 Angie K. Torres
Angie K. Torres Veronika Fleischhart
Veronika Fleischhart Nibaldo C. Inestrosa
Nibaldo C. Inestrosa- 1Facultad de Ciencias Biológicas, Pontificia Universidad Católica de Chile, Santiago, Chile
- 2Centro de Excelencia en Biomedicina de Magallanes (CEBIMA), Escuela de Medicina, Universidad de Magallanes, Punta Arenas, Chile
Mitochondria are key organelles for the optimal function of the cell. Among their many functions, they maintain protein homeostasis through their own proteostatic machinery, which involves proteases and chaperones that regulate protein import and folding inside mitochondria. In the early 2000s, the mitochondrial unfolded protein response (UPRmt) was first described in mammalian cells. This stress response is activated by the accumulation of unfolded/misfolded proteins within the mitochondrial matrix, which results in the transmission of a signal to the nucleus to increase the expression of proteases and chaperones to address the abnormal mitochondrial protein load. After its discovery, this retrograde signaling pathway has also been described in other organisms of different complexities, suggesting that it is a conserved stress response. Although there are some specific differences among organisms, the mechanism of this stress response is mostly similar and involves the transmission of a signal from mitochondria to the nucleus that induces chromatin remodeling to allow the binding of specific transcription factors to the promoters of chaperones and proteases. In the last decade, proteins and signaling pathways that could be involved in the regulation of the UPRmt, including the Wnt signaling pathway, have been described. This minireview aims to summarize what is known about the mechanism of the UPRmt and its regulation, specifically in mammals and C. elegans.
1 Introduction
Mitochondria are organelles with many functions, such as providing energy in the form of ATP and regulating calcium homeostasis, redox balance, and apoptosis (Harrington et al., 2023), being essential for maintaining cellular homeostasis. Interestingly, mitochondria contain nuclear-encoded and mitochondrial-encoded proteins that are assembled inside the organelle to form functional complexes within the mitochondrial matrix and the inner mitochondrial membrane (Annesley and Fisher, 2019). Thus, mitochondria have a proteostatic network composed of chaperones and proteases that ensure correct protein import and folding within them (Voos, 2009). In 2002, the mitochondrial unfolded protein response (UPRmt) was described for the first time as a specific stress response in the mitochondria of mammalian cells triggered by the accumulation of unfolded/misfolded proteins within the mitochondrial matrix (Zhao et al., 2002); this type of response had been previously described only in the endoplasmic reticulum, known as the endoplasmic reticulum unfolded protein response (UPRER).
The UPRmt involves retrograde signaling between mitochondria and the nucleus, which leads to the upregulation of several mitochondrial proteins, including antioxidant enzymes, mitochondrial import proteins, and mitochondrial chaperones and proteases, to decrease the unfolded/misfolded load (Zhao et al., 2002; Anderson and Haynes, 2020; Tran and Van Aken, 2020). The mechanism of the UPRmt in mammals has recently been described; however, it is still not fully understood. There are different axes of the UPRmt, with the canonical axis being the most studied (Munch, 2018); this axis involves the activation of the integrated stress response (ISR), which decreases the global translation rate, favoring the translation of specific stress-responsive proteins (Fiorese et al., 2016; Quiros et al., 2017; Anderson and Haynes, 2020). In the years after the UPRmt was first described in mammalian cells, this stress response was also described in the nematode C. elegans (C. elegans) (Yoneda et al., 2004), in yeast (Schleit et al., 2013) and in Drosophila melanogaster (Pareek and Pallanck, 2018); this suggests that the UPRmt is a conserved signaling pathway among eukaryotic organisms, including highly complex organisms such as humans and other mammals, and less complex organisms such as C. elegans and yeast. The mechanism of this stress response in C. elegans has been widely reported, more than the same response in mammals, mainly due to the ease of generating loss- or gain-of-function mutations in specific proteins in C. elegans (Haynes et al., 2007; Nargund et al., 2012); however, the mechanisms by which this stress response is regulated have not yet been fully described.
In this minireview, we will describe the mechanism of the UPRmt both in mammals and in C. elegans, as well as the similarities and differences among these species, and the different regulatory mechanisms described in recent years to provide a global view of what is known about the UPRmt today, providing a reference for future studies on the potential of this stress response as a new therapeutic target. The PubMed database was searched using the main keyword “mitochondrial unfolded protein response”, and articles describing the mechanism of the UPRmt and reporting more recent findings regarding the regulation of the UPRmt and its effect on diseases were selected.
2 The UPRmt in mammals
Regarding studies on the UPRmt in mammals, Zhao et al. (2002) showed that the transfection of COS-7 cells with a mutant misfolded form of ornithine trans-carbamylase (ΔOTC), a mitochondrial matrix protein involved in the urea cycle, results in the accumulation of this protein, inducing the upregulation of nuclear-encoded mitochondrial chaperones and proteases (Zhao et al., 2002). This work described for the first time the transcriptional UPRmt, a stress response triggered by the accumulation of misfolded proteins within the matrix that is currently known as the canonical UPRmt (Figure 1). In this stress response, stress signal transmission to the cytosol is thought to be driven through the processing of DAP3 binding cell death enhancer 1 (DELE1) by the protease Oma1 (Fessler et al., 2020; Guo et al., 2020). Oma1 constitutively cleaves the fusion dynamin-like GTPase L-OPA1 into small fragments, and this activity is increased under stress (Baker et al., 2014). Although the mechanism underlying stress-induced Oma1 activation is still not well characterized, it has been reported that stress signals are sensed through positively charged amino acids in the N-terminal region and that the transition to an active complex is associated with conformational changes involving the conserved C-terminal region (Baker et al., 2014). DELE1 is a 56 kDa protein that contains a mitochondrial targeting sequence (MTS) that allows it to be localized to the mitochondrial matrix in the absence of stress, where it has a short half-life due to its degradation by the protease Lonp1. However, during mitochondrial stress, DELE1 senses mitochondrial import deficiency since newly synthesized full-length DELE1 is cleaved by Oma1 in the N-terminal MTS, which produces short fragments (S-DELE1) that accumulate in the cytosol (Fessler et al., 2020; Guo et al., 2020). These fragments interact with and activate the kinase heme-regulated inhibitor (HRI), leading to the phosphorylation of eukaryotic translation initiation factor 2A (eIF2α), activating the ISR and increasing the expression of the transcription factors activating transcription factor 5 (ATF5), activating transcription factor 4 (ATF4) and C/EBP homologous protein (CHOP) (Fiorese et al., 2016; Fessler et al., 2020; Guo et al., 2020).
