
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cell Dev. Biol. , 12 June 2024
Sec. Cancer Cell Biology
Volume 12 - 2024 | https://doi.org/10.3389/fcell.2024.1399065
This article is part of the Research Topic Lipid Alterations in Cancer Development, Resistance and Recurrence View all 5 articles
 Fahad Khan1†
Fahad Khan1† Deena Elsori2†Meenakshi Verma3Shivam Pandey4Safia Obaidur Rab5Samra Siddiqui6Nadiyah M. Alabdallah7,8
Deena Elsori2†Meenakshi Verma3Shivam Pandey4Safia Obaidur Rab5Samra Siddiqui6Nadiyah M. Alabdallah7,8 Mohd Saeed9*
Mohd Saeed9* Pratibha Pandey10,11*
Pratibha Pandey10,11*Lipids, the primary constituents of the cell membrane, play essential roles in nearly all cellular functions, such as cell-cell recognition, signaling transduction, and energy provision. Lipid metabolism is necessary for the maintenance of life since it regulates the balance between the processes of synthesis and breakdown. Increasing evidence suggests that cancer cells exhibit abnormal lipid metabolism, significantly affecting their malignant characteristics, including self-renewal, differentiation, invasion, metastasis, and drug sensitivity and resistance. Prominent oncogenic signaling pathways that modulate metabolic gene expression and elevate metabolic enzyme activity include phosphoinositide 3-kinase (PI3K)/AKT, MAPK, NF-kB, Wnt, Notch, and Hippo pathway. Conversely, when metabolic processes are not regulated, they can lead to malfunctions in cellular signal transduction pathways. This, in turn, enables uncontrolled cancer cell growth by providing the necessary energy, building blocks, and redox potentials. Therefore, targeting lipid metabolism-associated oncogenic signaling pathways could be an effective therapeutic approach to decrease cancer incidence and promote survival. This review sheds light on the interactions between lipid reprogramming and signaling pathways in cancer. Exploring lipid metabolism as a target could provide a promising approach for creating anticancer treatments by identifying metabolic inhibitors. Additionally, we have also provided an overview of the drugs targeting lipid metabolism in cancer in this review.
Modifying cellular metabolism and energy production is a widely recognized characteristic of cancer cells (Hanahan and Weinberg, 2011). Various routes, processes, and proteins have a role in altering the cellular use of metabolites and chemicals to facilitate abnormal cell replication, spread from the original tumor, formation of new tumors, and evasion of the immune system. Comprehending the interaction between lipids, their metabolic processes, and associated signaling is paramount. Lipids consist of a wide variety of biomolecules with different compositions and functions, such as sterol and prenol lipids, fatty acyls, and glycerophospholipids, and they also play a significant role in tumors (Darwish et al., 2024). Lipids create the physical obstacles surrounding cellular organelles and defend the cell from the extracellular environment. Lipids can be used as substrates for biomass synthesis or stored for future oxidation to generate energy that supports cell movement and growth (Hopperton et al., 2014; Yao et al., 2016; Lin et al., 2017). Furthermore, lipids can directly bind to receptors, which allows them to trigger intricate signaling pathways that encourage cell growth and migration (Auciello et al., 2019; Wu et al., 2024). A high level of lipids or an imbalance between saturated and unsaturated fatty acids can disturb the body’s equilibrium and increase cellular strain. A characteristic of abnormal cell development and cancer progression involves alterations in lipid metabolism and signaling, which has only been recently recognized. The synthesis of phospholipids for cellular membranes is crucial in cancer cells and must be carefully regulated to meet other metabolic requirements. During stromal invasion, lipid compounds such as circulating free fatty acids (FFAs) might impact cancer cells. These molecules can supply extra substrates for cell development or significantly change cell signaling. When considering the changes that occur in the microenvironment as a consequence of obesity, these effects become much more significant (Boden and Shulman, 2002; Almoraie and Shatwan, 2024). Tumor cells can enhance the process of de novo lipogenesis, the absorption of fatty acids (FA), and the of fatty acid oxidation (FAO) to generate energy and accumulate lipids.
The crucial connection between the tumor microenvironment (TME) in facilitating and sustaining tumor growth is widely acknowledged. The complete population of immunological and stromal cells, in addition to cancer cells, must be considered when investigating the effects of lipids inside the TME (Wang JJ. et al., 2018; Corn et al., 2020). The cellular components and their interactions within the TME and the differences in cellular metabolism during different stages of cancer growth exhibit intricate and ever-changing characteristics (Yuan et al., 2022). In order to create more effective treatments, it is crucial to understand how different cell types affect cancer cell lipid metabolism or how lipids in the TME can affect them. Numerous research works have delineated the influence of lipid metabolism on the TME during the advancement of cancer (Yu X. et al., 2021). The interaction between a modified lipid metabolism and the TME can significantly influence other characteristics of cancer, such as through the involvement of lipid mediators such prostaglandin E2 (PGE2) and lysophosphatidic acid (LPA). As a result of their effects on tumor-promoting inflammation, angiogenesis, stromal cells, and immune cell compartment, these mechanisms can determine tumor progression and therapy resistance (Zelenay et al., 2015; Chiurchiù et al., 2018; Leuti et al., 2020). A better understanding of lipid metabolic reprogramming within the cancer milieu could discover new prognostic indicators and therapeutic targets for cancer treatment. Despite the importance of lipid metabolism in cancer, the link between lipid metabolism and carcinogenic signaling is not well understood. In a previous review, the authors presented studies on lipid metabolism in cancer progression and targeted only the main oncogenic pathways of deregulated lipid metabolism in cancer to a limited extent (Fu et al., 2020). A limited number of other publications highlight one or more cross-linked signaling pathways of dysregulated lipid metabolism in carcinogenesis processes in cancer (Hoxhaj and Manning, 2020; Tošić and Frank, 2021) and in a specific type of cancer (Hon et al., 2021). Moreover, the dysregulated mechanisms of cancer lipid metabolism have also been described, with no focus on the related cross-linked pathways or their therapeutic effects (Santos and Schulze, 2012; Sangineto et al., 2020). Thus, the objective of this review is focused on examining and combining several oncological pathways functionally associated with disrupted lipid metabolism. Ultimately, we proposed many potential therapeutic applications that specifically focus on addressing the modified lipid metabolism in cancer via targeting these signaling cascades and other potential targets.
Tumors possess remarkable adaptability, enabling them to flourish in unfavorable circumstances, ensuring their development and survival. The plasticity of cells is demonstrated through various metabolic phenotypes, which are influenced by genetic and epigenetic changes. These phenotypes are further impacted by the TME characteristics, including factors like nutrient and oxygen availability, pH levels, and cellular interactions (Strickaert et al., 2017). Lipid metabolism reprogramming is a critical factor in promoting the development and progression of tumors. Tumors can exhibit significant changes in their lipid profiles (Jin et al., 2023). A schematic illustration of abnormal lipid metabolism in cancer progression is presented in Figure 1.
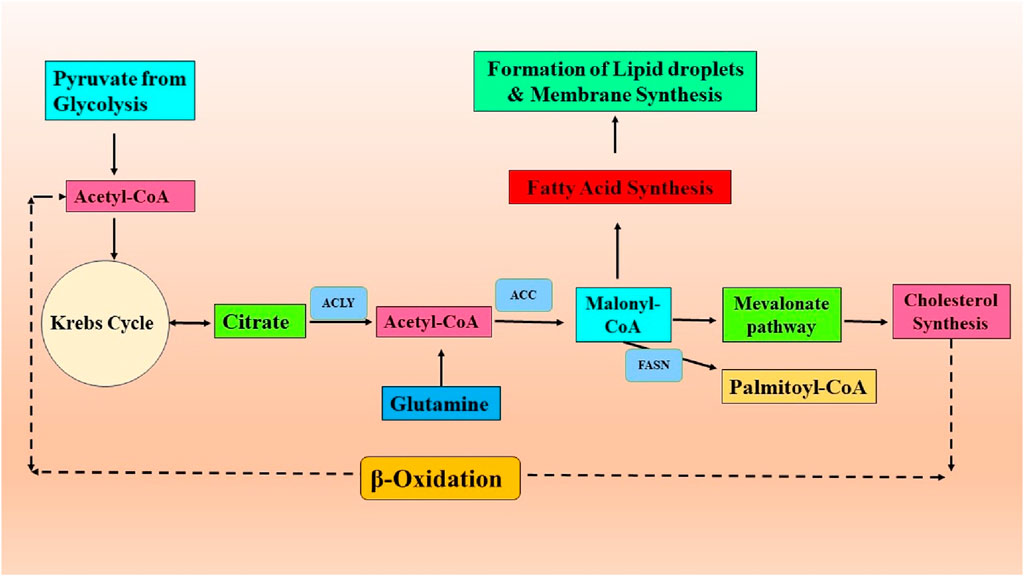
Figure 1. Lipid metabolic reprogramming in the process of cancer development.
Studies have long shown that certain cancers, including lung and breast cancer, exhibit an abnormal build-up of lipid droplets (LDs), which are associated with higher tumor grades (Medes et al., 1953). The aberration of lipid metabolism in cancer results from multiple intricate processes that contribute to the aggressiveness of tumors (Santos and Schulze, 2012). To begin with, cancer cells experience excessive stimulation of de novo lipid production or lipogenesis (Figure 1). More than 50 years ago, it was discovered that one of the critical characteristics of cancer cells is de novo lipid production (Medes et al., 1953). While non-transformed cells often obtain fatty acids from external food sources, cancer cells primarily rely on de novo fatty acid synthesis to produce lipids. This indicates that inhibiting this process could be an effective therapeutic strategy with few side effects. Lipogenesis necessitates the existence of acetyl-CoA, primarily obtained from the pyruvate derived from glucose, which enters the Krebs cycle (Figure 1). The Warburg effect in cancer can partially account for the excessive synthesis of fatty acids (Ocaña et al., 2020). Glucose provides carbon units for lipogenesis, although other carbon sources can be utilized if there is a shortfall in the glycolytic pathway. In low oxygen levels (hypoxia) conditions, glutamine can substitute glucose to generate α-ketoglutarate (αKG). This αKG molecule can then undergo reductive carboxylation through the action of isocitrate dehydrogenase 1 (IDH1) to make citrate. This process plays a crucial role in synthesizing lipids in cancer cells (Metallo et al., 2011). The somatic IDH1 R132H mutation, which generates the onco-metabolite 2-hydroxyglutarate, enhances de novo lipogenesis and FA oxidation in acute myeloid leukemia cells (Stuani et al., 2018). In contrast, acetate can also serve as a carbon source for lipid metabolism under low-nutrient circumstances. Acetate metabolism offers an alternative route for generating acetyl-coenzyme A (CoA) for lipogenesis, separate from converting citrate to acetyl-CoA. Cancer cells rely on certain members of the monocarboxylate transporter family to absorb external acetate. Once absorbed, acetate is transformed into acetyl-CoA by the action of acetyl-CoA synthetases (ACSS) (Walther et al., 2012; Schug et al., 2015). ACSS2 is overexpressed in human breast tumors, making them highly reliant on acetate for lipid production (Ligorio et al., 2021). These data emphasize the complex interconnection between glycolysis, glutaminolysis, acetate metabolism, and lipogenesis in cancer. An excessive amount of neutral lipids, such as triacylglycerols stored in LD, which build up in tumors to serve as a reserve of energy, are produced due to high levels of de novo FA synthesis. In addition, phospholipids are made to construct cancer cell membranes to meet the increased demand for cancer proliferation. Additionally, phospholipids are lipid messengers and intracellular signaling molecules in cancer, as reviewed by Cheng et al., 2016; Cheng et al., 2016). ATP citrate lyase (ACLY), acetyl-CoA carboxylase (ACC), and fatty acid synthase (FASN) are the most highly expressed lipogenic enzymes in several forms of cancer. Specifically, FASN has been identified as the crucial enzyme for lipogenesis, and its increased expression has been linked with an unfavorable prognosis in various cancer types (Kuhajda, 2006; Menendez and Lupu, 2007). Human malignancies, such as breast, colon, and prostate cancer, have elevated expression and activation of FASN, leading to an upsurge in the production of triacylglycerides (TG) deposited in lipid droplets (LDs). Therefore, targeting FASN in cancer is becoming increasingly intriguing (Mullen and Yet, 2015). Orlistat, a medication prescribed for treating obesity, has been found to specifically target FASN by blocking its thioesterase function, resulting in an anti-tumoral effect (Kridel et al., 2004). In addition, certain cancer types have been found to overexpress the pathway that produces mevalonate. This results in excessive cholesterol generation by converting acetyl-CoA by the enzyme 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) reductase. The presence of cholesterol in cancer cell membranes and abnormally elevated cholesterol levels in prostate cancer have been linked to the promotion of cancer growth (Xu et al., 2020). The sterol regulatory element-binding proteins (SREBP) transcription factors, which are triggered by the oncogenic PI3K/Akt/mTORC1 signaling cascade or cell cycle regulators, upregulate several enzymes involved in the fatty-acid and cholesterol-biosynthesis pathways in carcinoma (Ding et al., 2019). Monoacylglycerol lipase (MAGL) controls the amount of 2-arachidonoyl glycerol (2-AG) in the endocannabinoid system. Recent research has indicated that MAGL has a dual function, operating within the endocannabinoid system and exerting a significant influence on FFA levels to produce signaling lipids that promote tumor growth. MAGL, a serine hydrolase, is highly expressed in aggressive cancer cell lines, such as melanoma, ovarian, and breast cancer cell lines. It plays a role in the breakdown of monoacylglycerides into FFAs and glycerol (Nomura et al., 2010). Human prostate cancer cell lines that are not affected by androgens express higher levels of MAGL than androgen-dependent cell lines. Overexpression of MAGL in these cell lines has a dual effect on the endocannabinoid and fatty acid pathways, which promote the proliferation and aggressiveness of cancer cells (Nomura et al., 2011). MAGL is a constituent of a genetic signaling system associated with epithelial-mesenchymal transition (EMT) and the characteristics of stem cells in prostate cancer (Nomura et al., 2011).
Furthermore, in addition to the process of lipogenesis, it was noted that fatty acids (FAs), whether obtained from sources outside the cell or released from internal lipid reserves, can undergo oxidation within the mitochondria of cancer cells (Figure 1). In such circumstances, lipids serve as catalytic fuels, undergoing FAO or lipolysis to generate energy for cancer cells through ATP synthesis. In certain types of cancer, such as B cell lymphoma, the primary method of energy production is through mitochondrial FAO rather than relying on glycolysis (Caro et al., 2012). Three primary protein families participate in the cellular intake of fatty acids: the fatty acid translocase (FAT/CD 36/SR-B2) family, the membrane fatty acid binding protein (FABPm) family, and the fatty acid transport protein family (FATP) (Abumrad et al., 1999; Stremmel et al., 2001). Breast, ovarian, and colon malignancies have been shown to overexpress CD 36 (Lambrescu et al., 2024). There is strong evidence that CD 36 is involved in the aggressiveness of prostate cancer (Watt et al., 2019). Interestingly, studies have linked its overexpression to the epithelial-mesenchymal transition in hepatocellular carcinoma and, on the flip side, to leukemia survival (Farge et al., 2023; Deng et al., 2024). After entering the cytoplasm, FAs are attached to fatty acid-binding proteins (FABPs) and subsequently transformed into acyl-CoA. Acyl-CoA transportation into the mitochondria is facilitated by the protein carnitine palmitoyltransferase 1 (CPT1), which is frequently increased in cancer cells due to metabolic stress (Wang et al., 2018b). Carnitine palmitoyltransferase 1 (CPT1) is the primary enzyme that regulates the FAO. FAO occurs within the mitochondrial matrix and involves a cyclic catabolic process that produces nicotinamide adenine dinucleotide (NADH), flavin adenine dinucleotide (FADH2), NADPH, and ATP. In addition to mitochondria, FAO occurs in peroxisomes within cancer cells (Shen et al., 2020). Lipolysis and lipogenesis can co-occur in cancer cells (Carracedo et al., 2013). The cellular environment alters lipids’ metabolic process in cancer cells, namely, by the presence of adipocytes. Cancer-associated adipocytes serve as a significant supplier of external lipids for cancer cells. Research has shown a metabolic interaction between adipocytes and cancer cells, which causes the cancer cells to exhibit an aggressive phenotype (Mukherjee et al., 2022; Grunt et al., 2024). Cancer cells stimulate the release of FAs from nearby adipocytes through the secretion of soluble substances, including hormone-sensitive lipase and growth differentiation factor 15 (GDF15) (Bouche and Quail, 2023). The coexistence of cancer cells with adipocytes leads to the elevation of CD36 expression, resulting in an enhanced absorption of fatty acids by cancer cells. As a result, cancer cells absorb FAs generated by adipocytes. These fatty acids are then broken down in the mitochondria to provide the necessary energy for cancer cell growth, survival, spread, metastasis, and drug resistance (Kuo and Ann, 2018; Germain et al., 2020; Zakic et al., 2024). These findings emphasize the interaction between adipose tissue and cancer cells, which promotes cancer FAO and aggressiveness.
Lipid metabolic reprogramming is an important characteristic of tumors that contributes significantly to the development of cancer and resistance to therapy. Cancer cells rewire their metabolic pathways to produce a variety of essential signaling metabolites that influence signaling and epigenetic/transcriptional control, ultimately affecting cancer cell survival and proliferation in addition to energy and building blocks (Zhang et al., 2022). Gaining more insight into the mechanisms governing metabolic reprogramming in cancer could lead to the development of novel cancer targeting approaches. Recent research has indicated that dysregulated transcription factors have been detected in several types of human malignancies and have a substantial influence on lipid metabolism and signaling pathways in cancer. In this section, we examine the interplay between signaling and transcription factors in lipid metabolic reprogramming, and highlight the major transcription factors involved in lipid metabolic regulation.
ChREBP is a glucose-regulated transcription factor that plays a crucial role in regulating enzymes involved in fatty acid synthesis (Uyeda et al., 2002). In response to glucose, ChREBP translocates from the cytoplasm to the nucleus and forms a complex with MLX1 to bind to the carbohydrate response elements of lipogenic genes. Nevertheless, the function of ChREBP is inhibited by adenosine monophosphate (AMP), ketone molecules, and cAMP (Ortega-Prieto and Postic, 2019). The ChREBP/MLX heterodimer controls glucose and lipid metabolism by regulating the expression of specific enzymes involved in glycolysis (PKLR, FK, GLUT2, GLUT4), gluconeogenesis (G6PC), and lipogenesis (FASN, ACC1, SCD1, Elovl6) (Ishii et al., 2004). (ChREBP) ChREBP mutant mice exhibit elevated hepatic glucose storage and reduced hepatic fatty acid levels compared with wild-type mice (Iizuka et al., 2004). ChREBP plays a significant role in the development of various cancers (Figure 2). ChREBP knockdown via siRNA effectively suppresses aerobic glycolysis and lipid production in colon cancer cells. Furthermore, inhibition of ChREBP triggers the activation of p53, leading to the interruption of the cell cycle and a decrease in the formation of colon cancer in living organisms. This suggests that ChREBP has an oncogenic role (Lei et al., 2020a). MLXIPL mRNA showed positive connections with glycolytic and lipogenic genes in chromatin immunoprecipitation analysis of human hepatocellular carcinoma (HCC) and breast cancer. The expression of glucose transporter 1 is dramatically elevated in individuals with hepatocellular carcinoma (HCC) and there is a positive correlation between its expression and ChREBP. Conversely, a contrasting relationship was observed between the ChREBP protein and pyruvate dehydrogenase kinase 2 genes. These genes are responsible for the deactivation of acetyl-CoA formation (Lei et al., 2020b). Cancer cells exhibit a preference for the transformation of pyruvate into acetyl-CoA as opposed to the buildup of pyruvate (Contractor and Harris, 2012). Given the positive correlation between ChREBP protein and tumor malignancy, our findings indicate that the carcinogenic function of ChREBP/MLXIPL arises from the conversion of glucose into fat (Airley et al., 2014).
