 Wanyue Ding
Wanyue Ding Jingyu Chen2
Jingyu Chen2
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol. , 15 February 2024
Sec. Molecular and Cellular Pathology
Volume 12 - 2024 | https://doi.org/10.3389/fcell.2024.1287447
This article is part of the Research Topic Cellular Stress Response and Cardiovascular Aging Related Diseases View all articles
Mitochondrial dysfunction is one of the hallmarks of cardiovascular aging. The leakage of mitochondrial DNA (mtDNA) is increased in senescent cells, which are resistant to programmed cell death such as apoptosis. Due to its similarity to prokaryotic DNA, mtDNA could be recognized by cellular DNA sensors and trigger innate immune responses, resulting in chronic inflammatory conditions during aging. The mechanisms include cGAS-STING signaling, TLR-9 and inflammasomes activation. Mitochondrial quality controls such as mitophagy could prevent mitochondria from triggering harmful inflammatory responses, but when this homeostasis is out of balance, mtDNA-induced inflammation could become pathogenic and contribute to age-related cardiovascular diseases. Here, we summarize recent studies on mechanisms by which mtDNA promotes inflammation and aging-related cardiovascular diseases, and discuss the potential value of mtDNA in early screening and as therapeutic targets.
Cardiovascular diseases have long been the leading cause of death. It is estimated that there were 620 million patients with cardiovascular diseases worldwide in 2021, along with more than 20 million cardiovascular deaths (Lindstrom et al., 2022). Population aging has become an important factor worldwide contributing to the increasing burden of cardiovascular diseases. Age has long been identified as a significant risk factor for cardiovascular events. According to the statistics from American Heart Association, the overall prevalence of cardiovascular diseases (including coronary heart disease, heart failure, stroke, hypertension) in adults is approximately 48%, but it reaches around 78% in the population aged 60 to 79, and rises to about 90% in those aged 80 and above (Benjamin et al., 2019). Additionally, traditional risk factors such as hyperlipidemia and diabetes become more prevalent with advancing age. The normal physiology of the cardiovascular system is also significantly influenced by aging, including myocardial hypertrophy, increased arterial stiffness, and impaired endothelial function (Lakatta and Levy, 2003a; Lakatta and Levy, 2003b). Many physiological changes during the aging process contribute to impairment in cardiovascular system, among which age-related systemic chronic inflammation playing a significant role (Lettino et al., 2022).
Aging is characterized by systemic chronic inflammation, or described as “inflamm-aging,” which is manifested by senescent cells secreting inflammatory factors in the absence of acute infection, which also induces the senescence of normal cells in a paracrine manner (Li et al., 2023). The burden of senescent cells increases due to the weakened clearance of senescent cells, thus forming a vicious cycle of inflammation and aging, leading to cardiac and vascular dysfunction (Liberale et al., 2020). Inappropriate activation of chronic inflammation during aging involves complex mechanisms. Low-level toxin exposure derived from dysregulated microbiota and chronic infections (such as periodontitis) may participate in this process. Cellular senescence mechanisms also considered to play a key role, including telomere shortening, genome instability, protein catabolism defects, autophagy and mitophagy dysregulation, and mitochondrial dysfunction (Sanada et al., 2018).
The cardiovascular system is composed of terminally differentiated cells such as cardiomyocytes and vascular smooth muscle cells (VSMCs), which are rich in mitochondria and maintain the circulatory function under continuous metabolic and mechanical stress. Mitochondria is essential for maintaining the function of cardiovascular system (Abdellatif et al., 2023). Mitochondrial dysfunction is considered one of the hallmarks of cardiovascular aging. In addition to its functions in energy metabolism, mitochondria also maintain intracellular calcium homeostasis, redox balance, and act as a signaling center to regulate cellular behaviors such as mitophagy and apoptosis (Galluzzi et al., 2012).
There is a close connection between mitochondrial dysfunction and inflammation. As an organelle similar to prokaryotes, some structural components of mitochondria could directly induce inflammatory response as damage-associated molecular patterns (DAMPs) (Marchi et al., 2023). Mitochondrial DNA (mtDNA) with a circular double-stranded structure, can be leaked into the cytoplasm as a result of mitochondrial damage, and is recognized by pattern recognition receptors and exogenous DNA receptors, initiating inflammatory response (Wein and Sorek, 2022). Although this helps to clear injured cells to maintain tissue homeostasis, recent studies also suggested that it involved in aging-related cardiovascular diseases (Galluzzi et al., 2018; Harapas et al., 2022).
This review aims to summarize the mechanisms mtDNA initiating inflammatory responses and its role in aging-related cardiovascular diseases, and discusses potential therapeutic strategies of inhibiting mtDNA-mediated inflammation for aging-related cardiovascular diseases.
mtDNA is a small double-stranded circular molecule with a full length of 16,569 base pairs. It only includes 13 genes encoding oxidative phosphorylation-related proteins, ribosomal RNAs and transfer RNAs. It has also been reported in recent studies that mtDNA could be transcribed into non-coding RNAs (Gao et al., 2018). The remaining mitochondrial proteins rely on nuclear gene expression and then imported into the mitochondria (Calvo and Mootha, 2010). Since mitochondria are the site of reactive oxygen species (ROS) generation, mtDNA in a bare nucleoid form is susceptible to damage and accumulates with aging (Gredilla et al., 2010). Under physiological conditions, damaged mitochondria are cleared through mitochondrial quality control such as mitochondrial fission, fusion, and mitophagy (Picca et al., 2018). But when these mechanisms fail or mitochondria are irreversibly damaged, mtDNA will leak from the mitochondrial matrix and be recognized as DAMPs to trigger inflammatory response (Liu et al., 2022).
Although the mechanism of how mtDNA leaks into the cytoplasm or extracellular space has not been fully elucidated, the following mechanisms are considered to be involved. The mitochondrial permeability transition pore (mPTP) is a protein pore complex located at the contact point between the inner and outer membranes of mitochondria. The open of mPTP could result in the leakage of mtDNA into the cytoplasm (Yu et al., 2020). During apoptosis, BCL-2 Associated X (BAX) and BCL-2 Homologous Antagonist/Killer (BAK) also interact with mPTP subunits to regulate the open of mPTP (Rongvaux et al., 2014; White et al., 2014). Activated caspase-1 also involves in mitochondrial pore formation through activating the pore-forming protein gasdermin D (Huang et al., 2020). In addition, mitochondria-derived vesicles (MDVs) can also selectively regulate the transfer of damaged mitochondrial contents to lysosomes for degrading (Todkar et al., 2021). However, it is also reported that circulating mtDNA-containing MDVs could cause inflammatory responses (Picca et al., 2020).
Since the DNA of eukaryotic cells mainly locates in the nucleus or mitochondria, the DNA in the cytoplasm is usually recognized as exogenous from pathogens to initiate innate immune response (Civril et al., 2013). However, mtDNA leaking from mitochondria and accumulating in the cytoplasm, could also be recognized as powerful stimulator of inflammatory response through similar pathways (West et al., 2011). Type I interferon (IFN) response is an important signaling pathway against pathogenic infection, and Stimulator of Interferon Genes (STING) was identified as a protein mediating type I IFN response first (Ishikawa and Barber, 2008; Zhong et al., 2008; Sun et al., 2009). Although STING could be activated by cytoplasmic DNA, subsequent studies revealed that the ligands of STING are cyclic dinucleotides rather than cytoplasmic DNA, including cyclic diadenosine monophosphate (c-dAMP) and cyclic diguanylate monophosphate (c-dGMP) (Burdette et al., 2011). Cyclic GMP-AMP Synthase (cGAS) is the upstream DNA sensor of STING. In the presence of ATP and GTP, cGAS binds to and recognizes cytoplasmic DNA to catalyze the production of cyclic guanosine monophosphate-adenosine monophosphate (cGAMP), which could activate STING as its ligand (Sun et al., 2013; Wu et al., 2013).
