
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 12 October 2023
Sec. Stem Cell Research
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1270408
This article is part of the Research TopicNew Insights on Mechanisms that Regulate Male Germ Stem Cell Division and Fate in ChordataView all 4 articles
 Megan A. Gura1†
Megan A. Gura1† Myles A. Bartholomew1†
Myles A. Bartholomew1† Kimberly M. Abt1
Kimberly M. Abt1 Soňa Relovská2
Soňa Relovská2 Kimberly A. Seymour2
Kimberly A. Seymour2 Richard N. Freiman1,2*
Richard N. Freiman1,2*Prospermatogonia (ProSpg) link the embryonic development of male primordial germ cells to the healthy establishment of postnatal spermatogonia and spermatogonial stem cells. While these spermatogenic precursor cells undergo the characteristic transitions of cycling and quiescence, the transcriptional events underlying these developmental hallmarks remain unknown. Here, we investigated the expression and function of TBP-associated factor 4b (Taf4b) in the timely development of quiescent mouse ProSpg using an integration of gene expression profiling and chromatin mapping. We find that Taf4b mRNA expression is elevated during the transition of mitotic-to-quiescent ProSpg and Taf4b-deficient ProSpg are delayed in their entry into quiescence. Gene ontology, protein network analysis, and chromatin mapping demonstrate that TAF4b is a direct and indirect regulator of chromatin and cell cycle-related gene expression programs during ProSpg quiescence. Further validation of these cell cycle mRNA changes due to the loss of TAF4b was accomplished via immunostaining for proliferating cell nuclear antigen (PCNA). Together, these data indicate that TAF4b is a key transcriptional regulator of the chromatin and quiescent state of the developing mammalian spermatogenic precursor lineage.
Male fertility is dependent on a highly regulated series of developmental events that begin with primordial germ cell (PGC) specification in early fetal development and end with the exhaustion of an adult unipotent spermatogonial stem cell (SSC) population during old age (De Rooij and Grootegoed, 1998; Oatley and Brinster, 2008; Phillips et al., 2010). In mammals, PGC specification is achieved via BMP signals secreted from a small group of dorsally localized extra-embryonic cells to the posterior epiblast. This then induces the expression of the master transcription regulators PRDM1, PRDM14, and TFAP2C (Ohinata et al., 2005; Yamaji et al., 2008; Weber et al., 2010). While more heterogeneous in nature, several transcription factors mark and/or support adult SSC identity, including ID4, RHOX10, PAX7, and SALL4 (Oatley et al., 2011; Gassei and Orwig, 2013; Aloisio et al., 2014; Song et al., 2016; Fayomi and Orwig, 2018; Green et al., 2018; Shami et al., 2020). Prospermatogonia (ProSpg, also called ‘gonocytes’) are male germ cells that have differentiated past PGC specification and have the potential to differentiate into adult spermatogonia (Spg) or become part of the SSC pool which provides the long-term renewing capabilities of the testis. Several critical molecular events occur during ProSpg development. First, epigenetic marks that were erased in PGCs are re-established in ProSpg. Second, an initial pool of unipotent SSCs is thought to arise from the heterogeneous pool of developing ProSpg during this developmental window (Kubo et al., 2015; Hill et al., 2018). These events are necessary to initiate spermatogenesis in a timely manner and maintain it properly throughout adulthood. However, the underlying gene expression networks that correctly time and integrate these complex events are currently unknown.
Sequence-specific transcription factors are thought to recruit RNA Polymerase II (RNAPII) to specific core promoters via coactivators and the general transcription machinery. How the regulation of RNAPII transcription is achieved in ProSpg and how it is integrated with epigenetic reprogramming of the male germline genome at these developmental stages is unknown. We have identified a unique germ cell-enriched subunit of the general transcription factor TFIID, called TAF4b, that is required for proper long-term spermatogenesis in the mouse. Male mice lacking TAF4b become infertile after undergoing a first round of spermatogenesis; however, spermatogenesis can be re-established in the Taf4b-deficient testis following transplant of wildtype SSCs reflecting a functional adult SSC niche (Falender et al., 2005). More recent characterization of spermatogenesis in our Taf4b-deficient mice revealed that TAF4b is required for embryonic male germ cell development and helps regulate the delicate balance of SSC self-renewal and differentiation (Lovasco et al., 2015). In addition to studies of TAF4b in mice, human males who express a truncated version of the TAF4b protein are infertile owing to progressive azoospermia (Ayhan et al., 2014), and single nucleotide polymorphisms in human TAF4b have been linked to nonobstructive azoospermia (Xi et al., 2020). Thus, deciphering the developmental and molecular mechanisms of TAF4b in the mouse can improve our understanding and model important aspects of male reproductive development and fertility in humans.
To contextualize the progressive loss of Taf4b-deficient male germ cells during embryogenesis, we examined the spatial and temporal expression of TAF4b during embryonic testis development. We analyzed published RNA-seq data sets derived from E9.5-E18.5 Oct4-eGFP mice, in which germ cells were separated from somatic cell populations through fluorescence-activated cell sorting (FACS (Gura et al., 2020)). We determined that levels of Taf4b mRNA become progressively and significantly elevated in a germ cell-specific manner from embryonic day (E) 11.5 to E18.5. Germ cell-enriched TAF4b protein expression at E13.5 suggests it plays an important germ cell-specific role during ProSpg development (Gura et al., 2020). Mitotic stage (M) ProSpg are proliferative between E13.5 and E16.5, at which they then progress to transitional 1 (T1) ProSpg and enter a period of cellular quiescence. Soon after birth, T1 ProSpg become transitional 2 (T2) ProSpg by reentering the cell cycle until they become Spg at approximately postnatal day (P) 8 (McCarrey, 2013). Recent studies by Law et al., 2019, suggest that a subpopulation of T1 ProSpg acquires high Id4 expression, which marks a potential initial pool of SSCs (Law et al., 2019). However, the transcriptional logic underlying these ProSpg transitions remains unknown.
Here, we tested if and how TAF4b regulates critical transcriptional programs and cell states during early ProSpg development. First, we examined Taf4b expression in published single-cell (sc) RNA-seq data and confirmed it is enriched in quiescent T1 ProSpg. Next, we performed bulk RNA-seq on Taf4b-deficient quiescent ProSpg at E16.5 to reveal cell cycle and chromatin structure as top gene ontology (GO) categories modulated in the absence of TAF4b. For genomic mapping of TAF4b localization in T1 ProSpg, we employed cleavage under targets and release using nuclease (CUT&RUN), which identified 617 TAF4b peaks just upstream of numerous transcription start sites (TSSs). Distinctive GO categories from the CUT&RUN included RNA processing, chromatin modification, and cell cycle regulation. The most consistent DNA motifs within the TAF4b-bound promoters included Sp/Klf family and NFY binding sites; a remarkable similarity to our findings in embryonic mouse oocytes, and for the first time linking these ubiquitously-expressed transcription factors to the precise regulation of ProSpg development (Gura et al., 2022). At the cellular level, we determined that Taf4b-deficient ProSpg are delayed in their entry to quiescence, noting significant perturbation from controls at E14.5 and that these cells also display key cell cycle protein changes during ProSpg quiescence. Together, these data suggest that TAF4b is a dynamic and vital integrator of male germline transcription and chromatin states that promotes the timely quiescence of ProSpg required for further mammalian spermatogenic development.
Mice, homozygous for an Oct4-eGFP transgene (The Jackson Laboratory: B6; 129S4- Pou5f1tm2Jae/J), were mated for CUT&RUN collections. Mice, homozygous for an Oct4-eGFP transgene (The Jackson Laboratory: B6; 129S4-Pou5f1tm2Jae/J) and heterozygous for the Taf4b-deficiency mutation (in exon 12 of the 15 total exons of the Taf4b gene that disrupts the endogenous Taf4b gene), were mated for mRNA collections. Timed matings were estimated to begin at day 0.5 by evidence of a copulatory plug. The sex of the embryos was identified by confirming the presence or absence of testicular cords. Genomic DNA, isolated from tail biopsies using QIAGEN DNeasy Blood & Tissue Kits (Cat #: 69506), was used for PCR genotyping assays.
All animal protocols were reviewed and approved by Brown University Institutional Animal Care and Use Committee and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Gonads were dissected out of embryos into cold PBS.
Testes were gathered from embryonic mice (E13.5-E18.5), briefly fixed with 10% formalin and paraffin-embedded. The Molecular Pathology Core at Brown University completed tissue preparation and performed sectioning. After the serial sections were prepared, antigen retrieval was performed using a 0.1% antigen unmasking solution (Vector Laboratories, H-3300) in a steamer for 20 min. Samples were then permeabilized in 0.1% sodium citrate and Triton X-100 for 10 min, washed with 0.1% PBST (Triton X-100), and incubated in blocking buffer (PBS with 3% goat serum, 1% BSA, and 0.1% Triton X-100) for 1 hour. This was then followed by primary antibody incubation for 24 h at 4°C prepared in the blocking buffer. The samples were then washed in PBST, incubated with secondary antibody for 1 h at 37°C, washed in PBST, and mounted in DAPI-containing Vectashield Mounting Medium (Vector Laboratories H-1200, Burlingame, CA). Primary antibodies used were rat anti-TRA98 (1:100, abcam, ab82527), anti-KI67 rabbit mAb (1:100, Cell Signaling Technology D3B5, 9129S) and anti-PCNA (1:100, Cell Signaling Technology, 13110T). For immunofluorescence, the secondary antibodies used were Alexa Fluor 555-conjugated anti-rat IgG (1:500; abcam ab150158) and Alexa Fluor 488-conjugated anti-rabbit IgG (1:500; abcam ab150081). Images were all acquired at the same exposures and received the same immunofluorescence preparation. Visualization and counting were completed via blinded assessment of TRA98-expressing cells colocalized with MKI67 on a Zeiss Axio Imager M1 and Zeis software. A two-way ANOVA was used to determine significance using GraphPad Prism 9 version 9.3.1. for MacOS (GraphPad Software, Boston, Massachusetts United States, www.graphpad.com) with an n of 3-5 mice.
