 Maria Ladik1,2,3‡
Maria Ladik1,2,3‡ Marie Erard
Marie Erard Peter Vandenabeele
Peter Vandenabeele Franck B. Riquet
Franck B. Riquet
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Death, 05 April 2023
Sec. Non-Apoptotic Regulated Cell Death
Volume 2 - 2023 | https://doi.org/10.3389/fceld.2023.1127330
This article is part of the Research TopicReviews in Cell Death ResearchView all 6 articles
The formation of molecular complexes is a key feature of intracellular signaling pathways which governs to the initiation and execution of dedicated cellular processes. Tumor Necrosis Factor (TNF) and Reactive Oxygen Species (ROS) function as signaling molecules and are both involved in balancing cell fate decision between cell survival or cell demise. As master regulators of cell signaling, they are also instrumental in controlling various cellular processes towards tissue homeostasis, innate immunity and inflammation. Interestingly, TNF and ROS are interlinked and involved in regulating each other’s production via the engagement of molecular signaling complexes. This relationship calls for detailed reviewing of both TNF-induced and ROS-producing molecular complexes in the context of regulated cell death (RCD) modalities. Here, we outline biotechnological approaches that were used to investigate the TNF- and, concerning ROS, the NADPH oxidase-related molecular complexes with an emphasis on different regulated cell death modalities. This systematic review highlights how the cell death field has benefited from both biochemical and live-cell fluorescence imaging approaches. This knowledge and established workflows are highly generalizable, can be of a broader use for any protein-complex studies, and well suited for addressing new challenges in signaling dynamics. These will help understand molecular signaling complexes as ensembles organized into signaling platforms, most likely the key sites of signaling dynamics integration toward cell fate regulation.
Cell death plays an essential role in development, homeostasis, and inflammation (Pasparakis and Vandenabeele, 2015; van Loo and Bertrand, 2022). Despite the general recognition of the existence of cell death, the importance of the process remained largely underestimated for a long time mainly because dying cells were rarely observed. Only in the mid-19th century when the concept of histology was born, it allowed scientists to identify dying cells. Since the publication of such renowned works as the one written by Karl Vogt where he studied the disappearance of the notochord during the metamorphosis of amphibians, a large number of studies described the physiological importance of different types of cell death such as the best known one called apoptosis (Maghsoudi, Zakeri and Lockshin, 2012). For a long time, apoptosis was considered the only regulated type of cell death (Galluzzi et al., 2007). This cell death modality plays an important role during embryonic development and cell homeostasis (Montero and Hurlé, 2010; Herold, Rennekampff and Engeli, 2013; Gudipaty et al., 2018) and originate from either an intracellular signal or external stimulus like tumor necrosis factor (TNF).
Historically, apoptosis was considered as the standard regulated cell death (RCD) modality, whereas necrosis was thought to be accidental and uncontrolled, caused by abrupt changes in physicochemical environment such as temperature, osmotic pressure or pH. In the late 90s however, a regulated type of necrosis was identified in a fibrosarcoma cell line (L929), in which inhibition of caspases increased sensitivity to TNF–mediated necrosis (Vercammen et al., 1998; Degterev et al., 2005). Then it became clear that TNF signaling could be correlated to multiple cellular outcomes.
TNF treatment leads to TNF receptor 1 (TNFR1) trimerization and formation of complex I at the plasma membrane (Figure 1A). This supramolecular structure regulates a network of pro-survival and pro-inflammatory signaling pathways and can in turn lead to formation of a cytosolic signaling complex, complex II, facilitating apoptosis execution (Figures 1B, C). The TNF-mediated necrosis observation in L929 together with the pioneering finding of the lab of Jürg Tschopp reporting on the crucial role of receptor-interacting protein kinase 1 (RIPK1) in caspase-independent cell death laid the path to what has now become the most studied type of regulated necrosis (Holler et al., 2000), necroptosis via the formation of yet another signaling complex termed necrosome (Figure 1D). Many studies have since revealed the implication of necroptosis in multiple pathophysiological situations. This regulated cell death modality which appears to play an essential role during development (Kaiser et al., 2011; Oberst et al., 2011), is also associated with pathological situations such as inflammatory diseases (e.g., acute pancreatitis, septic shock), neurological diseases (e.g., stroke) or ischemia-reperfusion injury, and cardiac infarction (for review, (Jouan-Lanhouet et al., 2014; Khoury et al., 2020).
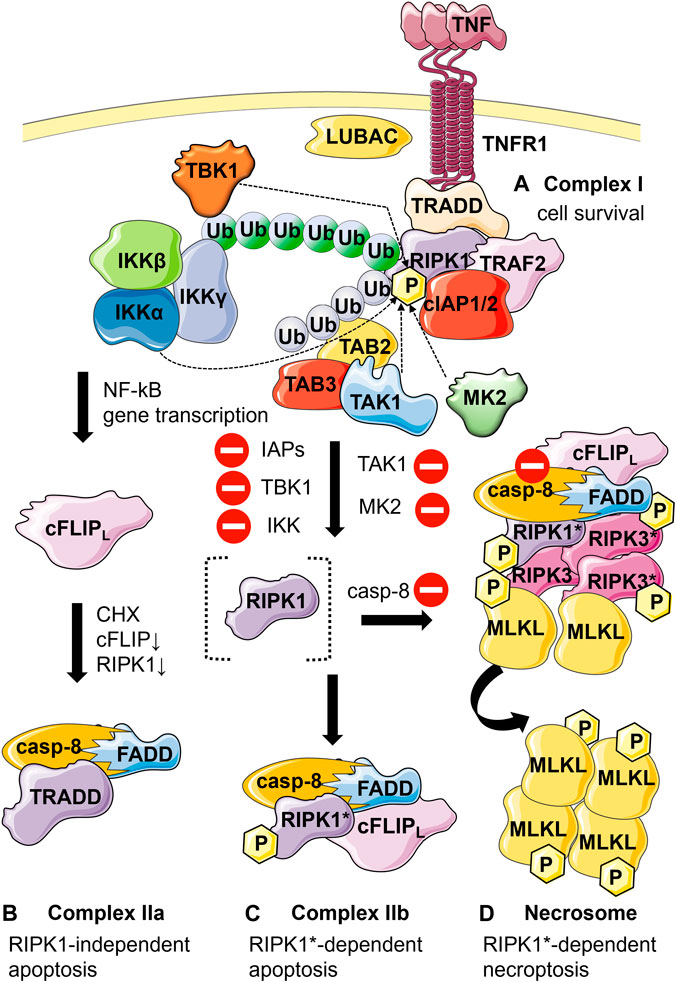
FIGURE 1. Schematic representation of the different TNF-mediated cell fates and respective associated molecular complexes. (A) Cell survival: Binding of TNF to TNFR1 leads to the recruitment of TNF receptor-associated protein with death domain (TRADD), receptor interacting protein kinase 1 (RIPK1), cellular inhibitor of apoptosis protein 1 and 2 (cIAP1/2), TNF receptor-associated factor 2 (TRAF2) to TNFR1 resulting in the formation of complex I. Ubiquitination of RIPK1 by cIAP1/2, TRAF2, and linear ubiquitin chain assembly complex (LUBAC) leads to the recruitment of transforming growth factor-β-activated kinase 1 (TAK1) through TAK1 binding proteins 2 and 3 (TAB2 and TAB3), as well as the recruitment of the inhibitor of NF-κB kinase (IKK) complex and the TANK-binding kinase 1 (TBK1). The IKK complex activates the nuclear factor-κB (NF-κB) signaling. Specific phosphorylation of RIPK1 in complex I at various sites by IKK, TBK1, mitogen-activated protein kinase-activated protein kinase 2 (MK2) or (indirectly) by TAK1 functions as a checkpoint keeping RIPK1 in a survival mode independent of its kinase activity. (B) RIPK1-independent apoptosis: When the levels of cellular FLICE-like inhibitory protein (cFLIP) (due to inhibition of translation by cycloheximide, CHX) and RIPK1 (resulting in reduced gene induction) are low TRADD interacts with Fas-associated protein with death domain (FADD) and pro-caspase-8, forming complex IIa. (C) RIPK1-dependent apoptosis: Alternatively, upon inhibition of IAPs, IKK, TBK1, MK2 and TAK1 (inhibitory symbols) RIPK1 becomes activated by autophosphorylation (RIPK1*). RIPK1* interacts with FADD, caspase-8 and cFLIPl (if present), forming complex IIb. Upon formation of complex IIa or IIb, caspase-8 gets activated through proximity-induced activation leading to apoptosis. (D) Necroptosis: Caspase-8 functions as a checkpoint for necroptosis induction. In the absence of active caspase-8 (inhibition symbol), RIPK1 and RIPK3 are no longer cleaved by caspase-8 and form the necrosome through their RIP homotypic interaction motif (RHIM). Autophosphorylation of the RIPK1 and RIPK3 proteins as well as the recruitment and RIPK3*-dependent phosphorylation of mixed lineage kinase domain-like protein (MLKL) results in plasma membrane permeabilization and necroptosis. P, phosphorylation. Blue ubiquitin chains, K63-linked ubiquitination; green ubiquitin chains, linear ubiquitination.
In the past decades, several major molecular players of the TNF signaling have been identified and their relationship to each other has been investigated. This indispensable information was mainly gained using classical biochemical approaches. These studies identified a number of molecular complexes and highlighted some essential protein-protein interactions (PPIs) as well as key protein activities implicated in cell fate decision, mainly at the cell population level. This will be reviewed here. However, cell death similarly to other cellular processes, has turned out to be a highly dynamic phenomenon governed by assembly and disassembly of macromolecular complexes in space and time, resulting in single-cell behaviors (cell survival, inflammatory cytokine production, apoptosis, necroptosis), that are driven by particular signaling pathways and lead to a heterogeneous response within a cell population. This representation has recently been complexified by the fact that an intricate game of cell death can occur within a cell, tipping the balance between different regulated cell death modalities (Bertheloot, Latz and Franklin, 2021). This changes the perspective on how to study molecular complexes and whether they should better be considered in the scope of molecular signaling platform.
Besides the TNFR complexes, another protein complex involved in cell death dynamics, and being part of such molecular signaling platform, will be discussed in this review: the NADPH oxidases, enzymes specifically dedicated to the production of ROS in various cells and tissues. Their sophisticated function depends on presence/absence of biological (microorganisms), chemical stimuli such as inflammatory mediators (Lambeth, 2004a; Begum et al., 2022), or even cytokines (TNF) (Kim et al., 2007a) and is tightly regulated at the level of PPIs and protein-lipids interactions.
In this context, the contribution of functional fluorescence live cell microscopy to the overall knowledge of protein complexes so far will be presented, along with the challenges and opportunities inherent to such approaches. We will first introduce the complexity of TNF signaling, as it is a well described example of how different cell fates are initiated at the molecular level (van Loo and Bertrand, 2022).
Binding of TNF to the extracellular domain of TNFR1 (Andera, 2009) leads to the recruitment of an adaptor protein TNFR-associated protein with death domain (TRADD) (Hsu, Xiong and Goeddel, 1995; Hsu, Shu, et al., 1996) that interacts with TNFR1 through its death domain (DD) (Figure 1A) and enables the recruitment of RIPK1 (Hsu, Huang, et al., 1996) owing to its scaffold function. TRADD, in turn, facilitates the recruitment of an E3 ubiquitin ligase TNF receptor associated factor 2 (TRAF2) that further mediates the signaling (Park et al., 2000) and ensures the recruitment of two other E3 ubiquitin ligases, the cellular inhibitor of apoptosis protein 1 and 2 (cIAP1/2) to the TNFR1 complex that is also known as complex I (Legler et al., 2003; Micheau and Tschopp, 2003). cIAPs and TRAF2 further promote K63-ubiquitination of RIPK1 thus stabilizing the TNFR1 complex (Mahoney et al., 2008). The group of Walczak identified another important player in stabilization of the TNFR1 signaling complex. Using a modified tandem affinity purification (TAP) approach three proteins were identified: heme-oxidized IRP2 Ub ligase 1 (HOIL-1), HOIL-1 interacting protein (HOIP) and SHANK-associated RH domain interactor (SHARPIN) that together form the linear ubiquitin chain assembly complex (LUBAC). The LUBAC complex assembles linear ubiquitin chains on RIPK1, thus stabilizing the complex and preventing TNF-induced cell death (Haas et al., 2009; Gerlach et al., 2011). K63-ubiquitination of RIPK1 by cIAP1 and 2 leads to further recruitment of transforming growth factor-β-activated kinase 1 (TAK1) that interacts with RIPK1 through TAK1 binding proteins 2 and 3 (TAB2 and TAB3) (Kanayama et al., 2004; Besse et al., 2007). Another protein complex that is recruited to ubiquitinated RIPK1 is the inhibitor of the NF-κB kinase complex (IKK) that associates with K63- and linearly ubiquitinated RIPK1 through IKKγ, also known as NF-κB essential modulator (NEMO) (Ea et al., 2006; Wu et al., 2006; Dynek et al., 2010; Gerlach et al., 2011). Recruitment of both TAK1 and IKK complexes to complex I leads to TAK1- and NEMO-dependent IKKβ phosphorylation and activation. The activated IKKβ phosphorylates inhibitor of NF-κB α (IκBα) followed by its ubiquitination and proteasomal breakdown. Degraded IκBα can no longer inhibit the nuclear factor-κB (NF-κB) transcription factor complex that subsequently translocates to the nucleus where it functions as a transcription factor for multiple genes involved in inflammation and cell survival (Amaya et al., 2014).
In case the cell survival pathway is abrogated, the TNF signaling further proceeds to induce apoptosis. The key player in this event is initiator caspase-8. The apoptotic pathway proceeds through the formation of one of two complexes: complex IIa (Figure 1B) or complex IIb (Figure 1C). The formation of complex IIa has been shown to depend on cellular FLICE-like inhibitory protein (cFLIP) expression (Wang, Du and Wang, 2008), an anti-apoptotic protein that regulates activation of caspase-8. It has been previously defined that human cFLIP is primarily expressed in two forms. The short form cFLIPS is shown to contain two death effector domains (DEDs), while the long form cFLIPL contains next to the two DEDs, a caspase-like domain where active cysteine residue is substituted by a tyrosine residue. The Wang group demonstrated that when the expression levels of cFLIP are low such as in case of TNF induction in combination with the translation inhibitor cycloheximide, complex IIa consisting of TRADD, Fas-associated protein with death domain (FADD), cFLIP and caspase-8 is formed inducing apoptotic signaling (Figure 1B) (Wang, Du and Wang, 2008; Ofengeim and Yuan, 2013). Ubiquitination of RIPK1 by IAPs promotes its survival activity (Bertrand et al., 2008). Alternatively, when following TNF stimulation RIPK1 ubiquitination is abrogated in the absence of functional cIAPs through genetic ablation or by using IAP antagonists such as Smac mimetics, RIPK1 gets recruited into the cytosolic 2 MDa complex IIb, also known as ripoptosome, that contains cFLIP, RIPK1, FADD and caspase-8 (Feoktistova et al., 2011; Tenev et al., 2011; Vanlangenakker et al., 2011). The survival function of RIPK1 is kept in check by phosphorylation by IKKs (Dondelinger et al., 2015; Dondelinger et al., 2019), mitogen-activated protein kinase-activated protein kinase 2 (MK2) (Dondelinger et al., 2017; Jaco et al., 2017; Menon et al., 2017), TBK1 (Lafont et al., 2018; Xu et al., 2018) and TAK1 (Geng et al., 2017). Inhibition of these kinases favors RIPK1-mediated apoptosis and necroptosis (Delanghe et al., 2020). The assembly of complex IIb is dependent on RIPK1 kinase activity and requires the interaction between RIPK1 and caspase-8 through the adapter protein FADD. FADD consists of a DD through which FADD interacts with RIPK1 and a DED that serves to recruit caspase-8 that contains tandem DEDs in its prodomain (Carrington et al., 2006). Upon recruitment to FADD, the prodomain of caspase-8 as well as FADD form filamentous high order structures that are required for the activation of caspase-8. Caspase-8 gets activated through proximity-induced activation (Siegel et al., 1998; Salvesen and Walsh, 2014). Activated caspase-8 finally cleaves a number of substrates such as effector caspase-3, and -7, thus inducing apoptosis.
