
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 12 March 2025
Sec. Signaling
Volume 13 - 2025 | https://doi.org/10.3389/fcell.2025.1559753
This article is part of the Research Topic Deciphering Signaling Pathway Interactions in Tissue Homeostasis View all articles
 Fabienne Brutscher*
Fabienne Brutscher* Konrad Basler*
Konrad Basler*The Toll/NF-κB pathway plays a central role in patterning the Drosophila embryo and in orchestrating the innate immune response against microbial infections. Both discoveries were associated with a Nobel Prize award and led to the recognition of the Toll-like receptor pathway in mammals, which has significant implications for diseases. Recent discoveries have revealed that the Toll/NF-κB pathway also maintains epithelial homeostasis of imaginal tissues during development: local Toll/NF-κB signaling activity monitors internal cellular fitness, and precancerous mutant cells can trigger systemic Toll/NF-κB pathway activation. However, this signaling can be exploited in diseases like cancer, in which Toll/NF-κB signaling is often co-opted or subverted. Various models have been proposed to explain how Toll/NF-κB signaling contributes to different types of cancer. Here we provide an overview of the functions of Toll/NF-κB signaling in imaginal tissue homeostasis with a focus on their misuse in pathological contexts, particularly their significance for tumor formation.
The Toll/NF-κB pathway is a central player in the Drosophila embryonic pattern formation (Nüsslein-Volhard and Wieschaus, 1980) and in the innate immune response against microbial infections (Lemaitre et al., 1996); both discoveries were associated with a Nobel Prize award and led to the recognition of the evolutionarily conserved Toll-like receptor (TLR) pathway in mammals and its pathological significance.
In Drosophila, there are nine Toll-related receptors (TRRs). The roles of Toll/NF-κB signaling in embryonic patterning and innate immunity are mainly based on the originally identified Toll receptor, Toll-1 (Tl-1) (Nüsslein-Volhard, 2022; Valanne et al., 2022). The eight additional TRRs are suggested to activate the Toll signaling cascade mostly in non-immune contexts during development and adulthood. For a comprehensive review see Anthoney et al. (2018). The homologous mammalian TLRs, of which there are ten in humans, recognize diverse microbial products thereby collectively regulating innate immunity against a broad spectrum of pathogens (Gay and Gangloff, 2007). In contrast to the Drosophila TRRs, mammalian TLRs are not considered to be involved in early mammalian development.
Drosophila TRRs bind an activated form of Spätzle (Spz), whereas mammalian TLRs directly recognize proteins, such as those from pathogens or released by injuries (Gay and Gangloff, 2007). Spz 1-6, which are members of the neurotrophin-protein family, are produced as pro-proteins. The pro-proteins are then cleaved through a serine-protease cascade in response to developmental cues during embryogenesis or extracellular stimuli, such as Gram-positive bacterial or fungal infection (Stathopoulos and Levine, 2002; Morisato and Anderson, 1994; Weber et al., 2003).
Both, Drosophila TRR and mammalian TLR activation trigger the canonical Toll/NF-κB signaling pathway (for simplicity hereafter referred to as the Toll pathway). Via the evolutionary conserved Toll intracellular (TIR) domain, the TRRs and TLRs recruit Myd88, which in turn activates the protein kinase Pelle or IRAK (in mammals). In Drosophila Pelle then phosphorylates the Drosophila l-κB factor Cactus, targeting it for proteasomal degradation (Horng and Medzhitov, 2001; Wu and Anderson, 1998; Sun et al., 2002). Consequently, the NF-κB transcription factors Dorsal (Dl) and Dif are no longer retained in the cytoplasm but can translocate to the nucleus and regulate the expression of distinct target genes (Bulet et al., 1999). Similarly, in mammals, the canonical signaling cascade downstream of TLR activation ultimately results in the nuclear translocation of NF-κB transcription factors to activate transcription (Kawai and Akira, 2007).
Pioneering experiments in Drosophila on the role of innate immune signaling in response to mutant cells during imaginal epithelial development revealed new Toll pathway functions: Internal cellular fitness is monitored by healthy cells that locally activate Toll signaling in adjacent mutant cells (Meyer et al., 2014). Additionally, systemic Toll pathway activation triggered by polarity-deficient epithelia maintains tissue homeostasis to prevent oncogenic transformation of imaginal tissues during larval development (Parisi et al., 2014). These findings paved the way for studies on Toll signaling in cancer, revealing its dual functionality. For example, activating the Toll pathway in polarity-deficient epithelial cancer clones mutant for scribble no longer protects tissue homeostasis, but instead promotes tumor growth (Katsukawa et al., 2018). Thus like in mammals (Perkins, 2004; Perkins and Gilmore, 2006), in Drosophila models, the consequences of Toll signaling in cancer are context-dependent and the underlying studies help to advance our understanding of the mammalian TLR pathway’s functionality.
Here we review the functions of Toll signaling in imaginal tissue homeostasis with a focus on their misuse in pathological contexts, specifically their contribution to tumor formation.
Cell competition describes the non-cell autonomous elimination of cells in a heterotypic setting. The eliminated cells, often referred to as “loser cells”, would otherwise persist if they were not intermingled with more competitive cells. Cell competition is crucial for monitoring cell fitness and maintaining tissue homeostasis (referred to as “canonical cell competition”) but can also be misused by precancerous cells to promote oncogenic clonal expansion (referred to as “super-competition”, see Section 3.1.2). For a comprehensive recent review on cell competition, see Nagata and Igaki (2024).
A classical paradigm of cell competition is the elimination of wing imaginal epithelial cells heterozygous for a mutation in a ribosomal protein gene (hereafter referred to as RpL14/+). The elimination requires local activation of Toll signaling in loser cells (Figure 1A) (Meyer et al., 2014). The signaling module downstream of TRR activation relies on the NF-κB transcription factors Dl and Dif to activate expression of the pro-apoptotic gene reaper (rpr) and induce apoptotic cell death (Meyer et al., 2014; Morata and Ballesteros-Arias, 2014). The mechanism of Toll pathway activation in RpL14/+ cells is still unclear, but may involve Spz as a ligand for the TRRs, Tl-3, and Tl-9 (Meyer et al., 2014) (Table 1). Adding an additional layer of complexity, a follow-up study demonstrates that activation of Toll signaling in this context is not exclusively dependent on locally produced Spz, but also on the systemic level of infection (Germani et al., 2018). This study shows that in a heterotypic population of cells with different levels of Toll signaling activity, cells with higher intrinsic Toll signaling activity were competitively eliminated (Germani et al., 2018) (Figure 1B). Taken together, both studies (Meyer et al., 2014; Germani et al., 2018) provide compelling evidence for a link between innate immunity and cell competition, where local activation of Toll signaling protects imaginal tissue homeostasis by promoting the elimination of “loser” cells.
