 Jian Kang
Jian Kang Stefano Gallucci
Stefano Gallucci Junqi Pan
Junqi Pan Jonathan S. Oakhill
Jonathan S. Oakhill Elaine Sanij1,2,3,4
Elaine Sanij1,2,3,4- 1St Vincent’s Institute of Medical Research, Melbourne, VIC, Australia
- 2Department of Medicine-St Vincent’s Hospital, University of Melbourne, Melbourne, VIC, Australia
- 3Department of Biochemistry and Molecular Biology, Monash University, Melbourne, VIC, Australia
- 4Division of Cancer Research, Peter MacCallum Cancer Centre, Melbourne, VIC, Australia
STK11 (serine-threonine kinase 11), also known as LKB1 (liver kinase B1) is a highly conserved master kinase that regulates cellular metabolism and polarity through a complex signaling network involving AMPK and 12 other AMPK-related kinases. Germline mutations in LKB1 have been causatively linked to Peutz-Jeghers Syndrome (PJS), an autosomal dominant hereditary disease with high cancer susceptibility. The identification of inactivating somatic mutations in LKB1 in different types of cancer further supports its tumor suppressive role. Deleterious mutations in LKB1 are frequently observed in patients with epithelial ovarian cancer. However, its inconsistent effects on tumorigenesis and cancer progression suggest that its functional impact is genetic context-dependent, requiring cooperation with other oncogenic lesions. In this review, we summarize the pleiotropic functions of LKB1 and how its altered activity in cancer cells is linked to oncogenic proliferation and growth, metastasis, metabolic reprogramming, genomic instability, and immune modulation. We also review the current mechanistic understandings of this master kinase as well as therapeutic implications with particular focus on the effects of LKB1 deficiency in ovarian cancer pathogenesis. Lastly, we discuss whether LKB1 deficiency can be exploited as an Achilles heel in ovarian cancer.
Introduction
The gene STK11 (serine-threonine kinase 11), also known as LKB1 (liver kinase B1) encodes a highly conserved serine threonine kinase. The full-length human LKB1 protein encompasses three domains including an N-terminal domain with nuclear localization signal, a central domain with kinase catalytic activity and a C-terminal domain with regulatory function (Hezel and Bardeesy, 2008). LKB1 is a constitutively active master kinase that regulates cellular metabolism and polarity through a complex signaling network. LKB1 forms a heterotrimeric complex with a pseudokinase STRAD (STE20-related adaptor) and a scaffolding protein CAB39 (calcium-binding protein 39, as known as MO25) (Baas et al., 2003; Boudeau et al., 2003). This interaction leads to its translocation from the nucleus to the cytoplasm where LKB1 phosphorylates AMPK (AMP-activated protein kinase) and 12 other AMPK-related kinases including MARK-1/2/3/4 (microtubule-associated protein/microtubule affinity regulating kinases-1/2/3/4), NUAK1/2 (NUA kinase family 1/2), SIK-1/2/3 (salt-inducible kinase-1/2/3), BRSK-1/2 (brain-specific kinase-1/2), and SNAK (sucrose non-fermenting protein-related kinase) (Jaleel et al., 2005; Lizcano et al., 2004). A recent study identified that LKB1 also phosphorylates and activates TSSK1B (testis-specific serine/threonine-protein kinase 1B), a member of the calcium/calmodulin-dependent protein kinase superfamily (Kim et al., 2024).
The signaling pathway through AMPK is one of the best characterized pathways downstream of LKB1. AMPK is an evolutionarily conserved protein in eukaryotic cells, composed of 3 subunits. The α subunit contains the catalytically active kinase domain while the β and γ subunits serve regulatory and localization roles. AMPK is a sensor of cellular energy homeostasis and can be activated by the increased ratios of AMP to ATP and ADP to ATP due to energy stresses (e.g., glucose restriction or hypoxia). LKB1 in complex with STRAD and CAB39, activates AMPK by phosphorylating Threonine 172 in the T-loop of the α kinase domain (Hawley et al., 2003). This leads to suppression of anabolic processes (e.g., synthesis of proteins, fatty acids and glycogen) and stimulation of catabolic processes (e.g., fatty acid oxidation, glycolysis and autophagy) to restore cell energy balance and regulate cell proliferation and growth (Gwinn et al., 2008; Shaw, 2009; Shaw et al., 2004). Moreover, the LKB1-AMPK pathway regulates epithelial tight junction assembly and cellular polarity by remodeling the actin cytoskeleton (Zhang et al., 2006; Zheng and Cantley, 2007).
Similarly, LKB1 can phosphorylate the T-loop of the catalytic subunit of 12 other AMPK-related kinases and increase their activities (Lizcano et al., 2004). These kinases mainly regulate cellular polarity and metabolism (Bright et al., 2008; Jaleel et al., 2005; Sun et al., 2020; Thirugnanam and Ramchandran, 2020; Timm et al., 2003; van de Vis et al., 2021). Unlike well-characterized LKB1-AMPK signaling pathway, how the biological effects of LKB1 are mediated by these kinases are less studied. Nevertheless, accumulating evidence supports that they are the major mediators of the functional impacts of LKB1 in cancer pathogenesis. In this review, we summarize the pleiotropic roles of LKB1 in cancer biology including oncogenic proliferation and growth, metastasis, metabolic reprogramming, genomic instability, and immune modulation. We particularly review the effects of LKB1 deficiency in pathogenesis of ovarian cancer and discuss whether LKB1 deficiency could be exploited as an Achilles heel in ovarian cancer.
LKB1 mutations in cancers
Germline mutations in LKB1 have been causatively linked to Peutz-Jeghers Syndrome (PJS), an autosomal dominant hereditary disease (Hemminki et al., 1998; Jenne et al., 1998). This disease is characterized by the formation of hamartomatous polyps in the gastrointestinal tract, mucocutaneous pigmentation, and high cancer susceptibility with more than 93% of patients developing malignancies by an average age of 43 years. These germline mutations eliminate the kinase activity of LKB1 (Hemminki et al., 1998; Jenne et al., 1998; Mehenni et al., 1998). The identification of inactivating somatic mutations in LKB1 in different types of cancer further supports its tumor suppressor role.
Analysis of the AACR project GENIE data set (v15.1) via cBioPortal shows genetic alterations of LKB1 in 3% of 160,994 cancer samples, of which the greatest prevalence of alterations is observed in non-small cell lung cancer (NSCLC) (13.46%), parathyroid cancer (11.76%), cervical cancer (8.64%), anal cancer (8.43%) and adrenocortical adenoma (6.9%) (Figure 1). The most common somatic mutations in LKB1 are missense mutations (2.07%), truncating mutations (0.21%), splice mutations (0.52%) and fusion mutations (0.21%). Notably, 30% of the mutations in NSCLC are missense mutations which disrupt its catalytic function and are predicted to be oncogenic (Gill et al., 2011; Granado-Martínez et al., 2020).
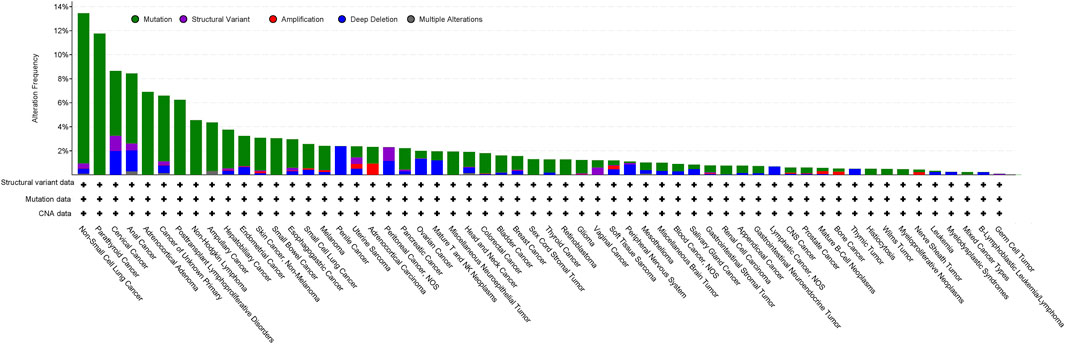
Figure 1. Analysis of the frequency of STK11/LKB1 mutations in cancer using the AACR project GENIE data set (v15.1) via cBioPortal (http://cbioportal.org) (access date 5th June 2024).
Beyond genetic mutations, LKB1 expression can be regulated by epigenetic mechanisms including promoter hypermethylation or small non-coding RNAs by targeting LKB1 3′UTR region and impairing LKB1 mRNA translation (Borzi et al., 2020). Posttranslational modifications of LKB1 including phosphorylation, ubiquitination, SUMOylation have been also reported to impact its protein abundance and function by regulation of intracellular localization, protein stability, conformation, and protein-protein interactions (Hu et al., 2023). Two recent reviews provide extensive overview of epigenetic and post-translational regulation of LKB1 expression (Borzi et al., 2020; Hu et al., 2023).
Functional impacts of LKB1 mutations in cancer pathogenesis
Tumorigenesis
The role of LKB1 in oncogenic transformation has been meticulously studied in mouse models. Homozygous loss of Lkb1 leads to mouse embryonic death, whereas mice with Lkb1 heterozygous deletion developed intestinal polyposis that recapitulate the histological changes found in patients with PJS (Bardeesy et al., 2002). Furthermore, mice with germline Lkb1 haploinsufficiency developed gastric harmartomas after 20 weeks of age and hepatocellular carcinoma after 30 weeks (Miyoshi et al., 2002; Nakau et al., 2002).
Somatic homozygous inactivation of Lkb1 in mouse endometria was found to promote the development of aggressive endometrial cancer much more potently than heterozygous endometrial Lkb1 inactivation (Contreras et al., 2010). A similar finding was reported in a genetically engineered mouse model of pancreatic cancer (Morton et al., 2010). However, more studies reported contradictory findings in which genetic manipulation of Lkb1 in mouse tissues did not promote prostate cancer (Hermanova et al., 2020), bladder cancer (Shorning et al., 2011), serous ovarian cancer (Tanwar et al., 2014), lung cancer (Ji et al., 2007), melanoma (Liu et al., 2012) or endometrial cancer (Cheng et al., 2014). While these findings suggest a tissue-specific role of LKB1 in tumorigenesis, increasing evidence from mouse models shows that LKB1 inactivation cooperates with other oncogenic events to promote tumorigenesis (Li and Zhu, 2020; Skoulidis and Heymach, 2019). Concurrent loss of Lkb1 and Tp53 inactivation promotes the development of osteosarcoma, lymphoma, sarcoma (Wei et al., 2005) and hepatic carcinoma in vivo (Takeda et al., 2006). Inactivation of LKB1 in mice carrying oncogenic Kras mutations leads to development of intraductal papillary mucinous neoplasm (a precursor of pancreatic cancer) (Collet et al., 2020), pancreatic cancer (Morton et al., 2010), lung adenocarcinoma (Ji et al., 2007; Murray et al., 2022; Rogers et al., 2017) and melanoma (Liu et al., 2012). Mice with concurrent loss of Pten and Lkb1 develop endometrial cancer (Cheng et al., 2014) bladder carcinoma (Shorning et al., 2011), lung squamous cell carcinoma (Xu et al., 2014), ovarian carcinoma (Tanwar et al., 2014), and prostate cancer (Hermanova et al., 2020). These findings suggest that LKB1 loss might occur as a secondary event that facilitate the transformation of oncogene-driven cancers.
