 Hans-Martin Herz1
Hans-Martin Herz1 Andreas Bergmann2*
Andreas Bergmann2*- 1Department of Hematology, St. Jude Children’s Research Hospital, Memphis, TN, United States
- 2Department of Molecular, Cell and Cancer Biology, UMass Chan Medical School, Worcester, MA, United States
We conducted an EMS mutagenesis screen on chromosome arm 2L to identify recessive suppressors of GMR-hid-induced apoptosis in the Drosophila eye. Through this screen, we recovered three alleles of the lysine demethylase gene Kdm5. Kdm5, a member of the JmjC-domain-containing protein family, possesses histone demethylase activity towards H3K4me3. Our data suggest that Kdm5 specifically regulates Hid-induced cell death during development, as we did not observe control of Reaper- or Grim-induced cell death by Kdm5. Interestingly, GMR-hid-induced apoptosis is suppressed independently of Kdm5’s demethylase activity. Our findings indicate that Rbf and dMyc are necessary for Kdm5 mosaics to suppress GMR-hid-induced cell death. Moreover, Kdm5 mosaics failed to suppress apoptosis induced by a mutant form of Hid that is resistant to inhibition by Erk-type MAPK activity. Additionally, Kdm5 dominantly enhances the wing phenotype of an activated MAPK mutant. These results collectively suggest that Kdm5 controls Hid-induced apoptosis by regulating the Rbf, dMyc, and MAPK pathways.
Introduction
Programmed cell death is an essential biological process during development and homeostasis of multi-cellular organisms. Apoptosis is the major form of cell death and accounts for more than 90% of all cell death (Newton et al., 2024). Apoptosis is evolutionarily conserved and is critical for the health of the organism. Alterations in the rate of apoptosis are linked to cancer, auto-immune diseases and neurodegeneration (Kayagaki et al., 2024). Therefore, a detailed understanding of the control of apoptosis is necessary to develop strategies for treatment of these diseases.
Control of apoptosis comes down to the control of caspases, a class of highly specialized Cys proteases which execute apoptosis when activated (Shalini et al., 2015). In living cells, caspases are inhibited by inhibitor-of-apoptosis proteins (IAPs) which directly bind to caspases and inhibit their activity (Gyrd-Hansen and Meier, 2010). In response to apoptosis-inducing signals, IAPs are degraded and release caspases for further activation. In Drosophila, the IAP-antagonists Reaper, Hid and Grim stimulate the ubiquitylation and degradation of Drosophila IAP1 (DIAP1) resulting in the release of the initiator caspase Dronc (Caspase-9-ortholog in Drosophila) from DIAP1 inhibition (Orme and Meier, 2009). Free Dronc is activated by incorporation into the apoptosome by the Apaf-1-like protein Dark (Dorstyn et al., 2018). Subsequently, the apoptosome activates effector caspases such as DrICE and Dcp-1 (caspase-3-orthologs in Drosophila) (Kumar, 2007) which execute the apoptotic process.
Expression of the IAP-antagonist Hid in the Drosophila compound eye using the GMR promoter (GMR-hid) causes a strong eye ablation phenotype due to massive apoptosis induced by Hid in the eye imaginal disc during larval development (Figure 1A) (Fan and Bergmann, 2008; Grether et al., 1995). We have used the strong eye ablation phenotype of GMR-hid in chemical (EMS) mutagenesis screens to identify genes involved in the control of Hid-induced apoptosis. Initially, in dominant modifier screens of the GMR-hid eye ablation phenotype, heterozygous mutants of diap1 (Goyal et al., 2000; Wilson et al., 2002) and of negative regulators of the EGFR/Ras/MAPK pathway such as argos and sprouty were recovered (Bergmann et al., 1998). Recovery of regulators of the EGFR/Ras/MAPK pathway revealed that Hid is negatively controlled both by MAPK phosphorylation and transcriptional repression (Bergmann et al., 1998; Bergmann et al., 2002; Kurada and White, 1998). However, mutations in the core components of the apoptotic pathway, dronc, dark and drICE were not recovered in the dominant/heterozygous modifier screens implying that they are not rate-limiting for the strong eye ablation phenotype of GMR-hid [reviewed in Xu et al. (2009)].
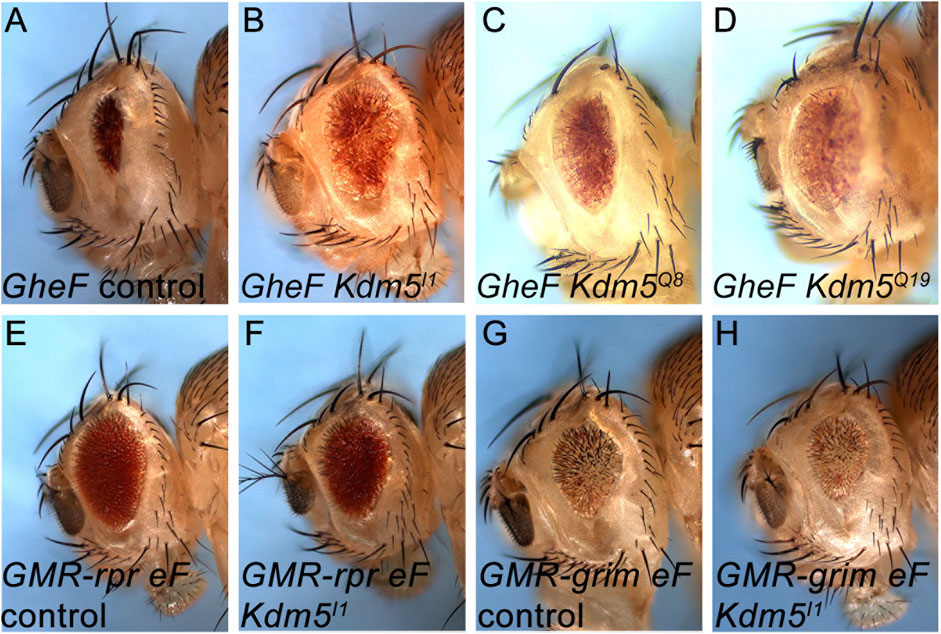
Figure 1. Identification of Kdm5 mutants as recessive suppressors of GMR-hid. (A) The unmodified GMR-hid ey-FLP (GheF) eye ablation phenotype. (B–D) Three alleles of Kdm5 behave as recessive suppressors of the GheF eye ablation phenotype in genetic mosaics. (E–H) Kdm5 mutants do not suppress GMR-reaper ey-FLP (GMR-rpr eF) (E, F) and GMR-grim ey-FLP (GMR-grim eF) (G, H) eye ablation phenotypes in genetic mosaics. Genotypes: (A) ey-FLP/+; FRT40/P[w+] FRT40; GMR-hid/+. (B) ey-FLP/+; Kdm5I1 FRT40/P[w+] FRT40; GMR-hid/+. (C) ey-FLP/+; Kdm5Q8 FRT40/P[w+] FRT40; GMR-hid/+. (D) ey-FLP/+; Kdm5Q19 FRT40/P[w+] FRT40; GMR-hid/+. (E) ey-FLP/+; ubi-GFP FRT40/CyO; GMR-rpr/+. (F) ey-FLP/+; Kdm5I1 FRT40/ubi-GFP FRT40; GMR-rpr/+. (G) ey-FLP/+; ubi GFP FRT40/CyO; GMR-grim/+. (H) ey-FLP/+; Kdm5I1 FRT40/ubi-GFP FRT40; GMR-grim/+.
Therefore, in a second-generation mutagenesis effort, we screened homozygous mutants for suppression of GMR-hid. Because we assumed that mutants of the core apoptotic components would be homozygous lethal (which was later confirmed for dronc and dark mutations), we performed the mutagenesis screens in genetic mosaics obtained by ey-FLP/FRT-induced mitotic recombination (Newsome et al., 2000; Xu and Rubin, 1993). We referred to this procedure as GheF (GMR-hid ey-FLP) screening (Xu et al., 2005) (see Supplementary Figure S1). We performed GheF screening for all four autosomal chromosome arms (2L, 2R, 3L and 3R). Using GheF screening, we recovered mutations of dronc (located on 3L), dark (2R) and drICE (3R) as strong suppressors of the GMR-hid-induced eye ablation phenotype (Srivastava et al., 2007; Xu et al., 2005; Xu et al., 2006); reviewed in Xu et al. (2009). Furthermore, we recovered mutants in additional genes such as D-cbl (3L), Uba1, vps25, ptc and cos2 (all 2R) as moderately strong suppressors of GMR-hid using GheF screening (Christiansen et al., 2012; Christiansen et al., 2013; Herz et al., 2006; Herz et al., 2009; Lee et al., 2008; Wang et al., 2008). These genes do not encode core components of the apoptotic pathway but can modify the outcome of the apoptotic process.
Here, we report the results of GheF screening for chromosome arm 2L. We recovered three mutant alleles of the gene lysine demethylase 5 (Kdm5), also known as little imaginal discs (lid), as moderately strong suppressors of GMR-hid in genetic mosaics. Kdm5 was initially identified as a Trithorax Group (TrxG) gene and encodes the Drosophila homolog of human Retinoblastoma-binding protein 2 (Rbp2) (Gildea et al., 2000). Rbp2 was identified as a Rb binding protein in a yeast two-hybrid screen and was shown to control Rb-mediated gene expression (Benevolenskaya et al., 2005; Defeo-Jones et al., 1991; Fattaey et al., 1993; Kim et al., 1994). After identification of Rbp2 as a JmjC-domain-containing histone demethylase, it was grouped as a member of the JARID1 family of histone demethylases (Klose et al., 2006). Kdm5 is the only JARID1 ortholog in Drosophila, while the human genome contains four, with Rbp2 classified as JARID1A.
Kdm5 encodes a protein with multiple domains (Figure 2A). Most notable is the JmjC domain which has lysine demethylase activity and was found to specifically demethylate trimethylated lysine 4 of histone H3 (H3K4me3) (Eissenberg et al., 2007; Lee et al., 2007; Lloret-Llinares et al., 2008; Secombe et al., 2007). In addition, Kdm5 contains a JmjN domain, an ARID (A/T-rich interaction domain), a Zn finger (ZF) and three PHDs (plant homeobox domains) (Figure 2A). The ARID is implicated in binding to A/T-rich DNA sequences, while the ZF and the PHDs are involved in DNA/chromatin interactions. The JmjN, ARID, ZF and PHD1 domains are required for the demethylase activity of Kdm5 (Li et al., 2010). PHD3 can bind to H3K4me3 (Li et al., 2010; Liu and Secombe, 2015) while PHD1 binds to unmethylated Lys4 of histone 3 (H3K4me0) (Li et al., 2010; Torres et al., 2015).
