
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 17 November 2023
Sec. Signaling
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1270980
This article is part of the Research Topic Myc as a Disease Target Beyond Cancer View all 16 articles
 Naoe Harafuji1†
Naoe Harafuji1† Chaozhe Yang1†
Chaozhe Yang1† Maoqing Wu1Girija Thiruvengadam1
Maoqing Wu1Girija Thiruvengadam1 Heather Gordish-Dressman1R. Griffin Thompson2P. Darwin Bell2
Heather Gordish-Dressman1R. Griffin Thompson2P. Darwin Bell2 Avi Z. Rosenberg3Claudia Dafinger4
Avi Z. Rosenberg3Claudia Dafinger4 Max C. Liebau5
Max C. Liebau5 Zsuzsanna Bebok2
Zsuzsanna Bebok2 Ljubica Caldovic6,7
Ljubica Caldovic6,7 Lisa M. Guay-Woodford1,6*
Lisa M. Guay-Woodford1,6*Autosomal recessive polycystic kidney disease (ARPKD; MIM#263200) is a severe, hereditary, hepato-renal fibrocystic disorder that leads to early childhood morbidity and mortality. Typical forms of ARPKD are caused by pathogenic variants in the PKHD1 gene, which encodes the fibrocystin/polyductin (FPC) protein. MYC overexpression has been proposed as a driver of renal cystogenesis, but little is known about MYC expression in recessive PKD. In the current study, we provide the first evidence that MYC is overexpressed in kidneys from ARPKD patients and confirm that MYC is upregulated in cystic kidneys from cpk mutant mice. In contrast, renal MYC expression levels were not altered in several Pkhd1 mutant mice that lack a significant cystic kidney phenotype. We leveraged previous observations that the carboxy-terminus of mouse FPC (FPC-CTD) is proteolytically cleaved through Notch-like processing, translocates to the nucleus, and binds to double stranded DNA, to examine whether the FPC-CTD plays a role in regulating MYC/Myc transcription. Using immunofluorescence, reporter gene assays, and ChIP, we demonstrate that both human and mouse FPC-CTD can localize to the nucleus, bind to the MYC/Myc P1 promoter, and activate MYC/Myc expression. Interestingly, we observed species-specific differences in FPC-CTD intracellular trafficking. Furthermore, our informatic analyses revealed limited sequence identity of FPC-CTD across vertebrate phyla and database queries identified temporal differences in PKHD1/Pkhd1 and CYS1/Cys1 expression patterns in mouse and human kidneys. Given that cystin, the Cys1 gene product, is a negative regulator of Myc transcription, these temporal differences in gene expression could contribute to the relative renoprotection from cystogenesis in Pkhd1-deficient mice. Taken together, our findings provide new mechanistic insights into differential mFPC-CTD and hFPC-CTD regulation of MYC expression in renal epithelial cells, which may illuminate the basis for the phenotypic disparities between human patients with PKHD1 pathogenic variants and Pkhd1-mutant mice.
The MYC proto-oncogene, encoding the MYC transcription factor, was first identified in patients with Burkitt’s lymphoma (Taub et al., 1982). MYC contributes to the regulation of multiple cellular signaling pathways involved in cell proliferation (Gearhart et al., 2007). Aberrant MYC expression induces malignant transformation of several tumor types (Dang, 2012; Gabay et al., 2014). In addition, MYC increases the expression of inflammatory and fibrotic factors, which may significantly contribute to the pathogenesis of cystic kidney diseases (Nevzorova et al., 2013; Karihaloo, 2015; Shen et al., 2017). MYC overexpression in renal epithelia has been reported in several mouse ADPKD models as well as in cpk mice (Cowley et al., 1991; Burtey et al., 2008; Kurbegovic and Trudel, 2013; Wu et al., 2013). Elevated MYC appears to be a signature of renal cystic disease and may define a causative pathway (Trudel, 2015; Parrot et al., 2019). However, the role of MYC activation in the initiation and progression of autosomal recessive polycystic kidney disease (ARPKD; MIM#263200) remains incompletely understood.
ARPKD is a hereditary hepato-renal fibrocystic disorder with an estimated incidence of 1 in 26,500 live births (Guay-Woodford et al., 2014; Alzarka et al., 2017). Pathogenic variants in the polycystic kidney and hepatic disease 1 (PKHD1) gene, located on chromosome 6p21.1, cause all typical forms of human ARPKD. The longest PKHD1 (MIM#606702) open reading frame (ORF) contains 67 exons, which encode a 4,074 amino acid protein called fibrocystin/polyductin (FPC) (Onuchic et al., 2002; Ward et al., 2002). Full length FPC is a single transmembrane (TM) domain protein predicted to have several immunoglobulin-like IPT/TIG conserved domains, two G8 domains, and multiple parallel beta-helix 1 (PbH1) repeats in a long extracellular segment (3,858 amino acids), and a short (192 amino acids) cytoplasmic C-terminal domain (CTD) (Supplementary Figure S5A) (Sharp et al., 2005; Wang et al., 2007). The PKHD1 mRNA is primarily expressed in the kidney, liver, lung, and pancreas (Onuchic et al., 2002; Ward et al., 2002; Xiong et al., 2002). In adult and fetal human tissues, FPC is expressed in renal collecting ducts, thick ascending limbs of loops of Henle, bile ducts, pancreatic ducts, epididymis, and testis (Ward et al., 2003; Menezes et al., 2004). Pathogenic sequence variants in the PKHD1 gene account for more than 80% of human ARPKD cases (Bergmann, 2017). Less than 1% of ARPKD patients have pathogenic sequence variants either in DZIP1L or CYS1 genes (Lu et al., 2017; Yang et al., 2021), and the molecular cause of ARPKD in remaining patients remains to be determined (Bergmann, 2017).
Pkhd1 is the mouse ortholog of PKHD1. The longest ORF of mouse Pkhd1 also contains 67 exons and encodes a 4,059 amino acid protein. The mouse and human FPC sequences are 73% identical overall but the CTD share only 55% identity (Nagasawa et al., 2002). Mouse FPC is also a single TM domain protein with 3,872 amino acid N-terminal segment and a short (187 amino acids) cytoplasmic CTD. Mouse FPC has the same numbers of conserved IPT/TIG and G8 domains as human FPC (Nagasawa et al., 2002; Bergmann, 2017). The intracellular domain of mouse FPC (mFPC-CTD), contains an 18-residue long ciliary targeting signal (CTS) that facilities delivery to the primary cilium (Follit et al., 2010). The mFPC-CTD, encoded by exons 65-67, undergoes Notch-like processing followed by regulated membrane-release and translocation to the nucleus (Kaimori et al., 2007), which is facilitated by the 25-residue long nuclear localization signal (NLS) (Hiesberger et al., 2006). Single particle electron microscopy analysis revealed that FPC-CTD forms a ring-like protein complex that binds to double stranded DNA, suggesting a role in gene expression regulation (Cameron Varano et al., 2017). Yet, the function of FPC-CTD in the nucleus remains poorly understood. Numerous rodent models of ARPKD with mutations and multiple exon deletions in Pkhd1 have been generated. However, these models express minimal or no renal disease (Katsuyama et al., 2000; Ward et al., 2002; Masyuk et al., 2004; Moser et al., 2005; Garcia-Gonzalez et al., 2007; Woollard et al., 2007; Gallagher et al., 2008; Kim et al., 2008; Williams et al., 2008; Hu et al., 2011; Outeda et al., 2017; Ishimoto et al., 2023).
The most widely studied mouse model of ARPKD, the cpk mouse carries a spontaneous insertion/deletion (indel) mutation in the Cys1 gene, encoding the cystin protein (Hou et al., 2002; Guay-Woodford, 2003; Nagao et al., 2012). The renal phenotype of cpk mice closely resembles human ARPKD. Mouse cystin, the product of the Cys1 gene, is a 145-amino acid, cilia-associated protein that is mainly expressed in mouse kidney and liver ductal epithelium as early as embryonic day 14.5 (Tao et al., 2009). Mouse cystin contains a predicted N-myristylation motif (MGSGSSR) and a NLS located in the first 27 amino acids at the N-terminus. Amino acids 28-35 of mouse cystin contain a cilium-targeting motif (AxEGG) that is required for cystin trafficking to the primary cilium (Tao et al., 2009). Our previous demonstration that cystin suppresses Myc transcription by binding to necdin, an activator of the Myc P1 promoter, links renal cystogenesis in cpk mice to Myc activation and enhanced MYC levels (Wu et al., 2013; Yang et al., 2021).
Human CYS1 encodes Cystin-1, a 158-amino acid protein (Fliegauf et al., 2003). Sequence comparison of human and mouse orthologs Cystin-1 and cystin shows 57% identity and 64% similarity (Fliegauf et al., 2003). Initial analysis of CYS1 expression in adult human tissues revealed high CYS1 mRNA abundance in the kidney and pancreas (Fliegauf et al., 2003). Subsequent RNA-seq analysis of human tissues consistently revealed high CYS1 mRNA levels in the kidney and lower expression in several other tissues including ovary, gall bladder, endometrium, pancreas, and lung (Fagerberg et al., 2014). The function of Cystin-1 is not understood, although we have reported the first genetic defect in human CYS1 that causes the renal ARPKD phenotype (Yang et al., 2021).
In the current study, we employed immunofluorescence imaging as well as bioinformatic, molecular, and biochemical analyses to comparatively evaluate the roles of human and mouse FPC-CTDs in the regulation of MYC expression in human and mouse renal epithelia.
All human studies were approved by the Institutional Review Board at the Children’s National Hospital or the University Hospital of Cologne. Human kidney samples were obtained from the NIDDK-funded UAB Childhood Cystic Kidney Disease Center Translational Resource at the University of Alabama at Birmingham and the University Hospital of Cologne. Kidney samples were obtained from patients ranging from 26 weeks of gestation age to 3 years of age (Supplementary Table S1).
All mouse experiments were approved by the Institutional Animal Care and Use Committees at Children’s National Research Institute, and experiments were carried out in accordance with relevant guidelines and regulations. Mouse colonies were maintained in the animal facility at Children’s National Research Institute. All mouse kidneys were harvested from 14-day-old, 10- and 12-month-old mutants and age-matched wild-type (WT) littermates. Genetic background information for all mouse lines used in this study is shown in Supplementary Table S2.
All antibodies used for this study are listed in the Supplementary Table S3, unless specified in the text.
Immunohistochemical staining for MYC was performed on formalin-fixed, paraffin embedded tissues using heat induced epitope retrieval solution (BOND Epitope Retrieval Solution 2, Leica Biosystems, Cat. No. AR9640) and an automated stainer (Bond-Max, Leica Biosystems). Tissues were incubated with anti-MYC antibody (Recombinant Anti-c-Myc antibody [Y69] - ChIP Grade, Abcam) at 1:25 dilution for 120 min.
Cultured cells and kidney tissues were collected, homogenized, and processed for immunoblotting as previously described (Wu et al., 2013; Dafinger et al., 2020). Immuno-reactive protein bands were visualized using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, Cat. No. 34095) and images were obtained with ChemiDoc Imaging System (Bio-Rad laboratory). Densitometry was performed using Image Lab software (Bio-Rad laboratory, Version 6.0).