Tumor growth and metastasis
As a tumor suppressor, LKB1 inactivation not only contributes to oncogenic transformation but also promotes tumor growth mainly through mTORC1-dependent and independent regulation of mRNA translation and growth control (Figure 2). LKB1 negatively regulates mTORC1 through phosphorylation of the mTORC1 upstream regulator TSC2 (TSC complex subunit 2) and the mTORC1 complex component RAPTOR (regulatory associated protein of MTOR complex 1) by AMPK. 4EBP1 (Eukaryotic translation initiation factor 4E-binding protein 1) and S6K1 (ribosomal S6 kinase) are the main effectors of mTORC1-dependent regulation of mRNA translation and growth control (Shackelford and Shaw, 2009). Studies in mouse models of invasive endometrial tumors show that loss of Lkb1 or concurrent loss of Pten and Lkb1 confer a strong dependence on mTOR signaling for tumor growth, rendering them hypersensitive to mTOR inhibition (Cheng et al., 2014; Contreras et al., 2010). This sensitivity was also observed in lung cancer cell lines with LKB1 and KRAS co-mutations, but the single mutation did not confer it. The mTOR inhibitor everolimus, has shown promising activity and safety profile in a phase II clinical trial (NCT02352844) in patients with solid tumors harboring hyperactive mTORC1 signaling due to TSC1/2, NF1/2 or LKB1 mutations (Table 1) (Devarakonda et al., 2021). Notably, one patient with LKB1 mutated advanced lung adenocarcinoma showed stable disease after receiving 10 mg of everolimus orally for 28 days, supporting the potential clinical utility of everolimus for treating LKB1 mutant tumors. AMPK also regulates mRNA translation through mTORC1-independent eEF2K (eukaryotic translation elongation factor 2 kinase) – eEF2 (eukaryotic translation elongation factor 2) pathway. eEF2 is a translation factor necessary for peptide translocation during the elongation phase of protein synthesis. Under metabolic stress, the energy sensor AMPK activates eEF2K which negatively regulates eEF2 activity through phosphorylation and thus reduces protein synthesis to preserve energy (Johanns et al., 2017).
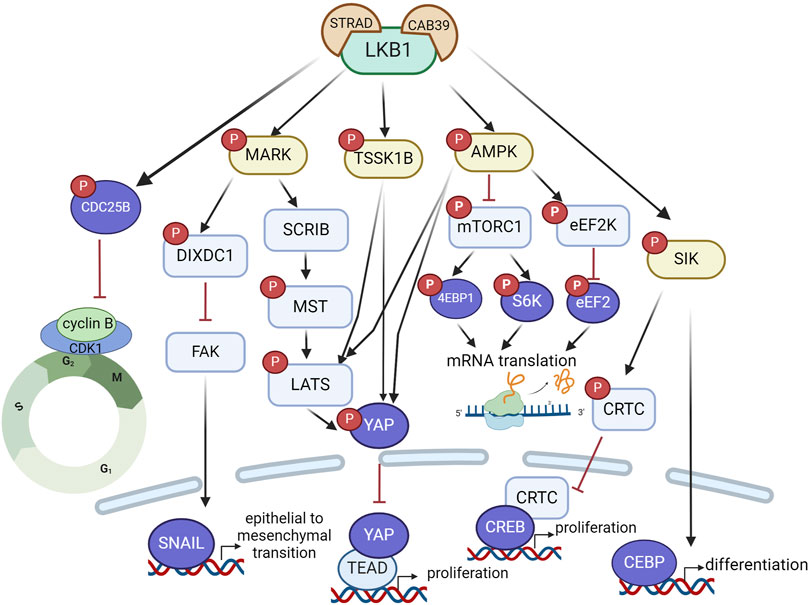
Figure 2. LKB1 functions as a tumor suppressor to restrain tumor growth and metastasis via a complex signaling network that regulate mRNA translation, protein synthesis, and transcriptional programs associated with cell proliferation, differentiation and epithelial mesenchymal transition. Created with BioRender.com.
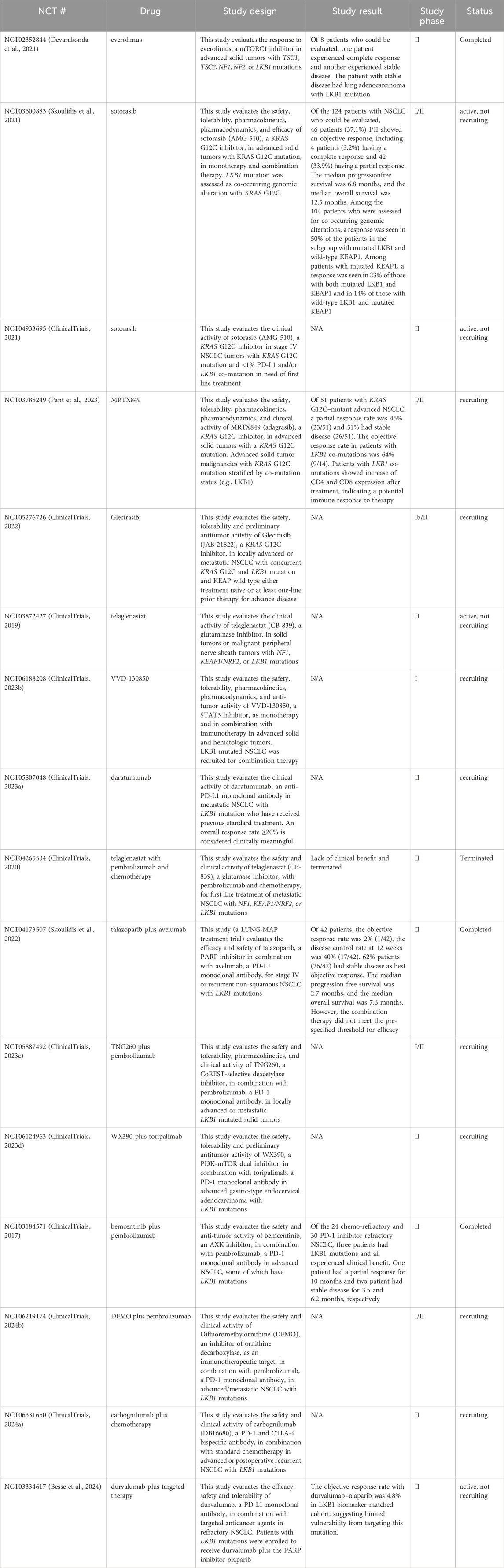
Table 1. The clinical trials in patients with LKB1-mutated tumors.
Hippo signaling is another critical pathway that mediates the effect of LKB1 on oncogenic growth control. This pathway is highly conserved in mammals where, canonically, the kinase MST1/2 phosphorylates LATS1/2 kinases, which in turn phosphorylate the transcriptional co-activator YAP, leading to its cytosolic retention and subsequent degradation. In the absence of phosphorylation, YAP translocates to the nucleus where it binds to TEAD (TEA-domain transcription factor) to activate a transcriptional program that drives tumor cell proliferation and growth. LKB1 promotes YAP phosphorylation through MARK, AMPK or TSSK1B, leading to YAP nuclear exclusion and proteasomal degradation and thus inactivation of its transcriptional activity (Figure 2). Loss of LKB1 in KRAS mutant lung cancer cells leads to YAP-mediated transcriptional activation, through regulation of localization of SCRIB, a scaffold protein involved in cell polarization, and Hippo kinases MST and LATS activity (Lenahan et al., 2023; Mohseni et al., 2014). Three LKB1 substrates, MARK1, 3 and 4, have also been found to control YAP-dependent transcription by complexing with LKB1, MST1, LATS1 and SCRIB (Mohseni et al., 2014). The LKB1-AMPK or LKB1-TSSK1B signaling pathways can phosphorylate YAP via LATS1/2 or directly phosphorylate YAP at a distinct site (Kim et al., 2024; Mo et al., 2015; Wang et al., 2015). Loss of YAP completely inhibited LKB1 deficient lung adenocarcinoma growth in vivo, indicating YAP as the critical mediator of oncogenic effects of LKB1 inactivation and YAP antagonism may represent a therapeutic approach for LKB1 mutant tumors.
SIKs are also reported to mediate the oncogenic effects of LKB1 inactivation in lung cancer. Knock-out of SIK1 and SIK3 increased tumor growth in a mouse model of oncogenic KRAS-driven lung cancer (Hollstein et al., 2019). Aberrant activation of CREB (CAMP responsive element binding protein) transcriptional activity through inactivation of SIK-CRTC (CREB regulated transcription coactivator) signaling underlines the aggressive phenotypes of LKB1-mutant lung cancer (Murray et al., 2019). LKB1 inactivation impairs SIKs phosphorylation of CRTCs, which leads to translocation of unphosphorylated CRTC into nucleus and activation of CREB-mediated transcription programs that promote cancer cell proliferation.
The tumor suppressor role of LKB1 is also linked to its effect on cell differentiation. In an oncogenic KRAS-driven mouse model of lung cancer where inactivation of LKB1 increases lung tumor burden, restoration of LKB1 activated the transcriptional program driven by CEBP (CCAAT enhancer binding protein), reinstated alveolar type II cell-like differentiation and consequently impeded proliferation and growth of lung tumors (Murray et al., 2022). Disruption of CEBP-driven lineage-specific transcriptional program following LKB1 inactivation led to loss of differentiated state and reversion to a progenitor-like state, which underscores the tumor suppressor role of LKB1 in lung cancer.
Beyond the effects on tumor growth, loss of LKB1 confers an invasive phenotype in various genetically engineered mouse models of cancer (Li et al., 2015). The process of cancer metastasis involves alterations in cell polarization and motility, cell detachment, and escape from anoikis, a programmed cell death induced by detachment from extracellular matrix. LKB1 regulates all these steps through AMPK-related proteins. LKB1 regulates cell motility through AMPK-mTOR-CREB (Pencik et al., 2023), cell polarization and microtubule organization through MARKs (McDonald, 2014), cell adhesion through FAK (focal adhesion kinase) (Kline et al., 2013) or NUAK (Zagórska et al., 2010) and anoikis via SIK (Cheng et al., 2009). A study of LKB1-dependent control of metastatic potential by Goodwin et al. defined a signaling pathway through MARK to regulate the expression level of SNAIL1, a critical transcriptional factor involved in induction of epithelial to mesenchymal transition (Goodwin et al., 2014). They showed that a scaffolding protein DIXDC1 (DIX domain containing 1) localizes to focal adhesions upon phosphorylation by MARK1. Loss of DIXDC1 results in upregulation of SNAIL1 in a FAK-dependent manner, leading to metastasis in mice (Goodwin et al., 2014). Taken together, LKB1 as a master kinase regulates cell growth and motility through complex signaling networks and its inactivation in cancer cells leads to a proliferative and metastatic phenotype (Figure 2). In the LKB1-driven signaling network, the AMPK-mTOR pathway is the most critical to target in LKB1-mutant tumors, especially due to the availability of clinically approved therapies.
Metabolic reprogramming
In addition to regulating transcriptional and translational activity, LKB1 plays a crucial role in controlling metabolic activity. Cancer cells primarily depend on aerobic glycolysis to produce ATP and metabolic intermediates to support their proliferation and growth (Figure 3). This is characterized by increased lactate production in the presence of abundant oxygen, known as the “Warburg effect”. Loss of LKB1 promotes a metabolic switch to glycolysis (Dupuy et al., 2013; Faubert et al., 2014; Li and Zhu, 2020). This metabolic reprogramming in LKB1-deficent cells is shown to be dependent on HIF-1α activation by mTORC1, at least in part through AMPK signaling (Faubert et al., 2014). In addition, loss of LKB1 in human papillomavirus-positive cervical cancer cells enhanced glycolysis by increasing the expression of HK2 (hexokinase 2), an enzyme that catalyzes the first step of glucose metabolism by phosphorylating glucose to glucose 6-phosphate (Zeng et al., 2017).