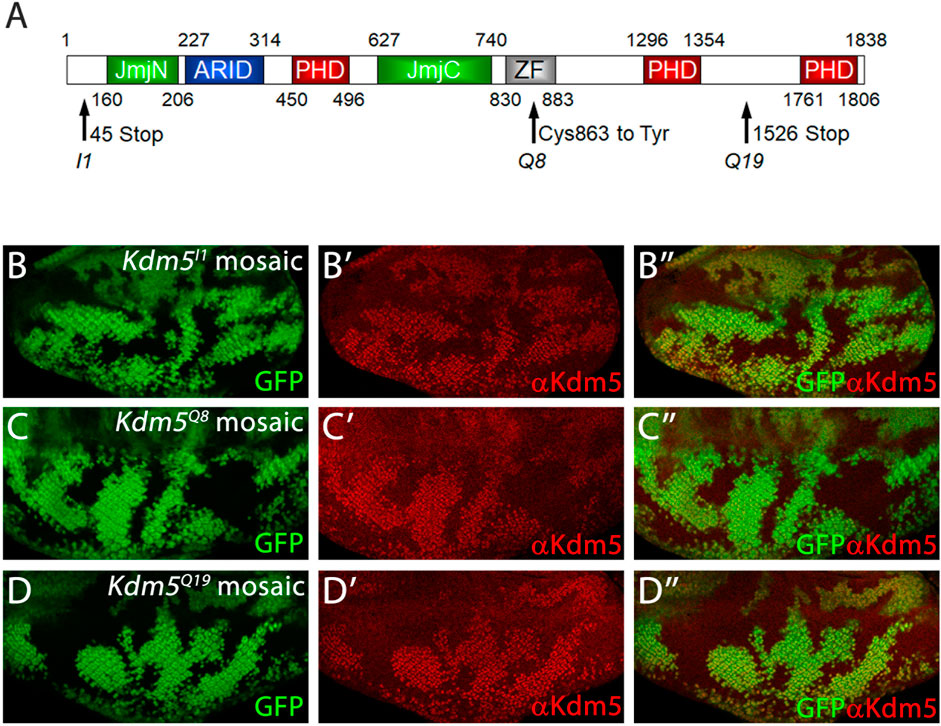
Figure 2. Mutant Kdm5 genes encode unstable Kdm5 proteins. (A) Schematic outline of the protein domains of Kdm5. JmjN = JumonjiN, ARID = AT-rich interaction domain (DNA binding), PHD = Plant Homeo Domain (protein interaction), JmjC = JumonjiC, ZF = Zink Finger. Molecular lesions of Kdm5I1, Kdm5Q8 and Kdm5Q19 are indicated by arrows. (B–D) Anti-Kdm5 antibody labeling of mosaic eye imaginal discs. Reduced Kdm5 protein levels are detected in Kdm5I1 (B–B”), Kdm5Q8 (C–C”) and Kdm5Q19 (D–D”) mutant clones (tissue). The absence of GFP identifies Kdm5 mutant clones. Genotypes: ey-FLP/+; Kdm5x FRT40/ubi-GFP FRT40 with x = I1 (B), Q8 (C) and Q19 (D).
H3K4me3 is enriched at promoter regions of transcriptionally active genes (Barski et al., 2007; Bernstein et al., 2005; Heintzman et al., 2007; Ng et al., 2003; Wirbelauer et al., 2005), and Kdm5 proteins were shown to colocalize with the H3K4me3 mark in those promoter regions (Liu and Secombe, 2015; Lloret-Llinares et al., 2012; Lopez-Bigas et al., 2008). Because the JmjC-domain of Kdm5 demethylates H3K4me3, a marker for gene activation, Kdm5 would be predicted to function as a silencer of gene expression by demethylating H3K4me3. However, this silencing function would be inconsistent with its original identification as a member of the TrxG gene family which is implicated in maintaining gene expression, for example, of homeotic genes. Consistently, Kdm5 has been shown to act as a transcriptional activator by maintaining Ubx expression in the haltere (Lloret-Llinares et al., 2008) and as co-activator of Drosophila Myc- (dMyc-) induced cell growth (Secombe et al., 2007). In the latter case, dMyc directly binds to the JmjC domain of Kdm5 and inhibits its catalytic demethylase activity. This enables the PHD3 of Kdm5 to bind to H3K4me3 and to recruit the Kdm5/dMyc complex to promoter regions of dMyc target genes such as Nop60B to stimulate dMyc-dependent transcription (Li et al., 2010; Liu and Secombe, 2015; Secombe et al., 2007). The interaction between Kdm5 and dMyc is also evolutionarily conserved for the mammalian proteins (Secombe et al., 2007). Another mechanism by which Kdm5 can act as an activator of gene expression and thus as a TrxG protein is through inhibition of the histone deacetylase Rpd3 (Lee et al., 2009). Therefore, Drosophila Kdm5 can act both as a transcriptional activator and transcriptional silencer. Consistent with this notion is also the observation that mammalian Rbp2/JARID1A can modulate both transcriptional activity and repression of Rb (Benevolenskaya et al., 2005). These opposite effects on control on transcription by Kdm5 are also confirmed by gene expression profiling analysis of Kdm5 mutants which revealed that of the deregulated genes about half are downregulated while the other half are upregulated (Drelon et al., 2018; Liu et al., 2014; Liu and Secombe, 2015).
As demonstrated first for dMyc-induced cell growth (Secombe et al., 2007) and consistent with its role as transcriptional activator, many of Kdm5’s functions are independent of the demethylase activity of the JmjC-domain of Kdm5 (Drelon et al., 2018; Hatch et al., 2021; Liu et al., 2014; Liu and Secombe, 2015). In fact, the demethylase activity of Kdm5 is not essential for development of Drosophila animals (Li et al., 2010). Flies defective for the JmjC-demethylase function of Kdm5 are phenotypically normal, although males, but not females, are short lived (Li et al., 2010). The survival of these demethylase-deficient animals can be potentially explained by a redundancy with another H3K4 demethylase gene in Drosophila, dKdm2, but nevertheless, global H3K4me3 levels are increased in demethylase-deficient Kdm5 flies (Li et al., 2010) suggesting that increased levels of H3K4me3 can be tolerated during development and also for normal survival of females. In contrast, a null allele of Kdm5 is 100% lethal (Drelon et al., 2018) suggesting that Kdm5 has essential functions during Drosophila development independently of its demethylase activity. The lethality of the null allele of Kdm5 was attributed to its role in larval growth and was mostly due to reduced proliferation of imaginal discs resulting in pupal lethality (Drelon et al., 2018). Apoptosis was also found to be increased in Kdm5 null mutant imaginal discs (Drelon et al., 2018), but the signal for control of apoptosis might be of non-autonomous origin coming from the prothoracic gland (Drelon et al., 2019). Kdm5 also regulates the expression of genes involved in mitochondrial morphology and physiology (Rogers et al., 2023) which is also a contributing factor for survival of the animals during development. Interestingly, restoring MAPK activity specifically in the prothoracic gland can restore viability in otherwise Kdm5 null mutant animals (Rogers et al., 2023) suggesting that Kdm5 also controls MAPK activation.
Here, we report the recovery of three EMS-induced alleles of Kdm5 as moderately strong suppressors of GMR-hid-induced eye ablation in genetic mosaics. Kdm5 specifically controls Hid-induced apoptosis and does not affect Reaper- and Grim-induced apoptosis. This function of Kdm5 is independent of the JmjC-demethylase activity. Furthermore, our data reveal that Kdm5 exerts its control over Hid-induced apoptosis through the activities of Rbf, dMyc, and MAPK. We present a model which incorporates these findings for the control of Hid-induced apoptosis.
Materials and methods
Identification of Kdm5 mutant alleles by GheF screening
The EMS mutagenesis screen for suppressors of GMR-hid in ey-FLP/FRT-induced mosaics for chromosome arm 2L is outlined in Supplementary Figure S1. Males of genotype y w ey-FLP; y+ FRT40A were incubated on tissue wipes soaked in 5% sucrose solution containing 25 mM Ethyl methanesulfonate (EMS) for 24 h. After that, they were allowed to recover for 3 h before being mated to virgin females of genotype y w ey-FLP; w+ FRT40A/CyO; GMR-hid at 25°C. 21,871 F1 offspring from this cross were screened for suppression of the GMR-hid-induced eye ablation phenotype. All dominant suppressors were discarded. 78 recessive suppressors were recovered, retested and balanced. One complementation group composed of three moderately strong suppressors of GMR-hid was identified by complementation crosses. Using several chromosomal deficiency stocks, the map position of this complementation group was determined at cytological interval 26A/B on chromosome arm 2L. Complementation crosses with existing Kdm5 mutants Kdm510424 and Kdm5k06801 (two P-element insertions in the first intron of the Kdm5 gene (Gildea et al., 2000)) identified the newly isolated complementation group as mutants of Kdm5. This was further confirmed by sequencing, rescue crosses and phenotypic analysis.
Drosophila husbandry and genetics
All crosses were performed on standard cornmeal-molasses medium (60 g/L cornmeal, 60 mL/L molasses, 23.5 g/L baker’s yeast, 6.5 g/L agar, 4 mL/L acid mix and 0.13% Tegosept). Genetic mosaics were induced in eye-antennal imaginal discs using the FLP/FRT mitotic recombination system (Xu and Rubin, 1993) using ey-FLP (Newsome et al., 2000) with GFP as genetic marker. Mutant clones are marked by loss of GFP. In the case of the MARCM (Lee and Luo, 2001) crosses in Figure 4G, mutant Kdm5 clones expressing the UAS-Kdm5JmjC* transgene are positively marked by GFP. The wing in Figure 3G is taken from the F1 offspring of the following cross: y w; Kdm5Q19 FRT40/CyO x y w Ubx-FLP; w+ FRT40/CyO. Non-CyO offspring was selected for imaging.
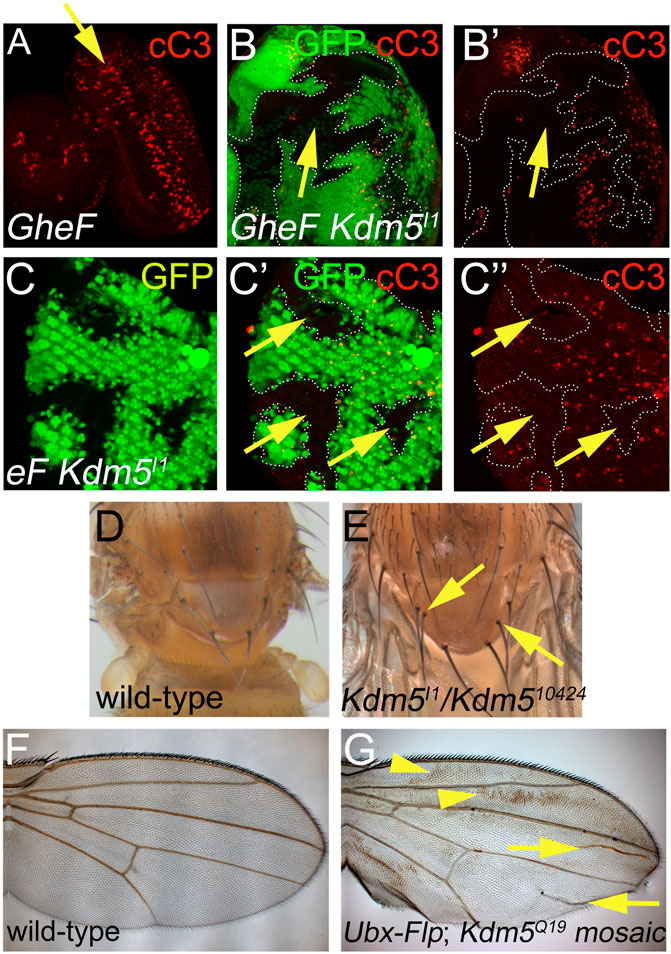
Figure 3. Cell death phenotypes of Kdm5 mutants. (A) GMR-hid induced cell death in 3rd instar larval eye imaginal discs visualized by cleaved Caspase 3 (cC3) labeling (red). A yellow arrow points to a wave of apoptosis induced by GMR-hid. (B, B’) Kdm5I1 mutant clones suppress GMR-hid-induced cell death in 3rd instar larval eye imaginal discs. White dotted lines outline the Kdm5 mutant clones with decreased activity of cleaved Caspase3 (cC3). The yellow arrow points to an area of interrupted cC3 labeling in a Kdm5 mutant clone. (C–C”) Kdm5I1controls developmental cell death in pupal eye discs at 30 h after puparium formation (APF). Kdm5I1 mutant clones outlined by white dotted lines show decreased Caspase 3 (cC3) activity. The yellow arrows point to areas of absent cC3 labeling in Kdm5 mutant clones. (D) Thorax of a wild-type fly with 4 scutellar bristles (macrochaetae). (E) Thorax of a Kdm5 mutant fly with two additional macrochaetae (yellow arrows). Adult flies of genotype Kdm5I1/Kdm510424 are very rare. We recovered only 5 flies of this genotype. All five have duplications of thoracic macrochaetae bristles. (F) A wing of a wild-type fly. (G) A Kdm5 mosaic wing displays a wing blemishing phenotype (yellow arrowheads) and additional wing vein material (yellow arrows). The penetrance of this phenotype is 100%. Genotypes: (A) ey-FLP/+; FRT40/P[w+] FRT40; GMR-hid/+. (B) ey-FLP/+; Kdm5I1 FRT40/P[w+] FRT40; GMR-hid/+. (C) ey-FLP/+; Kdm5I1 FRT40/ubi-GFP FRT40. (D) Canton S. (E) Kdm5I1/Kdm510424. (F) Canton S. (G) Ubx-FLP; Kdm5Q19 FRT40/ubi-GFP FRT40.