Caspase-8 functions as a checkpoint for RIPK1 kinase activity dependent apoptosis and necroptosis (Newton et al., 2019). Caspase-8 enzymatic activity is inhibited by using the pan-caspase inhibitor zVAD-fmk or in the presence of high expression levels of the short isoform cFLIPS that heterodimerizes with procaspase-8 and completely prevents its proteolytic activation (Vercammen et al., 1998; Schilling, Geserick and Leverkus, 2014), the cell switches towards necroptosis. Stimulation with a necroptosis trigger (TNF in combination with a Smac mimetic and zVAD-fmk) leads to the association between two major necroptotic players, RIPK1 and RIPK3, through their RIP homotypic interaction motif (RHIM) (He et al., 2009; Oerlemans et al., 2012). Depending on the nature of the stimulus, necroptosis will be initiated either by a RIPK1-RIPK3 hetero-interaction or by a RIPK3-RIPK3 homo-interaction (Orozco et al., 2014; Wu et al., 2014). This interaction initiates the nucleating core for the necrosome complex that consists of enzymatically inactive caspase-8, FADD, RIPK1 and RIPK3 (Cho et al., 2009; He et al., 2009) (Figure 1D). These RIP kinases are both auto- and trans-phosphorylated within the complex (Cho et al., 2009; He et al., 2009; Zhang et al., 2009; Vanden Berghe et al., 2014), leading to the formation of β-amyloid (Li et al., 2012) and β-strands (Wu et al., 2021) structures. The latter were recently reported to constitute a more stable structure and were suggested to favor a structural transformation, allowing the formation of RIPK3 homo-oligomers (Li et al., 2012; Rodriguez et al., 2016; Wu et al., 2021). Oligomerized RIPK3 further recruits and activates another important necroptotic player, the mixed lineage kinase domain-like protein (MLKL) (Figure 1D). Phosphorylation of the MLKL pseudokinase domain by RIPK3 unleashes the N-terminal four-helix bundle domain of MLKL that is coupled to the pseudokinase domain through a two-helix linker. The phosphorylated four-helix bundle domain further forms high-molecular weight oligomeric structures that translocate to the membrane and cause membrane disruption (Murphy et al., 2013; Dondelinger et al., 2014; Hildebrand et al., 2014; Liu et al., 2017). Samson et al. described precise choreography of these events. The phosphorylated MLKL oligomeric species assemble on presumed cytoplasmic necrosomes. Afterwards, these species co-traffic with tight junction proteins to the cell periphery via Golgi-microtubule-actin-dependent mechanisms and subsequently co-accumulate at the plasma membrane as heterogenous micro-size hotspots. Taken together, the study of Samson et al. identified MLKL trafficking and plasma membrane accumulation as two crucial necroptosis checkpoints (Samson et al., 2020).
In 2017, a couple of studies pointed out more complex functions of the protein that do not appear to be restricted to execution of cell death. The group of Wallach reported that MLKL is involved in endosomal trafficking thus assisting in its own extrusion from cells within extracellular vesicles (EVs) so as to antagonize cell death (Yoon et al., 2017). Similarly, the group of Doug Green demonstrated that activation of MLKL leads to shedding of damaged plasma membrane, an effect that is dependent on the endosomal sorting complexes required for transport III (ESCRT III) machinery. This process prolongs cell survival when MLKL activation is limited and results in sufficient time to generate stress signals via cytokines, chemokines and other regulatory molecules. This in turn promotes intercellular communication which will affect surrounding cells and induce antigenic cross-priming of CD8+ T-cells (Gong et al., 2017).
The information provided above demonstrates that the TNF pathway is a sequential process that involves spatially and temporally distinct signaling complexes whose assembly and disassembly leads to a dedicated outcome: survival or cell death. Beyond this concept, recent literature is proposing a more complex landscape in which emerging connectivity between different RCDs reveals how cell death pathways can be intertwined with consequences in infection and cancer (Bedoui, Herold and Strasser, 2020), and in stroke (Naito et al., 2020). The resulting intricacies and fine tuning of cell death effectors is tipping the balance between different cell fates (Bertheloot, Latz and Franklin, 2021), with unexpected findings regarding cells recovering from the brink of cell death (Gudipaty et al., 2018). Anastasis for apoptotic cells (Sun et al., 2017; Sun and Montell, 2017) or resuscitation for necroptotic cells (Gong et al., 2017), which in both cases questions where the point of no-return truly lies. Collectively these situations exemplify the necessary flexibility for molecular effector complexes to dynamically evolve and adapt in space and time towards survival purpose.
In this evolving paradigm in which the crosstalk is highlighted, we choose to briefly review a situation that has been lingering for years in the cell death field concerning the role of ROS in cell death (Blaser et al., 2016). A recent study from our laboratory discovered how the ROS scavenger butylate hydroxyanisole (BHA) is acting as a direct RIPK1 inhibitor, hence challenging a body of literature that has been claiming ROS involvement in PCDs on the extensive usage of this “ROS scavenger” (Delanghe et al., 2021a). To clarify the role of ROS and the different experimental strategies that are used for investigations, the second part of this review will focus on the NADPH oxidases, as a known source of ROS, and the implication of these multicomponent enzymatic complexes in various cell fates.
TNF, a master regulator in both cell survival and cell death, is also an important player in the regulation of cellular homeostasis and immunity (Blaser et al., 2016). Concerning the later one, TNF is known to stimulate the activity of the phagocyte NADPH oxidase (NOX) (Figure 2), a key enzyme of our innate immune system (Lambeth, 2004).
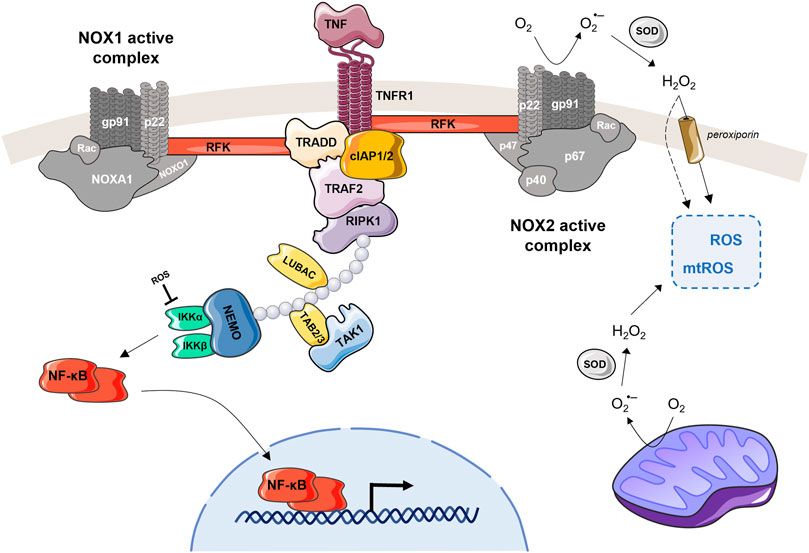
FIGURE 2. TNFR interacts with NOX1 or NOX2 and induce ROS production. TNF recognized by TNFR1 is able to activate the NOX1 or NOX2 complex. For NOX1 (on the left), the TNF-induced activation can be done through interaction of the cytosolic domain of TNFR1, TRADD and RFK, which then binds to the p22phox subunit. Recruitment of the NOX1 complex to TNFR1 can be also facilitated by RIPK and/or RAC1/RFK (Kim et al., 2007; Yazdanpanah et al., 2009; Park et al., 2012). In case of the NOX2 (on the right), the RFK is the main actor coupling the TNFR1 with p22phox subunit. The activated NOX2 complex converts extracellular O2 into superoxide anion, which is then transformed by SOD to H2O2. This extracellular H2O2 can freely pass through the plasma membrane, become part of the intracellular ROS pool (together with mtROS) and participate on cell signaling. The NOX1 complex, when TNF-activated, produces the ROS in the same manner. This figure also highlights NF-kB, which is involved in the regulation of TNF-induced ROS production (Morgan and Liu, 2011; Nakajima and Kitamura, 2013). When NF-kB is activated through the TNF-mediated cascade, it is translocated into the nucleus where it upregulates catalase and SOD expression levels. These antioxidants reduce the total amount of ROS in the cell, which then stimulates TNF and NOX2 expression leading to a new ROS production. Another feedback loop is that ROS produced by NOX1/2 can activate NF-kB by inhibiting IKK phosphatases.
The NADPH oxidases is a whole family of enzymes comprising of seven related isoforms, varying in the catalytic center: NOX1-5, DUOX1 and DUOX2. The NADPH oxidases are multicomponent complexes with a capacity to transport electrons across the plasma membrane and to generate superoxide and other downstream reactive oxygen species (ROS), which make them major oxidant generators in a human body (see the next paragraph below for more details about the NOX family). The main physiological functions of NOX family is the ROS production for antimicrobial host defense, but, they also strongly influence cellular signaling, regulation of gene expression and cell differentiation (Bedard and Krause, 2007).
Besides the NOXes, mitochondria are another important ROS source in mammals. During cellular respiration in mitochondria, the electrons released from the electron transport chain react with O2 to produce superoxide anions (Figure 2), which are further converted to H2O2 by superoxide dismutases (SODs) (Hamanaka and Chandel, 2010). Mitochondria-derived ROS (mtROS) can be delivered to bacteria-containing phagosomes via mitochondria-derived vesicles (Abuaita, Schultz and O’Riordan, 2018), and this way directly contribute to the bactericidal activity. While it was shown that mitochondria in macrophages can produce high amounts of mtROS, especially after stimulation, neutrophils contain only low numbers of active mitochondria (Dupré-Crochet, Erard and Nüβe, 2013). A crosstalk between NOXs-derived ROS and mitochondria may amplify ROS generation at different subcellular compartments to maintain sustained redox signaling. For example, NOX4-derived H2O2 in endothelial cells can increase mtROS production, which can be regulated by NOX2 siRNA (Kim et al., 2017; Fukai and Ushio-Fukai, 2020).
Although TNF has been known to potentiate the NADPH oxidase activity, for a long time it was considered that a second stimulus was necessary to actually activate the oxidase (Morgan, Kim and Liu, 2008). In laboratories, phorbol myristate acetate (PMA) is often chosen as this second stimulus. It acts through the activation of the protein kinase C (PKC) launching a phosphorylation cascade of several NOX subunits (el Benna, Faust and Babior, 1994) finally leading to the assembly of active enzyme complex. It has been proven that TNF is a powerful enabler of NOX2 activation in neutrophils (Elbim et al., 1993) and even in non-phagocytic cells such as HEK293 (Moe et al., 2006). Yazdanpanah et al. showed that physical interaction of TNF receptor 1 with the NADPH oxidase complex is dependent on riboflavin kinase (RFK). Immunoblotting analysis of anti-RFK immunoprecipitates from HeLa cell lysates showed the interaction of RFK with p22phox as well as with TNFR1 and TRADD, but not with RIPK1 (Yazdanpanah et al., 2009). Moreover, phagocytes lacking riboflavin or RFK activity display defective TNF-dependent NOX2 signaling, resulting in reduced ROS production and impaired innate immune responses against pathogens (Schramm et al., 2014).
TNF was also shown as an activator of the NOX1 in mouse fibroblasts inducing necrotic cell death (Kim et al., 2007). However, in case of NOX1, the mechanism explanations are more ambivalent. Morgan et al. suggested that in non-phagocytic cells, RIPK1 and TRADD together are able to recruit NOX1 protein organizer (NOXO1) and Rac1 to the membrane to join the p22phox subunit of NOX1 and form an active superoxide producing complex (Morgan, Kim and Liu, 2008). On the other hand, other studies support the role of RFK and claim that recruitment of the NOX1 complex to TNFR1 is also facilitated by RAC1/RFK (Chen and Goeddel, 2002; Ueyama, Geiszt and Leto, 2006; Yazdanpanah et al., 2009). Here, we propose a model of the TNF-NOX relation and highlight the role of RFK that interacts with TNF receptor 1 and membrane-bound subunits of the NADPH oxidase (Figure 2).
Production of superoxide by TNF-activated NOX1 further promotes sustained activation of c-Jun N-terminal kinase (JNK) leading to the necrotic cell death (Morgan, Kim and Liu, 2008). Moreover, induction of the JNK and ERK signaling pathways by NOX1 together with NOX2 was shown to play a role in M2 polarization and macrophage differentiation (Xu et al., 2016). Some cell surface receptors, including TNF receptors can prime the phagocytes for NOX2 activation (Nguyen, Green and Mecsas, 2017). Priming may include conformational changes or partial phosphorylation of the regulatory subunits, which renders the cells more susceptible to a secondary stimulus but does not lead to superoxide production.
On the one hand, ROS generation is induced by cytokines, as in case of TNF; on the other, ROS can stimulate proinflammatory cytokine production by activating NF-κB, the key transcription factor driving cell survival/cell death signaling (Morgan and Liu, 2011). In murine macrophages, H2O2 triggers TNF expression via activation of the p38 and JNK pathways (Nakao et al., 2008). In addition, H2O2 oxidizes the catalytic cysteines of MAPK-inactivating phosphatases, thus activating MAPKs such as p38. This positive feedback loop, in which TNF-induced ROS production subsequently triggers TNF expression, emphasizes the importance of proper ROS regulation in executing an adequate TNF-mediated innate response (Blaser et al., 2016).
Similar to TNF, some NADPH oxidase isoforms require sophisticated interplay of several proteins to perform its function. The first discovered member of the NOX family was the phagocyte NADPH oxidase (NOX2), which is often described as NOX prototype. NOX2 is expressed mainly in phagocytic cells, i.e., macrophages, neutrophils, which are involved in the microbial killing process. Phagocyte NADPH oxidase is a multi-component enzymatic complex composed of two membrane proteins [gp91phox (catalytic center) and p22phox], three cytosolic proteins (p47phox, p67phox and p40phox) and a GTPase Rac (Nauseef, 2019). In a resting state, the cytosolic subunits exist together in the cytosol as a complex (separated from the Rac protein) (Figure 3 left). Upon activation, p47phox is phosphorylated at specific sites (Sumimoto, 2008; El-Benna et al., 2009), inducing its conformational change and triggering migration of the ternary complex of the cytosolic subunits to the membrane. When the ternary complex reaches the membrane, it associates with gp91phox, p22phox and Rac forming the active complex able to produce superoxide anions (Figure 3 right). Except the natural process of phagocytosis, the NOX2 can be also activated by TNF or by using soluble stimuli, such as PMA as mentioned above.
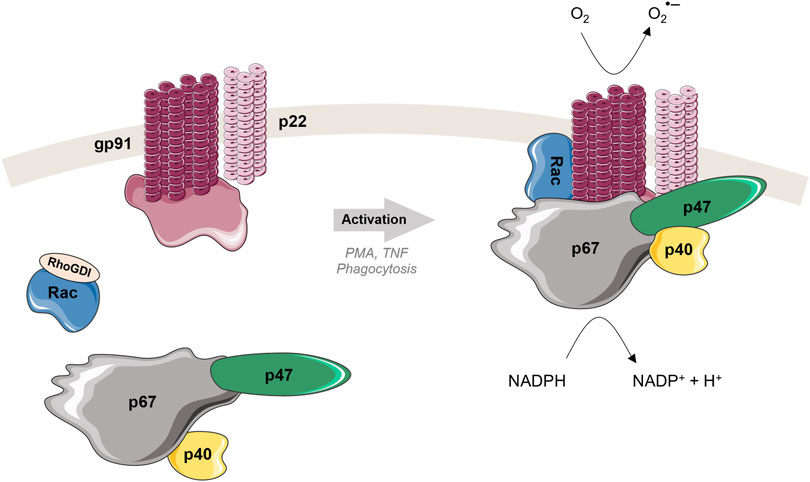
FIGURE 3. Structure and activation mechanism of phagocyte NADPH oxidase (NOX2). Left: In the resting state, cytosolic and membrane subunits are separated. The cytosolic subunits form a ternary complex in 1:1:1 ratio (Ziegler et al., 2019). Rac is in a GDP-bound form, localized in the cytosol separated from the other cytosolic subunits, and associated with RhoGDI. Right: Upon activation, the three cytosolic subunits, p47phox, p67phox and p40phox, migrate to the membrane to join two membrane subunits gp91phox and p22phox. Rac dissociates from RhoGDI, creates active GTP-bound form and relocates to the membrane as well. Once all subunits are in a good position, where all necessary interactions can take place, the active complex is formed. The NADPH substrate is oxidized, electrons are transferred by the catalytic subunit gp91phox through the membrane to the molecular oxygen, leading to the production of the superoxide radical anion (O2•-) and then associated ROS.