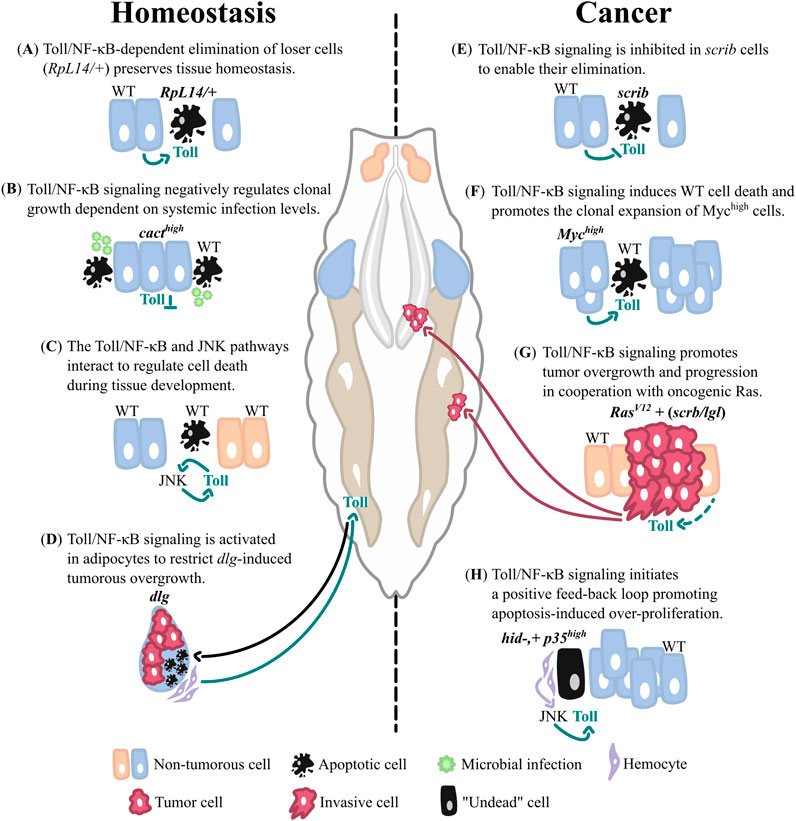
Figure 1. The functions of Toll/NF-κB signaling in imaginal tissue homeostasis and cancer. Schematic representations of the different contexts in which Toll signaling regulates tissue homeostasis and contributes to tumorous overgrowth in Drosophila imaginal tissues. The central scheme shows a Drosophila larva with the eye/antennal-(orange) and wing (blue) imaginal discs, the salivary glands (grey) and adipose tissue (brown). (A–D) The Toll pathway is critical for maintaining imaginal tissue homeostasis and protecting from oncogenic overgrowth: (A) Toll signaling is activated in RpL14/+ mutant cells to regulate loser cell elimination. (B) Toll pathway inhibition confers cells with a growth advantage over surrounding WT cells. (C) The Toll and JNK pathways interact to regulate developmental cell death. (D) The systemic activation of Toll signaling in adipocytes protects from tumorous overgrowth in response to tissue-wide loss of apicobasal polarity. (E–H) Toll signaling outputs are co-opted to promote cancer development (E) Toll signaling needs to be inhibited to enable the elimination of polarity-deficient cells and prevent their tumorous overgrowth. (F) Cells with an activating mutation in Myc induce Toll-dependent cell death in neighboring WT cells to promote their oncogenic expansion. (G) Toll signaling promotes tumor growth and progression in cooperation with oncogenic Ras. (H) Toll signaling is required in “undead” cells to initiate a feedback amplification loop and promote overgrowth of surrounding WT cells. Dashed lines represent uncertain connections, blunt arrows (┴) indicate inhibition while sharp arrows (→) indicate stimulation or cell movement to distant tissues (magenta arrow in (G)). Inhibition or stimulation of Toll signaling is shown by green arrows, stimulation of cell death in (D) is illustrated by a black arrow and stimulation of JNK signaling specifically by hemocytes is shown by a purple arrow in (H). WT, wild type.