Immunohistochemistry
Eye-antennal imaginal discs from third instar larvae or 26–30 h APF pupal eye discs were dissected using standard protocols and labeled with antibodies raised against the following antigens: anti-Kdm5 (a kind gift of Julie Secombe); cleaved Caspase-3 (cC3) (Cell Signaling Technology); H3K4me3, H3K4me2, H3K4me1 (all Abcam); anti-Diap1 (a kind gift of H.D. Ryoo), anti-Dronc, anti-DrICE (all kind gifts of P. Meier); anti-Hid (a kind gift of H. Steller); anti-Rbf (a kind gift of N. Dyson); anti-β-Gal, anti-dMyc, anti-p53 (all DSHB); anti-JNK (Cell Signaling Technology). Cy3 fluorescently-conjugated secondary antibodies were obtained from Jackson ImmunoResearch. In each experiment, multiple clones in 10–20 eye imaginal discs were analyzed. Images were captured using an Olympus Optical FV500 confocal microscope.
Results
Isolation of three Kdm5 alleles as recessive suppressors of GMR-hid in genetic mosaics
We performed an EMS mutagenesis screen on chromosome arm 2L to recover recessive suppressors of the GMR-hid-induced eye ablation phenotype in genetic mosaics obtained by ey-FLP/FRT-mediated mitotic recombination. The scheme of the GheF (GMR-hid ey-FLP) screen is shown in Supplementary Figure S1. We screened approximately 22,000 F1 offspring of EMS-treated males and GheF-bearing females, and recovered 78 suppressors in genetic mosaics. All of these suppressors are recessive as they do not suppress GMR-hid in a heterozygous, i.e., dominant manner.
By complementation analysis, we identified one complementation group consisting of three alleles, initially termed I1, Q8 and Q19, which suppressed GMR-hid moderately strong in ey-FLP/FRT-induced mosaics (Figures 1A–D). Deficiency mapping with overlapping deficiencies covering chromosome arm 2L identified cytological interval 26B1/2 as the chromosomal location of this complementation group. Complementation crosses with existing mutants of genes in this chromosomal location identified Kdm5 (formerly known as little imaginal discs (lid)) as the underlying gene of this complementation group. Consistently, a transgene encoding wild-type Kdm5 can restore the strong eye ablation phenotype of GMR-hid in Kdm5 mosaics (Figures 4A–C). Therefore, we refer to the newly isolated Kdm5 alleles as Kdm5I1, Kdm5Q8 and Kdm5Q19.
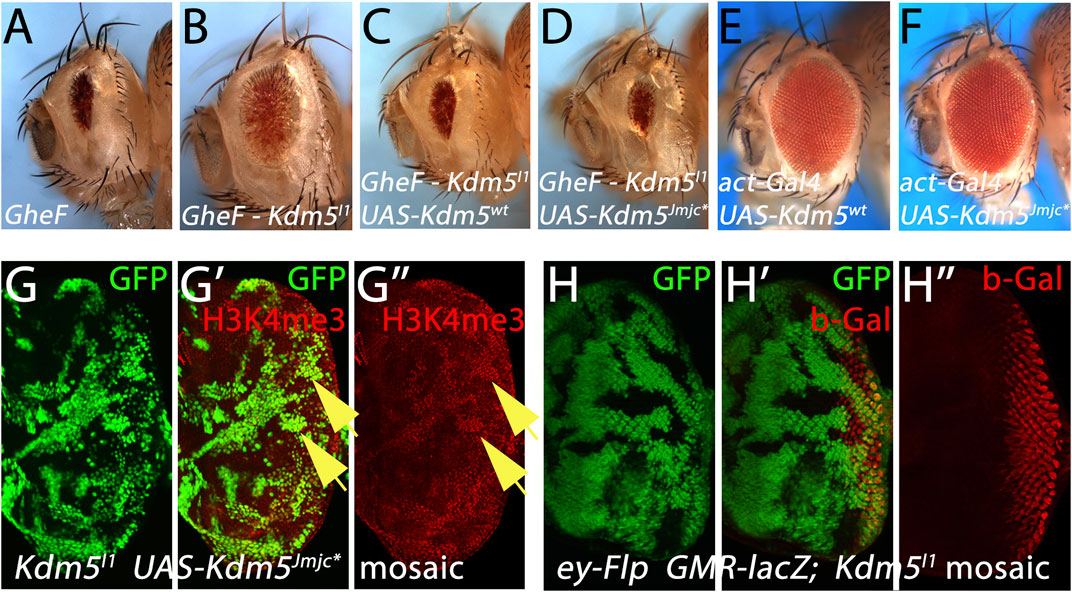
Figure 4. Kdm5 controls Hid-induced apoptosis independently of its JmjC demethylase activity. (A–D) Expression of UAS-transgenes encoding wild-type Kdm5 (UAS-Kdm5wt) (C) and a JmjC-domain mutant of Kdm5 (UAS-Kdm5JmjC*) (D) can revert the suppression of the GheF eye ablation phenotype by Kdm5I1 mosaics (B) back to the original GheF eye ablation phenotype (A). The Kdm5JmjC* transgene mutates two invariant residues in the JmjC domain rendering the demethylase activity of Kdm5 inactive. (E, F) Expression of UAS-Kdm5wt (E) and UAS-Kdm5JmjC* (F) under control of act-Gal4 (the same Gal4 driver used in (C,D)) does not cause an eye ablation phenotype. (G–G”) The UAS-Kdm5JmjC* transgene cannot rescue the enrichment of H3K4me3 marks in Kdm5I1 mutant clones (see yellow arrows as examples) and is thus catalytically inactive. In this experiment, Kdm5I1 mutant clones expressing UAS-Kdm5JmjC* were induced using the MARCM method. Hence, mutant clones expressing Kdm5JmjC* are positively marked by GFP. (H–H”) Kdm5 does not affect expression of transgenes from the GMR promoter. A GMR-lacZ reporter transgene does not show any changes of gene expression in Kdm5I1 mutant clones. Note that Kdm5I1 mutant clones are marked by absence of GFP in this experiment. Genotypes: (A) ey-FLP/+; FRT40/P[w+] FRT40; GMR-hid/+. (B) ey-FLP/act-Gal4; Kdm5I1 FRT40/P[w+] FRT40; GMR-hid/+. (C) ey-FLP/act-Gal4; Kdm5I1 FRT40 UAS-Kdm5wt/P[w+] FRT40; GMR-hid/+. (D) ey-FLP/act-Gal4; Kdm5I1 FRT40 UAS-Kdm5JmjC*/P[w+] FRT40; GMR-hid/+. (E) act-Gal4; UAS-Kdm5wt. (F) act-Gal4; UAS-Kdm5JmjC*. (G) hs-FLP UAS-CD8:GFP; Kdm5I1 FRT40 UAS-Kdm5JmjC*/P[tub-Gal80] FRT40; tub-Gal4/+. (H) ey-FLP GMR-lacZ/+; Kdm5I1 FRT40/ubi-GFP FRT40.
Interestingly, the eye ablation phenotype obtained by GMR-reaper and GMR-grim transgenes is not suppressed by Kdm5 mosaics (Figures 1E–H). This observation suggests that Kdm5 specifically controls Hid-induced apoptosis.
Phenotypic characterization of the new Kdm5 alleles
The three Kdm5 alleles isolated in this study have point mutations in the residues indicated in Figure 2A. Kdm5I1 has an early STOP codon at codon 45 (Figure 2A) and likely encodes a very strong, if not a null, loss-of-function mutant. Consistently, the suppression of GMR-hid by Kdm5I1 is the strongest of the three alleles. Immunolabeling of mosaic Kdm5I1 eye imaginal discs with anti-Kdm5 antibody confirms that no Kdm5 protein is produced in Kdm5I1 mutant clones (Figures 2B, B’). Kdm5Q8 has a point mutation changing Cys863 in the ZF domain to Tyr, and KdmQ19 has a premature STOP codon at codon 1,526 (Figure 2A). Using the anti-Kdm5-specific antibody, we found that the mutant Kdm5Q8 and Kdm5Q19 genes encode unstable proteins (Figures 2C, D’). In the case of Kdm5Q19, this may be due to nonsense-mediated RNA decay, while the point mutation in the ZF domain of Kdm5Q8 appears to render the protein unstable.
Previous work has shown that the JmjC-demethylase domain of Kdm5 demethylates H3K4me3 (Eissenberg et al., 2007; Lee et al., 2007; Lloret-Llinares et al., 2008; Secombe et al., 2007). Using our newly generated Kdm5 alleles, we confirmed that the global levels of H3K4me3 are increased in Kdm5 mutant clones of larval mosaic eye-antennal imaginal discs (Supplementary Figures S2A–A’’) suggesting that Kdm5 indeed possesses H3K4me3 demethylase activity and further confirming that the newly recovered alleles are Kdm5 mutants. Consistent with previous observations (Eissenberg et al., 2007; Lee et al., 2007; Lloret-Llinares et al., 2008; Secombe et al., 2007), our genetic analysis of Kdm5 mosaics does not demonstrate any demethylase activity towards H3K4me2 and H3K4me1 (Supplementary Figures S2B–C’’). These findings confirm that Kdm5 encodes a H3K4me3 demethylase consistent with previous reports (Eissenberg et al., 2007; Lee et al., 2007; Lloret-Llinares et al., 2008; Secombe et al., 2007).