Of seven members in the human family, NOX1, a non-phagocytic homolog of NOX2, also form a heterodimer with p22phox and requires only two cytosolic subunits, NOXO1 (homologous to p47phox) and NOXA1 (homologous to p67phox), for its function (Bedard and Krause, 2007). NOX1 is highly expressed in colon epithelial cells (Szanto et al., 2005) and in other locations such as smooth muscle, endothelial cells or tissues as uterus, placenta or prostate.
In a healthy organism, the NOX activity is tightly regulated. Any disbalance in the activity of the NADPH oxidase can lead to different pathologies. Chronic granulomatous disease (CGD), a genetic disorder is an example of insufficient NOX2 activity caused by mutations in any of the five subunits. CGD is characterized by severe and recurrent infections due to the inability of phagocytes to produce superoxide radical and kill invading bacteria and fungi (Stasia and Li, 2008). On the other hand, an excessive NOX activity causes an extreme oxidative stress leading first to damages of lipids and then of proteins and DNA. The end products of oxidative stress are involved in many diseases such as neurodegenerative disorders (Alzheimer’s or Parkinsons’s disease), pulmonary diseases (idiopathic pulmonary fibrosis or chronic obstructive pulmonary disorder) or cardiovascular diseases (hypertension or atherosclerosis) (Begum et al., 2022). NADPH oxidase activity has been also linked to regulation of insulin secretion. Generation of ROS modulates glucose-stimulated insulin secretion; having a positive effect upon acute ROS activation but a negative effect upon chronic ROS activation (Morgan et al., 2009). Increased expression of NOX isoforms was described in animal models of type 2 diabetes (Nakayama et al., 2005).
It should also be noted that chronic hyperglycemia itself can stimulate the expression and activation of NOX enzymes, leading to the excessive production of ROS, which then activates several pathways associated with diabetic tissue damage, including advanced glycation end products (AGEs) (Begum et al., 2022). Consequently, the AGEs activate several signaling networks causing stimulation of NF-κB activity, which induces the generation of proinflammatory cytokines, including IL-6 and TNF-α (Goldin et al., 2006).
Rheumatoid arthritis (RA) is another chronic inflammatory disease associated with high levels of ROS generated by NOX2 and accumulated in peripheral joints (Dang et al., 2006). In addition, proinflammatory cytokines such as TNF are secreted by macrophages, neutrophils, and T and B lymphocytes that reach the synovial membrane of the joints (Burmester, Feist and Dörner, 2014). Therapeutic inhibition of TNF signaling efficiently inhibits ROS production and reduces joint inflammation (Kennedy et al., 2011), pointing to the TNF-NOX cross talk.
Although elevated levels of ROS were reported for tumor necrosis factor receptor-associated periodic syndrome (TRAPS), the NADPH oxidase is not the source, as they were identified as mtROS (Bulua et al., 2011).
For the multiprotein NADPH oxidase isoforms as NOX 1 and NOX2, the regulation of their activities is mainly driven by modulation of protein-protein and protein-lipid interactions. For example, for NOX2, the p47phox, p67phox and p40phox cytosolic subunits form in the ternary complex in the cytosol in the resting state. Upon phagocytosis (main physiological trigger to activate NOX2), the phosphorylation of p47phox initiates the migration of the ternary complex and GTP-bound Rac to the membrane, where the active complex is formed (Li et al., 2009; Matute et al., 2009; Faure et al., 2013; Song et al., 2017) (Figure 3). As soon as the complex is formed and the p67phox is anchored to the gp91phox, Rac2 and p47phox detach from the phagosomal membrane. The detachment of p47phox is linked to the level of phosphoinositides (decrease of PI(3,4)P2 and accumulation of PI(3)P) in the phagosome membrane. The linkage of p40phox to PI(3)P sustains the NADPH oxidase activity. These observations emphasize the important role of protein-protein but also protein-lipid interactions in functioning of this membrane-bound proteins and enzymes (Valenta et al., 2020).
The superoxide anion radical produced by active NOX enzymes is converted to hydrogen peroxide (H2O2) by spontaneous dismutation or by SODs that are omnipresent in intracellular and even extracellular environment (Figure 2). H2O2 can be further converted in the highly reactive HOCl by myeloperoxidase and HO° by Fenton reactions (Dickinson and Chang, 2011). H2O2, less reactive than other ROS, has a longer lifetime (∼1 ms) and can diffuse on rather long distances at the cell scale and even across membranes by aquaporin channels, known as peroxiporins (Bienert et al., 2007) (Figure 2). As H2O2 is highly involved in redox signaling, regulation of its cytosolic concentrations is fundamental for cell survival and maintaining normal physiological conditions. In general, the effect of ROS, including H2O2, is all about balance. Anarchical chemical modifications due to high local levels of ROS that cannot be managed by the anti-oxidant machinery (e.g., SOD, catalase, etc.) leads in the end to a cell demise. In this saturation mode, ROS do not operate by fine tuning molecular structure of macromolecules anymore (disulfide bonds, oxidation of AA residue, etc.) to activate signaling cascades.
For better illustration, Figure 4 presents physiological ranges of H2O2, spanning from normal processes through adaptive ones (stress responses) to higher H2O2 concentrations evoking inflammatory responses, ultimately leading to growth arrest and cell death (Sies, 2017). One may note that there is no clear-cut frontier between these ranges. A moderate increase in the level of ROS will be beneficial for the inflammatory response during phagocytosis, in another situation, a similar amount of ROS will contribute to tumor promotion and progression, as they are involved in different signaling pathways and can induce DNA mutations (Perillo et al., 2020).
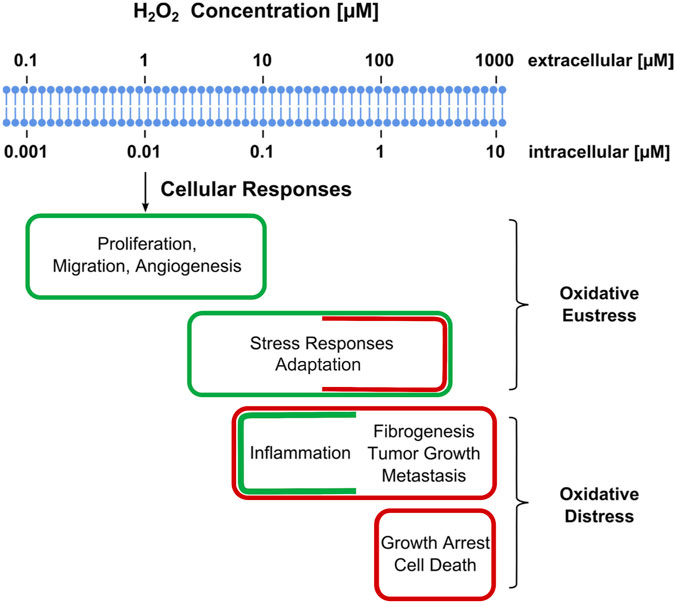
FIGURE 4. Estimated ranges of hydrogen peroxide concentrations involved in oxidative stress with regard to cellular responses. Adaptive stress responses are denoted as “oxidative eustress”. High H2O2 concentrations exceeding physiological ranges are denoted as “oxidative distress”. Assignment of a given H2O2 concentration to certain category may vary with cell type or with the duration of the exposure to H2O2. Adopted from (Sies, 2017).
Taken together, TNF-related signaling and NOX-mediated ROS production are highly complicated events (Vanden Berghe et al., 2010; Babu et al., 2015; Moghadam, Henneke and Kolter, 2021). The protein complexes associated to these events need to be investigated from different angles at the molecular level, which will provide more information and at the end improve our understanding of related physiological processes: ROS production, survival or demise of different cells at different life stages. The final approach usually includes the study of protein complex formation, composition, protein-protein interactions within the complex and spatiotemporal behavior. Numerous biochemical and biotechnological methods contributed to major discoveries that would not have been otherwise possible. The next sections will provide an overview of different techniques that were used for studying TNF- and NOX-related processes, and that are easily applicable to other similar protein systems.
Maybe the most popular method that falls within the conventional co-purification strategies used to assess PPIs is co-immunoprecipitation (Co-IP) (Figure 5A). Using this method enabled researchers to distinguish between two complexes: complex I and complex II and identify their composition. By stimulating a human fibrosarcoma cell line HT1080 with Flag-tagged human TNF (hTNF) for different time periods, and by subsequently using anti-Flag antibodies, Olivier Micheau and Jürg Tschopp defined that at earlier time points RIPK1, TRAF2, and TRADD co-immunoprecipitated with TNFR1, while caspase-8 and FADD did not. By immunoprecipitating caspase-8 following TNF stimulation in the same cells, they defined that four to 8 hours post-stimulation RIPK1, TRADD, and TRAF2 associated with caspase-8, coinciding with their disappearance from the primary complex, and confirming their dissociation from complex I (Micheau and Tschopp, 2003). Similarly, Dondelinger et al., 2013 demonstrated in MEF cells stimulated with hTNF in combination with TAK1 inhibitor or a Smac mimetic the formation of the complex IIb by immunoprecipitating caspase-8 and observed the RIPK1 recruitment (Dondelinger et al., 2013). The formation of the necrosome was indicated by multiple research groups. Sudan He and colleagues showed by means of immunoprecipitation against RIPK1 or Flag-RIPK3 the association with respectively RIPK3 or RIPK1 in HT-29 or HT-29 stably expressing Flag-RIPK3. The same group also demonstrated the association of RIPK1, Flag-RIPK3, FADD, and caspase-8 within the same complex by immunoprecipitating caspase-8 (He et al., 2009). Alternatively, Cho et al. showed RIPK1 and RIPK3 recruitment to the necrosome by isolating the complex either with caspase-8- or FADD-specific antibody from WT TNFR2+ Jurkat cells stimulated with TNF in combination with zVAD-fmk (Cho et al., 2009).
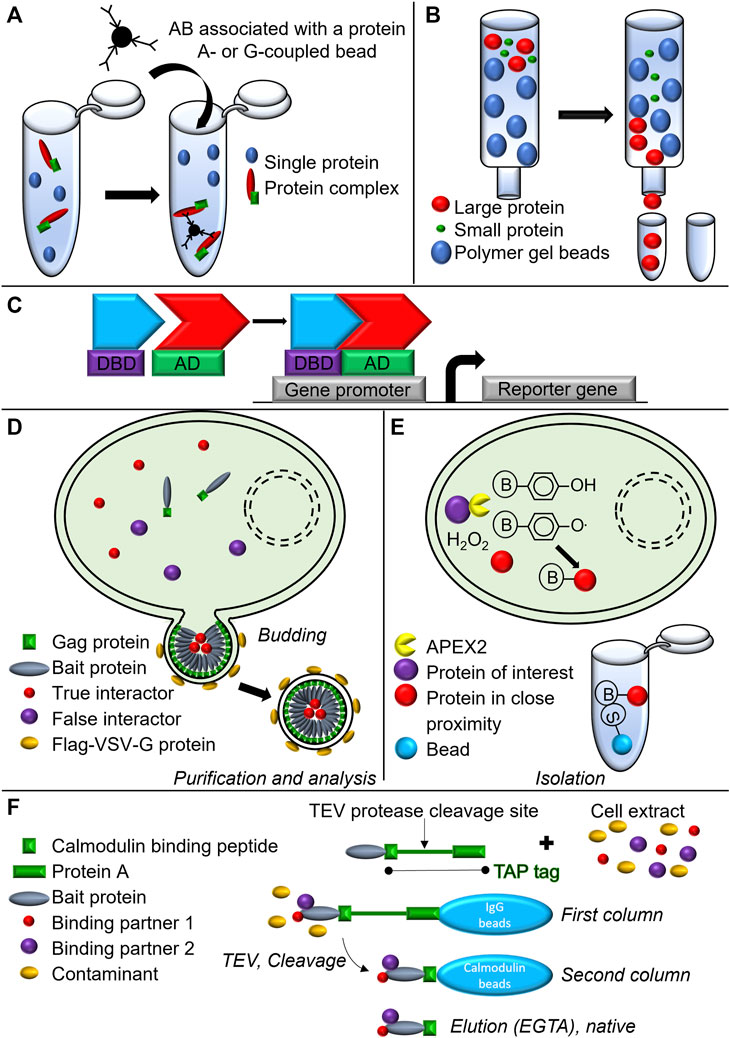
FIGURE 5. Schematic representation of biochemical and biotechnological approaches used to assess complex formation and composition. (A) Co-immunoprecipitation. (B) Gel filtration chromatography. (C) Yeast-two-hybrid assay. DBD, DNA binding domain; AD, Activating domain. (D) Virotrap. (E) APEX2-associated proteomics. S, Streptavidin; B, Biotin (F) TAP-TAG immunoprecipitation approach.
Another method to isolate protein complexes is the chromatographic gel filtration analysis (Lathe and Ruthven, 1955) (Figure 5B). By using this technique, combined with caspase-8 immunoprecipitation, Feoktistova et al. demonstrated in HaCaT cells that stably overexpressed cFLIPs (HaCaT-cFLIPs) and were stimulated with zVAD-fmk and an IAP antagonist, the formation of the large 2 MDa ripoptosome complex containing caspase-8, RIPK1, FADD, and cFLIPs (Figure 1C). Spontaneous cell death induced by cIAPs depletion in HaCaT-cFLIPs cells was shown to be RIPK3-dependent, although no RIPK3 was detected in the ripoptosome complex when performing immunoprecipitation (Feoktistova et al., 2011), suggesting the transient recruitment of the protein to the complex (Feoktistova et al., 2012).
Alternative methods to detect and even screen for PPIs use genetic systems, and the best-known is the yeast-two-hybrid assay (Gietz et al., 1997) (Figure 5C). A major example from the cell death field is the yeast-two-hybrid study that led to the discovery of RIPK1 as an interacting partner for Fas. A cDNA fragment comprising almost the entire cytoplasmic domain of human Fas was fused with the 3’ end of the coding region for the bacterial repressor LexA. The resulting gene fusion was further introduced in a reporter strain together with a transcriptional activator fusion protein library obtained from mRNA isolated from the Jurkat cell line. Following this approach, RIPK1 was discovered (Gietz et al., 1997). New biochemical approaches such as Virotrap (Figure 5D) and APEX2-associated proteomics (Figure 5E) have been introduced to the scientific community in order to get new insights into protein complex composition. While APEX2-associated proteomics was not used in the context of TNF signaling, Virotrap based approach was used in the context of TNF-induced NF-kB signal transduction pathway, identifying both, known and as well as novel interactor of TNF-induced inflammatory signaling (Van Quickelberghe et al., 2018). For a more detailed explanation of these techniques, we refer to the original research articles (Lam et al., 2015; Eyckerman et al., 2016; Titeca et al., 2017).
In addition to the latter, a modified TAP-TAG approach (Figure 5F) has been instrumental (as already mentioned in section 2.1) in revealing the presence of two previously unrecognized components: HOIL-1 and HOIP, which have been reported to form a linear Ub chain assembly complex (LUBAC). These findings highlighted how LUBAC stabilizes the TNFR signaling complex and is required for TNF-mediated gene induction process in the context of the survival complex (Haas et al., 2009).
