 Junko Tomikawa
Junko Tomikawa- Department of Maternal-Fetal Biology, National Research Institute for Child Health and Development, Tokyo, Japan
Mammalian genomic DNA is packed in a small nucleus, and its folding and organization in the nucleus are critical for gene regulation and cell fate determination. In interphase, chromosomes are compartmentalized into certain nuclear spaces and territories that are considered incompatible with each other. The regulation of gene expression is influenced by the epigenetic characteristics of topologically associated domains and A/B compartments within chromosomes (intrachromosomal). Previously, interactions among chromosomes detected via chromosome conformation capture-based methods were considered noise or artificial errors. However, recent studies based on newly developed ligation-independent methods have shown that inter-chromosomal interactions play important roles in gene regulation. This review summarizes the recent understanding of spatial genomic organization in mammalian interphase nuclei and discusses the potential mechanisms that determine cell identity. In addition, this review highlights the potential role of inter-chromosomal interactions in early mouse development.
Introduction
Development of the chromosome conformation capture (3C) assay, a proximity ligation assay used with PCR, was a breakthrough in chromatin biology (Dekker et al., 2002). Next-generation sequencing has led to the development of several 3C-derived approaches to assess contact frequencies between two genomic loci: circular 3C (4C)-seq to identify loci that interact with a single locus (Simonis et al., 2006; Wei et al., 2013) and high-throughput 3C (Hi-C) to map genome-wide interactions (Lieberman-Aiden et al., 2009; Duan et al., 2010). These methods marked an era of three-dimensional (3D) genome structural analysis of interphase nuclei. Recent developments in super-resolution microscopy and imaging techniques have facilitated high-throughput examination of chromatin conformation in single cells (Su et al., 2020; Takei et al., 2021). CRISPR/Cas9 is an adaptive immune system that cleaves exogenous gene elements in certain microorganisms. Endonuclease-deficient Cas9 facilitates targeted gene regulation through epigenetic editing and imaging of specific genomic loci in living cells. The combination of live imaging and CRISPR-based systems has improved the understanding of chromatin contact dynamics (Gu et al., 2018; Shaban and Seeber, 2020). Here, the recent advances in 3D genomics are summarized and future research directions are discussed.
Principles of genomic structure and nuclear organization
The mammalian genome contains a large amount of DNA with a total length of 2 m. Folding and organizing genomic DNA in the interphase nucleus is critical for gene regulation and cell fate determination. Proximity ligation-based genome-wide approaches such as Hi-C have drastically enhanced our understanding of the association between transcriptionally active/inactive states and 3D structural DNA patterns in the mammalian genome.
Hi-C revealed that the genome is classified into two spatial compartments: “A” compartment containing actively transcribed genes and “B” compartment containing transcriptionally silent domains (Lieberman-Aiden et al., 2009; Rao et al., 2014). The A compartment, located inside the cell nucleus, is gene-rich, marked by histone modifications for active transcription, and possesses high GC content. In contrast, the B compartment, located near the nuclear periphery, is gene-poor, condensed, and marked by histone modifications for gene silencing. Initially, the A/B compartments were considered to be approximately 1–10 Mb in size and comprise many topologically associating domains (TADs) (Lieberman-Aiden et al., 2009; Dixon et al., 2012), but recent high-resolution analysis has revealed that most compartments are less than 100 kb in size. The median compartment size is 12.5 kb (Harris et al., 2023). TADs are considered to be formed by active extrusion of chromatin loops (Sanborn et al., 2015; Fudenberg et al., 2016). Loop extrusion factors such as cohesin bind to chromatin fibers and chromatin loops gradually expand until they either drop out, encounter each other, or encounter extrusion barriers that define the TAD boundary (Nuebler et al., 2018). TADs regulate transcriptional activation by restricting enhancers to target promoters within the same loop. Genes within the same TAD synchronize their expression patterns and are often co-regulated during cellular differentiation (Szabo et al., 2019). Disruption of TAD boundaries can result in the aberrant expression of genes that should not be expressed, leading to developmental abnormalities and diseases (Giorgio et al., 2015; Lupiáñez et al., 2015).
Although TAD structure was thought to be relatively constant among different cell types across species, later studies adopting high-resolution analyses have revealed that there are sub-TADs within TADs and that many of sub-TADs change their structures during cell differentiation (Dixon et al., 2015; Ibn-Salem et al., 2017; Oudelaar et al., 2020; Georgiades et al., 2023). A/B compartments are cell type-specific and can switch over during cell differentiation and lineage commitment (Lieberman-Aiden et al., 2009; Nora et al., 2017; Rao et al., 2017; Schwarzer et al., 2017; Miura et al., 2019).
Chromosomal DNA is compartmentalized into constant intranuclear spaces called “chromosome territories” (Cremer and Cremer, 2010). Nuclear locations of chromosome territories in human and mouse cells are not random (Croft et al., 1999; Cremer et al., 2003). Gene-rich loci were more internally localized, whereas gene-poor loci were more peripherally localized. The distribution of chromosome territories is tissue-specific (Parada et al., 2004; Bolzer et al., 2005) and the territories are lost during mitosis and recovered after mitosis (Gerlich et al., 2003). Genomic DNA folds in the nuclear space in an orderly manner at multiple levels via different mechanisms.
Inter-chromosomal interactions revealed by proximity ligation-independent methods
Most genome-wide methods for determining chromatin structure are based on proximity ligation. Previously, inter-chromosomal interactions were considered artificial errors in Hi-C studies (Kalhor et al., 2012; Kaufmann et al., 2015; Bertero, 2021). Compared with the intrachromosomal interactions described in the previous section (e.g., TAD and compartment formation within a chromosome), the fundamental understanding of inter-chromosomal interactions remains limited (Figure 1). The nucleolus exhibits the best nuclear organization via inter-chromosomal interactions. In various eukaryotic species, including humans and mice, the nucleolus exhibits the largest nuclear organization via the coalescence of ribosomal DNA (rDNA) genes across multiple chromosomes (McStay, 2016; van Sluis et al., 2020; Mangan and McStay, 2021). However, such inter-chromosomal interactions in the nucleolus have rarely been detected in previous Hi-C studies. Yu and Lemos attempted to detect the long-range interactions among rDNAs by analyzing over 15 billion Hi-C reads from previous studies. However, the ratio of reads supporting such interactions was <1%, indicating the difficulty of recovering rDNA information from Hi-C data (Yu and Lemos, 2018). On the other hand, among the gene promoters bound by polycomb repressive complex 1 (PRC1) in mouse ES cells, an unusually strong intra- and inter-chromosomal spatial network was revealed by promoter-capture Hi-C techniques (Schoenfelder et al., 2015). Because the strongest spatial network was composed of the four Hox clusters, the clusters are proposed to act as central 3D nucleation points for PRC1-bound genes. This spatial network is considered to constrain genome organization and differentiation of ES cells (Joshi et al., 2015; Schoenfelder et al., 2015).