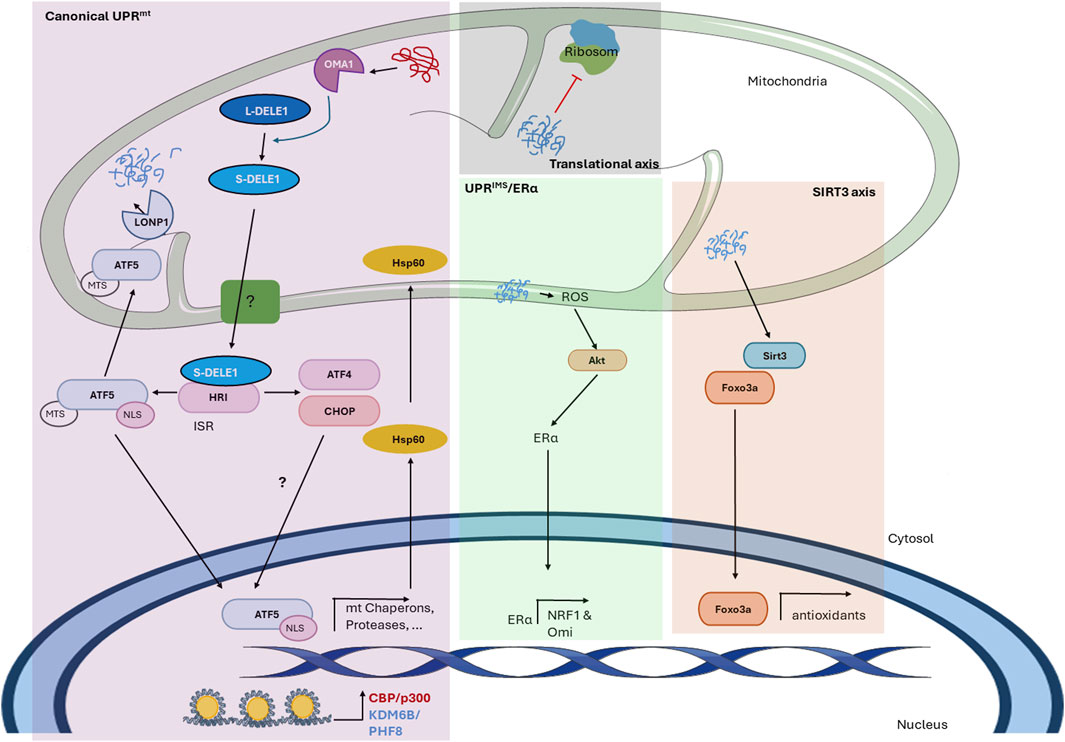
Figure 1. The mitochondrial unfolded protein response in mammals. The figure shows the mechanism of the UPRmt in mammals described thus far. The scheme shows the different axes of the mitochondrial-nuclear retrograde signaling pathway and the proteins involved. In the canonical UPRmt, the accumulation of abnormal proteins within the mitochondrial matrix activates the protease Oma1, which cleaves L-DELE1 into short fragments (S-DELE1) that are released into the cytosol. Once in the cytosol, S-DELE1 interacts with and activates the kinase HRI, activating the ISR and allowing the translation of ATF4, CHOP, and ATF5. The latter is translocated to the nucleus, where it binds to the promoters of UPRmt-related genes following chromatin remodeling by KDM6B and PHF8. CBP/p300 is also involved in epigenetic modification and the expression of UPRmt-related genes. In the translational axis, unfolded proteins reduce the mitochondrial translation rate locally without generating a global response. In the SIRT3 axis, unfolded proteins activate SIRT3, which induces the nuclear localization of the transcription factor FOXO3a to upregulate antioxidant enzyme expression. Finally, the UPRIMS/ERα is activated by misfolded protein accumulation in the IMS, leading to an increase in ROS levels, which in turn activates Akt kinase to phosphorylate and activate ERα. Undescribed proteins and mechanisms are shown with a question mark (?).
Although the order in which these transcription factors act is still unknown, ATF5 has been described as fundamental for UPRmt activation. ATF5 has an MTS and a nuclear localization signal (NLS) (Fiorese et al., 2016). Under normal conditions, ATF5 is imported into mitochondria for degradation, presumably by the protease Lonp1. Nevertheless, under stress conditions, the import of ATF5 is inhibited by an unknown mechanism, leading to its cytoplasmic accumulation and consequent nuclear translocation, where it binds to a specific UPRmt element to induce the expression of certain genes, including the mitochondrial chaperones Hsp60, Hsp10, and mtHsp70 and the proteases Lonp1 and ClpP (Nargund et al., 2015; Fiorese et al., 2016). Additionally, UPRmt-related genes contain CHOP-binding regions in their promoters, indicating the importance of CHOP in the expression of these genes (Zhao et al., 2002). CHOP induces ATF5 expression in the UPRER to induce apoptosis (Teske et al., 2013), and it has been reported that in HepG2 cells but not in other cells exposed to arsenite, ATF5 increases CHOP expression (Yamazaki et al., 2010), suggesting that these two transcription factors can regulate each other in a context-dependent manner. However, specifically in the UPRmt, the mechanism by which this process is regulated remains to be elucidated. Moreover, the precise role of ATF4 in this process is still unclear; however, it was identified as a regulatory factor that induces the expression of cytoprotective genes in response to mitochondrial stress, and it has also been proposed as a link between the UPRER and UPRmt (Quiros et al., 2017; Jiang et al., 2020). Moreover, a previous study demonstrated that heat shock transcription factor 1 (HSF1) is an important player in the UPRmt since under stress, HSF1 enters the nucleus and binds to the promoter of the chaperones Hsp60, Hsp10, and mtHsp70 but not to the promoter of the protease Lonp1 (Katiyar et al., 2020). However, how HSF-1 interacts with the canonical transcription factors ATF5, ATF4, and CHOP is still unknown.
It has been suggested that the UPRmt involves epigenetic modifications caused by the histone demethylases PHF8 and KDM6B since there is a positive correlation between the expression of these proteins and UPRmt gene expression. Furthermore, removal of lysine 27 trimethylation in histone 3 (H3K27me3) increases the expression of mitochondrial chaperones and proteases (Merkwirth et al., 2016). Moreover, the transcriptional coactivator CBP/p300 induces the acetylation of lysine 18 and 27 in histone 3 (H3K18Ac and H3K27Ac), probably after KDM6B and PHF8 exert their effect, and is indispensable for UPRmt-related gene expression (Li et al., 2021).
Along with the canonical UPRmt, different axes of the UPRmt have been described. There is a translational axis that decreases the mitochondrial translation rate locally to reduce the protein folding load and allow the handling of existing misfolded proteins (Munch and Harper, 2016). This translational axis is a local response and does not generate a cellular response since it is activated only when a few mitochondria are damaged (Munch, 2018). Moreover, the misfolded protein load in the mitochondrial matrix activates an antioxidant UPRmt axis driven by sirtuin 3 (SIRT3) (Papa and Germain, 2014; Munch, 2018). SIRT3 increases the nuclear localization of the transcription factor FOXO3a through its deacetylation, which increases the transcription of antioxidant enzymes such as superoxide dismutase 2 (SOD2) and catalase (Papa and Germain, 2014). Although it has been reported that Hsp10 and Lonp1 are substrates of SIRT3 deacetylation (Gibellini et al., 2014; Lu et al., 2015), there are contradictory findings regarding how the SIRT3 axis is related to canonical UPRmt-related gene expression (Gibellini et al., 2014; Papa and Germain, 2014; Lu et al., 2015; Chen et al., 2021; Wu et al., 2023). In addition, mitochondria have different compartments, and when misfolded protein accumulation occurs in the intermembrane space (IMS), another UPR, called the UPRIMS/ERα, is activated. This signaling pathway seems to be independent of the canonical UPRmt; however, these responses can act in parallel or complement each other. Protein aggregates in the IMS activate estrogen receptor alpha (ERα) in a ligand-independent manner through its phosphorylation at serine 167 (Papa and Germain, 2011). An increase in reactive oxygen species (ROS) production leads to the activation of the kinase AKT, which ultimately induces the activation of ERα and the transcription of nuclear respiratory factor 1 (NRF1) and the IMS protease Omi (Papa and Germain, 2011). Altogether, these UPRmt axes cope with the misfolded/unfolded load within mitochondria to maintain proper mitochondrial function.