Finally, another method, complementary to the classic biochemical approaches, which is very powerful in providing details about complex compositions is cross-linking mass spectrometry, which allows identification of proximal structural regions at amino acid level. It has been used for MLKL studies and revealed a role for MLKL PsKD (pseudokinase domain) in directing the transition of human MLKL from a basal monomeric state to a pro-necroptotic tetramer (Petrie et al., 2018). Furthermore, mass spectrometry combined with stoichiometry calculations has been used to generate an integrated model of the formation principles and architecture of the TNFR signaling complex (Ciuffa et al., 2022).
These methods and their combinations could allow in the future to further characterize necrosome composition with unrivaled precision.
Using the above-mentioned techniques made it possible to get insight into the composition and the structure of the complexes formed upon TNFR1 stimulation. One major limitation is that results come from the population average instead of individual cells and have a limited temporal resolution, providing only a snapshot without monitoring the entire process. Besides, sample preparation can become extremely labor-intensive and time-consuming just to get an insight on the spatial distribution of the complex formation. In addition, during sample preparation, native protein interactions can be lost and/or artificial ones can be generated.
At the present, an increasing number of studies make use of genetically encoded fluorescently labeled proteins that can be tracked/visualized in real-time while keeping the cellular context intact (Sun et al., 2012; Gong et al., 2017). The use of fluorescent proteins has provided additional information of spatiotemporal protein behavior and activity, complex formation and protein oligomerization, the dynamic events that have been assigned to multiple proteins participating in the TNF pathway. Some techniques also gain more attention as a non-invasive alternative to characterize the interacting partners within the complexes and their interacting domains. Additionally, with the rise of super-resolution microscopy, we are now able to monitor the protein (clustering) behavior and quantify the number of molecules per cluster at the single-molecule level (Große et al., 2016; Salvador-Gallego et al., 2016). In this section, we will describe several aspects of protein dynamics that are tackled by means of fluorescence microscopy.
The most obvious type of information provided by fluorescence microscopy is the protein localization and distribution. A renowned example from the necroptosis field for which localization and distribution were defined by fluorescence studies is MLKL. By creating a fusion between the N-terminal four-helix bundle domain of MLKL and the hormone-binding domain of estrogen receptor to induce dimer formation of the protein of interest, Chen et al., 2014 demonstrated that forced dimerization of MLKL N-terminal domain induced tetramers leading to necroptosis (Chen et al., 2014). Coupling the same fusion to enhanced green fluorescent protein (eGFP) demonstrated that in the non-stimulated condition the protein is uniformly distributed in the cytoplasm while it forms discrete dots in the vicinity of the plasma membrane in the necroptotic condition, providing a novel spatial distribution of MLKL during necroptosis (Chen et al., 2014). The recent study by Gong et al. confirmed the punctuate plasma-membrane-associated distribution of dimerizable N-terminal part (1—140aa) of hMLKL that was C-terminally coupled to the yellow fluorescent protein (YFP) variant Venus in MLKL-deficient MEF cells (Gong et al., 2017). Moreover, the same study showed that the ESCRT III component human charged multivesicular body protein 4B (hCHMP4B) coupled to the red fluorescent protein (RFP) variant mCherry rapidly translocated to the plasma membrane upon necroptosis induction and co-localized with MLKL in plasma membrane-associated puncta followed by damaged plasma membrane shedding. These experiments pointed out the involvement of the ESCRT III components in the delay of plasma membrane permeabilization and subsequent cell death following MLKL activation.
Another example from the necroptosis field whose activation results in puncta formation is RIPK3. Using human RIPK3 coupled at its N-terminus to mCherry in HeLa cells that do not express endogenous RIPK3, Sun et al. showed that in the unstimulated condition RIPK3 is uniformly distributed in the cytosol. Upon the necroptotic trigger, however, the protein forms discrete cytoplasmic puncta that are associated with necroptosis (Sun et al., 2012). The same study also showed that in the presence of the MLKL inhibitor necrosulfonamide (NSA), cell death was prevented but the RIPK3 puncta were still observed, although they failed to enlarge, suggesting that the compound prevented a certain step in necroptosis progression (Sun et al., 2012). Another study used murine RIPK3 coupled at its N-terminus to monomeric RFP (mRFP) in L929 cells. In contrast to the previous study (Sun et al., 2012), the punctuate behavior of RIPK3 was observed in less than 7% of the cells, including the dying ones. Expressing the same construct in L929 cells lacking MLKL expression caused massive RIPK3 puncta formation with formation of filamentous structures in most of the cells in the necroptotic condition (Chen et al., 2013). One of the possible reasons for the observed dissimilarity in RIPK3 behavior can be the use of different cellular models in these studies. It has been shown that various cell lines express different levels of RIPK3 suggesting that different cell lines may tolerate different RIPK3 levels, endogenous and exogenous, inducing a different response. Another possible explanation can be the fact that although necroptosis was inhibited in both studies due to the absence of MLKL activity, MLKL, though not active, was still present in HeLa cells while it was absent in the L929 cells. Upon NSA treatment, inactive MLKL could potentially still interact with RIPK3, preventing its massive oligomerization. This was not observed in MLKL-deficient L929 cells, where RIPK3 showed abnormal filament formation in the absence of MLKL suggesting that interaction between MLKL and RIPK3 regulates the extent of RIPK3 puncta formation (Sun et al., 2012; Chen et al., 2013). The studies exemplified here indicate that fluorescence imaging not only confirmed the biochemical studies showing that multimerization of RIPK3 and MLKL is associated with necroptosis but also went beyond, providing extra spatiotemporal information that is not easily achieved with conventional biochemical approaches.
MLKL and RIPK3 are not the only two proteins that by means of fluorescence live cell imaging have been shown to multimerize. Upon apoptosis induction, expression of a full-length caspase-8-eGFP fusion was shown to produce a diffuse distribution in the cytoplasm in transiently transfected HeLa cells treated with zVAD-fmk to block apoptosis. In contrast, a fusion of the DED-containing prodomain of caspase-8 showed a distinctive cytoplasmic filament network in the same condition (Siegel et al., 1998; Park et al., 2005). Upon apoptosis induction, the eGFP fusion of the full-length FADD that contains both DED and DD domain was also shown to form filaments similar to the DED domain of caspase-8 in transfected HeLa cells treated with zVAD-fmk. In contrast to the full-length protein, fusion of the DED domain of FADD with eGFP was also able to induce apoptosis but produced shorter, more numerous filaments. Overall, the fluorescence live cell imaging results indicated that proteins containing DED domain tend to form cytoplasmic filaments and suggested that assembly in molecular signaling platforms enhances the efficiency of caspase activation and is thus necessary for the initiation of the caspase cascade (Siegel et al., 1998). Another study described a possible explanation for the filament formation of caspase-8 and the possible length of caspase-8 chains that is potentially dependent on different parameters such concentration of procaspase-8 in the cell and stimulation strength. The latter appears to be one of the most crucial factors that influence the length of the chains. Weaker death receptor stimulation results in longer chains that potentially compensate for the low number of stimulated receptors. However, the length of the chains is limited to provide a safety control from spontaneous apoptosis as the unlimited size of the chains would lead to induction of cell death from only a little number of stimulated receptors (Schleich et al., 2012).
Filament formation is not a unique feature of DED domain-containing proteins but also other proteins such as caspase-2, a caspase activation and recruitment domain (CARD)-containing protein whose activity has been linked to TNF-induced apoptosis in endothelial cells (Espín et al., 2012). Using an eGFP fusion of caspase 2, it has been shown that the CARD domain is responsible for filamentous appearance of caspase-2 in NIH3T3 fibroblasts and COS cells. Moreover, the same fusion helped to determine the cellular localization of the protein that appeared to be nuclear at earlier time points where the fusion protein occurred as fiber- or dot-like structures. At later time points, however, caspase-2-eGFP fluorescence was more diffusely spread over the cytoplasm, suggesting the enzymatic processing of the protein releasing the active protein from its prodomain containing CARD (Colussi, Harvey and Kumar, 1998). In line with this, it has been previously shown by means of yeast-two-hybrid that dimerization of procaspase-2 is required for the autoproteolytic processing of the enzyme (Butt et al., 1998). This suggests that the formation of multiple procaspase-2 dimers within a larger multimer facilitates the autocatalytic processing of the protein and the propagation of the signaling cascade. Another study used the eGFP tagged caspase-2 prodomain (eGFP-PD) containing CARD and the Flag tagged CARD domain of RIP-associated ICH-1/CED-3 homologous protein with death domain (RAIDD) (RAIDD-CARD), an adaptor molecule containing both CARD and DD domain. This study showed that co-transfection of both domains caused a disruption of filamentous structures while dot-like structures containing both CARD domains appeared in the nucleus. As immunofluorescence analysis of RAIDD-CARD coupled to the Flag tag showed that RAIDD-CARD predominantly localized in the cytoplasm, the co-transfection experiment with the eGFP-PD indicated that it is caspase-2 prodomain that facilitates RAIDD-CARD localization to the nucleus (Shearwin-Whyatt, Harvey and Kumar, 2000). It is possible that through their interaction and translocation to the nucleus RAIDD and caspase-2 further form the PIDDosome complex by interacting with p53-induced protein with a death domain (PIDD) leading to apoptosis induced by genotoxic stress (Tinel and Tschopp, 2004).
Aside from inducing the survival or cell death response through the formation of complex I or complex II, TNF has been shown to generate a tempered and delayed inflammatory response in macrophages through inflammasome priming (Bezbradica, Coll and Schroder, 2017). One of the inflammasome components for which multimerization has been demonstrated by means of fluorescence imaging is the adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC). This protein contains both CARD and pyrin domain (PYD) and recruits pro-caspase-1 to the inflammasome complex through CARD-CARD interaction leading to caspase-1 activation. Using a fusion between ASC and eGFP (ASC-eGFP), the study showed the formation of large oligomers of 2 µm (only one per cell) in the cytoplasm of the THP-1 macrophages treated with Escherichia coli LPS. Since all cells containing these clusters exhibited biochemical and morphological characteristics of pyroptosis, another type of regulated cell death, these ASC oligomers were called pyroptosomes. By performing time-lapse confocal imaging it was revealed that the entire process of pyroptosome assembly takes less than 3 minutes (Fernandes-Alnemri et al., 2007). Incubation of cell lysates from THP-1-ASC-eGFP macrophages at 37°C in the presence of rhodamine-tagged zVAD-fmk to label caspase-1 revealed the co-localization of the ASC-eGFP signal with rhodamine indicating that ASC pyroptosomes contain activated caspase-1, suggesting their role in caspase-1 induction and showing their functional relevance (Fernandes-Alnemri et al., 2007).
Now, with respect to clustering, a convincing example is provided by the Bcl-2-associated X (Bax) protein and Bcl-2 homologous antagonist killer (Bak) protein whose pore-forming activity was used to illustrate the potential pore forming activity of MLKL (Dondelinger et al., 2014). Using cysteine linkage by disulphide bond formation and analytical ultracentrifugation, the molecular mechanisms involved in the oligomerization of Bax and Bak have been revealed (Dewson et al., 2008, 2009; Moldoveanu et al., 2013). However, the mechanisms of the mitochondrial membrane permeabilization remain unknown. By using super-resolution microscopy techniques (Figure 6A), single-molecule localization microscopy [SMLM, such as photo-activated localization microscopy (PALM) and direct stochastic optical reconstruction microscopy (dSTORM)], and stimulated emission depletion microscopy (STED), the researchers attempted to gain insight in these mechanisms.
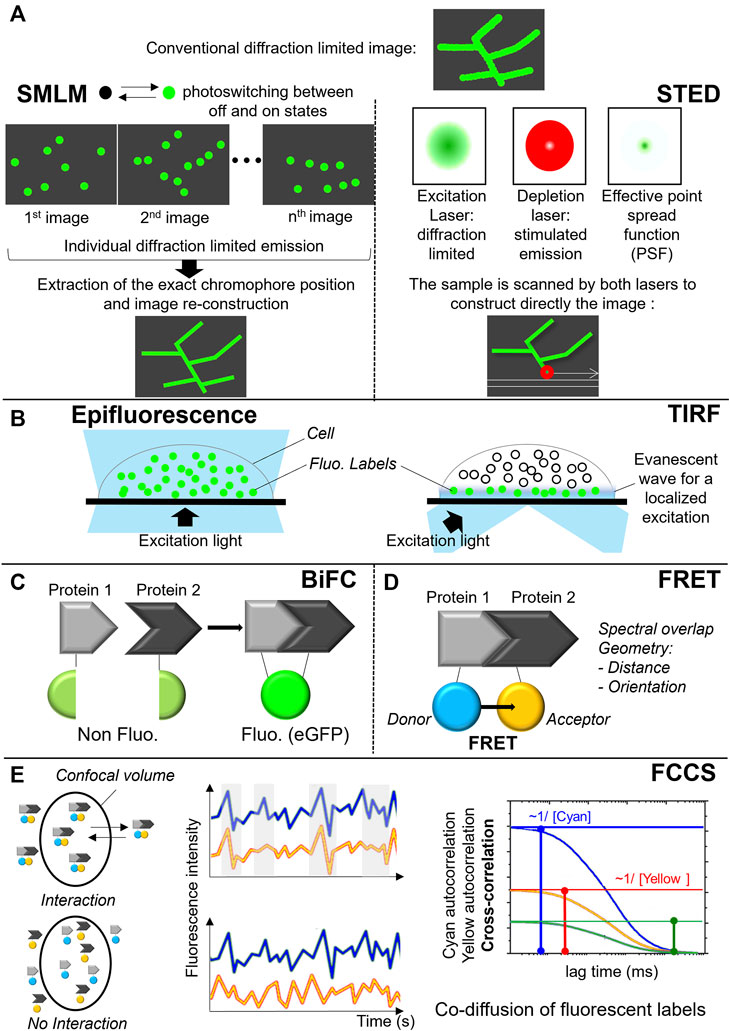
FIGURE 6. Fluorescent approaches to monitor protein localization and complex formation. (A) Super-resolution microscopy techniques: principle of single-molecule localization microscopy (PALM, dSTORM) on the left and stimulated emission depletion microscopy (STED) on the right. (B) Total Internal Reflection Fluorescence (TIRF). (C) Bimolecular fluorescence complementation assay (BiFC). (D) Förster resonance energy transfer (FRET). (E) Fluorescence cross-correlation spectroscopy (FCCS).
To produce a PALM or dSTORM images, only a small subset of the fluorescent labels is stochastically activated (i.e., switched on and off) during the measurement and their mean position are precisely calculated in each image. The same activation-deactivation process is repeated over thousands of times to ensure that all labels in a particular field are imaged. By superimposing the generated images containing the precise location of all labels, a super-resolved image is generated (Figure 6A) (Betzig et al., 2006; Hess, Girirajan and Mason, 2006). STED, on the other hand, uses a pair of synchronized laser pulses. The first pulse excites multiple fluorescent molecules, bringing the electrons from the ground state to an excited state. The excitation pulse is immediately followed by a highly energetic emission depletion pulse (STED pulse) that forces the excited electrons to relax into a higher vibrational level of the ground state, releasing a less energetic red shifted photon (stimulated emission). By arranging the STED pulse in a donut shape, only the excited fluorophores at the periphery of the excitation focus will be quenched by stimulated emission. In the center of the donut, where the intensity of the STED pulse is zero, the fluorescence remains unaffected and is detected (Figure 6A) (Hell and Wichmann, 1994; Klar and Hell, 1999).
By using PALM, the number of Bak molecules participating in mitochondrial clusters was estimated in MEF cells and varied from tens to several thousands. By defining the distribution of the Bak molecules within the mitochondrial membrane, it was suggested that Bak clusters do not form a pore but rather induce a mechanical tension in the membrane leading to its rupture (Nasu et al., 2016).
