 Rashmi Verma1†
Rashmi Verma1† Jyotsna Kailashiya
Jyotsna Kailashiya Rameshwar Nath Chaurasia
Rameshwar Nath Chaurasia Debabrata Dash
Debabrata Dash
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 25 February 2025
Sec. Signaling
Volume 13 - 2025 | https://doi.org/10.3389/fcell.2025.1534235
Introduction: Prion diseases are neurodegenerative disorders where infectious prion proteins (PrP) featuring an amyloidogenic amino acid sequence, PrP (106–126), accumulate in the brain leading to neuroinflammation while it can also access circulation by breaching the blood-brain barrier. Platelets are highly sensitive cells in blood, which have been widely employed as “peripheral” model for neurons. In addition to their stellar roles in hemostasis and thrombosis, platelets are also known to function as immune cells and possess necessary components of functional inflammasome. This study demonstrates that prion proteins drive inflammasome assembly in platelets and upregulate activity of caspase-1, a critical readout of functional inflammasomes.
Methods: Flow cytometric analysis was performed to measure intracellular ROS levels, caspase-1 activity, and platelet-monocyte/neutrophil interactions. Microscopy was used to assess the co-localization of NLRP3 and ASC.
Results: Inflammasome activation is fuelled by reactive oxygen species (ROS) generated in prion-stimulated platelets that eventually leads to formation of platelet-monocyte/neutrophil aggregates, which was prohibited by small-molecule inhibitors of either caspase-1 or ROS.
Discussion: Thus, in addition to their neurotoxic effects on neuronal cells and stimulation of pro-coagulant activity in platelets, prions also unleash an inflammatory response in the organism.
Prion disease is a neurodegenerative disorder caused by the scrapie form of prions (PrPsc), which induces abnormal folding of cellular prion (PrPc) leading to protease-resistant β-pleated sheets (Sandberg et al., 2011). In cerebrospinal fluid concentration of prion protein ranges from 1.5 ng/mL to 546 ng/mL (Vallabh et al., 2019) while prions in circulation are likely sourced from platelets (Robertson et al., 2006), endothelial cells (Simák et al., 2002) and lymphocytes (Prinz et al., 2003), which carry membrane-bound PrPc released into the bloodstream. PrP can cross the blood-brain barrier in both directions, i.e., either from blood-to-brain or brain-to-blood (Banks et al., 2009). The synthetic PrP (106–126) resembles PrPsc in several ways as it carries a protease-resistant amyloidogenic β-pleated sheet. Thus, PrP (106–126) has been extensively employed as a model for PrPsc-related research (Ettaiche et al., 2000; Gautam et al., 2022; Gu et al., 2002; Mallick et al., 2015; Melo et al., 2007).
Human platelets are anucleate blood cells essential for hemostasis; however, hyperactivity of platelets leads to pathological thrombus formation. Besides hemostatic activities, platelets also play a seminal role in innate and adaptive immune responses (Semple et al., 2011). Activated platelets interact with circulating neutrophils and monocytes that are critical mediators of inflammation and innate immunity, thereby driving an inflammatory phenotype. Platelets express numerous pattern recognition receptors, including toll-like receptors (TLRs) and nucleotide-binding oligomerization domain-like receptors (NLRs), which enable them to act as circulating cellular sensors that provide a unique link between hemostasis and inflammation (Zhang et al., 2015). NLRs recognize pathogen/damage-associated molecular patterns (PAMPs/DAMPs) and homeostasis-altering molecular patterns (HAMPs). Inflammation is directly linked with inflammasome assembly that produces pro-inflammatory cytokines, interleukin (IL)-1β and IL-18. NLRP3 inflammasome is a multimeric protein complex consisting of NLRs, apoptosis-associated speck-like protein (ASC), and pro-caspase-1. Inflammasome assembly instigates caspase-1 activation, which results in proteolytic cleavage of pro-IL-1β and pro-IL-18 into their active mature forms, IL-1β and IL-18, respectively.
Platelets possess all the components required for assembly of an inflammasome complex (Qiao et al., 2018). Of these, NLRP3 in platelets has received the greatest attention and has been linked to a variety of illnesses, such as sepsis (Cornelius et al., 2020), dengue (Hottz et al., 2013) and sickle cell disease (Vogel et al., 2018). Oxidative stress plays a significant role in promoting the activation of inflammasome complexes (Zhao and Zhao, 2020). Calcium overload has been reported to impair mitochondrial function and generate excessive ROS in sepsis resulting in inflammasome activation (Zhao et al., 2020). Epigallocatechin-3-Gallate (EGCG), a polyphenol and green tea extract known for its antioxidant properties, also inhibits prion-mediated neurotoxicity (Lee et al., 2015) and has recently been reported as an inhibitor for NLRP3 inflammasome (Zhang et al., 2021). NLPR3 also contributes to platelet activation, aggregation, and thrombus formation in vitro (Murthy et al., 2017) and regulate integrin outside-in signaling (Qiao et al., 2018).
We have previously reported that prion proteins unleash a prothrombotic state in platelets associated with raised intracellular calcium and microvesicle release (Mallick et al., 2015), which is restrained in the presence of fibrinogen (Gautam et al., 2022). It was reported that prion protein (100 µM) also activates NLRP3 inflammasome in microglial cells (Shi et al., 2012), which is linked to its neurotoxicity (Hafner-Bratkovič et al., 2012). Additionally, as platelets can be exposed to PrPC released from their α-granules upon stimulation (Starke et al., 2005), we queried whether prion might also play a role in activating the platelet inflammasome. In this study, we report that exposure to PrP fuels inflammasome assembly and caspase-1 activation in platelets mediated through an upsurge in ROS, that leads to formation of platelet-monocyte/neutrophil aggregates unleashing an inflammatory phenotype.