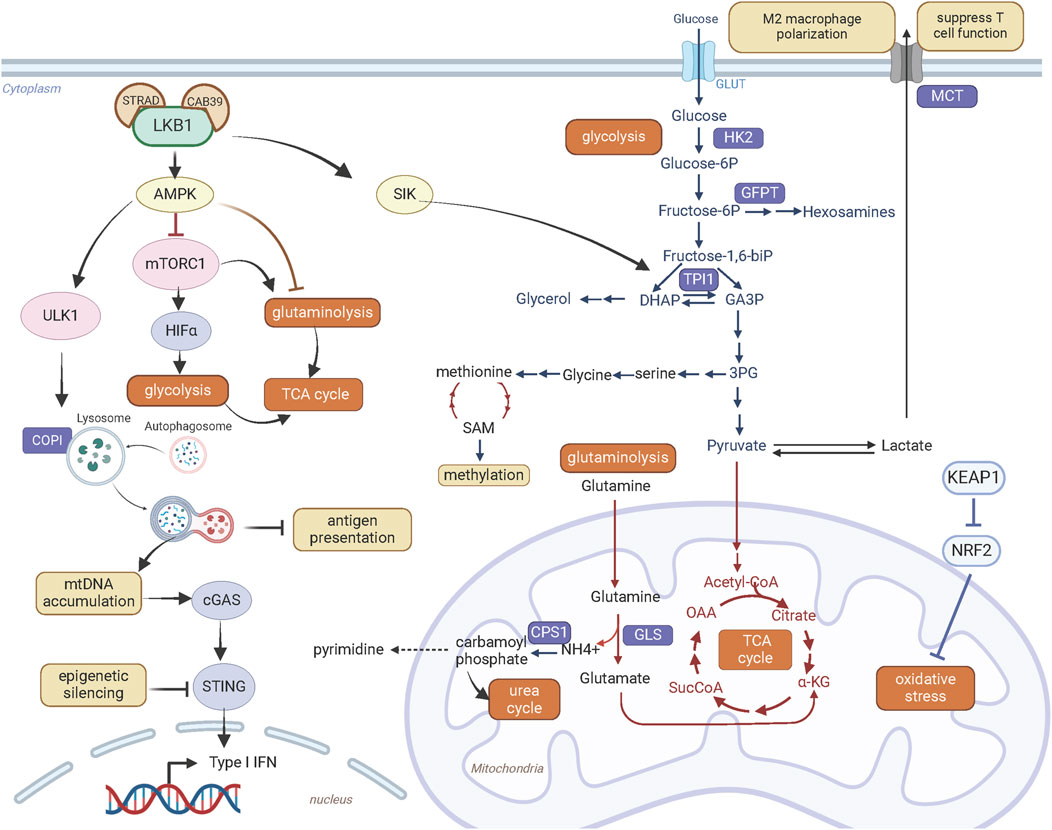
Figure 3. LKB1 promotes metabolic reprogramming and immune modulation. Created with BioRender.com.
The glycolysis pathway is linked to serine-glycine-one carbon pathway (Figure 3). The de novo synthesis of serine occurs through oxidation of the glycolysis intermediate 3-phosphoglycerate. LKB1 inactivation in KRAS mutant lung cancer cells enhances glycolysis and serine biosynthesis and protects cell survival from serine deprivation through deregulated AMPK-mTORC1 signaling (Kottakis et al., 2016). Elevated serine metabolism can increase abundance of SAM (S-adenosyl methionine) through methionine metabolic pathway. SAM transfers methyl groups to DNA, RNA, proteins or lipids catalyzed by DNA methyltransferases. The increased SAM content and DNA methylation with upregulated DNA methyltransferases suggest metabolic reprogramming coupled with epigenetic alterations contribute to oncogenic effects of LKB1 deficiency in KRAS mutant pancreatic cancer cells. Furthermore, these epigenetic alterations provide transcriptional plasticity of LKB1 mutant cells under metabolic stress and confer sensitivity to the DNA methyltransferase inhibitor Decitabine (Kottakis et al., 2016).
Loss of LKB1 is associated with a more aggressive tumor growth in genetically engineered mouse models of lung cancer harboring both Tp53 and Kras mutations. Interestingly, these three oncogenic mutations rarely co-occur in human lung cancer. A recent study by Stein et al. explained this species discrepancy from the effect of LKB1 on energy metabolism (Stein et al., 2023). They found that loss of LKB1 in KRAS and TP53 co-mutant human lung cancer cells decreased phosphorylation of TPI1 (Triosephosphate isomerase 1), a metabolic enzyme that catalyze the interconversion between glyceraldehyde-3-phosphate (GA3P) and dihydroxy-acetone phosphate (DHAP), at serine 21 through SIK (Figure 3). Decrease of TPI1 activity following LKB1 loss shifts energy balance away from energy production via glycolysis and towards glycerol lipid production and energy conservation. This metabolic alteration may exaggerate metabolic stresses experienced during tumorigenesis and is disadvantageous for cell proliferation and growth. In mouse lung cancer cells, instead, there is a substitution of an oxidizable cysteine for serine at residue 21 of TPI1. This mutation abrogates LKB1-SIK-mediated phosphorylation. Consequently, in the absence of TPI1-mediated energy regulation, loss of LKB1 improved the efficiency of energy production through glycolysis, underlying the synergy of these three oncogenic mutations in mouse lung tumor growth. Conversely, LKB1 deficiency in human lung cancer cells with TP53 and KRAS co-mutations have inferior metabolic state for cancer cell fitness through regulation of TPI1, providing a mechanistic explanation for the rare co-mutations of LKB1/KRAS/TP53 in human lung cancer.
Beside glucose, glutamine through glutaminolysis provides carbon source for synthesis of metabolites and energy production (Figure 3). This metabolic pathway converts glutamine by glutaminase to glutamate which is then catabolized to α-ketoglutarate via glutamate dehydrogenase 1 to fuel tricarboxylic acid cycle. LKB1 inactivation in KRAS mutant lung cancer cells promotes glutaminolysis and facilitates the formation of glutamine-derived succinate, fumarate and malate in the later stages of TCA cycle, thus promoting mitochondrial oxidative phosphorylation (Caiola et al., 2018). Elevated glutamine catabolism in LKB1 and KRAS co-mutant cells leads to accumulation of ammonia. CPS1 (Carbamoyl-phosphate synthase 1) uses ammonia and bicarbonate to produce carbamoyl phosphate in the mitochondria. Carbamoyl phosphate either enters urea cycle to convert highly toxic ammonia to urea for excretion or participates in metabolic pathways for the synthesis of pyrimidines (Figure 3). An increase of CPS1 expression and urea cycle metabolites in KRAS and LKB1 co-mutant lung cancer cells increases pyrimidine synthesis and benefits for tumor growth (Kim et al., 2017).
Moreover, elevated mitochondrial oxidative phosphorylation promotes reactive oxygen species (ROS) production. Tumors bearing LKB1 deletion have high levels of ROS, which provoke damages to cellular organelles and processes, and ultimately impair cellular fitness. In NSCLC, LKB1 is often co-mutated with KEAP1 deletion, defining an additional subgroup (Galan-Cobo et al., 2019). Loss of KEAP1 leads to accumulation of NRF2, a master antioxidant transcription factor that protects cells against oxidative stress (Figure 3). Galan-Cobo et al. (2019) characterized the metabolic phenotypes of KRAS/LKB1/KEAP1 co-mutant lung cancer. They defined addiction to glutamine metabolism in this subgroup of lung cancer confered high sensitivity to the glutaminase inhibitor CB-839 in vitro and in vivo.
However, with an increase in metabolic capacity, metabolic plasticity is reduced. This render LKB1 mutant cancer cells sensitive to metabolic stress caused by nutrient deprivation (Caiola et al., 2018). The dependence on glucose and glutamine metabolism thus can be leveraged to treat tumors with LKB1 mutations. The biguanide metformin is widely used to treat diabetes due to its ability to lower glucose, insulin, and IGF1 plasma levels and attenuate insulin resistance by increasing glucose uptake (Nasri and Rafieian-Kopaei, 2014). Metformin has been repurposed for anti-cancer treatments. In LKB1-deficient lung cancer cells, metformin induces apoptosis by inhibiting HIF-1α activity and promoting degradation of pro-survival proteins (Faubert et al., 2014; Huang et al., 2020; Luo et al., 2019). It also prevents resistance to cisplatin in LKB1 and KRAS co-mutated lung cancer cells by targeting tumor-initiating cells (Moro et al., 2018). A Phase II clinical trial, FAME, is currently underway to evaluate the effectiveness of metformin alone or in combination with a fasting-mimicking diet to improve standard chemotherapy outcomes for patients with LKB1-mutant lung adenocarcinoma (Vernieri et al., 2019). In addition, elevated glutamine metabolism supports targeting glutaminase as an approach to treat LKB1 mutant cancers (Sitthideatphaiboon et al., 2021). However, Telaglenastat (CB-839), a glutaminase inhibitor did not show clinical benefit in LKB1 mutant tumors either as a monotherapy or in combination with immunotherapy and chemotherapy (Table 1) (Skoulidis et al., 2020).
In addition, new metabolic vulnerabilities that have been identified through genetic approaches can be exploited to target LKB1 mutant tumors. Kim et al. (2013) identified that lung cancer cells with KRAS and LKB1 co-mutations rely on COPI (coatomer complex I)-dependent lysosome acidification, an essential step for lysosome maturation (Figure 3). As lysosomes degrade macromolecules and supply metabolic intermediates for TCA cycle, lysosomal function is coupled to mitochondrial health. Genetically or pharmacologically targeting COPI complex causes mitochondrial dysfunction and consequently cell death (Kim et al., 2013). Liu et al. (2013) also identified that DTYMK (deoxythymidylate kinase) is synthetically lethal with LKB1 deficiency in KRAS mutant lung cancer cells. DTYMK catalyzes the phosphorylation of thymidine monophosphate (dTMP) to thymidine diphosphate (dTDP) and thus contributes to dTTP biosynthesis. Its inhibition reduces the dTDP pool and leads to an accumulation of dUTP which is incorporated in DNA, causing DNA damage. LKB1-deleted cells were more dependent on the dTTP synthesis pathway due to the lower expression of DTYMK, and this defect in nucleotide metabolism renders LKB1 deficient cells sensitive to the inhibition of DTYMK. In addition, KRAS/LKB1 co-mutated cells were found to depend on an enzyme involved in the hexosamine biosynthesis, glutamine-fructose-6-phosphate transaminase 2 (GFPT2) (Kim et al., 2020). GFPT2 inhibition selectively impedes the tumor growth of this molecular subtype.
Overall, LKB1 is a master regulator of energy production, mainly through AMPK (Figure 3). LKB1 deficiency in cancer cells promotes metabolic reprogramming to support tumor growth and metastasis. These metabolic dependencies become potential vulnerabilities in LKB1 mutant tumors and provides new therapeutic intervention opportunities to develop personalized treatment for cancers with LKB1 mutations.
DNA damages and genomic instability
The constant insults from endogenous (e.g., nucleotide metabolism) and exogenous sources (e.g., UV or ionizing radiation, chemical or biological genotoxins) cause DNA damages. The DNA damage response (DDR) maintains genome stability and cellular viability, via a complex system of signaling pathways that include damage sensors, signal transducers and effectors of cell cycle checkpoints and DNA repair (Groelly et al., 2023). Defective DDR leads to genome instability, a hallmark of cancer, that predisposes cells to malignant transformation and progression driven by secondary events of activation of oncogenes and loss of tumor suppressor genes. LKB1 has been suggested to play a crucial role in homologous recombination (HR) repair in response to DNA double-strand breaks (DSBs). LKB1 inactivation hinders HR repair in response to irradiation and chemotherapeutic agents (Deng et al., 2021; Wang et al., 2016). Consequently, LKB1 deficient tumors exhibit high levels of genomic instability. In a KRAS-driven NSCLC mouse model, loss of Lkb1 increases tumor mutational burden compared with Tp53 mutation. Specifically, Lkb1 deficiency leads to an increase in the number of coding insertion-deletion and non-synonymous single nucleotide variations, reinforcing its important role in preserving genome stability (Wang et al., 2016).
LKB1 has been demonstrated to be a target of the protein kinase ATM, a central component of DDR that orchestrates responses to DSBs and stalled DNA replication forks (Sapkota et al., 2002; Wang et al., 2016). Phosphorylation of LKB1 at Thr363 by ATM was observed as early as 15 min after irradiation, supporting that it is a specific response to DNA DSBs (Sapkota et al., 2002). LKB1 forms DNA damage-induced nuclear foci in a complex with other DDR proteins including γ-H2AX, BRCA1 and ATM and facilitates the recruitment of RAD51 to chromatin for HR repair, providing molecular basis for the involvement of LKB1 in DNA repair (Wang et al., 2016). LKB1 deficiency causes DNA repair defects and confers sensitivity to cisplatin and poly-ADP ribose polymerase (PARP) inhibitors (Wang et al., 2016). As part of DDR, the mitotic checkpoint acts as a key surveillance mechanism of genome integrity by stalling cell division till DNA repair is completed. AZD1775, an inhibitor of the G2-M cell cycle checkpoint protein WEE1, in combination with cisplatin or radiation enhanced DNA damages through bypass of the G2-M checkpoint and improved survival of mice with Kras and Lkb1 co-mutant lung cancer (Richer et al., 2017). Similarly, CHK1 inhibitor AZD7762 was reported to synergize with the DNA-damaging drug gemcitabine in reducing cell viability and suppressing tumor growth of LKB1 deficient lung cancer (Liu et al., 2017). The effects of LKB1 on DDR and genome stability are thought to attribute to nucleus-localized LKB1. But the contribution of cytoplasmic LKB1 through metabolic and growth control cannot be excluded and warrants further investigation. More importantly, evaluation of the association of LKB1 mutation status with chemotherapy response will raise the potential of LKB1 as a therapeutic biomarker in cancer treatment.
