
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 27 March 2025
Sec. Cellular Biochemistry
Volume 13 - 2025 | https://doi.org/10.3389/fcell.2025.1566567
This article is part of the Research TopicMechanism Study of Bioactive Molecules Using Omics TechnologyView all 3 articles
 Ping Liu1,2†
Ping Liu1,2† Jun Zhang2,3†Jingdan Zhang4,5Yucheng Yuan2,3Zhiqing Liu2Sixian Chen1,2Kaifeng Chen2,3Li Dong5Zhiyuan Cheng3,6
Jun Zhang2,3†Jingdan Zhang4,5Yucheng Yuan2,3Zhiqing Liu2Sixian Chen1,2Kaifeng Chen2,3Li Dong5Zhiyuan Cheng3,6 Yinan Zhang1Meiyu Geng1,2
Yinan Zhang1Meiyu Geng1,2 Minjia Tan1,2,3,4,5*
Minjia Tan1,2,3,4,5* Wensi Zhao7*
Wensi Zhao7* Dong Xie7*
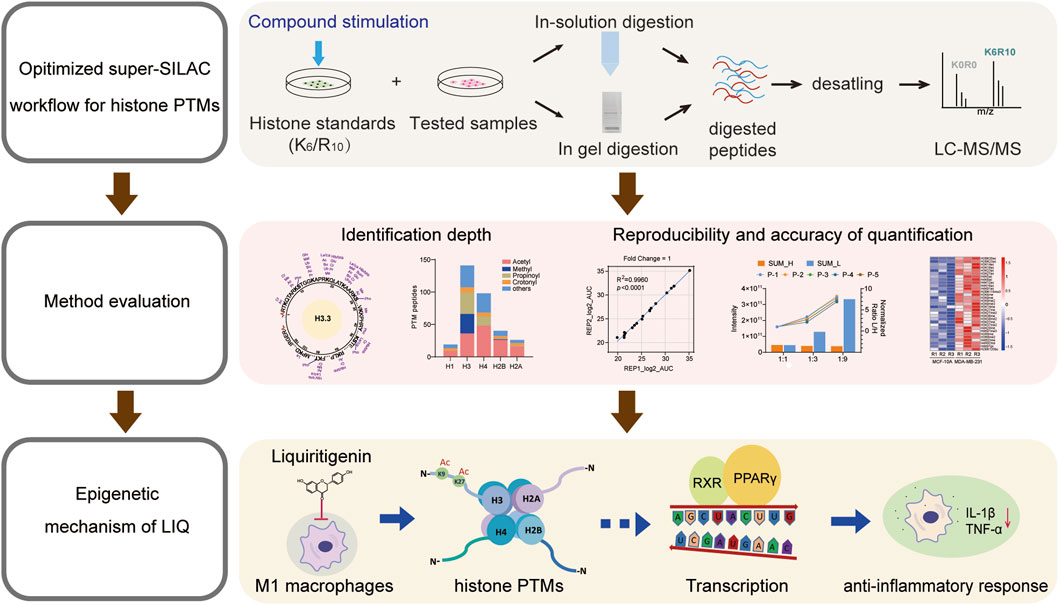
Dong Xie7*Liquiritigenin (LIQ) is a dihydroflavonone monomer compound with a planar ring structure that exhibits potent anti-inflammatory activity. The post-translational modifications (PTMs) of histones are closely associated with inflammatory diseases. To explore the relationships between the anti-inflammatory effects and epigenetic regulatory mechanisms of LIQ, we optimized the super stable isotope labeling by amino acids in cell culture (super-SILAC) method combined with a compound stimulation strategy. Moreover, we evaluated the identification coverage and demonstrated high reliability as well as reproducibility of the optimized method at both the peptide and cellular lysate levels, which are promising for elucidating disease pathology and drug mechanisms. We further applied the method to a system-wide characterization of histone PTMs in M1 macrophages treated with LIQ. The quantitative results showed that H4K5ac, H4K16ac, H3K9ac, H3K27ac, and H2BK12ac are significantly upregulated. Transcriptome analysis revealed that LIQ could exert anti-inflammatory effects by modulating the histone PTMs and regulating gene expressions through the peroxisome proliferator-activated receptor (PPAR) signaling pathway. Collectively, we provide a sensitive and universal strategy for research on the epigenetic mechanisms of natural products as well as facilitate epigenetic understanding of LIQ in inflammatory therapies.
Natural products with structural diversity often exhibit therapeutic activities against various diseases (Baker et al., 2007; Zhao et al., 2023). Approximately 25%–50% of commercially marketed drugs are derived from natural products (Kingston, 2011). Flavonoid compounds play important roles in anti-inflammatory activities through their planar ring structures, which make them promising lead compounds for inflammatory therapy (Theoharides et al., 2001; Wang et al., 2021; Li et al., 2023). Previous studies have revealed that most flavonoids and their derivative dihydroflavones exert anti-inflammatory effects by reducing the expressions of proinflammatory cytokines like IL-6, IL-8, TNF-α, and IL-1β through the NF-κB, MAPK, and JNK-STAT pathways (Serafini et al., 2010; Santangelo et al., 2007). Liquiritigenin (LIQ) is a dihydroflavonone monomer compound isolated from licorice that has been demonstrated to have potent anti-inflammatory effects through inhibition of the activation of NF-κB in macrophages as well as reduction of the production of inducible nitric oxide synthase (iNOS) and proinflammatory cytokines (Babu et al., 2023; Kim et al., 2008; Wang et al., 2015). Additionally, LIQ positively modulates the activity of sirtuin 3 (SIRT3), which is one of the most prominent deacetylases (Zhou et al., 2022). Nonetheless, the relationships between the anti-inflammatory effects of LIQ and regulation of its epigenetic mechanisms remain unclear.
Epigenetic regulation plays a crucial role in the regulation of gene expression. Epigenetic marks can alter the chromatin structure, thereby influencing gene expressions and cellular functions as well as participating in the pathogenesis of various diseases (Zhang and Cao, 2021; Zhu et al., 2021). Histone post-translational modifications (PTMs) are critical epigenetic regulatory factors that are regulated in a highly dynamic and complicated manner owing to the enzymes that catalyze the addition of specific PTMs (writers), reader proteins that recognize and bind specific domains (readers), and enzymes that remove PTMs (erasers) (Millán-Zambrano et al., 2022; Choudhary et al., 2014; Hyun et al., 2017). Small-molecule metabolites are closely associated with specific epigenetic modifications. For instance, various acyl-CoA donors can be catalyzed through a range of acylations by histone acetyltransferases (HATs) (Xie et al., 2024; Egger et al., 2024; He et al., 2023). Histone PTMs exert diverse effects on physiological and pathological processes. Typically, histone methylation and acetylation can impact histone–DNA binding affinity, alter chromatin accessibility, and regulate subsequent downstream gene transcriptions (Kuznetsova et al., 2020; Lawrence and Natoli, 2011; Sinha et al., 2023). In contrast, dysfunction of histone PTMs has been extensively studied in various diseases, including neurological disorders, inflammatory diseases, and cancers (Li et al., 2019; Netea et al., 2016; Noberini et al., 2018). Inhibition of H3K18/K27ac expression has been reported to significantly impact proliferation and metastasis of liver cancer cells (Cai et al., 2021). Histone deacetylase 3 (HDAC3) has been reported to be a crucial mediator of macrophage differentiation, activation, and polarization (Schultze, 2017; Van den Bossche et al., 2014). Comprehensive identification and quantitation of histone PTMs remain great challenges owing to several factors. First, histone PTMs exhibit a high degree of variability with more than 30 structurally distinct modifications, which are located on over 180 amino acid residues of histones (Millán-Zambrano et al., 2022; Zhao et al., 2024). Second, PTM crosstalk adds an additional layer of functional protein regulation and leads to expansion of the information content of a proteome. For instance, acetylation and methylation compete for the same site (H3K27) in embryonic stem cells (Huang et al., 2018; Ferrari et al., 2014). Third, histone PTMs are highly dynamic and reversible; histone PTMs with low abundances can further complicate identification and quantitation (Chan and Maze, 2020).
Mass spectrometry (MS)-based proteomics has advantages of high resolution, accuracy, and sensitivity, thereby providing the most accessible opportunity for global characterization of the histone PTM landscape (Cravatt et al., 2007). Among the MS-based quantitative strategies, stable isotope labeling by amino acids in cell culture (SILAC) is widely used in protein profiling and PTM analysis, where distinct isotopically encoded amino acids are introduced at the cellular level to reduce the experimental variability caused by sample preparation and improve quantitative accuracy (Kirchner and Selbach, 2012; Maile et al., 2015). Technological advancements from SILAC to super-SILAC have significantly expanded the quantitative capabilities of proteomics (Cuomo et al., 2011). The super-SILAC method utilizes a labeled mixture as an internal standard and is subsequently spiked into diverse samples for quantitative analysis (Noberini et al., 2016). This approach is beneficial for tissue samples and high-throughput sample sets. A standardized protocol for the super-SILAC method has been developed that utilizes treatment-naïve histone standard samples derived from various cell lines (Noberini and Bonaldi, 2017; Noberini et al., 2023). Tissue-specific histone propionylation and butyrylation were developed through derivatization methods in conjunction with super-SILAC, and compound stimulation has been suggested to increase the abundance of PTM peptides to facilitate further identification and quantitation (Vai et al., 2024).
In the present study, we optimized the super-SILAC method using multiple compound stimulations to increase the modification abundances of our histone standard samples. After assessing the identification depth, quantification reproducibility, and accuracy of our method at the synthetic peptide and whole-cell lysate levels, we applied the approach to M1 macrophages to elucidate the anti-inflammatory mechanism of LIQ. Quantitative transcriptomic results revealed the potential roles of histone PTMs in inflammatory processes and are expected to provide insights for further biological research on histone PTMs.