Prion peptide (106–126) (KTNMKHAGAAAAGAVVGGLG) was purchased from Biomatik, United States. Antibodies against NLRP3 (#NBP2-12446), and ASC (#NBP1-78977APC) were from Novus Biologicals, United States. Antibodies against IL-1β (#12242) and β-actin (#A2066) were procured from Cell Signaling Technology and Sigma, respectively. HRP-conjugated goat anti-rabbit and anti-mouse IgG were the products of Bangalore Genei. APC-Mouse anti-human CD41a (#559777), FITC-Mouse anti-human CD14 (#555397), FACS lysis solution and FACS Flow sheath fluid were from BD Biosciences. Calcein AM (#C3100MP) and goat anti-rabbit IgG (Alexa Flour 488-conjugated) (#A11008) were procured from Invitrogen. Permanox slides (#160005) were from ThermoFisher. The FAM-FLICA Caspase-1 Assay kit (#SKU 97) was from ImmunoChemistry Technologies. Fibrinogen (Fg, #4883), YVAD-CHO (#400011), thrombin receptor-activating peptide (TRAP, #S1820), 2′,7′-dichlorodihydrofluoresceindiacetate (H2DCFDA, #D6883), epigallocatechin gallate (EGCG, #E4143), N-acetyl L-cysteine (NAC, #A7250), IgG (#5006), ethylenediaminetetraacetic acid (EDTA), sodium orthovanadate, dimethylsulfoxide (DMSO), Triton X-100, prostaglandin E1 (PGE1), paraformaldehyde (PFA) were purchased from Sigma while bovine serum albumin (BSA) were from HIMedia. All reagents were of analytical grade. Type I deionized water (18.2 MΩ cm, Millipore) was used for preparation of solutions. Experiments were carried out strictly as per the guidelines of Institutional Ethical Committee.
Peripheral venous blood was collected from healthy volunteers (both male and female participants with age varying between 20 and 40 years) in acid citrate dextrose (trisodium citrate, 74.8 mM; citric acid, 38.06 mM; and dextrose, 136 mM). Platelets were isolated by differential centrifugation. Blood was centrifuged at 100 g for 20 min. PGE1 (1 µM) and EDTA (2 mM) were added to the supernatant (platelet-rich plasma, PRP). PRP was centrifuged at 800 g for 7 min. Cells were washed in buffer A (20 mM HEPES, 134 mM NaCl, 2.9 mM KCl, 1 mM MgCl2, 5 mM glucose, 0.34 mM NaH2PO4, 12 mM NaHCO3, 0.35% BSA, 1uM PGE1, pH 6.2) and centrifuged again at 800 g for 7 min. Finally, cells were suspended in buffer B (pH 7.4), which was the same as Buffer A but without PGE1 and BSA. Cell count was maintained at 2–4 × 108 cells/mL by an automated cell counter (Multisizer 4, Beckman Coulter). Leucocyte contamination was found to be less than 0.015%. All steps were executed under sterile conditions, and precautions were taken to maintain the cells in a resting state.
PrP (106–126) (KTNMKHMAGAAAAGAVVGGLG) was dissolved in 1 mL buffer B (please see above) at 0.5 mM stock concentration and stored at −20°C in aliquots. In the untreated control counterparts (resting platelets), buffer B substituted prion solution as the vehicle.
The activity of caspase-1 was determined using a fluorescent inhibitor of caspase-1 (FLICA), whose structure is represented by 5-carboxyfluorescein-Tyr-Val-Ala-Asp-fluoromethylketone (FAM-YVAD-FMK). FLICA is cell-permeant dye that covalently interacts with ‘active’ caspase-1 through YVAD sequence emitting green fluorescent signal while inhibiting further enzymatic activity. Unbound dye diffuses out of the cell during the wash steps. Thus, the degree of fluorescence reflects the extent of caspase-1activity. Staining was carried out as per the manufacturer’s protocol. In brief, prior to exposure to PrP (50 µM for 20 min at RT) platelets were incubated with either EGCG (5 µM) or NAC (1 mM) for 10 min at 37°C, followed by incubation with FLICA reagent for 30 min at 37°C in the dark. Stained cells were washed, followed by fixation. Cells were suspended in sheath fluid and analyzed on a flow cytometer (FACSCalibur, BD Biosciences).
Platelets were incubated with H2DCFDA for 30 min at RT in the dark. H2DCFDA enters the cell, is deacetylated by esterases, and oxidized by ROS to highly fluorescent 2′,7′- dichlorofluorescein (DCF). Platelets were pre-treated with either EGCG (5 µM) or NAC (1 mM) for 10 min at 37°C, followed by incubation with either PrP (50 µM) or buffer B (vehicle) for 20 min at RT. Population of stained cells were analyzed by flow cytometry (FACSCalibur, BD Biosciences). Data were evaluated using CellQuest Pro Software as described earlier (Chaurasia et al., 2022).
Platelets pre-treated with either prion (50 µM) or buffer B (vehicle) were allowed to adhere to Permanox slides for 30 min, washed thrice with 1X PBS, and fixed with 4% PFA for 10 min. Following three washes, platelets were permeabilized with 0.1% Triton X-100 for 1 min. Slides were blocked with 2% BSA for 1 h, washed and incubated with anti-NLRP3 antibody (1:50) overnight at 4°C, washed thrice, and then exposed to anti-rabbit Alexa Flour 488-conjugated secondary antibody (1:100) for 2 h at RT. Slides were rewashed and incubated with APC-conjugated rabbit anti-ASC antibody for 2 h at RT. Controls were processed identically except for the omission of primary antibodies. Slides were examined under Zeiss LSM 700 laser scanning confocal microscope with ×63 oil objective (numerical aperture, 1.40) and 1 AU pinhole size. Results were representative of three different experiments.
Platelet-monocyte/neutrophil (PMN) interaction was evaluated as described earlier (Ekhlak et al., 2023). Fresh human blood (20 μL) was added to a cocktail containing 10 μL each from APC-anti-CD41a (platelet-specific) and FITC-anti-CD14 (leucocyte-specific) antibodies and mixed gently. Then it was treated with either EGCG (5 µM) or YVAD (1 µM), followed by incubation with TRAP (2 μM) or PrP (50 µM) for 15–20 min at RT. RBCs were lysed with 800 μL FACS lysis solution (1X, BD Biosciences) for 10 min at RT. PMN interaction was analysed on a flow cytometer. Side scatter voltage was set at 350 with a threshold of 52 V. A dot plot of side scatter (SSC) versus log FITC-CD14 fluorescence was created using the CellQuest Pro software. Amorphous gates were drawn for monocyte (high fluorescence and low SSC) and neutrophil (low fluorescence and high SSC) populations. Fluorescence data were acquired from each sample using four-quadrant logarithmic amplification for 1,000 events in either neutrophil or monocyte gate and analysed with CellQuest Pro Software.