Kidney tissue samples from 14-day-old male mice were snap frozen, transferred to a gentleMACS M tubes (Miltenyi Biotec, Cat. No. 130-093-236, RRID:SCR_020269) in Buffer RLT plus 2-Mercaptoethanol (as per RNeasy Mini Kit instructions, Qiagen, Cat. No. 74104), and homogenized using a gentleMACS Dissociator (Miltenyi Biotec) per the manufacturer’s program RNA-02. Homogenized samples were transferred to microcentrifuge tubes for total RNA extraction using the RNeasy Mini Kit according to the manufacturer’s instructions. Total RNA from both 5-day-postconfluent mIMCD-3 cells stably expressing FPC-CTD and hTERT-immortalized human renal epithelial cells (hTERT-HRE) transiently transfected with FPC-CTD were isolated using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Isolated total RNA was treated with RQ1 RNase-Free DNase (Promega, Cat. No. M6101), and then repurified using the RNeasy Mini kit. RNA samples were reverse-transcribed using SuperScript III First-Strand Synthesis SuperMix (Thermo Fisher Scientific, Cat. No. 18080400) and oligo dT primers as described in the manufacturer’s instructions.
Quantitative RT-PCR was performed on a QuantStudio 7 Flex Real-Time PCR System (Thermo Fisher Scientific) using the default program. The PCR was performed with cDNA templates using Power SYBR Green PCR Master Mix (Thermo Fisher Scientific, Cat. No. 4368706) and mouse primers specific for sequences of Myc (forward: 5′- GCC CCC AAG GTA GTG ATC CT -3’; reverse: 5′- GTG CTC GTC TGC TTG AAT GG -3′). Peptidylprolyl isomerase A (PPIA) was used for normalization (forward: 5′- AGC ACT GGA GAG AAA GGA TT -3’; reverse: 5′- ATT ATG GCG TGT AAA GTC ACC A-3′) (Arensdorf and Rutkowski, 2013). Overexpression of FPC-CTD in cell lines was confirmed with Pkhd1 exon 66–67 specific primers (forward: 5′-CCA GAA GAC ATA TCT GAA TCC CAG GC-3’; reverse: 5′-AGC AAG AGA TCC TGG AAC ACA GGT-3′). Results were analyzed using QuantStudio Real-Time PCR Software and the ΔΔCt method (Livak and Schmittgen, 2001).
Protein sequences of FPC were collected from NCBI protein database (https://www.ncbi.nlm.nih.gov/protein/) using an advanced search with gene name, Pkhd1 and taxonomic groups Mammalia, Aves, Reptilia, Amphibia, Caecilians, and Fish. This resulted in 102 FPC sequences from mammals, birds, reptiles, amphibians, and fish (Supplementary Table S4). FPC sequence from each species was verified by protein alignment with human FPC; the protein sequences that were significantly shorter than human FPC sequence were removed. WebLogo 3 (Crooks et al., 2004) was used to visualize FPC sequence alignment (Supplementary Figure S1) that was generated with Clustal Omega (Sievers et al., 2011). The conservation scores of FPC amino acids were extracted from the WebLogo 3 raw data (Supplementary Table S5). Conserved domains in FPC were mapped by Conserved Domain Database (CDD) (Lu et al., 2020).
Prediction of nuclear localization signals (NLSs) were performed with the human and mouse FPC-CTD construct sequences using SeqNLS (Lin et al., 2012) with 0.86 as the cut-off score.
pcDNA5/FRT/TO (pcDNA5) was obtained from Thermo Fisher Scientific (Cat. No. V652020).
pcDNA5/FRT/TO-mPkhd1-CTD-V5 (pcDNA5-mFPC-CTD): the cytoplasmic tail of mouse FPC (Follit et al., 2010) expression construct was generated from pcDNA5/FRT/TO-mPkhd1 (full length) -V5 (gift from Dr. Feng Qian) with site-directed mutagenesis (SDM) using 5′- GCT AAC TGG ACA TGA TGC TTT GCT GCT GGT TTA AGA AAA GC -3′ and 5′- GCT TTT CTT AAA CCA GCA GCA AAG CAT CAT GTC CAG TTA GC -3′ primer set.
pcDNA5/FRT/TO-mPkhd1-CTDdelCTS-V5 (pcDNA5-mFPC-CTDdelCTS): the ciliary targeting sequence (Follit et al., 2010) deleted mFPC-CTD expression construct was made from mFPC-CTD by SDM with 5′- GCT AAC TGG ACA TGA TGC TTG ACA TAT CTG AAT CCC AGG CT -3′ and 5′- AGC CTG GGA TTC AGA TAT GTC AAG CAT CAT GTC CAG TTA GC -3′ primer set.
pcDNA5/FRT/TO-hPKHD1-CTD-V5 (pcDNA-hFPC-CTD): the expression construct containing the hFPC-CTD, comparable to mFPC-CTD (Follit et al., 2010), and fused to V5-tag was made by LifeSct LLC. The hFPC-CTD coding sequence was cloned between KpnI and NotI sites in pcDNA5/FRT/TO.
pcDNA5/FRT/TO-hPKHD1-CTDdelCTS-V5 (pcDNA-hFPC-CTDdelCTS): the ciliary targeting sequence deleted hFPC-CTD expression construct was made from hFPC-CTD by SDM using 5′- CCG TGG ACA GAA TGA CTG CCG AGA TTC CTG AAT CCC AGA C -3′ and 5′- GTC TGG GAT TCA GGA ATC TCG GCA GTC ATT CTG TCC ACG G -3′ primer set.
pGL4.22 [luc2CP/Puro] vector was purchased from Promega (Cat. No. E6771).
pRL-TK vector was purchased from promega (Cat. No. E2241).
pGL4.22-mouse Myc P1 (pGL4.22-mMyc P1): the mouse Myc P1 promoter (chr8:127735983-127736125, GRCm38/mm10 mouse genome assembly) construct was described previously (Wu et al., 2013).
pGL4.22-human MYC P1 (pGL4.22-hMYC P1): the human MYC P1 promoter (chr15:61985298-61985433, GRCh38/hg38 human genome assembly), which is comparable to the mouse Myc P1 promoter, was amplified by PCR from HEK293 genomic DNA and cloned into pGL4.22 vector at XhoI and HindIII sites using 5′- CCG CTC GAG GAG GGC GTG GGG GAA AAG A-3′ and 5′- CCC AAG CTT AGC CAG GGA CGG CCG G -3′ primer set. Sequence alignment of human and mouse MYC/Myc P1 promoter shown in Supplementary Figure S2 was created by Clustal Omega (Sievers et al., 2011).
Mouse TERT immortalized cortical collecting duct (mTERT-CCD) cells (Steele et al., 2010) were cultured in TERT culture medium [DMEM/F-12 medium (Thermo Fisher Scientific, Cat. No. 11330057) containing 5% heat-inactivated fetal bovine serum (FBS) (Atlanta Biologicals, Cat. No. S11050H), 1% penicillin/streptomycin (Thermo Fisher Scientific, Cat. No. 15140163), 1x Insulin-Transferrin-Selenium solution (Thermo Fisher scientific, Cat. No. 41400045), 0.2 μg/mL dexamethasone (Sigma-Aldrich, Cat. No. D8893) and 10 nM 3,3′,5-Triiodo-L-thyronine sodium salt (Sigma-Aldrich, Cat. No. T6397)] at 37°C in 5% CO2.
Mouse inner medullary collecting duct (mIMCD)-3 cells were purchased from American Type Culture Collection (ATCC, Cat. No. CRL-2123) and cultured in complete growth medium (CGM) [DMEM/F-12 medium (Thermo Fisher Scientific, Cat. No. 11330057) containing 10% heat-inactivated fetal bovine serum (Atlanta Biologicals, Cat. No. S11050H) and 1% penicillin/streptomycin (Thermo Fisher Scientific, Cat. No. 15140163)] at 37°C in 5% CO2.
mIMCD-3 mFPC-CTD stable cell lines were generated by transfection of either pcDNA5-mFPC-CTD or pcDNA5 into mIMCD-3 cells with Lipofectamine2000 transfection reagent (Thermo Fisher Scientific, Cat. No. 11668019). At 48 hrs post-transfection, cells with spontaneously integrated plasmids were selected with hygromycin (1 mg/mL) (Thermo Fisher Scientific, Cat. No. 10687010) for 1 week. The mFPC-CTD stably overexpressing cells and the control empty vector cells were then maintained in CGM with hygromycin (200 μg/mL).
Human kidney sections were minced and immediately placed in 1% collagenase type I (Sigma, Cat. No. C0130-1G) in DMEM/F12 (Thermo Fisher, Cat No. 11330032) and incubated on a rotator for 30 min at room temperature (RT). Renal epithelial cells and tubule fragments were then transferred to a conical tube containing DMEM/F12 and centrifuged at 750 g for 10 min. The supernatant was removed, and the tissue was resuspended in a complete medium, containing 0.2 mg/mL dexamethasone (Sigma, Cat. No. D8893-1 MG), 5% heat inactivated FBS (Hyclone, Cat. No. SH30396-03), 2 mM glutamate (Thermo Fisher, Cat. No. 25030081), 1x insulin-transferrin-sodium selenite (ITS) (Thermo Fisher, Cat. No. 1400045), 100 U/mL penicillin/streptomycin (Thermo Fisher, Cat. No. 15140122), and 10 nM triiodothyronine (Sigma, Cat. no. T6397-100 MG) in DMEM/F12. No antibiotics were added to the complete medium in preparation for the transduction with the hTERT lentiviral expression construct. Cells were maintained in a 37°C humidified incubator with 5% CO2.
Lenti-hTERT-Neo Virus (Cat. No. LV622), Lenti-p53 siRNA Virus (Cat. No. G219), and Polybrene (Cat. No. G062) were purchased from Applied Biological Materials (Richmond, BC, Canada). One day prior to transfection, primary human renal epithelial cells were plated at 20%–30% confluency in a 6 well plate. The next day, the complete medium was replaced with 1 mL of transfection medium, which contained 6 μg/mL of polybrene in the complete medium. Then, 1.54 × 108 transducing units (TU)/ml of Lenti-hTERT-Neo Virus and 1 × 106 TU/mL of Lenti-p53 siRNA Virus were added to the transfection medium at a multiplicity of infection (MOI) of 7. The plate was centrifuged at 200 g for 30 min and then placed back in the incubator at 37°C. After 24 h culture at 37°C, 1 mL of the complete medium was added to each 6 well and cultured for an additional 24 h. Cells immortalized with the hTERT gene then were selected at 48 h post transduction, using 800 μg/mL G418 in the complete medium.
Characterization of the hTERT-HRE cells was performed using immunoblotting of E-cadherin as an epithelial marker, ZO-1 as a tight junction marker, α-ENaC as a renal epithelial cell marker, AQP1 as a renal tubule cell marker, and Keratin 17/19 as an epithelial cell marker (Supplementary Figure S3A). In addition, RT-PCR was performed for PKHD1 as a renal epithelial marker (Supplementary Figure S3B).
