 Claire E. Senner1,2*
Claire E. Senner1,2* Miguel R. Branco
Miguel R. Branco Erica D. Watson
Erica D. Watson
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol., 27 June 2023
Sec. Developmental Epigenetics
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1209928
This article is part of the Research TopicIn Celebration of Women in Developmental EpigeneticsView all 11 articles
One-carbon metabolism, including the folate cycle, has a crucial role in fetal development though its molecular function is complex and unclear. The hypomorphic Mtrrgt allele is known to disrupt one-carbon metabolism, and thus methyl group availability, leading to several developmental phenotypes (e.g., neural tube closure defects, fetal growth anomalies). Remarkably, previous studies showed that some of the phenotypes were transgenerationally inherited. Here, we explored the genome-wide epigenetic impact of one-carbon metabolism in placentas associated with fetal growth phenotypes and determined whether specific DNA methylation changes were inherited. Firstly, methylome analysis of Mtrrgt/gt homozygous placentas revealed genome-wide epigenetic instability. Several differentially methylated regions (DMRs) were identified including at the Cxcl1 gene promoter and at the En2 gene locus, which may have phenotypic implications. Importantly, we discovered hypomethylation and ectopic expression of a subset of ERV elements throughout the genome of Mtrrgt/gt placentas with broad implications for genomic stability. Next, we determined that known spermatozoan DMRs in Mtrrgt/gt males were reprogrammed in the placenta with little evidence of direct or transgenerational germline DMR inheritance. However, some spermatozoan DMRs were associated with placental gene misexpression despite normalisation of DNA methylation, suggesting the inheritance of an alternative epigenetic mechanism. Integration of published wildtype histone ChIP-seq datasets with Mtrrgt/gt spermatozoan methylome and placental transcriptome datasets point towards H3K4me3 deposition at key loci. These data suggest that histone modifications might play a role in epigenetic inheritance in this context. Overall, this study sheds light on the mechanistic complexities of one-carbon metabolism in development and epigenetic inheritance.
It is well established that the vitamin folate (also known as folic acid) is important for fetal development. A highly recognisable example is increased risk of neural tube closure defects (e.g., spina bifida) in babies that result from maternal dietary folate deficiency (Emery et al., 1969). In fact, folic acid supplementation during pregnancy and folate fortification programmes improves pregnancy outcomes (MRC Vitamin Study Research Group, 1991; Gelineau-van Waes et al., 2008). Beyond the neural tube, other developmental defects [e.g., fetal growth restriction (Furness et al., 2008; Chen et al., 2018), congenital heart defects (Christensen et al., 2015)] and pregnancy disorders (Mislanova et al., 2011; Wu et al., 2015) are associated with dietary deficiency and/or mutations in key enzymes involved in its metabolism. Although well studied, the molecular role of folate metabolism during development is complex and not well understood. One-carbon metabolism, which includes the folate and methionine cycles, is required by all cells for thymidine synthesis and for methyl groups involved in a broad range of methylation reactions (Lin et al., 2022). As a result, it is hypothesised that rapidly proliferating cells in a developing fetus and placenta requires one-carbon metabolism for DNA synthesis and general epigenetic regulation. The specific genomic targets of one-carbon metabolism that drive developmental phenotypes remain unclear.
To explore the specific molecular role of one-carbon metabolism during development, we study a mouse model with a hypomorphic mutation in the methionine synthase reductase gene (Mtrrgt) (Padmanabhan et al., 2013). During one-carbon metabolism, folate metabolites are required to transmit methyl groups for the methylation of homocysteine by methionine synthase (MTR) to form methionine and tetrahydrofolate (Shane and Stokstad, 1985). Methionine acts as precursor for S-adenosylmethionine (SAM), which in turn serves as the sole methyl-donor for substrates involved in epigenetic regulation (e.g., DNA, histones, RNA) among other substrates (Ducker and Rabinowitz, 2017). Importantly, MTRR activates MTR through the reductive methylation of its vitamin B12 co-factor (Leclerc et al., 1998; Yamada et al., 2006; Elmore et al., 2007). The hypomorphic Mtrrgt mutation reduces Mtrr transcript expression to a level that is sufficient to diminish MTR activity by 60% of controls (Elmore et al., 2007; Padmanabhan et al., 2013). Consequently, the progression of one-carbon metabolism is disrupted by the Mtrrgt mutation as evidenced by plasma hyperhomocysteinemia (Elmore et al., 2007; Padmanabhan et al., 2013) and widespread changes in DNA methylation patterns (Padmanabhan et al., 2013; Bertozzi et al., 2021; Blake et al., 2021). Additionally, Mtrrgt/gt mice display several phenotypes similar to the clinical features of folate deficiency in humans (Krishnaswamy and Madhavan Nair, 2001) or human MTRR mutations (Schuh et al., 1984; Wilson et al., 1999) including macrocytic anemia (Padmanabhan et al., 2018) and neural tube closure defects (NTDs) (Padmanabhan et al., 2013; Wilkinson et al., 2021). Beyond this, other phenotypes have emerged in Mtrrgt/gt mice reflecting a broader influence of impaired one-carbon metabolism on development. These phenotypes include fetal growth defects (such as fetal growth restriction (FGR), fetal growth enhancement (FGE), or developmental delay) (Padmanabhan et al., 2013; Padmanabhan et al., 2017), complications during implantation (e.g., twinning, skewed implantation) (Padmanabhan et al., 2013; Wilkinson et al., 2021), haemorrhages, and/or congenital malformations (such as congenital heart defects and poor placentation) (Deng et al., 2008; Padmanabhan et al., 2013; Wilkinson et al., 2021). Therefore, the Mtrrgt mouse line is ideal for exploring the molecular consequences of defective one-carbon metabolism during growth and development.
Remarkably, the Mtrrgt mouse line is also a unique mammalian model of transgenerational epigenetic inheritance that occurs via the maternal grandparental lineage (Padmanabhan et al., 2013; Blake et al., 2021). Through highly controlled genetic pedigrees and embryo transfer experiments, we previously showed that an Mtrr+/gt genotype in male or female mice (i.e., the F0 generation) initiates multigenerational inheritance of developmental phenotypes in their wildtype (Mtrr+/+) grandprogeny (i.e., the F2–F4 generations) (Padmanabhan et al., 2013; Padmanabhan et al., 2017). This effect occurs through their F1 wildtype daughters (Padmanabhan et al., 2013). In general, the mechanism of epigenetic inheritance is not well understood. In the context of the Mtrrgt mouse line, we hypothesise that alterations in the epigenome of the F0 germline caused by abnormal one-carbon metabolism is inherited by the wildtype offspring of the next generation (and potentially beyond) to influence gene expression during development (Padmanabhan et al., 2013; Blake and Watson, 2016; Blake et al., 2021). Given the role of MTRR in one-carbon metabolism, and thus in cellular methylation, we initially focused on how the Mtrrgt mutation alters DNA methylation patterns across generations. Through a targeted analysis, we previously determined that developmental phenotypes at E10.5 in Mtrrgt/gt conceptuses or F2 Mtrr+/+ conceptuses derived by an F0 Mtrr+/gt maternal grandparent were associated with locus-specific changes in DNA methylation linked to gene misexpression (Padmanabhan et al., 2013; Blake et al., 2021). The effect was particularly striking in the placenta at key genes involved in the regulation of fetal growth and metabolism (Padmanabhan et al., 2013). It was also clear that epigenetic instability of DNA methylation occurs in mature spermatozoa from Mtrr+/gt and Mtrrgt/gt males as well as from F1 Mtrr+/+ male progeny of F0 Mtrr+/gt males (Blake et al., 2021), which otherwise display normal spermatogenesis and spermatozoa function (Blake et al., 2019). However, the extent to which these altered germline methylation patterns are recapitulated in (or inherited by) the somatic cells of the progeny and grandprogeny is currently not well understood in the Mtrrgt mouse line.
In this study, we use genome-wide approaches to investigate the global impact of one-carbon metabolism on the placental methylome in Mtrrgt/gt homozygous mice and in F2 Mtrr+/+ mice derived from F0 Mtrr+/gt maternal grandfathers. In doing so, we probe the underlying impact on fetal growth and whether the differentially methylated regions (DMRs) are functionally important and/or inherited. We reveal that the Mtrrgt/gt placental methylome is unstable with implications for phenotype establishment, transposable element regulation, and genetic stability. We also determine that specific DMRs observed in spermatozoa of Mtrrgt/gt males are reprogrammed in Mtrrgt/gt placentas and as a result, we explore other epigenetic mechanisms for inheritance including histone methylation (H3K4me3). We integrate published ChIP-seq datasets from wildtype embryonic and trophoblast lineages with our Mtrrgt/gt spermatozoan methylome and placental transcriptome datasets. Overall, these analyses delve into the mechanistic complexities of one-carbon metabolism during development and epigenetic inheritance of phenotype.
This research was regulated under the Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012 following ethical review by the University of Cambridge Animal Welfare and Ethical Review Body.
