 Alila Miramova1
Alila Miramova1 Anton Gartner
Anton Gartner Dmitri Ivanov
Dmitri Ivanov
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 01 July 2024
Sec. Cell Growth and Division
Volume 12 - 2024 | https://doi.org/10.3389/fcell.2024.1436563
This article is part of the Research TopicProceedings from ACDTM 2023: Cell Cycle, DNA Repair, and TelomeresView all 5 articles
Temozolomide (TMZ) is a methylating agent used as the first-line drug in the chemotherapy of glioblastomas. However, cancer cells eventually acquire resistance, necessitating the development of TMZ-potentiating therapy agents. TMZ induces several DNA base adducts, including O6-meG, 3-meA, and 7-meG. TMZ cytotoxicity stems from the ability of these adducts to directly (3-meA) or indirectly (O6-meG) impair DNA replication. Although TMZ toxicity is generally attributed to O6-meG, other alkylated bases can be similarly important depending on the status of various DNA repair pathways of the treated cells. In this mini-review we emphasize the necessity to distinguish TMZ-sensitive glioblastomas, which do not express methylguanine-DNA methyltransferase (MGMT) and are killed by the futile cycle of mismatch repair (MMR) of the O6-meG/T pairs, vs. TMZ-resistant MGMT-positive or MMR-negative glioblastomas, which are selected in the course of the treatment and are killed only at higher TMZ doses by the replication-blocking 3-meA. These two types of cells can be TMZ-sensitized by inhibiting different DNA repair pathways. However, in both cases, the toxic intermediates appear to be ssDNA gaps, a vulnerability also seen in BRCA-deficient cancers. PARP inhibitors (PARPi), which were initially developed to treat BRCA1/2-deficient cancers by synthetic lethality, were re-purposed in clinical trials to potentiate the effects of TMZ. We discuss how the recent advances in our understanding of the genetic determinants of TMZ toxicity might lead to new approaches for the treatment of glioblastomas by inhibiting PARP1 and other enzymes involved in the repair of alkylation damage (e.g., APE1).
Temozolomide (TMZ) is a chemotherapeutic drug well suited for the therapy of glioblastomas since it readily penetrates the blood-brain barrier (Kaina and Christmann, 2019). Therapeutically achievable plasma TMZ concentrations of 30–100 μM (Ostermann et al., 2004; Gupta et al., 2014) are sufficient to eliminate TMZ-sensitive cancers but are well tolerated by normal cells. TMZ belongs to the group of SN1 (first-order nucleophilic substitution) methylating agents, which include N-methyl-N-nitrosourea (MNU) and N-methyl-N’-nitro-N-nitrosoguanidine (MNNG). TMZ hydrolysis produces 5-(3-methyl 1-triazenyl) imidazole-4-carboxamide (MTIC), which has a half-life of only 2 min and releases a highly reactive methyldiazonium ion, which methylates nucleophilic N or O atoms of the DNA bases. The most toxic TMZ-induced DNA base adduct is O6-meG. Although TMZ induces several other base adducts, these are of minor importance in the killing of primary TMZ-sensitive tumors due to the high toxicity of O6-meG. O6-meG is capable of pairing not only with cytosine but also with thymine. O6-meG adducts mispaired with thymine during the first round of DNA replication after TMZ treatment are recognized by the mismatch repair (MMR) pathway. MMR corrects mismatched bases only in the newly synthesized strands. Thus, MMR will excise thymine while leaving the initial lesion, i.e., O6-meG, intact. Since O6-meG is left unrepaired, it continues to pair with thymine during gap filling. Repetitions of this process are referred to as “futile cycles” of MMR. The resultant persistent ssDNA gaps interfere with the second round of DNA replication (Stojic et al., 2004). Although DNA damage signaling leading to apoptosis induction was also proposed as a possible explanation for the MMR-induced cell death (Yoshioka et al., 2006), according to the “classical” model, replication fork progression across unrepaired nicks results in dsDNA breaks, which are highly toxic to the cell. Remarkably, the same model was initially proposed for the mechanism of cell killing by PARP inhibitors, which are currently tested in clinical trials as enhancers of TMZ (Table 1).
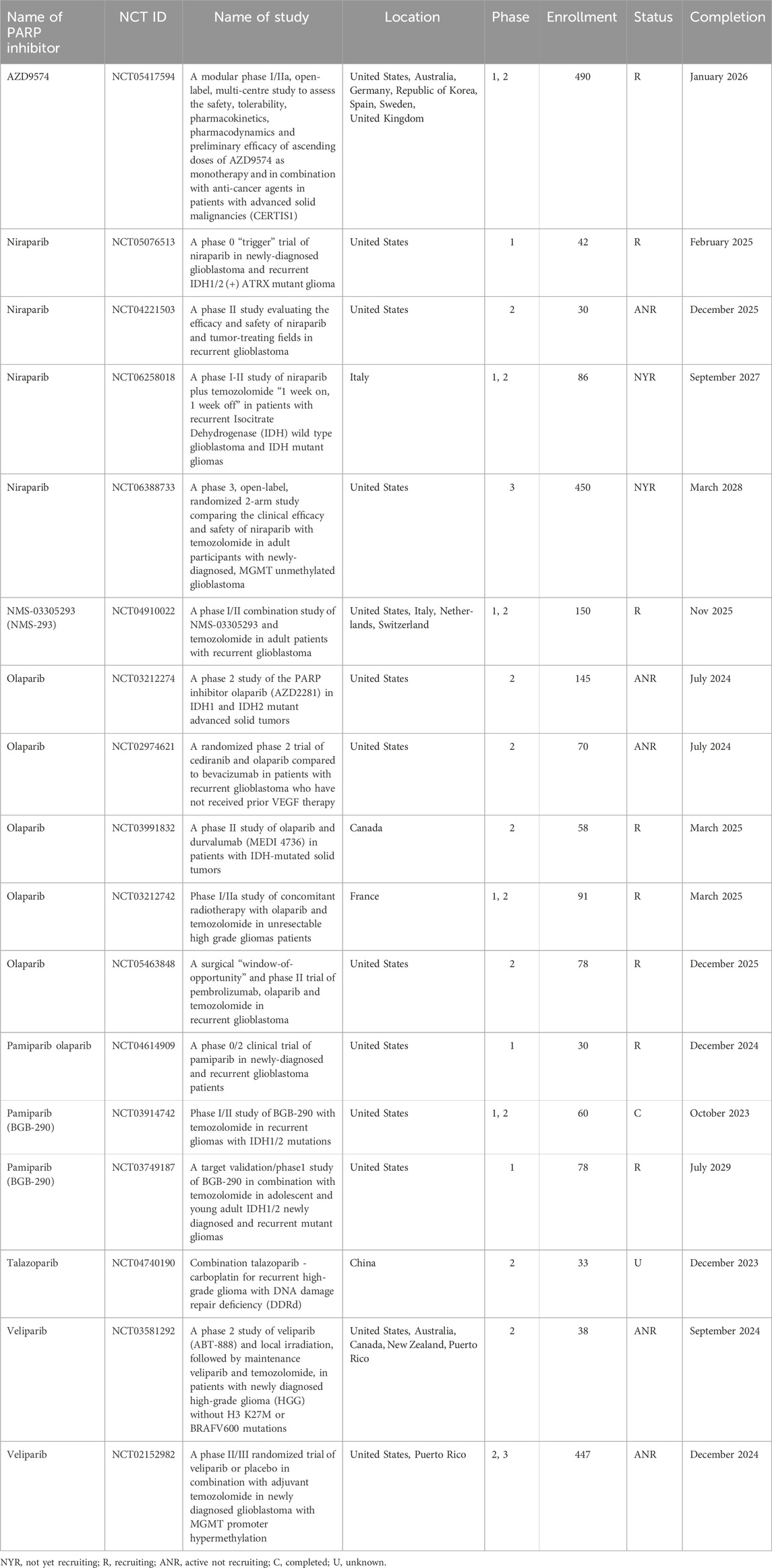
Table 1. Ongoing clinical trials testing PARP inhibitors in glioblastoma chemotherapy.
In recent years, a lot of research has been dedicated to PARP inhibitors (PARPi), which target poly (ADP-ribose) polymerase 1 (PARP1). PARP1 catalyzes the synthesis of poly (ADP-ribose) (PAR) chains using nicotinamide adenine dinucleotide (NAD+) as a substrate. PAR polymers attach to the PARP1 enzyme itself or other proteins that it physically interacts with, including histones and DNA repair factors. PARP1 binds to and gets activated primarily by ssDNA breaks and ssDNA gaps. PAR chains recruit the XRCC1 protein, a scaffold for DNA ligase 3 (LIG3), DNA polymerase β, and polynucleotide kinase 3’-phosphatase (PNKP), which repair the ssDNA breaks. In addition, PARP1 stimulates the repair of dsDNA breaks by recruiting MRE11, a nuclease responsible for end-resection in the homologous recombination (HR) repair pathway. PARP1 can also facilitate dsDNA break repair by non-homologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ). During DNA replication, PARP1 binds to replication forks and can promote fork reversal in response to replication stress. It can also affect chromatin structure to facilitate DNA repair (Ray Chaudhuri and Nussenzweig, 2017).