We have earlier demonstrated that, prion induces platelets to adopt a procoagulant state (Gautam et al., 2022; Mallick et al., 2015). In the present study, we further investigated the role of prions in transforming these cells to an inflammatory phenotype. Exposure of platelets to PrP (106–126) (50 µM) for 20 min brought about focal accumulations of NLRP3 and ASC, the key components of the inflammasome, in platelet cytosol (Figure 1A), reflective of the complex formation (Vogel et al., 2018)). This was associated with significant enhancement in caspase-1 activity (by 296%) in PrP-treated platelets (Figures 1B, C). Our previous study demonstrates that prion promotes platelet agglutination rather than aggregation (Mallick et al., 2015), this might be the reason behind the clustering of cells in PrP-treated platelets (Figure 1A).
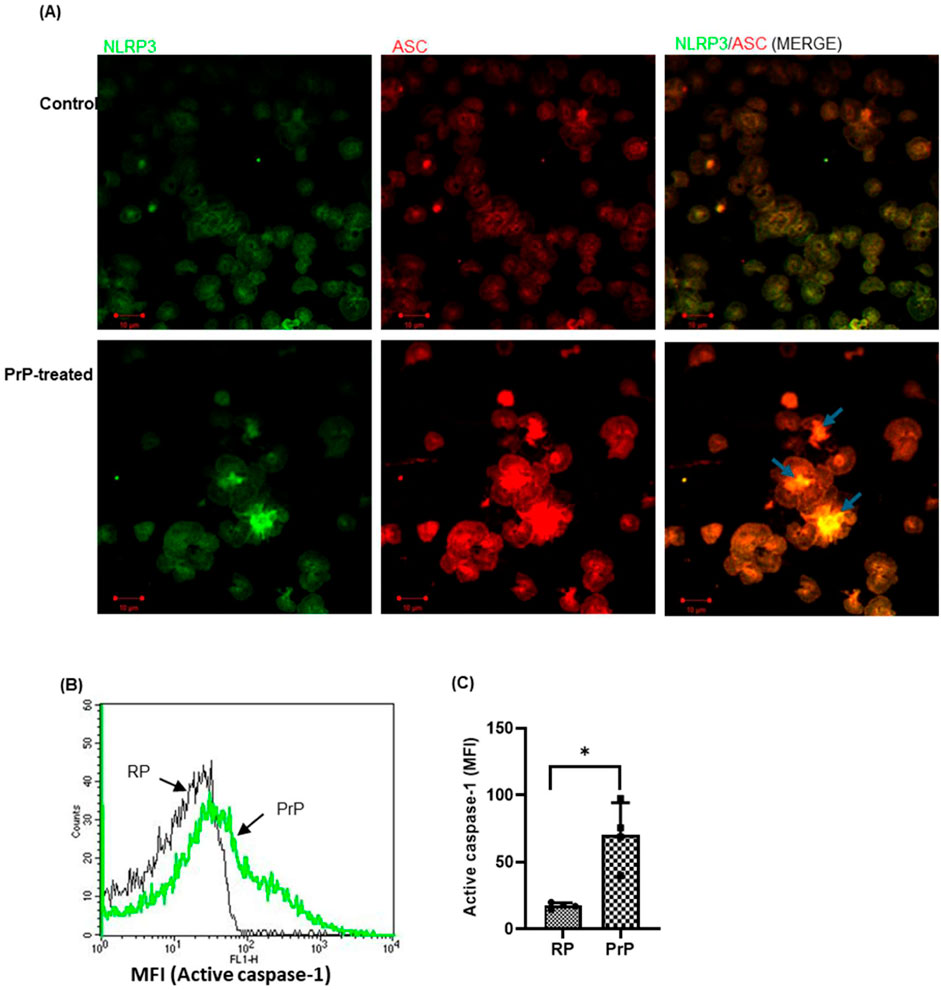
Figure 1. Prion promotes inflammasome assembly in platelets. (A) Confocal images representing inflammasome assembly in platelets treated either with prion or buffer B (Control). NLRP3 (green) and ASC (red) proteins were dispersed uniformly in control platelets while PrP treatment allowed focal clustering of these proteins prompting brighter visualization (scale bar, 10 µm). Arrows indicate co-localization of NLRP3 and ASC in PrP-treated platelets. RP, resting platelets. Images are representative of three independent experiments. (B, C) PrP (50 µM) provoked robust caspase-1 activation. The bar graph represents mean ± SEM (n = 4). *P < 0.05 as compared to RP, analysed by paired Student’s t-test.
We next investigated the underpinning molecular mechanism governing inflammasome activation in prion-challenged platelets. ROS is known to play a vital role in inflammasome activation (Abais et al., 2015). Level of platelet intracellular ROS was found to be augmented by 200% upon exposure to PrP (106–126). The rise was significantly blunted by 84% and 65%, respectively, upon pre-treatment of cells with either EGCG (5 µM) or NAC (1 mM), the known scavengers of ROS (Figures 2A, B). Next, we interrogated the role of ROS in driving prion-mediated inflammasome signaling. Prior exposure to either EGCG or NAC prohibited caspase-1 activity by 57% and 51%, respectively, thus implicating prion-ROS-inflammasome-caspase-1 signaling axis in transformation of resting platelets to functionally active inflammatory units (Figures 2C, D). It is noteworthy to state here that, PrP (106–126) induces ROS generation and caspase-1 activation in a dose-dependent manner evaluated at 20 μM and 50 µM prion concentrations (Supplementary Figure 3).
