 Shahnawaz A. Baba
Shahnawaz A. Baba Aran Zakeri
Aran Zakeri Jay S. Desgrosellier
Jay S. Desgrosellier- 1Department of Pathology, University of California, San Diego, La Jolla, CA, United States
- 2Moores Cancer Center, University of California, San Diego, La Jolla, CA, United States
Despite a critical role for tumor-initiating cancer stem cells (CSCs) in breast cancer progression, major questions remain about the properties and signaling pathways essential for their function. Recent discoveries highlighting mechanisms of CSC-resistance to the stress caused by chromosomal instability (CIN) may provide valuable new insight into the underlying forces driving stemness properties. While stress tolerance is a well-known attribute of CSCs, CIN-induced stress is distinctive since levels appear to increase during tumor initiation and metastasis. These dynamic changes in CIN levels may serve as a barrier constraining the effects of non-CSCs and shaping the stemness landscape during the early stages of disease progression. In contrast to most other stresses, CIN can also paradoxically activate pro-tumorigenic antiviral signaling. Though seemingly contradictory, this may indicate that mechanisms of CIN tolerance and pro-tumorigenic inflammatory signaling closely collaborate to define the CSC state. Together, these unique features may form the basis for a critical relationship between CIN and stemness properties.
Introduction
CIN is a widely appreciated hallmark of cancer (Hanahan and Weinberg, 2011). Defined by continuous chromosomal missegregation, CIN is distinct from aneuploidy, which represents a state of abnormal chromosome number (Holland and Cleveland, 2009). The combined effects of CIN and aneuploidy play a well-described role in tumor evolution by promoting karyotypic heterogeneity (Bakhoum and Compton, 2012; Gronroos and Lopez-Garcia, 2018) and is thoroughly reviewed elsewhere (Chen et al., 2024; Hosea et al., 2024). By altering gene dosage, CIN can promote amplification of new oncogenes and deletion of tumor suppressors that aid cancer evolution and resistance to therapy (Benner et al., 1991; Turner et al., 2017; Albertson, 2006; Lukow et al., 2021). However, inherent to all of these effects is the ability of cancer cells to first tolerate the stress induced by CIN. Thus, the ability to resist this stress must be an important feature of aggressive breast cancer cells. To distinguish the effects of CIN versus aneuploidy on breast cancer progression, approaches now exist to specifically measure CIN by quantifying chromosomal missegregations during anaphase (Bakhoum and Compton, 2012), or assessing enrichment for CIN gene signatures (Bakhoum et al., 2018; Carter et al., 2006). Studies using these approaches have shown that CIN levels are higher in metastatic lesions compared to matched primary disease, suggesting that this stress is induced during metastasis (Bakhoum et al., 2018). These high CIN levels may pose a significant barrier preventing metastatic outgrowth and selecting for the most CIN-resistant cells. Furthermore, distinguishing CIN’s effects from those due to aneuploidy revealed that CIN mediates unique biological responses. These include roles in tumor initiation/metastasis (Bakhoum et al., 2018), resistance to stress (Hong et al., 2022), and activation of pro-tumorigenic signaling pathways (Bakhoum et al., 2018; Hong et al., 2022), placing CIN at the intersection between stress tolerance and CSC properties (Figure 1). Together, this makes CIN’s potential function in defining cancer stemness an appealing target for further investigation.
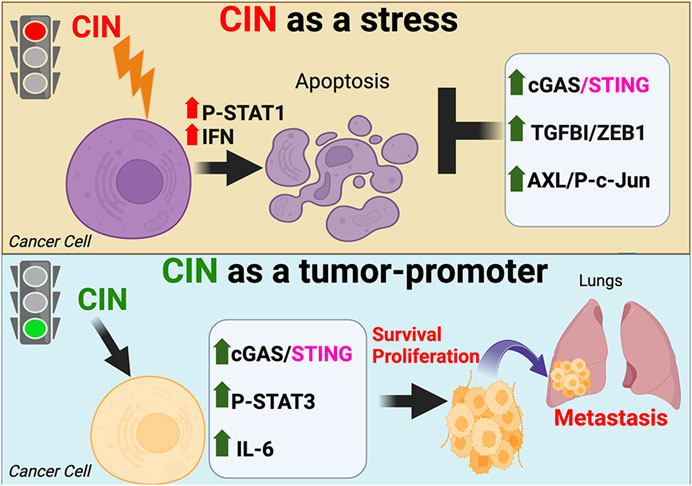
Figure 1. The dual effects of chromosomal instability (CIN) on cancer cells. CIN can paradoxically act as a lethal stress limiting the ability of tumor cells to transform or progress (top), or initiate pro-metastatic antiviral signaling that enhances cell survival and proliferation (bottom).
