent advances in understanding the role of the NF-κB signaling pathway in regulating pyroptosis during infection and inflammation.
1 Introduction
Programmed cell death (PCD) refers to strictly regulated cell death, including apoptosis, necroptosis, autophagy, pyroptosis, ferroptosis, and recently characterized cuproptosis (Bedoui et al., 2020; Tsvetkov et al., 2022). Pyroptosis is a pro-inflammatory PCD characterized by the formation of plasma membrane pores, initiated by the cleavage of Gasdermins (GSDMs) between their N- and C-terminal domains by activated Caspases (Shao, 2021). Initially, pyroptosis was reported as a result of Caspase-1 dependent cleavage of GSDMD, constituting the canonical pathway. Later, Caspase-4/-5/-11 activation by cytosolic LPS, and subsequent cleavage of GSDMD represents a non-canonical pathway (Wei Y. et al., 2022). Recent studies have uncovered additional pathways of pyroptosis, including the Caspases-8 mediated cleavage of GSDMD, Caspase-3-mediated cleavage of GSDME, as well as granzyme A-mediated cleavage of GSDMB (Shen et al., 2021; Zhang X. et al., 2020; Shi et al., 2015; Orning et al., 2018; Sarhan et al., 2018; Zhou et al., 2020; Kayagaki et al., 2015; Wang et al., 2017). The inflammatory caspases are normally activated through the assembly of inflammasome in response to stimulation such as the invaded bacteria and viruses (Fattinger et al., 2023; Zhao and Zhao, 2020; Miao et al., 2010; Rathinam and Fitzgerald, 2016; Pandeya et al., 2023). The assembly of inflammasome is initiated by the recognition of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) through the corresponding pattern recognition receptors (PRRs), also known as inflammasome sensors. This recognition leads to the recruitment of adaptor molecule ASC (apoptosis-associated speck-like protein containing a CARD) and pro-caspases (Rathinam and Fitzgerald, 2016). Release of inflammatory mediators is a crucial characteristic of pyroptosis.
The NF-κB transcription factors (p50/p105, p52/p100, RelA, c-Rel, RelB) and the pathways that control NF-κB activation are known to be involved in the regulation of immune responses and inflammation (Liu T. et al., 2017). Activation of NF-κB involves two major signaling pathways. The primary mechanism of canonical NF-κB activation is through the degradation of IκBα, which is triggered by its site-specific phosphorylation by a multi-subunit IκB kinase (IKK) complex. This complex becomes activated in response to injury, infection, inflammation and other stress conditions (Liu T. et al., 2017; Vallabhapurapu and Karin, 2009; Barnabei et al., 2021). The degradation of IκBα results in rapid and transient nuclear translocation of canonical NF-κB members, predominantly the p50/RelA and p50/c-Rel dimers (Liu T. et al., 2017; Hayden and Ghosh, 2008; Karin and Delhase, 2000). A second, non-canonical pathway, which is activated by a more limited set of stimuli, does not involve IκBα degradation. Instead, it relies on the processing of the NF-κB2 precursor protein p100 (Xiao et al., 2001a). A central signaling component of the non-canonical pathway is NF-κB-inducing kinase (NIK), which integrates signals and activates downstream IKKα. Activated IKKα triggers the processing of p100 into NF-κB2 p52, leading to the subsequent nuclear translocation of the noncanonical NF-κB complex p52/RelB (Sun, 2011).
Currently, much of our understanding of NF-κB signaling in pyroptosis has been acquired through the investigation of members of Toll-like receptors (TLRs), interleukin-1 receptors (IL-1Rs), and TNF receptors (TNFRs). These receptors play pivotal roles in sensing external stimuli and initiating downstream signaling cascades that ultimately regulate pyroptosis (Liu T. et al., 2017; Barnabei et al., 2021). Over the past decade, the DNA-sensing receptor cyclic GMP-AMP synthase (cGAS) and its downstream adaptor STING (stimulator of interferon genes) have been identified as central regulators of type I interferon (IFN) and NF-κB responses (Decout et al., 2021; Skopelja-Gardner et al., 2022; Domizio et al., 2022). cGAS detects cytosolic DNA from pathogens, DNA damage, or mitochondrial stress, activating STING to trigger immune responses. This pathway plays a pivotal role in cellular defense, mediating IFN production and NF-κB activation to combat infections and maintain homeostasis.
The NF-κB transcription factors have emerged as major regulators of PCD including apoptosis or necrosis (Dondelinger et al., 2015; Kucharczak et al., 2003; Karin and Lin, 2002). Recent evidence highlights the role of NF-κB signaling in regulating inflammasome activation, particularly the NLRP3, NLRC4, AIM2 (He Y. et al., 2016; Bezbradica et al., 2017; Wang J. et al., 2020; Chebly et al., 2022; Bombaci et al., 2022). Furthermore, NF-κB signaling exerts a bidirectional influence on inflammation mediated by inflammasomes (Kinoshita et al., 2015a). While inflammasome activation can trigger NF-κB activation and downstream proinflammatory responses, NF-κB signaling, in turn, regulates the assembly and activation of inflammasomes. Unsurprisingly, numerous studies have implicated NF-κB regulation as a key factor in the progression of diseases associated with pyroptosis (Wang et al., 2019; Chen X. et al., 2019; Cai et al., 2021; Xu et al., 2023; Chen et al., 2023). Therefore, a deeper understanding of the interplay between NF-κB activation and pyroptosis is crucial for developing therapeutic strategies for pyroptosis-related diseases.
2 Molecular mechanism of pyroptosis
Pyroptosis is a form of pro-inflammatory PCD, lacking characteristic features of apoptosis such as cell shrinkage, chromatin condensation, phospholipid externalization and exposure of phosphatidylserine on the outer surface of the cell, and Caspase-3/7 dependence. Cells undergoing pyroptosis appear flattened and swollen under the microscope, resembling deflated balloons. Other distinguishing features include cell membrane rupture and the release of large amounts of inflammatory cytokines and cellular contents. Inflammasome activation could lead to pyroptosis (Figure 1). The inflammasome is a protein complex that forms in response to different stimuli signals, including PAMPs from pathogens such as bacteria or viruses, and intrinsic signals DAMPs. Under certain conditions, the formation of the inflammasome requires assistance from NF-κB signaling, often referred to as “priming”. Priming triggers cells to adopt a heightened state of alertness, with increased expression of inflammasome-related proteins, preparing them for prompt response. Assembly of the inflammasomes leads to the activation of caspases, processing of pro-IL-1β and pro-IL-18 into their mature forms, and cleavage of gasdermins that lead to poration on the cell membrane (Yu P. et al., 2021; Martinon et al., 2002; Shi et al., 2017; Wei X. et al., 2022). Later studies revealed mechanisms of pyroptosis independent of inflammasomes. For example, Caspase-11, Caspase-3, Caspase-8, granzyme B, and granzyme A can directly cleave gasdermins, bypassing the inflammasome assembly (Zhou et al., 2020; Wang et al., 2017; Zhang Z. et al., 2020; Rathinam et al., 2019). Therefore, pyroptosis has been redefined as a type of the necrotic cell death mediated by gasdermin family proteins (Shi et al., 2017), including GSDMA, GSDMB, GSDMC, GSDMD, GSDME (DFNA5) and DFNB59. Recent findings reveal that GSDMD pore formation alone is insufficient to drive plasma membrane rupture (PMR), a crucial step in lytic cell death and the full release of DAMPs such as HMGB1 and the PMR marker lactate dehydrogenase (LDH). Emerging evidence identified the plasma membrane protein Ninjurin-1 (NINJ1) as a key regulator of PMR (Kayagaki et al., 2021; Degen et al., 2023; Han et al., 2024). Acting downstream of GSDMD activation, NINJ1 orchestrates the final steps of membrane disintegration during pyroptosis and other forms of lytic cell death. Structural and imaging studies have demonstrated that NINJ1 forms hydrophilic interfaces that encircle and detach membrane patches, promoting their fragmentation and ultimate loss (David et al., 2024). This mechanism is essential for complete PMR, enabling the release of LDH and other intracellular contents, thereby amplifying the inflammatory response.
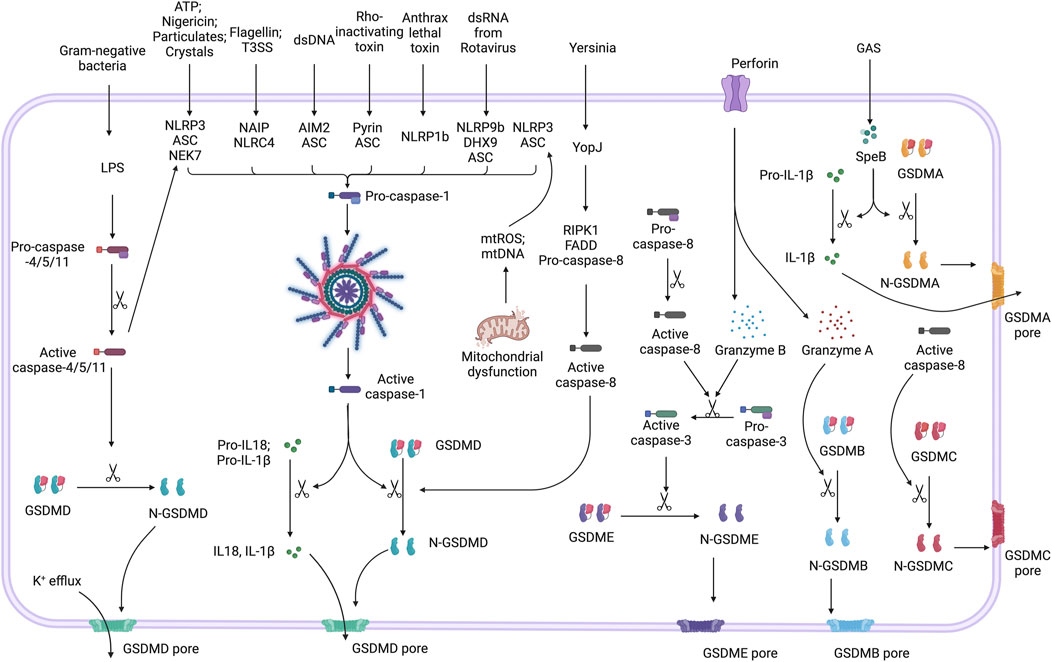
Figure 1. Mechanisms of pyroptosis pathways. Inflammasomes, such as NLRP3, NLRC4, AIM2, Pyrin, NLRP1b, and NLRP9b, are activated by stimuli including ATP, bacterial flagellin, dsDNA, Rho-inactivating toxins, anthrax lethal toxin, and viral dsRNA, respectively. Upon activation, these inflammasomes recruit and activate Caspase-1, a process known as canonical inflammasome activation. Active caspase-1 processes GSDMD into its active N-terminal form (N-GSDMD), which forms membrane pores. These pores lead to K+ efflux, cell lysis, and the release of inflammatory cytokines such as IL-1β and IL-18. Caspase-4, -5, and -11 directly activate GSDMD in response to LPS, and can indirectly amplify canonical inflammasome activation by triggering NLRP3 inflammasome pathway. Caspase-8, which is activated in response to Yersinia (via YopJ), also cleaves GSDMD. Caspase-3 activated by caspase-8 via apoptotic signaling or granzyme B delivered by perforin triggers the cleavage of GSDME, resulting in pore formation and pyroptotic-like cell death. Granzyme A directly cleaves GSDMB to form pores. Additionally, Group A Streptococcus (GAS) activates GSDMA as well as IL-1β through bacterial protease SpeB. The activation of GSDMC is associated with caspase-8 activity.
All gasdermins, except for DFNB59, share a similar structure characterized by two distinct and conserved domains connected through a hydrophobic linker. The N-terminal domain is capable of forming pores in membranes, while the hydrophobic linker facilitates interactions with lipid membranes. The C-terminal domain prevents oligomerization and pore-formation by the N-terminal domain. For gasdermins to become active, the C-terminal domain must be cleaved. Upon activation, the N-terminal gasdermin domains translocate to the plasma membrane, bind to phosphoinositides, and oligomerize to form membrane pores. These pores disrupt osmotic balance, leading to cell swelling and membrane rupture, ultimately causing the release of inflammatory cytokines and cell lysis. Notably, the pore-forming ability of gasdermin proteins has been demonstrated on artificial membranes in vitro (Ding et al., 2016). In contrast, DFNB59 has a highly truncated C-terminal domain, rendering it incapable of inhibiting the N-terminal domain. DFNB59 is associated with auditory function and stress-induced damage, mechanisms unrelated to pyroptosis (Zhang and Lieberman, 2020). Pyroptosis serves as a protective mechanism to eliminate harmful pathogens and alert neighboring cells. However, the release of cytokines during pyroptosis can amplify inflammatory responses, potentially leading to a cytokine storm and subsequent tissue damage (Wei Y. et al., 2022).
2.1 GSDMD-mediated pyroptosis
GSDMD is the first gasdermin to be associated with pyroptosis (He et al., 2015; Broz et al., 2020). Activated Caspase-1/4/5/11 cleaves GSDMD at the 272FLTD275 site (equivalent to 273LLSD276 in mouse GSDMD), inducing pyroptosis (Wang K. et al., 2020). In the canonical pathway, DAMPs and PAMPs trigger the activation of pattern recognition receptors (PRRs) such as the NOD-like receptors (NLRs) and the absent in melanoma 2 (AIM2)-like receptors (ALRs). Recognition of the agonists by their receptors normally lead to the assembly of inflammasome, which recruits and activates the corresponding caspases, leading to the processing of pro-cytokines and gasdermins, followed by pore formation on the cell membrane and lytic cell death.
The NLRP3 inflammasome is the most extensively studied pathway that function through GSDMD and have been extensively reviewed (Xu and Nunez, 2023; Li LR. et al., 2024). The NLRC4 inflammasome activation is triggered by selected PAMPs such as bacterial flagellin and the type 3 secretion system proteins, through its function partner NAIPs (Andrade and Zamboni, 2020). Cytosolic dsDNA, regardless of its sequence and origin (from foreign bacteria, fungi, viruses, or damaged cells), triggers AIM2 activation and formation of the inflammasome complex, which lead to Caspase-1 activation and the initiation of pyroptosis (Sharma et al., 2019). Activation of the pyrin inflammasome, as a result of reduced pyrin phosphorylation, leads to the recruitment and activation of Caspase-1 (Aubert et al., 2016; Ng et al., 2010; Kamanova et al., 2008; Xu et al., 2014). Bacillus anthracis activates NLRP1b inflammasome through anthrax lethal toxins, which facilitate ubiquitination and degradation of the N-terminal domain of NLRP1b. The remaining C-termini of NLRP1b recruit and activate Caspase-1 (Sandstrom et al., 2019; Chavarría-Smith and Vance, 2013). Rotavirus infection activates NLRP9b inflammasome via double-stranded RNA (dsRNA) in intestinal epithelial cells. The dsRNA interacts with DEAH-box helicase 9 (DHX9), which then binds to NLRP9b. Subsequently, NLRP9b forms inflammasome complexes with the adaptor protein ASC and activates Caspase-1 (Liu and Gack, 2020; Zhu et al., 2017).
In the non-canonical pyroptosis pathway, cytosolic LPS binds directly to pro-Caspase-4/5 in humans and pro-Caspase-11 in mice, resulting in autophosphorylation and activation (Yu P. et al., 2021). The active forms of Caspase-4/5 and Caspase-11 then trigger cleavage of GSDMD, ultimately leading to pyroptosis. LPS-induced pyroptosis allows potassium (K+) efflux through pores formed by GSDMD. This K+ efflux subsequently stimulates the activation of NLRP3 inflammasomes, leading to canonical pyroptosis mediated by Caspase-1. This connection suggests a coordination between Caspase-1-mediated canonical pyroptosis and Caspase-11-mediated non-canonical pyroptosis through the involvement of NLRP3 inflammasomes.
The involvement of Caspase-8 in the cleavage of GSDMD and initiation of pyroptosis was discovered more recently (Gram et al., 2019; Chen and Brodsky, 2023). Mouse GSDMD was cleaved by active caspase-8 at two sites, D276 and D88, and the cleavage at D276 generated the active P30 fragment as in the case of caspase-1 cleavage (Orning et al., 2018; Sarhan et al., 2018; Chen KW. et al., 2019). Activation of caspase-8 was RIPK1-dependent, mediated through its interaction with FAS Associated Death Domain Protein (FADD). Interestingly, GSDMD cleavage and pyroptosis occur upstream of NLRP3 activation, triggered caspase-1 activation and additional cleavage of GSDMD similarly as in the case of non-cononical activation (Chen and Brodsky, 2023).
2.2 GSDME-involved pyroptosis
GSDME can be cleaved by Caspase-3 and Caspase-8 downstream of the NLRP3 inflammasome (Wang C. et al., 2021). Caspase-3 was reported to cleave human GSDME at 267DMPD270, mouse GSDME at 267DMLD270, and zebrafish GSDME1 at 253SEVD256 (Wang et al., 2017). Previous studies revealed that Caspase-1 and/or Caspase-8 are critical for activating Caspase-3/GSDME-dependent cell death in the absence of GSDMD expression (Wei et al., 2023). In mice with the gain-of-function mutations in NLRP3, deficiency of GSDMD alone could not prevent secretion of IL-1β and IL-18 from macrophages in response to LPS or TNF-α stimulation (Wang C. et al., 2021). Sustained NLRP3 inflammasome activation induced Caspase-8/3 activation, GSDME cleavage, and IL-1β maturation in Gsdmd−/− macrophages. Aizawa et al. reported that Caspase-8 induced GSDME-dependent pyroptosis in Caspase-1/11-deficient macrophages (Aizawa et al., 2020). NLRP3 inflammasome activation recruited ACS, and subsequently Caspase-8. Activated Caspase-8 cleaved GSDME. Recently, Caspase-3/GSDME-mediated pyroptosis has been reported in human lung epithelial cells infected by SARS-CoV-2 (Planès et al., 2022). Interestingly, multiple SARS-CoV-2 3CL-(3C-like) proteases cleaved human NLRP1 at the Q333 site, triggering inflammasome assembly and cell death. At the same time, 3CL proteases also inactivate GSDMD, and alternatively, cells died via Caspase-3/GSDME mediated pyroptosis. The 3C protease from the EV71 enterovirus has been shown to cleave and inactivate GSDMD at the Q193 site (Lei et al., 2017).
The mechanisms of GSDME-mediated pyroptosis in tumor cells have been well studied (Hu et al., 2023). A variety of chemotherapeutic drugs kill tumor cells through both apoptosis and GSDME-mediated pyroptosis via Caspase-3 or Caspase-8 activation. For example, Zhang et al. reported doxorubicin (DOX)-induced pyroptosis via the Caspase-3/GSDME pathway in two breast cancer cell lines (MDA-MB-231 and T47D) (Zhang Z. et al., 2021). The GSDME-dependent pyroptosis is activated by the ROS/JNK signaling pathway. DOX treatment induced ROS production, which promoted the phosphorylation of JNK into p-JNK that initiated Caspase-3 activation. In parallel, Caspase-8 was also activated by ROS, which promoted the cleavage/activation of Caspase-3. Similarly, Shen et al. reported that DOX-induced pyroptosis occured through the ROS-JNK-Caspase 3-GSDME signaling in human tubular epithelial cells (Shen et al., 2021).