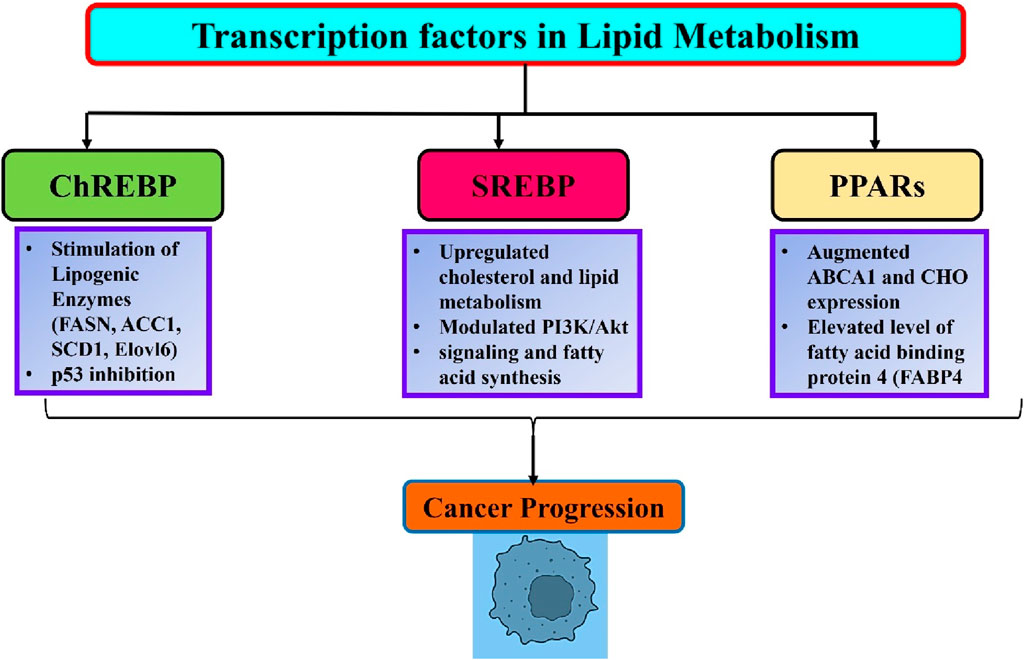
Figure 2. An overview of lipid metabolism related transcription factors and their role in cancer progression.
SREBP-1 functions as a transcription factor that controls lipid metabolism and reprograms tumor metabolism (Guo et al., 2014). SREBP controls the genes responsible for cholesterol production and fat breakdown (Horton et al., 2002). The levels and activity of SREBPs in normal tissues are closely regulated by endogenous sterol levels via a negative feedback mechanism. Nevertheless, cancer cells exploit the process of absorbing large amounts of glucose to produce fatty acids and cholesterol, which are essential for creating new cell membranes and lipid rafts (Vona et al., 2021). SREBP-1 has been recognized as a crucial factor in combining the flow of metabolism from glycolysis, which is influenced by PI3K/Akt signaling and fatty acid synthesis (Ricoult et al., 2016). SREBP-1 also controlled glutamine metabolism to promote lipid production (Figure 2) (Jhu et al., 2021). Moreover, research has demonstrated that SREBP-1 is upregulated in various types of malignancies and plays a role in promoting tumor growth (Cheng et al., 2018). Li CF. et al. (2017) found that by inhibiting the breakdown of SREBP, HCC progression can be reduced by regulating fatty acid (FA) and cholesterol metabolism. The anti-tumor capabilities of inhibiting the transfer of the SREBP-SCAP complex from the endoplasmic reticulum to the Golgi apparatus have been observed in many cancer cell lines, including HeLa, T98, and U2OS. Furthermore, the prevention of SREBP maturation not only hinders lipid metabolism but also disrupts the process of tubulin polymerization and construction of the mitotic spindle. This ultimately results in the reduced proliferation and migration of cancer cells (Gholkar et al., 2016). Overall, SREBP-1 is essential for the connection between oncogenic signaling and lipid metabolism (Figure 2).
PPARs, which are a subset of transcription factors, are part of the nuclear receptor family of proteins and have a significant impact on lipid metabolism. PPARs are composed of three primary isoforms, PPAR-α, PPAR-β, and PPAR-γ. These proteins exhibit varying levels of expression in various tissues, resulting in unique noncanonical activities in different cells. PPAR-α, which is present in adipose tissues, the liver, and the heart, controls fatty acid β-oxidation in fats. PPAR-β is widely expressed in several organs and plays a role in fat breakdown through β-oxidation (Brunetti et al., 2019). PPAR-γ expressed in adipose tissues promotes the development of adipose tissue and regulates fat production (Lodhi and Semenkovich, 2014; Ma et al., 2021). These proteins have been shown to control the function of other mitochondrial proteins, including carnitine palmitoyltransferase, citrate synthase, and cytochrome oxidases Hernandez-Quiles et al., 2021; Porcuna et al., 2021).
Moreover, significant connections between PPARs and tumors have been discovered in clinical and preclinical settings. The pan-cancer datasets of patients with 21 different types of cancer demonstrated that dysregulation of PPARs signaling disrupts several pathways related to lipid metabolism in tumor cells, which has a direct influence on the survival of patients (Chang and Lai, 2019. In addition, mice administered a PPARα agonist showed reduced body weight and a higher occurrence of liver cancer. Visible tumors and nodules with a diameter of approximately 11 mm were observed in the livers of mice treated with PPARα agonists (Peters et al., 1997). PPARδ promotes the formation of xenograft tumors derived from prostate cancer cells by modulating ATP-binding cassette transporter 1 (ABCA1) gene. ABCA1 is associated with efflux of cholesterol (CHO). PPARδ directly enhances the amount of ABCA1 mRNA and CHO expression in the cell membrane, leading to subsequent tumor growth (Her et al., 2013). PPARγ exhibits both tumor-suppressive and oncogenic effects in many types of cancers. PPARγ-mediated lipid production in lung cancer cells leads to a significant increase in mitochondrial reactive oxygen species stress, which plays a crucial role in suppressing tumor growth (Phan et al., 2017). Furthermore, pharmacological stimulation of PPARγ leads to an elevation in the production of fatty acid binding protein 4 (FABP4), which is followed by an increase in the amount of reactive oxygen species in lung cancer cells. The activation of PPARγ leads to the upregulation of FABP4 and lipoprotein lipase, which are associated with a more favorable prognosis in patients with lung and renal cancer (Hua et al., 2019). Conversely, the relationship between PPARγ and Nur77 has antagonistic effects on breast cancer. Nur77 engages PPARγ in the CD36 promoter and FABP4 to inhibit the transcription of these genes, thereby inhibiting the intake of fatty acids and the proliferation of cells. PPARγ directly interacts with Nur77 and enhances its ubiquitination of Nur77 through the action of the ubiquitin ligase Trim13, thereby exacerbating breast cancer (Yang et al., 2020). It is worth mentioning that dietary FAs have a direct binding effect on PPARs and imitate the actions of synthetic agonists that activate PPARs (Kliewer et al., 1997). PPARs detect fatty acid signals originating from dietary lipids, and act as lipid regulators to promote cancer progression (Figure 2). For example, a high-fat diet (HFD) stimulates the proliferation of tumor cells and facilitates the spread of colon cancer cells to the liver in mice. Nevertheless, deletion of PPARδ in mice effectively prevents the impact of a high-fat diet (HFD), as well as the expression of Nanog and CD44. This clearly indicates that PPARs play a crucial role in facilitating the tumor-promoting effect of HFD on cancer (Wang et al., 2019).
However, there are two more transcription factors of lipid metabolism FXR and LXR, their positive role in lipid metabolism reprogramming in cancer is still not validated yet. Therefore, we have discussed these two in detail, for more details please refer (Jeong et al., 2021).
Interestingly, some data suggest that cancer signaling controls lipid production, and conversely, lipogenesis plays a role in regulating oncogenic signaling. In general, complex crosstalk synchronizes anabolic activities, DNA replication, and the cell cycle to supply the building blocks required for cell expansion and multiplication. The majority of fatty acids produced by FASN are incorporated into phospho- and glycolipids, which are important components of cell membranes (Zhou et al., 2003; Hilvo et al., 2011). Research has revealed that FASN contributes to the development of membrane lipid rafts, which in turn promote the clustering of signal protein complexes inside these crucial membrane microdomains. In addition, FASN enhances the production of membrane RTKs, including EGFR, ERBB2, and hepatocyte growth factor receptor/c-MET, in breast, ovarian, and prostate cancers, as well as non-Hodgkin lymphoma (Menendez et al., 2004a; Uddin et al., 2010). It also triggers the attraction of adaptor proteins such as GRB2 to EGF-activated EGFR (Wagner et al., 2017). As a result, when exposed to FASN inhibitory medications, the expression of EGFR and ERBB2 as well as the clustering of macromolecular signaling complexes at the membrane are compromised (Grunt et al., 2009). In conclusion, FASN is essential for the synthesis and development of membrane-signaling complexes (Swinnen et al., 2003). FASN can facilitate signal transduction by supplying signaling lipids including diacylglycerol, phosphatidylinositol, and phosphatidylinositol 3,4,5-trisphosphate (PIP3) (Benjamin et al., 2015). These secondary messengers play essential roles in mitogenic and pro-survival signaling. Furthermore, lipids produced by FASN can be utilized for the post-translational lipidation of proteins, which is necessary for proper intracellular localization and functioning of several signaling proteins. Additionally, lipidomic analyses have demonstrated that an overactive FASN enzyme increases the amount of saturated fatty acids compared to unsaturated fatty acids in the membranes of cancerous cells. This leads to a “saturated” lipid profile, which affects signaling processes and provides resistance against oxidative stress and cytotoxic drugs (Rysman et al., 2010). Since blocking or downregulating FASN stops the production of newly synthesized lipids, the processes of signal creation, propagation, and transmission at the cell membrane as well as downstream signaling cascades gradually deteriorate (Tomek et al., 2011).
Cancer is characterized by significant modification of phosphatidyl inositides (PIs) in cell membranes. PIs play a crucial role in defining the characteristics and functionality of membranes and also act as regulators of membrane trafficking (Vicinanza et al., 2008; Ooms et al., 2015). PI (3,4,5) P3 and PI (4,5) P2 play a significant role in tumor cell migration and metastasis, among other factors. They control cellular processes by enlisting, stimulating, or restraining proteins in the plasma membrane to influence actin dynamics, resulting in changes in cellular motility and metastatic ability. Phosphoinositide 3-kinase (PI3K) facilitates the formation of phosphatidylinositol-3,4,5-trisphosphate [PI (3,4,5) P3] from its precursor PI (4,5) P2. Increased levels of PI (3,4,5) P3 promote the localization of guanine nucleotide exchange factors (such as Vav and Tiam) of Rho GTPases to the cell membrane. This leads to the rearrangement of the cytoskeleton, which in turn improves cell migration and metastasis (Fritz and Kaina, 2006). Nevertheless, in breast cancer cells, a decrease in the amount of PI (4,5) P2 in the plasma membrane increases the ability of the cells to move and spread to other parts of the body (Sengelaub et al., 2016). The PI3K/AKT/mTORC axis affects different elements of metabolism and influences lipid anabolic metabolism signaling. PI3Ks are triggered by receptors on the cell surface and then convert the inputs into the buildup of the signaling lipid PI (3,4,5) P3. This lipid, in turn, enables the activation of many effectors such as the serine/threonine kinase AKT. AKT can add a phosphate group to ATP-citrate lyase, which in turn triggers the activation of several genes that play a role in the production of cholesterol and fatty acids (Manning and Cantley, 2007).
Sphingolipid metabolites, such as ceramide and sphingosine, are important regulators of angiogenesis, metastasis, and cell survival (Milstien and Spiegel, 2006). The biologically active lipid sphingosine-1-phosphate (S1P) is produced by the enzymes SPHK1 and SPHK2, and can be irreversibly destroyed by S1P lyase (SGPL1) or dephosphorylated by sphingosine phosphatase (Le Stunff et al., 2002). Five cell surface G protein-coupled receptors, collectively referred to as S1PR1-5 are the main mediators of autocrine or paracrine signaling by which S1P functions (Pyne and Pyne, 2010). SP1 can also bind to intracellular targets, including NF-κB and HDAC1/2 (histone deacetylases 1/2) (Maceyka et al., 2012). Increased S1P levels in oral squamous cell carcinoma (OSCC) were shown by Patmanathan et al. (2016) to be a result of decreased SGPL1 levels and increased SPHK1 transcription. S1P increased cell motility and invasion and shielded OSCC cells from cisplatin-induced cell death. Moreover, it has been shown that S1PR1 is intimately related to the ongoing activation of signal transducer and activator of transcription-3 (STAT3) and the expression of IL-6 in both tumor cells and the surrounding tissues. These elements are important in the development of tumor malignancies and their spread to distant locations (Lee et al., 2010).
When FAs are incorporated into cancer cell membranes, the membrane phases separate, cell contact decreases, surface adhesion increases, and tissues invade (Mitra et al., 2014). In an animal cancer model, Le et al. (2009) showed that mice fed a high-fat diet (HD) and an excess of plasma FFAs had an early initiation of a considerable number of CTCs and an elevated incidence of lung metastasis. The polarized distribution of cellular content results from the separation of the cancer cell membrane into many phases, caused by the high concentration of polyunsaturated FFAs in the blood plasma. The exposed cells had a strong resemblance to circulating tumour cells (CTCs) obtained from animals fed a high-fat diet. A class of tiny intracellular proteins known as FABPs binds lipids. FABP1–10 are the ten isoforms or variations of FABPs. Attaching and storing FAs and directing them to proper locations inside the cell-the plasma membrane, nucleus, endoplasmic reticulum, mitochondria, and peroxisomes-are functions of FABPs (Furuhashi and Hotamisligil, 2008). Inactive components of cell membranes and lipids form lipid rafts that facilitate the recruitment of signaling proteins and the interaction of proteins to initiate signal transduction pathways. As such, FABPs, which act as lipid chaperones, may improve signaling associated with metastasis by interacting with other proteins when they attach to membrane rafts. L-FABP, or liver fatty acid-binding protein, binds particularly to hydrophobic substances such as cholesterol and bile acids, as well as ligands such as long-chain fatty acids. Ku et al. (2016) found that the AKT/mTOR/P70S6K/4EBP1 and Src/FAK/cdc42 pathways are activated when L-FABP interacts with VEGFR2 in membrane rafts. The hepatocellular carcinoma cells to grow new blood vessels and migrate was improved by this activation, which also increased VEGF-A expression (Ku et al., 2016). Moreover, many FABP isoforms are strongly associated with the development of cancer metastasis. Increased expression of FABP3 or FABP4 in non-small cell lung cancer (NSCLC) negatively correlates with a more advanced tumor node metastatic stage and reduces the overall survival of patients with NSCLC (Tang et al., 2016). FABP5 inhibits EGF-induced signaling pathways linked to metastasis and partially inhibits the degradation of EGFR by proteasomes, thereby promoting the spread of triple-negative breast cancer. Moreover, FABP7 participates in fatty acid metabolism and may be a useful marker of metastatic melanoma (Rodic et al., 2014).
The interplay between cell metabolism and epigenetic gene regulation is now widely recognized, and cancer cells take advantage of this molecular connection (Carrer and Wellen, 2015). Recently, it was discovered that ACC can regulate global histone acetylation. This establishes a direct connection between anabolic metabolism and gene regulation through epigenetics (Galdieri and Vancura, 2012). Furthermore, free acetate, when converted to acetyl-CoA by acetyl-CoA synthetase (ACSS), functions as an epigenetic metabolite and enhances lipid synthesis under low oxygen conditions (Gao et al., 2016). As previously demonstrated, ACLY plays a crucial role in histone acetylation (Wellen et al., 2009). Lipids can serve as carbon sources and produce acetyl-CoA, which is utilized for histone acetylation. Furthermore, experimental data showed that ACLY preserves DNA methylation patterns in adipocytes and controls DNMT1 expression (McDonnell et al., 2016). DNA methylation relies on the presence of a methyl donor. Methionine adenosyltransferase metabolizes and converts the important amino acid methionine into S-adenosylmethionine (SAM). Every cell can produce S-adenosylmethionine (SAM). This is necessary as a preliminary step for the methylation of various substances such as DNA, RNA, proteins, lipids, and other small molecules (Varela-Rey et al., 2014). This process produces a demethylated compound called S-adenosylhomocysteine (SAH), which plays a role in cellular sulfur metabolism. S-adenosylmethionine (SAM) plays a crucial role as a co-substrate that facilitates the transfer of methyl groups by DNMTs. This process is essential for methylation of CpG islands located in the promoter regions of genes. In addition to this crucial epigenetic regulation process, researchers have also detected the methylation of certain lysines in the nucleosomal core histone proteins H3 and H4, with variations including mono-, di-, and tri-methylation. The presence of methylated lysine residues in histone proteins can either activate or inhibit gene transcription. This effect depends on the number of methyl groups added and the overall structural conditions around methylated lysine (Du et al., 2015). Interestingly, it has been found that there is an unanticipated correlation between the SAM:SAH ratio and methylation of either histone H3 and phosphatase PP2A or the membrane phospholipid phosphatidylethanolamine (PE). Existing evidence suggests that PE can function as a sink for cellular methyl groups, leading to the consumption of S-adenosylmethionine (SAM). Additionally, PE produces S-adenosylhomocysteine (SAH), which can be utilized in the transsulfuration pathway to convert methionine into cysteine. However, in cells that are not part of the liver, methyl is taken up by histone H3 and phosphatase PP2A, instead of PE (Ye C. et al., 2017). These observations indicate a reciprocal relationship between the methylation of membrane lipids, epigenetic control of histone proteins, and phosphorylation-dependent cellular signaling.
Dysregulation of the epigenome and growth signals is also considered as hallmark of cancer. A recent study has uncovered the pathways by which oncogenic signaling controls the regulation of epigenetic genes. Growth-promoting ligands interact with certain receptor tyrosine kinases (RTKs) and initiate RAS activation, which subsequently promotes downstream signaling pathways such as PI3K-AKT-mTORC1. PI3K activity stimulates cellular metabolism through multiple mechanisms. For example, AKT boosts glycolysis by upregulating the expression of glucose transporter 1 (GLUT1) on the cell membrane, activating phosphofructokinase, and promoting the interaction of hexokinases 1 and 2 with mitochondria. In addition, AKT stimulates the transcription of ACLY, which generates acetyl-CoA via the mTORC1-SREBP-1c pathway (Muñoz-Pinedo et al., 2012). In addition, AKT phosphorylates ACLY, resulting in stimulation of its catalytic activity (Potapova et al., 2000). In summary, AKT signaling promotes glucose absorption and metabolism through glycolysis, leading to the generation of significant amounts of citrate derived from the citric acid (Krebs) cycle. Furthermore, it enhanced the production of ACLY and initiated the enzymatic degradation of citrate into acetyl-CoA by ACLY. Therefore, AKT promotes the synthesis of acetyl-CoA in the cytosol. Acetyl-CoA is a crucial component necessary for histone lysine acetyl transferases to modify nucleosomal core histone proteins by acetylation and for FASN to produce lipids. It is generally accepted that a complex regulatory network that functions in numerous directions connects lipid metabolism, oncogenic signaling, and cancer epigenetics (Kinnaird et al., 2016).