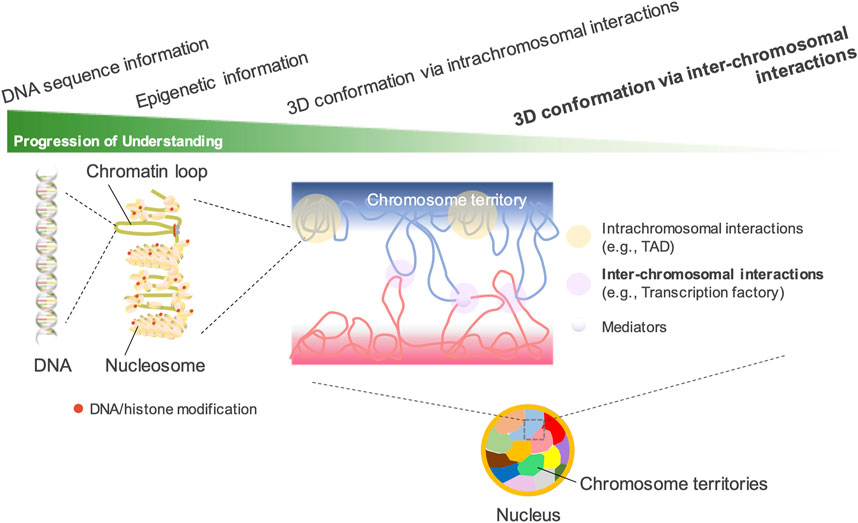
Figure 1. Understanding the spatial organization of the nuclear genome. In interphase nuclei, each chromosome occupies its own distinct territory, called “a chromosome territory.” In each territory, chromatin forms highly organized structures such as TADs and A/B compartments. Hi-C technology has greatly advanced the understanding of the 3D genomic structures configured by intrachromosomal interactions. However, the understanding of inter-chromosomal interactions has lagged far behind, as their presence can be detected by DNA fluorescence in situ hybridization but is difficult to detect by Hi-C. The existence of chromosome territories, which limit the interactions between different chromosomes, is another important reason.
In split-pool recognition of interactions by tag extension (SPRITE), the cross-linked nuclear lysate is first divided into wells of a 96-well plate, each containing a unique tag for ligation. After ligation, the samples are pooled and divided into the wells of a new 96-well plate containing different unique tags again. This process is repeated five or six times to generate more than one trillion barcodes (Quinodoz et al., 2018) (Figure 2). Therefore, SPRITE does not depend on the ligation of spatially close DNA fragments and facilitates the detection of interactions across larger distances in the genome. By applying this innovative approach to human cells, two major inter-chromosomal hubs were identified: nucleoli and nuclear speckles (Quinodoz et al., 2018). The loci clustered in the nucleoli exhibited low transcriptional activity and contained few genes. In contrast, different chromosomal loci with active gene transcription clustered in the nuclear speckles (Quinodoz et al., 2018, 2021). Furthermore, a group of genomic loci enriched in common super-enhancers is strongly involved in speckle-associated chromosomal interactions in a conserved manner across various cell types (Joo et al., 2023). This finding indicates the formation of transcriptionally active hubs in the nucleus. Similar results have been reported using another ligation-free method, genome architecture mapping (GAM) (Beagrie et al., 2017; Fiorillo et al., 2021) (Figure 2). A CRISPR-based live-cell imaging approach called 4D-CLING revealed that inter-chromosomal interactions are as common as intrachromosomal interactions (Maass et al., 2018). These new methods, not based on proximity ligation, show that inter-chromosomal interactions are common events in mammalian cells and occur at greater distances than intrachromosomal interactions.
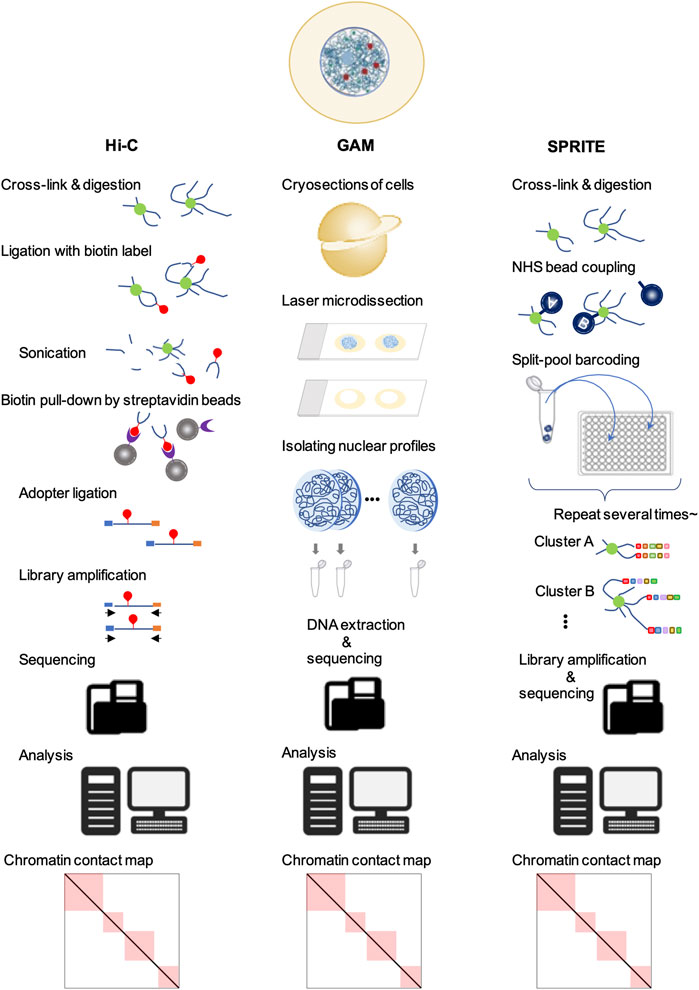
Figure 2. Techniques to study chromatin organization. In all of these methods, the first step is the fixation of cells for cross-linking. Next, in Hi-C, the DNA is digested, followed by different DNA regions that are in close spatial proximity and ligated with biotin (red). After end repair and adapter (blue and orange) ligation, the sequencing libraries are amplified, purified, and sequenced. Sequencing data are mapped to their genomic locations to yield genome-wide contact frequency matrices. In GAM, cells are cryosectioned into thin slices and the nuclei are isolated by laser microdissection. The DNA is extracted and sequenced. Analysis of locus co-occurrence in many sections allows proximity, including multi-way interactions, to be inferred without ligation. In SPRITE, chromatin is covalently coupled to N-hydroxysuccinimide (NHS) beads after fragmentation, followed by repeated pooling and splitting steps. The DNA fragments are barcoded sequentially. Next, the libraries are amplified and sequenced. The resulting sequencing data allow the detection of DNA fragment sequences in close proximity to each other.