Resistance to CIN-induced stress as a hallmark of CSCs
Although CIN is a hallmark of cancer (Hanahan and Weinberg, 2011), little is known about its relationship with stemness. Breast cancer CSCs bearing similarities to adult mammary stem cells (MaSCs) are important contributors to metastasis and disease progression (Al-Hajj et al., 2003; Lim et al., 2009; Prat and Perou, 2011; Malanchi et al., 2011). Stress tolerance is a key property associated with these CSCs, including resistance to exogenous stresses such as chemotherapy (Lytle et al., 2018) or hypoxia (Abd et al., 2023) as well as endogenous stresses caused by metabolic activity (Lee et al., 2015) or DNA damage (McGrail et al., 2018; Vitale et al., 2017). In addition to its role in tumor evolution, CIN is a significant stress that reduces cancer cell fitness (Gronroos and Lopez-Garcia, 2018). As a tumor-specific stress, all cancer cells exhibit some level of CIN resistance. In fact, CIN’s effects as a stress appear to outweigh any advantage posed by the increased karyotypic heterogeneity, as enhancing CIN levels negatively impacted glioblastoma CSCs (Godek et al., 2016). This emphasizes the importance of CIN tolerance in shaping stemness properties, as high CIN levels can tip even CSCs toward cell death. Consistent with this idea, recent studies have identified CIN as an endogenous stress more effectively tolerated by CSCs compared to non-stem cell types (Hong et al., 2022; Morel et al., 2017; Sun et al., 2022; Baba et al., 2023). While the acquisition of CIN is a normal part of aging, these cells are efficiently targeted for death and removal, and thus CIN is usually not found in healthy tissues (Barroso-Vilares et al., 2020; Chen et al., 2023). For this reason, discovery that normal mammary stem cells were better equipped to resist this stress during oncogenic transformation was somewhat unexpected (Morel et al., 2017). This may indicate that CIN tolerance is a conserved property of both normal stem cells and CSCs. In this same vein, it appears that the response to CIN may vary among different cell types, even in the same tissue, since altering the levels of CIN in mice predisposed to intestinal cancer showed striking differences in adenoma formation in distinct regions of the intestine (Hoevenaar et al., 2020). Thus, despite its role in cancer evolution, CIN-induced stress may be of more vital importance in shaping the heterogeneous tumor landscape, with CSCs characterized by enhanced resistance.
ZEB1 as a mechanism of CIN resistance
CIN is a universal feature of all cancer cells, however, recent studies have discovered enhanced CIN tolerance associated with CSCs (Hong et al., 2022; Morel et al., 2017; Sun et al., 2022; Baba et al., 2023) as well as normal MaSCs exposed to oncogenes (Morel et al., 2017). These cells possessed unique mechanisms underlying this behavior that provided a survival advantage over other non-stem cell types (Figure 2). By performing unbiased analysis of critical genes expressed by CSCs in an activated signaling state, ZEB1 and TGFBI (BIG-H3) were identified as critical for CIN tolerance (Sun et al., 2022). Further characterization revealed that these two independently identified genes were related as part of a TGFBI (BIG-H3)-ZEB1 signaling module (Sun et al., 2022), with TGFBI (BIG-H3) stimulating ZEB1 mRNA and protein levels (Sun et al., 2022). This presumably occurs through activation of Transforming Growth Factor-beta signaling, although the precise mechanism has yet to be elucidated. These findings were remarkably consistent with those from normal human MaSCs, where ZEB1 gene expression was identified as critical for resistance against CIN-induced stress caused by oncogenic HRAS (Morel et al., 2017). Further studies showed that these effects were due to ZEB1-mediated transcription of the methionine sulfoxide reductase MSRB3, which prevented DNA damage induced by reactive oxygen species (Morel et al., 2017). The stress caused by CIN served as a significant impediment to oncogenic transformation of normal human breast epithelial cells, while MaSCs expressing ZEB1-MSRB3 were more likely to be transformed since they could tolerate CIN (Morel et al., 2017). Similarly, TGFBI (BIG-H3) was also previously shown to mediate CIN resistance, as TGFBI knockout mice displayed enhanced levels of chromosomal aberrations and micronuclei (Zhang et al., 2009). Additionally, deletion of ZEB1 or TGFBI (BIG-H3) in breast cancer cells enhanced sensitivity to PARP inhibitors (Sun et al., 2022), suggesting that CIN resistance mechanisms may represent potential therapeutic vulnerabilities. Together, these findings highlight ZEB1 as a conserved mechanism of CIN tolerance present in both normal stem cells and CSCs.