In short, when mtDNA leaks into the cytoplasm, cGAS binds to mtDNA and catalyzes the synthesis of cGAMP. cGAMP acts as a ligand for STING to cause conformational changes and activate it (De Gaetano et al., 2021). The activated STING is transferred from the endoplasmic reticulum to the Golgi apparatus and recruited TANK Binding Kinase 1 (TBK1) and Inhibitor of Nuclear Transcription Factor-κB Kinase (IKK). Activated TBK1 and IKK phosphorylate downstream Interferon Regulatory Factor 3 (IRF3) and Inhibitor α of Nuclear Transcription Factor-κB (IκBα) respectively, following by activated IRF3 and Nuclear Transcription Factor-κB (NF-κB) nuclear translocation. These two transcription factors initiate type I IFN responses, and expression of pro-inflammatory cytokines, respectively (Kim et al., 2023).
Toll-like receptors (TLRs) are evolutionarily conserved pattern recognition receptors and play a crucial role in innate immune responses, especially the recognition of pathogens in extracellular matrix (Ospelt and Gay, 2010; Takeuchi and Akira, 2010). So far, 10 TLRs (TLR1-TLR10) have been identified in humans. TLRs are classified as a class I integrated transmembrane protein (Bell et al., 2003), The N-terminal domain constitutes the ectodomain, which serves as the recognition site for distinct pathogens-associated molecular patterns (PAMPs) and DAMPs to induce NF-κB activation (Vijay, 2018). TLR-9 is the first TLR proven to recognize DNA, which is mainly located in the endoplasmic reticulum and transported to lysosomes upon activation (Latz et al., 2004; Kim et al., 2008), and recognizes DNA hypomethylated CpG motifs (Barbalat et al., 2011). Nevertheless, TLR-9 is not highly specific for pathogen DNA recognition, and can also be activated by self-DNA (Marshak-Rothstein and Rifkin, 2007). But in any case, the low methylation feature of mitochondrial DNA makes it closer to exogenous DNA and can be recognized by TLR-9 (Bellizzi et al., 2013; Hong et al., 2013). There is still a lack of detailed elucidation on how mitochondrial DNA is transported into lysosomes and recognized by TLR-9, which may be related to mitophagy and MDVs transport (De Leo et al., 2016; Matheoud et al., 2016). TLR9 downstream signaling is transmitted through the adapter Myeloid Differentiation Primary Response Protein 88 (MyD88), which activates Mitogen-activated Protein Kinases and NF-κB to trigger inflammatory responses, or enhance type I IFN responses through IRF7 (Schiller et al., 2012).
The activation of inflammasome is an important in innate immune response to PAMPs or DAMPs. The inflammasome is a multi-subunit protein complex composed of receptor proteins, adapter proteins and caspase-1 (Guo et al., 2015). The receptor proteins are responsible for recognizing pathogens or stress signals, and then bind to adapter proteins and recruit procaspase-1 to convert into the active form of caspase-1. Its downstream effects are mainly initiating inflammatory responses by cleaving the N-terminal domain of Gasdermin D, making it an active form that could bind membrane phospholipids to form pores, and cleaving IL-1β and IL-18 precursors (Zheng et al., 2020).
It is known that multiple members of the NOD-like receptor family and PYHIN family can serve as receptor proteins for inflammasomes, including NOD, LRR and Pyrin domain-containing protein 1 (NLRP1), NLRP2, NLRP3, NLRP6, NLRP12, NLR family CARD domain-containing protein 4 (NLRC4), and Absent in Melanoma 2 (AIM2), Interferon-y Inducible Protein 16 (IFI16) (Atianand et al., 2013). Research evidence supports that mtDNA could be recognized as endogenous agonists of inflammasomes, and multiple receptor proteins including NLRP3, NLRC4, and AIM-2 have been reported to recognize mtDNA and activate the inflammasome (Nakahira et al., 2011; Jabir et al., 2015; Zhong et al., 2016). The structure of AIM-2 binding to mtDNA is relatively clear, which has a HIN200 domain at its C-terminus that could recognize and bind double-stranded DNA (Hornung et al., 2009). However, the activation of NLRP3 involves various regulatory factors, including K+ efflux, Ca2+ signaling, ROS, lysosome rupture (He et al., 2016). Therefore, although the complex of NLRP3 and NLRC4 appears to bind to mtDNA or oxidized mtDNA suggested by co-immunoprecipitation experiments, it is unclear whether they binds directly or other factors are required (West and Shadel, 2017).
According to data from Non-Communicable Disease Risk Factor Collaboration, the age-standardized prevalence of hypertension has exceeded 30% (Zhou et al., 2021). Hypertension increases the risk of cardiovascular events such as coronary heart disease and stroke (Carey et al., 2021). Endothelial dysfunction and vascular structural remodeling is important pathology in hypertension, in which inflammation plays an important role. Recent studies have found that mtDNA levels in the circulation and urine of hypertensive patients are elevated (Eirin et al., 2019), which is related to target organs damage including brain and kidney (Alé et al., 2017; Eirin et al., 2017), and it is proposed that mtDNA involves in the development of vascular pathology and hypertension.
Endothelial cells (ECs) and VSMCs are important components in regulating the contraction and relaxation function and structure of artery. Inflammatory mechanisms are involved in endothelial dysfunction, and mtDNA acts as an important stimuli of inflammatory activation (Simão et al., 2022). Mao Y et al. reported that in palmitic acid treated ECs, the expression levels of pro-inflammatory factors such as MCP1, IFN-γ, IL-1, and adhesion factors ICAM-1 were significantly increased, which enhanced the adhesion of monocytes to ECs. This process is caused by the leakage of mtDNA and activation of the cGAS-STING-IRF3 pathway in ECs. Knockdown of STING attenuated vascular inflammation and macrophage infiltration in high-fat diet fed mice (Mao et al., 2017). They also reported in another study that activation of the cGAS-STING-IRF3 pathway in ECs induced increased MST1 expression, leading to YAP inactivation and nuclear exclusion, thereby inhibiting endothelial cell proliferation, migration and vascular repair (Yuan et al., 2017). Consistent with previous reports, Huang LS et al. found that lipopolysaccharide activated Gasdermin D in ECs and resulted in mitochondrial pores formation and mtDNA leakage. Activated TBK1 phosphorylates LATS1, which subsequently leads to YAP1 degradation and inhibits ECs proliferation and vascular repair (Huang et al., 2020).
In addition, some studies have reported that mtDNA directly impairs endothelial cell-mediated vasodilation. McCarthy CG et al. found elevated circulating mtDNA levels in male spontaneously hypertensive rats (SHR), which were associated with downregulation of mitophagy in the aorta. The TLR-9 inhibitor ODN2088 lowered systolic blood pressure in SHR, while the TLR-9 agonist ODN2395 aggravated hypertension in Wistar-Kyoto rats and SHR rats. It was further found that ODN2395 treatment on mesenteric arteries reduced acetylcholine-induced relaxation and enhanced norepinephrine-induced contraction. This is related to the upregulated COX2 expression rather than the inhibition of eNOS (McCarthy et al., 2015). Consistent with previous reports, Echem C et al. further demonstrated that mtDNA treatment enhanced phenylephrine-induced vasoconstriction in male SHR, while inhibition of TLR-9 by ODN2088 reversed this phenomenon. The expression of IL-6 and TNF-α was also upregulated, whereas the expression and phosphorylation level of eNOS were not affected. Interestingly, Echem C et al. also found that the vasoconstrictive effect of mtDNA on SHR may be affected by gender. mtDNA did not enhance the contraction to phenylephrine of female SHR aorta, which may be related to the reduced phosphorylation level of ERK1/2 (Echem et al., 2019). However, Goulopoulou S et al. reported that mtDNA promotes vasoconstriction and preeclampsia in pregnant rats through TLR-9 and ERK1/2 phosphorylation (Goulopoulou et al., 2012). A recent study obtained umbilical cord serum and tissue from patients with preeclampsia and found that mtDNA leakage from trophoblast cells induced activation of NLRP3/caspase-1/IL-1β signaling in ECs, causing eNOS-related vasodilation dysfunction. The NLRP3 inhibitor INF39 or MCC950 can partially reverse vasodilation dysfunction (Lv et al., 2023) (Summarized in Figure 1).
