
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Death, 15 July 2024
Sec. Non-Apoptotic Regulated Cell Death
Volume 3 - 2024 | https://doi.org/10.3389/fceld.2024.1423805
This article is part of the Research TopicGrand Challenges in Cell Death ResearchView all articles
 Y. Shan1,2
Y. Shan1,2 B. Mollereau1*
B. Mollereau1*The exploration of multiple regulated cell death (RCD) pathways and the recognition that several cell death-related proteins, including caspases, serve non-canonical roles have significantly expanded and diversified cell death research. Caspases not only cleave cellular substrates, triggering apoptosis, but also impact essential processes such as cellular differentiation, proliferation, growth, and migration. These novel caspase-dependent regulatory networks are extensively studied during development, with Drosophila providing a diverse range of developmental models for investigating these phenomena. Moreover, recent insights into the non-canonical functions of cell death proteins have highlighted their pivotal role in cancer aggressiveness. Ultimately, understanding these non-canonical functions sheds light on the intricate connections between RCD pathways and their significance in promoting anti-oncogenic responses.
Following the discovery of the key genes responsible for regulated cell death (RCD) processes such as apoptosis, necroptosis, ferroptosis, and pyroptosis (Figure 1, Box 1), current research aims to decipher how these RCD pathways are precisely regulated in various cellular contexts, spanning from development to diseases. The thorough analysis of the RCD machinery opened a new era of research shedding light on alternative and non-canonical roles of cell death genes (Mollereau et al., 2012; Aram et al., 2017; Colon-Plaza and Su, 2022; Svandova et al., 2022; Dominguez and Fan, 2023). Non-canonical functions of caspases in cellular migration, proliferation and growth have been methodically studied in Drosophila, which offers a wide range of genetic tools and developing tissues to address these non-lethal functions. Extending on these fundamental works, the non-canonical roles of caspases and other cell death-related proteins and processes has been explored in human cellular and animal cancer models, with a focus on cancer initiation, inflammation or resistance to treatment (Ichim et al., 2022; Suthar and Bergmann, 2023). Interestingly, these non-canonical functions does not always confer cellular survival but can lead to intersections between different RCD pathways and ultimately to cell demise (Bedoui et al., 2020). In this review, we will first underscore the importance of developing animal models to study the physiological relevance of various regulated cell death modalities and their multifaceted regulation. We will then focus on recent studies that delve into the non-canonical engagement of cell death mechanisms, resulting in emergence of novel phenotypic traits in various cancer models of study. Finally, we will review the current understanding on the intersection between RCD pathways and their importance in the control of cancer progression. We apologize to the researchers whose work was not cited due to space limitation.
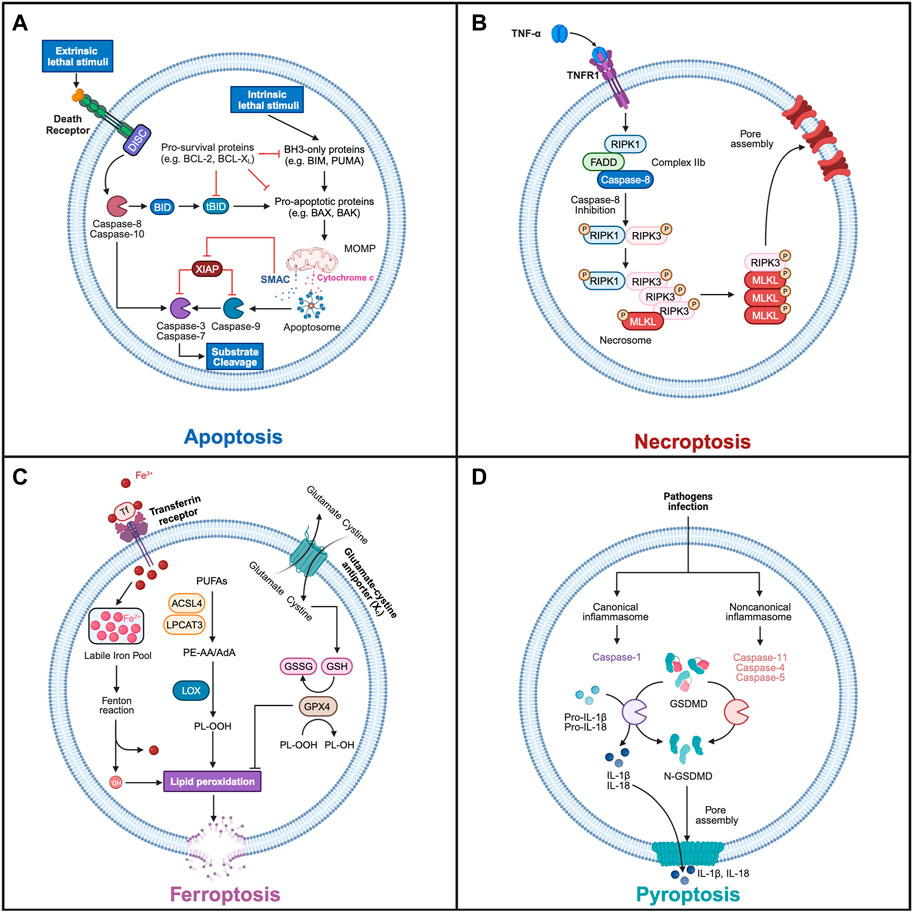
Figure 1. The main mechanisms of regulated cell death. (A) Apoptosis. The intrinsic stimuli trigger the activation of BH3-only proteins, which in turn activate pro-apoptotic proteins, such as BAX and BAK. This leads to their oligomerization at the mitochondrial outer membrane, inducing MOMP and the release of cytochrome c and second mitochondria-derived activator of caspase (SMAC) into the cytoplasm. This process can be inhibited by pro-survival BCL-2 family members. APAF1 binds to the released cytochrome c and recruits the initiator procaspase-9 to assemble the apoptosome, which can activate caspase-9, and then cleaves the downstream effector caspases (e.g., caspase-3, caspase-7), resulting in the proteolytic cleavage of many substrates. Moreover, the released SMAC binds to XIAP, preventing its inhibitory activity on caspase-9 and effector caspases. In extrinsic pathway, the activation of death receptors triggers the formation of the DISC, which can recruit and activate the initiator caspase-8. Activated caspase-8 then cleaves effector caspases, triggering subsequent substrate cleavage and apoptosis. In addition, Caspase-8 is able to cleave BH3-interacting domain death agonist (BID) into truncated BID (tBID), which induces intrinsic apoptosis by activating BAX and triggering MOMP. (B) Necroptosis. Upon TNFα binding to TNFR1, the inhibition of caspase-8 promotes the formation of necrosome where RIPK1 and RIPK3 interact by homotypic interaction motif (RHIM) and are activated by autophosphorylation or cross-phosphorylation. RIPK3 recruits and activates MLKL through phosphorylation, leading to its oligomerization and its translocation to the plasma membrane. This generates plasma membrane pores that disrupt its integrity and triggers necroptosis. (C) Ferroptosis. During ferroptosis, polyunsaturated fatty acids (PUFAs) undergo lipid peroxidation, which reacts with other membrane lipids leading to the cellular membrane’s rupture, promoting necrotic