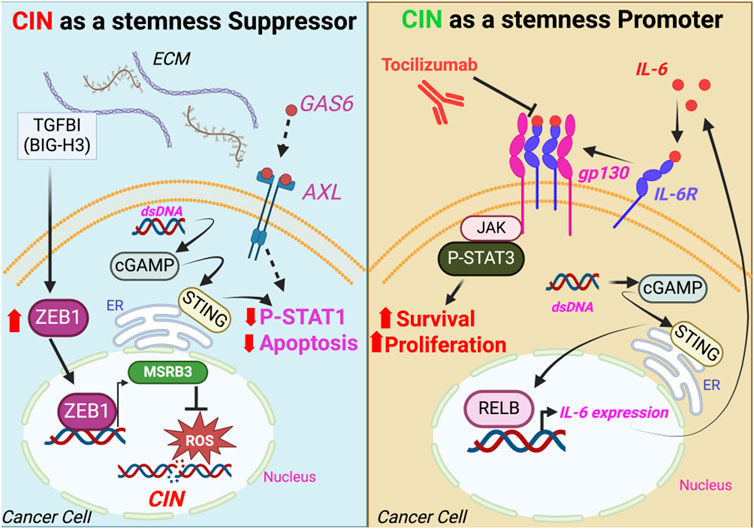
Figure 2. Signaling pathways mediating the CSC response to CIN. CSC resistance to the stress caused by CIN occurs through preventing DNA damage or inactivating STAT1-mediated cell death (left panel), while STING-induced cytokines such as IL-6 drive cell survival and proliferation via STAT3 activation (right panel). ECM, extracellular matrix; dsDNA, double-stranded DNA; ER, endoplasmic reticulum.
c-Jun/AXL as CSC-specific enhancers of CIN tolerance
Additional mechanisms involved in CSC resistance to CIN include a new role for the receptor tyrosine kinase AXL. Recent studies discovered AXL as a gene induced by activation of c-Jun/JNK stress signaling in CSCs, allowing these cells to tolerate chronic endogenous stress caused by CIN (Sun et al., 2022). This is consistent with previous roles described for AXL inhibition in blocking the DNA damage response and increasing sensitivity to PARP inhibitors (Ramkumar et al., 2021), as well as for JNK activation as necessary for CIN tolerance (Wong et al., 2014). In fact, both JUN and AXL were found to be enriched in CSCs (Sun et al., 2022) as well as cells with high-CIN (Bakhoum et al., 2018), suggesting that their expression might be part of the CIN response. This highlights a significant role for c-Jun/AXL signaling in mediating CIN tolerance in CSCs. These effects may be due to AXL’s ability to suppress STAT1-mediated cell death. STAT1 is required for CIN-induced cell death (Hong et al., 2022), which is deficient in aggressive cancer cells due to chronic inactivation of this pathway (Li et al., 2023). In other cell types, AXL has been shown to block STAT1-dependent responses (Rothlin et al., 2007), thus it is tempting to speculate it may similarly function to block the STAT1 tumor suppressor in CSCs, enhancing CIN tolerance. Future studies will explore how this new role for AXL may be related to its previously characterized functions in innate immunity (Bottai et al., 2016), including potential impacts on the STING/IL-6 pathway downstream of CIN (Bakhoum et al., 2018; Hong et al., 2022). Such findings may suggest similarities between AXL’s role in regenerating the epithelium after a viral infection (Fujino et al., 2019) and its effects downstream of CIN in cancer, such that AXL may maintain CSCs in a persistent activated state that underlies their metastatic potential. Despite similar impacts on CIN resistance, the c-Jun/AXL and TGFBI (BIG-H3)-ZEB1 pathways do not appear to crosstalk and instead may act in parallel, since only TGFBI (BIG-H3)-ZEB1 directly affected CIN levels. Taken together, these findings highlight important mechanisms for properly regulating the response to CIN-induced stress in CSCs. This raises the possibility that CIN tolerance pathways could be responsible for the aggressive nature of CSCs, underlying their role as key initiators of metastasis (Lawson et al., 2015).