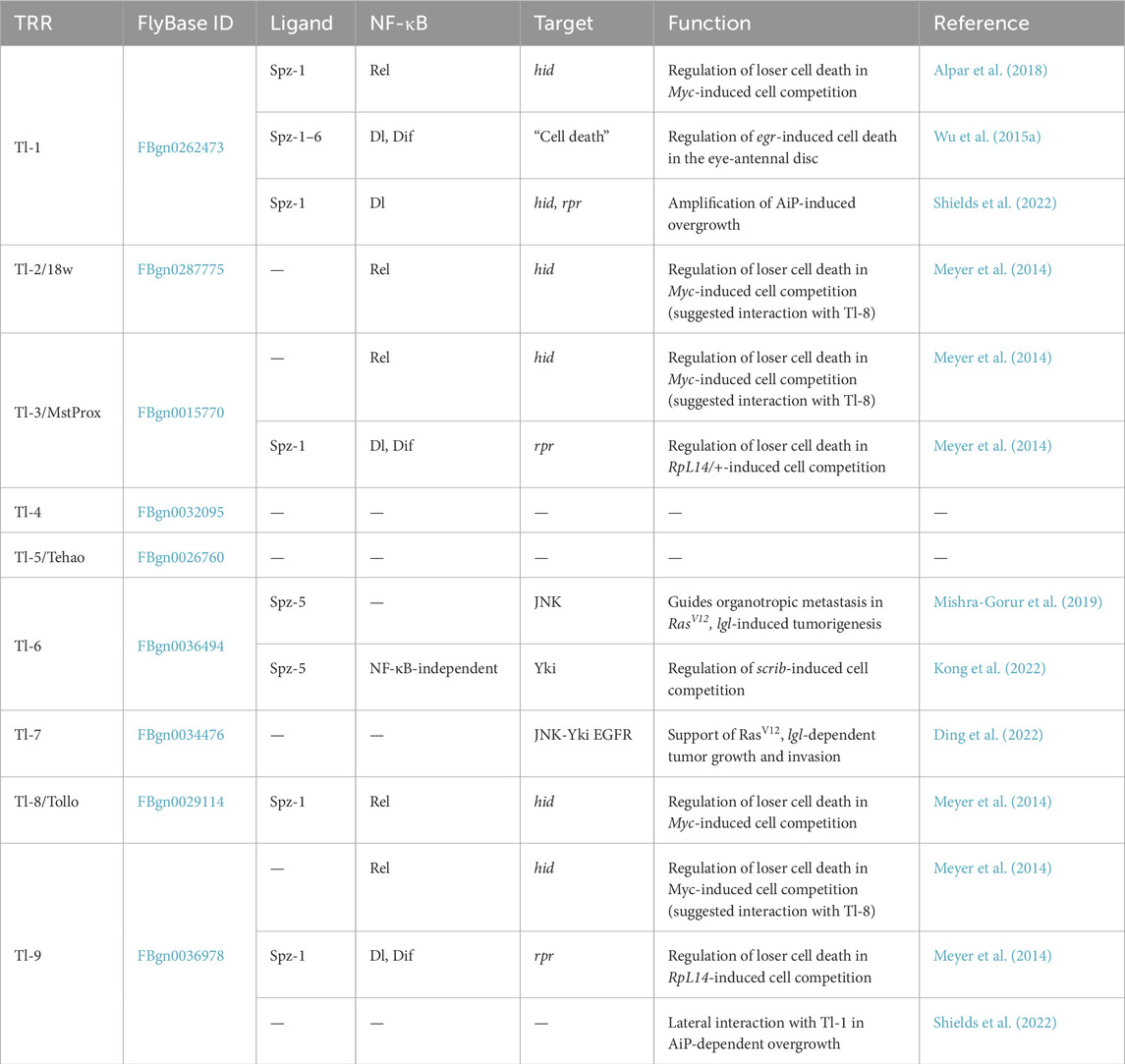
Table 1. TRR-Spz interactions and their function in imaginal tissue homeostasis and cancer.
Interestingly, recent findings suggest that TLR signaling may also be involved in the induction of loser cell apoptosis during cell competition in human cells (Zheng et al., 2021). The role of Toll signaling in cell competition may thus be conserved.
Robust development of imaginal epithelia occasionally requires that cells, i.e., excess or misplaced cells, are eliminated by cell death (Wolff and Ready, 1991; Raff, 1992). The evolutionarily conserved c-Jun N-terminal kinase (JNK) pathway is a fundamental regulator of cell death during development (Adachi-Yamada et al., 1999). In the context of cell competition, the contribution of the JNK pathway to Toll signaling-mediated apoptotic death of “loser” cells, is debated (Meyer et al., 2014; Katsukawa et al., 2018; Kodra et al., 2024). However, several studies have addressed the epistatic relationship between the Toll and JNK pathways in developmental cell death (Figure 1C) (Wu et al., 2015a; Li et al., 2020; Wu et al., 2015b).
Toll signaling is essential for egr-induced, JNK-dependent cell death in eye/antennal and wing imaginal epithelia (Wu et al., 2015a). The Toll pathway acts downstream of JNK-activated forkhead box transcription factor (FoxO) and was suggested to induce cell death independent of caspases (Wu et al., 2015a); the exact mechanism remains unclear. Elevated JNK signaling during development can directly induce Toll pathway activity, supposedly by regulating the expression of spz-6 in the epithelial peripodial membrane (Table 1) (Wu et al., 2015a). Interestingly, another study showed that ectopic Toll pathway activation in the wing imaginal epithelium can induce JNK signaling, leading to caspase-dependent cell death through an increase in reactive oxygen species (ROS) (Li et al., 2020). Based on these two examples of crosstalk, it is tempting to speculate there may be a positive feedback connection between the Toll and JNK pathways. There may also be a negative feedback mechanism to limit JNK-induced cell death in response to high levels of Toll pathway activity: The Toll pathway kinase Pelle can inhibit JNK- and caspase-dependent cell death through physical interaction with FoxO (Wu et al., 2015b). This interaction is independent of the NF-κB factor Dl (Wu et al., 2015b).
Thus, although the link between the Toll and JNK signaling pathways in developmental cell death has been repeatedly demonstrated (Wu et al., 2015a; Li et al., 2020; Wu et al., 2015b), further studies are needed to explain how the two pathways interact and crosstalk to modulate cell death during imaginal epithelial development.
The presence of tumorigenic cells in imaginal epithelia presents an internal threat to tissue integrity and organismal survival. Drosophila has emerged as a model to study not only the effects of local but also systemic signaling changes in response to tumors (Bilder et al., 2021). Defective apicobasal polarity and chromosomal instability (CIN) are two tumorigenic stimuli that trigger systemic activation of the innate immune system (Pastor-Pareja et al., 2008; Cordero et al., 2010; García-López et al., 2021; Liu et al., 2015).
Three epithelial polarity proteins, Lethal giant larvae (Lgl), Scribble (Scrib) and Discs large (Dlg) are established tumor suppressors that, if lost homozygous throughout an entire imaginal epithelium, can cause uncontrolled proliferation and amorphous overgrowth (Gateff, 1978; Bilder et al., 2000; Brumby and Richardson, 2003). To counteract the overgrowth, Drosophila mounts a systemic immune response that involves amplification and recruitment of circulating hemocytes to the side of tumor formation (Pastor-Pareja et al., 2008). Hemocyte-derived Eiger (Egr)/Tumor necrosis factor α (TNFα) thereupon induces JNK-dependent tumor cell death (Cordero et al., 2010).
Importantly, tumor-associated hemocytes (TAHs) also secrete the TRR ligand Spz which activates Toll signaling in adipose tissue, further restricting tumor overgrowth in imaginal epithelia (Figure 1D) (Parisi et al., 2014). Long-range Toll signaling from adipocytes is thought to contribute to tumor cell death by stimulating the expression of antimicrobial peptides (AMPs), i.e., Defensin (Parvy et al., 2019). The AMPs target tumor cells through the phosphatidylserine (PS)-rich domains they expose (Parvy et al., 2019).