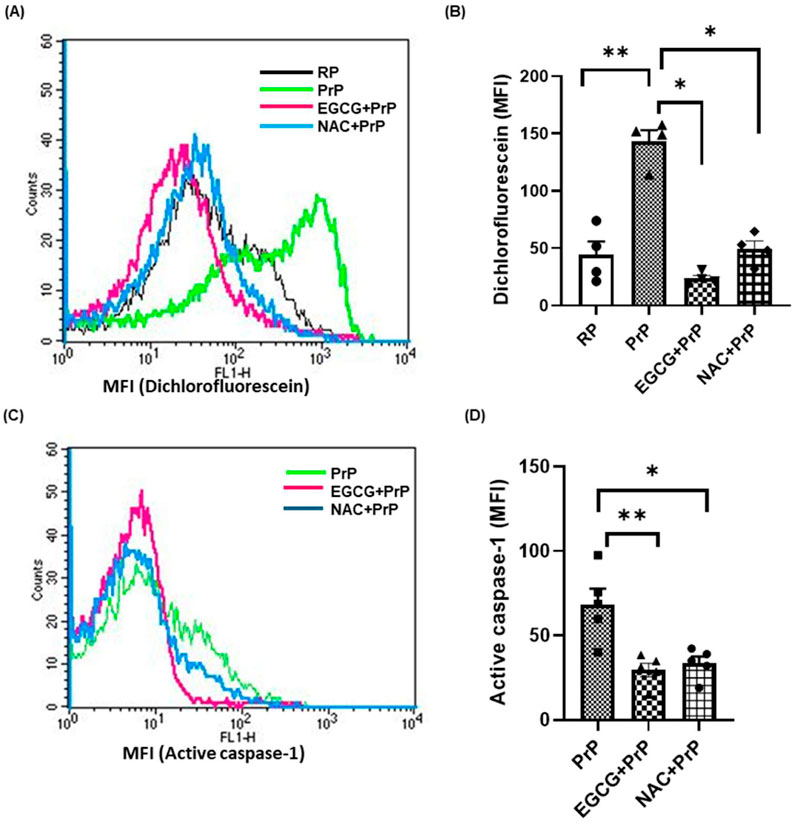
Figure 2. ROS plays a key role in inflammasome activation in platelets. (A, B) PrP (106–126) (50 µM) induced a significant rise in intracellular ROS in platelets, which was prevented upon pre-treatment with either EGCG (5 µM) or NAC (1 mM). (C, D) PrP-induced caspase-1 activity in platelets was significantly prohibited by either EGCG or NAC. RP, resting platelets treated with buffer B. Data are presented as mean ± SEM and are representatives of at least four different experiments. Statistical analysis was performed using repeated measures one-way ANOVA (paired), with Sidak’s multiple comparison test. *P < 0.05; **P < 0.01.
As prion induces NLRP3 inflammasome activation (signal 2) in platelets, it prompted us to ask whether it also instigates the priming signal (signal 1) that involves synthesis of pro-IL-1β and NLRP3. Although LPS has been shown to promote synthesis of pro-IL-1β in anucleate platelets (Brown et al., 2013; Lindemann et al., 2001), the necessity of signal-1 for inflammasome initiation in platelets has been a subject of debate (Hottz et al., 2013). We did not observe synthesis of either pro-IL-1β or NLRP3 in response to PrP (106–126) in platelets (Supplementary Figure 2), thus ruling out the induction of priming signal in these cells.
Platelets interaction with circulating monocytes and neutrophils is a sensitive index of the state of platelet activity (Cerletti et al., 2012; Ortiz-Muñoz et al., 2014). PMN interaction generates a pro-inflammatory phenotype by activating leucocytes and the emergence of neutrophil extracellular traps (NETs) (Margraf and Zarbock, 2019). Circulating amyloid-β, another β pleat-rich peptide, reportedly activates platelets leading to a localized inflammatory response by promoting NET formation (Canobbio et al., 2017). Since prion peptide upregulated NLRP3 inflammasome activity in platelets, we asked whether it would provoke interaction with leucocytes. Predictably, platelet agonist TRAP (2 µM) prompted a surge in platelet-monocyte as well as platelet-neutrophil aggregate formation, which was impaired by YVAD, a specific inhibitor of caspase-1 (Supplementary Figure 1), thus linking inflammasome to platelet-monocyte/neutrophil (PMN) interaction. Prion peptide, too, propelled platelet-monocyte and platelet-neutrophil interactions significantly by 178% and 39%, respectively (Figure 3). These prion-instigated events were forestalled either by the ROS-scavenger EGCG (by 31% and 29%, respectively), or by YVAD (by 33% and 32%, respectively) (Figure 3), thus implicating the prion-ROS-caspase-1 axis in evolution pro-inflammatory phenotype.

Figure 3. Prion prompts platelet-monocyte/neutrophil (PMN) aggregate formation. Flow cytometric analysis of platelet-monocyte (A–D) and platelet-neutrophil (F–I) interactions in whole blood stained with anti-CD41a-APC (specific for platelets) and anti-CD14-FITC (specific for neutrophils/monocytes), followed by treatment with PrP (106–126) (50 µM) in presence or absence of either EGCG (5 µM) or YVAD (1 µM), as indicated. Control platelets were treated with buffer B (vehicle) in place of PrP. Amorphous gates were drawn for monocyte (high fluorescence and low SSC) and neutrophil (low fluorescence and high SSC) populations. (E) (n = 7) and (J) (n = 6), bar diagrams showing the percentage of platelet-monocyte and platelet-neutrophil aggregate formation, respectively (mean ± SEM). *P < 0.05, **P < 0.01, as analysed by repeated measures one-way ANOVA (paired) with Dunnet’s multiple comparison test.
Several laboratories including ours have demonstrated high-affinity interaction between fibrinogen and β pleat-rich peptides such as prion and amyloid-β that prohibits action of these proteins on neuronal cells, as well as on platelets (Ahn et al., 2014; Ahn et al., 2010; Gautam et al., 2022; Sonkar et al., 2014). Keeping with above, we observed a 42% decline in prion-induced caspase-1 activity when platelets were pre-treated with fibrinogen (Figures 4A, B), reflective of impaired inflammasome activation. Concordantly, prion-mediated ROS generation, too, was restrained by 58% upon prior exposure of platelets to fibrinogen (Figures 4C–E). However, fibrinogen was unable to restrict prion-mediated PMN interaction (data not shown), which could be attributable to factors in blood interacting with either fibrinogen or PrP, thus precluding their association.