A potential CSC role for CIN-induced antiviral responses
While CSCs are capable of resisting many different forms of stress, CIN is unique, as it can activate pro-tumorigenic signaling pathways (Figure 2). Despite being a significant endogenous stress that limits transformation potential (Morel et al., 2017), studies using specific KIF2C constructs to modify CIN levels (Moore et al., 2005; Bakhoum et al., 2009) have shown that CIN can paradoxically enhance tumor progression and metastasis by inducing inflammatory signaling (Bakhoum et al., 2018; Hong et al., 2022; Li et al., 2023). These distinct responses to CIN act as two sides of the same coin, with CIN resistance representing an important prerequisite for any pro-tumor responses. Enhanced levels of CIN resulted in micronuclei, which ruptured to produce dsDNA in the cytoplasm (Bakhoum et al., 2018; Hong et al., 2022; Mackenzie et al., 2017). This was then sensed by the cGAS/STING pathway, initiating a chronic antiviral signaling response (Bakhoum et al., 2018; Hong et al., 2022). Of note, this did not activate the canonical downstream interferon/NFKB cell death, but instead initiated a tumor pro-survival and pro-proliferation response (Bakhoum et al., 2018; Hong et al., 2022), highlighting a crucial distinction between the CIN response and signaling due to a viral infection. While activation of cGAS/STING normally induces cell death due to interferon/TLR/STAT1, CIN-induced STING signaling resulted in enhanced survival and proliferation due to non-canonical NFKB (RelB) (Bakhoum et al., 2018) as well as IL-6/JAK2/STAT3 signaling (Hong et al., 2022). This may be due to inactivation of the STAT1 cell death response in aggressive cancer cells, leaving only the pro-tumorigenic response intact. While both responses are downstream of cGAS/STING, it appears likely that unique genes or pathways specific to aggressive cells may tip the balance toward the pro-tumorigenic response. Deciphering the specific mechanisms that provide this context in CSCs will likely furnish significant insight into what makes these cells so malignant. However, it is an unanswered question as to whether the pro-tumor antiviral signaling downstream of cGAS/STING acts in concert with CIN tolerance pathways or represents a parallel signaling module. Studies suggest that the chronic activation of STING may itself lead to tolerance (Hong et al., 2022). In summary, recent findings highlight an important role for CIN not only as a stress CSCs must tolerate, but as a potential cause of aggressive breast cancer cell properties.
An important caveat of these studies is that all studies to date were performed in stem-like claudin-low breast cancer cell lines. Thus, it remains to be determined whether luminal or basal-like breast cancer cells will respond in the same manner. This is particularly important since STING activation appeared to be uncoupled from CIN in ER+ patient breast cancers. Tumors with activated STING were associated with a robust immune response and overall positive prognosis, whereas high CIN levels were noted in ER+ cancers with low STING activation and poor prognosis (Parkes et al., 2021). Interestingly, there was no correlation between STING signaling and CIN in ER- patient cancers in this study. This may be partly due to the fact that only moderate CIN levels indicated a poor prognosis in Triple-Negative disease, whereas cancers with high levels of CIN had a better outcome (Jamal-Hanjani et al., 2015). More information is needed about the context in which the CIN/STING signaling axis occurs and what if any cell type-dependent co-factors are necessary to achieve this response.
Cooperation between CIN’s two roles in shaping CSC behavior
Although CIN’s role as both a cancer-specific stress and a tumor-promoter may appear contradictory, their cooperation may be critical for CSC properties. The ability of CSCs to tolerate high CIN levels would not only allow them to survive, but may also function as a prerequisite for any pro-tumor signaling response. By viewing these effects together, we may gain a more complete perspective of CIN’s role in cancer progression. Furthermore, since studies suggest that CIN levels may be dynamic during tumor initiation and metastasis, this vantage point may allow us to better identify the most critical stages and cell types affected by CIN. During metastasis, CIN levels have been observed to increase dramatically compared to matched primary tumors (Bakhoum et al., 2018). Similarly, our lab noted higher amounts of CIN in low-burden versus high-burden tumors from CSCs, suggesting that levels increase during the initial phases of tumor initiation and decrease as tumors become more established (Baba et al., 2023). Importantly, basal levels were similar between CSC and non-stem cell types prior to injecting into mice (Baba et al., 2023), consistent with an increase in CIN specific to early stage tumors. Since non-CSCs displayed reduced tumor-initiating ability, this may indicate that they are less able to tolerate this stress, with increased CIN serving as a critical barrier to the initiation of new tumors. Additionally, this suggests that CSCs possess the ability to tolerate high levels of CIN, underlying their role in initiating new tumors during the early stages of metastasis (Lawson et al., 2015). The differing effects of CIN in CSCs versus non-stem cells is consistent with studies showing that the oncogenic response to CIN varies in distinct tissues and cell types (Morel et al., 2017; Hoevenaar et al., 2020). Overall, these findings suggest that CIN may be induced during early tumor initiation, favoring a role for CSCs, whereas CIN appears to diminish as tumors grow and differentiate, allowing other cell types to become ascendant. Furthermore, the higher CIN levels tolerated by CSCs could also provide an advantage by inducing pro-tumor STING signaling, aiding their ability to survive and grow. Thus, this model of dynamic changes in CIN during tumor progression highlights how both pro-tumor and stress tolerance pathways may cooperate to enhance stemness and initiate new cancers.