Although all clinical PARPi bind to the PARP1 catalytic center to prevent NAD+ binding and PARylation, they differ in their effect on the adjacent allosteric regulatory domain and in the extent of catalytic inhibition. PARP1 auto-PARylation is required for its rapid release from a ssDNA break. Therefore, strong inhibitors, such as olaparib and talazoparib, tend to “trap” PARP1 on chromatin, resulting in a cytotoxic lesion, although they do not have an allosteric effect on PARP1. Conversely, an allosteric inhibitor veliparib, promotes PARP1 release by inducing changes in PARP1 structure. Although not currently used in clinic, the third type of inhibitors, the “reverse allosteric” inhibitors, trap PARP1 by affecting the inter-domain interactions (Zandarashvili et al., 2020). Overall, PARP1 trapping is associated with a higher cytotoxicity for tumors but also for normal cells (Pommier et al., 2016).
PARP inhibitors were initially discovered to be synthetic lethal in conjunction with HR deficiency, in particular with BRCA1 and BRCA2 mutations, which are common in breast and ovarian cancers. However, the identity of the relevant lesions that cause PARPi hypersensitivity of BRCA1/2-deficient cells is the subject of debate. According to the “classical” view, collapsed forks resulting from the absence of the BRCA1/2 fork protective function give rise to dsDNA breaks, which cannot be repaired by BRCA1/2 dependent HR. Since PARP1 plays a major role in ssDNA break repair, PARP1 inhibition leads to an accumulation of ssDNA breaks, which become converted to dsDNA breaks during the second cycle DNA replication (Simoneau et al., 2021), leading to PARP/BRCA synthetic lethality (Bryant et al., 2005; Farmer et al., 2005). Although ssDNA gaps might directly trigger the checkpoint response and block cell division, the ATR checkpoint pathway becomes more dramatically activated in the second S phase following PARPi treatment, presumably by the dsDNA breaks. ATR activation leads to the suppression of replication origin firing and G2/M arrest in the second cell cycle. Notably, BRCA1/2-deficient cells are unable to activate ATR in the second replication cycle (Simoneau et al., 2021). However, a recent report suggests that the critical defect resulting in PARPi hypersensitivity is the inability of BRCA1/2-deficient cells to suppress replication gaps (Cong et al., 2021; Cong and Cantor, 2022). In this scenario, ssDNA gaps rather than dsDNA breaks are the lethal lesion, and dsDNA break appearance might be indicative of the onset of apoptosis instead of being the primary cause of cell death (Cong et al., 2021; Panzarino et al., 2021). The major source of ssDNA gaps in unperturbed cells are Okazaki fragments. PARP1 is involved in the repair of unligated Okazaki fragments (Hanzlikova et al., 2018), and PARPi treatment results in the accumulation of nascent strand discontinuities (Vaitsiankova et al., 2022).
ssDNA gaps can be converted into dsDNA breaks via several mechanisms. For example, ssDNA might fold into secondary structures, such as hairpins, which are amenable to cleavage by nucleases creating fragile sites (Polleys et al., 2023; Whalen and Cantor, 2023). Extensive ssDNA regions can expose distant inverted repeats and lead to gross chromosomal rearrangements (Ait Saada et al., 2023). Post-replicative ssDNA gaps induced by PARPi and BRCA1/2 deficiency are filled by DNA polymerase Θ (POLQ) (Belan et al., 2022; Mann et al., 2022; Schrempf et al., 2022), and POLQ inhibitors act synergistically with PARPi in BRCA1/2 deficient cells (Zatreanu et al., 2021). However, translesion synthesis (TLS) polymerases can also suppress the gaps (Nayak et al., 2020). The relative contribution of different TLS polymerases vs. POLQ to sealing PARPi-induced gaps remains to be determined. Another recently proposed model suggests that PARPi sensitivity of BRCA2-deficient cells is mediated primarily by transcription/replication conflicts (TRCs) and R-loops. According to this model, PARP1 senses TRCs and pauses the replication fork until TRCs are resolved (Petropoulos et al., 2024). Interestingly, PARP1 trapping is not required for TRC induction and trapping reduces PARPi selectivity in killing BRCA-deficient but not normal cells.
Mutated versions of isocitrate dehydrogenase enzymes, cytoplasmic IDH1 and mitochondrial IDH2, are frequently found in gliomas. The mutant enzymes acquire a novel activity that reduces alpha-ketoglutarate (alpha-KG) to 2-hydroxyglutarate (2HG), which inhibits alpha-KG-dependent dioxygenases. These include lysine demethylases (KDMs), which are responsible for demethylating histone tails and chromatin remodeling. Similar to BRCA1/2 deficient cells, IDH1/2 mutant cells are sensitive to PARPi (Sulkowski et al., 2017; Sulkowski et al., 2020). However, unlike BRCA1/2 deficiency, IDH1/2 mutations do not result in genomic instability and accumulation of large deletions or insertions, suggesting that the HR pathway is functional. Instead, IDH1/2 mutants are characterized by increased heterochromatin leading to slowdown of replication forks, replication stress, and the accumulation of ssDNA gaps (Schvartzman et al., 2023). Thus, ssDNA gaps might be the underlying cause of PARPi sensitivity in both IDH1/2 and BRCA1/2 mutants. Several clinical trials are currently ongoing to test PARPi against IDH1/2 mutant gliomas, either as a monotherapy or in combination with TMZ (Fanucci et al., 2023; Sharma et al., 2023) (Table 1).
In conclusion, recent studies highlighted the importance of ssDNA gaps as the primary cytotoxic lesions induced by both BRCA deficiencies and the action of PARPi. The observation that ssDNA gaps are also implicated in the cell killing by TMZ strengthens the rationale for TMZ/PARPi combination therapy.
We will now examine the effect PARPi might have in TMZ-treated cells depending on the cell DNA repair capacity and the primary cytotoxic lesion induced by TMZ. The outcome of PARPi application is expected to differ depending on the status of three major DNA repair pathways, methylguanine-DNA methyltransferase (MGMT), MMR, and base excision repair (BER). MGMT directly repairs O6-meG lesions, reverting them to G. In MGMT-positive cells, PARP1 binds to and PARylates MGMT, stimulating its enzymatic activity. Thus, PARPi would reduce MGMT activity and sensitize cells to TMZ (Wu et al., 2021; Cropper et al., 2022). However, MGMT is not expressed in many gliomas, rendering these hypersensitive to very low (∼10 μM) TMZ concentrations.
In MGMT-negative cells, O6-meG adducts mispaired with thymine during DNA replication are recognized by the MMR pathway and, via the aforementioned “futile cycle” of MMR give rise to persistent gaps that stall replication forks. Single-stranded DNA breaks, which can be detected in an alkaline comet assay, appear during the first round of DNA replication and persist for many hours afterward. Stretches of ssDNA recruit RPA protein, which in turn binds to ATR kinase, which phosphorylates CHK1, triggering DNA damage checkpoint and cell cycle arrest before the second cell division after the TMZ treatment (Stojic et al., 2004). TMZ sensitivity of MGMT-negative cells appears to primarily depend on their ability to process stalled forks (Figure 1). The most TMZ- (and MNNG-) sensitive among MGMT-negative lines are deficient for Fanconi Anemia (FA) genes (MacLeod et al., 2019; Olivieri et al., 2020; Cheng et al., 2024). The reason for this is not well understood. A probable explanation could be that the FANCD2/FANCI dimer, the key effector of the FA pathway, protects stalled replication forks from nuclease-mediated degradation (Schlacher et al., 2012) and is likely required to rescue the forks colliding with sites of the ongoing O6-meG-induced futile cycle of mismatch repair. In addition, MMR-induced gaps might persist until mitosis and be resolved in a FANCD2/FANCI-dependent manner to allow for ordered chromosomal segregation. In mitosis FANCD2 specifically localizes to the sites where sister chromatid bridging occurs (Chan et al., 2009). FANCD2, in association with endonucleases (from prophase to metaphase) (Naim et al., 2013) and Bloom syndrome helicase (at anaphase) (Naim and Rosselli, 2009), resolves intermediates resulting from incomplete replication. Knockout of the PCNA ubiquitinating E3 enzyme RAD18 also significantly sensitizes MGMT-negative cells to TMZ, suggesting the involvement of TLS. TMZ induces PCNA mono-ubiquitination in MGMT-negative cells peaking at the time of the second round of DNA replication (Cheng et al., 2024). Based on a CRISPR screen in a RAD18 knockout line, the NHEJ pathway becomes more important for cell survival after TMZ treatment compared to wild type, indicating that ssDNA gaps are converted into dsDNA breaks when not processed by TLS. Interestingly, in combination with TMZ treatment, RAD18 deficiency increases the C>T substitutions resulting from O6-meG mispairing with thymine but abolishes the TMZ-mediated induction of all other types of single nucleotide variants (SNVs). These results suggest that translesion synthesis plays a role in filling in the ssDNA gaps generated by MMR (Cheng et al., 2024).