Inter-chromosomal interactions are involved in cell fate determination
Several studies have demonstrated the inter-chromosomal interactions between specific enhancers and target promoters. For example, within the Tead4 promoter interactomes investigated using 4C-seq, our group identified five genomic loci that enhance Tead4 promoter activity in a trophoblast lineage-specific manner in vitro (Tomikawa et al., 2020). These enhancers are located on chromosomes other than chromosome 6, where Tead4 is located. Particularly, the enhancer located on chromosome 19 contributes to the strong trophoblast lineage-specific expression of Tead4 at the blastocyst stage. Furthermore, inter-chromosomal enhancers promote Pax5 gene expression in a B cell-specific manner (Fujita et al., 2017).
The most extraordinary inter-chromosomal regulator is the multi-chromosomal enhancer acting on mouse olfactory receptor (OR) genes (Markenscoff-Papadimitriou et al., 2014; Monahan et al., 2017). This enhancer (the Greek island enhancer) consists of 63 enhancers on 18 chromosomes. Moreover, it acts as the key machinery for removing heterochromatin marks from a single OR gene stochastically selected for expression among more than 1,400 OR genes dispersed in multiple heterochromatic gene clusters (Magklara et al., 2011; Markenscoff-Papadimitriou et al., 2014; Monahan et al., 2019) (Figure 3).
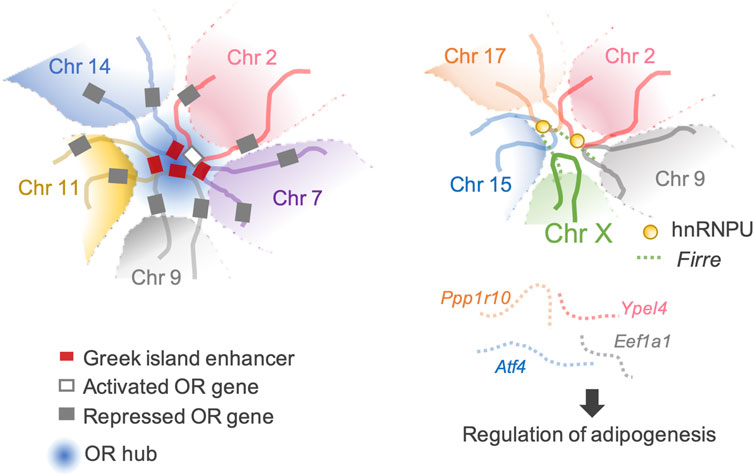
Figure 3. Transactivation of the target genes through inter-chromosomal genomic organization. (Left) An example of a typical OR hub. An OR gene can be activated through a concerted genomic organization that reduces the number of activating OR-specific enhancers from 63 available Greek Islands to 3–5 enhancer hubs. (Right) Firre-hnRNPU interplay mediates trans-chromosomal interactions, which promotes adipogenesis. Four genes involved in adipogenesis are organized together in spatial proximity and are co-expressed.
These studies indicate that interactions among different chromosomes affect gene expression. Moreover, inter-chromosomal interactions have important implications in normal cell fate determination. Although chromosome territories generally limit inter-chromosomal interactions, in exceptional cases, they allow interactions between certain genomic elements.
Long non-coding RNAs (lncRNAs) are involved in the formation of 3D chromatin structures
In addition to DNA and histones, RNA is a major component of the nucleus (Rinn and Chang, 2012). Transcribed RNA can be divided into mRNA and ncRNA. The number of known lncRNAs of approximately 200 bp or longer is rapidly increasing because of the accumulation of RNA-seq data (Mercer et al., 2009; Wang and Chang, 2011; Derrien et al., 2012). Although the approximate number of known human protein-coding genes is 20,000, the FANTOM-5 project has identified 28,000 human lncRNAs (Hon et al., 2017). However, the functions of most lncRNAs remain unknown. Moreover, their expression levels are low, and their sequences are not highly evolutionarily conserved (Necsulea et al., 2014; de Hoon et al., 2015). Many lncRNAs exhibit spatiotemporal expression patterns (Gupta et al., 2010; Cabili et al., 2011). Well-characterized lncRNAs modulate higher-order chromatin structures (Saxena and Carninci, 2011; Marchese and Huarte, 2014).
Xist is the master regulator of X chromosome inactivation (XCI), a dose compensation system that balances the expression levels of X-linked genes in male and female mammals. XCI is induced by the upregulation of Xist expression on the future inactivated X (Xi) chromosome (Mutzel and Schulz, 2020). Female mouse pluripotent ES cells possess two active X (Xa) chromosomes (Takahashi et al., 2018). In these cells, XCI is caused by the induction of differentiation, and Xist accumulates in the entire Xi chromosome in cis (Herzing et al., 1997; Hoki et al., 2009). Bacher et al. (2006) analyzed differentiated female mouse ES cells. They showed that Xi and Xa segregate into separate nuclear compartments after transient co-localization of the two X chromosomes at the beginning of the inactivation process. After XCI induction, PRC1 and PRC2 are transported to the Xi chromosome, where Xist accumulates. Mono-ubiquitination of lysine 119 on H2A (H2Aub119) and histone 3 lysine 27 trimethylation (H3K27me3) by PRC1 and PRC2, respectively, directly block transcription (Aranda et al., 2015), induce and maintain heterochromatinization of the Xi chromosome, and suppress gene expression (Almeida et al., 2017; Masui et al., 2023). Overall, Xist creates a heritably silent and heterochromatic nuclear territory with a 3D structure distinct from the Xa chromosome.