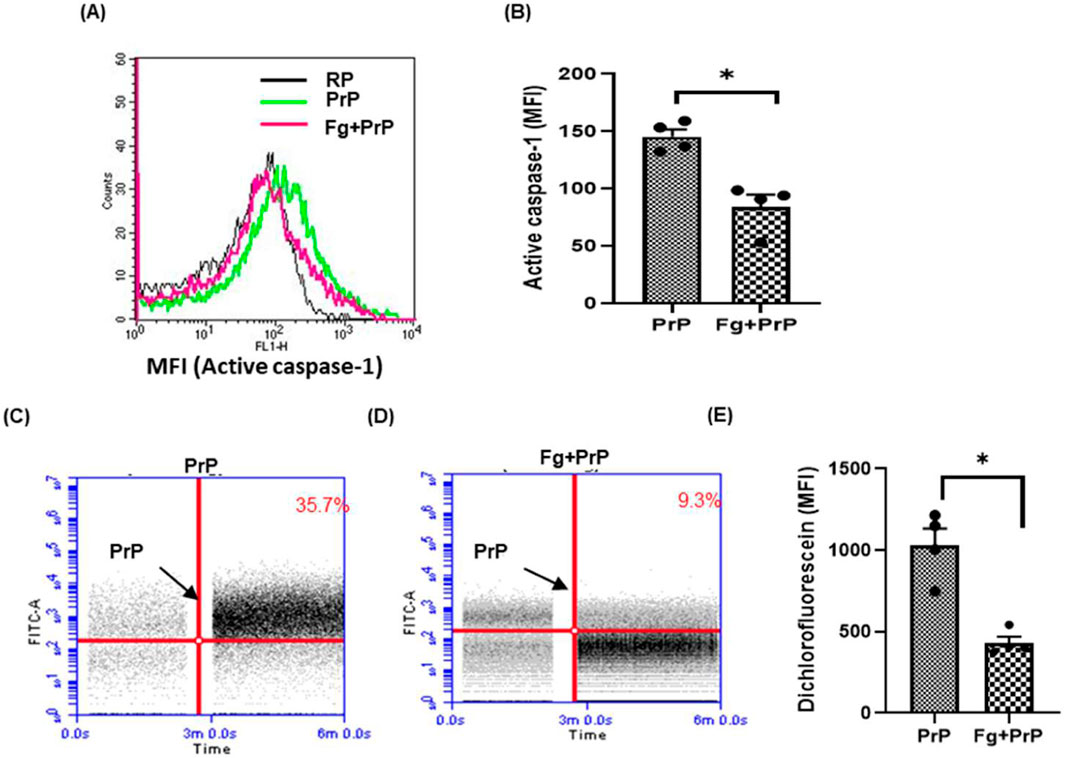
Figure 4. Fibrinogen prohibits prion-induced caspase-1 activation and generation of ROS. (A, B) prion-induced caspase-1 activity in the absence or presence of fibrinogen (Fg) (2 mg/mL). Fibrinogen was incubated for 10 min before adding PrP (106–126) (50 µM). (C–E) prion-induced surge in Intracellular ROS in platelet population in the absence or presence of fibrinogen, respectively. The bar graphs represent mean ± SEM from four independent experiments. RP, resting platelets treated with buffer B. *P < 0.05 as compared with PrP-treated platelets, analysis by paired Student’s t-test.
Platelets express approximately 2000 PrPc on their surface, while they also store them in their α-granules (Starke et al., 2005). Platelet activation triggers the release of stored PrPc from granules, augmenting prion surface count to as many as 4,500 molecules per platelet and raising potential for its interaction with nearby platelets (Holada et al., 1998). Prions can also cross the blood-brain barrier and enter vasculature, exposing themselves to blood cells in circulation (Banks et al., 2009). Diseases associated with protein misfolding such as prion disease, Alzheimer’s disease, type-2 diabetes, and Parkinson’s disease are marked by inflammatory responses (Shi et al., 2015). Misfolded proteins trigger cellular stress (Rao and Bredesen, 2004), which promotes inflammasome activation (Shi et al., 2015). Prion has also been shown to promote the transcription of DAMP receptors in neuronal cells (Carroll et al., 2018) and activate NLRP3 inflammasome in microglial cells (Shi et al., 2012). The effect of prion on platelets is not a novel concept as we have earlier identified its role in hemostasis (Gautam et al., 2022; Mallick et al., 2015; Shi et al., 2012). Beyond their primary contribution to hemostasis, platelets are also recognized as immune effector cells, which induce NLRP3 inflammasome activation in innate immune cells like monocytes (Rolfes et al., 2020). Previous research has demonstrated that inflammasome complex formation in activated platelets facilitates the progression of dengue and sickle cell disease (Hottz et al., 2013; Vogel et al., 2018). These findings inspired us to explore the role of misfolded prion protein in inflammasome signaling in platelets.
Oxidative stress plays a crucial role in the conversion of PrPc to PrPsc (Prasad and Bondy, 2019). Prion promotes ROS generation and Ca2+ metabolism in neuronal cells (De Mario et al., 2019) while increased calcium level provokes higher production of ROS (Görlach et al., 2015). In the CNS, oxidative stress and neuroinflammation are two of the main pathological hallmarks associated with multiple neurodegenerative diseases (Braidy et al., 2022). We have already demonstrated that prion instigates an increase in the level of intracellular free calcium in platelets (Mallick et al., 2015), which prompted us to examine the cytosolic oxidant status in these cells. In present study, prion was found to facilitate intracellular ROS production in platelets, which was inhibited in the presence of either of two ROS scavengers, EGCG and NAC. EGCG, known for its anti-oxidant property (Mokra et al., 2022), also inhibits NLRP3 inflammasome (Zhang et al., 2021). It has anti-prion activity (Barreca et al., 2018) and protects against prion protein-induced damage by regulating autophagy (Lee et al., 2015). We found that prion promotes inflammasome complex formation by generating active caspase-1, which subsequently triggers a pro-inflammatory response in platelets. ROS scavengers (NAC and EGCG) effectively attenuate caspase-1 activation, which underscores their potential in addressing inflammatory disorders. Inflammasome assembly was validated in our study from co-localization of NLRP3 and ASC proteins, and cleavage of pro-caspase-1 into its enzymatically active form.