CIN’s contribution to CSC immune evasion
In addition to affecting cancer cell behavior directly, CIN also impacts the tumor microenvironment (TME), primarily by regulating the recruitment and activation of immune cells. High levels of CIN appear to create an immunosuppressive TME due to reduced lymphocyte infiltration and defects in antigen presentation, causing decreased responsiveness to immunotherapies (Taylor et al., 2018; Davoli et al., 2017; Tripathi et al., 2019). This is somewhat unexpected since higher levels of CIN would be predicted to enhance the levels of neoantigens in cancer cells due to higher mutational burden. At least some of the effects of CIN on cancer immune evasion appear to involve cGAS/STING signaling (Li et al., 2023), which re-wires the immune landscape towards one that is pro-metastatic (Li et al., 2023). However, this too may be paradoxical, as CIN also appears to enhance susceptibility to anti-tumor immune responses by activating macrophages (Hayes et al., 2024). The role of CSCs in mediating CIN’s effects on the TME are still very much unclear. Given their scarcity, it seems unlikely that CSCs themselves produce an outsized response in the TME. Rather, it seems more probable that CIN contributes toward making CSCs particularly well-suited to immune evasion. Consistent with this possibility, CSCs evade the immune system by expressing high levels of PD-L1 and abrogating the anti-tumor effects of interferon signaling (Hsu et al., 2018), similar to the immunosuppression caused by CIN in cancer. This is may be to blame for the reported negative correlations between stemness and cancer immunity (Miranda et al., 2019). Thus, CIN may aid CSC immune evasion, enhancing their metastatic potential and leading to resistance to immune checkpoint blockade. CIN’s effects on immune regulation likely stem from increased cytokine secretion by cancer cells. Interleukin-6 (IL-6) has already been characterized as one such secreted signaling factor increased by CIN/STING signaling (Hong et al., 2022). While IL-6 has been shown to induce paracrine signaling in cancer cells resulting in increased proliferation and survival (Hong et al., 2022), it may also influence the immune microenvironment. Overall, there is still much to learn about the specific cancer cell-dependent and independent effects induced by CIN, especially with regard to CSCs and other cell type-dependent differences. Future studies will be needed to determine the role of CIN in mediating CSC immune evasion and prioritize the various pro-tumor cytokine signaling networks for their potential use as therapeutic targets.
Discussion
Taken together, these studies highlight CIN’s important contribution to promoting stemness features. Since CSCs have a disproportionate effect on cancer progression, there is a critical need for more effort in the field geared toward addressing the role that stresses, such as CIN, may play in modulating, and even driving CSC behavior. Toward this goal, there remain several critical unresolved questions that will serve as the basis for future studies. These include addressing how aggressive cancer cells, such as CSCs, can uniquely harness CIN to induce pro-tumorigenic cGAS/STING signaling relative to other cells. Recent studies indicate that CIN levels are similar in stem and non-stem cell types (Baba et al., 2023), suggesting that it is not sufficient for stemness. This may provide a rationale for future work to define factors that specify the unique CSC response to CIN. Central to this effort will be the elucidation of the sentinel antiviral response genes that underly any cell type or subtype-dependent differences in STING activity. This may include unique differences in the downstream genes expressed due to STING activation in CSCs. Identifying crucial CIN tolerance pathways is a key next step in these studies. This will address important questions regarding why aggressive cells lose sensitivity to interferon-induced STAT1-mediated cell death. While more tolerant of CIN, CSCs still have the capacity to undergo apoptosis due to elevated CIN levels (Godek et al., 2016), indicating that re-activating the pro-death response may be an attractive therapeutic approach. By targeting CIN tolerance pathways, this could re-sensitize aggressive cells to interferon-induced cell death, using the cell’s own CIN-induced STING response against it. This area of research may represent a promising new avenue with regard to potential new breast cancer therapies. By defining CIN’s role in stemness, and identifying the mechanisms that confer tolerance against this chronic insult, studies in this area may result in a major advance in our understanding of breast cancer stemness and shed light on the underlying cause of metastasis. This could potentially characterize aggressive cancers as less like a wound, and more similar to a chronic viral infection.