3 The UPRmt in Caenorhabditis elegans
Shortly after the UPRmt was described in mammalian cells, it was reported that the perturbation of protein handling in mitochondria resulting from an RNAi against a mitochondrial protease induces the expression of mitochondrial chaperones (Yoneda et al., 2004). The mechanism of the UPRmt in C. elegans has been largely described and, in general, is quite similar to that in mammals (Figure 2). Indeed, the signal produced by misfolded/unfolded accumulation in the matrix is transmitted to the cytosol by the release of short fragments, as in mammals; however, C. elegans does not have an Oma1 homolog (Kirstein-Miles and Morimoto, 2010). In C. elegans, the protease ClpP cleaves the abnormal proteins within the mitochondrial matrix into small fragments of approximately 20 residues that are released into the IMS through homodimers of the transporter HAF-1 in the inner mitochondrial membrane (IMM) (Haynes et al., 2010). The release of these peptides inhibits, by an unknown mechanism, the mitochondrial import of activating transcription factor associated with stress 1 (ATFS-1), an ATF5 homolog that is also the main transcription factor associated with the UPRmt in C. elegans. Like ATF5, ATFS-1 contains a weak MTS and an NLS (Haynes et al., 2010); when mitochondria are not perturbed, the MTS prevails, and ATFS-1 is imported into the mitochondria for degradation by the protease LON. However, under mitochondrial stress, the weak MTS allows the sensing of few changes, which leads to inhibition of its mitochondrial import and consequent accumulation in the nucleus, where it can bind to the promoters of UPRmt-associated genes (Nargund et al., 2012). Additionally, a nuclear complex formed by the homeobox domain transcription factor DVE-1 and ubiquitin-like protein UBL-5 binds to the promoters of mitochondrial chaperones and protease to facilitate the subsequent binding of ATFS-1 (Haynes et al., 2007). The binding of these proteins to promoters requires chromatin remodeling, as in mammals. The accumulation of abnormal proteins activates the histone methyltransferase MET-2 and the nuclear localization of the cofactor LIN-65 (Tian et al., 2016). MET-2 mono- or dimethylates H3K9 (H3K9me1/2), which results in general chromatin remodeling, leaving specific regions exposed, where DVE-1 and ATFS-1 can bind (Tian et al., 2016). Along with these proteins, the histone deacetylase HAD-1 and the two histone demethylases JMJD-3.1 and JMJD-1.2 are fundamental for UPRmt-related gene expression (Merkwirth et al., 2016; Shao et al., 2020), indicating the importance of chromatin changes and epigenetics in the UPRmt. Furthermore, as in mammals, the CBP/p300 homolog CBP-1 is necessary for UPRmt-related gene expression (Li et al., 2021). This coactivator is suggested to exert its effect between chromatin remodeling by JMJD-3.1 and JMJD-1.2 and the binding of ATFS-1 (Li et al., 2021).
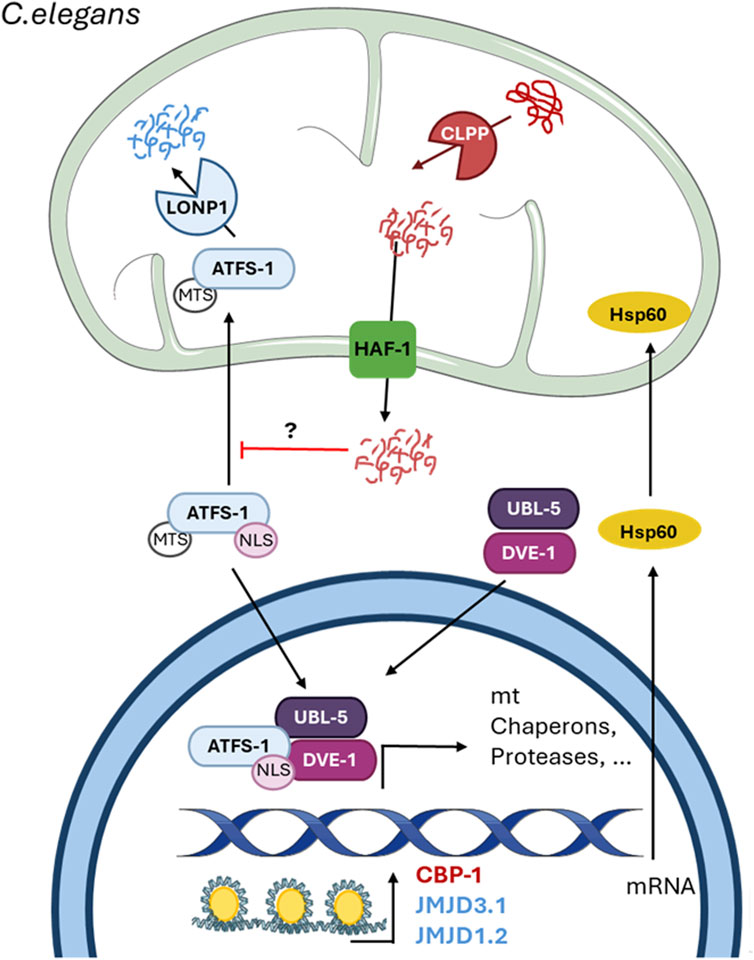
Figure 2. The mitochondrial unfolded protein response in C. elegans. The figure shows the mechanism of the UPRmt in C. elegans described thus far. The scheme shows the mitochondrial-nuclear retrograde signaling pathway and the proteins involved. The protease ClpP cleaves abnormal proteins inside the mitochondria into small fragments of less than 20 amino acids, which enter the cytosol through the HAF-1 transporter. The release of these peptides inhibits the mitochondrial import of the transcription factor ATFS-1 by an unknown mechanism and induces its nuclear translocation. In the nucleus, ATFS-1 forms a complex with UBL-5 and DVE-1 after chromatin remodeling by the methyl transferases JMJD3.1 and JMJD1.2. Additionally, CBP-1 acts upstream of these two methyltransferases but downstream of ATFS-1 to induce the expression of UPRmt-related genes. Undescribed proteins and mechanisms are shown with a question mark (?).
Just like UPRmt activation requires a decrease in global translation by the HRI-dependent activation of ISR in mammals, in C. elegans is also required a decrease in translation caused by phosphorylation of eIF2α, but in this case, by the kinase GCN2, which favors a better folding environment (Baker et al., 2012). These data support the idea that the UPRmt and its mechanism are conserved between mammals and C. elegans.