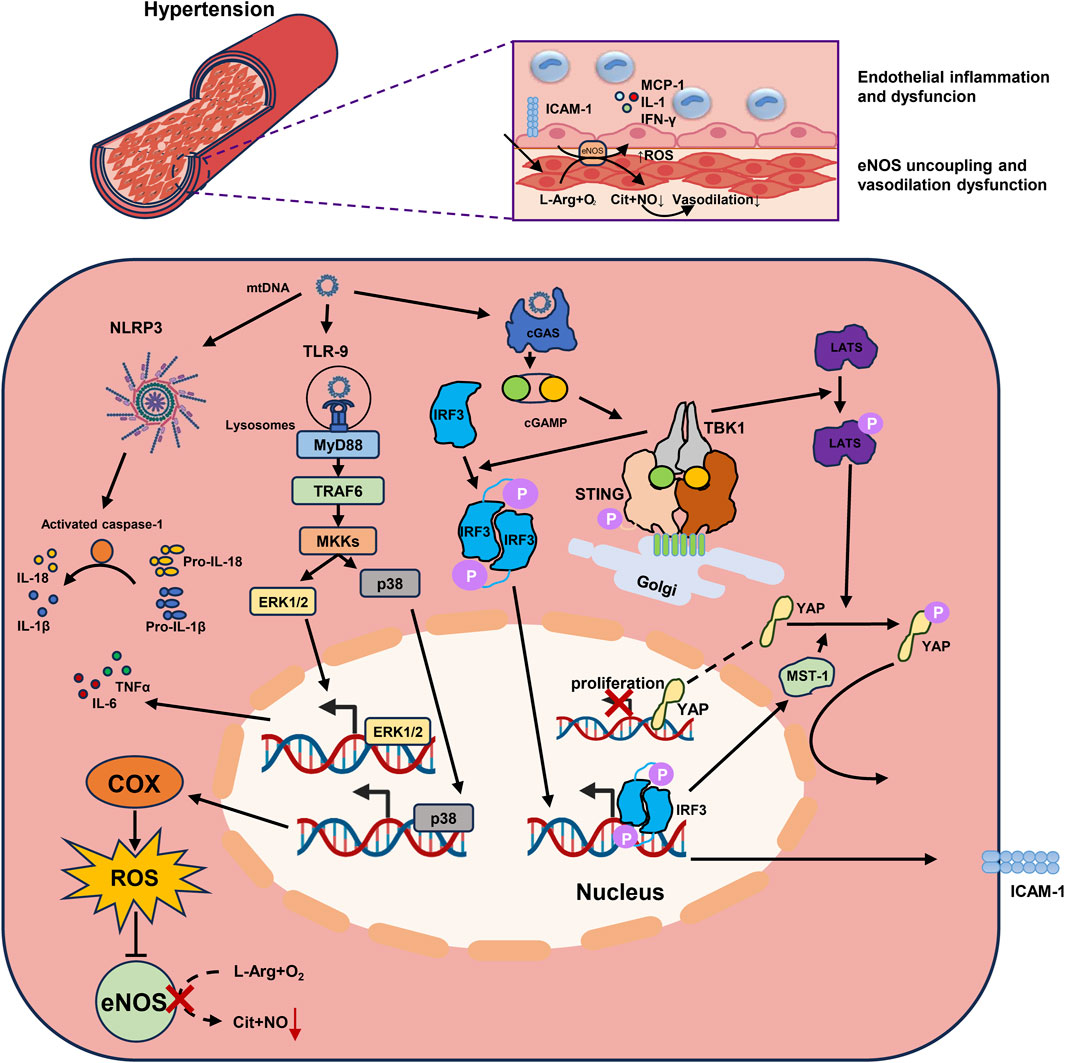
FIGURE 1. mtDNA promotes endothelial inflammation and dysfunction through multiple inflammatory mechanisms in hypertension. 1) mtDNA activates the cGAS-STING-TBK1 pathway and the downstream IRF3 in endothelial cells. The phosphorylated IRF3 homodimer binds to the ICAM-1 promoter, upregulates the its expression, and enhances adherence of monocytes to endothelial cells. 2) The LATS is also phosphorylated following the activated cGAS-STING-TBK1 pathway, promotes the phosphorylation of YAP and preventing its transport into the nucleus. IRF3 also participate in the phosphorylation of YAP by promoting MST-1 expression, and therefore impaired endothelial cells proliferation. 3) mtDNA transported into lysosomes could be recognized by TLR-9. TLR9 downstream signaling is conducted through the adapter MyD88, which activates ERK1/2 through TRAF6-MKKs to promote the expression of inflammatory factors. MKKs also activates p38 MAPK to promote the expression of COX, which resulting in eNOS uncoupling by generating ROS, and impaired vasodilation. 4) NLRP3 inflammasome in endothelial cells recognizes mtDNA and activates caspase-1 to cleave the precursors of IL-1β and IL-18 into their active forms. mtDNA, Mitochondrial DNA; cGAS, Cyclic GMP-AMP synthase; STING, Stimulator of Interferon Genes; TBK1, TANK Binding Kinase 1; IRF3, Interferon Regulatory Factor 3; ICAM-1, Intercellular Adhesion Molecule-1; LATS, Large tumor suppressor kinase; YAP, Yes-Associated Protein; MST-1, Mammalian Sterile 20-Like Kinase 1; TLR-9, Toll-like Receptors 9; MyD88, Myeloid Differentiation Primary Response Protein 88; ERK1/2, Extracellular signal-regulated kinases 1 and 2; TRAF6, TNF Receptor Associated Factor 6; p38 MAPK, P38 Mitogen Activated Protein Kinase; COX, Cyclo-oxygen-ase; eNOS, Endothelial Nitric Oxide Synthase; ROS, Reactive Oxygen Species; NLRP3, NOD-like receptor thermal protein domain associated protein 3.
Excessive proliferation of VSMCs and extracellular matrix synthesis are also important parts in vascular remodeling (Wang et al., 2018; Cai et al., 2021). However, there are few studies on the involvement of mtDNA regulating VSMCs function in hypertension. Arcidiacono MV et al. reported that STING was involved in the osteogenic phenotypic transformation of VSMCs in chronic kidney disease (Arcidiacono et al., 2019). Activation of the cGAS-STING pathway triggers the type I IFN response in VSMCs, resulting in their premature senescence and phenotype switching induced in a paracrine manner (Bi et al., 2021). It has also been reported that TLR-9 could be activated by oxidized hemoglobin-induced lipid peroxidation and leads to the proliferation of pulmonary VSMCs (Loomis et al., 2017). Further research is still required to clarify the role of mtDNA on the phenotype and function of VSMCs in hypertension.
Atherosclerosis is the pathological basis of many cardiovascular diseases such as coronary heart diseases and stroke. Its main feature is lipids depositing in the arterial intima and form into plaques, which leads to reduced blood flow. Plaques rupture can also cause thrombosis and acute complications (Gogulamudi et al., 2023). The pathology of atherosclerosis is complex, including foam cells forming, atherosclerotic plaque forming and rupture, calcification, and thrombus formation. Research evidence has shown that inflammation persist throughout the entire process of atherosclerosis (Wolf and Ley, 2019). mtDNA has also been reported to be involved in the inflammation of atherosclerotic lesions.
In the atherosclerotic lesions of ApoE deficient mice, the cGAMP levels in macrophages was increased and STING was activated, accompanied by the upregulated expression of TNF-α, CCL-2, IFN-β in artery. Bone marrow transplantation after knockout of STING in bone marrow-derived macrophages proved that mtDNA exacerbates the activation of macrophages in atherosclerotic plaques through the cGAS-STING-TBK1 pathway (Pham et al., 2021). Similarly, Liu Y et al. demonstrated that mtDNA induces the inflammatory response of bone marrow-derived macrophages through the STING/NFκB pathway in LDL receptor deficient mice, and proposed that a natural compound Aucubin could inhibit the expression of STING and alleviate atherosclerotic lesions (Liu et al., 2022). Interestingly, Li JL reported that mtDNA-induced inflammation may mediate the association between smoking and atherosclerosis progression. Exposure to e-cigarette smoke in ApoE deficient mice significantly increased mtDNA oxidative damage and upregulated TLR-9 expression in atherosclerotic plaques, as well as subsequent macrophage infiltration and inflammatory cytokines secretion. TLR-9 antagonist could reverse this process (Li et al., 2021). It is also reported that human antimicrobial peptide LL-37 could bind to mtDNA to form a complex, allowing it to escape degradation by DNase II. And the LL-37-mtDNA complex activated the inflammatory response through TLR-9 (Zhang et al., 2015).