The MCF-7, A549, MCF-10A, MDA-MB-231, THP1, and MDA-MB-468 cell lines used in this work were obtained from ATCC. The reagents used include DMEM for SILAC (cat. no. 88425, Thermo Fisher Scientific), dialyzed fetal bovine serum (FBS; cat. no. 30067334, GIBCO), 13C6-Lys (cat. no. 211204102, Silantes), 13C6-15N4-Arg (cat. no. 201604102, Silantes), protease inhibitor cocktail (cat. no. 6538282001, Roche), modified sequencing-grade trypsin (HLS TRY001N, HuaLishi Scientific), GSK-LSD1 dihydrochloride (GSK-LSD1·2HCl; S7574, Selleck), Jumonji histone demethylase inhibitor (JIB-04; S7281, Selleck), vorinostat or suberoylanilide hydroxamic acid (SAHA; HY-10221, MCE), nicotinamide (NAM; S1761, Beyotime), trichloroacetic acid (TCA; T0699, Sigma), Sep-Pak C18 cartridges (WAT023590, Waters), acetyl lysine antibody (ICP0380, Immunechem), methylated (ε-N) lysine antibody (ICP0501, Immunechem), horseradish peroxidase anti-histone H3 antibody (ab21054, Abcam), acetyl-histone H3 (Lys14) (D4B9) rabbit mAb (7627S, Cell Signaling), acetyl-histone H3 (Lys27) rabbit mAb (F0271, Selleck), anti-acetyl-histone H3 (Lys9) rabbit pAb (PTM-112, PTM Biolabs), anti-acetyl-histone H4 (Lys5) rabbit pAb (PTM-119, PTM Biolabs), acetyl-histone H4 (Lys16) (E2B8W) rabbit mAb (13534S, Cell Signaling), anti-histone H2B (acetyl K12) (Ab61228, Abcam), anti-histone H2A (acetyl K5) (Ab45152, Abcam), anti-monomethyl-histone H3 (Lys4) antibody (07-436, Millipore), trimethyl-histone H3 (Lys4) antibody (Cell Signaling, 9727S), dimethyl-histone H3 (Lys9) (D85B4) antibody (Cell Signaling, 4658S), trimethyl-histone H3 (Lys9) (D4W1U) antibody (Cell Signaling, 13969T), dimethyl-histone H3 (Lys27) rabbit mAb (F0329, Selleck), anti-trimethyl-histone H3 (Lys27) antibody (05-1951-S, Millipore), and dimethyl-histone H3 (Lys36) antibody (2901S, Cell Signaling).
The histone peptides were synthesized by Nanjing TGpeptide Biotechnology Co. Ltd. (Supplementary Table S1).
MCF-7, A549, MDA-MB-231, and MDA-MB-468 cells were cultured in DMEM containing 10% FBS (cat. no. 10091148, GIBCO) and 1% penicillin and streptomycin (CM-0525, Procell). The THP1 cells were cultured in RPMI 1640 medium containing 10% FBS and 1% penicillin and streptomycin solution. The MCF-10A cells were cultured with DMEM/F12 containing 5% horse serum (HS), 20 ng/mL of epidermal growth factor (EGF), 0.5 μg/mL of hydrocortisone, 10 μg/mL of insulin, 1% non-essential amino acids (NEAAs), as well as 1% penicillin and streptomycin. The THP1 cells were treated with 500 nM of phorbol myristate acetate (PMA) for 6 h, followed by treatment with 100 ng/mL of lipopolysaccharide (LPS) and 20 ng/mL of IFNγ for 12 h to activate the M1 type macrophages. Lastly, the M1-type cells were treated with LIQ for another 24 h. All cells were grown at 37°C supplied with 5% CO2.
For SILAC, the MCF-7 and A549 cells were labeled in DMEM with 13C6 lysine and 13C615N4 arginine containing 2 mM of L-glutamine, 1% penicillin and streptomycin, and 10% dialyzed FBS (cat. no. 04-011-1A, Biological Industry) for 6–8 passages. The labeling efficiency was determined by liquid chromatography mass spectrometry (LC-MS/MS) analysis before histone extraction. When the labeling efficiency was above 95%, the cells were treated with 2.0 μM of GSK-LSD1·2HCl and 0.1 μM of JIB-04 for another 7 d; on the sixth day, 5 μM of SAHA and 10 mM of NAM were added to the medium for 24 h. After 7 d, the cells were harvested for histone extraction.
The harvested cells were lysed using an extraction buffer (10 mM of HEPES at pH 7.0, 10 mM of KCl, 1.5 mM of MgCl2, 0.34 M of sucrose, and 1× protease inhibitor cocktail) with 0.5% nonidet P-40 (NP-40) on ice for 30 min. After centrifugation, the pellets were washed with the extraction buffer without NP-40, resuspended in 0.2 M of H2SO4, and incubated overnight at 4°C. The supernatant was collected for TCA precipitation, and the precipitate was washed with cold acetone. The histone precipitate was dried at room temperature and dissolved in ddH2O.
Histone was diluted in 100 mM of NH4HCO3. Then, sequence-grade trypsin was added at a trypsin-to-protein ratio of 1:50 and incubated overnight at 37°C. The sample was dried using SpeedVac. Lastly, the histone peptide sample was desalted using Sep-Pak C18 cartridges (Waters).
Histone was separated in 12% gel and stained with Coomassie brilliant blue. Then, the histone bands were cut and gel particles were washed with 50% ethanol to remove the blue color. Next, the gels were washed with water twice for 20 min each, and the gel bands were cut into 1 mm3 pieces. The gels were dehydrated in 100% acetonitrile (ACN) until they shrank into white pieces and were then dried using a SpeedVac. Later, the gels were rehydrated with 10 ng/μL of trypsin solution in 50 mM of NH4HCO3 and incubated overnight at 37°C. We then washed the gels in 50 μL of 50% ACN for 15 min, 75% ACN/0.1% TFA for 15 min, and 100% ACN for another 5 min. The three extracts were finally combined and dried using the SpeedVac. The tryptic peptides were desalted using Sep-Pak C18 cartridges.
The histone peptides were detected using the Orbitrap Ascend Tribrid™ mass spectrometer (Thermo Fisher Scientific) coupled with the Vanquish Neo high-performance liquid chromatography (HPLC) system (Thermo Fisher Scientific). The peptides were dissolved in 0.1% formic acid in water (v/v), centrifuged at 21,130g for 10 min, and the supernatant was injected into a self-made capillary liquid chromatography column filled with C18 resin. The flow phase A contained 0.1% formic acid in water (v/v), while the flow phase B included 0.1% formic acid, 90% ACN, and 10% water. The gradient elution steps were as follows: 0–7 min, 0%–5% B; 7–36 min, 5%–12% B; 36–101 min, 12%–30% B; 101–111 min, 30%–45% B; 111–120 min, 45%–80% B. Then, the eluted peptides were analyzed using the Ascend mass spectrometer, which was used for full MS scans from 300 to 1500 m/z with a resolution of 120,000. In the data-dependent mode, the precursor ions with charge states 1–5 were selected for higher energy collisional dissociation (HCD) fragmentation at 30% collision energy, and the secondary fragment ions were detected using Orbitrap with the dynamic exclusion time set to 10 s.
The histone PTMs were analyzed using Mascot software (version 2.3.01) and the UniProt human histone database. Trypsin/P was selected as the enzyme with five missed cleavages. The fixed modifications include label:13C(6)(K) and label:13C(6)15N(4) (R), while the variable modifications include oxidation (M), label:13C(6)(K) + methyl, label:13C(6)(K) + dimethyl, label:13C(6)(K) + trimethyl, label:13C(6)(K) + acetyl, label:13C(6)(K) + propionyl, label:13C(6)(K) + butyryl, label:13C(6)(K) + crotonyl, label:13C(6)(K) + malonyl, label:13C(6)(K) + succinyl, label:13C(6)(K) + GlyGly (ubiquitination), label:13C(6)(K) + lactyl, label:13C(6)(K) + hydroxyisobutyryl, label:13C(6)15N(4) (R) + dimethyl, label:13C(6)15N(4) (R) + trimethyl, and phospho (S/T). The peptide tolerance was 10 ppm and MS/MS tolerance was 0.02 Da. The analyses were conducted for five representative histone variants, including P0C0S8 (H2A1), P0C0S5 (H2A.Z), P62807 (H2B1C), P84243 (H3.3), and P62805 (H4).
A comprehensive wildcard database search was conducted using Byonic software (Protein Metrics Inc.) with the following parameter configurations. The precursor mass tolerance was set at 6 ppm, and the fragment ion mass tolerance was defined as 0.5 Da. Trypsin/P was designated as the digestion enzyme, and up to five missed cleavages were permitted. Data analyses were performed against the human histone sequence database derived from the UniProt database (version 2023-06-08, comprising 86 sequences). The fixed modifications include label:13C(6)(K) and label:13C(6)15N(4) (R); the variable modifications were label:13C(6)(K) + methyl, label:13C(6)(K) + dimethyl, label:13C(6)(K) + trimethyl, label:13C(6)(K) + acetyl, label:13C(6)(K) + propionyl, label:13C(6)(K) + butyryl, label:13C(6)(K) + crotonyl, label:13C(6)(K) + succinyl, label:13C(6)(K) + glutaryl, label:13C(6)(K) + GlyGly (ubiquitination), label:13C(6)(K) + lactyl, label:13C(6)(K) + hydroxyisobutyryl, label:13C(6)15N(4)(R) + methyl, label:13C(6)15N(4)(R) + dimethyl, and phospho (S/T). A Byonic score threshold of 250 was applied (Bern et al., 2009; Pirro et al., 2021; Riley et al., 2019), and only the peptide spectra with sequence lengths exceeding five residues were retained for analyses. For peptides identified with only heavy labels, we complemented their light-labeled data through in silico methods using Byonic. The valence states identified across unique peptides were integrated to ensure comprehensive representation. All quantitative analyses were conducted for six representative histone variants, including P0C0S8 (H2A1), P0C0S5 (H2A.Z), P62807 (H2B1C), P84243 (H3.3), P68431 (H3.1), and P62805 (H4). All spectra and ion-current integration regions were manually checked to ensure reliability prior to quantification.
The total RNA was extracted from the cells using the RNA extraction kit (Abclonal RK30120), and cDNA was obtained using the reverse transcription kit (Abclonal RK20429). Quantitative PCR (qPCR) was then performed using the SYBR Green Fast qPCR Mix (Abclonal RK21203) on the Bio-Rad system (Supplementary Table S2).
Appropriate proteins or histones were lysed with 2× loading buffer (100 mM of Tris-Cl, 4% SDS, 0.2% bromophenol blue, 20% glycerol, and 200 mM of DTT), and the solution was boiled at 99°C for denaturation. Then, approximately 2.0 μg of the protein or histone was separated using a polyacrylamide gel before transferring to a nitrocellulose membrane. The membrane was then blocked and immunoblotted with appropriate antibodies, before washing and incubating with peroxidase-conjugated secondary antibodies. Lastly, the membranes were washed and imaged by chemiluminescence (Clinx, chemiscope 6000).