To dissociate gonadal tissue into a single-cell suspension, embryonic gonads were harvested and placed in 0.25% Trypsin/EDTA and incubated at 37°C for 15 and 25 min for E14.5 and E16.5 gonads, respectively, as previously described (Gura et al., 2020). Eppendorf tubes were flicked to dissociate tissue halfway through and again at the end of the incubation. Trypsin was neutralized with FBS. Cells were pelleted at 1,500 RPM for 5 min, the supernatant was removed, and cells were resuspended in 100 μL PBS. The cell suspension was strained through a 35 μm mesh cap into a FACS tube (Gibco # 352235). Propidium iodide (1:500) was added to the cell suspension as a live/dead distinguishing stain. Fluorescence-activated cell sorting (FACS) was performed using a Becton Dickinson FACSAria III in the Flow Cytometry and Cell Sorting Core Facility at Brown University. A negative control of a non-transgenic mouse gonad was used for each experiment to establish an appropriate GFP signal baseline. Dead cells were discarded and the remaining cells were sorted into GFP+ and GFP− samples in PBS at 4°C for each embryo.
For RNA-seq analysis, GFP+ cells from each individual embryo were kept in separate tubes and were then spun down at 1,500 RPM for 5 min, had PBS removed, and were then resuspended in Trizol (ThermoFisher # 1556026). If samples had roughly less than 50 µL of PBS in the tube, Trizol was added immediately. The number of cells for each sample can be found in Supplementary Table S1. Samples were stored at −80°C.
For CUT&RUN, germ cells from all the gonads were pooled prior to FACS. Sorted germ cells were then spun down at 1,500 RPM for 5 min and were resuspended in 300 µL of PBS, then split into three Eppendorf tubes. These three tubes of germ cells were then used for CUT&RUN. The number of cells for each sample were as follows: Replicate 1 germ cell samples had approximately 56,000 cells per tube (obtained from 22 embryos) and Replicate 2 germ cell samples had approximately 131,000 cells per tube (obtained from 28 embryos).
All computational scripts regarding single cell RNA-seq (scRNA-seq) used in this publication are available to the public: https://github.com/mg859337/Gura_et_al._TAF4b_male_transcription. SRP194420, SRP158811, and SRP178196 were downloaded from NCBI SRA onto Brown University’s high-performance computing cluster at the Center for Computation and Visualization. We used Law et al. and Nguyen et al., which enriched for germ cells via FACS sorting (Law et al., 2019; Nguyen et al., 2020). We then combined the data with Tan et al., who collected the whole testis, thus establishing our comprehensive observation window from E12.5 to P7 (Tan et al., 2020). The fastq files were aligned using Cell Ranger (v 6.0.0) count and then aggregated using Cell Ranger aggr. The cloupe file created from Cell Ranger aggr was used as input for Loupe Cell Browser (v 5.0). We selected the germ cells by first assessing the log-normalized expression of germ cell marker Dazl within the complete 71,584 cell data set. Then, using unbiased clustering, we eliminated somatic cell clusters based on their mean Dazl expression being less than 0.85. The data set underwent further quality control and filtering based on fitting the metrics of 12.5–15.8 log2-normalized unique molecular identifiers, 10.6–12.8 log2-normalized features, and lastly, less than 24% mitochondrial transcript ratio (Quatredeniers et al., 2023).
Embryonic germ cells resuspended in Trizol were shipped to GENEWIZ (GENEWIZ Inc., NJ) on dry ice. Sample RNA extraction, sample QC, library preparation, sequencing, and initial bioinformatics were done at GENEWIZ. RNA was extracted following the Trizol Reagent User Guide (ThermoFisher Scientific). Glycogen was added (1 μL, 10 mg/mL) to the supernatant to increase RNA recovery. RNA was quantified using Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, CA, United States) and RNA integrity was checked with TapeStation (Agilent Technologies, Palo Alto, CA, United States) to see if the concentration met the requirements.
SMART-Seq v4 Ultra Low Input Kit for Sequencing was used for full-length cDNA synthesis and amplification (Clontech, Mountain View, CA), and Illumina Nextera XT Library Preparation Kit was used for library preparation. The sequencing libraries were multiplexed and clustered on a lane of a flowcell. After clustering, the flowcell was loaded onto an Illumina HiSeq 4000 according to manufacturer’s instructions. The samples were sequenced using a 2 × 150 Paired End (PE) configuration. Image analysis and base calling were conducted by the HiSeq Control Software (HCS) on the HiSeq instrument. Raw sequence data (.bcl files) generated from Illumina HiSeq were converted into fastq files and de-multiplexed using bcl2fastq (v 2.17). One mismatch was allowed for index sequence identification.
All computational scripts regarding RNA-seq used in this publication are available to the public: https://github.com/mg859337/Gura_et_al._TAF4b_male_transcription. All raw fastq files were initially processed on Brown University’s high-performance computing cluster. Reads were quality-trimmed and had adapters removed using Trim Galore! (v 0.5.0) with the parameters–nextera -q 10. Samples before and after trimming were analyzed using FastQC (v 0.11.5) for quality and then aligned to the Ensembl GRCm38 using HiSat2 (v 2.1.0) (Andrews, 2010; Pertea et al., 2016). Resulting sam files were converted to bam files using Samtools (v 1.9) (Li et al., 2009).
To obtain TPMs for each sample, StringTie (v 1.3.3b) was used with the optional parameters -A and -e. A gtf file for each sample was downloaded and, using RStudio (R v 4.0.2), TPMs of all samples were aggregated into one comma separated (csv) file using a custom R script. To create interactive Microsoft Excel files for exploring the TPMs of each dataset: the csv of aggregated TPMs was saved as an Excel spreadsheet (Supplementary Table S2). Colored tabs were added to set up different comparisons, and a flexible Excel function was created to adjust to gene name inputs. To explore the Excel files, please find the appropriate tab (named “Quick_Calc”) and type in the gene name of interest into the highlighted yellow boxes.
To obtain count tables, featurecounts (Subread v 1.6.2) was used (Liao et al., 2014). Metadata files for each dataset were created manually in Excel and saved as a csv. These count tables were used to create PCA plots by variance-stabilizing transformation (vst) of the data in DESeq2 (v 1.22.2) and plotting by ggplot2 (v 3.1.0) (Love et al., 2014; Wickham, 2016). DESeq2 was also used for differential gene expression analysis, where count tables and metadata files were used as input. We accounted for the litter batch effect in our mouse germ cells by setting it as a batch parameter in DESeq2. For the volcano plot, the output of DESeq2 was used and plotted using ggplot2. DEG lists were used for ClusterProfiler (v 3.16.1) input to create dotplots of significantly enriched gene ontology (GO) categories for all DEGs. The highest-confidence protein-protein interactions were identified using STRING, with unconnected proteins not shown in the images (Szklarczyk et al., 2019). Net plots were constructed using Bioconductor packages in R after normalization and QC. The top three GO processes category nets for both Up and Downregulated DEGs were cross-referenced with the TAF4b-bound gene TSS list.
The CUT&RUN performed in E16.5 germ cells followed the protocol in Hainer and Fazzio, 2019 (Hainer and Fazzio, 2019). CUT&RUN antibodies were as follows: polyclonal rabbit TAF4b (as previously described (Grive et al., 2016)), monoclonal rabbit H3K4me3 (EMD Millipore # 05-745R), rabbit IgG (ThermoFisher # 02–6102), pA-MNase was a generous gift from Dr. Thomas Fazzio, UMASS Med. For library preparation, the KAPA HyperPrep Kit (Roche Cat. No 07962363001) was used with New England Biolabs NEBNext Multiplex Oligos for Illumina (NEB #E7335). After library amplification through PCR, libraries were size-selected through gel extraction (∼150–650 bp) and cleaned up using the QIAGEN QIAquick Gel Extraction Kit (Cat. # 28704). CUT&RUN libraries in EB buffer were shipped to GENEWIZ (GENEWIZ Inc., NJ) on dry ice. Sample QC, sequencing, and initial bioinformatics were done at GENEWIZ.
The sequencing libraries were validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, United States), and quantified by using Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) as well as by quantitative PCR (KAPA Biosystems, Wilmington, MA, United States). The sequencing libraries were clustered on flowcells. After clustering, the flowcells were loaded onto the Illumina HiSeq instrument (4,000 or equivalent) according to manufacturer’s instructions. The samples were sequenced using a 2 × 150bp Paired End (PE) configuration. Raw sequence data (.bcl files) generated from Illumina HiSeq were converted into fastq files and de-multiplexed using bcl2fastq (v. 2.20). One mismatch was allowed for index sequence identification.
All computational scripts regarding CUT&RUN data analysis used in this publication are available at: https://github.com/mg859337/Gura_et_al._TAF4b_male_transcription and based on other CUT&RUN publications (Hainer and Fazzio, 2019). All raw fastq files were initially processed on Brown University’s high-performance computing cluster. Reads were quality-trimmed and had adapters removed using Trim Galore! (v 0.5.0) with the parameter -q 10 (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/). Samples before and after trimming were analyzed using FastQC (v 0.11.5) for quality and then aligned to the Ensembl GRCm39 using Bowtie2 (v 2.3.0). Fastq screen (v 0.13.0) was used to determine the percentage of reads uniquely mapped to the mouse genome in comparison to other species. Resulting sam files were converted to bam files, then unmapped, duplicated reads, and low quality mapped were removed using Samtools (v 1.9). Resulting bam files were split into size classes using a Unix script. For calling peaks, annotating peaks, and identifying coverage around TSSs, Homer (v 4.10) was used (Heinz et al., 2010). For gene track visualization, the final bam file before splitting into size classes was used as input to Integrative Genomics Viewer (IGV) (Robinson et al., 2011). A custom genome was created using a genome fasta and gtf file for Ensembl GRCm39.
Pie charts were created using data from Homer output and Venn diagrams were created using BioVenn. Dotplots of Promoter-TSS peaks were made using ClusterProfiler. TSS plots were created using the “tss” function of Homer and plotted using Microsoft Excel. All plots produced in RStudio were saved as an EPS file type and then opened in Adobe Illustrator in order to export a high-quality JPEG image. Excel file of the composite CUT&RUN replicates is presented as several unique tabs in Supplementary Table S3.