Cell death phenotypes of Kdm5 mutants
Because we identified the Kdm5 alleles as suppressors of the GMR-hid-induced eye ablation phenotype which is caused by massive apoptosis in the larval eye imaginal disc (Figure 3A (Fan and Bergmann, 2008; 2014), we examined whether Kdm5 affects apoptosis. First, the GMR-hid-induced apoptosis in larval eye imaginal discs is strongly suppressed autonomously in Kdm5 mutant clones (Figures 3B, B’), explaining the recovery of eye tissue in GMR-hid/Kdm5 mosaic flies.
We also examined whether Kdm5 has a role for normal developmentally occurring programmed cell death. In the pupal retina, all unspecified cells are removed by Hid-dependent apoptosis (Kurada and White, 1998). In Kdm5 mutant clones, this developmental apoptosis is strongly reduced (Figure 3C). Furthermore, while wild-type flies contain 4 scutellar bristles (macrochaetae) on the scutellum (Figure 3D), homozygous hypomorphic Kdm5I1/Kdm510424 survivors carry 1 to 2 additional macrochaetae (Figure 3E), a phenotype associated with reduced apoptosis (Kanuka et al., 2005) and also observed for dronc, dark and cytochrome c mutants (Lindblad et al., 2021; Mendes et al., 2006; Rodriguez et al., 1999). Finally, a wave of apoptosis occurs during wing maturation in newly eclosed flies (Kimura et al., 2004). When this apoptosis is blocked, a wing blemishing phenotype appears due to incomplete apoptosis as observed for H99 (deleting reaper, hid and grim), dronc, dark and drICE mutants (Chew et al., 2004; Link et al., 2007). In Kdm5 mosaic wings, a wing blemishing phenotype occurs in all animals tested (Figures 3F, G). Together, these data illustrate that Kdm5 is an important gene for the control of apoptosis during Drosophila development.
Kdm5 controls apoptosis independently of its demethylase activity
Although Kdm5 is not a transcription factor, its best characterized function is control of transcription, either as an activator or as a silencer (Secombe and Eisenman, 2007). The distinguishing feature between these two opposing functions is the requirement of the demethylase activity of the JmjC domain of Kdm5 (Secombe and Eisenman, 2007). Therefore, we examined if the JmjC demethylase activity of Kdm5 is required for the control of apoptosis. As expected, expression of a UAS-Kdm5wt rescue transgene can revert the suppression of GMR-hid by Kdm5 mosaics (Figures 4A–C). Interestingly, a UAS-Kdm5JmjC* transgene in which two critical residues in the demethylase domain have been mutated and thus generate a catalytic mutant (Secombe et al., 2007), can also revert the suppression of GMR-hid by Kdm5 mosaics (Figure 4D) suggesting that the JmjC demethylase activity of Kdm5 is not required for the suppression of GMR-hid. Expression of the UAS-Kdm5wt and UAS-Kdm5JmjC* transgenes under control of act-Gal4 [the same Gal4 driver used in Figures 4C, D)] does not cause an eye ablation phenotype (Figures 4E, F). This suggests that the reversal of GMR-hid suppression by Kdm5 mosaics observed with these UAS transgenes (Figures 4C, D) is not due to eye ablation caused by the expression of the UAS-Kdm5wt and UAS-Kdm5JmjC* alone.
To verify that the UAS-Kdm5JmjC* transgene indeed encodes a demethylase-defective variant, we found that the elevated H3K4me3 levels in Kdm5 mutant clones are not normalized by expression of the UAS-Kdm5JmjC* transgene (Figure 4G”) confirming that the Kdm5JmjC* transgene is indeed demethylase-deficient. These results suggest that Kdm5 controls Hid-induced apoptosis independently of its demethylase activity. The demethylase-independent function of Kdm5 implies that it may act as a transcriptional activator for control of Hid-induced apoptosis.
Kdm5 does not control the expression of apoptotic genes
To examine the mechanism by which Kdm5 controls Hid-induced apoptosis, we tested the expression levels of genes involved in apoptosis. First, although we already found that Kdm5 mosaics do not affect the GMR-reaper- and GMR-grim-induced eye ablation phenotypes (Figure 1), we nevertheless tested whether Kdm5 can act directly on the GMR promoter and thus controls hid expression from GMR-hid. However, expression of a GMR-lacZ reporter transgene is not affected in Kdm5 mosaics (Figure 4H”) suggesting that Kdm5 does not transcriptionally control the GMR promoter. Furthermore, the levels of all genes involved in and required for apoptosis (dronc, dark, drICE, hid, etc.) were not changed in Kdm5 mutant clones (Table 1).
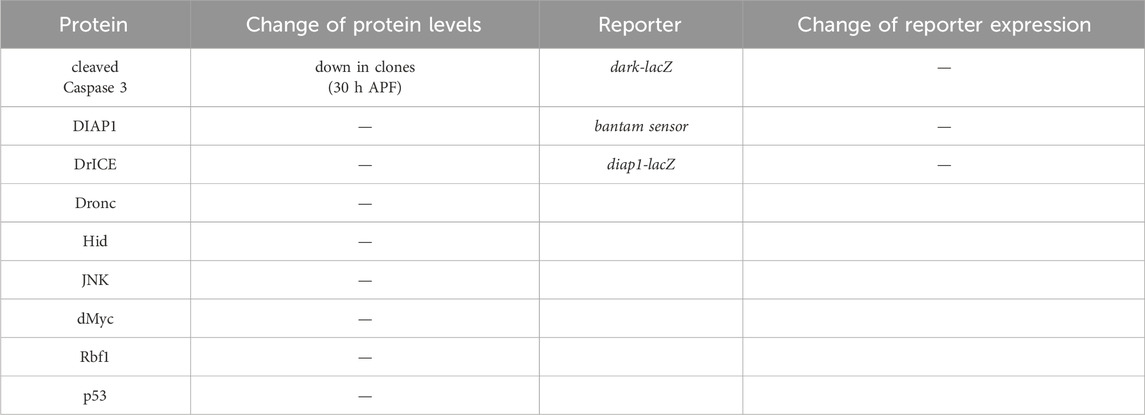
Table 1. Gene expression analysis of apoptotic genes in Kdm5 mutant clones. Listed are proteins and reporter genes which are directly or indirectly involved in the control of apoptosis. With the exception of cleaved Caspase 3, none of the proteins or reporter genes are deregulated in Kdm5 mutant clones in otherwise wild-type background in eye imaginal discs from 3rd instar larvae. Cleaved Caspase 3 antibody labeling was reduced in pupal eye discs at 26–30 h after puparium formation (APF) (see Figure 3C). The genotype used for the determination of the protein levels in the first and second columns was ey-FLP/+; Kdm5I1 FRT40/ubi-GFP FRT40. The genotype in the third and fourth column was ey-FLP/+; Kdm5I1 FRT40/ubi-GFP FRT40 plus the reporter transgene indicated.
Although the lack of a requirement of the demethylase domain indicates an activator function of Kdm5 for control of Hid-induced apoptosis, we also examined genes encoding negative regulators of apoptosis whose deregulation would require a silencing function of Kdm5 to suppress GMR-hid when Kdm5 is mutant. However, DIAP1 protein levels and expression of β-Gal from a diap1-lacZ reporter transgene are normal in Kdm5 mutant clones (Table 1). A Hid-specific inhibitor of apoptosis is the miRNA bantam, targeting the 3′UTR of the hid mRNA (Brennecke et al., 2003). However, a bantam sensor is unchanged in Kdm5 mutant clones (Table 1). These data suggest that Kdm5 is not directly controlling the gene expression or protein levels of apoptotic genes for control of Hid-induced apoptosis.
Kdm5 controls hid-induced cell death through Rbf and dMyc
To gain further insight into the role of Kdm5 for the control of Hid-induced apoptosis in Drosophila, we considered that the mammalian homolog of Kdm5, Rbp2 (JARID1A), was identified as a Rb binding protein (Defeo-Jones et al., 1991; Fattaey et al., 1993; Kim et al., 1994). The Drosophila homolog of Rb, Rbf, has been implicated in the control of Hid-induced apoptosis through inhibition of E2F1-dependent transcription (Moon et al., 2006). Therefore, we tested for a genetic interaction between Rbf and the ability of Kdm5 mosaics to suppress the GMR-hid eye ablation phenotype (Figures 5A, B). Indeed, heterozygosity of Rbf using two different Rbf alleles can revert the suppression of GMR-hid by Kdm5 mosaics back to the unmodified eye ablation phenotype (Figures 5A–D) suggesting that the suppression of the GMR-hid eye ablation phenotype by Kdm5 mosaics requires Rbf. The Rbf mutants to not modify the GMR-hid eye ablation phenotype on their own (i.e., in a Kdm5 wild-type background) (Figure 5H) suggesting that Rbf is rate-limiting for the suppression of GMR-hid by Kdm5. One function of Rbf is to negatively control the transcription factor E2F1 (Moon et al., 2006). Therefore, we also tested for a genetic interaction between E2F1 and Kdm5. Consistently, the suppressed eye ablation phenotype of GMR-hid by Kdm5 is even more strongly suppressed if E2F1 is heterozygous in this genetic background (Figure 5E). Heterozygous E2F1 mutants alone do not modify the GMR-hid phenotype (Figure 5I). These genetic interactions suggest that Kdm5 controls GMR-hid-induced apoptosis through negative regulation of Rbf.