While the significance of lipid metabolism concerning oncogenic pathways is acknowledged, the influence of lipid metabolism on the regulation of oncogenic signaling is gradually becoming apparent (Harayama and Riezman, 2018; Fu et al., 2020; Snaebjornsson et al., 2020). Cancer cells commonly exhibit excessive activation of oncogenic signaling pathways. Abnormal activation of critical pathways has been linked to changes in lipid metabolism in various types of cancer. In this subsequent part, we provide a concise overview of the latest significant breakthroughs in comprehending the role of lipid metabolism in promoting cancer growth and development through the modulation of diverse pathways.
Cancer cells frequently exhibit abnormal activation of lipid biosynthesis. Most cells in our body obtain the lipids they need by taking up fatty acids and lipoproteins from the bloodstream (Röhrig and Schulze, 2016). However, cancer cells activate de novo lipid biosynthesis to produce their lipids. This allows them to create cellular membranes and support rapid growth and multiplication (Menendez and Lupu, 2007). Sterols and fatty acids are both derived from cytosolic acetyl-CoA, which is synthesized either from citrate, an intermediate of the TCA cycle, through ATP citrate lyase (ACLY), or from acetate through acetyl-CoA synthetase. The PI3K–AKT pathway stimulates the production of new lipids through both post-translational and transcriptional mechanisms. AKT can facilitate the formation of new lipids by directly adding a phosphate group to ACLY, which enhances its activity and promotes the generation of cytosolic acetyl-CoA (Potapova et al., 2000; Berwick et al., 2002). This acetyl-CoA is then utilized to synthesize sterols, fatty acids, and protein acetylation reactions (Figure 3A). Previous studies have indicated that the AKT-ACLY pathway enhances tumor development and has a widespread impact on histone acetylation (Lee et al., 2014; Carrer et al., 2019). Besides its role in regulating cancer development through the PI3K-AKT pathway, ACLY is often excessively expressed in different human cancers. Blocking ACLY activity reduces the growth of cancer cells both in laboratory settings and living organisms, suggesting that this enzyme could be a promising target for cancer treatment (Hatzivassiliou et al., 2005; Zu et al., 2012). Due to its crucial involvement in lipid production, ACLY inhibitors, initially designed for metabolic conditions like hypercholesterolemia and type 2 diabetes, are now being explored as potential treatments for cancer (Granchi, 2018; Ray et al., 2019). The AKT signaling pathway also stimulates de novo lipid synthesis by activating the sterol regulatory element-binding protein (SREBP) family of transcription factors (SREBP1a, SREBP1c, and SREBP2). These transcription elements activate the expression of almost all enzymes involved in fatty acid and sterol synthesis, particularly ACLY (Porstmann et al., 2005; Li N. et al., 2023). The SREBPs are present as inactive transmembrane proteins in the endoplasmic reticulum (ER) and need to be transported to the Golgi apparatus for proteolytic processing to become active. The processing of SREBP results in the release of the N-terminal segment of the protein, which acts as the mature and active form. This segment then moves to the nucleus and starts the transcription process by binding to sterol response elements present in the promoter region related to lipid production and the production of NADPH, which is necessary for lipid synthesis. AKT triggers the activation of SREBP through a minimum of two subsequent pathways. Studies have demonstrated that mTORC1 promotes the processing and movement of SREBPs into the nucleus through various suggested mechanisms. This results in the stimulation of genes involved in lipid production and an elevation in the synthesis of new lipids in the liver in response to insulin or in cancer cells following the activation of oncogenic PI3K or Ras (Yecies et al., 2011; Owen et al., 2012; Ricoult et al., 2016). After processing, the fully developed active form of SREBP is directed towards ubiquitin-dependent degradation through GSK3-mediated phosphorylation (Kim et al., 2004; Sundqvist et al., 2005). Therefore, the activation of AKT signaling can enhance the processing of SREBP by activating mTORC1, and it can also increase the stability of the processed active SREBP by inhibiting GSK3. Recently, it has been reported that MYC collaborates with SREBP to stimulate lipogenesis and facilitate cancer proliferation (Gouw et al., 2019). Notably, many messenger RNAs stimulated by SREBP are controlled by the serine/arginine-rich (SR) protein family of splicing factors. Stimulation of mTORC1 signaling has been observed to promote the splicing of these mRNAs that encode lipogenic enzymes. This stimulation occurs through activating S6 kinase 1 (S6K), a downstream target of mTORC1, which phosphorylates and triggers SR protein kinase 2 (SRPK2). As a result, the SR proteins are phosphorylated and activated, leading to enhanced splicing of the mRNAs (Lee et al., 2017). Several factors and enzymes that play a role in the production of new lipids, such as SREBPs, SRPK2, and the lipogenic enzymes activated by SREBPs, have been observed to be increased in various types of cancer. These factors and enzymes are considered potential targets for cancer treatment. Collectively, these transcriptional programs, in conjunction with the aforementioned post-translational regulatory mechanisms, effectively amplify lipogenesis in cancer cells stimulated by oncogenic PI3K-Akt-mTOR signaling (Urbanski et al., 2018).
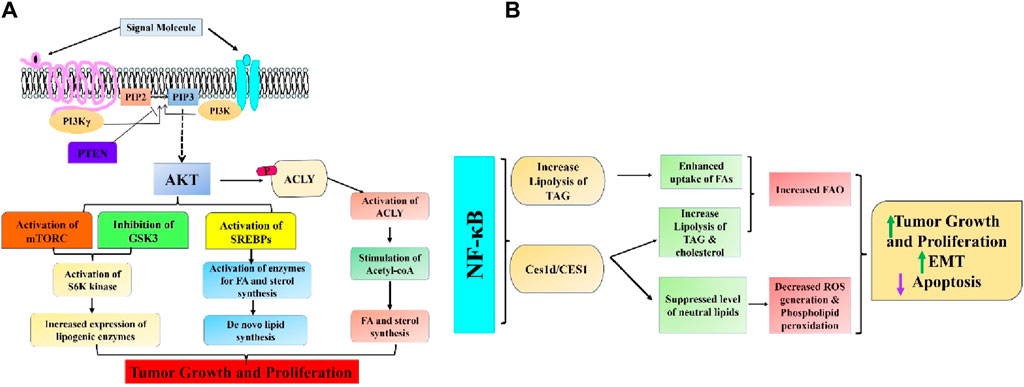
Figure 3. Intricate association between (A) PI3K/Akt/mTOR and (B) NF-κB signaling pathways and aberrant lipid metabolism in cancer.
NF-κB, a crucial transcription factor implicated in controlling the inflammatory response, is also a significant signaling pathway for cell survival that is frequently hyperactivated in numerous cancer types (Zinatizadeh et al., 2020). New research has demonstrated that NF-κB, through controlling metabolic programs affecting glycolysis, glutaminolysis, OXPHOS, and other metabolic pathways, is essential for tumor cells’ response to microenvironments deficient in nutrients. Preliminary findings in tumor cell lines indicated that NF-κB has a role in the development of colorectal cancer (CRC) by facilitating metabolic adaption to energy stress (Mauro et al., 2011). A recent study showed that the NF-κB-dependent adaptative mechanism is intrinsically connected to the regulation of lipid metabolism. This was supported by the observed changes in lipidomic profiles of RelA/RELA-deficient mice embryonic fibroblasts and human CRC cell lines (Capece et al., 2021). Recently, a study in Drosophila melanogaster revealed evidence of a historical link between nutrient imbalance and lipid metabolism. The link between these two factors is facilitated by NF-κB, a protein that has a vital function in the body’s reaction to fasting. NF-κB restrains the breakdown of triacylglycerol lipids by repressing the gene that encodes adipose triglyceride lipase (ATGL)/Brummer in the fat tissue. This metabolic process is recognized for maintaining cellular triacylglycerol reserves, the primary energy source in multicellular organisms (Molaei et al., 2019). The stimulation of NF-κB in human colorectal cancer cell lines during nutrient deprivation can enhance triacylglycerol lipolysis, which in turn mobilizes endogenous FFAs from lipid droplets and fuels FAO and OXPHOS during energy supply, in contrast to the result of this ancient adaptive mechanism in insects. Hence, the suppression of NF-κB hindered the breakdown of lipids during periods of low energy, reducing the flow of oxidative phosphorylation and the synthesis of ATP in the mitochondria, finally leading to metabolic crisis and cell death (Capece et al., 2021). Through integrating a metabolomic and transcriptome analysis, the researchers discovered carboxylesterase 1d (Ces1d), the mouse equivalent of human CES1. They found that Ces1d is a lipase regulated by NF-κB and has a crucial role in increasing the survival and adaptation of cancer cells to limited nutrition in aggressive human colorectal cancer (CRC). Furthermore, the metastasis-prone CRC consensus molecular subtype (CMS)4 exhibited higher CES1 expression and NF-κB activity, which were linked to inflammation, obesity, the epithelial-to-mesenchymal transition (EMT), and poorer clinical outcomes for CRC patients (Capece et al., 2021). Increased CES1 expression was also shown to be associated with reduced survival rate in overweight individuals with colorectal cancer (CRC) but not in non-overweight patients. This suggests that the NF-κB/CES1-dependent breakdown of fat may play a role in the clinical progression of CRC, specifically in obese patients. Ces1d/CES1 increased triacylglycerol and cholesterol ester lipolysis to transport endogenous FFAs and fuel FAO to fulfill the energy needs during nutrient deprivation. It prevented harmful neutral lipid accumulation that causes ROS generation and phospholipid peroxidation, which causes apoptosis and ferroptosis. Hence, the inhibition of Ces1d/CES1 through genetic or pharmacologic means resulted in the death of colorectal cancer (CRC) cells under conditions of nutrient deprivation in laboratory settings and reduced CRC growth in animal models. These findings indicate that CES1 could be a potential target for treating CRC. Ultimately, through modulating many oncogenic signaling pathways, NF-κB activation stimulates lipid metabolism and plays a vital role in cancer cell survival under metabolic stress (Figure 3B).
Cancer cells alter their intracellular lipid metabolism to optimize the use of available resources, in addition to being impacted by adipose tissue through paracrine and endocrine processes. Remarkably, STAT3 may fulfill various functions in this particular process (Tošić and Frank, 2021). Lipids serve multiple purposes for cancer cells in environments with limited nutrition: they can be used as an alternative energy source, a component of cell membranes to facilitate fast division and proliferation, a defense mechanism against excessive oxidative stress, and a building block for lipid-based hormones that promote cancer cells’ pathological growth (Vidavsky et al., 2019). Endogenous lipids have been linked to various cellular processes, including growth, differentiation, inflammation, autophagy, cell death, and membrane formation (Alizadeh et al., 2023). Therefore, changes in the levels of proteins that control the production, breakdown, and storage of lipids have been extensively documented in cancerous cells (Lodhi and Semenkovich, 2014). An example is when breast malignancies have increased HER2/neu, often followed by an amplification of the peroxisome proliferator-activated receptor gamma (PPARγ) binding protein (PBP) gene. This may be because these genes are located close to each other on chromosome 17q12–21 (Kauraniemi et al., 2001). Curiously, the STAT3 gene is also situated near these genetic locations, specifically at chromosome 17q 21.2. Both the overexpression of PBP and the activity of STAT3 boost the transcriptional activity of PPARγ, a significant lipid metabolism regulator and an essential mediator of adipogenesis and adipocyte differentiation (Wang C. et al., 2009; Kourtidis et al., 2009). Moreover, both the process of synthesizing fatty acids (controlled by the enzyme FASN) and the process of FAO are frequently increased in cancerous cells (Figure 4A) (Yoshii et al., 2015; Wang et al., 2018c). FASN-mediated de novo fatty acid synthesis and anabolic-driven growth, survival, and migration can be initiated by activating PI3K/AKT/mTOR and MAPK and by overexpressing HER/neu (Fhu and Ali, 2020). Additionally, it has been demonstrated that STAT3 activation increases the expression of FASN (Gao et al., 2020). The connection between FASN inhibition and trastuzumab resistance reversal in HER2-positive breast tumors has therapeutic potential (Vazquez-Martin et al., 2007). Furthermore, the breakdown of fatty acids (FA) through FAO promotes the growth of tumors by supplying ATP to meet the energy requirements of cells during metabolic stress caused by a lack of glucose (Qu et al., 2016). Activation of leptin, IL-6, or STAT3 through other mechanisms stimulates the production of carnitine palmitoyltransferase 1B (CPT1B), an enzyme that controls the rate of fatty acid oxidation (Koo et al., 2019). This pathway plays a key function in the self-renewal of breast cancer stem cells (BCSCs) and enhances their ability to survive chemotherapy. The process of FAO generates NADH and FADH2, which can decrease oxidative stress and increase ATP synthesis through the electron transport system (Koundouros and Poulogiannis, 2020). FAO also produces acetyl-CoA, which can be used for energy production via the Krebs cycle or for the generation of fatty acids and protein acetylation. These processes have the potential to stimulate tumor growth. Conforming to this correlation, the addition of acetyl-CoA partially counteracted the impacts of suppressing STAT3 on the growth and viability of breast cancer stem cells (BCSCs). Furthermore, the augmentation of FAO plays a crucial role in the emergence of chemoresistance in several malignant systems, such as AML and breast, lung, and stomach malignancies. Inhibiting this route hinders the abnormal proliferation of cancer cells and restores their sensitivity to chemotherapy (Li J. et al., 2013; He et al., 2019). Another possible strategy for utilizing the lipid properties mediated by STAT3 for therapeutic purposes is to differentiate between cancer cells and normal cells, which often have minimal STAT3 activity. The activation of STAT3 and subsequent malignant transformation of mammary epithelial cells leads to a substantial decrease in the cellular levels of N-acyl taurine and arachidonic acid (Tošić et al., 2021). Given the significance of both these compounds in plasma membrane modeling, an investigation was conducted to determine if this characteristic might be effectively utilized in innovative nanoparticle drug delivery systems for therapeutic purposes. Nanoparticles coated with poly-L-glutamic acid had a strong affinity for cells with aberrant STAT3 activity. When these particles were used to carry cytotoxic drugs, they specifically generated higher levels of apoptosis in these cells. A recent paper highlighted the significance of lipid architecture in determining the metastatic potential of cancer cells and its impact on cell survival and proliferation (Jin et al., 2020). This study discovered that cancer cells with a high ability to spread to other parts of the body had a larger amount of cholesterol, phosphatidylcholine (PC), and sphingomyelin (SM) in their cells. On the other hand, these cells have lower amounts of triacylglycerol. This finding aligns with the patterns observed in cellular TAG and SM levels after STAT3 activation in breast cancer cells, as reported by previous studies (Tošić et al., 2021). Jin et al. (2020) also demonstrated that various metabolic regulators significantly impacted the ability of breast cancer cells to develop brain metastases. These regulators include sterol regulatory element-binding protein 1 (SREBP1), which plays a crucial role in the synthesis of fatty acids (FA) and triglycerides (TAG), as well as the FA transporter CD36 and FA binding protein FABP6 (Jin et al., 2020). Interestingly, a correlation was observed between the activity of STAT3 and the mRNA expressions of SREBP1 and CD36 in several cellular and animal models. The expression of SREBP1, driven by high glucose, is dependent on the tyrosine phosphorylation of STAT3 (Guillet-Deniau et al., 2004), and this requirement is significantly reduced when STAT3 is silenced (Chen et al., 2018). Likewise, the excessive production of an always active version of STAT3 substantially increased the expression of CD36, and the signaling pathway, including JAK2/STAT3, was necessary for the induction of CD36 expression by IL13 (Yakubenko et al., 2013; Su et al., 2020). The data indicate that the ability of STAT3-driven malignancies to spread to other parts of the body may be partly due to its diverse impacts on lipid metabolism. However, additional research is required to understand these interesting connections’ underlying mechanisms fully. STAT3 was discovered to hinder cell ferroptosis by repressing the production of acyl-CoA synthetase long-chain family member 4 (ACSL4), an enzyme that promotes ferroptosis by increasing the presence of long polyunsaturated fatty acids in cell membranes (Ding et al., 2023). It was also demonstrated that STAT3 can collaborate with androgen receptor (AR) to stimulate the expression of cell cycle-related kinase (CCRK), thereby augmenting the tumorigenicity of liver cancer. Functionally, it has been demonstrated that CCRK enhances the presence of the fully developed version of SREBP1 in the nucleus through GSK3β/mTORC1 signaling, leading to an augmentation in the production and absorption of new lipids (Sun et al., 2018). STAT3 was discovered to enhance the intake of fatty acids (FA) in ovarian cancer, leading to the activation of FABP4, which in turn supported the proliferation of cancer cells (Yu et al., 2020).
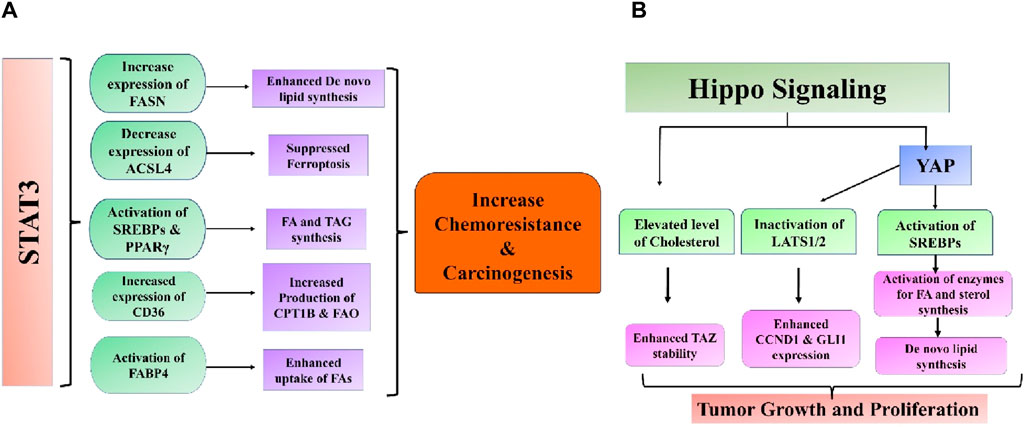
Figure 4. Intricate association between (A) STAT3 and (B) Hippo signaling pathways and aberrant lipid metabolism in cancer.