For MYC immunofluorescence staining, cells stably overexpressing mFPC-CTD were seeded onto coverslips in 6-well plates and cultured until confluent. For FPC-CTD localization assay, mTERT-CCD or hTERT-HRE cells were transiently transfected with pcDNA-mFPC-CTD and pcDNA-mFPC-CTDdelCTS, or pcDNA5-hFPC-CTD and pcDNA5-hFPC-CTDdelCTS, respectively. Forty-8 hrs. after transfection, cells were washed with PBS and fixed with 4% paraformaldehyde (PFA) for 10 min at RT and then permeabilized with 0.5% Triton X-100 in PBS for 5 min, followed by three washes with PBS before blocking with 1% BSA for 30 min. The cells were incubated with primary antibodies (anti-V5 or anti-Myc) overnight at 4°C followed by incubation with secondary antibody (Alexa Fluor 488 at 1:400 dilution) for 1 h at RT. The cells were then washed three times with PBS and mounted with ProLong Gold + DAPI (Life Technologies, Cat. No. P36935). Fluorescently labeled cells were analyzed on an Olympus FV1000 scanning laser confocal microscope configured with both an Argon Laser (5 mW, 488 nm), and a Green HeNe (10 mW, 543 nm) laser. Images were analyzed using Olympus FV10-ASW 3.0 Viewer software.
Cells were seeded in 24-well plate, grown to ∼90% confluence, and then transfected with pGL4.22-mMyc P1 or pGL4.22-hMYC P1): (0.3 µg/well) and pcDNA5-mFPC-CTD (0.6 μg or 1.2 μg/well) or pcDNA5-hFPC-CTD (0.6 µg/well) using Lipofectamine 2000. The differences in transfection efficiency were normalized by co-transfecting with 15 ng/well pRL plasmid that expressed Renilla luciferase (Promega) and adjusting the total amount of plasmid DNA to 1.5 ug/well by adding pcDNA5. The transfected cells were incubated for 48 h, lysed in 100 µL/well passive lysis buffer (Promega) and shaken for 20 min at RT. Firefly and Renilla luciferase activities were measured with Dual-Luciferase Reporter Assay System (Promega, Cat. No. E1910). The luminometer (FLUOstar OPTIMA, BMG LABTECH) was programmed using OPTIMA software to perform a 0 s delay, followed by a 5-s measurement period for each reporter assay. The 20 μL cell lysate was transferred into 96-well plate (Costar, Cat. No. 3912; white flat bottom), followed by the addition of 100 µL Luciferase Assay Reagent II and luminescence reading. After measurement of firefly luciferase activity, 100 µL of Stop&Glo reagent was added and quickly put back for reading of the Renilla luciferase activity. Data were collected from three independent transfections and processed using GraphPad Prism version 9.1.2 for Windows, GraphPad Software, San Diego, California United States of America, www.graphpad.com.
To determine the binding of FPC-CTD to the Myc P1 promoter, experiments were performed using Magna ChIP A/G Chromatin Immunoprecipitation Kit (MilliporeSigma, Cat. No. 17-10085), according to the manufacturer instructions and our previously published protocol (Wu et al., 2013). Because ChIP-grade anti-FPC-CTD antibodies were not available, we generated mIMCD-3 cells stably overexpressing mFPC-CTD-V5 and control cells stably transfected with empty vector and used ChIP-grade anti-V5 antibody (Abcam, Cat. No. ab15828) for immunoprecipitation. Cells were grown to 80%–90% confluence prior to experiments and processed according to the Magna ChIP A/G protocol. Following immunoprecipitation with anti-V5 antibody that recognized mFPC-CTD-V5 bound to the chromatin and subsequent protease digestion, we amplified Myc P1 using PCR primers specific to the full-length Myc P1 (forward primer: 5′- CGC TCG AGG AGA GAG GTG GGG AAG GGA GAA AG -3’; reverse primer: 5′- CCC AAG CTT AGT GAG GCG AGT CGG ACC CGG CA -3′) using the following PCR program: 94°C 3 min; 94°C 20 s, 62°C 20 s, 72°C 15 s, repeat for 40 cycles, 72°C 5 min, 10°C holding.
Gene expression profiles of PKHD1/Pkhd1, CYS1/Cys1, NDN/Ndn, and MYC/Myc across kidney developmental stages in humans and mice, were downloaded from Evo-devo mammalian organs portal (https://apps.kaessmannlab.org/evodevoapp/) (Cardoso-Moreira et al., 2019). RPKM (reads per kilo base of transcript per million mapped reads) values were normalized using the highest value as 1 and the lowest value as 0 for each gene.
Non-parametric Wilcoxon sign rank test was used for analysis of qRT-PCR data normalized to control samples (Figure 1C, and 2D). All other data were analyzed using either two-way Student’s t-test or nonparametric test with GraphPad Prism version 9.1.2 for Windows, GraphPad Software, San Diego, California United States, www.graphpad.com.
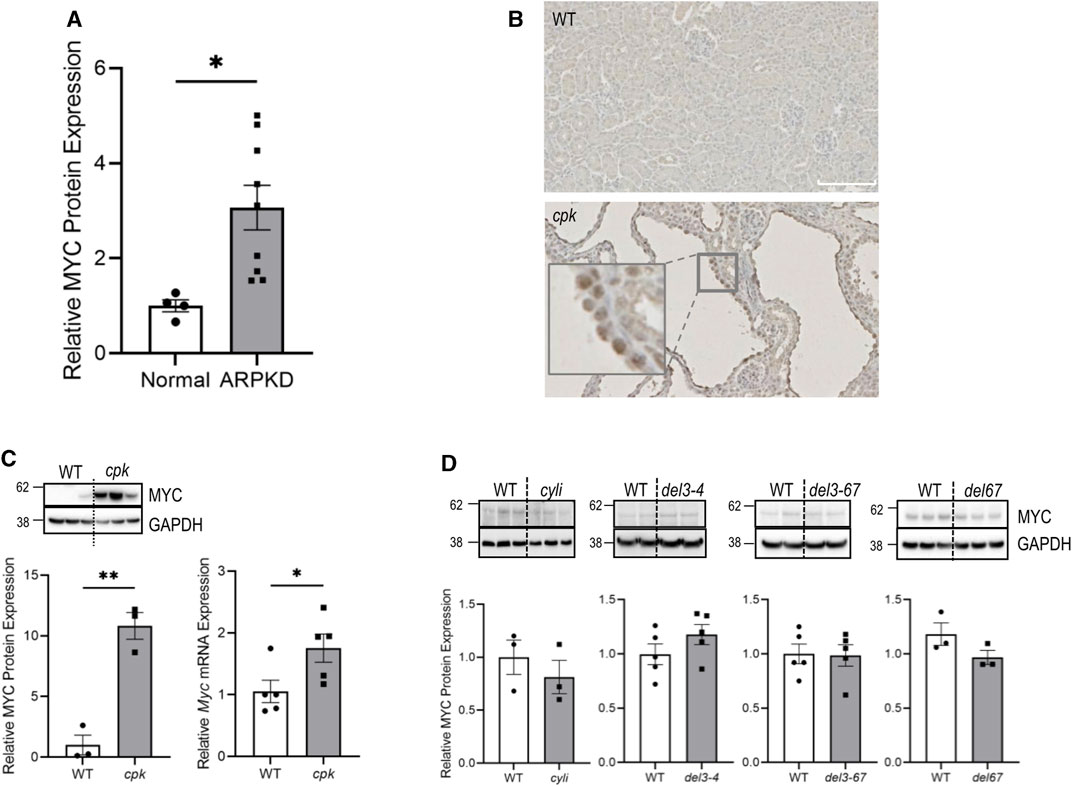
FIGURE 1. MYC/Myc overexpression is associated with renal cystic disease in human and mouse ARPKD. (A) Kidney lysates from normal human and ARPKD patients (Supplementary Table S1) were probed with anti-MYC and control anti-GAPDH antibodies. Relative MYC expression was normalized to GAPDH. Experiments were repeated twice independently. t-test *p < 0.05. Error bar indicates S.E.M. (B) Immunohistochemistry showing MYC expression (brown) in 14-day-old renal epithelial cells from wild-type (WT) and cpk mice. (C) Kidney lysates from 14-day-old WT and cpk mice were probed with anti-MYC and control anti-GAPDH antibodies. GAPDH was used for normalization (cpk mice, n = 6, t-test **p < 0.01). In parallel, Myc mRNA expression was quantified by qRT-PCR. The error bars indicate S.E.M. *indicates p < 0.05 (D) Kidney lysates from 14-day-old Pkhd1 mutant mice (except 1 month old for cyli) and WT littermates were probed with anti-MYC and control anti-GAPDH antibodies, respectively. No significant differences were observed between groups. cyli (n = 3), del3-4 (n = 5), del3-67 (n = 5) and del67 (n = 3). The error bars indicate S.E.M.
MYC overexpression is a signature feature of cystic renal epithelia in human ADPKD and various mouse PKD models (Trudel, 2015). However, MYC expression in ARPKD has not been reported. In the current study, we analyzed MYC expression in ARPKD kidneys by immunoblotting. While MYC expression was marginally detectable in adult kidneys and a kidney from an infant without kidney disease (Supplementary Figure S4), we observed higher MYC abundance in all kidneys from patients with defined pathogenic variants in PKHD1 (Figure 1A), with the highest MYC levels detected in kidneys from patients with PKHD1 truncating pathogenic variants (Supplementary Table S1), resulting in the loss of FPC-CTD (AR1, AR2, AR3, and AR8) (Figure 1A and Supplementary Figure S4).
We then evaluated MYC expression in mouse models of ARPKD. Using IHC, we confirmed increased nuclear expression of mouse MYC protein in dilated collecting ducts from cpk kidneys (Figure 1B). Quantitative analysis confirmed 11-fold higher levels of MYC protein and 1.8-fold higher levels of Myc mRNA in the kidneys from cpk mice compared to WT mice (Figure 1C). In contrast, MYC protein levels were not elevated in kidneys from four different Pkhd1 mutant mouse model lines (Pkhd1cyli, Pkhd1del3-4, Pkhd1del3-67, or Pkhd1del67) that did not exhibit a cystic kidney phenotype (Figure 1D). These data indicate an association between high MYC expression and the renal cystic phenotype in both human ARPKD and mice with an ARPKD-like kidney phenotype. Furthermore, the lack of enhanced MYC expression in Pkhd1 mutant mice without cystic kidney phenotype suggests differences in the function of mouse and human FPC-CTDs.
We note that in each of our mouse mutant lines, the predicted Pkhd1 translated products would be missing the FPC-CTD. Furthermore, while the mouse and human FPC sequences are 73% identical overall, the CTDs share only 55% identity (Nagasawa et al., 2002). Therefore, we analyzed phylogenetic conservation of FPC in vertebrates to better understand potential differences in the regulation of MYC expression in renal epithelia derived from patients with ARPKD and cpk and Pkhd1 mutant mice. We hypothesized that the low sequence conservation of the FPC-CTDs may contribute to functional differences among FPC orthologs; an important consideration given that mouse FPC undergoes Notch-like processing that releases the FPC-CTD, which can translocate to the nucleus.