Author contributions
SB: Writing–original draft, Writing–review and editing. AZ: Writing–review and editing. JD: Funding acquisition, Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by funding from the Tobacco-Related Disease Research Program [Grant #T32IR4741 (to JD)]; and the California Breast Cancer Research Program [Grant #B28IB5479 (to JD)].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abd, G. M., Laird, M. C., Ku, J. C., and Li, Y. (2023). Hypoxia-induced cancer cell reprogramming: a review on how cancer stem cells arise. Front. Oncol. 13, 1227884. doi:10.3389/fonc.2023.1227884
Albertson, D. G. (2006). Gene amplification in cancer. Trends Genet. 22, 447–455. doi:10.1016/j.tig.2006.06.007
Al-Hajj, M., Wicha, M. S., Benito-Hernandez, A., Morrison, S. J., and Clarke, M. F. (2003). Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 100, 3983–3988. doi:10.1073/pnas.0530291100
Baba, S. A., Sun, Q., Mugisha, S., Labhsetwar, S., Klemke, R., and Desgrosellier, J. S. (2023). Breast cancer stem cells tolerate chromosomal instability during tumor progression via c-Jun/AXL stress signaling. Heliyon 9, e20182. doi:10.1016/j.heliyon.2023.e20182
Bakhoum, S. F., and Compton, D. A. (2012). Chromosomal instability and cancer: a complex relationship with therapeutic potential. J. Clin. investigation 122, 1138–1143. doi:10.1172/JCI59954
Bakhoum, S. F., Ngo, B., Laughney, A. M., Cavallo, J. A., Murphy, C. J., Ly, P., et al. (2018). Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 553, 467–472. doi:10.1038/nature25432
Bakhoum, S. F., Thompson, S. L., Manning, A. L., and Compton, D. A. (2009). Genome stability is ensured by temporal control of kinetochore-microtubule dynamics. Nat. cell Biol. 11, 27–35. doi:10.1038/ncb1809
Barroso-Vilares, M., Macedo, J. C., Reis, M., Warren, J. D., Compton, D., and Logarinho, E. (2020). Small-molecule inhibition of aging-associated chromosomal instability delays cellular senescence. EMBO Rep. 21, e49248. doi:10.15252/embr.201949248
Benner, S. E., Wahl, G. M., and Von Hoff, D. D. (1991). Double minute chromosomes and homogeneously staining regions in tumors taken directly from patients versus in human tumor cell lines. Anticancer Drugs 2, 11–25. doi:10.1097/00001813-199102000-00002
Bottai, G., Raschioni, C., Székely, B., Di Tommaso, L., Szász, A. M., Losurdo, A., et al. (2016). AXL-associated tumor inflammation as a poor prognostic signature in chemotherapy-treated triple-negative breast cancer patients. NPJ Breast Cancer 2, 16033. doi:10.1038/npjbcancer.2016.33
Carter, S. L., Eklund, A. C., Kohane, I. S., Harris, L. N., and Szallasi, Z. (2006). A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat. Genet. 38, 1043–1048. doi:10.1038/ng1861
Chen, G., Li, Z., Iemura, K., and Tanaka, K. (2023). Oxidative stress induces chromosomal instability through replication stress in fibroblasts from aged mice. J. cell Sci. 136, jcs260688. doi:10.1242/jcs.260688
Chen, X., Agustinus, A. S., Li, J., DiBona, M., and Bakhoum, S. F. (2024). Chromosomal instability as a driver of cancer progression. Nat. Rev. Genet. doi:10.1038/s41576-024-00761-7
Davoli, T., Uno, H., Wooten, E. C., and Elledge, S. J. (2017). Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 355, eaaf8399. doi:10.1126/science.aaf8399
Fujino, N., Brand, O. J., Morgan, D. J., Fujimori, T., Grabiec, A. M., Jagger, C. P., et al. (2019). Sensing of apoptotic cells through Axl causes lung basal cell proliferation in inflammatory diseases. J. Exp. Med. 216, 2184–2201. doi:10.1084/jem.20171978