In addition, mtDNA could also affects the phenotype and function of VSMCs. Bi XJ et al. reported that oxidized mtDNA in ApoE deficient mouse model of chronic kidney disease triggered type I IFN response in VSMCs through the cGAS-STING pathway, inducing premature senescence and switching from a contractile phenotype to an inflammatory secretory phenotype. This not only aggravates inflammation in atherosclerotic lesions, but also leads to increased plaque vulnerability (Bi et al., 2021). In addition, previous studies reported that lipid-induced programmed cell death such as ferroptosis in VSMCs promoted calcified plaque formation in atherosclerotic lesions (Ma et al., 2021). On this basis, Chen ZD et al. recently reported that mtDNA triggers ferritinophagy-dependent ferroptosis of VSMCs by activating the cGAS-STING pathway. Oleoylethanolamide, an endogenous Peroxisome Proliferator-Activated Receptor α (PPARα) agonist, could attenuate reverse the ferroptosis of VSMCs and arterial intimal calcification (Chen et al., 2023) (Summarized in Figure 2).
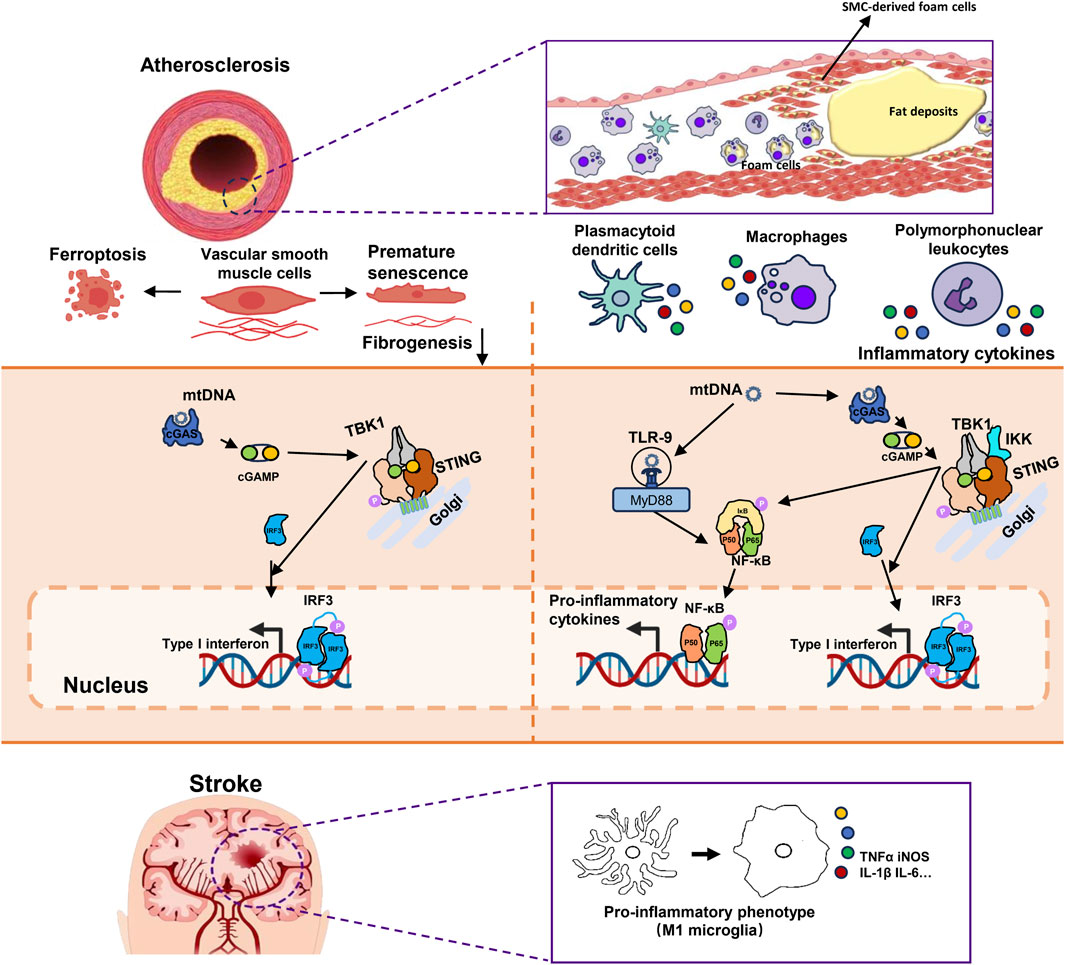
FIGURE 2. The mechanism of mtDNA promoting plaque vulnerability and inflammation in atherosclerosis and microglial M1 polarization in ischemic stroke. 1) mtDNA participates in the formation of atherosclerosis by affecting the phenotype and function of VSMCs. mtDNA activates the cGAS-STING-IRF3 pathway, triggering type I interferon responses in VSMCs which leads to premature senescence and reduced fibrogenesis ability. It also results in ferritinophagy-dependent ferroptosis of VSMCs and both phenotypes are associated with thinner fibrous caps and contribute to plaque instability. 2) The activation of the cGAS-STING-TBK1 pathway by mtDNA in immune cells leading to increased expression of inflammatory cytokines through both IRF3 and NF-κB. Additionally, NF-κB could also be activated by TLR-9. These processes aggravated the plaque inflammation. 3)Microglia are the main inflammatory cells in the central nervous system. In brain tissue after ischemia-reperfusion injury, mtDNA activates the cGAS-STING pathway in microglia. This activation recruits TBK1 and IKK. Activated TBK1 and IKK subsequently phosphorylate downstream IRF3 and IκBα, respectively. This is followed by the activation of IRF3 and NF-κB, leading to their nuclear translocation and initiating type I interferon responses. These responses can promote M1 polarization of microglia and the expression of pro-inflammatory cytokines. mtDNA, Mitochondrial DNA; VSMCs, Vascular smooth muscle cells; cGAS, Cyclic GMP-AMP synthase; STING, Stimulator of Interferon Genes; IRF3, Interferon Regulatory Factor 3; TBK1, TANK Binding Kinase 1; NF-κB, Nuclear Transcription Factor-κB; TLR-9, Toll-like receptors 9; IKK, Inhibitor of Nuclear Transcription Factor-κB Kinase; IκBα, Inhibitory Subunit of NF-κBα.
Stroke is the second leading cause of death worldwide. In 2019, there were 12.2 million incident cases of stroke, and 6.55 million deaths from stroke, accounting for 11.6% of the total deaths (Feigin et al., 2021). Age is one of the most significant risk factors for stroke, and it is reported 12.4% of men and 13.6% of women aged over 80 had stroke (Virani et al., 2021). Atherosclerosis is also an important precursor lesion that causes ischemic stroke, but unlike the situation in hypertension and atherosclerosis with continuous stimulation of external stress, stroke is an acute process of ischemic damage in brain due to cerebrovascular thromboembolism or hemorrhage. Inflammation is also involved in processes such as ischemia/reperfusion injury, blood-brain barrier disruption, and neural regeneration during recovery (Jayaraj et al., 2019).
In the central nervous system, microglia are the main cell type that promotes neuroinflammation. Previous studies have found that a large number of interferon-stimulated genes are upregulated in brain tissue with ischemia-reperfusion injury (McDonough et al., 2017). Studies have found that hypoxia and glucose deficiency can activate type I IFN response, inducing the activation of microglial cells in vitro (Minter et al., 2014). Liao, YJ et al. demonstrated that cGAS is activated by mtDNA in microglial cells, activating IRF3 and NF-κB to promote inflammation after cerebral ischemia/reperfusion (Liao et al., 2020). Consistent with previous reports, Jiang GL et al. reported that the cGAS-STING-IRF3 pathway was involved in the M1 polarization of microglia and the secretion of TNF-α in an ischemia-reperfusion model. Knockout of cGAS significantly attenuated microglia-mediated neuroinflammation, inhibited neuronal apoptosis and reduced infarct size (Jiang et al., 2021). Kong LQ et al. also reported that mtDNA leakage after middle cerebral artery occlusion promotes M1 polarization of microglial cells through cGAS-STING signaling, and verified the reduction of cerebral infarct size and recovery of neural function by knockout of STING (Kong et al., 2022) (Summarized in Figure 2).