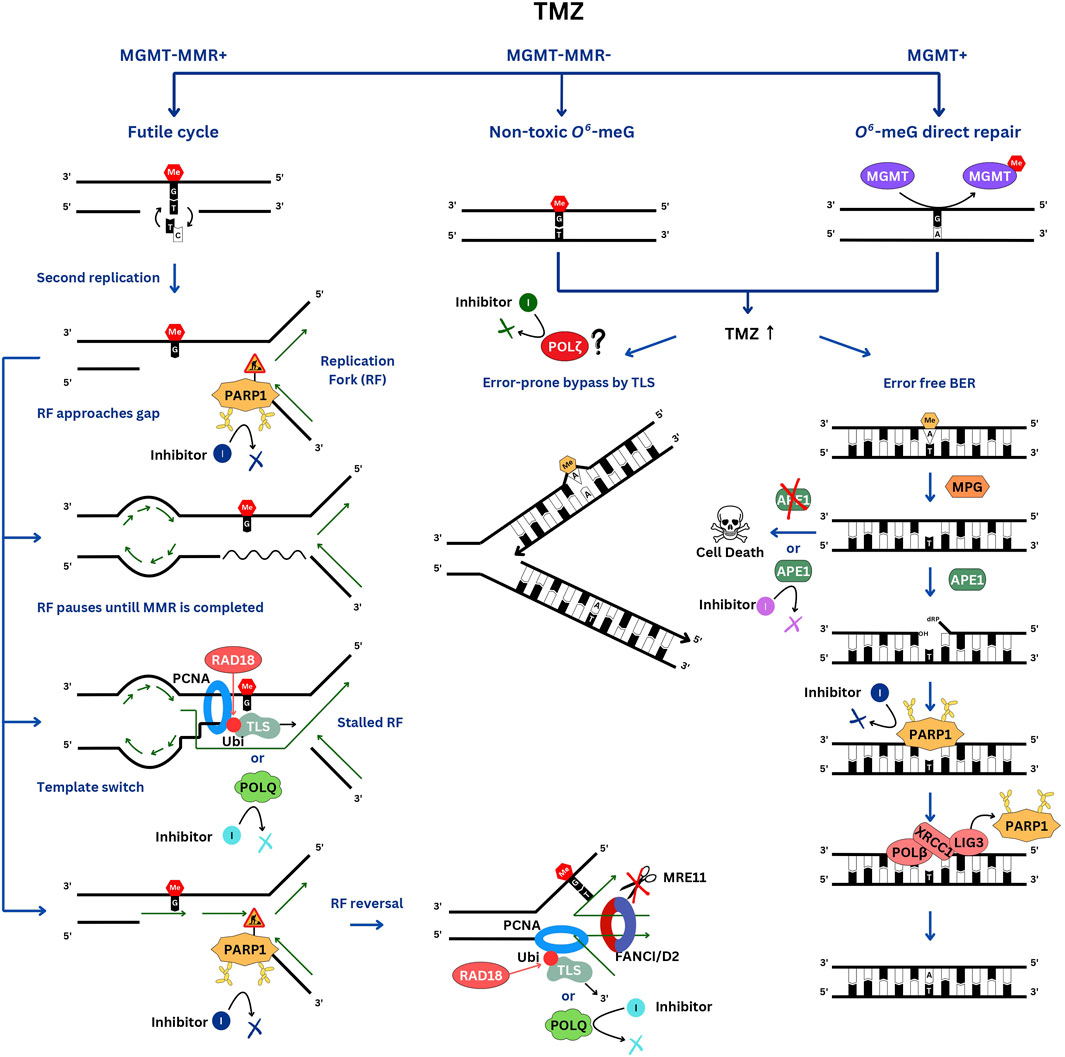
Figure 1. DNA repair pathways involved in TMZ resistance. In the TMZ-sensitive MGMT-negative glioblastomas, the futile cycle of O6-meG/T repair by MMR results in persistent ssDNA gaps, which interfere with the second S phase. When the replication fork encounters a gap, it pauses. PARP1 is activated by the ssDNA breaks and signals to slow down the fork. If the gap is not filled in a timely manner, the fork will have to be stabilized by reversal and protection from nucleases by the FANCI/D2 DNA clamp. Gaps can be skipped via re-priming and filled after the bulk of DNA replication is completed by the TLS polymerases, which are recruited to RAD18-ubiquitinated PCNA, and by template switching. In MGMT-positive cells, O6-meG is directly repaired, whereas in MMR-negative cells, replication of O6-meG/T leads to a C>T substitution without causing a gap. These cells are very resistant to TMZ. At high TMZ concentrations, 3-meA replication blocking lesion accumulates and is repaired by BER or by-passed by TLS. Of note, PARP1 is involved in TMZ resistance in both TMZ-sensitive and resistant cells but via different mechanisms.
Considering the crucial role of ssDNA breaks and gaps in TMZ cytotoxicity, PARP1 might be expected to be involved in their repair through its canonical function in the ssDNA break repair pathway. In addition, PARP1 is involved in the rescue of stalled forks. PARP1 is recruited to replication forks, restrains fork progression under replication stress and facilitates the recruitment of DNA translocases (HLTF, SHPRH, ZRANB3 and SMARCAL1) to reverse damaged forks (Liao et al., 2018; Ho et al., 2022). A reversal of the fork encountering a persistent gap caused by the ongoing futile MMR, might facilitate its bypass. However, PARP1 recruits MRE11 endonuclease (Bryant et al., 2009), which excessively degrades stalled forks in BRCA-mutant cells that are deficient in fork protection. Loss of PARP1 was shown to protect forks from MRE11-mediated degradation and promote cell survival in BRCA1- (Ray Chaudhuri et al., 2016) and BRCA2- (Ding et al., 2016) deficient cells. Thus, based on its various roles in replication fork remodeling, PARP1 might either promote cell survival or exacerbate TMZ toxicity.
There is limited evidence that PARP1 might play a role in the MMR pathway (Liu et al., 2011; Paul et al., 2023). Hypothetically, PARPi might inhibit MMR at an intermediate step, e.g., ssDNA gap, potentially enhancing TMZ sensitivity. In addition, MMR and PARP1 interact at G-quadruplexes (Cong et al., 2024; Isik et al., 2024). The question of PARP1 involvement in MMR remains underexplored and deserves further investigation.
Surprisingly, in the same CRISPR-based screens that revealed the critical role of the FA genes in TMZ/MNNG resistance, PARP1 depletion did not hyper-sensitize MGMT-negative glioblastomas to TMZ (MacLeod et al., 2019) and led to only a minor increase in MNNG sensitivity in RPE1 cells (Olivieri et al., 2020). Nevertheless, the non-trapping PARPi veliparib potentiated TMZ toxicity in MGMT-negative gliomas in vitro and in a mouse xenograft model (Gupta et al., 2014). The question of whether PARP inhibitors indeed increase TMZ sensitivity in MGMT-negative cells treated with low TMZ concentrations by inhibiting PARP1 [and not, e.g., other PARP proteins as was suggested in one recent study (Higuchi et al., 2020)] deserves careful investigation using PARP1 isogenic knockouts. In addition, it remains to be determined if PARP1 trapping is relevant to the action of PARPi in MGMT-negative cells treated with TMZ.
TMZ-treated glioblastomas acquire resistance and recur in almost 100% of cases. MMR inactivation, which abolishes futile cycles of O6-meG/T repair and the associated ssDNA gaps, leads to an extraordinary TMZ tolerance (1 mM TMZ and higher). A hypermutator phenotype associated with MMR deficiency was reported for recurrent gliomas in 60% of TMZ-treated patients (Johnson et al., 2014). Interestingly, TMZ resistance was found to readily build up in cell culture in the MGMT-negative but not in the MGMT-positive glioma lines upon as few as two 3-day cycles of TMZ treatment (Perazzoli et al., 2015). The in vitro acquisition of resistance was accompanied by the downregulation of MMR protein expression. However, the upregulation of MGMT in MGMT-negative lines was observed only rarely or not at all (Perazzoli et al., 2015; Yuan et al., 2020). Interestingly, PARPi prevented the emergence of TMZ resistance in vitro (Yuan et al., 2020). The rapid acquisition of TMZ resistance concomitant with MMR downregulation hints that an epigenetic mechanism might be responsible.
