 Xueting Wang1,2†
Xueting Wang1,2† Lu Liu3†Mengke Chen3
Lu Liu3†Mengke Chen3 Yun Quan3Jiaxin Zhang3
Yun Quan3Jiaxin Zhang3 Huiqiang Lou3Yisui Xia3
Huiqiang Lou3Yisui Xia3 Hongxiang Chen1,2*Wenya Hou3*
Hongxiang Chen1,2*Wenya Hou3*- 1Department of Dermatology, Huazhong University of Science and Technology Union Shenzhen Hospital, Shenzhen, China
- 2Guangdong Key Laboratory for Biomedical Measurements and Ultrasound Imaging, Nation Regional Key Technology Engineering Laboratory for Medical Ultrasound, School of Biomedical Engineering, Shenzhen University Medical School, Shenzhen, China
- 3Guangdong Key Laboratory for Genome Stability and Disease Prevention, Shenzhen University General Hospital and Medical School, Shenzhen, China
Mcm10 plays an essential role in the activation of replicative helicase CMG through the cell cycle-regulated interaction with the prototype MCM double hexamer in Saccharomyces cerevisiae. In this study, we reported that Mcm10 is phosphorylated by S-phase cyclin-dependent kinases (S-CDKs) at S66, which enhances Mcm10–-MCM association during the S phase. S66A single mutation or even deletion of whole N-terminus (a.a. 1–128) only causes mild growth defects. Nevertheless, S66 becomes indispensable in the absence of the Mcm10 C-terminus ((a.a. 463–571), the major MCM-binding domain. Using a two-degron strategy to efficiently deplete Mcm10, we show that mcm10-S66AΔC has a severe defect in proceeding into the S phase. Notably, both lethality and S-phase deficiency can be rescued by artificially tethering mcm10-S66AΔC to MCM. These findings illustrate how the Mcm10–MCM association is regulated as a crucial event in DNA replication initiation.
Introduction
DNA replication, the faithful duplication of genetic material, is a fundamental process crucial for the propagation of life. This intricate mechanism involves the coordinated action of numerous proteins orchestrating the unwinding of the double helix, the synthesis of new DNA strands, and the faithful transmission of genetic information to daughter cells (O’Donnell et al., 2013). Among the plethora of proteins involved, Mcm10 stands out for its indispensable but less understood role in the complex choreography of DNA replication. The significance of Mcm10 lies in its ability to act as a versatile coordinator, regulating various stages of DNA replication, including initiation (Douglas et al., 2018; Langston et al., 2023; Henrikus et al., 2024), progression (Langston et al., 2017; Looke et al., 2017), and termination (Campos et al., 2023), as well as replication-coupled nucleosome assembly (Zhao et al., 2023).
Mcm10 is among the minimal set of essential firing factors for reconstituted DNA synthesis in vitro (Yeeles et al., 2015), and it has been demonstrated to be crucial for the maturation of the helicase complex, which unwinds the DNA double helix, allowing the initiation of DNA synthesis in Saccharomyces cerevisiae (Kanke et al., 2012; van Deursen et al., 2012; Watase et al., 2012). Mcm10 binds multiple subunits of the Cdc45-MCM-GINS (CMG) holo-helicase, which is formed by the six Mcm2–7 ATPases, the Cdc45 protein, and the GINS complex (Bell and Labib, 2016; Costa and Diffley, 2022), thereby stimulating its helicase activity or bypassing blocks on lagging strand DNA during replication elongation (Langston et al., 2017; Looke et al., 2017). Mcm10 can also promote the progression of stalled forks, including those under conditions of topological stress during replication termination (Campos et al., 2023). On the other hand, Mcm10 also possesses strand annealing activity that can prevent fork regression caused by enzymes triggering fork reversal (Mayle et al., 2019).
During replication initiation, Mcm10 facilitates the remodeling of the MCM double hexamers (DHs) and subsequently triggers the activation of the assembled CMG helicase (Quan et al., 2015; Douglas et al., 2018; Lewis et al., 2022; Langston et al., 2023; Henrikus et al., 2024). Mcm10 specifically binds MCM DHs loaded on the chromatin through an intricate mode involving at least the N-terminal and C-terminal domains of Mcm10 with multiple Mcm2–7 subunits (Quan et al., 2015; Douglas and Diffley, 2016; Liu et al., 2020). Mcm10 N-terminus contributes to low-affinity interaction with MCM, and the C-terminus mediates high-affinity interaction (Quan et al., 2015). Moreover, their interaction is cell cycle-regulated with a relatively weak “G1-like” and strong “S-like” mode, as demonstrated in vivo and in vitro (Quan et al., 2015; Douglas and Diffley, 2016). However, the mechanistic details of how the Mcm10–MCM DH interaction is regulated have yet to be defined.
In this study, we investigate the cell cycle-regulated Mcm10–MCM interaction and elucidate its critical role in DNA replication initiation. Through a combination of in vitro and in vivo biochemical approaches, we show that Mcm10 is a novel substrate of S-phase cyclin-dependent kinases (S-CDKs). The phosphorylation of a single conserved site (S66) within the Mcm10 N-terminus enhances its association with the MCM complex. Phospho-mutation S66A alone does not exert any apparent effect on cell growth under normal conditions. However, the loss of both Mcm10 Ser66 phosphorylation and the C-terminal MCM-binding domain causes cell death, underscoring the synergistic role of phosphorylation and protein–protein interaction. Moreover, by utilizing the two-degron strategy along with cell cycle synchronization, we efficiently deplete Mcm10 protein in the late G1 phase and observe a severe S-phase defect in mcm10-S66AΔC. Intriguingly, both lethality and S-phase deficiency can be rescued by artificially tethering Mcm10-S66AΔC to MCM. These data provide insights into the cell cycle-regulated bivalent Mcm10–MCM interaction by S-CDKs and its essential role in orchestrating DNA replication although neither the Mcm10 N-terminus- nor C-terminus-mediated interaction alone is indispensable.
Results
Mcm10 phosphorylation facilitates its association with MCM
In previous studies, we reported a crucial role of cell cycle-regulated interaction of Mcm10 with MCM DHs in the remodeling and activation of the latter (Quan et al., 2015; Liu et al., 2020). Mcm10 is recruited to origins via low-affinity (without CDK and DDK activity, G1-like) and high-affinity (with CDK and DDK activity, S-phase-like) modes, as demonstrated in the in vitro reconstitution system (Douglas and Diffley, 2016). However, how the Mcm10–MCM DH interaction is regulated remains unknown. We noticed that the Mcm10 protein occasionally displays two forms migrating very closely on immunoblots (Figure 1A). Since the two bands were so close, we postulated that the slower-migrating one might be a phosphorylated form of Mcm10. To test this, we treated the cell lysates with λ phosphatase (λ PPase) prior to separation on a high-resolution polyacrylamide gel. The relatively slower-migrating band disappeared after λ PPase treatment (Figure 1A, lane 7). This result became clearer when only half of the samples were loaded (lane 6). The sensitivity of this slower-migrating band to λ phosphatase was specific because it was retained if PPase inhibitor cocktails (PhosphoSTOP) were added simultaneously (compare lanes 4–7). These results reveal that Mcm10 may undergo phosphorylation in vivo.