RNA was extracted from M1 macrophages treated with 30 μM of LIQ (three control and three LIQ groups) using TRIzol reagent according to manufacturer protocols (Invitrogen, CA, United States). Then, RNA purity and quantification were achieved using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific). The RNA integrity, library construction, transcriptome sequencing, and analysis were conducted by OE Biotech Co., Ltd. (Shanghai, China).
Statistical analyses were performed using GraphPad Prism software. All data were presented as mean ± standard error of the mean (SEM). Unpaired two-tailed Student’s t-test with equal variance was performed. The statistical details of each experiment are shown in the figure legends. The significance is indicated with symbols as follows: **p < 0.01 or ##p < 0.01, *p < 0.05, and not significant: p > 0.05.
HDACs are divided into the Rpd3/Hda1 and sirtuin families, which function through either zinc-dependent or NAD+-dependent mechanisms (Seto and Yoshida, 2014; Yang and Seto, 2008). Vorinostat (SAHA) is the first marketed inhibitor of the Rpd3/Hda1 family of HDACs, and NAM is an effective biochemical inhibitor of sirtuins (Anderson et al., 2003; Gallo et al., 2004; Bitterman et al., 2002). The synergistic effects of SAHA and NAM can significantly increase histone acetylation levels (Figure 1A). Similarly, lysine-specific demethylase (LSD) and Jumonji C (JMJC) are two evolutionarily conserved families of histone demethylases that have different reaction mechanisms. The LSD family is demethylated into monomethylated and dimethylated substrates by the flavin adenine dinucleotide (FAD)-dependent amine oxidation reactions, and the JMJC family id demethylated into mono-, di-, and tri-methylated residues through Fe (II) and α-ketoglutarate (Bitterman et al., 2002).
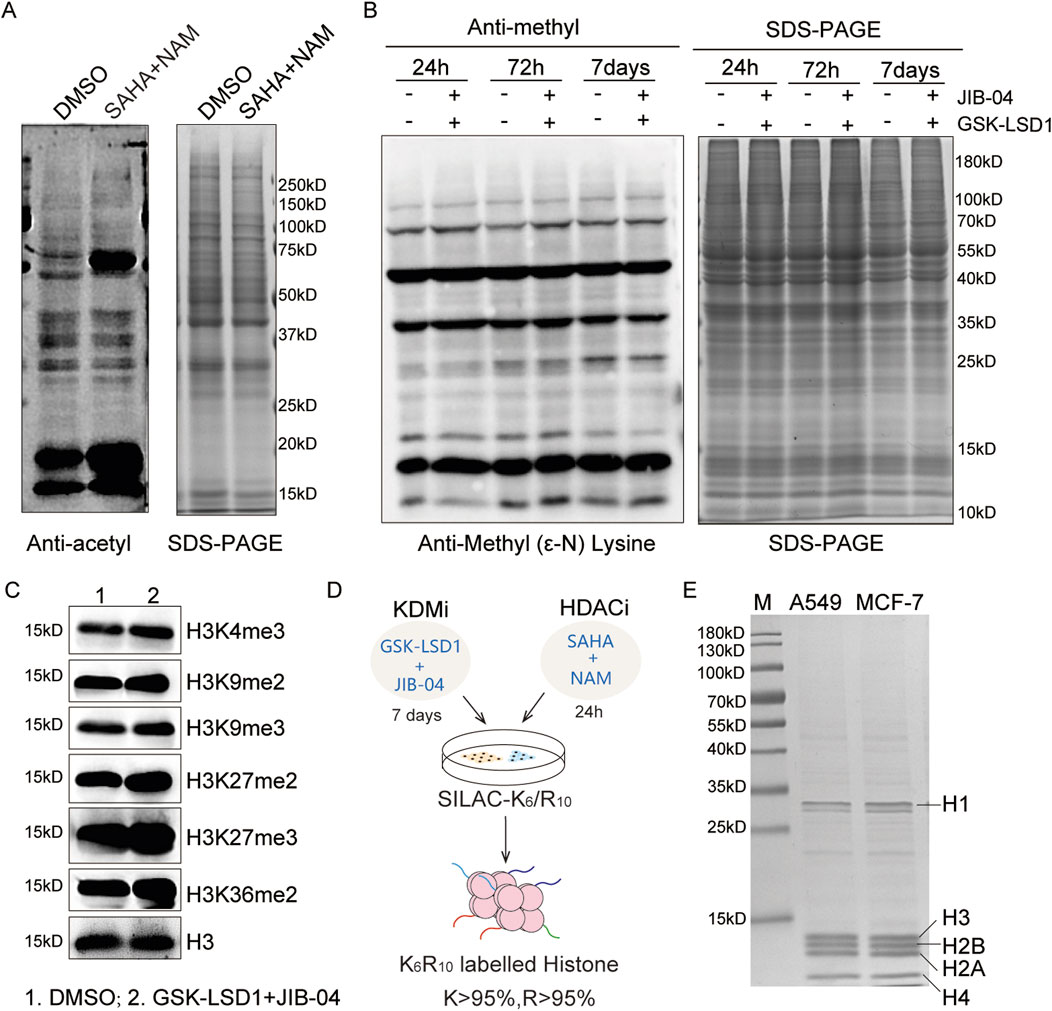
Figure 1. Optimization of the super-SILAC strategy: (A) immunoblot analysis of the global acetylation levels after treatments with SAHA (5.0 μM) and NAM (10 mM) for 24 h; (B, C) immunoblot analyses of the global and site-specific methylation levels following treatments with GSK-LSD1·2HCl (2.0 μM) and JIB-04 (0.1 μM); (D) workflow for the optimized super-SILAC strategy; (E) SDS-PAGE analysis of the purity of heavy histones extracted from A549 and MCF-7 cells.
We combined the two compounds GSK-LSD1·2HCl and JIB-04 to increase the histone methylation levels. Owing to the slower dynamics of methylation compared to acetylation, no significant changes were detected over a short period of time (Zee et al., 2010). We then explored the time-dependent changes in methylation levels, which showed that the histone methylation sites were upregulated after the 7-day treatment, especially at the H3K4me3, H3K9me3, H3K27me2, H3K27me3, and H3K36me2 modification sites (Figures 1B, C). Therefore, we employed GSK-LSD1·2HCl and JIB-04 to treat the cells for 7 d, where SAHA and NAM were added over the last 24 h to prepare the SILAC histone standard samples (Figure 1D). SDS-PAGE results indicate that the histone standard samples have high purity (Figure 1E). Based on increased levels of acetylation and methylation modifications, we obtained heavy-labeled histone mixture standards with labeling efficiencies of K > 95% and R > 95%.
To evaluate qualitative coverage, we utilized the in-gel digestion approach to collect H1, H2A, H2B, H3, and H4 of our histone standard samples. Following LC-MS/MS analysis, the data were analyzed against the UniProt human histone sequence database using Mascot software, which helped identify 12 modification types and 151 modification sites (Figure 2A; Supplementary Table S1). Among these, we found 59 histone PTM sites located on the N-terminal of H3, nine different modification sites on H3K18, and 10 different modification sites on H3K23. In addition, we identified a total of 123 acylation sites containing 42 acetylation sites and 81 other novel acylation sites (Figure 2B). The identified PTM peptides comprised 135 acetylated, 49 propionylated, and 26 crotonylated peptides (Figures 2C, D). Consequently, the diversity of the above modification sites and peptides can assist with subsequent qualitative and quantitative analyses.
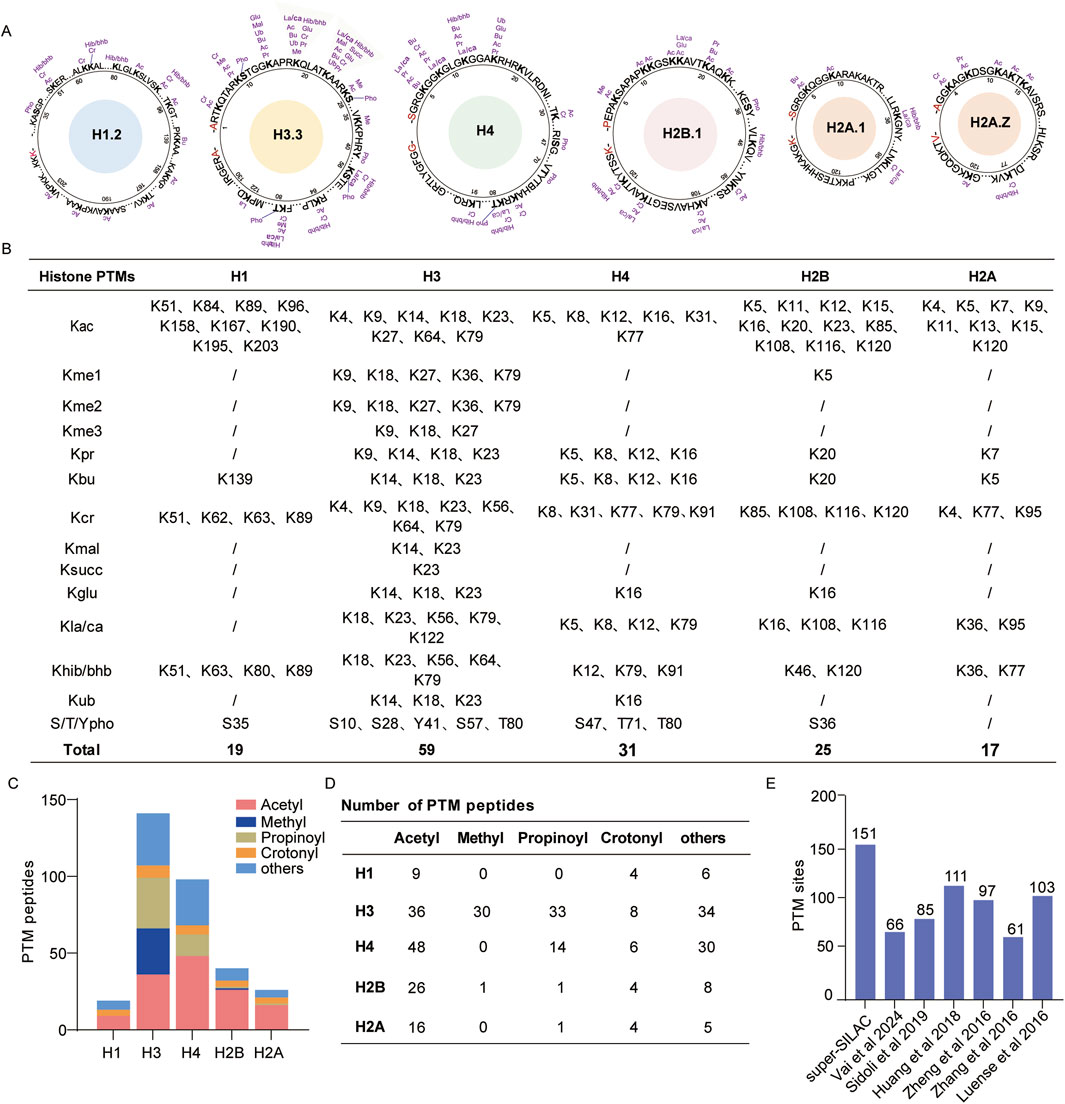
Figure 2. Deep identification of histone post-translational modifications (PTMs) by super-SILAC; (A) map of histone PTM sites identified by in-gel digestion; (B) list of identified histone PTM sites; (C, D) global distribution of histone PTM peptides; (E) number of histone PTM sites identified in this study and comparison with those of other histone PTM profiling studies. Abbreviations: lysine acetylation (Kac), monomethylation (Kme1), dimethylation (Kme2), trimethylation (Kme3), propionylation (Kpr), butyrylation (Kbu), crotonylation (Kcr), malonylation (Kmal), succinylation (Ksucc), ubiquitination (Kub), lactylation/carboxyethylation (Kla/Kca), 2-hydroxyisobutyrylation/β-hydroxybutyrylation (Khib/Kbhb), arginine monomethylation (Rme1), arginine dimethylation (Rme2), and serine/threonine/tyrosine phosphorylation (S/T/Ypho).