2.3 Other GSDMs involved in pyroptosis
Other gasdermins, including GSDMA, GSDMB and GSDMC are previously considered lacking the inflammatory Caspase (Caspase-1 and Caspase-4/5/11) cleavage site (Shi et al., 2017). However, recent study reveals that GSDMA in many bird, amphibians, and reptiles species contain caspase-1 cleavage sites such as YVAD or FASD in the linker and were cleaved by activated caspase-1 (Billman et al., 2024). Another protease that cleaves GSDMA is Streptococcal pyrogenic exotoxin B (SpeB), the virulence factor of group A Streptococcus (GAS), which directly cleaves GSDMA after Gln246 in keratinocytes (Deng et al., 2022; LaRock et al., 2022). In addition, recent studies have unveiled a correlation between the degradation of GSDMA and cisplatin resistance in tumor cells, suggesting that platinum-based drugs may induce GSDMA-mediated cell death in tumors through some unknown mechanism (Wang H. et al., 2024).
In airway epithelial cells, GSDMB has been shown to be cleaved by activated Caspase-1 after D236, releasing N-terminal fragments capable of inducing pyroptosis (Panganiban et al., 2018). Pyrotosis mediated by Granzyme A (GZMA)/GSDMB has been reported in tumor cells. GZMA released from cytotoxic lymphocytes and natural killer (NK) cells enter tumor cells via perforin. GSDMB is cleaved by cytosolic GZMA at Lys244 to release the N-terminal domain, which perforates the plasma membrane and leads to pyroptosis (Zhou et al., 2020). Research on granzymes has spanned over 30 years, and granzymes are involved in the elimination of virus-infected cells by immune cells. The discovery of GSDMB cleavage by GZMA offered a new mechanism for GZMA-mediated cell death. Interestingly, full-length GSDMB can promote oligomerization of Caspase-4 by a direct binding to its CARD domain, thereby facilitating cleavage of GSDMD and triggering non-canonical pyroptosis (Chen Q. et al., 2019).
GSDMC is expressed in multiple tissues, including the trachea, small intestine, colon, esophagus, skin, spleen, and vagina (Zhu et al., 2024). Both artificially truncated GSDMC-NT and the GSDMC-NT fragment that intracellularly cleaved by Caspase-8 have been demonstrated to provoke pyroptosis (Zhang JY. et al., 2021). A recent study by Hou et al. demonstrated that PD-L1 nuclear translocation in hypoxic tumor cells increases the production of GSDMC, which converted apoptosis to pyroptosis (Hou J. et al., 2020). In a hypoxic environment, phosphorylated STAT3 (Signal transducer and activator of transcription 3) interacts with PD-L1, leading to its nuclear translocation and subsequent upregulation of GSDMC transcription. As GSDMC levels increase, TNF promotes the cleavage of GSDMC by Caspase-8, producing the GSDMC-N-terminal fragment that drives pyroptosis. Additionally, the cellular metabolite α-ketoglutarate (α-KG) facilitates the assembly of the DR6 receptosome in tumor cells, forming a molecular platform that enables the efficient proteolysis of GSDMC by activated Caspase-8, thereby inducing pyroptosis.
3 NF-κB signalling pathway
3.1 NF-κB family members and regulators
NF-κB, IκB, and IKK are the main signaling nexus/hubs of the NF-κB signaling pathway. The NF-κB family consists of five main members: p65 (RelA), RelB, c-Rel, p105/p50 (NF-κB1) and p100/p52 (NF-κB2) (Figure 2). They share a conserved Rel Homology Domain (RHD) of over 300 amino acids at the N-terminus, which endows the ability of dimerization, nuclear translocation, and binding to specific sequences on DNA (Dorrington and Fraser, 2019; Yu et al., 2020; Zinatizadeh et al., 2021). Based on their structural characteristics at the C-terminus, the NF-κB family can be divided into NF-κB subfamily and Rel subfamily. NF-κB subfamily members, p105 and p100, possess long repetitive sequences, ankyrin repeats, at their C-terminus. While the Rel subfamily members, p65, RelB, and c-Rel, have a transcription-activation domain (TAD) at their C-terminus. The TAD is considered essential for the nuclear translocation of NF-κB, hence NF-κB subfamily homodimers are regarded as inhibitory regulators due to the lack of TAD. NF-κB subfamily could only activate gene transcription when forming heterodimers with the Rel subfamily members (Yu H. et al., 2021). In recent years, a sixth member of the NF-κB family has been discovered: RelAp43, a new splicing variant of RelA, lacking the TAD at its C-terminus. It is primarily activated by the rabies virus matrix protein, although its ability to interact with other NF-κB family members has been demonstrated in vitro, this perspective has not been widely accepted (Struzik and Szulc-Dąbrowska, 2019; Ben Khalifa et al., 2016; Luco et al., 2012).
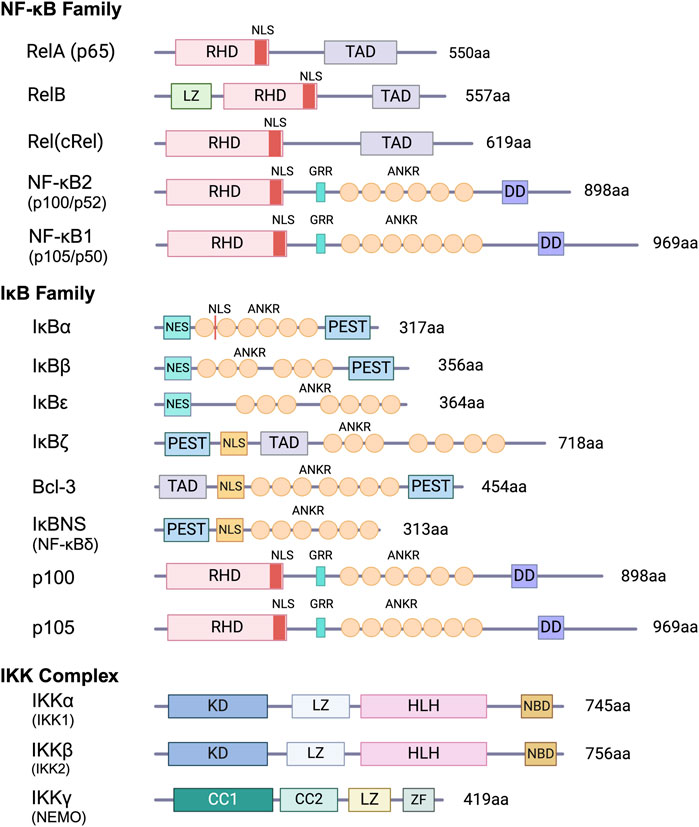
Figure 2. Schematic representation of NF-κB, IκB and IKK proteins. Key features of members of the NF-κB and IκB protein families and components of the IKK complex are shown. RHR, Rel homology region; NLS, nuclear localization sequence; ANKR, ankyrin repeats; DD, death domain; TAD, transactivation domain; PEST, region rich in proline, glutamate, serine. and threonine; LZ, leucine zipper; Kinase, kinase domain; HLH, helix-loop-helix region; NBD, NEMO-binding domain; CC, coiled-coil domain; ZF, zinc-finger.
Inhibitor of κB (IκB) serves as a key regulator in the NF-κB signaling pathway, as its name suggests, by inhibiting NF-κB. Its members include IκBα, IκBβ, IκBε, IκBγ, IκBz, Bcl-3, as well as the precursors of NF-κB subfamily, p105, and p100 (Zhang J. et al., 2023). Within 2 years of discovering NF-κB, Baltimore proposed the concept of IκB and identified IκBα, which remains the most extensively studied IκB to date (Baeuerle and Baltimore, 1988). IκB sequesters NF-κB in the cytoplasm, forming an inactive complex. Phosphorylation and degradation of IκB, which is typically mediated by the ubiquitin-proteasome system, are required for activating NF-κB, after that the free NF-κB, with the assistance of the nuclear transport receptor importin α/β, translocate through the nuclear pore and activates transcription (Hinz et al., 2012; Wang X. et al., 2020). The classical IκB proteins, including IκBα, IκBβ, and IκBε, are typically distributed in the cytoplasm. They contain six ankyrin repeats and are capable of binding to and covering the Rel Homology Domain (RHD), thereby exerting inhibitory effects. Non-classical IκB primarily resides in the nucleus (aka nuclear IκB) and is induced to increase as a target gene of NF-κB, which contains more than six ankyrin repeats (Wang X. et al., 2020).
IκB kinase (IKK) is primarily responsible for the phosphorylation of IκB, which was discovered nearly a decade after NF-κB. The IKK complex consists of active IKKα and IKKβ, and two molecules of the non-active IKKγ (NEMO) to form a tetramer (DiDonato et al., 1997; Rothwarf et al., 1998), while whether other components are involved is still under considerable controversy. IKKα and IKKβ are highly similar in sequence and structure, their N-terminal kinase domains need to be activated to phosphorylate IκB (Zhang J. et al., 2023). This activation mechanism may arise from autophosphorylation or upstream signals from kinases such as TAK1, MEKK3, and others (Wang et al., 2001; Mulero et al., 2019). In vitro recombination experiments have shown that IKKγ can only directly bind to IKKβ, while the binding of IKKγ to IKKα depends on IKKβ. Therefore, it is inferred that the non-enzymatic IKKγ plays a scaffold and regulatory role (Zandi and Karin, 2023). Although IKK possesses two enzymatically active subunits (α and β), studies showed that in specific pathways, the enzymatic activity of only one subunit is required (Israël, 2010). Other proteins that may participate in the IKK complex include NIK, MEKK1, IKAP (Cohen et al., 1998). It is worth noting that IKK does not always act as a positive regulator of NF-κB: in immune cells (T and B lymphocytes), IKK phosphorylates Bcl10, leading to inhibition of the NF-κB pathway (Abd-Ellah et al., 2018; Zhou et al., 2004).
3.2 Activation of NF-κB signaling pathway
During infection and oxidative stress, NF-κB regulates the transcription of inflammation-related genes (Capece et al., 2022; Xu G. et al., 2024). During the resting state, NF-κB is sequestered in the cytoplasm by IκB, preventing excessive gene transcription activity. Upon receiving specific stimuli, such as inflammatory cytokines or cellular stress, IKK phosphorylates and cleaves IκB, releasing NF-κB to enable nucleus translocation that initiates transcription of target genes (Anilkumar and Wright-Jin, 2024).
The NF-κB activation may proceed through the canonical and non-canonical pathways. Initially signaling mediated by IKKβ/NEMO/RelA was regarded as the canonical pathway. However, with the discovery that IKKβ can be partially replaced by IKKα and the involvement of RelA in the non-canonical pathway, this viewpoint has been challenged. It has been suggested that NEMO dependency to be used to define the canonical pathway (Liu T. et al., 2017). Despite this, research on the canonical pathway primarily focuses on IKKβ. Similarly, studies of RelA predominantly focused on the canonical pathway (Shih et al., 2011). Upon stimulation, ligand-loaded receptors recruit adaptor proteins such as TRADD, TRAF2, and RIK1 to activate IKKβ (Ma et al., 2024). The IκB proteins are then phosphorylated by IKK followed by ubiquitination and degradation, releasing the NF-κB dimer for translocation into the nucleus. The dimers corresponding to the classical pathway mainly consist of four types: RelA:RelA, RelA:p50, cRel:cRel, and cRel:p50.
The non-canonical pathway is mediated by IKKα and the NF-κB-inducing kinase (NIK) (Rodriguez et al., 2024). NIK (also known as MAP3K14) is the first identified component in the non-canonical pathway. Currently identified inducers of the non-canonical NF-κB pathway all converge at NIK. Therefore, the activation of NIK is defined as a key event in the non-canonical pathway, which normally lead to the RelB:p52 dimer. Under resting conditions, NIK remains at relatively low levels due to its degradation by TRAF3. Degradation of TRAF3 triggered by the stimulation signals results in the accumulation of NIK within the cell. However, since NIK requires de novo synthesis, it takes some time to reach the threshold required to trigger the non-canonical pathway, this may explain why the signaling response of this pathway is relatively slow. In the steady state, p52 primarily exists in the form of its precursor, p100. Both p100 and IκBα are phosphorylated by NIK (Xiao et al., 2001b). Subsequently, p100 is processed into p52, forming the RelB:p52 dimer. Phosphorylation of IKKα activates it. While p100 has been proposed to be a substrate for IKKα, IKKα did not induce the direct processing of p100. A potential explanation is the binding between IKKα and p100 requiring NIK (Yu et al., 2020; García-García et al., 2024). Although the role of IKKα has not been directly demonstrated, studies on transgenic mice have shown that mice with IKKα deficiency and NIK knockout have similar phenotypes, indicating the critical role of IKKα in the non-canonical NF-κB pathway. On the other hand, IKKα also functions as a negatively feedback regulator of non-canonical NF-κB activity by phosphorylating and degrading NIK, these contradictory results may be related to signal crosstalk (Razani et al., 2010; Gray et al., 2014). Other regulatory mechanisms mainly involve modulating the levels of NIK, such as TNAP, Zfp91, TBK1, and NLRP12 (Sun, 2011; Jin et al., 2012), but the specific regulatory mechanisms are not yet fully understood. OTUD1b has been reported to deubiquitinate TRAF3 to prevent its degradation, thus being regarded as a negative regulatory factor (Hu et al., 2013).
It is almost impossible to have completely independent signaling pathways in complex cells. Crosstalk within the NF-κB signaling pathways has been nicely-reviewed (Oeckinghaus et al., 2011). Here, we summarize the reports of NF-κB signaling pathway activation in inflammation over the past decade. The activation of NF-κB is thought to be part of a stress response as it is activated through the recognition of a variety of stimuli by receptors. These include, but are not limited to, the pattern recognition receptors like TLRs, cytokine receptors such as TNFR and IL-1R, and antigen receptors TCR (T cell receptor) and BCR (B cell Receptor) (Barnabei et al., 2021). Additionally, the cGAS-STING pathway has been identified as a central regulator of type I interferon (IFN) and NF-κB. In this review, we will focus on NF-κB signaling pathways mediated by TLRs, IL-1R,TNFR and cGAS-STING (Figure 3).
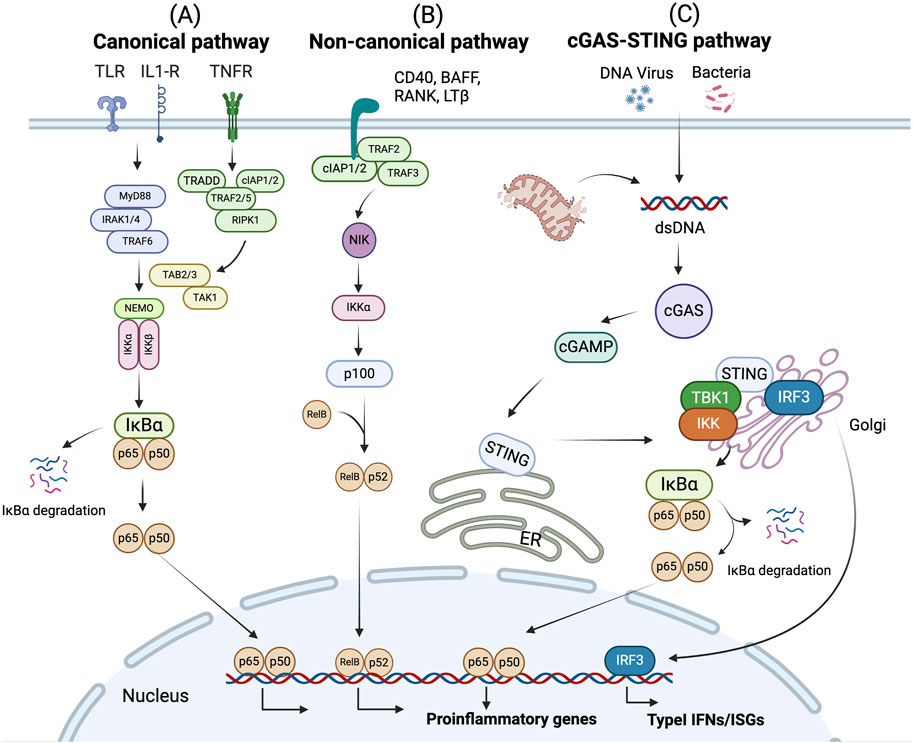
Figure 3. NF-κB signaling pathway. (A) The canonical pathway is induced by TLRs, TNFRs, and IL-1R. Activation of this cascade leads to the phosphorylation and degradation of inhibitory protein IκB. NF-κB p65 and p50 are activated by releasing from the IκB-containing complex, then translocating into nucleus. (B) The non-canonical pathway is dependent on the activation of p100 through BAFFR, CD40, RANK or LTβ. This cascade induces phosphorylation of NIK, which subsequently phosphorylates IKKα. Activated p100 releases p52. Then RelB/p52 heterodimer is activated and translocate to the nucleus. The activation of NF-κB signaling leads to the expression of proinflammatory genes. (C) cGAS recognizes dsDNA fragments from bacteria, viruses, mitochondria, etc., and catalytically generates 2′3′-cGAMP. STING proteins located in the ER are activated upon recognition of cGAMP. STING then translocates from the ER to Golgi. During this process, STING oligomers recruit TBK1, triggering TBK1 autophosphorylation. In turn, STING is phosphorylated by TBK1. Activated STING then recruits IRF3, facilitating the phosphorylation of IRF3 by TBK1. Activated IRF3 dimers then translocate to the nucleus to activate the transcription of type I IFNs, which in turn leads to the production of diverse IFN-stimulated genes (ISGs). Besides, STING induces NF-κB activity via TBK1-dependent and independent manners, resulting in the IκBα phosphorylation and subsequently translocation of p65/p50 to the nucleus, leading to the production of proinflammatory genes.
3.2.1 TLR-mediated NF-κB signalling
TLRs are pattern recognition receptors that recognize prototypical structures of microbes, including microbial surface components such as acylated lipopeptides, peptidoglycans, lipopolysaccharides and flagellin, and microbial nucleic acids such as dsRNA, ssRNA, or unmethylated DNA (Beutler et al., 2003). TLRs are transmembrane proteins characterized by the extracellular domains containing variable numbers of leucine-rich-repeat (LRR) motifs responsible for ligand recognition and a cytoplasmic Toll/interleukin 1 (IL-1) receptor (TIR) homology domain responsible for initiating intracellular signaling (Moresco et al., 2011). To date, 11 human TLRs and 13 mouse TLRs have been identified (Kawai and Akira, 2007a). The ligands of each TLR have previously been reviewed (Moresco et al., 2011; Fan Y. et al., 2023; Chen et al., 2021; Kawai and Akira, 2007b). There are two groups of TLRs. The group on cell surface (TLR1, TLR2, TLR4, TLR5, TLR6, and TLR11) mainly recognizes microbial envelope components, such as lipids and lipoproteins. The other group is intracellular TLRs (TLR3, TLR7, TLR8, and TLR9) that sense the nucleic acids derived from bacteria or viruses. TLRs are widely expressed in various immune cells (e.g., monocytes, macrophages, dendritic cells (DCs), B cells, T cells and neutrophils) as well as non immune-cells (e.g., fibroblast cells, epithelial cells and endothelial cells), and usually acts as the first responders to initiate pyroptosis, which play a crucial role in defending against pathogenic microbial infection through the induction of inflammatory cytokines (Kumar et al., 2009).