Analyzing single-cell RNA-seq data allowed us to examine the gene expression within ProSpg at finer resolution, which we reprocessed from published publicly available data sets (Tan et al., 2020; Gura et al., 2022; Quatredeniers et al., 2023). In our analysis, we examined the expression profile of Taf4b over time to evaluate what genes and biological processes are possibly modulated concurrently or downstream of TAF4b expression. After applying our computational workflow (see Methods), the complete data set consisted of 17,310 germ cells in our uniform manifold approximation and projection (UMAP) (Figure 1A). We then assessed the expression of Taf4b compared to its paralog Taf4a, which is ubiquitously expressed in somatic cells. Across all time points, Taf4a mRNA expression was notably lower than Taf4b mRNA expression (Figures 1B,C). When observing the expression of Taf4b mRNA over the E12.5-P7 time course, we detected that Taf4b expression peaks at P0 (Figure 1C). The dynamic range of Taf4b-expressing cells across time suggests Taf4b may act more akin to a dimmer switch than on/off regulation. Because previous experiments demonstrated that the Taf4b-deficient gonad exhibited changes in germ cell proliferation, we compared this newly characterized Taf4b expression profile to known proliferation markers, such as Mki67 (Sun and Kaufman, 2018). We saw an inverse expression pattern of Mki67 and Taf4b, where the significantly reduced expression of Mki67 between E16.5-P0 points to the characteristic quiescent period of T1 ProSpg (Figure 1D). The absence of Mki67 transcripts helps illustrate quiescence because its mRNA is expressed during S phase up until mitotic exit. Plk1 mRNA, another marker of cell proliferation, is decreased similarly from E16.5 to P2 (Figure 1E) (Liu et al., 2017). The combined decreased markers of proliferation and increased Taf4b expression led us to hypothesize that Taf4b may play a role in the cell cycle transitions during ProSpg development.
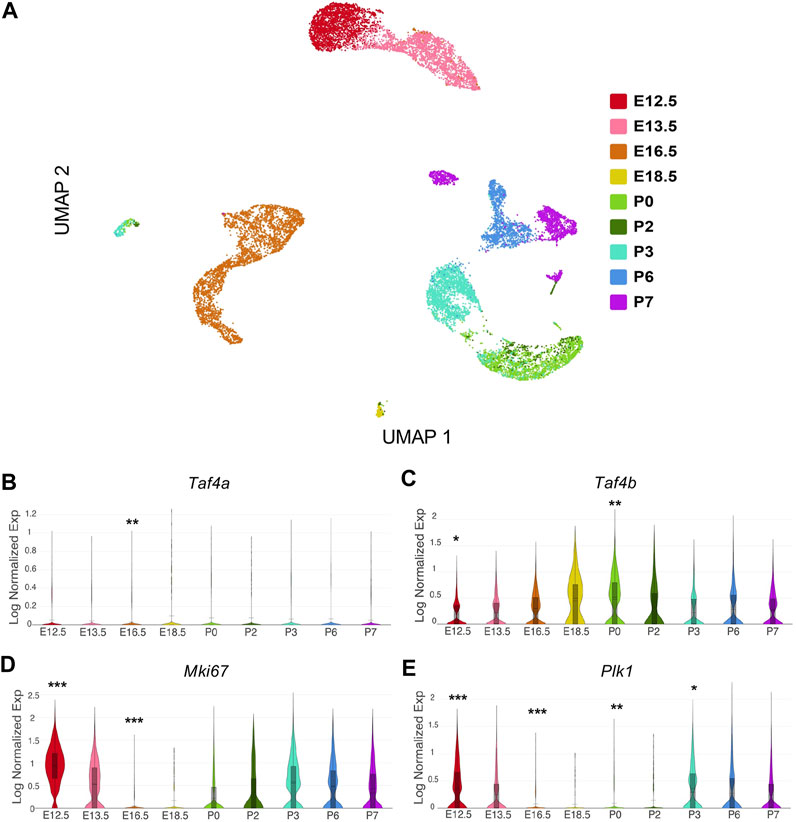
FIGURE 1. Characterizing Taf4b mRNA expression across male germ cell development in mouse. (A) UMAP of E12.5-P7 germ cells colored by time point. (B–E) Expression of Taf4a, Taf4b, Mki67, and Plk1 across the developmental time course. p-values are denoted (*** = p< 0.001, ** = p< 0.01, * = p< 0.05) and are adjusted using the Benjamini-Hochberg correction method for multiple tests.
We were intrigued to observe a clear increase in Taf4b mRNA during the ProSpg transition to quiescence. To better understand the potential function of TAF4b during this transition, we compared global mRNA differences between Taf4b-wildtype, -heterozygous, and -deficient ProSpg at E16.5. We first generated mouse embryos that were transgenic for Oct4-eGFP and either wildtype, heterozygous, or deficient for Taf4b, then sorted for GFP+ germ cells from the male embryonic testis. This provided germ cell-isolated samples from all three genotypes to perform bulk RNA-seq. Our ProSpg RNA-seq samples consisted of 7 Taf4b-wildtype, 9 Taf4b-heterozygous, and 8 Taf4b-deficient E16.5 embryos from six different litters (Supplementary Table S1). Although Taf4b-heterozygous male mice do not display overt signs of impaired spermatogenesis (Falender et al., 2005), including them as a unique hypomorphic and genotypic group was important. A PCA plot of these samples indicates that samples cluster primarily by genotype. While Taf4b-wildtype (WT) replicates display the tightest clustering, they do overlap with the Taf4b-heterozygous (Het) replicates, indicating a shared transcriptional profile (Figure 2A). This was expected as both genotypes are fertile. In contrast, the Taf4b-deficient (Def) replicates were much more dispersed and non-overlapping with the other two genotypes, suggesting large-scale transcriptional dysregulation. There were three Def samples that clustered more closely with the WT and Het samples. However, we completed the downstream analysis with all replicates because there were subtle but statistically significant transcriptional differences in these three samples compared to WT/HET. Overall, we detected 4551 DEGs between the Taf4b-wildtype and -deficient ProSpg; 3820 were defined as protein-coding with an adjusted p-value <0.05 (Figure 2B). Some genes noted to be dysregulated were Mki67 and Plk1 (Liu et al., 2017; Sun and Kaufman, 2018). These markers of proliferation should decrease during this quiescent time point but are significantly upregulated in the Taf4b-deficient ProSpg (Figure 2B). Using gene ontology (GO) of the DEGs, we determined which biological processes are disrupted in the absence of Taf4b. This GO analysis indicated cell cycle regulation is the primary disruption driven by Taf4b ablation, followed by chromatin modification and gland development. (Figures 2C,D).
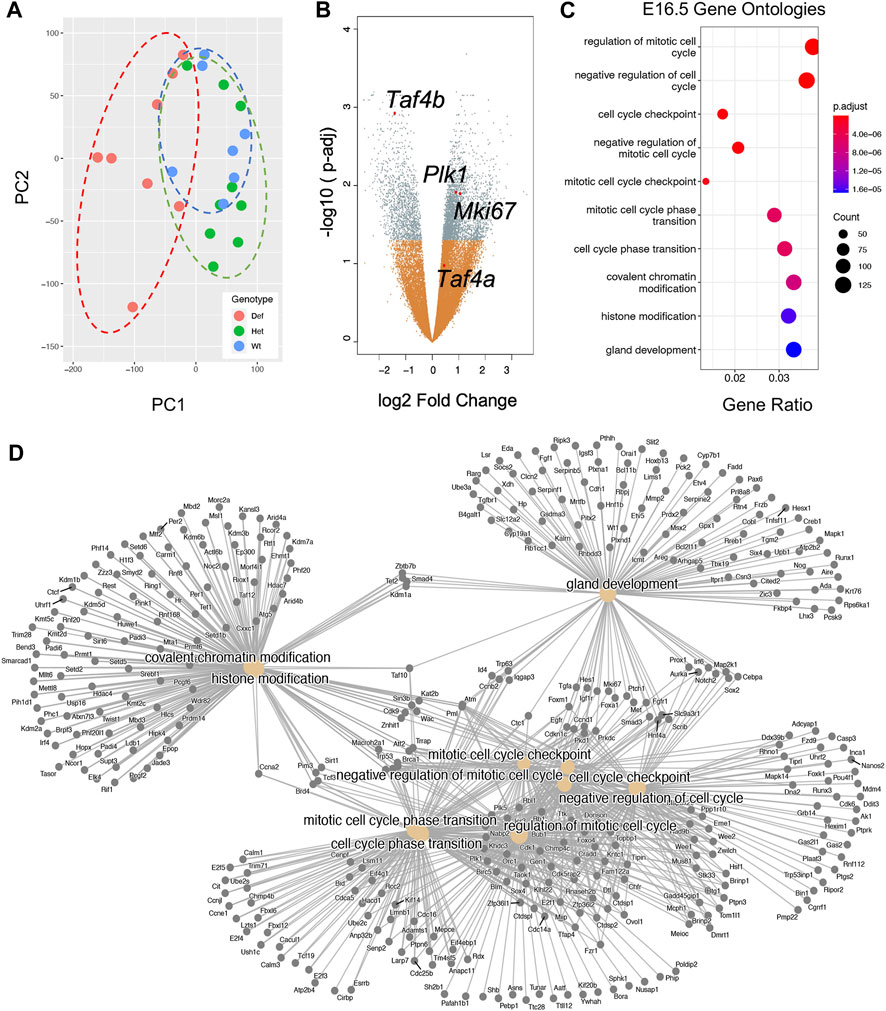
FIGURE 2. E16.5 ProSpg RNA-seq experiment reveals significant regulation of mitotic gene programs. (A) PCA plot of the E16.5 samples labeled based on Taf4b genotype as indicated by color and dotted lines. (B) Volcano plot of genes; the 4551 significant genes (p-adj <0.05) are labeled in grey, DEGs of interest plus Taf4b are specified and nonsignificant gene changes are labeled in orange. (C) Dotplot of GO biological process analysis of all 3820 protein-coding DEGs from (B). (D) Category netplot showing the relationships between the top ten GO biological processes from (C) and the individual DEGs within each ontology.