We compared our results with the findings of other proteomics-based histone PTM profiling studies. Vai et al. (2024) identified 66 histone PTM sites in clinical breast cancer tissue with the optimized histone in-gel derivatization procedure. Sidoli et al. (2019) identified 85 histone PTM sites using the SILAC method in the murine T lymphoblast cell line EL4. Huang et al. (2018) identified 111 histone PTM sites in six cancer cell lines using in-gel digestion. Zheng et al. (2016) identified 97 histone PTM sites through in-gel digestion in mouse embryonic stem cells and neural progenitor cells. Luense et al. (2016) obtained 103 histone PTM sites in human germ cells using propionylation labeling and trypsin digestion. Compared with these results, our optimized super-SILAC strategy shows deeper peptide coverage and more histone modification sites (Figure 2E). The proposed compound stimulation strategy enhances the abundances of various histone PTMs, especially those with low abundances, which allows an in-depth profiling dataset of the histone PTMs.
We initially synthesized five histone-modified peptides, namely, H3K18ac, H3K23ac, H3K27ac, H3K23pr, and H3K18bu/K23ac, to evaluate the quantitative reproducibility and accuracies of our histone standard samples. The purity of these peptides was verified by LC-MS/MS, and the results show that the heavy-labeled histone standard peptide and light-labeled synthesized peptide exhibit high-quality and nearly consistent retention times of the MS/MS spectra (Figures 3A, B). Based on the purities of the five synthesized peptides, we combined them into a mixture with specific proportions to ensure approximately equal quantitative areas under the curve (AUCs) in the extracted ion chromatograms (XICs); then, the histone standard peptides were mixed with above synthesized peptide mixture at ratios of 1:1, 1:3, and 1:9 (Figure 3B). The quantitative results show high correlation (R > 0.99) between the replications in different groups, demonstrating good reproducibility (Figure 3C; Supplementary Table S2). Furthermore, we evaluated the normalized ratio and absolute relative error of each peptide, whose results show consistent quantitative trends with the mixing ratios (Figure 3D) and less than 30% error compared to the theoretical values (Figure 3E). The coefficient of variation (CV) values of all groups were below 20%, indicating relatively low variability (Figure 3F). Thus, our histone standard samples demonstrate effective reproducibility and high accuracies for quantitative analysis at the peptide level.
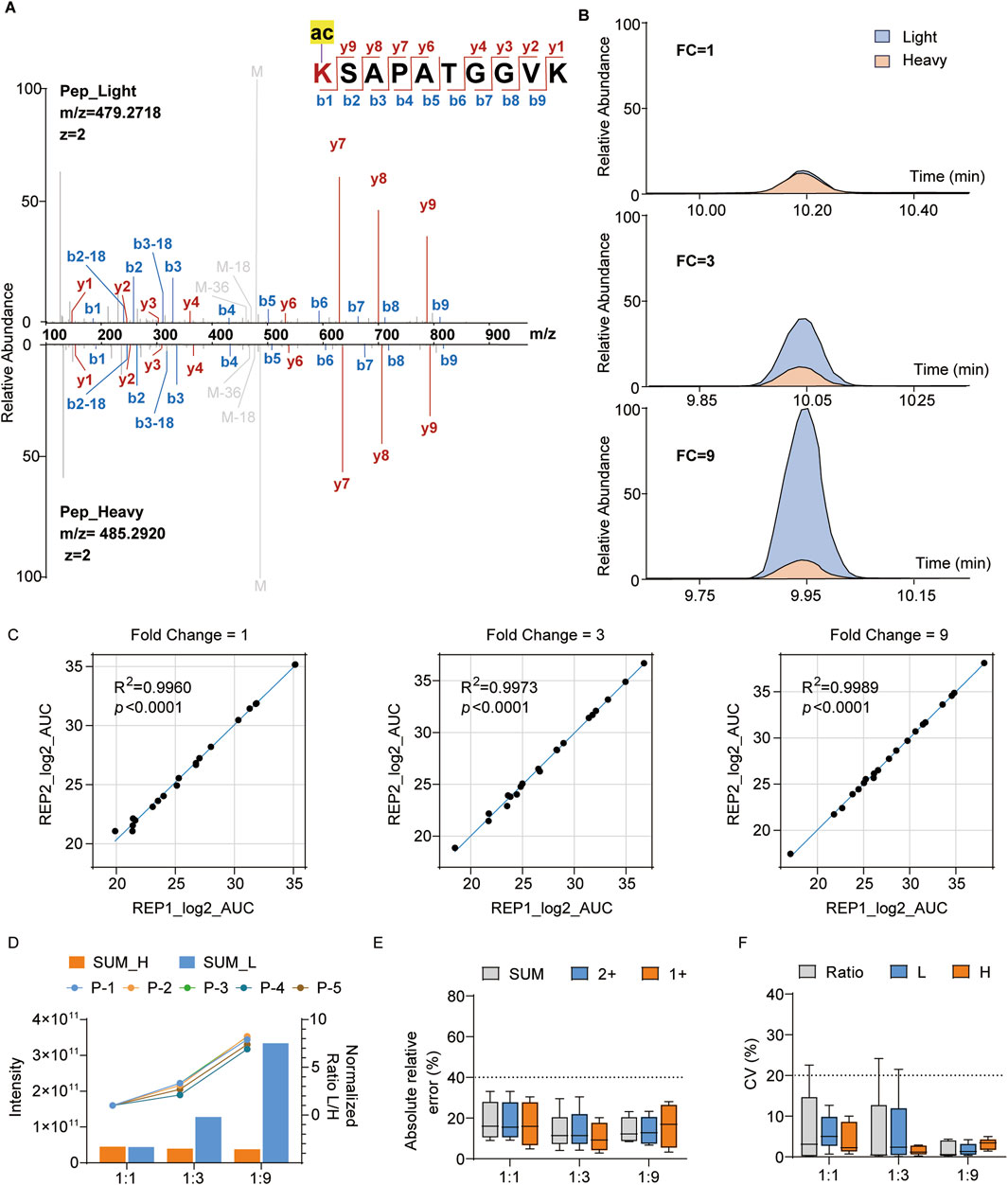
Figure 3. Quantification of the synthesized histone modification peptides with heavy histone standards: (A) representative MS/MS spectrum of the light-labeled synthesized peptides and heavy-labeled histone peptides with the sequence KSAPATGGVK (H3K27ac); (B) relative abundances of the representative peptides in (A) at different mixing ratios (blue: light-labeled peptide; yellow: heavy-labeled peptide); (C) correlation analysis between two replicates at different mixing ratios (REP1: replicate 1, REP2: replicate 2); (D) quantification intensities of the peptides and normalized ratios across different groups; (E, F) absolute relative errors and coefficient of variation (CV) values for different groups.
It is widely acknowledged that histone PTMs play crucial regulatory roles in cancer. Extensively studied histone marks, such as H3K9me3, H3K9ac, and H3K14ac, that are associated with the activation of gene expression (Liu et al., 2023) exhibit consistent increasing trends in Luminal A-like, Luminal B-like, and triple-negative breast cancer, suggesting that they could be used as potential biomarkers (Noberini et al., 2019). Similarly, our WB results show consistent upregulation of these sites in triple-negative breast cancer cells (MDA-MB-468 and MDA-MB-231) relative to non-tumorigenic breast epithelial cells (MCF-10A) (Figure 4A). Some authors have systematically analyzed histone PTMs in breast cancer cell lines through the super-SILAC method (Cuomo et al., 2011; Noberini et al., 2018; Noberini et al., 2019); however, their works have only focused on histone PTMs of H3 and H4. Therefore, we aimed to systematically assess the differences between cancer cells and ordinary epithelial cells to further evaluate our approach in a complex sample system.