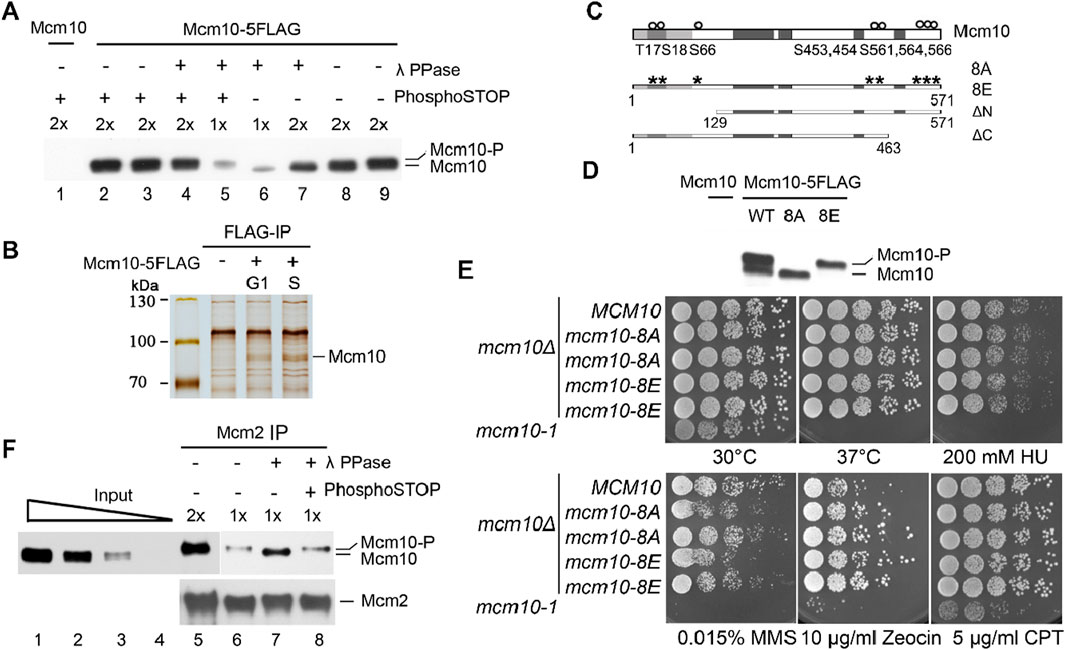
Figure 1. The phosphorylation of Mcm10 may facilitate its association with MCM. (A) Mcm10 undergoes phosphorylation in vivo. Yeast cells carrying 5FLAG-tagged Mcm10 (Strain QY317, Table 1) at its endogenous locus were grown to the exponential phase. Cell lysates were resolved by a high-resolution 8% PAGE containing SDS. Cells with untagged Mcm10 were applied as a control. λ phosphatase (PPase) or PPase inhibitor cocktails (PhosphoSTOP) were added to the lysates before SDS-PAGE. (B, C) Mapping of Mcm10 phosphorylation sites by immunoprecipitation-coupled mass spectrometry analysis (IP/MS). Yeast cells carrying 5FLAG-tagged Mcm10 (Strain QY317, Table 1) at its endogenous locus were cultured and arrested in G1 by α-factor. Cells were released for 0 min (G1) and 40 min (S) before collection. Cell lysates were incubated with anti-FLAG M2 affinity gel. Bound fractions were eluted by 0.5 μg/μL FLAG peptides and resolved by 8% PAGE containing SDS. Untagged Mcm10 was subjected to the same procedure as a control. The Mcm10 bands were excised and analyzed by MS/MS. (C) Putative phosphorylation sites. (D) Mutations of all eight putative phosphorylation sites to A or E abrogate the shift of Mcm10. The alanine (mcm10-8A) or glutamic acid (mcm10-8E) substitutions of all eight putative phosphorylation sites (S or T) are marked as asterisks. Darker boxes represent relatively conserved domains. (E) Phospho-mutants of MCM10, 8A, and 8E show nearly wild-type growth under normal or stressed conditions. mcm10-8A/E mutant strains were obtained by plasmid shuffling on plates containing 5-FOA. Wild-type MCM10 was cloned and expressed in the pRS316 vector to allow the mcm10Δ mutant to grow. The pRS316-MCM10 plasmid was removed from 5-FOA plates because its URA3 expression converted 5-FOA into a toxin. Rescue plasmids expressing MCM10 or its mutants were tested to examine their ability to support cell growth. Five-fold serial dilutions of log phase cells were spotted on the indicated plates and incubated for 2°days at 30°C, unless otherwise stated, before being photographed. (F) Phosphorylated Mcm10 may preferentially bind to Mcm2. Mcm2 was immunoprecipitated by anti-Mcm2 antibodies. The input and immunoprecipitation (IP) fractions were resolved by a high-resolution gel. The IP fractions were subjected to λ-PPase and/or PhosphoSTOP treatment to verify the phosphorylation form. The amounts loaded were titrated (1x and 2x) to achieve good separation.
To investigate the physiological role of Mcm10 phosphorylation, we first mapped post-translational modification sites on Mcm10. Endogenous Mcm10-5FLAG protein was precipitated from cells synchronized in G1 (by α-factor) or S phase (release for 40 min) (Figure 1B). Liquid chromatography–mass spectrometry/mass spectrometry (LC–MS/MS) revealed eight possible phosphorylation sites near the Mcm10 N-terminus (T17, S18, and S66 cluster) and C-terminus (S453, 454, 561, 564, and 566) (Figure 1C). To investigate the possible role of Mcm10 phosphorylation, we mutated all these sites to alanine (mcm10-8A) or glutamic acid (mcm10-8E). Since MCM10 is essential for cell viability, the mcm10 mutants were constructed via plasmid shuffling. WT MCM10 was cloned and expressed on a pRS316/URA3 single-copy vector to allow the growth of mcm10Δ. The mutant mcm10 allele in a second vector, pRS313/HIS3, was also introduced. The pRS316-MCM10 plasmid can be eliminated on 5-fluoroorotic acid (5-FOA) plates because it expresses URA3, which converts 5-FOA to a toxin. Thus, the growth on 5-FOA plates reflects the physiological function of the copy of the mcm10 mutant expressed on pRS313. Five-fold serial dilution of log phase cells was spotted on SC-His plates in the presence or absence of 5-FOA. Mcm10-8A and mcm10-8E proteins migrated as non-phosphorylated and phosphorylated forms, respectively (Figure 1D). This indicates that the observed shift is due to phosphorylation among these sites. Both mutants grew as well as the wild-type (WT) regardless of being under normal or various stress conditions (Figure 1E), suggesting that Mcm10 phosphorylation has no apparent effect on overall cell growth under the tested conditions.
Next, we examined whether Mcm10 phosphorylation affects this interaction using co-immunoprecipitation (CoIP). Endogenous Mcm2 was precipitated with anti-Mcm2 antibodies. Phosphorylated Mcm10 seemed to be enriched in the Mcm2-bound fraction compared to the input samples (Figure 1F, lanes 6 and 7). To confirm this, we treated Mcm2 precipitates with λ PPase and observed a PPase inhibitor-sensitive shift of Mcm10 to the faster-migrating form (Figure 1F, compare lanes 7–9). These data suggest that phosphorylation may facilitate the Mcm10–MCM interaction in vivo although recombinant Mcm10 binds directly to Mcm2, as shown in the previous in vitro pull-down assays (Quan et al., 2015).