In MMR-negative cells O6-meG is rendered non-toxic since, when left unrepaired it doesn’t affect the replication process and only results in C>T substitutions (Figure 1). Instead, other DNA base adducts accumulate in cells, which become exposed to higher TMZ concentrations. In these cells, TMZ toxicity is mediated mainly by a replication-blocking 3-meA, which is either removed by BER or bypassed by TLS. In addition, 7-meG, although non-toxic and non-mutagenic by itself, also triggers BER and might lead to the accumulation of toxic repair intermediates. BER is a preferred repair pathway for cell survival, since it is error-free and, if completed prior to DNA replication, precludes replication problems. During BER, 3-meA base is first excised by the N-methylpurine DNA glycosylase (MPG, also known as alkyl adenine DNA glycosylase, AAG), resulting in an abasic site, which is then cleaved by the apurinic/apyrimidinic site endonuclease (APE1). PARP1 efficiently binds to BER intermediates with a 5’-deoxyribose phosphate (5’-dRP) end, which are generated by the APE1 cleavage (Cistulli et al., 2004). Interestingly, PARP1 and the PAR-binding chromatin remodeler ALC1/CHD1L also promote chromatin accessibility to APE1 (Hewitt et al., 2021; Verma et al., 2021). Methyl methanesulfonate (MMS) and PARPi sensitivity of the ALC1−/− cells is partially rescued by inhibiting BER via MPG depletion (Hewitt et al., 2021). Therefore, PARP1 activation might be triggered not only by ssDNA breaks but also by the abasic sites resulting from the excision of the alkylated bases by the MPG. However, PARP1 is not essential for BER (Ray Chaudhuri and Nussenzweig, 2017) and in this context, PARP1 trapping is more detrimental to cell survival than inhibiting its catalytic activity. PARPi olaparib induces more PARP1 trapped on chromatin than veliparib, which makes it more efficient in potentiating TMZ cytotoxicity in TMZ-resistant cells (Murai et al., 2014). XRCC1 plays a dual role of releasing PARP1 from chromatin, thus limiting its activity, and recruiting LIG3 and POLB to complete BER. XRCC1−/− PARP1−/− double knockout cell line is more resistant to MMS, which induces mainly 3-meA and 7-meG, than either PARP1−/− or XRCC1−/− single knockouts. Thus, it appears that promoting the release of PARP1 from the DNA is a more important aspect of XRCC1 function than serving as a scaffold for LIG3 and POLB (Demin et al., 2021). A likely explanation for the toxicity associated with XRCC1 deficiency is excessive PARP1 activity leading to NAD+ depletion and cell death by an “energetic catastrophe,” which is prevented in XRCC1−/− PARP1−/− double knockout. Compared to TMZ, MMS induces more 3-meA and 7-meG and less O6-meG, thus mimicking TMZ effects in MGMT-positive or MMR-deficient cells. Remarkably, in the MGMT-positive MCF-7 cell line, XRCC1−/−, PARP1−/− double knockout is also much more resistant to TMZ than XRCC1−/− and PARP−/− singles, similar to what was observed with MMS (Hirota et al., 2022). However, in MGMT-negative TK6 cells, the differences in TMZ sensitivities among the three knockouts are very modest (Hirota et al., 2022), consistent with 3-meA playing only a minor role in inducing MGMT-negative MMR-proficient cell lethality compared to O6-meG.
The response to the experimental methylating agent MMS was the subject of detailed studies in the past. As opposed to TMZ, MMS belongs to the group of SN2 (second-order nucleophilic substitution) methylating agents and leads to 3-meA and 7-meG and less O6-meG. MMS is not used in the contemporary clinical practice. Compared to MMS, TMZ displays superior pharmacokinetic properties, e.g., TMZ is readily absorbed in the intestinal tract, penetrates the blood-brain barrier, and is a prodrug converted into an active intermediate with the clinically suitable kinetics.
Based on early research in yeast, MMS was considered a radiomimetic, inducing dsDNA breaks that are repaired by HR. However, it was later demonstrated that the dsDNA breaks, which had been observed following MMS treatment, were an experimental artefact caused by in vitro handling of the DNA samples and the requirement of HR proteins for MMS resistance in budding yeast most likely stems from the necessity to protect the stalled replication forks (Lundin et al., 2005). Although BER is the optimal repair pathway of 3-meA, upon treatment with high concentrations of MMS, it may become overwhelmed by the number of lesions, and replication forks may start encountering unrepaired 3-meA adducts, causing them to stall. In Xenopus cell-free extracts, it was found that replication of MMS-damaged DNA results in the accumulation of ssDNA gaps behind the fork, which are then either filled in by TLS or repaired via template switching (TS) in a RAD51-dependent manner (Hashimoto et al., 2010). Without fork-protecting RAD51, MMS-induced ssDNA gaps behind replication forks were greatly enhanced and generated via the MRE11-mediated degradation of the native strand in the stalled forks (Hashimoto et al., 2010). This result suggests that fork protection might play an important role in MMS resistance and, by inference, in resistance of MGMT-positive or MMR-deficient lines to TMZ. However, FA pathway deficiencies do not sensitize MGMT-positive glioblastomas to TMZ (MacLeod et al., 2019). Similarly, RAD18-mediated TLS doesn’t appear to play a role in either MGMT-positive or MMR-deficient glioblastoma lines (Cheng et al., 2024). Most likely, the fork encounter with the MMR-generated gaps necessitates a different kind of response compared to a small replication-blocking adduct, like 3-meA.
Similar to the lack of FA pathway requirement, BRCA1 doesn’t appear to be important for fork protection with regard to resistance to 3-meA. Although BRCA1 knockout cells display hypersensitivity to MMS, this is reversed by 53BP1 deficiency (Cong et al., 2021). According to the “traditional” point of view, inactivation of 53BP1 would facilitate resection of dsDNA breaks and HR, implicating dsDNA breaks as the MMS-induced lethal lesion. However, an alternative hypothesis postulates that 53BP1 deficiency restores Okazaki fragment processing (OFP), which is defective in BRCA1 mutants (Cong et al., 2021). In this scenario, MMS induces additional gaps, which synergize with the endogenous ones caused by the OFP defect. MMS also leads to PARP1 activation via BER-induced abasic sites (Hewitt et al., 2021). Importantly, the loss of 53BP1 rescues HR, PARPi hypersensitivity, and embryonic lethality of mice with mutant BRCA1 alleles but not the defects of interstrand crosslink repair or replication fork stabilization (Bunting et al., 2010; Bunting et al., 2012; Li et al., 2016). It is worth noting that the rescue is not complete since the 53BP1−/− double knockout mice with null rather than mutated BRCA1 alleles, while viable, still display an HR defect and PARPi sensitivity (Chen et al., 2020). PARPi resistance of BRCA1/53BP1 double deficient cells can be reversed by depletion of LIG3, which leads to the accumulation of PARPi-induced ssDNA gaps (Paes Dias et al., 2021). Interestingly, ssDNA gaps in PARPi-treated BRCA1-deficient cells appear to be different from the gaps in BRCA1/53BP1 double deficient cells with depleted LIG3, since only the latter but not the former are suppressed by the inhibition of MRE11. This observation highlights the general trend that although all ssDNA gaps might activate PARP1, they differ in their origin and the response they elicit.
In Xenopus extracts (Hashimoto et al., 2010), as well as in yeast (Lopes et al., 2006), MMS has little effect on the ssDNA gaps directly at the forks, which result from the uncoupling of the leading and lagging strand synthesis or uncoupling of the replicative helicase and polymerase. It is also worth noting that only very few reversed forks were observed in MMS-treated yeast (Prado, 2018), presumably because 3-meA lesions are easily bypassed, and fork reversal might be very transient. Nevertheless, knockdown of a DNA translocase with fork-reversing activity, ZRANB3, was reported to sensitize human cells to MMS (Weston et al., 2012), suggesting that fork reversal might still contribute to MMS resistance. Overall, it appears that in the case of 3-meA, ssDNA gap repair behind the fork via TLS and TS might be more important for lesion tolerance compared to fork reversal or fork protection. In yeast, TS is regulated by a Rad5, a bi-functional enzyme with PCNA poly-ubiquitin ligase and DNA translocase activities. Two RAD5 orthologues, HLTF and SHPRH, exist in human cells. MMS treatment was reported to result in the degradation of HLTF and an enhanced SHPRH association with Rad18 and TLS polymerase κ, which is capable of more error-free bypass of MMS-induced lesions than a competing polymerase η (Lin et al., 2011). However, in a more recent study, knockout of HLTF resulted in an increased MMS mutagenesis, while knockout of SHPRH did not change the mutation frequency and increased MMS resistance (Seelinger et al., 2020). Since both studies determined the rate of MMS-induced mutations using MMS-treated plasmids transfected into human cells, the number of analyzed mutations was generally small. With the advent of an affordable whole genome sequencing (WGS), it will be important to re-investigate the error rates of different TLS polymerases bypassing MMS- and TMZ-induced DNA base adducts in vivo and their contribution to TMZ resistance. Interestingly, a recent report uncovered that upregulation of TLS by DNA polymerase ζ suppresses MMS hypersensitivity in yeast deficient for a replisome component Ctf4, which is normally involved in TS (Dolce et al., 2022).