CIN, which is often associated with tumorigenesis in mammals and flies (Nowak et al., 2002; Dekanty et al., 2012; Barrio et al., 2023; Gerlach and Herranz, 2020), also induces local and systemic immune responses: ROS-triggered local activation of Toll signaling in CIN-affected cells is suggested to induce a systemic immune response and to mediate the effective JNK-mediated death of transformed epithelial cells (Liu et al., 2015).
The Toll pathway can also be hijacked and its output used to promote tumor growth and progression rather than tissue integrity.
Mammalian TLR signaling is often mis-regulated in pathological contexts, including in different human cancers (Gilmore et al., 2002). The below studies suggest that also in Drosophila, Toll signaling can drive overgrowth, tumor formation and progression towards malignancy.
A tissue-wide homozygous loss-of-function mutation in tumor suppressors, such as in the polarity gene dlg, systemically activates Toll signaling in adipocytes to limit tumorous overgrowth, as discussed in Section 2.3 (Parisi et al., 2014). However, when polarity-deficient cells, such as those homozygous mutant for scrib, are surrounded by wild type (WT) tissue, they are competitively eliminated via localized activation of the JNK pathway (Brumby and Richardson, 2003; Agrawal et al., 1995; Igaki et al., 2009; Pagliarini and Xu, 2003; Snigdha et al., 2021). Additionally, interaction of Spz-5/Tl-6 at competitive cell boundaries is proposed to activate the Hippo pathway independent of NF-κB transcription factors, which further supports the elimination of scrib cells (Table 1) (Kong et al., 2022).
Interestingly, Toll signaling must be actively inhibited in scrib cells for their effective competitive elimination (Figure 1E) (Katsukawa et al., 2018). In contrast, ectopic activation of Toll signaling drives oncogenic expansion of these polarity-deficient cells (Katsukawa et al., 2018). Mechanistically, WT cells surrounding scrib mutants secrete the protease inhibitor Serpin 5 (Spn-5), which suppresses Spz-mediated Toll signaling in scrib cells. In the absence of Spn-5, scrib cells survive and overgrow due to Toll-mediated activation of the JNK and Yki signaling pathways.
Thus, immune signaling is suppressed in polarity deficient cells, as ectopic Toll pathway activation impairs their effective elimination and promotes tumor growth. Further experiments are necessary to investigate whether tumor-initiating alterations, when acquired clonally rather than through tissue-wide loss of apicobasal polarity, fail to trigger or remain unaffected by systemic Toll pathway activation.
During tumor development, an activating mutation in a growth-promoting gene, such as Myc (hereafter: oncogenic Myc or Myc) provides cells with a competitive growth advantage, also referred to as super-competition (Moreno and Basler, 2004; De La Cova et al., 2004).
Cells with elevated Myc expression are suggested to remain isolated from systemic tumor-suppressive immune signaling (Alpar et al., 2018), but they locally induce Toll signaling in surrounding WT “loser” cells (Figure 1F) (Meyer et al., 2014; Morata and Ballesteros-Arias, 2014). In the context of Myc super-competition, the Toll pathway is suggested to mediate loser cell death via activation of the pro-apoptotic genes hid and rpr (Meyer et al., 2014; Alpar et al., 2018). Crucial for the activation of the Toll pathway in WT loser cells is the increased expression of Spz and Spz-processing enzymes (SPE) in the Myc-expressing winner cells (Alpar et al., 2018). Activated Spz induces Toll signaling in WT loser cells mainly via TRRs Tl-1 and Tl-8 (Alpar et al., 2018). These receptors are downregulated in oncogenic Myc-expressing cells, pointing to a mechanism that ensures specificity in killing WT loser cells (Alpar et al., 2018).
In contrast to RpL14/+-induced cell competition, in the context of oncogenic Myc-induced super-competition, Toll-induced death of loser cells does not protect tissue integrity, but instead promotes the expansion of tumor cells.
It appears paradoxical that Toll signaling has opposite functions in two competitive contexts, both induced by the presence of tumor cells: Toll pathway activation induces the death of WT cells surrounding Myc cells but induces over-proliferation in polarity deficient cells. However, common to both contexts is that the activation of Toll signaling ultimately accelerates tumor growth by promoting the expansion of tumorous cells.
The growth-controlling small GTPase, Ras, is frequently linked to mammalian cancer (Sanchez-Vega et al., 2018). Cells with an activating mutation in Ras (hereafter: oncogenic Ras or RasV12) are resistant to apoptotic stimuli (Kurada and White, 1998; Bergmann et al., 1998). However, the acquisition of additional mutations are necessary to accelerate tumor growth and initiate the progression towards malignancy (Hanahan and Weinberg, 2000).
Recent findings in the eye/antennal imaginal epithelium show that ectopic Toll pathway activation is sufficient to promote overgrowth in tissues expressing oncogenic Ras (Figure 1G) (Brutscher et al., 2024; Dillard et al., 2024). It is suggested that Toll signaling mediates overgrowth by repressing differentiation and increasing proliferation in cells predisposed by oncogenic Ras (Brutscher et al., 2024). Brutscher et al. hypothesize that induction of caspase expression in response to Toll signaling activity, as seen in WT cells, may contribute to RasV12-related tumorigenic overgrowth through apoptosis-induced proliferation (Brutscher et al., 2024).
Toll signaling is activated in malignant tumor cells with concomitant activation of oncogenic Ras and loss of apicobasal polarity. Even though the underlying mechanism is not yet fully elucidated, elevated expression of upstream components of the Toll pathway, such as PGRP-SA, are suggested to contribute to local Toll pathway activation in malignant tumor cells (Dillard et al., 2024). Interestingly, the level of Toll signaling appears heterogeneous in the imaginal epithelium (Dillard et al., 2024). The authors suggest it may account for spatially variable functions of Toll signaling within the tumor epithelium, which promote either overgrowth or tumor cell invasion through variable levels of JNK signaling activation (Dillard et al., 2024). While Toll signaling-induced overgrowth in cooperation with RasV12 is independent of the JNK pathway (Brutscher et al., 2024), the strong activation of JNK signaling downstream of Toll is suggested to play an important role during tumor progression (discussed in Section 3.2) (Dillard et al., 2024).