The Mcm10 S66 phospho-mutant is synthetic lethal with its C-terminus truncation
Phosphorylation sites were enriched in the Mcm10 N-terminus (Mcm10-N, a.a., 1–128) and C-terminus (Mcm10-C, a.a., 461–571), which mediate interactions with MCM. Such a multivalent interaction mode prompted us to examine the synthetic effect of these factors on cell growth. Correlating with the relatively greater contribution of Mcm10-C than Mcm10-N to interactions with MCM, the mcm10ΔC allele showed much weaker growth than mcm10ΔN (Figure 2A, lines 4 and 2). Interestingly, the deletion of both Mcm10-N and Mcm10-C resulted in minimal growth (line 7). We then combined phosphorylation mutations with either mcm10ΔN or mcm10ΔC. Mutating five C-terminal phosphorylation sites displayed synthetic sickness with mcm10ΔN (lines 1 and 3), whereas mutating three N-terminal sites in mcm10ΔC caused cell death (lines 5 and 6). These results indicate that phosphorylation is important for the proper function of Mcm10 in vivo. Next, we determined which phosphorylation site(s) are indispensable for the growth of mcm10ΔC. Through serial mutation analysis, we demonstrated that mutation of the S66 residue alone is synthetic lethal with mcm10ΔC (Figure 2B, lines 5, 7, and 9). Moreover, mimicking both non-phosphorylated (A) and phosphorylated (E or D) mutations displayed similar phenotypes. This may be explained by either of two possibilities. First, neither S66D nor S66E may function as phospho-mimics. Second, reversible phosphorylation of S66 is important for the survival of mcm10ΔC. The mcm10-S66ΔC mutants were expressed at a comparable level relative to WT in vivo in these experiments, thus excluding the possibility that the lethality could be due to a failure in the expression of mcm10 alleles (Figure 2D). These data suggest that a single phosphorylation site in the Mcm10 N-terminus (S66) becomes indispensable in the absence of the C-terminus, the major MCM-interaction motif.
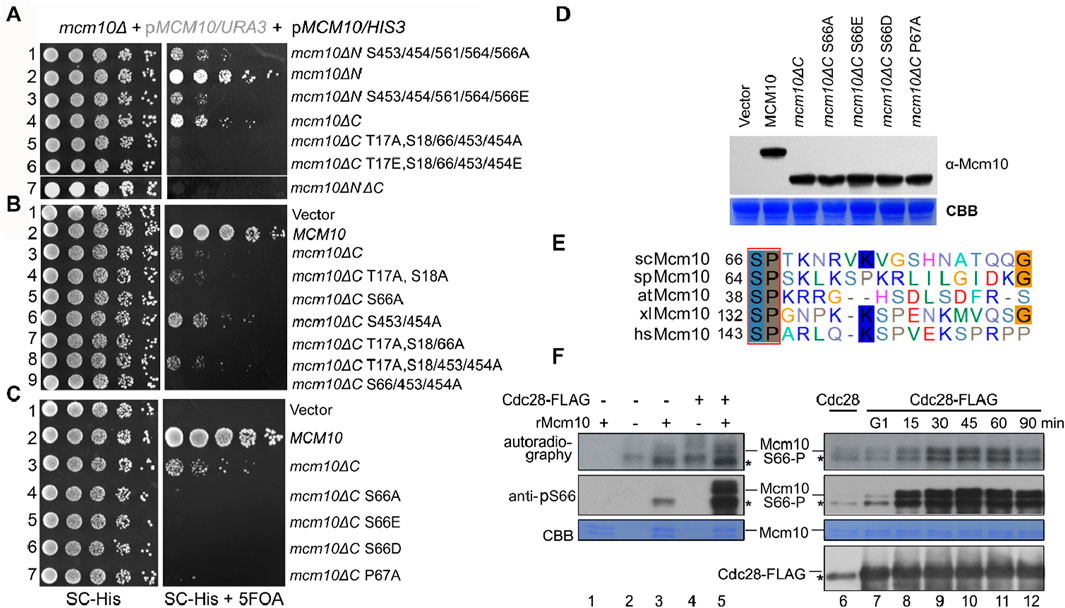
Figure 2. Mcm10 S66 is a substrate of S-CDKs and becomes indispensable in the absence of the Mcm10 C-terminus (A) mcm10 phosphorylation mutants show a synergistic defect with loss of the N- or C-terminal interaction region. Plasmid shuffling and serial dilution analyses were conducted similarly to the methods shown in Figure 1E. (B, C) Mapping of the critical phosphorylation site(s) required for the viability of mcm10ΔC. S66, a putative CDK target, is the sole phosphorylation site required for the growth of mcm10ΔC. (D) All mcm10ΔC derivative inviable alleles are expressed at a comparable level to wild-type MCM10. Whole-cell extracts were prepared from log-phase cells and immunoblotted by an anti-FLAG antibody. The membrane was then stained with Coomassie brilliant blue (CBB) to show the exact loading. (E) The CDK target site in Mcm10 may be conserved from yeast to human. The amino acid sequence of Mcm10 from different organisms was aligned using ClustalW. (F) In vitro kinase assays. Cdc28-5FLAG was immunoprecipitated from asynchronized (left panel) or synchronized cells released from G1 arrest for the indicated time (right panel) and then incubated with purified recombinant Mcm10 proteins in the presence of γ-32P-ATP. Mcm10 phosphorylation was detected by autoradiography (top panel) or anti-pS66 immunoblots (middle panel). Nonspecific bands were labeled by “*”.
Interestingly, S66 is followed by a conserved canonical “P-X-K-X-R” motif recognized by CDKs (Figure 2E). Moreover, mcm10-P67AΔC phenocopied mcm10-S66AΔC (Figure 2C, line 7), implicating that Mcm10 S66 may be a substrate of CDKs. To test this, we first conducted a kinase assay using purified Cdc28-FLAG from asynchronized yeast cells. In the presence of [γ-32P]-ATP, purified recombinant Mcm10 was phosphorylated by Cdc28, as indicated by both autoradiography and immunoblots using antibodies specific to phosphorylated S66 (pS66) (Figure 2F, lane 5). To examine which CDKs phosphorylate Mcm10, we next purified Cdc28 kinase from cells after release from G1 and repeated the in vitro kinase assays. S66 phosphorylation was barely detectable by CDKs from G1 cells (Figure 2F, lanes 6–7) and significantly increased by CDKs from S cells (compare lanes 7–12). Meanwhile, all reactions contained similar amounts of Cdc28. These data suggest that Mcm10 S66 might be a target of S-CDKs.
To validate these in vitro observations, we next probed Mcm10 S66 phosphorylation during the cell cycle in vivo. After collecting synchronized G1- and S-phase (release from α-factor for 60 min) cells, we prepared spheroplasts and fractionated soluble proteins into non-chromatin-bound (non-Chr) and chromatin-bound (Chr) through a sucrose cushion, as described previously (Quan et al., 2015; Liu et al., 2020). Both fractions were subjected to Mcm10-IP. The protein level of Cdc45, which is heavily increased upon S-phase entry, can be used as an indicator of the cell cycle stage (Figure 3A, lanes 1–2). In agreement with previous results (Quan et al., 2015), both Mcm2 and Cdc45 co-precipitated with Mcm10 exclusively in Chr fraction (Figure 3A, compare lanes 7–8 with 5–6), demonstrating a successful chromatin fractionation. However, as shown by anti-pS66 immunoblotting (IB), Mcm10-S66 phosphorylation was clearly detected in both non-Chr and Chr fractions (Figure 3A, compare lanes 6 and 8). This indicates that S66 phosphorylation occurs prior to Mcm10 recruitment to chromatin. In terms of timing, S66 was phosphorylated only in S cells but not in G1 cells (Figure 3A, compare lanes 5 and 6). Consistent with in vitro kinase activities shown in Figure 2F, these data confirm that Mcm10 is a bona fide substrate of S-CDKs.