TLR signaling pathways lead to the activation of NF-κB and IRF3 to induce the expression of various genes encoding inflammatory cytokines, the inactive pro-forms of inflammatory cytokines such as IL-1β and IL-18, and type I interferons and chemokines (Liu T. et al., 2017). Upon binding by specific ligands, TLRs recruit a set of adaptor proteins with TIR domains by homophilic interaction of their TIR domains. TLR signaling depends critically on a total of four adaptor proteins—myeloid differentiation primary response protein 88 (MyD88), TIR-associated protein (TIRAP, also known as MyD88-adaptor like protein, MAL), Toll-receptor-associated activator of interferon (TRIF), and Toll-receptor-associated molecule (TRAM), which directly bind to activated TLRs and recruit downstream signaling components (Moresco et al., 2011). Ligand binding leads to dimerization of TLR, and the dimerized cytosolic TIR domains (Latz et al., 2007) recruit MyD88 and TRIF (some TLRs require adaptor TIRAP or TRAM), respectively, to stimulate the assembly of a large supramolecular complex called Myddosome or Triffosome (O'Neill and Bowie, 2007; Fitzgerald and Kagan, 2020). Depending on the components of those large oligomeric signaling complexes, TLR signaling pathways can be mainly classified as either MyD88-dependent pathways, which leads to the activation of NF-κB, IFN-responsive factors (IRFs), and the mitogen-activated protein kinases (MAPK) pathways, inducing the expression of pro-inflammatory cytokines (E.g., TNF, IL-1β, IL-8, IL-18, IL-6, IL-12 and IL-8 et al.); or TRIF-dependent pathways, which mainly activate IRFs to mediate the expression of type I IFNs (IFNa or IFNβ) (Duan et al., 2022; Balka and De Nardo, 2019). All TLRs, except TLR3, utilize a MyD88-dependent pathway resulting in the production of cytokines through NF-κB, and also trigger MAPK cascades that lead to activation of AP-1 (activator protein 1) and cAMP (cyclic AMP) response element-binding protein (CREB) (Moresco et al., 2011). TLR3 and TLR4 utilize the TRIF-dependent pathway to activate NF-κB and additionally activate IRF3 to induce type I IFNs (Hu et al., 2015).
3.2.2 IL-1R family-mediated NF-κB signaling
IL-1R family members are receptors for endogenous alarm mediators sensing the integrity of barriers or the fitness of cells. The first biochemical characterization of an IL-1 receptor (IL-1R) was reported in 1985 (Dower et al., 1985). The IL-1R family now consists of 10 members that are receptors or co-receptors for presently 11 ligands of the IL-1 family of cytokines (Boraschi et al., 2018). All members of the IL-1R family are characterized by an intracellular TIR domain (except for IL-1R2 which lacks the TIR domain) that are homologous to other members of the IL-1R and TLR families, and three extracellular immunoglobulin domains that bind specific IL-1 related cytokines (Boraschi et al., 2018). Among those IL-1R families, only IL-1R1, IL-1R4, IL-1R5 and IL-1R6 have the capability to bind cytokines of the IL-1 family and initiate signaling, while IL-1R3 and IL-1R7 are essential in forming the signaling receptor complexes (Zarezadeh Mehrabadi et al., 2022) (Li et al., 2021). Upon ligand binding, the accessory co-receptor (IL-1R3 or IL-1R7) is recruited, which leads to the recruitment of MyD88 to the receptor complex through TIR:TIR domain interactions, forming myddosome. IL-1Rs interact directly with MyD88 and the signaling of all IL-1Rs is MyD88 dependent. The formation of myddosome leads to the recruitment of IL-1R-associated kinases (IRAKs). The IRAK family contains four members, IRAK-1, IRAK-2, IRAK-3 (also known as IRAK-M), and IRAK-4, and the different roles of them in the signaling of TLRs and IL-Rs have been reviewed (Flannery and Bowie, 2010). Next, effector TRAF6 is recruited to the C-terminal domains of either oligomeric IRAK-1 or IRAK-2 and activated via K63 autoubiquitination. TRAF6 subsequently ubiquitinates and activates TGFβ-associated kinase (TAK1), which is responsible for phosphorylating/activating IKK-β. TRAF6 and TAK1 both facilitate the degradation of IKKγ. This allows the release of the NF-κB dimer and its translocation to the nucleus (Liu T. et al., 2017).
3.2.3 TNFR family-mediated NF-κB signaling
TNFα is expressed by multiple cells of the innate and adaptive immune systems, including macrophages, monocytes, B lymphocytes and T lymphocytes, and by nonimmune cells, such as endothelial cells and fibroblasts (MacEwan, 2002). As an inflammatory factor, it plays a prominent role in the interconnection of different types of programmed cell death, including apoptosis, necrosis and pyroptosis (Frank and Vince, 2019). There are two forms of TNF, membrane-bound TNF (mTNF, 26 KDa) and soluble TNF (sTNF, 17 KDa) which lacks the transmembrane domain (Richter et al., 2012; Grell et al., 1995). TNF signaling is initiated by the interaction between TNFα and its membrane receptors TNFR1 or TNFR2 (Jang et al., 2021). TNFR1 is expressed at low level across all human tissues and can respond to both mTNFa and sTNFa, whereas expression of TNFR2 is restricted to particular cells, including oligodendrocytes, astrocytes, T cells, myocytes, thymocytes, endothelial cells and in human mesenchymal stem cells, and TNFR2 binds only with mTNFα (Grell et al., 1995; Cabal-Hierro and Lazo, 2012). TNFR1 and TNFR2 have similar extracellular structures where TNFα binds, but distinct intracellular structures for binding of adaptor proteins (Tartaglia et al., 1991). Both TNFR1 and 2 signaling pathways lead to the activation of NF-κB (Ting and Bertrand, 2016). TNFR1 contains an intracellular death domain (DD) to recruit the DD-containing proteins, enabling strong activation of proinflammatory pathways and cytotoxic signaling. TNFR2 does not have a DD. It recruits the TNFR associated factor 1 (TRAF1) and 2 (TRAF2) with the help of a short peptide motif and activates the alternative NF-κB pathway (Hsu et al., 1995; Rothe et al., 1995; Medler and Wajant, 2019).
TNFR1 mediated signaling pathways have been well characterized. Binding of TNFα to TNFR1 leads to the formation of two different TNF signaling complexes, referred to as complexes I and complex II (including IIa, IIb, and IIc), which result in distinct cellular responses (Brenner et al., 2015). The functional outcome of complex I signaling is the induction of inflammation, cell survival and proliferation, and the immune defense against pathogens (Brenner et al., 2015). During the formation of complex I, activated TNFR1 undergoes conformational change in the DD, followed by the recruitment of TRADD, and subsequently receptor-interacting serine/threonine-protein kinase 1 (RIPK1), TNFR-associated factor 2 or 5 (TRAF2/5), cellular inhibitor of apoptosis protein 1 or 2 (cIAP1/2), and the linear ubiquitin chain assembly complex (LUBAC) (Jang et al., 2021). Formation of the Complex I triggers the activation of NF-κB or AP-1 family transcription factors, which control the expression of antiapoptotic proteins that prevent the initiation of the cell death processes (Haas et al., 2009). The NF-κB pathway activated by the formation of TNF Complex I relies on the ubiquitination of RIPK1. The ubiquitinated RIPK1 recruits the transforming growth factor-beta (TGF-β)-activated kinase 1 (TAK1) complex, consisting of TAK-binding protein 1 (TAB1), 2 (TAB2) and 3 (TAB3), and the IKK complex constituted of NEMO, IKKα, and IKKβ(144). The IKK complex is essential for the phosphorylation of IκBα, which lead to its degradation and release of NF-κB to translocate to the nucleus to initiate transcription. NF-κB can induce the transcription of anti-apoptotic genes such as cIAP-1, cIAP-2, cFLIP, TRAF1 and TRAF2 (Micheau and Tschopp, 2003), which are also components of TNF Complexes.
While the assembly of complex I occurs on the plasma membrane, complex II is assembled in the cytoplasm. Formation of the Complex II (also known as death-inducing signaling complex, DISC) triggers cell death processes (apoptosis, necroptosis or pyroptosis) after the internalization of the receptor (Micheau and Tschopp, 2003). Based on their components, they can be further sub-grouped into complex IIa, IIb and IIc (Holbrook et al., 2019). Complex IIa consists of TRADD, RIPK1, TRAF2, cIAP1/2, pro-Caspase-8, and Fas-associated protein with death domain (FADD). Complex IIb contains all components in complex IIa plus RIPK3 (Wang et al., 2008). Both Complex IIa and IIb (known as apoptosome) activate Caspase-8 and downstream Caspase-3 to trigger apoptosis or induce GSDME-mediated pyroptosis (Micheau and Tschopp, 2003; Zhai et al., 2022). Complex IIc (also known as necrosome) is composed of RIPK1 and RIPK3, which induces necroptosis through a Caspase-8/RIPK3/MLKL (mixed lineage kinase domain-like protein) pathway (Gough and Myles, 2020; Vandenabeele et al., 2010).
TNFR2 can activate both canonical and non-canonical NF-κB signaling, leading to numerous changes in gene expression that drive inflammation, cell proliferation and cell survival (Borghi et al., 2016). Binding of TNF to TNFR2 triggers the recruitment of TRAF2 along with TRAF1, TRAF3, cIAP1, and cIAP2 to activate the canonical NF-κB pathway (Jang et al., 2021; Rodríguez et al., 2011). TRAF2 also acts as a negative regulator of TNFR2-induced non-canonical NF-κB signaling. Degradation of TRAF2 is a fundamental step in TNFR2-induced activation of the non-canonical NF-κB pathway (Borghi et al., 2016; Ruspi et al., 2014; Vallabhapurapu et al., 2008). The cIAP1/2–TRAF2–TRAF3 signaling complex normally targets NIK for degradation and keeps it at a low level under unstimulated conditions. In the absence of TRAF2, NIK accumulates and phosphorylates IKKa, which in turn phosphorylates the NF-κB p100 subunit (Sun, 2017). Phosphorylates NF-κB p100 is also ubiquitinated by the SCF-beta-TRCP ubiquitin ligase complex at K48 and is subsequently processed by the proteasome to NF-κB p52. Then p52, together with another transcriptionally competent NF-κB subunit RelB, translocate into the nucleus, and initiates gene transcription (Borghi et al., 2016; Ruspi et al., 2014).
3.2.4 cGAS-STING mediated NF-κB signaling
Over the past decade, significant progress has been made in understanding how nucleic acid recognition is coupled to immune gene expression, particularly through the DNA-sensing receptor cGAS and its downstream effector, STING (Motwani et al., 2019; Du et al., 2023). The detection of mislocalized DNA in the cytosolic by cGAS has been recognized as a pivotal mechanism in cellular responses to pathogen invasion, DNA damage, and mitochondrial stress (Decout et al., 2021; Skopelja-Gardner et al., 2022; Domizio et al., 2022). cGAS-mediated STING activation drives IFN responses and also activates NF-κB signaling pathway.
Activation of the cGAS-STING pathway begins with the recognition of double-stranded DNA (dsDNA), either of foreign or mislocalized self-origin, by cGAS (Decout et al., 2021; Chen et al., 2016). Upon binding dsDNA, cGAS is activated to catalyze the synthesis of the second messenger 2′3′-cyclic GMP-AMP (cGAMP). The cGAMP binds to the endoplasmic-reticulum (ER)-membrane located adaptor protein STING, inducing a conformational change that activates STING. Once activated, STING traffics from the ER to the ER-Golgi intermediate compartment and the Golgi apparatus. During this process, STING recruits and activates TANK-binding kinase 1 (TBK1), which phosphorylates both IRF3 and IKKε. The phosphorylation of IRF3 results in its dimerization and nuclear translocation, where it drives the expression of type I IFNs. Concurrently, activated IKKε phosphorylates IκB, which is subsequently ubiquitinated and degraded via the proteasome, releasing NF-κB to enable nuclear localization (Zhang L. et al., 2023). This dual activation of IRF3 and NF-κB amplifies the immune response, enabling robust inflammatory and antiviral signaling.
The critical role of cGAS-STING-mediated NF-κB activation in promoting inflammation and immune regulation is well established (Zhang L. et al., 2023; Balka et al., 2020). Studies have demonstrated that AIM2-deficient cells, which bypass rapid pyroptotic elimination, rely on the cGAS-STING-TBK1-IRF3/NF-κB axis to produce pro-inflammatory cytokines such as C-X-C motif chemokine 10 (CXCL10), TNF-α, and IFN-β (Baatarjav et al., 2022). In a cerebral venous sinus thrombosis model, cGAS–STING was found to activate the NF-kB cascade and downstream NLRP3 inflammasome (Ding et al., 2022). Function of cGAS-STING in pyroptosis has been reviewed recently (Liu et al., 2024).
4 Regulation of inflammasome through NF-κB
4.1 NLRP3
NLRP3 inflammasome activation normally occurs in two stages, involving a priming signal (signal I) and an activation signal (signal II) (Figure 4). In most cell types, including macrophages, minimal levels of NLRP3 were observed before inflammasome activation, and pro-IL-1β was not constitutively expressed (He Y. et al., 2016). Signal I upregulated the transcription of NLRP3 and pro-IL-1 (Garlanda et al., 2013; McKee and Coll, 2020). NLRP3 is regulated at the transcriptional level, by TLR and cytokine stimulation through activating the NF-κB pathway (Bauernfeind et al., 2009). It has been reported that NF-κB regulates NLRP3 transcription by binding directly to the NLRP3 promoter region (Qiao et al., 2012). Recent research has added Sphingosine 1-phosphate (S1P)/S1P Receptor (S1PR) signaling as an additional inducer for NLRP3 priming (Hou L. et al., 2020). While the precise mechanism behind S1P/S1PR signaling mediated NLRP3 inflammasome priming remains unclear, S1PR has been shown to activate the NF-κB pathway under acute pancreatitis conditions (Yang et al., 2022).
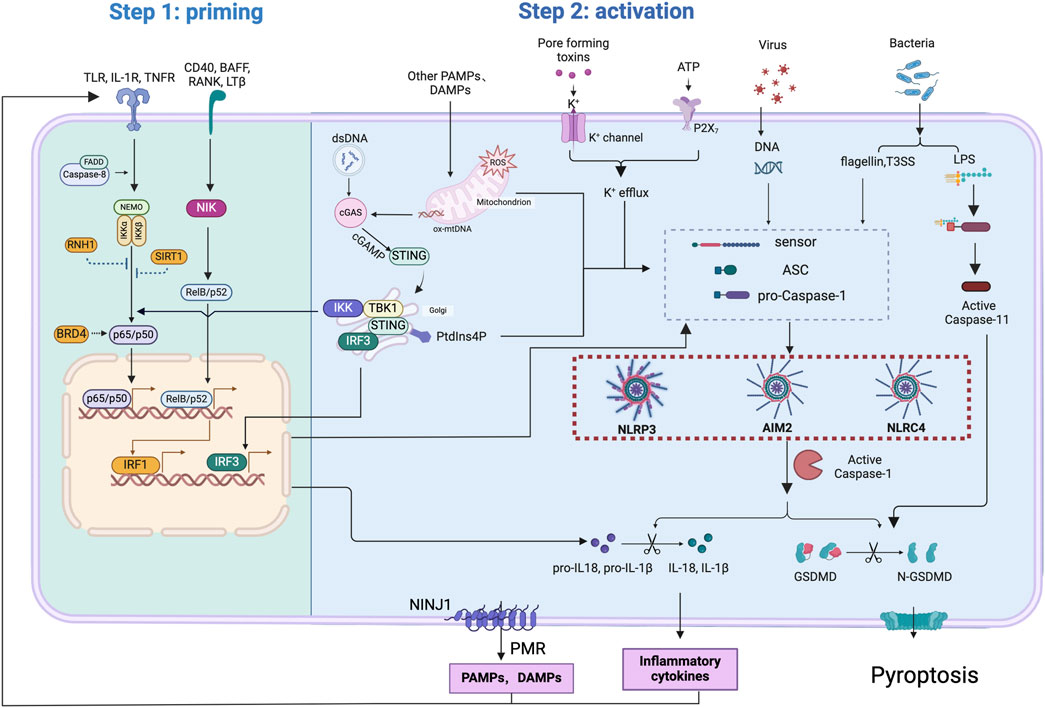
Figure 4. Mechanisms involved in the regulation of inflammasome activation and pyroptosis through NF-κB signaling. NF-κB signaling primes inflammasome activation by canonical NF-κB pathway (through receptors such as TLR,TNFR, and IL-1R), non-canonical pathway (through receptors such as CD40, BAFF, RANK, LTβ) or cGAS-STING pathway. FADD and caspase-8 contribute to canonical NF-κB activation. Binding of transcription factor, including p65/p50,RelB/p52, IRF1 and IRF3,on nuclear DNA drives the expression of inflammasome components, including NLRP3, NLRC4, AIM2, CASPASE11, GSDMD, pro-IL-1β, and pro-IL-18. Inhibitors like RNH1 and SIRT1 negatively regulate canonical NF-κB activation. PAMPs and DAMPs, released through plasma membrane rupture caused by activation of NINJ1 or GSDMD pores, act as priming signals activating NF-κB pathway.
Several other factors regulate NLRP3 activation through modulating the NF-κB pathway. Leucine rich repeat protein ribonuclease inhibitor (RNH1) plays a role in attenuating NLRP3 inflammasome activation and is involved in reducing the priming signal. RNH1 knockout cells exhibited reduced levels of IκBα and increased levels of phospho-IκBα, indicating enhanced NF-κB activation (Bombaci et al., 2022). Inhibition of bromodomain-containing protein 4 (BRD4), which reduces NF-κB signaling through the suppression of IκBα phosphorylation and degradation, results in the upregulation of NLRP3 at the transcriptional level by enhancing the activity of the NLRP3 promoter (Tan et al., 2020). NEK7 has been identified as a mediator of increased expression of Caspase-1, NLRP3, and GSDMD in inflammatory bowel disease (IBD). In NEK7 knockdown intestinal epithelial cells, ATP/LPS-induced activation of the NLRP3 inflammasome was abolished. NEK7 itself is regulated by the NF-κB signaling pathway, with p65 promoting the transcription of NEK7 through binding to its corresponding binding site within the NEK7 promoter, while p65 inhibition blocks this effect (Chen X. et al., 2019).
FAS-associated death domain protein (FADD) and Caspase-8 were found to contribute to NLRP3 inflammasome priming (Lemmers et al., 2007). Caspase-8 was recruited to the complex containing IKKαβ in response to TLR4 stimulation (Lemmers et al., 2007), This interaction is pivotal for the initiation of NF-κB signaling. Macrophages derived from mice deficient in Caspase-8 exhibited reduced pro-IL-β and TNF levels upon priming and decreased NLRP3 activation (Allam et al., 2014). FADD has been identified as an upstream regulator of Caspase-8, as supported by the observation that FADD-depleted macrophages exhibit significantly reduced processing of mature Caspase-8 when subjected to canonical NLRP3 inflammasome stimuli. Accordingly, FADD deficiency impaired NLRP3 activation by LPS or C. rodential infection (Gurung et al., 2014). Both FADD and Caspase-8 were required for the transcriptional upregulation of NLRP3 and pro-IL-1β in an NF-κB dependent manner. Moreover, deletion of both Caspase-8 and NEMO, the NF-κB essential modulator, resulted in the transcriptional downregulation of NLRP3, TNF-α and IL-1β expression after LPS treatment in hepatocytes (Boaru et al., 2015).