To understand phylogenetic changes in Pkhd1, we queried the NCBI protein database for PKHD1 orthologs and collected FPC protein sequences of 66 mammalian, 27 bird, 5 reptile, 3 amphibian, and 1 fish (Supplementary Table S4). The large variance in the number of FPC proteins in each class may in part reflect the number of sequenced vertebrate genomes in the NCBI data base. However, while genomic data are available for multiple fish, a PKHD1 ortholog was identified only in the genome of Latimeria chalumnae (a coelacanth).
After alignment of 102 vertebrate FPC protein sequences, we used the WebLogo 3 entropy scores to evaluate FPC conservation across species (Supplementary Figure S1 and Supplementary Table S5). First, we compared the WebLogo 3 entropy scores for each of the conserved domains in the extracellular portion of FPC: five IPT domains, three TIG domains, two G8 domains, two PbH1 domains, and the TM domain with entropy scores from three regions of FPC that do not correspond to any conserved domains (Supplementary Figure S5B). Since the average WebLogo 3 entropy scores were similar (Supplementary Figure S5B), we then compared the WebLogo 3 entropy scores of FPC extracellular and cytoplasmic domains. Consistent with different protein sequence identities between human and mouse extracellular and cytoplasmic portions of FPC (Nagasawa et al., 2002) the WebLogo 3 entropy scores of vertebrate FPC extracellular domain were higher than entropy scores of FPC-cytoplasmic domains (Supplementary Figure S5C). This analysis supports the hypothesis that lower sequence conservation of mouse and human FPC cytoplasmic domain compared to extracellular domain may be functionally significant and potentially contribute to the phenotypic variability observed between PKHD1 vs Pkhd1 mutants.
Both human and mouse, FPC-CTD are encoded by PKHD1/Pkhd1 exons 65, 66 and 67 (Supplementary Figure S5D). Overall, human and mouse FPC-CTD share 55% sequence identity, (Nagasawa et al., 2002). However, the CTS, localized in the human and mouse FPC-CTDs, are highly conserved (Figure 2A, red and blue highlights). Prior studies have experimentally validated one NLS in the mouse FPC-CTD (Figure 2A, purple highlight) (Hiesberger et al., 2006). However, using the web-based NLS prediction tool, SeqNLS (Lin and Hu, 2013), we identified two NLSs in mouse FPC-CTD (score >0.86), one of which overlapped with the experimentally identified NLS (Figure 2A, red bold text for predicted and purple highlight for experimentally validated NLS) (Hiesberger et al., 2006). On the other hand, human FPC-CTD had only one predicted NLS (score >0.86) (Figure 2A, red bold text). Interestingly, the two mouse, and the one predicted human NLS showed low sequence similarity.
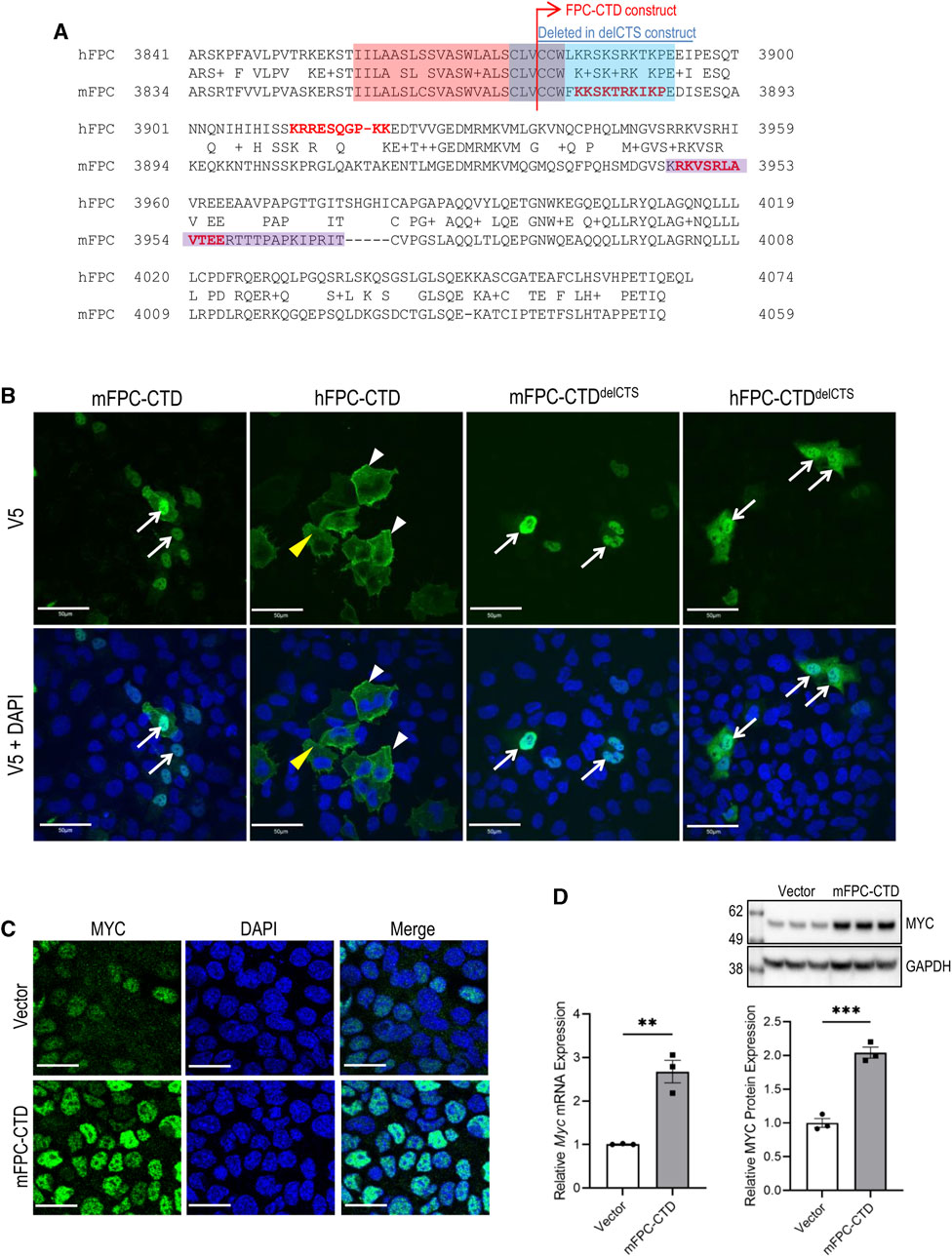
FIGURE 2. Subcellular localization of mFPC-CTD and hFPC-CTD and regulation of MYC expression by mFPC-CTD. (A) The alignments of human and mouse FPC-CTD. The red highlight indicates TM and blue highlight indicates the CTS. Predicted nuclear localization signals (NLSs) are shown in the red bold typeface. The purple highlight indicates experimentally tested NLS (Hiesberger et al., 2006). Blue line above the alignment indicates amino acids deleted from FPC-CTD to generate the FPC-CTDdelCTS constructs. (B) Transient transfection of V5-tagged mFPC-CTD, mFPC-CTDdelCTS, hFPC-CTD, and hFPC-CTDdelCTS, in mTERT-CCD or hTERT-HRE cells. Immunofluorescent staining was performed using anti-V5 antibody. White arrows—nucleus; yellow arrowheads—cytosol; white arrowheads—cell membrane; scale bars = 50 µM. (C) Immunofluorescent staining showing increased MYC expression in mIMCD-3 cells stably expressing mFPC-CTD. Green—MYC; blue—DAPI; scale bars = 20 µM. (D) Mouse Myc mRNA and MYC protein levels were increased in mIMCD-3 cells stably expressing mFPC-CTD. Myc mRNA expression was quantified by qRT-PCR. MYC protein expression was analysed with anti-MYC and normalized to GAPDH expression (N = 2, n = 3, t-test **p < 0.01, ***p < 0.001). Error bar indicates S.E.M.
While it has been determined that the mouse FPC-CTD translocates to the nucleus (Hiesberger et al., 2006), the intracellular trafficking of human FPC-CTD has not been determined and the nuclear function of FPC-CTD is not fully understood. Therefore, we compared intracellular localization of human and mouse FPC-CTD and tested their functions in the nucleus, concentrating on MYC/Myc regulation. Sequences the human and mouse FPC-CTD included the V5 tag and both the mouse and human FPC-CTDs contained the intracellular portion of the CTS (Figure 2A, blue highlight). The NLS sequences are shown in Figure 2A (red bold text).
By immunofluorescence, the mFPC-CTD localized primarily to the nucleus and was essentially absent from the cytoplasm (Figure 2B, left column). In contrast, the hFPC-CTD was largely excluded from the nucleus, localized to the cytoplasm and decorated the cell membrane (Figure 2B, second column from left). We suspected that nuclear trafficking of the hFPC-CTD construct with only one NLS was confounded by the CTS. Therefore, we deleted the CTS from both human and mouse constructs to generate plasmids expressing hFPC-CTDdelCTS and mFPC-CTDdelCTS respectively. Overexpression of these proteins showed strong nuclear localization for both human and mouse FPC-CTDs, although a fraction of hFPC-CTDdelCTS was retained in the cytoplasm (Figure 2B, right two columns).
We then investigated whether mFPC-CTD can regulate Myc expression in mIMCD-3 cells stably expressing the intact mFPC-CTD mouse construct. mIMCD-3 cells stably expressing the empty vector (pcDNA5) served as a control. This experimental approach allowed evaluation of the effect of mFPC-CTD on Myc expression in non-proliferating cells, 5 days post-confluence. The intensity of MYC immunostaining was higher in mIMCD-3 cells expressing mFPC-CTD than in the control cells (Figure 2C). Both Myc mRNA and MYC protein levels were higher in mIMCD-3 cells stably expressing mFPC-CTD, compared to control cells (Figure 2D). Taken together, our data provide the first evidence that overexpression of mFPC-CTD enhances MYC expression in cultured renal epithelial cells.
Efforts to perform these experiments with hFPC were confounded by the inability to generate an hTERT-HRE cell line that stably overexpressed either hFPC-CTD or hFPC-CTDdelCTS. We observed that expression of hFPC-CTD was silenced after several passages of hTERT-HRE cells under selection, suggesting that stably expressed hFPC-CTD may be cytotoxic.
The mechanisms that govern Myc transcription are complex and involve multiple promoters (P0, P1, P2, and P3) and transcription start sites (Battey et al., 1983; Bentley and Groudine, 1986; Ray et al., 1987). The P1 and P2 promoters are the predominant Myc regulatory elements (Albert et al., 2001). In disease states, Myc overexpression primarily is driven from the P1 promoter (Wierstra and Alves, 2008). Therefore, we cloned the mouse Myc P1 and human MYC P1 promoter into a pGL4.22 reporter gene construct (Figure 3A). Co-transfection of mIMCD-3 cells with constructs expressing mFPC-CTD and Myc P1 promoter driven reporter gene showed dose-dependent activation of the Myc P1 promoter by mFPC-CTD (Figure 3B). Myc P1 promoter activation in mTERT-CCD cells was not affected by deletion of the CTS (Figure 3C). In comparison, co-transfection of hTERT-HRE cells with constructs expressing hFPC-CTD and MYC P1 reporter plasmid showed only minimal, though statistically significant activation of the MYC P1 promoter (Figure 3D). However, deletion of the CTS from the hFPC-CTD, which enhanced its nuclear localization, resulted in a 1.6-fold activation of the MYC P1 promoter (Figure 3D). These data demonstrate that both human and mouse FPC-CTDs can activate the Myc/MYC P1 promoter in cultured cells. Furthermore, we tested hFPC-CTD activation of MYC P1 promoter in mIMCD-3 cell line and found comparable activation in this mouse line as in the human cell line, hTERT-HRE (Supplementary Figure S6). However, we note that hFPC-CTD nuclear trafficking is regulated by the CTS.