Godek, K. M., Venere, M., Wu, Q., Mills, K. D., Hickey, W. F., Rich, J. N., et al. (2016). Chromosomal instability affects the tumorigenicity of glioblastoma tumor-initiating cells. Cancer Discov. 6, 532–545. doi:10.1158/2159-8290.CD-15-1154
Gronroos, E., and Lopez-Garcia, C. (2018). Tolerance of chromosomal instability in cancer: mechanisms and therapeutic opportunities. Cancer Res. 78, 6529–6535. doi:10.1158/0008-5472.CAN-18-1958
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. doi:10.1016/j.cell.2011.02.013
Hayes, B. H., Wang, M., Zhu, H., Phan, S. H., Dooling, L. J., Andrechak, J. C., et al. (2024). Chromosomal instability induced in cancer can enhance macrophage-initiated immune responses that include anti-tumor IgG. eLife 12. doi:10.7554/eLife.88054
Hoevenaar, W. H. M., Janssen, A., Quirindongo, A. I., Ma, H., Klaasen, S. J., Teixeira, A., et al. (2020). Degree and site of chromosomal instability define its oncogenic potential. Nat. Commun. 11, 1501. doi:10.1038/s41467-020-15279-9
Holland, A. J., and Cleveland, D. W. (2009). Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat. Rev. Mol. Cell Biol. 10, 478–487. doi:10.1038/nrm2718
Hong, C., Schubert, M., Tijhuis, A. E., Requesens, M., Roorda, M., van den Brink, A., et al. (2022). cGAS-STING drives the IL-6-dependent survival of chromosomally instable cancers. Nature 607, 366–373. doi:10.1038/s41586-022-04847-2
Hosea, R., Hillary, S., Naqvi, S., Wu, S., and Kasim, V. (2024). The two sides of chromosomal instability: drivers and brakes in cancer. Signal Transduct. Target Ther. 9, 75. doi:10.1038/s41392-024-01767-7
Hsu, J. M., Xia, W., Hsu, Y. H., Chan, L. C., Yu, W. H., Cha, J. H., et al. (2018). STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat. Commun. 9, 1908. doi:10.1038/s41467-018-04313-6
Jamal-Hanjani, M., A’Hern, R., Birkbak, N. J., Gorman, P., Grönroos, E., Ngang, S., et al. (2015). Extreme chromosomal instability forecasts improved outcome in ER-negative breast cancer: a prospective validation cohort study from the TACT trial. Ann. Oncol. 26, 1340–1346. doi:10.1093/annonc/mdv178
Lawson, D. A., Bhakta, N. R., Kessenbrock, K., Prummel, K. D., Yu, Y., Takai, K., et al. (2015). Single-cell analysis reveals a stem-cell program in human metastatic breast cancer cells. Nature 526, 131–135. doi:10.1038/nature15260
Lee, E., Yang, J., Ku, M., Kim, N. H., Park, Y., Park, C. B., et al. (2015). Metabolic stress induces a Wnt-dependent cancer stem cell-like state transition. Cell Death Dis. 6, e1805. doi:10.1038/cddis.2015.171
Li, J., Hubisz, M. J., Earlie, E. M., Duran, M. A., Hong, C., Varela, A. A., et al. (2023). Non-cell-autonomous cancer progression from chromosomal instability. Nature 620, 1080–1088. doi:10.1038/s41586-023-06464-z
Lim, E., Vaillant, F., Wu, D., Forrest, N. C., Pal, B., Hart, A. H., et al. (2009). Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 15, 907–913. doi:10.1038/nm.2000
Lukow, D. A., Sausville, E. L., Suri, P., Chunduri, N. K., Wieland, A., Leu, J., et al. (2021). Chromosomal instability accelerates the evolution of resistance to anti-cancer therapies. Dev. cell 56, 2427–2439.e4. doi:10.1016/j.devcel.2021.07.009
Lytle, N. K., Barber, A. G., and Reya, T. (2018). Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer 18, 669–680. doi:10.1038/s41568-018-0056-x
Mackenzie, K. J., Carroll, P., Martin, C. A., Murina, O., Fluteau, A., Simpson, D. J., et al. (2017). cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. doi:10.1038/nature23449
Malanchi, I., Santamaria-Martínez, A., Susanto, E., Peng, H., Lehr, H. A., Delaloye, J. F., et al. (2011). Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481, 85–89. doi:10.1038/nature10694