4 The UPRmt in disease
It has been reported that the UPRmt is activated in different diseases in which mitochondrial dysfunction seems to be a key player, such as cardiac disease (Smyrnias et al., 2019), kidney disease (Liu et al., 2023), mitochondrial disease (Suarez-Rivero et al., 2022b), cancer (Inigo and Chandra, 2022) and neurodegenerative diseases (Beck et al., 2016; Cooper et al., 2017). For instance, in Alzheimer’s disease (AD), the two main toxic proteins that accumulate in AD, amyloid-β (Aβ) peptide and tau protein, impair mitochondrial function in the early stages of the disease (Torres et al., 2021; Bartman et al., 2024). Additionally, mitochondrial diseases are caused either by pathological mutations in mitochondrial DNA (mtDNA) or nuclear DNA affecting OXPHOS complexes, which are inherited maternally or in an autosomal recessive way, respectively (Gropman et al., 2024). In both cases, the UPRmt is activated to compensate for mitochondrial dysfunction; however, at some points, this response is no longer enough to decrease mitochondrial damage (Suarez-Rivero et al., 2022a).
The activation of the UPRmt has beneficial effects on increasing longevity (Xin et al., 2022) and improving mitochondrial function since it maintains ATP production, reduces ROS levels, and decreases apoptosis (Svagusa et al., 2020; Lu et al., 2022). Thus, it has been proposed that the activation of the UPRmt could be a promising therapeutic approach for various diseases, although it seems paradoxical that inducing mitochondrial stress in the presence of mitochondrial dysfunction could be beneficial (Suarez-Rivero et al., 2022a). Antibiotics, mainly doxycycline, activate the UPRmt; however, the chronic use of antibiotics is still controversial (Suarez-Rivero et al., 2021). Therefore, recent studies have shown that different compounds could be safer therapeutic agents for several diseases (Table 1). Despite favorable outcomes, contradictory evidence indicates that overactivation or prolonged activation of the UPRmt could be detrimental (Lu et al., 2022), indicating the importance of proper balance in the activation of the UPRmt.
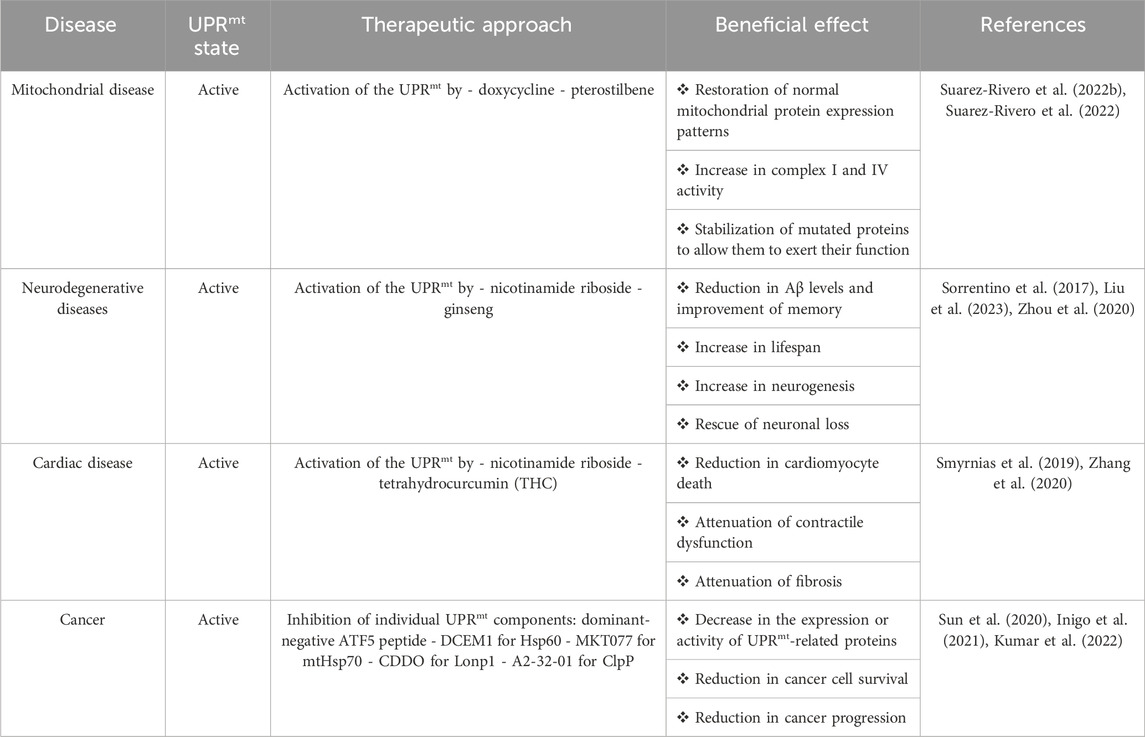
Table 1. The UPRmt in diseases.
The activation of the UPRmt is related to cancer progression (Keerthiga et al., 2021). ATF5, Hsp60, mtHsp70, Lonp1, and Clpp are upregulated in cancer, favoring tumor growth (Deng and Haynes, 2017; Inigo and Chandra, 2022). ATF5 induces the upregulation of antiapoptotic proteins such as Bcl-2 and MCL1, promoting tumor cell growth, and the upregulation of integrin-α2 and integrin-β1, which favors cancer cell invasion (Nukuda et al., 2016; Wang et al., 2022). Hsp60 is involved in preventing apoptosis by inhibiting mitochondrial permeability transition pore opening and stabilizing the protein survivin (Ghosh et al., 2010; Kim et al., 2019). mtHsp70 reduces p53 activity, promoting tumor cell survival, and regulates PI3K/AKT signaling to induce epithelial-mesenchymal transition of tumor cells (Wadhwa et al., 2002; Na et al., 2016). Lonp1 induces tumor metabolic reprogramming and promotes inflammatory cytokine production generating an immunosuppressive tumor environment (Quiros et al., 2014; Kuo et al., 2020). Finally, ClpP stabilizes OXPHOS complexes, maintaining ATP production, and regulates Src/PI3K/AKT signaling, favoring proliferation and invasion (Seo et al., 2016; Luo et al., 2020). Indeed, research on therapeutic approaches related to the UPRmt in cancer have focused on the inhibition of the UPRmt, specifically on targeting individual UPRmt-associated proteins (Table 1) (Inigo et al., 2021).
5 UPRmt regulation
Although the mechanism by which the UPRmt is regulated is still not fully understood, some reports suggest that different proteins and signaling pathways could be involved in this process. In C. elegans, the SUMO protease ubiquitin-like protease 4 (ULP-4) regulates DVE-1 and ATFS-1 when the UPRmt is induced. ULP-4 deSUMOylates DVE-1 to allow its accumulation in the nucleus, and deSUMOylates ATFS-1 to stabilize it and increases its transcriptional activity (Gao et al., 2019). These data suggest the posttranslational regulation of UPRmt-related transcription factors, which could also occur in mammals since, for example, ATF5 can also be SUMOylated and acetylated in other contexts (Liu et al., 2011; Yuan et al., 2018). However, this phenomenon has not yet been studied in mammals. In mammals, the protein GrpEL1, a nucleotide exchanger that controls the conversion of mtHsp70-ADP to mtHsp70-ATP, is also a regulator of the UPRmt. When this stress response is activated, GrpEL1 forms a complex with mtHsp70 to promote its function and reduce the aggregation of proteins in mitochondria (Ma et al., 2022). Additionally, a recent study suggested that the UPRmt is linked to and dependent on mitophagy, with FUN14 domain-containing protein 1 (FUNDC1) acting upstream of its activation, inducing this stress response by decreasing the mtDNA content (Ji et al., 2022), which increases the misfolded protein load. Moreover, recently, it was shown that the activation of the UPRmt, in addition to the release of short DELE1 fragments, requires the release of mitochondrial ROS (mtROS) as signaling molecules into the cytosol (Sutandy et al., 2023). Once in the cytosol, mtROS oxidize the chaperone HSP40 (DNAJA1), which increases its interaction with cytosolic HSP70 to drive the translocation of HSF-1 to the nucleus to activate the transcription of mitochondrial chaperones and proteases (Sutandy et al., 2023).