cell death. Ferroptosis can be initiated by endocytosis of ferric iron (Fe3+) by the transferrin receptor (TfR). Fe3+ is then reduced to ferric iron (Fe2+) increasing the cellular labile iron pool (LIP) before entering the Fenton reaction to produce Fe3+ and hydroxy radical (.OH). This reactive oxygen species in turn promotes non-enzymatic peroxidation of membrane lipids, such as arachidonic acid (AA)- and adrenic acid (AdA)-containing, phosphatidylethanolamine (PE) and phosphatidylcholine (PC). Lipid peroxidation can also be induced by enzymatic reactions in which free PUFAs are first linked to CoA by Acyl-CoA synthetase long-chain family member 4 (ACSL4), generating PE-AA and PE-AdA, which are then incorporated into PC/PE by lysophosphatidylcholine acyltransferase 3 (LPCAT3). The phospholipids (PL), PE-AA and PE-AdA, are peroxidated into PL-OOH by the lipoxygenase (LOX). Ferroptosis is inhibited by glutathione Peroxidase 4 (GPX4) that reduces lipid peroxide to lipid alcohol by oxidizing glutathione (GSH) into its oxidized form (GSSG). This protective pathway requires the effective production of GSH, which depends on the import of extracellular cystine by the cystine-glutamate antiporter system Xc-, which is then sequentially converted into GSH. (D) Pyroptosis. The infection of pathogens induces the formation of canonical and noncanonical inflammasomes, which subsequently activate inflammatory caspases and cleave GSDMD. The cleaved GSDMD (N-terminal fragment) binds to cell membrane phospholipids, assembles into an oligomeric pore and results in the membrane rupture. The activation of the inflammatory caspase-1 can also promote the maturation of proinflammatory cytokines such as interleukin-1 beta (IL-1β) and interleukin-18 (IL-18), that in turn are released through GSDMD pore to propagate proinflammatory responses. Schematics were generated with Biorender.
Box 1| regulated cell death
Apoptosis is the most studied and best understood regulated cell death modality (Figure 1A). It is induced by intrinsic or extrinsic death stimuli, which ultimately leads to the activation of caspase cysteine protease, the cleavage of hundreds cellular substrates and cell demise (Green, 2022; Ichim, et al., 2022).
In intrinsic apoptosis pathway, mitochondrial outer membrane permeabilization (MOMP) is induced mainly by pro-apoptotic BAX and BAK proteins, and lately described also for BOK and truncated BID (tBID). This leads to the cytoplasmic release of mitochondrial intermembrane space proteins, including cytochrome c and second mitochondria-derived activator of caspases (SMAC). Pro-survival BCL-2 family members (e.g., BCL-2, MCL1, BCL-xL) inhibit, while pro-apoptotic members (including BAX, BAK, BID, BIM, or PUMA) promote MOMP (Kalkavan and Green, 2018). Upon release of cytochrome c, apoptotic protease-activating factor 1 (APAF1) assembles into a potent caspase-activating complex known as the apoptosome, which recruits and activates the initiator procaspase-9 (Bao and Shi, 2007; Riedl and Salvesen, 2007). Activated caspase-9, in turn, executes the cleavage and activation of downstream effector caspases (e.g., caspase-3, caspase-7), which leads to the cleavage of cellular substrates. In addition, the MOMP-dependent release of SMAC acts as an antagonist to X-linked inhibitor of apoptosis protein (XIAP), counteracting its inhibitory effects on caspase-3 and subsequent apoptosis.
In extrinsic apoptosis, the death receptors (e.g., FAS or TNFR) triggers the formation of the death-inducing signaling complex (DISC), leading to the recruitment and activation of the initiator caspase-8 (Tummers and Green, 2017). Activated caspase-8 then cleaves effector caspases, triggering cellular demise. Caspase-8 also has the ability to promote intrinsic apoptosis by cleaving BH3-interacting domain death agonist (BID), which activates BAX and triggers subsequent MOMP.
The identification and characterization of the necroptosis pathway came from the study of tumor necrosis factor receptor 1 (TNFR1) signaling complexes. Depending on the experimental context, TNFR1 activation induces the formation of three distinct signaling complexes that lead to several cellular outcomes, including cell survival, proliferation, or cell death (apoptosis or necroptosis) (Vandenabeele et al., 2010). Upon TNFα binding to TNFR1, the assembly of cIAPs, RIPK1, TRADD and TRAFs, forms the TNFR1 signaling complex, referred to as complex I. This complex triggers the activation of NF-kB and AP1 transcription factors, promoting cellular survival and proliferation. The deubiquitylation of RIPK1 by CYLD permits the association of RIPK1 with FADD, caspase-8, with or without TRADD forming the complexes IIa or IIb, respectively, which induces caspase-8-mediated apoptosis. When caspase-8 is inhibited, the complex IIa or IIb trigger necroptosis (Figure 1B and Tenev et al., 2011; Tummers and Green, 2017). In the necrosome, RIPK3 phosphorylates mixed lineage kinase domain like pseudokinase (MLKL), leading to its translocation to the plasma membrane and its oligomerization. This generates a pore that disrupts the integrity of the plasma membrane and triggers necrotic cell death (Sun et al., 2012; Murphy et al., 2013).