Immune modulations
Recent studies have shown that, in addition to its direct effects on tumor cells, LKB1 plays an important role in regulating the tumor microenvironment. LKB1 inactivation in lung adenocarcinoma is associated with an immunosuppressive phenotype. It is characterized by suppression of T cell infiltration, particularly the number and function of effector CD8+ T cells (Koyama et al., 2016; Skoulidis et al., 2015), inhibition of PD-L1 expression and IFN-γ signaling, increase of recruitment of granulocytic myeloid-derived suppressor cells (Li et al., 2021) and secretion of immunosuppressive cytokines such as IL-6 (Koyama et al., 2016). Loss of LKB1 in KRAS-mutant lung adenocarcinoma leads to a distinct immune profile compared to TP53 null tumors (Biton et al., 2018; Skoulidis et al., 2015). Tumors with KRAS and TP53 co-mutations exhibit an adaptive immune response with immune activation evidenced by enhanced T-cell infiltration and upregulation of expression of cell intrinsic immune checkpoint signals such as PD-1/PD-L1. In contrast, tumors with KRAS/LKB1 co-mutations show a cold immune microenvironment with lack of immune system engagement. This poor immune surveillance underlies the low sensitivity of immunotherapies in lung adenocarcinoma patients with LKB1 deficiency. LKB1 mutation status is associated with intrinsic resistance to immunotherapy checkpoint blockade (ICB) in KRAS-mutant non-small cell lung cancer (NSCLC) (Koyama et al., 2016; Skoulidis et al., 2018). Several retrospective studies have evaluated the predictive potential of LKB1 status on ICB response but the results remain inconclusive (Pons-Tostivint et al., 2021).
The immunosuppressive phenotype of LKB1 deficient lung cancer is associated with the regulatory role of LKB1 in metabolism. As discussed above, loss of LKB1 leads to a metabolic switch to aerobic glycolysis and increase of lactate secretion (Figure 3). Upregulation of MCT4 (also known as solute carrier family 16 member 4 or SLC16A4), a major lactate transporter, contributes to the immunosuppressive phenotype including M2 macrophage polarization and T cell hypofunction observed in a Lkb1-deficient Kras-driven lung cancer mouse model (Qian et al., 2023). The in vivo evidence supports that suppression of glycolysis by depletion of MCT4 transporter or blocking lactate receptor GPR81 can abrogate M2 macrophage polarization, partially restore T cell function, and overcome PD-1 inhibitor resistance. Further studies are required to establish the clinical relevance of lactate metabolism in immunotherapy resistance of human LKB1 mutant lung tumors.
Restricted antigen presentation to MHC complexes has been attributed to low T cell infiltration in LKB1and KRAS co-mutant tumors. Deng et al reported LKB1 loss increased autophagic flux and reduced proteasomal degradation of antigen peptides, thereby suppressing antigen presentation (Deng et al., 2021). Inhibiting autophagy by targeting ULK1 restored antigen presentation through enhancing immunoproteasome activity and synergized with PD-1 inhibition to promote anti-tumor immunity.
STING is a key regulator of the innate immune response by linking detection of aberrant cytoplasmic double-strand DNA by cGAS to activation of innate immune signaling that facilitate T-cell recruitment (Figure 3). LKB1 inactivation in KRAS mutant lung cancer cells causes cytoplasmic accumulation of mitochondrial DNA due to defects in autophagy and mitochondrial dysfunction, which triggers activation of STING signaling. However, STING expression is suppressed in LKB1 mutant cells through epigenetic silencing via the methyltransferases EZH2 and DNMT, consequently disrupting cytoplasmic mitochondrial DNA sensing (Kitajima et al., 2019). Reinduction of LKB1 restored STING expression and rescued chemokine production that promote T-cell recruitment. To develop therapeutic approaches to restore and activate STING signaling and enhance immunogenicity in KRAS and LKB1 co-mutant lung cancer, Kitajima et al. (2022) performed a drug screen and identified inhibition of MPS1 to activate cGAS-STING pathway signaling. MPS1 (also known as TTK protein kinase, TTK) is a critical regulator of the mitotic spindle assembly checkpoint. Abrogation of the mitotic spindle assembly checkpoint by MPS1 inhibition disrupts chromosome segregation, increases chromosome instability and generates micronuclei in cytoplasm, which are recognized by the cGAS-STING pathway. Furthermore, they designed a sequential treatment of an epigenetic inhibitor decitabine to restore STING expression followed by pulse treatment with the MPS1 inhibitor BAY-1217389 to activate the STING pathway. This sequential combination therapy enhanced T cell recruitment and improved sensitivity to PD-1 blockade in mice harboring Kras and Lkb1 mutated lung cancer (Kitajima et al., 2022).
Collectively, loss of LKB1 not only alters intracellular signaling and cellular functions but also modifies the extracellular microenvironment, enabling cancer cells to evade cancer immunosurveillance thus promoting intrinsic resistance to immunotherapy. Several new ICBs in combination with targeted therapies or chemotherapies are currently tested in clinic for tumors with LKB1 mutations (Table 1). Further understanding of the impacts of LKB1 deficiency on cancer cell immunogenicity and tumor immune infiltration will facilitate development of new strategies to increase the clinical efficacy of immunotherapy in patients with LKB1 mutated cancers.
LKB1 in ovarian cancer
Ovarian cancer
Annually 300,000 women throughout the world are diagnosed with ovarian cancer. Sadly, late-stage diagnosis occurs in 58% of cases with the 5-year survival rate of only 30% post-metastasis, in contrast to 92% when the disease is detected early, which strengthens the importance of timely screening and early intervention (Arora et al., 2024). Despite its prevalence and impact, early diagnosis remains challenging due to vague clinical symptoms and the lack of preventative screening methods (Arora et al., 2024).
Ovarian cancer comprises four subtypes: epithelial, germ cell, small cell carcinomas, and sex-cord stromal tumors. Epithelial ovarian cancer (EOC), the most prevalent (90% of cases), have five histological types which are categorized as type I or type II(Hollis, 2023). Type I tumors, including low-grade serous ovarian cancer, endometrioid, clear-cell, and mucinous carcinomas, are generally confined to the ovary and not invasive. These tumors are often identified at an early stage with good prognosis. In contrast, type II tumors, notably high-grade serous ovarian cancer (HGSOC), which accounts for 70%–80% of ovarian cancer cases, are highly proliferative and aggressive with high chromosomal instability (Vang et al., 2009). The most common mutations in HGSOC are TP53 (90%), BRCA1/2 and other HR genes (50%) and genes involved in PI3K and RAS pathways (45%) (Yan et al., 2017).
Owing to the difficulty in early diagnosis and lack of preventative screening, the site and cell of origin of serous carcinoma are still under debate. Both ovarian surface epithelium (OSE) and fallopian tube epithelial (FTE) cells have been considered as the origin for serous ovarian epithelial cancer. Nevertheless, recent genomic studies support HGSOC is mostly originated from the epithelium of distal fallopian tube where a peculiar lesion called serous tubal intra-epithelial carcinoma (STIC) forms as a precursor of HGSOC (Kuhn et al., 2016; Kurman and Shih, 2016; Labidi-Galy et al., 2017).
The standard treatment for ovarian cancer includes surgical debulking, systemic chemotherapy and targeted therapy (Arora et al., 2024). However, recurrence following surgery and chemotherapy is common. 90% of HGSOC acquire resistance to chemotherapy within 2-3 years. HR-deficiency is a key determinant of sensitivity to chemotherapy and PARP inhibitors. The introduction of PARP inhibitors like olaparib, rucaparib, and niraparib, as maintenance therapy has dramatically improved the progression-free survival of HR-deficient HGSOC. However, 30%–50% of cases that initially respond to PARP inhibitors develop resistance leading to incurable disease (Wakefield et al., 2019). Mechanisms of resistance to PARP inhibitor include restoration of HR activity mostly through reversion mutations, stabilization of stalled DNA replication forks, metabolic alterations, and increased drug efflux (Burdett et al., 2023; Wakefield et al., 2019). No effective therapeutic options exist for resistant HGSOC, making the search for new therapies a critical unmet clinical need.
LKB1 expression and mutations in ovarian cancer
To characterize LKB1 genetic alteration in ovarian cancer, we analyzed a TCGA ovarian serous cystadenocarcinoma dataset (N = 182) and identified homozygous and heterozygous gene deletions in 4.4% (N = 8) and 85.2% (N = 155) of samples, respectively (Figure 4A). Similarly, genomic analysis of a small cohort of 75 HGSOC cases by Tanwar et al. (2014) identified copy number loss in 31% (23/75), allelic imbalance in 64% (48/75) and copy number gain in only 1/75 of samples. In a cohort study of 62 Chinese patients with epithelial ovarian cancer, deleterious germline LKB1 mutations have been found in 5.3% (Li et al., 2019).
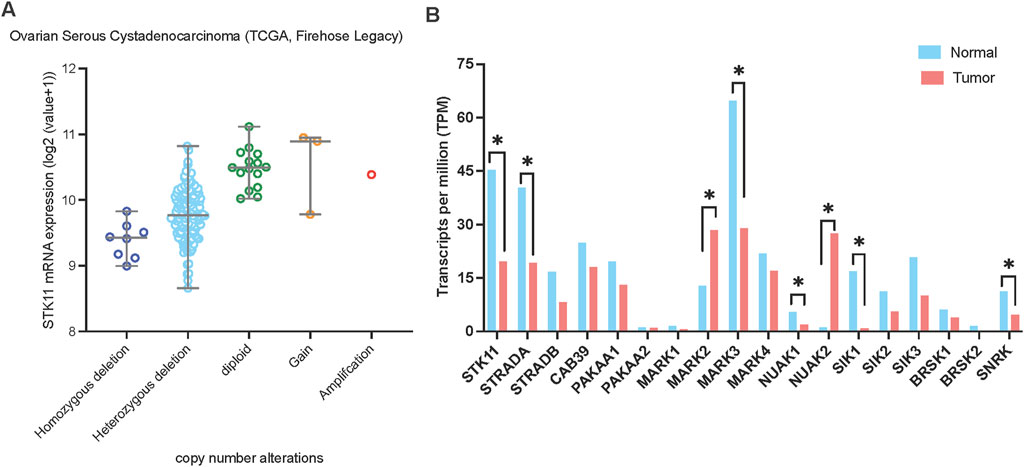
Figure 4. LKB1 copy number alterations and mRNA expression in ovarian cancer. (A) LKB1 mRNA expression levels in ovarian serous cystadenocarcinoma correlate with LKB1 copy number alterations. The data was assessed from cBioPortal (www.cbioportal.org). (B) The mRNA expression level of the genes involved in LKB1 signaling in ovarian cancer. The data was assessed from GEPIA on 5th June 2024.
Moreover, two studies examined LKB1 protein expression in human epithelial ovarian cancer. George et al reported that LKB1 expression was predominantly reduced in 71% of HGSOC (213/298), compared to 47% of clear-cell carcinoma (15/32), 29% of mucinous carcinoma (8/28) and 22% of endometrioid carcinoma (6/27), assessed by immunohistochemical analysis of a tissue microarray (George et al., 2016). In the HGSOC cohort, there was no significant correlation between LKB1 protein expression and progression-free survival or overall survival. Notably, only a third of the samples with reduced LKB1 protein expression had a gene copy number deletion, suggesting additional mechanisms of LKB1 silencing exist in HGSOC. They also examined 15 cases of STIC, the precursor of HGSOC for LKB1 expression. A significant decrease of LKB1 protein expression was observed in 13/15 cases, suggesting loss of LKB1 activity as an early event in HGSOC development. The other study of LKB1 protein expression conducted by Tanwar et al reported that 54% of HGSOC samples (N = 92) showed absence of LKB1 expression (Tanwar et al., 2014). These studies suggest that the loss or reduced expression of LKB1 is a common alteration in HGSOC.