After observing global transcriptional dysregulation with 1,474 downregulated genes and 3077 upregulated genes, we sought to parse out what direction particular biological processes are due to Taf4b ablation. Of the 1,259 downregulated genes that were protein-coding, the top three biological process gene ontology categories were cilium movement, negative regulation of the immune system process, and cell killing (Figures 3A,C). From the upregulated genes, 2,561 were protein-coding, and we saw that the biological process categories are primarily related to cell cycle, DNA repair, and covalent chromatin modification (Figures 3B,D). Surprisingly, there were almost twice as many upregulated genes than downregulated genes in the absence of TAF4b, many of which encoded critical components of the cell cycle and chromatin regulatory machinery.
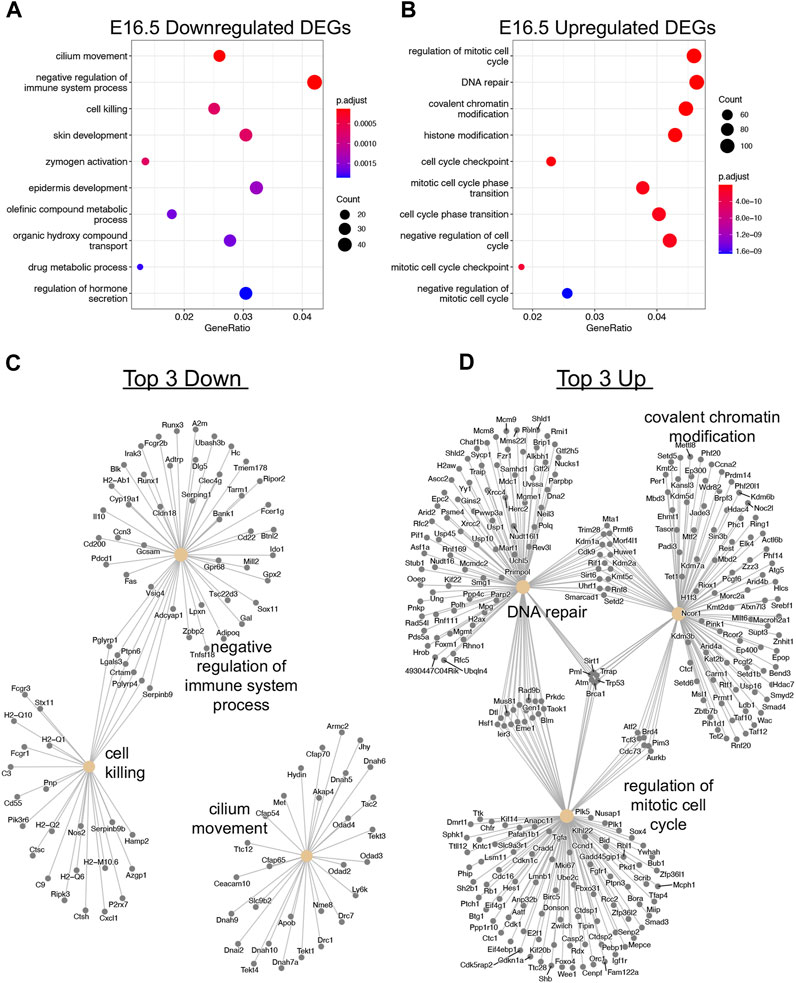
FIGURE 3. Transcriptional dysregulation of cell cycle and chromatin modification genes. (A) Dotplot of GO biological process analysis of 1,259 protein-coding Down DEGs. (B) Dotplot of GO biological process analysis of 2,561 protein-coding Up DEGs. (C) A category netplot composed of the top three downregulated biological processes from (A) and the individual DEGs within each ontology. (D) A category netplot composed of the top three upregulated biological processes from (B) and the individual DEGs within each ontology.
Mitotic ProSpg enter a characteristic period of quiescence between E13.5 and E16.5. Based upon our scRNA-seq and bulk RNA-seq data, we hypothesized that TAF4b modulated some aspect of ProSpg quiescence. We performed immunofluorescence on Taf4b-wildtype and Taf4b-deficient testis sections to detect TRA98 (a germ cell marker) and MKi67 (a marker of cell proliferation) from E13.5 to E18.5 (Grive et al., 2014; Sun and Kaufman, 2018). Representative images of each genotype and time point are shown (Figure 4A), as well as the quantification of the MKi67- quiescent wildtype and Taf4b-deficient ProSpg (indicated by the TRA98-only signal) at each timepoint (Figure 4B). At E13.5, the majority of Taf4b-deficient M ProSpg were still proliferating as indicated by the nuclear localization of MKi67 (Figure 4A). At E14.5, there is a significant reduction of ProSpg entering quiescence in the Taf4b-deficient compared to wildtype controls (Figure 4B). Interestingly, the percentage of quiescent cells at both E16.5 to E18.5 did not differ significantly between controls and Taf4b-deficient Pro-Spg. These data indicate that although Taf4b-defcient ProSpg can largely achieve quiescence, they struggle with its timely entry and this transition. We also compared staining for the proliferating cell nuclear antigen (PCNA), which is a DNA replication and repair protein, across matched Taf4b-wildtype and Taf4b-deficient testis sections (Figure 4C). At E16.5, there is a significant increase in the percent of Taf4b-deficient ProSpg expressing PCNA compared to matched wildtype controls and this difference is non-significant at E18.5 (Figure 4D). Taken together, these data indicate that Taf4b-deficient ProSpg are altered in their timing of ProSpg quiescence entry.
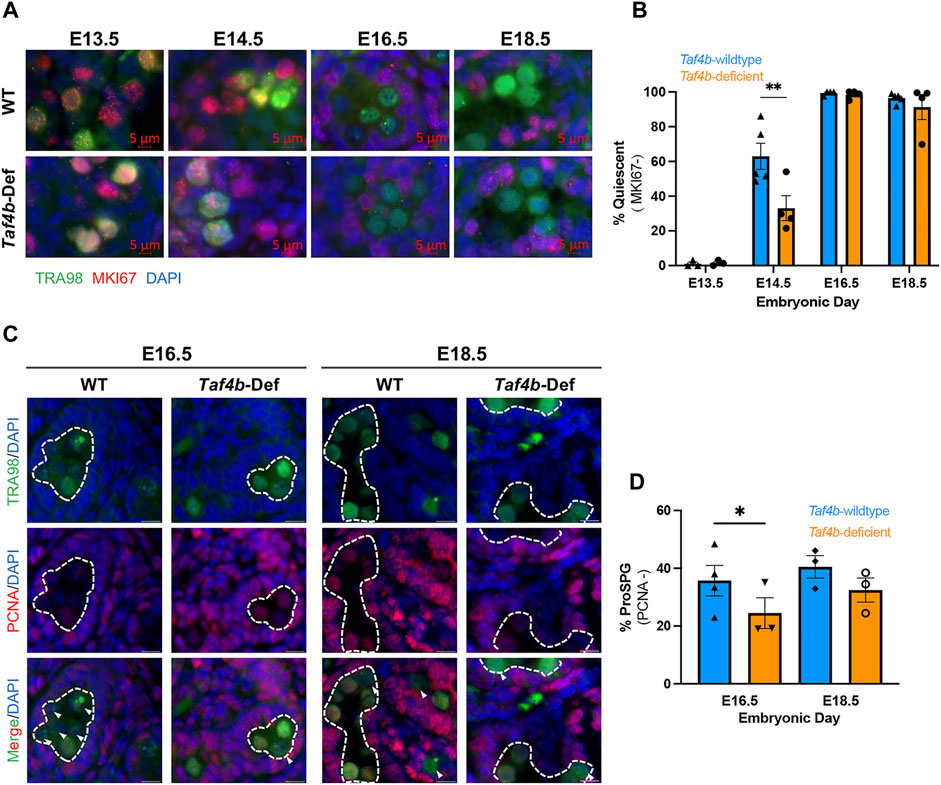
FIGURE 4. Taf4b-deficient ProSpg have increased MKI67 at E14.5. (A) Immunofluorescence of E13.5-E18.5 testis sections comparing Taf4b-wildtype (TAF4b +/+) to Taf4b-deficient ProSpg (TAF4b −/−). Antibodies for TRA98 (green), MKI67 (red) were used, and DAPI labels DNA. (B) Quantification of MKI67-/TRA98+ cells in Taf4b-wildtype (blue) and Taf4b-deficient (orange) testis sections. Taf4b-deficient ProSpg have significantly fewer MKI67- ProSpg than wildtype at E14.5 (n = 3-5 mice, ** = p-adj< 0.01 as determined by two-way ANOVA). (C) Immunofluorescence of E16.5-E18.5 testis sections comparing Taf4b-wildtype (TAF4b +/+) to Taf4b-deficient ProSpg (TAF4b −/−). Antibodies for TRA98 (green), PCNA (red) were used, and DAPI (blue) labels DNA. (D) Quantification of PCNA−/TRA98+ cells in Taf4b-wildtype (blue) and Taf4b-deficient (orange) testis sections. Taf4b-deficient ProSpg have significantly fewer PCNA− ProSpg than wildtype at E16.5 (n = 3-4 mice, * = p-value< 0.05 as determined by paired t-test).