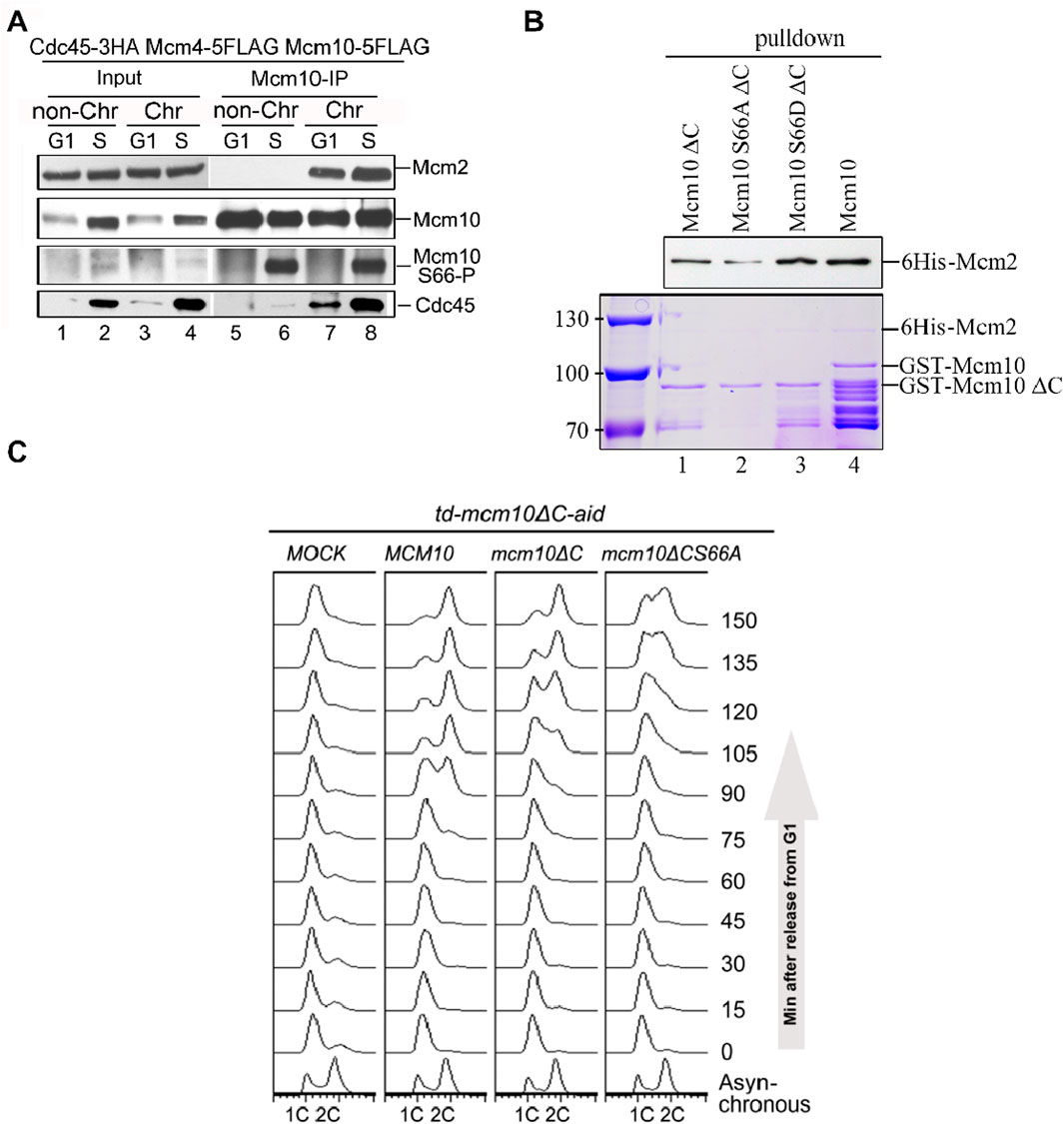
Figure 3. The Mcm10–Mcm2 interaction is an essential requirement of replication initiation. (A) Mcm10-pS66 occurs prior to being loaded onto chromatin in the S phase. Mcm10-FLAG was precipitated from either non-chromatin or chromatin-associated fractions and then subjected to Mcm10-IP with the anti-Mcm10 antibody. (B) Mcm10-S66 phospho-mutations affect the interaction with Mcm2 in vitro. GST pull-down assays were performed using affinity-purified 6-His-Mcm2 along with GST-tagged Mcm10 and its mutant proteins. The bound proteins were separated by SDS-PAGE and detected either by immunoblotting against anti-His antibodies or CBB staining. (C) mcm10 mutants are defective in S-phase entry and progression. Endogenous Mcm10 protein was depleted by the two-degron strategy as above. The td-mcm10ΔC-aid cells bearing a serial of mcm10 mutant plasmids were released from α-factor synchronization. The ability of each mcm10 allele to support DNA synthesis was monitored by flow cytometry.
Coinciding with the appearance of S66 phosphorylation, the Mcm10–Mcm2 interaction was relatively increased during the S phase (Figure 3A, compare lanes 7 and 8), which is reminiscent of the preferential binding of phosphorylated Mcm10 with Mcm2, as shown in Figure 1F. To test whether S66 phosphorylation contributes to the enhanced Mcm10–Mcm2 association, we conducted pulldown assays using affinity-purified recombinant proteins. As shown in Figure 3B, Mcm10-S66D∆C displayed a relatively stronger Mcm2-binding than Mcm10-S66A∆C and Mcm10-∆C (compare lanes 1–3). These in vivo and in vitro biochemical data suggest that although the Mcm10–MCM interaction is mainly mediated by the Mcm10 C-terminus, it can be enhanced by the phosphorylation of S66 at its N-terminus during the S phase. Together with the genetic results shown in Figure 2, which show that loss of both interaction motifs (mcm10-S66A∆C) causes cell death, we postulated that the lethality of mcm10-S66AΔC may be due to the essential function of Mcm10 in DNA replication. To test it, we measured cell cycle progression by flow cytometry. Since the mcm10-S66AΔC mutant is lethal, we tested its ability to support DNA synthesis in the Mcm10 conditional knockout background. Since very small residual Mcm10 can support its essential function, we used a previously developed two-degron system, temperature-induced degron (td) and auxin-induced degron (aid), in the mcm10ΔC allele, which had been shown to deplete endogenous Mcm10 protein efficiently. Under this condition, the DNA content did not change after release from G1 for over 150 min (Figure 3C, Mock). Next, we introduced a set of mcm10 alleles to test whether they could support DNA synthesis after depletion. DNA increased more slowly to 2C content in mcm10ΔC than in WT (Figure 3C, 90–120 min). For mcm10-S66AΔC cells, the S phase started approximately 30 min later than mcm10ΔC and failed to reach 2C even after 150 min. These data indicate that the lethality of mcm10-S66AΔC is likely due to a failure in DNA synthesis. These data provide genetic evidence that MCM-binding mediated by both Mcm10 C-terminus and S66 phosphorylation defines an indispensable role of Mcm10 in replication.