If replication is initiated before BER repair of methylated bases is complete, replication fork-BER conflicts could ensue. It is presumed that an encounter of the replication fork with the BER-induced ssDNA gaps will result in fork collapse and dsDNA breaks, although, to our knowledge, this issue has yet to be investigated. In order to differentiate the direct effects of 3-meA from BER-induced gaps, it will be important to perform experiments with a set of triple knockouts of DNA repair genes in MSH2−/− MPG−/− genetic background. The use of these cell lines in combination with WGS will allow evaluating the role of TLS and TS in TMZ resistance and mutagenesis. Similar experiments performed with a MMS-treated set of yeast double deletion mutants with mag1 (yeast orthologue of MPG) more than a decade ago, suggested that the yeast translocase Rad5, TLS polymerases η and ζ, as well as human polymerases κ, and ι, when expressed in yeast, can all mediate replication through 3-meA (Johnson et al., 2007).
Overall, interfering with the BER pathway appears to be the most efficient route to re-sensitize MMR-deficient cells to TMZ and thus prevent the build-up of TMZ resistance through MMR inactivation. In MMR-deficient cells treated with high TMZ concentrations, the effect of PARPi would be primarily to impede the completion of BER by blocking access to the transient ssDNA breaks, which are formed in the process, and the potential role of PARPi in the inhibition of replication fork rescue is likely to be of secondary importance. PARP inhibition leads to synergistic cytotoxicity with the TMZ treatment in cultured MMR-deficient cells, which is even more pronounced than in MMR-proficient cells (Gupta et al., 2014). However, in vivo, the PARPi/TMZ combination therapy faces problems with not achieving an effective concentration of PARPi in the brain (Gupta et al., 2014) and due to hematological toxicity, which ensues when the drugs are used together (Gueble et al., 2022). A new generation of PARPi with improved blood-brain barrier penetrance, high PARP1 inhibition potency, enhanced PARP1 vs. PARP2 selectivity, and reduced toxicity to normal cells are currently being tested, and might overcome this limitation (Gueble et al., 2022).
Since BER is activated in response to 3-meA and 7-meG, blocking it at an intermediate step would result in toxic intermediates, such as abasic sites or ssDNA breaks. The second step of BER, the conversion of the abasic site into ssDNA break, which is catalyzed by the APE1 endonuclease, appears to represent an Achilles’ heel of this repair pathway. MMS treatment of yeast deficient in APE1 orthologues, APN1 and APN2, leads to dsDNA breaks even without DNA replication (Ma et al., 2011). A combination of TMZ with an APE1 inhibitor should result in the accumulation of 3-meA and 7-meG-induced abasic sites in tumor clones that managed to overcome O6-meG toxicity via MMR inactivation. Elimination of these clones would prevent the build-up of TMZ resistance. Indeed, in an exceptional case, TMZ treatment cured a patient of glioblastoma, carrying an APE1 gene rendered inactive by a translocation (Wheeler et al., 2021). It is worth noting that APE1 inhibition would be equally expected to sensitize MGMT-positive gliomas to TMZ.
Several substances were reported to act as APE1 inhibitors. Methoxyamine forms a stable adduct with an abasic site and renders it refractory to cleavage by APE1. It enhances TMZ cytotoxicity in MGMT-positive but not in MGMT-negative glioblastomas in vitro, but the required concentration (20 mM) is very high (Montaldi and Sakamoto-Hojo, 2013), and clinical studies were unsuccessful (Malfatti et al., 2023). Lucanthone was initially developed as an anti-schistosome agent and a topoisomerase II inhibitor but was later shown to inhibit recombinant APE1 protein in vitro (Luo and Kelley, 2004; Naidu et al., 2011) and at micromolar concentrations sensitizes cultured cells to TMZ (Luo and Kelley, 2004). Lucanthone is able to cross the blood-brain barrier and has been tested as an adjuvant for brain tumor radiotherapy (Del Rowe et al., 1999). However, it likely has several targets besides APE1 and was reported to inhibit autophagy in gliomas (Radin et al., 2022; Radin et al., 2024). Thus, the mechanism of lucanthone action remains uncertain.
High-throughput screens for the inhibitors of the APE1 abasic site endonuclease activity identified several compounds [reviewed in Malfatti et al. (2023)]. Two of them (Madhusudan et al., 2005; Rai et al., 2012) became commercially available, however, recent studies cast doubt on their specificity and efficacy (Xue and Demple, 2022; Pidugu et al., 2023). As of today, none of the compounds, which were isolated in the screens, advanced to clinical trials (Malfatti et al., 2023). The most recent effort using high-throughput X-ray crystallography screen of the fragment library binding to APE1 crystals resulted in the development of two lead compounds targeting the APE1 endonuclease domain with IC50 values below the micromolar range (Das et al., 2023). This new approach might facilitate the development of novel APE1 inhibitors to be tested in clinical trials.
As of today, our knowledge of TMZ response is mostly limited to BER in TMZ-resistant MMR-deficient or MGMT-positive cells. The mechanism of cell death triggered by “futile MMR cycles” in primary TMZ-sensitive glioblastomas remains largely enigmatic. While it appears that ssDNA gaps are generated and TLS is involved in both TMZ-sensitive and TMZ-resistant cells, the identity of a TLS polymerase is likely to be different since RAD18-dependent PCNA ubiquitination plays an important role in the former but not in the latter (Cheng et al., 2024). It is possible but remains to be tested if MMR-generated gaps are filled in by a Y-family polymerase (η, κ or ι) while 3-meA-induced gaps are dealt with by the polymerase ζ, which can be recruited to chromatin in a ubi-PCNA-independent manner via REV1. A recently developed REV1 inhibitor, JH-RE-06, targets REV1/polymerase zeta complex formation (Wojtaszek et al., 2019). Given that JH-RE-06 synergizes with PARPi to kill BRCA-deficient cells (Taglialatela et al., 2021), it might be worth testing in combination with TMZ. Overall, finding new approaches to re-sensitize TMZ-resistant cells might be a better strategy than to hyper-sensitize primary TMZ-sensitive glioblastomas, which already withstand only very low TMZ concentrations. If inactivation of MMR doesn’t increase the odds of tumor survival in combination therapy, because cells are rendered hypersensitive to 3-meA by the second drug, the problem of TMZ resistance may be solved. In this regard, targeting APE1 holds a great promise. Alternatively, since WRN helicase plays an essential role in MMR-deficient cells, treatment with a WRN inhibitor (Baltgalvis et al., 2024) might eliminate the emerging TMZ-resistant MMR-deficient glioblastoma cells.
There are certain parallels between BRCA deficiencies and TMZ treatment. Both are currently believed to induce ssDNA gaps, which underlie sensitivity to PARPi. While the origin of the gaps is obviously different, certain features of the cellular response to the gaps might be universal, such as PARP1 signaling or post-replicative fill-in by TLS or POLQ. Thus, TLS and POLQ inhibitors, which were reported to synergize with PARPi in targeting BRCA-deficient cells (Taglialatela et al., 2021; Zatreanu et al., 2021), might potentiate TMZ as well, especially as a three-drug combination with PARPi. Future studies will shed light on the feasibility of this approach.
AM: Visualization, Writing–review and editing. AG: Writing–review and editing. DI: Conceptualization, Supervision, Writing–original draft, Writing–review and editing.
The authors declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Korean Institute for Basic Science (IBS-R022-A2-2023, IBS-R022-D1-2024).