To distinguish which DEGs identified in our E16.5 bulk RNA-seq experiment were direct targets of TAF4b, we performed Cleavage Under Targets and Release Using Nuclease (CUT&RUN), a genome mapping technique to identify enrichment of specific proteins and histone modifications in the genome (Skene and Henikoff, 2017; Hainer and Fazzio, 2019). We isolated two replicates of wildtype Oct4-eGFP E16.5 male germ cells using FACS and examined the genomic localization of TAF4b. H3K4me3 served as a positive control and marked active promoters, and IgG served as a negative control. CUT&RUN data analysis using the program Homer identified 64,891 H3K4me3 peaks and 7,861 TAF4b peaks in Replicate 1 and 1,730 H3K4me3 peaks and 848 TAF4b peaks in Replicate 2. The differences in the number of peaks called can be attributed to stochastic differences in sample preparation and starting material. However, it is necessary to note that these differences did not affect the assay’s specificity because we still see the same top motifs and the greater than 80% overlap between each replicate. We also found that 73% and 88% of TAF4b peaks were classified as localizing to promoters/transcription start site (“promoter-TSS”) for Replicate 1 and 2, respectively (Figure 5A). Importantly, 617 TAF4b peaks overlapped between the two replicates allowing us to perform GO analysis of the shared TAF4b-bound gene promoter-TSSs between the two replicates. These top five categories include mRNA processing, proteasomal degredation, RNA splicing, covalent chromatin modification, and negative regulation of the cell cycle (Figure 5B). When plotting the enrichment profile of TAF4b relative to TSSs, we found the highest TAF4b enrichment was located just upstream of the TSS (Figure 5C). This initial chromatin mapping of TAF4b indicates that the majority of the binding sites for TAF4b are just upstream of the TSS which is consistent with where TFIID is known to bind to core promoters.
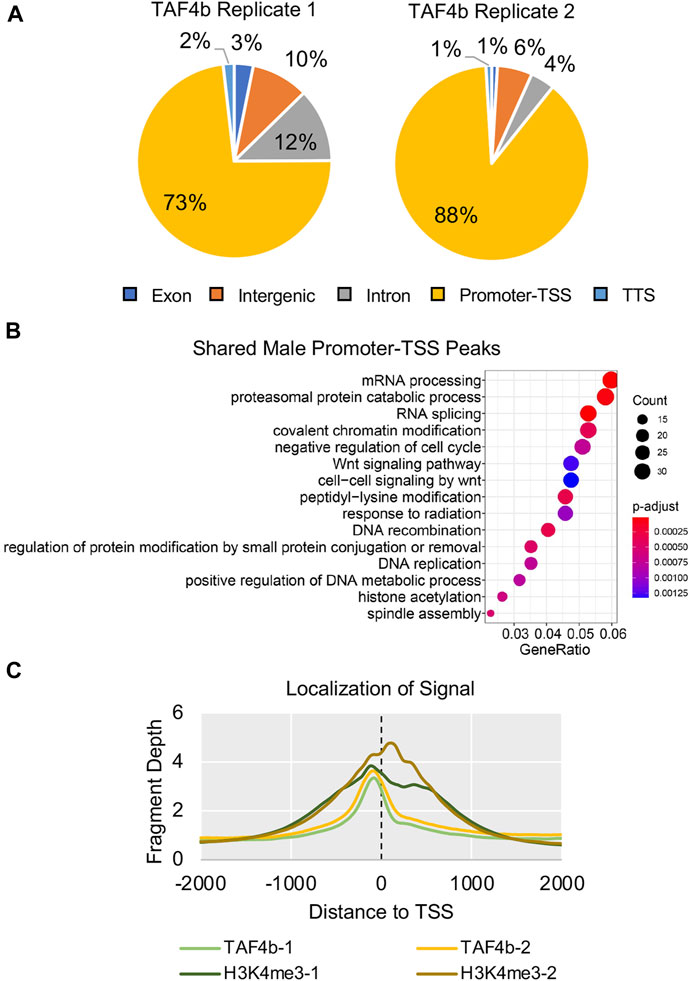
FIGURE 5. Genome-wide Occupancy of H3K4me3 and TAF4b in E16.5 ProSpg. CUT& RUN of E16.5 ProSpg using antibodies for H3K4me3 and TAF4b. (A) Pie charts of TAF4b peak locations across the E16.5 ProSpg genome in two independent CUT&RUN replicates. (B) GO biological process dotplot for the shared CUT&RUN peaks categorized as “promoter-TSS”. (C) Average enrichment of TAF4b and H3K4me3 signal near TSSs (dotted line) for each replicate.
We then explored the motifs associated with TAF4b-enriched peaks within our CUT&RUN data and found consistent enrichment of CCAAT and GC motifs, which are associated with the NFY complex and the Sp/Klf transcription factor family, respectively (Figures 6A,B) (Suske, 2017). Importantly, these motifs were shared between the two CUT&RUN replicates. We then compared DEGs from our RNA-seq experiment to direct targets of TAF4b from the CUT&RUN to identify a subset of genes directly modulated by TAF4b at E16.5. When comparing our DEGs to the “promoter-TSS” peaks of TAF4b, we found 876 DEGs that had at least one peak near their TSS. GO analysis of these 876 TAF4b bound and differentially expressed genes reveals that the most notable biological process affected is “regulation of the mitotic cell cycle” (Figure 6C). A volcano plot of these TAF4b-bound E16.5 DEGs revealed far more upregulated DEGs (674 genes) than downregulated DEGs (202 genes) in Taf4b-deficient ProSpg (Figure 6D). As examples of TAF4b-bound DEGs, we present TSS gene tracks for Nuclear Transcription Factor Y Subunit Alpha (Nfya), Protein Regulator of cytokinesis 1 (Prc1), H2A histone family member X (H2afx), Kruppel-like factor 6 (Klf6), and Cyclin Dependent Kinase 20 (Cdk20), with TAF4b and H3K4me3 enrichment compared with control IgG (Figure 6E). Nfya is a component of the NFY transcription factor complex and interestingly TAF4b peaks were located between the TSS of Nfya and another gene Oard1 which was not a DEG. Prc1 encodes a microtubule-binding protein that plays a role in mitosis and binds to Plk1 another critical mitosis gene (Hernández-Ortega et al., 2019). H2afx encodes an essential DNA damage response protein and has been previously found to be increased in Taf4b-deficient oocytes (Grive et al., 2016). Klf6 encodes a transcription factor that plays a role in several developmental processes such as cell proliferation and differentiation (Hu et al., 2021). Cdk20, decreased in Taf4b-deficient ProSpg, encodes a positive regulator of the cell cycle by activating CDK2 and Cyclin D (Lai et al., 2020). A protein-protein interaction network of these 876 TAF4b-bound DEGs revealed Cdk1 and other cell cycle-related genes as a major group of associated genes and interestingly the entire NFY complex (encoded by Nfya, Nfyb, and Nfyc) was present (Supplementary Figure S1).
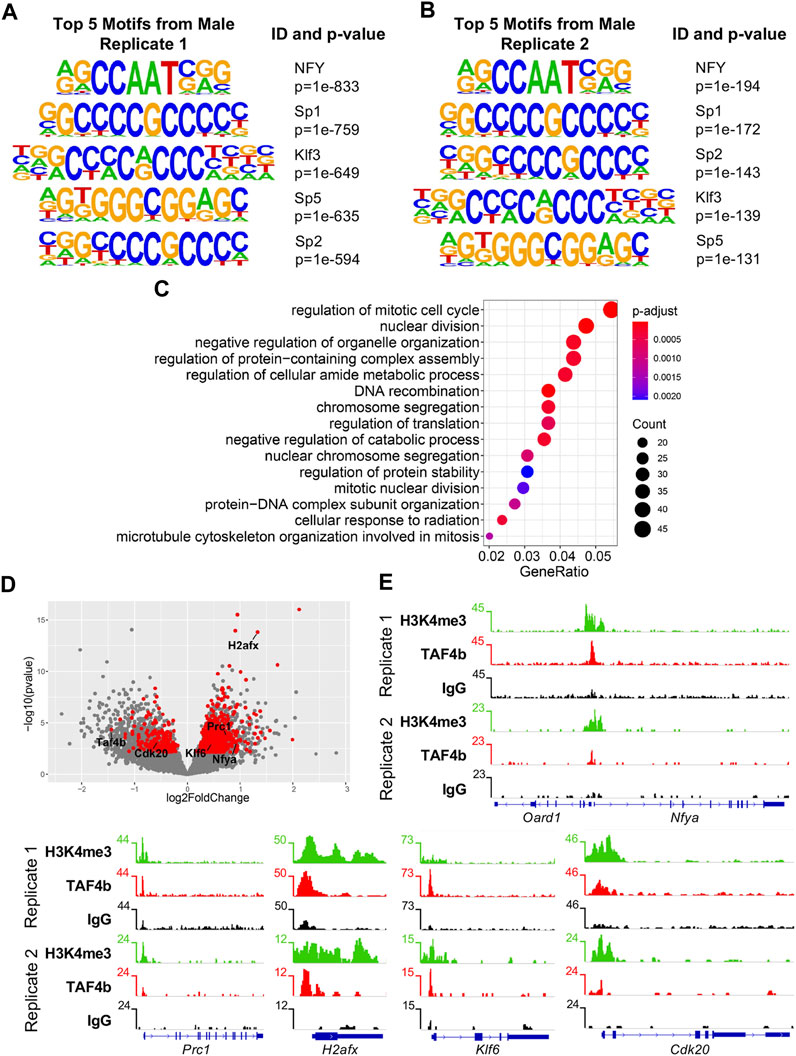
FIGURE 6. CUT&RUN and RNA-seq integration identify TAF4b bound and modulated cell cycle genes. (A) Top five TAF4b motifs from “promoter-TSS” peaks in Top five TAF4b motifs from “promoter-TSS” peaks in male Replicate 1. (B) Top five TAF4b motifs from “promoter-TSS” peaks in male Replicate 2. (C) Biological process GO dotplot of the 876 genes that are DEGs and had a TAF4b promoter-TSS peak in at least one of the two germ cell samples. (D) Volcano plot of the 876 DEGs that had at least one TAF4b promoter peak (red dots). (E) Gene track of Oard1 and Nfya, in which Nfya was a DEG that had a TAF4b promoter-TSS called in both replicates. Gene tracks of Prc1, H2afx, Klf6, and Cdk20, which were DEGs (labeled red dots in D) that had a TAF4b promoter-TSS called in both replicates.