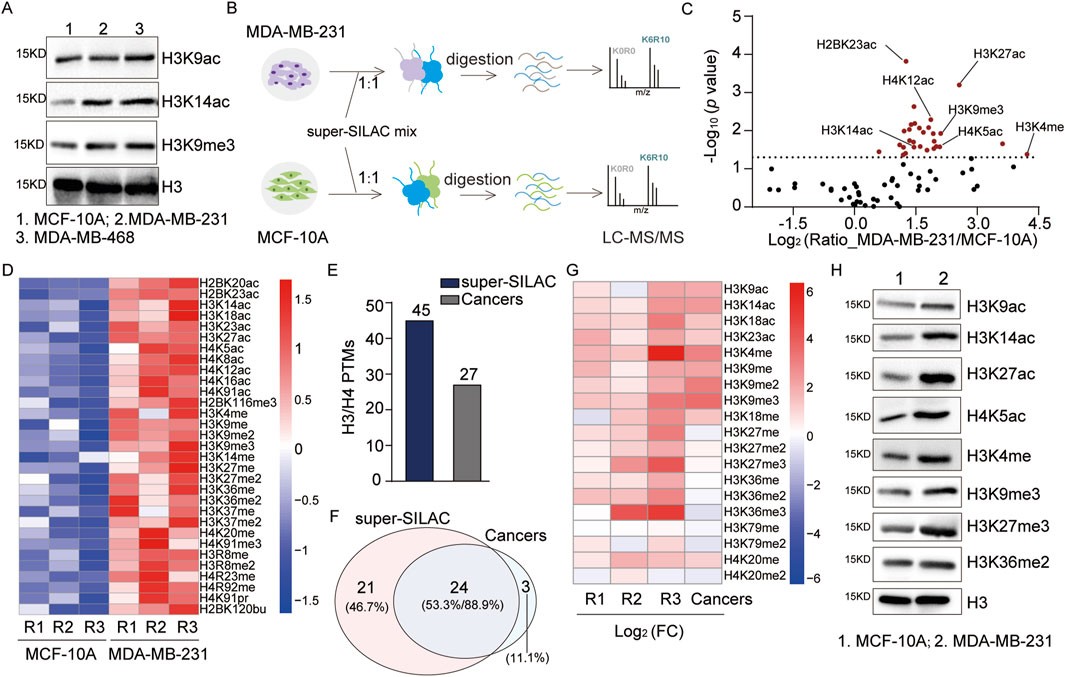
Figure 4. Systematic analysis of the histone PTMs in breast cancer cells: (A) immunoblot analyses of the levels of H3K9ac, H3K14ac, and H3K9me3 in breast cancer and non-tumorigenic breast epithelial cells; (B) workflow for the systematic analysis of histone PTM sites in breast cancer cells; (C) scatter plot analysis of the differential histone PTM sites (red dots p < 0.05); (D) heatmap analysis of the quantitative sites with p-values <0.05 in MCF-10A and MDA-MB-231 groups based on three replicates (R1: replicate 1, R2: replicate 2, R3: replicate 3); (E, F) comparative analysis of the numbers and overlaps of H3/H4 PTM sites identified in this study with those from a previous study by Noberini et al. (2018); (G) comparative heatmap analysis of the quantitative H3/H4 PTM sites identified by Noberini et al. (2018); (H) immunoblot analysis of specific histone PTM sites in breast cancer and non-tumorigenic breast epithelial cells.
By applying the optimized super-SILAC method, we extracted histones from MCF-10A and MDA-MB-231 cell lines as well as performed quantitative analyses of the histone PTMs (Figure 4B). Compared to non-tumorigenic breast epithelial cells, a total of 82 histone PTM sites were quantified (Figure 4C; Supplementary Table S3A), which contained 31 significantly upregulated sites (p < 0.05) (Figure 4D). In addition to previously reported upregulated sites, such as H3K4me, H3K9ac, and H4K14ac, we quantified two uncharacterized sites, namely, H4K91pr and H2BK120bu, which were also significantly upregulated (p < 0.05) (Figure 4D). Focusing on H3 and H4, we quantitatively analyzed 45 sites that encompass the majority of sites identified by Noberini et al. (2018) (Figures 4E, F; Supplementary Table S3B). The comparison of overlapping identified sites showed consistent quantitative results for most sites (Figure 4G). For further validation, we assessed the variation trends of the PTM sites via WB, which were consistent with the above quantitative results (Figure 4H). In conclusion, our optimized super-SILAC method for quantitative analysis demonstrates high reliability at both the peptide and cellular lysate levels. These results highlight the potential of our histone standard samples in elucidating disease pathology and drug mechanisms.
Histone PTMs, especially acetylation, enable regulation of the transcriptional repression of macrophage inflammatory responses (Shi et al., 2024). The THP1 cells, which are commonly used to model macrophage differentiation, were induced into the M0, M1, and M2 phenotypes; their acetylation levels were assessed by pan-acetylation antibodies, and the results revealed a significant downregulation of acetylation in M1 macrophages and marked upregulation in M2 macrophages (Figure 5A).
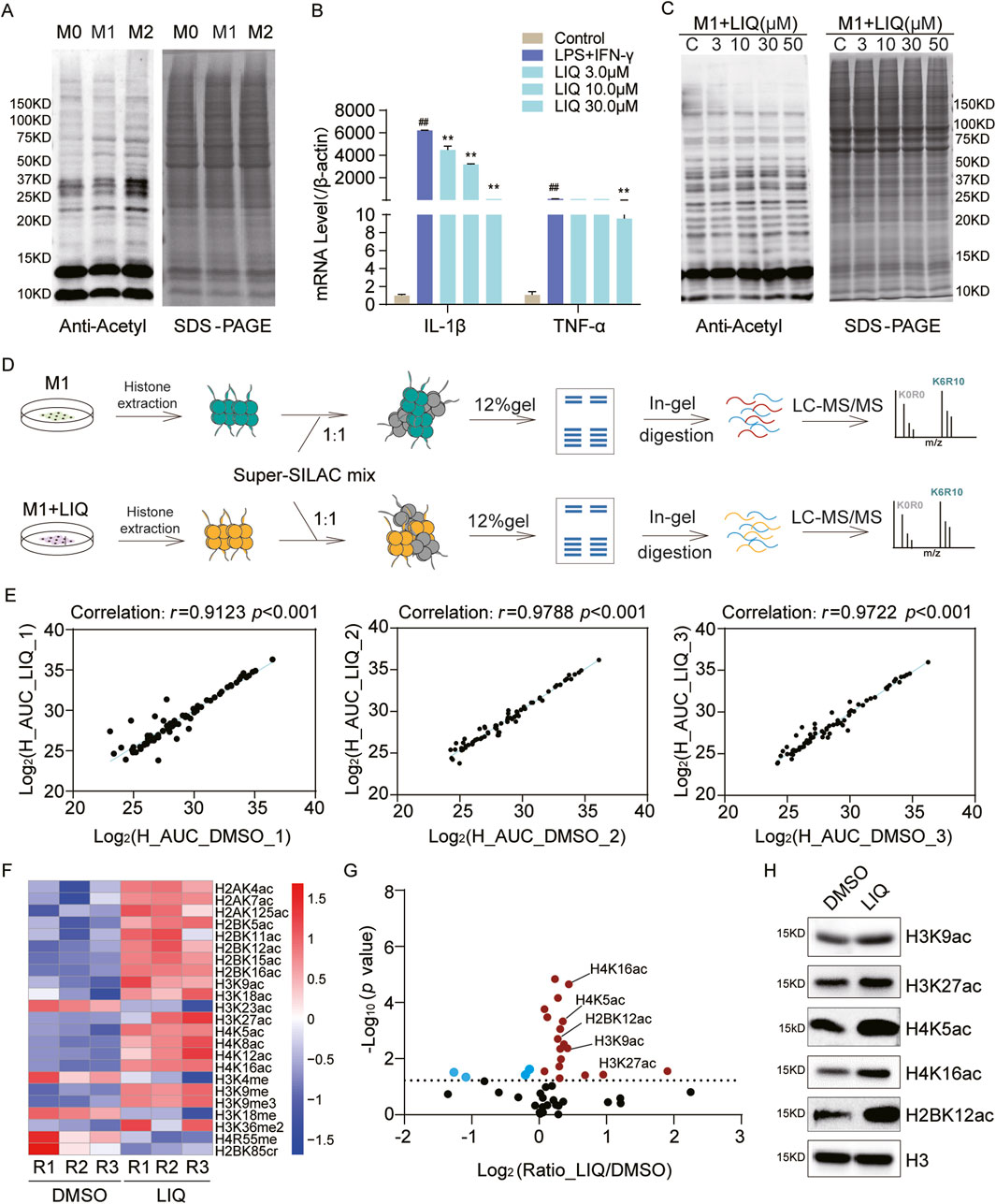
Figure 5. Systematic analysis of histone PTMs regulated by liquiritigenin (LIQ) in macrophages; (A) immunoblot analysis of the global acetylation levels in M0, M1, and M2 phenotype macrophages. (B) RT-qPCR analysis of the IL-1β and TNFα mRNA expressions following LIQ treatment from three replicates; mean ± standard error of the mean (SEM) (n = 3), ##p < 0.01 compared with the control group, **p < 0.01 compared with the lipopolysaccharide + IFN-γ group; (C) immunoblot analysis of the global acetylation levels in M1 macrophages treated with LIQ; (D) workflow for the identification of LIQ-mediated histone PTM sites in M1 macrophages; (E) correlation analysis of the heavy peptides between dimethyl sulfoxide (DMSO) and LIQ groups based on three replicates; (F) heatmap analysis of the quantitative histone PTM sites with p < 0.05 across the replicates; (G) scatter plot analysis of the differential histone PTM sites, where red dots represent up-regulated sites and blue dots represent down-regulated sites (p < 0.05); (H) immunoblot analysis of the specific histone PTM sites after LIQ treatment.
LIQ is a dihydroflavonone monomer found in licorice and has been widely demonstrated to exert natural anti-inflammatory effects (Babu et al., 2023; Tu et al., 2019; Yu et al., 2015). To explore the epigenetic regulatory mechanisms of LIQ, we treated M1 macrophages with varying concentrations of LIQ (3, 10, 30, and 50 μM), with dimethyl sulfoxide (DMSO) as the control. The qPCR results showed that LIQ treatment significantly inhibited the expressions of inflammatory factors (IL-1β and TNF-α) in M1 macrophages (Figure 5B). Furthermore, statistical analysis of the inflammatory factors revealed significant downregulation of IL-1β, IL-R1, NOS2, and TNF (Supplementary Figure S1C), indicating potential anti-inflammatory effects of LIQ. WB revealed that LIQ treatment upregulated histone acetylation levels in a concentration-dependent manner (Figure 5C), suggesting that LIQ may modulate inflammatory responses through the regulation of histone PTMs.
We next applied super-SILAC to identify LIQ-mediated histone PTMs in M1 macrophages (Figure 5D; Supplementary Table S4). Comparisons of the DMSO and LIQ groups across three biological replicates revealed a high degree of correlation (r > 0.91) (Figure 5E). A total of 49 histone PTM sites were quantified, which were mapped to 15 PTM types. We found that most of the acetylation sites and several methylation sites were upregulated in the LIQ-treated groups (p < 0.05) (Figure 5F). Scatter plot analysis revealed significant upregulation of these sites across all three replicates (p < 0.05), such as H3K9ac, H3K27ac, H4K5ac, H4K16ac, and H2BK12ac (Figure 5G). WB analysis confirmed consistency with the MS analysis results (Figure 5H). The above findings highlight that LIQ can potentially regulate epigenetic modification alterations to participate in anti-inflammatory mechanisms.