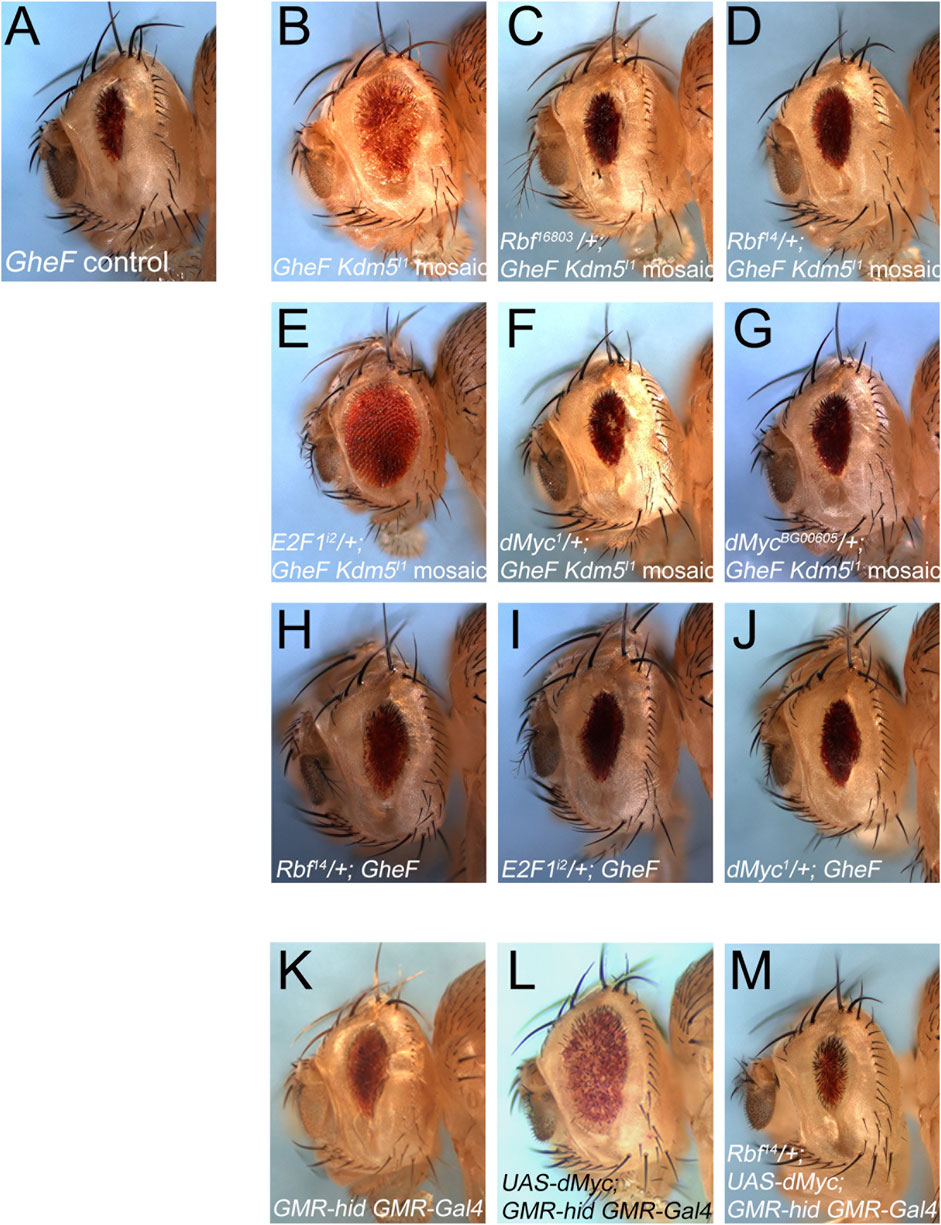
Figure 5. Kdm5 controls Hid-induced apoptosis through Rbf/E2F1 and dMyc. (A) The unmodified GheF eye ablation phenotype. (B) Kdm5I1 mosaics suppress the GheF eye ablation phenotype. (C, D) Two independent Rbf mutant alleles can dominantly revert the suppression of GheF-induced eye ablation by Kdm5I1 mosaics back to the original GheF eye ablation phenotype (compare to (A)) with 100% penetrance. (E) An E2F1 mutant allele dominantly suppresses even further the GheF eye ablation phenotype by Kdm5I1 mosaics (compare to (B)) with 100% penetrance. (F, G) Two independent dMyc mutants can dominantly revert the suppression of GheF-induced eye ablation by Kdm5I1 mosaics back to the original GheF eye ablation phenotype (compare to (A)) with 100% penetrance. (H–J) The Rbf14, E2F1i2 and dMyc1 mutants used in panels (D–F) do not modify the GheF eye ablation phenotype. (K) The unmodified GMR-hid GMR-Gal4 eye ablation phenotype. (L) Expression of UAS-dMyc by GMR-Gal4 suppresses the GMR-hid eye ablation phenotype. (M) Rbf14 can dominantly revert the suppression of GMR-hid by dMyc overexpression back to the original GMR-hid eye ablation phenotype with 100% penetrance. Genotypes: (A) ey-FLP/+; FRT40/P[w+] FRT40; GMR-hid/+. (B) ey-FLP/+; Kdm5I1 FRT40/P[w+] FRT40; GMR-hid/+. (C) ey-FLP/Rbf16803; Kdm5I1 FRT40/P[w+] FRT40; GMR-hid/+. (D) ey-FLP/Rbf14; Kdm5I1 FRT40/P[w+] FRT40; GMR-hid/+. (E) ey-FLP/+; Kdm5I1 FRT40/P[w+] FRT40; GMR-hid/E2F1i2. (F) ey-FLP/dMyc1; Kdm5I1FRT40/P[w+] FRT40; GMR-hid/+. (G) ey-FLP/dMycBG00605; Kdm5I1FRT40/P[w+] FRT40; GMR-hid/+. (H) ey-FLP/Rbf14; P[w+] FRT40/+; GMR-hid/+. (I) ey-FLP/+; P[w+] FRT40/+; GMR-hid/E2F1i2. (J) ey-FLP/dMyc1; GMR-hid/+. (K) GMR-hid GMR-Gal4/+. (L) GMR-hid GMR-Gal4/UAS-dMyc. (M) Rbf14/+; GMR-hid GMR-Gal4/UAS-dMyc.
Another factor which was shown to interact with Kdm5 both genetically and physically is Drosophila Myc (dMyc) (Secombe et al., 2007). In this context, it is interesting to note that this interaction results in inhibition of the demethylase activity of Kdm5 (Secombe et al., 2007) which is not required for the suppression of GMR-hid (Figure 4D). Therefore, we performed similar genetic interaction experiments as with Rbf and E2F1. Indeed, heterozygously, dMyc mutants revert the suppression of GMR-hid by Kdm5 back to the original eye ablation phenotype (Figures 5F, G) suggesting that dMyc is required for the suppression of GMR-hid by Kdm5. Heterozygous dMyc mutants alone do not suppress the GMR-hid eye ablation phenotype (Figure 5J) suggesting that dMyc is rate-limiting for the suppression of GMR-hid by Kdm5. This genetic interaction implies that Kdm5 and dMyc have an antagonistic relationship in this context.
Finally, to examine if Rbf/E2F1 and dMyc cooperate for the control of GMR-hid induced cell death, we took advantage of the observation that overexpression of dMyc can suppress the GMR-hid eye ablation phenotype (Figures 5K, L). This suppression is reverted back to the original GMR-hid eye ablation phenotype by heterozygosity of Rbf (Figure 5M) suggesting that dMyc requires Rbf to suppress GMR-hid. Together, these genetic interaction data demonstrate that Kdm5 regulates GMR-hid through inhibition of Rbf and dMyc, and that Rbf and dMyc cooperate in this context.
Kdm5 regulates GMR-hid-induced eye ablation through control of MAPK signaling
While E2F1 and potentially dMyc can directly control endogenous hid expression (Moon et al., 2005), this type of regulation does not explain the suppression of GMR-hid by Kdm5 as hid expression from GMR-hid is independent of the control of endogenous hid. This consideration would imply a post-translational control of Hid directly or indirectly by Kdm5. We have previously shown that Erk-type MAPK, encoded by the rolled (rl) gene, can post-translationally control Hid function by inhibitory phosphorylation (Bergmann et al., 1998). This type of regulation by MAPK on GMR-hid would also explain the inability of Kdm5 mosaics to suppress GMR-reaper and GMR-grim (Figure 1) which are not subject to control by MAPK (Bergmann et al., 1998; Kurada and White, 1998).
Therefore, we examined if Kdm5 mosaics can suppress a GMR-hid mutant in which the five MAPK phosphorylation sites of Hid have been mutated (GMR-hidAla5) and is thus unresponsive to inhibitory MAPK phosphorylation (Bergmann et al., 1998). Indeed, Kdm5 mosaics are not able to suppress the eye ablation phenotype of GMR-hidAla5 (Figures 6A–C). As a positive control, to demonstrate that the very strong GMR-hidAla5 eye ablation phenotype is in fact suppressible, we tested another mutant, S2, which was recovered in the same GheF screen of chromosome arm 2L and which displays a similar moderate suppression of GMR-hidwt in mosaics as Kdm5 (Supplementary Figure S1). S2 is a single allele recovered in the GheF 2L screen and the underlying gene mutated in S2 is unknown. Indeed, S2 mosaics can suppress the GMR-hidAla5 eye ablation phenotype (Figure 6D) suggesting that the GMR-hidAla5 eye ablation phenotype is suppressible and that the inability of Kdm5 to suppress GMR-hidAla5 might be because of its unresponsive nature to MAPK phosphorylation. The MAPK-dependent suppression of GMR-hidwt by Kdm5 suggests that Kdm5 negatively regulates MAPK activity and therefore that Kdm5 mutants have increased MAPK activity.
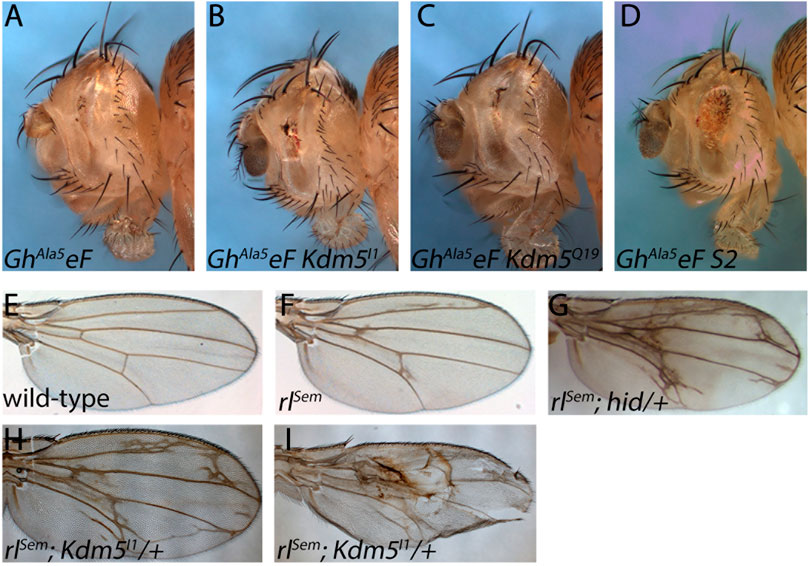
Figure 6. The suppression of GMR-hid by Kdm5 is sensitive to MAPK activity. (A) A GMR-hid transgene in which the five MAPK phosphorylation sites of Hid have been mutated to unphosphorylatable Ala residues (GMR-hidAla5) and is unresponsive to MAPK phosphorylation causes a very strong eye ablation phenotype. GhAla5eF = GMR-hidAla5 ey-FLP. (B, C) Kdm5 mosaics of Kdm5I1 and Kdm5Q19 are unable to suppress the GMR-hidAla5 eye ablation phenotype. (D) An unrelated suppressor of GMR-hid, S2 (see Supplementary Figure S1) is able to suppress GMR-hidAla5 in genetic mosaics. (E) A wing of a wild-type fly. (F) A wing of a heterozygous gain-of-function mutant of MAPK, encoded by the rolled gene, rolledSevenmaker (rlSem/+). (G) Heterozygous hid mutants dominantly enhance the rlSem wing phenotype. Note the extra wing vein material. (H, I) Heterozygosity of Kdm5 dominantly enhances the rlSem wing phenotype with a lot of extra wing vein material similar to rlSem/+; hid/+ (G). In extreme cases, wing blistering is observed (I). Genotypes: (A) GMR-hidAla5 ey-FLP; ubi-GFP FRT40/CyO. (B) GMR-hidAla5 ey-FLP; Kdm5I1 FRT40/ubi-GFP FRT40. (C) GMR-hidAla5 ey-FLP; Kdm5Q19 FRT40/ubi-GFP FRT40. (D) GMR-hidAla5 ey-FLP; S2 FRT40/ubi-GFP FRT40. (E) Canton S. (F) rlSem/+. (G) rlSem/+; hidWR+X1/+. (H, I) rlSem/Kdm5I1.