However, some studies found that AIM2 inflammasomes and cGAS was activated by nuclear DNA in stroke models according to the close colocalization of DAPI and 53BP1 (Li et al., 2020). Gamdzyk M also reported that cytosolic DNA derived from retrotransposon LINE-1 activated cGAS-STING signaling and promoted apoptosis of neurons (Gamdzyk et al., 2020). However, the role of mtDNA in apoptosis and loss of neurons has been rarely reported and remains to be further explored.
Cell-free mitochondrial DNA (cf-mtDNA) are considered to be released after cell death or transported through MDVs into circulation. Cosentino N et al. reported that increased cf-mtDNA levels could be detected in more than 91% of 466 patients admitted with confirmed ST-segment elevation myocardial infarction (Cosentino et al., 2021). However, as a non-specific biomarker, cf-mtDNA may be more suitable for screening of metabolic diseases and inflammatory states. Ueda K et al. found that cf-mtDNA levels were higher in smokers, and could predict the risk of atherosclerosis (Ueda et al., 2023). Padilla-Sánchez S D et al. reported that cf-mtDNA levels increased with age and body mass index in healthy adults (Padilla-Sánchez et al., 2020). Alvarado-Vásquez N also reported that cf-mtDNA was associated with endothelial dysfunction in patients with prediabetes (Alvarado-Vásquez, 2015). These suggest the role of cf-mtDNA in reflecting inflammation and metabolic diseases risk.
As for therapeutics, reducing mtDNA leakage appears to be an attractive strategy. Increased burden of senescent cells, which are resistant to apoptosis, is associated with chronic inflammation, and a class of drugs called senolytics could selectively clear senescent cells (Kirkland and Tchkonia, 2020). Iske J et al. recently reported that the senolytics treatment of Dasatinib and Quercetin cleared senescent cells, reduced cf-mtDNA and inhibited inflammation in experimental animals (Iske et al., 2020). In addition, some studies have also raised the possibility of regulating mPTP to inhibit mtDNA leakage. Cyclosporin A has been reported to bind to mitochondrial cyclophilin D to inhibit the opening of mPTP and inhibit the inflammation mediated by mtDNA leakage (Tanveer et al., 1996; Xiao et al., 2018; Liu et al., 2019). Also, substantial efforts have been devoted to the development of compounds that inhibit signaling molecules including cGAS, STING, and TLR-9, and to explore their therapeutic value in inflammatory diseases, which have been detailed elsewhere (Krieg, 2006; Decout et al., 2021).
In summary, mitochondrial dysfunction is a hallmark of cardiovascular aging, and mtDNA leakage is associated with a chronic inflammation and promotes age-related cardiovascular diseases. Reducing the leakage of mtDNA or inhibiting inflammatory signals seems to be attractive therapeutic strategies. The specific mechanisms remain to be further explored to solve the gap in research and development and therapeutic applications.
WD: Conceptualization, Writing–original draft, Writing–review and editing. JC: Writing–review and editing. LZ: Writing–review and editing. SW: Writing–review and editing, Visualization. XC: Visualization, Writing–review and editing. HC: Funding acquisition, Supervision, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Scientific and Technological Breakthrough Project of Heilongjiang Province (2022ZXJ02C01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdellatif, M., Rainer, P. P., Sedej, S., and Kroemer, G. (2023). Hallmarks of cardiovascular ageing. Nat. Rev. Cardiol. 16. doi:10.1038/s41569-023-00881-3
Alé, A., Zhang, Y., Han, C., and Cai, D. (2017). Obesity-associated extracellular mtDNA activates central TGFβ pathway to cause blood pressure increase. Am. J. Physiol. Endocrinol. Metab. 312 (3), E161–E174. doi:10.1152/ajpendo.00337.2016
Alvarado-Vásquez, N. (2015). Circulating cell-free mitochondrial DNA as the probable inducer of early endothelial dysfunction in the prediabetic patient. Exp. Gerontol. 69, 70–78. doi:10.1016/j.exger.2015.05.010
Arcidiacono, M. V., Carrillo-López, N., Panizo, S., Castro-Grattoni, A. L., Valcheva, P., Ulloa, C., et al. (2019). Barley-ß-glucans reduce systemic inflammation, renal injury and aortic calcification through ADAM17 and neutral-sphingomyelinase2 inhibition. Sci. Rep. 9 (1), 17810. doi:10.1038/s41598-019-54306-8
Atianand, M. K., Rathinam, V. A., and Fitzgerald, K. A. (2013). SnapShot: inflammasomes. Cell. 153 (1), 272–272.e1. doi:10.1016/j.cell.2013.03.009
Barbalat, R., Ewald, S. E., Mouchess, M. L., and Barton, G. M. (2011). Nucleic acid recognition by the innate immune system. Annu. Rev. Immunol. 29, 185–214. doi:10.1146/annurev-immunol-031210-101340
Bell, J. K., Mullen, G. E. D., Leifer, C. A., Mazzoni, A., Davies, D. R., and Segal, D. M. (2003). Leucine-rich repeats and pathogen recognition in Toll-like receptors. Trends Immunol. 24 (10), 528–533. doi:10.1016/s1471-4906(03)00242-4
Bellizzi, D., D’Aquila, P., Scafone, T., Giordano, M., Riso, V., Riccio, A., et al. (2013). The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 20 (6), 537–547. doi:10.1093/dnares/dst029
Benjamin, E. J., Muntner, P., Alonso, A., Bittencourt, M. S., Callaway, C. W., Carson, A. P., et al. (2019). Heart disease and stroke statistics-2019 update: a report from the American heart association. Circulation 139 (10), e56–e528. doi:10.1161/CIR.0000000000000659
Bi, X., Du, C., Wang, X., Wang, X.-Y., Han, W., Wang, Y., et al. (2021). Mitochondrial damage-induced innate immune activation in vascular smooth muscle cells promotes chronic kidney disease-associated plaque vulnerability. Adv. Sci. (Weinh) 8 (5), 2002738. doi:10.1002/advs.202002738
Burdette, D. L., Monroe, K. M., Sotelo-Troha, K., Iwig, J. S., Eckert, B., Hyodo, M., et al. (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478 (7370), 515–518. doi:10.1038/nature10429
Cai, Z., Gong, Z., Li, Z., Li, L., and Kong, W. (2021). Vascular extracellular matrix remodeling and hypertension. Antioxid. Redox Signal 34 (10), 765–783. doi:10.1089/ars.2020.8110
Calvo, S. E., and Mootha, V. K. (2010). The mitochondrial proteome and human disease. Annu. Rev. Genomics Hum. Genet. 11, 25–44. doi:10.1146/annurev-genom-082509-141720
Carey, R. M., Wright, J. T., Taler, S. J., and Whelton, P. K. (2021). Guideline-driven management of hypertension: an evidence-based update. Circ. Res. 128 (7), 827–846. doi:10.1161/CIRCRESAHA.121.318083
Chen, Z., Sun, X., Li, X., and Liu, N. (2023). Oleoylethanolamide alleviates hyperlipidaemia-mediated vascular calcification via attenuating mitochondrial DNA stress triggered autophagy-dependent ferroptosis by activating PPARα. Biochem. Pharmacol. 208, 115379. doi:10.1016/j.bcp.2022.115379
Civril, F., Deimling, T., de Oliveira Mann, C. C., Ablasser, A., Moldt, M., Witte, G., et al. (2013). Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498 (7454), 332–337. doi:10.1038/nature12305