The ability of systemic innate immune activation to protect epithelia from oncogenic expansion of abnormally developed cells, as in response to loss of apicobasal polarity alone (Parisi et al., 2014) (discussed in Section 2.3), is impaired in the context of malignant tumor cells. Malignant tumor cells are, for example, induced by concomitant activation of oncogenic Ras and loss of apicobasal polarity, e.g., RasV12,scrib.
In RasV12,scrib-transformed cells, as part of the systemic immune response, TAH-derived Egr no longer induces tumor cell death as in response to polarity-deficient epithelia (Cordero et al., 2010; Parvy et al., 2019), but promotes malignancy (Cordero et al., 2010). It is therefore tempting to speculate that systemic Toll activation in adipocytes of larvae bearing RasV12, scrib tumors in the eye/antennal imaginal epithelium (Parisi et al., 2014) no longer promotes tumor cell death, but also contributes to tumor progression.
While the effect of systemic Toll pathway activation on malignant tumors remains elusive, recent research addressed the tumor-autonomous function of Toll signaling in the progression of malignant tumors induced in the eye/antennal imaginal epithelium. There is cumulative evidence that the Toll pathway promotes tumor progression through activation of JNK signaling (Figure 1G) (Dillard et al., 2024; Mishra-Gorur et al., 2019; Ding et al., 2022), a pathway well established to trigger tumor cell invasion through activation of matrix metalloproteinase gene mmp1 (Uhlirova and Bohmann, 2006). The EMT factors, Snail and Twist have been suggested to mediate the activation of the JNK pathway downstream of Toll signaling in RasV12,scrib cells (Dillard et al., 2024). Another study shows that Tl-7-dependent endocytosis of Egr facilitates JNK pathway activation to promote the invasion of RasV12, lgl cells (Ding et al., 2022). Interestingly, in this model, Toll signaling is suggested to accelerate tumor growth through positive regulation of Epidermal growth factor receptor (EGFR) levels, although the mechanism behind this is still unclear (Ding et al., 2022).
RasV12,scrib tumor cells, derived from the eye/antennal epithelium, frequently invade distal regions of the brain, i.e., the ventral nerve cord and occasionally even target tissues that are located further away from the tumor origin (Pagliarini and Xu, 2003; Igaki et al., 2006). Spz was found to be expressed in target tissues, such as, for example, in the larval salivary gland and adipose tissue, and act as a chemoattractant, guiding tumor cell invasion through interaction with Tl-6 in RasV12,lgl cells (Mishra-Gorur et al., 2019).
In contrast to the eye/antennal imaginal epithelium, in the wing imaginal epithelium Toll signaling was reported to inhibit tumor growth and progression (Snigdha et al., 2021). The Toll pathway target and negative regulator Cactus accumulates in malignant wing imaginal tumor cells after concomitant activation of oncogenic Ras and impaired apicobasal polarity (scribRNAi) (Snigdha et al., 2021). Cactus was suggested to be regulated by Yki and to promote JNK-dependent tumor growth and progression (Snigdha et al., 2021). It remains to be seen whether the upregulated Cactus levels in this context indeed inhibit Toll signaling, but still trigger JNK activation and promote tumor progression.
As discussed in the previous chapters, the Toll pathway regulates cell death in various contexts through the upregulation of pro-apoptotic genes. The subsequent apoptotic cascade involves the activation of the initiator caspase Dronc, which promotes the activation of the effector caspases DrICE and Dcp-1 to induce cell death.
If the apoptotic cascade cannot be completed because the effector caspase activity is inhibited by expression of the baculoviral anti-apoptosis protein P35, then the cells remain trapped in an “undead”-like state (Clem et al., 1991; Hay et al., 1994). In these “undead” cells, the activity of Dronc triggers apoptosis-induced compensatory over-proliferation (AiP) of surrounding WT cells. AiP involves the recruitment of circulating hemocytes and activation of JNK signaling in “undead” cells (recently reviewed in (Diwanji and Bergmann, 2019; Fogarty and Bergmann, 2017)). Intriguingly, AiP is suggested to contribute to various forms of cancer (Ryoo and Bergmann, 2012).
A recent study suggests Toll signaling may regulate AiP (Figure 1H) (Shields et al., 2022). Tl-9 was upregulated downstream of JNK in “undead” (hid-, p35high) cells and was required for AiP in surrounding WT cells. Interaction of Tl-9 with Tl-1 induces Toll signaling and increases the expression of the pro-apoptotic genes hid and rpr. This in turn establishes a feedback amplification loop promoting AiP through the generation of ROS, recruitment of circulating hemocytes and activation of JNK signaling in “undead” cells. Similarly, Tl-9, has also been reported to induce pro-apoptotic gene expression in the context of RpL14/+-induced cell competition (Meyer et al., 2014) suggesting similarities between the local innate immune responses to unfit and undead cells. The heterologous interaction of TRRs, as seen here with Tl-9 and Tl-1, may provide specificity and complexity to the signaling outcome downstream of canonical Tl-1/NF-κB signaling in different contexts and presents an interesting avenue for future research.
The Toll pathway plays important but opposing roles during developmental tissue homeostasis and cancer. Different cellular contexts and hence the integration with different signaling pathways, appears to determine the overall response to Toll signaling activity. To preserve tissue homeostasis or protect from oncogenic transformation, Toll signaling eliminates abnormally developed cells through the induction of apoptosis. In contrast, in the context of cancer, Toll signaling promotes tumor growth and progression, thus serving as an oncogenic factor. Tumor cells evade the Toll-related homeostatic, i.e., growth-inhibiting functions and hijack the Toll-induced target effectors to their own advantage. We discussed different models underlying Toll-dependent tissue overgrowth: a) The Toll pathway can indirectly induce tumorous overgrowth through the selective killing of WT cells adjacent to tumor cells; b) In contrast, it can also promote overgrowth by directly increasing the proliferation of tumor cells. Depending on the genetic context, different signaling pathways have been described to mediate the Toll-dependent proliferative response; c) Toll signaling promotes overgrowth through fueling a feedback amplification loop in the context of apoptosis-induced proliferation.
It will be interesting to further explore how Toll signaling can both promote and inhibit growth. The interaction of multiple TRRs, as proposed in (Shields et al., 2022) or the different TRR-Spz interactions (Table 1) may help define signaling specificity in different cellular contexts. Similarly, in mammals, different TLRs have different functions on cancer cells and can promote or suppress tumor growth, such as by differentially regulating cancer cell apoptosis and proliferation (Yu et al., 2013).