The restoration of the Mcm10–CMG interaction rescues the lethality and replication defects in mcm10ΔC-S66A
If the failure/delay in S-phase entry is solely caused by the compromised Mcm10–MCM interaction in mcm10-S66AΔC, it should be suppressed by reinforcing this interaction. We adopted an in vivo GFP trap strategy to achieve this (Figure 4A). If we add a GFP tag to one protein and a GBP (GFP binding protein) tag to another protein, these two proteins can be tethered to each other through strong affinity between the GFP and GBP pair. In Figure 4B, we introduced a pRS313/HIS3 plasmid expressing each mcm10 allele with or without a GBP tag at the C-terminus by plasmid shuffling. Control experiments showed that Mcm10-GBP or Mcm2-GFP supported WT growth (Figure 4B, lines 1 and 5). The co-expression of Mcm10-GBP and Mcm2-GFP also displayed normal growth (Figure 4B, line 13), indicating that the dissociation of Mcm10 and Mcm2 is not important for normal growth. Notably, strains expressing mcm10-S66AΔC-GBP or mcm10-S66DΔC-GBP became viable depending on the presence of Mcm2-GFP (compare lines 3, 4, 7, 8, 15, and 16). However, mcm10 (129–463) (deleting both N- and C-terminus) and the temperature-sensitive allele mcm10-1 could not be rescued by fusing with Mcm2 (Figure 4C, compare lines 4–8). These results suggest that the lethality of mcm10-S66ΔC is very likely due to the loss of its association with Mcm2. Since multiple Mcm2–7 subunits are partners of Mcm10, we also checked whether tethering the interaction of the defective mutant of Mcm10 with other partners has a similar effect as Mcm2. As shown side by side in Figure 4B, mcm10-S66ΔC-GBP survived in the presence of Mcm4-GFP and Mcm2-GFP. Moreover, tethering mcm10-S66ΔC to Cdc45, a subunit of the CMG complex, could rescue their lethality as well (Figure 4D). However, tethering Mcm10 to Pol3 had no rescue effect at all (Figure 4E). Pol3 is a subunit of DNA Pol δ, which is involved in replication progression but not in initiation. Therefore, we propose that the lethality of these mcm10 mutations is attributable to defective interactions with MCM or its activator Cdc45 during replication initiation. Furthermore, flow cytometry profiles showed that the mcm10-S66AΔC mutant could gradually proceed into the S phase and reach 2C content dependent on fusion with Mcm2 (Figure 4F). These results suggest that the restoration of Mcm10–CMG interactions can suppress the lethality and replication deficiency of mcm10-S66ΔC. Hence, we propose that S-CDK-regulated Mcm10 N-terminus-mediated interaction, along with Mcm10 C-terminus-mediated interaction with MCM, defines an indispensable step in DNA replication initiation in budding yeast.
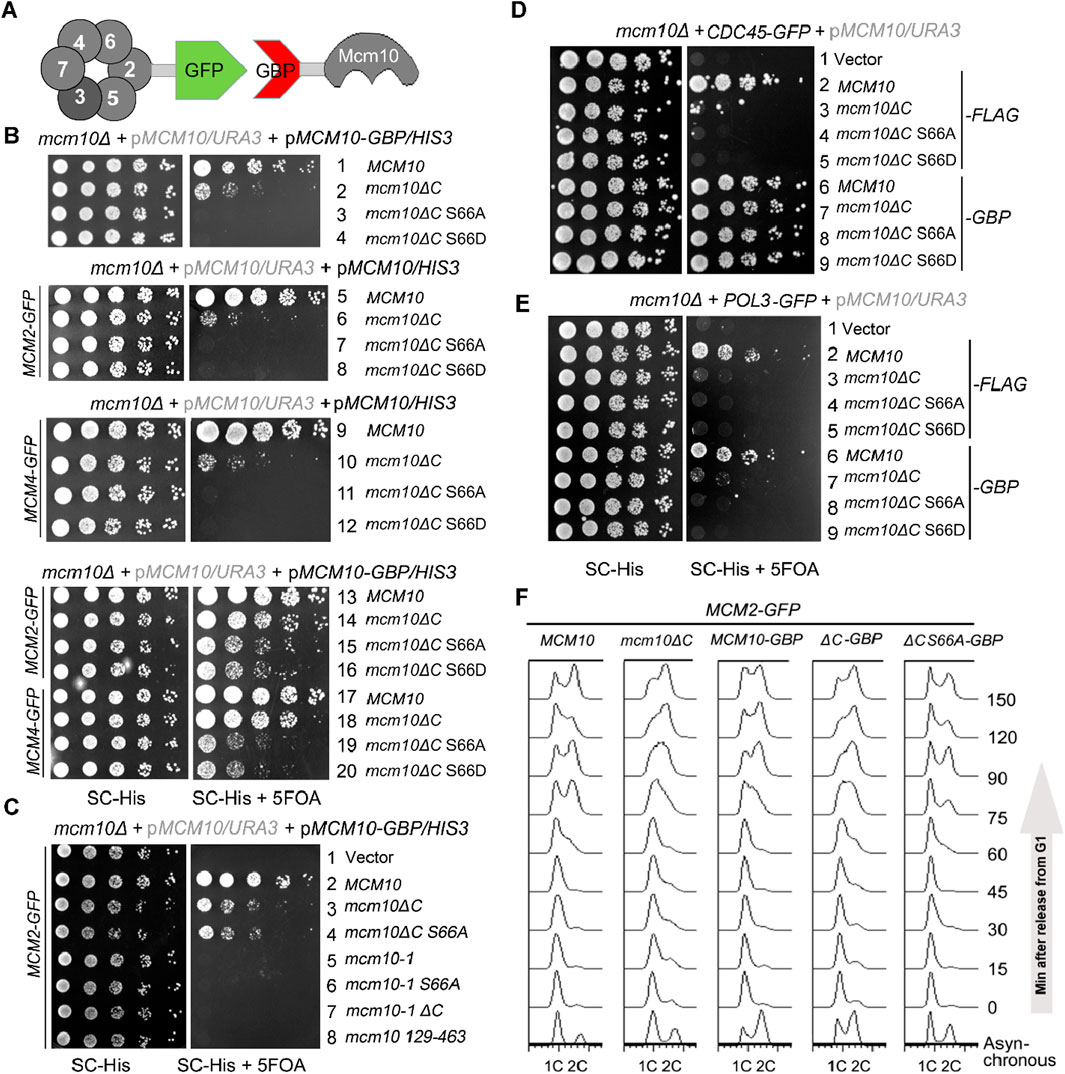
Figure 4. The lethality of mcm10ΔC-S66 mutants can be suppressed by restoring their interaction with either Mcm2 or Mcm4. (A) An in vivo GFP trap strategy to enforce the Mcm10–MCM interaction. Mcm2 or Mcm4 was tagged with GFP, while Mcm10 or its mutant form was fused with GBP. Interactions between any two proteins might be restored artificially via the GFP–GBP pair through an in vivo GFP trap experiment. (B, C) In vivo GFP trap of Mcm10 with either Mcm2 or Mcm4 suppresses the lethality of mcm10-S66ΔC alleles. Either Mcm10-GBP or Mcm2-GFP with a single tag showed no effect on the growth of wild-type or mcm10 alleles. Plasmid shuffling and serial dilution assays were carried out, as shown in Figure 2. (D, E) In vivo GFP trap of Mcm10 with either Cdc45 (D) or Pol2 (E). Mcm10 mutants carrying a 5FLAG tag were applied as negative controls, which showed no effect on the growth in either Cdc45-GFP or Pol3-GFP cells. Plasmid shuffling and serial dilution assays were carried out, as shown above. (F) In vivo GFP trap of Mcm10 with Mcm2 partially also suppresses the S-phase defect of mcm10-S66ΔC alleles. Cells were synchronized at G1 with the α-factor and released into the S phase. Cell cycle progression was monitored by flow cytometry analysis of the DNA content for each mcm10ΔC allele.
Discussion
CMG maturation is characterized by a cascade of events: origin DNA melting (Lewis et al., 2022), dissociation of the transient dimeric CMG (Quan et al., 2015; Liu et al., 2020), and exclusion of the lagging strand from the central channel of the CMG ring (Henrikus et al., 2024). Mcm10 is instrumental in facilitating these structural transitions (Yeeles et al., 2015; Douglas et al., 2018). Moreover, Mcm10 has been demonstrated to stimulate the helicase activity of the recombinant CMG complex in vitro (Langston et al., 2017; Langston and O’Donnell, 2019; Langston et al., 2023) and induce CMG remodeling under stress (Wasserman et al., 2019), suggesting its direct effect on the CMG complex. Our results lead us to posit that the interaction between Mcm10–MCM/CMG is a critical determinant for the orchestration of CMG remodeling, particularly throughout the CMG maturation during initiation. Subsequent investigations might explore whether S-CDK-regulated Mcm10–MCM association is involved in the complete separation of the splayed dimeric CMG, especially considering that Mcm10 binds to the N-terminal domains of Mcm2, Mcm4, and Mcm6 (Quan et al., 2015; Douglas and Diffley, 2016), which are posited as the interfaces of MCM double hexamers (Li et al., 2015). Additionally, phosphorylation of MCM10 has been reported in human cells (Izumi et al., 2000) and Xenopus (Chadha et al., 2016). With the recent paradigm shift in the understanding of initiation mechanisms in metazoa (Cvetkovic et al., 2023; Lim et al., 2023; Xia et al., 2023; Terui et al., 2024), further studies are warranted to elucidate whether the Mcm10–MCM interaction is subject to cell cycle-dependent regulation in higher eukaryotes.
