 YingChu Gu
YingChu Gu ZeRui Wu
ZeRui Wu Heng Xie2
Heng Xie2 Ye Gu
Ye Gu
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Death , 05 February 2024
Sec. Non-Apoptotic Regulated Cell Death
Volume 3 - 2024 | https://doi.org/10.3389/fceld.2024.1337724
Periprosthetic osteolysis is a difficult-to-treat complication of arthroplasty. The pathological mechanisms of periprosthetic osteolysis are mainly weakened function of osteoblasts and excessive activation of osteoclasts. Many studies have demonstrated that the imbalance between the formation of bone by osteoblasts and the absorption of bone by osteoclasts is the direct cause of osteolytic diseases. Autophagy, as an important self-protective cellular mechanism, has significant effects on the regulation of osteoblast function, such as osteoblast differentiation, proliferation, and apoptosis. Osteoblasts, which play an important role in maintaining bone homeostasis, have attracted increasing attention in recent years. Up till now, Several signaling pathways have been proved to regulate autophagy of osteoblasts, including the AMPK, NF-κB, FoxO3 and other signaling pathways. This article reviews the recent progress in understanding osteoblast autophagy and mitophagy in the context of periprosthetic osteolysis and the signaling pathways which are involved in these processes. By summarizing previous studies describing the mechanism underlying osteoblast autophagy, we wish to contribute new therapeutic ideas and potential therapeutic targets for periprosthetic osteolysis.
As the world ages, the number of patients receiving total joint replacement (TJR) is increasing. As the preferred treatment for end-stage bone and joint diseases, TJR effectively relieves pain and re-establishes joint function, which vastly improving patients’ quality of life (Talmo et al., 2010). According to data from 2000 to 2014, the number of total hip arthroplasty (THA) procedures is expected to increase by 71%–635,000 by 2030, and the number of total knee arthroplasty (TKA) procedures is expected to increase by 85%–1,260,000 (Sloan et al., 2018). However, increasing research shows that an increase in the number of TJRs inevitably leads to a series of postoperative complications, such as infection, periprosthetic fracture, instability/dislocation, neurovascular injury, periprosthetic osteolysis (PPOL), and so on (Talmo et al., 2012). Most complications can be prevented through effective preoperative examination and high-quality postoperative care, but PPOL is a difficult problem for doctors and patients (Purdue et al., 2007). Among all reported joint replacement operations, the hazards posed by PPOL are common and serious. According to data from the National Joint Replacement Registration System of the United Kingdom, PPOL most often leads to failed hip and knee revision surgeries, with approximately 48.1% of hips needing hip revision and 39.6% of knees needing revision (Blom et al., 2021). Although PPOL has been studied for more than 50 years, its pathogenesis has not been fully clarified. Although some therapeutic methods have led to positive results in experimental animal models, few cases have been successfully treated in clinical practice. Therefore, more research and investment are still needed for PPOL prevention and treatment. Over time, wear debris generated on the surface of a prosthetic joint is the main factor limiting the survival of joint implants. Considerable research-based evidence has shown the promoting effect of chronic low-grade inflammation on PPOL. Many experiments have repeatedly shown that after artificial prosthesis wear particles contact immune cells, the latter express proinflammatory factors, chemokines and other substances. This cellular response results in chronic aseptic inflammation of surrounding tissue (Ollivere et al., 2012). In addition, many osteoclasts are generated due to inflammation, and the osteogenic capacity of osteoblasts is reduced due to inflammation. Multiple factors promote each other to form the pathophysiological basis of PPOL; however, the main cause is local bone metabolism imbalance caused by the functional disorder of osteoblasts and osteoclasts (Noordin and Masri, 2012).
Osteoblasts adhere to the bone surface after differentiation from mesenchymal stem cells (MSCs). Their functions include mainly generating bone matrix and regulating osteoclast functions. They promote mineralization of bone matrix by synthesizing type I colloidin (Kim et al., 2020). In an osteolytic microenvironment influenced by wear particles, the osteoblast phenotype is changed, with the expression of corresponding genes changing, diminishing the osteogenic capacity of osteoblasts (Vermes et al., 2001). The literature suggests that the damage to osteoblast osteogenic ability induced by wear particles and the reduction in collagen synthesis are direct causes of PPOL occurrence and development. Besides, the inflammation and oxidative stress induced by wear particles can directly injure osteoblasts and thus lead to a reduction in their number (O'Neill et al., 2013).
Autophagy can help cells survive nutritional stress at critical times of development. When cells are in a state of stress or their internal nutrient and energy needs are increased or reduced by other autophagy-inducing factors, the autophagy in cells is activated and contributes to cell adaptation to the new environment (Dikic and Elazar, 2018). In addition, autophagy plays a “housekeeping” role by eliminating misfolded proteins, damaged organelles and intracellular pathogens (Kim and Lee, 2014). Autophagy can be further activated under stress conditions such as nutrient or energy deficiency to degrade cytoplasmic substances into metabolites that can be used for biosynthesis or energy production to promote cell survival. Therefore, Moderate autophagy has a protective effect on cells, while excessive autophagy may be harmful and cause irreversible cell damage (Maiuri et al., 2007). Autophagy is regulated by a group of genes which called autophagy-related genes (ATGs). Autophagy is mediated after the formation of an “autophagosome”, which is an isolated double membrane transiently formed organelle (also known as a phagocyte). Excess or potentially damaging cytosol is isolated in an autophagosome and delivered to a lysosome, where it is degraded (Mizushima et al., 2008). Autophagy is an effective means by which osteoblasts protect themselves in harmful environments and plays a key role in maintaining the balance of bone metabolism, osteoblast proliferation and differentiation and bone formation (di Giacomo et al., 2020). However, excessive autophagic flux leads to the death of osteoblasts, and inhibiting excessive autophagic flux is thus a potential strategy to treat osteolysis (Ma et al., 2021). Proanthocyanidins can be used to treat PPOL in mouse skulls caused by tricalcium phosphate (TCP) wear particles by inhibiting autophagy and oxidative stress (Lin et al., 2019). Zhiwei Zhang et al. used nano alumina (Al) particles and bortezomib (BTZ) to reduce the rate of osteoblast autophagy induced by titanium (Ti) particles and thereby attenuate cell apoptosis, inflammation and osteolysis (Zhang Z. et al., 2020). Liu N, Jia QiYan and others also proved that inhibition of autophagy can be an efficient treatment for peri-implant osteolysis induced by stress granules (Liu N. et al., 2016; Yan et al., 2018).
Classical autophagy is a process involving the sequential and selective activation of multiple ATGs proteins. Initiation of the autophagy cascade is physiologically inhibited by MTORC1, which can mediate the inactivation phosphorylation of ATG13 and ULK1. The complex containing ULK1 and ATG13 is central to initiating autophagy (Klionsky et al., 2021). AMPK can counteract the inhibition of MTORC1 by phosphorylating ULK1 and Beclin1. ULK1 triggers an autophagy cascade by promoting Beclin1 to form complexes with different proteins (Galluzzi et al., 2017). In addition, multiple signaling pathways and transcription factors have been shown to regulate ATGs expression. These different signaling pathways together constitute a complex autophagy network.
Therefore, an in-depth understanding of the mechanisms of autophagy is needed for developing treatments for PPOL. This article mainly reviews the effects of osteoblast autophagy in PPOL and related signal transduction pathways with the aim of suggesting new ideas and potential therapeutic targets for PPOL. Figure 1 shows the major effects of autophagy on osteoblasts in PPOL. Table 1 lists important studies from the past decade on autophagy in osteoblasts during PPOL. Table 2 summarizes some key autophagy genes or signaling pathways that play an important role in regulating the function of osteoblasts.
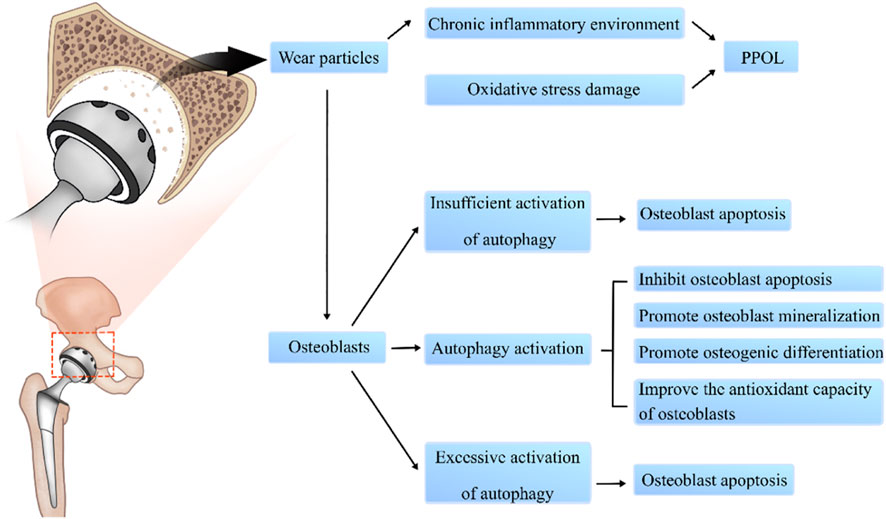
FIGURE 1. The role of autophagy in osteoblasts during PPOL.
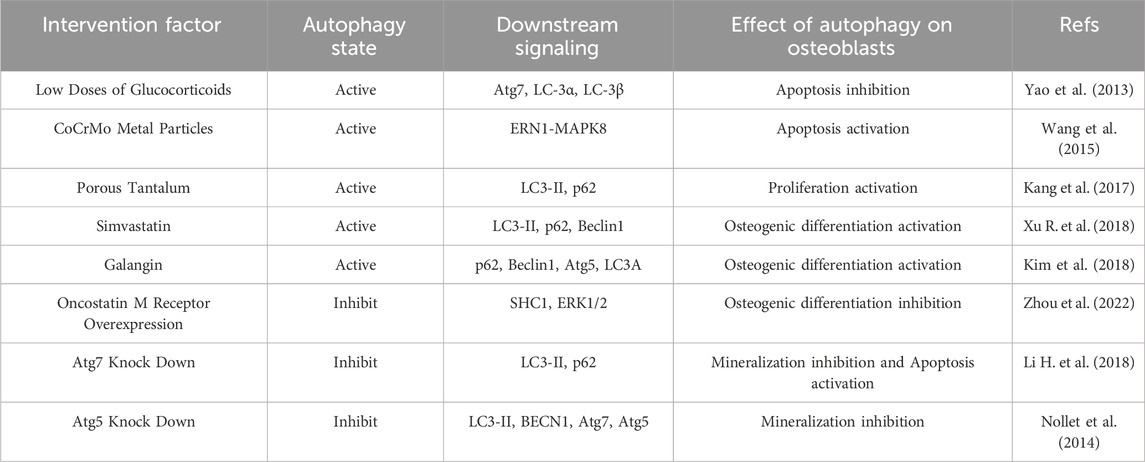
TABLE 1. Summary of important studies from the past decade on autophagy in osteoblasts during osteolysis.
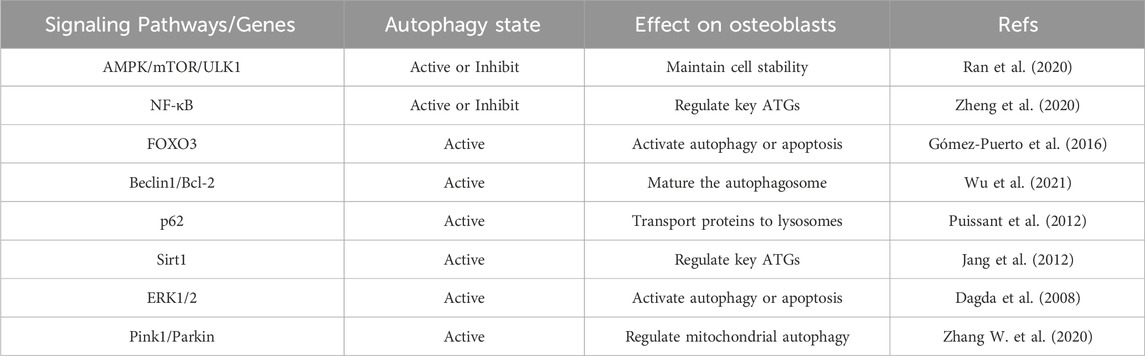
TABLE 2. Summary of important studies in the past decade on osteoblast autophagy in the context of osteolysis.