MtrrGt(XG334)Byg (MGI:3526159) mouse line, referred to as the Mtrrgt mouse line, was generated when a β-geo gene-trap (gt) vector was inserted into intron 9 of the Mtrr gene in 129P2Ola/Hsd embryonic stem cells (ESCs) (Elmore et al., 2007; Padmanabhan et al., 2013). Mtrrgt ECSs were injected into C57Bl/6J blastocysts and upon germline transmission, the Mtrrgt allele was backcrossed into the C57Bl/6J genetic background for at least eight generations (Padmanabhan et al., 2013). Mtrr+/+ and Mtrr+/gt mice were generated from Mtrr+/gt intercrosses. Mtrrgt/gt mice were generated by Mtrrgt/gt intercrosses. Since the Mtrrgt allele has a multigenerational effect (Padmanabhan et al., 2013; Bertozzi et al., 2021; Blake et al., 2021), C57Bl/6J mice from The Jackson Laboratories (www.jaxmice.jax.org) were used as controls and were bred in-house and maintained separately from the Mtrrgt mouse line. The effects of the maternal grandpaternal Mtrrgt allele were determined by the following pedigree: F0 Mtrr+/gt males were mated to C57Bl/6J females. The resulting F1 Mtrr+/+ females were mated to C57Bl/6J males to generate F2 Mtrr+/+ conceptuses. Genotyping for Mtrr+ and Mtrrgt alleles was performed using PCR on DNA extracted from ear tissue or yolk sac using a three-primer reaction resulting in a wildtype band at 252 bp and a mutant band at 383 bp (Padmanabhan et al., 2013). Primer sequences: primer a (5′-GAGATTGGGTCCCTCTTCCAC), primer b (5′-GCTGCGCTTCTGAATCCACAG), and primer c (5′-CG ACTTCCGGAGCGGATCTC) (Padmanabhan et al., 2013). All mice were housed in a temperature-and humidity-controlled environment with a 12 h light-dark cycle. All mice were fed a normal chow diet (Rodent No. 3 chow, Special Diet Services) ad libitum from weaning, which included (per kg of diet): 1.6 g choline, 2.73 mg folic acid, 26.8 μg vitamin B12, 3.4 g methionine, 51.3 mg zinc.
Noon of the day that the vaginal plug was detected was defined as embryonic (E) day 0.5. Mice were euthanized by cervical dislocation. Fetuses and placentas were dissected in cold 1x phosphate buffered saline (PBS) at E10.5 using a Zeiss SteReo Discovery V8 microscope, scored for phenotypes, and photographed. Fetuses and placentas were weighed and measured separately and snap frozen in liquid nitrogen (stored at −80°C). Both male and female placentas were assessed since no phenotypic sexual dimorphism was identified at E10.5 (Padmanabhan et al., 2017).
Conceptuses were rigorously scored for gross phenotypes during dissection and allocated to the phenotypic categories that were previously defined, including phenotypically normal (PN), fetal growth enhancement (FGE), fetal growth restriction (FGR), developmental delay, severe abnormalities (e.g., congenital heart defects, neural tube closure defects, hemorrhages, skewed conceptus orientation, twinning, etc.), and resorption (Padmanabhan et al., 2013; Wilkinson et al., 2021). Notably, conceptuses with >1 phenotype were counted once and classified by the most severe phenotype observed. Only PN, FGR, and FGE conceptuses were assessed in this study. Phenotype parameters are defined below.
Fetuses and placentas met all developmental milestones appropriate for the developmental stage according to e-Mouse Atlas Project (https://www.emouseatlas.org/emap/home.html). PN fetuses at E10.5 contained 30–39 somite pairs and had crown-rump lengths that were within two standard deviations (sd) from the mean of C57Bl/6J fetuses at E10.5, putting them within the normal range for growth. All PN conceptuses lacked abnormalities identified via gross assessment.
Conceptuses with FGR and FGE lacked abnormalities identified via gross dissection and met the staging criteria for E10.5 (i.e., 30–39 somite pairs). Yet, the fetuses displayed crown-rump lengths that were ≥2 sd below (for FGR) or above (for FGE) the mean crown-rump length for C57Bl/6J fetuses (Padmanabhan et al., 2013). Conceptus size was unaffected by litter size in all pedigrees and stages assessed (Padmanabhan et al., 2017).
Whole placentas at E10.5 were homogenized using a MagNA Lyser Instrument (Roche) and incubated on an Eppendorf ThermoMixer at 1,000 rpm at 56°C for 10 min. Genomic DNA was extracted using a QIAamp Fast DNA Tissue kit (Qiagen) following the manufacturer’s instructions. MeDIP-Seq was carried out as described previously (Ficz et al., 2011). Briefly, genomic DNA was sonicated to yield 150–600 bp fragments, and adaptors for paired-end sequencing (Illumina) were ligated using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs). Immunoprecipitations were carried out using 500 ng DNA per sample, 1.25 μg anti-5mC antibody (Eurogentec Cat# BI-MECY-0100, RRID:AB_2616058) or mouse immunoglobulin G (IgG) control and 10 μL Dynabeads coupled with M-280 sheep anti-mouse antibody (Invitrogen). Pulled down DNA was amplified for 12 cycles (meDIP) or 15 cycles (IgG control) with adapter-specific indexed primers. Final clean-up and size selection was carried out with AMPure-XP SPRI beads (Beckman Coulter). Libraries were quantified and assessed using the Kapa Library Quantification Kit (Kapa Biosystems) and Bioanalyzer 2100 System (Agilent). Indexed libraries were sequenced (50-bp paired-end) on an Illumina HiSeq 2500 sequencer. Raw fastq data were trimmed with TrimGalore (v0.6.6), using default parameters, and unique reads mapped to the Mus musculus GRCm38 genome assembly using Bowtie2 (v2.4.1). Data analysis was carried out using SeqMonk software (www.bioinformatics.babraham.ac.uk).
Whole male placentas at E10.5 were homogenized using lysing matrix D beads. RNA library preparation and sequencing was performed by Cambridge Genomic Services, Department of Pathology, University of Cambridge. The concentration and purity of RNA was determined by a SpectroStar spectrophotometer (BMG LABTECH) and the RNA integrity was determined by an Agilent Tapestation Bioanalyzer (Aligent Technologies LDA United Kingdom Ltd.). Libraries were prepared using 200 ng of total RNA and TruSeq stranded mRNA Library Preparation kit (Illumina). A unique index sequence was added to each RNA library to allow for multiplex sequencing. Libraries were pooled and sequenced on the Illumina NextSeq500 platform with 75 bp single-end reads. Sequencing was performed in duplicate to provide >18 million reads per sample. To monitor sequencing quality control, 1% PhiX Control (Illumina) spike-in was used. Quality control of Fastq files was performed using FastQC and fastq_screen. Sequences were trimmed with Trim Galore! and aligned to GRCm38 mouse genome using STAR aligner. Alignments were processed using custom ClusterFlow (v0.5dev) pipelines and assessed using MultiQC (0.9. dev0). Gene quantification was determined with HTSeq- Counts (v0.6.1p1). Additional quality control was performed with rRNA and mtRNA counts script, feature counts (v 1.5.0- p2) and qualimap (v2.2). Differential gene expression was performed with DESeq2 package (v1.22.2, R v3.5.2). Read counts were normalised on the estimated size factors.
To include transposon-derived reads that do not map uniquely, the meDIP-seq datasets were re-aligned using the default settings of bowtie2 to assign reads with multiple equally best alignments to one of those locations at random. Average methylation levels over pro-viral, full-length elements were generated after merging Repeatmasker annotations for RLTR4_Mm and RLTR4_MM-int elements. RNA-seq data was analysed using SQuIRE (Yang et al., 2019), which assigns multimapping reads using an expectation-maximisation algorithm and provides both subfamily-level and single copy-level information. Differential expression analysis was performed using SQuIRE’s Call function.
First, we analysed the extent to which impaired one-carbon metabolism affected the placental methylome and ascertained whether there was an impact on fetal growth. Our initial focus was on Mtrrgt/gt placentas of conceptuses derived from Mtrrgt/gt intercrosses (Figure 1A). We carried out high-throughput sequencing of immunoprecipitated methylated DNA (meDIP-seq) from C57Bl/6J control and Mtrrgt/gt placentas at E10.5. Mtrrgt/gt placentas were divided into two phenotypic groups including those from fetuses that were phenotypically normal (PN) or were FGR based on crown-rump length (Padmanabhan et al., 2013; Wilkinson et al., 2021). Placentas from C57Bl/6J mice were controls since the Mtrrgt allele was backcrossed into the C57Bl/6J genetic background (Padmanabhan et al., 2013). However, we previously identified four regions of structural variation between C57Bl/6J and the Mtrrgt line (Blake et al., 2021). To avoid false discovery of changes in DNA methylation during the meDIP-seq data analysis, these regions were excluded bioinformatically along with the 20 Mb region of 129P2Ola/Hsd genomic sequence surrounding the gene-trapped Mtrr allele (Bertozzi et al., 2021; Blake et al., 2021), that remained after eight backcrosses (Padmanabhan et al., 2013).