Ferroptosis is a regulated form of necrosis, that clearly differentiates itself from other cell death pathways (Figure 1C; Dixon, 2017). It is characterized by an iron-dependent process of lipid peroxidation. In ferroptosis, ferrous iron (Fe2+) participates in the Fenton reaction with hydrogen peroxide, resulting in the generation of hydroxyl radicals (.OH). These radicals, oxidize lipids and in particular polyunsaturated fatty acids (PUFAs), leading to the accumulation of toxic peroxidated lipids within cellular membranes and subsequent induction of necrotic cell death (Stockwell et al., 2017). Key players in ferroptosis include Acyl-CoA synthetase long-chain family member 4 (ACSL4) and Lysophosphatidylcholine Acyltransferase 3 (LPCAT3), which promote the synthesis of membrane lipids that are particularly susceptible to peroxidation (Doll et al., 2017). To inhibit lipid peroxidation, endogenous mechanisms are mobilized, with a central role played by glutathione peroxidase 4 (GPX4) (Dixon, 2017; Ingold et al., 2017). GPX4 works as a defense against ferroptosis by detoxifying peroxidated lipids. Furthermore, GPX4 depends on the availability of glutathione, which is synthesized from cysteine, glutamine, and glycine and inhibits ferroptosis. Importantly, glutathione levels are maintained by the xc− transporter that imports cystine into the cytoplasm and exports glutamate. Interestingly, FSP1 acts independently of glutathione and in parallel to GPX4 to inhibit ferroptosis (Bersuker et al., 2019; Doll et al., 2019). Several factors promote ferroptosis, including inhibitors targeting GPX4, disruptions in glutathione synthesis, inhibition of the xc− transporter to perturb cysteine supply, and disturbances in iron homeostasis. Conversely, endogenous inhibitors of ferroptosis include molecules such as glutathione, ubiquinone, vitamin E, and selenium.
Pyroptosis is an extremely immunogenic form of cell death, characterized by cell swelling, lysis of plasma membrane and release of proinflammatory cytokines (Yuan et al., 2016; Newton et al., 2021). Pyroptosis involves the inflammatory caspases, (caspase-1 and caspase-11, the mouse homologue of human caspase-4 and -5), which promote the cleavage of gasdermin D (GSDMD) and the subsequent formation of membrane pores (Kayagaki and Dixit, 2019). Caspase-1 also cleaves and activates pro-inflammatory cytokine interleukin-1β (IL-1β) and pro-inflammatory cytokine interleukin-18 (IL-18). Cleaved IL-1β discriminates between caspase-1–dependent pyroptosis and necrosis. Pyroptosis is induced by pathogens (bacteria or virus) and is predominantly observed in immune cells such as macrophages, monocytes, and dendritic cells.
Macroautophagy (referred to as autophagy), is a process where various cellular materials, ranging from proteins to entire organelles, are broken down through lysosomal degradation (Boland et al., 2018; Klionsky et al., 2021). Originally identified in yeast, continued research across different models has detailed the formation and breakdown processes of the autophagosome, a unique organelle specific to autophagy (Lamb et al., 2013). Autophagy plays multiple roles within cells, from helping them withstand starvation to maintaining cellular health by managing and disposing of faulty proteins and organelles. The perturbation or loss of autophagy is associated with neuronal cell death in neurodegenerative diseases (Mollereau et al., 2023). The current view is that mild induction of autophagy promotes cell survival, while uncontrolled autophagy results in cell demise. While there is a good understanding on the autophagy role in supporting cell survival, the specifics surrounding how it might cause cell death are less clear (Chen et al., 2023).
Autophagic cell death has been initially identified as a type 2 cell death, distinguished by its distinct structural appearance. This involves the presence of numerous autophagic vacuoles, like autophagosomes and autolysosomes, in the cell cytoplasm. On a mechanistic level, the process of autophagy-dependent cell death is managed by the various components of the autophagy machinery, including ATG genes and complexes of autophagic proteins (Galluzzi et al., 2018). Finally, the machinery of autophagy interacts extensively with many, possibly all, forms of regulated cell death but the underlying mechanisms are still poorly understood (Napoletano et al., 2019).
Drosophila models have played crucial roles in investigating alternative and non-canonical functions of apoptosis across vital biological processes, including cell differentiation, proliferation, and migration, which are the focus of excellent reviews (Mollereau, et al., 2012; Aram, et al., 2017; Colon-Plaza and Su, 2022; Svandova, et al., 2022; Dominguez and Fan, 2023). Among these functions, the non-canonical involvement of caspases in regulating cell differentiation and apoptosis-induced proliferation (AiP) has been the subject of comprehensive reviews elsewhere (Domingos and Steller, 2007; Fuchs and Steller, 2011; Mollereau, et al., 2012; Diwanji and Bergmann, 2019; Dominguez and Fan, 2023). In this section, we will specifically examine the impact of caspases on cell migration and growth. Early work from Montell and colleagues proposed a non-apoptotic role of the apoptotic machinery in border cell migration during Drosophila oogenesis (Geisbrecht and Montell, 2004). Indeed, they showed that Drosophila inhibitor of apoptosis 1 (DIAP) inhibition did not result in apoptosis but rather in a Dronc (orthologous to mammalian caspase-2/9)- and Drosophila Apaf-1 (dApaf-1)-dependent inhibition of border cell migration. Subsequently, Arama and colleagues expanded upon these findings through studies of irradiation-induced cell migration and invasion in the Drosophila wing imaginal disc (Gorelick-Ashkenazi et al., 2018). Following irradiation, DNA damage pathways are activated, which in turn induce both apoptosis by p53 via the upstream Mre11–Rad50–NBS1 (MNR) complex/Ataxia Telangiectasia Mutated (ATM) kinase/Checkpoint kinase 2 (CHK2) pathways and cell migration by the Rad9–Hus1–Rad1 (9–1–1)/Ataxia Telangiectasia and Rad3-related kinase (ATR) pathway. Importantly, they showed that effector caspase inhibition triggered extensive, long-distance cell migration. In particular, they proposed that the effector caspase, Drosophila caspase-1 (Dcp-1, orthologous to human caspase-7), inhibits irradiation-induced cell migration. Recently, Milan and colleagues proposed an alternative model for the regulation of migration by caspases during chromosomal instability-induced cell invasion in the Drosophila wing imaginal disc (Barrio et al., 2023). Chromosomal instability followed by apoptosis was induced by depleting the spindle assembly checkpoints bub3 or rod with RNAi. They showed that in this context, the reduction but not full inhibition of apoptosis resulted in sublethal induction of the IAP antagonists, Reaper, Hid, Grim and the subsequent caspase activation, which promoted local cell migration. In this paradigm, caspases induce local cell migration by enhancing DNA damage. Interestingly, these results obtained on cell migration induction by caspases in Drosophila (Barrio, et al., 2023) are supported by mammalian cellular studies showing a role of caspase-3 and caspase-8 in DNA damage and cell migration (reviewed in Castillo Ferrer et al., 2021). These differences between Arama and Milan studies may stem from the different migration paradigms, local versus long distance, which were induced by distinct DNA damage stimuli in Drosophila -i.e., by tissue specific depletion of spindle check proteins, or at the whole organism level by irradiation, respectively. A significant advantage of using Drosophila in these investigations is the capability to live-track cell migration within a tissue context. Thus, further research in caspase-deficient and irradiated mice, which includes tracking labeled tumor cells, is needed to determine the conservation in mammalian of this dual regulation of cellular migration by caspases. Furthermore, pinpointing the exact caspase substrates responsible for regulating cell migration and invasiveness will be a key step towards comprehending the precise function and conservation of caspases, dependent on the cellular and tissue environment.