To further characterize the genetic interaction between rl/MAPK and Kdm5, we took advantage of the wing vein phenotype caused by a weak gain-of-function mutant of MAPK called rlSevenmaker (rlSem) (Brunner et al., 1994) (Figures 6E, F). The wing vein phenotype of rlSem is strongly enhanced by heterozygosity of Kdm5 (Figure 6H) further supporting that Kdm5 negatively regulates MAPK. In strong cases, we also observed a wing blistering phenotype which was not observed in rlSem wings alone (Figure 6I). Heterozygous Kdm5 wings do not show any specific wing vein phenotype (data not shown). Interestingly, heterozygosity of hid also strongly enhances the rlSem wing vein phenotype (Figure 6G) demonstrating the strong genetic link between Kdm5 and hid through MAPK signaling. In summary, these genetic interaction studies demonstrate that Kdm5 regulates Hid through control of MAPK activity.
Discussion
In this paper, we report the recovery of three alleles of the histone demethylase gene Kdm5 as moderately strong suppressors of GMR-hid-induced apoptosis. To our knowledge, these are the first EMS-induced alleles of Kdm5. We identified the mutations of these alleles and found that they either produce truncated proteins or unstable proteins, or both (Figure 2). Kdm5 is a nuclear protein and although it is not a transcription factor, all evidence points to an essential role of Kdm5 in transcriptional control, both as an activator and a silencer. The silencing function of Kdm5 is mediated through demethylation of H3K4me3 by the JmjC domain (Secombe and Eisenman, 2007). However, the demethylase activity of the JmjC domain of Kdm5 is not involved in the control of Hid-induced apoptosis (Figure 4D). Therefore, other domains of Kdm5 are involved in the control of Hid-induced apoptosis. A good candidate for such a domain is the ZF domain, as the Kdm5Q8 allele changes a conserved Cys residue to Tyr. However, this mutation causes protein instability (Figure 2) and the general instability of Kdm5Q8 protein might cause the suppression, and not the specific mutation in the ZF domain.
The control of apoptosis is very specific to the IAP antagonist Hid, as both the GMR-reaper- and GMR-grim-induced eye ablation phenotypes are not affected by Kdm5 mosaics (Figure 1). Consistently, Kdm5 does not control expression of transgenes from the GMR promoter and of core components of the apoptotic pathway such as dronc, dark and diap1 which would otherwise also affect Reaper- and Grim-induced apoptosis. The phenotypic characterization of the new Kdm5 alleles supports the notion that Kdm5 is at least partially required for apoptosis. Consistently, it was reported that overexpression of Kdm5 can ectopically induce apoptosis in a dMyc-expressing context (Secombe et al., 2007) and that JARID1A−/− mice show a decrease in apoptosis of hematopoietic stem cells (Klose et al., 2007). However, it has also been reported that wing imaginal discs of homozygous Kdm5 mutant larvae have actually increased levels of apoptosis (Drelon et al., 2018). Nevertheless, this increased apoptosis in wing imaginal discs is caused non-autonomously by signaling defects in the mutant prothoracic gland (Drelon et al., 2019). We would also like to emphasize that Kdm5 only controls Hid-induced apoptosis. Thus, other types of apoptosis, such as Reaper- or Grim-induced apoptosis which are not under Kdm5 control, may be the cause of this ectopic apoptosis seen in Kdm5 mutants.
While we were unable to identify a specific target gene of Kdm5 that is involved in the control of apoptosis (see Table 1), we established genetic interactions between Kdm5 on one hand and Rbf/E2F1 and dMyc on the other hand for the control of Hid-induced apoptosis (Figure 5). The mammalian ortholog of Drosophila Kdm5, Rbp2/JARID1A was originally identified as an interacting protein with Rb, and that interaction is conserved in Drosophila (Benevolenskaya et al., 2005; Defeo-Jones et al., 1991; Fattaey et al., 1993; Kim et al., 1994; Secombe et al., 2007). Both Drosophila Kdm5 and mammalian JARID1A can form a protein complex with the corresponding Myc orthologs, which in Drosophila controls Myc-induced growth (Drelon et al., 2018; Secombe et al., 2007). Interestingly, dMyc binds to the JmjC domain of Kdm5 and inhibits its demethylase activity (Secombe et al., 2007). This inhibition converts the silencer function of Kdm5 into a transcriptional (co-) activator (Secombe and Eisenman, 2007; Secombe et al., 2007). Given that the demethylase function of Kdm5 is not required for the control of Hid-induced apoptosis (Figure 4) and that dMyc is rate-limiting for the control of Hid-induced apoptosis by Kdm5 (Figure 5), it is possible that a similar inhibition of Kdm5’s demethylase activity by dMyc occurs for the control of Hid-induced apoptosis by Kdm5.
Putting all these data together, we propose the following model of Kdm5 action for the control of Hid-induced apoptosis. Kdm5 forms protein complexes with both Rbf and dMyc (Figure 7). dMyc inhibits the demethylase function of Kdm5 and thus maintains high levels of H3K4me3 at promoter regions of dMyc target genes. At the same time, the interaction between Rbf and Kdm5 blocks Rbf’s ability to inhibit E2F1 which can now also bind to enhancer/promoter regions of target genes (Figure 7). Our genetic analysis suggests that dMyc and E2F1 synergistically control Hid-induced apoptosis (Figure 5) and thus may act on the promoter region of the same genes. However, while E2F1 has been shown to be present at the promoter region of endogenous hid (Moon et al., 2005), it is unlikely that the cooperative control by Rbf and dMyc occurs on the endogenous hid gene. First, all our assays were performed with the heterologous GMR promoter which is not controlled by Rbf and dMyc. Second, the GMR promoter is not affected by Kdm5 (Figure 4). Third, expression of endogenous hid is not altered in Kdm5 mutant clones (Table 1). These considerations suggest that Kdm5 regulates the expression of another gene(s) for the control of the apoptotic activity of Hid.
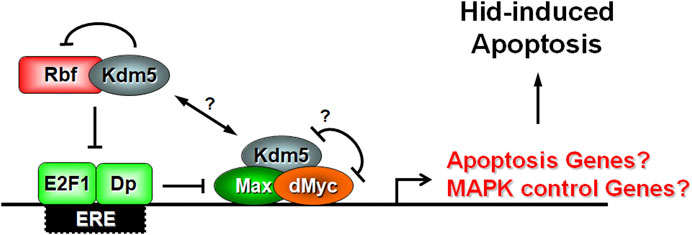
Figure 7. Model of Kdm5’s function for control of the apoptotic activity of Hid. Our genetic interaction data and previously published biochemical data suggest that Kdm5 interacts with both Rbf and dMyc. The interaction between Kdm5 and Rbf is inhibitory. The genetic interaction between Kdm5 and dMyc reveals a potential mutually inhibitory relationship, although it is unclear if dMyc directly inhibits the JmjC-dependent histone demethylase activity of Kdm5 as it does in another context (Secombe et al., 2007). Our genetic interaction studies also indicate an inhibitory relationship between E2F1 and dMyc for the control of Hid-induced apoptosis. Combined, these interactions may control the expression of a negative regulator of RTK/MAPK signaling which in turn regulates the ability of Hid to induce apoptosis through MAPK-dependent phosphorylation. We cannot exclude that Kdm5 also regulates the expression of a specific apoptotic gene. Max is a transcriptional co-activator of dMyc. Dp is a transcriptional co-activator of E2F1. Question marks (?) indicate uncertainty.
While we cannot exclude that another apoptotic gene is regulated by Kdm5, E2F1 and dMyc, our genetic analysis points towards control of MAPK activity (Figure 6). We showed previously that in contrast to Reaper and Grim, Hid is negatively controlled by phosphorylation of five Ser or Thr residues by Erk-type MAPK (Bergmann et al., 1998). A phosphorylation-defective mutant of Hid under GMR-control (GMR-hidAla5) cannot be suppressed by Kdm5 mosaics (Figure 6) strongly suggesting that Kdm5 controls MAPK phosphorylation activity for regulation of Hid. This may occur through regulation of a gene which directly or indirectly controls MAPK activity (Figure 7). Regulation of MAPK activity by Kdm5 is not unprecedented. Drelon et al. (2019) showed that Kdm5 regulates MAPK activity through transcriptional control of the receptor tyrosine kinase (RTK) torso in the prothoracic gland (Drelon et al., 2019). While there is no known role of Torso for development of imaginal discs in Drosophila larvae, other RTKs such as the Drosophila EGFR receptor (DER) have very established roles, especially for eye development (Malartre, 2016). However, while Drelon et al. (2019) demonstrated that Kdm5 is required for MAPK activity in prothoracic glands, our data suggest that Kdm5 negatively controls MAPK activity for the control of Hid-induced apoptosis in imaginal discs. This discrepancy may be caused by tissue-specific differences for the control of MAPK activity by Kdm5 and may be the result of the control of different target genes in these tissues. Therefore, instead of torso in the prothoracic gland, genes that negatively control DER and thus MAPK activity such as argos or sprouty may be controlled by Kdm5 for control of Hid-induced apoptosis. Future work will reveal the target gene(s) of Kdm5 that control MAPK activity for regulation of Hid-induced apoptosis in Drosophila.
Kdm5 is not the only JmjC-domain-containing protein that regulates Hid activity. The Drosophila ortholog of the phosphatidylserine receptor, dPSR, has also been implicated in controlling Hid activity (Krieser et al., 2007). dPSR is a JmjC-domain-only protein, and its mammalian ortholog JMJD6 demethylates histone H3 at Arg2 and histone H4 at Arg3 (Chang et al., 2007). Initially, PSR was classified as the phosphatidylserine receptor for engulfment of apoptotic cells (Fadok et al., 2000). However, the function of dPSR in engulfment in Drosophila is uncertain. dPSR mutant macrophages (hemocytes) can engulf apoptotic cells and dPSR protein is nuclear (Krieser et al., 2007) which is inconsistent with the predicted localization of an engulfment receptor at the cell surface. Interestingly, further characterization of dPSR revealed that it inhibits the ability of Hid to induce apoptosis (Krieser et al., 2007) suggesting that Kdm5 and dPSR may have antagonistic activities for the control of the apoptotic activity of Hid. Whether Kdm5 and dPSR directly interact to exert this antagonistic function is unknown. It is also unknown whether dPSR requires a functional JmjC domain for this activity.
Given the irreversible effect of apoptosis, it is not surprising that apoptosis is a highly regulated process. Apoptosis is controlled at transcriptional, post-transcriptional (alternative splicing) and post-translational (phosphorylation, ubiquitylation, etc.) levels. Here, we also add epigenetic control as an additional control mechanism of apoptosis. Resistance to apoptosis is a hallmark of many cancers and is often accompanied by epigenetic deregulation (Chakraborty et al., 2024; Ozyerli-Goknar and Bagci-Onder, 2021). Thus, it is crucial to better understand the epigenetic mechanisms that suppress apoptosis in various cancers and other diseases to help advance the development of targeted therapies.
We have performed recessive mutagenesis screens for the control of Hid-induced apoptosis in genetic mosaics using GheF screening for all autosomal chromosome arms. These screens yielded mutations in the core components of the apoptotic pathway, Dronc, Dark and Drice (Wu et al., 2016; Xu et al., 2005; Xu et al., 2006; Xu et al., 2009). In addition, we recovered mutations in genes that modify the outcome of the apoptotic pathway such as Uba1 affecting ubiquitylation and D-cbl, regulating MAPK activity (Lee et al., 2008; Wang et al., 2008). We also recovered mutations in vps25, cos2 and ptc which affect apoptosis non-autonomously through control of the Notch and Hedgehog signaling pathways (Christiansen et al., 2012; Christiansen et al., 2013; Herz et al., 2006; Herz et al., 2009). Here, we added epigenetic control of apoptosis by the recovery of Kdm5 mutants as an additional layer of regulation. In summary, these screens underscore the importance of unbiased mutagenesis screens to understand the control of a biological process, in this case apoptosis, at all possible levels. This detailed understanding will also have important implications for understanding and treatment of diseases in human patients.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Ethics statement
The manuscript presents research on animals that do not require ethical approval for their study.