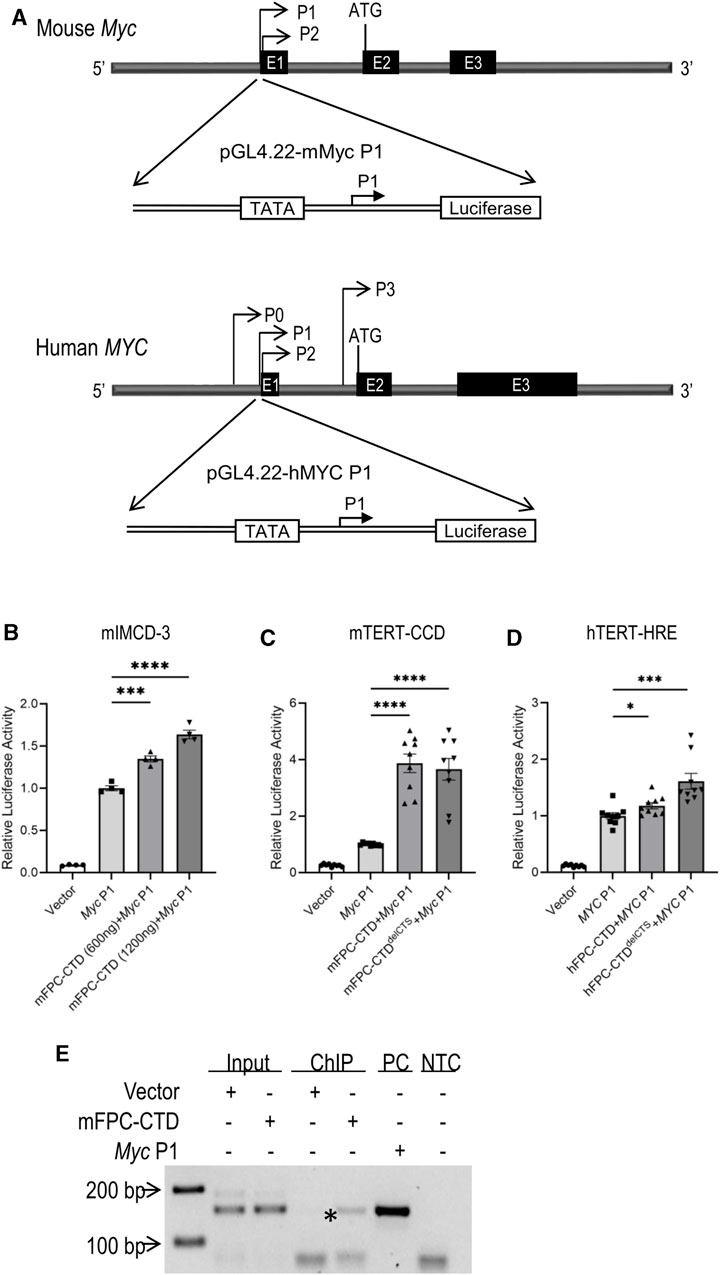
FIGURE 3. Activation of the Myc/MYC P1 promoter by m/hFPC-CTDs and binding of mFPC-CTD to the Myc P1 promoter (A) Schema of mouse (upper) and human (lower) Myc/MYC P1 luciferase reporter assay constructs. (B) The overexpression of mFPC-CTD increased Myc P1 promoter activity in mIMCD-3 cells in a dose-dependent manner. (C) Overexpression of mFPC-CTD and mFPC-CTDdelCTS increased Myc P1 promoter activity in mTERT-CCD cells. (D) hFPC-CTDdelCTS increased the MYC P1 promoter activity ∼1.6 fold in hTERT-HRE cells while hFPC-CTD only increased activity ∼1.1 fold. Experiments were repeated 3 times independently (N = 3) with 3 technical replicates (n = 3). The data were statistically analysed by combining all technical replicates (total n = 9) and *p < 0.05, ***p < 0.001, ****p < 0.0001. Error bar indicates S.E.M. (E) ChIP assay showing FPC-CTD binding to the endogenous Myc P1 promoter in mIMCD-3 cells. Asterisk indicates mFPC-CTD immunoprecipitation with endogenous Myc P1 promoter. PC indicates PCR amplification product using Myc P1 plasmid DNA as a template. NTC indicates negative, no template control. Experiments were repeated independently twice.
To confirm the binding of mFPC-CTD to the Myc P1 promoter, we performed ChIP assays. We found that mFPC-CTD-V5 bound to the endogenous Myc P1 promoter in non-proliferating cells (Figure 3E, asterisk). As noted above, corresponding ChIP experiments with hFPC-CTD were confounded by our inability to generate an appropriate hTERT-HRE cell line.
Considering the observed differences in the nuclear trafficking and function of human and mouse FPC-CTDs, we sought to better understand how species-specific regulation of MYC/Myc expression may contribute to the divergent renal phenotypes in human ARPKD and the Pkhd1 mouse models. Therefore, we analyzed cystogene expression patterns in human and mouse kidneys during intrauterine and postnatal development using the Evo-devo mammalian organs portal (http://evodevoapp.kaessmannlab.org) (Cardoso-Moreira et al., 2019). We specifically focused on genes that are known to be mutated in human ARPKD (PKHD1/Pkhd1), in cpk mice (CYS1/Cys1), as well as necdin (NDN/Ndn), which we have previously shown regulates Myc expression (Wu et al., 2013). We normalized the kidney developmental stages of mouse and human during pre- and postnatal development. RPKM of PKHD1/Pkhd1, CYS1/Cys1, NDN/Ndn, and MYC/Myc genes were graphed at the corresponding developmental stages of human and mouse kidneys (Figure 4). These analyses demonstrated different timing of PKHD1/Pkhd1 and CYS1/Cys1 mRNA expression peaks during human and mouse kidney development (Figure 4, top panels). In the fetal human kidney, PKHD1 mRNA levels progressively increase and reach maximal expression prior to birth, whereas the progressive increase in CYS1 mRNA lags, reaching maximum expression in the post-natal period. Conversely, in the mouse kidney, maximum expression of Cys1 mRNA precedes the peak of Pkhd1 mRNA expression. Expression patterns of NDN/Ndn mRNA during human and mouse kidney development were similar (Figure 4, middle panels), with expression peaking in early developmental stages (6–8 weeks post conception and e12.5–14.5 in human and mouse kidney, respectively) and decreasing thereafter. Expression of MYC/Myc mRNA also peaked during early nephrogenesis and gradually decreased thereafter in both human and mouse kidneys (Figure 4, bottom panels).
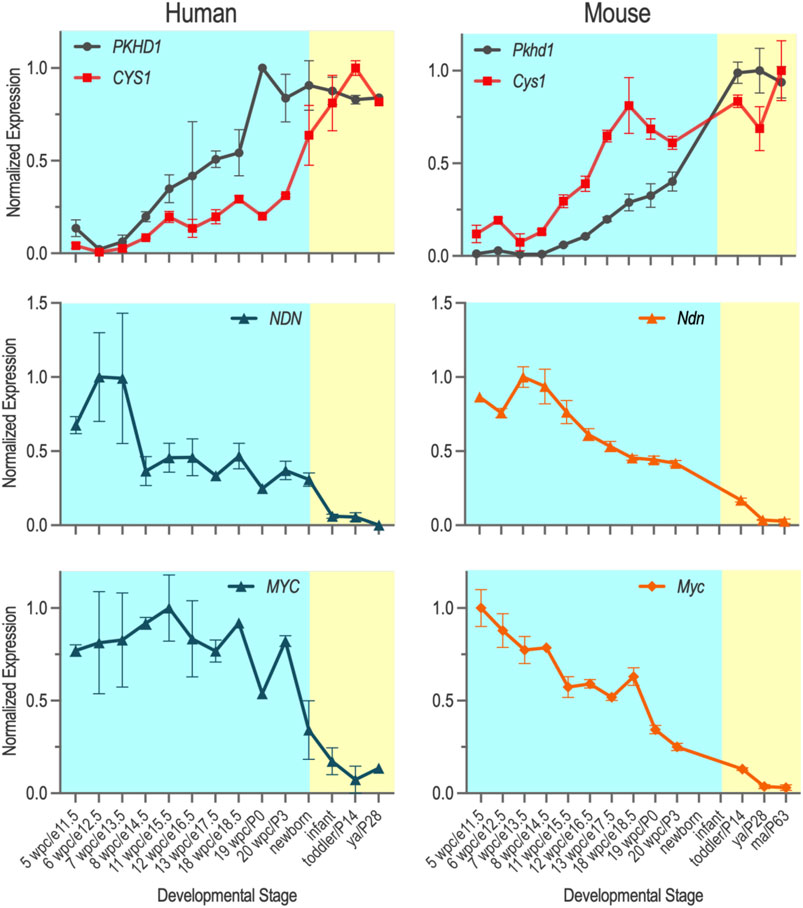
FIGURE 4. Temporal expression of PKHD1/Pkhd1, CYS1/Cys1, CYS1/Cys1 and MYC/Myc mRNAs during pre- and post-natal kidney development in human and mouse were obtained from Evo-devo mammalian organs (https://apps.kaessmannlab.org/evodevoapp/), normalized, and graphed using GraphPad Prism. Y-axis shows normalized gene expression levels. X-axis shows kidney developmental stages that correspond to each other in humans and mice (human/mouse).
With the assumption that mRNA expression serves as an appropriate proxy for protein levels, the species-specific differences in the PKHD1/Pkhd1 and CYS1/Cys1 developmental expression patterns and the observation that cystin may be protective of MYC/Myc activation suggest a mouse-specific renoprotective mechanism in mice lacking functional FPC. Therefore, we hypothesized that limiting cystin protein in kidneys from Pkhd1 mutant mice may evoke dilatation of renal tubules and/or collecting ducts. To address this hypothesis, we crossed the cpk allele into mice that are homozygous for the Pkhd1cyli mutation (cyli), an indel in Pkhd1 exon 48 that causes premature termination of protein translation (Yang et al., 2023). Kidneys from 10- and 12- month-old cyli/cyli;cpk/+ mice on a mixed genetic background (D.B/11Ei; C57BL/6J, Supplementary Table S2) had mild tubular dilations that were absent in kidneys from age-matched cyli/cyli;+/+ mice (Supplementary Figure S7).