In the NOX field, the arrangement of NOX2 in nanoclusters was shown using dSTORM combined with selective illumination of the cell membrane by total internal reflection fluorescence (TIRF) microscopy (Figure 6B). The number of these clusters increased during frustrated phagocytosis on IgG-coated coverslips after 10 min. This result indicated that NOX was delivered at the phagosomal membrane. It has also been shown that this NOX2 delivery required p47phox (Joly et al., 2020).
Two other studies using STED defined the assembly of Bax clusters in HeLa and U2OS cells that appeared to be ring and arc-shaped. In addition, the data suggested that both rings and arcs of Bax oligomers are able to perforate membranes, thus leading to mitochondrial outer membrane permeabilization (Große et al., 2016; Salvador-Gallego et al., 2016). These studies demonstrate the strength of the novel microscopy methodologies to define the protein localization and distribution at the single-molecule level (Hedde and Nienhaus, 2010). Indeed, as any other microscopy methods, super-resolution microscopy has also its drawbacks. Jacquemet et al. provided useful guidelines to super-resolution imaging techniques covering their advantages and their limitations as well (Jacquemet et al., 2020).
Overall, the examples provided above emphasize the significant contribution of fluorescence microscopy approaches to determine the spatiotemporal behavior and distribution of the fluorescently tagged proteins involved in cell death. Combination of fluorescence microscopy with the biochemical analysis showed that the formation of higher order molecular structures is common for cell death proteins and is necessary for further signal propagation resulting in cell death, thus providing strong evidence for the biological relevance of these molecular signaling platforms.
Another important application of fluorescence microscopy is studying protein-protein interactions within signaling complexes and protein clusters. Three techniques can be used to monitor protein-protein interactions are bimolecular fluorescence complementation assay (BiFC) (Figure 6C), Förster resonance energy transfer (FRET) (Figure 6D) or fluorescence cross-correlation spectroscopy (FCCS) (Figure 6E). In the case of BiFC, the interacting proteins of interest are coupled to complementary parts of a fluorescent protein such as eGFP. Interaction of the proteins within live cells brings both parts of the fluorescent protein together allowing this reporter protein to adopt its native three-dimensional structure and emit a fluorescent signal (Figure 6C) (Hu, Chinenov and Kerppola, 2002). FRET microscopy, in contrast to BiFC, uses two fluorophores, a donor and a acceptor, whose respective emission and absorption spectra sufficiently overlap and forming a FRET pair. The excited donor fluorophore can transfer its energy to the acceptor fluorophore nearby through non-radiative dipole-dipole coupling. Besides the spectral overlap of the donor and the acceptor, the efficiency of the energy transfer depends on geometrical parameters: the relative orientation of the fluorophores and their distance that need to be below 10 nm. Depending on the FRET pair, an efficiency of 50% is usually observed for distances around 5 nm, which is fully consistent with the expected distance between FPs fused to the interacting protein of interest. FRET is able to probe PPIs but also to access to geometrical parameters related to the interaction (Figure 6D) (Padilla-Parra and Tramier, 2012).
In FCCS, the fluctuations of the fluorescence intensities of FP-tagged proteins diffusing in and out a confocal volume are analyzed. If the fluctuations are correlated, the proteins of interest are interacting (Figure 6E). A cross-correlation function, which has an amplitude with a non-zero value proves the co-diffusion of proteins of interest. From these amplitudes, the concentrations of both partners, being the fraction of proteins in interactions, can be calculated. This strategy can be used as quantitative approach for estimation of Kd in living cells (Hwang and Wohland, 2007).By using the BiFC assay, the importance of the CARD domain of caspase-1 in pyroptosome assembly has been pointed out (Proell et al., 2013). In this study, the N- and C-terminal part of Venus were coupled to the CARD domain of caspase-1 and the CARD domain of ASC, respectively. Co-expression of both domains in HEK293T cells resulted in the formation of foci that resembled the pyroptosomes described by Fernandes-Almenri et al., 2007 (Fernandes-Alnemri et al., 2007). This indicated an active role of the caspase-1 CARD in productive platform formation. Additionally, it was indicated that the CARD domain plays an important role in self-assembly of ASC. The N- and C-terminal part of Venus were coupled to either the full-length protein or to its separate domains (PYD or CARD). Expression of the full-length protein coupled to either the C- or N-terminal part of Venus led to the formation of large foci in the HEK293T cells. This was not observed when only the PYD domains or CARD domains coupled to either the C- or N-terminal part of Venus were introduced in the same cells. Their expression resulted in restoration of the fluorescent signal but remained diffused without forming larger foci, indicating that these domains are not sufficient to induce pyroptosome formation (Proell et al., 2013). The inability of the separate ASC domains to oligomerize observed by BiFC was, however, not supported by two other independent studies using fluorescently tagged PYD or CARD domain of ACS. These studies showed that the domains, when expressed separately in COS-7 or HEK293T cells, form filament-like aggregates similarly to what has been shown for caspase-2 and RAIDD (Masumoto, Taniguchi and Sagara, 2001; Sahillioglu et al., 2014). The observed discrepancy between the studies can be ascribed to the different methodologies used in the above-mentioned studies. However as always, differences 1) in abundance of tagged proteins of interest, 2) in cellular models, and 3) in triggers used do have an impact on the results. Finally, one should note that BiFC works in a non-reversible manner.
Another study using FRET measured via flow cytometry in HEK293T cells showed that the RHDLL sequence within the FADD DED domain is important for the protein self-assembly. This sequence is homologous to an RXDLL motif conserved among many DED-containing proteins (Muppidi et al., 2006). A recent study re-examining FADD self-assembly confirmed previous findings by showing interactions between the DED domains of FADD by quantitative sensitized emission FRET measurements in HeLa cells expressing FADD or its truncated versions coupled at their N-terminus to either CFP or YFP FRET pair (Wang et al., 2013). In addition, by measuring FRET between the CFP and YFP tagged DED containing a mutation in Phe25, the in vitro studies suggesting the dependency of FADD self-association on this residue were confirmed (Carrington et al., 2006; Wang et al., 2013). Moreover, the same study showed that the expression of CFP- and YFP-tagged N-terminal part of FADD that does not contain phosphorylatable C-terminal tail produced the highest FRET measured in the study. This indicates that the C-terminal tail of FADD that gets phosphorylated on Ser194 can greatly regulate the strength of FADD self-association (Wang et al., 2013).
The cytosolic complex of NOX2 composed of p40phox, p47phox and p67phox was recently extensively monitored in live cells by FRET measured via the variations of the donor fluorescence lifetime (Fluorescence Lifetime Imaging or FLIM) and FCCS (Ziegler et al., 2019). The subunits were tagged at their N and C termini with various combinations of cyan, yellow and red fluorescent proteins (FP). Their pairwise interactions were explored. For all combination of labelling with FPs at the C- or N- termini, FRET was observed at various efficiencies except for the NN-termini labelling of p47phox and p67phox, which showed no FRET. Nevertheless, p47phox and p67phox labeled at their C- or N- termini co-diffused as shown by FCCS proving their interaction whatever the FPs position. This is an example of the complementarity of FRET-FLIM and FCCS approaches. Indeed, the FRET efficiency strongly depends on the geometry of the interaction that influences the distance between the FPs whereas the FCCS only rely on their co-diffusion. In the p47phox and p67phox interactions, the N- termini are too far to observe FRET giving indirectly a topological information on their relative 3D organization. From those experiments the authors deduced not only specific interactions between p47phox and p67phox and p40phox and p67phox but also a 1:1:1 stoichiometry in the NOX2 complex in live cells. From the FCCS experiments they also deduced that nearly 100% of p47phox and p67phox were present in complexes in living cells and that the apparent Kd of this interaction was in a range of few hundred nM. Furthermore, combining the topological information obtained by FRET data in live cells with small-angle X-ray scattering (SAXS) models and published partial crystal structures of isolated subunits in vitro, the authors built a 3D model of the cytosolic complex valid and consistent with the current knowledge of the activation mechanism. In particular, the study revealed an elongated shape of the ternary complex and a flexible hinge between the two SH3 domains of p47phox and the rest of the complex. The flexible hinge is probably required to establish the initial contact with phospholipids and p22phox at the membrane and then to bend the complex in order to bring p67phox closer to NOX2 (Ziegler et al., 2019). This study also provides new insights on the relative arrangement of the functional domains and on their interaction interfaces, which could support the design of innovative therapeutic agents aiming at the disruption of the complex.
The examples summarized in this section provide a proof of applicability of fluorescence microscopy in monitoring protein-protein interaction within various molecular complexes that have been demonstrated to play a key role in various regulated cell death modalities but also in inflammation.
The recent advancement in microscopic techniques makes it possible to not only define the spatiotemporal characteristics of the protein of interest but also investigate the protein complex composition event from a structural point of view in the living cells in real time. Over the last 15 years, these techniques have become complementary to biochemistry techniques, which are also constantly evolving thanks to the progress of analytical methods such as mass spectrometry. In parallel, during the two last years, since the success of AlphaFold2 (Jumper et al., 2021) at the CASP141 (Critical Assessment of Techniques for Protein Structure Prediction) competition in 2020, and the publication of RoseTTAFold in 2022 (Baek et al., 2021), the panel of efficient modelling strategies to explore protein structures and their interactions rapidly expand. These new tools based on deep learning methods predict the structure of a single chain protein from its primary sequence with very high accuracy. Most of them are accessible to the whole community through simplified interfaces designed for the end user and by making the code open-access. Continued efforts are now focused on the accurate prediction of protein complexes (Evans et al., 2021; Bryant et al., 2022; Bryant, Pozzati and Elofsson, 2022; Yu et al., 2023) and tremendous progress in modeling those complexes have been shown at the CASP152 competition in autumn 2022. However, prediction of asymmetric complexes composed of protein units containing intrinsically disordered regions or existing in several states (resting, activated at different levels) as NADPH oxidases or TNF complex remains a challenge, at least for now.
While considering opting for fluorescence microscopy to study dynamic events of the proteins of interest, several aspects should be kept in mind. First of all, although fluorescent proteins are generally innocuous tags, they can potentially affect target protein structure, function, or localization. To avoid these issues both biochemical and immuno-labelling approaches to analyze the endogenous protein behavior should be applied to confirm the results obtained by fluorescence microscopy techniques. Secondly, most of the described examples have been performed upon overexpression that may result in a non-physiological outcome thus leading to a biased interpretation. A possible solution would be to ensure the expression of the fluorescently tagged proteins at levels close to endogenous. A well-known strategy is using lentiviral vectors that allow a sustained protein expression through stable integration of the transgene in the host genome. Nowadays, genome editing tools such as clustered regulated interspaced short palindromic repeats - associated protein-9 nuclease (CRISPR-Cas9) are gaining attention in terms of fluorescent labelling of endogenous proteins allowing the fluorescent protein expression at endogenous levels without further perturbation of the intracellular environment (Bukhari and Müller, 2019; Zhong et al., 2021). Nevertheless, an expected drawback could concern the monitoring of the fluorescent signal in condition of low signal to noise ratio, because of the low expression level of the proteins of interest. Continued improvements of fluorescent proteins in term of brightness, maturation rate, photostability and monomeric properties (Fabritius et al., 2018; Campbell et al., 2020; Legault et al., 2022) will improve the detection of protein of interest expressed at endogenous level.
Alternative strategy to visualize endogenous proteins without interfering their native state is using FP-tagged nanobodies. Nanobodies were introduced as a completely new class of antibodies missing the light chain, and with a molecular weight of ∼13 kDa, they are the smallest intact antigen binding fragments. A nanobody against an endogenous protein, genetically coupled to an FP and introduced into living cells as DNA-encoded expression constructs provide a new class of imaging tools (Rothbauer et al., 2006; Wagner and Rothbauer, 2021).
In the future combination of genome editing technologies together with super-resolution microscopy approaches (Nasu et al., 2016; Salvador-Gallego et al., 2016) will further enable researchers to spy on protein behavior at the single-molecule level. A very often used method, correlative light-electron microscopy (CLEM) combines the advantage of fluorescent and electron microscopy and enables the analysis of targeted fluorescent probe at the ultrastructural level (Grabenbauer, 2012). Nevertheless its utility, CLEM still requires fixation of the sample, and the quality of the CLEM image relies on the ability of the sample to maintain native organization during fixation and subsequent labelling. Thus, CLEM or any other electron microscopy technique does not allow live-cell imaging (yet). Presumably, this will remain the major challenge for future–to be able to investigate and image the protein complexes in their native environment at the single-molecule level in various cellular compartments. Advent of new technologies combined with existing super-resolution fluorescence microscopy methods (Sochacki et al., 2014) may soon help to achieve this goal and push the frontiers of our knowledge of protein complexes and many biological processes, including cell death.
FR, ME, and HV conceptualized, wrote and edited the manuscript. ML participated on the manuscript writing. ML, HV, and ME designed figures. PV read and edited the manuscript.