McGrail, D. J., Lin, C. C. J., Dai, H., Mo, W., Stephan, C., Davies, P., et al. (2018). Defective replication stress response is inherently linked to the cancer stem cell phenotype. Cell Rep. 23, 2095–2106. doi:10.1016/j.celrep.2018.04.068
Miranda, A., Hamilton, P. T., Zhang, A. W., Pattnaik, S., Becht, E., Mezheyeuski, A., et al. (2019). Cancer stemness, intratumoral heterogeneity, and immune response across cancers. Proc. Natl. Acad. Sci. U. S. A. 116, 9020–9029. doi:10.1073/pnas.1818210116
Moore, A. T., Rankin, K. E., von Dassow, G., Peris, L., Wagenbach, M., Ovechkina, Y., et al. (2005). MCAK associates with the tips of polymerizing microtubules. J. Cell Biol. 169, 391–397. doi:10.1083/jcb.200411089
Morel, A. P., Ginestier, C., Pommier, R. M., Cabaud, O., Ruiz, E., Wicinski, J., et al. (2017). A stemness-related ZEB1-MSRB3 axis governs cellular pliancy and breast cancer genome stability. Nat. Med. 23, 568–578. doi:10.1038/nm.4323
Parkes, E. E., Humphries, M. P., Gilmore, E., Sidi, F. A., Bingham, V., Phyu, S. M., et al. (2021). The clinical and molecular significance associated with STING signaling in breast cancer. NPJ Breast Cancer 7, 81. doi:10.1038/s41523-021-00283-z
Prat, A., and Perou, C. M. (2011). Deconstructing the molecular portraits of breast cancer. Mol. Oncol. 5, 5–23. doi:10.1016/j.molonc.2010.11.003
Ramkumar, K., Stewart, C. A., Cargill, K. R., Della Corte, C. M., Wang, Q., Shen, L., et al. (2021). AXL inhibition induces DNA damage and replication stress in non-small cell lung cancer cells and promotes sensitivity to ATR inhibitors. Mol. Cancer Res. 19, 485–497. doi:10.1158/1541-7786.MCR-20-0414
Rothlin, C. V., Ghosh, S., Zuniga, E. I., Oldstone, M. B., and Lemke, G. (2007). TAM receptors are pleiotropic inhibitors of the innate immune response. Cell 131, 1124–1136. doi:10.1016/j.cell.2007.10.034
Sun, Q., Wang, Y., Officer, A., Pecknold, B., Lee, G., Harismendy, O., et al. (2022). Stem-like breast cancer cells in the activated state resist genetic stress via TGFBI-ZEB1. NPJ Breast Cancer 8, 5. doi:10.1038/s41523-021-00375-w
Taylor, A. M., Shih, J., Ha, G., Gao, G. F., Zhang, X., Berger, A. C., et al. (2018). Genomic and functional approaches to understanding cancer aneuploidy. Cancer cell 33, 676–689.e3. doi:10.1016/j.ccell.2018.03.007
Tripathi, R., Modur, V., Senovilla, L., Kroemer, G., and Komurov, K. (2019). Suppression of tumor antigen presentation during aneuploid tumor evolution contributes to immune evasion. Oncoimmunology 8, 1657374. doi:10.1080/2162402X.2019.1657374
Turner, K. M., Deshpande, V., Beyter, D., Koga, T., Rusert, J., Lee, C., et al. (2017). Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125. doi:10.1038/nature21356
Vitale, I., Manic, G., De Maria, R., Kroemer, G., and Galluzzi, L. (2017). DNA damage in stem cells. Mol. cell 66, 306–319. doi:10.1016/j.molcel.2017.04.006
Wong, H. W., Shaukat, Z., Wang, J., Saint, R., and Gregory, S. L. (2014). JNK signaling is needed to tolerate chromosomal instability. Cell Cycle 13, 622–631. doi:10.4161/cc.27484
Keywords: chromosomal instability, cancer stem cell, stress tolerance, inflammation, breast cancer, metastasis
Citation: Baba SA, Zakeri A and Desgrosellier JS (2024) Chromosomal instability as an architect of the cancer stemness landscape. Front. Cell Dev. Biol. 12:1450614. doi: 10.3389/fcell.2024.1450614
Received: 17 June 2024; Accepted: 04 September 2024;
Published: 13 September 2024.
Edited by:
Monia Orciani, Marche Polytechnic University, ItalyReviewed by:
Udayan Bhattacharya, NewYork-Presbyterian, United StatesCopyright © 2024 Baba, Zakeri and Desgrosellier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jay S. Desgrosellier, amRlc2dyb3NlbGxpZXJAdWNzZC5lZHU=