In a recent study, the non-apoptotic roles of the initiator caspases Dronc was investigated in a Drosophila model of oncogenic transformation, which relies on the simultaneous activation of EGFR and JAK/Stat pathways (Xu et al., 2022). Baena-Lopez and colleagues showed that non-lethal caspase activation is detected within the transformed cells and that caspase activation mitigates tumor growth and malignant cell transformation in the tumor by repressing c-Jun N-terminal Kinase (JNK) signaling. Interestingly, they also found that caspase-dependent JNK signaling affects the tumor environment by limiting the number of tumor-associated macrophages with tumor-promoting properties. Collectively, these results suggest that non-lethal activation of the Drosophila caspase-2/9 limits tumor development by inhibiting their growth, transformation and by acting on their tumor environment. Here, Dronc activation is dependent on dApaf-1, but is apparently independent of Hid, Reaper and Grim. Collectively, these studies shed light on the non-canonical role of caspases on various vital process such as cellular migration, growth and malignant transformation. Further investigation on the conservation of these regulatory networks in mammalian cancer cell models will demonstrate their significance in oncogenesis.
Non-canonical functions of cell death genes are best studied in cells submitted to sublethal stresses, which induce a preconditioning protective response. Various preconditioning stimuli, such as those targeting the mitochondria or the endoplasmic reticulum (ER), have been shown to induce protective responses in cellular and animal models, although the exact mechanisms are not well understood (Mendes et al., 2009; Fouillet et al., 2012; Mollereau, 2013; Hetz and Mollereau, 2014; Mollereau et al., 2014; Mollereau, 2015; Mollereau et al., 2016). In a series of elegant experiments, researchers used sublethal doses of a novel class of drugs called BH3 mimetics that inactivate the Bcl-2 family of anti-apoptotic proteins, to induce MOMP in a limited number of mitochondria (a phenomenon hereafter named minority MOMP) (Figure 2). This was not associated with apoptosis but rather triggered non-lethal caspase activation associated with DNA damage, mutagenesis and transformation of non-cancerous cells (Ichim et al., 2015; Ichim and Tait, 2016). In a follow-up study, melanoma cells engineered to express low-level of pro-apoptotic protein tBID and therefore undergo minority MOMP, triggered a JNK/AP1-dependent transcriptional program responsible for enhanced cancer cell aggressiveness (Berthenet et al., 2020). Interestingly, a recent study by Green and colleagues further investigated apoptosis-induced emergence of drug-tolerant persister lung carcinoma cells in response to BH3 mimetics treatment (Kalkavan et al., 2022; Kalkavan et al., 2023; Killarney et al., 2023). In contrast to previous studies, they found that BH3 mimetics induce sublethal cytochrome c release, leading to caspase-independent activation of heme-regulated inhibitor kinase (HRI) and engagement of the integrated stress response (ISR). This resulted in the synthesis of ATF4, leading to a drug-tolerant persister phenotype. This caspase-independent mechanism seems to contradict the previously observed role of caspases in promoting tumor aggressiveness. The contradictory findings between these studies could be attributed to differences in the types of cancer cells used and/or the varying concentrations of BH3 mimetics employed. Higher doses of BH3 mimetics have been associated with caspase-dependent DNA damage (Ichim, et al., 2015), which was not observed with lower doses (Kalkavan, et al., 2022). Consequently, different signaling pathways and responses may be engaged in the context of minority MOMP depending on the BH3 mimetic concentration.
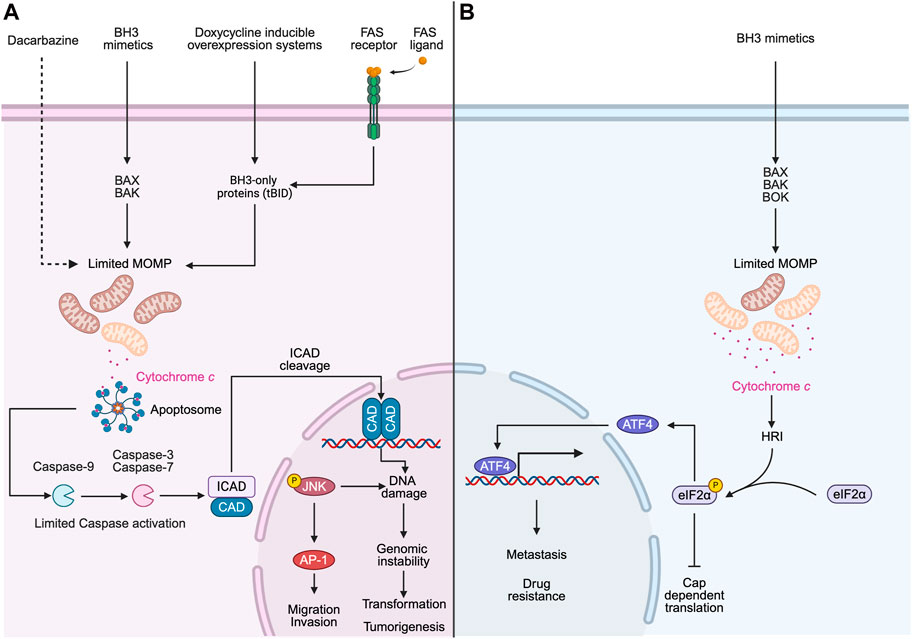
Figure 2. Minority MOMP promotes cancer cell aggressiveness. (A) Limited MOMP can be induced under several non-lethal stimuli (e.g., Dacarbazine, BH3-mimetics, doxycycline inducible overexpression of tBID and FAS ligand), and this limited MOMP triggers the release of cytochrome c to the cytosol and the activation of caspases, which leads to the cleavage of inhibitor of caspase-activated Dnase (ICAD) and the subsequent activation of caspase-activated Dnase (CAD) and DNA damage. This caspase-dependent activation is not sufficient to induce apoptosis but promotes genomic instability, oncogenic transformation and tumorigenesis. In addition, minority MOMP activates the JNK-AP1 transcriptional axis, which enhances melanoma cancer cell migration and invasion. (B) Limited MOMP would allow release of sublethal cytochrome c into the cytosol, where it binds directly to heme-regulated inhibitor (HRI) kinase, and promotes the downstream phosphorylation of Eukaryotic Initiation Factor 2α (eIF2α). When eIF2α is phosphorylated, Activating Transcription Factor 4 (ATF4) is preferentially translated while global protein translation is suppressed. ATF4 initiates the transcription of downstream genes, which contributes to cancer aggressiveness and drug resistance. Schematics were generated with Biorender.