LKB1 in early serous ovarian tumorigenesis
To understand the functional impacts of LKB1 loss in the aetiology of serous ovarian cancer, Tanwar et al. (2014) conditionally deleted Lkb1 in mouse OSE cells. This led to abnormal papillary growth and widespread shedding of surface epithelial cells. Simultaneous deletion of Pten and Lkb1 led to the development of high-grade papillary serous carcinomas with increased mTOR activity. In a complementary approach, George et al. (2016) used a series of primary FTE cell lines harboring dominant negative form of TP53 (TP53-R175H) or an immortalized FTE cell line with three genetic modification including expressing TERT, a dominant negative TP53 and Rb loss, to study the role of LKB1 in serous tumorigenesis. Interestingly, loss of LKB1 in TP53 mutant primary and immortal FTE cells led to premature cellular senescence and resulted in a G2/M cell cycle arrest. Overexpression of cyclin E1 bypassed LKB1-induced senescence, re-initiated cell proliferation and promoted anchorage-independent growth of FTE cells. Consistent with its role in control cell polarity, defect of apical to basal polarity was also observed in LKB1 deficient primary FTE cells and a significant destabilized epithelial integrity in LKB1-TP53 co-mutated cells. These findings reinforce the concept that the loss of LKB1 is an early genetic alteration that co-operates with other oncogenic events, to affect cell polarity and differentiation and promote oncogenic transformation.
To test this concept, we examined a TCGA data set (Table 2) and identified 5.1% (16/311) of serous ovarian cancer with LKB1 deletion. LKB1 deletion co-occurs with common HGSOC genetic alterations, particularly with CCNE1 mutations, but are not detected in tumors with BRCA1 or BRCA2 mutations. Further analysis of clinical genomic data will provide more understanding of synergy of LKB1 deficiency and other oncogenes/tumor suppressors in ovarian tumorigenesis.
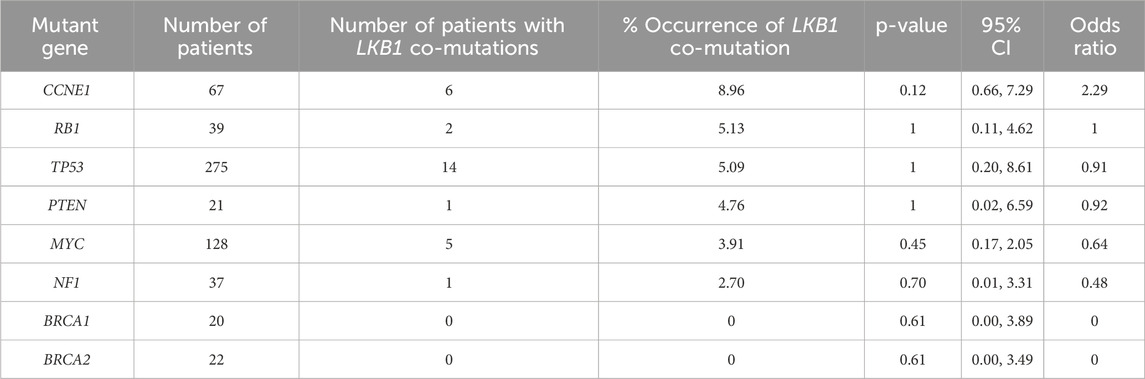
Table 2. Frequency of LKB1 deep deletion co-occurring with common mutations in HGSOC. Analysis of 311 ovarian cancer serous cystadenocarcinoma in TCGA dataset via http://www.cbioportal.org (access date 27th September 2024). Statistical analysis was performed using Fisher’s exact test.
LKB1 in ovarian cancer progression
In contrast to the established tumor suppressor role in serous ovarian cancer initiation, there is no consensus on LKB1’s role in ovarian cancer progression. The Shepherd group exploited a three-dimensional model system of spheroid formed by spontaneous cell aggregation in suspension, to study the role of LKB1 during metastatic progression of EOC (Tomas and Shepherd, 2023). They reported that depletion of LKB1 by siRNA or CRISPR-Cas9 did not affect proliferation of EOC cells in adherent 2D culture conditions. However, LKB1 depletion significantly impaired anchorage-independent growth of EOC cells and reduced viability of the spheroids, implicating LKB1 loss abrogates EOC metastatic potential (Buensuceso et al., 2020; Peart et al., 2015). They further injected EOC cells directly into the peritoneal space of immunodeficient mice and demonstrated that loss of LKB1 improved survival, reduced tumor burden and hindered disease spreading in this orthotopic mouse xenograft model of metastasis (Buensuceso et al., 2020). Their studies suggest that functional LKB1 signaling is required to maintain dormancy of EOC cells in spheroids growth and protect from anoikis during the process of dissemination to other organs.
Studies from the Shepherd group also demonstrated that depletion of AMPK by siRNA in EOC spheroids had no effect on cell viability, suggesting the protective effect of LKB1 in EOC spheroids is independent of AMKP signaling. One substrate of LKB1, NUAK1 has been reported to promote EOC spheroid formation through fibronectin expression (Fritz et al., 2020) and its expression is associated with poor prognosis in ovarian cancer (Phippen et al., 2016). Loss of LKB1 significantly decreased NUAK1 expression in adherent EOC cells and spheroids (Fritz et al., 2020), suggesting that NUAK1 may mediate the biological effects of LKB1 on spheroid formation, anoikis-resistance and metastatic potential. Loss of LKB1 or NUAK1 in EOC spheroid cells leads to accumulation of reactive oxygen species and subsequently induces activation of NF-kB signaling as an adaptive response to oxidative stress (Buensuceso et al., 2022). Taken together, their studies suggest that an intact LKB1 signaling is required for EOC metastasis.
To gain further insights into the functional impact of LKB1 on ovarian cancer progression, we analyzed the mRNA expression levels of LKB1 and its major downstream 13 kinase in ovarian tumor tissues compared with normal tissues using ovarian cancer datasets from TCGA and GTEx projects through GEPIA (Tang et al., 2017) (Figure 4B). Most genes showed downregulation at the mRNA level in ovarian tumor tissues except MARK2 and NUAK2. Overall, the data suggests that LKB1 signaling is suppressed in ovarian tumors. Furthermore, a recent study reported that one LKB1 substrate, MARK3 has a tumor suppressor role in HGSOC (Machino et al., 2022). Activation of MARK3 under metabolic stress phosphorylates CDC25B and induces cell cycle G2/M phase arrest (Figure 2). Depletion of LKB1 abrogated MARK3 activation, which may contribute to increased cell proliferation.
Collectively, the contradictory data of the role of LKB1 in ovarian cancer cell survival and invasiveness suggests its effects could be genetic context-dependent through interaction with other oncogenic signaling. It is also possible that the requirement for LKB1 signaling is varied at different stages of cancer progression from primary tumor growth, distal metastasis to tumor recurrence and adjusted by different metabolic stresses and microenvironment. This complexity thus confers the heterogenous phenotypes of LKB1 deficient ovarian tumors.
Conclusion and perspectives
LKB1 has been identified as a gene responsible for PJS, a rare autosomal dominant disease with an increased predisposition to malignant tumors in multiple tissues. While Lkb1 knockout mice are predisposed to developing cancer, LKB1 loss alone is not sufficient to initiate tumorigenesis. Accumulating evidence supports that LKB1 loss might occur as a secondary oncogenic lesion that facilitate transformation by at least one constitutively active oncogene. As a tumor suppressor, LKB1 phosphorylates its target substrates and regulates their activities to exert its biological functions primarily impacting on cell growth, metabolism and polarity. While AMPK as the canonical downstream kinase regulated by LKB1 has been well studied, the roles of other target kinases in mediating LKB1 functional impacts in cancer biology are still less defined. Further characterization of these substrates is crucial to understand the multi-functions of LKB1.
Deletion of LKB1 is a common genetic alteration in epithelial ovarian cancer. Studies in human FTE cell lines and mouse OSE cells suggest that loss of LKB1 co-occur with other oncogenic mutations including TP53 inactivation, overexpression of cyclin E1 or loss of PTEN during ovarian transformation despite lack of mechanistic investigation. The studies in an ovarian cancer spheroid model system, however, suggest that LKB1 function is required for anchorage-independent growth and metastasis potential in ovarian cancer cells. It should be noted that these ovarian cancer cell lines have heterogenous genomic characteristics and LKB1 deficiency may have distinct functional impacts via interactions with various oncogenic mutations. To advance our understanding of LKB1 signaling in ovarian cancer, particularly HGSOC, several areas warrant further investigations: (i) The evaluation of the prevalence of LKB1 mutations, and co-occurring genomic alterations, as well as the impact on prognosis, response to therapy and survival outcomes will help define the molecular subtypes of LKB1 mutant ovarian cancer. (ii) In addition to commercially available cell lines, HGSOC patient-derived cell lines may provide more clinically relevant in vitro models to study the biology of LKB1 (Pishas et al., 2021). (iii) Given LKB1 is a master kinase which primarily regulates cell metabolism and cell growth, deciphering signal transduction and metabolic alterations is required to determine the functional impact of LKB1 deficiency, in synergy with the common genetic mutations in ovarian cancer cells. (iv) Comprehensive profiling of the tumor microenvironment of LKB1 mutant ovarian cancer using patient samples and syngeneic mouse ovarian cancer models may provide new mechanistic insight into the relative low response rate of EOC to immune therapy. Altogether, such studies will identify therapeutic vulnerabilities and enable developing personalized treatments for patients with LKB1 mutant ovarian tumors.
Author contributions
JK: Writing–review and editing, Writing–original draft, Supervision, Project administration, Funding acquisition, Conceptualization. SG: Writing–review and editing, Writing–original draft. JP: Writing–review and editing, Writing–original draft. JO: Writing–review and editing, Writing–original draft. ES: Writing–review and editing, Writing–original draft, Supervision, Project administration, Funding acquisition, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by NHMRC ideas grant (#1184873) and Tour de Cure Foundation (to ES and JK). ES received support from the Victorian Cancer Agency (Research Fellowship MCRF19007). JK received support from the 5Point Foundation (Christine Martin Fellowship).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Arora, T., Mullangi, S., Vadakekut, E. S., and Lekkala, M. R. (2024). “Epithelial Ovarian cancer,” in StatPearls (StatPearls Publishing).