Experimental procedures
Strains and plasmids
The strains and plasmids used in this study are listed in Tables 1, 2, respectively. Mutants were generated using recombination-mediated cassette exchange or tetrad dissection, as previously described (Quan et al., 2015). All the constructs were confirmed by DNA sequencing.
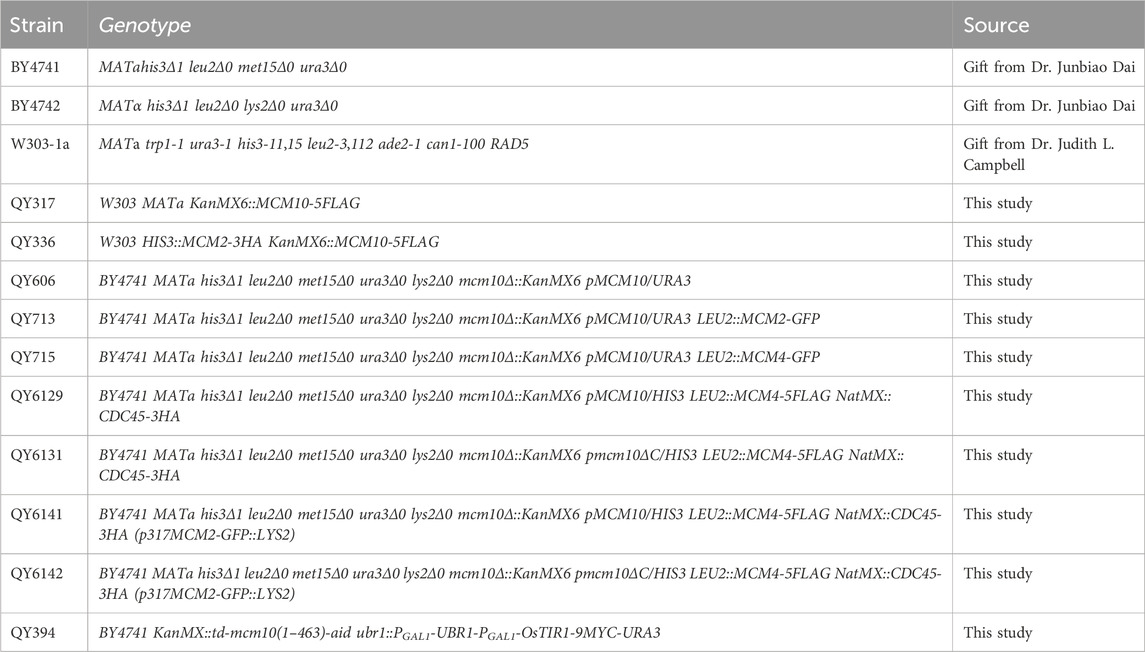
Table 1. Strains used in this study.
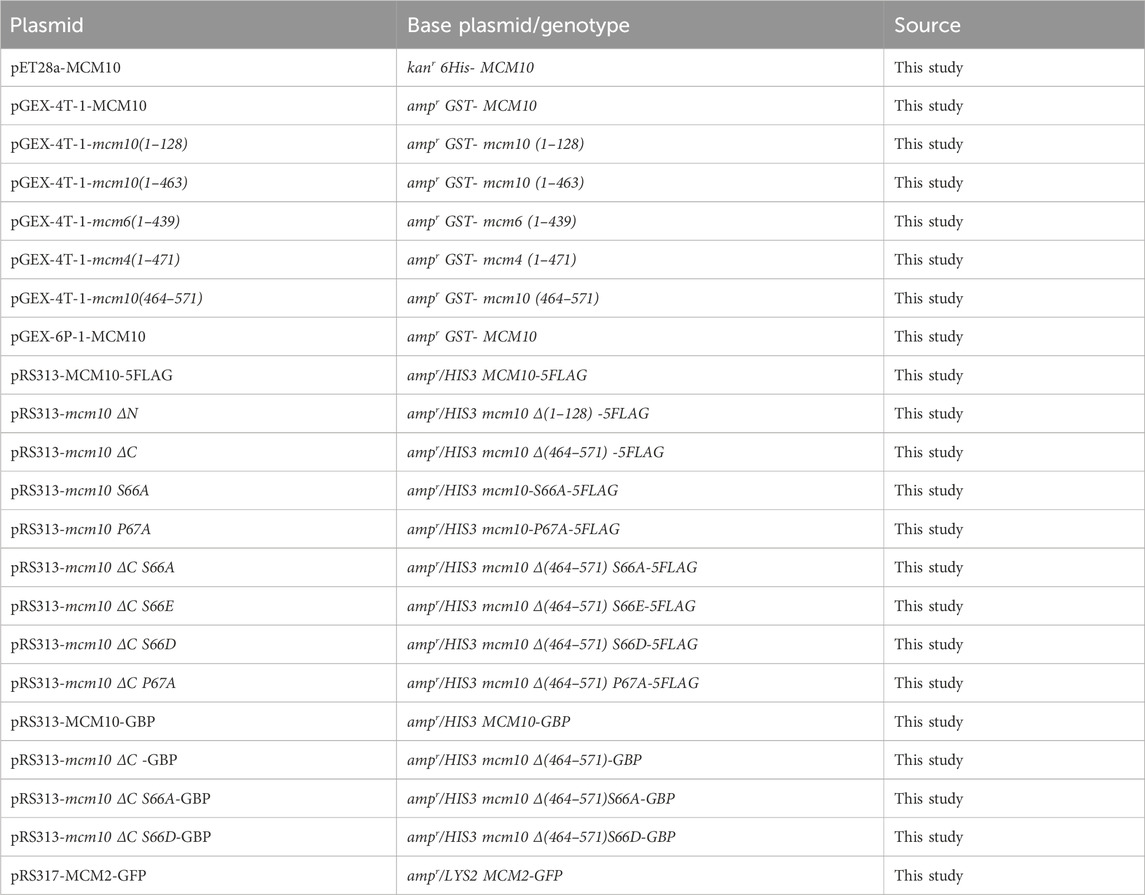
Table 2. Plasmids used in this study.
Cell synchronization and flow cytometry analysis
A total of 7.5 μg/mL of α factor was added for cell synchronization in the G1 phase. G1 arrested cells were released by filter washing twice in a fresh medium and continued growth for the indicated time. Samples were collected and fixed with 70% ethanol and then processed for flow cytometry using a BD Biosciences FACSVerse machine.