The apoptosis of osteoblasts induced by wear particles is recognized as a cause for the occurrence and progression of osteolysis near a prosthesis, mainly due to chronic inflammation and oxidative stress induced by the wear particles (Zhang Y. et al., 2022). This process may be reversed by enhancing the antiapoptotic ability of osteoblasts. All cells respond quickly to the external environment or conditions by regulating autophagic flux to survive stress conditions. Survival stress mainly includes a high sugar level, a low oxygen level, inflammation, hunger, hormone activity, and oxidative stress (Lacarrière-Keïta et al., 2022). Jia J et al. demonstrated that autophagy may contribute to osteoblast survival under stress conditions after glucocorticoid exposure. Lower concentrations of glucocorticoids can activate autophagy in osteoblasts, while higher concentrations of glucocorticoids induce apoptosis (Jia et al., 2011). Xia et al. showed that certain metabolic stressors can activate autophagy in osteoblasts. Autophagy may be a mechanism by which bone cells repair damaged organelles or cell membranes. However, sustained high levels of stress can lead to the accumulation of autophagosomes and cell death (Xia et al., 2010). Therefore, the activation of autophagy induced by stress factors alone is attenuated but not enough to overcome sustained high stress levels. Can we enhance the antiapoptotic capacity in osteoblasts under survival stress through artificial autophagy regulation? Recent reports suggest that β-ecdysterone regulates autophagy in osteoblasts. Specifically, β-ecdysterone reverses dexamethasone-induced osteoblast apoptosis by promoting autophagy in osteoblasts through the AMPK/mTOR pathway. Moreover, β-ecdysone-induced autophagy activation has been associated with the promotion of osteoblast differentiation and fracture healing (Tang et al., 2021). Similarly, studies have shown that paeoniflorin inhibits dexamethasone-induced osteoblast apoptosis by enhancing autophagic flux in osteoblasts by regulating the AKT/mTOR signaling pathway (Yang et al., 2021).
However, other studies have shown that the application of autophagy inhibitors attenuated CoCrMo metal particle-induced osteolysis near prostheses. CoCrMo metal granule-induced autophagy in osteoblasts may increase the expression of the apoptotic proteins BAX and CASP3, thereby exacerbating osteolysis (Wang et al., 2015). In some cases where autophagy upregulation is uncontrolled, overactivation of autophagy can be lethal to cells. This may be due to stress exceeding the extent to which autophagy can compensate, or to the inability of cells to survive non-specific degradation of large amounts of cytoplasmic contents. Overexpression of Atg1 in Drosophila is sufficient to induce high levels of autophagy, which has the ability to inhibit cell growth and induce apoptosis (Scott et al., 2007). Bcl-2, as an anti-apoptotic protein, can interact with the evolutionarily conserved autophagy protein Beclin1. Blocking the binding of Bcl-2 to Beclin1 induces excessive autophagy and promotes apoptosis. Bcl-2 acts not only as an anti-apoptotic protein, but also as an anti-autophagy protein through its inhibitory interaction with Beclin1 (Pattingre et al., 2005). Therefore, the regulation of autophagy can increase antiapoptotic activity in osteoblasts, showing certain potential research value for the treatment of PPOL. Autophagy has two sides: it increases the antiapoptotic capacity of normal cells by degrading damaged organelles and recycling energy-related molecules, but excessive self-phagocytosis induces osteoblast apoptosis.
BMSCs are the main sources of osteoblast differentiation. They can differentiate into bone, cartilage, fat, nerve and muscle cells (Ning et al., 2022). MC3T3-E1 precursor osteoblasts are the main cells used in osteogenic differentiation studies performed in vitro (Ren et al., 2023). The development of the chronic inflammatory environment induced by wear particles can inhibit the differentiation of BMSCs into osteoblasts and promote the differentiation and proliferation of osteoclasts. This effect shifts the bone balance toward bone resorption and ultimately leads to increased osteolysis (Deng et al., 2021).
Activation of autophagy may reverse the inhibitory effect of abrasive particles on osteogenic differentiation. Resveratrol induces osteogenic differentiation of MSCs by promoting autophagy and synergizing with osteogenesis-inducible factors (Vidoni et al., 2019). Strontium (Sr) at a concentration of 3 mM activated the AMPK/mTOR signaling pathway to promote the differentiation of MC3T3-E1 cells into osteoblasts, and inhibition of autophagy counteracted this effect (Cheng et al., 2019). During the differentiation of BMSCs into osteoblasts, inhibition of OSMR expression activates the ERK1/2 signaling pathway and induces autophagy, thus facilitating this process (Zhou et al., 2022).
Many scholars support that autophagy activation promotes the differentiation of osteoblasts, which contributes to alleviating osteolysis near prostheses. However, it has also been reported that the effect of autophagy on osteoblast differentiation is bidirectional. As a traditional Chinese medicine formulation, QiangGuYin activates the AKT/mTOR pathway and inhibits autophagy in osteoblasts by inhibiting the expression of CIP-1, thereby attenuating osteoporosis (Yuan et al., 2022). AMPK is a major pathway leading to autophagy activation. Notably, the preliminary activation of AMPK is necessary for osteoblast differentiation. The continuous activation of the AMPK or autophagy pathway in the late stage of osteoblast differentiation has inhibitory effects (Xi et al., 2016). Pantovic A et al. demonstrated that osteogenic differentiation of BMSCs requires the initial activation of the AMPK/mTOR autophagy pathway and later activation of the Akt/mTOR autophagy pathway (Pantovic et al., 2013). Therefore, the regulation of autophagy plays a very important role in the differentiation of osteoblasts, and further study of its regulatory mechanism may lead to a new treatment for osteolysis.
Ti granule-induced oxidative stress injury in osteoblasts has been demonstrated (Wei et al., 2009). Ti particles mediate mitochondrial damage in osteoblasts. When mitochondria are damaged, they produce too much excessive reactive oxygen species (ROS). Among the ROS targets are mitochondria themselves, and a vicious cycle develops between ROS and mitochondria. The main effect of oxidative stress after bone injury is inhibited function and activity of osteoblasts and bone cells and inhibited bone formation. Moreover, oxidative stress promotes osteoclast proliferation and enhances bone resorption (Hameister et al., 2020). These discoveries indicated that ROS overexpression exacerbates wear granule-induced osteolysis, and maintaining nromal mitochondrial function is necessary for the function and activity of osteoblasts. As an important mechanism contributing to mitochondrial quality maintenance, mitochondrial autophagy can mediate the degradation of damaged mitochondria and reduce the production of ROS, thereby protecting other normal cells and maintaining mitochondrial homeostasis (Shi et al., 2023).
Although not effective in all cells, resveratrol has been shown to function as an autophagy activator in osteoblasts. Xuhao Yang et al. found that the application of resveratrol to osteoporotic rats enhanced mitophagy in osteoblasts by increasing the expression of Sirt1, thereby attenuating the oxidative stress-induced damage to osteoblasts induced by glucocorticoids. (Yang et al., 2019). Bartolome A et al. demonstrated that autophagy inhibition in osteoblasts exacerbated oxidative stress damage in a high-glucose environment (Bartolomé et al., 2013). Low doses of glucocorticoids promoted autophagy in osteoblasts in response to hormone-induced oxidative stress damage. When the hormone dose was increased, autophagy attenuated the increase in ROS levels, and osteoblasts underwent apoptosis (Zhang et al., 2018).
In addition to the activation of mitochondrial autophagy, autophagy can also directly regulate cellular antioxidant defense mechanisms. The most relevant interaction between autophagy and oxidative stress response mechanisms is achieved through Nrf2 signaling. When autophagy is activated, the accumulated p62 can compete with Keap1 to activate Nrf2. This results in a reversal of Keap1 binding to Nrf2. The continuous accumulation of non-ubiquitinated Nrf2 can mediate the activation of antioxidant gene transcription (Lee et al., 2020). Besides, the activation of Nrf2 can also have a positive effect on the autophagy process. NDP52 is a cargo recognition protein regulated by NRF2. NDP52 acts in a similar way to p62, mainly mediating the degradation of goods in lysosomes (Jo et al., 2014). Moreover, NRF2 can regulate other autophagy related genes, such as ULK1, p62, ATG7, ATG4 and so on (Pajares et al., 2016). SIRT1 has also been proved to activate antioxidant defense mechanisms by mediating the deacetylation of Nrf2 in vitro (Ding et al., 2016). Foxo3 also shows a similar role as a transcription factor, which can activate antioxidant defense mechanisms by regulating the level of autophagy in cells. Therefore, there is a close relationship between the signaling pathway of autophagy and Reduction-Oxidation.
In conclusion, autophagy activation may confer self-protection to osteoblasts in response to oxidative stress injury. However, the level of autophagy induced by survival stress is limited, and cells struggle to mitigate progressively enhanced oxidative stress damage. Therefore, targeted regulation of autophagy to increase the antioxidative capacity of osteoblasts is important for improving the prognosis of osteolysis.
The mineralization of osteoblasts has great effects in the prognosis of osteolysis near a prosthesis. Osteoblast mineralization is a complex process involving many signaling pathways and cellular processes (Hasegawa et al., 2022). Autophagy has been recently identified as a necessary condition for the mineralization of osteoblast. Osteoblast mineralization itself can induce autophagy. Autophagosomes can carry secreted apatite crystals to the extracellular space. Atg5-deficient transgenic mice showed a significant decrease in bone mass that was mainly attributed to a decreased number of osteoblasts and thus decreased mineralization (Nollet et al., 2014). As an autophagy-activating pathway, the AMPK pathway promotes the differentiation and mineralization of osteoblasts by inducing autophagy. The application of AMPK pathway inhibitors reverses this effect (Li Y. et al., 2018). Vitro studies prove that kaempferol enhances the mineralization ability of MC3T3-E1 cells by promoting autophagy (Kim et al., 2016). Vivo studies suggest that glucosamine induces osteoblast differentiation and mineralization by promoting autophagy, thereby improving the prognosis of osteoporotic mice (Su et al., 2022). However, other studies show that rapamycin-induced autophagy significantly reduces the activity and mineralization of osteoblasts. While overexpression of RUNX2 reverses this result by inhibiting ATGs expression (Ren et al., 2022). Therefore, autophagy exerts different effects on osteoblast mineralization according to the different pathways it activates and the degree to which they are activated. Autogenerated autophagy in cells challenged with environmental stress is generally considered beneficial, but excessive autophagic flux induced by certain pathological factors, including external stimuli, may impair cell activity and function.
In conclusion, autophagosomes are carriers of osteoblast mineralized crystals and are necessary conditions for osteoblast mineralization. Unfortunately, the mechanism by which autophagy regulates osteoblast mineralization remains unclear, mainly due to the complexity of the mineralization process and poorly characterized autophagy signaling pathways.
AMPK is an AMP-dependent protein kinase. It is a heterotrimeric complex composed by α, β and γ subunits. AMPK activation increases the catabolism (ATP production) pathway activation rate and reduces the anabolic (ATP utilization) pathway activation rate. Thus, AMPK has great effects in whole-body energy metabolism (Carling, 2017). In decades, AMPK has been identified as the guardian of mitochondrial homeostasis (Friis et al., 2014). AMPK induces autophagy through two different mechanisms: inhibiting the activation of mammalian rapamycin target protein (mTOR) and inducing the direct phosphorylation of mammalian autophagy initiation kinase (ULK1). In addition, mTOR and ULK1 interact. Therefore, AMPK, mTOR and ULK1 activate or inhibit each other to form a triangular network (Alers et al., 2012). AMPK is phosphorylated when an organism has a low energy level or is hungry. Activation of AMPK can induce autophagy by inhibiting mTOR activity in two ways: (1) AMPK enhances the activity of TSC1/TSC2 complex by mediating the phosphorylation of TSC2. The TSC1/TSC2 complex inactivates RHEB by hydrolyzing GTP. Inactivated RHEB can not induce the activation of mTORC1. (2) AMPK can block RAPTOR binding to mTOR or mTOR substrates and inhibiting the activation of mTOR signaling pathways by directly phosphorylating RAPTOR. The inactivation of mTOR reduces the inhibitory effect of mTOR on ULK1. Then AMPK binds to ULK1 and induces autophagy (Li and Chen, 2019). In addition, AMPK directly phosphorylates multiple sites in ULK1 to increase autophagic flux (Kim et al., 2011). In conclusion, both AMPK and mTOR regulate ULK1 function. An increasing number of reports have shown two-way regulation among AMPK, mTOR and ULK1, which means that AMPK, ULK1 and mTOR form a signal triplet. They regulate each other to maintain dynamic autophagy in balance (Wong et al., 2013). Figure 2 shows a schematic diagram of the AMPK/mTOR/ULK1 signaling pathway.