The role of IKKβ in inflammasome activation is controversial. Firstly, Nanda and co-workers have reported a pivotal role for IKKβ in the transcriptional upregulation of inflammasome components (Nanda et al., 2021). Inhibition of IKKβ resulted in decreased inflammasome activation, which is consistent with other studies (Bauernfeind et al., 2009; Boaru et al., 2015; Unterreiner et al., 2021; Schroder and Tschopp, 2010). Intriguingly, their research also unveiled the involvement of IKKβ in the assembly of the NLRP3 inflammasome through recruiting the NLRP3 inflammasome to the trans-Golgi network, where the assembly occurs. Furthermore, IKKβ directly binds to NLRP3, facilitating NLRP3 binding to phosphatidylinositol-4-phosphate (PI4P) on the trans-Golgi network, thereby promoting oligomerization and inflammasome assembly (Asare et al., 2022; Schmacke et al., 2022). Contrary to the observations mentioned above, Greten et al. reported that inhibition of IKKβ, either pharmacologically or genetically, enhanced Caspase-1 dependent IL-1β production in macrophages and related inflammation upon LPS challenge (Greten et al., 2007). Despite a decrease in pro-IL-1β mRNA synthesis under these conditions, efficient pro-IL-1β processing by casaspe-1 leads to even higher IL-1β secretion. It is speculated that during LPS challenge, NF-κB contributed to the upregulation of transcriptions of several genes encoding inhibitors of Caspase-1 activity, thus reducing IL-1β processing. The use of prolonged LPS treatment alone in the latter study, in contrast to the combined use of LPS and NLRP3 stimuli to rapidly activate the NLRP3 inflammasome, might explain the contradictory observation mentioned above.
Additionally, inhibitors targeting TBK1/IKKε have been found to amplify NLRP3 inflammasome responses, including IL-1β and IL-18 secretion, Caspase-1 cleavage, and ASC speck formation. Interestingly, the inhibition of TBK1/IKKε on NLRP3 inflammasome responses does not seem to rely on transcriptional mechanisms but rather through posttranslational regulation, mediated by the phosphorylation of the serine 3 (S5 in humans) on NLRP3 (Fischer et al., 2021). Conversely, a more recent study highlightes an alternative role for TBK1, demonstrating that TBK1 activation enhances NF-κB signling, as evidenced by increased phosphorylation of NF-κB p65. This activation is associated with elevated protein levels of key NLRP3 inflammasome components (NLRP3, ASC, and caspase-1) and increased production of cleaved IL-1β in microglia (Liao et al., 2024).
Moreover, previous research has demonstrated that IKK/NF-κB pathway plays a role in promoting autophagy by upregulating the expression of the autophagy receptor p62, which enhanced the clearance of damaged mitochondria through mitophagy via a Parkin-ubiquitin-dependent mechanism (Zhong et al., 2016). Deletion of p62 specifically in myeloid cells lead to the abnormal accumulation of damaged mitochondria and excessive production of IL-1β, resulting in increased sensitivity to endotoxin-induced shock. A truncating variant of p50 (NFKB1R157*/R157*) has been identified in patients with necrotizing fasciitis or severe soft tissue inflammations (Nurmi et al., 2024). Autophagy was impaired in patients’ cells, causing increased release of mtROS and mtDNA from damaged mitochondria, which enhances NLRP3 inflammasome activation. These findings collectively suggest that while NF-κB may mediate the priming signal of NLRP3 inflammasome activation, the induction of mitophagy or autophage by NF-κB may function as an autoregulatory mechanism to restrain the pro-inflammatory function.
Recent studies have increasingly highlighted the activation of the NF-κB/NLRP3 pathway in various disease models. Molecules and chemicals that modulate this pathway are being investigated for their therapeutic potential (Li Y. et al., 2024; Xiong et al., 2025; Zhao P. et al., 2024; Hao et al., 2024; Alad et al., 2024; Wei et al., 2024). One notable study examined the role of acetyl-CoA synthetase 2 (ACSS2) in the pathogenesis of acute kidney injury (AKI). Mice deficient in ACSS2 showed reduced NLRP3 inflammasome activation in renal tubular epithelial cells. These effects are regulated by the transcription factor KLF5, which mediates NF-κB activation (Lu et al., 2024). In cardiovascular diseases, the NF-κB/NLRP3 pathway is frequently implicated, especially upon activation by oxidative stress or ROS (Xu Z. et al., 2024; Antar et al., 2024). However, the detailed mechanisms remain poorly understood. In a cadmium (Cd) induced chronic kidney injury model, ROS was found to promote NLRP3 inflammasome activation by inhibiting SIRT1 expression and deacetylase activity. This led to decreased SIRT1–p65 interactions, increased acetylated p65 levels, and subsequent NF-κB activation (Dong et al., 2024a). A similar regulatory role of SIRT1 has been observed in shortwave blue light (SWBL)-induced cataracts. SWBL irradiation suppressed SIRT1 expression, promoted nuclear translocation of NF-κB and activation of the NLRP3 inflammasome, leading to lens epithelial cells pyroptosis and cataract formation (Ji et al., 2024). Furthermore, under hypoxic conditions, NF-κB signaling is regulated by HIF-1α, which acts as a positive regulator of NLRP3 inflammasome actuvation and ASC oligomerization in human dental pulp fibroblasts (Wang D. et al., 2024).
4.2 NLRC4
Much less is known about the role of NF-κB in the activation of the NLRC4 inflammasome. In intestinal epithelial cells, the activation of the NLRC4 inflammasome and subsequent pyroptosis induced by flagellin from the gram-positive bacterium Clostridioides difficile is mediated by the NF-κB pathway (Chebly et al., 2022; Keestra-Gounder and Nagao, 2023). Unlike in macrophages, inhibition of IKKα in intestinal epithelial cells leads to decreased expression of pro-casapase-1 and interleukin genes, suggesting a role of NF-κB in NLRC4 activation in this context (Chebly et al., 2022). Further evidence supporting a role of the NF-κB pathway in NLRC4 inflammasome activation is provided by findings that TLR4 inhibition in HK-2 cells (an immortalized proximal tubule epithelial cell line) reduced NLRC4 inflammasome activation, as indicated by decreased protein levels of NLRC4, Caspase-1, ASC, and IL-1β (Dong et al., 2024b). Moreover, NF-κB signaling has been demonstrated to negatively regulate NLRC4-induced cell death. Pretreatment of macrophages with NF-κB activators blocks the NLRC4/Caspase-8-dependent cell death, likely through the initiation of transcriptional anti-apoptotic responses (Lee et al., 2018). Anaplasma Phagocytophilum has been reported to activate the NLRC4 inflammasome in macrophages, mediated by RIPK2 through activating NF-κB signaling upon infection, which promoted COX2 expression. COX2 contributed to the production of PGE2, which activated the NLRC4 inflammasome. (Wang et al., 2016). However, we found the evidence provided by the authors on the involvement of NLRC4 activation is not convincing. Experiments using cells deficient in NLRC4 should be included as controls.
4.3 AIM2
A few studies reported a role of the NF-κB pathway in regulating the AIM2 inflammasome activity, however, the exact mechanism was not clearly defined. A correlation, rather thatn causation, was normally reported. In human papilloma virus (HPV)-infected cervical cancer cells, Sirtuin 1 (SIRT1) enabled HPV-infected cervical cancer cells to continue growing by nullifying AIM2 inflammasome-mediated immunity. Mechanistically, SIRT1 repressed the NF-κB-driven transcription of the AIM2 gene by destabilizing the RELB mRNA (So et al., 2018). In an LPS induced acute lung injury model, HMGB1 functioned through the TLR2/TLR4 dependent NF-κB signaling pathways to regulate AIM2 inflammasome activation (Wang J. et al., 2020). The mRNA levels of AIM2, ASC and Caspase-1 are upregulated upon administration of recombinant HMGB1. Additionally, ozanimod, a sphingosine 1-phosphate (S1P) receptor modulator, has been shown to suppress intracerebral hemorrhage (ICH) and neuroinflammation by inhibiting the SIRT3/NF-κB/AIM2 pathway. This inhibition is evidenced by the downregulation of AIM2 expression level, less cleaved Caspase-1, and reduced levels of NF-κB signaling pathways, as indicated by decreased p65 levels (Li et al., 2023). Similarly, Nicorandil, an ATP-sensitive potassium channel opener, was reported to protect against neuroinflammation in ischemic stroke by suppressing the NF-κB/AIM2/GSDMD pathway (Zhao C. et al., 2024). Treatment with channel moculators was found to affect inflammatory cytokine secretion and pyroptosis-related protein expression, including AIM2, while at the same time lead to changes of the level of p-NF-κB p65, NF-κB p65, and p-IκBα. But a direct regulation of NF-kB on AIM2 is lacking. Moreover, Pseudorabies virus (PRV) infection enhances mRNA levels and protein expression of pro-IL-1β by targeting the TLR-NF-κB axis. The AIM2 inflammasome, activated by PRV genomic DNA, is essential for mature IL-1β secretion both in vivo and in vitro. AIM2-deficient mice displayed heightened susceptibility to PRV infection, indicating a critical role of AIM2-mediated inflammatory responses in host defense against PRV infection. Notably, multiple TLR receptors (including TLR2,3,4 and 5), participate in pro-IL-1β upregulation, all capable of activating NF-κB upon PRV infection (Zhou et al., 2023). While a direct role of NF-κB on AIM2 was not reported, the two pathways are clearly integrated function collaboratively to fend off virus infection.
5 Regulation of pyroptosis through NF-κB
Many studies have highlighted the connection between NF-κB signaling and pyroptosis in the pathogenesis of various diseases, including IBD, diabetic kidney disease (DKD), osteoarthritis (OA), myocardial infarction, rheumatoid arthritis, and neurodegenerative diseases (Yu H. et al., 2021; Yang et al., 2021; Xu et al., 2020; Lei et al., 2018; Erdem et al., 2024), making NF-κB inhibitors potential therapeutic agents for relevant diseases. Recent research has shed light on mechanisms by which NF-κB signaling regulates pyroptosis in different cell types/context.
The role of NF-κB in tumorigenesis involves both inhibition and promotion of gasdermin-mediated pyroptosis. On one hand, NF-κB activation may facilitate pyroptosis and demonstrate anti-tumor effects. For instance, increased NF-κB expression, induced by metformin-triggered AMPK/SIRT1 signaling, activated Bax and promoted cytochrome c release, subsequently initiated Caspase-3 activation and GSDME cleavage, resulting in pyroptosis in various cancer cells (Zheng et al., 2020). Additionally, polyphyllin VI (PPVI), a primary saponin derived from traditional Chinese medicine, exhibited potent anti-tumor effects through the stimulation of cytosolic ROS production, which activated the NF-κB/NLRP3/GSDMD pathway and led to pyroptosis in non-small cell lung cancer (NSCLC) (Teng et al., 2020). GALNT6 knockdown inhibited pancreatic ductal adenocarcinoma (PDAC) cell growth through promoting NF-κB phosphorylation and subsequently stimulating the expression of NLRP3, GSDMD and GSDME to augment pyroptosis (Ding et al., 2023). On the other hand, inhibition of NF-κB signaling has also been found to promote tumor cell pyroptosis and impede tumor development. For instance, the piperlongumine analogue L50377 suppressed cell growth and promoted pyroptosis, through ROS-mediated NF-κB inhibition in NSCLC (Li et al., 2019). Tanshinone II A also prevented NF-κB activation and enhanced the miR-145/Caspase-3/GSDMD pathway mediated pyroptosis in Hela cells, thereby repressing cervical cancer progression (Tong et al., 2020). Moreover, the tumor suppressor DRD2 restricts NF-κB signaling, triggering GSDME mediated pyroptosis and exerting anti-tumor effects in breast cancer (Tan et al., 2021). The seemingly controversial conclusions could reflect the complexity of NF-κB signaling in different contexts.
In adipose tissue from LPS treated or obese mice, the transcription of multiple inflammasome components, including GSDMD, was significantly enhanced. The GSDMD promoter contains two NF-κB binding sites, and the transcription of GSDMD was blocked when either one of the two binding sites was mutated. Inhibition of NF-κB using melatonin was found to inhibit inflammasome activation and GSDMD induced pyroptosis (Liu Z. et al., 2017). Interferon regulatory factor 1 (IRF1) has been found to play a compensatory role with IRF2 in regulating GSDMD gene expression and pyroptosis in EA. hy926 endothelial cells (Kayagaki et al., 2019). Recent findings further elucidate that IRF1 was crucial in upregulating the transcription of GSDMD through binding at the −526/-515 site in GSDMD promotor. IRF1 is transcriptionally regulated by the p52/RelB heterodimer, which is activated by the non-canonical NF-κB signaling pathways mediated by NIK. Downregulation of GSDMD expression was observed in IRF1-mutant or NIK siRNA transfected endothelial cells. Silencing NIK or knockdown of p100/p52 impeded pyroptosis mediated by NLRP3 signals, underscoring the significance of the noncanonical NF-κB signaling in NLRP3 inflammasome-mediated pyroptosis in endothelial cells (Fan X. et al., 2023). Overall, the regulation of GSDMD expression through NF-κB signaling appears to occur at the transcription level in specific cells, likely attributable to the low basal levels of GSDMD in those cells.
In macrophages, TLR-mediated priming signals redirect cell death from apoptosis towards pyroptosis during wild-type Yersinia pseudotuberculosis infection, and this process depends upon an intact Yesinia T3SS (Bergsbaken and Cookson, 2007). Bergsbaken and Cookson found that activation of TLR-mediated signaling prior to infection simultaneously rendered macrophages susceptible to killing by YopJ deficient Yersinia and induced Caspase-1 dependent pyroptosis in macrophages. Inhibition of the TLR/NF-κB pathway has been proven to suppress pyroptosis (Bergsbaken and Cookson, 2007). Wang et al. showed that TLR4/NF-κB signaling induces GSDMD-mediated pyroptosis in tubular epithelial cells and contributed to the progression of diabetic kidney disease. In vivo, treatment of db/db mice (mice under diabetic conditions) with TLR4 inhibitor decreased the cleavage of Caspase-1 and GSDMD in renal cortex tissue, protected tubular cell injury by suppressing pyroptosis (Wang et al., 2019). In vitro, TLR4 or NF-κB inhibition led to a significant decrease of phosphorylated-p65, Caspase-1 p20 and GSDMD-NT in high glucose stimulated HK-2 cells, indicating that NF-κB was the downstream signal molecule of TLR4 under high glucose conditions and the activation of pyroptosis in HK-2 cells was markedly inhibited by TLR4/NF-κB signaling inhibition.
The inflammatory cytokines, like IL-1β, IL-33 and TNFα also play a role in regulating pyroptosis through NF-κB signaling pathway. The biological effects of IL-1β are mediated through IL-1R. In nucleus pulposus cells, IL-1β treatment induced NLRP3 inflammasome priming and activation, and formed a positive feedback loop through NLRP3 inflammasome activation via the IL-1β/IL-1R/NF-κB pathway (Chen et al., 2020). Similarly, Cai et al. found that cadmium induced NLRP3 inflammasome-dependent cerebral pyroptosis through the TRAF6/NF-κB pathway. And this NLRP3 inflammasome-dependent cerebral pyroptosis contributes to aggravating the inflammatory response through activating the IL-1β/IL-1R/IκBα/NF-κB/NLRP3 inflammasome feedback loop in the brain of swine (Cai et al., 2021). In alveolar macrophages, LPS-TLR4 signaling not only induced IL-1β maturation and release through NLRP3 activation, but also upregulated IL-1R1 expression on alveolar macrophage surface through MyD88 and NF-κB dependent pathway. At the same time, IL-1β was able to increase ASC speck formation and Caspase-1 activation in the alveolar macrophage in WT and Nlrp3−/− mice, but not in the alveolar macrophage from IL-1R1−/− mice. This indicates an important role of IL-1β/IL-1R1 signaling in mediating alveolar macrophage pyroptosis (He X. et al., 2016). IL-1β released from pyroptosis could lead to further activation of inflammasomes in neighboring cells, thereby promoting additional pyroptosis (Zhou et al., 2021). During pulmonary ischemia-reperfusion injury (IRI), monocytes trigger pyroptosis in human pulmonary microvascular endothelial cells (HPMECs) via IL- 1β secretion (Zhou et al., 2021). In vivo, monocyte depletion attenuated IRI-induced pulmonary edema, cytokine production and pyroptosis activation. In vitro, activation of monocytes (U937 cells) was detected under hypoxia-reoxygenation (H/R) conditions, as indicated by the increased levels of inflammatory factors (IL-1β, IL-6, IL-8, IL-18, and TNF-α) and NLRP3 protein. In a co-culture experiment of U937 cells and HPMECs In under H/R conditions, HPMEC pyroptosis was reduced when NLRP3 or IL-1β was inhibited in monocytes, or knockdown of IL-1R, and inhibition of the NF-κB pathway in HPMECs, suggesting that the IL-1R/NF-κB/NLRP3 pathway mediated HPMEC pyroptosis.
Other members of the IL-1 family, most prominently IL-1α and IL-33, are constitutively produced by non-immune cells, frequently by barrier cells such as keratinocytes or endothelial cells (Martin, 2016). The precursor forms of IL-1α and IL-33 are already biologically active if released into the tissue. A recent research revealed that IL-33 can induce macrophage pyroptosis in septic mice via the NF-κB/p38 MAPK signaling pathway (Ke and Cai, 2021). In the sepsis model of IL-33−/− transgenic mice, there was significant reduction of the NF-κB/p38 MAPK signaling, mitigation of macrophage pyroptosis, and a reduction in the mortality rate of septic mice. IL-33 is considered crucial for human mucosal epithelial allergic responses. A recent study characterized the effects of Dermatophagoides pteronyssinus 1 (DerP1), one of the most common allergens from mites that causes ocular allergies, in mice and human corneal epithelial cells (HCECs). In vivo, DerP1 triggered the activation of the NLRP3 inflammasome, release of inflammatory cytokines and IL-33, in mouse corneas. In vitro, pyroptotic bodies were found in the HCECs after sensitization with DerP1. Deficiency of IL-33 attenuated DerP1 induce pyroptosis in HCECs, indicating that the IL-33 feedback signaling contributed to pyroptosis (Ran et al., 2023).
Recent investigations have uncovered the involvement of TNF/TNFR signaling in the regulation of pyroptotic cell death, which occurs in the absence of X-linked inhibitor of apoptosis (XIAP). XIAP is a member of the IAP (inhibitor of apoptosis proteins) family, initially identified for its ability to inhibit caspases that execute apoptosis. XIAP regulates cell death triggered by various stimuli and mediates inflammatory signaling (Witt et al., 2023; Knop et al., 2019; Shemin et al., 1989). It has been proved that XIAP is a strong stimulator of NF-κB (Hofer-Warbinek et al., 2000). In the absence of XIAP, pathogen ligands or TNFα stimulated the extrinsic apoptotic Caspase-8 activation in a TNF/TNFR signaling dependent manner in dendritic cells, macrophages and neutrophils (Lawlor et al., 2017; Lawlor et al., 2015; Vince et al., 2012; Yabal et al., 2014; Wicki et al., 2016). Activated Caspase-8 can process precursor IL-1β directly in addition to inducing NLRP3 inflammasome assembly, leading to Caspase-1 activation and subsequent processing of relevant substrates. Recently, Hughes et al. reported that two human patients with XIAP deficiency suffered from inflammatory bowel disease, exhibiting heightened Caspase-8-activation and increased IL-1β and GSDMD processing in peripheral blood mononuclear cells (PBMCs) and inflamed colonic mucosae (Hughes et al., 2023). Using a XIAP deficient model, they demonstrated that extrinsic Caspase-8 activation led to the processing of multiple downstream apoptotic and pyroptotic effectors, which act redundantly to cause both excess cell death and inflammatory IL-1β release. Moreover, upon anti-TNF therapy with immune suppression for the patient, the levels of cleaved Caspase-8 and GSDMD detected in patient were substantially reduced (Hughes et al., 2023). Also in the absence of XIAP, TNFR2-specific activation (triggered by TNC-sc (mu)TNF80) led to a NF-κB driven transcriptional profile similar as TNFR1 activation with the exception of upregulation of NLRP3 and Caspase-11. Activation and upregulation of the canonical inflammasome upon loss of XIAP were mediated by RIPK1 kinase activity and ROS production (Knop et al., 2019).