Whole-cell extracts and immunoblotting
Whole-cell extracts (WCEs) of 100 OD600 units of asynchronized or synchronized cells were prepared by glass bead beating (Mini-Beadbeater-16, BioSpec, United States) in lysis buffer [45 mM HEPES, pH 7.2, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.2% NP-40, 1 mM PMSF, 2 mM DTT, 1× Protease Inhibitor Cocktail tablet (Roche), and 1× PhosSTOP tablet (Roche)]. Protein fractions were separated by SDS polyacrylamide gel electrophoresis (PAGE) and transferred to a PVDF membrane. Each protein was probed with the antibody specifically indicated in each figure by Western blotting. The antibodies used in this study are anti-Cdc45 (gift from Dr. Karim Labib), mouse anti-FLAG M2-specific monoclonal antibody (1:2000, Sigma), mouse anti-HA 16B12 (1:1000, Millipore), polyclonal anti-GST (glutathione transferase) (1:1000, OriGene), anti-6-His antibodies (1:1000, OriGene), anti-tubulin (1:10,000, MBL), and anti-Rad53 (1:1000, Abcam); protein-G-agarose (GE Healthcare) and NHS-activated agarose resins (GE Healthcare) were also used. HRP-conjugated anti-rabbit or anti-mouse IgG was used as the secondary antibody (1:10,000, Sigma). Anti-pS66 (Mcm10 S66 phosphorylation) was developed in rabbits against oligopeptides IEVPQ{pS}PTKNRVKVC (GenScript), {pS} stands for the phosphorylated Ser residue (GenScript).
Phosphatase treatment
Mcm10-5FLAG in whole-cell extracts or co-immunoprecipitated with Mcm2 was treated with 200 U λ phosphatase (New England Biolabs) in the presence or absence of PhosphoSTOP (Roche) at 30°C for 20 min.
Immunoprecipitation
Immunoprecipitation (IP) analysis was performed using strains co-expressing the tagged versions of each protein at a physiological level, as indicated in each figure. IP was carried out as described previously. Input (IN) corresponding to approximately 100 μg total protein was analyzed in parallel with immunoprecipitates. Proteins were analyzed by mass spectrometry or Western blotting using the indicated antibodies.
Affinity purification and mass spectrometry
Mcm10-5FLAG was precipitated from whole-cell extracts using M2 affinity gel (Sigma). Nonspecific bound proteins were removed by washing with 0.5 μg/μL FLAG peptide. The bound fraction was boiled in an equal volume of 2x SDS loading buffer and resolved on an 8% SDS-PAGE and silver staining. An untagged strain was subjected to the same procedure as a control. The bands specific to the Mcm10-5FLAG sample were cut and digested by trypsin (NEB), followed by mass spectrometry analysis (Q Exactive™ Hybrid Quadrupole-Orbitrap Mass Spectrometer, Thermo Fisher Scientific).
Protein expression and purification
Full-length and truncated forms of pGEX4T-1-MCM10, pGEX-4T-1-mcm6(1–439), pGEX-4T-1-mcm4(1–471), pET28a-MCM10, pET28a-MCM2, and pET28a-mcm2(1–299) constructs used in the biochemical experiments were expressed in E. coli BL21 (DE3) RIL codon-plus (Stratagene) and purified using affinity tags, followed by conventional column chromatography.
Preparation of antibodies and Mcm10 agarose beads
To produce polyclonal antibodies specific to Mcm10 or Mcm2, the purified full-length protein was used to immunize rabbits. Polyclonal antibodies were affinity-purified. Mcm10 beads were prepared by immobilizing purified Mcm10 protein to NHS-activated agarose beads, as recommended by the manufacturer (GE Healthcare), which was used for an efficient in vitro pull-down assay.
Preparation of antibodies
Antibodies specific to Mcm10 S66 phosphorylation were developed in rabbits against oligopeptides IEVPQ{pS}PTKNRVKVC. Antibodies were affinity-purified using non-phosphorylated oligopeptides to remove the antibody reacting with the non-phosphorylated polypeptide (GenScript).
In vitro pull-down assay
Approximately 10 pmol of each protein was mixed with glutathione-Sepharose 4B (GE Healthcare Life Sciences), or anti-Mcm10 agarose beads made in this study were mixed in 100 μL of the binding buffer (50 mM HEPES-NaOH, pH 7.6, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 1 mM PMSF, 1 μg/μL BSA, and 0.1% Triton X-100) and incubated for 1 h at 4°C. The beads were washed at least three times prior to Western blotting and/or Coomassie staining.
In vitro kinase assay
Cdc28-5FLAG from WCE of 50 OD600 units of asynchronized or G1-released cells was precipitated with 10 μL M2 affinity gel (Sigma) and washed five times with lysis buffer and once with kinase buffer [20 mM HEPES/KOH, pH 7.2, 80 mM β-glycerophosphate, 10 mM MgCl, 20 mM EGTA, 100 μM ATP, protease inhibitor tablets (EDTA free, Roche), and PhosphoSTOP (Roche)]. Each reaction mixture (20 μL) contained 10 μL Cdc28-5Flag-bound beads, 2 μg GST-Mcm10, and 2 μCi γ-32P-ATP. After incubation at 30°C for 30 min, the reaction products were separated by 8% SDS-PAGE and transferred onto a PVDF membrane.
Construction of mcm10 alleles
The viability of various mcm10 alleles was determined by plasmid shuffling since MCM10 is an essential gene. Wild-type MCM10 was cloned and expressed in the pRS316/URA3 vector to allow mcm10Δ mutants to grow. The pRS316-MCM10 plasmid was removed on 5-FOA plates because it expresses URA3, which caused cells toxic to 5-FOA. The ability to support cell growth was tested for various mcm10 alleles expressed in the pRS313/HIS3 vector under a range of genetic backgrounds, as indicated in each figure. Five-fold serial dilution of log phase cells was spotted on SC-His plates in the presence or absence of 5-FOA and incubated for 2 days at the indicated temperature before photography.
In vivo GFP trap assay (protein tethering assay)
The interaction between mcm10 mutants and Mcm2–7 was restored through a tethering strategy using the in vivo GFP trap (Quan et al., 2015). In Mcm10–Mcm2–7 tethering experiments, each mcm10 allele was fused to GBP, while the Mcm2 or Mcm4 subunit was tagged with GFP. To ensure specifically targeted protein tethering, the omission of one of the GFP/GBP pairs was included as controls in all tethering assays.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author contributions
XW: funding acquisition, investigation, methodology, resources, and writing–original draft. LL: investigation, methodology, resources, validation, and writing–review and editing. MC: data curation, project administration, and writing–review and editing. YQ: investigation, resources, and writing–review and editing. JZ: funding acquisition, investigation, resources, and writing–review and editing. HL: supervision and writing–review and editing. YX: resources and writing–review and editing. HC: conceptualization, resources, and writing–review and editing. WH: supervision, writing–original draft, and writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Natural Science Foundation of China [32101039 to WH; 32200422 to XW; 32300074 to JZ]; China Postdoctoral Science Foundation [2022M722183 to XW]; Natural science foundation of Guangdong province of China [2022A1515011208 to WH]; Shenzhen Basic Research Project (Natural Science Foundation) [JCYJ20220530142004009 to XW]; Shenzhen Excellent Scientific and Technological Innovation Talent Training Project (Doctoral Startup Project) [RCBS20221008093347106 to XW]; Shenzhen Medical Research Fund (SMRF) [A2301020].