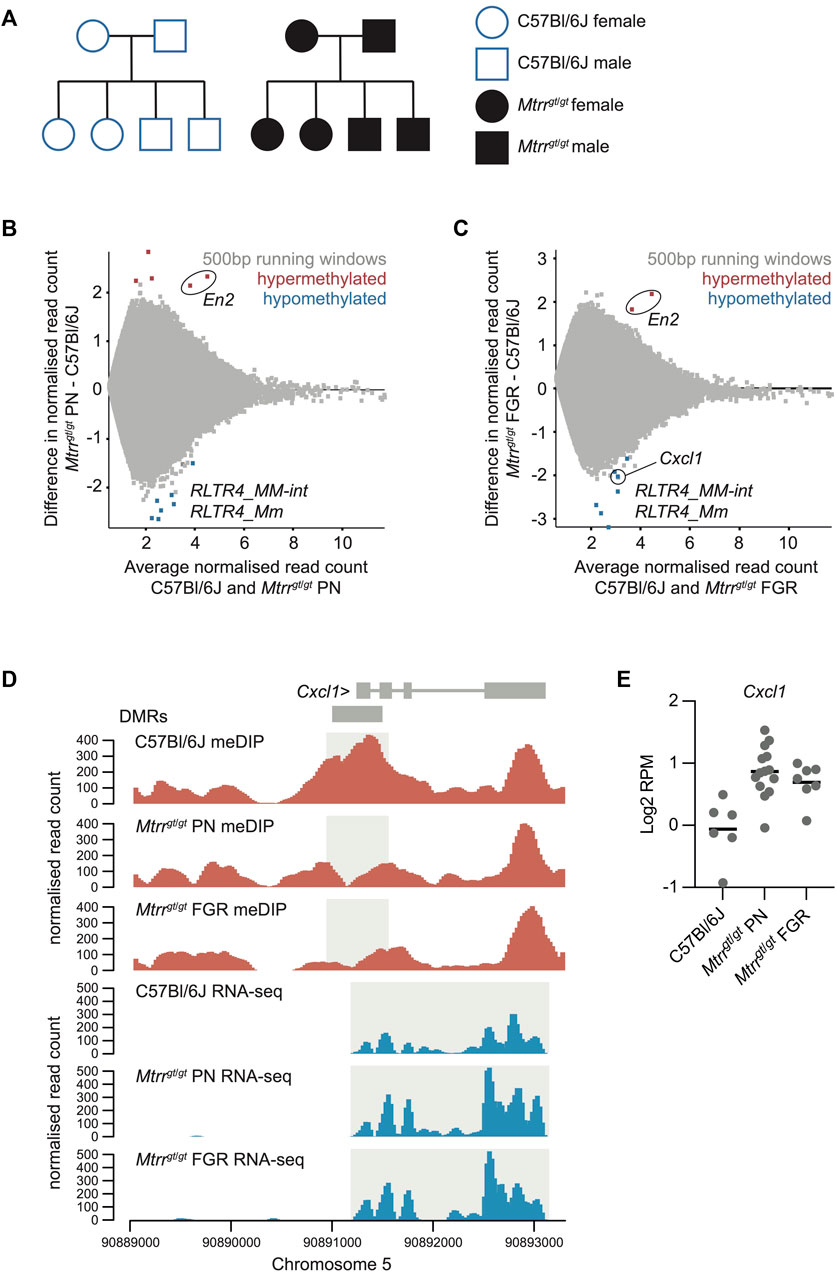
FIGURE 1. Analysis of the Mtrrgt/gt placental methylome. (A) Schematization of the C57Bl6J control pedigree (blue outline, white fill) and Mtrrgt/gt pedigree (black outline, black fill) used in this study. Square, males; Circles, females. (B,C) MA plot of log2 normalized meDIP-seq read counts of 500 bp contiguous regions in (B) C57Bl/6J placentas and Mtrrgt/gt placentas from phenotypically normal (PN) fetuses, and (C) C57Bl/6J placentas and Mtrrgt/gt placentas associated with fetal growth restriction (FGR). Hypermethylated (red) and hypomethylated (blue) differentially methylated 500 bp regions (DMR) were identified using EdgeR. (D) Data tracks showing normalized meDIP-seq (red) and RNA-seq (blue) reads across the Cxcl1 locus on mouse chromosome 5 in C57Bl/6J, Mtrrgt/gt PN and Mtrrgt/gt FGR placentas. DMR and transcript expression are highlighted in light grey. (E) Graph showing Cxcl1 transcript expression (log2RPM) ascertained by RNA-seq in C57Bl/6J, Mtrrgt/gt PN, and Mtrrgt/gt FGR placentas at E10.5. In all cases data was normalized to the largest data store. For meDIP-seq: C57Bl/6J, N = 8 placentas; Mtrrgt/gt PN, N = 7 placentas; Mtrrgt/gt FGR, N = 7 placentas. For RNA-seq: C57Bl/6J, N = 6 placentas; Mtrrgt/gt PN, N = 14 placentas; Mtrrgt/gt FGR, N = 7 placentas.
At the global level, we found that the distribution of meDIP-seq reads across different genomic features were not significantly different between C57Bl/6J control and Mtrrgt/gt placentas even when phenotypic severity was considered (Supplementary Figure S1A). Furthermore, meDIP-seq datasets from individual placentas did not cluster by Mtrr genotype or fetal growth phenotype when data store similarity tools were implemented (Supplementary Figure S1B). As global DNA methylation patterns were similar between experimental groups, we next ascertained differences in DNA methylation at individual loci compared to control placentas. DMRs were defined using the EdgeR function embedded within Seqmonk software (www.bioinformatics.babraham.ac.uk) with default settings (p < 0.05, with multiple testing correction) assessing 500 bp contiguous regions. The resulting DMRs were further filtered for regions that displayed a log2 fold change (FC) > 1 in DNA methylation compared to controls. Only a few DMRs were present in Mtrrgt/gt placentas (i.e., PN: 13 DMRs; FGR: 9 DMRs), though both hyper- and hypomethylated regions were observed (Figures 1B, C; Supplementary File S1). The low number of DMRs caused by the Mtrrgt allele suggested that DNA methylation changes were subtle regardless of fetal growth phenotypes or were “hidden” by our analysis of whole placentas as individual cell types might be differently affected.
Despite the low number of total DMRs, three key findings emerged (explored further below). Firstly, only one placental DMR associated with the misexpression of a protein-coding gene (Cxcl1; Figures 1D, E). Secondly, we identified two hypermethylated DMRs located within the En2 gene in Mtrrgt/gt placentas that were common to PN and FGR conceptuses (Figures 1B, C). Since the En2 DMRs were also identified in mature spermatozoa from Mtrrgt/gt males and in Mtrrgt/gt embryos at E10.5 (Blake et al., 2021), they were flagged for further analysis in the context of development and epigenetic inheritance. Lastly, of the 12 hypomethylated DMRs that were identified (i.e., 3 shared DMRs in PN and FGR placentas, 5 DMRs in PN placentas only, 4 DMRs in FGR placentas only), ten were associated with endogenous retroviruses (ERVs; Supplementary File S1). Strikingly, the majority of these DMRs (7/10) overlapped with ERV1 elements of the RLTR4 subclass (Figures 1B, C) with implications for genetic stability. Further exploration into the importance of these findings was explored below.
To explore whether altered placental DNA methylation caused by the Mtrrgt/gt genotype had a gene regulatory effect, we carried out RNA-seq on Mtrrgt/gt placentas at E10.5 associated with PN and FGR fetuses. Using DESeq, the RNA-seq data was assessed for differentially expressed genes that were within 2 kb of a DMR (identified in Mtrrgt/gt placentas) and had transcript levels with a log2FC > 0.6 compared to control placentas. The Cxcl1 gene [chemokine (C-X-C motif) ligand 1] was the only dysregulated gene identified in this context. We observed that hypomethylation at the DMR located in the promoter of Cxcl1 was associated with a modest upregulation of Cxcl1 transcripts (Figures 1D, E). This finding exemplifies canonical regulation of a gene by DNA methylation. Since Mtrrgt/gt placentas from both PN and FGR fetuses displayed hypomethylation at the Cxcl1 DMR and upregulation of Cxcl1 transcripts (Figures 1D, E), these molecular changes were likely insufficient to drive the fetal growth phenotype. Yet, CXCL1 is important for decidual angiogenesis to promote maternal blood flow into the implantation site (Ma et al., 2021). Therefore, dysregulation of Cxcl1 mRNA in Mtrrgt/gt placentas might have implications for fetoplacental development beyond fetal growth.
The only two hypermethylated DMRs identified in Mtrrgt/gt placentas were found within the En2 gene. Therefore, their functional importance was explored. These two 500 bp DMRs were in fact contiguous and represented one single 1 kb region in the single En2 intron (Figure 2A). The En2 gene encodes a homeobox transcription factor that, when knocked out in mice, leads to autism-spectrum disease-like behaviours (Cheh et al., 2006; Brielmaier et al., 2012; Provenzano et al., 2014) that are accompanied by cerebellar foliation defects (Joyner et al., 1991) and loss of GABAergic interneurons in somatosensory and visual cortical areas (Sgado et al., 2013; Allegra et al., 2014). Indeed, En2 mRNA is expressed in multiple regions of the developing brain (Davis et al., 1988) and is involved in neurogenesis (Lee et al., 1997). Low levels of En2 transcripts were reported by RNA-seq in the ectoplacental cone (Bastian et al., 2021) (a population of trophoblast progenitor cells in the mouse placenta). However, our RNA-seq data from whole C57Bl/6J control placentas at E10.5 showed that En2 transcripts were very lowly expressed (Figure 2B) and thus, En2 might be considered as an unexpressed gene in the placenta at this developmental stage. Importantly, hypermethylation of the En2 DMR in Mtrrgt/gt placentas was not associated with a change in En2 transcript levels (Figure 2B) indicating that the En2 DMR is an unlikely regulator of En2 gene expression in the placenta.