Interestingly, the physiological role of minority MOMP has also been explored in the context of the immune response following infection (Brokatzky et al., 2019). The authors showed that virus, bacteria and protozoans pathogens all elicited minority MOMP, which in turn promoted the secretion of immune cytokines in epithelial cells. These results support a non-apoptotic role of the apoptotic machinery in the immune response against infection.
Another example of cellular adaptation to stress is observed in confined migration, a mechanical stress that cancer cells encounter during the process of metastasis. Confined cell migration, induced by forced passage through microporous membranes, was found to stimulate resistance to anoikis, an apoptotic cell death modality triggered by loss of cell adhesion and subsequent detachment (Fanfone et al., 2022). This resistance to anoikis was linked to the inhibitory role of apoptosis proteins (IAPs) and was associated with enhanced cell motility and evasion from natural killer cell-mediated immune surveillance. These findings demonstrate how cancer cells can survive and even become more aggressive and metastatic in stressful environments by employing the apoptotic machinery for cellular protection.
In conclusion, these studies shed light on the complex mechanisms underlying cancer cell resistance to stress and reveal how the apoptotic machinery can be hijacked for promoting cancer aggressiveness and enhanced metastatic potential. Further investigations are needed to fully understand the extent of these responses and their implications in tumor growth and metastasis in both in vitro and in vivo models.
The non-canonical roles of cell death genes are not limited in response to non-lethal stresses or in developing tissues but they also regulate the intersection between RCD pathways during the execution of cell death. Indeed, it has been proposed that the intersections between different RCD pathways is a general rule in several cell types and tissues in which the execution of cell death involves two or more RCD pathways in a cell death continuum (Napoletano et al., 2019).
The necroptosis was discovered while studying FAS-induced apoptosis in T cells in which caspases where inhibited (Kawahara et al., 1998; Holler et al., 2000; Bedoui, et al., 2020). Although the inhibition of caspases inhibited apoptosis, T cells did not survive and undergo necroptosis. Further characterization showed that caspase-8 acts as inhibitor of necroptosis, which is unleashed upon the addition of caspase inhibitors and the induction of TNFR1/FAS (Varfolomeev et al., 1998). Hence, upon stimulation of TNFR1 when both caspase-8 activity and NF-KB are inhibited, the necrosome consisting of the receptor-interacting protein kinases (RIPK1, RIPK3) and FADD is formed. This signaling complex leads to the activation of MLKL and the subsequent pore formation at the plasma membrane. Thus, necroptosis is a RCD program activated upon apoptosis inhibition in immune cells and in other cells such as epithelial and cancer cells. It was proposed that necroptosis is a back-up/safe cell death program to eliminate infected cells in which apoptosis is blocked by the pathogens (Bedoui, et al., 2020). Interestingly, the intersection between apoptosis and regulated necrosis pathways exists also in Drosophila. Indeed, it has been observed during Drosophila spermatogenesis that spontaneous germ cell necrosis, is negatively regulated by the apoptotic machinery (Yacobi-Sharon et al., 2013; Napoletano et al., 2017). In this form of necrosis that is distinct from necroptosis, germ cell necrosis depends on Drosophila p53B isoform, mitochondrial (Omi/HtraA2) and lysosomal factors (Cathepsin D). In conditions in which caspases are inhibited by the expression of caspase inhibitors (diap1 or p35), germ cell necrosis is enhanced (Yacobi-Sharon, et al., 2013). This suggests a physiological role of non-lethal activation of caspases during spermatogenesis, limiting an aberrant germ cell necrosis. Interestingly, Drosophila genome does not carry RIPK or MLKL, indicating that although the intersection between apoptosis and necrosis is present in flies, the molecular mechanisms have diverged during evolution.
Another well documented RCD crosstalk is between apoptosis and pyroptosis (Bedoui, et al., 2020). An example was reported in macrophages infected with Yersinia pseudotuberculosis, which normally die by apoptosis. In situations where caspases are inhibited, the infected macrophages still die by activating pyroptosis (Orning et al., 2018). The interplay between apoptosis and pyroptosis is regulated by caspase-8, which upon its activation, notably restricts the occurrence of pyroptosis in mice intestine. Thus, caspase-8 acts as a molecular switch controlling the activation of apoptosis, pyroptosis and necroptosis signaling pathways (Fritsch et al., 2019). Interestingly, caspase-3 can also promote the cleavage of the GSDMD-related protein GSDME (also known as DFNA5, deafness, autosomal dominant 5) into its linker form GSDME-N, which perforates membranes, forms pores and induces pyroptosis in cancer cells exposed to TNF or chemotherapy drugs (Wang et al., 2017). Thus, the crosstalk between apoptosis and pyroptosis involves both initiator and effector caspases.
In a recent study, Weindel et al. investigated the role of mitochondria in the mutual regulation of pyroptosis and necroptosis (Poltorak, 2022; Weindel et al., 2022). Their aim was to understand how leucine-rich repeat kinase 2 (LRRK2) gain of function (Lrrk2G2019S allele) affects cell death processes in response to bacterial infection (Figure 3). LRRK2 is a large protein with various domains, including a kinase domain similar to RIPKs (Zhang et al., 2010). Since LRRK2 is frequently mutated in Parkinson’s disease, previous research focused on the central nervous system. In this study, the researchers used the observation that LRRK2-deficient mice are susceptible to Mycobacterium tuberculosis infection (Weindel et al., 2020). They discovered that when Lrrk2G2019S macrophages were infected with bacteria, a non-canonical form of pyroptosis was triggered. In this process, GSDMD targeted the outer mitochondrial membrane instead of the plasma membrane. This unusual event resulted in mitochondrial depolarization and the release of mitochondrial reactive oxygen species (ROS) and damage-associated molecular pattern molecules (DAMPs), leading to the activation of RIPK1/RIPK3 and subsequent necroptosis. These findings highlight the crucial role of mitochondria in the transition from pyroptosis to necroptosis in macrophages exposed to bacterial infection.