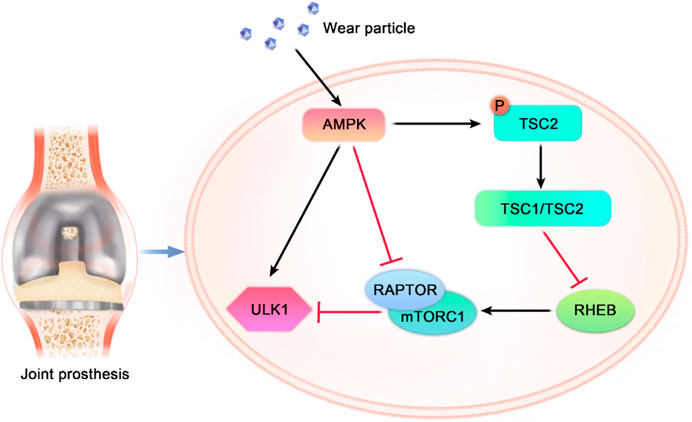
FIGURE 2. Mutual regulatory mechanism among AMPK, mTOR and ULK1. Exogenous stimuli such as wear particles or drugs activate AMPK. Activated AMPK directly phosphorylates ULK1 to promote autophagy. AMPK inhibits autophagy by phosphorylating RAPTOR, blocking the binding of RAPTOR to mTOR. In addition, AMPK phosphorylates TSC2 to enhance the GTPase activity of the TSC1/TSC2 complex. The TSC1/TSC2 complex inactivates RHEB, resulting in the loss of RHEB activation of mTORC, thereby inhibiting autophagy. TSC1: tuberous sclerosis 1, TSC2: tuberous sclerosis 2, RHEB: Ras homolog-enriched in brain protein, RAPTOR: regulatory-associated protein of mTOR, and mTORC1: mTOR complex 1.
The ability of AMPK to regulate autophagy can affect the function and activity of osteoblasts in osteolytic diseases. Negative pressure drainage at a wound site enhances autophagy in osteoblasts by activating AMPK and regulating ULK1 function, thus promoting bone regeneration (Zhang S. et al., 2022). Through the osteogenic differentiation of human dental pulp MSCs, knocking down AMPK prevented early mTOR inhibition and weakened autophagic flux, which hindered osteoblast differentiation. The inhibition of Akt reduces the mTOR activation rate, enhances autophagic flux and promotes osteoblast differentiation (Pantovic et al., 2013). Inhibition of Akt reduces mTOR activation and enhances autophagy, which can promote osteoblast differentiation (Lu et al., 2021).
Under physiological conditions, autophagic flux in osteoblasts remains at the basal level. When cells encounter excessive harmful stimuli such as wear particles or oxidative stress and inflammation-inducing stimuli, autophagic flux is increased to enhance self-protection. However, typically, the autophagic flux in osteoblasts is insufficient to attenuate the response to foreign stimuli, resulting in osteolysis. The AMPK signaling pathway is a main pathway that enhances autophagic flux in osteoblasts challenged with wear particles. The upregulation of AMPK leads to inhibited mTOR activation and promoted ULK1 activation, both of which increase autophagic flux in osteoblasts and thus confer self-protection to osteoblasts challenged with harmful stimuli. Therefore, targeted activation of AMPK to promote autophagy has become a potential treatment for osteolysis near prostheses.
Nuclear factor κB (NF-κB) family members and the retroviral oncoprotein v-Rel constitute the NF-κB/Rel protein family, which has an important position in inflammation and the immune response (Sun, 2017). NF-κB is formed by the dimerization of five subunits, namely, NF-κB1 (p50), NF-κB2 (p52), RELA (also known as p65), RELB and c-REL (Hayden and Ghosh, 2008). NF-κB activation is mediated through two main signaling pathways: the classical and nonclassical NF-κB signaling pathways. The classical signaling pathway mediates the activation of NF-κB1 (p50), RELA and c-Rel, while the nonclassical signaling pathway mediates the activation of NF-κB2 (p52) and RELB (Sun, 2011). As the NF-κB pathway is activated by interleukin-1 (IL-1) and tumor necrosis factor-α (TNFα) and the NF-κB pathway exerts effects on the expression of other proinflammatory genes, the NF-κB pathway is considered a typical proinflammatory signaling pathway (Lawrence, 2009). Moreover, the antiapoptotic effect of NF-κB shows that it plays a crucial role in the occurrence and development of tumors (Hoesel and Schmid, 2013). Complex crosstalk between the NF-κB signaling pathway and components of the autophagy pathway has been reported (Verzella et al., 2020). Depending on the different inducers, NF-κB can positively or negatively regulate autophagy activation. Mojgan Djavaheri-Mergny et al. showed that TNFA-induced NFkB activation leads to inhibition of autophagy in cell lines derived from three types of cancer (Ewing sarcoma, breast cancer and acute promyelocytic leukemia) (Djavaheri-Mergny et al., 2007). Role of hypoxia et al. showed that pharmacologically activated NF-κB under hypoxia conditions can induce autophagy in stromal fibroblasts (Martinez-Outschoorn et al., 2010). Studies by Mathieu Nivon et al. have shown that NF-κB can activate autophagy during the recovery phase of cellular heat shock response and increase cell survival by clearing irreversibly damaged proteins (Nivon et al., 2009). NF-κB can also affect autophagy by inducing or inhibiting the expression of multiple ATGs. NF-κB and its family members p65/RelA recognize and bind the promoter of Beclin1, promoting the expression of the Beclin1 gene and enhancing autophagy. Interestingly, NF-κB exerts the opposite regulatory effect on Beclin1. Activation of NF-κB upregulates the expression of A20, which inhibits Beclin1 ubiquitination and thus inhibits autophagy (Trocoli and Djavaheri-Mergny, 2011). In addition, NF-κB influence autophagy by regulating Bnip3. Under physiological conditions, p65 inhibits Bnip3 transcription and reduces autophagy by preventing E2F1 from binding to the Bnip3 promoter. The activity of NF-κB decreases under hypoxic conditions, which leads to increased Bnip3 expression and autophagy induction (Shaw et al., 2008). It has also been suggested that starvation can induce multiple sites phosphorylation of Bcl-2 by JNK1 signaling pathway and mediate the dissociation of Bcl-2/Beclin1 complex. The NF-κB signaling pathway can inhibit the dissociation of the Bcl-2/Beclin 1 complex by inhibiting the JNK1 signaling pathway, thereby inhibiting autophagy (Wei et al., 2008) The regulatory effect of NF-κB on autophagy is briefly shown in Figure 3.
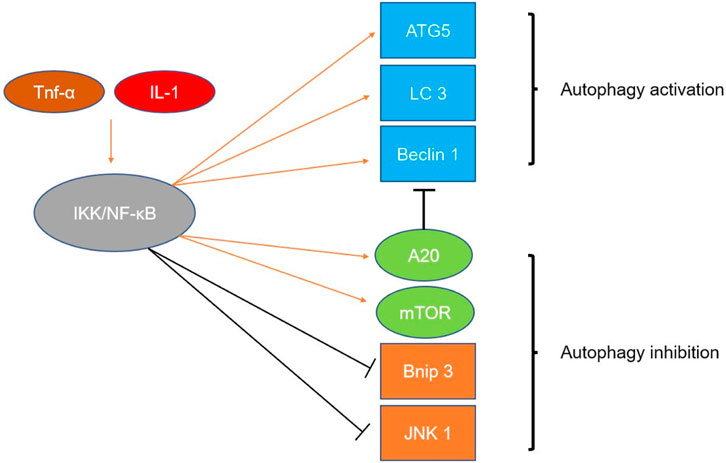
FIGURE 3. Mechanism by which NF-κB regulates autophagy. Inflammatory factors such as Tnf-α and IL-1 can activate the IKK/NF-κB pathway. Activation of the NF-κB pathway can upregulate the expression and activity of autophagy-related genes, such as Beclin1, ATG5, and LC3. Activation of the NF-κB pathway also upregulates A20 expression, and upregulation of A20 inhibits Beclin1 ubiquitination and limits autophagy. Activation of the NF-κB pathway also enhances the activity of mTOR, thereby inhibiting autophagy. NF-κB activation inhibits autophagy by inhibiting Bnip3 transcription. NF-κB can inhibit the dissociation of the Bcl-2/Beclin1 complex by inhibiting the JNK1 signaling pathway, thereby inhibiting autophagy.
The NF-κB signaling pathway also promotes autophagy in osteoblasts and has significant effects in regulating the function and activity of osteoblasts (Zhang et al., 2021). Without drug stimulation or other interventions, osteoblasts exhibit a certain level of basic autophagic flux. SN50, a blocking agent of the NF-κB pathway, exerts a significant effect on autophagy in osteoblasts only when administered at a certain concentration (25 mg/mL). At this dose, it significantly inhibited autophagic flux in osteoblasts by blocking the NF-κB pathway (Qin et al., 2016). Tnf-α promoted osteoblast autophagy and reduced osteoblast apoptosis in avascular necrotic tissue in the femoral head by activating the P38 MAPK/NF-κB signaling pathway (Zheng et al., 2020). Low concentrations of lipopolysaccharide (LPS) promoted osteoblast autophagy and osteoblast proliferation and inhibited osteoblast apoptosis by activating the NF-κB signaling pathway, thereby accelerating fracture healing (Xu M. X. et al., 2018). However, other studies have shown that diosgenin enhances the differentiation and proliferation of osteoblasts by inhibiting osteoblast autophagy through the ASPP2/NF-κβ pathway (Zhu et al., 2017).
As a classical inflammatory pathway, the NF-κB pathway engages in complicated crosstalk with autophagy and inflammatory pathway components. Interference with NF-κB activity under different conditions may exert different effects on osteoblast activity and function. The regulatory effect on autophagy mediated by NF-κB suggests a mechanism that links inflammation and immune signaling to cell death and self-phagocytosis, but the exact mechanism is unclear. The upregulation of IL-1 and TNF-α induced by wear particles exerts a direct activating effect on the NF-κB pathway. Therefore, the regulation of autophagy by NF-κB plays an important role in the development and outcome of PPOL. Although NF-κB knockout inhibits apoptosis by reducing inflammation, it also abrogates the protective effect of NF-κB-mediated autophagy. Therefore, the regulation of the NF-κB pathway is important to the prognosis of osteolysis near a prosthesis.
In mammals, four members of the FoxO family are expressed: Foxo1, FoxO3, Foxo4 and Foxo6. FoxOs were initially identified as downstream regulators of the insulin pathway. FoxO can control several processes critical to cell homeostasis by regulating multiple targeted genes, including cellular energy production, resistance to oxidative stress, and cell viability and proliferation (Link, 2019). The activity of the FoxO transcription factor can be inhibited by insulin and growth factor signals. Acute loss of IR/IGF1R in the muscles of diabetic rats left FoxOs uninhibited, and the mitochondrial respiration mediated by complex I was thus strongly inhibited (Bhardwaj et al., 2021). Although the FoxO family shows the ability to induce autophagy, the role played by FoxO3 is the most important to autophagy. FoxO3 has long been considered to play a key role in the molecular basis of longevity (Morris et al., 2015). In response to cell survival stress, FoxO3 is transferred to the nucleus and induces the expression of multiple ATGs, including LC3b, Gabarapl1, PI3KII, Ulk1, Atg12, Beclin1, Atg4b, and Bnip3, to promote autophagy. TLR4 (Toll-like receptor 4) inhibits autophagy in microglia through the PI3K-FoxO3 pathway, thereby reducing the phagocytosis rate (Lee et al., 2019). Recently, FoxO3 was shown to downregulate apoptosis by upregulating autophagy in osteoblasts (Fitzwalter and Thorburn, 2018). In MSCs, FoxO3 needs to be activated to induce autophagy, which attenuates any increase in ROS levels caused by increased mitochondrial respiration during osteoblast differentiation and reduces the osteoblast apoptosis rate. Gomez Bodo et al. found that during the osteogenic differentiation of human BMSCs, mitochondrial metabolism is accelerated to produce enough energy to drive differentiation, which leads to an increase in ROS levels and FoxO3 activation. This process depends mainly on Ser294 phosphorylation in FoxO3 induced by mitogen-activated protein kinase 8 (MAPK8). These findings show that FoxO3 activation is important for reducing ROS levels by activating autophagy (Gómez-Puerto et al., 2016).
What’s more, it is reported that FoxO3a is part of the autophagy homeostasis feedback monitoring loop. In cases when normal autophagy of cells is inhibited, FoxO3a induces apoptosis if appropriate levels of autophagy are not re-established within a certain period. The relationship between FoxO3a and autophagy and apoptosis is briefly shown in Figure 4. The transcription factor FoxO3a is both a regulator and a substrate of autophagy. Under physiological conditions, FoxO3a is degraded by basal autophagy. The destruction of autophagy leads to the accumulation of FoxO3a, which leads to the upregulation of BBC3/PUMA, thus inducing apoptosis (Fitzwalter et al., 2018). FoxO3a, as a coinducer of autophagy and apoptosis, is associated with autophagy and apoptosis. FoxO3a induces autophagy by reversely activating autophagy-related genes when autophagy levels are inhibited. However, under the continuous inhibition of autophagy, FoxO3a can induce cell apoptosis as well (Fitzwalter and Thorburn, 2018). In summary, FoxO3a is essential for the regulation of autophagy homeostasis and the regulation of normal physiological functions of cells.