Previous studies have demonstrated an association between MYC expression and renal cyst development in both human ADPKD and mouse PKD models (Trudel, 2015). MYC is overexpressed in cystic renal epithelial cells derived from ADPKD kidneys (Lanoix et al., 1996). Gene expression profiling studies demonstrated that the genes and pathways regulated by MYC are upregulated in kidneys from ADPKD patients (Husson et al., 2004; Song et al., 2009). Similar observations have been reported in mouse models of PKD (Trudel et al., 1998; Burtey et al., 2008; Kurbegovic and Trudel, 2013). Causality between MYC overexpression and renal cystogenesis is further suggested by observations in the SBM mouse model, in which a Myc transgene is driven by a β-globin promoter and SV40 enhancer (Trudel et al., 1991). Cystic kidney disease developed in transgenic mice overexpressing MYC, whereas mice that spontaneously lost the transgene did not develop renal cysts (Trudel et al., 1994). Additionally, cpk mice treated with Myc-antisense oligo exhibited reduced MYC protein expression, fewer renal cysts and improved renal function (Ricker et al., 2002).
In this study, we provide the first evidence that MYC is overexpressed in kidneys from patients with PKHD1-related ARPKD and confirm previous observations that MYC is upregulated in cystic renal epithelial cells from cpk kidneys (Ricker et al., 2002). In contrast, renal MYC expression levels were not altered in any of the Pkhd1 mutant mice that lack a significant renal cystic phenotype. Our findings extend the proposition that MYC upregulation is a driver of the renal cystogenesis (Kurbegovic and Trudel, 2020).
Loss of functional FPC has different phenotypic consequences in human and mouse kidneys. In patients with pathogenic PKHD1 sequence variants, even partial loss of FPC function can result in dramatic renal cystic disease (Cordido et al., 2021), suggesting that human FPC functions to maintain tubular integrity. In the absence of FPC, expression of MYC is aberrantly high leading to renal epithelial cell proliferation and cystogenesis. On the other hand, in mice lacking functional FPC, we show that Myc expression is not elevated and renal cysts are absent. A simple explanation could be that human PKHD1 gene is important for kidney development and MYC homeostasis, while mouse Pkhd1 plays a minimal role in nephrogenesis and Myc transcriptional regulation. This thesis is supported by a novel mouse line that was engineered to delete exon 67, which encodes most of the C-terminus, including the nuclear localization signal. Homozygous Pkhd1del67 mice do not have a cystic phenotype (Outeda et al., 2017). In addition, a recent report describes a new model derived from the Pkhd1del67 line such that exons 3-67 are deleted. Similar to homozygous Pkhd1del67 mice, homozygous Pkhd1del3-67 mutants do not express a renal cystic phenotype (Ishimoto et al., 2023). But is the explanation for the mouse-human phenotypic disparity so simple?
We and others have shown that mouse FPC-CTD is proteolytically cleaved through Notch-like processing (Kaimori et al., 2007; Follit et al., 2010; Cameron Varano et al., 2017). The FPC-CTD traffics into the nucleus and binds to double stranded DNA as a member of a ring-structure protein complex (Kaimori et al., 2007; Follit et al., 2010; Cameron Varano et al., 2017). However, it is not clear how FPC-CTD regulates gene expression, or which genes are regulated. To address this question and to explain the phenotypic disparity across species, we hypothesized that nuclear functions of hFPC-CTD and mFPC-CTD differ, particularly with respect to transcriptional regulation of MYC/Myc expression. As our experimental model, we employed in vitro overexpression to compare the nuclear trafficking and function of the human and mouse FPC-CTD.
Using immunofluorescence, reporter gene assays, and ChIP, we demonstrate that the mFPC-CTD traffics into the nucleus, binds to the Myc P1 promoter, and when overexpressed can activate Myc expression. In reporter gene assays, hFPC-CTD and mFPC-CTD have comparable functions; both activate MYC/Myc P1 promoter. However, we observed differences in cellular trafficking of these intracellular FPC fragments. While mFPC-CTD largely localized to the nucleus, hFPC-CTD remained associated with plasma membrane and localized in the cytoplasm, with translocation into nucleus only upon removal of CTS. This indicates that hFPC-CTD has the functional ability to activate MYC P1 promoter similar to mFPC-CTD, but the nuclear transport of these two proteins differs, suggesting that intracellular transport may in part explain the species-specific differences in function.
Our experimental findings regarding the FPC-CTD nuclear function raises a new conundrum about recessive PKD pathogenesis. We show that in ARPKD loss of function of human FPC-CTD (patients AR1, AR2, AR3, and AR8: Supplementary Table S1) is associated with aberrant overexpression of MYC in the cystic kidneys, suggesting that FPC functions as a negative regulator of MYC to prevent cystogenesis, as was previously shown for the pro-proliferative STAT3 (Dafinger et al., 2020). But our reporter assays indicate that hFPC-CTD upregulates MYC. It is important to note that this apparent paradox may reflect the difference between in vivo mechanisms where FPC-CTD-associated proteins may dictate specific regulatory function and reductionist reporter assays demonstrating the activation of a specific promoter by an overexpressed protein. Further studies will be necessary to decipher MYC/Myc transcriptional activation and inhibition during kidney development and how FPC-CTD, or the lack of it may contribute to MYC/Myc expression regulation.
While our studies provide novel information about the nuclear trafficking and function of human and mouse FPC-CTDs, a recent study identified mitochondrial targeting sequences in both mFPC-CTD and hFPC-CTD and demonstrated the trafficking of these proteins into the mitochondria (Walker et al., 2022). These developments imply that understanding both the nuclear and mitochondrial functions of human and mouse FPC-CTDs will be necessary to better define their roles in kidney cystogenesis.
Another key to human-mouse phenotype paradox may involve the CYS1/Cys1 gene. In the mouse, cystin negatively regulates MYC expression through binding to mouse NDN and preventing its activation of Myc expression (Wu et al., 2013). While the mechanism of negative regulation is not completely understood, cystin could either compete with mouse NDN for binding to the Myc P1 promoter, or alternatively, cystin and mouse NDN could form a complex that binds to the Myc P1 promoter and inhibits its activity (Wu et al., 2013). Our data mining revealed that Cys1 expression is upregulated before Pkhd1 during mouse kidney development. In cpk mice, cystogenesis is initiated in the distal portion of developing proximal tubules at e16.5–17.5 and continues after birth (Preminger et al., 1982; Nidess et al., 1984; Avner et al., 1987). The activation of Cys1 expression before Pkhd1 in the developing mouse kidney and suppression of Myc expression by cystin could explain the absence of renal cystic phenotype in mice with mutant Pkhd1.
To better understand FPC function during development, we extended our study to analyze gene orthologs encoding FPC across phyla. These analyses suggest that the CTD may be evolutionary innovation associated with vertebrate transition from aquatic to terrestrial life. We found a Pkhd1 orthologue in only one fish genome, the coelacanth L. chalumnae. This observation is consistent with a previous study that suggested the Pkhd1 paralogue, Pkhd1l1, is the ancestral gene because Pkhd1l1 gene is present in the Fugu (puffer fish) genome, but Pkhd1 is not (Hogan et al., 2003). PKHD1L1 and PKHD1 are similar; both encode proteins that have a large extracellular segment with similar arrangement of conserved structural domains (Supplementary Figure S5A) and 41.5% protein sequence identity (Hogan et al., 2003). However, their cytoplasmic segments are quite different: human and mouse PKHD1L1-encoded proteins have very short cytoplasmic tails, eight and six amino acids, respectively, while hFPC-CTD and mFPC-CTD have 192 and 184 amino acids (Hogan et al., 2003).
In addition, our informatic analysis revealed higher sequence conservation of the FPC extracellular than intracellular domain across phyla, which may in part explain the difference in nuclear trafficking between mFPC-CTD and hFPC-CTD. The mFPC-CTD has two predicted canonical NLSs, one of which has been experimentally validated (Hiesberger et al., 2006). In contrast, hFPC-CTD has only one predicted NLS. The difference in the number of NLSs in human and mouse FPC-CTD could explain the differences in the distribution of hFPC-CTD and mFPC-CTD in the nucleus versus other subcellular compartments, e.g., mitochondria. This finding raises an intriguing possibility that will require further study.
Finally, we used data mining to compare the temporal expression of PKHD1/Pkhd1 and CYS1/Cys1 in developing human and mouse kidneys respectively. In human kidneys, maximal expression of PKHD1 preceded CYS1. However, during mouse kidney development, Cys1 expression is upregulated before Pkhd1 (Figure 4). Assuming similar regulation of MYC/Myc gene expression by human and mouse cystin in association with NDN, our data suggest that 1) MYC/Myc expression can be activated by human and mouse NDN during early kidney development and factors that regulate MYC/Myc expression in later stages of kidney development differ in humans and mice; and 2) during middle to late stages of mouse nephrogenesis, Myc gene expression is downregulated by cystin in the mouse kidney but much less than in the human kidney. Therefore, the differences in the temporal expression pattens of CYS1/Cys1 could contribute to the relative renoprotection from cystogenesis in Pkhd1-deficient mice.
In summary, we provide the first report of elevated MYC levels in PKHD1-deficient human kidneys. In contrast, we show that MYC abundance is unaltered in non-cystic kidneys from Pkhd1-deficient mice. We demonstrate several key differences between human and mouse that may explain the relative renoprotection in Pkhd1-deficient kidneys: 1) differences in the number of NLS in the FPC-CTD; 2) differential impact of the human and mouse CTS on intracellular trafficking and subcellular distribution of the FPC-CTD; and 3) differences in the temporal expression of PKHD1/Pkhd1 and CYS1/Cys1 during nephrogenesis. In addition, we observed that reduced cystin levels in Pkhd1-deficient mice lead to renal tubular dilatation, suggesting that in mice both cystin and FPC-CTD are necessary to maintain renal tubular architecture. Given the limited sequence identity of human vs mouse FPC-CTD, we speculate that the cytosolic cleavage peptides may have different protein interacting partner(s) and these protein complexes may differentially regulate MYC/Myc expression in vivo. Taken together, our data extend previous observations and indicate that MYC dysregulation is a central driver of renal cystogenesis in both ARPKD and ADPKD.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The studies involving humans were approved by Institutional Review Board at the Children’s National Hospital or the University Hospital of Cologne. Human kidney samples were obtained from the NIDDK-funded UAB Childhood Cystic Kidney Disease Center Translational Resource at the University of Alabama at Birmingham and from the University Hospital of Cologne. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants legal guardians/next of kin. The animal study was approved by Institutional Animal Care and Use Committees at Children’s National Research Institute. The study was conducted in accordance with the local legislation and institutional requirements.
NH: Writing–original draft, Investigation, Visualization, Project administration, Validation, Conceptualization, Data curation, Formal Analysis. CY: Conceptualization, Data curation, Investigation, Project administration, Resources, Visualization, Writing–original draft, Validation. MW: Conceptualization, Data curation, Investigation, Writing–original draft. GT: Investigation, Validation, Writing–original draft, Conceptualization, Resources. HG-D: Formal Analysis, Writing–original draft. RT: Investigation, Resources, Writing–original draft. PB: Methodology, Project administration, Resources, Supervision, Writing–original draft, Conceptualization. AR: Investigation, Methodology, Project administration, Supervision, Writing–original draft, Conceptualization, Visualization. CD: Investigation, Writing–original draft. ML: Project administration, Resources, Supervision, Writing–review and editing, Conceptualization, Data curation, Visualization. ZB: Project administration, Writing–review and editing, Data curation. LC: Conceptualization, Data curation, Methodology, Project administration, Supervision, Visualization, Writing–review and editing, Formal Analysis, Investigation, Validation. LG-W: Conceptualization, Data curation, Funding acquisition, Methodology, Project administration, Resources, Supervision, Writing–review and editing, Validation, Visualization.