Additionally, NF-κB signaling plays a key role in regulating noncanonical pyroptosis. The noncanonical pyroptosis pathway involves the direct activation of Caspase-11 by cytosolic LPS. Like NLRP3, the activation of Caspase-11 also requires an initial increase in the level of its inactive precursor, pro-Caspase-11. The mechanisms controlling the transcriptional priming for the Caspase-11 display diversity (Agnew et al., 2021; Downs et al., 2020). Caspase-11 expression is initially undetectable in resting macrophages but can be transcriptionally regulated via NF-κB pathways. Induction of pro-Caspase-11 expression by LPS or IFN-γ occurred through TLR4/TRIF signaling and subsequent binding of NF-κB and STAT to the Caspase-11 promoter (Schauvliege et al., 2002). Consistent with this conclusion, a reduction in pro-Caspase-11 levels has been observed when the NF-κB signaling pathway is inhibited through various methods in LPS-treated cells (Kobori et al., 2004; Hu et al., 2024; Appleton et al., 1987). Therefore, we speculate that various other molecules that are capable of activating the NF-κB signaling pathway may also upregulate Caspase-11 expression.
The crosstalk between the NF-κB pathway and PANoptosis (a combined mechanism of pyroptosis, apoptosis, and necroptosis) through the cGAS-STING pathway in the context of lung inflammation and acute respiratory distress syndrome (ARDS) has been reported (Messaoud-Nacer et al., 2022). Activation of STING by diABZI, a synthetic agonist, triggers NF-κB signaling and TBK1/IRF3 phosphorylation, leading to the production of pro-inflammatory cytokines (TNF-α, IL-6) and type I interferons. This initiates cell death through PANoptosis mechanisms, including necroptosis via MLKL phosphorylation, apoptosis through Caspase-3 activation, and pyroptosis mediated by Gasdermin D cleavage following inflammasome activation (NLRP3, AIM2). Cell death releases self-dsDNA, which is sensed by cGAS to produce 2′3′-cGAMP, resulting in STING hyperactivation and an amplified inflammatory response. This feedback loop, marked by increased DNA sensor expression (IFI204, DDX41) and inflammasome activity, sustains NF-κB-driven cytokine production and enhances PANoptosis. The interplay between STING signaling and PANoptosis underscores the potent inflammatory potential of this pathway, with implications for therapeutic strategies targeting STING in infection or cancer that must balance immune activation with the risk of excessive inflammation and ARDS.
6 Inflammasome modulates inflammation via NF-κB
NLPR3 activation has been shown to trigger NF-κB activation (Cai et al., 2024; Kinoshita et al., 2015b). Kinoshita and colleagues observed reduced NF-κB activation and cytokine production in response to Staphylococcus aureus infection in NLRP3 knocking down THP-1 cells. Notably, this effect is not affected by a Caspase-1 inhibitor. In addition, the NF-κB activation observed is not mediated through an autocrine mechanism in response to newly synthesized IL-1β. Although the precise mechanism by which NLRP3 regulates NF-κB activation is not elucidated, these findings highlight a role of NLRP3 in inducing inflammation in the innate immune system (Kinoshita et al., 2015a).
NLRC4, much like NLRP3, plays a crucial role in regulating various innate immune pathways, including the NF-κB pathway. NLRC4 was found to cooperate with ASC to activate NF-κB (Masumoto et al., 2003). Activation of NF-κB by cytosolic flagellin is evidenced by events including p65 phosphorylation, nuclear translocation and IκB-α degradation. Interestingly, Caspase-1 is shown dispensable for NF-κB activation in this condition, but crucial for robust NOS2 expression. This is achieved through cleavage of the chromatin regulator PARP1 and enhanced chromatin accessibility of the NF-κB binding sites on the Nos2 promoter (Buzzo et al., 2017).
Elevated glucose levels have been shown to upregulate NLRC4 expression and induces its phosphorylation on Ser533, both in diabetic mice and macrophages (Zhang et al., 2019). Furthermore, high glucose levels lead to increased NLRC4-dependent NF-κB activation, resulting in the induction of a senescence-associated secretory phenotype (Zhang et al., 2019; Chien et al., 2011; Salminen et al., 2012). In the context of septic shock, silencing NLRC4 has been found to decrease levels of TNF-α and IL-6, two key pro-inflammatory cytokines mediated by the NF-κB pathway (Wang SS. et al., 2021). However, the precise mechanisms behind NLRC4-dependent NF-κB activation remains unclear. Further investigation is needed to determine whether the release of IL-1β following NLRC4 inflammasome activation initiates a feedback loop that activates the NF-κB pathway (Bent et al., 2018; Dey et al., 2014).
The AIM2 inflammasome plays a protective role by reducing pro-inflammatory response in cardiomyocytes through regulating NF-κB activation. AIM2-deficient cells exhibit increased NF-κB activation, as evidenced by increased IκBα degradation, p65 phosphorylation and acetylation, following stimulation with IFN-γ and LPS (Furrer et al., 2016). Mechanistically, AIM2 indirectly restricts NF-κB p65-dependent pro-inflammatory cytokine transcription, primarily by blocking STAT1 phosphorylation. The inhibitory effect of AIM2 is attenuated in the absence of STAT1.
Many inflammasomes, including NLRP3, NLRC4, and AIM2 require the adaptor protein ASC for inflammasome assembly and activation. However, the role of ASC in NF-κB activity appears more complex. High doses of ASC transfection inhibit RIP2 and Caspase-1 induced NF-κB activity (Sarkar et al., 2006), while other studies suggest that ASC mediates NF-κB activation, which requires the catalytic activity of Caspase-8 (Hasegawa et al., 2005). This dual role of ASC in regulating NF-κB signaling may reflect a balance between the interaction of ASC with its many binding/functional partners.
7 Conclusion
The ever-expanding collection of studies has significantly advanced our understanding of pyroptosis regulation. Among the myriad of pathways influencing pyroptosis, the intricate interplay between pyroptosis and NF-κB signaling stands out as a critical axis in orchestrating inflammatory responses and governing cell death mechanisms. While much is known about NF-κB’s role in driving the production of pro-inflammatory cytokines and inflammasome components, questions persist regarding the context-dependent activation of specific NF-κB subunits and their downstream targets in pyroptosis. Additionally, how NF-κB signaling interacts with other cellular pathways to fine-tune pyroptotic responses under varying physiological and pathological conditions remains incompletely elucidated.
Understanding these regulatory mechanisms holds profound implications for the development of targeted therapeutic strategies. Dysregulated pyroptosis and NF-κB activation contribute to the progression of numerous inflammatory and autoimmune disorders, as well as infectious and malignant diseases. Thus, a better understanding of NF-κB’s multifaceted role in pyroptosis could unlock new avenues for mitigating excessive inflammatory responses, controlling tissue damage, and improving outcomes in inflammatory pathologies.
Future research should prioritize identifying the specific NF-κB components involved in pyroptosis regulation, delineating the spatiotemporal dynamics of NF-κB activation in different cellular contexts, and unraveling the feedback mechanisms that balance pyroptosis induction and resolution. Furthermore, integrating systems biology approaches with in vivo models of disease may offer holistic insights into the interplay between NF-κB signaling and pyroptosis. By bridging these knowledge gaps, we can advance not only our fundamental understanding of these pathways but also their translational potential in the development of innovative anti-inflammatory therapies.
Author contributions
LY: Writing–original draft, Writing–review and editing. YaZ: Writing–original draft, Writing–review and editing. ZC: Writing–original draft, Writing–review and editing. YuZ: Writing–original draft, Writing–review and editing. ZL: Conceptualization, Funding acquisition, Resources, Writing–original draft, Writing–review and editing. YW: Conceptualization, Funding acquisition, Resources, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The research was supported by the National Institutes of Health (R01 HL142640 and R01GM132443 to YW and ZL, and R01HL146744 and R01HL167767 to ZL). The funder was not involved in the design and execution of the study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Generative AI statement
The author(s) declare that no Generative AI was used in the creation of this manuscript.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abd-Ellah, A., Voogdt, C., Krappmann, D., Möller, P., and Marienfeld, R. B. (2018). GSK3β modulates NF-κB activation and RelB degradation through site-specific phosphorylation of BCL10. Sci. Rep. 8 (1), 1352. doi:10.1038/s41598-018-19822-z
Agnew, A., Nulty, C., and Creagh, E. M. (2021). Regulation, activation and function of caspase-11 during health and disease. Int. J. Mol. Sci. 22 (4), 1506. doi:10.3390/ijms22041506
Aizawa, E., Karasawa, T., Watanabe, S., Komada, T., Kimura, H., Kamata, R., et al. (2020). GSDME-dependent incomplete pyroptosis permits selective IL-1α release under caspase-1 inhibition. iScience 23 (5), 101070. doi:10.1016/j.isci.2020.101070
Alad, M., Grant, M. P., Epure, L. M., Shih, S. Y., Merle, G., Im, H. J., et al. (2024). Short link N modulates inflammasome activity in intervertebral discs through interaction with CD14. Biomolecules 14 (10), 1312. doi:10.3390/biom14101312
Allam, R., Lawlor, K. E., Yu, E. C., Mildenhall, A. L., Moujalled, D. M., Lewis, R. S., et al. (2014). Mitochondrial apoptosis is dispensable for NLRP3 inflammasome activation but non-apoptotic caspase-8 is required for inflammasome priming. EMBO Rep. 15 (9), 982–990. doi:10.15252/embr.201438463
Andrade, W. A., and Zamboni, D. S. (2020). NLRC4 biology in immunity and inflammation. J. Leukoc. Biol. 108 (4), 1117–1127. doi:10.1002/JLB.3MR0420-573R
Anilkumar, S., and Wright-Jin, E. (2024). NF-κB as an inducible regulator of inflammation in the central nervous system. Cells 13 (6), 485. doi:10.3390/cells13060485
Antar, S. A., Abdo, W., Helal, A. I., Abduh, M. S., Hakami, Z. H., Germoush, M. O., et al. (2024). Coenzyme Q10 mitigates cadmium cardiotoxicity by downregulating NF-κB/NLRP3 inflammasome axis and attenuating oxidative stress in mice. Life Sci. 348, 122688. doi:10.1016/j.lfs.2024.122688
Appleton, C. P., Hatle, L. K., and Popp, R. L. (1987). Superior vena cava and hepatic vein Doppler echocardiography in healthy adults. J. Am. Coll. Cardiol. 10 (5), 1032–1039. doi:10.1016/s0735-1097(87)80343-1
Asare, Y., Shnipova, M., Živković, L., Schlegl, C., Tosato, F., Aronova, A., et al. (2022). IKKβ binds NLRP3 providing a shortcut to inflammasome activation for rapid immune responses. Signal Transduct. Target Ther. 7 (1), 355. doi:10.1038/s41392-022-01189-3
Aubert, D. F., Xu, H., Yang, J., Shi, X., Gao, W., Li, L., et al. (2016). A burkholderia type VI effector deamidates Rho GTPases to activate the pyrin inflammasome and trigger inflammation. Cell Host Microbe 19 (5), 664–674. doi:10.1016/j.chom.2016.04.004
Baatarjav, C., Komada, T., Karasawa, T., Yamada, N., Sampilvanjil, A., Matsumura, T., et al. (2022). dsDNA-induced AIM2 pyroptosis halts aberrant inflammation during rhabdomyolysis-induced acute kidney injury. Cell Death Differ. 29 (12), 2487–2502. doi:10.1038/s41418-022-01033-9
Baeuerle, P. A., and Baltimore, D. (1988). IκB: a specific inhibitor of the NF-κB transcription factor. Science 242 (4878), 540–546. doi:10.1126/science.3140380
Balka, K. R., and De Nardo, D. (2019). Understanding early TLR signaling through the Myddosome. J. Leukoc. Biol. 105 (2), 339–351. doi:10.1002/jlb.Mr0318-096r
Balka, K. R., Louis, C., Saunders, T. L., Smith, A. M., Calleja, D. J., D'Silva, D. B., et al. (2020). TBK1 and IKKε act redundantly to mediate STING-induced NF-κB responses in myeloid cells. Cell Rep. 31 (1), 107492. doi:10.1016/j.celrep.2020.03.056
Barnabei, L., Laplantine, E., Mbongo, W., Rieux-Laucat, F., and Weil, R. (2021). NF-κB: at the borders of autoimmunity and inflammation. Front. Immunol. 12, 716469. doi:10.3389/fimmu.2021.716469
Bauernfeind, F. G., Horvath, G., Stutz, A., Alnemri, E. S., MacDonald, K., Speert, D., et al. (2009). Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J. Immunol. 183 (2), 787–791. doi:10.4049/jimmunol.0901363
Bedoui, S., Herold, M. J., and Strasser, A. (2020). Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 21 (11), 678–695. doi:10.1038/s41580-020-0270-8
Ben Khalifa, Y., Luco, S., Besson, B., Sonthonnax, F., Archambaud, M., Grimes, J. M., et al. (2016). The matrix protein of rabies virus binds to RelAp43 to modulate NF-κB-dependent gene expression related to innate immunity. Sci. Rep. 6 (1), 39420. doi:10.1038/srep39420
Bent, R., Moll, L., Grabbe, S., and Bros, M. (2018). Interleukin-1 beta-A friend or foe in malignancies? Int. J. Mol. Sci. 19 (8), 2155. doi:10.3390/ijms19082155
Bergsbaken, T., and Cookson, B. T. (2007). Macrophage activation redirects yersinia-infected host cell death from apoptosis to caspase-1-dependent pyroptosis. PLoS Pathog. 3 (11), e161. doi:10.1371/journal.ppat.0030161
Beutler, B., Hoebe, K., Du, X., and Ulevitch, R. J. (2003). How we detect microbes and respond to them: the Toll-like receptors and their transducers. J. Leukoc. Biol. 74 (4), 479–485. doi:10.1189/jlb.0203082
Bezbradica, J. S., Coll, R. C., and Schroder, K. (2017). Sterile signals generate weaker and delayed macrophage NLRP3 inflammasome responses relative to microbial signals. Cell Mol. Immunol. 14 (1), 118–126. doi:10.1038/cmi.2016.11
Billman, Z. P., Kovacs, S. B., Wei, B., Kang, K., Cissé, O. H., and Miao, E. A. (2024). Caspase-1 activates gasdermin A in non-mammals. Elife 12. doi:10.7554/eLife.92362
Boaru, S. G., Borkham-Kamphorst, E., Van de Leur, E., Lehnen, E., Liedtke, C., and Weiskirchen, R. (2015). NLRP3 inflammasome expression is driven by NF-κB in cultured hepatocytes. Biochem. Biophys. Res. Commun. 458 (3), 700–706. doi:10.1016/j.bbrc.2015.02.029
Bombaci, G., Sarangdhar, M. A., Andina, N., Tardivel, A., Yu, E. C., Mackie, G. M., et al. (2022). LRR-protein RNH1 dampens the inflammasome activation and is associated with COVID-19 severity. Life Sci. Alliance 5 (6), e202101226. doi:10.26508/lsa.202101226
Boraschi, D., Italiani, P., Weil, S., and Martin, M. U. (2018). The family of the interleukin-1 receptors. Immunol. Rev. 281 (1), 197–232. doi:10.1111/imr.12606
Borghi, A., Verstrepen, L., and Beyaert, R. (2016). TRAF2 multitasking in TNF receptor-induced signaling to NF-κB, MAP kinases and cell death. Biochem. Pharmacol. 116, 1–10. doi:10.1016/j.bcp.2016.03.009
Brenner, D., Blaser, H., and Mak, T. W. (2015). Regulation of tumour necrosis factor signalling: live or let die. Nat. Rev. Immunol. 15 (6), 362–374. doi:10.1038/nri3834
Broz, P., Pelegrin, P., and Shao, F. (2020). The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 20 (3), 143–157. doi:10.1038/s41577-019-0228-2
Buzzo, C. L., Medina, T., Branco, L. M., Lage, S. L., Ferreira, L. C., Amarante-Mendes, G. P., et al. (2017). Epigenetic regulation of nitric oxide synthase 2, inducible (Nos2) by NLRC4 inflammasomes involves PARP1 cleavage. Sci. Rep. 7, 41686. doi:10.1038/srep41686
Cabal-Hierro, L., and Lazo, P. S. (2012). Signal transduction by tumor necrosis factor receptors. Cell Signal 24 (6), 1297–1305. doi:10.1016/j.cellsig.2012.02.006
Cai, G., Song, X., Luo, H., Dai, G., Zhang, H., Jiang, D., et al. (2024). NLRP3 blockade by MCC950 suppressed osteoclastogenesis via NF-κB/c-Fos/NFATc1 signal pathway and alleviated bone loss in diabetes mellitus. Mol. Cell Endocrinol. 594, 112382. doi:10.1016/j.mce.2024.112382
Cai, J., Guan, H., Jiao, X., Yang, J., Chen, X., Zhang, H., et al. (2021). NLRP3 inflammasome mediated pyroptosis is involved in cadmium exposure-induced neuroinflammation through the IL-1β/IkB-α-NF-κB-NLRP3 feedback loop in swine. Toxicology 453, 152720. doi:10.1016/j.tox.2021.152720
Capece, D., Verzella, D., Flati, I., Arboretto, P., Cornice, J., and Franzoso, G. (2022). NF-κB: blending metabolism, immunity, and inflammation. Trends Immunol. 43 (9), 757–775. doi:10.1016/j.it.2022.07.004