Interestingly, in yeast, mitochondria trigger a UPRmt-like response before the UPRmt is activated in response to mitochondrial precursor protein accumulation, which is an immediate response (Poveda-Huertes et al., 2020). This early response is mediated by the nuclear HMG-box domain-containing transcription factor Rox1, which translocates to mitochondria, maintaining mitochondrial import, the membrane potential, and translation (Poveda-Huertes et al., 2020). However, whether this early UPRmt-like response occurs in mammals or C. elegans is not known.
Regarding signaling pathways, there are some reports in C. elegans showing non-autonomous regulation through different pathways. One of these pathways is the follicle-stimulating hormone G protein-coupled receptor (FSHR1)/sphingosine kinase (SPHK-1) pathway, in which FSHR activates this stress response in neurons and promotes the stress-induced association of SPHK-1 with intestinal mitochondria (Kim and Sieburth, 2020). Additionally, ROS produced in GABAergic neurons act as signaling molecules by oxidizing the GABAA receptor UNC-49 (Pohl et al., 2023). This oxidation of UNC-49 increases its channel activity in muscle cells, which induces the activation of the UPRmt in intestinal cells via an unknown mechanism, suggesting that other tissues may be involved in the neuronal-intestinal regulation of the UPRmt (Pohl et al., 2023). Moreover, it has been proposed that the Wnt signaling pathway may also be involved in the regulation of this stress response. Wnt signaling is a key pathway during development but is also important for proper adult neuronal function (Inestrosa et al., 2021). There are two pathways of Wnt signaling, the β-catenin-independent or non-canonical signaling and β-catenin-dependent or canonical signaling pathway, which regulate the expression of Wnt target genes (Inestrosa and Arenas, 2010; Inestrosa et al., 2021). Preliminary results from our laboratory indicate that mitochondrial chaperones and proteases involved in the UPRmt have Wnt-responsive elements in their promoters (Torres et al., 2022a; b), and the modulation of Wnt signaling, both in C. elegans and in primary hippocampal neuronal culture, regulates the expression of UPRmt-associated proteins (Torres et al., 2022a; b; Torres et al., 2023). These data suggest that Wnt signaling may have a direct effect on the expression of UPRmt genes, which could be Wnt target genes.
6 Conclusion
The UPRmt, which is involved in the mitochondrial stress response, is a key signaling pathway for maintaining the protective function of mitochondria upon protein accumulation. This stress response has been described in yeast, nematodes, and mammals, suggesting that it is an essential protective mechanism for survival among eukaryotic organisms. Indeed, it has been described as a compensatory response that reduces mitochondrial damage in several diseases; however, at some point, the degree of mitochondrial dysfunction reaches a critical level, and endogenous activation of the UPRmt is insufficient for countering it. Although the mechanism underlying the UPRmt has been described over the years, the regulation of this stress response has been less studied. Thus, more information about how to safely modulate the UPRmt while avoiding the detrimental effects that could result from its long-term activation is needed. This information is essential for the development of new drug-based therapeutic approaches for chronic diseases such as mitochondrial diseases, cancer, and AD.
Author contributions
AT: Conceptualization, Writing–original draft, Writing–review and editing. VF: Writing–review and editing. NI: Conceptualization, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by a PhD fellowship from ANID N°21211882 of the Chilean Government to AT, which is gratefully acknowledged. The Vice Rectory of Research of the Pontifical Catholic University (PUC) and the Faculty of Biological Sciences of PUC to NI.
Acknowledgments
We would like to thank Dra. Paulina Villaseca for her assistance with reviewing and editing the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Anderson, N. S., and Haynes, C. M. (2020). Folding the mitochondrial UPR into the integrated stress response. Trends Cell. Biol. 30 (6), 428–439. doi:10.1016/j.tcb.2020.03.001
Annesley, S. J., and Fisher, P. R. (2019). Mitochondria in health and disease. Cells 8 (7), 680. doi:10.3390/cells8070680
Baker, B. M., Nargund, A. M., Sun, T., and Haynes, C. M. (2012). Protective coupling of mitochondrial function and protein synthesis via the eIF2α kinase GCN-2. PLoS Genet. 8 (6), e1002760. doi:10.1371/journal.pgen.1002760
Baker, M. J., Lampe, P. A., Stojanovski, D., Korwitz, A., Anand, R., Tatsuta, T., et al. (2014). Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 33 (6), 578–593. doi:10.1002/embj.201386474
Bartman, S., Coppotelli, G., and Ross, J. M. (2024). Mitochondrial dysfunction: a key player in brain aging and diseases. Curr. Issues Mol. Biol. 46 (3), 1987–2026. doi:10.3390/cimb46030130
Beck, J. S., Mufson, E. J., and Counts, S. E. (2016). Evidence for mitochondrial UPR gene activation in familial and sporadic alzheimer's disease. Curr. Alzheimer Res. 13 (6), 610–614. doi:10.2174/1567205013666151221145445
Chen, H., Zhang, D. M., Zhang, Z. P., Li, M. Z., and Wu, H. F. (2021). SIRT3-mediated mitochondrial unfolded protein response weakens breast cancer sensitivity to cisplatin. Genes. Genomics 43 (12), 1433–1444. doi:10.1007/s13258-021-01145-5
Cooper, J. F., Machiela, E., Dues, D. J., Spielbauer, K. K., Senchuk, M. M., and Van Raamsdonk, J. M. (2017). Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson's disease models. Sci. Rep. 7 (1), 16441. doi:10.1038/s41598-017-16637-2
Deng, P., and Haynes, C. M. (2017). Mitochondrial dysfunction in cancer: potential roles of ATF5 and the mitochondrial UPR. Semin. Cancer Biol. 47, 43–49. doi:10.1016/j.semcancer.2017.05.002
Fessler, E., Eckl, E. M., Schmitt, S., Mancilla, I. A., Meyer-Bender, M. F., Hanf, M., et al. (2020). A pathway coordinated by DELE1 relays mitochondrial stress to the cytosol. Nature 579 (7799), 433–437. doi:10.1038/s41586-020-2076-4
Fiorese, C. J., Schulz, A. M., Lin, Y. F., Rosin, N., Pellegrino, M. W., and Haynes, C. M. (2016). The transcription factor ATF5 mediates a mammalian mitochondrial UPR. Curr. Biol. 26 (15), 2037–2043. doi:10.1016/j.cub.2016.06.002
Gao, K., Li, Y., Hu, S., and Liu, Y. (2019). SUMO peptidase ULP-4 regulates mitochondrial UPR-mediated innate immunity and lifespan extension. Elife 8, e41792. doi:10.7554/eLife.41792