It has been observed that de novo lipogenesis is regulated by crucial elements of the hippo cell signaling system. Within the endoplasmic reticulum (ER), LATS2 interacts with SREBP-2, causing it to be sequestered and preventing its transport from the ER to Golgi (Lee et al., 2022). Furthermore, YAP can serve as a coactivator with SREBP-1c and SREBP-2 in the nucleus to enhance the expression of genes linked with lipid synthesis (Shimano and Sato, 2017; Shi et al., 2019). The Hippo signaling pathway serves as a key upstream regulator of lipid production and a mediator for liver maintenance, crucial for the liver’s recovery from metabolic stress damage. Lipids can regulate the Hippo signaling pathway, potentially leading to the development of liver diseases such as nonalcoholic steatohepatitis (NASH) progressing to HCC or fibrosis (Wang et al., 2020). Non-alcoholic fatty liver disease (NAFLD) is marked by excessive fat buildup leading to elevated cholesterol levels and is recognized as a significant contributor to the sustenance and survival of cancer cells (Novak et al., 2013). High cholesterol levels have been identified to reduce TAZ phosphorylation through LATS1/2 by activating RhoA, which inhibits LATS1/2. TAZ expression increased in fibrotic NASH livers compared to normal or non-fibrotic ones. The study also indicated that high cholesterol levels could enhance TAZ stability, leading to the promotion of NASH fibrosis (Wang et al., 2016). Inactivating LATS1/2 may promote the progression from NASH to HCC, as indicated by another study. Within NASH-HCC, the junction protein JCAD linked to obesity interacts with LATS2, hindering its kinase activity by binding to the kinase domain but not affecting the connection between LATS2 and MOB1. By inhibiting kinase activity, YAP phosphorylation level, cellular location, and activity are directly affected, leading to the expression of target genes, including CCND1 and GLI2, which stimulate cell proliferation (Rinella, 2015; Ye J. et al., 2017). Lipid uptake is a crucial activity that influences lipid availability to cells and is increased in various malignancies, especially in metastasis-initiating cells and NAFLD. Fatty acid uptake occurs through fatty acid translocase CD36 and members of the FABP family (Pascual et al., 2017; Feng et al., 2022). Consistently, increased CD36 expression has been seen in NAFLD patients and cells that initiate metastasis. CD36 facilitates the intake of oxidized LDL, which can provide FFAs by enhancing lipophagy, a specialized type of autophagy that degrades lipid droplets. Lysophosphatidic acid (LPA) levels increase due to lipophagy, activating YAP (Tian et al., 2019). SAV1 deletion upregulates CD36 mRNA expression in PTEN-deficient livers by inhibiting YAP, an upstream negative regulator. CRABP2, a lipid transporter belonging to the FABP family, is suggested to influence the Hippo signaling pathway in triple-negative breast cancer through its interaction with estrogen receptor alpha (ER). The association between CRABP2 and LATS1 inhibits the ubiquitination of LATS1, leading to the activation of Hippo signaling in ER-positive tumors. This process helps to prevent the invasion and spread of cancer cells. Ubiquitination of LATS1 enhances the invasion and metastasis of ER-negative malignancy. It is not apparent how ER expression influences the function of CRABP2. The correlation between lipid absorption and the Hippo signaling pathway is intricate yet crucial in advancing aggressive cancer (Feng et al., 2019) (Figure 4B).
It has also been found that Wnt signaling is involved with the process of cancer lipogenesis (El-Sahli et al., 2019). Glutamine can be redirected to reductive carboxylation to produce α-ketoglutarate, which generates acetyl-CoA for de novo lipogenesis. De novo lipogenesis provides high energy levels and building blocks for critical cellular components to expand cells. Deviant lipid levels in cells are associated with the advancement of cancer in several types of malignancies. In breast cancer, there is an increased buildup of FFAs, cholesterol esters, and phospholipids, which have been proven to enhance tumor growth and invasion by cancer cells. Tumors rich in cholesterol ester were linked to higher rates of breast tumor growth and tissue death. Linoleic acid was demonstrated to stimulate breast cancer cell movement through plasminogen activator inhibitor-1 (PAI-1) and SMAD4 (Byon et al., 2009). Yao et al. (2018) discovered in a recent study that canonical Wnt signaling via MYC facilitates the transformation of triacylglycerol into phospholipids and enhances the presence of unsaturated fatty acyl groups in phospholipids. Cancer cells heavily rely on lipid metabolism remodeling to incorporate unsaturated fatty acids into their cell membranes for maintenance, energy storage, and signaling purposes. Unsaturated fatty acids are associated with the stem-like traits in ovarian cancer (Mukherjee et al., 2017). Reducing β-catenin in breast cancer cells led to a decrease in critical lipogenic enzymes such as citrate carriers, ACC, and FASN. This highlights the involvement of the Wnt/β-catenin pathway in de novo lipid synthesis (Vergara et al., 2017). Mounting data has emphasized the importance of lipid metabolism in cancer stem cell survival and upkeep. Subtle discrepancies exist in the lipid metabolism changes between CSC and non-CSC groups. In breast cancer, the enzyme carnitine palmitoyltransferase I (CPT1), responsible for fatty acid β-oxidation, is found to be higher in the CSC (cancer stem cell) population compared to the non-CSC population (Wang Y. et al., 2018). Stearoyl-CoA desaturases (SCDs) could be the crucial link between Wnt signaling and lipid metabolism in these CSCs. SCDs speed up the process of lipid desaturation, and there is increasing evidence pointing to SCDs as a characteristic feature of CSCs. The relationship between the Wnt pathway and SCDs is firmly established. β-catenin has been shown to enhance the sterol regulatory element binding protein 1 (SREBP-1)-associated expression of the primary types of SCD (SCD1 and SCD2) in CSCs. Suppression of SCD increased the levels of β-catenin expression, which was reversed by adding monounsaturated fatty acids (MUFAs) produced by SCDs (Lai et al., 2017). In another investigation, MUFAs were essential for Wnt ligand synthesis and secretion. SCD1 was demonstrated to control the Hippo/YAP pathway, a crucial cancer stem cell-related oncogenic pathway, partly through Wnt signaling. Simultaneous blocking of Wnt and YAP has been demonstrated to slow down the progression of triple-negative breast cancer in both mesenchymal and epithelial forms (Rios-Esteves and Resh, 2013; Sulaiman et al., 2018). Collective research indicates that Wnt pathway may play a role in lipid metabolic changes in cancer cells, and targeting both pathways could be more efficient in addressing CSCs (Figure 5A).
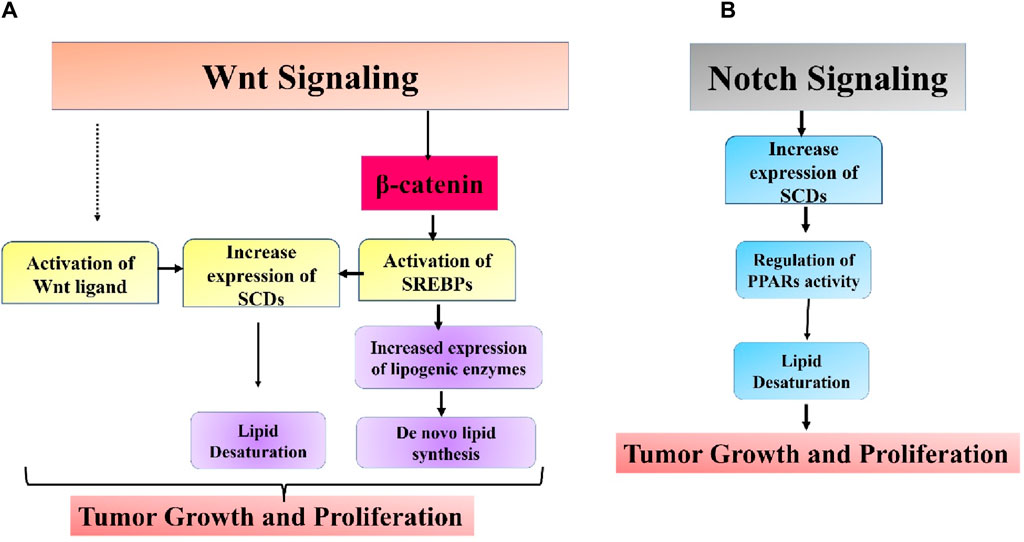
Figure 5. Intricate association between (A) Wnt and (B) Notch signaling pathways and aberrant lipid metabolism in cancer.
The Notch pathway is a highly conserved mechanism for transmitting signals strongly associated with biological processes like tumor spread and immune evasion (Pandey et al., 2023a; Pandey et al., 2023b). The Notch signaling system can control the expression of peroxisome proliferator-activated receptor α and lipid oxidation genes to maintain lipid and redox equilibrium in lipid metabolism. Targeting SCD1-dependent lipid desaturation in colon cancer preferentially kills colon CSCs by blocking Notch signaling (Yu Y. et al., 2021). A recent study found that Notch controls lipid metabolism, which is associated with the beginning of tumor formation. Notch signaling-induced changes in lipid metabolism are crucial for the development of liposarcomas. Adipocytes that activate the Notch pathway experience dedifferentiation, leading to lipid metabolic failure and the eventual development of liposarcoma. Blocking Notch signaling can decrease lipids’ absorption and fatty acid oxidation. A possible mechanism was proposed involving Notch-induced suppression of PPARγ ligands synthesis, which are crucial regulators of lipid balance in adipocytes. Administering a synthetic PPARγ supplement partially reversed the transformation of adipocytes produced by Notch activation (Bi et al., 2016). Endothelial cells have been found to control the production of endothelial lipase, an enzyme associated with fatty acid transport through the vessel wall through the Notch signaling pathway (Jabs et al., 2018). The overall impact of Notch signaling in cancer remains mainly uninvestigated, and additional research is needed to ascertain if other molecular pathways could play a role in Notch-induced changes in lipid metabolism in cancer (Figure 5B).
FASN inhibitors have been extensively studied, and one of the compounds that has progressed to clinical testing is TVB-2640. TVB-2640 has notable anticancer effects in several types of carcinoma, such as lung, ovarian, and breast cancers (Falchook et al., 2021). C75, a chemical that targets FASN, enhances fatty acid oxidation and induces cell death during S phase in breast cancer cells (Menendez et al., 2004b). The FAS inhibitor C75 reduced lipid droplet formation, PGE2 synthesis, and cell proliferation in CACO-2 cells, indicating that lipid droplets serve as regulatory reservoirs for prostaglandin formation and play a role in mediating inflammatory pathways crucial for cancer cell metabolism (Accioly et al., 2008). Lipid metabolism was investigated in a transgenic mouse model of medulloblastoma, including a mutated smoothened allele that disrupts the sonic hedgehog pathway (Bhatia et al., 2011). Approximately two-thirds of the mice acquired MB. The tumor area showed higher levels of FASN expression, increased production of new fatty acids, and reduced fatty acid breakdown. The increased levels of FASN were likewise associated with the levels of the transcription factors E2F1 and MYCN (Bhatia et al., 2011). Animals with medulloblastoma, when treated with the fatty acid synthesis inhibitor C75, exhibited improved survival, reduced lipid synthesis, and decreased expression of proliferation markers MYCN and cyclin D2. Acetyl CoA carboxylase was also identified as being improperly controlled in this mouse model (Bhatia et al., 2011).
Cerulenin was the first naturally occurring, non-competitive inhibitor of FAS that has been found. Cerulenin undergoes covalent bonding with a cysteine residue located in the active site of the ketoacyl synthase (KS) domain of the fungal FAS enzyme. This interaction leads to substantial alterations in the conformation of the active site (Mallick et al., 2023). Cerulenin inhibits the activation of estrogen receptors in ovarian cancer, hinders the spread of colon cancer to the liver, and triggers cancer cell death in both laboratory and living organisms (Menendez et al., 2004a; Shiragami et al., 2013). Orlistat is an FDA-approved medication for managing obesity authorized in 1999. Orlistat triggers endoplasmic reticulum stress, leading to increased apoptotic cell death in prostate, breast, and colon cancer cells (Menendez et al., 2005; Little et al., 2007). Preclinical studies are currently being conducted on the utilization of ACC inhibitors. Soraphen A has demonstrated anti-cancer effects by blocking the ACC enzyme in cancer cells. Beckers et al. (2007) discovered that Soraphen A effectively suppressed fatty acid production in LNCaP and PC-3M prostate cancer cells at extremely low concentrations, leading to an enhanced process of β-oxidation of fatty acids. This resulted in a reduction in phospholipid levels within cancer cells, ultimately hindering their growth (Beckers et al., 2007). ND-646 is an allosteric inhibitor of ACC that hinders the formation of non-small cell lung carcinoma in both laboratory settings and living organisms (Svensson et al., 2016). TOFA, an allosteric inhibitor of ACC, triggers cell death by releasing pro-apoptotic proteins from the mitochondria in breast, colon, lung, and ovarian cancers (Wang D. et al., 2009; Guseva et al., 2011; Li S. et al., 2013). A939572, an SCD1 inhibitor, triggers endoplasmic reticulum stress and induces apoptotic death in renal cell carcinoma (RCC) (von Roemeling et al., 2013). A939572 inhibits phosphorylation of the PI3K/Akt pathway and markedly reduces the viability of lung cancer cells in living organisms (She et al., 2019). Another potential strategy involves targeting the cholesterol synthesis. Statins are inhibitors of β-hydroxy β-methylglutaryl-CoA reductase (HMGCR), and have demonstrated promising results in laboratory and living organism studies. Statins hinder lipid metabolism and promote cell survival in different types of cancers, such as colon, pancreas, liver, breast, prostate, bladder, lung, and skin cancers (Apostolova et al., 2016; Ahmadi et al., 2020). Statin use has been shown to lower the occurrence and reappearance of multiple types of cancer in 3129 human epidemiologic studies, including bladder, breast, colon, kidney, lung, skin, pancreatic, and prostate cancers (Cauley et al., 2006; Cao et al., 2023; Takada et al., 2023; Wang et al., 2023; Zhang et al., 2024). Simvastatin, a cholesterol synthesis inhibitor, blocks the Akt pathway and induces apoptosis in prostate cancer cells (Zhuang et al., 2005). A preliminary phase II trial in NSCLC investigated the possibility of simvastatin enhancing the effectiveness of gefitinib in a specific subgroup of gefitinib-resistant patients (Han et al., 2011). Furthermore, a recent study conducted in patients with locally advanced breast cancer indicated that the combination of simvastatin and neoadjuvant chemotherapy resulted in enhanced therapeutic responses (Yulian and Siregar, 2021). Nevertheless, a phase III clinical trial conducted on patients with advanced gastric cancer found no significant benefit of combining simvastatin with capecitabine-cisplatin compared to a placebo in combination with capecitabine-cisplatin (Kim et al., 2019).
Another strategy for targeting lipid uptake focused on CD36 and FABPs. ABT-510, a synthetic analog of thrombospondin-1, is presently undergoing phase I clinical trials for the management of glioblastomas, melanomas, and renal cell carcinomas (Ebbinghaus et al., 2007; Markovic et al., 2007; Nabors et al., 2010). ABT-510 binds to CD36 and triggers the expression of the death receptor FAS (Isenberg et al., 2008). Al-Jameel et al. (2019) developed a potent recombinant FABP5 inhibitor, dmrFABP5. FABP5 interacts with and carries medium- and long-chain fatty acids in the nuclei of cancer cells. dmrFABP5 is produced by substituting three amino acids in FABP5, resulting in loss of its capacity to bind fatty acids. dmrFABP5 significantly inhibited the proliferation, migration, and invasion of prostate cancer cells (Al-Jameel et al., 2019). When exposed to chemotherapeutic drugs such as Docetaxel or Cabazitaxel, two advanced synthetic inhibitors, Stony Brook fatty acid-binding protein inhibitor 102 (SBFI-102) and SBFI-103, were found to effectively decrease tumor growth in animal experiments conducted in a living organism (Carbonetti et al., 2020). EI-05, a FABP5 activator, boosts lipid droplet formation and interferon-β production, thereby enhancing the anti-tumor function of macrophages under inflammatory conditions. Administering EI-05 effectively suppresses the development of breast cancer cells (Rao et al., 2015). However, there is vast description of the therapeutic drugs targeting lipid metabolism for the treatment of cancer, so we have summarized these drugs on the basis of targets in Table 1.
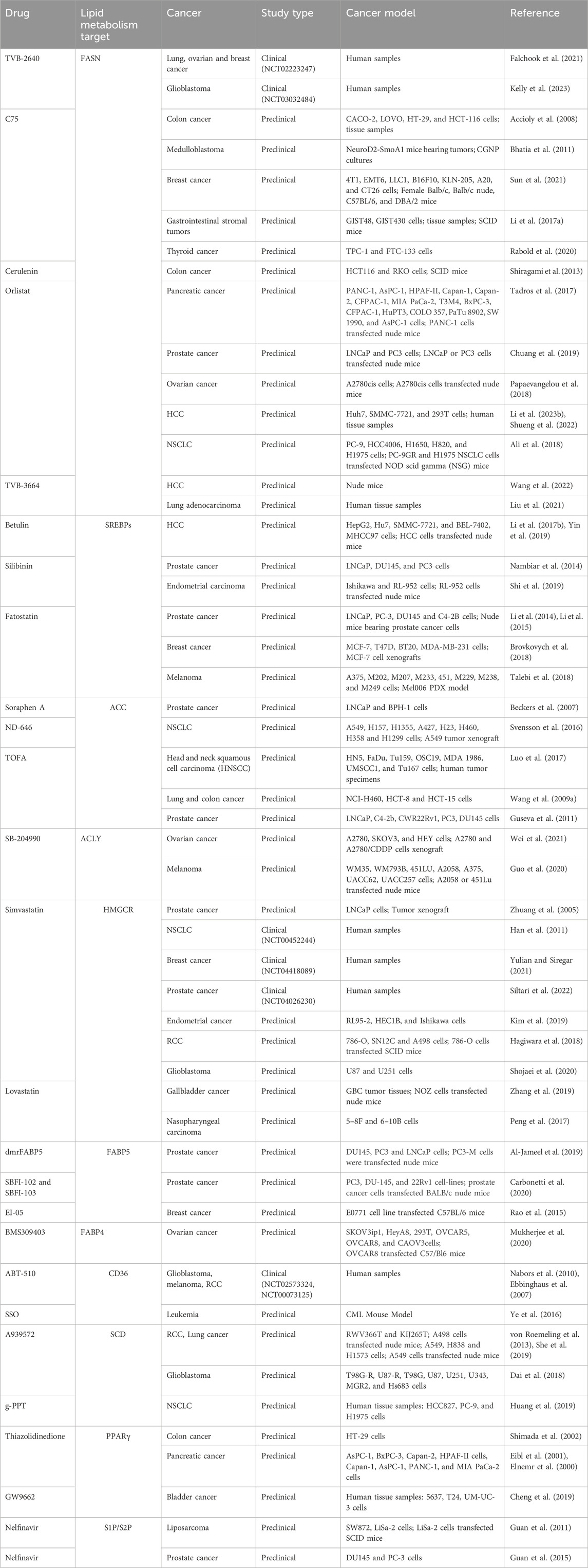
Table 1. Therapeutic drugs targeting key molecules of altered lipid metabolism in different types of carcinoma.
It is widely accepted that altered lipid metabolism plays a role in cancer metabolism. We explored significant and developing processes in lipid metabolism that promote cancer growth and advancement, as well as prospective targets of oncogenic signaling pathways. Here we have summarized the potential therapeutic strategies targeting the crosstalk between oncogenic signaling and lipid metabolism to inhibit cancer progression and metastasis. Dysregulated lipid metabolism plays a crucial role in tumor metabolic adaptability and contributes to therapeutic resistance, metastatic dissemination, and secondary tumor growth. The local TME is crucial because lipids from nearby tissues play a significant role in both primary and secondary tumor formation, emphasizing the importance of lipids in cancer progression. Tumors experience a change in lipid metabolism, which is closely associated with the development of malignancy and the advancement of the tumor. Lipid metabolic reprogramming is closely linked to oncogenic signaling in cancer cells. The activation of oncogenic signals can induce the expression of genes linked to lipid metabolism, which subsequently controls lipid metabolism in cancer cells. Lipid metabolic by-products can increase the activation of cancer-causing pathways, leading to increased cancer cell growth, invasion, and spread to distant parts of the body. There have been significant advancements in the identification of therapeutic targets for lipid metabolism in cancer therapy. The interaction between lipid metabolism and important signaling mediators in the environment of cancer is a highly interesting topic of study. An in-depth understanding of how dysregulated lipid metabolism influences cancer progression is crucial for the development of innovative cancer therapy strategies. It is clear that focusing on a single component or pathway of lipid metabolism is unlikely to lead to more effective cancer treatment. Developing treatments that can impede tumor growth and boost the body’s ability to fight tumors is a highly promising strategy for lipid-based medicinal research. Little information is available regarding how oncogenic signaling pathways indirectly influence the cancer microenvironment or the lipid metabolism it activates. Investigating the relationship between modifying medicines and lipid metabolism will be a promising area of future cancer research.