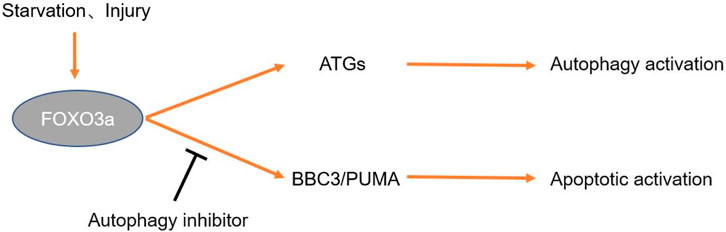
FIGURE 4. Mechanism by which FoxO3a regulates autophagy. Exogenous stimuli such as hunger and trauma can induce upregulation of FoxO3a expression. FoxO3a induces autophagy by regulating the expression of ATGs. If autophagy inhibition persists, FoxO3a activates the BBC3/PUMA pathway to induce apoptosis.
When osteoblasts are injured by oxidative stress, the increase in intracellular ROS induces the activation of FoxO3. Activated FoxO3 induces the expression of a variety of ATGs, thereby promoting osteoblast autophagy to remove organelles damaged by oxidative stress and reducing the osteoblast apoptosis rate by recycling energy-related components and correcting misaligned structures. FoxO3 knockout inhibits the self-purification of cells. However, when oxidative stress damage induced by excessive ROS levels exceeds the phagocytosis induced by Foxo activation, apoptosis is unabated. Oxidative stress injury induced by wear particles has a significant influence in the pathogenesis of PPOL. Therefore, treatment targeting FoxO3 to activate autophagy may increase treatment effectiveness and improve the prognosis of PPOL.
Beclin1 can form complexes with different proteins, which have different regulatory effects on autophagy. Some studies have shown that Beclin1 forms complexes with class III PI3Ks (hVps34, hVps15) (Hurley and Young, 2017). The Beclin1-hVps34-hVps15 core complex regulates the process of autophagy by forming distinct complexes with Atg14L, UVRAG and Rubicon. The Beclin1–hVps34–hVps15-Atg14L complex facilitates autophagosome nucleation, and the Beclin1–hVps34–hVps15-UVRAG complex facilitates autophagosome maturation. In addition, the Beclin1–hVps34–hVps15-Rubicon complex inhibits autophagy (Xu and Qin, 2019). The binding of Beclin1 and Bcl-2 also regulates autophagy. The combination of Bcl-2/Bcl-xl and Beclin1 inhibits autophagy. Bcl-2/Bcl-xl can bind to Beclin1 in the BH3 domain under physiological conditions. The dynamic binding of the bcl2-Beclin1 complex can regulate autophagy within a normal level (Kang et al., 2011). The deletion of Beclin1 reduces autophagic flux until it reaches an abnormally low level and leads to cell death finally. In contrast, the loss of Bcl-2 induces overactive autophagy, which may also lead to cell death. c-Jun N-terminal protein kinase 1 (JNK1) phosphorylates Bcl-2, causing the dissociation of the Bcl 2–Beclin1 complex and inducing autophagy (Wei et al., 2008). Overexpression of death-inducing kinase (DAPK) phosphorylates the BH3 domain of Beclin1 and promotes the dissociation of Beclin1 from Bcl-xl, thereby inducing autophagy (Levin-Salomon et al., 2014). ROCK1 can also phosphorylate T119 in Beclin1, resulting in the dissociation of Beclin1 from Bcl-2. The mice showed autophagy inhibition after ROCK1 knockout (Gurkar et al., 2013). Mst1 induces phosphorylation at Thr108 of Beclin1 under stress conditions. On the contrary, Phosphorylation at Thr108 promotes the binding of Beclin1 to Bcl-2/Bcl-xl and stabilizes the Beclin1/Bcl-2 homodimer. The stabilized dimer inhibits the PI3K kinase activity of the Atg14 L–Beclin1–Vps34 complex and ultimately inhibits autophagy (Maejima et al., 2013). Figure 5 shows the composition of the Beclin1 complex and the regulatory mechanism it mediates in conjunction with Bcl-2.
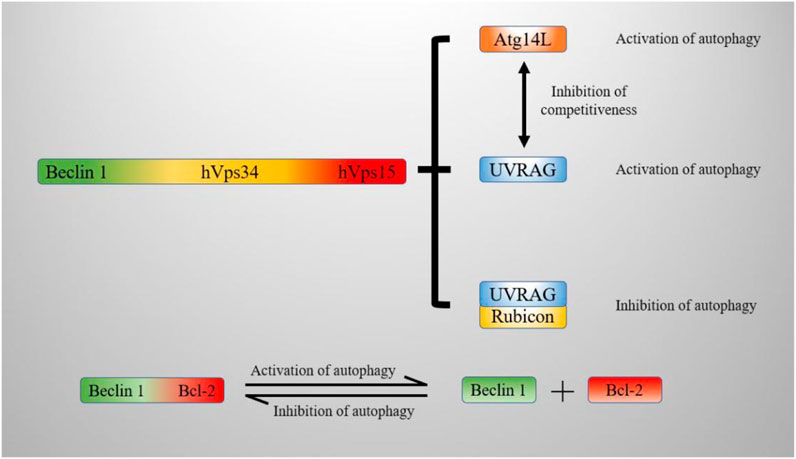
FIGURE 5. Mechanism by which Beclin1 regulates autophagy. The Beclin1-hVps34-hVps15 core complex promotes autophagy by binding to Atg14L and UVRAG. The binding of the complex to Rubicon decreases autophagic flux. The combination of Beclin1 and Bcl-2 inhibits autophagy; the dissociation of Beclin1 from Bcl-2 promotes autophagy. hVps34: Vacuolar sorting protein VPS34, hVps15: Vacuolar sorting protein VPS15, Atg14 L: Autophagy-related protein 14 L, UVRAG: UV radiation resistance-associated gene protein, Rubicon: RUBCN protein, Bcl-2: B-cell leukemia/lymphoma-2 protein.
Beclin1 has been identified as an autophagic marker gene because of its key role in autophagy, as well as in autophagy of osteoblasts. PTH1-34 enhances the autophagic activity of osteoblasts by regulating the expression of Beclin1, thereby increasing the proliferation and mineralization of osteoblasts and reducing bone loss (Wu et al., 2021). Many drugs currently used to regulate autophagy in osteoblasts lead to changes in Beclin1 expression. Studies have shown that the expression of Beclin1 in the osteoblasts of osteoporotic rats treated with high-dose resveratrol was increased. Resveratrol treatment also increased the expression of Beclin1 mRNA in dexamethasone (DEX)-treated osteoblasts (Yang et al., 2019).
In summary, the dynamic balance mediated by Beclin1 and Bcl-2 binding is important in maintaining autophagy homeostasis under physiological conditions. The accelerated autophagy induced by wear particles may be ameliorated by regulating Beclin1 and Bcl-2 phosphorylation levels. Beclin1 is also a key component in autophagy maturation and nucleation processes. Therefore, regulating autophagy in osteoblasts by interfering with Beclin1 expression may be important for the treatment of osteolysis near a prosthesis.
The sirtuin gene family is a highly conserved subfamily of genes that are also class III histone deacetylases (Brachmann et al., 1995). Sirt1 is mainly expressed in the nucleus of most cell types. Sirt1 is involved in many cellular processes, such as cell senescence, endocrine signal transduction, balanced glucose levels, aging and longevity (Tang, 2016). The regulation of macroautophagy by Sirt1 has been extensively studied. Although Sirt1 is not an important part of the autophagy mechanism, in some cases, the activation of Sirt1 is an important step in activating autophagy (Wang et al., 2021). Sirt1 is able to deacetylate key components of the autophagy activating network (such as the products of the ATGs Atg5, Atg7 and Atg8). Sirt1 also induces key autophagy genes by activating members of the FoxO transcription factor family (Ng and Tang, 2013). Hariharan et al. have shown that Sirt1 deacetylation of FoxO1 is needed for hunger-induced cardiac autophagy and that FoxO1 enhances autophagy flux by activating Rab7 (Hariharan et al., 2010). The upregulation of Sirt1 alleviates MN-induced microglial autophagy dysfunction by regulating FoxO3 deacetylation. The activation of FoxO3 to LC3B promoter depends on the deacetylation of FoxO3 by Sirt1 (Yan et al., 2023). Therefore, Sirt1 can enhance autophagy by mediating the deacetylation of the FoxO family. The bone mass of Sirt1-knockout mice was reported to be decreased, and the proportion of osteoblasts and osteoclasts was unbalanced. Therefore, Sirt1 has been confirmed to be a direct osteoblast-regulating factor in many studies. The bone formation ability of Sirt1-knockout mice was weakened, which proved a role for Sirt1 in new bone formation. In addition to bone loss caused by Sirt1 deficiency, the tibias of anorexic mice showed increased levels of bone marrow fat (Louvet et al., 2020). The upregulation of Sirt1 induced by seventeen β-estradiol (17β-E2) promoted autophagy through the AMPK/mTOR pathway and inhibited apoptosis by activating FoxO3a in osteoblasts (Aquilano et al., 2013).
As an important regulatory factors of cellular defenses against oxidative stress and cell survival, Sirt1 maintains cell activity by activating autophagy. Sirt1 interferes with autophagy mainly by regulating key pathways in the autophagy network, which is closely related to its role in clearing senile cells, recycling energy-related molecules and inhibiting apoptosis. In addition, Sirt1 may exert an important effect on mitochondrial function. Sirt1 is activated mainly in cells with damaged mitochondria and participates in the regulation of mitophagy. Sirt1 plays a protective role by clearing damaged mitochondria and recycling energy-related molecules. Mitochondrial autophagy is a process by which cells selectively remove damaged mitochondria. Selective removal of damaged mitochondria prevents excessive accumulation of ROS in cells, which is essential for the functional integrity and cell survival of the entire mitochondrial network. Some studies have shown that Sirt1 can mediate PINK1/Parkin-dependent mitochondrial autophagy. Quercetin can enhance mitochondrial autophagy by activating Sirt1-PINK1/Parkin pathways in tubular epithelial cells in vivo and in vitro, thereby effectively alleviating cellular senescence in tubular epithelial cells and renal interstitial fibrosis (Liu et al., 2020).
Therefore, Sirt1 may play more roles than currently recognized, and therefore, continued research on Sirt1 regulation of autophagy in osteoblasts is both a reasonable and necessary endeavor. Similar to FoxO3, Sirt1 is a key factor in cellular defenses against oxidative stress damage, and the activation of Sirt1 enhances FoxO3 clearance of ROS. The synergistic regulatory effects of Sirt1 and FoxO3 on autophagy and oxidative stress in osteolysis near prostheses need to be further studied. The regulatory effect of Sirt1 on autophagy is briefly shown in Figure 6.
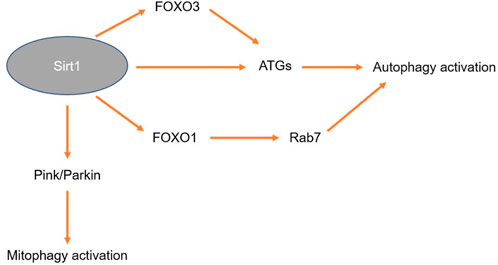
FIGURE 6. Mechanism by which Sirt1 regulates autophagy. Activati+on of Sirt1 upregulates the expression of FoxO3, thereby inducing the expression of ATGs. Sirt1 itself can also induce the expression of ATGs. Sirt1 can activate the expression of FoxO1, upregulate the expression of Rab7 and induce autophagy. Sirt1 can also activate the PINK1/Parkin signaling pathway to induce mitochondrial autophagy.
P62 (sqstm1) is a ubiquitin-binding protein, which is mainly involved in physiological processes such as signal transduction, oxidative stress and autophagy (Komatsu et al., 2007). P62 is a multifunctional protein composed of a variety of different interacting domains (Lin et al., 2013). Through its C-terminal UBA domain or LIR domain, P62 transports ubiquitin to lysosomes for degradation (Myeku and Figueiredo-Pereira, 2011). During autophagy, the degradation of autophagosome cargo in lysosomes leads to a decrease in sqstm1 levels. In contrast, autophagy inhibitor proteins stabilize sqstm1 levels (Katsuragi et al., 2015). P62 functions as an adapter by binding ubiquitinated protein aggregates and trafficking them to lysosomes. The transcription of P62 is mediated by various signaling pathways, such as the oxidative stress response-related (Nrf2), Ras/MAPK, and JNK/c-Jun pathways (Puissant et al., 2012). It is reported that autophagy in osteoblast can be mediated by regulating P62 function. Cannabinoid receptor type 2 (CNR2) induces osteogenic differentiation by activating P62-mediated autophagy (Xu et al., 2020). In rats with type 2 diabetes (T2DM), exercise downregulated long-chain noncoding rnameg3 (meg3) and activated P62-mediated autophagy to promote bone formation (Chen X. et al., 2021). P62 participates in the regulation of the ubiquitin–proteasome system (UPS) through the action of the KEAP1-NrF2 pathway (Liu W. J. et al., 2016). P62 is the integration center of various cellular functions, including the formation of autophagosomes and the transfer of ubiquitin proteins to lysosomes. P62 is a multidomain protein with multiple domain. The role of some interaction domains in P62 is still poorly understood, thus P62 deserves further study.