When integrating our CUT&RUN “promoter-TSS” peaks with TAF4b DEGs, we again found an enrichment for SP/KLF transcription factor binding motifs (Figure 7A). However when we looked at the integrated data we no longer observed NFY in the top five overall binding motifs. The surprising absence of NFY sites in this analysis prompted us to further parse these data into TAF4b peaks whose levels of mRNA decreased (Downregulated DEGs) or increased (Upregulated DEGs) in TAF4b-deficient ProSpg. We first looked at the occupancy of TAF4b and noted that it centered around the −50 bp region upstream of the TSS, further supporting a role for TAF4b in core promoter recognition and transcription initiation. It was also observed that there was a slight difference in the median being approximately 5 bases futher upsteam in the Up DEGs as compared to the Down DEGs (Figure 7B). Interestingly, we discovered that NFY is actually the top Down DEG motif while Sp1 is the top Up DEG motif (Figures 7C,D). The differences in top motifs between decreased and increased mRNA levels may allude to the multiple modes of transcriptional modulation by TAF4b. As follow-up, we mapped TAF4b targets to the GO analysis from bulk RNA-seq to determine which GO categories were directly modulated. Most notable in this analysis were the categories of cell cycle regulation, covalent chromatin modification, and DNA repair which had increased mRNA expression levels in the absence of TAF4b and also contained 7 genes that had TAF4b-bound promoters (Figure 7E). Finally, we quantified the overall fraction of CUT&RUN peaks that were also DEGs and found significantly more mRNAs that were increased in the absence of TAF4b to contain a TAF4b-bound promoter (Figure 7F). Overall, these data suggest that TAF4b specifically controls the levels of cell cycle and chromatin regulating gene expression programs required for ProSpg to properly navigate the transition from cycling to quiescence during embryonic mammalian testes development.
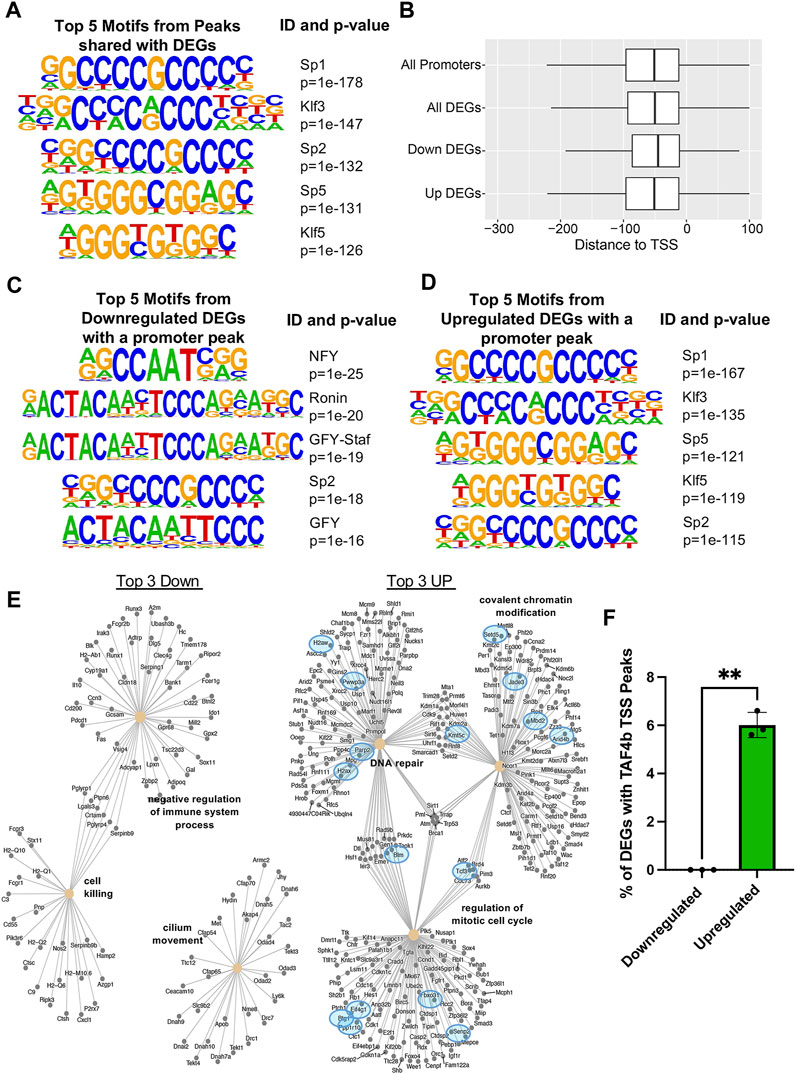
FIGURE 7. Sp/Klf motif enrichment at cell cycle and chromatin regulating TAF4b bound gene promoters. (A) Top five motifs enriched at TAF4b “promoter-TSS” peaks for genes that were also DEGs, the promoter ID, and the associated p-value. (B) Boxplots (no outliers included) of promoter TAF4b peaks relative to the TSS with a median of −53 bp, all TAF4b “promoter-TSS” peaks for genes that were also DEGs with a median of −51 bp, TAF4b “promoter-TSS” peaks for genes that were only Downregulated DEGs with a median of −49 bp and TAF4b “promoter-TSS” peaks for genes that were only Upregulated DEGs with a median of −53 bp. (C) Top five TAF4b associated binding motifs from “promoter-TSS” peaks in Down DEGs and (D) Up DEGs. (E) Category netplots composed of the top three downregulated (left) and upregulated (right) biological processes from (Figures 3C,D) along with DEGs that contained TAF4b peaks at their TSS (blue circles). (F) Comparison of TAF4b target DEG percentages from top three upregulated and downregulated GO categories. Significance determined by Welch’s t-test, ** = p-value <0.01.
Soon after sex determination in mammals, embryonic female and male germ cells receive distinct signals to initiate a sex-specific germ cell differentiation program (Capel, 2019). However, it is unclear how these newly defined germ cells integrate external signals to execute precise and sex-specific gene expression programs and cell cycle states required for germ cell development. We have discovered that the embryonic germline-enriched TFIID subunit, TAF4b, is required for male and female germ cell development and maintenance in mice. Here, we identify many ProSpg genes whose expression are affected by the loss of TAF4b and whose core promoter regions display TAF4b occupancy just upstream of the TSS. TAF4b enrichment at ProSpg core promoters correlates with the binding sites for Sp1 and NFY, two well-characterized and ubiquitous promoter-proximal and sequence-specific transcription factors. The integration of these high throughput genomic sequencing and mapping data indicate that TAF4b fine tunes male-specific cell cycle gene expression required for the timely entry of ProSpg into quiescence via complex and unexpected mechanisms.
Several notable regulators of post-transcriptional gene expression play similar developmental functions as TAF4b when ProSpg transition towards quiescence. The most notable is the RNA-binding protein DND1 which displays intriguing parallels with the functions of TAF4b shown here. DND1 deficiency in mice leads to defects of prospermatogonial entry into quiescence and increased germ cell loss (Cook et al., 2011). However, the Ter mutation in the Dnd1 gene on the 129 genetic background strikingly results in spontaneous teratoma formation at E16.5 (Cook et al., 2011). While Taf4b-deficient testes display similar kinetics in male germ cell loss and fertility disruption, our Taf4b-deficiency presented here is on a C57Bl/6 background where we might not expect to see these teratomas (Falender et al., 2005). Remarkably, TAF4b and DND1 share common regulation of mitotic cell cycle and chromatin modification gene expression, suggesting they are two distinct modes of regulating a common ProSpg quiescence and survival program ((Ruthig et al., 2019), this study). A second RNA-binding protein, NANOS2, is also responsible for ProSpg development and has been recently shown to be bound to and work with DND1 in regulating specific RNA loading into the CNOT deadenylase complex (Suzuki et al., 2016; Ruthig et al., 2019). Interestingly, Nanos2 is an upregulated DEG, and Dnd1 is a downregulated DEG in E16.5 Taf4b-deficient ProSpg. The developmental similarities of perturbed quiescence, increased germ cell loss, and infertility in Nanos2, Dnd1, and Taf4b mouse mutants suggest that regulating the gene expression of prospermatogonia transitions is critical to properly set up the future spermatogonia that arise from this lineage in the postnatal testis. The dysregulation of Nanos2 and Dnd1 in the absence of TAF4b also indicates cross-talk between these crucial gene expression regulators and the sensitivity of germline development.
There are also several key transcription factors that direct cell cycle progression and transcription during prospermatogonial development. The master regulator of the G1 phase of the cell cycle, RB1, is required for the timely mitotic arrest of early prospermatogonia (Spiller et al., 2010). A recent germ cell-specific knockout approach for RB1 linked this cell cycle defect to disrupting the metabolic nature of these developing prospermatogonia and their corresponding ability to become SSCs (Du et al., 2021). While the timing of RB1 and TAF4b functions in promoting quiescence are similar, RB1 deficiency leads to much more severe cell cycle defects than Taf4b-deficiency, suggesting they play more complementary roles in SSC development (Du et al., 2021). RHOX10 is a homeodomain family sequence-specific DNA-binding protein found in the reproductive homeobox cluster of the X chromosome and is required for prospermatogonial and SSC development. A recent genomic study using similar tools implemented here uncovered a network of transcription factors that are direct targets of RHOX10, most notably DMRT1 and ZBTB16 (Tan et al., 2021). While there are minor differences in the timing and cells used in these two studies, Tan et al., 2021 uncovered a critical CCAAT box bound by RHOX10 in the Dmrt1 promoter and predicted it to be a binding site for the NFY transcription factor, which was the most correlated binding site at TAF4b bound core promoters (Tan et al., 2021). It will be interesting to determine how these transcriptional regulators work together and separately to promote prospermatogonial development required for the proper establishment of SSCs and longterm mammalian spermatogenesis.