Chavarría-Smith, J., and Vance, R. E. (2013). Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PLoS Pathog. 9 (6), e1003452. doi:10.1371/journal.ppat.1003452
Chebly, H., Marvaud, J. C., Safa, L., Elkak, A. K., Kobeissy, P. H., Kansau, I., et al. (2022). Clostridioides difficile flagellin activates the intracellular NLRC4 inflammasome. Int. J. Mol. Sci. 23 (20), 12366. doi:10.3390/ijms232012366
Chen, F., Jiang, G., Liu, H., Li, Z., Pei, Y., Wang, H., et al. (2020). Melatonin alleviates intervertebral disc degeneration by disrupting the IL-1β/NF-κB-NLRP3 inflammasome positive feedback loop. Bone Res. 8, 10. doi:10.1038/s41413-020-0087-2
Chen, F., Zou, L., Williams, B., and Chao, W. (2021). Targeting toll-like receptors in sepsis: from bench to clinical trials. Antioxid. Redox Signal 35 (15), 1324–1339. doi:10.1089/ars.2021.0005
Chen, J., Li, Q., Hong, Y., Zhou, X., Yu, C., Tian, X., et al. (2023). Inhibition of the NF-κB signaling pathway alleviates pyroptosis in bladder epithelial cells and neurogenic bladder fibrosis. Int. J. Mol. Sci. 24 (13), 11160. doi:10.3390/ijms241311160
Chen, K. W., and Brodsky, I. E. (2023). Yersinia interactions with regulated cell death pathways. Curr. Opin. Microbiol. 71, 102256. doi:10.1016/j.mib.2022.102256
Chen, K. W., Demarco, B., Heilig, R., Shkarina, K., Boettcher, A., Farady, C. J., et al. (2019b). Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 38 (10), e101638. doi:10.15252/embj.2019101638
Chen, Q., Shi, P., Wang, Y., Zou, D., Wu, X., Wang, D., et al. (2019c). GSDMB promotes non-canonical pyroptosis by enhancing caspase-4 activity. J. Mol. Cell Biol. 11 (6), 496–508. doi:10.1093/jmcb/mjy056
Chen, Q., Sun, L., and Chen, Z. J. (2016). Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 17 (10), 1142–1149. doi:10.1038/ni.3558
Chen, X., Liu, G., Yuan, Y., Wu, G., Wang, S., and Yuan, L. (2019a). NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. 10 (12), 906. doi:10.1038/s41419-019-2157-1
Chien, Y., Scuoppo, C., Wang, X., Fang, X., Balgley, B., Bolden, J. E., et al. (2011). Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev. 25 (20), 2125–2136. doi:10.1101/gad.17276711
Cohen, L., Henzel, W. J., and Baeuerle, P. A. (1998). IKAP is a scaffold protein of the IkappaB kinase complex. Nature 395 (6699), 292–296. doi:10.1038/26254
David, L., Borges, J. P., Hollingsworth, L. R., Volchuk, A., Jansen, I., Garlick, E., et al. (2024). NINJ1 mediates plasma membrane rupture by cutting and releasing membrane disks. Cell 187 (9), 2224–2235.e16. doi:10.1016/j.cell.2024.03.008
Decout, A., Katz, J. D., Venkatraman, S., and Ablasser, A. (2021). The cGAS-STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21 (9), 548–569. doi:10.1038/s41577-021-00524-z
Degen, M., Santos, J. C., Pluhackova, K., Cebrero, G., Ramos, S., Jankevicius, G., et al. (2023). Structural basis of NINJ1-mediated plasma membrane rupture in cell death. Nature 618 (7967), 1065–1071. doi:10.1038/s41586-023-05991-z
Deng, W., Bai, Y., Deng, F., Pan, Y., Mei, S., Zheng, Z., et al. (2022). Author Correction: Streptococcal pyrogenic exotoxin B cleaves GSDMA and triggers pyroptosis. Nature. 608 (7923), E28. doi:10.1038/s41586-022-05109-x
Dey, N., Sinha, M., Gupta, S., Gonzalez, M. N., Fang, R., Endsley, J. J., et al. (2014). Caspase-1/ASC inflammasome-mediated activation of IL-1β-ROS-NF-κB pathway for control of Trypanosoma cruzi replication and survival is dispensable in NLRP3-/- macrophages. PLoS One 9 (11), e111539. doi:10.1371/journal.pone.0111539
DiDonato, J. A., Hayakawa, M., Rothwarf, D. M., Zandi, E., and Karin, M. (1997). A cytokine-responsive IkappaB kinase that activates the transcription factor NF-kappaB. Nature 388 (6642), 548–554. doi:10.1038/41493
Ding, J., Wang, K., Liu, W., She, Y., Sun, Q., Shi, J., et al. (2016). Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535 (7610), 111–116. doi:10.1038/nature18590
Ding, M., Liu, J., Lv, H., Zhu, Y., Chen, Y., Peng, H., et al. (2023). Knocking down GALNT6 promotes pyroptosis of pancreatic ductal adenocarcinoma cells through NF-κB/NLRP3/GSDMD and GSDME signaling pathway. Front. Oncol. 13, 1097772. doi:10.3389/fonc.2023.1097772
Ding, R., Li, H., Liu, Y., Ou, W., Zhang, X., Chai, H., et al. (2022). Activating cGAS-STING axis contributes to neuroinflammation in CVST mouse model and induces inflammasome activation and microglia pyroptosis. J. Neuroinflammation 19 (1), 137. doi:10.1186/s12974-022-02511-0
Domizio, J. D., Gulen, M. F., Saidoune, F., Thacker, V. V., Yatim, A., Sharma, K., et al. (2022). The cGAS-STING pathway drives type I IFN immunopathology in COVID-19. Nature 603 (7899), 145–151. doi:10.1038/s41586-022-04421-w
Dondelinger, Y., Jouan-Lanhouet, S., Divert, T., Theatre, E., Bertin, J., Gough, P. J., et al. (2015). NF-κB-Independent role of ikkα/ikkβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60 (1), 63–76. doi:10.1016/j.molcel.2015.07.032
Dong, W., Luo, M., Li, Y., Chen, X., Li, L., and Chang, Q. (2024b). MICT ameliorates hypertensive nephropathy by inhibiting TLR4/NF-κB pathway and down-regulating NLRC4 inflammasome. PLoS One 19 (7), e0306137. doi:10.1371/journal.pone.0306137
Dong, W., Zhang, K., Wang, X., Li, J., Zou, H., Yuan, Y., et al. (2024a). SIRT1 alleviates Cd nephrotoxicity through NF-κB/p65 deacetylation-mediated pyroptosis in rat renal tubular epithelial cells. Sci. Total Environ. 929, 172392. doi:10.1016/j.scitotenv.2024.172392
Dorrington, M. G., and Fraser, I. D. (2019). NF-κB signaling in macrophages: dynamics, crosstalk, and signal integration. Front. Immunol. 10, 705. doi:10.3389/fimmu.2019.00705
Dower, S. K., Kronheim, S. R., March, C. J., Conlon, P. J., Hopp, T. P., Gillis, S., et al. (1985). Detection and characterization of high affinity plasma membrane receptors for human interleukin 1. J. Exp. Med. 162 (2), 501–515. doi:10.1084/jem.162.2.501
Downs, K. P., Nguyen, H., Dorfleutner, A., and Stehlik, C. (2020). An overview of the non-canonical inflammasome. Mol. Asp. Med. 76, 100924. doi:10.1016/j.mam.2020.100924
Du, Y., Hu, Z., Luo, Y., Wang, H. Y., Yu, X., and Wang, R. F. (2023). Function and regulation of cGAS-STING signaling in infectious diseases. Front. Immunol. 14, 1130423. doi:10.3389/fimmu.2023.1130423
Duan, T., Du, Y., Xing, C., Wang, H. Y., and Wang, R. F. (2022). Toll-like receptor signaling and its role in cell-mediated immunity. Front. Immunol. 13, 812774. doi:10.3389/fimmu.2022.812774
Erdem, M., Erdem, S., Alver, A., Kiran, T. R., and Karahan, S. C. (2024). β2-adrenoceptor agonist formoterol attenuates NLRP3 inflammasome activation and GSDMD-mediated pyroptosis in microglia through enhancing IκBα/NF-κB inhibition, SQSTM1/p62-dependent selective autophagy and ESCRT-III-mediated plasma membrane repair. Mol. Cell Neurosci. 130, 103956. doi:10.1016/j.mcn.2024.103956
Fan, X., Li, Q., Wang, Y., Zhang, D. M., Zhou, J., Chen, Q., et al. (2023b). Non-canonical NF-κB contributes to endothelial pyroptosis and atherogenesis dependent on IRF-1. Transl. Res. 255, 1–13. doi:10.1016/j.trsl.2022.11.001
Fan, Y., Guan, B., Xu, J., Zhang, H., Yi, L., and Yang, Z. (2023a). Role of toll-like receptor-mediated pyroptosis in sepsis-induced cardiomyopathy. Biomed. Pharmacother. 167, 115493. doi:10.1016/j.biopha.2023.115493
Fattinger, S. A., Maurer, L., Geiser, P., Bernard, E. M., Enz, U., Ganguillet, S., et al. (2023). Gasdermin D is the only Gasdermin that provides protection against acute Salmonella gut infection in mice. Proc. Natl. Acad. Sci. U. S. A. 120 (48), e2315503120. doi:10.1073/pnas.2315503120
Fischer, F. A., Mies, L. F. M., Nizami, S., Pantazi, E., Danielli, S., Demarco, B., et al. (2021). TBK1 and IKKε act like an OFF switch to limit NLRP3 inflammasome pathway activation. Proc. Natl. Acad. Sci. U. S. A. 118 (38), e2009309118. doi:10.1073/pnas.2009309118
Fitzgerald, K. A., and Kagan, J. C. (2020). Toll-like receptors and the control of immunity. Cell 180 (6), 1044–1066. doi:10.1016/j.cell.2020.02.041
Flannery, S., and Bowie, A. G. (2010). The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem. Pharmacol. 80 (12), 1981–1991. doi:10.1016/j.bcp.2010.06.020
Frank, D., and Vince, J. E. (2019). Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 26 (1), 99–114. doi:10.1038/s41418-018-0212-6
Furrer, A., Hottiger, M. O., and Valaperti, A. (2016). Absent in Melanoma 2 (AIM2) limits pro-inflammatory cytokine transcription in cardiomyocytes by inhibiting STAT1 phosphorylation. Mol. Immunol. 74, 47–58. doi:10.1016/j.molimm.2016.04.009
García-García, V. A., Alameda, J. P., Fernández-Aceñero, M. J., Navarro, M., García-Escudero, R., Page, A., et al. (2024). Nuclear versus cytoplasmic IKKα signaling in keratinocytes leads to opposite skin phenotypes and inflammatory responses, and a different predisposition to cancer. Oncogene. doi:10.1038/s41388-024-03203-0
Garlanda, C., Dinarello, C. A., and Mantovani, A. (2013). The interleukin-1 family: back to the future. Immunity 39 (6), 1003–1018. doi:10.1016/j.immuni.2013.11.010
Gough, P., and Myles, I. A. (2020). Tumor necrosis factor receptors: pleiotropic signaling complexes and their differential effects. Front. Immunol. 11, 585880. doi:10.3389/fimmu.2020.585880
Gram, A. M., Booty, L. M., and Bryant, C. E. (2019). Chopping GSDMD: caspase-8 has joined the team of pyroptosis-mediating caspases. EMBO J. 38 (10), e102065. doi:10.15252/embj.2019102065
Gray, C. M., Remouchamps, C., McCorkell, K. A., Solt, L. A., Dejardin, E., Orange, J. S., et al. (2014). Noncanonical NF-κB signaling is limited by classical NF-κB activity. Sci. Signal. 7 (311), ra13–ra. doi:10.1126/scisignal.2004557
Grell, M., Douni, E., Wajant, H., Löhden, M., Clauss, M., Maxeiner, B., et al. (1995). The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83 (5), 793–802. doi:10.1016/0092-8674(95)90192-2
Greten, F. R., Arkan, M. C., Bollrath, J., Hsu, L. C., Goode, J., Miething, C., et al. (2007). NF-kappaB is a negative regulator of IL-1beta secretion as revealed by genetic and pharmacological inhibition of IKKbeta. Cell 130 (5), 918–931. doi:10.1016/j.cell.2007.07.009
Gurung, P., Anand, P. K., Malireddi, R. K., Vande Walle, L., Van Opdenbosch, N., Dillon, C. P., et al. (2014). FADD and caspase-8 mediate priming and activation of the canonical and noncanonical Nlrp3 inflammasomes. J. Immunol. 192 (4), 1835–1846. doi:10.4049/jimmunol.1302839
Haas, T. L., Emmerich, C. H., Gerlach, B., Schmukle, A. C., Cordier, S. M., Rieser, E., et al. (2009). Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36 (5), 831–844. doi:10.1016/j.molcel.2009.10.013
Han, J. H., Karki, R., Malireddi, R. K. S., Mall, R., Sarkar, R., Sharma, B. R., et al. (2024). NINJ1 mediates inflammatory cell death, PANoptosis, and lethality during infection conditions and heat stress. Nat. Commun. 15 (1), 1739. doi:10.1038/s41467-024-45466-x
Hao, Q. Y., Yan, J., Wei, J. T., Zeng, Y. H., Feng, L. Y., Que, D. D., et al. (2024). Prevotella copri promotes vascular calcification via lipopolysaccharide through activation of NF-κB signaling pathway. Gut Microbes 16 (1), 2351532. doi:10.1080/19490976.2024.2351532
Hasegawa, M., Imamura, R., Kinoshita, T., Matsumoto, N., Masumoto, J., Inohara, N., et al. (2005). ASC-mediated NF-kappaB activation leading to interleukin-8 production requires caspase-8 and is inhibited by CLARP. J. Biol. Chem. 280 (15), 15122–15130. doi:10.1074/jbc.M412284200
Hayden, M. S., and Ghosh, S. (2008). Shared principles in NF-kappaB signaling. Cell 132 (3), 344–362. doi:10.1016/j.cell.2008.01.020
He, W. T., Wan, H., Hu, L., Chen, P., Wang, X., Huang, Z., et al. (2015). Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res. 25 (12), 1285–1298. doi:10.1038/cr.2015.139
He, X., Qian, Y., Li, Z., Fan, E. K., Li, Y., Wu, L., et al. (2016b). TLR4-Upregulated IL-1β and IL-1RI promote alveolar macrophage pyroptosis and lung inflammation through an autocrine mechanism. Sci. Rep. 6, 31663. doi:10.1038/srep31663
He, Y., Hara, H., and Núñez, G. (2016a). Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41 (12), 1012–1021. doi:10.1016/j.tibs.2016.09.002
Hinz, M., Arslan, S. Ç., and Scheidereit, C. (2012). It takes two to tango: IκBs, the multifunctional partners of NF-κB. Immunol. Rev. 246 (1), 59–76. doi:10.1111/j.1600-065X.2012.01102.x
Hofer-Warbinek, R., Schmid, J. A., Stehlik, C., Binder, B. R., Lipp, J., and de Martin, R. (2000). Activation of NF-kappa B by XIAP, the X chromosome-linked inhibitor of apoptosis, in endothelial cells involves TAK1. J. Biol. Chem. 275 (29), 22064–22068. doi:10.1074/jbc.M910346199
Holbrook, J., Lara-Reyna, S., Jarosz-Griffiths, H., and McDermott, M. (2019). Tumour necrosis factor signalling in health and disease. F1000Res 8. doi:10.12688/f1000research.17023.1
Hou, J., Zhao, R., Xia, W., Chang, C. W., You, Y., Hsu, J. M., et al. (2020a). PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 22 (10), 1264–1275. doi:10.1038/s41556-020-0575-z
Hou, L., Yang, L., Chang, N., Zhao, X., Zhou, X., Dong, C., et al. (2020b). Macrophage sphingosine 1-phosphate receptor 2 blockade attenuates liver inflammation and fibrogenesis triggered by NLRP3 inflammasome. Front. Immunol. 11, 1149. doi:10.3389/fimmu.2020.01149
Hsu, H., Xiong, J., and Goeddel, D. V. (1995). The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 81 (4), 495–504. doi:10.1016/0092-8674(95)90070-5
Hu, A., Xiao, F., Wu, W., Xu, H., and Su, J. (2024). LincRNA-EPS inhibits caspase-11 and NLRP3 inflammasomes in gingival fibroblasts to alleviate periodontal inflammation. Cell Prolif. 57 (1), e13539. doi:10.1111/cpr.13539
Hu, H., Brittain, G. C., Chang, J.-H., Puebla-Osorio, N., Jin, J., Zal, A., et al. (2013). OTUD7B controls non-canonical NF-κB activation through deubiquitination of TRAF3. Nature 494 (7437), 371–374. doi:10.1038/nature11831
Hu, W., Jain, A., Gao, Y., Dozmorov, I. M., Mandraju, R., Wakeland, E. K., et al. (2015). Differential outcome of TRIF-mediated signaling in TLR4 and TLR3 induced DC maturation. Proc. Natl. Acad. Sci. U. S. A. 112 (45), 13994–13999. doi:10.1073/pnas.1510760112
Hu, Y., Liu, Y., Zong, L., Zhang, W., Liu, R., Xing, Q., et al. (2023). The multifaceted roles of GSDME-mediated pyroptosis in cancer: therapeutic strategies and persisting obstacles. Cell Death Dis. 14 (12), 836. doi:10.1038/s41419-023-06382-y
Hughes, S. A., Lin, M., Weir, A., Huang, B., Xiong, L., Chua, N. K., et al. (2023). Caspase-8-driven apoptotic and pyroptotic crosstalk causes cell death and IL-1β release in X-linked inhibitor of apoptosis (XIAP) deficiency. Embo J. 42 (5), e110468. doi:10.15252/embj.2021110468
Israël, A. (2010). The IKK complex, a central regulator of NF-kappaB activation. Cold Spring Harb. Perspect. Biol. 2 (3), a000158. doi:10.1101/cshperspect.a000158
Jang, D. I., Lee, A. H., Shin, H. Y., Song, H. R., Park, J. H., Kang, T. B., et al. (2021). The role of tumor necrosis factor alpha (TNF-α) in autoimmune disease and current TNF-α inhibitors in therapeutics. Int. J. Mol. Sci. 22 (5), 2719. doi:10.3390/ijms22052719
Ji, Z., Zhang, D., Wang, Y., Liu, X., Wang, M., Zhu, X., et al. (2024). The role of the SIRT1/NF-κB/NLRP3 pathway in the pyroptosis of lens epithelial cells under shortwave blue light radiation. Exp. Eye Res. 246, 110019. doi:10.1016/j.exer.2024.110019
Jin, J., Xiao, Y., Chang, J.-H., Yu, J., Hu, H., Starr, R., et al. (2012). The kinase TBK1 controls IgA class switching by negatively regulating noncanonical NF-κB signaling. Nat. Immunol. 13 (11), 1101–1109. doi:10.1038/ni.2423
Kamanova, J., Kofronova, O., Masin, J., Genth, H., Vojtova, J., Linhartova, I., et al. (2008). Adenylate cyclase toxin subverts phagocyte function by RhoA inhibition and unproductive ruffling. J. Immunol. 181 (8), 5587–5597. doi:10.4049/jimmunol.181.8.5587
Karin, M., and Delhase, M. (2000). The I kappa B kinase (IKK) and NF-kappa B: key elements of proinflammatory signalling. Semin. Immunol. 12 (1), 85–98. doi:10.1006/smim.2000.0210
Karin, M., and Lin, A. (2002). NF-kappaB at the crossroads of life and death. Nat. Immunol. 3 (3), 221–227. doi:10.1038/ni0302-221
Kawai, T., and Akira, S. (2007a). TLR signaling. Semin. Immunol. 19 (1), 24–32. doi:10.1016/j.smim.2006.12.004
Kawai, T., and Akira, S. (2007b). Signaling to NF-kappaB by toll-like receptors. Trends Mol. Med. 13 (11), 460–469. doi:10.1016/j.molmed.2007.09.002