LIQ treatment resulted in changes to the histone PTM landscape in macrophages, suggesting that it could affect the expressions of certain genes. Volcano plot analysis and principal component analysis (PCA) of the altered genes revealed significant differences between the LIQ-treated and control groups (Figure 6A; Supplementary Figure S1A; Supplementary Table S5A). A total of 473 differentially expressed genes (DEGs) were identified based on a significance threshold of p < 0.05 and fold change >2.0 (Supplementary Table S5B). Among these, 157 genes were significantly upregulated, whereas 316 genes were significantly downregulated (Figure 6B; Supplementary Figure S1B). Gene ontology (GO) molecular function analysis revealed that LIQ affected the structural constituents of the ribosomes, rRNA binding, and histone deacetylase binding (false detection rate (FDR) <0.05). In addition, these DEGs were involved in biological processes such as translation, protein localization, inflammatory responses, and miRNA metabolism. The GO cellular components analysis showed that these genes were primarily located in the ribosomes and protein complexes (FDR <0.05) (Figure 6C).
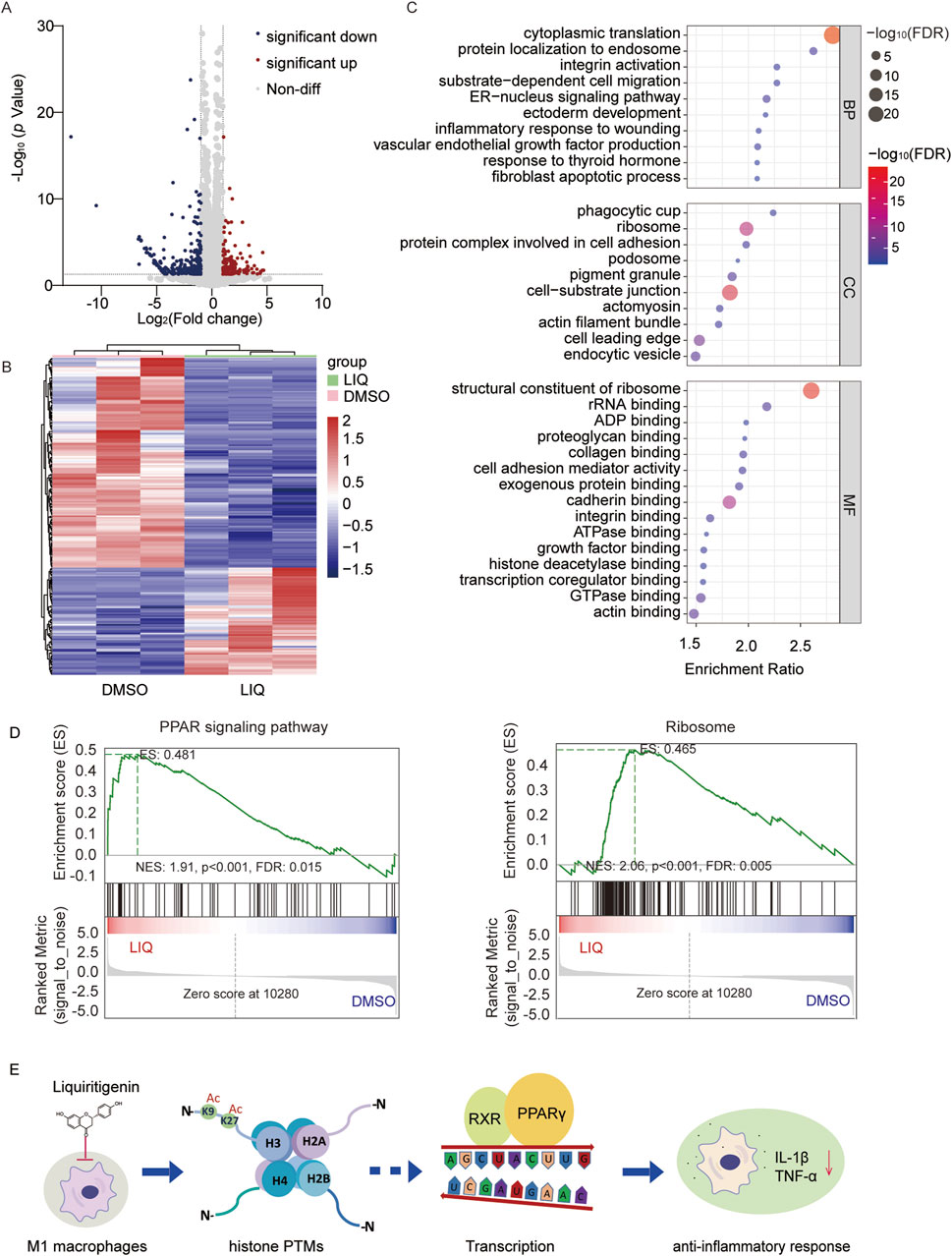
Figure 6. LIQ regulates gene expressions through the peroxisome proliferator-activated receptor (PPAR) signaling pathway in anti-inflammatory processes. (A) Volcano plot of the genes identified in M1 macrophages following LIQ treatment; the red and blue dots represent significantly upregulated and downregulated genes, respectively, while the gray dots represent non-differential genes; representative differentially expressed genes (-log10 p = 0–30, p < 0.05) are displayed, and all significant data are shown in Supplementary Figure S2. (B) Cluster analysis of the differentially expressed genes (fold change >2.0 and p < 0.01) in the DMSO and LIQ groups. (C) Gene Ontology (GO) analysis of the related genes with p < 0.05, showing top-10 biological processes (BPs) and cellular components (CCs) as well as top-15 molecular functions (MFs). (D) Gene set enrichment analysis (GSEA) through comparisons with the KEGG database for the PPAR signaling pathway and ribosome enrichment in the LIQ groups (FDR <0.05). (E) Proposed mechanisms underlying the anti-inflammatory effects of LIQ.
Gene set enrichment analysis (GSEA) was used to identify the significantly enriched pathways, such as PPAR signaling and ribosome pathways (FDR <0.05) (Figure 6D). The PPAR signaling pathway is reportedly involved in anti-inflammatory responses. Moreover, activation of the PPARs may influence acetylase activity, and changes to the acetylation levels could in turn modulate PPAR signaling (Zeng et al., 2022). Considering the upregulation of histone acetylation by LIQ, we also examined the genes related to deacetylases. The results showed significant downregulation of deacetylases, such as HDAC4, HDAC5, HDAC8, HDAC10, SIRT2, SIRT3, and SIRT7, indicating that their genes may influence epigenetic regulation by mediating alterations in deacetylase upon LIQ treatment (Supplementary Figure S1D). In summary, transcriptome analysis revealed that LIQ may exert anti-inflammatory effects by modulating the histone PTMs and regulating gene expressions through the PPAR signaling pathway (Figure 6E).
Given their planar ring structures, dihydroflavonones exhibit a broad spectrum of biological activities, encompassing anti-inflammatory and antitumor effects. LIQ is a dihydroflavonone monomer compound derived from natural products. However, the relationships between its anti-inflammatory effects and epigenetic regulatory mechanisms remain unclear (Hu et al., 2017; Chagas et al., 2022). In the present study, we optimized the super-SILAC method through compound stimulations and found that LIQ could exert anti-inflammatory activity by regulating the histone PTMs and gene expressions through the PPAR signaling pathway in M1 macrophages. The optimized method provides a powerful tool for the epigenetic study of natural products and offers insights into the epigenetic mechanisms of LIQ.
Compared with previous research, we improved the abundances of the PTM peptides through compound stimulations. Based on quantitative analyses at the peptide and cellular lysate levels, we demonstrated high accuracy and reproducibility of our histone standard samples. Moreover, the optimized method was applicable to both cell and tissue sample types. Notably, the quantitative accuracy of high-abundance modifications was relatively higher than that of low-abundance modifications at the synthesized peptide level. At the cellular lysate level, the shared identified sites between the findings of Noberini et al. (2018) and our results showed consistent trends for most sites, and discrepancies were observed only in a few sites, such as H4K20me3 and H4K16ac, which could be attributed to variations in the cell line cultures or MS detection method. Notably, the widespread upregulation highlights aberrant epigenetic regulation in breast cancer cells, potentially driving the expressions of oncogenes. The underlying mechanisms of this phenomenon merit further exploration.
Histone acetylation and methylation are associated with transcriptional activation and chromatin structure alterations (Shahbazian and Grunstein, 2007). We found that LIQ upregulated the acetylation and methylation sites, such as H3K9ac, H3K27ac, H4K5ac, H4K16ac, and H2BK12ac, in M1 macrophages. Thus, we hypothesized that LIQ exerted anti-inflammatory function through epigenetic regulation. Through transcriptome analysis, we found that LIQ significantly affected the PPAR signaling pathway, which is involved in lipid metabolism and inflammatory responses (Daynes and Jones, 2002; Ahmadian et al., 2013; Han et al., 2017; Maréchal et al., 2018). PPAR activation affects deacetylase activity, enhancing H3 acetylation and promoting macrophage polarization (Liu et al., 2020; Wang et al., 2022); alteration of the acetylation level thus regulates PPAR signaling, which is cooperatively involved in the regulation of gene expression (Zeng et al., 2022). Furthermore, H3K27ac directly affects the expression of PPARγ by enhancing its transcriptional activity (Duan et al., 2022). The increase in H3K9ac/14ac positively regulates the transcriptional activity of PPARγ (Małodobra-Mazur et al., 2024), and PPARγ activation is an indispensable factor in maintaining the acetylation levels of H3K9 and H3K27 (Yuan et al., 2021). All of the above evidence further confirms that the anti-inflammatory mechanisms of LIQ are closely related to histone epigenetic regulation and the PPAR signaling pathway. In addition, we found that the XIST and CD200 genes were silenced by LIQ, and several transcription factors like the RXRG, KLF4, FOXP3, and ERG were significantly regulated (p < 0.05, fold change >2.0) (Supplementary Table S5). Thus, the detailed mechanisms by which LIQ silences XIST, CD200, and their transcription factors need further investigation. In summary, the findings of this study advance our understanding of the anti-inflammatory mechanisms of dihydroflavonones in macrophages and provide a strategy for future research on the epigenetic mechanisms of natural products.