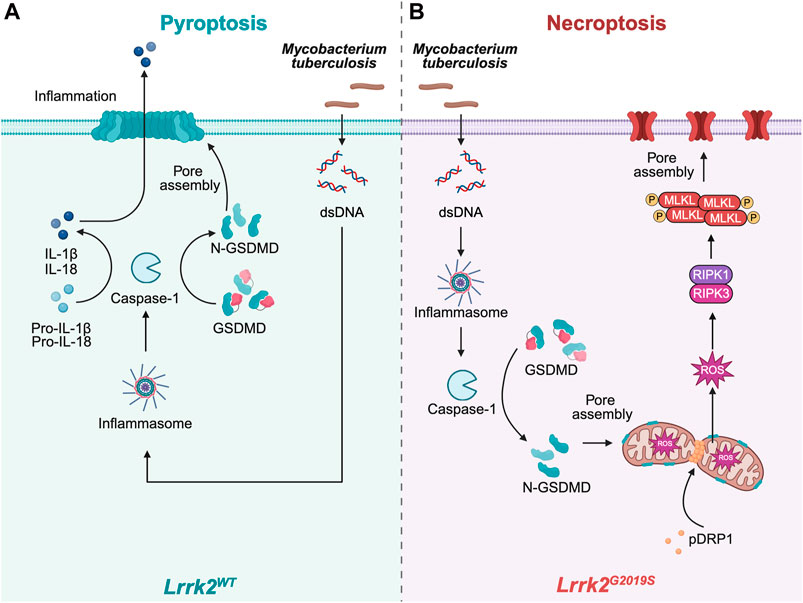
Figure 3. Mitochondrial targeting of GSDMD mediates necroptosis in Lrrk2G2019S mutants. (A) Upon bacterial infection (e.g.,: Mycobacterium tuberculosis), inflammasome activation in Lrrk2 wild type (WT) macrophages proceeds canonically, which triggers the activation of Caspase-1. This leads to the cleavage of GSDMD into N-GSDMD as well as of proinflammatory cytokines. N-GSDMD pores are assembled in the plasma membrane to allow the release of proinflammatory cytokines and execute pyroptosis. (B) However, in Lrrk2G2019S macrophages, mitochondria are fragmented and prone to depolarization in response to cellular stress due to the increased mitochondrial fission by phosphorylated Dynamin-1-like protein (pDRP1). Instead of forming pores at the plasma membrane, GSDMD is recruited to the mitochondria to assemble pores, resulting in release of mtROS and activation of RIPK1/RIPK3, which finally elicit necroptosis. Schematics were generated with Biorender.
The characterization of multiple RCD pathways and the understanding of the non-canonical roles of the cell death machinery led to a paradigm shift in RCD research, with an exponential expansion and diversification in recent years. From studies in animal models and human cancer cells, it is now clear that the activation of RCD pathways is rarely linear and does not always lead to cellular demise. Depending on the cellular context, RCD can lead to the activation of non-lethal responses during development and to increased cancer aggressiveness.
This diversification of RCD proteins functions and the intersection between RCD pathways has raised several grand challenges for research on various pathologies including cancer and neurodegenerative diseases. Some of them are listed below:
1. Elucidating the multifunctional nature of numerous proteins and organelles (mitochondria, lipid droplet, lysosomes, ER.) originally linked to apoptosis, revealing their involvement in non-apoptotic processes that are indispensable in both normal and cancer cells. Unraveling the intricate mechanisms governing these non-apoptotic functions poses an immense challenge that lies ahead.
2. Uncovering novel canonical and non-canonical pathways that elicit alternative RCD modalities in cancer cells refractory to conventional apoptotic stimuli. This bears significant implications for the advancement of efficient anti-cancer therapeutics.
3. Enhancing our mechanistic understanding of regulated forms of necrosis, such as necroptosis, pyroptosis and ferroptosis, which have emerged as a promising strategy for eliminating apoptosis-resistant cancer cells. Deciphering the intricate anti-cancer immune responses evoked by distinct forms of regulated necrosis holds the key to unlocking innovative therapeutic avenues.
4. The burgeoning evidence incriminating diverse RCD modalities for a large spectrum of pathologies, encompassing neurodegenerative disorders and cancer. A comprehensive mechanistic comprehension of this causal interplay is instrumental in identifying and targeting pivotal regulators to impede the relentless progression of these debilitating diseases.
YS: Writing–original draft, Writing–review and editing. BM: Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. YS is supported by the Chinese Scholarship Council. We thank the Fondation ARC, Ligue contre le cancer and INCA (PLBIO23-188) grants to BM.
We are grateful to Gabriel Ichim and Eli Arama for critical reading of the manuscript. Figures were generated with Biorender.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aram, L., Yacobi-Sharon, K., and Arama, E. (2017). CDPs: caspase-dependent non-lethal cellular processes. Cell Death Differ. 24, 1307–1310. doi:10.1038/cdd.2017.111
Bao, Q., and Shi, Y. (2007). Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ. 14, 56–65. doi:10.1038/sj.cdd.4402028
Barrio, L., Gaspar, A. E., Muzzopappa, M., Ghosh, K., Romao, D., Clemente-Ruiz, M., et al. (2023). Chromosomal instability-induced cell invasion through caspase-driven DNA damage. Curr. Biol. 33, 4446–4457.e5. doi:10.1016/j.cub.2023.09.004
Bedoui, S., Herold, M. J., and Strasser, A. (2020). Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. Cell Biol. 21, 678–695. doi:10.1038/s41580-020-0270-8
Bersuker, K., Hendricks, J. M., Li, Z., Magtanong, L., Ford, B., Tang, P. H., et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. doi:10.1038/s41586-019-1705-2
Berthenet, K., Castillo Ferrer, C., Fanfone, D., Popgeorgiev, N., Neves, D., Bertolino, P., et al. (2020). Failed apoptosis enhances melanoma cancer cell aggressiveness. Cell Rep. 31, 107731. doi:10.1016/j.celrep.2020.107731
Boland, B., Yu, W. H., Corti, O., Mollereau, B., Henriques, A., Bezard, E., et al. (2018). Promoting the clearance of neurotoxic proteins in neurodegenerative disorders of ageing. Nat. Rev. Drug Discov. 17, 660–688. doi:10.1038/nrd.2018.109
Brokatzky, D., Dorflinger, B., Haimovici, A., Weber, A., Kirschnek, S., Vier, J., et al. (2019). A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J. 38, e100907. doi:10.15252/embj.2018100907