Baas, A. F., Boudeau, J., Sapkota, G. P., Smit, L., Medema, R., Morrice, N. A., et al. (2003). Activation of the tumour suppressor kinase LKB1 by the STE20-like pseudokinase STRAD. Embo J. 22 (12), 3062–3072. doi:10.1093/emboj/cdg292
Bardeesy, N., Sinha, M., Hezel, A. F., Signoretti, S., Hathaway, N. A., Sharpless, N. E., et al. (2002). Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature 419 (6903), 162–167. doi:10.1038/nature01045
Besse, B., Pons-Tostivint, E., Park, K., Hartl, S., Forde, P. M., Hochmair, M. J., et al. (2024). Biomarker-directed targeted therapy plus durvalumab in advanced non-small-cell lung cancer: a phase 2 umbrella trial. Nat. Med. 30 (3), 716–729. doi:10.1038/s41591-024-02808-y
Biton, J., Mansuet-Lupo, A., Pécuchet, N., Alifano, M., Ouakrim, H., Arrondeau, J., et al. (2018). TP53, STK11, and EGFR mutations predict tumor immune profile and the response to anti-PD-1 in lung adenocarcinoma. Clin. Cancer Res. 24 (22), 5710–5723. doi:10.1158/1078-0432.Ccr-18-0163
Borzi, C., Galli, G., Ganzinelli, M., Signorelli, D., Vernieri, C., Garassino, M. C., et al. (2020). Beyond LKB1 mutations in non-small cell lung cancer: defining LKB1less phenotype to optimize patient selection and treatment. Pharm. (Basel) 13 (11), 385. doi:10.3390/ph13110385
Boudeau, J., Baas, A. F., Deak, M., Morrice, N. A., Kieloch, A., Schutkowski, M., et al. (2003). MO25alpha/beta interact with STRADalpha/beta enhancing their ability to bind, activate and localize LKB1 in the cytoplasm. Embo J. 22 (19), 5102–5114. doi:10.1093/emboj/cdg490
Bright, N. J., Carling, D., and Thornton, C. (2008). Investigating the regulation of brain-specific kinases 1 and 2 by phosphorylation. J. Biol. Chem. 283 (22), 14946–14954. doi:10.1074/jbc.M710381200
Buensuceso, A., Fritz, J. L., Collins, O., Valdés, Y. R., Borrelli, M. J., DiMattia, G. E., et al. (2022). Loss of LKB1-NUAK1 signalling enhances NF-κB activity in a spheroid model of high-grade serous ovarian cancer. Sci. Rep. 12 (1), 3011. doi:10.1038/s41598-022-06796-2
Buensuceso, A., Ramos-Valdes, Y., DiMattia, G. E., and Shepherd, T. G. (2020). AMPK-independent LKB1 activity is required for efficient epithelial ovarian cancer metastasis. Mol. Cancer Res. 18 (3), 488–500. doi:10.1158/1541-7786.Mcr-19-0530
Burdett, N. L., Willis, M. O., Alsop, K., Hunt, A. L., Pandey, A., Hamilton, P. T., et al. (2023). Multiomic analysis of homologous recombination-deficient end-stage high-grade serous ovarian cancer. Nat. Genet. 55 (3), 437–450. doi:10.1038/s41588-023-01320-2
Caiola, E., Falcetta, F., Giordano, S., Marabese, M., Garassino, M. C., Broggini, M., et al. (2018). Co-occurring KRAS mutation/LKB1 loss in non-small cell lung cancer cells results in enhanced metabolic activity susceptible to caloric restriction: an in vitro integrated multilevel approach. J. Exp. Clin. Cancer Res. 37 (1), 302. doi:10.1186/s13046-018-0954-5
Cheng, H., Liu, P., Wang, Z. C., Zou, L., Santiago, S., Garbitt, V., et al. (2009). SIK1 couples LKB1 to p53-dependent anoikis and suppresses metastasis. Sci. Signal 2 (80), ra35. doi:10.1126/scisignal.2000369
Cheng, H., Liu, P., Zhang, F., Xu, E., Symonds, L., Ohlson, C. E., et al. (2014). A genetic mouse model of invasive endometrial cancer driven by concurrent loss of Pten and Lkb1 Is highly responsive to mTOR inhibition. Cancer Res. 74 (1), 15–23. doi:10.1158/0008-5472.Can-13-0544
ClinicalTrials (2017). Bemcentinib (BGB324) in combination with pembrolizumab in patients with advanced NSCLC. Available at: https://clinicaltrials.gov/study/NCT03184571 (Accessed September 24, 2023).
ClinicalTrials (2019). Testing whether cancers with specific mutations respond better to glutaminase inhibitor, telaglenastat hydrochloride, anti-cancer treatment, BeGIN study. Available at: https://ClinicalTrials.gov/show/NCT03872427 (Accessed September 10, 2024).
ClinicalTrials (2020). KEAPSAKE: a study of telaglenastat (CB-839) with standard-of-care chemoimmunotherapy in 1L KEAP1/NRF2-mutated, nonsquamous NSCLC (keapsake). Available at: https://clinicaltrials.gov/study/NCT04265534 (Accessed September 21, 2024).
ClinicalTrials (2021). A study of sotorasib (AMG 510) in participants with stage IV NSCLC whose tumors harbor a KRAS p.G12C mutation in need of first-line treatment (CodeBreaK201). Available at: https://clinicaltrials.gov/study/NCT04933695 (Accessed September 11, 2024).
ClinicalTrials (2022). A study of JAB-21822 in advanced or metastatic NSCLC with KRAS p.G12C and STK11 Co-mutation and wild-type KEAP1. Available at: https://clinicaltrials.gov/study/NCT05276726 (Accessed September 28, 2024).
ClinicalTrials (2023b). A first-in-human (FIH) study to evaluate the safety and tolerability of VVD-130850 in participants with advanced solid and hematologic tumors. Available at: https://clinicaltrials.gov/study/NCT06188208 (Accessed September 19, 2024).
ClinicalTrials (2023c). Study of TNG260 and an anti-PD antibody in STK11 mutated solid tumors. Available at: https://clinicaltrials.gov/study/NCT05887492 (Accessed September 08, 2024).
ClinicalTrials (2023d). A study of WX390 combined with toripalimab in patients with advanced gastric-type endocervical adenocarcinoma with STK11 mutations. Available at: https://clinicaltrials.gov/study/NCT06124963 (Accessed September 11, 2024).
ClinicalTrials (2024a). A single-arm pilot study of first-line treatment with carbognilumab combined with chemotherapy in patients with STK11-mutated advanced or postoperative recurrent non-small cell lung cancer. Available at: https://clinicaltrials.gov/study/NCT06331650 (Accessed September 03, 2024).
ClinicalTrials (2024b). Targeting ODC as an immunotherapeutic target in STK11 (LKB1) pathway-deficient NSCLC (DFMO). Available at: https://clinicaltrials.gov/study/NCT06219174 (Accessed September 08, 2024).
Collet, L., Ghurburrun, E., Meyers, N., Assi, M., Pirlot, B., Leclercq, I. A., et al. (2020). Kras and Lkb1 mutations synergistically induce intraductal papillary mucinous neoplasm derived from pancreatic duct cells. Gut 69 (4), 704–714. doi:10.1136/gutjnl-2018-318059
Contreras, C. M., Akbay, E. A., Gallardo, T. D., Haynie, J. M., Sharma, S., Tagao, O., et al. (2010). Lkb1 inactivation is sufficient to drive endometrial cancers that are aggressive yet highly responsive to mTOR inhibitor monotherapy. Dis. Model Mech. 3 (3-4), 181–193. doi:10.1242/dmm.004440
Deng, J., Thennavan, A., Dolgalev, I., Chen, T., Li, J., Marzio, A., et al. (2021). ULK1 inhibition overcomes compromised antigen presentation and restores antitumor immunity in LKB1-mutant lung cancer. Nat. Cancer 2 (5), 503–514. doi:10.1038/s43018-021-00208-6
Devarakonda, S., Pellini, B., Verghese, L., Park, H., Morgensztern, D., Govindan, R., et al. (2021). A phase II study of everolimus in patients with advanced solid malignancies with TSC1, TSC2, NF1, NF2 or STK11 mutations. J. Thorac. Dis. 13 (7), 4054–4062. doi:10.21037/jtd-21-195
Dupuy, F., Griss, T., Blagih, J., Bridon, G., Avizonis, D., Ling, C., et al. (2013). LKB1 is a central regulator of tumor initiation and pro-growth metabolism in ErbB2-mediated breast cancer. Cancer Metab. 1 (1), 18. doi:10.1186/2049-3002-1-18
Faubert, B., Vincent, E. E., Griss, T., Samborska, B., Izreig, S., Svensson, R. U., et al. (2014). Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1α. Proc. Natl. Acad. Sci. U. S. A. 111 (7), 2554–2559. doi:10.1073/pnas.1312570111
Fritz, J. L., Collins, O., Saxena, P., Buensuceso, A., Ramos Valdes, Y., Francis, K. E., et al. (2020). A novel role for NUAK1 in promoting ovarian cancer metastasis through regulation of fibronectin production in spheroids. Cancers (Basel) 12 (5), 1250. doi:10.3390/cancers12051250
Galan-Cobo, A., Sitthideatphaiboon, P., Qu, X., Poteete, A., Pisegna, M. A., Tong, P., et al. (2019). LKB1 and KEAP1/NRF2 pathways cooperatively promote metabolic reprogramming with enhanced glutamine dependence in KRAS-mutant lung adenocarcinoma. Cancer Res. 79 (13), 3251–3267. doi:10.1158/0008-5472.CAN-18-3527
George, S. H., Milea, A., Sowamber, R., Chehade, R., Tone, A., and Shaw, P. A. (2016). Loss of LKB1 and p53 synergizes to alter fallopian tube epithelial phenotype and high-grade serous tumorigenesis. Oncogene 35 (1), 59–68. doi:10.1038/onc.2015.62
Gill, R. K., Yang, S. H., Meerzaman, D., Mechanic, L. E., Bowman, E. D., Jeon, H. S., et al. (2011). Frequent homozygous deletion of the LKB1/STK11 gene in non-small cell lung cancer. Oncogene 30 (35), 3784–3791. doi:10.1038/onc.2011.98
Goodwin, J. M., Svensson, R. U., Lou, H. J., Winslow, M. M., Turk, B. E., and Shaw, R. J. (2014). An AMPK-independent signaling pathway downstream of the LKB1 tumor suppressor controls Snail1 and metastatic potential. Mol. Cell 55 (3), 436–450. doi:10.1016/j.molcel.2014.06.021
Granado-Martínez, P., Garcia-Ortega, S., González-Sánchez, E., McGrail, K., Selgas, R., Grueso, J., et al. (2020). STK11 (LKB1) missense somatic mutant isoforms promote tumor growth, motility and inflammation. Commun. Biol. 3 (1), 366. doi:10.1038/s42003-020-1092-0
Groelly, F. J., Fawkes, M., Dagg, R. A., Blackford, A. N., and Tarsounas, M. (2023). Targeting DNA damage response pathways in cancer. Nat. Rev. Cancer 23 (2), 78–94. doi:10.1038/s41568-022-00535-5
Gwinn, D. M., Shackelford, D. B., Egan, D. F., Mihaylova, M. M., Mery, A., Vasquez, D. S., et al. (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 30 (2), 214–226. doi:10.1016/j.molcel.2008.03.003
Hawley, S. A., Boudeau, J., Reid, J. L., Mustard, K. J., Udd, L., Mäkelä, T. P., et al. (2003). Complexes between the LKB1 tumor suppressor, STRAD alpha/beta and MO25 alpha/beta are upstream kinases in the AMP-activated protein kinase cascade. J. Biol. 2 (4), 28. doi:10.1186/1475-4924-2-28
Hemminki, A., Markie, D., Tomlinson, I., Avizienyte, E., Roth, S., Loukola, A., et al. (1998). A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 391 (6663), 184–187. doi:10.1038/34432