Kayagaki, N., Kornfeld, O. S., Lee, B. L., Stowe, I. B., O'Rourke, K., Li, Q., et al. (2021). NINJ1 mediates plasma membrane rupture during lytic cell death. Nature 591 (7848), 131–136. doi:10.1038/s41586-021-03218-7
Kayagaki, N., Lee, B. L., Stowe, I. B., Kornfeld, O. S., O'Rourke, K., Mirrashidi, K. M., et al. (2019). IRF2 transcriptionally induces GSDMD expression for pyroptosis. Sci. Signal 12 (582), eaax4917. doi:10.1126/scisignal.aax4917
Kayagaki, N., Stowe, I. B., Lee, B. L., O'Rourke, K., Anderson, K., Warming, S., et al. (2015). Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 526 (7575), 666–671. doi:10.1038/nature15541
Ke, J., and Cai, G. (2021). Effect of IL-33 on pyroptosis of macrophages in mice with sepsis via NF-κB/p38 MAPK signaling pathway. Acta Cir. Bras. 36 (5), e360501. doi:10.1590/acb360501
Keestra-Gounder, A. M., and Nagao, P. E. (2023). Inflammasome activation by Gram-positive bacteria: mechanisms of activation and regulation. Front. Immunol. 14, 1075834. doi:10.3389/fimmu.2023.1075834
Kinoshita, T., Imamura, R., Kushiyama, H., and Suda, T. (2015a). NLRP3 mediates NF-κB activation and cytokine induction in microbially induced and sterile inflammation. PLoS One 10 (3), e0119179. doi:10.1371/journal.pone.0119179
Kinoshita, T., Imamura, R., Kushiyama, H., and Suda, T. (2015b). NLRP3 mediates NF-κB activation and cytokine induction in microbially induced and sterile inflammation. PLoS One 10 (3), e0119179. doi:10.1371/journal.pone.0119179
Knop, J., Spilgies, L. M., Rufli, S., Reinhart, R., Vasilikos, L., Yabal, M., et al. (2019). TNFR2 induced priming of the inflammasome leads to a RIPK1-dependent cell death in the absence of XIAP. Cell. Death. Dis. 10 (700). doi:10.1038/s41419-019-1938-x
Kobori, M., Yang, Z., Gong, D., Heissmeyer, V., Zhu, H., Jung, Y. K., et al. (2004). Wedelolactone suppresses LPS-induced caspase-11 expression by directly inhibiting the IKK complex. Cell Death Differ. 11 (1), 123–130. doi:10.1038/sj.cdd.4401325
Kucharczak, J., Simmons, M. J., Fan, Y., and Gélinas, C. (2003). To be, or not to be: NF-kappaB is the answer--role of Rel/NF-kappaB in the regulation of apoptosis. Oncogene 22 (56), 8961–8982. doi:10.1038/sj.onc.1207230
Kumar, H., Kawai, T., and Akira, S. (2009). Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun. 388 (4), 621–625. doi:10.1016/j.bbrc.2009.08.062
LaRock, D. L., Johnson, A. F., Wilde, S., Sands, J. S., Monteiro, M. P., and LaRock, C. N. (2022). Group A Streptococcus induces GSDMA-dependent pyroptosis in keratinocytes. Nature 605 (7910), 527–531. doi:10.1038/s41586-022-04717-x
Latz, E., Verma, A., Visintin, A., Gong, M., Sirois, C. M., Klein, D. C., et al. (2007). Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat. Immunol. 8 (7), 772–779. doi:10.1038/ni1479
Lawlor, K. E., Feltham, R., Yabal, M., Conos, S. A., Chen, K. W., Ziehe, S., et al. (2017). XIAP loss triggers RIPK3- and caspase-8-driven IL-1β activation and cell death as a consequence of TLR-MyD88-induced cIAP1-TRAF2 degradation. Cell Rep. 20 (3), 668–682. doi:10.1016/j.celrep.2017.06.073
Lawlor, K. E., Khan, N., Mildenhall, A., Gerlic, M., Croker, B. A., D'Cruz, A. A., et al. (2015). RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun. 6, 6282. doi:10.1038/ncomms7282
Lee, B. L., Mirrashidi, K. M., Stowe, I. B., Kummerfeld, S. K., Watanabe, C., Haley, B., et al. (2018). ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Sci. Rep. 8 (1), 3788. doi:10.1038/s41598-018-21998-3
Lei, Q., Yi, T., and Chen, C. (2018). NF-κB-Gasdermin D (GSDMD) Axis couples oxidative stress and NACHT, LRR and PYD domains-containing protein 3 (NLRP3) inflammasome-mediated cardiomyocyte pyroptosis following myocardial infarction. Med. Sci. Monit. 24, 6044–6052. doi:10.12659/msm.908529
Lei, X., Zhang, Z., Xiao, X., Qi, J., He, B., and Wang, J. (2017). Enterovirus 71 inhibits pyroptosis through cleavage of gasdermin D. J. Virol. 91 (18). doi:10.1128/jvi.01069-17
Lemmers, B., Salmena, L., Bidère, N., Su, H., Matysiak-Zablocki, E., Murakami, K., et al. (2007). Essential role for caspase-8 in Toll-like receptors and NFkappaB signaling. J. Biol. Chem. 282 (10), 7416–7423. doi:10.1074/jbc.M606721200
Li, L. R., Chen, L., and Sun, Z. J. (2024a). Igniting hope: harnessing NLRP3 inflammasome-GSDMD-mediated pyroptosis for cancer immunotherapy. Life Sci. 354, 122951. doi:10.1016/j.lfs.2024.122951
Li, Q., Chen, L., Dong, Z., Zhao, Y., Deng, H., Wu, J., et al. (2019). Piperlongumine analogue L50377 induces pyroptosis via ROS mediated NF-κB suppression in non-small-cell lung cancer. Chem. Biol. Interact. 313, 108820. doi:10.1016/j.cbi.2019.108820
Li, S., Jiang, L., Beckmann, K., Højen, J. F., Pessara, U., Powers, N. E., et al. (2021). A novel anti-human IL-1R7 antibody reduces IL-18-mediated inflammatory signaling. J. Biol. Chem. 296, 100630. doi:10.1016/j.jbc.2021.100630
Li, X., Zhang, H., Zheng, W., Sun, J., Wang, L., and He, Z. (2023). Ozanimod-dependent activation of SIRT3/NF-κB/AIM2 pathway attenuates secondary injury after intracerebral hemorrhage. Mol. Neurobiol. 60 (3), 1117–1131. doi:10.1007/s12035-022-03137-2
Li, Y., Zhuang, Y., Chen, Y., Wang, G., Tang, Z., Zhong, Y., et al. (2024b). Euphorbia factor L2 alleviated gouty inflammation by specifically suppressing both the priming and activation of NLRP3 inflammasome. Int. Immunopharmacol. 138, 112598. doi:10.1016/j.intimp.2024.112598
Liao, Q., Yang, Y., Li, Y., Zhang, J., Fan, K., Guo, Y., et al. (2024). Targeting TANK-binding kinase 1 attenuates painful diabetic neuropathy via inhibiting microglia pyroptosis. Cell Commun. Signal 22 (1), 368. doi:10.1186/s12964-024-01723-6
Liu, G., and Gack, M. U. (2020). Distinct and orchestrated functions of RNA sensors in innate immunity. Immunity 53 (1), 26–42. doi:10.1016/j.immuni.2020.03.017
Liu, J., Zhou, J., Luan, Y., Li, X., Meng, X., Liao, W., et al. (2024). cGAS-STING, inflammasomes and pyroptosis: an overview of crosstalk mechanism of activation and regulation. Cell Commun. Signal 22 (1), 22. doi:10.1186/s12964-023-01466-w
Liu, T., Zhang, L., Joo, D., and Sun, S. C. (2017a). NF-κB signaling in inflammation. Signal Transduct. Target Ther. 2, 17023. doi:10.1038/sigtrans.2017.23
Liu, Z., Gan, L., Xu, Y., Luo, D., Ren, Q., Wu, S., et al. (2017b). Melatonin alleviates inflammasome-induced pyroptosis through inhibiting NF-κB/GSDMD signal in mice adipose tissue. J. Pineal Res. 63 (1). doi:10.1111/jpi.12414
Lu, J., Hou, Y., Liu, S. X., Jin, B., Liu, J., Li, N., et al. (2024). Acetyl-CoA synthetase 2 induces pyroptosis and inflammation of renal epithelial tubular cells in sepsis-induced acute kidney injury by upregulating the KLF5/NF-κB pathway. Cell Commun. Signal 22 (1), 187. doi:10.1186/s12964-024-01556-3
Luco, S., Delmas, O., Vidalain, P.-O., Tangy, F., Weil, R., and Bourhy, H. (2012). RelAp43, a member of the NF-κB family involved in innate immune response against Lyssavirus infection. PLoS Pathog. 8 (12), e1003060. doi:10.1371/journal.ppat.1003060
Ma, M., Jiang, W., and Zhou, R. (2024). DAMPs and DAMP-sensing receptors in inflammation and diseases. Immunity 57 (4), 752–771. doi:10.1016/j.immuni.2024.03.002
MacEwan, D. J. (2002). TNF ligands and receptors--a matter of life and death. Br. J. Pharmacol. 135 (4), 855–875. doi:10.1038/sj.bjp.0704549
Martin, S. J. (2016). Cell death and inflammation: the case for IL-1 family cytokines as the canonical DAMPs of the immune system. Febs J. 283 (14), 2599–2615. doi:10.1111/febs.13775
Martinon, F., Burns, K., and Tschopp, J. (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10 (2), 417–426. doi:10.1016/s1097-2765(02)00599-3
Masumoto, J., Dowds, T. A., Schaner, P., Chen, F. F., Ogura, Y., Li, M., et al. (2003). ASC is an activating adaptor for NF-kappa B and caspase-8-dependent apoptosis. Biochem. Biophys. Res. Commun. 303 (1), 69–73. doi:10.1016/s0006-291x(03)00309-7
McKee, C. M., and Coll, R. C. (2020). NLRP3 inflammasome priming: a riddle wrapped in a mystery inside an enigma. J. Leukoc. Biol. 108 (3), 937–952. doi:10.1002/jlb.3mr0720-513r
Medler, J., and Wajant, H. (2019). Tumor necrosis factor receptor-2 (TNFR2): an overview of an emerging drug target. Expert Opin. Ther. Targets 23 (4), 295–307. doi:10.1080/14728222.2019.1586886
Messaoud-Nacer, Y., Culerier, E., Rose, S., Maillet, I., Rouxel, N., Briault, S., et al. (2022). STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS). Cell Death Dis. 13 (3), 269. doi:10.1038/s41419-022-04664-5
Miao, E. A., Leaf, I. A., Treuting, P. M., Mao, D. P., Dors, M., Sarkar, A., et al. (2010). Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11 (12), 1136–1142. doi:10.1038/ni.1960
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114 (2), 181–190. doi:10.1016/s0092-8674(03)00521-x
Moresco, E. M., LaVine, D., and Beutler, B. (2011). Toll-like receptors. Curr. Biol. 21 (13), R488–R493. PubMed PMID: 21741580. doi:10.1016/j.cub.2011.05.039
Motwani, M., Pesiridis, S., and Fitzgerald, K. A. (2019). DNA sensing by the cGAS-STING pathway in health and disease. Nat. Rev. Genet. 20 (11), 657–674. doi:10.1038/s41576-019-0151-1
Mulero, M. C., Huxford, T., and Ghosh, G. (2019). NF-κB, IκB, and IKK: integral components of immune system signaling. Struct. Immunol. 1172, 207–226. doi:10.1007/978-981-13-9367-9_10
Nanda, S. K., Prescott, A. R., Figueras-Vadillo, C., and Cohen, P. (2021). IKKβ is required for the formation of the NLRP3 inflammasome. EMBO Rep. 22 (10), e50743. doi:10.15252/embr.202050743
Ng, J., Hirota, S. A., Gross, O., Li, Y., Ulke-Lemee, A., Potentier, M. S., et al. (2010). Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139 (2), 542–552. doi:10.1053/j.gastro.2010.04.005
Nurmi, K., Silventoinen, K., Keskitalo, S., Rajamaki, K., Kouri, V. P., Kinnunen, M., et al. (2024). Truncating NFKB1 variants cause combined NLRP3 inflammasome activation and type I interferon signaling and predispose to necrotizing fasciitis. Cell Rep. Med. 5 (4), 101503. doi:10.1016/j.xcrm.2024.101503
Oeckinghaus, A., Hayden, M. S., and Ghosh, S. (2011). Crosstalk in NF-κB signaling pathways. Nat. Immunol. 12 (8), 695–708. doi:10.1038/ni.2065
O'Neill, L. A., and Bowie, A. G. (2007). The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7 (5), 353–364. doi:10.1038/nri2079
Orning, P., Weng, D., Starheim, K., Ratner, D., Best, Z., Lee, B., et al. (2018). Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362 (6418), 1064–1069. doi:10.1126/science.aau2818
Pandeya, A., Zhang, Y., Cui, J., Yang, L., Li, J., Zhang, G., et al. (2023). Inflammasome activation and pyroptosis mediate coagulopathy and inflammation in Salmonella systemic infection. Microbiol. Res. 275, 127460. doi:10.1016/j.micres.2023.127460
Panganiban, R. A., Sun, M., Dahlin, A., Park, H. R., Kan, M., Himes, B. E., et al. (2018). A functional splice variant associated with decreased asthma risk abolishes the ability of gasdermin B to induce epithelial cell pyroptosis. J. Allergy Clin. Immunol. 142 (5), 1469–1478. doi:10.1016/j.jaci.2017.11.040
Planès, R., Pinilla, M., Santoni, K., Hessel, A., Passemar, C., Lay, K., et al. (2022). Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol. Cell 82 (13), 2385–2400.e9. doi:10.1016/j.molcel.2022.04.033
Qiao, Y., Wang, P., Qi, J., Zhang, L., and Gao, C. (2012). TLR-induced NF-κB activation regulates NLRP3 expression in murine macrophages. FEBS Lett. 586 (7), 1022–1026. doi:10.1016/j.febslet.2012.02.045
Ran, S., Shu, Q., and Gao, X. (2023). Dermatophagoides pteronyssinus 1 (DerP1) may trigger NLRP3-mediated corneal epithelial cell pyroptosis by elevating interleukin-33 expression levels. Curr. Eye Res. 48 (12), 1100–1111. doi:10.1080/02713683.2023.2250583
Rathinam, V. A., and Fitzgerald, K. A. (2016). Inflammasome complexes: emerging mechanisms and effector functions. Cell 165 (4), 792–800. doi:10.1016/j.cell.2016.03.046
Rathinam, V. A. K., Zhao, Y., and Shao, F. (2019). Innate immunity to intracellular LPS. Nat. Immunol. 20 (5), 527–533. doi:10.1038/s41590-019-0368-3
Razani, B., Zarnegar, B., Ytterberg, A. J., Shiba, T., Dempsey, P. W., Ware, C. F., et al. (2010). Negative feedback in noncanonical NF-kappaB signaling modulates NIK stability through IKKalpha-mediated phosphorylation. Sci. Signal. 3 (123), ra41–ra. doi:10.1126/scisignal.2000778
Richter, C., Messerschmidt, S., Holeiter, G., Tepperink, J., Osswald, S., Zappe, A., et al. (2012). The tumor necrosis factor receptor stalk regions define responsiveness to soluble versus membrane-bound ligand. Mol. Cell Biol. 32 (13), 2515–2529. doi:10.1128/mcb.06458-11
Rodriguez, B. N., Huang, H., Chia, J. J., and Hoffmann, A. (2024). The noncanonical NFκB pathway: regulatory mechanisms in health and disease. WIREs Mech. Dis. 16, e1646. doi:10.1002/wsbm.1646
Rodríguez, M., Cabal-Hierro, L., Carcedo, M. T., Iglesias, J. M., Artime, N., Darnay, B. G., et al. (2011). NF-kappaB signal triggering and termination by tumor necrosis factor receptor 2. J. Biol. Chem. 286 (26), 22814–22824. doi:10.1074/jbc.M111.225631
Rothe, M., Sarma, V., Dixit, V. M., and Goeddel, D. V. (1995). TRAF2-mediated activation of NF-kappa B by TNF receptor 2 and CD40. Science 269 (5229), 1424–1427. doi:10.1126/science.7544915
Rothwarf, D. M., Zandi, E., Natoli, G., and Karin, M. (1998). IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395 (6699), 297–300. doi:10.1038/26261
Ruspi, G., Schmidt, E. M., McCann, F., Feldmann, M., Williams, R. O., Stoop, A. A., et al. (2014). TNFR2 increases the sensitivity of ligand-induced activation of the p38 MAPK and NF-κB pathways and signals TRAF2 protein degradation in macrophages. Cell Signal 26 (4), 683–690. doi:10.1016/j.cellsig.2013.12.009
Salminen, A., Kauppinen, A., and Kaarniranta, K. (2012). Emerging role of NF-κB signaling in the induction of senescence-associated secretory phenotype (SASP). Cell Signal 24 (4), 835–845. doi:10.1016/j.cellsig.2011.12.006
Sandstrom, A., Mitchell, P. S., Goers, L., Mu, E. W., Lesser, C. F., and Vance, R. E. (2019). Functional degradation: a mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science. 364 (6435), eaau1330. doi:10.1126/science.aau1330
Sarhan, J., Liu, B. C., Muendlein, H. I., Li, P., Nilson, R., Tang, A. Y., et al. (2018). Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc. Natl. Acad. Sci. U. S. A. 115 (46), E10888–E10897. doi:10.1073/pnas.1809548115
Sarkar, A., Duncan, M., Hart, J., Hertlein, E., Guttridge, D. C., and Wewers, M. D. (2006). ASC directs NF-kappaB activation by regulating receptor interacting protein-2 (RIP2) caspase-1 interactions. J. Immunol. 176 (8), 4979–4986. doi:10.4049/jimmunol.176.8.4979
Schauvliege, R., Vanrobaeys, J., Schotte, P., and Beyaert, R. (2002). Caspase-11 gene expression in response to lipopolysaccharide and interferon-gamma requires nuclear factor-kappa B and signal transducer and activator of transcription (STAT) 1. J. Biol. Chem. 277 (44), 41624–41630. doi:10.1074/jbc.M207852200
Schmacke, N. A., O'Duill, F., Gaidt, M. M., Szymanska, I., Kamper, J. M., Schmid-Burgk, J. L., et al. (2022). IKKβ primes inflammasome formation by recruiting NLRP3 to the trans-Golgi network. Immunity 55 (12), 2271–2284.e7. doi:10.1016/j.immuni.2022.10.021
Schroder, K., and Tschopp, J. (2010). The inflammasomes. Cell 140 (6), 821–832. doi:10.1016/j.cell.2010.01.040
Shao, F. (2021). Gasdermins: making pores for pyroptosis. Nat. Rev. Immunol. 21 (10), 620–621. doi:10.1038/s41577-021-00602-2
Sharma, B. R., Karki, R., and Kanneganti, T. D. (2019). Role of AIM2 inflammasome in inflammatory diseases, cancer and infection. Eur. J. Immunol. 49 (11), 1998–2011. doi:10.1002/eji.201848070
Shemin, D., Clark, D. D., and Chazan, J. A. (1989). Unexplained pleural effusions in the peritoneal dialysis population. Perit. Dial. Int. 9 (2), 143. doi:10.1177/089686088900900213
Shen, X., Wang, H., Weng, C., Jiang, H., and Chen, J. (2021). Caspase 3/GSDME-dependent pyroptosis contributes to chemotherapy drug-induced nephrotoxicity. Cell Death Dis. 12 (2), 186. doi:10.1038/s41419-021-03458-5
Shi, J., Gao, W., and Shao, F. (2017). Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem. Sci. 42 (4), 245–254. doi:10.1016/j.tibs.2016.10.004
Shi, J., Zhao, Y., Wang, K., Shi, X., Wang, Y., Huang, H., et al. (2015). Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526 (7575), 660–665. doi:10.1038/nature15514
Shih, V. F.-S., Tsui, R., Caldwell, A., and Hoffmann, A. (2011). A single NFκB system for both canonical and non-canonical signaling. Cell Res. 21 (1), 86–102. doi:10.1038/cr.2010.161