Protein phosphatase 2A (PP2A) is a serine/threonine phosphatase that regulates phosphorylation in the body (Dzulko et al., 2020). As a trimer, PP2A is composed of catalytic subunit C, structural subunit A and regulatory subunit B. Its function depends mainly on the regulation of catalytic subunit C (Mazhar et al., 2020). On this basis, the diversity of PP2A holoenzyme compositions leads to a variety of possibilities for its regulation in cells. The literature confirms that PP2A has significant influence in the incidence of tumors, respiratory diseases and nervous system diseases. The regulation of PP2Ac methylation levels attenuates MN-induced abnormal autophagy in MN2a cells through the action of the mTORC1/ULK1 signaling pathway (Xu et al., 2021).
ULK1 is a serine/threonine kinase upstream of the autophagy core mechanism, which is a direct substrate for mTORC1. The reversible phosphorylation of ULK1 is a central signaling mechanism that regulates famine-induced autophagy. When the amino acid level is reduced, the activity of mTORC1 is inhibited, and ULK1 is activated simultaneously, thus upregulating the autophagy level (Jung et al., 2009). Wong et al. found that starvation induced autophagy faster and more strongly than mTORC1 inhibitors. This is because starvation can not only block mTORC1 activity but also activate PP2Ac to dephosphorylate ULK1 (Xu et al., 2021). Under physiological conditions, PP2Ac binds to Alpha4 as a complex, which has no regulatory effect on ULK1. Starvation can induce the release of PP2Ac from an inactive complex containing Alpha4 to synthesize an active regulatory complex with PRL65 and B55α, thereby mediating the dephosphorylation of ULK1 (Wong et al., 2015). The regulatory effect of PP2Ac on autophagy is briefly shown in Figure 7

FIGURE 7. Mechanism by which PP2ac regulates autophagy. Starvation induces dissociation of PP2Ac from the Alpha4 complex. PRL65 and B55α are synthesized as active regulatory complexes that mediate the dephosphorylation of ULK1 and upregulation of autophagy levels. Starvation also upregulates autophagy levels by inhibiting the mTORC1 pathway and reducing ULK1 phosphorylation.
The expression of PP2Ac was significantly increased in macrophages infected with Mycobacterium bovis. Inhibition of PP2Ac by nilotinib conferred protection through autophagy onto macrophages by activating the AMPK signaling pathway, thus increasing the antimicrobial function and survivability of the cells (Hussain et al., 2019). Taurine blocked mitophagy by inhibiting PP2Ac methylation, thereby reducing excessive polarization of macrophages into the proinflammatory type. VDAC1 and PINK1 are mediators of PP2Ac methylation-induced mitophagy and are needed to maintain PP2Ac hypermethylation (Meng et al., 2021). In decades, many scholars have proved that PP2A is related to orthopedic diseases. Okamura H et al. found that a reduction in PP2A levels promoted osteoblast differentiation by regulating Osterix and other bone-related transcription factors (Okamura et al., 2017). PP2ac can coordinate with the mTOR/ULK1 pathway to induce rapid autophagy activation during cell starvation. In addition, PP2ac is also closely related to a variety of autophagy signaling pathways, such as PINK1/Parkin and AMPK. It also plays an important role in bone metabolic diseases. PP2ac deserves further study as a potential regulator of osteoblast function.
The object of autophagy can be a large number of non-selective proteins or a single cell component that is highly regulated, depending on the inducible factor. As a selective autophagy, mitophagy has been extensively studied. Mitophagy regulates the quality and quantity of mitochondria by selectively degrading mitochondria. Specific mitochondrial outer membrane receptors or ubiquitin molecules coupled to mitochondrial surface proteins mediate the formation of the autophagosome around the mitochondria (Onishi et al., 2021). Mitochondrial autophagy mediates variety important processes such as cell growth and development, apoptosis and inflammation. Mitophagy also plays a protective role against cell proliferation and differentiation in its role as an “intracellular steward” (Lou et al., 2020).
Parkin is composed of 465 amino acids and is encoded by the second largest human gene after antinutrophin. In 1998, Parkin was identified as the pathogenic gene underlying autosomal recessive juvenile Parkinson’s disease (AR-JP) (Pickrell and Youle, 2015). PINK1 is another pathogenic gene associated with early-onset Parkinson’s disease, including AR-JP (Singleton et al., 2013). PINK1 and Parkin exhibit different molecular functions: one is a protein kinase, the other is a ubiquitin ligase, but they have the same pathogenic effect. PINK1/Parkin, as a classical mitochondrial self-regulation pathway, has been widely studied by scholars (Nardin et al., 2016). PINK1, as a surveillance molecule, constantly monitors the health of mitochondria under physiological conditions and responds quickly when mitochondria are damaged. In healthy mitochondria, PINK1 is rapidly degraded by the UPS during its passage through mitochondrial membranes. In contrast, damaged mitochondria cause PINK1 accumulation due to the loss of membrane potential. This leads to rapid recruitment and activation of Parkin and subsequent initiation of mitophagy (Nguyen et al., 2016). Under physiological conditions, Parkin is maintained in a “closed” state through multiple intramolecular interactions, making its inhibitory state the basal condition. PINK1 activates Parkin by directly phosphorylating Ser65 in the Parkin Ubl domain (Bingol and Sheng, 2016). Mutations in both Parkin and PINK led to significant inhibition of mitochondrial protein renewal and mitophagy. The accumulation of damaged mitochondria is the main cause of Parkinson’s disease (Wang et al., 2016). In decades, the PINK1/Parkin pathway has been proved to have great influence in osteoblast function. Parkin activates the β-catenin signaling pathway to enhance autophagy and promote the differentiation of BMSCs into osteoblasts. In contrast, self-regulation of Parkin expression leads to accelerated osteoblastic differentiation, causing steatosis of MSCs and reduced expression of type I collagen (Zhang W. et al., 2020). VK2 reversed the inhibition of dexamethasone-induced autophagy by upregulating the expression of Parkin and other autophagy and mitochondrial markers, thereby increasing osteoblast function (Chen et al., 2020). These findings suggest that in-depth study of the PINK1/Parkin activation mechanism may lead to a better understanding and new ideas for autophagy-related cytoprotection.
To date, the pathological mechanism underlying PPOL has not been fully clarified. The function and metabolic changes in many cells of bone tissue are involved. Osteoblasts, derived from BMSCs, are important sources of bone cells that represent the final differentiation state of osteoblast cell lines. Therefore, changes in the function and activity of osteoblasts have an indispensable influence on the occurrence and development of PPOL. Autophagy, as a self-protective and self-purification process, plays a substantial role in maintaining the function and activity of osteoblasts. A large amount of credible evidence suggests that autophagy is involved in the development and differentiation of osteoblasts and the maintenance of bone homeostasis. Activation of autophagy in response to environmental stress or foreign stimuli enhances osteoblast activity and inhibits osteoblast apoptosis. Activation of autophagy is indispensable in the early stages of BMSC differentiation into osteoblasts. Moreover, the activation of autophagy alleviates the oxidative stress damage induced by Ti particles and increases the antioxidative capacity of osteoblasts. Since autophagosomes are vehicles for secreting apatite crystals into the extracellular space, autophagy is identified as necessary for the initiation of osteoblast mineralization. Impaired autophagy causes osteoblast failure to clear damaged organelles or remain viable in a nutrient-deficient state, which leads to osteoblast apoptosis and disrupts the bone-remodeling balance. In contrast, the overactivation of autophagy leads to irreversible osteoblast self-degradation, which causes dysregulated bone homeostasis. Therefore, autophagy regulatory mechanisms have been proven to increase the activity and function of osteoblasts and thus play important roles in metabolic diseases of bone. In this review, we summarized and discussed several signaling pathways that have been shown to regulate autophagy in osteoblasts under physiological conditions and during pathological changes in bone metabolism. AMPK, mTOR and ULK1 regulate each other to maintain autophagy stability and prevent excessive autophagy activation. Crosstalk between the NF-κB signaling pathway and autophagy pathway components is complex. Activation of NF-κB signaling may exert opposite effects on autophagy in different cells or diseased tissue. NF-κB mainly plays a role in affecting downstream autophagy genes, such as Binp3, Beclin1, and mTOR. FoxO3 mainly mediates autophagy initiated by nutrient deficiency. FoxO3 is activated during cell starvation to mediate autophagy. However, continuous inhibition of autophagy can induce FOXO3 to initiate apoptosis. Beclin1 plays a significant role in the generation and regulation of autophagy, as it is a key component of autophagosome formation. Beclin1 induces autophagosome nucleation and maturation by forming complexes with different molecules. Moreover, its dynamic binding with Bcl-2 is crucial for the stability of autophagic flux. Sirt1 affects autophagy mainly through its regulation of key components in the autophagy induction network; these components include Atg5, Atg7 and Atg8. Sirt1 also induces the expression of autophagy pathway components by activating FoxO family members. However, the autophagic gene network is constructed by multiple signaling pathways, and multiple signaling pathway components engage in crosstalk. Unfortunately, research on autophagy mechanisms to date has been insufficient, and more in-depth research is still needed. The autophagy signaling pathway in osteoblasts reviewed in this paper is only part of the autophagic gene network, and the activation and action modes of each signaling pathway may not be limited to those discussed herein. Future research should focus on the crosstalk among different autophagic signaling pathway components so that we can better understand the complex autophagic gene network.
Most studies have focused on the effect of autophagy inhibitors on osteolysis. 3-MA and LY294002 have been shown to inhibit osteoclast generation and reduce the expression of pro-inflammatory factors in vitro, thus playing a certain therapeutic role in PPOL (Chen W. et al., 2021). 3-MA can also reduce CoCrMo granulose-induced osteoblast apoptosis by inhibiting autophagy (Wang et al., 2015). However, due to the complexity of the autophagy network, the role of autophagy in PPOL has been controversial and no clear conclusions have been drawn so far. Inhibition or activation of autophagy alone may be difficult to achieve a therapeutic effect because autophagy does not work consistently in different cells. And autophagy also plays different roles at different times in the same cell. For example, autophagy is an essential condition in the mineralization process of osteoblasts, but excessive autophagy can mediate apoptosis of osteoblasts. Therefore, enhancing the precision of autophagy control or maintaining appropriate autophagy strength may be helpful for future PPOL treatment. Due to the single mechanism and effect of most drugs, it is difficult to accurately regulate the intensity of autophagy. And the long-term safety of some drugs remains to be determined. Further research on the regulation of key genes in autophagy may be helpful. How to regulate key genes in the autophagy process of specific cells, instead of interfering with the overall level of autophagy, is our next research direction.
PPOL, a common and difficult-to-treat complication of joint replacement surgery, affects the majority of patients. Improving the function and activity of osteoblasts has great significance in improving the prognosis of PPOL. Autophagy regulation has been proven to reduce the osteoblast apoptosis rate and increase osteoblast differentiation and mineralization. In summary, we hope to provide new research directions and therapeutic targets for the treatment of PPOL and improve prognosis by summarizing the latest information on autophagy signaling pathways. However, the autophagy gene network is composed of multiple signaling pathways, and crosstalk among these signaling pathways is complex. To date, our understanding of the mechanism of autophagy is still insufficient. The autophagy signaling pathways in osteoblasts reviewed in this paper constitute only a portion of the autophagy gene network, and the activation and action modes of each signaling pathway may involve more mechanisms than are described herein. In future studies, we will focus on analyzing the crosstalk between various autophagy signaling pathway components. Through drug intervention or gene-targeted regulation, the autophagic flux in osteoblasts in the wear particle-influenced microenvironment has been shown to be regulated within a stable range. We should not only pay full attention to the positive effects of autophagy on osteoblasts but also prevent the damage caused by the excessive activation of autophagy, which is the focus of our subsequent research.