In addition, the multifaceted responses to Toll signaling activity during tissue homeostasis and in different cancer contexts may also depend on the nuclear concentration of Dorsal, as suggested in (Dillard et al., 2024). Similarly, this relationship defines the dorsoventral patterning during Drosophila embryogenesis (Roth et al., 1989).
The Drosophila larva provides a powerful in vivo platform to not only study the effects of local, but also systemic Toll signaling activity on tissue homeostasis and cancer. Therefore, it will be interesting to explore whether and how local and systemic Toll signaling activity in response to developmental or cancerous cues interact to mediate signaling specificity.
Given the high degree of conservation with mammalian TLR/NF-κB signaling, the insights into the functions of Toll signaling in tissue homeostasis and cancer from Drosophila research hold great potential. They may help drive future progress in deciphering the complexity of TLR/NF-κB signaling in different cancer contexts, which is critical for the development of therapeutics targeting TLR/NF-κB signaling.
FB: Conceptualization, Investigation, Visualization, Writing–original draft. KB: Project administration, Resources, Supervision, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adachi-Yamada, T., Fujimura-Kamada, K., Nishida, Y., and Matsumoto, K. (1999). Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature 400 (6740), 166–169. doi:10.1038/22112
Agrawal, N., Kango, M., Mishra, A., and Sinha, P. (1995). Neoplastic transformation and aberrant cell–cell interactions in genetic mosaics of lethal(2)giant larvae (lgl), a tumor suppressor gene of Drosophila. Dev. Biol. 172 (1), 218–229. doi:10.1006/dbio.1995.0017
Alpar, L., Bergantiños, C., and Johnston, L. A. (2018). Spatially restricted regulation of spätzle/toll signaling during cell competition. Dev. Cell 46 (6), 706–719. doi:10.1016/j.devcel.2018.08.001
Anthoney, N., Foldi, I., and Hidalgo, A. (2018). Toll and toll-like receptor signalling in development. Dev. Camb. 145 (9), dev156018. doi:10.1242/dev.156018
Barrio, L., Gaspar, A. E., Muzzopappa, M., Ghosh, K., Romao, D., Clemente-Ruiz, M., et al. (2023). Chromosomal instability-induced cell invasion through caspase-driven DNA damage. Curr. Biol. 33 (20), 4446–4457.e5. doi:10.1016/j.cub.2023.09.004
Bergmann, A., Agapite, J., McCall, K., and Steller, H. (1998). The Drosophila gene hid is a direct molecular target of ras-dependent survival signaling. Cell 95 (3), 331–341. doi:10.1016/s0092-8674(00)81765-1
Bilder, D., Li, M., and Perrimon, N. (2000). Cooperative regulation of cell polarity and growth by Drosophila tumor suppressors. Science. 289 (5476), 113–116. doi:10.1126/science.289.5476.113
Bilder, D., Ong, K., Hsi, T. C., Adiga, K., and Kim, J. (2021). Tumour-host interactions through the lens of Drosophila. Nat. Rev. Cancer 21 (11), 687–700. doi:10.1038/s41568-021-00387-5
Brumby, A. M., and Richardson, H. E. (2003). Scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J. 22 (21), 5769–5779. doi:10.1093/emboj/cdg548
Brutscher, F., Germani, F., Hausmann, G., Jutz, L., and Basler, K. (2024). Activation of the innate immune system accelerates growth in cooperation with oncogenic Ras. bioRxiv. Available at: https://www.biorxiv.org/content/10.1101/2024.05.23.595555v1 (Accessed May 29, 2024).
Bulet, P., Hetru, C., Dimarcq, J. L., and Hoffmann, D. (1999). Antimicrobial peptides in insects; structure and function. Dev. Comp. Immunol. 23 (4–5), 329–344. doi:10.1016/s0145-305x(99)00015-4
Clem, R. J., Fechheimer, M., and Miller, L. K. (1991). Prevention of apoptosis by a baculovirus gene during infection of insect cells. Science 254 (5036), 1388–1390. doi:10.1126/science.1962198
Cordero, J. B., Macagno, J. P., Stefanatos, R. K., Strathdee, K. E., Cagan, R. L., and Vidal, M. (2010). Oncogenic ras diverts a host TNF tumor suppressor activity into tumor promoter. Dev. Cell 18 (6), 999–1011. doi:10.1016/j.devcel.2010.05.014
Dekanty, A., Barrio, L., Muzzopappa, M., Auer, H., and Milán, M. (2012). Aneuploidy-induced delaminating cells drive tumorigenesis in Drosophila epithelia. Proc. Natl. Acad. Sci. 109 (50), 20549–20554. doi:10.1073/pnas.1206675109
De La Cova, C., Abril, M., Bellosta, P., Gallant, P., and Johnston, L. A. (2004). Drosophila myc regulates organ size by inducing cell competition. Cell 117 (1), 107–116. doi:10.1016/s0092-8674(04)00214-4
Dillard, C., Reis, J. T., Jain, A., Le Borgne, R., Jasper, H., and Rusten, T. E. (2024). Oncogenic Ras-driven Dorsal/NF-κB signaling contributes to tumorigenesis in a Drosophila carcinoma model. bioRxiv. Available at: https://www.biorxiv.org/content/10.1101/2024.05.08.593126v2 (Accessed May 23, 2024).
Ding, X., Li, Z., Lin, G., Li, W., and Xue, L. (2022). Toll-7 promotes tumour growth and invasion in Drosophila. Cell Prolif. 55 (2), e13188. doi:10.1111/cpr.13188
Diwanji, N., and Bergmann, A. (2019). “Two sides of the same coin – compensatory proliferation in regeneration and cancer,” in The Drosophila model in cancer (Springer International Publishing), 65–85.