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by NIH grants (R01DK121530), PKD Foundation, and The Moran Family Foundation to LG-W.
The authors thank current and former members of the Guay-Woodford and PB laboratories for experimental support, review, and critical evaluation of the reported studies. We also thank the Baltimore PKD Center for the del3-4 and del67 mouse lines, Feng Qian for the pcDNA5/FRT/TO-mPkhd1 plasmid, Gregory G. Germino for del3-67 mouse, and Trenton R. Schoeb for cyli/cyli; cpk/+ renal histopathological analyses. Finally, we thank Marie Trudel for critically reviewing the manuscript and providing insightful suggestions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1270980/full#supplementary-material
Albert, T., Wells, J., Funk, J. O., Pullner, A., Raschke, E. E., Stelzer, G., et al. (2001). The chromatin structure of the dual c-myc promoter P1/P2 is regulated by separate elements. J. Biol. Chem. 276 (23), 20482–20490. doi:10.1074/jbc.M100265200
Alzarka, B., Morizono, H., Bollman, J. W., Kim, D., and Guay-Woodford, L. M. (2017). Design and implementation of the hepatorenal fibrocystic disease core center clinical database: a centralized Resource for characterizing autosomal recessive polycystic kidney disease and other hepatorenal fibrocystic diseases. Front. Pediatr. 5, 80. doi:10.3389/fped.2017.00080
Arensdorf, A. M., and Rutkowski, D. T. (2013). Endoplasmic reticulum stress impairs IL-4/IL-13 signaling through C/EBPβ-mediated transcriptional suppression. J. Cell Sci. 126 (Pt 17), 4026–4036. doi:10.1242/jcs.130757
Avner, E. D., Studnicki, F. E., Young, M. C., Sweeney, W. E., Piesco, N. P., Ellis, D., et al. (1987). Congenital murine polycystic kidney disease. I. The ontogeny of tubular cyst formation. Pediatr. Nephrol. 1 (4), 587–596. doi:10.1007/BF00853593
Battey, J., Moulding, C., Taub, R., Murphy, W., Stewart, T., Potter, H., et al. (1983). The human c-myc oncogene: structural consequences of translocation into the IgH locus in Burkitt lymphoma. Cell 34 (3), 779–787. doi:10.1016/0092-8674(83)90534-2
Bentley, D. L., and Groudine, M. (1986). Novel promoter upstream of the human c-myc gene and regulation of c-myc expression in B-cell lymphomas. Mol. Cell Biol. 6 (10), 3481–3489. doi:10.1128/mcb.6.10.3481
Bergmann, C. (2017). Genetics of autosomal recessive polycystic kidney disease and its differential diagnoses. Front. Pediatr. 5, 221. doi:10.3389/fped.2017.00221
Burtey, S., Riera, M., Ribe, E., Pennekamp, P., Passage, E., Rance, R., et al. (2008). Overexpression of PKD2 in the mouse is associated with renal tubulopathy. Nephrol. Dial. Transpl. 23 (4), 1157–1165. doi:10.1093/ndt/gfm763
Cameron Varano, A., Harafuji, N., Dearnaley, W., Guay-Woodford, L., and Kelly, D. F. (2017). Preparation of disease-related protein assemblies for single particle electron microscopy. Methods Mol. Biol. 1647, 185–196. doi:10.1007/978-1-4939-7201-2_12
Cardoso-Moreira, M., Halbert, J., Valloton, D., Velten, B., Chen, C., Shao, Y., et al. (2019). Gene expression across mammalian organ development. Nature 571 (7766), 505–509. doi:10.1038/s41586-019-1338-5
Cordido, A., Vizoso-Gonzalez, M., and Garcia-Gonzalez, M. A. (2021). Molecular pathophysiology of autosomal recessive polycystic kidney disease. Int. J. Mol. Sci. 22 (12), 6523. doi:10.3390/ijms22126523
Cowley, B. D., Chadwick, L. J., Grantham, J. J., and Calvet, J. P. (1991). Elevated proto-oncogene expression in polycystic kidneys of the C57BL/6J (cpk) mouse. J. Am. Soc. Nephrol. 1 (8), 1048–1053. doi:10.1681/ASN.V181048
Crooks, G. E., Hon, G., Chandonia, J. M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14 (6), 1188–1190. doi:10.1101/gr.849004
Dafinger, C., Mandel, A. M., Braun, A., Göbel, H., Burgmaier, K., Massella, L., et al. (2020). The carboxy-terminus of the human ARPKD protein fibrocystin can control STAT3 signalling by regulating SRC-activation. J. Cell Mol. Med. 24 (24), 14633–14638. doi:10.1111/jcmm.16014
Fagerberg, L., Hallström, B. M., Oksvold, P., Kampf, C., Djureinovic, D., Odeberg, J., et al. (2014). Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell Proteomics 13 (2), 397–406. doi:10.1074/mcp.M113.035600
Fliegauf, M., Frohlich, C., Horvath, J., Olbrich, H., Hildebrandt, F., and Omran, H. (2003). Identification of the human CYS1 gene and candidate gene analysis in Boichis disease. Pediatr. Nephrol. 18 (6), 498–505. doi:10.1007/s00467-003-1141-1
Follit, J. A., Li, L., Vucica, Y., and Pazour, G. J. (2010). The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J. Cell Biol. 188 (1), 21–28. doi:10.1083/jcb.200910096
Gabay, M., Li, Y., and Felsher, D. W. (2014). MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 4 (6), a014241. doi:10.1101/cshperspect.a014241
Gallagher, A. R., Esquivel, E. L., Briere, T. S., Tian, X., Mitobe, M., Menezes, L. F., et al. (2008). Biliary and pancreatic dysgenesis in mice harboring a mutation in Pkhd1. Am. J. Pathol. 172 (2), 417–429. doi:10.2353/ajpath.2008.070381
Garcia-Gonzalez, M. A., Menezes, L. F., Piontek, K. B., Kaimori, J., Huso, D. L., Watnick, T., et al. (2007). Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum. Mol. Genet. 16 (16), 1940–1950. doi:10.1093/hmg/ddm141
Gearhart, J., Pashos, E. E., and Prasad, M. K. (2007). Pluripotency redux--advances in stem-cell research. N. Engl. J. Med. 357 (15), 1469–1472. doi:10.1056/NEJMp078126
Guay-Woodford, L. M. (2003). Murine models of polycystic kidney disease: molecular and therapeutic insights. Am. J. Physiol. Ren. Physiol. 285 (6), F1034–F1049. doi:10.1152/ajprenal.00195.2003
Guay-Woodford, L. M., Bissler, J. J., Braun, M. C., Bockenhauer, D., Cadnapaphornchai, M. A., Dell, K. M., et al. (2014). Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J. Pediatr. 165 (3), 611–617. doi:10.1016/j.jpeds.2014.06.015
Hiesberger, T., Gourley, E., Erickson, A., Koulen, P., Ward, C. J., Masyuk, T. V., et al. (2006). Proteolytic cleavage and nuclear translocation of fibrocystin is regulated by intracellular Ca2+ and activation of protein kinase C. J. Biol. Chem. 281 (45), 34357–34364. doi:10.1074/jbc.M606740200
Hogan, M. C., Griffin, M. D., Rossetti, S., Torres, V. E., Ward, C. J., and Harris, P. C. (2003). PKHDL1, a homolog of the autosomal recessive polycystic kidney disease gene, encodes a receptor with inducible T lymphocyte expression. Hum. Mol. Genet. 12 (6), 685–698. doi:10.1093/hmg/ddg068
Hou, X., Mrug, M., Yoder, B. K., Lefkowitz, E. J., Kremmidiotis, G., D'Eustachio, P., et al. (2002). Cystin, a novel cilia-associated protein, is disrupted in the cpk mouse model of polycystic kidney disease. J. Clin. Invest. 109 (4), 533–540. doi:10.1172/JCI14099
Hu, B., He, X., Li, A., Qiu, Q., Li, C., Liang, D., et al. (2011). Cystogenesis in ARPKD results from increased apoptosis in collecting duct epithelial cells of Pkhd1 mutant kidneys. Exp. Cell Res. 317 (2), 173–187. doi:10.1016/j.yexcr.2010.09.012
Husson, H., Manavalan, P., Akmaev, V. R., Russo, R. J., Cook, B., Richards, B., et al. (2004). New insights into ADPKD molecular pathways using combination of SAGE and microarray technologies. Genomics 84 (3), 497–510. doi:10.1016/j.ygeno.2004.03.009
Ishimoto, Y., Menezes, L. F., Zhou, F., Yoshida, T., Komori, T., Qiu, J., et al. (2023). A novel ARPKD mouse model with near-complete deletion of the Polycystic Kidney and Hepatic Disease 1 (Pkhd1) genomic locus presents with multiple phenotypes but not renal cysts. Kidney Int. 104, 611–616. doi:10.1016/j.kint.2023.05.027
Kaimori, J. Y., Nagasawa, Y., Menezes, L. F., Garcia-Gonzalez, M. A., Deng, J., Imai, E., et al. (2007). Polyductin undergoes notch-like processing and regulated release from primary cilia. Hum. Mol. Genet. 16 (8), 942–956. doi:10.1093/hmg/ddm039
Karihaloo, A. (2015). “Role of inflammation in polycystic kidney disease,” in Polycystic kidney disease.
Katsuyama, M., Masuyama, T., Komura, I., Hibino, T., and Takahashi, H. (2000). Characterization of a novel polycystic kidney rat model with accompanying polycystic liver. Exp. Anim. 49 (1), 51–55. doi:10.1538/expanim.49.51
Kim, I., Fu, Y., Hui, K., Moeckel, G., Mai, W., Li, C., et al. (2008). Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J. Am. Soc. Nephrol. 19 (3), 455–468. doi:10.1681/ASN.2007070770
Kurbegovic, A., and Trudel, M. (2013). Progressive development of polycystic kidney disease in the mouse model expressing Pkd1 extracellular domain. Hum. Mol. Genet. 22 (12), 2361–2375. doi:10.1093/hmg/ddt081
Kurbegovic, A., and Trudel, M. (2020). The master regulators Myc and p53 cellular signaling and functions in polycystic kidney disease. Cell Signal 71, 109594. doi:10.1016/j.cellsig.2020.109594
Lanoix, J., D'Agati, V., Szabolcs, M., and Trudel, M. (1996). Dysregulation of cellular proliferation and apoptosis mediates human autosomal dominant polycystic kidney disease (ADPKD). Oncogene 13 (6), 1153–1160.