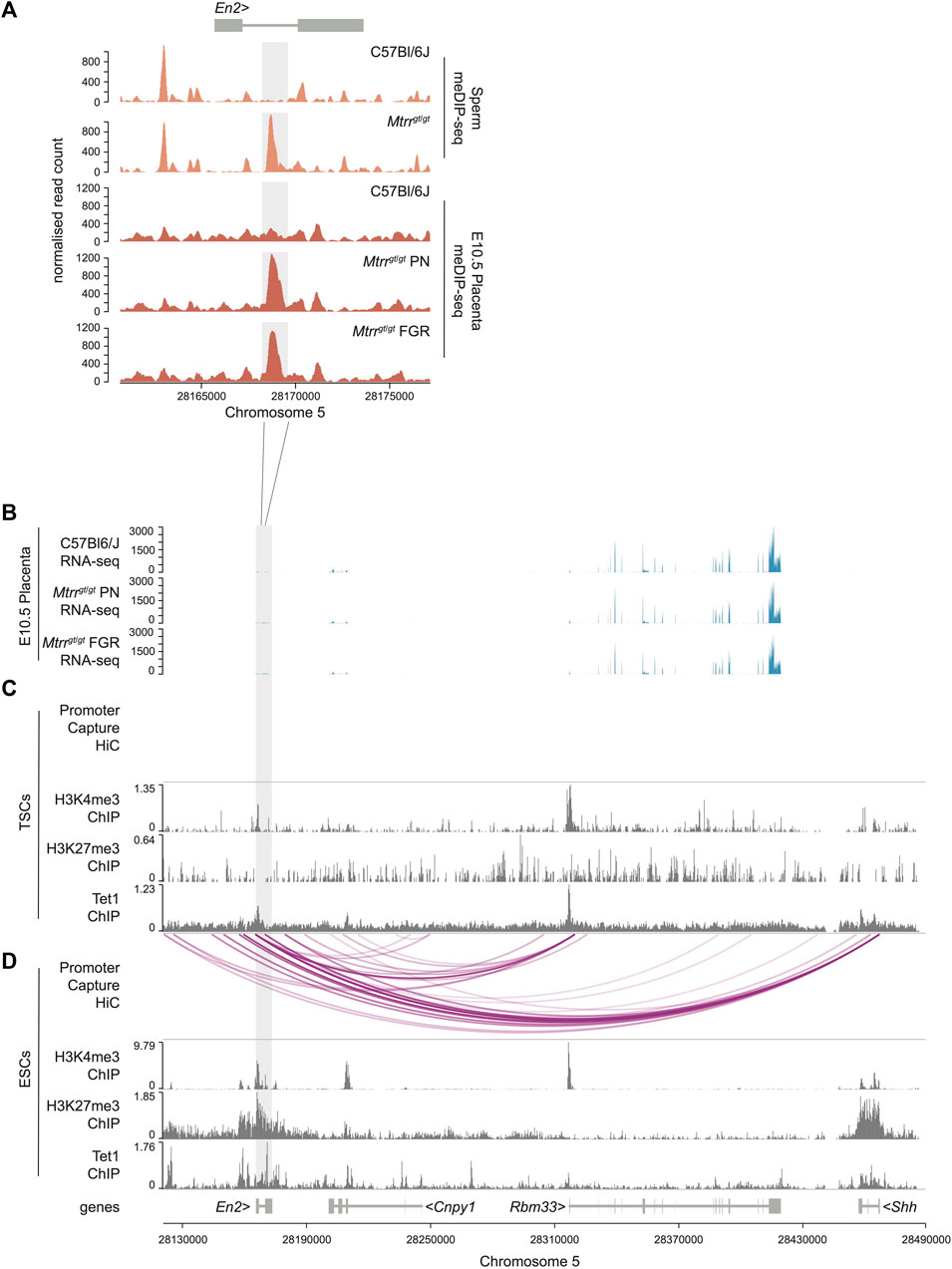
FIGURE 2. En2 DMR as a potential regulator of developmentally important genes in ESCs and not TSCs. (A) Data tracks showing normalized meDIP-seq read counts across the En2 gene in spermatozoa from C57BL/6J and Mtrrgt/gt mice (orange) and placentas from C57Bl/6J and Mtrrgt/gt conceptuses at E10.5 (red). Placentas were associated with either phenotypically normal (PN) or fetal growth restricted (FGR) fetuses. The En2 DMR is highlighted in light grey. (B) RNA-seq data tracks showing gene expression (blue) in the genomic region near to the En2 DMR (light grey) in placentas at E10.5 from C57Bl/6J and Mtrrgt/gt conceptuses. Placentas from PN and FGR fetuses were considered. (C,D) Data tracks showing complete promoter capture HiC-based interactions (purple lines) and H3K27me3, H3K4me3 and Tet1 ChIP-seq peaks (dark grey) at the En2 locus and downstream genes within (C) wildtype mouse trophoblast stem cells (TSCs) and (D) wildtype mouse embryonic stem cells (ESCs). See also Supplementary File S3 for data sources. For sperm meDIP-seq: C57Bl/6J, N = 8 males; Mtrrgt/gt, N = 8 males. For placenta meDIP-seq: C57Bl/6J, N = 8 placentas; Mtrrgt/gt PN, N = 7 placentas; Mtrrgt/gt FGR, N = 7 placentas.
To investigate a broader regulatory role of the En2 DMR in the placenta, we explored histone methylation (e.g., H3K4me3 and H3K27me3) enrichment and potential interactions of the DMR with neighbouring genes. To do this, we analysed published H3K4me3 and H3K27me3 ChIP-seq datasets and promoter capture Hi-C datasets from wildtype mouse trophoblast stem cells (TSCs) (Schoenfelder et al., 2018). TSCs are an in vitro model of undifferentiated trophoblast cells of the placenta (Tanaka et al., 1998), and these datasets represent the most suitable available for analysis. No enrichment of H3K4me3 or H3K27me3 modifications was evident at the En2 DMR in TSCs and no complete DMR-promoter interactions were evident in TSCs within the genomic region assessed (Figures 2B, C). Accordingly, genes downstream of the En2 DMR were also expressed at normal levels in Mtrrgt/gt placentas (Figure 2B) indicating that hypermethylation of this region had little to no effect on cis regulation of gene expression in the placenta.
The hypermethylated En2 DMR is prevalent in different tissue types including mature spermatozoa of Mtrrgt/gt males and Mtrrgt/gt embryos and placentas at E10.5 (Blake et al., 2021; this study). Given that the En2 gene is important for the development of embryonic lineages [e.g., neurogenesis (Lee et al., 1997)], the potential regulatory importance of the En2 DMR was explored outside of the placenta. Additional ChIP-seq and promoter capture Hi-C datasets from wildtype mouse ESCs (Schoenfelder et al., 2018) were analysed in the region proximal to the En2 DMR. The data revealed that the En2 DMR had hallmarks of a regulatory locus in ESCs since it was bivalently marked by the enrichment of repressive H3K27me3 and active H3K4me3 modifications (Figure 2D) in a manner that poises this region for activation upon cell differentiation (Macrae et al., 2023). The genomic region defined by the En2 DMR in ESCs was also enriched for the DNA demethylating enzyme TET1 (Figure 2D), which typically co-localises with polycomb complexes and contributes to keeping unmethylated enhancers and promoters methylation-free (Parry et al., 2021). Furthermore, promoter capture Hi-C experiments in ESCs (Schoenfelder et al., 2018) revealed a potential interaction of the En2 DMR with the promoters of nearby genes, including Cnpy1 (canopy FGF signalling regulator 1), Rbm33 (RNA binding motif 33), and the developmental regulator Shh (sonic hedgehog) (Figure 2D). These data contrasted the TSCs data, which showed no such DMR-promoter interactions (Figure 2C). Therefore, we hypothesised that ectopic hypermethylation of the En2 DMR specifically within Mtrrgt/gt embryos might affect expression of surrounding genes with developmental consequences. Further analysis of the developmental role of the En2 DMR in the embryo is required, particularly in the context of abnormal one-carbon metabolism.
We identified ten hypomethylated DMRs in Mtrrgt/gt placentas at E10.5 that were associated with ERV elements (Supplementary File S1). Specifically, seven of these overlapped with RLTR4 elements of the ERV1 subfamily (separately annotated as RLTR4_Mm and RLTR4_MM-int for the LTRs and internal region, respectively; Figures 1B, C). RLTR4 elements are relatively young retrotransposons that are closely related to murine leukemia virus and that, at least in some mouse strains, remain transpositionally active (Maksakova et al., 2006). Some transposable elements (e.g., IAPs) are highly methylated and resistant to epigenetic reprogramming to avoid genomic transposition (Kobayashi et al., 2012). It is unclear whether this is the case for RLTR4 elements. The RLTR4 elements that associated with placental DMRs in this study typically displayed a pro-viral, full-length configuration, rather than being solo LTRs or other isolated fragments. Furthermore, six out of seven of the RLTR4-associated DMRs mapped to two discrete genomic loci on mouse chromosomes 11 or 18, including in regions that were intragenic (and antisense) to Camk2b or were upstream of the gene Pik3c3, respectively (Figures 3A, B). Remarkably, a loss of DNA methylation at these DMRs corresponded with ectopic expression of the RLTR4 element in Mtrrgt/gt placentas, independent of the fetal growth phenotype (Figures 3A, B). However, no expression changes in the associated protein-coding genes were observed (Figures 3A, B). Therefore, the genomic regions demarcated by these DMRs appear to require methylation to repress the ERV element activity and not to regulate cis gene expression in the placenta.