FK: Conceptualization, Validation, Visualization, Writing–original draft, Writing–review and editing. DE: Conceptualization, Validation, Writing–original draft, Writing–review and editing. MV: Conceptualization, Supervision, Validation, Visualization, Writing–review and editing. SP: Formal Analysis, Investigation, Validation, Visualization, Writing–review and editing. SR: Formal Analysis, Investigation, Supervision, Validation, Visualization, Writing–review and editing. SS: Formal Analysis, Investigation, Validation, Visualization, Writing–review and editing. NMA: Data curation, Supervision, Formal Analysis, Validation, Resources, Visualization, Writing–review and editing. MS: Conceptualization, Project administration, Supervision, Visualization, Writing–review and editing. PP: Conceptualization, Investigation, Project administration, Supervision, Visualization, Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors are thankful to the Deanship of Research and Graduate Studies, King Khalid University, Abha, Saudi Arabia, for financially supporting this work through the Large Research Group Project under Grant no. R.G.P.2/44/45.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abumrad, N., Coburn, C., and Ibrahimi, A. (1999). Membrane proteins implicated in long-chain fatty acid uptake by mammalian cells: CD36, FATP and FABPm. Biochim. Biophys. Acta 1441 (1), 4–13. doi:10.1016/s1388-1981(99)00137-7
Accioly, M. T., Pacheco, P., Maya-Monteiro, C. M., Carrossini, N., Robbs, B. K., Oliveira, S. S., et al. (2008). Lipid bodies are reservoirs of cyclooxygenase-2 and sites of prostaglandin-E2 synthesis in colon cancer cells. Cancer Res. 68 (6), 1732–1740. doi:10.1158/0008-5472.CAN-07-1999
Ahmadi, M., Amiri, S., Pecic, S., Machaj, F., Rosik, J., Łos, M. J., et al. (2020). Pleiotropic effects of statins: a focus on cancer. Biochim. Biophys. Acta Mol. Basis Dis. 1866 (12), 165968. doi:10.1016/j.bbadis.2020.165968
Airley, R. E., McHugh, P., Evans, A. R., Harris, B., Winchester, L., Buffa, F. M., et al. (2014). Role of carbohydrate response element-binding protein (ChREBP) in generating an aerobic metabolic phenotype and in breast cancer progression. Br. J. Cancer 110 (3), 715–723. doi:10.1038/bjc.2013.765
Ali, A., Levantini, E., Teo, J. T., Goggi, J., Clohessy, J. G., Wu, C. S., et al. (2018). Fatty acid synthase mediates EGFR palmitoylation in EGFR mutated non-small cell lung cancer. EMBO Mol. Med. 10 (3), e8313. doi:10.15252/emmm.201708313
Alizadeh, J., Kavoosi, M., Singh, N., Lorzadeh, S., Ravandi, A., Kidane, B., et al. (2023). Regulation of autophagy via carbohydrate and lipid metabolism in cancer. Cancers (Basel) 15 (8), 2195. doi:10.3390/cancers15082195
Al-Jameel, W., Gou, X., Jin, X., Zhang, J., Wei, Q., Ai, J., et al. (2019). Inactivated FABP5 suppresses malignant progression of prostate cancer cells by inhibiting the activation of nuclear fatty acid receptor PPARγ. Genes Cancer 10 (3-4), 80–96. doi:10.18632/genesandcancer.192
Almoraie, N. M., and Shatwan, I. M. (2024). The potential effects of dietary antioxidants in obesity: a comprehensive review of the literature. Healthc. (Basel) 12 (4), 416. doi:10.3390/healthcare12040416
Apostolova, S. N., Toshkova, R. A., Momchilova, A. B., and Tzoneva, R. D. (2016). Statins and alkylphospholipids as new anticancer agents targeting lipid metabolism. Anticancer Agents Med. Chem. 16 (12), 1512–1522. doi:10.2174/1871520616666160624093955
Auciello, F. R., Bulusu, V., Oon, C., Tait-Mulder, J., Berry, M., Bhattacharyya, S., et al. (2019). A stromal lysolipid-autotaxin signaling Axis promotes pancreatic tumor progression. Cancer Discov. 9 (5), 617–627. doi:10.1158/2159-8290.CD-18-1212
Beckers, A., Organe, S., Timmermans, L., Scheys, K., Peeters, A., Brusselmans, K., et al. (2007). Chemical inhibition of acetyl-CoA carboxylase induces growth arrest and cytotoxicity selectively in cancer cells. Cancer Res. 67 (17), 8180–8187. doi:10.1158/0008-5472.CAN-07-0389
Benjamin, D. I., Li, D. S., Lowe, W., Heuer, T., Kemble, G., and Nomura, D. K. (2015). Diacylglycerol metabolism and signaling is a driving force underlying FASN inhibitor sensitivity in cancer cells. ACS Chem. Biol. 10 (7), 1616–1623. doi:10.1021/acschembio.5b00240
Berwick, D. C., Hers, I., Heesom, K. J., Moule, S. K., and Tavare, J. M. (2002). The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J. Biol. Chem. 277 (37), 33895–33900. doi:10.1074/jbc.M204681200
Bhatia, B., Hsieh, M., Kenney, A. M., and Nahlé, Z. (2011). Mitogenic Sonic hedgehog signaling drives E2F1-dependent lipogenesis in progenitor cells and medulloblastoma. Oncogene 30 (4), 410–422. doi:10.1038/onc.2010.454
Bi, P., Yue, F., Karki, A., Castro, B., Wirbisky, S. E., Wang, C., et al. (2016). Notch activation drives adipocyte dedifferentiation and tumorigenic transformation in mice. J. Exp. Med. 213 (10), 2019–2037. doi:10.1084/jem.20160157
Boden, G., and Shulman, G. I. (2002). Free fatty acids in obesity and type 2 diabetes: defining their role in the development of insulin resistance and beta-cell dysfunction. Eur. J. Clin. Invest. 32 (Suppl. 3), 14–23. doi:10.1046/j.1365-2362.32.s3.3.x
Bouche, C., and Quail, D. F. (2023). Fueling the tumor microenvironment with cancer-associated adipocytes. Cancer Res. 83 (8), 1170–1172. doi:10.1158/0008-5472.CAN-23-0505
Brovkovych, V., Izhar, Y., Danes, J. M., Dubrovskyi, O., Sakallioglu, I. T., Morrow, L. M., et al. (2018). Fatostatin induces pro- and anti-apoptotic lipid accumulation in breast cancer. Oncogenesis 7 (8), 66. doi:10.1038/s41389-018-0076-0
Brunetti, L., Loiodice, F., Piemontese, L., Tortorella, P., and Laghezza, A. (2019). New approaches to cancer therapy: combining fatty acid amide hydrolase (FAAH) inhibition with peroxisome proliferator-activated receptors (PPARs) activation. J. Med. Chem. 62 (24), 10995–11003. doi:10.1021/acs.jmedchem.9b00885
Byon, C. H., Hardy, R. W., Ren, C., Ponnazhagan, S., Welch, D. R., McDonald, J. M., et al. (2009). Free fatty acids enhance breast cancer cell migration through plasminogen activator inhibitor-1 and SMAD4. Lab. Invest. 89 (11), 1221–1228. doi:10.1038/labinvest.2009.97
Cao, Z., Yao, J., He, Y., Lou, D., Huang, J., Zhang, Y., et al. (2023). Association between statin exposure and incidence and prognosis of prostate cancer: a meta-analysis based on observational studies. Am. J. Clin. Oncol. 46 (7), 323–334. doi:10.1097/COC.0000000000001012
Capece, D., D'Andrea, D., Begalli, F., Goracci, L., Tornatore, L., Alexander, J. L., et al. (2021). Enhanced triacylglycerol catabolism by carboxylesterase 1 promotes aggressive colorectal carcinoma. J. Clin. Invest. 131 (11), e137845. doi:10.1172/JCI137845
Carbonetti, G., Converso, C., Clement, T., Wang, C., Trotman, L. C., Ojima, I., et al. (2020). Docetaxel/cabazitaxel and fatty acid binding protein 5 inhibitors produce synergistic inhibition of prostate cancer growth. Prostate 80 (1), 88–98. doi:10.1002/pros.23921
Caro, P., Kishan, A. U., Norberg, E., Stanley, I. A., Chapuy, B., Ficarro, S. B., et al. (2012). Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 22 (4), 547–560. doi:10.1016/j.ccr.2012.08.014
Carracedo, A., Cantley, L. C., and Pandolfi, P. P. (2013). Cancer metabolism: fatty acid oxidation in the limelight. Nat. Rev. Cancer 13 (4), 227–232. doi:10.1038/nrc3483
Carrer, A., Trefely, S., Zhao, S., Campbell, S. L., Norgard, R. J., Schultz, K. C., et al. (2019). Acetyl-CoA metabolism supports multistep pancreatic tumorigenesis. Cancer Discov. 9 (3), 416–435. doi:10.1158/2159-8290.CD-18-0567
Carrer, A., and Wellen, K. E. (2015). Metabolism and epigenetics: a link cancer cells exploit. Curr. Opin. Biotechnol. 34, 23–29. doi:10.1016/j.copbio.2014.11.012
Cauley, J. A., McTiernan, A., Rodabough, R. J., LaCroix, A., Bauer, D. C., Margolis, K. L., et al. (2006). Statin use and breast cancer: prospective results from the Women's Health Initiative. J. Natl. Cancer Inst. 98 (10), 700–707. doi:10.1093/jnci/djj188
Chang, W. H., and Lai, A. G. (2019). The pan-cancer mutational landscape of the PPAR pathway reveals universal patterns of dysregulated metabolism and interactions with tumor immunity and hypoxia. Ann. N. Y. Acad. Sci. 1448 (1), 65–82. doi:10.1111/nyas.14170
Chen, J., Yue, J., Liu, Y., Liu, J., Jiao, K., Teng, M., et al. (2018). Blocking of STAT-3/SREBP1-mediated glucose-lipid metabolism is involved in dietary phytoestrogen-inhibited ovariectomized-induced body weight gain in rats. J. Nutr. Biochem. 61, 17–23. doi:10.1016/j.jnutbio.2018.06.009
Cheng, C., Geng, F., Cheng, X., and Guo, D. (2018). Lipid metabolism reprogramming and its potential targets in cancer. Cancer Commun. (Lond). 38 (1), 27. doi:10.1186/s40880-018-0301-4
Cheng, M., Bhujwalla, Z. M., and Glunde, K. (2016). Targeting phospholipid metabolism in cancer. Front. Oncol. 6, 266. doi:10.3389/fonc.2016.00266
Cheng, S., Qian, K., Wang, Y., Wang, G., Liu, X., Xiao, Y., et al. (2019). PPARγ inhibition regulates the cell cycle, proliferation and motility of bladder cancer cells. J. Cell Mol. Med. 23 (5), 3724–3736. doi:10.1111/jcmm.14280
Chiurchiù, V., Leuti, A., and Maccarrone, M. (2018). Bioactive lipids and chronic inflammation: managing the fire within. Front. Immunol. 9, 38. doi:10.3389/fimmu.2018.00038
Chuang, H. Y., Lee, Y. P., Lin, W. C., Lin, Y. H., and Hwang, J. J. (2019). Fatty acid inhibition sensitizes androgen-dependent and -independent prostate cancer to radiotherapy via FASN/NF-κB pathway. Sci. Rep. 9 (1), 13284. doi:10.1038/s41598-019-49486-2
Contractor, T., and Harris, C. R. (2012). p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 72 (2), 560–567. doi:10.1158/0008-5472.CAN-11-1215
Corn, K. C., Windham, M. A., and Rafat, M. (2020). Lipids in the tumor microenvironment: from cancer progression to treatment. Prog. Lipid Res. 80, 101055. doi:10.1016/j.plipres.2020.101055
Dai, S., Yan, Y., Xu, Z., Zeng, S., Qian, L., Huo, L., et al. (2018). SCD1 confers temozolomide resistance to human glioma cells via the akt/gsk3β/β-catenin signaling Axis. Front. Pharmacol. 8, 960. doi:10.3389/fphar.2017.00960
Darwish, A., Pammer, M., Gallyas, F., Vígh, L., Balogi, Z., and Juhász, K. (2024). Emerging lipid targets in glioblastoma. Cancers (Basel) 16 (2), 397. doi:10.3390/cancers16020397
Deng, T., Zhao, J., Tong, Y., Chen, Z., He, B., Li, J., et al. (2024). Crosstalk between endothelial progenitor cells and HCC through periostin/CCL2/CD36 supports formation of the pro-metastatic microenvironment in HCC. Oncogene 43, 944–961. doi:10.1038/s41388-024-02960-2
Ding, K., Liu, C., Li, L., Yang, M., Jiang, N., Luo, S., et al. (2023). Acyl-CoA synthase ACSL4: an essential target in ferroptosis and fatty acid metabolism. Chin. Med. J. Engl. 136 (21), 2521–2537. doi:10.1097/CM9.0000000000002533
Ding, X., Zhang, W., Li, S., and Yang, H. (2019). The role of cholesterol metabolism in cancer. Am. J. Cancer Res. 9 (2), 219–227.
Du, J., Johnson, L. M., Jacobsen, S. E., and Patel, D. J. (2015). DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 16 (9), 519–532. doi:10.1038/nrm4043
Ebbinghaus, S., Hussain, M., Tannir, N., Gordon, M., Desai, A. A., Knight, R. A., et al. (2007). Phase 2 study of ABT-510 in patients with previously untreated advanced renal cell carcinoma. Clin. Cancer Res. 13 (22 Pt 1), 6689–6695. doi:10.1158/1078-0432.CCR-07-1477
Eibl, G., Wente, M. N., Reber, H. A., and Hines, O. J. (2001). Peroxisome proliferator-activated receptor gamma induces pancreatic cancer cell apoptosis. Biochem. Biophys. Res. Commun. 287 (2), 522–529. doi:10.1006/bbrc.2001.5619
Elnemr, A., Ohta, T., Iwata, K., Ninomia, I., Fushida, S., Nishimura, G., et al. (2000). PPARgamma ligand (thiazolidinedione) induces growth arrest and differentiation markers of human pancreatic cancer cells. Int. J. Oncol. 17 (6), 1157–1164. doi:10.3892/ijo.17.6.1157
El-Sahli, S., Xie, Y., Wang, L., and Liu, S. (2019). Wnt signaling in cancer metabolism and immunity. Cancers (Basel) 11 (7), 904. doi:10.3390/cancers11070904
Falchook, G., Infante, J., Arkenau, H. T., Patel, M. R., Dean, E., Borazanci, E., et al. (2021). First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine 34, 100797. doi:10.1016/j.eclinm.2021.100797
Farge, T., Nakhle, J., Lagarde, D., Cognet, G., Polley, N., Castellano, R., et al. (2023). CD36 drives metastasis and relapse in acute myeloid leukemia. Cancer Res. 83 (17), 2824–2838. doi:10.1158/0008-5472.CAN-22-3682
Feng, X., Zhang, M., Wang, B., Zhou, C., Mu, Y., Li, J., et al. (2019). CRABP2 regulates invasion and metastasis of breast cancer through hippo pathway dependent on ER status. J. Exp. Clin. Cancer Res. 38 (1), 361. doi:10.1186/s13046-019-1345-2
Feng, Y., Sun, W., Sun, F., Yin, G., Liang, P., Chen, S., et al. (2022). Biological mechanisms and related natural inhibitors of CD36 in nonalcoholic fatty liver. Drug Des. Devel Ther. 16, 3829–3845. doi:10.2147/DDDT.S386982
Fhu, C. W., and Ali, A. (2020). Fatty acid synthase: an emerging target in cancer. Molecules 25 (17), 3935. doi:10.3390/molecules25173935
Fritz, G., and Kaina, B. (2006). Rho GTPases: promising cellular targets for novel anticancer drugs. Curr. Cancer Drug Targets 6 (1), 1–14. doi:10.2174/156800906775471752
Fu, Y., Zou, T., Shen, X., Nelson, P. J., Li, J., Wu, C., et al. (2020). Lipid metabolism in cancer progression and therapeutic strategies. MedComm 2 (1), 27–59. doi:10.1002/mco2.27
Furuhashi, M., and Hotamisligil, G. S. (2008). Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat. Rev. Drug Discov. 7 (6), 489–503. doi:10.1038/nrd2589
Galdieri, L., and Vancura, A. (2012). Acetyl-CoA carboxylase regulates global histone acetylation. J. Biol. Chem. 287 (28), 23865–23876. doi:10.1074/jbc.M112.380519
Gao, P., Wang, L. L., Liu, J., Dong, F., Song, W., Liao, L., et al. (2020). Dihydroartemisinin inhibits endothelial cell tube formation by suppression of the STAT3 signaling pathway. Life Sci. 242, 117221. doi:10.1016/j.lfs.2019.117221
Gao, X., Lin, S. H., Ren, F., Li, J. T., Chen, J. J., Yao, C. B., et al. (2016). Acetate functions as an epigenetic metabolite to promote lipid synthesis under hypoxia. Nat. Commun. 7, 11960. doi:10.1038/ncomms11960
Germain, N., Dhayer, M., Boileau, M., Fovez, Q., Kluza, J., and Marchetti, P. (2020). Lipid metabolism and resistance to anticancer treatment. Biol. (Basel) 9 (12), 474. doi:10.3390/biology9120474
Gholkar, A. A., Cheung, K., Williams, K. J., Lo, Y. C., Hamideh, S. A., Nnebe, C., et al. (2016). Fatostatin inhibits cancer cell proliferation by affecting mitotic microtubule spindle assembly and cell division. J. Biol. Chem. 291 (33), 17001–17008. doi:10.1074/jbc.C116.737346
Gouw, A. M., Margulis, K., Liu, N. S., Raman, S. J., Mancuso, A., Toal, G. G., et al. (2019). The MYC oncogene cooperates with sterol-regulated element-binding protein to regulate lipogenesis essential for neoplastic growth. Cell Metab. 30 (3), 556–572. doi:10.1016/j.cmet.2019.07.012
Granchi, C. (2018). ATP citrate lyase (ACLY) inhibitors: an anti-cancer strategy at the crossroads of glucose and lipid metabolism. Eur. J. Med. Chem. 157, 1276–1291. doi:10.1016/j.ejmech.2018.09.001
Grunt, T. W., Wagner, R., Grusch, M., Berger, W., Singer, C. F., Marian, B., et al. (2009). Interaction between fatty acid synthase- and ErbB-systems in ovarian cancer cells. Biochem. Biophys. Res. Commun. 385 (3), 454–459. doi:10.1016/j.bbrc.2009.05.085
Grunt, T. W., Wagner, R., Ries, A., Berghoff, A. S., Preusser, M., Grusch, M., et al. (2024). Targeting endogenous fatty acid synthesis stimulates the migration of ovarian cancer cells to adipocytes and promotes the transport of fatty acids from adipocytes to cancer cells. Int. J. Oncol. 64 (3), 24. doi:10.3892/ijo.2024.5612
Guan, M., Fousek, K., Jiang, C., Guo, S., Synold, T., Xi, B., et al. (2011). Nelfinavir induces liposarcoma apoptosis through inhibition of regulated intramembrane proteolysis of SREBP-1 and ATF6. Clin. Cancer Res. 17 (7), 1796–1806. doi:10.1158/1078-0432.CCR-10-3216
Guan, M., Su, L., Yuan, Y. C., Li, H., and Chow, W. A. (2015). Nelfinavir and nelfinavir analogs block site-2 protease cleavage to inhibit castration-resistant prostate cancer. Sci. Rep. 5, 9698. doi:10.1038/srep09698
Guillet-Deniau, I., Pichard, A. L., Koné, A., Esnous, C., Nieruchalski, M., Girard, J., et al. (2004). Glucose induces de novo lipogenesis in rat muscle satellite cells through a sterol-regulatory-element-binding-protein-1c-dependent pathway. J. Cell Sci. 117 (Pt 10), 1937–1944. doi:10.1242/jcs.01069