We are grateful to Orlando Scharer for the many suggestions and for carefully reading the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ait Saada, A., Guo, W., Costa, A. B., Yang, J., Wang, J., and Lobachev, K. S. (2023). Widely spaced and divergent inverted repeats become a potent source of chromosomal rearrangements in long single-stranded DNA regions. Nucleic Acids Res. 51, 3722–3734. doi:10.1093/nar/gkad153
Baltgalvis, K. A., Lamb, K. N., Symons, K. T., Wu, C. C., Hoffman, M. A., Snead, A. N., et al. (2024). Chemoproteomic discovery of a covalent allosteric inhibitor of WRN helicase. Nature 629, 435–442. doi:10.1038/s41586-024-07318-y
Belan, O., Sebald, M., Adamowicz, M., Anand, R., Vancevska, A., Neves, J., et al. (2022). POLQ seals post-replicative ssDNA gaps to maintain genome stability in BRCA-deficient cancer cells. Mol. Cell 82, 4664–4680.e9. doi:10.1016/j.molcel.2022.11.008
Bryant, H. E., Petermann, E., Schultz, N., Jemth, A. S., Loseva, O., Issaeva, N., et al. (2009). PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 28, 2601–2615. doi:10.1038/emboj.2009.206
Bryant, H. E., Schultz, N., Thomas, H. D., Parker, K. M., Flower, D., Lopez, E., et al. (2005). Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 434, 913–917. doi:10.1038/nature03443
Bunting, S. F., Callen, E., Kozak, M. L., Kim, J. M., Wong, N., Lopez-Contreras, A. J., et al. (2012). BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 46, 125–135. doi:10.1016/j.molcel.2012.02.015
Bunting, S. F., Callen, E., Wong, N., Chen, H. T., Polato, F., Gunn, A., et al. (2010). 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141, 243–254. doi:10.1016/j.cell.2010.03.012
Chan, K. L., Palmai-Pallag, T., Ying, S., and Hickson, I. D. (2009). Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760. doi:10.1038/ncb1882
Chen, J., Li, P., Song, L., Bai, L., Huen, M. S. Y., Liu, Y., et al. (2020). 53BP1 loss rescues embryonic lethality but not genomic instability of BRCA1 total knockout mice. Cell Death Differ. 27, 2552–2567. doi:10.1038/s41418-020-0521-4
Cheng, X., An, J., Lou, J., Gu, Q., Ding, W., Droby, G. N., et al. (2024). Trans-lesion synthesis and mismatch repair pathway crosstalk defines chemoresistance and hypermutation mechanisms in glioblastoma. Nat. Commun. 15, 1957. doi:10.1038/s41467-024-45979-5
Cistulli, C., Lavrik, O. I., Prasad, R., Hou, E., and Wilson, S. H. (2004). AP endonuclease and poly(ADP-ribose) polymerase-1 interact with the same base excision repair intermediate. DNA Repair (Amst) 3, 581–591. doi:10.1016/j.dnarep.2003.09.012
Cong, K., and Cantor, S. B. (2022). Exploiting replication gaps for cancer therapy. Mol. Cell 82, 2363–2369. doi:10.1016/j.molcel.2022.04.023
Cong, K., MacGilvary, N., Lee, S., MacLeod, S. G., Calvo, J., Peng, M., et al. (2024). FANCJ promotes PARP1 activity during DNA replication that is essential in BRCA1 deficient cells. Nat. Commun. 15, 2599. doi:10.1038/s41467-024-46824-5
Cong, K., Peng, M., Kousholt, A. N., Lee, W. T. C., Lee, S., Nayak, S., et al. (2021). Replication gaps are a key determinant of PARP inhibitor synthetic lethality with BRCA deficiency. Mol. Cell. 81, 3227. doi:10.1016/j.molcel.2021.07.015
Cropper, J. D., Alimbetov, D. S., Brown, K. T. G., Likhotvorik, R. I., Robles, A. J., Guerra, J. T., et al. (2022). PARP1-MGMT complex underpins pathway crosstalk in O(6)-methylguanine repair. J. Hematol. Oncol. 15, 146. doi:10.1186/s13045-022-01367-4
Das, D., Duncton, M. A. J., Georgiadis, T. M., Pellicena, P., Clark, J., Sobol, R. W., et al. (2023). A new drug discovery platform: application to DNA polymerase eta and apurinic/apyrimidinic endonuclease 1. Int. J. Mol. Sci. 24, 16637. doi:10.3390/ijms242316637
Del Rowe, J. D., Bello, J., Mitnick, R., Sood, B., Filippi, C., Moran, J., et al. (1999). Accelerated regression of brain metastases in patients receiving whole brain radiation and the topoisomerase II inhibitor, lucanthone. Int. J. Radiat. Oncol. Biol. Phys. 43, 89–93. doi:10.1016/s0360-3016(98)00374-5
Demin, A. A., Hirota, K., Tsuda, M., Adamowicz, M., Hailstone, R., Brazina, J., et al. (2021). XRCC1 prevents toxic PARP1 trapping during DNA base excision repair. Mol. Cell 81, 3018–3030.e5. doi:10.1016/j.molcel.2021.05.009
Ding, X., Ray Chaudhuri, A., Callen, E., Pang, Y., Biswas, K., Klarmann, K. D., et al. (2016). Synthetic viability by BRCA2 and PARP1/ARTD1 deficiencies. Nat. Commun. 7, 12425. doi:10.1038/ncomms12425
Dolce, V., Dusi, S., Giannattasio, M., Joseph, C. R., Fumasoni, M., and Branzei, D. (2022). Parental histone deposition on the replicated strands promotes error-free DNA damage tolerance and regulates drug resistance. Genes Dev. 36, 167–179. doi:10.1101/gad.349207.121
Fanucci, K., Pilat, M. J., Shyr, D., Shyr, Y., Boerner, S., Li, J., et al. (2023). Multicenter phase II trial of the PARP inhibitor olaparib in recurrent IDH1- and IDH2-mutant glioma. Cancer Res. Commun. 3, 192–201. doi:10.1158/2767-9764.CRC-22-0436
Farmer, H., McCabe, N., Lord, C. J., Tutt, A. N., Johnson, D. A., Richardson, T. B., et al. (2005). Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 434, 917–921. doi:10.1038/nature03445
Gueble, S. E., Vasquez, J. C., and Bindra, R. S. (2022). The role of PARP inhibitors in patients with primary malignant central nervous system tumors. Curr. Treat. Options Oncol. 23, 1566–1589. doi:10.1007/s11864-022-01024-5
Gupta, S. K., Mladek, A. C., Carlson, B. L., Boakye-Agyeman, F., Bakken, K. K., Kizilbash, S. H., et al. (2014). Discordant in vitro and in vivo chemopotentiating effects of the PARP inhibitor veliparib in temozolomide-sensitive versus -resistant glioblastoma multiforme xenografts. Clin. Cancer Res. 20, 3730–3741. doi:10.1158/1078-0432.CCR-13-3446
Hanzlikova, H., Kalasova, I., Demin, A. A., Pennicott, L. E., Cihlarova, Z., and Caldecott, K. W. (2018). The importance of poly(ADP-ribose) polymerase as a sensor of unligated Okazaki fragments during DNA replication. Mol. Cell 71, 319–331. doi:10.1016/j.molcel.2018.06.004
Hashimoto, Y., Ray Chaudhuri, A., Lopes, M., and Costanzo, V. (2010). Rad51 protects nascent DNA from Mre11-dependent degradation and promotes continuous DNA synthesis. Nat. Struct. Mol. Biol. 17, 1305–1311. doi:10.1038/nsmb.1927
Hewitt, G., Borel, V., Segura-Bayona, S., Takaki, T., Ruis, P., Bellelli, R., et al. (2021). Defective ALC1 nucleosome remodeling confers PARPi sensitization and synthetic lethality with HRD. Mol. Cell 81, 767–783.e11. doi:10.1016/j.molcel.2020.12.006
Higuchi, F., Nagashima, H., Ning, J., Koerner, M. V. A., Wakimoto, H., and Cahill, D. P. (2020). Restoration of temozolomide sensitivity by PARP inhibitors in mismatch repair deficient glioblastoma is independent of base excision repair. Clin. Cancer Res. 26, 1690–1699. doi:10.1158/1078-0432.CCR-19-2000
Hirota, K., Ooka, M., Shimizu, N., Yamada, K., Tsuda, M., Ibrahim, M. A., et al. (2022). XRCC1 counteracts poly(ADP ribose)polymerase (PARP) poisons, olaparib and talazoparib, and a clinical alkylating agent, temozolomide, by promoting the removal of trapped PARP1 from broken DNA. Genes Cells 27, 331–344. doi:10.1111/gtc.12929