Firre, another lncRNA that modulates local higher-order chromatin structures, is predominantly expressed on the Xa chromosome in females and localizes at an approximately 5 Mb locus, flanking its transcription site on the X chromosome. Allele-specific deletion of the Firre locus showed that Firre RNA transcribed from the Xa chromosome specifically helps anchor the PRC1 and PRC2 complexes to the Xi chromosome to maintain H3K27me3 levels and affects the nuclear localization of the Xi chromosome, which normally localizes in the vicinity of the nucleolus (Fang et al., 2020). CCCTC-binding factor (CTCF)-binding sites are located across the Firre locus. CTCF specifically binds to the Firre locus on the Xi chromosome but not to the Xa chromosome (Hacisuleyman et al., 2014; Yang et al., 2015). CTCF affects the nucleolar association of genomic loci, and the extent of CTCF binding across the Firre locus of the Xi chromosome is reduced by Xa chromosome-specific Firre deletion (Yang et al., 2015; Fang et al., 2020). These data suggest potential cooperation between Firre RNA and CTCF in maintaining Xi chromosome location. CTCF binding sites facilitate inter-chromosomal interactions (Botta et al., 2010). Firre interacts with chromosomes 2, 9, 15, and 17, which overlap with known genes including Slc25a12, Ypel4, Eef1a1, Atf4, and Ppp1r10. Four of these genes (Ypel4, Eef1a1, Atf4, and Ppp1r10) play regulatory roles in adipogenesis (Rubi et al., 2004; Seo et al., 2009; Lee et al., 2011; Nukitrangsan et al., 2011) (Figure 3). LncRNAs tether genes involved in similar biological processes in close proximity, facilitate spatiotemporal co-regulation, and serve as nuclear organization factors. Proper localization of Firre requires physical interactions with the heterogeneous nuclear ribonucleoprotein U (hnRNPU) to maintain multi-chromosomal nuclear interactions (Figure 3). Genetic deletion of Firre results in the loss of nuclear proximity of several inter-chromosomal loci to the Firre locus (Hacisuleyman et al., 2014). Similarly, many lncRNAs remain in the nucleus and interact with the chromatin to regulate their spatial structure and function (Tsai et al., 2010; Rinn and Guttman, 2014; Somarowthu et al., 2015; Zhao et al., 2020; Mattick et al., 2023).
LncRNAs are important for establishing higher-order 3D genomic structures in the nucleus. Exploring the roles of lncRNAs and their functions in dynamic assembly with other macromolecules will enhance our understanding of cellular development and evolutionary biology.
Differentiation potential and frequency of inter-chromosomal interactions
In 2017, two groups successively reported the time-course changes of chromatin structures before and after zygotic genome activation (ZGA) examined using low-input Hi-C methods. Although their data resolution was limited, these studies showed that TAD structures are obscure in mouse zygotes and 2-cell stage embryos, which are totipotent, and are gradually established during embryonic development between 4-cell and 7.5 days post-coitum (dpc) stages (Du et al., 2017; Ke et al., 2017). Single-nucleus Hi-C analysis further revealed that TADs are formed but in an immature state in these totipotent cells (Flyamer et al., 2017; Gassler et al., 2017; Tomikawa and Miyamoto, 2021). Sperm nuclei have unique features, such as a small nuclear volume, highly condensed DNA, and loss of approximately 90% of histones. However, the TADs and A/B compartments observed in mouse sperm were similar to those observed in somatic cells. The notable difference between the 3D genome structures of sperm and somatic cells is that inter-TAD and inter-chromosomal interactions are frequently observed in sperm (Battulin et al., 2015; Ke et al., 2017; Wang et al., 2019). A comparison of the ratio of inter-chromosomal interactions among all chromosomal interactions at different stages (sperm, oocyte, zygote, 2-cell, 4-cell, 8-cell, 3.5 dpc, and 7.5 dpc) revealed that it was most abundant in sperm (∼11%) and gradually decreased during development from the zygote (9.5%) to the 8-cell stage (3%). Interestingly, it transiently increased to 6% at 3.5 dpc (blastocysts) and decreased again to 3% at 7.5 dpc (Ke et al., 2017). The occurrence of pluripotent cells in the inner cell mass may be involved in the transient increase of inter-chromosomal interactions in blastocyst. The observed dynamics of inter-chromosomal interactions suggests their potentially critical role in organizing 3D chromatin structures in the nuclei during early development. TADs are almost completely absent in early Drosophila embryos before ZGA and are re-established after ZGA (Hug et al., 2017). Although TADs are immature in totipotent cells in mice, ZGA occurs in these cells in a genome-wide manner (Kigami et al., 2003; Eckersley-Maslin et al., 2018). The pharmacological inhibition of ZGA did not interfere with the formation of TAD structures (Du et al., 2017; Ke et al., 2017). These observations suggest that TAD formation and ZGA are regulated independently. The abundance of inter-chromosomal interactions at the zygotes and 2-cell stages suggests the possibility that the 3D genomic architectures defined by such interactions play regulatory roles in essential developmental events, such as ZGA initiation and acquisition of totipotency. The progress of chromatin research for totipotent cells has lagged behind that for somatic cells, owing to the absence of a faithful in vitro model for totipotent cells and the experimental difficulty in acquiring large numbers of totipotent embryos.
Current limitations and future challenges
To date, 3C-based methods, such as Hi-C (Fullwood et al., 2009; Lieberman-Aiden et al., 2009; Rao et al., 2014; Mumbach et al., 2016), and new methods, such as GAM (Beagrie et al., 2017) and SPRITE (Quinodoz et al., 2018), have provided insights into the genome-wide 3D spatial structure and physical proximity of DNA in the interphase nuclei. Mammalian genomes are characterized by DNA loops (Rao et al., 2014), TADs (Dixon et al., 2012; Nora et al., 2012), A/B compartments (Dekker and Mirny, 2016; Dixon et al., 2016), and other higher-order structures. These 3D structures play critical roles in the regulation of genomic functions. However, the extent to which the results obtained using the above-mentioned molecular methods are faithful to the actual 3D structure of the genome in vivo needs to be further carefully validated, as these methods indirectly quantify the proximity between two or more genomic loci via molecular manipulation steps and sequencing. Conversely, microscopy-based methods can directly image the target’s appearance. Microscopic techniques have advanced rapidly over the last few years (Jerkovic and Cavalli, 2021). Moreover, it is now possible to simultaneously visualize thousands of DNA loci, hundreds of different RNA molecules, and several types of protein and histone modifications, thereby enabling high-throughput chromatin structural and functional analyses in thousands of single cells (Su et al., 2020; Takei et al., 2021). These optical microscopy methods have been accompanied by the development of electron microscopy, which allows the study of genome structure at nanometer and kilobase resolutions. The application of such advanced imaging technologies provides powerful information about 3D genome organization that complements molecular techniques, such as Hi-C and SPRITE, thereby enabling the study of genome structure and function in previously unthinkable ways.