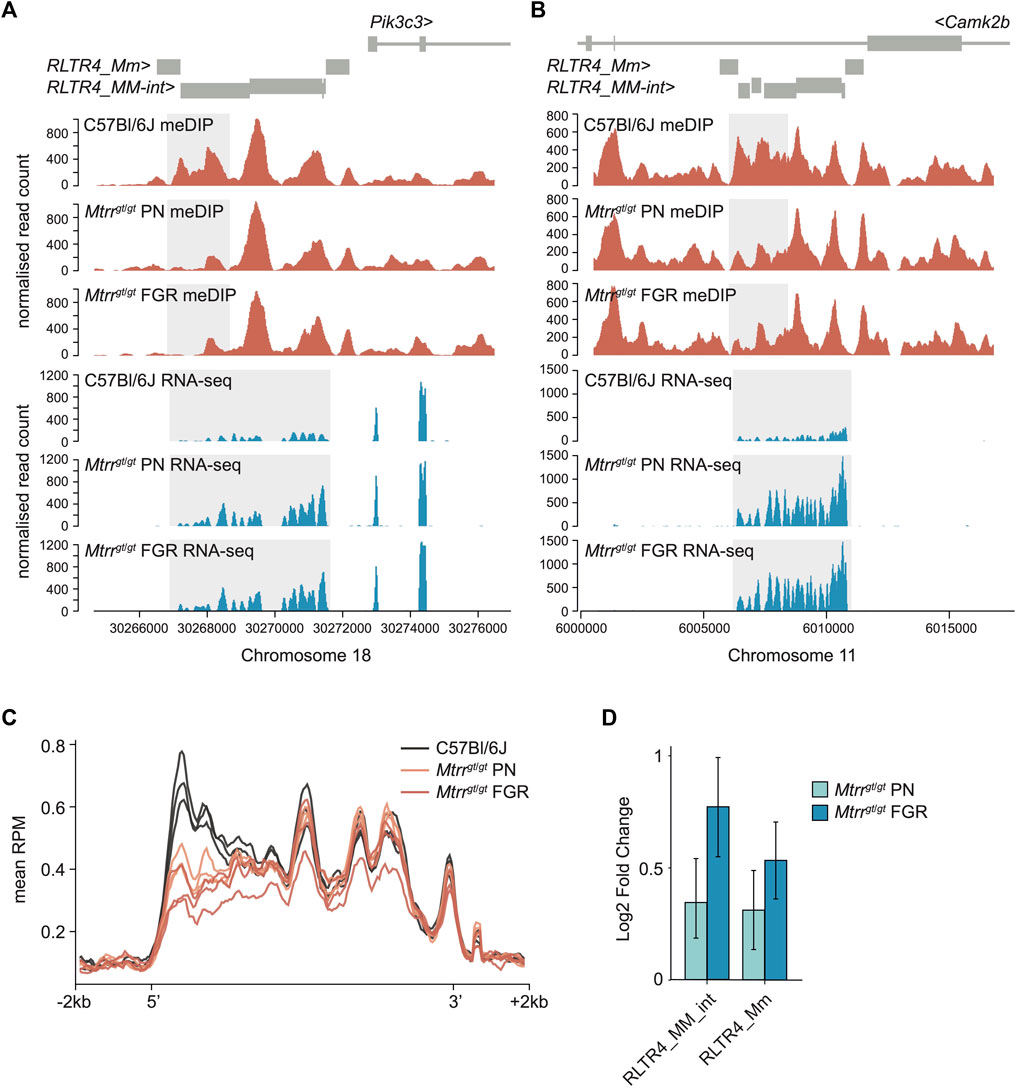
FIGURE 3. Analysis of DNA methylation and transcript expression at ERV subfamily in Mtrrgt/gt placentas. (A,B) Data tracks showing normalized meDIP-seq (red) and RNA-seq (blue) reads across full-length ERVs comprising RLTR4_Mm and RLTR4_MM-int elements on mouse (A) chromosome 18 associated with the Pik3c3 gene and (B) chromosome 11 associated with the Camk2b gene in placentas of C57Bl/6J and Mtrrgt/gt conceptuses at E10.5. Placentas from phenotypically normal (PN) and fetal growth restricted (FGR) fetuses were assessed. Differentially methylated region (DMR) and transcript expression are highlighted in light grey. (C) Graph representing the average meDIP-seq reads mapping across all full-length RLTR4 elements ± 2 kb in the genome in individual placentas from C57Bl/6J (black), Mtrrgt/gt PN (orange) and Mtrrgt/gt FGR (red) fetuses at E10.5. (D) Enrichment of RLTR4_Mm and RLTR4_MM-int expression in placentas from Mtrrgt/gt PN (light blue) and Mtrrgt/gt FGR (dark blue) fetuses at E10.5 relative to C57Bl/6J control placentas as determined by RNA-seq. For meDIP-seq: C57Bl/6J, N = 8 placentas; Mtrrgt/gt PN, N = 7 placentas; Mtrrgt/gt FGR, N = 7 placentas. For RNA-seq: C57Bl/6J, N = 6 placentas; Mtrrgt/gt PN, N = 14 placentas; Mtrrgt/gt FGR, N = 7 placentas.
Due to their repetitive nature and evolutionary young age, the mapability of short sequencing reads to RLTR4 elements is low. Therefore, to fully appreciated the dysregulation of DNA methylation at RLTR4 elements in Mtrrgt/gt placentas, the placental meDIP-seq and RNA-seq datasets from C57Bl/6J and Mtrrgt/gt placentas at E10.5 were re-mapped to include non-unique reads by using random assignment (bowtie2) for meDIP-seq data and an expectation-maximisation algorithm [SQuIRE (Yang et al., 2019)] for RNA-seq data. When considered globally, the remapped data revealed consistent DNA hypomethylation at the 5′ end of RLTR4 full-length elements in Mtrrgt/gt placentas at E10.5 (Figure 3C). This pattern of DNA hypomethylation was associated with transcript enrichment of global RLTR4_Mm and RLTR4_MM-int elements in Mtrrgt/gt placentas compared to controls (Figure 3D). Differential expression analysis of individual elements uncovered significant upregulation of twenty-four RLTR4_Mm or RLTR4_MM-int elements that converged upon 15 full-length loci (Supplementary File S2). None of these methylation changes associated with altered expression of nearby protein-coding genes in Mtrrgt/gt placentas. Furthermore, methylation and transcriptional dysregulation at RLTR4_Mm and RLTR4_Mm-int elements was unlikely to regulate fetal growth since the RLTR4 elements were similarly affected in Mtrrgt/gt placentas associated with PN and FGR fetuses (Figure 3D). Overall, these data reinforced the hypothesis that epigenetic instability is inherent to the Mtrrgt mouse line (Padmanabhan et al., 2013; Blake et al., 2021) with implications for genetic stability and phenotype establishment beyond FGR.
In our previous study of epigenetic inheritance in the Mtrrgt mouse line (Blake et al., 2021), we found that a small number of candidate DMRs identified in mature spermatozoa (obtained from the cauda epididymis and vas deferens) were not recapitulated in embryos or placentas at E10.5 when interrogated by bisulfite pyrosequencing. Here, we aimed to validate this finding on a genome-wide scale by comparing our spermatozoa (Blake et al., 2021) and placenta meDIP-seq datasets from control and Mtrrgt/gt mice. First, we harmonised DMR calling between datasets by reanalysing the spermatozoa meDIP-seq datasets according to our analysis of the placenta meDIP-seq data. Hypermethylated spermatozoa DMRs in Mtrrgt/gt males that were previously identified in very highly methylated regions in control spermatozoa and described as false positives (Blake et al., 2021), were also clearly identifiable by the current analysis. Accordingly, we screened out these DMRs using whole genome bisulphite sequencing data to quantify absolute methylation levels across all DMRs (Sun et al., 2018). Similar to our candidate-based approach (Blake et al., 2021), DNA methylation patterns in nearly all genomic regions identified as spermatozoa DMRs in Mtrrgt/gt males were normal in Mtrrgt/gt placentas at E10.5 compared to control placentas (Figures 4A–C). This finding occurred regardless of the Mtrrgt/gt fetal growth phenotype. The only exception was the common hypermethylated En2 DMR that appeared in both spermatozoa and placentas (Figures 4B, C). Conversely, the placental DMRs that overlap with RLTR4 elements were normally methylated in sperm of Mtrrgt/gt males (relative to control spermatozoa) suggesting that germline transposon silencing is maintained and unlikely to play a key role in epigenetic inheritance mechanisms in the Mtrrgt mouse line. Overall, these results indicated that placenta and spermatozoa DMRs in Mtrrgt/gt mice were tissue-specific and that most of the spermatozoa DMRs were effectively reprogrammed in the pre-implantation embryo or during placental development, notwithstanding the shared Mtrrgt/gt genotype of the parental and offspring generations. These data might negate DNA methylation as a mechanistic factor in epigenetic inheritance within the Mtrrgt mouse line. Instead, altered heritability of other epigenetic factors, such as histone modifications or small non-coding RNA content in germ cells, might be an alternative or additional mechanism.