Hermanova, I., Zúñiga-García, P., Caro-Maldonado, A., Fernandez-Ruiz, S., Salvador, F., Martín-Martín, N., et al. (2020). Genetic manipulation of LKB1 elicits lethal metastatic prostate cancer. J. Exp. Med. 217 (6), e20191787. doi:10.1084/jem.20191787
Hezel, A. F., and Bardeesy, N. (2008). LKB1; linking cell structure and tumor suppression. Oncogene 27 (55), 6908–6919. doi:10.1038/onc.2008.342
Hollis, R. L. (2023). Molecular characteristics and clinical behaviour of epithelial ovarian cancers. Cancer Lett. 555, 216057. doi:10.1016/j.canlet.2023.216057
Hollstein, P. E., Eichner, L. J., Brun, S. N., Kamireddy, A., Svensson, R. U., Vera, L. I., et al. (2019). The AMPK-related kinases SIK1 and SIK3 mediate key tumor-suppressive effects of LKB1 in NSCLC. Cancer Discov. 9 (11), 1606–1627. doi:10.1158/2159-8290.Cd-18-1261
Hu, L., Liu, M., Tang, B., Li, Q., Pan, B. S., Xu, C., et al. (2023). Posttranslational regulation of liver kinase B1 in human cancer. J. Biol. Chem. 299 (4), 104570. doi:10.1016/j.jbc.2023.104570
Huang, S., He, T., Yang, S., Sheng, H., Tang, X., Bao, F., et al. (2020). Metformin reverses chemoresistance in non-small cell lung cancer via accelerating ubiquitination-mediated degradation of Nrf2. Transl. Lung Cancer Res. 9 (6), 2337–2355. doi:10.21037/tlcr-20-1072
Jaleel, M., McBride, A., Lizcano, J. M., Deak, M., Toth, R., Morrice, N. A., et al. (2005). Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 579 (6), 1417–1423. doi:10.1016/j.febslet.2005.01.042
Jenne, D. E., Reimann, H., Nezu, J., Friedel, W., Loff, S., Jeschke, R., et al. (1998). Peutz-Jeghers syndrome is caused by mutations in a novel serine threonine kinase. Nat. Genet. 18 (1), 38–43. doi:10.1038/ng0198-38
Ji, H., Ramsey, M. R., Hayes, D. N., Fan, C., McNamara, K., Kozlowski, P., et al. (2007). LKB1 modulates lung cancer differentiation and metastasis. Nature 448 (7155), 807–810. doi:10.1038/nature06030
Johanns, M., Pyr Dit Ruys, S., Houddane, A., Vertommen, D., Herinckx, G., Hue, L., et al. (2017). Direct and indirect activation of eukaryotic elongation factor 2 kinase by AMP-activated protein kinase. Cell Signal 36, 212–221. doi:10.1016/j.cellsig.2017.05.010
Kim, C.-L., Lim, S.-B., Choi, S.-H., Kim, D. H., Sim, Y. E., Jo, E.-H., et al. (2024). The LKB1–TSSK1B axis controls YAP phosphorylation to regulate the Hippo–YAP pathway. Cell Death and Dis. 15 (1), 76. doi:10.1038/s41419-024-06465-4
Kim, H. S., Mendiratta, S., Kim, J., Pecot, C. V., Larsen, J. E., Zubovych, I., et al. (2013). Systematic identification of molecular subtype-selective vulnerabilities in non-small-cell lung cancer. Cell 155 (3), 552–566. doi:10.1016/j.cell.2013.09.041
Kim, J., Hu, Z., Cai, L., Li, K., Choi, E., Faubert, B., et al. (2017). CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546 (7656), 168–172. doi:10.1038/nature22359
Kim, J., Lee, H. M., Cai, F., Ko, B., Yang, C., Lieu, E. L., et al. (2020). The hexosamine biosynthesis pathway is a targetable liability in KRAS/LKB1 mutant lung cancer. Nat. Metab. 2 (12), 1401–1412. doi:10.1038/s42255-020-00316-0
Kitajima, S., Ivanova, E., Guo, S., Yoshida, R., Campisi, M., Sundararaman, S. K., et al. (2019). Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 9 (1), 34–45. doi:10.1158/2159-8290.Cd-18-0689
Kitajima, S., Tani, T., Springer, B. F., Campisi, M., Osaki, T., Haratani, K., et al. (2022). MPS1 inhibition primes immunogenicity of KRAS-LKB1 mutant lung cancer. Cancer Cell 40 (10), 1128–1144.e8. doi:10.1016/j.ccell.2022.08.015
Kline, E. R., Shupe, J., Gilbert-Ross, M., Zhou, W., and Marcus, A. I. (2013). LKB1 represses focal adhesion kinase (FAK) signaling via a FAK-LKB1 complex to regulate FAK site maturation and directional persistence. J. Biol. Chem. 288 (24), 17663–17674. doi:10.1074/jbc.M112.444620
Kottakis, F., Nicolay, B. N., Roumane, A., Karnik, R., Gu, H., Nagle, J. M., et al. (2016). LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature 539 (7629), 390–395. doi:10.1038/nature20132
Koyama, S., Akbay, E. A., Li, Y. Y., Aref, A. R., Skoulidis, F., Herter-Sprie, G. S., et al. (2016). STK11/LKB1 deficiency promotes neutrophil recruitment and proinflammatory cytokine production to suppress T-cell activity in the lung tumor microenvironment. Cancer Res. 76 (5), 999–1008. doi:10.1158/0008-5472.Can-15-1439
Kuhn, E., Wang, T. L., Doberstein, K., Bahadirli-Talbott, A., Ayhan, A., Sehdev, A. S., et al. (2016). CCNE1 amplification and centrosome number abnormality in serous tubal intraepithelial carcinoma: further evidence supporting its role as a precursor of ovarian high-grade serous carcinoma. Mod. Pathol. 29 (10), 1254–1261. doi:10.1038/modpathol.2016.101
Kurman, R. J., and Shih, M. (2016). The dualistic model of ovarian carcinogenesis: revisited, revised, and expanded. Am. J. Pathol. 186 (4), 733–747. doi:10.1016/j.ajpath.2015.11.011
Labidi-Galy, S. I., Papp, E., Hallberg, D., Niknafs, N., Adleff, V., Noe, M., et al. (2017). High grade serous ovarian carcinomas originate in the fallopian tube. Nat. Commun. 8 (1), 1093. doi:10.1038/s41467-017-00962-1
Lenahan, S. M., Sarausky, H. M., Deming, P., and Seward, D. J. (2023). STK11 loss leads to YAP1-mediated transcriptional activation in human KRAS-driven lung adenocarcinoma cell lines. Cancer Gene Ther. 31, 1–8. doi:10.1038/s41417-023-00687-y
Li, N., Huang, D., Lu, N., and Luo, L. (2015). Role of the LKB1/AMPK pathway in tumor invasion and metastasis of cancer cells (Review). Oncol. Rep. 34 (6), 2821–2826. doi:10.3892/or.2015.4288
Li, R., Salehi-Rad, R., Crosson, W., Momcilovic, M., Lim, R. J., Ong, S. L., et al. (2021). Inhibition of granulocytic myeloid-derived suppressor cells overcomes resistance to immune checkpoint inhibition in LKB1-deficient non-small cell lung cancer. Cancer Res. 81 (12), 3295–3308. doi:10.1158/0008-5472.Can-20-3564
Li, T. T., and Zhu, H. B. (2020). LKB1 and cancer: the dual role of metabolic regulation. Biomed. Pharmacother. 132, 110872. doi:10.1016/j.biopha.2020.110872
Li, W., Shao, D., Li, L., Wu, M., Ma, S., Tan, X., et al. (2019). Germline and somatic mutations of multi-gene panel in Chinese patients with epithelial ovarian cancer: a prospective cohort study. J. Ovarian Res. 12 (1), 80. doi:10.1186/s13048-019-0560-y
Liu, W., Monahan, K. B., Pfefferle, A. D., Shimamura, T., Sorrentino, J., Chan, K. T., et al. (2012). LKB1/STK11 inactivation leads to expansion of a prometastatic tumor subpopulation in melanoma. Cancer Cell 21 (6), 751–764. doi:10.1016/j.ccr.2012.03.048
Liu, Y., Li, Y., Wang, X., Liu, F., Gao, P., Quinn, M. M., et al. (2017). Gemcitabine and Chk1 inhibitor AZD7762 synergistically suppress the growth of lkb1-deficient lung adenocarcinoma. Cancer Res. 77 (18), 5068–5076. doi:10.1158/0008-5472.Can-17-0567
Liu, Y., Marks, K., Cowley, G. S., Carretero, J., Liu, Q., Nieland, T. J., et al. (2013). Metabolic and functional genomic studies identify deoxythymidylate kinase as a target in LKB1-mutant lung cancer. Cancer Discov. 3 (8), 870–879. doi:10.1158/2159-8290.Cd-13-0015
Lizcano, J. M., Göransson, O., Toth, R., Deak, M., Morrice, N. A., Boudeau, J., et al. (2004). LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J. 23 (4), 833–843. doi:10.1038/sj.emboj.7600110
Luo, Z., Chen, W., Wu, W., Luo, W., Zhu, T., Guo, G., et al. (2019). Metformin promotes survivin degradation through AMPK/PKA/GSK-3β-axis in non-small cell lung cancer. J. Cell Biochem. 120 (7), 11890–11899. doi:10.1002/jcb.28470
Machino, H., Kaneko, S., Komatsu, M., Ikawa, N., Asada, K., Nakato, R., et al. (2022). The metabolic stress-activated checkpoint LKB1-MARK3 axis acts as a tumor suppressor in high-grade serous ovarian carcinoma. Commun. Biol. 5 (1), 39. doi:10.1038/s42003-021-02992-4
McDonald, J. A. (2014). Canonical and noncanonical roles of Par-1/MARK kinases in cell migration. Int. Rev. Cell Mol. Biol. 312 169–199. doi:10.1016/B978-0-12-800178-3.00006-3
Mehenni, H., Gehrig, C., Nezu, J., Oku, A., Shimane, M., Rossier, C., et al. (1998). Loss of LKB1 kinase activity in Peutz-Jeghers syndrome, and evidence for allelic and locus heterogeneity. Am. J. Hum. Genet. 63 (6), 1641–1650. doi:10.1086/302159
Miyoshi, H., Nakau, M., Ishikawa, T. O., Seldin, M. F., Oshima, M., and Taketo, M. M. (2002). Gastrointestinal hamartomatous polyposis in Lkb1 heterozygous knockout mice. Cancer Res. 62 (8), 2261–2266.
Mo, J. S., Meng, Z., Kim, Y. C., Park, H. W., Hansen, C. G., Kim, S., et al. (2015). Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 17 (4), 500–510. doi:10.1038/ncb3111
Mohseni, M., Sun, J., Lau, A., Curtis, S., Goldsmith, J., Fox, V. L., et al. (2014). A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat. Cell Biol. 16 (1), 108–117. doi:10.1038/ncb2884
Moro, M., Caiola, E., Ganzinelli, M., Zulato, E., Rulli, E., Marabese, M., et al. (2018). Metformin enhances cisplatin-induced apoptosis and prevents resistance to cisplatin in Co-mutated KRAS/LKB1 NSCLC. J. Thorac. Oncol. 13 (11), 1692–1704. doi:10.1016/j.jtho.2018.07.102
Morton, J. P., Jamieson, N. B., Karim, S. A., Athineos, D., Ridgway, R. A., Nixon, C., et al. (2010). LKB1 haploinsufficiency cooperates with Kras to promote pancreatic cancer through suppression of p21-dependent growth arrest. Gastroenterology 139 (2), 586–597. doi:10.1053/j.gastro.2010.04.055
Murray, C. W., Brady, J. J., Han, M., Cai, H., Tsai, M. K., Pierce, S. E., et al. (2022). LKB1 drives stasis and C/EBP-mediated reprogramming to an alveolar type II fate in lung cancer. Nat. Commun. 13 (1), 1090. doi:10.1038/s41467-022-28619-8
Murray, C. W., Brady, J. J., Tsai, M. K., Li, C., Winters, I. P., Tang, R., et al. (2019). An LKB1-SIK Axis suppresses lung tumor growth and controls differentiation. Cancer Discov. 9 (11), 1590–1605. doi:10.1158/2159-8290.Cd-18-1237
Nakau, M., Miyoshi, H., Seldin, M. F., Imamura, M., Oshima, M., and Taketo, M. M. (2002). Hepatocellular carcinoma caused by loss of heterozygosity in Lkb1 gene knockout mice. Cancer Res. 62 (16), 4549–4553.
Nasri, H., and Rafieian-Kopaei, M. (2014). Metformin: current knowledge. J. Res. Med. Sci. 19 (7), 658–664.