Castillo Ferrer, C., Berthenet, K., and Ichim, G. (2021). Apoptosis - fueling the oncogenic fire. Febs J. 288, 4445–4463. doi:10.1111/febs.15624
Chen, F., Cai, X., Kang, R., Liu, J., and Tang, D. (2023). Autophagy-dependent ferroptosis in cancer. Antioxid. Redox Signal 39, 79–101. doi:10.1089/ars.2022.0202
Colon-Plaza, S., and Su, T. T. (2022). Non-apoptotic role of apoptotic caspases in the Drosophila nervous system. Front. Cell Dev. Biol. 10, 839358. doi:10.3389/fcell.2022.839358
Diwanji, N., and Bergmann, A. (2019). Two sides of the same coin - compensatory proliferation in regeneration and cancer. Adv. Exp. Med. Biol. 1167, 65–85. doi:10.1007/978-3-030-23629-8_4
Doll, S., Freitas, F. P., Shah, R., Aldrovandi, M., da Silva, M. C., Ingold, I., et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. doi:10.1038/s41586-019-1707-0
Doll, S., Proneth, B., Tyurina, Y. Y., Panzilius, E., Kobayashi, S., Ingold, I., et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 13, 91–98. doi:10.1038/nchembio.2239
Domingos, P. M., and Steller, H. (2007). Pathways regulating apoptosis during patterning and development. Curr. Opin. Genet. Dev. 17, 294–299. doi:10.1016/j.gde.2007.05.009
Dominguez, D., and Fan, Y. (2023). Non-lethal roles of the initiator caspase Dronc in Drosophila. Front. Cell Death 2. doi:10.3389/fceld.2023.1184041
Fanfone, D., Wu, Z., Mammi, J., Berthenet, K., Neves, D., Weber, K., et al. (2022). Confined migration promotes cancer metastasis through resistance to anoikis and increased invasiveness. Elife 11, e73150. doi:10.7554/eLife.73150
Fouillet, A., Levet, C., Virgone, A., Robin, M., Dourlen, P., Rieusset, J., et al. (2012). ER stress inhibits neuronal death by promoting autophagy. Autophagy 8, 915–926. doi:10.4161/auto.19716
Fritsch, M., Gunther, S. D., Schwarzer, R., Albert, M. C., Schorn, F., Werthenbach, J. P., et al. (2019). Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575, 683–687. doi:10.1038/s41586-019-1770-6
Fuchs, Y., and Steller, H. (2011). Programmed cell death in animal development and disease. Cell 147, 742–758. doi:10.1016/j.cell.2011.10.033
Galluzzi, L., Vitale, I., Aaronson, S. A., Abrams, J. M., Adam, D., Agostinis, P., et al. (2018). Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541. doi:10.1038/s41418-017-0012-4
Geisbrecht, E. R., and Montell, D. J. (2004). A role for Drosophila IAP1-mediated caspase inhibition in Rac-dependent cell migration. Cell 118, 111–125. doi:10.1016/j.cell.2004.06.020
Gorelick-Ashkenazi, A., Weiss, R., Sapozhnikov, L., Florentin, A., Tarayrah-Ibraheim, L., Dweik, D., et al. (2018). Caspases maintain tissue integrity by an apoptosis-independent inhibition of cell migration and invasion. Nat. Commun. 9, 2806. doi:10.1038/s41467-018-05204-6
Green, D. R. (2022). The future of death. Cold Spring Harb. Perspect. Biol. 14, a041111. doi:10.1101/cshperspect.a041111
Hetz, C., and Mollereau, B. (2014). Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 15, 233–249. doi:10.1038/nrn3689
Holler, N., Zaru, R., Micheau, O., Thome, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1, 489–495. doi:10.1038/82732
Ichim, G., Gibert, B., Adriouch, S., Brenner, C., Davoust, N., Desagher, S., et al. (2022). Keeping cell death alive: an introduction into the French cell death research network. Biomolecules 12, 901. doi:10.3390/biom12070901
Ichim, G., Lopez, J., Ahmed, S. U., Muthalagu, N., Giampazolias, E., Delgado, M. E., et al. (2015). Limited mitochondrial permeabilization causes DNA damage and genomic instability in the absence of cell death. Mol. Cell 57, 860–872. doi:10.1016/j.molcel.2015.01.018
Ichim, G., and Tait, S. W. (2016). A fate worse than death: apoptosis as an oncogenic process. Nat. Rev. Cancer 16, 539–548. doi:10.1038/nrc.2016.58
Ingold, I., Berndt, C., Schmitt, S., Doll, S., Poschmann, G., Buday, K., et al. (2017). Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172, 409–422. doi:10.1016/j.cell.2017.11.048
Kalkavan, H., Chen, M. J., Crawford, J. C., Quarato, G., Fitzgerald, P., Tait, S. W. G., et al. (2022). Sublethal cytochrome c release generates drug-tolerant persister cells. Cell 185 (185), 3356–3374.e22. doi:10.1016/j.cell.2022.07.025
Kalkavan, H., and Green, D. R. (2018). MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 25, 46–55. doi:10.1038/cdd.2017.179
Kalkavan, H., Ruhl, S., Shaw, J. J. P., and Green, D. R. (2023). Non-lethal outcomes of engaging regulated cell death pathways in cancer. Nat. Cancer 4, 795–806. doi:10.1038/s43018-023-00571-6
Kawahara, A., Ohsawa, Y., Matsumura, H., Uchiyama, Y., and Nagata, S. (1998). Caspase-independent cell killing by Fas-associated protein with death domain. J. Cell Biol. 143, 1353–1360. doi:10.1083/jcb.143.5.1353
Kayagaki, N., and Dixit, V. M. (2019). Rescue from a fiery death: a therapeutic endeavor. Science 366, 688–689. doi:10.1126/science.aaw1177
Killarney, S. T., Tait, S. W. G., Green, D. R., and Wood, K. C. (2023). Sublethal engagement of apoptotic pathways in residual cancer. Trends Cell Biol. 34, 225–238. doi:10.1016/j.tcb.2023.07.005
Klionsky, D. J., Abdel-Aziz, A. K., Abdelfatah, S., Abdellatif, M., Abdoli, A., Abel, S., et al. 2021. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition)(1). Autophagy;17:1–382.