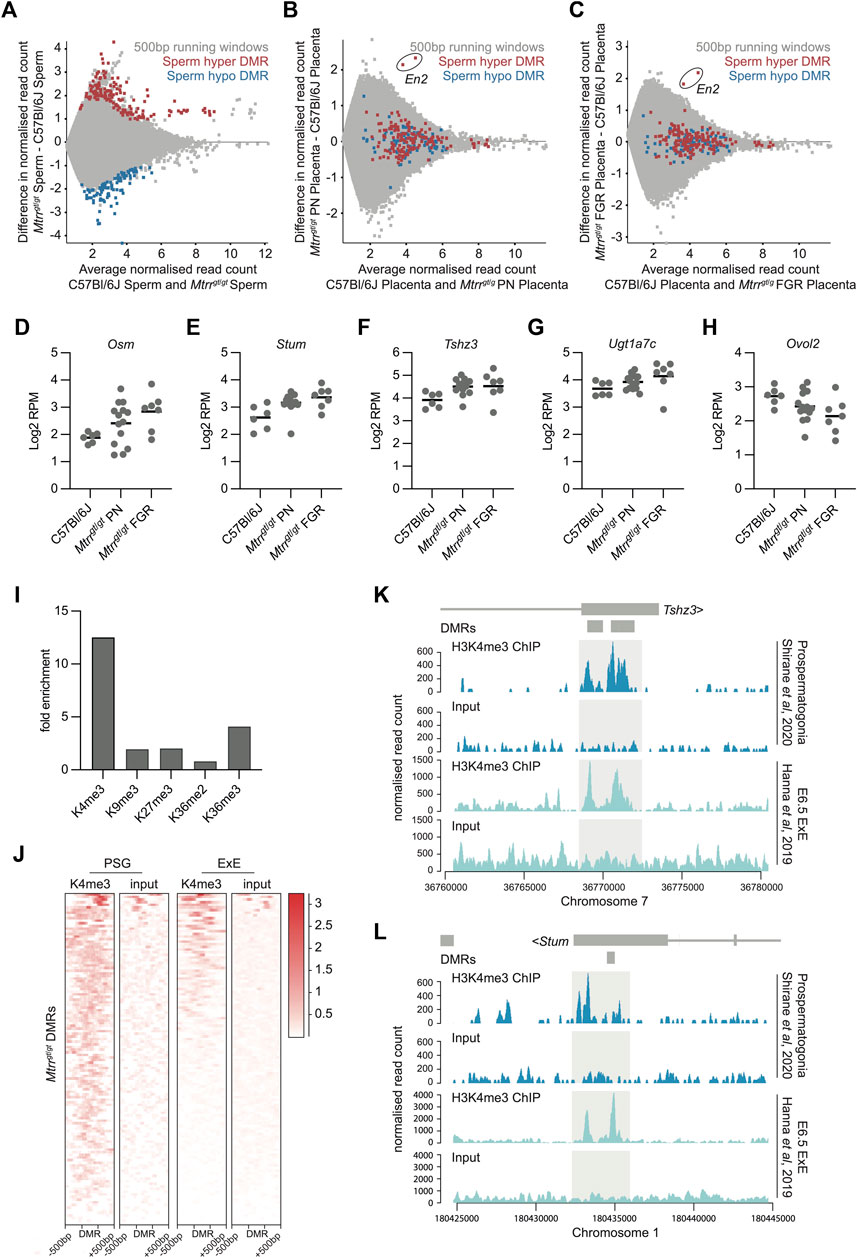
FIGURE 4. Spermatozoa DMRs in Mtrrgt/gt males were normalized in Mtrrgt/gt placentas yet associated with transcriptional dysregulation. (A) MA plot of log2 normalized meDIP-seq read counts of 500 bp contiguous regions in spermatozoa from C57Bl/6J and Mtrrgt/gt males. Hypermethylated (red) and hypomethylated (blue) differentially methylated 500 bp regions (DMRs) were identified using EdgeR. (B) MA plot of log2 normalized meDIP-seq read counts of 500 bp contiguous regions in placentas from C57Bl/6J and Mtrrgt/gt phenotypically normal fetuses at E10.5. The genomic regions where spermatozoa DMRs from Mtrrgt/gt males were identified are highlighted on the placenta data. Hypermethylated spermatozoa DMRs (red), hypomethylated spermatozoa DMRs (blue). The En2 DMRs are indicated. (C) MA plot of log2 normalized meDIP-seq read counts of 500 bp contiguous regions in placentas from C57Bl/6J fetuses and Mtrrgt/gt fetal growth restricted (FGR) fetuses at E10.5. The genomic regions where spermatozoa DMRs from Mtrrgt/gt males were identified are highlighted on the placenta data. Hypermethylated spermatozoa DMRs (red), hypomethylated spermatozoa DMRs (blue). The En2 DMRs are indicated. (D–H) Graphs showing placental transcript expression (log2RPM) of genes that were associated with spermatozoa DMRs including (D) Osm, (E) Stum, (F) Tshz3, (G) Ugt1a7c, and (H) Ovol2. Data was ascertained by RNA-seq of placentas from C57Bl/6J and Mtrrgt/gt conceptuses at E10.5. Placentas from phenotypically normal (PN) and fetal growth restricted (FGR) fetuses were assessed. (I) Enrichment for specific histone modifications in wildtype prospermatogonia ascertained by ChIP-seq at the 500 bp regions defined as DMRs in spermatozoa of Mtrrgt/gt males. Enrichment determined relative to the baseline genome. (J) Probe alignment plot showing H3K4me3 enrichment ascertained by ChIP-seq from wildtype prospermatogonia and extraembryonic ectoderm (ExE) at E6.5 compared to input controls in regions identified as spermatozoa DMRs (± 500 bp) in Mtrrgt/gt males. (K,L) Data tracks showing normalized H3K4me3 ChIP-seq reads and input controls for prospermatogonia (dark blue) and extraembryonic ectoderm (ExE) at E6.5 (light blue) in the regions surround the (K) Tshz3 and (L) Stum sperm DMRs from Mtrrgt/gt males. Light grey boxes highlight H3K4me3 peaks. Small dark grey boxes indicate the DMRs. See also Supplementary File S3 for data sources. For spermatozoa meDIP-seq: C57Bl/6J, N = 8 males; Mtrrgt/gt, N = 8 males. For placenta meDIP-seq: C57Bl/6J, N = 8 placentas; Mtrrgt/gt PN, N = 7 placentas; Mtrrgt/gt FGR, N = 7 placentas. For RNA-seq: C57Bl/6J, N = 6 placentas; Mtrrgt/gt PN, N = 14 placentas; Mtrrgt/gt FGR, N = 7 placentas.
Previously, our locus-specific analysis showed that some genes associated with spermatozoa DMRs were misexpressed in somatic tissues despite reprogramming of DNA methylation at these sites (Blake et al., 2021). Therefore, we questioned the extent to which this association occurred in the wider placental genome. Using DESeq, the Mtrrgt/gt placental RNA-seq dataset was assessed for differentially expressed genes that were within 2 kb of a sperm DMR from Mtrrgt/gt males and had transcript levels with a log2FC > 0.6 compared to control placentas. Five misexpressed genes (i.e., Stum (mechanosensory transducer mediator; membrane protein), Tshz3 (teashirt zinc finger family member 3; transcription factor), Ovol2 (ovo like zinc finger 2; transcription factor), Osm (oncostatin m; cytokine), and Ugt1a7c (UDP glucouronosyltransferase 1 family, polypeptide A7C; enzyme in glucouronidation pathway) met these criteria but only in Mtrrgt/gt placentas with FGR (Figures 4D–H). The occurrence of transcriptional disruption despite normal DNA methylation reinforced our hypothesis that abnormal one-carbon metabolism influences other epigenetic mechanisms.
Next, we explored whether histone modifications were present in the developing germline at regions demarcated by spermatozoa DMRs to better understand the broader epigenetic context of these regions. To do this, ChIP-seq datasets were analysed for histone mark enrichment in developing wildtype male germ cells (i.e., prospermatogonia) (Shirane et al., 2020) at specific genomic regions defined by spermatozoa DMRs from Mtrrgt/gt males. First, we found that 103 out of 252 DMRs (40.9%) overlapped with an H3K4me3 peak in wildtype prospermatogonia, the majority which were located within gene bodies (Supplementary Table S1). This value represented a 12-fold enrichment compared to the baseline genome (i.e., only 3.3% of 500 bp regions across the whole genome overlapped with H3K4me3 peaks) and was substantially more enriched than the other histone modifications at the same locations (Figure 4I). Since H3K4me3 is typically associated with active transcription (Howe et al., 2017), it was an ideal candidate to further explore as an underlying inherited epigenetic mark associated with transcriptional disruption in the placenta. Therefore, we assessed whether wildtype trophoblast progenitor cells at E6.5 (i.e., extraembryonic ectoderm) displayed H3K4me3 enrichment at genomic locations identified as spermatozoa DMRs using a published ChIP-seq dataset (Hanna et al., 2019). Indeed, a substantial subset of these genomic regions was also enriched for H3K4me3 in extraembryonic ectoderm (Figure 4J). Remarkably, four out of five dysregulated genes in Mtrrgt/gt placentas that were associated with a spermatozoa DMR in Mtrrgt/gt males (i.e., Stum, Tshz3, Ovol2, Ugta7c) were among those enriched for H3K4me3 in both prospermatogonia and extraembryonic ectoderm (Figure 4K–L; Supplementary Figure S2). We infer from these data that the Mtrrgt allele potentially disrupts histone marks, such as H3K4me3, in developing and/or mature germ cells leading to altered patterns of the same histone mark in the early conceptus with implications for gene regulation. Indeed, we found that 59.0% of H3K4me3 peaks identified in extraembryonic ectoderm were also found in prospermatogonia (using MACS peak calling function embedded in SeqMonk software). This finding more broadly supports a role for H3K4me3 in inheritance of epimutations from germ cells to the placenta. Future mechanistic experiments should focus on multigenerational patterns of H3K4me3 in the Mtrrgt mouse line.
Our previous locus-specific analyses in F2 Mtrr+/+ placentas indicated significant alteration of DNA methylation patterns caused by either a maternal grandfather or maternal grandmother Mtrrgt allele (Padmanabhan et al., 2013). Here, a transgenerational mechanism was explored in the Mtrr+gt maternal grandfather pedigree (Figure 5A) using a genome-wide approach to identify the locations of spermatozoa DMRs from F0 Mtrr+/gt males (Blake et al., 2021) and determine whether the placental methylome and transcriptome was altered in these regions two generations later in the F2 Mtrr+/+ grandprogeny. The following matings were performed to generate this pedigree (Figure 5A): F0 Mtrr+/gt males were mated with C57Bl/6J control females, and the resulting F1 Mtrr+/+ females were selected for mating with C57Bl/6J males to generate F2 Mtrr+/+ conceptuses. F2 Mtrr+/+ conceptuses were rigorously phenotyped at E10.5, and the placentas from PN and FGE fetuses were examined. First, the broader methylome of whole F2 Mtrr+/+ placentas was assessed via meDIP-seq. When compared with C57Bl/6J controls, there were no significant differences in the distribution of meDIP reads across genomic features (Supplementary Figure S1C) and no clustering of biological replicates according to phenotype or pedigree (Supplementary Figure S1D) indicating similar global methylation among experimental groups. Few DMRs were identified by the meDIP-seq analysis including one hypermethylated DMR in F2 Mtrr+/+ placentas from PN fetuses (Figure 5B) and 11 DMRs (10 hypomethylated, 1 hypermethylated) in FGE-associated F2 Mtrr+/+ placentas (Figure 5C). Closer analysis revealed that 9 out of 10 of the hypomethylated DMRs from F2 Mtrr+/+ FGE placentas were clustered in two locations on chromosomes 14 and 17, which are frequently susceptible to mapping artefacts in our datasets and so were excluded. The remaining three placental DMRs from F2 Mtrr+/+ placentas were in nondescript genomic regions (Supplementary File S1). Importantly, the RLTR4 elements identified in Mtrrgt/gt placentas (Figure 3) exhibited normal levels of DNA methylation and transcript expression in the F2 Mtrr+/+ placentas relative to control placentas (Supplementary Figure S3). This result suggested that the changes in DNA methylation described in Mtrrgt/gt placentas are intrinsically associated with the Mtrrgt allele and are unlikely to be transgenerationally inherited or caused by genetic differences between the C57Bl/6J control and Mtrrgt mouse lines.