The original contributions presented in this study are included in the article/Supplementary Material, and any further inquiries may be directed to the corresponding authors.
Ethical approval was not required for the studies on humans in accordance with local legislation and institutional requirements because only commercially available established cell lines were used in this work.
PL: investigation, methodology, project administration, supervision, visualization, writing–original draft, and writing–review and editing. JuZ: investigation, methodology, software, visualization, writing–original draft, and writing–review and editing. JiZ: investigation, methodology, and writing–review and editing. YY: data curation, visualization, and writing–review and editing. ZL: investigation, methodology, and writing–review and editing. SC: investigation, methodology, and writing–review and editing. KC: investigation, supervision, and writing–review and editing. LD: investigation and writing–review and editing. ZC: Data curation, writing–review and editing. YZ: resources, supervision, and writing–review and editing. MG: project administration and writing–review and editing. MT: funding acquisition, resources, supervision, and writing–review and editing. WZ: methodology, project administration, software, and writing–review and editing. DX: project administration, writing–review and editing, funding acquisition.
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key Research and Development Program of China (no. 2024YFC2815903), Natural Science Foundation of China (nos. 22225702 and 82272943), Program of Shanghai Academic Research Leader (no. 22XD1420900), and Science and Technology Commission of Shanghai Municipality (no. 21Y11913400).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2025.1566567/full#supplementary-material
Ahmadian, M., Suh, J. M., Hah, N., Liddle, C., Atkins, A. R., Downes, M., et al. (2013). PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med. 19, 557–566. doi:10.1038/nm.3159
Anderson, R. M., Bitterman, K. J., Wood, J. G., Medvedik, O., and Sinclair, D. A. (2003). Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185. doi:10.1038/nature01578
Babu, V., Kapkoti, D. S., Binwal, M., Bhakuni, R. S., Shanker, K., Singh, M., et al. (2023). Liquiritigenin, isoliquiritigenin rich extract of glycyrrhiza glabra roots attenuates inflammation in macrophages and collagen-induced arthritis in rats. Inflammopharmacology 31, 983–996. doi:10.1007/s10787-023-01152-w
Baker, D. D., Chu, M., Oza, U., and Rajgarhia, V. (2007). The value of natural products to future pharmaceutical discovery. Nat. Prod. Rep. 24, 1225–1244. doi:10.1039/b602241n
Bern, M., Phinney, B. S., and Goldberg, D. (2009). Reanalysis of tyrannosaurus rex mass spectra. J. Proteome Res. 8, 4328–4332. doi:10.1021/pr900349r
Bitterman, K. J., Anderson, R. M., Cohen, H. Y., Latorre-Esteves, M., and Sinclair, D. A. (2002). Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 277, 45099–45107. doi:10.1074/jbc.M205670200
Cai, L. Y., Chen, S. J., Xiao, S. H., Sun, Q. J., Ding, C. H., Zheng, B. N., et al. (2021). Targeting p300/CBP attenuates hepatocellular carcinoma progression through epigenetic regulation of metabolism. Cancer Res. 81, 860–872. doi:10.1158/0008-5472.Can-20-1323
Chagas, M., Behrens, M. D., Moragas-Tellis, C. J., Penedo, G. X. M., Silva, A. R., and GonçALVES-De-Albuquerque, C. F. (2022). Flavonols and flavones as potential anti-inflammatory, antioxidant, and antibacterial compounds. Oxid. Med. Cell. Longev. 2022, 9966750. doi:10.1155/2022/9966750
Chan, J. C., and Maze, I. (2020). Nothing is yet set in (Hi)stone: novel post-translational modifications regulating chromatin function. Trends Biochem. Sci. 45, 829–844. doi:10.1016/j.tibs.2020.05.009
Choudhary, C., Weinert, B. T., Nishida, Y., Verdin, E., and Mann, M. (2014). The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell. Biol. 15, 536–550. doi:10.1038/nrm3841
Cravatt, B. F., Simon, G. M., and Yates, J. R. (2007). The biological impact of mass-spectrometry-based proteomics. Nature, 450, 991–1000. doi:10.1038/nature06525
Cuomo, A., Moretti, S., Minucci, S., and Bonaldi, T. (2011). SILAC-based proteomic analysis to dissect the “histone modification signature” of human breast cancer cells. Amino Acids 41, 387–399. doi:10.1007/s00726-010-0668-2
Daynes, R. A., and Jones, D. C. (2002). Emerging roles of PPARs in inflammation and immunity. Nat. Rev. Immunol. 2, 748–759. doi:10.1038/nri912
Duan, P., Wang, H., Yi, X., Zhang, H., Chen, H., and Pan, Z. (2022). C/EBPα regulates the fate of bone marrow mesenchymal stem cells and steroid-induced avascular necrosis of the femoral head by targeting the PPARγ signalling pathway. Stem Cell. Res. Ther. 13, 342. doi:10.1186/s13287-022-03027-3
Egger, A. S., Rauch, E., Sharma, S., Kipura, T., Hotze, M., Mair, T., et al. (2024). Linking metabolism and histone acetylation dynamics by integrated metabolic flux analysis of Acetyl-CoA and histone acetylation sites. Mol. Metab. 90, 102032. doi:10.1016/j.molmet.2024.102032
Ferrari, K. J., Scelfo, A., Jammula, S., Cuomo, A., Barozzi, I., StüTZER, A., et al. (2014). Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell. 53, 49–62. doi:10.1016/j.molcel.2013.10.030
Gallo, C. M., Smith, D. L., and Smith, J. S. (2004). Nicotinamide clearance by Pnc1 directly regulates Sir2-mediated silencing and longevity. Mol. Cell. Biol. 24, 1301–1312. doi:10.1128/mcb.24.3.1301-1312.2004
Han, L., Shen, W. J., Bittner, S., Kraemer, F. B., and Azhar, S. (2017). PPARs: regulators of metabolism and as therapeutic targets in cardiovascular disease. Part I: PPAR-α. Future Cardiol. 13, 259–278. doi:10.2217/fca-2016-0059
He, W., Li, Q., and Li, X. (2023). Acetyl-CoA regulates lipid metabolism and histone acetylation modification in cancer. Biochim. Biophys. Acta Rev. Cancer 1878, 188837. doi:10.1016/j.bbcan.2022.188837
Huang, X., Yan, J., Zhang, M., Wang, Y., Chen, Y., Fu, X., et al. (2018). Targeting epigenetic crosstalk as a therapeutic strategy for EZH2-aberrant solid tumors. Cell. 175, 186–199. doi:10.1016/j.cell.2018.08.058
Hu, C., Zhou, Z., Xiang, Y., Song, X., Wang, H., Tao, K., et al. (2017). Design, synthesis and anti-inflammatory activity of dihydroflavonol derivatives. Med. Chem. Res. 27, 194–205. doi:10.1007/s00044-017-2054-z
Hyun, K., Jeon, J., Park, K., and Kim, J. (2017). Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 49, e324. doi:10.1038/emm.2017.11
Kim, Y. W., Zhao, R. J., Park, S. J., Lee, J. R., Cho, I. J., Yang, C. H., et al. (2008). Anti-inflammatory effects of liquiritigenin as a consequence of the inhibition of NF-kappaB-dependent iNOS and proinflammatory cytokines production. Br. J. Pharmacol. 154, 165–173. doi:10.1038/bjp.2008.79
Kingston, D. G. (2011). Modern natural products drug discovery and its relevance to biodiversity conservation. J. Nat. Prod. 74, 496–511. doi:10.1021/np100550t
Kirchner, M., and Selbach, M. (2012). In vivo quantitative proteome profiling: planning and evaluation of SILAC experiments. Methods Mol. Biol. 893, 175–199. doi:10.1007/978-1-61779-885-6_13
Kuznetsova, T., Prange, K. H. M., Glass, C. K., and De Winther, M. P. J. (2020). Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat. Rev. Cardiol. 17, 216–228. doi:10.1038/s41569-019-0265-3
Lawrence, T., and Natoli, G. (2011). Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 11, 750–761. doi:10.1038/nri3088
Li, J., Ahn, J. H., and Wang, G. G. (2019). Understanding histone H3 lysine 36 methylation and its deregulation in disease. Cell. Mol. Life Sci. 76, 2899–2916. doi:10.1007/s00018-019-03144-y
Li, J., Zhao, R., Miao, P., Xu, F., Chen, J., Jiang, X., et al. (2023). Discovery of anti-inflammatory natural flavonoids: diverse scaffolds and promising leads for drug discovery. Eur. J. Med. Chem. 260, 115791. doi:10.1016/j.ejmech.2023.115791
Liu, X., Guo, C., Leng, T., Fan, Z., Mai, J., Chen, J., et al. (2023). Differential regulation of H3K9/H3K14 acetylation by small molecules drives neuron-fate-induction of glioma cell. Cell. Death Dis. 14, 142. doi:10.1038/s41419-023-05611-8
Liu, Y., Luo, S., Zhan, Y., Wang, J., Zhao, R., Li, Y., et al. (2020). Increased expression of PPAR-γ modulates monocytes into a M2-like phenotype in SLE patients: an implicative protective mechanism and potential therapeutic strategy of systemic lupus erythematosus. Front. Immunol. 11, 579372. doi:10.3389/fimmu.2020.579372
Luense, L. J., Wang, X., Schon, S. B., Weller, A. H., Lin Shiao, E., Bryant, J. M., et al. (2016). Comprehensive analysis of histone post-translational modifications in mouse and human male germ cells. Epigenetics Chromatin 9, 24. doi:10.1186/s13072-016-0072-6
Maile, T. M., Izrael-Tomasevic, A., Cheung, T., Guler, G. D., Tindell, C., Masselot, A., et al. (2015). Mass spectrometric quantification of histone post-translational modifications by a hybrid chemical labeling method. Mol. Cell. Proteomics 14, 1148–1158. doi:10.1074/mcp.O114.046573
Małodobra-Mazur, M., Ołdakowska, M., and Dobosz, T. (2024). Exploring PPAR gamma and PPAR alpha's regulation role in metabolism via epigenetics mechanism. Biomolecules 14, 1445. doi:10.3390/biom14111445
MaréCHAL, L., Laviolette, M., Rodrigue-Way, A., Sow, B., Brochu, M., Caron, V., et al. (2018). The CD36-pparγ pathway in metabolic disorders. Int. J. Mol. Sci. 19, 1529. doi:10.3390/ijms19051529
MilláN-Zambrano, G., Burton, A., Bannister, A. J., and Schneider, R. (2022). Histone post-translational modifications - cause and consequence of genome function. Nat. Rev. Genet. 23, 563–580. doi:10.1038/s41576-022-00468-7
Netea, M. G., Joosten, L. A., Latz, E., Mills, K. H., Natoli, G., Stunnenberg, H. G., et al. (2016). Trained immunity: a program of innate immune memory in health and disease. Science 352, aaf1098. doi:10.1126/science.aaf1098
Noberini, R., and Bonaldi, T. (2017). A super-SILAC strategy for the accurate and multiplexed profiling of histone posttranslational modifications. Methods Enzymol. 586, 311–332. doi:10.1016/bs.mie.2016.09.036
Noberini, R., Longhi, E., and Bonaldi, T. (2023). A super-SILAC approach for profiling histone posttranslational modifications. Methods Mol. Biol. 2603, 87–102. doi:10.1007/978-1-0716-2863-8_7
Noberini, R., Osti, D., Miccolo, C., Richichi, C., Lupia, M., Corleone, G., et al. (2018). Extensive and systematic rewiring of histone post-translational modifications in cancer model systems. Nucleic Acids Res. 46, 3817–3832. doi:10.1093/nar/gky224
Noberini, R., Restellini, C., Savoia, E. O., Raimondi, F., Ghiani, L., Jodice, M. G., et al. (2019). Profiling of epigenetic features in clinical samples reveals novel widespread changes in cancer. Cancers (Basel) 11, 723. doi:10.3390/cancers11050723
Noberini, R., Uggetti, A., Pruneri, G., Minucci, S., and Bonaldi, T. (2016). Pathology tissue-quantitative mass spectrometry analysis to profile histone post-translational modification patterns in patient samples. Mol. Cell. Proteomics 15, 866–877. doi:10.1074/mcp.M115.054510
Pirro, M., Mohammed, Y., De Ru, A. H., Janssen, G. M. C., Tjokrodirijo, R. T. N., Madunić, K., et al. (2021). Oxonium ion guided analysis of quantitative proteomics data reveals site-specific O-glycosylation of anterior gradient protein 2 (AGR2). Int. J. Mol. Sci. 22, 5369. doi:10.3390/ijms22105369
Riley, N. M., Hebert, A. S., Westphall, M. S., and Coon, J. J. (2019). Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat. Commun. 10, 1311. doi:10.1038/s41467-019-09222-w
Santangelo, C., Varì, R., Scazzocchio, B., Di Benedetto, R., Filesi, C., and Masella, R. (2007). Polyphenols, intracellular signalling and inflammation. Ann. Ist. Super. Sanita 43, 394–405.