YG: Writing–original draft. ZW: Writing–review and editing. HX: Writing–review and editing. TF: Writing–review and editing. QW: Writing–review and editing. YG: Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work is supported by grants from Key R&D Project of Science and Technology Department of Jiangsu Province (No. BE2021673), Project Leader: YG; Science and Technology Development Project of Suzhou Science and Technology Bureau (Nos SYSD202202 and SYSD2020013), Project Leader: YG; Clinical key disease diagnosis and treatment project of Suzhou Health Commission (No. LCZX201824), Project Leader: YG; Science and Technology Planning Project of Changshu Municipal Health Commission (No. csws201907), Project Leader: YG; Science and Technology Development Project of Changshu Science and Technology Bureau (Nos CS202119, CS201817, and CS202217), Project Leader: YG, QW, and FT; Horizontal research Project of Soochow University (No. H200833), Project Leader: YG; Science and technology development plan project of Suzhou Science and Technology Bureau (No. SKY2022019), Project Leader: YG.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alers, S., Löffler, A. S., Wesselborg, S., and Stork, B. (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol. Cell Biol. 32 (1), 2–11. doi:10.1128/MCB.06159-11
Aquilano, K., Baldelli, S., Pagliei, B., and Ciriolo, M. R. (2013). Extranuclear localization of SIRT1 and PGC-1α: an insight into possible roles in diseases associated with mitochondrial dysfunction. Curr. Mol. Med. 13 (1), 140–154. doi:10.2174/156652413804486241
Bartolomé, A., López-Herradón, A., Portal-Núñez, S., García-Aguilar, A., Esbrit, P., Benito, M., et al. (2013). Autophagy impairment aggravates the inhibitory effects of high glucose on osteoblast viability and function. Biochem. J. 455 (3), 329–337. doi:10.1042/BJ20130562
Bhardwaj, G., Penniman, C. M., Jena, J., Suarez Beltran, P. A., Foster, C., Poro, K., et al. (2021). Insulin and IGF-1 receptors regulate complex I-dependent mitochondrial bioenergetics and supercomplexes via FoxOs in muscle. J. Clin. Invest 131 (18), e146415. doi:10.1172/JCI146415
Bingol, B., and Sheng, M. (2016). Mechanisms of mitophagy: PINK1, parkin, USP30 and beyond. Free Radic. Biol. Med. 100, 210–222. doi:10.1016/j.freeradbiomed.2016.04.015
Blom, A. W., Hunt, L. P., Matharu, G. S., Reed, M., and Whitehouse, M. R. (2021). The effect of surgical approach in total knee replacement on outcomes. An analysis of 875,166 elective operations from the National Joint Registry for England, Wales, Northern Ireland and the Isle of Man. Knee 31, 144–157. doi:10.1016/j.knee.2021.04.009
Brachmann, C. B., Sherman, J. M., Devine, S. E., Cameron, E. E., Pillus, L., and Boeke, J. D. (1995). The SIR2 gene family, conserved from bacteria to humans, functions in silencing, cell cycle progression, and chromosome stability. Genes Dev. 9 (23), 2888–2902. doi:10.1101/gad.9.23.2888
Carling, D. (2017). AMPK signalling in health and disease. Curr. Opin. Cell Biol. 45, 31–37. doi:10.1016/j.ceb.2017.01.005
Chen, L., Shi, X., Weng, S. J., Xie, J., Tang, J. H., Yan, D-Y., et al. (2020). Vitamin K2 can rescue the dexamethasone-induced downregulation of osteoblast autophagy and mitophagy thereby restoring osteoblast function in vitro and in vivo. Front. Pharmacol. 11, 1209. doi:10.3389/fphar.2020.01209
Chen, W., Xian, G., Gu, M., Pan, B., Wu, X., Ye, Y., et al. (2021). Autophagy inhibitors 3-MA and LY294002 repress osteoclastogenesis and titanium particle-stimulated osteolysis. Biomater. Sci. 9 (14), 4922–4935. doi:10.1039/d1bm00691f
Chen, X., Yang, K., Jin, X., Meng, Z., Liu, B., Yu, H., et al. (2021). Bone autophagy: a potential way of exercise-mediated Meg3/P62/runx2 pathway to regulate bone formation in T2DM mice. Diabetes Metab. Syndr. Obes. 14, 2753–2764. doi:10.2147/DMSO.S299744
Cheng, Y., Huang, L., Wang, Y., Huo, Q., Shao, Y., Bao, H., et al. (2019). Strontium promotes osteogenic differentiation by activating autophagy via the the AMPK/mTOR signaling pathway in MC3T3‑E1 cells. Int. J. Mol. Med. 44 (2), 652–660. doi:10.3892/ijmm.2019.4216
Dagda, R. K., Zhu, J., Kulich, S. M., and Chu, C. T. (2008). Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson's disease. Autophagy 4 (6), 770–782. doi:10.4161/auto.6458
Deng, Z., Zhang, R., Li, M., Wang, S., Fu, G., Jin, J., et al. (2021). STAT3/IL-6 dependent induction of inflammatory response in osteoblast and osteoclast formation in nanoscale wear particle-induced aseptic prosthesis loosening. Biomater. Sci. 9 (4), 1291–1300. doi:10.1039/d0bm01256d
di Giacomo, V., Cataldi, A., and Sancilio, S. (2020). Biological factors, metals, and biomaterials regulating osteogenesis through autophagy. Int. J. Mol. Sci. 21 (8), 2789. doi:10.3390/ijms21082789
Dikic, I., and Elazar, Z. (2018). Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 19 (6), 349–364. doi:10.1038/s41580-018-0003-4
Ding, Y. W., Zhao, G. J., Li, X. L., Hong, G. L., Li, M. F., Qiu, Q. M., et al. (2016). SIRT1 exerts protective effects against paraquat-induced injury in mouse type II alveolar epithelial cells by deacetylating NRF2 in vitro. Int. J. Mol. Med. 37 (4), 1049–1058. doi:10.3892/ijmm.2016.2503
Djavaheri-Mergny, M., Amelotti, M., Mathieu, J., Besançon, F., Bauvy, C., and Codogno, P. (2007). Regulation of autophagy by NFkappaB transcription factor and reactives oxygen species. Autophagy 3 (4), 390–392. doi:10.4161/auto.4248
Dzulko, M., Pons, M., Henke, A., Schneider, G., and Krämer, O. H. (2020). The PP2A subunit PR130 is a key regulator of cell development and oncogenic transformation. Biochim. Biophys. Acta Rev. Cancer 1874 (2), 188453. doi:10.1016/j.bbcan.2020.188453
Fitzwalter, B. E., and Thorburn, A. (2018). FOXO3 links autophagy to apoptosis. Autophagy 14 (8), 1467–1468. doi:10.1080/15548627.2018.1475819
Fitzwalter, B. E., Towers, C. G., Sullivan, K. D., Andrysik, Z., Hoh, M., Ludwig, M., et al. (2018). Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev. Cell 44 (5), 555–565. doi:10.1016/j.devcel.2018.02.014
Friis, R. M. N., Glaves, J. P., Huan, T., Li, L., Sykes, B. D., and Schultz, M. C. (2014). Rewiring AMPK and mitochondrial retrograde signaling for metabolic control of aging and histone acetylation in respiratory-defective cells. Cell Rep. 7 (2), 565–574. doi:10.1016/j.celrep.2014.03.029
Galluzzi, L., Baehrecke, E. H., Ballabio, A., Boya, P., Bravo-San Pedro, J. M., Cecconi, F., et al. (2017). Molecular definitions of autophagy and related processes. EMBO J. 36 (13), 1811–1836. doi:10.15252/embj.201796697
Gómez-Puerto, M. C., Verhagen, L. P., Braat, A. K., Lam, E. W. F., Coffer, P. J., and Lorenowicz, M. J. (2016). Activation of autophagy by FOXO3 regulates redox homeostasis during osteogenic differentiation. Autophagy 12 (10), 1804–1816. doi:10.1080/15548627.2016.1203484
Gurkar, A. U., Chu, K., Raj, L., Bouley, R., Lee, S. H., Kim, Y. B., et al. (2013). Identification of ROCK1 kinase as a critical regulator of Beclin1-mediated autophagy during metabolic stress. Nat. Commun. 4, 2189. doi:10.1038/ncomms3189
Hameister, R., Kaur, C., Dheen, S. T., Lohmann, C. H., and Singh, G. (2020). Reactive oxygen/nitrogen species (ROS/RNS) and oxidative stress in arthroplasty. J. Biomed. Mater Res. B Appl. Biomater. 108 (5), 2073–2087. doi:10.1002/jbm.b.34546
Hariharan, N., Maejima, Y., Nakae, J., Paik, J., Depinho, R. A., and Sadoshima, J. (2010). Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ. Res. 107 (12), 1470–1482. doi:10.1161/CIRCRESAHA.110.227371
Hasegawa, T., Hongo, H., Yamamoto, T., Abe, M., Yoshino, H., Haraguchi-Kitakamae, M., et al. (2022). Matrix vesicle-mediated mineralization and osteocytic regulation of bone mineralization. Int. J. Mol. Sci. 23 (17), 9941. doi:10.3390/ijms23179941
Hayden, M. S., and Ghosh, S. (2008). Shared principles in NF-kappaB signaling. Cell 132 (3), 344–362. doi:10.1016/j.cell.2008.01.020
Hoesel, B., and Schmid, J. A. (2013). The complexity of NF-κB signaling in inflammation and cancer. Mol. Cancer 12, 86. doi:10.1186/1476-4598-12-86
Hurley, J. H., and Young, L. N. (2017). Mechanisms of autophagy initiation. Annu. Rev. Biochem. 86, 225–244. doi:10.1146/annurev-biochem-061516-044820
Hussain, T., Zhao, D., Shah, S. Z. A., Sabir, N., Wang, J., Liao, Y., et al. (2019). PP2Ac modulates AMPK-mediated induction of autophagy in Mycobacterium bovis-infected macrophages. Int. J. Mol. Sci. 20 (23), 6030. doi:10.3390/ijms20236030
Jang, S. Y., Kang, H. T., and Hwang, E. S. (2012). Nicotinamide-induced mitophagy: event mediated by high NAD+/NADH ratio and SIRT1 protein activation. J. Biol. Chem. 287 (23), 19304–19314. doi:10.1074/jbc.M112.363747
Jia, J., Yao, W., Guan, M., Dai, W., Shahnazari, M., Kar, R., et al. (2011). Glucocorticoid dose determines osteocyte cell fate. FASEB J. 25 (10), 3366–3376. doi:10.1096/fj.11-182519
Jo, C., Gundemir, S., Pritchard, S., Jin, Y. N., Rahman, I., and Johnson, G. V. (2014). Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat. Commun. 5, 3496. doi:10.1038/ncomms4496
Jung, C. H., Jun, C. B., Ro, S. H., Kim, Y. M., Otto, N. M., Cao, J., et al. (2009). ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 20 (7), 1992–2003. doi:10.1091/mbc.e08-12-1249
Kang, C., Wei, L., Song, B., Chen, L., Liu, J., Deng, B., et al. (2017). Involvement of autophagy in tantalum nanoparticle-induced osteoblast proliferation. Int. J. Nanomedicine 12, 4323–4333. doi:10.2147/IJN.S136281
Kang, R., Zeh, H. J., Lotze, M. T., and Tang, D. (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18 (4), 571–580. doi:10.1038/cdd.2010.191
Katsuragi, Y., Ichimura, Y., and Komatsu, M. (2015). p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282 (24), 4672–4678. doi:10.1111/febs.13540
Kim, I. R., Kim, S. E., Baek, H. S., Kim, B. J., Kim, C. H., Chung, I. K., et al. (2016). The role of kaempferol-induced autophagy on differentiation and mineralization of osteoblastic MC3T3-E1 cells. BMC Complement. Altern. Med. 16 (1), 333. doi:10.1186/s12906-016-1320-9