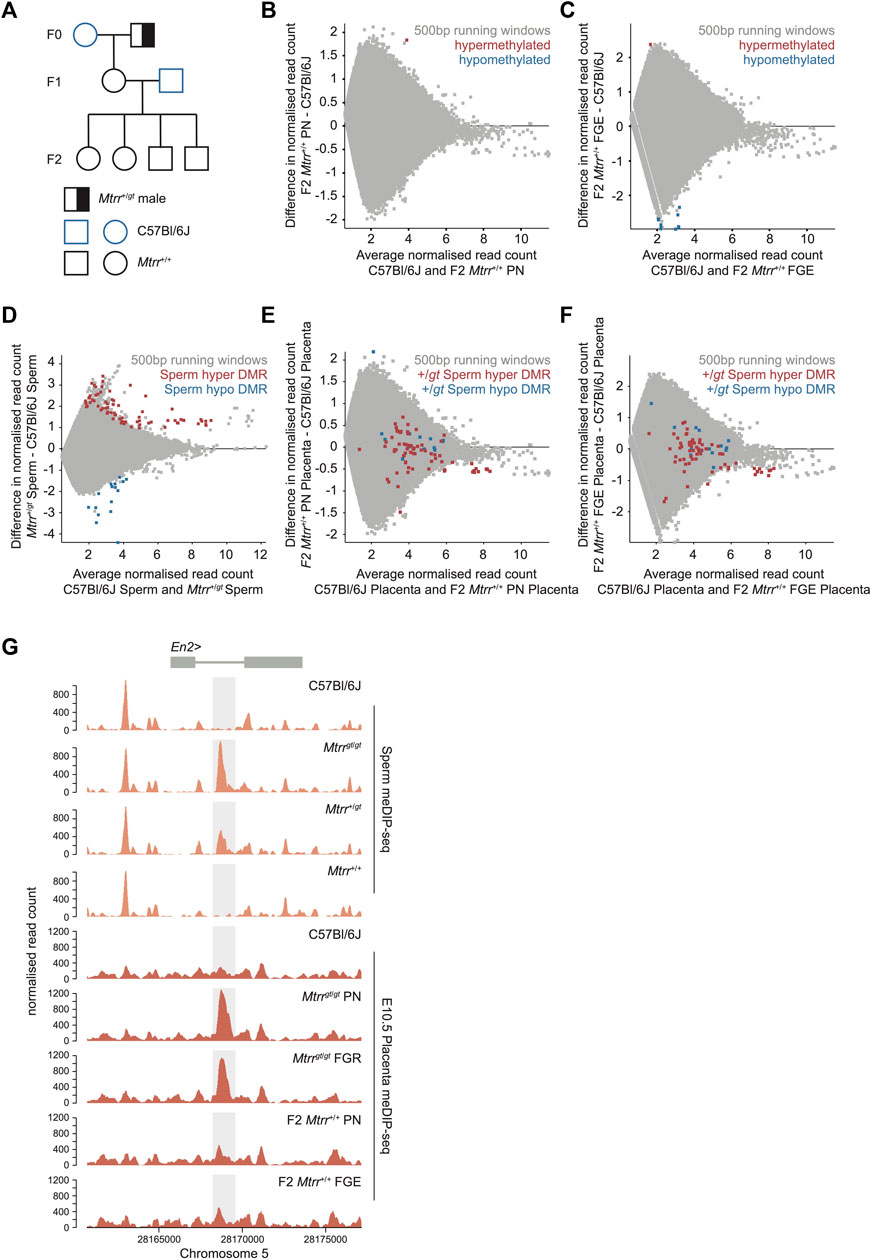
FIGURE 5. Spermatozoa DNA methylation patterns are not transgenerationally inherited in the Mtrrgt mouse line. (A) Mtrr+/gt maternal grandfather pedigree used in this study. Squares, males; circles females; blue outline, C57Bl/6J mouse line; black outline, Mtrrgt mouse line; white fill, Mtrr+/+; half black/half white fill; Mtrr+/gt. F0, parental generation; F1, first filial generation; F2, second filial generation. (B) MA plot of log2 normalized meDIP-seq read counts of 500 bp contiguous regions in placentas at E10.5 from C57Bl/6J conceptuses and F2 Mtrr+/+ conceptuses derived from F0 Mtrr+/gt maternal grandfathers. Placentas from phenotypically normal (PN) fetuses were assessed. Hypermethylated (red) and hypomethylated (blue) differentially methylated regions (DMRs) were determined relative to control placentas using EdgeR. (C) MA plot of log2 normalized meDIP-seq read counts of 500 bp contiguous regions in placentas at E10.5 from C57Bl/6J conceptuses and F2 Mtrr+/+ fetal growth enhanced (FGE) conceptuses derived from F0 Mtrr+/gt maternal grandfathers. Hypermethylated (red) and hypomethylated (blue) DMRs were determined using EdgeR. (D) MA plot of log2 normalized meDIP-seq read counts of 500 bp contiguous regions in spermatozoa from C57Bl/6J and F0 Mtrr+/gt males. Hypermethylated DMRs (red), hypomethylated DMRs (blue). (E) MA plot of log2 normalized meDIP-seq read counts of 500 bp contiguous regions in placentas from C57Bl/6J and F2 Mtrr+/+ PN fetuses at E10.5. The genomic regions where spermatozoa DMRs from F0 Mtrr+/gt males were identified are highlighted on the placenta data. Hypermethylated spermatozoa DMRs (red), hypomethylated spermatozoa DMRs (blue). (F) MA plot of log2 normalized meDIP-seq read counts of 500 bp contiguous regions in placentas at E10.5 from C57Bl/6J fetuses and F2 Mtrr+/+ FGE fetuses. The genomic regions where spermatozoa DMRs from F0 Mtrr+/gt males were identified are highlighted on the placenta data. Hypermethylated sperm spermatozoa m DMRs (red), hypomethylated spermatozoa DMRs (blue). (G) Data tracks across the En2 gene showing normalized meDIP-seq read counts in spermatozoa (orange) from C57BL/6J males and Mtrrgt/gt, Mtrr+/gt and Mtrr+/+ males together with meDIP read counts in placentas at E10.5 (red) associated with C57Bl/6J fetuses, Mtrrgt/gt PN and FGR fetuses, and F2 Mtrr+/+ PN and FGE fetuses. The En2 DMR is highlighted in light grey. In all cases data was normalized to the largest data store. For spermatozoa meDIP-seq: C57Bl/6J, N = 8 males, F0 Mtrr+/gt, N = 8 males. For placenta meDIP-seq: C57Bl/6J, N = 8 placentas; F2 Mtrr+/+ PN, N = 8 placentas; F2 Mtrr+/+ FGE, N = 3 placentas.
When the meDIP-seq datasets from spermatozoa of F0 Mtrr+/gt males (Figure 5D) (Blake et al., 2021) were compared to placentas of F2 Mtrr+/+ conceptuses (PN and FGE) at E10.5, there was no DMR overlap (Figures 5E, F). This finding reinforces our hypothesis that specific DMRs in the Mtrrgt mouse line are not inherited from germline to somatic cells over multiple generations. This was even the case at the En2 DMR, which was present in spermatozoa from F0 Mtrr+/gt males and not in F2 Mtrr+/+ placentas (Figure 5G). In this context, the spermatozoa and placenta methylome data revealed that the dosage of the Mtrrgt allele in mice correlated with the degree of hypermethylation at the En2 DMR (Figure 5G). Therefore, the En2 locus was particularly responsive to Mtrr-driven disruption of one-carbon metabolism. Altogether, these data further separate the transmission of specific differential methylation patterns via the germline from fetal growth phenotype inheritance in the Mtrrgt mouse line.
Despite its well-studied role in development and disease, the molecular function of one-carbon metabolism is complex and not well understood. Here, we used the Mtrrgt mouse line to explore the epigenetic role of one-carbon metabolism by assessing the placental methylome in association with fetal growth phenotypes. In doing so, we identified several genomic regions in Mtrrgt/gt placentas with altered DNA methylation including in a gene promoter that conceivably regulates Cxcl1 gene expression in a canonical manner, in a presumptive developmental regulatory region located within the En2 gene, and in a subset of RLTR4 transposable elements. While unlikely to underlie the fetal growth phenotypes, it is possible that these DNA methylation changes are functionally relevant in other tissue types and/or for driving other phenotypes. For instance, CXC chemokine expression from peripheral blood mononuclear cells correlates with folate and homocysteine levels in human subjects (Holven et al., 2002). Alternatively, knocking out the mouse gene Mtfhr to disrupt folate metabolism causes cerebellar patterning defects that are associated with downregulation of En2 gene expression (Chen et al., 2005). Ultimately, our findings support widespread epigenetic instability in the Mtrrgt mouse line.