Lin, J. R., and Hu, J. (2013). SeqNLS: nuclear localization signal prediction based on frequent pattern mining and linear motif scoring. PLoS One 8 (10), e76864. doi:10.1371/journal.pone.0076864
Lin, J. R., Mondal, A. M., Liu, R., and Hu, J. (2012). Minimalist ensemble algorithms for genome-wide protein localization prediction. BMC Bioinforma. 13, 157. doi:10.1186/1471-2105-13-157
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 (4), 402–408. doi:10.1006/meth.2001.1262
Lu, H., Galeano, M. C. R., Ott, E., Kaeslin, G., Kausalya, P. J., Kramer, C., et al. (2017). Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 49 (7), 1025–1034. doi:10.1038/ng.3871
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48 (D1), D265-D268–d268. doi:10.1093/nar/gkz991
Masyuk, T. V., Huang, B. Q., Masyuk, A. I., Ritman, E. L., Torres, V. E., Wang, X., et al. (2004). Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycystic kidney disease. Am. J. Pathol. 165 (5), 1719–1730. doi:10.1016/s0002-9440(10)63427-x
Menezes, L. F., Cai, Y., Nagasawa, Y., Silva, A. M., Watkins, M. L., Da Silva, A. M., et al. (2004). Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int. 66 (4), 1345–1355. doi:10.1111/j.1523-1755.2004.00844.x
Moser, M., Matthiesen, S., Kirfel, J., Schorle, H., Bergmann, C., Senderek, J., et al. (2005). A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD). Hepatology 41 (5), 1113–1121. doi:10.1002/hep.20655
Nagao, S., Kugita, M., Yoshihara, D., and Yamaguchi, T. (2012). Animal models for human polycystic kidney disease. Exp. Anim. 61 (5), 477–488. doi:10.1538/expanim.61.477
Nagasawa, Y., Matthiesen, S., Onuchic, L. F., Hou, X., Bergmann, C., Esquivel, E., et al. (2002). Identification and characterization of Pkhd1, the mouse orthologue of the human ARPKD gene. J. Am. Soc. Nephrol. 13 (9), 2246–2258. doi:10.1097/01.asn.0000030392.19694.9d
Nevzorova, Y. A., Hu, W., Cubero, F. J., Haas, U., Freimuth, J., Tacke, F., et al. (2013). Overexpression of c-myc in hepatocytes promotes activation of hepatic stellate cells and facilitates the onset of liver fibrosis. Biochim. Biophys. Acta 1832 (10), 1765–1775. doi:10.1016/j.bbadis.2013.06.001
Nidess, R., Koch, W. E., Fried, F. A., McFarland, E., and Mandell, J. (1984). Development of the embryonic murine kidney in normal and congenital polycystic kidney disease: characterization of a proximal tubular degenerative process as the first observable light microscopic defect. J. Urol. 131 (1), 156–162. doi:10.1016/s0022-5347(17)50250-5
Onuchic, L. F., Furu, L., Nagasawa, Y., Hou, X., Eggermann, T., Ren, Z., et al. (2002). PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am. J. Hum. Genet. 70 (5), 1305–1317. doi:10.1086/340448
Outeda, P., Menezes, L., Hartung, E. A., Bridges, S., Zhou, F., Zhu, X., et al. (2017). A novel model of autosomal recessive polycystic kidney questions the role of the fibrocystin C-terminus in disease mechanism. Kidney Int. 92 (5), 1130–1144. doi:10.1016/j.kint.2017.04.027
Parrot, C., Kurbegovic, A., Yao, G., Couillard, M., Cote, O., and Trudel, M. (2019). c-Myc is a regulator of the PKD1 gene and PC1-induced pathogenesis. Hum. Mol. Genet. 28 (5), 751–763. doi:10.1093/hmg/ddy379
Preminger, G. M., Koch, W. E., Fried, F. A., McFarland, E., Murphy, E. D., and Mandell, J. (1982). Murine congenital polycystic kidney disease: a model for studying development of cystic disease. J. Urol. 127 (3), 556–560. doi:10.1016/s0022-5347(17)53911-7
Ray, D., Meneceur, P., Tavitian, A., and Robert-Lezenes, J. (1987). Presence of a c-myc transcript initiated in intron 1 in Friend erythroleukemia cells and in other murine cell types with no evidence of c-myc gene rearrangement. Mol. Cell Biol. 7 (2), 940–945. doi:10.1128/mcb.7.2.940
Ricker, J. L., Mata, J. E., Iversen, P. L., and Gattone, V. H. (2002). c-myc antisense oligonucleotide treatment ameliorates murine ARPKD. Kidney Int. 61 (1 Suppl. l), S125–S131. doi:10.1046/j.1523-1755.2002.0610s1125.x
Sharp, A. M., Messiaen, L. M., Page, G., Antignac, C., Gubler, M. C., Onuchic, L. F., et al. (2005). Comprehensive genomic analysis of PKHD1 mutations in ARPKD cohorts. J. Med. Genet. 42 (4), 336–349. doi:10.1136/jmg.2004.024489
Shen, Y., Miao, N., Wang, B., Xu, J., Gan, X., Xu, D., et al. (2017). c-Myc promotes renal fibrosis by inducing integrin αv-mediated transforming growth factor-β signaling. Kidney Int. 92 (4), 888–899. doi:10.1016/j.kint.2017.03.006
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7, 539. doi:10.1038/msb.2011.75
Song, X., Di Giovanni, V., He, N., Wang, K., Ingram, A., Rosenblum, N. D., et al. (2009). Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum. Mol. Genet. 18 (13), 2328–2343. doi:10.1093/hmg/ddp165
Steele, S. L., Wu, Y., Kolb, R. J., Gooz, M., Haycraft, C. J., Keyser, K. T., et al. (2010). Telomerase immortalization of principal cells from mouse collecting duct. Am. J. Physiol. Ren. Physiol. 299 (6), F1507–F1514. doi:10.1152/ajprenal.00183.2010
Tao, B., Bu, S., Yang, Z., Siroky, B., Kappes, J. C., Kispert, A., et al. (2009). Cystin localizes to primary cilia via membrane microdomains and a targeting motif. J. Am. Soc. Nephrol. 20 (12), 2570–2580. doi:10.1681/ASN.2009020188
Taub, R., Kirsch, I., Morton, C., Lenoir, G., Swan, D., Tronick, S., et al. (1982). Translocation of the c-myc gene into the immunoglobulin heavy chain locus in human Burkitt lymphoma and murine plasmacytoma cells. Proc. Natl. Acad. Sci. U. S. A. 79 (24), 7837–7841. doi:10.1073/pnas.79.24.7837
Trudel, M. (2015). “c-Myc signalling in the genetic mechanism of polycystic kidney disease,” in Polycystic kidney disease.
Trudel, M., Barisoni, L., Lanoix, J., and D'Agati, V. (1998). Polycystic kidney disease in SBM transgenic mice: role of c-myc in disease induction and progression. Am. J. Pathol. 152 (1), 219–229.
Trudel, M., Chretien, N., and D'Agati, V. (1994). Disappearance of polycystic kidney disease in revertant c-myc transgenic mice. Mamm. Genome 5 (3), 149–152. doi:10.1007/BF00352345
Trudel, M., D'Agati, V., and Costantini, F. (1991). C-myc as an inducer of polycystic kidney disease in transgenic mice. Kidney Int. 39 (4), 665–671. doi:10.1038/ki.1991.80
Walker, R., Yao, Q., Xu, H., Maranto, A., Swaney, K., Ramachandran, S., et al. (2022). Fibrocystin/Polyductin releases a C-terminal fragment that translocates into mitochondria and prevents cystogenesis. Res. Square. [PREPRINT (Version 1). doi:10.21203/rs.3.rs-2016158/v1
Wang, S., Zhang, J., Nauli, S. M., Li, X., Starremans, P. G., Luo, Y., et al. (2007). Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol. Cell Biol. 27 (8), 3241–3252. doi:10.1128/MCB.00072-07
Ward, C. J., Hogan, M. C., Rossetti, S., Walker, D., Sneddon, T., Wang, X., et al. (2002). The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 30 (3), 259–269. doi:10.1038/ng833
Ward, C. J., Yuan, D., Masyuk, T. V., Wang, X., Punyashthiti, R., Whelan, S., et al. (2003). Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 12 (20), 2703–2710. doi:10.1093/hmg/ddg274
Wierstra, I., and Alves, J. (2008). The c-myc promoter: still MysterY and challenge. Adv. Cancer Res. 99, 113–333. doi:10.1016/S0065-230X(07)99004-1
Williams, S. S., Cobo-Stark, P., James, L. R., Somlo, S., and Igarashi, P. (2008). Kidney cysts, pancreatic cysts, and biliary disease in a mouse model of autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 23 (5), 733–741. doi:10.1007/s00467-007-0735-4
Woollard, J. R., Punyashtiti, R., Richardson, S., Masyuk, T. V., Whelan, S., Huang, B. Q., et al. (2007). A mouse model of autosomal recessive polycystic kidney disease with biliary duct and proximal tubule dilatation. Kidney Int. 72 (3), 328–336. doi:10.1038/sj.ki.5002294
Wu, M., Yang, C., Tao, B., Bu, S., and Guay-Woodford, L. M. (2013). The ciliary protein cystin forms a regulatory complex with necdin to modulate Myc expression. PLoS One 8 (12), e83062. doi:10.1371/journal.pone.0083062
Xiong, H., Chen, Y., Yi, Y., Tsuchiya, K., Moeckel, G., Cheung, J., et al. (2002). A novel gene encoding a TIG multiple domain protein is a positional candidate for autosomal recessive polycystic kidney disease. Genomics 80 (1), 96–104. doi:10.1006/geno.2002.6802
Yang, C., Harafuji, N., Caldovic, L., Yu, W., Boddu, R., Bhattacharya, S., et al. (2023). Pkhd1(cyli/cyli) mice have altered renal Pkhd1 mRNA processing and hormonally sensitive liver disease. J. Mol. Med. Berl. 101 (9), 1141–1151. doi:10.1007/s00109-023-02351-2
Keywords: ARPKD, MYC, FPC, cystin, PKHD1, Cys1
Citation: Harafuji N, Yang C, Wu M, Thiruvengadam G, Gordish-Dressman H, Thompson RG, Bell PD, Rosenberg AZ, Dafinger C, Liebau MC, Bebok Z, Caldovic L and Guay-Woodford LM (2023) Differential regulation of MYC expression by PKHD1/Pkhd1 in human and mouse kidneys: phenotypic implications for recessive polycystic kidney disease. Front. Cell Dev. Biol. 11:1270980. doi: 10.3389/fcell.2023.1270980
Received: 01 August 2023; Accepted: 30 October 2023;
Published: 17 November 2023.
Edited by:
Jonathan R. Whitfield, Vall d’Hebron Institute of Oncology (VHIO), SpainReviewed by:
Robin Maser, University of Kansas Medical Center, United StatesCopyright © 2023 Harafuji, Yang, Wu, Thiruvengadam, Gordish-Dressman, Thompson, Bell, Rosenberg, Dafinger, Liebau, Bebok, Caldovic and Guay-Woodford. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa M. Guay-Woodford, Z3VheXdvb2RmbEBjaG9wLmVkdQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.