Acknowledgments
The authors thank Dr. K. Labib for generously sharing Cdc45 antibodies.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bell, S. P., and Labib, K. (2016). Chromosome duplication in Saccharomyces cerevisiae. Genetics 203 (3), 1027–1067. doi:10.1534/genetics.115.186452
Campos, L. V., Van Ravenstein, S. X., Vontalge, E. J., Greer, B. H., Heintzman, D. R., Kavlashvili, T., et al. (2023). RTEL1 and MCM10 overcome topological stress during vertebrate replication termination. Cell Rep. 42 (2), 112109. doi:10.1016/j.celrep.2023.112109
Chadha, G. S., Gambus, A., Gillespie, P. J., and Blow, J. J. (2016). Xenopus Mcm10 is a CDK-substrate required for replication fork stability. Cell Cycle 15 (16), 2183–2195. doi:10.1080/15384101.2016.1199305
Costa, A., and Diffley, J. F. X. (2022). The initiation of eukaryotic DNA replication. Annu. Rev. Biochem. 91, 107–131. doi:10.1146/annurev-biochem-072321-110228
Cvetkovic, M. A., Passaretti, P., Butryn, A., Reynolds-Winczura, A., Kingsley, G., Skagia, A., et al. (2023). The structural mechanism of dimeric DONSON in replicative helicase activation. Mol. Cell 83 (22), 4017–4031.e9. doi:10.1016/j.molcel.2023.09.029
Douglas, M. E., Ali, F. A., Costa, A., and Diffley, J. F. X. (2018). The mechanism of eukaryotic CMG helicase activation. Nature 555 (7695), 265–268. doi:10.1038/nature25787
Douglas, M. E., and Diffley, J. F. X. (2016). Recruitment of Mcm10 to sites of replication initiation requires direct binding to the minichromosome maintenance (MCM) complex. J. Biol. Chem. 291 (11), 5879–5888. doi:10.1074/jbc.M115.707802
Henrikus, S. S., Gross, M. H., Willhoft, O., Pühringer, T., Lewis, J. S., McClure, A. W., et al. (2024). Unwinding of a eukaryotic origin of replication visualized by cryo-EM. Nat. Struct. Mol. Biol. 31, 1265–1276. doi:10.1038/s41594-024-01280-z
Izumi, M., Yanagi, K., Mizuno, T., Yokoi, M., Kawasaki, Y., Moon, K. Y., et al. (2000). The human homolog of Saccharomyces cerevisiae Mcm10 interacts with replication factors and dissociates from nuclease-resistant nuclear structures in G(2) phase. Nucleic Acids Res. 28 (23), 4769–4777. doi:10.1093/nar/28.23.4769
Kanke, M., Kodama, Y., Takahashi, T. S., Nakagawa, T., and Masukata, H. (2012). Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO J. 31 (9), 2182–2194. doi:10.1038/emboj.2012.68
Langston, L. D., Georgescu, R. E., and O'Donnell, M. E. (2023). Mechanism of eukaryotic origin unwinding is a dual helicase DNA shearing process. Proc. Natl. Acad. Sci. U. S. A. 120 (52), e2316466120. doi:10.1073/pnas.2316466120
Langston, L. D., Mayle, R., Schauer, G. D., Yurieva, O., Zhang, D., Yao, N. Y., et al. (2017). Mcm10 promotes rapid isomerization of CMG-DNA for replisome bypass of lagging strand DNA blocks. Elife 6, e29118. doi:10.7554/eLife.29118
Langston, L. D., and O’Donnell, M. E. (2019). An explanation for origin unwinding in eukaryotes. Elife 8, e46515. doi:10.7554/eLife.46515
Lewis, J. S., Gross, M. H., Sousa, J., Henrikus, S. S., Greiwe, J. F., Nans, A., et al. (2022). Mechanism of replication origin melting nucleated by CMG helicase assembly. Nature 606 (7916), 1007–1014. doi:10.1038/s41586-022-04829-4
Li, N., Zhai, Y., Zhang, Y., Li, W., Yang, M., Lei, J., et al. (2015). Structure of the eukaryotic MCM complex at 3.8 A. Nature 524 (7564), 186–191. doi:10.1038/nature14685
Lim, Y., Tamayo-Orrego, L., Schmid, E., Tarnauskaite, Z., Kochenova, O. V., Gruar, R., et al. (2023). In silico protein interaction screening uncovers DONSON's role in replication initiation. Science 381 (6664), eadi3448. doi:10.1126/science.adi3448
Liu, L., Zhang, Y., Zhang, J., Wang, J. H., Cao, Q., Li, Z., et al. (2020). Characterization of the dimeric CMG/pre-initiation complex and its transition into DNA replication forks. Cell Mol. Life Sci. 77 (15), 3041–3058. doi:10.1007/s00018-019-03333-9
Looke, M., Maloney, M. F., and Bell, S. P. (2017). Mcm10 regulates DNA replication elongation by stimulating the CMG replicative helicase. Genes Dev. 31 (3), 291–305. doi:10.1101/gad.291336.116
Mayle, R., Langston, L., Molloy, K. R., Zhang, D., Chait, B. T., and O'Donnell, M. E. (2019). Mcm10 has potent strand-annealing activity and limits translocase-mediated fork regression. Proc. Natl. Acad. Sci. U. S. A. 116 (3), 798–803. doi:10.1073/pnas.1819107116
O’Donnell, M., Langston, L., and Stillman, B. (2013). Principles and concepts of DNA replication in bacteria, archaea, and eukarya. Cold Spring Harb. Perspect. Biol. 5 (7), a010108. doi:10.1101/cshperspect.a010108
Quan, Y., Xia, Y., Liu, L., Cui, J., Li, Z., Cao, Q., et al. (2015). Cell-cycle-regulated interaction between Mcm10 and double hexameric mcm2-7 is required for helicase splitting and activation during S phase. Cell Rep. 13 (11), 2576–2586. doi:10.1016/j.celrep.2015.11.018
Terui, R., Berger, S. E., Sambel, L. A., Song, D., and Chistol, G. (2024). Single-molecule imaging reveals the mechanism of bidirectional replication initiation in metazoa. Cell 187 (15), 3992–4009.e25. doi:10.1016/j.cell.2024.05.024
van Deursen, F., Sengupta, S., De Piccoli, G., Sanchez-Diaz, A., and Labib, K. (2012). Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO J. 31 (9), 2195–2206. doi:10.1038/emboj.2012.69
Wasserman, M. R., Schauer, G. D., O'Donnell, M. E., and Liu, S. (2019). Replication fork activation is enabled by a single-stranded DNA gate in CMG helicase. Cell 178 (3), 600–611. doi:10.1016/j.cell.2019.06.032
Watase, G., Takisawa, H., and Kanemaki, M. T. (2012). Mcm10 plays a role in functioning of the eukaryotic replicative DNA helicase, Cdc45-Mcm-GINS. Curr. Biol. 22 (4), 343–349. doi:10.1016/j.cub.2012.01.023
Xia, Y., Sonneville, R., Jenkyn-Bedford, M., Ji, L., Alabert, C., Hong, Y., et al. (2023). DNSN-1 recruits GINS for CMG helicase assembly during DNA replication initiation in Caenorhabditis elegans. Science 381 (6664), eadi4932. doi:10.1126/science.adi4932
Yeeles, J. T., Deegan, T. D., Janska, A., Early, A., and Diffley, J. F. X. (2015). Regulated eukaryotic DNA replication origin firing with purified proteins. Nature 519 (7544), 431–435. doi:10.1038/nature14285
Keywords: cell cycle, DNA replication, Mcm10, phosphorylation, protein–protein interaction
Citation: Wang X, Liu L, Chen M, Quan Y, Zhang J, Lou H, Xia Y, Chen H and Hou W (2024) S-CDK-regulated bipartite interaction of Mcm10 with MCM is essential for DNA replication. Front. Cell Dev. Biol. 12:1420033. doi: 10.3389/fcell.2024.1420033
Received: 19 April 2024; Accepted: 27 August 2024;
Published: 19 September 2024.
Edited by:
Song-Tao Liu, University of Toledo, United StatesReviewed by:
Helmut Pospiech, University Hospital Düsseldorf, GermanyYing Wai Chan, The University of Hong Kong, Hong Kong SAR, China
Copyright © 2024 Wang, Liu, Chen, Quan, Zhang, Lou, Xia, Chen and Hou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenya Hou, d2VueWEuaG91QHN6dS5lZHUuY24=; Hongxiang Chen, aG9uZ3hpYW5nY2hlbkBob3RtYWlsLmNvbQ==
†These authors have contributed equally to this work