In a recent study, blocking inflammasome activation was reported to attenuate proliferation and invasion of circulating pro-inflammatory monocytes in cases of stroke (Baek, 2024). PrPsc activates microglia and astrocytes to release pro-inflammatory cytokines, namely, IL-1β, IL-8, TNF-α, and IL-6, unleashing an inflammatory phenotype. Activated platelets engage with leucocytes through numerous surface receptors that include P-selectin on platelets ligating with P-selectin glycoprotein ligand-1 (PSGL-1) expressed on leucocyte membrane. Similarly, CD40L, GPIIb/IIIa and GPIb, respectively, allow platelets and platelet-derived extracellular vesicles (PEVs) to interact with CD40, ICAM-1 and Mac-1 receptors present on leucocytes. In recent years, PEVs have emerged as a significant source of non-coding regulatory RNAs in the blood (Tao et al., 2017), which are capable of binding to leucocytes (Fendl et al., 2018). We have earlier demonstrated significant shedding of PEVs from prion-challenged platelets (Mallick et al., 2015). Here we demonstrate that prion promotes platelet-monocyte/neutrophil interaction, which is repressed either by EGCG or YVAD, the inhibitor of caspase-1.
Pro-thrombotic attributes of platelet-monocyte/neutrophil interaction have already been recognized (Ghasemzadeh and Hosseini, 2013). Thrombus is a dynamic structure composed of platelets, RBCs, leucocytes, fibrin, and von Willebrand Factor, with platelets and fibrin playing seminal roles in its formation and stability. However, the sizeable presence of prions within the thrombus milieu sourced from platelets cannot be disregarded. Research into this area could lead to newer insights into role of prions linked to inflammation and thrombogenesis. In conclusion, we found that prion stimulates ROS production in platelets leading to activation of caspase-1 and platelet-monocyte/neutrophil aggregate formation, thus implicating a prion-ROS-inflammasome-caspase-1 signaling axis in transformation of resting platelets to functionally active inflammatory units. However, considerable caution must be used in drawing such a conclusion due to limitations like only one type of prion fragment being employed in the study and experiments being performed only ex vivo on isolated platelets and whole blood.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Fresh venous human blood was collected from healthy volunteers under informed consent strictly as per the recommendation of the Institutional Ethical Committee of Banaras Hindu University (Approval No. Dean/2015-16/EC/76). The study methodologies confirmed to the standards set by the Declaration of Helsinki. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
RV: Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing–original draft, Writing–review and editing. JK: Formal Analysis, Investigation, Writing–review and editing. AM: Data curation, Formal Analysis, Writing–review and editing. RC: Formal Analysis, Writing–review and editing. DD: Conceptualization, Formal Analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by a J. C. Bose National Fellowship (JBR/2023/000004), and grants received by D. Dash from the Department of Biotechnology (DBT) (BT/PR48671/CMD/150/41/2023), Government of India, the Indian Council of Medical Research (ICMR) Small Extramural Grant (EMDR/SG/12/2023-2835), ICMR Extramural Grant under CAR (71/4/2018-BMS/CAR) and the Institute of Eminence (IoE) Faculty Incentive Grant (OH-31-IOE/6031) from Banaras Hindu University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2025.1534235/full#supplementary-material
Abais, J. M., Xia, M., Zhang, Y., Boini, K. M., and Li, P. L. (2015). Redox regulation of NLRP3 inflammasomes: ROS as trigger or effector? Antioxid. Redox Signal 22 (13), 1111–1129. doi:10.1089/ars.2014.5994
Ahn, H. J., Glickman, J. F., Poon, K. L., Zamolodchikov, D., Jno-Charles, O. C., Norris, E. H., et al. (2014). A novel Aβ-fibrinogen interaction inhibitor rescues altered thrombosis and cognitive decline in Alzheimer's disease mice. J. Exp. Med. 211 (6), 1049–1062. doi:10.1084/jem.20131751
Ahn, H. J., Zamolodchikov, D., Cortes-Canteli, M., Norris, E. H., Glickman, J. F., and Strickland, S. (2010). Alzheimer's disease peptide beta-amyloid interacts with fibrinogen and induces its oligomerization. Proc. Natl. Acad. Sci. U. S. A. 107 (50), 21812–21817. doi:10.1073/pnas.1010373107
Banks, W. A., Robinson, S. M., Diaz-Espinoza, R., Urayama, A., and Soto, C. (2009). Transport of prion protein across the blood-brain barrier. Exp. Neurol. 218 (1), 162–167. doi:10.1016/j.expneurol.2009.04.025
Barreca, M. L., Iraci, N., Biggi, S., Cecchetti, V., and Biasini, E. (2018). Pharmacological agents targeting the cellular prion protein. Pathogens 7 (1), 27. doi:10.3390/pathogens7010027
Braidy, N., Cig, B., Jiang, L. H., and Shu, X. (2022). Editorial: the role of astrocyte signalling pathways in ageing-induced neurodegenerative pathologies. Front. Physiol. 13, 1002428. doi:10.3389/fphys.2022.1002428
Brown, G. T., Narayanan, P., Li, W., Silverstein, R. L., and McIntyre, T. M. (2013). Lipopolysaccharide stimulates platelets through an IL-1β autocrine loop. J. Immunol. 191 (10), 5196–5203. doi:10.4049/jimmunol.1300354
Canobbio, I., Visconte, C., Momi, S., Guidetti, G. F., Zarà, M., Canino, J., et al. (2017). Platelet amyloid precursor protein is a modulator of venous thromboembolism in mice. Blood 130 (4), 527–536. doi:10.1182/blood-2017-01-764910