Ghosh, J. C., Siegelin, M. D., Dohi, T., and Altieri, D. C. (2010). Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 70 (22), 8988–8993. doi:10.1158/0008-5472.CAN-10-2225
Gibellini, L., Pinti, M., Beretti, F., Pierri, C. L., Onofrio, A., Riccio, M., et al. (2014). Sirtuin 3 interacts with Lon protease and regulates its acetylation status. Mitochondrion 18, 76–81. doi:10.1016/j.mito.2014.08.001
Gropman, A. L., Uittenbogaard, M. N., and Chiaramello, A. E. (2024). Challenges and opportunities to bridge translational to clinical research for personalized mitochondrial medicine. Neurotherapeutics 21 (1), e00311. doi:10.1016/j.neurot.2023.e00311
Guo, X., Aviles, G., Liu, Y., Tian, R., Unger, B. A., Lin, Y. T., et al. (2020). Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature 579 (7799), 427–432. doi:10.1038/s41586-020-2078-2
Harrington, J. S., Ryter, S. W., Plataki, M., Price, D. R., and Choi, A. M. K. (2023). Mitochondria in health, disease, and aging. Physiol. Rev. 103 (4), 2349–2422. doi:10.1152/physrev.00058.2021
Haynes, C. M., Petrova, K., Benedetti, C., Yang, Y., and Ron, D. (2007). ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev. Cell. 13 (4), 467–480. doi:10.1016/j.devcel.2007.07.016
Haynes, C. M., Yang, Y., Blais, S. P., Neubert, T. A., and Ron, D. (2010). The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell. 37 (4), 529–540. doi:10.1016/j.molcel.2010.01.015
Inestrosa, N. C., and Arenas, E. (2010). Emerging roles of Wnts in the adult nervous system. Nat. Rev. Neurosci. 11 (2), 77–86. doi:10.1038/nrn2755
Inestrosa, N. C., Tapia-Rojas, C., Cerpa, W., Cisternas, P., and Zolezzi, J. M. (2021). WNT signaling is a key player in alzheimer's disease. Handb. Exp. Pharmacol. 269, 357–382. doi:10.1007/164_2021_532
Inigo, J. R., and Chandra, D. (2022). The mitochondrial unfolded protein response (UPR(mt)): shielding against toxicity to mitochondria in cancer. J. Hematol. Oncol. 15 (1), 98. doi:10.1186/s13045-022-01317-0
Inigo, J. R., Kumar, R., and Chandra, D. (2021). Targeting the mitochondrial unfolded protein response in cancer: opportunities and challenges. Trends Cancer 7 (12), 1050–1053. doi:10.1016/j.trecan.2021.08.008
Ji, H., Wang, J., Muid, D., Song, W., Jiang, Y., and Zhou, H. (2022). FUNDC1 activates the mitochondrial unfolded protein response to preserve mitochondrial quality control in cardiac ischemia/reperfusion injury. Cell. Signal 92, 110249. doi:10.1016/j.cellsig.2022.110249
Jiang, D., Cui, H., Xie, N., Banerjee, S., Liu, R. M., Dai, H., et al. (2020). ATF4 mediates mitochondrial unfolded protein response in alveolar epithelial cells. Am. J. Respir. Cell. Mol. Biol. 63 (4), 478–489. doi:10.1165/rcmb.2020-0107OC
Katiyar, A., Fujimoto, M., Tan, K., Kurashima, A., Srivastava, P., Okada, M., et al. (2020). HSF1 is required for induction of mitochondrial chaperones during the mitochondrial unfolded protein response. FEBS Open Bio 10 (6), 1135–1148. doi:10.1002/2211-5463.12863
Keerthiga, R., Pei, D. S., and Fu, A. (2021). Mitochondrial dysfunction, UPR(mt) signaling, and targeted therapy in metastasis tumor. Cell. Biosci. 11 (1), 186. doi:10.1186/s13578-021-00696-0
Kim, S., and Sieburth, D. (2020). FSHR-1/GPCR regulates the mitochondrial unfolded protein response in Caenorhabditis elegans. Genetics 214 (2), 409–418. doi:10.1534/genetics.119.302947
Kim, W., Ryu, J., and Kim, J. E. (2019). CCAR2/DBC1 and Hsp60 positively regulate expression of survivin in neuroblastoma cells. Int. J. Mol. Sci. 20 (1), 131. doi:10.3390/ijms20010131
Kirstein-Miles, J., and Morimoto, R. I. (2010). Caenorhabditis elegans as a model system to study intercompartmental proteostasis: interrelation of mitochondrial function, longevity, and neurodegenerative diseases. Dev. Dyn. 239 (5), 1529–1538. doi:10.1002/dvdy.22292
Kumar, R., Chaudhary, A. K., Woytash, J., Inigo, J. R., Gokhale, A. A., Bshara, W., et al. (2022). A mitochondrial unfolded protein response inhibitor suppresses prostate cancer growth in mice via HSP60. J. Clin. Invest. 132 (13). doi:10.1172/JCI149906
Kuo, C. L., Chou, H. Y., Chiu, Y. C., Cheng, A. N., Fan, C. C., Chang, Y. N., et al. (2020). Mitochondrial oxidative stress by Lon-PYCR1 maintains an immunosuppressive tumor microenvironment that promotes cancer progression and metastasis. Cancer Lett. 474, 138–150. doi:10.1016/j.canlet.2020.01.019
Li, T. Y., Sleiman, M. B., Li, H., Gao, A. W., Mottis, A., Bachmann, A. M., et al. (2021). The transcriptional coactivator CBP/p300 is an evolutionarily conserved node that promotes longevity in response to mitochondrial stress. Nat. Aging 1 (2), 165–178. doi:10.1038/s43587-020-00025-z
Liu, D. X., Qian, D., Wang, B., Yang, J. M., and Lu, Z. (2011). p300-Dependent ATF5 acetylation is essential for Egr-1 gene activation and cell proliferation and survival. Mol. Cell. Biol. 31 (18), 3906–3916. doi:10.1128/MCB.05887-11
Liu, Y., Zhang, L., Zhang, S., Liu, J., Li, X., Yang, K., et al. (2023). ATF5 regulates tubulointerstitial injury in diabetic kidney disease via mitochondrial unfolded protein response. Mol. Med. 29 (1), 57. doi:10.1186/s10020-023-00651-4
Lu, H., Wang, X., Li, M., Ji, D., Liang, D., Liang, C., et al. (2022). Mitochondrial unfolded protein response and integrated stress response as promising therapeutic targets for mitochondrial diseases. Cells 12 (1), 20. doi:10.3390/cells12010020
Lu, Z., Chen, Y., Aponte, A. M., Battaglia, V., Gucek, M., and Sack, M. N. (2015). Prolonged fasting identifies heat shock protein 10 as a Sirtuin 3 substrate: elucidating a new mechanism linking mitochondrial protein acetylation to fatty acid oxidation enzyme folding and function. J. Biol. Chem. 290 (4), 2466–2476. doi:10.1074/jbc.M114.606228
Luo, J., Zeng, B., Tao, C., Lu, M., and Ren, G. (2020). ClpP regulates breast cancer cell proliferation, invasion and apoptosis by modulating the Src/PI3K/Akt signaling pathway. PeerJ 8, e8754. doi:10.7717/peerj.8754
Ma, C., Gao, B., Wang, Z., You, W., Yu, Z., Shen, H., et al. (2022). GrpEL1 regulates mitochondrial unfolded protein response after experimental subarachnoid hemorrhage in vivo and in vitro. Brain Res. Bull. 181, 97–108. doi:10.1016/j.brainresbull.2022.01.014
Merkwirth, C., Jovaisaite, V., Durieux, J., Matilainen, O., Jordan, S. D., Quiros, P. M., et al. (2016). Two conserved histone demethylases regulate mitochondrial stress-induced longevity. Cell. 165 (5), 1209–1223. doi:10.1016/j.cell.2016.04.012