Kim, J., Kundu, M., Viollet, B., and Guan, K. L. (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13 (2), 132–141. doi:10.1038/ncb2152
Kim, J. H., Kang, H. M., Yu, S. B., Song, J. M., Kim, C. H., Kim, B. J., et al. (2018). Cytoprotective effect of flavonoid-induced autophagy on bisphosphonate mediated cell death in osteoblast. J. Cell Biochem. 119 (7), 5571–5580. doi:10.1002/jcb.26728
Kim, J. M., Lin, C., Stavre, Z., Greenblatt, M. B., and Shim, J. H. (2020). Osteoblast-osteoclast communication and bone homeostasis. Cells 9 (9), 2073. doi:10.3390/cells9092073
Kim, K. H., and Lee, M. S. (2014). Autophagy--a key player in cellular and body metabolism. Nat. Rev. Endocrinol. 10 (6), 322–337. doi:10.1038/nrendo.2014.35
Klionsky, D. J., Petroni, G., Amaravadi, R. K., Baehrecke, E. H., Ballabio, A., Boya, P., et al. (2021). Autophagy in major human diseases. EMBO J. 40 (19), e108863. doi:10.15252/embj.2021108863
Komatsu, M., Waguri, S., Koike, M., Sou, Y. S., Ueno, T., Hara, T., et al. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131 (6), 1149–1163. doi:10.1016/j.cell.2007.10.035
Lacarrière-Keïta, C., Nassari, S., and Jean, S. (2022). Autophagy in cell fate decisions: knowledge gained from Drosophila. Genome 65 (12), 573–584. doi:10.1139/gen-2022-0069
Lawrence, T. (2009). The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 1 (6), a001651. doi:10.1101/cshperspect.a001651
Lee, D. H., Park, J. S., Lee, Y. S., Han, J., Kwon, S. W., Han, D. H., et al. (2020). SQSTM1/p62 activates NFE2L2/NRF2 via ULK1-mediated autophagic KEAP1 degradation and protects mouse liver from lipotoxicity. Autophagy 16 (11), 1949–1973. doi:10.1080/15548627.2020.1712108
Lee, J. W., Nam, H., Kim, L. E., Jeon, Y., Min, H., Ha, S., et al. (2019). TLR4 (toll-like receptor 4) activation suppresses autophagy through inhibition of FOXO3 and impairs phagocytic capacity of microglia. Autophagy 15 (5), 753–770. doi:10.1080/15548627.2018.1556946
Levin-Salomon, V., Bialik, S., and Kimchi, A. (2014). DAP-kinase and autophagy. Apoptosis 19 (2), 346–356. doi:10.1007/s10495-013-0918-3
Li, H., Li, D., Ma, Z., Qian, Z., Kang, X., Jin, X., et al. (2018). Defective autophagy in osteoblasts induces endoplasmic reticulum stress and causes remarkable bone loss. Autophagy 14 (10), 1726–1741. doi:10.1080/15548627.2018.1483807
Li, Y., Su, J., Sun, W., Cai, L., and Deng, Z. (2018). AMP-activated protein kinase stimulates osteoblast differentiation and mineralization through autophagy induction. Int. J. Mol. Med. 41 (5), 2535–2544. doi:10.3892/ijmm.2018.3498
Li, Y., and Chen, Y. (2019). AMPK and autophagy. Adv. Exp. Med. Biol. 1206, 85–108. doi:10.1007/978-981-15-0602-4_4
Lin, K., Chen, J. H., Fang, Z. H., Ye, C. L., Han, C. J., Yan, M., et al. (2019). The effect of procyanidin on periprosthetic osteolysis caused by TCP wear particles in the mouse calvaria and its mechanism, 35 (3), 250–255. doi:10.12047/j.cjap.5765.2019.053
Lin, X., Li, S., Zhao, Y., Ma, X., Zhang, K., He, X., et al. (2013). Interaction domains of p62: a bridge between p62 and selective autophagy. DNA Cell Biol. 32 (5), 220–227. doi:10.1089/dna.2012.1915
Link, W. (2019). Introduction to FOXO biology. Methods Mol. Biol. 1890, 1–9. doi:10.1007/978-1-4939-8900-3_1
Liu, N., Meng, J., Wang, Z., Zhou, G., Shi, T., and Zhao, J. (2016). Autophagy mediated TiAl₆V₄ particle-induced peri-implant osteolysis by promoting expression of TNF-α. Biochem. Biophys. Res. Commun. 473 (1), 133–139. doi:10.1016/j.bbrc.2016.03.065
Liu, W. J., Ye, L., Huang, W. F., Guo, L. J., Xu, Z. G., Wu, H. L., et al. (2016). p62 links the autophagy pathway and the ubiqutin-proteasome system upon ubiquitinated protein degradation. Cell Mol. Biol. Lett. 21, 29. doi:10.1186/s11658-016-0031-z
Liu, T., Yang, Q., Zhang, X., Qin, R., Shan, W., Zhang, H., et al. (2020). Quercetin alleviates kidney fibrosis by reducing renal tubular epithelial cell senescence through the SIRT1/PINK1/mitophagy axis. Life Sci. 257, 118116. doi:10.1016/j.lfs.2020.118116
Lou, G., Palikaras, K., Lautrup, S., Scheibye-Knudsen, M., Tavernarakis, N., and Fang, E. F. (2020). Mitophagy and neuroprotection. Trends Mol. Med. 26 (1), 8–20. doi:10.1016/j.molmed.2019.07.002
Louvet, L., Leterme, D., Delplace, S., Miellot, F., Marchandise, P., Gauthier, V., et al. (2020). Sirtuin 1 deficiency decreases bone mass and increases bone marrow adiposity in a mouse model of chronic energy deficiency. Bone 136, 115361. doi:10.1016/j.bone.2020.115361
Lu, D. G., Lu, M. J., Yao, S. H., Lin, J. J., Luo, S., Wei, J. H., et al. (2021). Long non-coding RNA TUG1 promotes the osteogenic differentiation of bone marrow mesenchymal stem cells by regulating the AMPK/mTOR/autophagy pathway. Biomed. Res. 42 (6), 239–246. doi:10.2220/biomedres.42.239
Ma, B., Guan, G., Lv, Q., and Yang, L. (2021). Curcumin ameliorates palmitic acid-induced saos-2 cell apoptosis via inhibiting oxidative stress and autophagy. Evid. Based Complement. Altern. Med. 2021, 5563660. doi:10.1155/2021/5563660
Maejima, Y., Kyoi, S., Zhai, P., Liu, T., Li, H., Ivessa, A., et al. (2013). Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat. Med. 19 (11), 1478–1488. doi:10.1038/nm.3322
Maiuri, M. C., Zalckvar, E., Kimchi, A., and Kroemer, G. (2007). Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8 (9), 741–752. doi:10.1038/nrm2239
Martinez-Outschoorn, U. E., Trimmer, C., Lin, Z., Whitaker-Menezes, D., Chiavarina, B., Zhou, J., et al. (2010). Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle 9 (17), 3515–3533. doi:10.4161/cc.9.17.12928
Mazhar, S., Leonard, D., Sosa, A., Schlatzer, D., Thomas, D., and Narla, G. (2020). Challenges and reinterpretation of antibody-based research on phosphorylation of Tyr307 on PP2Ac. Cell Rep. 30 (9), 3164–3170. doi:10.1016/j.celrep.2020.02.012
Meng, L., Lu, C., Wu, B., Lan, C., Mo, L., Chen, C., et al. (2021). Taurine antagonizes macrophages M1 polarization by mitophagy-glycolysis switch blockage via dragging SAM-PP2Ac transmethylation. Front. Immunol. 12, 648913. doi:10.3389/fimmu.2021.648913
Mizushima, N., Levine, B., Cuervo, A. M., and Klionsky, D. J. (2008). Autophagy fights disease through cellular self-digestion. Nature 451 (7182), 1069–1075. doi:10.1038/nature06639
Morris, B. J., Willcox, D. C., Donlon, T. A., and Willcox, B. J. (2015). FOXO3: a major gene for human longevity--A mini-review. Gerontology 61 (6), 515–525. doi:10.1159/000375235
Myeku, N., and Figueiredo-Pereira, M. E. (2011). Dynamics of the degradation of ubiquitinated proteins by proteasomes and autophagy: association with sequestosome 1/p62. J. Biol. Chem. 286 (25), 22426–22440. doi:10.1074/jbc.M110.149252
Nardin, A., Schrepfer, E., and Ziviani, E. (2016). Counteracting PINK/parkin deficiency in the activation of mitophagy: a potential therapeutic intervention for Parkinson's disease. Curr. Neuropharmacol. 14 (3), 250–259. doi:10.2174/1570159x13666151030104414
Ng, F., and Tang, B. L. (2013). Sirtuins' modulation of autophagy. J. Cell Physiol. 228 (12), 2262–2270. doi:10.1002/jcp.24399
Nguyen, T. N., Padman, B. S., and Lazarou, M. (2016). Deciphering the molecular signals of PINK1/parkin mitophagy. Trends Cell Biol. 26 (10), 733–744. doi:10.1016/j.tcb.2016.05.008
Ning, K., Yang, B., Chen, M., Man, G., Liu, S., Wang, D. E., et al. (2022). Functional heterogeneity of bone marrow mesenchymal stem cell subpopulations in physiology and pathology. Int. J. Mol. Sci. 23 (19), 11928. doi:10.3390/ijms231911928
Nivon, M., Richet, E., Codogno, P., Arrigo, A. P., and Kretz-Remy, C. (2009). Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy 5 (6), 766–783. doi:10.4161/auto.8788
Nollet, M., Santucci-Darmanin, S., Breuil, V., Al-Sahlanee, R., Cros, C., Topi, M., et al. (2014). Autophagy in osteoblasts is involved in mineralization and bone homeostasis. Autophagy 10 (11), 1965–1977. doi:10.4161/auto.36182
Noordin, S., and Masri, B. (2012). Periprosthetic osteolysis: genetics, mechanisms and potential therapeutic interventions. Can. J. Surg. 55 (6), 408–417. doi:10.1503/cjs.003711
Okamura, H., Yoshida, K., Morimoto, H., Teramachi, J., Ochiai, K., Haneji, T., et al. (2017). Role of protein phosphatase 2A in osteoblast differentiation and function. J. Clin. Med. 6 (3), 23. doi:10.3390/jcm6030023
Ollivere, B., Wimhurst, J. A., Clark, I. M., and Donell, S. T. (2012). Current concepts in osteolysis. J. Bone Jt. Surg. Br. 94 (1), 10–15. doi:10.1302/0301-620X.94B1.28047
O'Neill, S. C., Queally, J. M., Devitt, B. M., Doran, P. P., and O'Byrne, J. M. (2013). The role of osteoblasts in peri-prosthetic osteolysis. Bone Jt. J. 95-B (8), 1022–1026. doi:10.1302/0301-620X.95B8.31229
Onishi, M., Yamano, K., Sato, M., Matsuda, N., and Okamoto, K. (2021). Molecular mechanisms and physiological functions of mitophagy. EMBO J. 40 (3), e104705. doi:10.15252/embj.2020104705
Pajares, M., Jiménez-Moreno, N., García-Yagüe, Á. J., Escoll, M., de Ceballos, M. L., Van Leuven, F., et al. (2016). Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12 (10), 1902–1916. doi:10.1080/15548627.2016.1208889
Pantovic, A., Krstic, A., Janjetovic, K., Kocic, J., Harhaji-Trajkovic, L., Bugarski, D., et al. (2013). Coordinated time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy controls osteogenic differentiation of human mesenchymal stem cells. Bone 52 (1), 524–531. doi:10.1016/j.bone.2012.10.024
Pattingre, S., Tassa, A., Qu, X., Garuti, R., Liang, X. H., Mizushima, N., et al. (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122 (6), 927–939. doi:10.1016/j.cell.2005.07.002
Pickrell, A. M., and Youle, R. J. (2015). The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron 85 (2), 257–273. doi:10.1016/j.neuron.2014.12.007
Puissant, A., Fenouille, N., and Auberger, P. (2012). When autophagy meets cancer through p62/SQSTM1. Am. J. Cancer Res. 2 (4), 397–413.