Schultze, J. L. (2017). Chromatin remodeling in monocyte and macrophage activation. Adv. Protein Chem. Struct. Biol. 106, 1–15. doi:10.1016/bs.apcsb.2016.09.001
Serafini, M., Peluso, I., and Raguzzini, A. (2010). Flavonoids as anti-inflammatory agents. Proc. Nutr. Soc. 69, 273–278. doi:10.1017/s002966511000162x
Seto, E., and Yoshida, M. (2014). Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6, a018713. doi:10.1101/cshperspect.a018713
Shahbazian, M. D., and Grunstein, M. (2007). Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 76, 75–100. doi:10.1146/annurev.biochem.76.052705.162114
Shi, W., Cassmann, T. J., Bhagwate, A. V., Hitosugi, T., and Ip, W. K. E. (2024). Lactic acid induces transcriptional repression of macrophage inflammatory response via histone acetylation. Cell. Rep. 43, 113746. doi:10.1016/j.celrep.2024.113746
Sidoli, S., Lopes, M., Lund, P. J., Goldman, N., Fasolino, M., Coradin, M., et al. (2019). A mass spectrometry-based assay using metabolic labeling to rapidly monitor chromatin accessibility of modified histone proteins. Sci. Rep. 9, 13613. doi:10.1038/s41598-019-49894-4
Sinha, K. K., Bilokapic, S., Du, Y., Malik, D., and Halic, M. (2023). Histone modifications regulate pioneer transcription factor cooperativity. Nature 619, 378–384. doi:10.1038/s41586-023-06112-6
Theoharides, T. C., Alexandrakis, M., Kempuraj, D., and Lytinas, M. (2001). Anti-inflammatory actions of flavonoids and structural requirements for new design. Int. J. Immunopathol. Pharmacol. 14, 119–127.
Tu, C., Ma, Y., Song, M., Yan, J., Xiao, Y., and Wu, H. (2019). Liquiritigenin inhibits IL-1β-induced inflammation and cartilage matrix degradation in rat chondrocytes. Eur. J. Pharmacol. 858, 172445. doi:10.1016/j.ejphar.2019.172445
Vai, A., Noberini, R., Ghirardi, C., Rodrigues De Paula, D., Carminati, M., Pallavi, R., et al. (2024). Improved mass spectrometry-based methods reveal abundant propionylation and tissue-specific histone propionylation profiles. Mol. Cell. Proteomics 23, 100799. doi:10.1016/j.mcpro.2024.100799
Van Den Bossche, J., Neele, A. E., Hoeksema, M. A., and De Winther, M. P. (2014). Macrophage polarization: the epigenetic point of view. Curr. Opin. Lipidol. 25, 367–373. doi:10.1097/mol.0000000000000109
Wang, R., Zhang, C. Y., Bai, L. P., Pan, H. D., Shu, L. M., Kong, A. N., et al. (2015). Flavonoids derived from liquorice suppress murine macrophage activation by up-regulating heme oxygenase-1 independent of Nrf2 activation. Int. Immunopharmacol. 28, 917–924. doi:10.1016/j.intimp.2015.03.040
Wang, S., Lin, Y., Gao, L., Yang, Z., Lin, J., Ren, S., et al. (2022). PPAR-γ integrates obesity and adipocyte clock through epigenetic regulation of Bmal1. Theranostics 12, 1589–1606. doi:10.7150/thno.69054
Wang, X., Cao, Y., Chen, S., Lin, J., Bian, J., and Huang, D. (2021). Anti-inflammation activity of flavones and their structure-activity relationship. J. Agric. Food Chem. 69, 7285–7302. doi:10.1021/acs.jafc.1c02015
Xie, J., Yu, Z., Zhu, Y., Zheng, M., and Zhu, Y. (2024). Functions of coenzyme A and acyl-CoA in post-translational modification and human disease. Front. Biosci. Landmark Ed. 29, 331. doi:10.31083/j.fbl2909331
Yang, X. J., and Seto, E. (2008). The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell. Biol. 9, 206–218. doi:10.1038/nrm2346
Yu, J. Y., Ha, J. Y., Kim, K. M., Jung, Y. S., Jung, J. C., and Oh, S. (2015). Anti-Inflammatory activities of licorice extract and its active compounds, glycyrrhizic acid, liquiritin and liquiritigenin, in BV2 cells and mice liver. Molecules 20, 13041–13054. doi:10.3390/molecules200713041
Yuan, H., Suzuki, S., Hirata-Tsuchiya, S., Sato, A., Nemoto, E., Saito, M., et al. (2021). Pparγ-induced global H3K27 acetylation maintains osteo/cementogenic abilities of periodontal ligament fibroblasts. Int. J. Mol. Sci. 22, 8646. doi:10.3390/ijms22168646
Zee, B. M., Levin, R. S., Dimaggio, P. A., and Garcia, B. A. (2010). Global turnover of histone post-translational modifications and variants in human cells. Epigenetics Chromatin 3, 22. doi:10.1186/1756-8935-3-22
Zeng, Z., Lyu, T., and Lyu, Y. (2022). LoSWEET14, a sugar transporter in lily, is regulated by transcription factor LoABF2 to participate in the ABA signaling pathway and enhance tolerance to multiple abiotic stresses in tobacco. Int. J. Mol. Sci. 23, 15093. doi:10.3390/ijms232315093
Zhang, Q., and Cao, X. (2021). Epigenetic remodeling in innate immunity and inflammation. Annu. Rev. Immunol. 39, 279–311. doi:10.1146/annurev-immunol-093019-123619
Zhao, W. S., Chen, K. F., Liu, M., Jia, X. L., Huang, Y. Q., Hao, B. B., et al. (2023). Investigation of targets and anticancer mechanisms of covalently acting natural products by functional proteomics. Acta Pharmacol. Sin. 44, 1701–1711. doi:10.1038/s41401-023-01072-z
Zhao, W., Chen, K., Zhang, J., Zhang, M., Guo, J., Xie, D., et al. (2024). Multi-step HPLC fractionation enabled in-depth and unbiased characterization of histone PTMs. J. Chromatogr. A 1736, 465368. doi:10.1016/j.chroma.2024.465368
Zheng, S., Sun, M., Zhang, K., Gu, J., Guo, Z., Tian, S., et al. (2016). Profiling post-translational modifications of histones in neural differentiation of embryonic stem cells using liquid chromatography-mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1017-1018, 36–44. doi:10.1016/j.jchromb.2016.02.022
Zhou, M., Dai, Y., Ma, Y., Yan, Y., Hua, M., Gao, Q., et al. (2022). Protective effects of liquiritigenin against cisplatin-induced nephrotoxicity via NRF2/SIRT3-mediated improvement of mitochondrial function. Molecules 27, 3823. doi:10.3390/molecules27123823
Keywords: histone post-translational modifications, super-SILAC, liquiritigenin, inflammation, epigenetics, PPAR
Citation: Liu P, Zhang J, Zhang J, Yuan Y, Liu Z, Chen S, Chen K, Dong L, Cheng Z, Zhang Y, Geng M, Tan M, Zhao W and Xie D (2025) Quantitation of global histone post-translational modifications reveal anti-inflammatory epigenetic mechanisms of liquiritigenin based on the optimized super-SILAC strategy. Front. Cell Dev. Biol. 13:1566567. doi: 10.3389/fcell.2025.1566567
Received: 25 January 2025; Accepted: 24 February 2025;
Published: 27 March 2025.
Edited by:
Zhongwei Xu, Logistics University of People’s Armed Police Force, ChinaReviewed by:
Liang Qiao, Fudan University, ChinaCopyright © 2025 Liu, Zhang, Zhang, Yuan, Liu, Chen, Chen, Dong, Cheng, Zhang, Geng, Tan, Zhao and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Minjia Tan, bWp0YW5Ac2ltbS5hYy5jbg==; Wensi Zhao, emhhb193ZW5zaUB0b25namkuZWR1LmNu; Dong Xie, eGllZG9uZ0B0b25namkuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.