Carroll, J. A., Race, B., Williams, K., and Chesebro, B. (2018). Toll-like receptor 2 confers partial neuroprotection during prion disease. PLoS One 13 (12), e0208559. doi:10.1371/journal.pone.0208559
Cerletti, C., Tamburrelli, C., Izzi, B., Gianfagna, F., and de Gaetano, G. (2012). Platelet-leukocyte interactions in thrombosis. Thromb. Res. 129 (3), 263–266. doi:10.1016/j.thromres.2011.10.010
Chaurasia, S. N., Ekhlak, M., Kushwaha, G., Singh, V., Mallick, R. L., and Dash, D. (2022). Notch signaling functions in noncanonical juxtacrine manner in platelets to amplify thrombogenicity. Elife 11, e79590. doi:10.7554/eLife.79590
Cornelius, D. C., Travis, O. K., Tramel, R. W., Borges-Rodriguez, M., Baik, C. H., Greer, M., et al. (2020). NLRP3 inflammasome inhibition attenuates sepsis-induced platelet activation and prevents multi-organ injury in cecal-ligation puncture. PLoS One 15 (6), e0234039. doi:10.1371/journal.pone.0234039
De Mario, A., Peggion, C., Massimino, M. L., Norante, R. P., Zulian, A., Bertoli, A., et al. (2019). The link of the prion protein with Ca2+ metabolism and ROS production, and the possible implication in aβ toxicity. Int. J. Mol. Sci. 20 (18), 4640. doi:10.3390/ijms20184640
Ekhlak, M., Kulkarni, P. P., Singh, V., Chaurasia, S. N., Mohapatra, S. K., Chaurasia, R. N., et al. (2023). Necroptosis executioner MLKL plays pivotal roles in agonist-induced platelet prothrombotic responses and lytic cell death in a temporal order. Cell Death Differ. 30 (8), 1886–1899. doi:10.1038/s41418-023-01181-6
Ettaiche, M., Pichot, R., Vincent, J. P., and Chabry, J. (2000). In vivo cytotoxicity of the prion protein fragment 106-126. J. Biol. Chem. 275 (47), 36487–36490. doi:10.1074/jbc.C000579200
Fendl, B., Eichhorn, T., Weiss, R., Tripisciano, C., Spittler, A., Fischer, M. B., et al. (2018). Differential interaction of platelet-derived extracellular vesicles with circulating immune cells: roles of TAM receptors, CD11b, and phosphatidylserine. Front. Immunol. 9, 2797. doi:10.3389/fimmu.2018.02797
Gautam, D., Kailashiya, J., Tiwari, A., Chaurasia, R. N., Annarapu, G. K., Guchhait, P., et al. (2022). Fibrinogen mitigates prion-mediated platelet activation and neuronal cell toxicity. Front. Cell Dev. Biol. 10, 834016. doi:10.3389/fcell.2022.834016
Ghasemzadeh, M., and Hosseini, E. (2013). Platelet-leukocyte crosstalk: linking proinflammatory responses to procoagulant state. Thromb. Res. 131 (3), 191–197. doi:10.1016/j.thromres.2012.11.028
Görlach, A., Bertram, K., Hudecova, S., and Krizanova, O. (2015). Calcium and ROS: a mutual interplay. Redox Biol. 6, 260–271. doi:10.1016/j.redox.2015.08.010
Gu, Y., Fujioka, H., Mishra, R. S., Li, R., and Singh, N. (2002). Prion peptide 106-126 modulates the aggregation of cellular prion protein and induces the synthesis of potentially neurotoxic transmembrane PrP. J. Biol. Chem. 277 (3), 2275–2286. doi:10.1074/jbc.M104345200
Hafner-Bratkovič, I., Benčina, M., Fitzgerald, K. A., Golenbock, D., and Jerala, R. (2012). NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL-1β and neuronal toxicity. Cell Mol. Life Sci. 69 (24), 4215–4228. doi:10.1007/s00018-012-1140-0
Holada, K., Mondoro, T. H., Muller, J., and Vostal, J. G. (1998). Increased expression of phosphatidylinositol-specific phospholipase C resistant prion proteins on the surface of activated platelets. Br. J. Haematol. 103 (1), 276–282. doi:10.1046/j.1365-2141.1998.01032.x
Hottz, E. D., Lopes, J. F., Freitas, C., Valls-de-Souza, R., Oliveira, M. F., Bozza, M. T., et al. (2013). Platelets mediate increased endothelium permeability in dengue through NLRP3-inflammasome activation. Blood 122 (20), 3405–3414. doi:10.1182/blood-2013-05-504449
Lee, J. H., Moon, J. H., Kim, S. W., Jeong, J. K., Nazim, U. M., Lee, Y. J., et al. (2015). EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells. Oncotarget 6 (12), 9701–9717. doi:10.18632/oncotarget.3832
Lindemann, S., Tolley, N. D., Dixon, D. A., McIntyre, T. M., Prescott, S. M., Zimmerman, G. A., et al. (2001). Activated platelets mediate inflammatory signaling by regulated interleukin 1beta synthesis. J. Cell Biol. 154 (3), 485–490. doi:10.1083/jcb.200105058
Mallick, R. L., Kumari, S., Singh, N., Sonkar, V. K., and Dash, D. (2015). Prion protein fragment (106-126) induces prothrombotic state by raising platelet intracellular calcium and microparticle release. Cell Calcium 57 (4), 300–311. doi:10.1016/j.ceca.2015.02.002
Margraf, A., and Zarbock, A. (2019). Platelets in inflammation and resolution. J. Immunol. 203 (9), 2357–2367. doi:10.4049/jimmunol.1900899
Melo, J. B., Agostinho, P., and Oliveira, C. R. (2007). Prion protein aggregation and neurotoxicity in cortical neurons. Ann. N. Y. Acad. Sci. 1096, 220–229. doi:10.1196/annals.1397.088
Mokra, D., Adamcakova, J., and Mokry, J. (2022). Green tea polyphenol (-)-Epigallocatechin-3-Gallate (EGCG): a time for a New player in the treatment of respiratory diseases? Antioxidants (Basel) 11 (8), 1566. doi:10.3390/antiox11081566
Murthy, P., Durco, F., Miller-Ocuin, J. L., Takedai, T., Shankar, S., Liang, X., et al. (2017). The NLRP3 inflammasome and bruton's tyrosine kinase in platelets co-regulate platelet activation, aggregation, and in vitro thrombus formation. Biochem. Biophys. Res. Commun. 483 (1), 230–236. doi:10.1016/j.bbrc.2016.12.161