Pant, S., Yaeger, R., Spira, A. I., Pelster, M., Sabari, J. K., Hafez, N., et al. (2023). KRYSTAL-1: activity and safety of adagrasib (MRTX849) in patients with advanced solid tumors harboring a KRASG12C mutation. J. Clin. Oncol. 41 (36_Suppl. l), 425082. doi:10.1200/JCO.2023.41.36_suppl.425082
Peart, T., Ramos Valdes, Y., Correa, R. J., Fazio, E., Bertrand, M., McGee, J., et al. (2015). Intact LKB1 activity is required for survival of dormant ovarian cancer spheroids. Oncotarget 6 (26), 22424–22438. doi:10.18632/oncotarget.4211
Pencik, J., Philippe, C., Schlederer, M., Atas, E., Pecoraro, M., Grund-Gröschke, S., et al. (2023). STAT3/LKB1 controls metastatic prostate cancer by regulating mTORC1/CREB pathway. Mol. Cancer 22 (1), 133. doi:10.1186/s12943-023-01825-8
Phippen, N. T., Bateman, N. W., Wang, G., Conrads, K. A., Ao, W., Teng, P.-n., et al. (2016). NUAK1 (ARK5) is associated with poor prognosis in ovarian cancer. Front. Oncol. 6, 213. doi:10.3389/fonc.2016.00213
Pishas, K. I., Cowley, K. J., Pandey, A., Hoang, T., Beach, J. A., Luu, J., et al. (2021). Phenotypic consequences of slc25a40-ABCB1 fusions beyond drug resistance in high-grade serous ovarian cancer. Cancers (Basel) 13 (22), 5644. doi:10.3390/cancers13225644
Pons-Tostivint, E., Lugat, A., Fontenau, J. F., Denis, M. G., and Bennouna, J. (2021). STK11/LKB1 modulation of the immune response in lung cancer: from biology to therapeutic impact. Cells 10 (11), 3129. doi:10.3390/cells10113129
Qian, Y., Galan-Cobo, A., Guijarro, I., Dang, M., Molkentine, D., Poteete, A., et al. (2023). MCT4-dependent lactate secretion suppresses antitumor immunity in LKB1-deficient lung adenocarcinoma. Cancer Cell 41 (7), 1363–1380.e7. doi:10.1016/j.ccell.2023.05.015
Richer, A. L., Cala, J. M., O'Brien, K., Carson, V. M., Inge, L. J., and Whitsett, T. G. (2017). WEE1 kinase inhibitor AZD1775 has preclinical efficacy in LKB1-deficient non–small cell lung cancer. Cancer Res. 77 (17), 4663–4672. doi:10.1158/0008-5472.Can-16-3565
Rogers, Z. N., McFarland, C. D., Winters, I. P., Naranjo, S., Chuang, C.-H., Petrov, D., et al. (2017). A quantitative and multiplexed approach to uncover the fitness landscape of tumor suppression in vivo. Nat. Methods 14 (7), 737–742. doi:10.1038/nmeth.4297
Sapkota, G. P., Deak, M., Kieloch, A., Morrice, N., Goodarzi, A. A., Smythe, C., et al. (2002). Ionizing radiation induces ataxia telangiectasia mutated kinase (ATM)-mediated phosphorylation of LKB1/STK11 at Thr-366. Biochem. J. 368 (Pt 2), 507–516. doi:10.1042/bj20021284
Shackelford, D. B., and Shaw, R. J. (2009). The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer 9 (8), 563–575. doi:10.1038/nrc2676
Shaw, R. J. (2009). LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol. (Oxf) 196 (1), 65–80. doi:10.1111/j.1748-1716.2009.01972.x
Shaw, R. J., Bardeesy, N., Manning, B. D., Lopez, L., Kosmatka, M., DePinho, R. A., et al. (2004). The LKB1 tumor suppressor negatively regulates mTOR signaling. Cancer Cell 6 (1), 91–99. doi:10.1016/j.ccr.2004.06.007
Shorning, B. Y., Griffiths, D., and Clarke, A. R. (2011). Lkb1 and Pten synergise to suppress mTOR-mediated tumorigenesis and epithelial-mesenchymal transition in the mouse bladder. PLoS One 6 (1), e16209. doi:10.1371/journal.pone.0016209
Sitthideatphaiboon, P., Galan-Cobo, A., Negrao, M. V., Qu, X., Poteete, A., Zhang, F., et al. (2021). STK11/LKB1 mutations in NSCLC are associated with KEAP1/NRF2-dependent radiotherapy resistance targetable by glutaminase inhibition. Clin. Cancer Res. 27 (6), 1720–1733. doi:10.1158/1078-0432.Ccr-20-2859
Skoulidis, F., Byers, L. A., Diao, L., Papadimitrakopoulou, V. A., Tong, P., Izzo, J., et al. (2015). Co-occurring genomic alterations define major subsets of KRAS-mutant lung adenocarcinoma with distinct biology, immune profiles, and therapeutic vulnerabilities. Cancer Discov. 5 (8), 860–877. doi:10.1158/2159-8290.Cd-14-1236
Skoulidis, F., Goldberg, M. E., Greenawalt, D. M., Hellmann, M. D., Awad, M. M., Gainor, J. F., et al. (2018). STK11/LKB1 mutations and PD-1 inhibitor resistance in KRAS-mutant lung adenocarcinoma. Cancer Discov. 8 (7), 822–835. doi:10.1158/2159-8290.Cd-18-0099
Skoulidis, F., and Heymach, J. V. (2019). Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 19 (9), 495–509. doi:10.1038/s41568-019-0179-8
Skoulidis, F., Li, B. T., Dy, G. K., Price, T. J., Falchook, G. S., Wolf, J., et al. (2021). Sotorasib for lung cancers with KRAS p.G12C mutation. N. Engl. J. Med. 384 (25) 2371–2381. doi:10.1056/NEJMoa2103695
Skoulidis, F., Neal, J. W., Akerley, W. L., Paik, P. K., Papagiannakopoulos, T., Reckamp, K. L., et al. (2020). A phase II randomized study of telaglenastat, a glutaminase (GLS) inhibitor, versus placebo, in combination with pembrolizumab (Pembro) and chemotherapy as first-line treatment for KEAP1/NRF2-mutated non-squamous metastatic non-small cell lung cancer (mNSCLC). J. Clin. Oncol. 38 (15_Suppl. l), TPS9627. doi:10.1200/JCO.2020.38.15_suppl.TPS9627
Skoulidis, F., Redman, M. W., Suga, J. M., Baghdadi, T. A., Villano, J. L., Goldberg, S. B., et al. (2022). A phase II study of talazoparib plus avelumab in patients with stage IV or recurrent nonsquamous non–small cell lung cancer bearing pathogenic STK11 genomic alterations (SWOG S1900C, LUNG-MAP sub-study, NCT04173507). J. Clin. Oncol. 40 (16_Suppl. l), 9060. doi:10.1200/JCO.2022.40.16_suppl.9060
Stein, B. D., Ferrarone, J. R., Gardner, E. E., Chang, J. W., Wu, D., Hollstein, P. E., et al. (2023). LKB1-Dependent regulation of TPI1 creates a divergent metabolic liability between human and mouse lung adenocarcinoma. Cancer Discov. 13 (4), 1002–1025. doi:10.1158/2159-8290.Cd-22-0805
Sun, Z., Jiang, Q., Li, J., and Guo, J. (2020). The potent roles of salt-inducible kinases (SIKs) in metabolic homeostasis and tumorigenesis. Signal Transduct. Target Ther. 5 (1), 150. doi:10.1038/s41392-020-00265-w
Takeda, H., Miyoshi, H., Kojima, Y., Oshima, M., and Taketo, M. M. (2006). Accelerated onsets of gastric hamartomas and hepatic adenomas/carcinomas in Lkb1+/-p53-/- compound mutant mice. Oncogene 25 (12), 1816–1820. doi:10.1038/sj.onc.1209207
Tang, Z., Li, C., Kang, B., Gao, G., Li, C., and Zhang, Z. (2017). GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 45 (W1), W98–W102. doi:10.1093/nar/gkx247
Tanwar, P. S., Mohapatra, G., Chiang, S., Engler, D. A., Zhang, L., Kaneko-Tarui, T., et al. (2014). Loss of LKB1 and PTEN tumor suppressor genes in the ovarian surface epithelium induces papillary serous ovarian cancer. Carcinogenesis 35 (3), 546–553. doi:10.1093/carcin/bgt357
Thirugnanam, K., and Ramchandran, R. (2020). SNRK: a metabolic regulator with multifaceted role in development and disease. Vessel Plus 4, 26. doi:10.20517/2574-1209.2020.18
Timm, T., Li, X. Y., Biernat, J., Jiao, J., Mandelkow, E., Vandekerckhove, J., et al. (2003). MARKK, a Ste20-like kinase, activates the polarity-inducing kinase MARK/PAR-1. Embo J. 22 (19), 5090–5101. doi:10.1093/emboj/cdg447
Tomas, E., and Shepherd, T. G. (2023). Insights into high-grade serous carcinoma pathobiology using three-dimensional culture model systems. J. Ovarian Res. 16 (1), 70. doi:10.1186/s13048-023-01145-x
van de Vis, R. A. J., Moustakas, A., and van der Heide, L. P. (2021). NUAK1 and NUAK2 fine-tune TGF-β signaling. Cancers (Basel) 13 (13), 3377. doi:10.3390/cancers13133377
Vang, R., Shih Ie, M., and Kurman, R. J. (2009). Ovarian low-grade and high-grade serous carcinoma: pathogenesis, clinicopathologic and molecular biologic features, and diagnostic problems. Adv. Anat. Pathol. 16 (5), 267–282. doi:10.1097/PAP.0b013e3181b4fffa
Vernieri, C., Signorelli, D., Galli, G., Ganzinelli, M., Moro, M., Fabbri, A., et al. (2019). Exploiting FAsting-mimicking diet and MEtformin to improve the efficacy of platinum-pemetrexed chemotherapy in advanced LKB1-inactivated lung adenocarcinoma: the FAME trial. Clin. Lung Cancer 20 (3), e413–e417. doi:10.1016/j.cllc.2018.12.011
Wakefield, M. J., Nesic, K., Kondrashova, O., and Scott, C. L. (2019). Diverse mechanisms of PARP inhibitor resistance in ovarian cancer. Biochim. Biophys. Acta Rev. Cancer 1872 (2), 188307. doi:10.1016/j.bbcan.2019.08.002
Wang, W., Xiao, Z.-D., Li, X., Aziz, K. E., Gan, B., Johnson, R. L., et al. (2015). AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat. Cell Biol. 17 (4), 490–499. doi:10.1038/ncb3113
Wang, Y. S., Chen, J., Cui, F., Wang, H., Wang, S., Hang, W., et al. (2016). LKB1 is a DNA damage response protein that regulates cellular sensitivity to PARP inhibitors. Oncotarget 7 (45), 73389–73401. doi:10.18632/oncotarget.12334
Wei, C., Amos, C. I., Stephens, L. C., Campos, I., Deng, J. M., Behringer, R. R., et al. (2005). Mutation of Lkb1 and p53 genes exert a cooperative effect on tumorigenesis. Cancer Res. 65 (24), 11297–11303. doi:10.1158/0008-5472.Can-05-0716
Xu, C., Fillmore, C. M., Koyama, S., Wu, H., Zhao, Y., Chen, Z., et al. (2014). Loss of Lkb1 and Pten leads to lung squamous cell carcinoma with elevated PD-L1 expression. Cancer Cell 25 (5), 590–604. doi:10.1016/j.ccr.2014.03.033
Yan, S., Frank, D., Son, J., Hannan, K. M., Hannan, R. D., Chan, K. T., et al. (2017). The potential of targeting ribosome biogenesis in high-grade serous ovarian cancer. Int. J. Mol. Sci. 18 (1), 210. doi:10.3390/ijms18010210
Zagórska, A., Deak, M., Campbell, D. G., Banerjee, S., Hirano, M., Aizawa, S., et al. (2010). New roles for the LKB1-NUAK pathway in controlling myosin phosphatase complexes and cell adhesion. Sci. Signal 3 (115), ra25. doi:10.1126/scisignal.2000616
Zeng, Q., Chen, J., Li, Y., Werle, K. D., Zhao, R. X., Quan, C. S., et al. (2017). LKB1 inhibits HPV-associated cancer progression by targeting cellular metabolism. Oncogene 36 (9), 1245–1255. doi:10.1038/onc.2016.290
Zhang, L., Li, J., Young, L. H., and Caplan, M. J. (2006). AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc. Natl. Acad. Sci. U. S. A. 103 (46), 17272–17277. doi:10.1073/pnas.0608531103
Keywords: LKB1, STK11, cancer biology, ovarian cancer, metabolism, immunotherapy, targeted therapy, cancer signaling
Citation: Kang J, Gallucci S, Pan J, Oakhill JS and Sanij E (2024) The role of STK11/LKB1 in cancer biology: implications for ovarian tumorigenesis and progression. Front. Cell Dev. Biol. 12:1449543. doi: 10.3389/fcell.2024.1449543
Received: 15 June 2024; Accepted: 11 October 2024;
Published: 31 October 2024.
Edited by:
Vihandha Wickramasinghe, Peter MacCallum Cancer Centre, AustraliaReviewed by:
Melissa Gilbert-Ross, Emory University, United StatesMuhammad Babar Khawar, Chinese Academy of Sciences (CAS), China
Copyright © 2024 Kang, Gallucci, Pan, Oakhill and Sanij. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian Kang, amthbmdAc3ZpLmVkdS5hdQ==