Fogarty, C. E., and Bergmann, A. (2017). Killers creating new life: caspases drive apoptosis-induced proliferation in tissue repair and disease. Cell Death Differ. 24 (8), 1390–1400. doi:10.1038/cdd.2017.47
García-López, L., Adrados, I., Ferres-Marco, D., and Dominguez, M. (2021). A blueprint for cancer-related inflammation and host innate immunity. Cells 10 (11), 3211. doi:10.3390/cells10113211
Gateff, E. (1978). Malignant neoplasms of genetic origin in Drosophila melanogaster. Science. 200 (4349), 1448–1459. doi:10.1126/science.96525
Gay, N. J., and Gangloff, M. (2007). Structure and function of toll receptors and their ligands. Annu. Rev. Biochem. 76, 141–165. doi:10.1146/annurev.biochem.76.060305.151318
Gerlach, S. U., and Herranz, H. (2020). Genomic instability and cancer: lessons from Drosophila. Open Biol. 10 (6), 200060. doi:10.1098/rsob.200060
Germani, F., Hain, D., Sternlicht, D., Moreno, E., and Basler, K. (2018). The Toll pathway inhibits tissue growth and regulates cell fitness in an infection-dependent manner. eLife 7, e39939. doi:10.7554/eLife.39939
Gilmore, T., Gapuzan, M. E., Kalaitzidis, D., and Starczynowski, D. (2002). Rel/NF-κB/IκB signal transduction in the generation and treatment of human cancer. Cancer Lett. 181 (1), 1–9. doi:10.1016/s0304-3835(01)00795-9
Hanahan, D., and Weinberg, R. A. (2000). The hallmarks of cancer. Cell 100 (1), 57–70. doi:10.1016/s0092-8674(00)81683-9
Hay, B. A., Wolff, T., and Rubin, G. M. (1994). Expression of baculovirus P35 prevents cell death in Drosophila. Development 120 (8), 2121–2129. doi:10.1242/dev.120.8.2121
Horng, T., and Medzhitov, R. (2001). Drosophila MyD88 is an adapter in the Toll signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 98 (22), 12654–12658. doi:10.1073/pnas.231471798
Igaki, T., Pagliarini, R. A., and Xu, T. (2006). Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr. Biol. 16 (11), 1139–1146. doi:10.1016/j.cub.2006.04.042
Igaki, T., Pastor-Pareja, J. C., Aonuma, H., Miura, M., and Xu, T. (2009). Intrinsic tumor suppression and epithelial maintenance by endocytic activation of eiger/TNF signaling in Drosophila. Dev. Cell 16 (3), 458–465. doi:10.1016/j.devcel.2009.01.002
Katsukawa, M., Ohsawa, S., Zhang, L., Yan, Y., and Igaki, T. (2018). Serpin facilitates tumor-suppressive cell competition by blocking toll-mediated Yki activation in Drosophila. Curr. Biol. 28 (11), 1756–1767. doi:10.1016/j.cub.2018.04.022
Kawai, T., and Akira, S. (2007). Signaling to NF-kappaB by toll-like receptors. Trends Mol. Med. 13 (11), 460–469. doi:10.1016/j.molmed.2007.09.002
Kodra, A. L., Singh, A. S., Cova, C. de la, Ziosi, M., and Johnston, L. A. (2024). The Drosophila TNF Eiger contributes to Myc super-competition independent of JNK activity. bioRxiv. Available at: https://www.biorxiv.org/content/10.1101/2021.04.06.438672v3 (Accessed November 18, 2024).
Kong, D., Zhao, S., Xu, W., Dong, J., and Ma, X. (2022). Fat body-derived Spz5 remotely facilitates tumor-suppressive cell competition through Toll-6-α-Spectrin axis-mediated Hippo activation. Cell Rep. 39 (12), 110980. doi:10.1016/j.celrep.2022.110980
Kurada, P., and White, K. (1998). Ras promotes cell survival in Drosophila by downregulating hid expression. Cell 95 (3), 319–329. doi:10.1016/s0092-8674(00)81764-x
Lemaitre, B., Nicolas, E., Michaut, L., Reichhart, J. M., and Hoffmann, J. A. (1996). The dorsoventral regulatory gene cassette spatzle/Toll/Cactus controls the potent antifungal response in Drosophila adults. Cell 86 (6), 973–983. doi:10.1016/s0092-8674(00)80172-5
Li, Z., Wu, C., Ding, X., Li, W., and Xue, L. (2020). Toll signaling promotes JNK-dependent apoptosis in Drosophila. Cell Div. 15 (1), 7. doi:10.1186/s13008-020-00062-5
Liu, D., Shaukat, Z., Saint, R. B., and Gregory, S. L. (2015). Chromosomal instability triggers cell death via local signalling through the innate immune receptor Toll. Oncotarget 6 (36), 38552–38565. doi:10.18632/oncotarget.6035
Meyer, S. N., Amoyel, M., Bergantiños, C., de la Cova, C., Schertel, C., Basler, K., et al. (2014). An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science 346 (6214), 1258236. doi:10.1126/science.1258236
Mishra-Gorur, K., Li, D., Ma, X., Yarman, Y., Xue, L., and Xu, T. (2019). Spz/Toll-6 signal guides organotropic metastasis in Drosophila. Dis. Model Mech. 12 (10), dmm039727. doi:10.1242/dmm.039727
Morata, G., and Ballesteros-Arias, L. (2014). Developmental biology. Death to the losers. Science 346 (6214), 1181–1182. doi:10.1126/science.aaa2345
Moreno, E., and Basler, K. (2004). dMyc transforms cells into super-competitors. Cell 117 (1), 117–129. doi:10.1016/s0092-8674(04)00262-4
Morisato, D., and Anderson, K. V. (1994). The spätzle gene encodes a component of the extracellular signaling pathway establishing the dorsal-ventral pattern of the Drosophila embryo. Cell 76 (4), 677–688. doi:10.1016/0092-8674(94)90507-x
Nagata, R., and Igaki, T. (2024). Cell competition: emerging signaling and unsolved questions. FEBS Lett. 598 (4), 379–389. doi:10.1002/1873-3468.14822
Nowak, M. A., Komarova, N. L., Sengupta, A., Jallepalli, P. V., Shih, I. M., Vogelstein, B., et al. (2002). The role of chromosomal instability in tumor initiation. Proc. Natl. Acad. Sci. 99 (25), 16226–16231. doi:10.1073/pnas.202617399