HV was supported by a PhD fellowship from MESRI. Research in the Vandenabeele unit is supported by the FWO (research grants G.0C76.18N, G.0B71.18N, G.0B96.20N, G.0A93.22N, EOS MODEL-IDI Grant (30826052), and EOS CD-INFLADIS (40007512)), grants from the Special Research Fund UGent (Methusalem grant BOF16/MET_V/007, BOF22/MET_V/007, iBOF ATLANTIS grant 20/IBF/039), grants from the Foundation against Cancer (F/2016/865, F/2020/1505), CRIG and GGIG consortia, and VIB. We thank the VIB for training, support, and availability of core facilities.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The handling editor BM declared a past co-authorship with the author FR.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1https://www.predictioncenter.org/casp14/
2https://www.predictioncenter.org/casp15/
Abuaita, B. H., Schultz, T. L., and O’Riordan, M. X. (2018). Mitochondria-derived vesicles deliver antimicrobial reactive oxygen species to control phagosome-localized Staphylococcus aureus. Cell Host Microbe 24 (5), 625–636. doi:10.1016/j.chom.2018.10.005
Amaya, M., Keck, F., Bailey, C., and Narayanan, A. (2014). The role of the IKK complex in viral infections. Pathogens Dis. 72 (1), 32–44. doi:10.1111/2049-632X.12210
Andera, L. (2009). Signaling activated BY the death receptors OF the TNFR family. Biomed. Pap. 153 (3), 173–180. doi:10.5507/bp.2009.029
Babu, D., Leclercq, G., Goossens, V., Vanden Berghe, T., Van Hamme, E., Vandenabeele, P., et al. (2015). Mitochondria and NADPH oxidases are the major sources of TNF-α/cycloheximide-induced oxidative stress in murine intestinal epithelial MODE-K cells. Cell. Signal. 27 (6), 1141–1158. doi:10.1016/j.cellsig.2015.02.019
Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., et al. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science 373 (6557), 871–876. doi:10.1126/science.abj8754
Betzig, E., Patterson, G. H., Sougrat, R., Lindwasser, O. W., Olenych, S., Bonifacino, J. S., et al. (2006). Imaging intracellular fluorescent proteins at Nanometer resolution. Science 313 (5793), 1642–1645. doi:10.1126/science.1127344
Bedard, K., and Krause, K.-H. (2007). The NOX family of ROS-generating NADPH oxidases: Physiology and Pathophysiology. Physiol. Rev. 87 (1), 245–313. doi:10.1152/physrev.00044.2005
Bedoui, S., Herold, M. J., and Strasser, A. (2020). Emerging connectivity of programmed cell death pathways and its physiological implications. Nat. Rev. Mol. cell Biol. 21 (11), 678–695. doi:10.1038/s41580-020-0270-8
Begum, R., et al. (2022). NADPH oxidase family proteins: Signaling dynamics to disease management. Cell. Mol. Immunol. 19 (6), 660–686. doi:10.1016/s0021-9258(17)31534-x
Vanden Berghe, T., Vanlangenakker, N., Parthoens, E., Deckers, W., Devos, M., Festjens, N., et al. (2010). Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 17 (6), 922–930. doi:10.1038/cdd.2009.184
Bertheloot, D., Latz, E., and Franklin, B. S. (2021). Necroptosis, pyroptosis and apoptosis: An intricate game of cell death. Cell. Mol. Immunol. 18 (5), 1106–1121. doi:10.1038/s41423-020-00630-3
Bertrand, M. J., Milutinovic, S., Dickson, K. M., Ho, W. C., Boudreault, A., and Durkin, J. (2008). cIAP1 and cIAP2 Facilitate Cancer Cell Survival by Functioning as E3 Ligases that Promote RIP1 Ubiquitination. Molecular Cell 30 (6), 689–700. doi:10.1016/j.molcel.2008.05.014
Besse, A., Lamothe, B., Campos, A. D., Webster, W. K., Maddineni, U., Lin, S. C., et al. (2007). TAK1-dependent signaling requires functional interaction with TAB2/TAB3. J. Biol. Chem. 282 (6), 3918–3928. doi:10.1074/jbc.M608867200
Bezbradica, J. S., Coll, R. C., and Schroder, K. (2017). Sterile signals generate weaker and delayed macrophage NLRP3 inflammasome responses relative to microbial signals. Cell. Mol. Immunol. 14 (1), 118–126. doi:10.1038/cmi.2016.11
Bienert, G. P., Moller, A. L. B., Kristiansen, K. A., Schulz, A., Moller, I. M., Schjoerring, J. K., et al. (2007). Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 282 (2), 1183–1192. doi:10.1074/jbc.M603761200
Blaser, H., et al. (2016). TNF and ROS crosstalk in inflammation. Trends Cell Biol., 249–261. doi:10.1016/j.tcb.2015.12.002
Bryant, P., Pozzati, G., Zhu, W., Shenoy, A., Kundrotas, P., and Elofsson, A. (2022). Predicting the structure of large protein complexes using AlphaFold and Monte Carlo tree search. Nat. Commun. 13 (1), 6028. doi:10.1038/s41467-022-33729-4
Bryant, P., Pozzati, G., and Elofsson, A. (2022). Improved prediction of protein-protein interactions using AlphaFold2. Nat. Commun. 13 (1), 1265. doi:10.1038/s41467-022-28865-w
Bukhari, H., and Müller, T. (2019). Endogenous fluorescence tagging by CRISPR. Trends Cell Biol. 29 (11), 912–928. doi:10.1016/j.tcb.2019.08.004
Bulua, A. C., Simon, A., Maddipati, R., Pelletier, M., Park, H., Kim, K. Y., et al. (2011). Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208 (3), 519–533. doi:10.1084/jem.20102049
Burmester, G. R., Feist, E., and Dörner, T. (2014). Emerging cell and cytokine targets in rheumatoid arthritis. Nat. Rev. Rheumatol. 10 (2), 77–88. doi:10.1038/nrrheum.2013.168
Butt, A. J., Harvey, N. L., Parasivam, G., and Kumar, S. (1998). Dimerization and Autoprocessing of the Nedd2 (Caspase-2) precursor requires both the prodomain and the Carboxyl-terminal regions. J. Biol. Chem. 273 (12), 6763–6768. doi:10.1074/jbc.273.12.6763
Colussi, P. A., Harvey, N. L., and Kumar, S. (1998). Prodomain-dependent nuclear localization of the caspase-2 (Nedd2) precursor. A novel function for a caspase prodomain. J. Biol. Chem. 273 (38), 24535–24542. doi:10.1074/jbc.273.38.24535
Campbell, B. C., Nabel, E. M., Murdock, M. H., Lao-Peregrin, C., Tsoulfas, P., Blackmore, M. G., et al. (2020). mGreenLantern: a bright monomeric fluorescent protein with rapid expression and cell filling properties for neuronal imaging. Proc. Natl. Acad. Sci. 117 (48), 30710–30721. doi:10.1073/pnas.2000942117
Carrington, P. E., Sandu, C., Wei, Y., Hill, J. M., Morisawa, G., Huang, T., et al. (2006). The structure of FADD and its mode of interaction with procaspase-8. Mol. Cell 22 (5), 599–610. doi:10.1016/j.molcel.2006.04.018
Chen, G., and Goeddel, D. V. (2002). TNF-R1 signaling: A Beautiful pathway. Science 296 (5573), 1634–1635. doi:10.1126/science.1071924
Chen, W., Zhou, Z., Li, L., Zhong, C. Q., Zheng, X., Wu, X., et al. (2013). Diverse sequence Determinants control human and mouse receptor interacting protein 3 (RIP3) and mixed lineage kinase domain-like (MLKL) interaction in necroptotic signaling. J. Biol. Chem. 288 (23), 16247–16261. doi:10.1074/jbc.M112.435545
Chen, X., Li, W., Ren, J., Huang, D., He, W. T., Song, Y., et al. (2014). Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res. 24 (1), 105–121. doi:10.1038/cr.2013.171
Chhibber-Goel, J., Coleman-Vaughan, C., Agrawal, V., Sawhney, N., Hickey, E., Powell, J. C., et al. (2016). γ-Secretase activity is required for regulated intramembrane proteolysis of tumor necrosis factor (TNF) receptor 1 and TNF-mediated pro-apoptotic signaling. J. Biol. Chem. 291 (11), 5971–5985. doi:10.1074/jbc.M115.679076
Cho, Y., Challa, S., Moquin, D., Genga, R., Ray, T. D., Guildford, M., et al. (2009). Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and Virus-induced inflammation. Cell 137 (6), 1112–1123. doi:10.1016/j.cell.2009.05.037
Ciuffa, R., Uliana, F., Mannion, J., Mehnert, M., Tenev, T., Marulli, C., et al. (2022). Novel biochemical, structural, and systems insights into inflammatory signaling revealed by contextual interaction proteomics. Proc. Natl. Acad. Sci. 119 (40), e2117175119. doi:10.1073/pnas.2117175119
Dang, P. M.-C., Stensballe, A., Boussetta, T., Raad, H., Dewas, C., Kroviarski, Y., et al. (2006). A specific p47phox -serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. investigation 116 (7), 2033–2043. doi:10.1172/JCI27544
Degterev, A., Huang, Z., Boyce, M., Li, Y., Jagtap, P., Mizushima, N., et al. (2005). Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1 (2), 112–119. doi:10.1038/nchembio711
Delanghe, T., Dondelinger, Y., and Bertrand, M. J. M. (2020). RIPK1 Kinase-Dependent Death: A Symphony of Phosphorylation Events. Trends in Cell Biology 30 (3), 189–200. doi:10.1016/j.tcb.2019.12.009
Delanghe, T., Huyghe, J., Lee, S., Priem, D., Van Coillie, S., Gilbert, B., et al. (2021a). Antioxidant and food additive BHA prevents TNF cytotoxicity by acting as a direct RIPK1 inhibitor. Cell Death Dis. 12 (7), 699. doi:10.1038/s41419-021-03994-0
Dewson, G., Kratina, T., Czabotar, P., Day, C. L., Adams, J. M., and Kluck, R. M. (2009). Bak activation for apoptosis involves oligomerization of dimers via their alpha6 helices. Mol. Cell 36 (4), 696–703. doi:10.1016/j.molcel.2009.11.008
Dewson, G., Kratina, T., Sim, H. W., Puthalakath, H., Adams, J. M., Colman, P. M., et al. (2008). To trigger apoptosis, Bak Exposes its BH3 domain and Homodimerizes via BH3:Groove interactions. Mol. Cell 30 (3), 369–380. doi:10.1016/j.molcel.2008.04.005
Dickinson, B. C., and Chang, C. J. (2011). Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 7 (8), 504–511. doi:10.1038/nchembio.607
Dondelinger, Y., Aguileta, M. A., Goossens, V., Dubuisson, C., GrootjanS, S., Dejardin, E., et al. (2013). RIPK3 contributes to TNFR1-mediated RIPK1 kinase-dependent apoptosis in conditions of cIAP1/2 depletion or TAK1 kinase inhibition. Cell Death Differ. 20 (10), 1381–1392. doi:10.1038/cdd.2013.94
Dondelinger, Y., Declercq, W., Montessuit, S., Roelandt, R., Goncalves, A., Bruggeman, I., et al. (2014). MLKL Compromises plasma membrane Integrity by binding to phosphatidylinositol phosphates. Cell Rep. 7 (4), 971–981. doi:10.1016/j.celrep.2014.04.026
Dondelinger, Y., Delanghe, T., Rojas-Rivera, D., Priem, D., Delvaeye, T., Bruggeman, I., et al. (2017). MK2 phosphorylation of RIPK1 regulates TNF-mediated cell death. Nat. Cell Biol. 19 (10), 1237–1247. doi:10.1038/ncb3608
Dondelinger, Y., Jouan-Lanhouet, S., Divert, T., Theatre, E., Bertin, J., Gough, P. J., et al. (2015). NF-κB-Independent role of IKKα/IKKβ in preventing RIPK1 kinase-dependent apoptotic and necroptotic cell death during TNF signaling. Mol. Cell 60 (1), 63–76. doi:10.1016/j.molcel.2015.07.032
Dondelinger, Y., Delanghe, T., Priem, D., Wynosky-Dolfi, M. A., Sorobetea, D., Rojas-Rivera, D., et al. (2019). Serine 25 phosphorylation inhibits RIPK1 kinase-dependent cell death in models of infection and inflammation. Nature Communications 10 (1), 1729. doi:10.1038/s41467-019-09690-0
Dupré-Crochet, S., Erard, M., and Nüβe, O. (2013). ROS production in phagocytes: Why, when, and where? J. Leukoc. Biol. 94, 657–670. doi:10.1189/jlb.1012544
Dynek, J. N., Goncharov, T., Dueber, E. C., Fedorova, A. V., Izrael-Tomasevic, A., Phu, L., et al. (2010). c-IAP1 and UbcH5 promote K11-linked polyubiquitination of RIP1 in TNF signalling. EMBO J. 29 (24), 4198–4209. doi:10.1038/emboj.2010.300
Ea, C.-K., Deng, L., Xia, Z. P., Pineda, G., and Chen, Z. J. (2006). Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol. Cell 22 (2), 245–257. doi:10.1016/j.molcel.2006.03.026
El Benna, J., Faust, L. P., and Babior, B. M. (1994). The phosphorylation of the respiratory burst oxidase component p47phox during neutrophil activation. Phosphorylation of sites recognized by protein kinase C and by proline-directed kinases. J. Biol. Chem. 269 (38), 23431–23436.
El-Benna, J., et al. (2009). p47phox, the phagocyte NADPH oxidase/NOX2 organizer: Structure, phosphorylation and implication in diseases. Exp. Mol. Med. 41. doi:10.3858/emm.2009.41.4.058
Elbim, C., Chollet-Martin, S., Bailly, S., Hakim, J., and Gougerot-Pocidalo, M. A. (1993). Priming of polymorphonuclear neutrophils by tumor necrosis factor alpha in whole blood: Identification of two polymorphonuclear neutrophil subpopulations in response to formyl-peptides. Blood 82 (2), 633–640. doi:10.1182/blood.V82.2.633.633
Espín, R., et al. (2012). TNF receptors regulate vascular homeostasis in zebrafish through a caspase-8, caspase-2 and P53 apoptotic program that bypasses caspase-3’. Dis. Models Mech. doi:10.1242/dmm.010249
Evans, R., et al. (2021). Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. doi:10.1101/2021.10.04.463034
Eyckerman, S., Titeca, K., Van Quickelberghe, E., Cloots, E., Verhee, A., Samyn, N., et al. (2016). Trapping mammalian protein complexes in viral particles. Nat. Commun. 7 (1), 11416. doi:10.1038/ncomms11416
Fabritius, A., Ng, D., Kist, A. M., Erdogan, M., Portugues, R., and Griesbeck, O. (2018). Imaging-based screening platform Assists protein Engineering. Cell Chem. Biol. 25 (12), 1554–1561. doi:10.1016/j.chembiol.2018.08.008
Faure, M. C., Sulpice, J. C., Delattre, M., Lavielle, M., Prigent, M., Cuif, M. H., et al. (2013). The recruitment of p47phox and Rac2G12V at the phagosome is transient and phosphatidylserine dependent. Biol. Cell 105 (11), 501–518. doi:10.1111/boc.201300010
Feng, S., Yang, Y., Mei, Y., Ma, L., Zhu, D. e., Hoti, N., et al. (2007). Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell. Signal. 19 (10), 2056–2067. doi:10.1016/j.cellsig.2007.05.016
Feoktistova, M., Geserick, P., Kellert, B., Dimitrova, D. P., Langlais, C., Hupe, M., et al. (2011). cIAPs block ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex Differentially regulated by cFLIP isoforms. Mol. Cell 43 (3), 449–463. doi:10.1016/j.molcel.2011.06.011
Feoktistova, M., Geserick, P., Panayotova-Dimitrova, D., and Leverkus, M. (2012). Pick your poison: The Ripoptosome, a cell death platform regulating apoptosis and necroptosis. Cell Cycle 11 (3), 460–467. doi:10.4161/cc.11.3.19060
Fernandes-Alnemri, T., Wu, J., Yu, J. W., Datta, P., Miller, B., JankoWski, W., et al. (2007). The pyroptosome: A supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ. 14 (9), 1590–1604. doi:10.1038/sj.cdd.4402194
Fukai, T., and Ushio-Fukai, M. (2020). Cross-talk between NADPH oxidase and mitochondria: Role in ROS signaling and angiogenesis. Cells 9 (8), 1849. doi:10.3390/cells9081849
Galluzzi, L., Maiuri, M. C., Vitale, I., ZiscHka, H., Castedo, M., ZitvogeL, L., et al. (2007). Cell death modalities: Classification and pathophysiological implications. Cell Death Differ. 14 (7), 1237–1243. doi:10.1038/sj.cdd.4402148
Gerlach, B., Cordier, S. M., Schmukle, A. C., Emmerich, C. H., Rieser, E., Haas, T. L., et al. (2011). Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471 (7340), 591–596. doi:10.1038/nature09816