Author contributions
JT: Writing–original draft, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by JSPS KAKENHI (Grant Numbers 20K06605 and 23K05730 to JT).
Acknowledgments
I am sincerely grateful to Dr. Kazuhiko Nakabayashi for helpful suggestions and discussions.
Conflict of interest
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Almeida, M., Pintacuda, G., Masui, O., Koseki, Y., Gdula, M., Cerase, A., et al. (2017). PCGF3/5-PRC1 initiates Polycomb recruitment in X chromosome inactivation. Science 356, 1081–1084. doi:10.1126/science.aal2512
Aranda, S., Mas, G., and Di Croce, L. (2015). Regulation of gene transcription by Polycomb proteins. Sci. Adv. 1, e1500737. doi:10.1126/sciadv.1500737
Bacher, C. P., Guggiari, M., Brors, B., Augui, S., Clerc, P., Avner, P., et al. (2006). Transient colocalization of X-inactivation centres accompanies the initiation of X inactivation. Nat. Cell Biol. 8, 293–299. doi:10.1038/ncb1365
Battulin, N., Fishman, V. S., Mazur, A. M., Pomaznoy, M., Khabarova, A. A., Afonnikov, D. A., et al. (2015). Comparison of the three-dimensional organization of sperm and fibroblast genomes using the Hi-C approach. Genome Biol. 16, 77. doi:10.1186/s13059-015-0642-0
Beagrie, R. A., Scialdone, A., Schueler, M., Kraemer, D. C. A., Chotalia, M., Xie, S. Q., et al. (2017). Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524. doi:10.1038/nature21411
Bertero, A. (2021). RNA biogenesis instructs functional inter-chromosomal genome architecture. Front. Genet. 12, 645863. doi:10.3389/fgene.2021.645863
Bolzer, A., Kreth, G., Solovei, I., Koehler, D., Saracoglu, K., Fauth, C., et al. (2005). Three-dimensional maps of all chromosomes in human male fibroblast nuclei and prometaphase rosettes. PLoS Biol. 3, e157. doi:10.1371/journal.pbio.0030157
Botta, M., Haider, S., Leung, I. X. Y., Lio, P., and Mozziconacci, J. (2010). Intra- and inter-chromosomal interactions correlate with CTCF binding genome wide. Mol. Syst. Biol. 6, 426. doi:10.1038/msb.2010.79
Cabili, M. N., Trapnell, C., Goff, L., Koziol, M., Tazon-Vega, B., Regev, A., et al. (2011). Integrative annotation of human large intergenic noncoding RNAs reveals global properties and specific subclasses. Genes Dev. 25, 1915–1927. doi:10.1101/gad.17446611
Cremer, M., Küpper, K., Wagler, B., Wizelman, L., von Hase, J., Weiland, Y., et al. (2003). Inheritance of gene density-related higher order chromatin arrangements in normal and tumor cell nuclei. J. Cell Biol. 162, 809–820. doi:10.1083/jcb.200304096
Cremer, T., and Cremer, M. (2010). Chromosome territories. Cold Spring Harb. Perspect. Biol. 2, a003889. doi:10.1101/cshperspect.a003889
Croft, J. A., Bridger, J. M., Boyle, S., Perry, P., Teague, P., and Bickmore, W. A. (1999). Differences in the localization and morphology of chromosomes in the human nucleus. J. Cell Biol. 145, 1119–1131. doi:10.1083/jcb.145.6.1119
de Hoon, M., Shin, J. W., and Carninci, P. (2015). Paradigm shifts in genomics through the FANTOM projects. Mamm. Genome 26, 391–402. doi:10.1007/s00335-015-9593-8
Dekker, J., and Mirny, L. (2016). The 3D genome as moderator of chromosomal communication. Cell 164, 1110–1121. doi:10.1016/j.cell.2016.02.007
Dekker, J., Rippe, K., Dekker, M., and Kleckner, N. (2002). Capturing chromosome conformation. Science 295, 1306–1311. doi:10.1126/science.1067799
Derrien, T., Johnson, R., Bussotti, G., Tanzer, A., Djebali, S., Tilgner, H., et al. (2012). The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 22, 1775–1789. doi:10.1101/gr.132159.111
Dixon, J. R., Gorkin, D. U., and Ren, B. (2016). Chromatin domains: the unit of chromosome organization. Mol. Cell 62, 668–680. doi:10.1016/j.molcel.2016.05.018
Dixon, J. R., Jung, I., Selvaraj, S., Shen, Y., Antosiewicz-Bourget, J. E., Lee, A. Y., et al. (2015). Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336. doi:10.1038/nature14222
Dixon, J. R., Selvaraj, S., Yue, F., Kim, A., Li, Y., Shen, Y., et al. (2012). Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380. doi:10.1038/nature11082
Du, Z., Zheng, H., Huang, B., Ma, R., Wu, J., Zhang, X., et al. (2017). Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 547, 232–235. doi:10.1038/nature23263
Duan, Z., Andronescu, M., Schutz, K., McIlwain, S., Kim, Y. J., Lee, C., et al. (2010). A three-dimensional model of the yeast genome. Nature 465, 363–367. doi:10.1038/nature08973
Eckersley-Maslin, M. A., Alda-Catalinas, C., and Reik, W. (2018). Dynamics of the epigenetic landscape during the maternal-to-zygotic transition. Nat. Rev. Mol. Cell Biol. 19, 436–450. doi:10.1038/s41580-018-0008-z
Fang, H., Bonora, G., Lewandowski, J. P., Thakur, J., Filippova, G. N., Henikoff, S., et al. (2020). Trans- and cis-acting effects of Firre on epigenetic features of the inactive X chromosome. Nat. Commun. 11, 6053. doi:10.1038/s41467-020-19879-3
Fiorillo, L., Musella, F., Conte, M., Kempfer, R., Chiariello, A. M., Bianco, S., et al. (2021). Publisher Correction: comparison of the Hi-C, GAM and SPRITE methods using polymer models of chromatin. Nat. Methods 18, 1266. doi:10.1038/s41592-021-01251-y
Flyamer, I. M., Gassler, J., Imakaev, M., Brandão, H. B., Ulianov, S. V., Abdennur, N., et al. (2017). Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114. doi:10.1038/nature21711