The precise molecular mechanism of TAF4b in promoting ProSpg transitions remains unknown, but several interesting new twists are revealed here. TFIID was first discovered as a ubiquitous multiprotein core promoter binding and coactivator complex consisting of the TATA-box binding protein and 14 TBP-associated factors (TAFs) required for Sp1-dependent transcriptional activation in vitro (Pugh, 1990). However, determining Sp1 as the top binding motif at the cell cycle and chromatin modification gene promoters bound by TAF4b in ProSpg was surprising, given this unique cell type-specificity. Moreover, mRNA expression levels of these gene programs primarily increase in the absence of TAF4b, suggesting TAF4b plays some role in limiting their expression as ProSpg transitions to quiescence. Future studies will determine whether TAF4b works in a chromatin modification capacity along with its canonical function in the TFIID complex. As TAF4b regulates chromatin modification genes, we also plan on determining if changes in chromatin accessibility are a strong driver of our observed gene expression changes. This non-canonical role of TAF4b may contribute to proper silencing and timely repression of specific genes during spermatogenesis. We also know now that other members of the TFIID complex have been found in chromatin-modifying complexes like that of the Spt/Ada/Gcn5 acetyltransferase (SAGA) complex. Members such as TAF5L, TAF6L, and TAF9B are just some with documented dual activity (Soffers and Workman, 2020). Another possibility is that as later T2 ProSpg return to proliferation quickly after birth, TAF4b helps mark the cell cycle and chromatin modifying genes for future activation during later stages of postnatal germ cell expansion. Lastly, the TATA-box that marks a subset of TFIID-dependent promoters is usually found between −25 bp and −30 bp upstream of the TSS, and our peaks of TAF4b binding center around the −50 bp region suggesting that TAF4b might extend the upstream boundary of the TFIID footprint at the core promoter by 20 bp (Sloutskin et al., 2021). Future studies of ProSpg core promoter architecture and TAF4b′s transcriptional mechanism with individual SP/KlF and NFY family members will be required to understand these novel aspects of ProSpg transcription regulation.
While we focus here on TAF4b’s role in promoting the early development of the male germline, we have recently reported that at identical embryonic time points in ovarian development, TAF4b plays a similar critical role in early oocyte differentiation (Gura et al., 2022). As female and male embryonic germ cells express unique sets of genes and navigate different biological processes, i.e., meiotic initiation vs. mitotic arrest, it is surprising that the same transcription factor would have distinct functions in the early life of these future gametes. We propose that one common function of TAF4b, and its associated regulatory complex, is to promote the embryonic germ cell identity and survival after PGC specification, analogous to the germ cell licensing function of DAZL (Gill et al., 2011). In addition, TAF4b has likely evolved sex-specific functions that allow it to promote context-specific transcription events in both male and female developing germ cells. One commonality of these two populations is that even though they enter meiosis at different times, they both exit the mitotic cell cycle at the end of PGC development, and TAF4b, along with many other unknown regulatory proteins, helps direct these transitions. Finally, genome-wide epigenetic modifications are erased in both female and male germ cell populations at the end of PGC development. In the context of a more open genome, TAF4b and its related regulators could help maintain the appropriate chromatin environment for controlling transcription-coupled DNA repair mechanisms unique and critical to the germline genome of both sexes and, ultimately, the next-generation.
The male mouse E16.5 RNA-sequencing data are available from NCBI GEO under accession number GSE188351. The male mouse E16.5 CUT&RUN sequencing data are available from NCBI GEO under accession number GSE188701. The sequencing datasets accessed in this research are from the follow accession numbers: the scRNA-seq mouse data from E12.5 to P7 male sorted Oct4-eGFP gonads used for Figure 1 was obtained through NCBI GEO: Q9 GSE119045, GSE124904, and GSE130593.
This study was approved by Brown University IACUC protocol #21-02-0005. The primary method of euthanasia is CO2 inhalation and the secondary method used is cervical dislocation both as per American Veterinary Medical Association (AVMA) guidelines on euthanasia. The study was conducted in accordance with the local legislation and institutional requirements.
MG: Conceptualization, Formal Analysis, Funding acquisition, Validation, Writing–review and editing, Data curation, Investigation, Methodology, Resources, Software, Visualization, Writing–original draft. MB: Conceptualization, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Resources, Software, Validation, Visualization, Writing–original draft, Writing–review and editing. KA: Writing–review and editing, Data curation, Investigation, Methodology, Software. SR: Data curation, Investigation, Methodology, Writing–review and editing. KS: Data curation, Investigation, Methodology, Writing–review and editing, Conceptualization, Project administration, Resources, Visualization. RF: Conceptualization, Project administration, Writing–review and editing, Formal Analysis, Funding acquisition, Supervision, Validation.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The Brown University Flow Cytometry and Sorting Facility has received generous support in part by the National Institutes of Health (NCRR Grant No. 1S10RR021051) and the Division of Biology and Medicine, Brown University. As some of our insights were gained by reprocessing publicly available scRNA-seq datasets, we greatly appreciate both the researchers that generated and shared the data initially and the respective repositories for making them available. We are grateful to the Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD) for their generous support through awards 1F31HD097933 to MG, 1F31HD105340 to KA, and 1R01HD091848 to RF. We thank the generous support of the Howard Hughes Medical Institute for the Gilliam Fellowship to MB. We also thank the US-Israel Binational Science Foundation (BSF) for their support to RF (2019285). We thank the Rhode Island Foundation and the Advance-CTR at Brown University supported by the NIH IDeA-CTR grant (U54GM115677) for support for these studies and its publication.
We thank the Center for Computation and Visualization at Brown University for computational resources for scRNA-seq, RNA-seq, and CUT&RUN data analysis. We thank Kevin Carlson and the Brown University Flow Cytometry and Sorting Facility for expertise completing the flow sorting. We also thank Joseph Aguilera and Abigail Brown for their assistance with ATAC-seq data analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1270408/full#supplementary-material
SUPPLEMENTARY TABLE S1 | Cell numbers for RNA-seq samples.
SUPPLEMENTARY TABLE S2 | Output for E16.5 ProSpg RNA-seq.
SUPPLEMENTARY TABLE S3 | TAF4b and H3K4me3 CUT&RUN in E16.5 ProSpg.
SUPPLEMENTARY FIGURE S1 | Protein-protein interactions of TAF4b bound DEGs in E16.5 ProSpg.
Aloisio, G. M., Nakada, Y., Saatcioglu, H. D., Peña, C. G., Baker, M. D., Tarnawa, E. D., et al. (2014). PAX7 expression defines germline stem cells in the adult testis. J. Clin. Invest. 124, 3929–3944. doi:10.1172/JCI75943
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/.
Ayhan, Ö., Balkan, M., Guven, A., Hazan, R., Atar, M., Tok, A., et al. (2014). Truncating mutations in TAF4B and ZMYND15 causing recessive azoospermia.J. Med. Genet. 51, 239–244. doi:10.1136/jmedgenet-2013-102102
Capel, B. (2019). Women in reproductive science: to be or not to be a testis. Reproduction 158 (6), F101–f11. doi:10.1530/rep-19-0151
Cook, M. S., Munger, S. C., Nadeau, J. H., and Capel, B. (2011). Regulation of male germ cell cycle arrest and differentiation by DND1 is modulated by genetic background. Development 138, 23–32. doi:10.1242/dev.057000
De Rooij, D. G., and Grootegoed, J. A. (1998). Spermatogonial stem cells. Curr. Opin. Cell Biol. 10, 694–701. doi:10.1016/S0955-0674(98)80109-9
Du, G., Oatley, M. J., Law, N. C., Robbins, C., Wu, X., and Oatley, J. M. (2021). Proper timing of a quiescence period in precursor prospermatogonia is required for stem cell pool establishment in the male germline. Development 148, dev194571. doi:10.1242/dev.194571
Falender, A. E., Freiman, R. N., Geles, K. G., Lo, K. C., Hwang, K. S., Lamb, D. J., et al. (2005). Maintenance of spermatogenesis requires TAF4b, a gonad-specific subunit of TFIID. Genes Dev. 19, 794–803. doi:10.1101/gad.1290105
Fayomi, A. P., and Orwig, K. E. (2018). Spermatogonial stem cells and spermatogenesis in mice, monkeys and men. Stem Cell Res. 29, 207–214. doi:10.1016/j.scr.2018.04.009
Gassei, K., and Orwig, K. E. (2013). SALL4 expression in gonocytes and spermatogonial clones of postnatal mouse testes. PLoS One 8, e53976. doi:10.1371/journal.pone.0053976
Gill, M. E., Hu, Y.-C., Lin, Y., and Page, D. C. (2011). Licensing of gametogenesis, dependent on RNA binding protein DAZL, as a gateway to sexual differentiation of fetal germ cells. Proc. Natl. Acad. Sci. 108, 7443–7448. doi:10.1073/pnas.1104501108
Green, C. D., Ma, Q., Manske, G. L., Shami, A. N., Zheng, X., Marini, S., et al. (2018). A comprehensive roadmap of murine spermatogenesis defined by single-cell RNA-seq. Dev. Cell 46, 651–667. doi:10.1016/j.devcel.2018.07.025