Skopelja-Gardner, S., An, J., and Elkon, K. B. (2022). Role of the cGAS-STING pathway in systemic and organ-specific diseases. Nat. Rev. Nephrol. 18 (9), 558–572. doi:10.1038/s41581-022-00589-6
So, D., Shin, H. W., Kim, J., Lee, M., Myeong, J., Chun, Y. S., et al. (2018). Cervical cancer is addicted to SIRT1 disarming the AIM2 antiviral defense. Oncogene 37 (38), 5191–5204. doi:10.1038/s41388-018-0339-4
Struzik, J., and Szulc-Dąbrowska, L. (2019). Manipulation of non-canonical NF-κB signaling by non-oncogenic viruses. Archivum Immunol. Ther. Exp. 67 (1), 41–48. doi:10.1007/s00005-018-0522-x
Sun, S. C. (2011). Non-canonical NF-κB signaling pathway. Cell Res. 21 (1), 71–85. doi:10.1038/cr.2010.177
Sun, S. C. (2017). The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 17 (9), 545–558. doi:10.1038/nri.2017.52
Tan, Y., Sun, R., Liu, L., Yang, D., Xiang, Q., Li, L., et al. (2021). Tumor suppressor DRD2 facilitates M1 macrophages and restricts NF-κB signaling to trigger pyroptosis in breast cancer. Theranostics 11 (11), 5214–5231. doi:10.7150/thno.58322
Tan, Y. F., Wang, M., Chen, Z. Y., Wang, L., and Liu, X. H. (2020). Inhibition of BRD4 prevents proliferation and epithelial-mesenchymal transition in renal cell carcinoma via NLRP3 inflammasome-induced pyroptosis. Cell Death Dis. 11 (4), 239. doi:10.1038/s41419-020-2431-2
Tartaglia, L. A., Weber, R. F., Figari, I. S., Reynolds, C., Palladino, M. A., and Goeddel, D. V. (1991). The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc. Natl. Acad. Sci. U. S. A. 88 (20), 9292–9296. doi:10.1073/pnas.88.20.9292
Teng, J. F., Mei, Q. B., Zhou, X. G., Tang, Y., Xiong, R., Qiu, W. Q., et al. (2020). Polyphyllin VI induces caspase-1-mediated pyroptosis via the induction of ROS/NF-κB/NLRP3/GSDMD signal Axis in non-small cell lung cancer. Cancers (Basel) 12 (1), 193. doi:10.3390/cancers12010193
Ting, A. T., and Bertrand, M. J. M. (2016). More to life than NF-κB in TNFR1 signaling. Trends Immunol. 37 (8), 535–545. doi:10.1016/j.it.2016.06.002
Tong, W., Guo, J., and Yang, C. (2020). Tanshinone II A enhances pyroptosis and represses cell proliferation of HeLa cells by regulating miR-145/GSDMD signaling pathway. Biosci. Rep. 40 (4). doi:10.1042/bsr20200259
Tsvetkov, P., Coy, S., Petrova, B., Dreishpoon, M., Verma, A., Abdusamad, M., et al. (2022). Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375 (6586), 1254–1261. doi:10.1126/science.abf0529
Unterreiner, A., Rubert, J., Kauffmann, M., Fruhauf, A., Heiser, D., Erbel, P., et al. (2021). Pharmacological inhibition of IKKβ dampens NLRP3 inflammasome activation after priming in the human myeloid cell line THP-1. Biochem. Biophys. Res. Commun. 545:177–82. doi:10.1016/j.bbrc.2021.01.051
Vallabhapurapu, S., and Karin, M. (2009). Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733. doi:10.1146/annurev.immunol.021908.132641
Vallabhapurapu, S., Matsuzawa, A., Zhang, W., Tseng, P. H., Keats, J. J., Wang, H., et al. (2008). Nonredundant and complementary functions of TRAF2 and TRAF3 in a ubiquitination cascade that activates NIK-dependent alternative NF-kappaB signaling. Nat. Immunol. 9 (12), 1364–1370. doi:10.1038/ni.1678
Vandenabeele, P., Declercq, W., Van Herreweghe, F., and Vanden Berghe, T. (2010). The role of the kinases RIP1 and RIP3 in TNF-induced necrosis. Sci. Signal 3 (115), re4. doi:10.1126/scisignal.3115re4
Vince, J. E., Wong, W. W., Gentle, I., Lawlor, K. E., Allam, R., O'Reilly, L., et al. (2012). Inhibitor of apoptosis proteins limit RIP3 kinase-dependent interleukin-1 activation. Immunity 36 (2), 215–227. doi:10.1016/j.immuni.2012.01.012
Wang, C., Deng, L., Hong, M., Akkaraju, G. R., Inoue, J.-i., and Chen, Z. J. (2001). TAK1 is a ubiquitin-dependent kinase of MKK and IKK. Nature 412 (6844), 346–351. doi:10.1038/35085597
Wang, C., Yang, T., Xiao, J., Xu, C., Alippe, Y., Sun, K., et al. (2021a). NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci. Immunol. 6 (64), eabj3859. doi:10.1126/sciimmunol.abj3859
Wang, D., Wang, M., Sun, S., Zhang, C., Song, Y., Li, J., et al. (2024b). Hypoxia-induced NLRP3 inflammasome activation via the HIF-1α/NF-κB signaling pathway in human dental pulp fibroblasts. BMC Oral Health 24 (1), 1156. doi:10.1186/s12903-024-04936-w
Wang, H., Wang, H., Chen, J., Liu, P., and Xiao, X. (2024a). Overexpressed FAM111B degrades GSDMA to promote esophageal cancer tumorigenesis and cisplatin resistance. Cell Oncol. (Dordr) 47 (1), 343–359. doi:10.1007/s13402-023-00871-0
Wang, J., Li, R., Peng, Z., Hu, B., Rao, X., and Li, J. (2020a). HMGB1 participates in LPS-induced acute lung injury by activating the AIM2 inflammasome in macrophages and inducing polarization of M1 macrophages via TLR2, TLR4, and RAGE/NF-κB signaling pathways. Int. J. Mol. Med. 45 (1), 61–80. doi:10.3892/ijmm.2019.4402
Wang, K., Sun, Q., Zhong, X., Zeng, M., Zeng, H., Shi, X., et al. (2020b). Structural mechanism for GSDMD targeting by autoprocessed caspases in pyroptosis. Cell 180 (5), 941–955. doi:10.1016/j.cell.2020.02.002
Wang, L., Du, F., and Wang, X. (2008). TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133 (4), 693–703. doi:10.1016/j.cell.2008.03.036
Wang, S. S., Yan, C. S., and Luo, J. M. (2021b). NLRC4 gene silencing-dependent blockade of NOD-like receptor pathway inhibits inflammation, reduces proliferation and increases apoptosis of dendritic cells in mice with septic shock. Aging (Albany NY) 13 (1), 1440–1457. doi:10.18632/aging.202379
Wang, X., Peng, H., Huang, Y., Kong, W., Cui, Q., Du, J., et al. (2020c). Post-translational modifications of IκBα: the state of the art. Front. Cell Dev. Biol. 8, 574706. doi:10.3389/fcell.2020.574706
Wang, X., Shaw, D. K., Hammond, H. L., Sutterwala, F. S., Rayamajhi, M., Shirey, K. A., et al. (2016). The prostaglandin E2-EP3 receptor Axis regulates anaplasma phagocytophilum-mediated NLRC4 inflammasome activation. PLoS Pathog. 12 (8), e1005803. doi:10.1371/journal.ppat.1005803
Wang, Y., Gao, W., Shi, X., Ding, J., Liu, W., He, H., et al. (2017). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547 (7661), 99–103. doi:10.1038/nature22393
Wang, Y., Zhu, X., Yuan, S., Wen, S., Liu, X., Wang, C., et al. (2019). TLR4/NF-κB signaling induces GSDMD-related pyroptosis in tubular cells in diabetic kidney disease. Front. Endocrinol. (Lausanne) 10, 603. doi:10.3389/fendo.2019.00603
Wei, W., Heng, Y. Y., Wu, F. F., Dong, H. Y., Zhang, P. F., Li, J. X., et al. (2024). Sodium Tanshinone IIA Sulfonate alleviates vascular senescence in diabetic mice by modulating the A20-NFκB-NLRP3 inflammasome-catalase pathway. Sci. Rep. 14 (1), 17665. doi:10.1038/s41598-024-68169-1
Wei, X., Xie, F., Zhou, X., Wu, Y., Yan, H., Liu, T., et al. (2022b). Role of pyroptosis in inflammation and cancer. Cell Mol. Immunol. 19 (9), 971–992. doi:10.1038/s41423-022-00905-x
Wei, Y., Lan, B., Zheng, T., Yang, L., Zhang, X., Cheng, L., et al. (2023). GSDME-mediated pyroptosis promotes the progression and associated inflammation of atherosclerosis. Nat. Commun. 14 (1), 929. doi:10.1038/s41467-023-36614-w
Wei, Y., Yang, L., Pandeya, A., Cui, J., Zhang, Y., and Li, Z. (2022a). Pyroptosis-induced inflammation and tissue damage. J. Mol. Biol. 434 (4), 167301. doi:10.1016/j.jmb.2021.167301
Wicki, S., Gurzeler, U., Wei-Lynn Wong, W., Jost, P. J., Bachmann, D., and Kaufmann, T. (2016). Loss of XIAP facilitates switch to TNFα-induced necroptosis in mouse neutrophils. Cell Death Dis. 7 (10), e2422. doi:10.1038/cddis.2016.311
Witt, A., Goncharov, T., Lee, Y. M., Kist, M., Dohse, M., Eastham, J., et al. (2023). XIAP deletion sensitizes mice to TNF-induced and RIP1-mediated death. Cell Death Dis. 14 (4), 262. doi:10.1038/s41419-023-05793-1
Xiao, G., Harhaj, E. W., and Sun, S. C. (2001a). NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 7 (2), 401–409. doi:10.1016/s1097-2765(01)00187-3
Xiao, G., Harhaj, E. W., and Sun, S.-C. (2001b). NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. cell 7 (2), 401–409. doi:10.1016/s1097-2765(01)00187-3
Xiong, S., Xu, C., Yang, C., Luo, H., Xie, J., Xia, B., et al. (2025). FuKe QianJin capsule alleviates endometritis via inhibiting inflammation and pyroptosis through modulating TLR4/NF-κB/NLRP3 pathway. J. Ethnopharmacol. 337 (Pt 3), 118962. doi:10.1016/j.jep.2024.118962
Xu, G., Dong, F., Su, L., Tan, Z.-X., Lei, M., Li, L., et al. (2024a). The role and therapeutic potential of nuclear factor κB (NF-κB) in ischemic stroke. Biomed. and Pharmacother. 171, 116140. doi:10.1016/j.biopha.2024.116140
Xu, H., Yang, J., Gao, W., Li, L., Li, P., Zhang, L., et al. (2014). Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513 (7517), 237–241. doi:10.1038/nature13449
Xu, J., and Nunez, G. (2023). The NLRP3 inflammasome: activation and regulation. Trends Biochem. Sci. 48 (4), 331–344. doi:10.1016/j.tibs.2022.10.002
Xu, X., Qin, Z., Zhang, C., Mi, X., Zhang, C., Zhou, F., et al. (2023). TRIM29 promotes podocyte pyroptosis in diabetic nephropathy through the NF-kB/NLRP3 inflammasome pathway. Cell Biol. Int. 47 (6), 1126–1135. doi:10.1002/cbin.12006
Xu, Z., Li, J., Su, B., Gao, H., Ren, M., Lin, Y., et al. (2024b). A role of ROS-dependent defects in mitochondrial dynamic and autophagy in carbon black nanoparticle-mediated myocardial cell damage. Free Radic. Biol. Med. 220, 249–261. doi:10.1016/j.freeradbiomed.2024.04.241
Xu, Z., Liu, R., Huang, L., Xu, Y., Su, M., Chen, J., et al. (2020). CD147 aggravated inflammatory bowel disease by triggering NF-κB-Mediated pyroptosis. Biomed. Res. Int. 2020, 5341247. doi:10.1155/2020/5341247
Yabal, M., Müller, N., Adler, H., Knies, N., Groß, C. J., Damgaard, R. B., et al. (2014). XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 7 (6), 1796–1808. doi:10.1016/j.celrep.2014.05.008
Yang, J., Tang, X., Li, B., and Shi, J. (2022). Sphingosine 1-phosphate receptor 2 mediated early stages of pancreatic and systemic inflammatory responses via NF-kappa B activation in acute pancreatitis. Cell Commun. Signal 20 (1), 157. doi:10.1186/s12964-022-00971-8
Yang, P., Feng, W., Li, C., Kou, Y., Li, D., Liu, S., et al. (2021). LPS induces fibroblast-like synoviocytes RSC-364 cells to pyroptosis through NF-κB mediated dual signalling pathway. J. Mol. Histol. 52 (4), 661–669. doi:10.1007/s10735-021-09988-8
Yu, H., Lin, L., Zhang, Z., Zhang, H., and Hu, H. (2020). Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct. Target. Ther. 5 (1), 209. doi:10.1038/s41392-020-00312-6
Yu, H., Yao, S., Zhou, C., Fu, F., Luo, H., Du, W., et al. (2021b). Morroniside attenuates apoptosis and pyroptosis of chondrocytes and ameliorates osteoarthritic development by inhibiting NF-κB signaling. J. Ethnopharmacol. 266, 113447. doi:10.1016/j.jep.2020.113447
Yu, P., Zhang, X., Liu, N., Tang, L., Peng, C., and Chen, X. (2021a). Pyroptosis: mechanisms and diseases. Signal Transduct. Target. Ther. 6 (1), 128. doi:10.1038/s41392-021-00507-5
Zandi, E., and Karin, M. (2023). Bridging the gap: composition, regulation, and physiological function of the IkappaB kinase complex. Mol. Cell. Biol. 19, 4547–4551. doi:10.1128/mcb.19.7.4547
Zarezadeh Mehrabadi, A., Aghamohamadi, N., Khoshmirsafa, M., Aghamajidi, A., Pilehforoshha, M., Massoumi, R., et al. (2022). The roles of interleukin-1 receptor accessory protein in certain inflammatory conditions. Immunology 166 (1), 38–46. doi:10.1111/imm.13462
Zhai, Z., Yang, F., Xu, W., Han, J., Luo, G., Li, Y., et al. (2022). Attenuation of rheumatoid arthritis through the inhibition of tumor necrosis factor-induced caspase 3/gasdermin E-mediated pyroptosis. Arthritis Rheumatol. 74 (3), 427–440. doi:10.1002/art.41963
Zhang, J., Zhang, R., Li, W., Ma, X.-C., Qiu, F., and Sun, C.-P. (2023a). IκB kinase β (IKKβ): structure, transduction mechanism, biological function, and discovery of its inhibitors. Int. J. Biol. Sci. 19 (13), 4181–4203. doi:10.7150/ijbs.85158
Zhang, J. Y., Zhou, B., Sun, R. Y., Ai, Y. L., Cheng, K., Li, F. N., et al. (2021b). The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 31 (9), 980–997. doi:10.1038/s41422-021-00506-9
Zhang, L., Wei, X., Wang, Z., Liu, P., Hou, Y., Xu, Y., et al. (2023b). NF-κB activation enhances STING signaling by altering microtubule-mediated STING trafficking. Cell Rep. 42 (3), 112185. doi:10.1016/j.celrep.2023.112185
Zhang, P., Wang, Q., Nie, L., Zhu, R., Zhou, X., Zhao, P., et al. (2019). Hyperglycemia-induced inflamm-aging accelerates gingival senescence via NLRC4 phosphorylation. J. Biol. Chem. 294 (49), 18807–18819. doi:10.1074/jbc.RA119.010648
Zhang, X., Zhang, P., An, L., Sun, N., Peng, L., Tang, W., et al. (2020a). Miltirone induces cell death in hepatocellular carcinoma cell through GSDME-dependent pyroptosis. Acta Pharm. Sin. B 10 (8), 1397–1413. doi:10.1016/j.apsb.2020.06.015
Zhang, Z., and Lieberman, J. (2020). Lighting a fire on the reef. Sci. Immunol. 5 (54), eabf0905. doi:10.1126/sciimmunol.abf0905
Zhang, Z., Zhang, H., Li, D., Zhou, X., Qin, Q., and Zhang, Q. (2021a). Caspase-3-mediated GSDME induced Pyroptosis in breast cancer cells through the ROS/JNK signalling pathway. J. Cell Mol. Med. 25 (17), 8159–8168. doi:10.1111/jcmm.16574
Zhang, Z., Zhang, Y., Xia, S., Kong, Q., Li, S., Liu, X., et al. (2020b). Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature 579 (7799), 415–420. doi:10.1038/s41586-020-2071-9
Zhao, C., Fu, X., Yang, Z., Zhang, Q., and Zhao, Y. (2024b). ATP-sensitive potassium channel opener, Nicorandil, inhibits NF-κB/AIM2/GSDMD pathway activation to protect against neuroinflammation in ischemic stroke. Neurochem. Int. 179, 105810. doi:10.1016/j.neuint.2024.105810
Zhao, C., and Zhao, W. (2020). NLRP3 inflammasome-A key player in antiviral responses. Front. Immunol. 11, 211. doi:10.3389/fimmu.2020.00211
Zhao, P., Ning, J., Huang, J., and Huang, X. (2024a). Mechanism of Resveratrol on LPS/ATP-induced pyroptosis and inflammatory response in HT29 cells. Autoimmunity 57 (1), 2427094. doi:10.1080/08916934.2024.2427094
Zheng, Z., Bian, Y., Zhang, Y., Ren, G., and Li, G. (2020). Metformin activates AMPK/SIRT1/NF-κB pathway and induces mitochondrial dysfunction to drive caspase3/GSDME-mediated cancer cell pyroptosis. Cell Cycle 19 (10), 1089–1104. doi:10.1080/15384101.2020.1743911
Zhong, Z., Umemura, A., Sanchez-Lopez, E., Liang, S., Shalapour, S., Wong, J., et al. (2016). NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell 164 (5), 896–910. doi:10.1016/j.cell.2015.12.057
Zhou, H., Wertz, I., O'Rourke, K., Ultsch, M., Seshagiri, S., Eby, M., et al. (2004). Bcl10 activates the NF-κB pathway through ubiquitination of NEMO. Nature 427 (6970), 167–171. doi:10.1038/nature02273
Zhou, P., Guo, H., Li, Y., Liu, Q., Qiao, X., Lu, Y., et al. (2021). Monocytes promote pyroptosis of endothelial cells during lung ischemia-reperfusion via IL-1R/NF-κB/NLRP3 signaling. Life Sci. 276, 119402. doi:10.1016/j.lfs.2021.119402
Zhou, Q., Zhang, L., Lin, Q., Liu, H., Ye, G., Liu, X., et al. (2023). Pseudorabies virus infection activates the TLR-NF-κb Axis and AIM2 inflammasome to enhance inflammatory responses in mice. J. Virol. 97 (3), e0000323. doi:10.1128/jvi.00003-23
Zhou, Z., He, H., Wang, K., Shi, X., Wang, Y., Su, Y., et al. (2020). Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 368 (6494), eaaz7548. doi:10.1126/science.aaz7548
Zhu, C., Xu, S., Jiang, R., Yu, Y., Bian, J., and Zou, Z. (2024). The gasdermin family: emerging therapeutic targets in diseases. Signal Transduct. Target Ther. 9 (1), 87. doi:10.1038/s41392-024-01801-8
Zhu, S., Ding, S., Wang, P., Wei, Z., Pan, W., Palm, N. W., et al. (2017). Nlrp9b inflammasome restricts rotavirus infection in intestinal epithelial cells. Nature 546 (7660), 667–670. doi:10.1038/nature22967
Keywords: pyroptosis, NF-κB, inflammasome, infection, inflammation
Citation: Yang L, Zhang Y, Chai Z, Zhou Y, Li Z and Wei Y (2025) Regulation of pyroptosis by NF-κB signaling. Front. Cell Death 3:1503799. doi: 10.3389/fceld.2024.1503799
Received: 29 September 2024; Accepted: 19 December 2024;
Published: 07 January 2025.
Edited by:
Patrick Legembre, University of Limoges, FranceReviewed by:
Bernhard Ryffel, Centre National de la Recherche Scientifique (CNRS), FranceChristelle Vincent-Fabert, UMR7276 Contrôle des réponses immunes B et des lymphoproliférations (CRIBL), France
Copyright © 2025 Yang, Zhang, Chai, Zhou, Li and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yinan Wei, eWluYW4ud2VpQHRhbXUuZWR1; Zhenyu Li, emxpMjFAdGFtdS5lZHU=