Ortiz-Muñoz, G., Mallavia, B., Bins, A., Headley, M., Krummel, M. F., and Looney, M. R. (2014). Aspirin-triggered 15-epi-lipoxin A4 regulates neutrophil-platelet aggregation and attenuates acute lung injury in mice. Blood 124 (17), 2625–2634. doi:10.1182/blood-2014-03-562876
Prasad, K. N., and Bondy, S. C. (2019). Oxidative and inflammatory events in prion diseases: can they Be therapeutic targets? Curr. Aging Sci. 11 (4), 216–225. doi:10.2174/1874609812666190111100205
Prinz, M., Huber, G., Macpherson, A. J., Heppner, F. L., Glatzel, M., Eugster, H. P., et al. (2003). Oral prion infection requires normal numbers of Peyer's patches but not of enteric lymphocytes. Am. J. Pathol. 162 (4), 1103–1111. doi:10.1016/s0002-9440(10)63907-7
Qiao, J., Wu, X., Luo, Q., Wei, G., Xu, M., Wu, Y., et al. (2018). NLRP3 regulates platelet integrin αIIbβ3 outside-in signaling, hemostasis and arterial thrombosis. Haematologica 103 (9), 1568–1576. doi:10.3324/haematol.2018.191700
Rao, R. V., and Bredesen, D. E. (2004). Misfolded proteins, endoplasmic reticulum stress and neurodegeneration. Curr. Opin. Cell Biol. 16 (6), 653–662. doi:10.1016/j.ceb.2004.09.012
Robertson, C., Booth, S. A., Beniac, D. R., Coulthart, M. B., Booth, T. F., and McNicol, A. (2006). Cellular prion protein is released on exosomes from activated platelets. Blood 107 (10), 3907–3911. doi:10.1182/blood-2005-02-0802
Rolfes, V., Ribeiro, L. S., Hawwari, I., Böttcher, L., Rosero, N., Maasewerd, S., et al. (2020). Platelets fuel the inflammasome activation of innate immune cells. Cell Rep. 31 (6), 107615. doi:10.1016/j.celrep.2020.107615
Sandberg, M. K., Al-Doujaily, H., Sharps, B., Clarke, A. R., and Collinge, J. (2011). Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 470 (7335), 540–542. doi:10.1038/nature09768
Semple, J. W., Italiano, J. E., and Freedman, J. (2011). Platelets and the immune continuum. Nat. Rev. Immunol. 11 (4), 264–274. doi:10.1038/nri2956
Shi, F., Kouadir, M., and Yang, Y. (2015). NALP3 inflammasome activation in protein misfolding diseases. Life Sci. 135, 9–14. doi:10.1016/j.lfs.2015.05.011
Shi, F., Yang, L., Kouadir, M., Yang, Y., Wang, J., Zhou, X., et al. (2012). The NALP3 inflammasome is involved in neurotoxic prion peptide-induced microglial activation. J. Neuroinflammation 9, 73. doi:10.1186/1742-2094-9-73
Simák, J., Holada, K., D'Agnillo, F., Janota, J., and Vostal, J. G. (2002). Cellular prion protein is expressed on endothelial cells and is released during apoptosis on membrane microparticles found in human plasma. Transfusion 42 (3), 334–342. doi:10.1046/j.1537-2995.2002.00072.x
Sonkar, V. K., Kulkarni, P. P., and Dash, D. (2014). Amyloid β peptide stimulates platelet activation through RhoA-dependent modulation of actomyosin organization. Faseb J. 28 (4), 1819–1829. doi:10.1096/fj.13-243691
Starke, R., Harrison, P., Mackie, I., Wang, G., Erusalimsky, J. D., Gale, R., et al. (2005). The expression of prion protein (PrP(C)) in the megakaryocyte lineage. J. Thromb. Haemost. 3 (6), 1266–1273. doi:10.1111/j.1538-7836.2005.01343.x
Tao, S. C., Guo, S. C., and Zhang, C. Q. (2017). Platelet-derived extracellular vesicles: an emerging therapeutic approach. Int. J. Biol. Sci. 13 (7), 828–834. doi:10.7150/ijbs.19776
Vallabh, S. M., Nobuhara, C. K., Llorens, F., Zerr, I., Parchi, P., Capellari, S., et al. (2019). Prion protein quantification in human cerebrospinal fluid as a tool for prion disease drug development. Proc. Natl. Acad. Sci. U. S. A. 116 (16), 7793–7798. doi:10.1073/pnas.1901947116
Vogel, S., Arora, T., Wang, X., Mendelsohn, L., Nichols, J., Allen, D., et al. (2018). The platelet NLRP3 inflammasome is upregulated in sickle cell disease via HMGB1/TLR4 and Bruton tyrosine kinase. Blood Adv. 2 (20), 2672–2680. doi:10.1182/bloodadvances.2018021709
Zhang, S., Zhang, S., Hu, L., Zhai, L., Xue, R., Ye, J., et al. (2015). Nucleotide-binding oligomerization domain 2 receptor is expressed in platelets and enhances platelet activation and thrombosis. Circulation 131 (13), 1160–1170. doi:10.1161/circulationaha.114.013743
Zhang, Z., Zhang, X., Bi, K., He, Y., Yan, W., Yang, C. S., et al. (2021). Potential protective mechanisms of green tea polyphenol EGCG against COVID-19. Trends Food Sci. Technol. 114, 11–24. doi:10.1016/j.tifs.2021.05.023
Zhao, C., and Zhao, W. (2020). NLRP3 inflammasome-A key player in antiviral responses. Front. Immunol. 11, 211. doi:10.3389/fimmu.2020.00211
Keywords: prion, platelets, NLRP3 inflammasome, platelet-monocyte/neutrophil interaction, ROS, EGCG, caspase-1
Citation: Verma R, Kailashiya J, Mukherjee A, Chaurasia RN and Dash D (2025) Prion protein fragment (106–126) activates NLRP3 inflammasome and promotes platelet-monocyte/neutrophil interactions, potentially contributing to an inflammatory state. Front. Cell Dev. Biol. 13:1534235. doi: 10.3389/fcell.2025.1534235
Received: 25 November 2024; Accepted: 10 February 2025;
Published: 25 February 2025.
Edited by:
Bilal ÇİĞ, Ahi Evran University Medicine Faculty Department of Physiology, TürkiyeReviewed by:
Ulrike Resch, Medical University of Vienna, AustriaCopyright © 2025 Verma, Kailashiya, Mukherjee, Chaurasia and Dash. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Debabrata Dash, ZGRhc2guYmlvY2hlbUBnbWFpbC5jb20=
†ORCID: Rashmi Verma, orcid.org/0009-0002-0011-4895, Debabrata Dash, orcid.org/0009-0008-3595-8928
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.