Ho, Y. C., Ku, C. S., Tsai, S. S., Shiu, J. L., Jiang, Y. Z., Miriam, H. E., et al. (2022). PARP1 recruits DNA translocases to restrain DNA replication and facilitate DNA repair. PLoS Genet. 18, e1010545. doi:10.1371/journal.pgen.1010545
Isik, E., Shukla, K., Pospisilova, M., Konig, C., Andrs, M., Rao, S., et al. (2024). MutSβ-MutLβ-FANCJ axis mediates the restart of DNA replication after fork stalling at cotranscriptional G4/R-loops. Sci. Adv. 10, eadk2685. doi:10.1126/sciadv.adk2685
Johnson, B. E., Mazor, T., Hong, C., Barnes, M., Aihara, K., McLean, C. Y., et al. (2014). Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193. doi:10.1126/science.1239947
Johnson, R. E., Yu, S. L., Prakash, S., and Prakash, L. (2007). A role for yeast and human translesion synthesis DNA polymerases in promoting replication through 3-methyl adenine. Mol. Cell Biol. 27, 7198–7205. doi:10.1128/MCB.01079-07
Kaina, B., and Christmann, M. (2019). DNA repair in personalized brain cancer therapy with temozolomide and nitrosoureas. DNA Repair (Amst). 78, 128–141. doi:10.1016/j.dnarep.2019.04.007
Li, M., Cole, F., Patel, D. S., Misenko, S. M., Her, J., Malhowski, A., et al. (2016). 53BP1 ablation rescues genomic instability in mice expressing ‘RING-less’ BRCA1. EMBO Rep. 17, 1532–1541. doi:10.15252/embr.201642497
Liao, H., Ji, F., Helleday, T., and Ying, S. (2018). Mechanisms for stalled replication fork stabilization: new targets for synthetic lethality strategies in cancer treatments. EMBO Rep. 19, e46263. doi:10.15252/embr.201846263
Lin, J. R., Zeman, M. K., Chen, J. Y., Yee, M. C., and Cimprich, K. A. (2011). SHPRH and HLTF act in a damage-specific manner to coordinate different forms of postreplication repair and prevent mutagenesis. Mol. Cell 42, 237–249. doi:10.1016/j.molcel.2011.02.026
Liu, Y., Kadyrov, F. A., and Modrich, P. (2011). PARP-1 enhances the mismatch-dependence of 5’-directed excision in human mismatch repair in vitro. DNA Repair (Amst). 10, 1145–1153. doi:10.1016/j.dnarep.2011.08.012
Lopes, M., Foiani, M., and Sogo, J. M. (2006). Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell 21, 15–27. doi:10.1016/j.molcel.2005.11.015
Lundin, C., North, M., Erixon, K., Walters, K., Jenssen, D., Goldman, A. S., et al. (2005). Methyl methanesulfonate (MMS) produces heat-labile DNA damage but no detectable in vivo DNA double-strand breaks. Nucleic Acids Res. 33, 3799–3811. doi:10.1093/nar/gki681
Luo, M., and Kelley, M. R. (2004). Inhibition of the human apurinic/apyrimidinic endonuclease (APE1) repair activity and sensitization of breast cancer cells to DNA alkylating agents with lucanthone. Anticancer Res. 24, 2127–2134.
Ma, W., Westmoreland, J. W., Gordenin, D. A., and Resnick, M. A. (2011). Alkylation base damage is converted into repairable double-strand breaks and complex intermediates in G2 cells lacking AP endonuclease. PLoS Genet. 7, e1002059. doi:10.1371/journal.pgen.1002059
MacLeod, G., Bozek, D. A., Rajakulendran, N., Monteiro, V., Ahmadi, M., Steinhart, Z., et al. (2019). Genome-wide CRISPR-cas9 screens expose genetic vulnerabilities and mechanisms of temozolomide sensitivity in glioblastoma stem cells. Cell Rep. 27, 971–986. doi:10.1016/j.celrep.2019.03.047
Madhusudan, S., Smart, F., Shrimpton, P., Parsons, J. L., Gardiner, L., Houlbrook, S., et al. (2005). Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 33, 4711–4724. doi:10.1093/nar/gki781
Malfatti, M. C., Bellina, A., Antoniali, G., and Tell, G. (2023). Revisiting two decades of research focused on targeting APE1 for cancer therapy: the pros and cons. Cells 12, 1895. doi:10.3390/cells12141895
Mann, A., Ramirez-Otero, M. A., De Antoni, A., Hanthi, Y. W., Sannino, V., Baldi, G., et al. (2022). POLtheta prevents MRE11-NBS1-CtIP-dependent fork breakage in the absence of BRCA2/RAD51 by filling lagging-strand gaps. Mol. Cell 82, 4218–4231 e4218. doi:10.1016/j.molcel.2022.09.013
Montaldi, A. P., and Sakamoto-Hojo, E. T. (2013). Methoxyamine sensitizes the resistant glioblastoma T98G cell line to the alkylating agent temozolomide. Clin. Exp. Med. 13, 279–288. doi:10.1007/s10238-012-0201-x
Murai, J., Zhang, Y., Morris, J., Ji, J., Takeda, S., Doroshow, J. H., et al. (2014). Rationale for poly(ADP-ribose) polymerase (PARP) inhibitors in combination therapy with camptothecins or temozolomide based on PARP trapping versus catalytic inhibition. J. Pharmacol. Exp. Ther. 349, 408–416. doi:10.1124/jpet.113.210146
Naidu, M. D., Agarwal, R., Pena, L. A., Cunha, L., Mezei, M., Shen, M., et al. (2011). Lucanthone and its derivative hycanthone inhibit apurinic endonuclease-1 (APE1) by direct protein binding. PLoS One 6, e23679. doi:10.1371/journal.pone.0023679
Naim, V., and Rosselli, F. (2009). The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 11, 761–768. doi:10.1038/ncb1883
Naim, V., Wilhelm, T., Debatisse, M., and Rosselli, F. (2013). ERCC1 and MUS81-EME1 promote sister chromatid separation by processing late replication intermediates at common fragile sites during mitosis. Nat. Cell Biol. 15, 1008–1015. doi:10.1038/ncb2793
Nayak, S., Calvo, J. A., Cong, K., Peng, M., Berthiaume, E., Jackson, J., et al. (2020). Inhibition of the translesion synthesis polymerase REV1 exploits replication gaps as a cancer vulnerability. Sci. Adv. 6, eaaz7808. doi:10.1126/sciadv.aaz7808
Olivieri, M., Cho, T., Alvarez-Quilon, A., Li, K., Schellenberg, M. J., Zimmermann, M., et al. (2020). A genetic map of the response to DNA damage in human cells. Cell 182, 481–496. doi:10.1016/j.cell.2020.05.040
Ostermann, S., Csajka, C., Buclin, T., Leyvraz, S., Lejeune, F., Decosterd, L. A., et al. (2004). Plasma and cerebrospinal fluid population pharmacokinetics of temozolomide in malignant glioma patients. Clin. Cancer Res. 10, 3728–3736. doi:10.1158/1078-0432.CCR-03-0807
Paes Dias, M., Tripathi, V., van der Heijden, I., Cong, K., Manolika, E. M., Bhin, J., et al. (2021). Loss of nuclear DNA ligase III reverts PARP inhibitor resistance in BRCA1/53BP1 double-deficient cells by exposing ssDNA gaps. Mol. Cell 81, 4692–4708.e9. doi:10.1016/j.molcel.2021.09.005
Panzarino, N. J., Krais, J. J., Cong, K., Peng, M., Mosqueda, M., Nayak, S. U., et al. (2021). Replication gaps underlie BRCA deficiency and therapy response. Cancer Res. 81, 1388–1397. doi:10.1158/0008-5472.CAN-20-1602
Paul, S., Chatterjee, S., Sinha, S., Dash, S. R., Pradhan, R., Das, B., et al. (2023). Veliparib (ABT-888), a PARP inhibitor potentiates the cytotoxic activity of 5-fluorouracil by inhibiting MMR pathway through deregulation of MSH6 in colorectal cancer stem cells. Expert Opin. Ther. Targets 27, 999–1015. doi:10.1080/14728222.2023.2266572
Perazzoli, G., Prados, J., Ortiz, R., Caba, O., Cabeza, L., Berdasco, M., et al. (2015). Temozolomide resistance in glioblastoma cell lines: implication of MGMT, MMR, P-glycoprotein and CD133 expression. PLoS One 10, e0140131. doi:10.1371/journal.pone.0140131
Petropoulos, M., Karamichali, A., Rossetti, G. G., Freudenmann, A., Iacovino, L. G., Dionellis, V. S., et al. (2024). Transcription-replication conflicts underlie sensitivity to PARP inhibitors. Nature 628, 433–441. doi:10.1038/s41586-024-07217-2
Pidugu, L. S., Servius, H. W., Sevdalis, S. E., Cook, M. E., Varney, K. M., Pozharski, E., et al. (2023). Characterizing inhibitors of human AP endonuclease 1. PLoS One 18, e0280526. doi:10.1371/journal.pone.0280526