Grive, K. J., Gustafson, E. A., Seymour, K. A., Baddoo, M., Schorl, C., Golnoski, K., et al. (2016). TAF4b regulates oocyte-specific genes essential for meiosis. PLoS Genet. 12, 10061288–e1006218. doi:10.1371/journal.pgen.1006128
Grive, K. J., Seymour, K. A., Mehta, R., and Freiman, R. N. (2014). TAF4b promotes mouse primordial follicle assembly and oocyte survival. Dev. Biol. 392 (1), 42–51. doi:10.1016/j.ydbio.2014.05.001
Gura, M. A., Mikedis, M. M., Seymour, K. A., De Rooij, D. G., Page, D. C., and Freiman, R. N. (2020). Dynamic and regulated TAF gene expression during mouse embryonic germ cell development. PLoS Genet. 16, e1008515–e1008528. doi:10.1371/journal.pgen.1008515
Gura, M. A., Relovská, S., Abt, K. M., Seymour, K. A., Wu, T., Kaya, H., et al. (2022). TAF4b transcription networks regulating early oocyte differentiation. Dev 149, dev200074–15. doi:10.1242/dev.200074
Hainer, S. J., and Fazzio, T. G. (2019). High-resolution chromatin profiling using CUT&RUN. Curr. Protoc. Mol. Biol. 126, e85–e22. doi:10.1002/cpmb.85
Heinz, S., Benner, C., Spann, N., Bertolino, E., Lin, Y. C., Laslo, P., et al. (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. doi:10.1016/j.molcel.2010.05.004
Hernández-Ortega, S., Sánchez-Botet, A., Quandt, E., Masip, N., Gasa, L., Verde, G., et al. (2019). Phosphoregulation of the oncogenic protein regulator of cytokinesis 1 (PRC1) by the atypical CDK16/CCNY complex. Exp. Mol. Med. 51, 1–17. doi:10.1038/s12276-019-0242-2
Hill, P. W. S., Leitch, H. G., Requena, C. E., Sun, Z., Amouroux, R., Roman-Trufero, M., et al. (2018). Epigenetic reprogramming enables the transition from primordial germ cell to gonocyte. Nature 555 (7696), 392–396. doi:10.1038/nature25964
Hu, K., Zheng, Q. K., Ma, R. J., Ma, C., Sun, Z. G., and Zhang, N. (2021). Krüppel-like factor 6 splice variant 1: an oncogenic transcription factor involved in the progression of multiple malignant tumors. Front. Cell Dev. Biol. 9, 661731–661739. doi:10.3389/fcell.2021.661731
Kubo, N., Toh, H., Shirane, K., Shirakawa, T., Kobayashi, H., Sato, T., et al. (2015). DNA methylation and gene expression dynamics during spermatogonial stem cell differentiation in the early postnatal mouse testis. BMC Genomics 16 (1), 624. doi:10.1186/s12864-015-1833-5
Lai, L., Shin, G. Y., and Qiu, H. (2020). The role of cell cycle regulators in cell survival — dual functions of cyclin-dependent. Int. J. Mol. Sci. 21, 1–14. doi:10.3390/ijms21228504
Law, N. C., Oatley, M. J., and Oatley, J. M. (2019). Developmental kinetics and transcriptome dynamics of stem cell specification in the spermatogenic lineage. Nat. Commun. 10, 2787–2814. doi:10.1038/s41467-019-10596-0
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi:10.1093/bioinformatics/btp352
Liao, Y., Smyth, G. K., and Shi, W. (2014). FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. doi:10.1093/bioinformatics/btt656
Liu, Z., Sun, Q., and Wang, X. (2017). PLK1, A potential target for cancer therapy. Transl. Oncol. 10 (1), 22–32. doi:10.1016/j.tranon.2016.10.003
Lovasco, L. A., Gustafson, E. A., Seymour, K. A., De Rooij, D. G., and Freiman, R. N. (2015). TAF4b is required for mouse spermatogonial stem cell development. Stem Cells 33, 1267–1276. doi:10.1002/stem.1914
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. doi:10.1186/s13059-014-0550-8
McCarrey, J. R. (2013). Toward a more precise and informative nomenclature describing fetal and neonatal male germ cells in rodents. Biol. Reprod. 89, 47. doi:10.1095/biolreprod.113.110502
Nguyen, D. H., Soygur, B., Peng, S. P., Malki, S., Hu, G., and Laird, D. J. (2020). Apoptosis in the fetal testis eliminates developmentally defective germ cell clones. Nat. Cell Biol. 22, 1423–1435. doi:10.1038/s41556-020-00603-8
Oatley, J. M., and Brinster, R. L. (2008). Regulation of spermatogonial stem cell self-renewal in mammals. Annu. Rev. Cell Dev. Biol. 24, 263–286. doi:10.1146/annurev.cellbio.24.110707.175355
Oatley, M. J., Kaucher, A. V., Racicot, K. E., and Oatley, J. M. (2011). Inhibitor of DNA binding 4 is expressed selectively by single spermatogonia in the male germline and regulates the self-renewal of spermatogonial stem cells in mice. Biol. Reprod. 85, 347–356. doi:10.1095/biolreprod.111.091330
Ohinata, Y., Payer, B., O’Carroll, D., Ancelin, K., Ono, Y., Sano, M., et al. (2005). Blimp1 is a critical determinant of the germ cell lineage in mice. Nature 436, 207–213. doi:10.1038/nature03813
Pertea, M., Kim, D., Pertea, G. M., Leek, J. T., and Salzberg, S. L. (2016). Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 11, 1650–1667. doi:10.1038/nprot.2016.095
Phillips, B. T., Gassei, K., and Orwig, K. E. (2010). Spermatogonial stem cell regulation and spermatogenesis. Philos. Trans. R. Soc. L B Biol. Sci. 365, 1663–1678. doi:10.1098/rstb.2010.0026
Pugh, B. F. (1990). Mechanism of transcriptional activation by Sp1: evidence for coactivators. Cell 61 (7), 1187–1197. doi:10.1016/0092-8674(90)90683-6
Quatredeniers, M., Serafin, A. S., Benmerah, A., Rausell, A., Saunier, S., and Viau, A. (2023). Meta-analysis of single-cell and single-nucleus transcriptomics reveals kidney cell type consensus signatures. Sci. Data 10 (1), 361. doi:10.1038/s41597-023-02209-9
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi:10.1038/nbt.1754
Ruthig, V. A., Friedersdorf, M. B., Garness, J. A., Munger, S. C., Bunce, C., Keene, J. D., et al. (2019). The RNA-binding protein DND1 acts sequentially as a negative regulator of pluripotency and a positive regulator of epigenetic modifiers required for germ cell reprogramming. Development 146, dev175950. doi:10.1242/dev.175950
Shami, A. N., Zheng, X., Munyoki, S. K., Ma, Q., Manske, G. L., Green, C. D., et al. (2020). Single-cell RNA sequencing of human, macaque, and mouse testes uncovers conserved and divergent features of mammalian spermatogenesis. Dev. Cell 54, 529–547. doi:10.1016/j.devcel.2020.05.010
Skene, P. J., and Henikoff, S. (2017). An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 6, e21856. doi:10.7554/eLife.21856
Sloutskin, A., Shir-Shapira, H., Freiman, R. N., and Juven-Gershon, T. (2021). The core promoter is a regulatory hub for developmental gene expression. Front. Cell Dev. Biol. 9, 666508. doi:10.3389/fcell.2021.666508
Soffers, J. H. M., and Workman, J. L. (2020). The SAGA chromatin-modifying complex: the sum of its parts is greater than the whole. Genes Dev. 34 (19-20), 1287–1303. doi:10.1101/gad.341156.120
Song, H.-W., Bettegowda, A., Lake, B. B., Zhao, A. H., Skarbrevik, D., Babajanian, E., et al. (2016). The homeobox transcription factor RHOX10 drives mouse spermatogonial stem cell establishment. Cell Rep. 17, 149–164. doi:10.1016/j.celrep.2016.08.090
Spiller, C. M., Wilhelm, D., and Koopman, P. (2010). Retinoblastoma 1 protein modulates XY germ cell entry into G1/G0 arrest during fetal development in mice. Biol. Reprod. 82, 433–443. doi:10.1095/biolreprod.109.078691
Sun, X., and Kaufman, P. D. (2018). Ki-67: more than a proliferation marker. Chromosoma 127 (2), 175–186. doi:10.1007/s00412-018-0659-8
Suske, G. (2017). NF-Y and SP transcription factors — new insights in a long-standing liaison. Biochimica Biophysica Acta (BBA) - Gene Regul. Mech. 1860 (5), 590–597. doi:10.1016/j.bbagrm.2016.08.011
Suzuki, A., Niimi, Y., Shinmyozu, K., Zhou, Z., Kiso, M., and Saga, Y. (2016). Dead end1 is an essential partner of NANOS2 for selective binding of target RNAs in male germ cell development. EMBO Rep. 17, 37–46. doi:10.15252/embr.201540828
Szklarczyk, D., Gable, A. L., Lyon, D., Junge, A., Wyder, S., Huerta-Cepas, J., et al. (2019). STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47, D607–D613. doi:10.1093/nar/gky1131
Tan, K., Song, H.-W., and Wilkinson, M. F. (2021). RHOX10 drives mouse spermatogonial stem cell establishment through a transcription factor signaling cascade. Cell Rep. 36, 109423. doi:10.1016/j.celrep.2021.109423
Tan, K., Song, H. W., and Wilkinson, M. F. (2020). Single-cell RNAseq analysis of testicular germ and somatic cell development during the perinatal period. Dev 147, dev183251. doi:10.1242/dev.183251
Weber, S., Eckert, D., Nettersheim, D., Gillis, A. J. M., Schäfer, S., Kuckenberg, P., et al. (2010). Critical function of AP-2 gamma/TCFAP2C in mouse embryonic germ cell maintenance. Biol. Reprod. 82, 214–223. doi:10.1095/biolreprod.109.078717
Wickham, H. (2016). “ggplot2: elegant graphics for data analysis,” in Wiley interdisciplinary reviews: Computational statistics. 2nd ed (Springer-Verlag). Available: ggplot.org.
Xi, Q., Zhang, H., Zhang, X., Jiang, Y., Wang, R., Liu, R., et al. (2020). Analysis of TATA-box binding protein associated factor 4b gene mutations in a Chinese population with nonobstructive azoospermia. Med. (United States) 99, e20561–e20566. doi:10.1097/MD.0000000000020561
Keywords: transcription, spermatogenesis, prospermatogonia, quiescence, TAF4B, spermatogonial stem cells, male fertility
Citation: Gura MA, Bartholomew MA, Abt KM, Relovská S, Seymour KA and Freiman RN (2023) Transcription and chromatin regulation by TAF4b during cellular quiescence of developing prospermatogonia. Front. Cell Dev. Biol. 11:1270408. doi: 10.3389/fcell.2023.1270408
Received: 31 July 2023; Accepted: 26 September 2023;
Published: 12 October 2023.
Edited by:
Rafael Henrique Nóbrega, São Paulo State University, BrazilReviewed by:
Jianqiang Bao, University of Science and Technology of China, ChinaCopyright © 2023 Gura, Bartholomew, Abt, Relovská, Seymour and Freiman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Richard N. Freiman, cmljaGFyZF9mcmVpbWFuQGJyb3duLmVkdQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.