Munch, C. (2018). The different axes of the mammalian mitochondrial unfolded protein response. BMC Biol. 16 (1), 81. doi:10.1186/s12915-018-0548-x
Munch, C., and Harper, J. W. (2016). Mitochondrial unfolded protein response controls matrix pre-RNA processing and translation. Nature 534 (7609), 710–713. doi:10.1038/nature18302
Na, Y., Kaul, S. C., Ryu, J., Lee, J. S., Ahn, H. M., Kaul, Z., et al. (2016). Stress chaperone mortalin contributes to epithelial-mesenchymal transition and cancer metastasis. Cancer Res. 76 (9), 2754–2765. doi:10.1158/0008-5472.CAN-15-2704
Nargund, A. M., Fiorese, C. J., Pellegrino, M. W., Deng, P., and Haynes, C. M. (2015). Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol. Cell. 58 (1), 123–133. doi:10.1016/j.molcel.2015.02.008
Nargund, A. M., Pellegrino, M. W., Fiorese, C. J., Baker, B. M., and Haynes, C. M. (2012). Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337 (6094), 587–590. doi:10.1126/science.1223560
Nukuda, A., Endoh, H., Yasuda, M., Mizutani, T., Kawabata, K., and Haga, H. (2016). Role of ATF5 in the invasive potential of diverse human cancer cell lines. Biochem. Biophys. Res. Commun. 474 (3), 509–514. doi:10.1016/j.bbrc.2016.04.131
Papa, L., and Germain, D. (2011). Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J. Cell. Sci. 124 (Pt 9), 1396–1402. doi:10.1242/jcs.078220
Papa, L., and Germain, D. (2014). SirT3 regulates the mitochondrial unfolded protein response. Mol. Cell. Biol. 34 (4), 699–710. doi:10.1128/MCB.01337-13
Pareek, G., and Pallanck, L. J. (2018). Inactivation of Lon protease reveals a link between mitochondrial unfolded protein stress and mitochondrial translation inhibition. Cell. Death Dis. 9 (12), 1168. doi:10.1038/s41419-018-1213-6
Pohl, F., Germann, A. L., Mao, J., Hou, S., Bakare, B., Kong Thoo Lin, P., et al. (2023). UNC-49 is a redox-sensitive GABA(A) receptor that regulates the mitochondrial unfolded protein response cell nonautonomously. Sci. Adv. 9 (44), eadh2584. doi:10.1126/sciadv.adh2584
Poveda-Huertes, D., Matic, S., Marada, A., Habernig, L., Licheva, M., Myketin, L., et al. (2020). An early mtUPR: redistribution of the nuclear transcription factor Rox1 to mitochondria protects against intramitochondrial proteotoxic aggregates. Mol. Cell. 77 (1), 180–188. doi:10.1016/j.molcel.2019.09.026
Quiros, P. M., Espanol, Y., Acin-Perez, R., Rodriguez, F., Barcena, C., Watanabe, K., et al. (2014). ATP-dependent Lon protease controls tumor bioenergetics by reprogramming mitochondrial activity. Cell. Rep. 8 (2), 542–556. doi:10.1016/j.celrep.2014.06.018
Quiros, P. M., Prado, M. A., Zamboni, N., D'Amico, D., Williams, R. W., Finley, D., et al. (2017). Multi-omics analysis identifies ATF4 as a key regulator of the mitochondrial stress response in mammals. J. Cell. Biol. 216 (7), 2027–2045. doi:10.1083/jcb.201702058
Schleit, J., Johnson, S. C., Bennett, C. F., Simko, M., Trongtham, N., Castanza, A., et al. (2013). Molecular mechanisms underlying genotype-dependent responses to dietary restriction. Aging Cell. 12 (6), 1050–1061. doi:10.1111/acel.12130
Seo, J. H., Rivadeneira, D. B., Caino, M. C., Chae, Y. C., Speicher, D. W., Tang, H. Y., et al. (2016). The mitochondrial unfoldase-peptidase complex ClpXP controls bioenergetics stress and metastasis. PLoS Biol. 14 (7), e1002507. doi:10.1371/journal.pbio.1002507
Shao, L. W., Peng, Q., Dong, M., Gao, K., Li, Y., Li, Y., et al. (2020). Histone deacetylase HDA-1 modulates mitochondrial stress response and longevity. Nat. Commun. 11 (1), 4639. doi:10.1038/s41467-020-18501-w
Smyrnias, I., Gray, S. P., Okonko, D. O., Sawyer, G., Zoccarato, A., Catibog, N., et al. (2019). Cardioprotective effect of the mitochondrial unfolded protein response during chronic pressure overload. J. Am. Coll. Cardiol. 73 (14), 1795–1806. doi:10.1016/j.jacc.2018.12.087
Sorrentino, V., Romani, M., Mouchiroud, L., Beck, J. S., Zhang, H., D’Amico, D., et al. (2017). Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 552 (7684), 187–193. doi:10.1038/nature25143
Suarez-Rivero, J. M., Pastor-Maldonado, C. J., Povea-Cabello, S., Alvarez-Cordoba, M., Villalon-Garcia, I., Talaveron-Rey, M., et al. (2021). Mitochondria and antibiotics: for good or for evil? Biomolecules 11 (7), 1050. doi:10.3390/biom11071050
Suarez-Rivero, J. M., Pastor-Maldonado, C. J., Povea-Cabello, S., Alvarez-Cordoba, M., Villalon-Garcia, I., Talaveron-Rey, M., et al. (2022a). Activation of the mitochondrial unfolded protein response: a new therapeutic target? Biomedicines 10 (7), 1611. doi:10.3390/biomedicines10071611
Suarez-Rivero, J. M., Pastor-Maldonado, C. J., Povea-Cabello, S., Alvarez-Cordoba, M., Villalon-Garcia, I., Talaveron-Rey, M., et al. (2022b). UPR(mt) activation improves pathological alterations in cellular models of mitochondrial diseases. Orphanet J. Rare Dis. 17 (1), 204. doi:10.1186/s13023-022-02331-8
Sutandy, F. X. R., Gossner, I., Tascher, G., and Munch, C. (2023). A cytosolic surveillance mechanism activates the mitochondrial UPR. Nature 618 (7966), 849–854. doi:10.1038/s41586-023-06142-0
Sun, X., Jefferson, P., Zhou, Q., Angelastro, J. M., and Greene, L. A. (2020). Dominant-negative ATF5 compromises cancer cell survival by targeting CEBPB and CEBPD. Mol. Cancer Res. 18 (2), 216–228. doi:10.1158/1541-7786.MCR-19-0631
Svagusa, T., Martinic, M., Martinic, M., Kovacevic, L., Sepac, A., Milicic, D., et al. (2020). Mitochondrial unfolded protein response, mitophagy and other mitochondrial quality control mechanisms in heart disease and aged heart. Croat. Med. J. 61 (2), 126–138. doi:10.3325/cmj.2020.61.126
Teske, B. F., Fusakio, M. E., Zhou, D., Shan, J., McClintick, J. N., Kilberg, M. S., et al. (2013). CHOP induces activating transcription factor 5 (ATF5) to trigger apoptosis in response to perturbations in protein homeostasis. Mol. Biol. Cell. 24 (15), 2477–2490. doi:10.1091/mbc.E13-01-0067
Tian, Y., Garcia, G., Bian, Q., Steffen, K. K., Joe, L., Wolff, S., et al. (2016). Mitochondrial stress induces chromatin reorganization to promote longevity and UPR(mt). Cell. 165 (5), 1197–1208. doi:10.1016/j.cell.2016.04.011
Torres, A. K., Cicali, K. A., Fleishhart, V., Mira, R. G., Tapia-Rojas, C., and Inestrosa, N. C. (2023). “Conserved regulation of mtUPR protein expression mediated by Wnt/β-catenin signaling in vitro and in the nematode Caenorhabditis elegans,” in XXXV annual meeting society of cell Biology of Chile; abstract N° 207 (Puerto Varas, Chile: Society of Cell Biology of Chile).