Author contributions
H-MH: Conceptualization, Data curation, Formal Analysis, Investigation, Methodology, Validation, Writing–review and editing. AB: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Resources, Supervision, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by a grant from the National Institute of General Medical Sciences (NIGMS) under award number R35GM118330 to AB.
Acknowledgments
We would like to thank Zhihong Chen and Clare Bolduc for technical assistance and all members of the Bergmann lab for insightful comments and advice during this work. We are grateful to Julie Secombe, Georg Halder, David Stein, Pascal Meier, Hermann Steller, Hyung Don Ryoo, Hugo Bellen, Nick Dyson, Damian Brunner, Steve Cohen, John Abrams, Steve Hou and Barry Dickson for providing antibodies and fly stocks. We would like to thank the Bloomington Drosophila Stock Center (supported by NIH grant 5P40OD018537-10) for fly stocks and the Developmental Studies Hybridoma Bank (DSHB) for antibodies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Author disclaimer
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fceld.2024.1471050/full#supplementary-material
References
Abbott, M. K., and Lengyel, J. A. (1991). Embryonic head involution and rotation of male terminalia require the Drosophila locus head involution defective. Genetics 129, 783–789. doi:10.1093/genetics/129.3.783
Barski, A., Cuddapah, S., Cui, K., Roh, T. Y., Schones, D. E., Wang, Z., et al. (2007). High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837. doi:10.1016/j.cell.2007.05.009
Benevolenskaya, E. V., Murray, H. L., Branton, P., Young, R. A., and Kaelin, W. G. (2005). Binding of pRB to the PHD protein RBP2 promotes cellular differentiation. Mol. Cell 18, 623–635. doi:10.1016/j.molcel.2005.05.012
Bergmann, A., Agapite, J., McCall, K., and Steller, H. (1998). The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell 95, 331–341. doi:10.1016/s0092-8674(00)81765-1
Bergmann, A., Tugentman, M., Shilo, B. Z., and Steller, H. (2002). Regulation of cell number by MAPK-dependent control of apoptosis: a mechanism for trophic survival signaling. Dev. Cell 2, 159–170. doi:10.1016/s1534-5807(02)00116-8
Bernstein, B. E., Kamal, M., Lindblad-Toh, K., Bekiranov, S., Bailey, D. K., Huebert, D. J., et al. (2005). Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120, 169–181. doi:10.1016/j.cell.2005.01.001
Brennecke, J., Hipfner, D. R., Stark, A., Russell, R. B., and Cohen, S. M. (2003). Bantam encodes a developmentally regulated microRNA that controls cell proliferation and regulates the proapoptotic gene hid in Drosophila. Cell 113, 25–36. doi:10.1016/s0092-8674(03)00231-9
Brunner, D., Oellers, N., Szabad, J., Biggs, W. H., Zipursky, S. L., and Hafen, E. (1994). A gain-of-function mutation in Drosophila MAP kinase activates multiple receptor tyrosine kinase signaling pathways. Cell 76, 875–888. doi:10.1016/0092-8674(94)90362-x
Chakraborty, S., Nandi, P., Mishra, J., Niharika, , Roy, A., Manna, S., et al. (2024). Molecular mechanisms in regulation of autophagy and apoptosis in view of epigenetic regulation of genes and involvement of liquid-liquid phase separation. Cancer Lett. 587, 216779. doi:10.1016/j.canlet.2024.216779
Chang, B., Chen, Y., Zhao, Y., and Bruick, R. K. (2007). JMJD6 is a histone arginine demethylase. Science 318, 444–447. doi:10.1126/science.1145801
Chen, P., Nordstrom, W., Gish, B., and Abrams, J. M. (1996). grim, a novel cell death gene in Drosophila. Genes Dev. 10, 1773–1782. doi:10.1101/gad.10.14.1773
Chew, S. K., Akdemir, F., Chen, P., Lu, W. J., Mills, K., Daish, T., et al. (2004). The apical caspase dronc governs programmed and unprogrammed cell death in Drosophila. Dev. Cell 7, 897–907. doi:10.1016/j.devcel.2004.09.016
Christiansen, A. E., Ding, T., and Bergmann, A. (2012). Ligand-independent activation of the Hedgehog pathway displays non-cell autonomous proliferation during eye development in Drosophila. Mech. Dev. 129, 98–108. doi:10.1016/j.mod.2012.05.009
Christiansen, A. E., Ding, T., Fan, Y., Graves, H. K., Herz, H. M., Lindblad, J. L., et al. (2013). Non-cell autonomous control of apoptosis by ligand-independent Hedgehog signaling in Drosophila. Cell death Differ. 20, 302–311. doi:10.1038/cdd.2012.126
Defeo-Jones, D., Huang, P. S., Jones, R. E., Haskell, K. M., Vuocolo, G. A., Hanobik, M. G., et al. (1991). Cloning of cDNAs for cellular proteins that bind to the retinoblastoma gene product. Nature 352, 251–254. doi:10.1038/352251a0
Dorstyn, L., Akey, C. W., and Kumar, S. (2018). New insights into apoptosome structure and function. Cell death Differ. 25, 1194–1208. doi:10.1038/s41418-017-0025-z
Drelon, C., Belalcazar, H. M., and Secombe, J. (2018). The histone demethylase KDM5 is essential for larval growth in Drosophila. Genetics 209, 773–787. doi:10.1534/genetics.118.301004
Drelon, C., Rogers, M. F., Belalcazar, H. M., and Secombe, J. (2019). The histone demethylase KDM5 controls developmental timing in Drosophila by promoting prothoracic gland endocycles. Development 146, dev182568. doi:10.1242/dev.182568
Du, W., Vidal, M., Xie, J. E., and Dyson, N. (1996). RBF, a novel RB-related gene that regulates E2F activity and interacts with cyclin E in Drosophila. Genes Dev. 10, 1206–1218. doi:10.1101/gad.10.10.1206
Eissenberg, J. C., Lee, M. G., Schneider, J., Ilvarsonn, A., Shiekhattar, R., and Shilatifard, A. (2007). The trithorax-group gene in Drosophila little imaginal discs encodes a trimethylated histone H3 Lys4 demethylase. Nat. Struct. Mol. Biol. 14, 344–346. doi:10.1038/nsmb1217
Fadok, V. A., Bratton, D. L., Rose, D. M., Pearson, A., Ezekewitz, R. A., and Henson, P. M. (2000). A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 405, 85–90. doi:10.1038/35011084
Fan, Y., and Bergmann, A. (2008). Distinct mechanisms of apoptosis-induced compensatory proliferation in proliferating and differentiating tissues in the Drosophila eye. Dev. Cell 14, 399–410. doi:10.1016/j.devcel.2008.01.003
Fan, Y., and Bergmann, A. (2014). Multiple mechanisms modulate distinct cellular susceptibilities toward apoptosis in the developing Drosophila eye. Dev. Cell 30, 48–60. doi:10.1016/j.devcel.2014.05.007
Fattaey, A. R., Helin, K., Dembski, M. S., Dyson, N., Harlow, E., Vuocolo, G. A., et al. (1993). Characterization of the retinoblastoma binding proteins RBP1 and RBP2. Oncogene 8, 3149–3156.
Gildea, J. J., Lopez, R., and Shearn, A. (2000). A screen for new trithorax group genes identified little imaginal discs, the Drosophila melanogaster homologue of human retinoblastoma binding protein 2. Genetics 156, 645–663. doi:10.1093/genetics/156.2.645
Goyal, L., McCall, K., Agapite, J., Hartwieg, E., and Steller, H. (2000). Induction of apoptosis by Drosophila reaper, hid and grim through inhibition of IAP function. EMBO J. 19, 589–597. doi:10.1093/emboj/19.4.589
Grether, M. E., Abrams, J. M., Agapite, J., White, K., and Steller, H. (1995). The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 9, 1694–1708. doi:10.1101/gad.9.14.1694
Gyrd-Hansen, M., and Meier, P. (2010). IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat. Rev. Cancer 10, 561–574. doi:10.1038/nrc2889
Hatch, H. A. M., Belalcazar, H. M., Marshall, O. J., and Secombe, J. (2021). A KDM5-Prospero transcriptional axis functions during early neurodevelopment to regulate mushroom body formation. Elife 10, e63886. doi:10.7554/eLife.63886
Heintzman, N. D., Stuart, R. K., Hon, G., Fu, Y., Ching, C. W., Hawkins, R. D., et al. (2007). Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39, 311–318. doi:10.1038/ng1966
Herz, H. M., Chen, Z., Scherr, H., Lackey, M., Bolduc, C., and Bergmann, A. (2006). vps25 mosaics display non-autonomous cell survival and overgrowth, and autonomous apoptosis. Development 133, 1871–1880. doi:10.1242/dev.02356
Herz, H. M., Woodfield, S. E., Chen, Z., Bolduc, C., and Bergmann, A. (2009). Common and distinct genetic properties of ESCRT-II components in Drosophila. PLoS One 4, e4165. doi:10.1371/journal.pone.0004165
Kanuka, H., Kuranaga, E., Takemoto, K., Hiratou, T., Okano, H., and Miura, M. (2005). Drosophila caspase transduces Shaggy/GSK-3beta kinase activity in neural precursor development. EMBO J. 24, 3793–3806. doi:10.1038/sj.emboj.7600822
Kayagaki, N., Webster, J. D., and Newton, K. (2024). Control of cell death in health and disease. Annu. Rev. Pathol. 19, 157–180. doi:10.1146/annurev-pathmechdis-051022-014433
Kim, Y. W., Otterson, G. A., Kratzke, R. A., Coxon, A. B., and Kaye, F. J. (1994). Differential specificity for binding of retinoblastoma binding protein 2 to RB, p107, and TATA-binding protein. Mol. Cell Biol. 14, 7256–7264. doi:10.1128/mcb.14.11.7256
Kimura, K., Kodama, A., Hayasaka, Y., and Ohta, T. (2004). Activation of the cAMP/PKA signaling pathway is required for post-ecdysial cell death in wing epidermal cells of Drosophila melanogaster. Development 131, 1597–1606. doi:10.1242/dev.01049
Klose, R. J., Kallin, E. M., and Zhang, Y. (2006). JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet. 7, 715–727. doi:10.1038/nrg1945
Klose, R. J., Yan, Q., Tothova, Z., Yamane, K., Erdjument-Bromage, H., Tempst, P., et al. (2007). The retinoblastoma binding protein RBP2 is an H3K4 demethylase. Cell 128, 889–900. doi:10.1016/j.cell.2007.02.013
Krieser, R. J., Moore, F. E., Dresnek, D., Pellock, B. J., Patel, R., Huang, A., et al. (2007). The Drosophila homolog of the putative phosphatidylserine receptor functions to inhibit apoptosis. Development 134, 2407–2414. doi:10.1242/dev.02860