Polleys, E. J., Del Priore, I., Haber, J. E., and Freudenreich, C. H. (2023). Structure-forming CAG/CTG repeats interfere with gap repair to cause repeat expansions and chromosome breaks. Nat. Commun. 14, 2469. doi:10.1038/s41467-023-37901-2
Pommier, Y., O’Connor, M. J., and de Bono, J. (2016). Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 8, 362ps317. doi:10.1126/scitranslmed.aaf9246
Prado, F. (2018). Homologous recombination: to fork and beyond. Genes (Basel) 9, 603. doi:10.3390/genes9120603
Radin, D. P., Shifman, S., Outhwaite, I. R., Sharma, A., Bases, R., Seeliger, M. A., et al. (2024). Lucanthone, a potential PPT1 inhibitor, perturbs stemness, reduces tumor microtube formation, and slows the growth of temozolomide-resistant gliomas in vivo. J. Pharmacol. Exp. Ther. 389, 51–60. doi:10.1124/jpet.123.002021
Radin, D. P., Smith, G., Moushiaveshi, V., Wolf, A., Bases, R., and Tsirka, S. E. (2022). Lucanthone targets lysosomes to perturb glioma proliferation, chemoresistance and stemness, and slows tumor growth in vivo. Front. Oncol. 12, 852940. doi:10.3389/fonc.2022.852940
Rai, G., Vyjayanti, V. N., Dorjsuren, D., Simeonov, A., Jadhav, A., Wilson, D. M., et al. (2012). Synthesis, biological evaluation, and structure-activity relationships of a novel class of apurinic/apyrimidinic endonuclease 1 inhibitors. J. Med. Chem. 55, 3101–3112. doi:10.1021/jm201537d
Ray Chaudhuri, A., Callen, E., Ding, X., Gogola, E., Duarte, A. A., Lee, J. E., et al. (2016). Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 535, 382–387. doi:10.1038/nature18325
Ray Chaudhuri, A., and Nussenzweig, A. (2017). The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 18, 610–621. doi:10.1038/nrm.2017.53
Schlacher, K., Wu, H., and Jasin, M. (2012). A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116. doi:10.1016/j.ccr.2012.05.015
Schrempf, A., Bernardo, S., Arasa Verge, E. A., Ramirez Otero, M. A., Wilson, J., Kirchhofer, D., et al. (2022). POLθ processes ssDNA gaps and promotes replication fork progression in BRCA1-deficient cells. Cell Rep. 41, 111716. doi:10.1016/j.celrep.2022.111716
Schvartzman, J. M., Forsyth, G., Walch, H., Chatila, W., Taglialatela, A., Lee, B. J., et al. (2023). Oncogenic IDH mutations increase heterochromatin-related replication stress without impacting homologous recombination. Mol. Cell 83, 2347–2356.e8. doi:10.1016/j.molcel.2023.05.026
Seelinger, M., Sogaard, C. K., and Otterlei, M. (2020). The human RAD5 homologs, HLTF and SHPRH, have separate functions in DNA damage tolerance dependent on the DNA lesion type. Biomolecules 10, 463. doi:10.3390/biom10030463
Sharma, N., Mallela, A. N., Shi, D. D., Tang, L. W., Abou-Al-Shaar, H., Gersey, Z. C., et al. (2023). Isocitrate dehydrogenase mutations in gliomas: a review of current understanding and trials. Neurooncol Adv. 5, vdad053. doi:10.1093/noajnl/vdad053
Simoneau, A., Xiong, R., and Zou, L. (2021). The trans cell cycle effects of PARP inhibitors underlie their selectivity toward BRCA1/2-deficient cells. Genes Dev. 35, 1271–1289. doi:10.1101/gad.348479.121
Stojic, L., Mojas, N., Cejka, P., Di Pietro, M., Ferrari, S., Marra, G., et al. (2004). Mismatch repair-dependent G2 checkpoint induced by low doses of SN1 type methylating agents requires the ATR kinase. Genes Dev. 18, 1331–1344. doi:10.1101/gad.294404
Sulkowski, P. L., Corso, C. D., Robinson, N. D., Scanlon, S. E., Purshouse, K. R., Bai, H., et al. (2017). 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci. Transl. Med. 9, eaal2463. doi:10.1126/scitranslmed.aal2463
Sulkowski, P. L., Oeck, S., Dow, J., Economos, N. G., Mirfakhraie, L., Liu, Y., et al. (2020). Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 582, 586–591. doi:10.1038/s41586-020-2363-0
Taglialatela, A., Leuzzi, G., Sannino, V., Cuella-Martin, R., Huang, J. W., Wu-Baer, F., et al. (2021). REV1-Polzeta maintains the viability of homologous recombination-deficient cancer cells through mutagenic repair of PRIMPOL-dependent ssDNA gaps. Mol. Cell 81, 4008–4025. doi:10.1016/j.molcel.2021.08.016
Vaitsiankova, A., Burdova, K., Sobol, M., Gautam, A., Benada, O., Hanzlikova, H., et al. (2022). PARP inhibition impedes the maturation of nascent DNA strands during DNA replication. Nat. Struct. Mol. Biol. 29, 329–338. doi:10.1038/s41594-022-00747-1
Verma, P., Zhou, Y., Cao, Z., Deraska, P. V., Deb, M., Arai, E., et al. (2021). ALC1 links chromatin accessibility to PARP inhibitor response in homologous recombination-deficient cells. Nat. Cell Biol. 23, 160–171. doi:10.1038/s41556-020-00624-3
Weston, R., Peeters, H., and Ahel, D. (2012). ZRANB3 is a structure-specific ATP-dependent endonuclease involved in replication stress response. Genes Dev. 26, 1558–1572. doi:10.1101/gad.193516.112
Whalen, J. M., and Cantor, S. B. (2023). Unveiling the toxicity of single-stranded DNA gaps through a yeast model. Nat. Struct. Mol. Biol. 30, 870–872. doi:10.1038/s41594-023-01031-6
Wheeler, D. A., Takebe, N., Hinoue, T., Hoadley, K. A., Cardenas, M. F., Hamilton, A. M., et al. (2021). Molecular features of cancers exhibiting exceptional responses to treatment. Cancer Cell 39, 38–53.e7. doi:10.1016/j.ccell.2020.10.015
Wojtaszek, J. L., Chatterjee, N., Najeeb, J., Ramos, A., Lee, M., Bian, K., et al. (2019). A small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell 178, 152–159. doi:10.1016/j.cell.2019.05.028
Wu, S., Li, X., Gao, F., de Groot, J. F., Koul, D., and Yung, W. K. A. (2021). PARP-mediated PARylation of MGMT is critical to promote repair of temozolomide-induced O6-methylguanine DNA damage in glioblastoma. Neuro Oncol. 23, 920–931. doi:10.1093/neuonc/noab003
Xue, Z., and Demple, B. (2022). Knockout and inhibition of Ape1: roles of Ape1 in base excision DNA repair and modulation of gene expression. Antioxidants (Basel) 11, 1817. doi:10.3390/antiox11091817
Yoshioka, K., Yoshioka, Y., and Hsieh, P. (2006). ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts. Mol. Cell 22, 501–510. doi:10.1016/j.molcel.2006.04.023
Yuan, A. L., Meode, M., Tan, M., Maxwell, L., Bering, E. A., Pedersen, H., et al. (2020). PARP inhibition suppresses the emergence of temozolomide resistance in a model system. J. Neurooncol 148, 463–472. doi:10.1007/s11060-020-03561-1
Zandarashvili, L., Langelier, M. F., Velagapudi, U. K., Hancock, M. A., Steffen, J. D., Billur, R., et al. (2020). Structural basis for allosteric PARP-1 retention on DNA breaks. Science 368, eaax6367. doi:10.1126/science.aax6367
Keywords: temozolomide, chemotherapy, glioblastoma, PARP inhibitor, ssDNA gaps, mismatch repair
Citation: Miramova A, Gartner A and Ivanov D (2024) How to sensitize glioblastomas to temozolomide chemotherapy: a gap-centered view. Front. Cell Dev. Biol. 12:1436563. doi: 10.3389/fcell.2024.1436563
Received: 22 May 2024; Accepted: 17 June 2024;
Published: 01 July 2024.
Edited by:
Andrew J. Deans, University of Melbourne, AustraliaReviewed by:
Alessandro A. Sartori, University of Zurich, SwitzerlandCopyright © 2024 Miramova, Gartner and Ivanov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dmitri Ivanov, cGV0ZXJzYnVyZzAxQGlicy5yZS5rcg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.