Cosentino, N., Campodonico, J., Moltrasio, M., Lucci, C., Milazzo, V., Rubino, M., et al. (2021). Mitochondrial biomarkers in patients with ST-elevation myocardial infarction and their potential prognostic implications: a prospective observational study. J. Clin. Med. 10 (2), 275. doi:10.3390/jcm10020275
Decout, A., Katz, J. D., Venkatraman, S., and Ablasser, A. (2021). The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21 (9), 548–569. doi:10.1038/s41577-021-00524-z
De Gaetano, A., Solodka, K., Zanini, G., Selleri, V., Mattioli, A. V., Nasi, M., et al. (2021). Molecular mechanisms of mtDNA-mediated inflammation. Cells 10 (11), 2898. doi:10.3390/cells10112898
De Leo, M. G., Staiano, L., Vicinanza, M., Luciani, A., Carissimo, A., Mutarelli, M., et al. (2016). Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell. Biol. 18 (8), 839–850. doi:10.1038/ncb3386
Echem, C., Costa, T. J. da, Oliveira, V., Giglio Colli, L., Landgraf, M. A., Rodrigues, S. F., et al. (2019). Mitochondrial DNA: a new driver for sex differences in spontaneous hypertension. Pharmacol. Res. 144, 142–150. doi:10.1016/j.phrs.2019.04.008
Eirin, A., Herrmann, S. M., Saad, A., Abumoawad, A., Tang, H., Lerman, A., et al. (2019). Urinary mitochondrial DNA copy number identifies renal mitochondrial injury in renovascular hypertensive patients undergoing renal revascularization: a Pilot Study. Acta Physiol. (Oxf) 226 (3), e13267. doi:10.1111/apha.13267
Eirin, A., Saad, A., Woollard, J. R., Juncos, L. A., Calhoun, D. A., Tang, H., et al. (2017). Glomerular hyperfiltration in obese african American hypertensive patients is associated with elevated urinary mitochondrial-DNA copy number. Am. J. Hypertens. 30 (11), 1112–1119. doi:10.1093/ajh/hpx103
Feigin, V. L., Stark, B. A., Johnson, C. O., Roth, G. A., Bisignano, C., Abady, G. G., et al. (2021). Global, regional, and national burden of stroke and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet Neurology 20 (10), 795–820. doi:10.1016/s1474-4422(21)00252-0
Galluzzi, L., Kepp, O., Trojel-Hansen, C., and Kroemer, G. (2012). Mitochondrial control of cellular life, stress, and death. Circ. Res. 111 (9), 1198–1207. doi:10.1161/CIRCRESAHA.112.268946
Galluzzi, L., Yamazaki, T., and Kroemer, G. (2018). Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell. Biol. 19 (11), 731–745. doi:10.1038/s41580-018-0068-0
Gamdzyk, M., Doycheva, D. M., Araujo, C., Ocak, U., Luo, Y., Tang, J., et al. (2020). cGAS/STING pathway activation contributes to delayed neurodegeneration in neonatal hypoxia-ischemia rat model: possible involvement of LINE-1. Mol. Neurobiol. 57 (6), 2600–2619. doi:10.1007/s12035-020-01904-7
Gao, S., Tian, X., Chang, H., Sun, Y., Wu, Z., Cheng, Z., et al. (2018). Two novel lncRNAs discovered in human mitochondrial DNA using PacBio full-length transcriptome data. Mitochondrion 38, 41–47. doi:10.1016/j.mito.2017.08.002
Gogulamudi, V. R., Durrant, J. R., Adeyemo, A. O., Ho, H. M., Walker, A. E., and Lesniewski, L. A. (2023). Advancing age increases the size and severity of spontaneous atheromas in mouse models of atherosclerosis. Geroscience 45, 1913–1931. Published online April 22, 2023. doi:10.1007/s11357-023-00776-8
Goulopoulou, S., Matsumoto, T., Bomfim, G. F., and Webb, R. C. (2012). Toll-like receptor 9 activation: a novel mechanism linking placenta-derived mitochondrial DNA and vascular dysfunction in pre-eclampsia. Clin. Sci. (Lond). 123 (7), 429–435. doi:10.1042/CS20120130
Gredilla, R., Bohr, V. A., and Stevnsner, T. (2010). Mitochondrial DNA repair and association with aging--an update. Exp. Gerontol. 45 (7-8), 478–488. doi:10.1016/j.exger.2010.01.017
Guo, H., Callaway, J. B., and Ting, J. P.-Y. (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat. Med. 21 (7), 677–687. doi:10.1038/nm.3893
Harapas, C. R., Idiiatullina, E., Al-Azab, M., Hrovat-Schaale, K., Reygaerts, T., Steiner, A., et al. (2022). Organellar homeostasis and innate immune sensing. Nat. Rev. Immunol. 22 (9), 535–549. doi:10.1038/s41577-022-00682-8
He, Y., Hara, H., and Núñez, G. (2016). Mechanism and regulation of NLRP3 inflammasome activation. Trends Biochem. Sci. 41 (12), 1012–1021. doi:10.1016/j.tibs.2016.09.002
Hong, E. E., Okitsu, C. Y., Smith, A. D., and Hsieh, C.-L. (2013). Regionally specific and genome-wide analyses conclusively demonstrate the absence of CpG methylation in human mitochondrial DNA. Mol. Cell. Biol. 33 (14), 2683–2690. doi:10.1128/MCB.00220-13
Hornung, V., Ablasser, A., Charrel-Dennis, M., Bauernfeind, F., Horvath, G., Caffrey, D. R., et al. (2009). AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458 (7237), 514–518. doi:10.1038/nature07725
Huang, L. S., Hong, Z., Wu, W., Xiong, S., Zhong, M., Gao, X., et al. (2020). mtDNA activates cGAS signaling and suppresses the YAP-mediated endothelial cell proliferation program to promote inflammatory injury. Immunity 52 (3), 475–486. doi:10.1016/j.immuni.2020.02.002
Ishikawa, H., and Barber, G. N. (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455 (7213), 674–678. doi:10.1038/nature07317
Iske, J., Seyda, M., Heinbokel, T., Maenosono, R., Minami, K., Nian, Y., et al. (2020). Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat. Commun. 11 (1), 4289. doi:10.1038/s41467-020-18039-x
Jabir, M. S., Hopkins, L., Ritchie, N. D., Ullah, I., Bayes, H. K., Li, D., et al. (2015). Mitochondrial damage contributes to Pseudomonas aeruginosa activation of the inflammasome and is downregulated by autophagy. Autophagy 11 (1), 166–182. doi:10.4161/15548627.2014.981915
Jayaraj, R. L., Azimullah, S., Beiram, R., Jalal, F. Y., and Rosenberg, G. A. (2019). Neuroinflammation: friend and foe for ischemic stroke. J. Neuroinflammation 16 (1), 142. doi:10.1186/s12974-019-1516-2
Jiang, G.-L., Yang, X.-L., Zhou, H.-J., Long, J., Liu, B., Zhang, L.-M., et al. (2021). cGAS knockdown promotes microglial M2 polarization to alleviate neuroinflammation by inhibiting cGAS-STING signaling pathway in cerebral ischemic stroke. Brain Res. Bull. 171, 183–195. doi:10.1016/j.brainresbull.2021.03.010
Kim, J., Kim, H.-S., and Chung, J. H. (2023). Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp. Mol. Med. 55 (3), 510–519. doi:10.1038/s12276-023-00965-7
Kim, Y.-M., Brinkmann, M. M., Paquet, M.-E., and Ploegh, H. L. (2008). UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature 452 (7184), 234–238. doi:10.1038/nature06726
Kirkland, J. L., and Tchkonia, T. (2020). Senolytic drugs: from discovery to translation. J. Intern Med. 288 (5), 518–536. doi:10.1111/joim.13141
Kong, L., Li, W., Chang, E., Wang, W., Shen, N., Xu, X., et al. (2022). mtDNA-STING Axis mediates microglial polarization via IRF3/NF-κB signaling after ischemic stroke. Front. Immunol. 13, 860977. doi:10.3389/fimmu.2022.860977