Lamb, C. A., Yoshimori, T., and Tooze, S. A. (2013). The autophagosome: origins unknown, biogenesis complex. Nat. Rev. Mol. Cell Biol. 14, 759–774. doi:10.1038/nrm3696
Mendes, C. S., Levet, C., Chatelain, G., Dourlen, P., Fouillet, A., Dichtel-Danjoy, M. L., et al. (2009). ER stress protects from retinal degeneration. Embo J. 28, 1296–1307. doi:10.1038/emboj.2009.76
Mollereau, B. (2013). Establishing links between endoplasmic reticulum-mediated hormesis and cancer. Mol. Cell Biol. 33, 2372–2374. doi:10.1128/MCB.00315-13
Mollereau, B. (2015). Cooling-induced ER stress is good for your brain. EBioMedicine 2, 482–483. doi:10.1016/j.ebiom.2015.05.008
Mollereau, B., Hayflick, S. J., Escalante, R., Mauthe, M., Papandreou, A., Iuso, A., et al. (2023). A burning question from the first international BPAN symposium: is restoration of autophagy a promising therapeutic strategy for BPAN? Autophagy 19, 3234–3239. doi:10.1080/15548627.2023.2247314
Mollereau, B., Manie, S., and Napoletano, F. (2014). Getting the better of ER stress. J. Cell Commun. Signal 8, 311–321. doi:10.1007/s12079-014-0251-9
Mollereau, B., Perez-Garijo, A., Bergmann, A., Miura, M., Gerlitz, O., Ryoo, H. D., et al. (2012). Compensatory proliferation and apoptosis-induced proliferation: a need for clarification. Cell Death Differ. 20, 181. doi:10.1038/cdd.2012.82
Mollereau, B., Rzechorzek, N. M., Roussel, B. D., Sedru, M., Van den Brink, D., Bailly-Maitre, B., et al. (2016). Adaptive preconditioning in neurological diseases - therapeutic insights from proteostatic perturbations. Brain Res. Mar. 1648, 603–616. doi:10.1016/j.brainres.2016.02.033
Murphy, J. M., Czabotar, P. E., Hildebrand, J. M., Lucet, I. S., Zhang, J. G., Alvarez-Diaz, S., et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39, 443–453. doi:10.1016/j.immuni.2013.06.018
Napoletano, F., Baron, O., Vandenabeele, P., Mollereau, B., and Fanto, M. (2019). Intersections between regulated cell death and autophagy. Trends Cell Biol. 29, 323–338. doi:10.1016/j.tcb.2018.12.007
Napoletano, F., Gibert, B., Yacobi-Sharon, K., Vincent, S., Favrot, C., Mehlen, P., et al. (2017). p53-dependent programmed necrosis controls germ cell homeostasis during spermatogenesis. PLoS Genet. 13, e1007024. doi:10.1371/journal.pgen.1007024
Newton, K., Dixit, V. M., and Kayagaki, N. (2021). Dying cells fan the flames of inflammation. Science 374, 1076–1080. doi:10.1126/science.abi5934
Orning, P., Weng, D., Starheim, K., Ratner, D., Best, Z., Lee, B., et al. (2018). Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362, 1064–1069. doi:10.1126/science.aau2818
Poltorak, A. (2022). Cell death: all roads lead to mitochondria. Curr. Biol. 32, R891–R894. doi:10.1016/j.cub.2022.07.025
Riedl, S. J., and Salvesen, G. S. (2007). The apoptosome: signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 8, 405–413. doi:10.1038/nrm2153
Stockwell, B. R., Friedmann Angeli, J. P., Bayir, H., Bush, A. I., Conrad, M., Dixon, S. J., et al. (2017). Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285. doi:10.1016/j.cell.2017.09.021
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148, 213–227. doi:10.1016/j.cell.2011.11.031
Suthar, K. P., and Bergmann, A. (2023). Something new under the sun: caspases promote cell survival under moderate heat stress. EMBO Rep. 24, e57276. doi:10.15252/embr.202357276
Svandova, E., Lesot, H., Sharpe, P., and Matalova, E. (2022). Making the head: caspases in life and death. Front. Cell Dev. Biol. 10, 1075751. doi:10.3389/fcell.2022.1075751
Tenev, T., Bianchi, K., Darding, M., Broemer, M., Langlais, C., Wallberg, F., et al. (2011). The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol. Cell 43, 432–448. doi:10.1016/j.molcel.2011.06.006
Tummers, B., and Green, D. R. (2017). Caspase-8: regulating life and death. Immunol. Rev. 277, 76–89. doi:10.1111/imr.12541
Vandenabeele, P., Galluzzi, L., Vanden Berghe, T., and Kroemer, G. (2010). Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat. Rev. Mol. Cell Biol. 11, 700–714. doi:10.1038/nrm2970
Varfolomeev, E. E., Schuchmann, M., Luria, V., Chiannilkulchai, N., Beckmann, J. S., Mett, I. L., et al. (1998). Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9, 267–276. doi:10.1016/s1074-7613(00)80609-3
Wang, Y., Gao, W., Shi, X., Ding, J., Liu, W., He, H., et al. (2017). Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103. doi:10.1038/nature22393
Weindel, C. G., Bell, S. L., Vail, K. J., West, K. O., Patrick, K. L., and Watson, R. O. (2020). LRRK2 maintains mitochondrial homeostasis and regulates innate immune responses to Mycobacterium tuberculosis. Elife. 9, e51071. doi:10.7554/eLife.51071
Weindel, C. G., Martinez, E. L., Zhao, X., Mabry, C. J., Bell, S. L., Vail, K. J., et al. (2022). Mitochondrial ROS promotes susceptibility to infection via gasdermin D-mediated necroptosis. Cell 185, 3214–3231.e23. doi:10.1016/j.cell.2022.06.038
Xu, D. C., Wang, L., Yamada, K. M., and Baena-Lopez, L. A. (2022). Non-apoptotic activation of Drosophila caspase-2/9 modulates JNK signaling, the tumor microenvironment, and growth of wound-like tumors. Cell Rep. 39, 110718. doi:10.1016/j.celrep.2022.110718
Yacobi-Sharon, K., Namdar, Y., and Arama, E. (2013). Alternative germ cell death pathway in Drosophila involves HtrA2/omi, lysosomes, and a caspase-9 counterpart. Dev. Cell 25, 29–42. doi:10.1016/j.devcel.2013.02.002
Yuan, J., Najafov, A., and Py, B. F. (2016). Roles of caspases in necrotic cell death. Cell 167, 1693–1704. doi:10.1016/j.cell.2016.11.047
Keywords: apoptosis, necrosis, cancer, migration, Drosophila
Citation: Shan Y and Mollereau B (2024) Non-canonical functions of regulated cell death machinery regulate cellular growth, invasion and the interplay between cell death modalities. Front. Cell. Death 3:1423805. doi: 10.3389/fceld.2024.1423805
Received: 26 April 2024; Accepted: 12 June 2024;
Published: 15 July 2024.
Edited by:
Zhenggang Liu, National Institutes of Health (NIH), United StatesReviewed by:
Lawrence M. Schwartz, University of Massachusetts Amherst, United StatesCopyright © 2024 Shan and Mollereau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: B. Mollereau, YmVydHJhbmQubW9sbGVyZWF1QGVucy1seW9uLmZy
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.