Guo, D., Bell, E. H., Mischel, P., and Chakravarti, A. (2014). Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 20 (15), 2619–2626. doi:10.2174/13816128113199990486
Guo, W., Ma, J., Yang, Y., Guo, S., Zhang, W., Zhao, T., et al. (2020). ATP-citrate lyase epigenetically potentiates oxidative phosphorylation to promote melanoma growth and adaptive resistance to MAPK inhibition. Clin. Cancer Res. 26 (11), 2725–2739. doi:10.1158/1078-0432.CCR-19-1359
Guseva, N. V., Rokhlin, O. W., Glover, R. A., and Cohen, M. B. (2011). TOFA (5-tetradecyl-oxy-2-furoic acid) reduces fatty acid synthesis, inhibits expression of AR, neuropilin-1 and Mcl-1 and kills prostate cancer cells independent of p53 status. Cancer Biol. Ther. 12 (1), 80–85. doi:10.4161/cbt.12.1.15721
Hagiwara, N., Watanabe, M., Iizuka-Ohashi, M., Yokota, I., Toriyama, S., Sukeno, M., et al. (2018). Mevalonate pathway blockage enhances the efficacy of mTOR inhibitors with the activation of retinoblastoma protein in renal cell carcinoma. Cancer Lett. 431, 182–189. doi:10.1016/j.canlet.2018.05.025
Han, J. Y., Lee, S. H., Yoo, N. J., Hyung, L. S., Moon, Y. J., Yun, T., et al. (2011). A randomized phase II study of gefitinib plus simvastatin versus gefitinib alone in previously treated patients with advanced non-small cell lung cancer. Clin. Cancer Res. 17 (6), 1553–1560. doi:10.1158/1078-0432.CCR-10-2525
Hanahan, D., and Weinberg, R. A. (2011). Hallmarks of cancer: the next generation. Cell 144 (5), 646–674. doi:10.1016/j.cell.2011.02.013
Harayama, T., and Riezman, H. (2018). Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 19 (5), 281–296. doi:10.1038/nrm.2017.138
Hatzivassiliou, G., Zhao, F., Bauer, D. E., Andreadis, C., Shaw, A. N., Dhanak, D., et al. (2005). ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell 8 (4), 311–321. doi:10.1016/j.ccr.2005.09.008
He, W., Liang, B., Wang, C., Li, S., Zhao, Y., Huang, Q., et al. (2019). MSC-regulated lncRNA MACC1-AS1 promotes stemness and chemoresistance through fatty acid oxidation in gastric cancer. Oncogene 38 (23), 4637–4654. doi:10.1038/s41388-019-0747-0
Her, N. G., Jeong, S. I., Cho, K., Ha, T. K., Han, J., Ko, K. P., et al. (2013). PPARδ promotes oncogenic redirection of TGF-β1 signaling through the activation of the ABCA1-Cav1 pathway. Cell Cycle 12 (10), 1521–1535. doi:10.4161/cc.24636
Hernandez-Quiles, M., Broekema, M. F., and Kalkhoven, E. (2021). PPARgamma in metabolism, immunity, and cancer: unified and diverse mechanisms of action. Front. Endocrinol. (Lausanne) 12, 624112. doi:10.3389/fendo.2021.624112
Hilvo, M., Denkert, C., Lehtinen, L., Müller, B., Brockmöller, S., Seppänen-Laakso, T., et al. (2011). Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 71 (9), 3236–3245. doi:10.1158/0008-5472.CAN-10-3894
Hon, K. W., Zainal Abidin, S. A., Othman, I., and Naidu, R. (2021). The crosstalk between signaling pathways and cancer metabolism in colorectal cancer. Front. Pharmacol. 12, 768861. doi:10.3389/fphar.2021.768861
Hopperton, K. E., Duncan, R. E., Bazinet, R. P., and Archer, M. C. (2014). Fatty acid synthase plays a role in cancer metabolism beyond providing fatty acids for phospholipid synthesis or sustaining elevations in glycolytic activity. Exp. Cell Res. 320 (2), 302–310. doi:10.1016/j.yexcr.2013.10.016
Horton, J. D., Goldstein, J. L., and Brown, M. S. (2002). SREBPs: activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109 (9), 1125–1131. doi:10.1172/JCI15593
Hoxhaj, G., and Manning, B. D. (2020). The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 20 (2), 74–88. doi:10.1038/s41568-019-0216-7
Hua, T. N. M., Kim, M. K., Vo, V. T. A., Choi, J. W., Choi, J. H., Kim, H. W., et al. (2019). Inhibition of oncogenic Src induces FABP4-mediated lipolysis via PPARγ activation exerting cancer growth suppression. EBioMedicine 41, 134–145. doi:10.1016/j.ebiom.2019.02.015
Huang, Q., Wang, Q., Li, D., Wei, X., Jia, Y., Zhang, Z., et al. (2019). Co-administration of 20(S)-protopanaxatriol (g-PPT) and EGFR-TKI overcomes EGFR-TKI resistance by decreasing SCD1 induced lipid accumulation in non-small cell lung cancer. J. Exp. Clin. Cancer Res. 38 (1), 129. doi:10.1186/s13046-019-1120-4
Iizuka, K., Bruick, R. K., Liang, G., Horton, J. D., and Uyeda, K. (2004). Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc. Natl. Acad. Sci. U. S. A. 101 (19), 7281–7286. doi:10.1073/pnas.0401516101
Isenberg, J. S., Yu, C., and Roberts, D. D. (2008). Differential effects of ABT-510 and a CD36-binding peptide derived from the type 1 repeats of thrombospondin-1 on fatty acid uptake, nitric oxide signaling, and caspase activation in vascular cells. Biochem. Pharmacol. 75 (4), 875–882. doi:10.1016/j.bcp.2007.10.025
Ishii, S., Iizuka, K., Miller, B. C., and Uyeda, K. (2004). Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. U. S. A. 101 (44), 15597–15602. doi:10.1073/pnas.0405238101
Jabs, M., Rose, A. J., Lehmann, L. H., Taylor, J., Moll, I., Sijmonsma, T. P., et al. (2018). Inhibition of endothelial Notch signaling impairs fatty acid transport and leads to metabolic and vascular remodeling of the adult heart. Circulation 137 (24), 2592–2608. doi:10.1161/CIRCULATIONAHA.117.029733
Jeong, D. W., Lee, S., and Chun, Y. S. (2021). How cancer cells remodel lipid metabolism: strategies targeting transcription factors. Lipids Health Dis. 20 (1), 163. doi:10.1186/s12944-021-01593-8
Jhu, J. W., Yan, J. B., Lin, Z. H., Lin, S. C., and Peng, I. C. (2021). SREBP1-Induced glutamine synthetase triggers a feedforward loop to upregulate SREBP1 through Sp1 O-GlcNAcylation and augments lipid droplet formation in cancer cells. Int. J. Mol. Sci. 22 (18), 9814. doi:10.3390/ijms22189814
Jin, H. R., Wang, J., Wang, Z. J., Xi, M. J., Xia, B. H., Deng, K., et al. (2023). Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics. J. Hematol. Oncol. 16 (1), 103. doi:10.1186/s13045-023-01498-2
Jin, X., Demere, Z., Nair, K., Ali, A., Ferraro, G. B., Natoli, T., et al. (2020). A metastasis map of human cancer cell lines. Nature 588 (7837), 331–336. doi:10.1038/s41586-020-2969-2
Kauraniemi, P., Bärlund, M., Monni, O., and Kallioniemi, A. (2001). New amplified and highly expressed genes discovered in the ERBB2 amplicon in breast cancer by cDNA microarrays. Cancer Res. 61 (22), 8235–8240.
Kelly, W., Diaz Duque, A. E., Michalek, J., Konkel, B., Caflisch, L., Chen, Y., et al. (2023). Phase II investigation of TVB-2640 (denifanstat) with bevacizumab in patients with first relapse high-grade astrocytoma. Clin. Cancer Res. 29 (13), 2419–2425. doi:10.1158/1078-0432.CCR-22-2807
Kim, J. S., Turbov, J., Rosales, R., Thaete, L. G., and Rodriguez, G. C. (2019). Combination simvastatin and metformin synergistically inhibits endometrial cancer cell growth. Gynecol. Oncol. 154 (2), 432–440. doi:10.1016/j.ygyno.2019.05.022
Kim, K. H., Song, M. J., Yoo, E. J., Choe, S. S., Park, S. D., and Kim, J. B. (2004). Regulatory role of glycogen synthase kinase 3 for transcriptional activity of ADD1/SREBP1c. J. Biol. Chem. 279 (50), 51999–52006. doi:10.1074/jbc.M405522200
Kinnaird, A., Zhao, S., Wellen, K. E., and Michelakis, E. D. (2016). Metabolic control of epigenetics in cancer. Nat. Rev. Cancer 16 (11), 694–707. doi:10.1038/nrc.2016.82
Kliewer, S. A., Sundseth, S. S., Jones, S. A., Brown, P. J., Wisely, G. B., Koble, C. S., et al. (1997). Fatty acids and eicosanoids regulate gene expression through direct interactions with peroxisome proliferator-activated receptors alpha and gamma. Proc. Natl. Acad. Sci. U. S. A. 94 (9), 4318–4323. doi:10.1073/pnas.94.9.4318
Koo, Y. D., Lee, J. S., Lee, S. A., Quaresma, P. G. F., Bhat, R., Haynes, W. G., et al. (2019). SUMO-specific protease 2 mediates leptin-induced fatty acid oxidation in skeletal muscle. Metabolism 95, 27–35. doi:10.1016/j.metabol.2019.03.004
Koundouros, N., and Poulogiannis, G. (2020). Reprogramming of fatty acid metabolism in cancer. Br. J. Cancer 122 (1), 4–22. doi:10.1038/s41416-019-0650-z
Kourtidis, A., Srinivasaiah, R., Carkner, R. D., Brosnan, M. J., and Conklin, D. S. (2009). Peroxisome proliferator-activated receptor-gamma protects ERBB2-positive breast cancer cells from palmitate toxicity. Breast Cancer Res. 11 (2), R16. doi:10.1186/bcr2240
Kridel, S. J., Axelrod, F., Rozenkrantz, N., and Smith, J. W. (2004). Orlistat is a novel inhibitor of fatty acid synthase with antitumor activity. Cancer Res. 64 (6), 2070–2075. doi:10.1158/0008-5472.can-03-3645
Ku, C. Y., Liu, Y. H., Lin, H. Y., Lu, S. C., and Lin, J. Y. (2016). Liver fatty acid-binding protein (L-FABP) promotes cellular angiogenesis and migration in hepatocellular carcinoma. Oncotarget 7 (14), 18229–18246. doi:10.18632/oncotarget.7571
Kuhajda, F. P. (2006). Fatty acid synthase and cancer: new application of an old pathway. Cancer Res. 66 (12), 5977–5980. doi:10.1158/0008-5472.CAN-05-4673
Kuo, C. Y., and Ann, D. K. (2018). When fats commit crimes: fatty acid metabolism, cancer stemness and therapeutic resistance. Cancer Commun. (Lond). 38 (1), 47. doi:10.1186/s40880-018-0317-9
Lai, K. K. Y., Kweon, S. M., Chi, F., Hwang, E., Kabe, Y., Higashiyama, R., et al. (2017). Stearoyl-CoA desaturase promotes liver fibrosis and tumor development in mice via a Wnt positive-signaling loop by stabilization of low-density lipoprotein-receptor-related proteins 5 and 6. Gastroenterology 152 (6), 1477–1491. doi:10.1053/j.gastro.2017.01.021
Lambrescu, I. M., Gaina, G. F., Ceafalan, L. C., and Hinescu, M. E. (2024). Inside anticancer therapy resistance and metastasis. Focus on CD36. J. Cancer 15 (6), 1675–1686. doi:10.7150/jca.90457
Le, T. T., Huff, T. B., and Cheng, J. X. (2009). Coherent anti-Stokes Raman scattering imaging of lipids in cancer metastasis. BMC Cancer 9, 42. doi:10.1186/1471-2407-9-42
Lee, G., Zheng, Y., Cho, S., Jang, C., England, C., Dempsey, J. M., et al. (2017). Post-transcriptional regulation of de novo lipogenesis by mTORC1-S6K1-SRPK2 signaling. Cell 171 (7), 1545–1558. doi:10.1016/j.cell.2017.10.037
Lee, H., Deng, J., Kujawski, M., Yang, C., Liu, Y., Herrmann, A., et al. (2010). STAT3-induced S1PR1 expression is crucial for persistent STAT3 activation in tumors. Nat. Med. 16 (12), 1421–1428. doi:10.1038/nm.2250
Lee, J. V., Carrer, A., Shah, S., Snyder, N. W., Wei, S., Venneti, S., et al. (2014). Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 20 (2), 306–319. doi:10.1016/j.cmet.2014.06.004
Lee, U., Cho, E. Y., and Jho, E. H. (2022). Regulation of Hippo signaling by metabolic pathways in cancer. Biochim. Biophys. Acta Mol. Cell Res. 1869 (4), 119201. doi:10.1016/j.bbamcr.2021.119201
Lei, Y., Hu, Q., and Gu, J. (2020a). Expressions of carbohydrate response element binding protein and glucose transporters in liver cancer and clinical significance. Pathol. Oncol. Res. 26 (2), 1331–1340. doi:10.1007/s12253-019-00708-y
Lei, Y., Zhou, S., Hu, Q., Chen, X., and Gu, J. (2020b). Carbohydrate response element binding protein (ChREBP) correlates with colon cancer progression and contributes to cell proliferation. Sci. Rep. 10 (1), 4233. doi:10.1038/s41598-020-60903-9
Le Stunff, H., Galve-Roperh, I., Peterson, C., Milstien, S., and Spiegel, S. (2002). Sphingosine-1-phosphate phosphohydrolase in regulation of sphingolipid metabolism and apoptosis. J. Cell Biol. 158 (6), 1039–1049. doi:10.1083/jcb.200203123
Leuti, A., Fazio, D., Fava, M., Piccoli, A., Oddi, S., and Maccarrone, M. (2020). Bioactive lipids, inflammation and chronic diseases. Adv. Drug Deliv. Rev. 159, 133–169. doi:10.1016/j.addr.2020.06.028
Li, C. F., Fang, F. M., Chen, Y. Y., Liu, T. T., Chan, T. C., Yu, S. C., et al. (2017a). Overexpressed fatty acid synthase in gastrointestinal stromal tumors: targeting a progression-associated metabolic driver enhances the antitumor effect of imatinib. Clin. Cancer Res. 23 (16), 4908–4918. doi:10.1158/1078-0432.CCR-16-2770
Li, J., Zhao, S., Zhou, X., Zhang, T., Zhao, L., Miao, P., et al. (2013a). Inhibition of lipolysis by mercaptoacetate and etomoxir specifically sensitize drug-resistant lung adenocarcinoma cell to paclitaxel. PLoS One 8 (9), e74623. doi:10.1371/journal.pone.0074623
Li, N., Li, X., Ding, Y., Liu, X., Diggle, K., Kisseleva, T., et al. (2023a). SREBP regulation of lipid metabolism in liver disease, and therapeutic strategies. Biomedicines 11 (12), 3280. doi:10.3390/biomedicines11123280
Li, N., Zhou, Z. S., Shen, Y., Xu, J., Miao, H. H., Xiong, Y., et al. (2017b). Inhibition of the sterol regulatory element-binding protein pathway suppresses hepatocellular carcinoma by repressing inflammation in mice. Hepatology 65 (6), 1936–1947. doi:10.1002/hep.29018
Li, S., Qiu, L., Wu, B., Shen, H., Zhu, J., Zhou, L., et al. (2013b). TOFA suppresses ovarian cancer cell growth in vitro and in vivo. Mol. Med. Rep. 8 (2), 373–378. doi:10.3892/mmr.2013.1505
Li, X., Chen, Y. T., Hu, P., and Huang, W. C. (2014). Fatostatin displays high antitumor activity in prostate cancer by blocking SREBP-regulated metabolic pathways and androgen receptor signaling. Mol. Cancer Ther. 13 (4), 855–866. doi:10.1158/1535-7163.MCT-13-0797
Li, X., Wu, J. B., Chung, L. W., and Huang, W. C. (2015). Anti-cancer efficacy of SREBP inhibitor, alone or in combination with docetaxel, in prostate cancer harboring p53 mutations. Oncotarget 6 (38), 41018–41032. doi:10.18632/oncotarget.5879
Li, Y., Yang, W., Zheng, Y., Dai, W., Ji, J., Wu, L., et al. (2023b). Targeting fatty acid synthase modulates sensitivity of hepatocellular carcinoma to sorafenib via ferroptosis. J. Exp. Clin. Cancer Res. 42 (1), 6. doi:10.1186/s13046-022-02567-z
Ligorio, F., Pellegrini, I., Castagnoli, L., Vingiani, A., Lobefaro, R., Zattarin, E., et al. (2021). Targeting lipid metabolism is an emerging strategy to enhance the efficacy of anti-HER2 therapies in HER2-positive breast cancer. Cancer Lett. 511, 77–87. doi:10.1016/j.canlet.2021.04.023
Lin, H., Patel, S., Affleck, V. S., Wilson, I., Turnbull, D. M., Joshi, A. R., et al. (2017). Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 19 (1), 43–54. doi:10.1093/neuonc/now128
Little, J. L., Wheeler, F. B., Fels, D. R., Koumenis, C., and Kridel, S. J. (2007). Inhibition of fatty acid synthase induces endoplasmic reticulum stress in tumor cells. Cancer Res. 67 (3), 1262–1269. doi:10.1158/0008-5472.CAN-06-1794
Liu, Y., Gao, G. F., Minna, J. D., Williams, N. S., and Westover, K. D. (2021). Loss of wild type KRAS in KRASMUT lung adenocarcinoma is associated with cancer mortality and confers sensitivity to FASN inhibitors. Lung Cancer 153, 73–80. doi:10.1016/j.lungcan.2020.12.032
Lodhi, I. J., and Semenkovich, C. F. (2014). Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. 19 (3), 380–392. doi:10.1016/j.cmet.2014.01.002
Luo, J., Hong, Y., Lu, Y., Qiu, S., Chaganty, B. K., Zhang, L., et al. (2017). Acetyl-CoA carboxylase rewires cancer metabolism to allow cancer cells to survive inhibition of the Warburg effect by cetuximab. Cancer Lett. 384, 39–49. doi:10.1016/j.canlet.2016.09.020
Ma, S., Zhou, B., Yang, Q., Pan, Y., Yang, W., Freedland, S. J., et al. (2021). A transcriptional regulatory loop of master regulator transcription factors, PPARG, and fatty acid synthesis promotes esophageal adenocarcinoma. Cancer Res. 81 (5), 1216–1229. doi:10.1158/0008-5472.CAN-20-0652
Maceyka, M., Harikumar, K. B., Milstien, S., and Spiegel, S. (2012). Sphingosine-1-phosphate signaling and its role in disease. Trends Cell Biol. 22 (1), 50–60. doi:10.1016/j.tcb.2011.09.003
Mallick, R., Bhowmik, P., and Duttaroy, A. K. (2023). Targeting fatty acid uptake and metabolism in cancer cells: a promising strategy for cancer treatment. Biomed. Pharmacother. 167, 115591. doi:10.1016/j.biopha.2023.115591
Manning, B. D., and Cantley, L. C. (2007). AKT/PKB signaling: navigating downstream. Cell 129 (7), 1261–1274. doi:10.1016/j.cell.2007.06.009
Markovic, S. N., Suman, V. J., Rao, R. A., Ingle, J. N., Kaur, J. S., Erickson, L. A., et al. (2007). A phase II study of ABT-510 (thrombospondin-1 analog) for the treatment of metastatic melanoma. Am. J. Clin. Oncol. 30 (3), 303–309. doi:10.1097/01.coc.0000256104.80089.35
Mauro, C., Leow, S. C., Anso, E., Rocha, S., Thotakura, A. K., Tornatore, L., et al. (2011). NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 13 (10), 1272–1279. doi:10.1038/ncb2324
McDonnell, E., Crown, S. B., Fox, D. B., Kitir, B., Ilkayeva, O. R., Olsen, C. A., et al. (2016). Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. 17 (6), 1463–1472. doi:10.1016/j.celrep.2016.10.012
Medes, G., Thomas, A., and Weinhouse, S. (1953). Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer Res. 13 (1), 27–29.