Krieg, A. M. (2006). Therapeutic potential of Toll-like receptor 9 activation. Nat. Rev. Drug Discov. 5 (6), 471–484. doi:10.1038/nrd2059
Lakatta, E. G., and Levy, D. (2003a). Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part I: aging arteries: a “set up” for vascular disease. Circulation 107 (1), 139–146. doi:10.1161/01.cir.0000048892.83521.58
Lakatta, E. G., and Levy, D. (2003b). Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: Part II: the aging heart in health: links to heart disease. Circulation 107 (2), 346–354. doi:10.1161/01.cir.0000048893.62841.f7
Latz, E., Schoenemeyer, A., Visintin, A., Fitzgerald, K. A., Monks, B. G., Knetter, C. F., et al. (2004). TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol. 5 (2), 190–198. doi:10.1038/ni1028
Lettino, M., Mascherbauer, J., Nordaby, M., Ziegler, A., Collet, J. P., Derumeaux, G., et al. (2022). Cardiovascular disease in the elderly: proceedings of the European society of cardiology-cardiovascular round table. Eur. J. Prev. Cardiol. 29 (10), 1412–1424. doi:10.1093/eurjpc/zwac033
Li, J., Huynh, D. L., Cornwell, W. D., Tang, M.-S., Simborio, H., Huang, J., et al. (2021). Electronic cigarettes induce mitochondrial DNA damage and trigger TLR9 (Toll-Like receptor 9)-mediated atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 41 (2), 839–853. doi:10.1161/ATVBAHA.120.315556
Li, Q., Cao, Y., Dang, C., Han, B., Han, R., Ma, H., et al. (2020). Inhibition of double-strand DNA-sensing cGAS ameliorates brain injury after ischemic stroke. EMBO Mol. Med. 12 (4), e11002. doi:10.15252/emmm.201911002
Li, X., Li, C., Zhang, W., Wang, Y., Qian, P., and Huang, H. (2023). Inflammation and aging: signaling pathways and intervention therapies. Signal Transduct. Target Ther. 8 (1), 239. doi:10.1038/s41392-023-01502-8
Liao, Y., Cheng, J., Kong, X., Li, S., Li, X., Zhang, M., et al. (2020). HDAC3 inhibition ameliorates ischemia/reperfusion-induced brain injury by regulating the microglial cGAS-STING pathway. Theranostics 10 (21), 9644–9662. doi:10.7150/thno.47651
Liberale, L., Montecucco, F., Tardif, J.-C., Libby, P., and Camici, G. G. (2020). Inflamm-ageing: the role of inflammation in age-dependent cardiovascular disease. Eur. Heart J. 41 (31), 2974–2982. doi:10.1093/eurheartj/ehz961
Lindstrom, M., DeCleene, N., Dorsey, H., Fuster, V., Johnson, C. O., LeGrand, K. E., et al. (2022). Global burden of cardiovascular diseases and risks collaboration, 1990-2021. J. Am. Coll. Cardiol. 80 (25), 2372–2425. doi:10.1016/j.jacc.2022.11.001
Liu, H., Liu, X., Zhou, J., and Li, T. (2022a). Mitochondrial DNA is a vital driving force in ischemia-reperfusion injury in cardiovascular diseases. Oxid. Med. Cell. Longev. 2022, 6235747. doi:10.1155/2022/6235747
Liu, R., Xu, F., Bi, S., Zhao, X., Jia, B., and Cen, Y. (2019). Mitochondrial DNA-induced inflammatory responses and lung injury in thermal injury murine model: protective effect of cyclosporine-A. J. Burn Care and Res. 40 (3), 355–360. doi:10.1093/jbcr/irz029
Liu, Y., Zhang, Y., Zhu, H., Shen, W., Chen, Z., Bai, J., et al. (2022b). Aucubin administration suppresses STING signaling and mitigated high-fat diet-induced atherosclerosis and steatohepatosis in LDL receptor deficient mice. Food Chem. Toxicol. 169, 113422. doi:10.1016/j.fct.2022.113422
Loomis, Z., Eigenberger, P., Redinius, K., Lisk, C., Karoor, V., Nozik-Grayck, E., et al. (2017). Hemoglobin induced cell trauma indirectly influences endothelial TLR9 activity resulting in pulmonary vascular smooth muscle cell activation. PLoS One 12 (2), e0171219. doi:10.1371/journal.pone.0171219
Lv, Z., Lv, D.-Y., Meng, J.-Y., Sha, X.-Y., Qian, X.-Y., Chen, Y.-S., et al. (2023). Trophoblastic mitochondrial DNA induces endothelial dysfunction and NLRP3 inflammasome activation: implications for preeclampsia. Int. Immunopharmacol. 114, 109523. doi:10.1016/j.intimp.2022.109523
Ma, W.-Q., Sun, X.-J., Zhu, Y., and Liu, N.-F. (2021). Metformin attenuates hyperlipidaemia-associated vascular calcification through anti-ferroptotic effects. Free Radic. Biol. Med. 165, 229–242. doi:10.1016/j.freeradbiomed.2021.01.033
Mao, Y., Luo, W., Zhang, L., Wu, W., Yuan, L., Xu, H., et al. (2017). STING-IRF3 triggers endothelial inflammation in response to free fatty acid-induced mitochondrial damage in diet-induced obesity. Arterioscler. Thromb. Vasc. Biol. 37 (5), 920–929. doi:10.1161/ATVBAHA.117.309017
Marchi, S., Guilbaud, E., Tait, S. W. G., Yamazaki, T., and Galluzzi, L. (2023). Mitochondrial control of inflammation. Nat. Rev. Immunol. 23 (3), 159–173. doi:10.1038/s41577-022-00760-x
Marshak-Rothstein, A., and Rifkin, I. R. (2007). Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu. Rev. Immunol. 25, 419–441. doi:10.1146/annurev.immunol.22.012703.104514
Matheoud, D., Sugiura, A., Bellemare-Pelletier, A., Laplante, A., Rondeau, C., Chemali, M., et al. (2016). Parkinson’s disease-related proteins PINK1 and parkin repress mitochondrial antigen presentation. Cell. 166 (2), 314–327. doi:10.1016/j.cell.2016.05.039
McCarthy, C. G., Wenceslau, C. F., Goulopoulou, S., Ogbi, S., Baban, B., and Sullivan, J. C. (2015). Circulating mitochondrial DNA and Toll-like receptor 9 are associated with vascular dysfunction in spontaneously hypertensive rats. Cardiovasc. Res. 107 (1), 119–130. doi:10.1093/cvr/cvv137
McDonough, A., Lee, R. V., Noor, S., Lee, C., Le, T., Iorga, M., et al. (2017). Ischemia/reperfusion induces interferon-stimulated gene expression in microglia. J. Neurosci. 37 (34), 8292–8308. doi:10.1523/JNEUROSCI.0725-17.2017
Minter, M. R., Zhang, M., Ates, R. C., Taylor, J. M., and Crack, P. J. (2014). Type-1 interferons contribute to oxygen glucose deprivation induced neuro-inflammation in BE(2)M17 human neuroblastoma cells. J. Neuroinflammation 11, 43. doi:10.1186/1742-2094-11-43
Nakahira, K., Haspel, J. A., Rathinam, V. A. K., Lee, S.-J., Dolinay, T., Lam, H. C., et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 12 (3), 222–230. doi:10.1038/ni.1980
Ospelt, C., and Gay, S. (2010). TLRs and chronic inflammation. Int. J. Biochem. Cell. Biol. 42 (4), 495–505. doi:10.1016/j.biocel.2009.10.010
Padilla-Sánchez, S. D., Navarrete, D., Caicedo, A., and Teran, E. (2020). Circulating cell-free mitochondrial DNA levels correlate with body mass index and age. Biochimica Biophysica Acta (BBA) - Mol. Basis Dis. 1866 (12), 165963. doi:10.1016/j.bbadis.2020.165963
Pham, P. T., Fukuda, D., Nishimoto, S., Kim-Kaneyama, J.-R., Lei, X.-F., Takahashi, Y., et al. (2021). STING, a cytosolic DNA sensor, plays a critical role in atherogenesis: a link between innate immunity and chronic inflammation caused by lifestyle-related diseases. Eur. Heart J. 42 (42), 4336–4348. doi:10.1093/eurheartj/ehab249
Picca, A., Guerra, F., Calvani, R., Coelho-Júnior, H. J., Landi, F., Bernabei, R., et al. (2020). Extracellular vesicles and damage-associated molecular patterns: a pandora’s box in health and disease. Front. Immunol. 11, 601740. doi:10.3389/fimmu.2020.601740