Fudenberg, G., Imakaev, M., Lu, C., Goloborodko, A., Abdennur, N., and Mirny, L. A. (2016). Formation of chromosomal domains by loop extrusion. Cell Rep. 15, 2038–2049. doi:10.1016/j.celrep.2016.04.085
Fujita, T., Kitaura, F., Yuno, M., Suzuki, Y., Sugano, S., and Fujii, H. (2017). Locus-specific ChIP combined with NGS analysis reveals genomic regulatory regions that physically interact with the Pax5 promoter in a chicken B cell line. DNA Res. 24, 537–548. doi:10.1093/dnares/dsx023
Fullwood, M. J., Liu, M. H., Pan, Y. F., Liu, J., Xu, H., Mohamed, Y. B., et al. (2009). An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 462, 58–64. doi:10.1038/nature08497
Gassler, J., Brandão, H. B., Imakaev, M., Flyamer, I. M., Ladstätter, S., Bickmore, W. A., et al. (2017). A mechanism of cohesin-dependent loop extrusion organizes zygotic genome architecture. EMBO J. 36, 3600–3618. doi:10.15252/embj.201798083
Georgiades, E., Harrold, C. L., Roberts, N., Kassouf, M., Riva, S. G., Sanders, E., et al. (2023) Active regulatory elements recruit cohesin to establish cell-specific chromatin domains. doi:10.1101/2023.10.13.562171
Gerlich, D., Beaudouin, J., Kalbfuss, B., Daigle, N., Eils, R., and Ellenberg, J. (2003). Global chromosome positions are transmitted through mitosis in mammalian cells. Cell 112, 751–764. doi:10.1016/s0092-8674(03)00189-2
Giorgio, E., Robyr, D., Spielmann, M., Ferrero, E., Di Gregorio, E., Imperiale, D., et al. (2015). A large genomic deletion leads to enhancer adoption by the lamin B1 gene: a second path to autosomal dominant adult-onset demyelinating leukodystrophy (ADLD). Hum. Mol. Genet. 24, 3143–3154. doi:10.1093/hmg/ddv065
Gu, B., Swigut, T., Spencley, A., Bauer, M. R., Chung, M., Meyer, T., et al. (2018). Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 359, 1050–1055. doi:10.1126/science.aao3136
Gupta, R. A., Shah, N., Wang, K. C., Kim, J., Horlings, H. M., Wong, D. J., et al. (2010). Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. doi:10.1038/nature08975
Hacisuleyman, E., Goff, L. A., Trapnell, C., Williams, A., Henao-Mejia, J., Sun, L., et al. (2014). Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 21, 198–206. doi:10.1038/nsmb.2764
Harris, H. L., Gu, H., Olshansky, M., Wang, A., Farabella, I., Eliaz, Y., et al. (2023). Chromatin alternates between A and B compartments at kilobase scale for subgenic organization. Nat. Commun. 14, 3303. doi:10.1038/s41467-023-38429-1
Herzing, L. B., Romer, J. T., Horn, J. M., and Ashworth, A. (1997). Xist has properties of the X-chromosome inactivation centre. Nature 386, 272–275. doi:10.1038/386272a0
Hoki, Y., Kimura, N., Kanbayashi, M., Amakawa, Y., Ohhata, T., Sasaki, H., et al. (2009). A proximal conserved repeat in the Xist gene is essential as a genomic element for X-inactivation in mouse. Development 136, 139–146. doi:10.1242/dev.026427
Hon, C.-C., Ramilowski, J. A., Harshbarger, J., Bertin, N., Rackham, O. J. L., Gough, J., et al. (2017). An atlas of human long non-coding RNAs with accurate 5’ ends. Nature 543, 199–204. doi:10.1038/nature21374
Hug, C. B., Grimaldi, A. G., Kruse, K., and Vaquerizas, J. M. (2017). Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 169, 216–228. doi:10.1016/j.cell.2017.03.024
Ibn-Salem, J., Muro, E. M., and Andrade-Navarro, M. A. (2017). Co-regulation of paralog genes in the three-dimensional chromatin architecture. Nucleic Acids Res. 45, 81–91. doi:10.1093/nar/gkw813
Jerkovic, I., and Cavalli, G. (2021). Understanding 3D genome organization by multidisciplinary methods. Nat. Rev. Mol. Cell Biol. 22, 511–528. doi:10.1038/s41580-021-00362-w
Joo, J., Cho, S., Hong, S., Min, S., Kim, K., Kumar, R., et al. (2023). Probabilistic establishment of speckle-associated inter-chromosomal interactions. Nucleic Acids Res. 51, 5377–5395. doi:10.1093/nar/gkad211
Joshi, O., Wang, S.-Y., Kuznetsova, T., Atlasi, Y., Peng, T., Fabre, P. J., et al. (2015). Dynamic reorganization of extremely long-range promoter-promoter interactions between two states of pluripotency. Cell Stem Cell 17, 748–757. doi:10.1016/j.stem.2015.11.010
Kalhor, R., Tjong, H., Jayathilaka, N., Alber, F., and Chen, L. (2012). Genome architectures revealed by tethered chromosome conformation capture and population-based modeling. Nat. Biotechnol. 30, 90–98. doi:10.1038/nbt.2057
Kaufmann, S., Fuchs, C., Gonik, M., Khrameeva, E. E., Mironov, A. A., and Frishman, D. (2015). Inter-chromosomal contact networks provide insights into mammalian chromatin organization. PLoS ONE 10, e0126125. doi:10.1371/journal.pone.0126125
Ke, Y., Xu, Y., Chen, X., Feng, S., Liu, Z., Sun, Y., et al. (2017). 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell 170, 367–381. doi:10.1016/j.cell.2017.06.029
Kigami, D., Minami, N., Takayama, H., and Imai, H. (2003). MuERV-L is one of the earliest transcribed genes in mouse one-cell embryos. Biol. Reproduction 68, 651–654. doi:10.1095/biolreprod.102.007906
Lee, E. K., Lee, M. J., Abdelmohsen, K., Kim, W., Kim, M. M., Srikantan, S., et al. (2011). miR-130 suppresses adipogenesis by inhibiting peroxisome proliferator-activated receptor gamma expression. Mol. Cell Biol. 31, 626–638. doi:10.1128/MCB.00894-10
Lieberman-Aiden, E., van Berkum, N. L., Williams, L., Imakaev, M., Ragoczy, T., Telling, A., et al. (2009). Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293. doi:10.1126/science.1181369