Purdue, P. E., Koulouvaris, P., Potter, H. G., Nestor, B. J., and Sculco, T. P. (2007). The cellular and molecular biology of periprosthetic osteolysis. Clin. Orthop. Relat. Res. 454, 251–261. doi:10.1097/01.blo.0000238813.95035.1b
Qin, H., Xu, H. Z., and Gong, Y. Q. (2016). Mechanism of NF-κB signaling pathway and autophagy in the regulation of osteoblast differentiation. Mol. Membr. Biol. 33 (6-8), 138–144. doi:10.1080/09687688.2017.1400601
Ran, D., Ma, Y., Liu, W., Luo, T., Zheng, J., Wang, D., et al. (2020). TGF-β-activated kinase 1 (TAK1) mediates cadmium-induced autophagy in osteoblasts via the AMPK/mTORC1/ULK1 pathway. Toxicology 442, 152538. doi:10.1016/j.tox.2020.152538
Ren, C., Xu, Y., Liu, H., Wang, Z., Ma, T., Li, Z., et al. (2022). Effects of runt-related transcription factor 2 (RUNX2) on the autophagy of rapamycin-treated osteoblasts. Bioengineered 13 (3), 5262–5276. doi:10.1080/21655979.2022.2037881
Ren, M. S., Xie, H. H., Ding, Y., Li, Z. H., and Liu, B. (2023). Er-xian decoction drug-containing serum promotes Mc3t3-e1 cell proliferation and osteogenic differentiation via regulating BK channel. J. Ethnopharmacol. 302 (Pt A), 115887. doi:10.1016/j.jep.2022.115887
Scott, R. C., Juhász, G., and Neufeld, T. P. (2007). Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 17 (1), 1–11. doi:10.1016/j.cub.2006.10.053
Shaw, J., Yurkova, N., Zhang, T., Gang, H., Aguilar, F., Weidman, D., et al. (2008). Antagonism of E2F-1 regulated Bnip3 transcription by NF-kappaB is essential for basal cell survival. Proc. Natl. Acad. Sci. U. S. A. 105 (52), 20734–20739. doi:10.1073/pnas.0807735105
Shi, Y., Liu, Q., Chen, W., Wang, R., Wang, L., Liu, Z. Q., et al. (2023). Protection of Taohong Siwu Decoction on PC12 cells injured by oxygen glucose deprivation/reperfusion via mitophagy-NLRP3 inflammasome pathway in vitro. J. Ethnopharmacol. 301, 115784. doi:10.1016/j.jep.2022.115784
Singleton, A. B., Farrer, M. J., and Bonifati, V. (2013). The genetics of Parkinson's disease: progress and therapeutic implications. Mov. Disord. 28 (1), 14–23. doi:10.1002/mds.25249
Sloan, M., Premkumar, A., and Sheth, N. P. (2018). Projected volume of primary total joint arthroplasty in the U.S., 2014 to 2030. J. Bone Jt. Surg. Am. 100 (17), 1455–1460. doi:10.2106/JBJS.17.01617
Su, W., Lv, C., Huang, L., Zheng, X., and Yang, S. (2022). Glucosamine delays the progression of osteoporosis in senile mice by promoting osteoblast autophagy. Nutr. Metab. (Lond). 19 (1), 75. doi:10.1186/s12986-022-00688-y
Sun, S. C. (2011). Non-canonical NF-κB signaling pathway. Cell Res. 21 (1), 71–85. doi:10.1038/cr.2010.177
Sun, S. C. (2017). The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 17 (9), 545–558. doi:10.1038/nri.2017.52
Talmo, C. T., Aghazadeh, M., and Bono, J. V. (2012). Perioperative complications following total joint replacement. Clin. Geriatr. Med. 28 (3), 471–487. doi:10.1016/j.cger.2012.05.006
Talmo, C. T., Robbins, C. E., and Bono, J. V. (2010). Total joint replacement in the elderly patient. Clin. Geriatr. Med. 26 (3), 517–529. doi:10.1016/j.cger.2010.04.002
Tang, B. L. (2016). Sirt1 and the mitochondria. Mol. Cells 39 (2), 87–95. doi:10.14348/molcells.2016.2318
Tang, Y., Mo, Y., Xin, D., Xiong, Z., Zeng, L., Luo, G., et al. (2021). Regulation of osteoblast autophagy based on PI3K/AKT/mTOR signaling pathway study on the effect of β-ecdysterone on fracture healing. J. Orthop. Surg. Res. 16 (1), 719. doi:10.1186/s13018-021-02862-z
Trocoli, A., and Djavaheri-Mergny, M. (2011). The complex interplay between autophagy and NF-κB signaling pathways in cancer cells. Am. J. Cancer Res. 1 (5), 629–649. PMID: 21994903.
Vermes, C., Glant, T. T., Hallab, N. J., Fritz, E. A., Roebuck, K. A., and Jacobs, J. J. (2001). The potential role of the osteoblast in the development of periprosthetic osteolysis: review of in vitro osteoblast responses to wear debris, corrosion products, and cytokines and growth factors. J. Arthroplasty 16 (8 Suppl. 1), 95–100. doi:10.1054/arth.2001.28719
Verzella, D., Pescatore, A., Capece, D., Vecchiotti, D., Ursini, M. V., Franzoso, G., et al. (2020). Life, death, and autophagy in cancer: NF-κB turns up everywhere. Cell Death Dis. 11 (3), 210. doi:10.1038/s41419-020-2399-y
Vidoni, C., Ferraresi, A., Secomandi, E., Vallino, L., Gardin, C., Zavan, B., et al. (2019). Autophagy drives osteogenic differentiation of human gingival mesenchymal stem cells. Cell Commun. Signal 17 (1), 98. doi:10.1186/s12964-019-0414-7
Wang, B., Abraham, N., Gao, G., and Yang, Q. (2016). Dysregulation of autophagy and mitochondrial function in Parkinson's disease. Transl. Neurodegener. 5, 19. doi:10.1186/s40035-016-0065-1
Wang, L., Xu, C., Johansen, T., Berger, S. L., and Dou, Z. (2021). SIRT1 - a new mammalian substrate of nuclear autophagy. Autophagy 17 (2), 593–595. doi:10.1080/15548627.2020.1860541
Wang, Z., Liu, N., Liu, K., Zhou, G., Gan, J., Wang, Z., et al. (2015). Autophagy mediated CoCrMo particle-induced peri-implant osteolysis by promoting osteoblast apoptosis. Autophagy 11 (12), 2358–2369. doi:10.1080/15548627.2015.1106779
Wei, X., Zhang, X., Flick, L. M., Drissi, H., Schwarz, E. M., and O'Keefe, R. J. (2009). Titanium particles stimulate COX-2 expression in synovial fibroblasts through an oxidative stress-induced, calpain-dependent, NF-kappaB pathway. Am. J. Physiol. Cell Physiol. 297 (2), C310–C320. doi:10.1152/ajpcell.00597.2008
Wei, Y., Pattingre, S., Sinha, S., Bassik, M., and Levine, B. (2008). JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol. Cell 30 (6), 678–688. doi:10.1016/j.molcel.2008.06.001
Wong, P. M., Feng, Y., Wang, J., Shi, R., and Jiang, X. (2015). Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 6, 8048. doi:10.1038/ncomms9048
Wong, P. M., Puente, C., Ganley, I. G., and Jiang, X. (2013). The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy 9 (2), 124–137. doi:10.4161/auto.23323
Wu, H., Xue, Y., Zhang, Y., Wang, Y., and Hou, J. (2021). PTH1-34 promotes osteoblast formation through Beclin1-dependent autophagic activation. J. Bone Min. Metab. 39 (4), 572–582. doi:10.1007/s00774-021-01212-7
Xi, G., Rosen, C. J., and Clemmons, D. R. (2016). IGF-I and IGFBP-2 stimulate AMPK activation and autophagy, which are required for osteoblast differentiation. Endocrinology 157 (1), 268–281. doi:10.1210/en.2015-1690
Xia, X., Kar, R., Gluhak-Heinrich, J., Yao, W., Lane, N. E., Bonewald, L. F., et al. (2010). Glucocorticoid-induced autophagy in osteocytes. J. Bone Min. Res. 25 (11), 2479–2488. doi:10.1002/jbmr.160
Xu, A., Yang, Y., Shao, Y., Wu, M., and Sun, Y. (2020). Activation of cannabinoid receptor type 2-induced osteogenic differentiation involves autophagy induction and p62-mediated Nrf2 deactivation. Cell Commun. Signal 18 (1), 9. doi:10.1186/s12964-020-0512-6
Xu, H. D., and Qin, Z. H. (2019). Beclin 1, bcl-2 and autophagy. Adv. Exp. Med. Biol. 1206, 109–126. doi:10.1007/978-981-15-0602-4_5
Xu, M. X., Sun, X. X., Li, W., Xie, G., Yang, Q., Qu, Z. W., et al. (2018). LPS at low concentration promotes the fracture healing through regulating the autophagy of osteoblasts via NF-κB signal pathway. Eur. Rev. Med. Pharmacol. Sci. 22 (6), 1569–1579. doi:10.26355/eurrev_201803_14561
Xu, R., Shi, G., Xu, L., Gu, Q., Fu, Y., Zhang, P., et al. (2018). Simvastatin improves oral implant osseointegration via enhanced autophagy and osteogenesis of BMSCs and inhibited osteoclast activity. J. Tissue Eng. Regen. Med. 12 (5), 1209–1219. doi:10.1002/term.2652
Xu, Y., Wei, L., Tang, S., Shi, Q., Wu, B., Yang, X., et al. (2021). Regulation PP2Ac methylation ameliorating autophagy dysfunction caused by Mn is associated with mTORC1/ULK1 pathway. Food Chem. Toxicol. 156, 112441. doi:10.1016/j.fct.2021.112441
Yan, D., Yang, Y., Lang, J., Wang, X., Huang, Y., Meng, J., et al. (2023). SIRT1/FOXO3-mediated autophagy signaling involved in manganese-induced neuroinflammation in microglia. Ecotoxicol. Environ. Saf. 256, 114872. doi:10.1016/j.ecoenv.2023.114872
Yan, J. Q., Zhang, Y., Liu, F. S., Cai, T. T., Tong, K. Q., Zhu, C., et al. (2018). TCP wear particles causes injury of periprosthetic osteocytes in the mouse calvaria, 34 (1), 83–87. doi:10.12047/j.cjap.5608.2018.021
Yang, L., Liu, S., Mu, S., Guo, R., Zhou, L., and Fu, Q. (2021). Paeoniflorin attenuates dexamethasone-induced apoptosis of osteoblast cells and promotes bone formation via regulating AKT/mTOR/Autophagy signaling pathway. Evid. Based Complement. Altern. Med. 2021, 6623464. doi:10.1155/2021/6623464
Yang, X., Jiang, T., Wang, Y., and Guo, L. (2019). The role and mechanism of SIRT1 in resveratrol-regulated osteoblast autophagy in osteoporosis rats. Sci. Rep. 9 (1), 18424. doi:10.1038/s41598-019-44766-3
Yao, W., Dai, W., Jiang, J. X., and Lane, N. E. (2013). Glucocorticoids and osteocyte autophagy. Bone 54 (2), 279–284. doi:10.1016/j.bone.2013.01.034
Yuan, Y., Sun, J., Zhou, H., Wang, S., He, C., Chen, T., et al. (2022). The effect of QiangGuYin on osteoporosis through the AKT/mTOR/autophagy signaling pathway mediated by CKIP-1. Aging (Albany NY) 14 (2), 892–906. doi:10.18632/aging.203848
Zhang, L., Sun, Y., Xu, W., Geng, Y., Su, Y., Wang, Q., et al. (2021). Baicalin inhibits Salmonella typhimurium-induced inflammation and mediates autophagy through TLR4/MAPK/NF-κB signalling pathway. Basic Clin. Pharmacol. Toxicol. 128 (2), 241–255. doi:10.1111/bcpt.13497
Zhang, S., Liu, Y., and Liang, Q. (2018). Low-dose dexamethasone affects osteoblast viability by inducing autophagy via intracellular ROS. Mol. Med. Rep. 17 (3), 4307–4316. doi:10.3892/mmr.2018.8461
Zhang, W., Hou, W., Chen, M., Chen, E., Xue, D., Ye, C., et al. (2020). Upregulation of parkin accelerates osteoblastic differentiation of bone marrow-derived mesenchymal stem cells and bone regeneration by enhancing autophagy and β-catenin signaling. Front. Cell Dev. Biol. 8, 576104. doi:10.3389/fcell.2020.576104
Zhang, Z., Fu, X., Xu, L., Hu, X., Deng, F., Yang, Z., et al. (2020). Nanosized alumina particle and proteasome inhibitor bortezomib prevented inflammation and osteolysis induced by titanium particle via autophagy and NF-κB signaling. Sci. Rep. 10 (1), 5562. doi:10.1038/s41598-020-62254-x
Zheng, L. W., Wang, W. C., Mao, X. Z., Luo, Y. H., Tong, Z. Y., and Li, D. (2020). TNF-α regulates the early development of avascular necrosis of the femoral head by mediating osteoblast autophagy and apoptosis via the p38 MAPK/NF-κB signaling pathway. Cell Biol. Int. 44 (9), 1881–1889. doi:10.1002/cbin.11394
Zhang, S., Xie, Y., Yan, F., Zhang, Y., Yang, Z., Chen, Z., et al. (2022). Negative pressure wound therapy improves bone regeneration by promoting osteogenic differentiation via the AMPK-ULK1-autophagy axis. Autophagy 18 (9), 2229–2245. doi:10.1080/15548627.2021.2016231
Zhang, Y., Yan, M., Niu, W., Mao, H., Yang, P., Xu, B., et al. (2022). Tricalcium phosphate particles promote pyroptotic death of calvaria osteocytes through the ROS/NLRP3/Caspase-1 signaling axis in amouse osteolysis model. Int. Immunopharmacol. 107, 108699. doi:10.1016/j.intimp.2022.108699
Zhou, J., Yang, J., Dong, Y., Shi, Y., Zhu, E., Yuan, H., et al. (2022). Oncostatin M receptor regulates osteoblast differentiation via extracellular signal-regulated kinase/autophagy signaling. Stem Cell Res. Ther. 13 (1), 278. Published 2022 Jun 28. doi:10.1186/s13287-022-02958-1
Keywords: wear particles, periprosthetic osteolysis, osteoblasts, autophagy, signaling pathways
Citation: Gu Y, Wu Z, Xie H, Fang T, Wang Q and Gu Y (2024) Regulatory signaling pathways of osteoblast autophagy in periprosthetic osteolysis. Front. Cell. Death 3:1337724. doi: 10.3389/fceld.2024.1337724
Received: 13 November 2023; Accepted: 16 January 2024;
Published: 05 February 2024.
Edited by:
Michael Joseph Morgan, Northeastern State University, United StatesReviewed by:
Prakash P. Praharaj, University of Texas Southwestern Medical Center, United StatesCopyright © 2024 Gu, Wu, Xie, Fang, Wang and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ye Gu, ZWR3aW5ndXllQDEyNi5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.