Kumar, S. (2007). Caspase function in programmed cell death. Cell death Differ. 14, 32–43. doi:10.1038/sj.cdd.4402060
Kurada, P., and White, K. (1998). Ras promotes cell survival in Drosophila by downregulating hid expression. Cell 95, 319–329. doi:10.1016/s0092-8674(00)81764-x
Lee, N., Erdjument-Bromage, H., Tempst, P., Jones, R. S., and Zhang, Y. (2009). The H3K4 demethylase lid associates with and inhibits histone deacetylase Rpd3. Mol. Cell Biol. 29, 1401–1410. doi:10.1128/MCB.01643-08
Lee, N., Zhang, J., Klose, R. J., Erdjument-Bromage, H., Tempst, P., Jones, R. S., et al. (2007). The trithorax-group protein Lid is a histone H3 trimethyl-Lys4 demethylase. Nat. Struct. Mol. Biol. 14, 341–343. doi:10.1038/nsmb1216
Lee, T., and Luo, L. (2001). Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 24, 251–254. doi:10.1016/s0166-2236(00)01791-4
Lee, T. V., Ding, T., Chen, Z., Rajendran, V., Scherr, H., Lackey, M., et al. (2008). The E1 ubiquitin-activating enzyme Uba1 in Drosophila controls apoptosis autonomously and tissue growth non-autonomously. Development 135, 43–52. doi:10.1242/dev.011288
Li, L., Greer, C., Eisenman, R. N., and Secombe, J. (2010). Essential functions of the histone demethylase lid. PLoS Genet. 6, e1001221. doi:10.1371/journal.pgen.1001221
Lindblad, J. L., Tare, M., Amcheslavsky, A., Shields, A., and Bergmann, A. (2021). Non-apoptotic enteroblast-specific role of the initiator caspase Dronc for development and homeostasis of the Drosophila intestine. Sci. Rep. 11, 2645. doi:10.1038/s41598-021-81261-0
Link, N., Chen, P., Lu, W. J., Pogue, K., Chuong, A., Mata, M., et al. (2007). A collective form of cell death requires homeodomain interacting protein kinase. J. Cell Biol. 178, 567–574. doi:10.1083/jcb.200702125
Liu, X., Greer, C., and Secombe, J. (2014). KDM5 interacts with Foxo to modulate cellular levels of oxidative stress. PLoS Genet. 10, e1004676. doi:10.1371/journal.pgen.1004676
Liu, X., and Secombe, J. (2015). The histone demethylase KDM5 activates gene expression by recognizing chromatin context through its PHD reader motif. Cell Rep. 13, 2219–2231. doi:10.1016/j.celrep.2015.11.007
Lloret-Llinares, M., Carre, C., Vaquero, A., de Olano, N., and Azorin, F. (2008). Characterization of Drosophila melanogaster JmjC+N histone demethylases. Nucleic Acids Res. 36, 2852–2863. doi:10.1093/nar/gkn098
Lloret-Llinares, M., Perez-Lluch, S., Rossell, D., Moran, T., Ponsa-Cobas, J., Auer, H., et al. (2012). dKDM5/LID regulates H3K4me3 dynamics at the transcription-start site (TSS) of actively transcribed developmental genes. Nucleic Acids Res. 40, 9493–9505. doi:10.1093/nar/gks773
Lopez-Bigas, N., Kisiel, T. A., DeWaal, D. C., Holmes, K. B., Volkert, T. L., Gupta, S., et al. (2008). Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol. Cell 31, 520–530. doi:10.1016/j.molcel.2008.08.004
Malartre, M. (2016). Regulatory mechanisms of EGFR signalling during Drosophila eye development. Cell Mol. Life Sci. 73, 1825–1843. doi:10.1007/s00018-016-2153-x
Mendes, C. S., Arama, E., Brown, S., Scherr, H., Srivastava, M., Bergmann, A., et al. (2006). Cytochrome c-d regulates developmental apoptosis in the Drosophila retina. EMBO Rep. 7, 933–939. doi:10.1038/sj.embor.7400773
Moon, N. S., Di Stefano, L., and Dyson, N. (2006). A gradient of epidermal growth factor receptor signaling determines the sensitivity of rbf1 mutant cells to E2F-dependent apoptosis. Mol. Cell Biol. 26, 7601–7615. doi:10.1128/MCB.00836-06
Moon, N. S., Frolov, M. V., Kwon, E. J., Di Stefano, L., Dimova, D. K., Morris, E. J., et al. (2005). Drosophila E2F1 has context-specific pro- and antiapoptotic properties during development. Dev. Cell 9, 463–475. doi:10.1016/j.devcel.2005.08.015
Newsome, T. P., Asling, B., and Dickson, B. J. (2000). Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development 127, 851–860. doi:10.1242/dev.127.4.851
Newton, K., Strasser, A., Kayagaki, N., and Dixit, V. M. (2024). Cell death. Cell 187, 235–256. doi:10.1016/j.cell.2023.11.044
Ng, H. H., Robert, F., Young, R. A., and Struhl, K. (2003). Targeted recruitment of Set1 histone methylase by elongating Pol II provides a localized mark and memory of recent transcriptional activity. Mol. Cell 11, 709–719. doi:10.1016/s1097-2765(03)00092-3
Oh, S. W., Kingsley, T., Shin, H. H., Zheng, Z., Chen, H. W., Chen, X., et al. (2003). A P-element insertion screen identified mutations in 455 novel essential genes in Drosophila. Genetics 163, 195–201. doi:10.1093/genetics/163.1.195
Orme, M., and Meier, P. (2009). Inhibitor of apoptosis proteins in Drosophila: gatekeepers of death. Apoptosis 14, 950–960. doi:10.1007/s10495-009-0358-2
Ozyerli-Goknar, E., and Bagci-Onder, T. (2021). Epigenetic deregulation of apoptosis in cancers. Cancers (Basel) 13, 3210. doi:10.3390/cancers13133210
Pierce, S. B., Yost, C., Britton, J. S., Loo, L. W., Flynn, E. M., Edgar, B. A., et al. (2004). dMyc is required for larval growth and endoreplication in Drosophila. Development 131, 2317–2327. doi:10.1242/dev.01108
Rodriguez, A., Oliver, H., Zou, H., Chen, P., Wang, X., and Abrams, J. M. (1999). Dark is a Drosophila homologue of Apaf-1/CED-4 and functions in an evolutionarily conserved death pathway. Nat. Cell Biol. 1, 272–279. doi:10.1038/12984
Rogers, M. F., Marshall, O. J., and Secombe, J. (2023). KDM5-mediated activation of genes required for mitochondrial biology is necessary for viability in Drosophila. Development 150, dev202024. doi:10.1242/dev.202024
Ryoo, H. D., Bergmann, A., Gonen, H., Ciechanover, A., and Steller, H. (2002). Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nat. Cell Biol. 4, 432–438. doi:10.1038/ncb795
Schreiber-Agus, N., Stein, D., Chen, K., Goltz, J. S., Stevens, L., and DePinho, R. A. (1997). Drosophila Myc is oncogenic in mammalian cells and plays a role in the diminutive phenotype. Proc. Natl. Acad. Sci. U. S. A. 94, 1235–1240. doi:10.1073/pnas.94.4.1235
Secombe, J., and Eisenman, R. N. (2007). The function and regulation of the JARID1 family of histone H3 lysine 4 demethylases: the Myc connection. Cell Cycle 6, 1324–1328. doi:10.4161/cc.6.11.4269
Secombe, J., Li, L., Carlos, L., and Eisenman, R. N. (2007). The Trithorax group protein Lid is a trimethyl histone H3K4 demethylase required for dMyc-induced cell growth. Genes Dev. 21, 537–551. doi:10.1101/gad.1523007
Shalini, S., Dorstyn, L., Dawar, S., and Kumar, S. (2015). Old, new and emerging functions of caspases. Cell death Differ. 22, 526–539. doi:10.1038/cdd.2014.216
Srivastava, M., Scherr, H., Lackey, M., Xu, D., Chen, Z., Lu, J., et al. (2007). ARK, the Apaf-1 related killer in Drosophila, requires diverse domains for its apoptotic activity. Cell death Differ. 14, 92–102. doi:10.1038/sj.cdd.4401931
Torres, I. O., Kuchenbecker, K. M., Nnadi, C. I., Fletterick, R. J., Kelly, M. J., and Fujimori, D. G. (2015). Histone demethylase KDM5A is regulated by its reader domain through a positive-feedback mechanism. Nat. Commun. 6, 6204. doi:10.1038/ncomms7204
Wang, Y., Werz, C., Xu, D., Chen, Z., Li, Y., Hafen, E., et al. (2008). Drosophila cbl is essential for control of cell death and cell differentiation during eye development. PLoS One 3, e1447. doi:10.1371/journal.pone.0001447
White, K., Tahaoglu, E., and Steller, H. (1996). Cell killing by the Drosophila gene reaper. Science 271, 805–807. doi:10.1126/science.271.5250.805
Wilson, R., Goyal, L., Ditzel, M., Zachariou, A., Baker, D. A., Agapite, J., et al. (2002). The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat. Cell Biol. 4, 445–450. doi:10.1038/ncb799
Wirbelauer, C., Bell, O., and Schubeler, D. (2005). Variant histone H3.3 is deposited at sites of nucleosomal displacement throughout transcribed genes while active histone modifications show a promoter-proximal bias. Genes Dev. 19, 1761–1766. doi:10.1101/gad.347705
Wu, Y., Lindblad, J. L., Garnett, J., Kamber Kaya, H. E., Xu, D., Zhao, Y., et al. (2016). Genetic characterization of two gain-of-function alleles of the effector caspase DrICE in Drosophila. Cell death Differ. 23, 723–732. doi:10.1038/cdd.2015.144
Xu, D., Li, Y., Arcaro, M., Lackey, M., and Bergmann, A. (2005). The CARD-carrying caspase Dronc is essential for most, but not all, developmental cell death in Drosophila. Development 132, 2125–2134. doi:10.1242/dev.01790
Xu, D., Wang, Y., Willecke, R., Chen, Z., Ding, T., and Bergmann, A. (2006). The effector caspases drICE and dcp-1 have partially overlapping functions in the apoptotic pathway in Drosophila. Cell death Differ. 13, 1697–1706. doi:10.1038/sj.cdd.4401920
Xu, D., Woodfield, S. E., Lee, T. V., Fan, Y., Antonio, C., and Bergmann, A. (2009). Genetic control of programmed cell death (apoptosis) in Drosophila. Fly. (Austin) 3, 78–90. doi:10.4161/fly.3.1.7800
Keywords: Drosophila, cell death, Hid, Kdm5, Rb, Myc, MAPK
Citation: Herz H-M and Bergmann A (2024) The histone demethylase Kdm5 controls Hid-induced cell death in Drosophila. Front. Cell. Death 3:1471050. doi: 10.3389/fceld.2024.1471050
Received: 16 August 2024; Accepted: 31 October 2024;
Published: 20 November 2024.
Edited by:
Bertrand Mollereau, Université de Lyon, FranceReviewed by:
Madhuri Kango-Singh, University of Dayton, United StatesHyung Don Ryoo, New York University, United States
Copyright © 2024 Herz and Bergmann. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andreas Bergmann, QW5kcmVhcy5iZXJnbWFubkB1bWFzc21lZC5lZHU=