Lupiáñez, D. G., Kraft, K., Heinrich, V., Krawitz, P., Brancati, F., Klopocki, E., et al. (2015). Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025. doi:10.1016/j.cell.2015.04.004
Maass, P. G., Barutcu, A. R., Weiner, C. L., and Rinn, J. L. (2018). Inter-chromosomal contact properties in live-cell imaging and in Hi-C. Mol. Cell 70, 188–189. doi:10.1016/j.molcel.2018.03.021
Magklara, A., Yen, A., Colquitt, B. M., Clowney, E. J., Allen, W., Markenscoff-Papadimitriou, E., et al. (2011). An epigenetic signature for monoallelic olfactory receptor expression. Cell 145, 555–570. doi:10.1016/j.cell.2011.03.040
Mangan, H., and McStay, B. (2021). Human nucleoli comprise multiple constrained territories, tethered to individual chromosomes. Genes Dev. 35, 483–488. doi:10.1101/gad.348234.121
Marchese, F. P., and Huarte, M. (2014). Long non-coding RNAs and chromatin modifiers: their place in the epigenetic code. Epigenetics 9, 21–26. doi:10.4161/epi.27472
Markenscoff-Papadimitriou, E., Allen, W. E., Colquitt, B. M., Goh, T., Murphy, K. K., Monahan, K., et al. (2014). Enhancer interaction networks as a means for singular olfactory receptor expression. Cell 159, 543–557. doi:10.1016/j.cell.2014.09.033
Masui, O., Corbel, C., Nagao, K., Endo, T. A., Kezuka, F., Diabangouaya, P., et al. (2023). Polycomb repressive complexes 1 and 2 are each essential for maintenance of X inactivation in extra-embryonic lineages. Nat. Cell Biol. 25, 134–144. doi:10.1038/s41556-022-01047-y
Mattick, J. S., Amaral, P. P., Carninci, P., Carpenter, S., Chang, H. Y., Chen, L.-L., et al. (2023). Long non-coding RNAs: definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 24, 430–447. doi:10.1038/s41580-022-00566-8
McStay, B. (2016). Nucleolar organizer regions: genomic “dark matter” requiring illumination. Genes Dev. 30, 1598–1610. doi:10.1101/gad.283838.116
Mercer, T. R., Dinger, M. E., and Mattick, J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10, 155–159. doi:10.1038/nrg2521
Miura, H., Takahashi, S., Poonperm, R., Tanigawa, A., Takebayashi, S.-I., and Hiratani, I. (2019). Single-cell DNA replication profiling identifies spatiotemporal developmental dynamics of chromosome organization. Nat. Genet. 51, 1356–1368. doi:10.1038/s41588-019-0474-z
Monahan, K., Horta, A., and Lomvardas, S. (2019). LHX2-and LDB1-mediated trans interactions regulate olfactory receptor choice. Nature 565, 448–453. doi:10.1038/s41586-018-0845-0
Monahan, K., Schieren, I., Cheung, J., Mumbey-Wafula, A., Monuki, E. S., and Lomvardas, S. (2017). Cooperative interactions enable singular olfactory receptor expression in mouse olfactory neurons. Elife 6, e28620. doi:10.7554/eLife.28620
Mumbach, M. R., Rubin, A. J., Flynn, R. A., Dai, C., Khavari, P. A., Greenleaf, W. J., et al. (2016). HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nat. Methods 13, 919–922. doi:10.1038/nmeth.3999
Mutzel, V., and Schulz, E. G. (2020). Dosage sensing, threshold responses, and epigenetic memory: a systems biology perspective on random X-chromosome inactivation. BioEssays 42, 1900163. doi:10.1002/bies.201900163
Necsulea, A., Soumillon, M., Warnefors, M., Liechti, A., Daish, T., Zeller, U., et al. (2014). The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505, 635–640. doi:10.1038/nature12943
Nora, E. P., Goloborodko, A., Valton, A.-L., Gibcus, J. H., Uebersohn, A., Abdennur, N., et al. (2017). Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944. doi:10.1016/j.cell.2017.05.004
Nora, E. P., Lajoie, B. R., Schulz, E. G., Giorgetti, L., Okamoto, I., Servant, N., et al. (2012). Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385. doi:10.1038/nature11049
Nuebler, J., Fudenberg, G., Imakaev, M., Abdennur, N., and Mirny, L. A. (2018). Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. U.S.A. 115, E6697–E6706. doi:10.1073/pnas.1717730115
Nukitrangsan, N., Okabe, T., Toda, T., Inafuku, M., Iwasaki, H., Yanagita, T., et al. (2011). Effect of peucedanum japonicum thunb on the expression of obesity-related genes in mice on a high-fat diet. J. Oleo Sci. 60, 527–536. doi:10.5650/jos.60.527
Oudelaar, A. M., Beagrie, R. A., Gosden, M., de Ornellas, S., Georgiades, E., Kerry, J., et al. (2020). Dynamics of the 4D genome during in vivo lineage specification and differentiation. Nat. Commun. 11, 2722. doi:10.1038/s41467-020-16598-7
Parada, L. A., McQueen, P. G., and Misteli, T. (2004). Tissue-specific spatial organization of genomes. Genome Biol. 5, R44. doi:10.1186/gb-2004-5-7-r44
Quinodoz, S. A., Jachowicz, J. W., Bhat, P., Ollikainen, N., Banerjee, A. K., Goronzy, I. N., et al. (2021). RNA promotes the formation of spatial compartments in the nucleus. Cell 184, 5775–5790.e30. doi:10.1016/j.cell.2021.10.014
Quinodoz, S. A., Ollikainen, N., Tabak, B., Palla, A., Schmidt, J. M., Detmar, E., et al. (2018). Higher-order inter-chromosomal hubs shape 3D genome organization in the nucleus. Cell 174, 744–757. doi:10.1016/j.cell.2018.05.024
Rao, S. S. P., Huang, S.-C., Glenn St Hilaire, B., Engreitz, J. M., Perez, E. M., Kieffer-Kwon, K.-R., et al. (2017). Cohesin loss eliminates all loop domains. Cell 171, 305–320. doi:10.1016/j.cell.2017.09.026
Rao, S. S. P., Huntley, M. H., Durand, N. C., Stamenova, E. K., Bochkov, I. D., Robinson, J. T., et al. (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. doi:10.1016/j.cell.2014.11.021