Geng, J., Ito, Y., Shi, L., Amin, P., Chu, J., Ouchida, A. T., et al. (2017). Regulation of RIPK1 activation by TAK1-mediated phosphorylation dictates apoptosis and necroptosis. Nature Communications 8 (1), 359. doi:10.1038/s41467-017-00406-w
Gietz, R. D., Triggs-Raine, B., Robbins, A., Graham, K. C., and Woods, R. A. (1997). Identification of proteins that interact with a protein of interest: Applications of the yeast two-hybrid system. Mol. Cell. Biochem. 172 (1–2), 67–79. doi:10.1023/A:1006859319926
Goldin, A., Beckman, J. A., Schmidt, A. M., and Creager, M. A. (2006). Advanced glycation end products: Sparking the development of diabetic vascular injury. Circulation 114 (6), 597–605. doi:10.1161/CIRCULATIONAHA.106.621854
Gong, Y.-N., Guy, C., Olauson, H., Becker, J. U., Yang, M., Fitzgerald, P., et al. (2017). ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169 (2), 286–300. doi:10.1016/j.cell.2017.03.020
Grabenbauer, M. (2012). Correlative light and electron microscopy of GFP. Methods Cell Biol. 111, 117–138. doi:10.1016/B978-0-12-416026-2.00007-8
Große, L., Wurm, C. A., Bruser, C., Neumann, D., Jans, D. C., and Jakobs, S. (2016). Bax assembles into large ring-like structures remodeling the mitochondrial outer membrane in apoptosis. EMBO J. 35 (4), 402–413. doi:10.15252/embj.201592789
Gudipaty, S. A., Conner, C. M., Rosenblatt, J., and Montell, D. J. (2018). Unconventional ways to live and Die: Cell death and survival in development, homeostasis, and disease. Annu. Rev. cell Dev. Biol. 34, 311–332. doi:10.1146/annurev-cellbio-100616-060748
Haas, T. L., Emmerich, C. H., Gerlach, B., Schmukle, A. C., Cordier, S. M., Rieser, E., et al. (2009). Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 36 (5), 831–844. doi:10.1016/j.molcel.2009.10.013
Hamanaka, R. B., and Chandel, N. S. (2010). Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem. Sci. 35 (9), 505–513. doi:10.1016/j.tibs.2010.04.002
He, S., Wang, L., Miao, L., Wang, T., Du, F., Zhao, L., et al. (2009). Receptor interacting protein kinase-3 determines cellular necrotic response to TNF-alpha. Cell 137 (6), 1100–1111. doi:10.1016/j.cell.2009.05.021
Hedde, P. N., and Nienhaus, G. U. (2010). Optical imaging of nanoscale cellular structures. Biophys. Rev. 2 (4), 147–158. doi:10.1007/s12551-010-0037-0
Hell, S. W., and Wichmann, J. (1994). Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 19 (11), 780–782. doi:10.1364/OL.19.000780
Herold, C., Rennekampff, H. O., and Engeli, S. (2013). Apoptotic pathways in adipose tissue. Apoptosis 18 (8), 911–916. doi:10.1007/s10495-013-0848-0
Hess, S. T., Girirajan, T. P. K., and Mason, M. D. (2006). Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophysical J. 91 (11), 4258–4272. doi:10.1529/biophysj.106.091116
Hildebrand, J. M., Tanzer, M. C., Lucet, I. S., Young, S. N., Spall, S. K., Sharma, P., et al. (2014). Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. 111 (42), 15072–15077. doi:10.1073/pnas.1408987111
Holler, N., ZaRu, R., Micheau, O., ThoMe, M., Attinger, A., Valitutti, S., et al. (2000). Fas triggers an alternative, caspase-8–independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 1 (6), 489–495. doi:10.1038/82732
Hsu, H., Huang, J., Shu, H. B., Baichwal, V., and Goeddel, D. V. (1996). TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 4 (4), 387–396. doi:10.1016/S1074-7613(00)80252-6
Hsu, H., Shu, H.-B., Pan, M. G., and Goeddel, D. V. (1996). TRADD–TRAF2 and TRADD–FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 84 (2), 299–308. doi:10.1016/S0092-8674(00)80984-8
Hsu, H., Xiong, J., and Goeddel, D. V. (1995). The TNF receptor 1-associated protein TRADD signals cell death and NF-κB activation. Cell 81 (4), 495–504. doi:10.1016/0092-8674(95)90070-5
Hu, C.-D., Chinenov, Y., and Kerppola, T. K. (2002). Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 9 (4), 789–798. doi:10.1016/S1097-2765(02)00496-3
Hwang, L. C., and Wohland, T. (2007). Recent Advances in fluorescence cross-correlation spectroscopy. Cell Biochem. Biophysics 49 (1), 1–13. doi:10.1007/s12013-007-0042-5
Jaco, I., Annibaldi, A., Lalaoui, N., Wilson, R., Tenev, T., and Laurien, L. (2017). MK2 Phosphorylates RIPK1 to Prevent TNF-Induced Cell Death. Molecular Cell 66 (5), 698–710.e5. doi:10.1016/j.molcel.2017.05.003
Jacquemet, G., Carisey, A. F., Hamidi, H., Henriques, R., and Leterrier, C. (2020). The cell biologist’s guide to super-resolution microscopy. J. Cell Sci. 133 (11), jcs240713. doi:10.1242/jcs.240713
Joly, J., Hudik, E., Lecart, S., Roos, D., Verkuijlen, P., Wrona, D., et al. (2020). Membrane dynamics and organization of the phagocyte NADPH oxidase in PLB-985 cells. Front. Cell Dev. Biol. 8, 608600. doi:10.3389/fcell.2020.608600
Jouan-Lanhouet, S., Riquet, F., Duprez, L., Vanden Berghe, T., Takahashi, N., and Vandenabeele, P. (2014). Necroptosis, in vivo detection in experimental disease models. Seminars Cell & Dev. Biol. 35, 2–13. doi:10.1016/j.semcdb.2014.08.010
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., et al. (2021). Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. doi:10.1038/s41586-021-03819-2
Kaiser, W. J., Upton, J. W., Long, A. B., Livingston-Rosanoff, D., Daley-Bauer, L. P., Hakem, R., et al. (2011). RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature 471 (7338), 368–372. doi:10.1038/nature09857
Kanayama, A., Seth, R. B., Sun, L., Ea, C. K., Hong, M., Shaito, A., et al. (2004). TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol. Cell 15 (4), 535–548. doi:10.1016/j.molcel.2004.08.008
Kennedy, A., Chang, T. C., Biniecka, M., O'Sullivan, J. N., Heffernan, E., et al. (2011). Tumor necrosis factor blocking therapy alters joint inflammation and hypoxia. Arthritis & Rheumatism 63 (4), 923–932. doi:10.1002/art.30221
Khoury, M. K., Gupta, K., Franco, S. R., and Liu, B. (2020). Necroptosis in the Pathophysiology of disease. Am. J. pathology 190 (2), 272–285. doi:10.1016/j.ajpath.2019.10.012
Kim, Y.-M., Kim, S. J., Tatsunami, R., Yamamura, H., Fukai, T., and Ushio-Fukai, M. (2017). ROS-induced ROS release orchestrated by Nox4, Nox2, and mitochondria in VEGF signaling and angiogenesis. Am. J. Physiology-Cell Physiology 312 (6), C749-C764–C764. doi:10.1152/ajpcell.00346.2016
Kim, Y.-S., Morgan, M. J., Choksi, S., and Liu, Z. G. (2007a). TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell 26 (5), 675–687. doi:10.1016/j.molcel.2007.04.021
Klar, T. A., and Hell, S. W. (1999). Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 24 (14), 954–956. doi:10.1364/OL.24.000954
Lafont, E., Draber, P., Rieser, E., Rieser, M., Kupka, S., and de Miguel, D. (2018). TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nature Cell Biology 20 (12), 1389–1399. doi:10.1038/s41556-018-0229-6
Legault, S., Fraser-Halberg, D. P., McAnelly, R. L., Eason, M. G., Thompson, M. C., and Chica, R. A. (2022). Generation of bright monomeric red fluorescent proteins via computational design of enhanced chromophore packing. Chem. Sci. 13 (5), 1408–1418. doi:10.1039/D1SC05088E
Lam, S. S., Martell, J. D., Kamer, K. J., Deerinck, T. J., Ellisman, M. H., Mootha, V. K., et al. (2015). Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 12 (1), 51–54. doi:10.1038/nmeth.3179
Lambeth, J. D. (2004a). NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4 (3), 181–189. doi:10.1038/nri1312
Lathe, G. H., and Ruthven, C. R. (1955). The separation of substances on the basis of their molecular weights, using columns of starch and water. Biochem. J. 60 (4). http://www.ncbi.nlm.nih.gov/pubmed/13249976.
Legler, D. F., Micheau, O., Doucey, M. A., Tschopp, J., and Bron, C. (2003). Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity 18 (5), 655–664. doi:10.1016/S1074-7613(03)00092-X
Li, J., McQuade, T., Siemer, A. B., Napetschnig, J., Moriwaki, K., Hsiao, Y. S., et al. (2012). The RIP1/RIP3 necrosome forms a functional amyloid signaling complex required for programmed necrosis. Cell 150 (2), 339–350. doi:10.1016/j.cell.2012.06.019
Li, X. J., et al. (2009). A fluorescently tagged C-terminal fragment of p47 phox detects NADPH oxidase dynamics during phagocytosis. Mol. Biol. Cell 20. doi:10.1091/mbc.E08-06-0620
Lin, Y., Devin, A., Rodriguez, Y., and Liu, Z. G. (1999). Cleavage of the death domain kinase RIP by Caspase-8 prompts TNF-induced apoptosis. Genes & Dev. 13 (19), 2514–2526. doi:10.1101/gad.13.19.2514
Liu, S., Liu, H., Johnston, A., Hanna-Addams, S., Reynoso, E., Xiang, Y., et al. (2017). MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. Proc. Natl. Acad. Sci. 114 (36), E7450-E7459. doi:10.1073/pnas.1707531114
Mahoney, D. J., Cheung, H. H., Mrad, R. L., Plenchette, S., Simard, C., EnwErE, E., et al. (2008). Both cIAP1 and cIAP2 regulate TNFalpha-mediated NF-kappaB activation. Proc. Natl. Acad. Sci. 105 (33), 11778–11783. doi:10.1073/pnas.0711122105
Maghsoudi, N., Zakeri, Z., and Lockshin, R. A. (2012). Programmed cell death and apoptosis--where it came from and where it is going: From Elie Metchnikoff to the control of caspases. Exp. Oncol. 34 (3), 146–152. http://www.ncbi.nlm.nih.gov/pubmed/23069998.
Menon, M. B., Gropengießer, J., Fischer, J., Novikova, L., Deuretzbacher, A., and Lafera, J. (2017). p38MAPK/MK2-dependent phosphorylation controls cytotoxic RIPK1 signalling in inflammation and infection. Nature Cell Biology 19 (10), 1248–1259. doi:10.1038/ncb3614
Masumoto, J., Taniguchi, S., and Sagara, J. (2001). Pyrin N-terminal Homology domain- and caspase recruitment domain-dependent oligomerization of ASC. Biochem. Biophysical Res. Commun. 280 (3), 652–655. doi:10.1006/bbrc.2000.4190
Matute, J. D., et al. (2009). A new genetic subgroup of chronic granulomatous disease with autosomal recessive mutations in p40 phox and selective defects in neutrophil NADPH oxidase activity. Blood 114. doi:10.1182/blood-2009-07-231498
Micheau, O., and Tschopp, J. (2003). Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114 (2), 181–190. doi:10.1016/S0092-8674(03)00521-X
Moe, K. T., Aulia, S., Jiang, F., Chua, Y. L., Koh, T. H., Wong, M. C., et al. (2006). Differential upregulation of Nox homologues of NADPH oxidase by tumor necrosis factor-? In human aortic smooth muscle and embryonic kidney cells. J. Cell. Mol. Med. 10 (1), 231–239. doi:10.1111/j.1582-4934.2006.tb00304.x
Moghadam, Z. M., Henneke, P., and Kolter, J. (2021). From Flies to men: ROS and the NADPH oxidase in phagocytes. Front. Cell Dev. Biol. 9, 628991. doi:10.3389/fcell.2021.628991
Moldoveanu, T., Grace, C. R., Llambi, F., Nourse, A., Fitzgerald, P., Gehring, K., et al. (2013). BID-induced structural changes in BAK promote apoptosis. Nat. Struct. Mol. Biol. 20 (5), 589–597. doi:10.1038/nsmb.2563
Montero, J. A., and Hurlé, J. M. (2010). Sculpturing digit shape by cell death. Apoptosis 15 (3), 365–375. doi:10.1007/s10495-009-0444-5
Morgan, D., RebElato, E., Abdulkader, F., Graciano, M. F. R., Oliveira-Emilio, H. R., Hirata, A. E., et al. (2009). Association of NAD(P)H oxidase with glucose-induced insulin secretion by pancreatic β-cells. Endocrinology 150 (5), 2197–2201. doi:10.1210/en.2008-1149
Morgan, M. J., Kim, Y. S., and Liu, Z. G. (2008). TNFalpha and reactive oxygen species in necrotic cell death. Cell Res. 18 (3), 343–349. doi:10.1038/cr.2008.31
Morgan, M. J., and Liu, Z. (2011). Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 21 (1), 103–115. doi:10.1038/cr.2010.178
Muppidi, J. R., Lobito, A. A., RaMaswaMy, M., Yang, J. K., Wang, L., Wu, H., et al. (2006). Homotypic FADD interactions through a conserved RXDLL motif are required for death receptor-induced apoptosis. Cell Death Differ. 13 (10), 1641–1650. doi:10.1038/sj.cdd.4401855
Murphy, J. M., Czabotar, P. E., Hildebrand, J. M., Lucet, I. S., Zhang, J. G., Alvarez-Diaz, S., et al. (2013). The pseudokinase MLKL mediates necroptosis via a molecular Switch mechanism. Immunity 39 (3), 443–453. doi:10.1016/j.immuni.2013.06.018
Naito, M. G., Xu, D., Amin, P., Lee, J., Wang, H., Li, W., et al. (2020). Sequential activation of necroptosis and apoptosis cooperates to mediate vascular and neural pathology in stroke. Proc. Natl. Acad. Sci. 117 (9), 4959–4970. doi:10.1073/pnas.1916427117
Nakajima, S., and Kitamura, M. (2013). Bidirectional regulation of NF-κB by reactive oxygen species: A role of unfolded protein response. Free Radic. Biol. Med. 65, 162–174. doi:10.1016/j.freeradbiomed.2013.06.020
Nakao, N., Kurokawa, T., Nonami, T., Tumurkhuu, G., Koide, N., and Yokochi, T. (2008). Hydrogen peroxide induces the production of tumor necrosis factor-α in RAW 264.7 macrophage cells via activation of p38 and stress-activated protein kinase. Innate Immun. 14 (3), 190–196. doi:10.1177/1753425908093932
Nakayama, M., Inoguchi, T., Sonta, T., Maeda, Y., Sasaki, S., Sawada, F., et al. (2005). Increased expression of NAD(P)H oxidase in islets of animal models of Type 2 diabetes and its improvement by an AT1 receptor antagonist. Biochem. Biophysical Res. Commun. 332 (4), 927–933. doi:10.1016/j.bbrc.2005.05.065
Nasu, Y., Benke, A., Arakawa, S., Yoshida, G. J., Kawamura, G., Manley, S., et al. (2016). In situ Characterization of Bak clusters responsible for cell death using single molecule localization microscopy. Sci. Rep. 6 (1), 27505. doi:10.1038/srep27505
Newton, K., Wickliffe, K. E., Dugger, D. L., Maltzman, A., Roose-Girma, M., Dohse, M., et al. (2019). Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 574 (7778), 428–431. doi:10.1038/s41586-019-1548-x
Nauseef, W. M. (2019). The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol. 60, 130–140. doi:10.1016/j.coi.2019.05.006
Nguyen, G. T., Green, E. R., and Mecsas, J. (2017). Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial Resistance. Front. Cell. Infect. Microbiol. 7, 373. doi:10.3389/fcimb.2017.00373
Oberst, A., Dillon, C. P., Weinlich, R., McCormick, L. L., Fitzgerald, P., Pop, C., et al. (2011). Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature 471 (7338), 363–367. doi:10.1038/nature09852
Oerlemans, M. I. F. J., Liu, J., Arslan, F., den Ouden, K., van Middelaar, B. J., Doevendans, P. A., et al. (2012). Inhibition of RIP1-dependent necrosis prevents adverse cardiac remodeling after myocardial ischemia–reperfusion in vivo. Basic Res. Cardiol. 107 (4), 270. doi:10.1007/s00395-012-0270-8
Ofengeim, D., and Yuan, J. (2013). Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat. Rev. Mol. Cell Biol. 14 (11), 727–736. doi:10.1038/nrm3683