Picca, A., Mankowski, R. T., Burman, J. L., Donisi, L., Kim, J.-S., Marzetti, E., et al. (2018). Mitochondrial quality control mechanisms as molecular targets in cardiac ageing. Nat. Rev. Cardiol. 15 (9), 543–554. doi:10.1038/s41569-018-0059-z
Rongvaux, A., Jackson, R., Harman, C. C. D., Li, T., West, A. P., de Zoete, M. R., et al. (2014). Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA. Cell. 159 (7), 1563–1577. doi:10.1016/j.cell.2014.11.037
Sanada, F., Taniyama, Y., Muratsu, J., Otsu, R., Shimizu, H., Rakugi, H., et al. (2018). Source of chronic inflammation in aging. Front. Cardiovasc Med. 5, 12. doi:10.3389/fcvm.2018.00012
Schiller, M., Parcina, M., Heyder, P., Foermer, S., Ostrop, J., Leo, A., et al. (2012). Induction of type I IFN is a physiological immune reaction to apoptotic cell-derived membrane microparticles. J. Immunol. 189 (4), 1747–1756. doi:10.4049/jimmunol.1100631
Simão, V. A., Ferder, L., Manucha, W., and Chuffa, L. G. A. (2022). Epigenetic mechanisms involved in inflammaging-associated hypertension. Curr. Hypertens. Rep. 24 (11), 547–562. doi:10.1007/s11906-022-01214-4
Sun, L., Wu, J., Du, F., Chen, X., and Chen, Z. J. (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339 (6121), 786–791. doi:10.1126/science.1232458
Sun, W., Li, Y., Chen, L., Chen, H., You, F., Zhou, X., et al. (2009). ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. U. S. A. 106 (21), 8653–8658. doi:10.1073/pnas.0900850106
Takeuchi, O., and Akira, S. (2010). Pattern recognition receptors and inflammation. Cell. 140 (6), 805–820. doi:10.1016/j.cell.2010.01.022
Tanveer, A., Virji, S., Andreeva, L., Totty, N. F., Hsuan, J. J., Ward, J. M., et al. (1996). Involvement of cyclophilin D in the activation of a mitochondrial pore by Ca2+ and oxidant stress. Eur. J. Biochem. 238 (1), 166–172. doi:10.1111/j.1432-1033.1996.0166q.x
Todkar, K., Chikhi, L., Desjardins, V., El-Mortada, F., Pépin, G., and Germain, M. (2021). Selective packaging of mitochondrial proteins into extracellular vesicles prevents the release of mitochondrial DAMPs. Nat. Commun. 12 (1), 1971. doi:10.1038/s41467-021-21984-w
Ueda, K., Sakai, C., Ishida, T., Morita, K., Kobayashi, Y., Horikoshi, Y., et al. (2023). Cigarette smoke induces mitochondrial DNA damage and activates cGAS-STING pathway: application to a biomarker for atherosclerosis. Clin. Sci. (Lond). 137 (2), 163–180. doi:10.1042/CS20220525
Vijay, K. (2018). Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int. Immunopharmacol. 59, 391–412. doi:10.1016/j.intimp.2018.03.002
Virani, S. S., Alonso, A., Aparicio, H. J., Benjamin, E. J., Bittencourt, M. S., Callaway, C. W., et al. (2021). Heart disease and stroke statistics-2021 update: a report from the American heart association. Circulation 143 (8), e254–e743. doi:10.1161/CIR.0000000000000950
Wang, D., Uhrin, P., Mocan, A., Waltenberger, B., Breuss, J. M., Tewari, D., et al. (2018). Vascular smooth muscle cell proliferation as a therapeutic target. Part 1: molecular targets and pathways. Biotechnol. Adv. 36 (6), 1586–1607. doi:10.1016/j.biotechadv.2018.04.006
Wein, T., and Sorek, R. (2022). Bacterial origins of human cell-autonomous innate immune mechanisms. Nat. Rev. Immunol. 22 (10), 629–638. doi:10.1038/s41577-022-00705-4
West, A. P., and Shadel, G. S. (2017). Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat. Rev. Immunol. 17 (6), 363–375. doi:10.1038/nri.2017.21
West, A. P., Shadel, G. S., and Ghosh, S. (2011). Mitochondria in innate immune responses. Nat. Rev. Immunol. 11 (6), 389–402. doi:10.1038/nri2975
White, M. J., McArthur, K., Metcalf, D., Lane, R. M., Cambier, J. C., Herold, M. J., et al. (2014). Apoptotic caspases suppress mtDNA-induced STING-mediated type I IFN production. Cell. 159 (7), 1549–1562. doi:10.1016/j.cell.2014.11.036
Wolf, D., and Ley, K. (2019). Immunity and inflammation in atherosclerosis. Circ. Res. 124 (2), 315–327. doi:10.1161/CIRCRESAHA.118.313591
Wu, J., Sun, L., Chen, X., Du, F., Shi, H., Chen, C., et al. (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339 (6121), 826–830. doi:10.1126/science.1229963
Xiao, Z., Jia, B., Zhao, X., Bi, S., and Meng, W. (2018). Attenuation of lipopolysaccharide-induced acute lung injury by cyclosporine-A via suppression of mitochondrial DNA. Med. Sci. Monit. 24, 7682–7688. doi:10.12659/MSM.909909
Yu, C.-H., Davidson, S., Harapas, C. R., Hilton, J. B., Mlodzianoski, M. J., Laohamonthonkul, P., et al. (2020). TDP-43 triggers mitochondrial DNA release via mPTP to activate cGAS/STING in ALS. Cell. 183 (3), 636–649. doi:10.1016/j.cell.2020.09.020
Yuan, L., Mao, Y., Luo, W., Wu, W., Xu, H., Wang, X. L., et al. (2017). Palmitic acid dysregulates the Hippo-YAP pathway and inhibits angiogenesis by inducing mitochondrial damage and activating the cytosolic DNA sensor cGAS-STING-IRF3 signaling mechanism. J. Biol. Chem. 292 (36), 15002–15015. doi:10.1074/jbc.M117.804005
Zhang, Z., Meng, P., Han, Y., Shen, C., Li, B., Hakim, M. A., et al. (2015). Mitochondrial DNA-LL-37 complex promotes atherosclerosis by escaping from autophagic recognition. Immunity 43 (6), 1137–1147. doi:10.1016/j.immuni.2015.10.018
Zheng, D., Liwinski, T., and Elinav, E. (2020). Inflammasome activation and regulation: toward a better understanding of complex mechanisms. Cell. Discov. 6 (1), 36–22. doi:10.1038/s41421-020-0167-x
Zhong, B., Yang, Y., Li, S., Wang, Y.-Y., Li, Y., Diao, F., et al. (2008). The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29 (4), 538–550. doi:10.1016/j.immuni.2008.09.003
Zhong, Z., Umemura, A., Sanchez-Lopez, E., Liang, S., Shalapour, S., Wong, J., et al. (2016). NF-κB restricts inflammasome activation via elimination of damaged mitochondria. Cell. 164 (5), 896–910. doi:10.1016/j.cell.2015.12.057
Zhou, B., Carrillo-Larco, R. M., Danaei, G., Riley, L. M., Paciorek, C. J., Stevens, G. A., et al. (2021). Worldwide trends in hypertension prevalence and progress in treatment and control from 1990 to 2019: a pooled analysis of 1201 population-representative studies with 104 million participants. Lancet 398 (10304), 957–980. doi:10.1016/S0140-6736(21)01330-1
Keywords: cardiovasuclar diseases, inflammation, mitochondrial DNA, senescence, innate immunity
Citation: Ding W, Chen J, Zhao L, Wu S, Chen X and Chen H (2024) Mitochondrial DNA leakage triggers inflammation in age-related cardiovascular diseases. Front. Cell Dev. Biol. 12:1287447. doi: 10.3389/fcell.2024.1287447
Received: 01 September 2023; Accepted: 23 January 2024;
Published: 15 February 2024.
Edited by:
Hua Zhu, The Ohio State University, United StatesReviewed by:
Carlos García Padilla, University of Jaén, SpainCopyright © 2024 Ding, Chen, Zhao, Wu, Chen and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hong Chen, enl5a3h5Y2hAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.