Rinn, J., and Guttman, M. (2014). RNA Function. RNA and dynamic nuclear organization. Science 345, 1240–1241. doi:10.1126/science.1252966
Rinn, J. L., and Chang, H. Y. (2012). Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 81, 145–166. doi:10.1146/annurev-biochem-051410-092902
Rubi, B., del Arco, A., Bartley, C., Satrustegui, J., and Maechler, P. (2004). The malate-aspartate NADH shuttle member Aralar1 determines glucose metabolic fate, mitochondrial activity, and insulin secretion in beta cells. J. Biol. Chem. 279, 55659–55666. doi:10.1074/jbc.M409303200
Sanborn, A. L., Rao, S. S. P., Huang, S.-C., Durand, N. C., Huntley, M. H., Jewett, A. I., et al. (2015). Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U. S. A. 112, E6456–E6465. doi:10.1073/pnas.1518552112
Saxena, A., and Carninci, P. (2011). Long non-coding RNA modifies chromatin: epigenetic silencing by long non-coding RNAs. Bioessays 33, 830–839. doi:10.1002/bies.201100084
Schoenfelder, S., Sugar, R., Dimond, A., Javierre, B.-M., Armstrong, H., Mifsud, B., et al. (2015). Polycomb repressive complex PRC1 spatially constrains the mouse embryonic stem cell genome. Nat. Genet. 47, 1179–1186. doi:10.1038/ng.3393
Schwarzer, W., Abdennur, N., Goloborodko, A., Pekowska, A., Fudenberg, G., Loe-Mie, Y., et al. (2017). Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56. doi:10.1038/nature24281
Seo, J., Fortuno, E. S., Suh, J. M., Stenesen, D., Tang, W., Parks, E. J., et al. (2009). Atf4 regulates obesity, glucose homeostasis, and energy expenditure. Diabetes 58, 2565–2573. doi:10.2337/db09-0335
Shaban, H. A., and Seeber, A. (2020). Monitoring the spatio-temporal organization and dynamics of the genome. Nucleic Acids Res. 48, 3423–3434. doi:10.1093/nar/gkaa135
Simonis, M., Klous, P., Splinter, E., Moshkin, Y., Willemsen, R., de Wit, E., et al. (2006). Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C). Nat. Genet. 38, 1348–1354. doi:10.1038/ng1896
Somarowthu, S., Legiewicz, M., Chillón, I., Marcia, M., Liu, F., and Pyle, A. M. (2015). HOTAIR forms an intricate and modular secondary structure. Mol. Cell 58, 353–361. doi:10.1016/j.molcel.2015.03.006
Su, J.-H., Zheng, P., Kinrot, S. S., Bintu, B., and Zhuang, X. (2020). Genome-Scale imaging of the 3D organization and transcriptional activity of chromatin. Cell 182, 1641–1659. doi:10.1016/j.cell.2020.07.032
Szabo, Q., Bantignies, F., and Cavalli, G. (2019). Principles of genome folding into topologically associating domains. Sci. Adv. 5, eaaw1668. doi:10.1126/sciadv.aaw1668
Takahashi, S., Kobayashi, S., and Hiratani, I. (2018). Epigenetic differences between naïve and primed pluripotent stem cells. Cell. Mol. Life Sci. 75, 1191–1203. doi:10.1007/s00018-017-2703-x
Takei, Y., Yun, J., Zheng, S., Ollikainen, N., Pierson, N., White, J., et al. (2021). Integrated spatial genomics reveals global architecture of single nuclei. Nature 590, 344–350. doi:10.1038/s41586-020-03126-2
Tomikawa, J., and Miyamoto, K. (2021). Structural alteration of the nucleus for the reprogramming of gene expression. FEBS J. 289, 7221–7233. doi:10.1111/febs.15894
Tomikawa, J., Takada, S., Okamura, K., Terao, M., Ogata-Kawata, H., Akutsu, H., et al. (2020). Exploring trophoblast-specific Tead4 enhancers through chromatin conformation capture assays followed by functional screening. Nucleic Acids Res. 48, 278–289. doi:10.1093/nar/gkz1034
Tsai, M.-C., Manor, O., Wan, Y., Mosammaparast, N., Wang, J. K., Lan, F., et al. (2010). Long noncoding RNA as modular scaffold of histone modification complexes. Science 329, 689–693. doi:10.1126/science.1192002
van Sluis, M., van Vuuren, C., Mangan, H., and McStay, B. (2020). NORs on human acrocentric chromosome p-arms are active by default and can associate with nucleoli independently of rDNA. Proc. Natl. Acad. Sci. U. S. A. 117, 10368–10377. doi:10.1073/pnas.2001812117
Wang, K. C., and Chang, H. Y. (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914. doi:10.1016/j.molcel.2011.08.018
Wang, Y., Wang, H., Zhang, Y., Du, Z., Si, W., Fan, S., et al. (2019). Reprogramming of meiotic chromatin architecture during spermatogenesis. Mol. Cell 73, 547–561. doi:10.1016/j.molcel.2018.11.019
Wei, Z., Gao, F., Kim, S., Yang, H., Lyu, J., An, W., et al. (2013). Klf4 organizes long-range chromosomal interactions with the Oct4 locus in reprogramming and pluripotency. Cell Stem Cell 13, 36–47. doi:10.1016/j.stem.2013.05.010
Yang, F., Deng, X., Ma, W., Berletch, J. B., Rabaia, N., Wei, G., et al. (2015). The lncRNA Firre anchors the inactive X chromosome to the nucleolus by binding CTCF and maintains H3K27me3 methylation. Genome Biol. 16, 52. doi:10.1186/s13059-015-0618-0
Yu, S., and Lemos, B. (2018). The long-range interaction map of ribosomal DNA arrays. PLoS Genet. 14, e1007258. doi:10.1371/journal.pgen.1007258
Keywords: 3D nuclear organization, Hi-C, lncRNA, totipotent cells, epigenetics
Citation: Tomikawa J (2024) Potential roles of inter-chromosomal interactions in cell fate determination. Front. Cell Dev. Biol. 12:1397807. doi: 10.3389/fcell.2024.1397807
Received: 08 March 2024; Accepted: 23 April 2024;
Published: 07 May 2024.
Edited by:
Argyris Papantonis, University Medical Center Göttingen, GermanyReviewed by:
Mayra Furlan-Magaril, National Autonomous University of Mexico, MexicoCopyright © 2024 Tomikawa. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junko Tomikawa, dG9taWthd2EtakBuY2NoZC5nby5qcA==