Orozco, S., Yatim, N., Werner, M. R., Tran, H., Gunja, S. Y., Tait, S. W. G., et al. (2014). RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 21 (10), 1511–1521. doi:10.1038/cdd.2014.76
Padilla-Parra, S., and Tramier, M. (2012). FRET microscopy in the living cell: Different approaches, strengths and weaknesses. BioEssays 34 (5), 369–376. doi:10.1002/bies.201100086
Park, K.-J., Lee, C. H., Kim, A., Jeong, K. J., Kim, C. H., and Kim, Y. S. (2012). Death receptors 4 and 5 activate Nox1 NADPH oxidase through riboflavin kinase to induce reactive oxygen species-mediated apoptotic cell death. J. Biol. Chem. 287 (5), 3313–3325. doi:10.1074/jbc.M111.309021
Park, M.-Y., Ryu, S. W., Kim, K. D., Lim, J. S., Lee, Z. W., and Kim, E. (2005). Fas-associated factor-1 mediates chemotherapeutic-induced apoptosis via death effector filament formation. Int. J. Cancer 115 (3), 412–418. doi:10.1002/ijc.20857
Park, Y. C., Ye, H., Hsia, C., Segal, D., Rich, R. L., Liou, H. C., et al. (2000). A novel mechanism of TRAF signaling revealed by structural and functional Analyses of the TRADD–TRAF2 interaction. Cell 101 (7), 777–787. doi:10.1016/S0092-8674(00)80889-2
Pasparakis, M., and Vandenabeele, P. (2015). Necroptosis and its role in inflammation. Nature 517 (7534), 311–320. doi:10.1038/nature14191
Perillo, B., Di Donato, M., Pezone, A., Di Zazzo, E., Giovannelli, P., Galasso, G., et al. (2020). ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 52 (2), 192–203. doi:10.1038/s12276-020-0384-2
Petrie, E. J., Sandow, J. J., Jacobsen, A. V., Smith, B. J., Griffin, M. D. W., Lucet, I. S., et al. (2018). Conformational switching of the pseudokinase domain promotes human MLKL tetramerization and cell death by necroptosis. Nat. Commun. 9 (1), 2422. doi:10.1038/s41467-018-04714-7
Proell, M., Gerlic, M., Mace, P. D., Reed, J. C., and Riedl, S. J. (2013). The CARD plays a critical role in ASC foci formation and inflammasome signalling. Biochem. J. 449 (3), 613–621. doi:10.1042/BJ20121198
Rodriguez, D. A., Weinlich, R., Brown, S., Guy, C., Fitzgerald, P., Dillon, C. P., et al. (2016). Characterization of RIPK3-mediated phosphorylation of the activation loop of MLKL during necroptosis. Cell Death Differ. 23 (1), 76–88. doi:10.1038/cdd.2015.70
Rothbauer, U., Zolghadr, K., Tillib, S., Nowak, D., Schermelleh, L., Gahl, A., et al. (2006). Targeting and tracing antigens in live cells with fluorescent nanobodies. Nat. Methods 3 (11), 887–889. doi:10.1038/nmeth953
Sahillioglu, A. C., Sumbul, F., Ozoren, N., and Haliloglu, T. (2014). Structural and dynamics aspects of ASC speck assembly. Structure 22 (12), 1722–1734. doi:10.1016/j.str.2014.09.011
Salvador-Gallego, R., Mund, M., Cosentino, K., Schneider, J., Unsay, J., Schraermeyer, U., et al. (2016). Bax assembly into rings and arcs in apoptotic mitochondria is linked to membrane pores. EMBO J. 35 (4), 389–401. doi:10.15252/embj.201593384
Salvesen, G. S., and Walsh, C. M. (2014). Functions of caspase 8: The identified and the mysterious. Seminars Immunol. 26 (3), 246–252. doi:10.1016/j.smim.2014.03.005
Samson, A. L., Zhang, Y., Geoghegan, N. D., Gavin, X. J., Davies, K. A., Mlodzianoski, M. J., et al. (2020a). MLKL trafficking and accumulation at the plasma membrane control the kinetics and threshold for necroptosis. Nat. Commun. 11 (1), 3151. doi:10.1038/s41467-020-16887-1
Schilling, R., Geserick, P., and Leverkus, M. (2014). Characterization of the ripoptosome and its components. Methods Enzym. 545, 83–102. doi:10.1016/B978-0-12-801430-1.00004-4
Schleich, K., Warnken, U., Fricker, N., Ozturk, S., Richter, P., Kammerer, K., et al. (2012). Stoichiometry of the CD95 death-inducing signaling complex: Experimental and modeling evidence for a death effector domain chain model. Mol. Cell 47 (2), 306–319. doi:10.1016/j.molcel.2012.05.006
Schramm, M., Wiegmann, K., Schramm, S., Gluschko, A., Herb, M., Utermohlen, O., et al. (2014). Riboflavin (vitamin B 2) deficiency impairs NADPH oxidase 2 (Nox2) priming and defense against Listeria monocytogenes. Eur. J. Immunol. 44 (3), 728–741. doi:10.1002/eji.201343940
Shearwin-Whyatt, L. M., Harvey, N. L., and Kumar, S. (2000). Subcellular localization and CARD-dependent oligomerization of the death adaptor RAIDD. Cell Death Differ. 7 (2), 155–165. doi:10.1038/sj.cdd.4400632
Siegel, R. M., Martin, D. A., Zheng, L., Ng, S. Y., Bertin, J., Cohen, J., et al. (1998). Death-effector filaments: Novel cytoplasmic structures that recruit caspases and trigger apoptosis. J. Cell Biol. 141 (5), 1243–1253. doi:10.1083/jcb.141.5.1243
Sies, H. (2017). Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 11, 613–619. doi:10.1016/j.redox.2016.12.035
Sochacki, K. A., Shtengel, G., van Engelenburg, S. B., Hess, H. F., and Taraska, J. W. (2014). Correlative super-resolution fluorescence and metal-replica transmission electron microscopy. Nat. Methods 11 (3), 305–308. doi:10.1038/nmeth.2816
Song, Z. M., Bouchab, L., Hudik, E., Le Bars, R., Nusse, O., and Dupre-Crochet, S. (2017). Phosphoinositol 3-phosphate acts as a timer for reactive oxygen species production in the phagosome. J. Leukoc. Biol. 101 (5), 1155–1168. doi:10.1189/jlb.1A0716-305R
Stasia, M. J., and Li, X. J. (2008). Genetics and immunopathology of chronic granulomatous disease. Seminars Immunopathol. 30 (3), 209–235. doi:10.1007/s00281-008-0121-8
Sumimoto, H. (2008). Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 275 (13), 3249–3277. doi:10.1111/j.1742-4658.2008.06488.x
Sun, G., Guzman, E., Balasanyan, V., Conner, C. M., Wong, K., Zhou, H. R., et al. (2017). A molecular signature for anastasis, recovery from the brink of apoptotic cell death. J. cell Biol. 216 (10), 3355–3368. doi:10.1083/jcb.201706134
Sun, G., and Montell, D. J. (2017). Q&A: Cellular near death experiences-what is anastasis? BMC Biol. 15 (1), 92. doi:10.1186/s12915-017-0441-z
Sun, L., Wang, H., Wang, Z., He, S., Chen, S., Liao, D., et al. (2012). Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148 (1–2), 213–227. doi:10.1016/j.cell.2011.11.031
Szanto, I., Rubbia-Brandt, L., Kiss, P., Steger, K., Banfi, B., Kovari, E., et al. (2005). Expression ofNOX1, a superoxide-generating NADPH oxidase, in colon cancer and inflammatory bowel disease. J. Pathology 207 (2), 164–176. doi:10.1002/path.1824
Tenev, T., Bianchi, K., Darding, M., Broemer, M., Langlais, C., Wallberg, F., et al. (2011). The ripoptosome, a signaling platform that assembles in response to genotoxic stress and Loss of IAPs. Mol. Cell 43 (3), 432–448. doi:10.1016/j.molcel.2011.06.006
Tinel, A., and Tschopp, J. (2004). The PIDDosome, a protein complex implicated in activation of caspase-2 in response to genotoxic stress. Science 304 (5672), 843–846. doi:10.1126/science.1095432
Titeca, K., Van Quickelberghe, E., Samyn, N., De Sutter, D., Verhee, A., Gevaert, K., et al. (2017). Analyzing trapped protein complexes by Virotrap and SFINX. Nat. Protoc. 12 (5), 881–898. doi:10.1038/nprot.2017.014
Ueyama, T., Geiszt, M., and Leto, T. L. (2006). Involvement of Rac1 in activation of multicomponent Nox1-and Nox3-based NADPH oxidases. Mol. Cell. Biol. 26 (6), 2160–2174. doi:10.1128/MCB.26.6.2160-2174.2006
Valenta, H., et al. (2020). The NADPH oxidase and the phagosome. Mol. Cell. Biol. Phagocytosis, 153–177. doi:10.1007/978-3-030-40406-2_9
van Loo, G., and Bertrand, M. J. M. (2022). Death by TNF: A road to inflammation. Nat. Rev. Immunol., 1–15. doi:10.1038/s41577-022-00792-3
Van Quickelberghe, E., De Sutter, D., van Loo, G., Eyckerman, S., and Gevaert, K. (2018). A protein-protein interaction map of the TNF-induced NF-κB signal transduction pathway. Sci. Data 5 (1), 180289. doi:10.1038/sdata.2018.289
Vanden Berghe, T., Linkermann, A., Jouan-Lanhouet, S., Walczak, H., and Vandenabeele, P. (2014). Regulated necrosis: The expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 15 (2), 135–147. doi:10.1038/nrm3737
Vanlangenakker, N., Vanden Berghe, T., Bogaert, P., Laukens, B., Zobel, K., Deshayes, K., et al. (2011). cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 18 (4), 656–665. doi:10.1038/cdd.2010.138
Vercammen, D., BeyaeRt, R., Denecker, G., Goossens, V., Van Loo, G., Declercq, W., et al. (1998). Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J. Exp. Med. 187 (9), 1477–1485. doi:10.1084/jem.187.9.1477
Wagner, T. R., and Rothbauer, U. (2021). Nanobodies – little helpers unravelling intracellular signaling. Free Radic. Biol. Med. 176, 46–61. doi:10.1016/j.freeradbiomed.2021.09.005
Wang, L., Du, F., and Wang, X. (2008). TNF-Α induces two distinct caspase-8 activation pathways. Cell 133 (4), 693–703. doi:10.1016/j.cell.2008.03.036
Wang, S., Chen, Y., Wu, Q., and Hua, Z. C. (2013). Detection of Fas-associated death domain and its variants’ self-association by fluorescence resonance energy transfer in living cells. Mol. imaging 12 (2), 111–120. http://www.ncbi.nlm.nih.gov/pubmed/23415399.
Wu, C.-J., Conze, D. B., Li, T., Srinivasula, S. M., and Ashwell, J. D. (2006). Sensing of Lys 63-linked polyubiquitination by NEMO is a key event in NF-kappaB activation [corrected]. Nat. Cell Biol. 8 (4), 398–406. doi:10.1038/ncb1384
Wu, X.-N., Yang, Z. H., Wang, X. K., Zhang, Y., Wan, H., Song, Y., et al. (2014). Distinct roles of RIP1–RIP3 hetero- and RIP3–RIP3 homo-interaction in mediating necroptosis. Cell Death Differ. 21 (11), 1709–1720. doi:10.1038/cdd.2014.77
Wu, X., Hu, H., Dong, X. Q., Zhang, J., Wang, J., Schwieters, C. D., et al. (2021). The amyloid structure of mouse RIPK3 (receptor interacting protein kinase 3) in cell necroptosis. Nat. Commun. 12 (1), 1627. doi:10.1038/s41467-021-21881-2
Xu, Q., Choksi, S., Qu, J., Jang, J., Choe, M., Banfi, B., et al. (2016). NADPH oxidases are essential for macrophage differentiation. J. Biol. Chem. 291 (38), 20030–20041. doi:10.1074/jbc.M116.731216
Yazdanpanah, B., Wiegmann, K., Tchikov, V., Krut, O., Pongratz, C., Schramm, M., et al. (2009). Riboflavin kinase couples TNF receptor 1 to NADPH oxidase. Nature 460 (7259), 1159–1163. doi:10.1038/nature08206
Yoon, S., Kovalenko, A., Bogdanov, K., and Wallach, D. (2017). MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity 47 (1), 51–65. doi:10.1016/j.immuni.2017.06.001
Yu, D., Chojnowski, G., Rosenthal, M., and Kosinski, J. (2023). AlphaPulldown—A python package for protein–protein interaction screens using AlphaFold-multimer. Bioinforma. Ed. by L. Cowen 39 (1), btac749. doi:10.1093/bioinformatics/btac749
Zhang, D.-W., Shao, J., Lin, J., Zhang, N., Lu, B. J., Lin, S. C., et al. (2009). RIP3, an energy Metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 325 (5938), 332–336. doi:10.1126/science.1172308
Zhong, H., Ceballos, C. C., Massengill, C. I., Muniak, M. A., Ma, L., Qin, M., et al. (2021). High-fidelity, efficient, and reversible labeling of endogenous proteins using CRISPR-based designer exon insertion. eLife 10, e64911. doi:10.7554/eLife.64911
Ziegler, C. S., Bouchab, L., Tramier, M., Durand, D., Fieschi, F., Dupre-Crochet, S., et al. (2019). Quantitative live-cell imaging and 3D modeling reveal critical functional features in the cytosolic complex of phagocyte NADPH oxidase. J. Biol. Chem. 294, 3824–3836. doi:10.1074/jbc.RA118.006864
ASC apoptosis-associated speck-like protein containing a CARD
Bak Bcl-2 homologous antagonist killer
Bax Bcl-2-associated X protein
BHA butylate hydroxyanisole
BiFC bimolecular fluorescence complementation assay
CARD caspase activation and recruitment domain
cFLIP cellular FLICE-like inhibitory protein
cIAP1/2 cellular inhibitor of apoptosis protein 1 and 2
CRISPR-Cas9 clustered regulated interspaced short palindromic repeats - associated protein-9 nuclease
Co-IP co-immunoprecipitation
CFP cyan fluorescent protein
DD death domain
DED death effector domain
dSTORM direct stochastic optical reconstruction microscopy
ESCRT III endosomal sorting complexes required for transport III
eGFP enhanced green fluorescent protein
EV extracellular vesicle
FADD Fas-associated protein with death domain
FRET Förster resonance energy transfer
HOIL-1 heme-oxidized IRP2 Ub ligase1
HOIP HOIL-1 interacting protein
hCHMP4B human charged multivesicular body protein 4B
HEK293 human embryonic kidney 293
hTNF human TNF
IκBα inhibitor of NF-κB α
IKK inhibitor of the NF-κB kinase complex
Kd dissociation constant
LUBAC linear ubiquitin chain assembly complex
MK2 mitogen-activated protein kinase-activated protein kinase 2
MLKL mixed lineage kinase domain-like protein
mRFP monomeric RFP
NADPH nicotinamide adenine dinucleotide phosphate
NSA necrosulfonamide
NEMO NF-κB essential modulator
NF-κB nuclear factor-κB
PIDD p53-induced protein with a death domain
PI(3,4)P2 phosphatidylinositol 3,4-bisphosphate
PI(3)P phosphatidylinositol-3-phosphate
PALM photoactivated localization microscopy
PLAD pre-ligand-binding assembly domain
PKC protein kinase C
PMA phorbol myristate acetate
PPI protein-protein interaction
PYD pyrin domain
RIPK1/3 receptor-interacting protein kinase 1 and 3
RFP red fluorescent protein
RHIM RIP homotypic interaction motif
RAIDD RIP-associated ICH-1/CED-3 homologous protein with death domain
SHARPIN SHANK-associated RH domain interactor
STED stimulated emission depletion microscopy
TAK1 transforming growth factor-β-activated kinase 1
TBK1 TANK binding kinase 1
TAB2/3 TAK1 binding protein 2 and 3
TAP tandem affinity purification
TIRF total internal reflection fluorescence
TNFR1 TNF receptor 1
TRAF2 TNF receptor associated factor 2
TRADD TNFR-associated protein with death domain
TRPM7 transient receptor potential melastatin related 7
TNF tumor necrosis factor
YFP yellow fluorescent protein.
Keywords: cell death, TNF, NOX, protein complex, oxidative stress, fluorescence, microscopy
Citation: Ladik M, Valenta H, Erard M, Vandenabeele P and Riquet FB (2023) From TNF-induced signaling to NADPH oxidase enzyme activity: Methods to investigate protein complexes involved in regulated cell death modalities. Front. Cell. Death 2:1127330. doi: 10.3389/fceld.2023.1127330
Received: 19 December 2022; Accepted: 03 March 2023;
Published: 05 April 2023.
Edited by:
Bertrand Mollereau, Université de Lyon, FranceReviewed by:
Maria Tanzer, Max Planck Institute of Biochemistry, GermanyCopyright © 2023 Ladik, Valenta, Erard, Vandenabeele and Riquet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Franck B. Riquet, ZnJhbmNrLnJpcXVldEB1bml2LWxpbGxlLmZy
†Present address: Hana Valenta, Laboratory for Nanobiology, Department of Chemistry, KU Leuven, Leuven, Belgium
‡These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.