Torres, A. K., Jara, C., Park-Kang, H. S., Polanco, C. M., Tapia, D., Alarcon, F., et al. (2021). Synaptic mitochondria: an early target of amyloid-β and tau in alzheimer's disease. J. Alzheimers Dis. 84 (4), 1391–1414. doi:10.3233/JAD-215139
Torres, A. K., Tapia-Rojas, C., and Inestrosa, N. C. (2022a). “Activation of Wnt/β-catenin signaling as a new regulatory mechanism of the mtUPR-related protein expression,” in 13th Targeting mitochondria meeting (Berlin, Germany: World Mitochondrial Society).
Torres, A. K., Tapia-Rojas, C., and Inestrosa, N. C. (2022b). “Canonical Wnt signaling regulates the expression of mtUPR-related proteins in neuronal cells by a mechanism that requires β-catenin-dependent transcription,” in 2022 neuroscience meeting (San Diego, CA, USA: Society for Neuroscience).
Tran, H. C., and Van Aken, O. (2020). Mitochondrial unfolded protein-related responses across kingdoms: similar problems, different regulators. Mitochondrion 53, 166–177. doi:10.1016/j.mito.2020.05.009
Voos, W. (2009). Mitochondrial protein homeostasis: the cooperative roles of chaperones and proteases. Res. Microbiol. 160 (9), 718–725. doi:10.1016/j.resmic.2009.08.003
Wadhwa, R., Yaguchi, T., Hasan, M. K., Mitsui, Y., Reddel, R. R., and Kaul, S. C. (2002). Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp. Cell. Res. 274 (2), 246–253. doi:10.1006/excr.2002.5468
Wang, G., Fan, Y., Cao, P., and Tan, K. (2022). Insight into the mitochondrial unfolded protein response and cancer: opportunities and challenges. Cell. Biosci. 12 (1), 18. doi:10.1186/s13578-022-00747-0
Wu, L., Yan, X., Sun, R., Ma, Y., Yao, W., Gao, B., et al. (2023). Sirt3 restricts tumor initiation via promoting LONP1 deacetylation and K63 ubiquitination. J. Transl. Med. 21 (1), 81. doi:10.1186/s12967-023-03925-x
Xin, N., Durieux, J., Yang, C., Wolff, S., Kim, H. E., and Dillin, A. (2022). The UPRmt preserves mitochondrial import to extend lifespan. J. Cell. Biol. 221 (7), e202201071. doi:10.1083/jcb.202201071
Yamazaki, T., Ohmi, A., Kurumaya, H., Kato, K., Abe, T., Yamamoto, H., et al. (2010). Regulation of the human CHOP gene promoter by the stress response transcription factor ATF5 via the AARE1 site in human hepatoma HepG2 cells. Life Sci. 87 (9-10), 294–301. doi:10.1016/j.lfs.2010.07.006
Yoneda, T., Benedetti, C., Urano, F., Clark, S. G., Harding, H. P., and Ron, D. (2004). Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J. Cell. Sci. 117 (Pt 18), 4055–4066. doi:10.1242/jcs.01275
Yuan, Y., Gaither, K., Kim, E., Liu, E., Hu, M., Lengel, K., et al. (2018). SUMO2/3 modification of activating transcription factor 5 (ATF5) controls its dynamic translocation at the centrosome. J. Biol. Chem. 293 (8), 2939–2948. doi:10.1074/jbc.RA117.001151
Zhang, B., Tan, Y., Zhang, Z., Feng, P., Ding, W., Wang, Q., et al. (2020). Novel PGC-1alpha/ATF5 axis partly activates UPR(mt) and mediates cardioprotective role of Tetrahydrocurcumin in pathological cardiac hypertrophy. Oxid. Med. Cell Longev. 2020, 9187065. doi:10.1155/2020/9187065
Zhao, Q., Wang, J., Levichkin, I. V., Stasinopoulos, S., Ryan, M. T., and Hoogenraad, N. J. (2002). A mitochondrial specific stress response in mammalian cells. EMBO J. 21 (17), 4411–4419. doi:10.1093/emboj/cdf445
Zhou, Q., Zhu, L., Qiu, W., Liu, Y., Yang, F., Chen, W., et al. (2020). Nicotinamide riboside enhances mitochondrial proteostasis and adult neurogenesis through activation of mitochondrial unfolded protein response signaling in the brain of ALS SOD1(G93A) mice. Int. J. Biol. Sci. 16 (2), 284–297. doi:10.7150/ijbs.38487
Keywords: Caenorhabditis elegans, mitochondria, UPRmt, stress, misfolded protein, wnt signaling
Citation: Torres AK, Fleischhart V and Inestrosa NC (2024) Mitochondrial unfolded protein response (UPRmt): what we know thus far. Front. Cell Dev. Biol. 12:1405393. doi: 10.3389/fcell.2024.1405393
Received: 22 March 2024; Accepted: 06 May 2024;
Published: 31 May 2024.
Edited by:
Anne Chiaramello, George Washington University, United StatesReviewed by:
José Antonio Sánchez Alcázar, Universidad Pablo de Olavide, SpainCopyright © 2024 Torres, Fleischhart and Inestrosa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nibaldo C. Inestrosa, bmluZXN0cm9zYUBiaW8ucHVjLmNs