Nüsslein-Volhard, C. (2022). The Toll gene in Drosophila pattern formation. Trends Genet. 38 (3), 231–245. doi:10.1016/j.tig.2021.09.006
Nüsslein-Volhard, C., and Wieschaus, E. (1980). Mutations affecting segment number and polarity in drosophila. Nature 287 (5785), 795–801. doi:10.1038/287795a0
Pagliarini, R. A., and Xu, T. (2003). A genetic screen in Drosophila for metastatic behavior. Science. 302 (5648), 1227–1231. doi:10.1126/science.1088474
Parisi, F., Stefanatos, R. K., Strathdee, K., Yu, Y., and Vidal, M. (2014). Transformed epithelia trigger non-tissue-autonomous tumor suppressor response by adipocytes via activation of toll and eiger/TNF signaling. Cell Rep. 6 (5), 855–867. doi:10.1016/j.celrep.2014.01.039
Parvy, J. P., Yu, Y., Dostalova, A., Kondo, S., Kurjan, A., Bulet, P., et al. (2019). The antimicrobial peptide defensin cooperates with tumour necrosis factor to drive tumour cell death in Drosophila. eLife 8, e45061. doi:10.7554/eLife.45061
Pastor-Pareja, J. C., Ming, W., and Tian, X. (2008). An innate immune response of blood cells to tumors and tissue damage in Drosophila. DMM Dis. Models Mech. 1 (2–3), 144–154. doi:10.1242/dmm.000950
Perkins, N. D. (2004). NF-kappaB: tumor promoter or suppressor? Trends Cell Biol. 14 (2), 64–69. doi:10.1016/j.tcb.2003.12.004
Perkins, N. D., and Gilmore, T. D. (2006). Good cop, bad cop: the different faces of NF-kappaB. Cell Death Differ. 13 (5), 759–772. doi:10.1038/sj.cdd.4401838
Raff, M. C. (1992). Social controls on cell survival and cell death. Nature 356 (6368), 397–400. doi:10.1038/356397a0
Roth, S., Stein, D., and Nüsslein-Volhard, C. (1989). A gradient of nuclear localization of the dorsal protein determines dorsoventral pattern in the Drosophila embryo. Cell 59 (6), 1189–1202. doi:10.1016/0092-8674(89)90774-5
Ryoo, H. D., and Bergmann, A. (2012). The role of apoptosis-induced proliferation for regeneration and cancer. Cold Spring Harb. Perspect. Biol. 4 (8), a008797. doi:10.1101/cshperspect.a008797
Sanchez-Vega, F., Mina, M., Armenia, J., Chatila, W. K., Luna, A., La, K. C., et al. (2018). Oncogenic signaling pathways in the cancer genome atlas. Cell 173 (2), 321–337.e10. doi:10.1016/j.cell.2018.03.035
Shields, A., Amcheslavsky, A., Brown, E., Lee, T. V., Nie, Y., Tanji, T., et al. (2022). Toll-9 interacts with Toll-1 to mediate a feedback loop during apoptosis-induced proliferation in Drosophila. Cell Rep. 39 (7), 110817. doi:10.1016/j.celrep.2022.110817
Snigdha, K., Singh, A., and Kango-Singh, M. (2021). Yorkie-Cactus (IκBα)-JNK axis promotes tumor growth and progression in Drosophila. Oncogene 40 (24), 4124–4136. doi:10.1038/s41388-021-01831-4
Stathopoulos, A., and Levine, M. (2002). Dorsal gradient networks in the Drosophila embryo. Dev. Biol. 246 (1), 57–67. doi:10.1006/dbio.2002.0652
Sun, H., Bristow, B. N., Qu, G., and Wasserman, S. A. (2002). A heterotrimeric death domain complex in toll signaling. Proc. Natl. Acad. Sci. U. S. A. 99 (20), 12871–12876. doi:10.1073/pnas.202396399
Uhlirova, M., and Bohmann, D. (2006). JNK- and Fos-regulated Mmp1 expression cooperates with Ras to induce invasive tumors in Drosophila. EMBO J. 25 (22), 5294–5304. doi:10.1038/sj.emboj.7601401
Valanne, S., Vesala, L., Maasdorp, M. K., Salminen, T. S., and Rämet, M. (2022). The Drosophila toll pathway in innate immunity: from the core pathway toward effector functions. J. Immunol. 209 (10), 1817–1825. doi:10.4049/jimmunol.2200476
Weber, A. N. R., Tauszig-Delamasure, S., Hoffmann, J. A., Lelièvre, E., Gascan, H., Ray, K. P., et al. (2003). Binding of the Drosophila cytokine Spätzle to Toll is direct and establishes signaling. Nat. Immunol. 4 (8), 794–800. doi:10.1038/ni955
Wolff, T., and Ready, D. F. (1991). Cell death in normal and rough eye mutants of Drosophila. Development 113 (3), 825–839. doi:10.1242/dev.113.3.825
Wu, C., Chen, C., Dai, J., Zhang, F., Chen, Y., Li, W., et al. (2015a). Toll pathway modulates TNF-induced JNK-dependent cell death in Drosophila. Open Biol. 5 (7), 140171. doi:10.1098/rsob.140171
Wu, C., Chen, Y., Wang, F., Chen, C., Zhang, S., Li, C., et al. (2015b). Pelle modulates dFoxO-mediated cell death in Drosophila. PLOS Genet. 11 (10), e1005589. doi:10.1371/journal.pgen.1005589
Wu, L. P., and Anderson, K. V. (1998). Regulated nuclear import of Rel proteins in the Drosophila immune response. Nature 392 (6671), 93–97. doi:10.1038/32195
Yu, L., Wang, L., and Chen, S. (2013). Dual character of Toll-like receptor signaling: pro-tumorigenic effects and anti-tumor functions. Biochimica Biophysica Acta (BBA) - Rev. Cancer 1835 (2), 144–154. doi:10.1016/j.bbcan.2012.10.006
Keywords: Drosophila, innate immune signaling, Toll/NF-κB pathway, cell competition, tissue homeostasis, cell death, cancer
Citation: Brutscher F and Basler K (2025) Functions of Drosophila Toll/NF-κB signaling in imaginal tissue homeostasis and cancer. Front. Cell Dev. Biol. 13:1559753. doi: 10.3389/fcell.2025.1559753
Received: 13 January 2025; Accepted: 19 February 2025;
Published: 12 March 2025.
Edited by:
John Hancock, University of the West of England, United KingdomReviewed by:
Mojgan Padash Barmchi, University of Oklahoma, United StatesCopyright © 2025 Brutscher and Basler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabienne Brutscher, ZmFiaWVubmUuYnJ1dHNjaGVyQHV6aC5jaA==; Konrad Basler, a29ucmFkLmJhc2xlckBtbHMudXpoLmNo
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.