Menendez, J. A., and Lupu, R. (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7 (10), 763–777. doi:10.1038/nrc2222
Menendez, J. A., Mehmi, I., Atlas, E., Colomer, R., and Lupu, R. (2004a). Novel signaling molecules implicated in tumor-associated fatty acid synthase-dependent breast cancer cell proliferation and survival: role of exogenous dietary fatty acids, p53-p21WAF1/CIP1, ERK1/2 MAPK, p27KIP1, BRCA1, and NF-κB. Int. J. Oncol. 24 (3), 591–608. doi:10.3892/ijo.24.3.591
Menendez, J. A., Oza, B. P., Atlas, E., Verma, V. A., Mehmi, I., and Lupu, R. (2004b). Inhibition of tumor-associated fatty acid synthase activity antagonizes estradiol- and tamoxifen-induced agonist transactivation of estrogen receptor (ER) in human endometrial adenocarcinoma cells. Oncogene 23 (28), 4945–4958. doi:10.1038/sj.onc.1207476
Menendez, J. A., Vellon, L., and Lupu, R. (2005). Antitumoral actions of the anti-obesity drug orlistat (XenicalTM) in breast cancer cells: blockade of cell cycle progression, promotion of apoptotic cell death and PEA3-mediated transcriptional repression of Her2/neu (erbB-2) oncogene. Ann. Oncol. 16 (8), 1253–1267. doi:10.1093/annonc/mdi239
Metallo, C. M., Gameiro, P. A., Bell, E. L., Mattaini, K. R., Yang, J., Hiller, K., et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481 (7381), 380–384. doi:10.1038/nature10602
Milstien, S., and Spiegel, S. (2006). Targeting sphingosine-1-phosphate: a novel avenue for cancer therapeutics. Cancer Cell 9 (3), 148–150. doi:10.1016/j.ccr.2006.02.025
Mitra, R., Goodman, O. B., and Le, T. T. (2014). Enhanced detection of metastatic prostate cancer cells in human plasma with lipid bodies staining. BMC Cancer 14, 91. doi:10.1186/1471-2407-14-91
Molaei, M., Vandehoef, C., and Karpac, J. (2019). NF-κB shapes metabolic adaptation by attenuating foxo-mediated lipolysis in Drosophila. Dev. Cell 49 (5), 802–810. doi:10.1016/j.devcel.2019.04.009
Mukherjee, A., Bilecz, A. J., and Lengyel, E. (2022). The adipocyte microenvironment and cancer. Cancer Metastasis Rev. 41 (3), 575–587. doi:10.1007/s10555-022-10059-x
Mukherjee, A., Chiang, C. Y., Daifotis, H. A., Nieman, K. M., Fahrmann, J. F., Lastra, R. R., et al. (2020). Adipocyte-induced FABP4 expression in ovarian cancer cells promotes metastasis and mediates carboplatin resistance. Cancer Res. 80 (8), 1748–1761. doi:10.1158/0008-5472.CAN-19-1999
Mukherjee, A., Kenny, H. A., and Lengyel, E. (2017). Unsaturated fatty acids maintain cancer cell stemness. Cell Stem Cell 20 (3), 291–292. doi:10.1016/j.stem.2017.02.008
Mullen, G. E., and Yet, L. (2015). Progress in the development of fatty acid synthase inhibitors as anticancer targets. Bioorg Med. Chem. Lett. 25 (20), 4363–4369. doi:10.1016/j.bmcl.2015.08.087
Muñoz-Pinedo, C., El Mjiyad, N., and Ricci, J. E. (2012). Cancer metabolism: current perspectives and future directions. Cell Death Dis. 3 (1), e248. doi:10.1038/cddis.2011.123
Nabors, L. B., Fiveash, J. B., Markert, J. M., Kekan, M. S., Gillespie, G. Y., Huang, Z., et al. (2010). A phase 1 trial of ABT-510 concurrent with standard chemoradiation for patients with newly diagnosed glioblastoma. Arch. Neurol. 67 (3), 313–319. doi:10.1001/archneurol.2010.16
Nambiar, D. K., Deep, G., Singh, R. P., Agarwal, C., and Agarwal, R. (2014). Silibinin inhibits aberrant lipid metabolism, proliferation and emergence of androgen-independence in prostate cancer cells via primarily targeting the sterol response element binding protein 1. Oncotarget 5 (20), 10017–10033. doi:10.18632/oncotarget.2488
Nomura, D. K., Lombardi, D. P., Chang, J. W., Niessen, S., Ward, A. M., Long, J. Z., et al. (2011). Monoacylglycerol lipase exerts dual control over endocannabinoid and fatty acid pathways to support prostate cancer. Chem. Biol. 18 (7), 846–856. doi:10.1016/j.chembiol.2011.05.009
Nomura, D. K., Long, J. Z., Niessen, S., Hoover, H. S., Ng, S. W., and Cravatt, B. F. (2010). Monoacylglycerol lipase regulates a fatty acid network that promotes cancer pathogenesis. Cell 140 (1), 49–61. doi:10.1016/j.cell.2009.11.027
Novak, A., Binnington, B., Ngan, B., Chadwick, K., Fleshner, N., and Lingwood, C. A. (2013). Cholesterol masks membrane glycosphingolipid tumor-associated antigens to reduce their immunodetection in human cancer biopsies. Glycobiology 23 (11), 1230–1239. doi:10.1093/glycob/cwt059
Ocaña, M. C., Martínez-Poveda, B., Quesada, A. R., and Medina, M. Á. (2020). Glucose favors lipid anabolic metabolism in the invasive breast cancer cell line MDA-MB-231. Biol. (Basel). 9 (1), 16. doi:10.3390/biology9010016
Ooms, L. M., Binge, L. C., Davies, E. M., Rahman, P., Conway, J. R., Gurung, R., et al. (2015). The inositol polyphosphate 5-phosphatase PIPP regulates AKT1-dependent breast cancer growth and metastasis. Cancer Cell 28 (2), 155–169. doi:10.1016/j.ccell.2015.07.003
Ortega-Prieto, P., and Postic, C. (2019). Carbohydrate sensing through the transcription factor ChREBP. Front. Genet. 10, 472. doi:10.3389/fgene.2019.00472
Owen, J. L., Zhang, Y., Bae, S. H., Farooqi, M. S., Liang, G., Hammer, R. E., et al. (2012). Insulin stimulation of SREBP-1c processing in transgenic rat hepatocytes requires p70 S6-kinase. Proc. Natl. Acad. Sci. U. S. A. 109 (40), 16184–16189. doi:10.1073/pnas.1213343109
Pandey, P., Khan, F., Choi, M., Singh, S. K., Kang, H. N., Park, M. N., et al. (2023b). Review deciphering potent therapeutic approaches targeting Notch signaling pathway in breast cancer. Biomed. Pharmacother. 164, 114938. doi:10.1016/j.biopha.2023.114938
Pandey, P., Khan, F., Singh, M., Verma, A., Kumar, H., Mazumder, A., et al. (2023a). Study deciphering the crucial involvement of Notch signaling pathway in human cancers. Endocr. Metab. Immune Disord. Drug Targets 22, 1241–1253. doi:10.2174/0118715303261691231107113548
Papaevangelou, E., Almeida, G. S., Box, C., deSouza, N. M., and Chung, Y. L. (2018). The effect of FASN inhibition on the growth and metabolism of a cisplatin-resistant ovarian carcinoma model. Int. J. Cancer 143 (4), 992–1002. doi:10.1002/ijc.31392
Pascual, G., Avgustinova, A., Mejetta, S., Martín, M., Castellanos, A., Attolini, C. S., et al. (2017). Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541 (7635), 41–45. doi:10.1038/nature20791
Patmanathan, S. N., Johnson, S. P., Lai, S. L., Panja Bernam, S., Lopes, V., Wei, W., et al. (2016). Aberrant expression of the S1P regulating enzymes, SPHK1 and SGPL1, contributes to a migratory phenotype in OSCC mediated through S1PR2. Sci. Rep. 6, 25650. doi:10.1038/srep25650
Peng, Y., He, G., Tang, D., Xiong, L., Wen, Y., Miao, X., et al. (2017). Lovastatin inhibits cancer stem cells and sensitizes to chemo- and photodynamic therapy in nasopharyngeal carcinoma. J. Cancer 8 (9), 1655–1664. doi:10.7150/jca.19100
Peters, J. M., Cattley, R. C., and Gonzalez, F. J. (1997). Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis 18 (11), 2029–2033. doi:10.1093/carcin/18.11.2029
Phan, A. N. H., Vo, V. T. A., Hua, T. N. M., Kim, M. K., Jo, S. Y., Choi, J. W., et al. (2017). PPARγ sumoylation-mediated lipid accumulation in lung cancer. Oncotarget 8 (47), 82491–82505. doi:10.18632/oncotarget.19700
Porcuna, J., Mínguez-Martínez, J., and Ricote, M. (2021). The PPARα and PPARγ epigenetic landscape in cancer and immune and metabolic disorders. Int. J. Mol. Sci. 22 (19), 10573. doi:10.3390/ijms221910573
Porstmann, T., Griffiths, B., Chung, Y. L., Delpuech, O., Griffiths, J. R., Downward, J., et al. (2005). PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 24 (43), 6465–6481. doi:10.1038/sj.onc.1208802
Potapova, I. A., El-Maghrabi, M. R., Doronin, S. V., and Benjamin, W. B. (2000). Phosphorylation of recombinant human ATP:citrate lyase by cAMP-dependent protein kinase abolishes homotropic allosteric regulation of the enzyme by citrate and increases the enzyme activity. Allosteric activation of ATP: citrate lyase by phosphorylated sugars. Biochemistry 39 (5), 1169–1179. doi:10.1021/bi992159y
Pyne, N. J., and Pyne, S. (2010). Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 10 (7), 489–503. doi:10.1038/nrc2875
Qu, Q., Zeng, F., Liu, X., Wang, Q. J., and Deng, F. (2016). Fatty acid oxidation and carnitine palmitoyltransferase I: emerging therapeutic targets in cancer. Cell Death Dis. 7 (5), e2226. doi:10.1038/cddis.2016.132
Rabold, K., Aschenbrenner, A., Thiele, C., Boahen, C. K., Schiltmans, A., Smit, J. W. A., et al. (2020). Enhanced lipid biosynthesis in human tumor-induced macrophages contributes to their protumoral characteristics. J. Immunother. Cancer 8 (2), e000638. doi:10.1136/jitc-2020-000638
Rao, E., Singh, P., Zhai, X., Li, Y., Zhu, G., Zhang, Y., et al. (2015). Inhibition of tumor growth by a newly-identified activator for epidermal fatty acid binding protein. Oncotarget 6 (10), 7815–7827. doi:10.18632/oncotarget.3485
Ray, K. K., Bays, H. E., Catapano, A. L., Lalwani, N. D., Bloedon, L. T., Sterling, L. R., et al. (2019). Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N. Engl. J. Med. 380 (11), 1022–1032. doi:10.1056/NEJMoa1803917
Ricoult, S. J., Yecies, J. L., Ben-Sahra, I., and Manning, B. D. (2016). Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 35 (10), 1250–1260. doi:10.1038/onc.2015.179
Rinella, M. E. (2015). Nonalcoholic fatty liver disease: a systematic review. JAMA 313 (22), 2263–2273. doi:10.1001/jama.2015.5370
Rios-Esteves, J., and Resh, M. D. (2013). Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep. 4 (6), 1072–1081. doi:10.1016/j.celrep.2013.08.027
Rodic, S., Mihalcioiu, C., and Saleh, R. R. (2014). Detection methods of circulating tumor cells in cutaneous melanoma: a systematic review. Crit. Rev. Oncol. Hematol. 91 (1), 74–92. doi:10.1016/j.critrevonc.2014.01.007
Röhrig, F., and Schulze, A. (2016). The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 16 (11), 732–749. doi:10.1038/nrc.2016.89
Rysman, E., Brusselmans, K., Scheys, K., Timmermans, L., Derua, R., Munck, S., et al. (2010). De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 70 (20), 8117–8126. doi:10.1158/0008-5472.CAN-09-3871
Sangineto, M., Villani, R., Cavallone, F., Romano, A., Loizzi, D., and Serviddio, G. (2020). Lipid metabolism in development and progression of hepatocellular carcinoma. Cancers (Basel) 12 (6), 1419. doi:10.3390/cancers12061419
Santos, C. R., and Schulze, A. (2012). Lipid metabolism in cancer. FEBS J. 279 (15), 2610–2623. doi:10.1111/j.1742-4658.2012.08644.x
Schug, Z. T., Peck, B., Jones, D. T., Zhang, Q., Grosskurth, S., Alam, I. S., et al. (2015). Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27 (1), 57–71. doi:10.1016/j.ccell.2014.12.002
Sengelaub, C. A., Navrazhina, K., Ross, J. B., Halberg, N., and Tavazoie, S. F. (2016). PTPRN2 and PLCβ1 promote metastatic breast cancer cell migration through PI(4,5)P2-dependent actin remodeling. EMBO J. 35 (1), 62–76. doi:10.15252/embj.201591973
She, K., Fang, S., Du, W., Fan, X., He, J., Pan, H., et al. (2019). SCD1 is required for EGFR-targeting cancer therapy of lung cancer via re-activation of EGFR/PI3K/AKT signals. Cancer Cell Int. 19, 103. doi:10.1186/s12935-019-0809-y
Shen, S., Faouzi, S., Souquere, S., Roy, S., Routier, E., Libenciuc, C., et al. (2020). Melanoma persister cells are tolerant to BRAF/MEK inhibitors via ACOX1-mediated fatty acid oxidation. Cell Rep. 33 (8), 108421. doi:10.1016/j.celrep.2020.108421
Shi, Z., Zhou, Q., Gao, S., Li, W., Li, X., Liu, Z., et al. (2019). Silibinin inhibits endometrial carcinoma via blocking pathways of STAT3 activation and SREBP1-mediated lipid accumulation. Life Sci. 217, 70–80. doi:10.1016/j.lfs.2018.11.037
Shimada, T., Kojima, K., Yoshiura, K., Hiraishi, H., and Terano, A. (2002). Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut 50 (5), 658–664. doi:10.1136/gut.50.5.658
Shimano, H., and Sato, R. (2017). SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat. Rev. Endocrinol. 13 (12), 710–730. doi:10.1038/nrendo.2017.91
Shiragami, R., Murata, S., Kosugi, C., Tezuka, T., Yamazaki, M., Hirano, A., et al. (2013). Enhanced antitumor activity of cerulenin combined with oxaliplatin in human colon cancer cells. Int. J. Oncol. 43 (2), 431–438. doi:10.3892/ijo.2013.1978
Shojaei, S., Koleini, N., Samiei, E., Aghaei, M., Cole, L. K., Alizadeh, J., et al. (2020). Simvastatin increases temozolomide-induced cell death by targeting the fusion of autophagosomes and lysosomes. FEBS J. 287 (5), 1005–1034. doi:10.1111/febs.15069
Shueng, P. W., Chan, H. W., Lin, W. C., Kuo, D. Y., and Chuang, H. Y. (2022). Orlistat resensitizes sorafenib-resistance in hepatocellular carcinoma cells through modulating metabolism. Int. J. Mol. Sci. 23 (12), 6501. doi:10.3390/ijms23126501
Siltari, A., Riikonen, J., Koskimäki, J., Pakarainen, T., Ettala, O., Boström, P., et al. (2022). Randomised double-blind phase 3 clinical study testing impact of atorvastatin on prostate cancer progression after initiation of androgen deprivation therapy: study protocol. BMJ Open 12 (4), e050264. doi:10.1136/bmjopen-2021-050264
Snaebjornsson, M. T., Janaki-Raman, S., and Schulze, A. (2020). Greasing the wheels of the cancer machine: the role of lipid metabolism in cancer. Cell Metab. 31 (1), 62–76. doi:10.1016/j.cmet.2019.11.010
Stremmel, W., Pohl, L., Ring, A., and Herrmann, T. (2001). A new concept of cellular uptake and intracellular trafficking of long-chain fatty acids. Lipids 36 (9), 981–989. doi:10.1007/s11745-001-0809-2
Strickaert, A., Saiselet, M., Dom, G., De Deken, X., Dumont, J. E., Feron, O., et al. (2017). Cancer heterogeneity is not compatible with one unique cancer cell metabolic map. Oncogene 36 (19), 2637–2642. doi:10.1038/onc.2016.411
Stuani, L., Riols, F., Millard, P., Sabatier, M., Batut, A., Saland, E., et al. (2018). Stable isotope labeling highlights enhanced fatty acid and lipid metabolism in human acute myeloid leukemia. Int. J. Mol. Sci. 19 (11), 3325. doi:10.3390/ijms19113325
Su, T., Huang, C., Yang, C., Jiang, T., Su, J., Chen, M., et al. (2020). Apigenin inhibits STAT3/CD36 signaling axis and reduces visceral obesity. Pharmacol. Res. 152, 104586. doi:10.1016/j.phrs.2019.104586
Sulaiman, A., McGarry, S., Li, L., Jia, D., Ooi, S., Addison, C., et al. (2018). Dual inhibition of Wnt and Yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol. Oncol. 12 (4), 423–440. doi:10.1002/1878-0261.12167
Sun, H., Yang, W., Tian, Y., Zeng, X., Zhou, J., Mok, M. T. S., et al. (2018). An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat. Commun. 9 (1), 5214. doi:10.1038/s41467-018-07402-8
Sun, P., Zhang, X., Wang, R. J., Ma, Q. Y., Xu, L., Wang, Y., et al. (2021). PI3Kα inhibitor CYH33 triggers antitumor immunity in murine breast cancer by activating CD8+T cells and promoting fatty acid metabolism. J. Immunother. Cancer 9 (8), e003093. doi:10.1136/jitc-2021-003093
Sundqvist, A., Bengoechea-Alonso, M. T., Ye, X., Lukiyanchuk, V., Jin, J., Harper, J. W., et al. (2005). Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell Metab. 1 (6), 379–391. doi:10.1016/j.cmet.2005.04.010
Svensson, R. U., Parker, S. J., Eichner, L. J., Kolar, M. J., Wallace, M., Brun, S. N., et al. (2016). Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 22 (10), 1108–1119. doi:10.1038/nm.4181
Swinnen, J. V., Van Veldhoven, P. P., Timmermans, L., De Schrijver, E., Brusselmans, K., Vanderhoydonc, F., et al. (2003). Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Biochem. Biophys. Res. Commun. 302 (4), 898–903. doi:10.1016/s0006-291x(03)00265-1
Tadros, S., Shukla, S. K., King, R. J., Gunda, V., Vernucci, E., Abrego, J., et al. (2017). De novo lipid synthesis facilitates gemcitabine resistance through endoplasmic reticulum stress in pancreatic cancer. Cancer Res. 77 (20), 5503–5517. doi:10.1158/0008-5472.CAN-16-3062
Takada, K., Kashiwagi, S., Iimori, N., Kouhashi, R., Yabumoto, A., Goto, W., et al. (2023). Impact of oral statin therapy on clinical outcomes in patients with cT1 breast cancer. BMC Cancer 23 (1), 224. doi:10.1186/s12885-023-10631-w
Talebi, A., Dehairs, J., Rambow, F., Rogiers, A., Nittner, D., Derua, R., et al. (2018). Sustained SREBP-1-dependent lipogenesis as a key mediator of resistance to BRAF-targeted therapy. Nat. Commun. 9 (1), 2500. doi:10.1038/s41467-018-04664-0
Tang, Z., Shen, Q., Xie, H., Zhou, X., Li, J., Feng, J., et al. (2016). Elevated expression of FABP3 and FABP4 cooperatively correlates with poor prognosis in non-small cell lung cancer (NSCLC). Oncotarget 7 (29), 46253–46262. doi:10.18632/oncotarget.10086
Tian, Y., Yang, B., Qiu, W., Hao, Y., Zhang, Z., Yang, B., et al. (2019). ER-residential Nogo-B accelerates NAFLD-associated HCC mediated by metabolic reprogramming of oxLDL lipophagy. Nat. Commun. 10 (1), 3391. doi:10.1038/s41467-019-11274-x
Tomek, K., Wagner, R., Varga, F., Singer, C. F., Karlic, H., and Grunt, T. W. (2011). Blockade of fatty acid synthase induces ubiquitination and degradation of phosphoinositide-3-kinase signaling proteins in ovarian cancer. Mol. Cancer Res. 9 (12), 1767–1779. doi:10.1158/1541-7786.MCR-10-0467
Tošić, I., and Frank, D. A. (2021). STAT3 as a mediator of oncogenic cellular metabolism: pathogenic and therapeutic implications. Neoplasia 23 (12), 1167–1178. doi:10.1016/j.neo.2021.10.003
Tošić, I., Heppler, L. N., Egusquiaguirre, S. P., Boehnke, N., Correa, S., Costa, D. F., et al. (2021). Lipidome-based targeting of STAT3-driven breast cancer cells using poly-l-glutamic acid-coated layer-by-layer nanoparticles. Mol. Cancer Ther. 20 (4), 726–738. doi:10.1158/1535-7163.MCT-20-0505
Uddin, S., Hussain, A. R., Ahmed, M., Bu, R., Ahmed, S. O., Ajarim, D., et al. (2010). Inhibition of fatty acid synthase suppresses c-Met receptor kinase and induces apoptosis in diffuse large B-cell lymphoma. Mol. Cancer Ther. 9 (5), 1244–1255. doi:10.1158/1535-7163.MCT-09-1061
Urbanski, L. M., Leclair, N., and Anczuków, O. (2018). Alternative-splicing defects in cancer: splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip. Rev. RNA 9 (4), e1476. doi:10.1002/wrna.1476
Uyeda, K., Yamashita, H., and Kawaguchi, T. (2002). Carbohydrate responsive element-binding protein (ChREBP): a key regulator of glucose metabolism and fat storage. Biochem. Pharmacol. 63 (12), 2075–2080. doi:10.1016/s0006-2952(02)01012-2
Varela-Rey, M., Iruarrizaga-Lejarreta, M., Lozano, J. J., Aransay, A. M., Fernandez, A. F., Lavin, J. L., et al. (2014). S-adenosylmethionine levels regulate the schwann cell DNA methylome. Neuron 81 (5), 1024–1039. doi:10.1016/j.neuron.2014.01.037
Vazquez-Martin, A., Colomer, R., Brunet, J., and Menendez, J. A. (2007). Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int. J. Oncol. 31 (4), 769–776. doi:10.3892/ijo.31.4.769
Vergara, D., Stanca, E., Guerra, F., Priore, P., Gaballo, A., Franck, J., et al. (2017). β-Catenin knockdown affects mitochondrial biogenesis and lipid metabolism in breast cancer cells. Front. Physiol. 8, 544. doi:10.3389/fphys.2017.00544
Vicinanza, M., D'Angelo, G., Di Campli, A., and De Matteis, M. A. (2008). Function and dysfunction of the PI system in membrane trafficking. EMBO J. 27 (19), 2457–2470. doi:10.1038/emboj.2008.169
Vidavsky, N., Kunitake, JAMR, Diaz-Rubio, M. E., Chiou, A. E., Loh, H. C., Zhang, S., et al. (2019). Mapping and profiling lipid distribution in a 3D model of breast cancer progression. ACS Cent. Sci. 5 (5), 768–780. doi:10.1021/acscentsci.8b00932
Vona, R., Iessi, E., and Matarrese, P. (2021). Role of cholesterol and lipid rafts in cancer signaling: a promising therapeutic opportunity? Front. Cell Dev. Biol. 9, 622908. doi:10.3389/fcell.2021.622908
von Roemeling, C. A., Marlow, L. A., Wei, J. J., Cooper, S. J., Caulfield, T. R., Wu, K., et al. (2013). Stearoyl-CoA desaturase 1 is a novel molecular therapeutic target for clear cell renal cell carcinoma. Clin. Cancer Res. 19 (9), 2368–2380. doi:10.1158/1078-0432.CCR-12-3249
Wagner, R., Stübiger, G., Veigel, D., Wuczkowski, M., Lanzerstorfer, P., Weghuber, J., et al. (2017). Multi-level suppression of receptor-PI3K-mTORC1 by fatty acid synthase inhibitors is crucial for their efficacy against ovarian cancer cells. Oncotarget 8 (7), 11600–11613. doi:10.18632/oncotarget.14591
Walther, J. L., Metallo, C. M., Zhang, J., and Stephanopoulos, G. (2012). Optimization of 13C isotopic tracers for metabolic flux analysis in mammalian cells. Metab. Eng. 14 (2), 162–171. doi:10.1016/j.ymben.2011.12.004
Wang, C., Xu, C., Sun, M., Luo, D., Liao, D. F., and Cao, D. (2009a). Acetyl-CoA carboxylase-alpha inhibitor TOFA induces human cancer cell apoptosis. Biochem. Biophys. Res. Commun. 385 (3), 302–306. doi:10.1016/j.bbrc.2009.05.045
Wang, D., Dai, S., Lou, D., Wang, T., Wang, S., and Zheng, Z. (2023). Association between statins exposure and risk of skin cancer: an updated meta-analysis. Int. J. Dermatol 62 (11), 1332–1344. doi:10.1111/ijd.16816
Wang, D., Fu, L., Wei, J., Xiong, Y., and DuBois, R. N. (2019). PPARδ mediates the effect of dietary fat in promoting colorectal cancer metastasis. Cancer Res. 79 (17), 4480–4490. doi:10.1158/0008-5472.CAN-19-0384
Wang, D., Zhou, Y., Lei, W., Zhang, K., Shi, J., Hu, Y., et al. (2009b). Signal transducer and activator of transcription 3 (STAT3) regulates adipocyte differentiation via peroxisome-proliferator-activated receptor gamma (PPARgamma). Biol. Cell 102 (1), 1–12. doi:10.1042/BC20090070
Wang, H., Zhou, Y., Xu, H., Wang, X., Zhang, Y., Shang, R., et al. (2022). Therapeutic efficacy of FASN inhibition in preclinical models of HCC. Hepatology 76 (4), 951–966. doi:10.1002/hep.32359
Wang, J. J., Lei, K. F., and Han, F. (2018a). Tumor microenvironment: recent advances in various cancer treatments. Eur. Rev. Med. Pharmacol. Sci. 22 (12), 3855–3864. doi:10.26355/eurrev_201806_15270
Wang, T., Fahrmann, J. F., Lee, H., Li, Y. J., Tripathi, S. C., Yue, C., et al. (2018b). JAK/STAT3-Regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 27 (1), 136–150. doi:10.1016/j.cmet.2017.11.001
Wang, T., Fahrmann, J. F., Lee, H., Li, Y. J., Tripathi, S. C., Yue, C., et al. (2018c). JAK/STAT3-Regulated fatty acid β-oxidation is critical for breast cancer stem cell self-renewal and chemoresistance. Cell Metab. 27 (6), 1357. doi:10.1016/j.cmet.2018.04.018
Wang, X., Cai, B., Yang, X., Sonubi, O. O., Zheng, Z., Ramakrishnan, R., et al. (2020). Cholesterol stabilizes TAZ in hepatocytes to promote experimental non-alcoholic steatohepatitis. Cell Metab. 31 (5), 969–986. doi:10.1016/j.cmet.2020.03.010
Wang, X., Zheng, Z., Caviglia, J. M., Corey, K. E., Herfel, T. M., Cai, B., et al. (2016). Hepatocyte TAZ/WWTR1 promotes inflammation and fibrosis in nonalcoholic steatohepatitis. Cell Metab. 24 (6), 848–862. doi:10.1016/j.cmet.2016.09.016
Wang, Y., Chen, Y., Guan, L., Zhang, H., Huang, Y., Johnson, C. H., et al. (2018d). Carnitine palmitoyltransferase 1C regulates cancer cell senescence through mitochondria-associated metabolic reprograming. Cell Death Differ. 25 (4), 735–748. doi:10.1038/s41418-017-0013-3
Watt, M. J., Clark, A. K., Selth, L. A., Haynes, V. R., Lister, N., Rebello, R., et al. (2019). Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 11 (478), eaau5758. doi:10.1126/scitranslmed.aau5758
Wei, X., Shi, J., Lin, Q., Ma, X., Pang, Y., Mao, H., et al. (2021). Targeting ACLY attenuates tumor growth and acquired cisplatin resistance in ovarian cancer by inhibiting the PI3K-AKT pathway and activating the AMPK-ROS pathway. Front. Oncol. 11, 642229. doi:10.3389/fonc.2021.642229
Wellen, K. E., Hatzivassiliou, G., Sachdeva, U. M., Bui, T. V., Cross, J. R., and Thompson, C. B. (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324 (5930), 1076–1080. doi:10.1126/science.1164097
Wu, Y., Pu, X., Wang, X., and Xu, M. (2024). Reprogramming of lipid metabolism in the tumor microenvironment: a strategy for tumor immunotherapy. Lipids Health Dis. 23 (1), 35. doi:10.1186/s12944-024-02024-0
Xu, H., Zhou, S., Tang, Q., Xia, H., and Bi, F. (2020). Cholesterol metabolism: new functions and therapeutic approaches in cancer. Biochim. Biophys. Acta Rev. Cancer 1874 (1), 188394. doi:10.1016/j.bbcan.2020.188394
Yakubenko, V. P., Hsi, L. C., Cathcart, M. K., and Bhattacharjee, A. (2013). From macrophage interleukin-13 receptor to foam cell formation: mechanisms for αMβ2 integrin interference. J. Biol. Chem. 288 (4), 2778–2788. doi:10.1074/jbc.M112.381343
Yang, P. B., Hou, P. P., Liu, F. Y., Hong, W. B., Chen, H. Z., Sun, X. Y., et al. (2020). Blocking PPARγ interaction facilitates Nur77 interdiction of fatty acid uptake and suppresses breast cancer progression. Proc. Natl. Acad. Sci. U. S. A. 117 (44), 27412–27422. doi:10.1073/pnas.2002997117
Yao, C. H., Fowle-Grider, R., Mahieu, N. G., Liu, G. Y., Chen, Y. J., Wang, R., et al. (2016). Exogenous fatty acids are the preferred source of membrane lipids in proliferating fibroblasts. Cell Chem. Biol. 23 (4), 483–493. doi:10.1016/j.chembiol.2016.03.007
Yao, Y., Sun, S., Wang, J., Fei, F., Dong, Z., Ke, A. W., et al. (2018). Canonical Wnt signaling remodels lipid metabolism in zebrafish hepatocytes following Ras oncogenic insult. Cancer Res. 78 (19), 5548–5560. doi:10.1158/0008-5472.CAN-17-3964
Ye, C., Sutter, B. M., Wang, Y., Kuang, Z., and Tu, B. P. (2017a). A metabolic function for phospholipid and histone methylation. Mol. Cell 66 (2), 180–193. doi:10.1016/j.molcel.2017.02.026
Ye, H., Adane, B., Khan, N., Sullivan, T., Minhajuddin, M., Gasparetto, M., et al. (2016). Leukemic stem cells evade chemotherapy by metabolic adaptation to an adipose tissue niche. Cell Stem Cell 19 (1), 23–37. doi:10.1016/j.stem.2016.06.001
Ye, J., Li, T. S., Xu, G., Zhao, Y. M., Zhang, N. P., Fan, J., et al. (2017b). JCAD promotes progression of nonalcoholic steatohepatitis to liver cancer by inhibiting LATS2 kinase activity. Cancer Res. 77 (19), 5287–5300. doi:10.1158/0008-5472.CAN-17-0229
Yecies, J. L., Zhang, H. H., Menon, S., Liu, S., Yecies, D., Lipovsky, A. I., et al. (2011). Akt stimulates hepatic SREBP1c and lipogenesis through parallel mTORC1-dependent and independent pathways. Cell Metab. 14 (1), 21–32. doi:10.1016/j.cmet.2011.06.002
Yin, F., Feng, F., Wang, L., Wang, X., Li, Z., and Cao, Y. (2019). SREBP-1 inhibitor Betulin enhances the antitumor effect of Sorafenib on hepatocellular carcinoma via restricting cellular glycolytic activity. Cell Death Dis. 10 (9), 672. doi:10.1038/s41419-019-1884-7
Yoshii, Y., Furukawa, T., Saga, T., and Fujibayashi, Y. (2015). Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: overview and application. Cancer Lett. 356 (2 Pt A), 211–216. doi:10.1016/j.canlet.2014.02.019
Yu, C., Niu, X., Du, Y., Chen, Y., Liu, X., Xu, L., et al. (2020). IL-17A promotes fatty acid uptake through the IL-17A/IL-17RA/p-STAT3/FABP4 axis to fuel ovarian cancer growth in an adipocyte-rich microenvironment. Cancer Immunol. Immunother. 69 (1), 115–126. doi:10.1007/s00262-019-02445-2
Yu, X., Mi, S., Ye, J., and Lou, G. (2021a). Aberrant lipid metabolism in cancer cells and tumor microenvironment: the player rather than bystander in cancer progression and metastasis. J. Cancer 12 (24), 7498–7506. doi:10.7150/jca.64833
Yu, Y., Kim, H., Choi, S., Yu, J., Lee, J. Y., Lee, H., et al. (2021b). Targeting a lipid desaturation enzyme, SCD1, selectively eliminates colon cancer stem cells through the suppression of Wnt and NOTCH signaling. Cells 10 (1), 106. doi:10.3390/cells10010106
Yuan, Y., Li, H., Pu, W., Chen, L., Guo, D., Jiang, H., et al. (2022). Cancer metabolism and tumor microenvironment: fostering each other? Sci. China Life Sci. 65 (2), 236–279. doi:10.1007/s11427-021-1999-2
Yulian, E. D., Siregar, N. C., and Bajuadjir, B. (2021). Combination of simvastatin and FAC improves response to neoadjuvant chemotherapy in locally advanced breast cancer. Cancer Res. Treat. 53 (4), 1072–1083. doi:10.4143/crt.2020.1024
Zakic, T., Kalezic, A., Drvendzija, Z., Udicki, M., Ivkovic Kapicl, T., Srdic Galic, B., et al. (2024). Breast cancer: mitochondria-centered metabolic alterations in tumor and associated adipose tissue. Cells 13 (2), 155. doi:10.3390/cells13020155
Zelenay, S., van der Veen, A. G., Böttcher, J. P., Snelgrove, K. J., Rogers, N., Acton, S. E., et al. (2015). Cyclooxygenase-dependent tumor growth through evasion of immunity. Cell 162 (6), 1257–1270. doi:10.1016/j.cell.2015.08.015
Zhang, M., Wei, T., Zhang, X., and Guo, D. (2022). Targeting lipid metabolism reprogramming of immunocytes in response to the tumor microenvironment stressor: a potential approach for tumor therapy. Front. Immunol. 13, 937406. doi:10.3389/fimmu.2022.937406
Zhang, X., Lou, D., Fu, R., Wu, F., Zheng, D., and Ma, X. (2024). Association between statins types with incidence of liver cancer: an updated meta-analysis. Curr. Med. Chem. 31 (6), 762–775. doi:10.2174/0929867330666230701000400
Zhang, Y., Liu, Y., Duan, J., Wang, H., Zhang, Y., Qiao, K., et al. (2019). Cholesterol depletion sensitizes gallbladder cancer to cisplatin by impairing DNA damage response. Cell Cycle 18 (23), 3337–3350. doi:10.1080/15384101.2019.1676581
Zhou, W., Simpson, P. J., McFadden, J. M., Townsend, C. A., Medghalchi, S. M., Vadlamudi, A., et al. (2003). Fatty acid synthase inhibition triggers apoptosis during S phase in human cancer cells. Cancer Res. 63 (21), 7330–7337.
Zhuang, L., Kim, J., Adam, R. M., Solomon, K. R., and Freeman, M. R. (2005). Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J. Clin. Invest. 115 (4), 959–968. doi:10.1172/JCI19935
Zinatizadeh, M. R., Schock, B., Chalbatani, G. M., Zarandi, P. K., Jalali, S. A., and Miri, S. R. (2020). The Nuclear Factor Kappa B (NF-kB) signaling in cancer development and immune diseases. Genes Dis. 8 (3), 287–297. doi:10.1016/j.gendis.2020.06.005
Keywords: cancer, lipid metabolism, cell signaling, oncogenic proteins, anticancer therapy
Citation: Khan F, Elsori D, Verma M, Pandey S, Obaidur Rab S, Siddiqui S, Alabdallah NM, Saeed M and Pandey P (2024) Unraveling the intricate relationship between lipid metabolism and oncogenic signaling pathways. Front. Cell Dev. Biol. 12:1399065. doi: 10.3389/fcell.2024.1399065
Received: 11 March 2024; Accepted: 28 May 2024;
Published: 12 June 2024.
Edited by:
Francesca Pagliari, German Cancer Research Center (DKFZ), GermanyReviewed by:
Parmanand Malvi, University of Alabama at Birmingham, United StatesCopyright © 2024 Khan, Elsori, Verma, Pandey, Obaidur Rab, Siddiqui, Alabdallah, Saeed and Pandey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohd Saeed, bW8uc2FlZWRAdW9oLmVkdS5zYQ==; Pratibha Pandey, c2h1a2xhLnByYXRpYmhhMTk4NUBnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.