Our previous locus-specific analyses indicated that Mtrrgt/gt placentas or wildtype placentas exposed to a maternal grandparental Mtrrgt allele are epigenetically unstable (Padmanabhan et al., 2013; Bertozzi et al., 2021; Blake et al., 2021). Yet, we identified fewer placenta DMRs by meDIP-seq than were expected despite using standard analysis parameters that yielded many spermatozoa DMRs in Mtrrgt/gt mice (e.g., 13 placenta DMRs vs. 252 spermatozoa DMRs). It is possible that placental DNA methylation is less sensitive than male germ cells to impaired one-carbon metabolism. DNA in trophoblast cells is globally hypomethylated compared to other cell types (Senner et al., 2012) and changes in DNA methylation might be less striking in this context. The use of whole placentas that contain multiple cell types (e.g., trophoblast cell subtypes, fetal vascular endothelium, and maternal decidua and immune cells) with their own DNA methylation and transcriptional signatures (Vento-Tormo et al., 2018; Andrews et al., 2023) might confound our analysis to some extent. The placental DMRs that we identified are likely present throughout the tissue, while other undetected DMRs may be confined to a single cell type and not appreciated in our analysis. Assaying the placenta at earlier developmental time points when the trophoblast progenitor population is more homogeneous may be informative. Alternatively, single cell-based sequencing methods may uncover additional DMRs in Mtrrgt/gt placentas at E10.5 that correlate with cell-type specific transcriptional dysregulation and phenotypes.
Transposable elements, which make up ∼40% of the mammalian genome (Goodier and Kazazian, 2008), are heavily methylated to suppress transposition causing deleterious mutation. In this study, we observed hypomethylation and ectopic expression of several RLTR4 elements in Mtrrgt/gt placentas, which might have profound consequences to genomic stability during development. While whole genome sequencing revealed that de novo mutation rates are similar in control and Mtrrgt/gt mice (Blake et al., 2021), it is still possible that increased transposition might occur in this context, generating structural variation with implications for phenotype inheritance. Since spermatozoa from Mtrrgt/gt males showed normal RLTR4 DNA methylation, we propose that DNA methylation was poorly maintained in early embryogenesis or in placenta development to cause hypomethylation at these sites. Although RLTR4 elements were the only transposons identified by meDIP-seq in this study, DNA methylation patterns of variably methylated intracisternal A particle (VM-IAP) retrotransposons are also considerably shifted in Mtrrgt/gt mice as determined by bisulfite pyrosequencing (Bertozzi et al., 2021). KRAB-ZPFs are known to regulate VM-IAPs (Bertozzi et al., 2020), and mechanistically, the Mtrrgt locus contains a cluster of 129P2Ola/Hsd-derived KRAB zinc finger proteins (ZFPs) in an otherwise C57Bl/6J background because of the mutagenesis process (Bertozzi et al., 2021). However, the KRAB-ZFP clusters within the Mtrr locus do not appear to regulate RLTR4 expression (Wolf et al., 2020). Others have shown that paternal Mthfr deficiency in mice causes hypomethylation of L1Md subfamily of LINE-1 retrotransposons (Karahan et al., 2021). Altogether, these data highlight the importance of one-carbon metabolism in maintaining epigenetic stability at early developmental stages when deleterious transposition events could have profound consequences.
While the mechanistic understanding of epigenetic inheritance remains in its infancy, several candidate epigenetic factors have been identified (e.g., chromatin modifications, small non-coding RNA content in germ cells) (Blake and Watson, 2016; Hanna et al., 2019). Our data provides genome-wide evidence that nearly all spermatozoa DMRs caused by the Mtrrgt allele were epigenetically reprogrammed in the placenta and were not transgenerationally inherited. This contrasts with another study that demonstrates transgenerational inheritance of directed epimutations of DNA methylation in mouse obesity genes along with an obesity phenotype, despite evidence that these epimutations are reprogrammed in primordial germ cells (Takahashi et al., 2023). The lack of DMR inheritance in the Mtrrgt mouse line suggests that there might be paradigm-specific effects. However, there are clues that spermatozoa DMRs caused by an Mtrrgt allele might still play a role in epigenetic inheritance since they are associated locus-specific disruption of transcription in Mtrrgt/gt placentas (this study) and in F2 Mtrr+/+ embryos and adult livers (Blake et al., 2021) despite being reprogrammed to normal tissue-specific methylation levels. This association evokes a role for other epigenetic mechanisms aside from DNA methylation in epigenetic inheritance mechanisms. We observed enrichment for the activating H3K4me3 histone mark in developing wildtype male germ cells and trophoblast specifically at genomic locations defined by spermatozoa DMRs in Mtrrgt/gt mice including loci associated with Mtrrgt/gt placental gene misexpression. Others have shown a similar association in a mouse model of paternal Mthfr deficiency (Karahan et al., 2021).
We propose a model whereby impaired one-carbon metabolism alters H3K4me3 deposition in developing male germ cells, which then drives the changes in DNA methylation through modified access of DNA methyltransferases. In the pre-implantation embryo when DNA methylation is reprogrammed, a subset of abnormal H3K4me3 marks may persist, driving further changes in establishing aberrant de novo DNA methylation patterns or in gene expression in early cell lineages of the placenta and/or embryo. The most drastic epigenetic changes likely lead to altered lineage decisions and developmental phenotypes. Since a wide spectrum of phenotypes are observed in Mtrrgt mouse line, disrupting one-carbon metabolism might cause stochastic epigenetic changes across the genome, affecting different cell types in different individuals. The most striking data that reinforces a potential role for histone H3K4me in epigenetic inheritance comes from a study whereby wildtype mice were fed a folate-deficient diet. Mature spermatozoa from folate-deficient males displayed alterations in histone H3K4me3 patterns specifically at developmental genes and putative enhancers, a subset of which were retained in the F1 8-cell embryos and were associated with gene misexpression (Lismer et al., 2020). We did not observe any discernable changes in H3K4me3 enrichment in spermatozoa or 8-cell embryos derived from folate-deficient males specifically within the genomic regions identified as spermatozoa DMRs from Mtrrgt/gt males. This may be due to the differences in the mouse models employed, with our genetic approach causing a more severe metabolic effect than dietary deficiency. Regardless, this finding suggests that genomic hotspots regulated by one-carbon metabolism are unlikely and that the epigenome is differently or stochastically affected in these models. To fully understand the mechanisms involved in epigenetic inheritance, histone methylation should be explored as an inherited epigenetic mechanism in the Mtrrgt mouse model.
Overall, this study together with our previously published work (Padmanabhan et al., 2013; Bertozzi et al., 2021; Blake et al., 2021) indicate that one-carbon metabolism is required for the maintenance of epigenetic stability in the placenta and the germline. The widespread effect of disrupting one-carbon metabolism on the epigenome provides some explanation towards the complex molecular role of folate metabolism during development. Instability of the epigenome can alter transcriptional pathways and genomic stability, with substantial downstream effects on developmental outcome. Single-cell sequencing technology and a broader analysis of epigenetic mechanisms (e.g., histone marks and chromatin structure) together with DNA methylation will enable the identification of complex epigenome-phenotype relationships that persist over multiple generations in context of the Mtrrgt mouse line.
The original data presented in the study are deposited in the GEO repository, accession number GSE233482. The published datasets presented in this study can be found in online repositories. The names of the repository and the accession numbers can be found in Supplementary File S3.
The animal study was reviewed and approved by University of Cambridge Animal Welfare and Ethical Review Body.
CS and EW conceptualised the study and designed the experiments. EW performed the dissections and phenotyped the conceptuses. CS and ZD generated the placenta meDIP libraries. MP, CS, and MB designed and performed the bioinformatics analyses. CS, MB, and EW collected and analysed the data, and interpreted the results. EW and CS wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a Next Generation Fellowship from the Centre for Trophoblast Research (to CS) and the Lister Institute for Preventative Medicine (to EW). ZD was funded by a Wellcome Trust DTP studentship in Developmental Mechanisms (222275/Z/20/Z).
RNA library preparation and sequencing was performed by Cambridge Genomics Services, Department of Pathology, University of Cambridge, Cambridge, United Kingdom. Next generation sequencing of the meDIP libraries was performed at The Babraham Institute, Cambridge, United Kingdom.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1209928/full#supplementary-material
Allegra, M., Genovesi, S., Maggia, M., Cenni, M. C., Zunino, G., Sgado, P., et al. (2014). Altered GABAergic markers, increased binocularity and reduced plasticity in the visual cortex of Engrailed-2 knockout mice. Front. Cell. Neurosci. 8, 163. doi:10.3389/fncel.2014.00163
Andrews, S., Krueger, C., Mellado-Lopez, M., Hemberger, M., Dean, W., Perez-Garcia, V., et al. (2023). Mechanisms and function of de novo DNA methylation in placental development reveals an essential role for DNMT3B. Nat. Commun. 14, 371. doi:10.1038/s41467-023-36019-9
Bastian, F. B., Roux, J., Niknejad, A., Comte, A., Fonseca Costa, S. S., De Farias, T. M., et al. (2021). The bgee suite: Integrated curated expression atlas and comparative transcriptomics in animals. Nucleic Acids Res. 49, D831–D847. doi:10.1093/nar/gkaa793
Bertozzi, T. M., Becker, J. L., Blake, G. E. T., Bansal, A., Nguyen, D. K., Fernandez-Twinn, D. S., et al. (2021). Variably methylated retrotransposons are refractory to a range of environmental perturbations. Nat. Genet. 53, 1233–1242. doi:10.1038/s41588-021-00898-9
Bertozzi, T. M., Elmer, J. L., Macfarlan, T. S., and Ferguson-Smith, A. C. (2020). KRAB zinc finger protein diversification drives mammalian interindividual methylation variability. Proc. Natl. Acad. Sci. U. S. A. 117, 31290–31300. doi:10.1073/pnas.2017053117
Blake, G. E. T., Hall, J., Petkovic, G. E., and Watson, E. D. (2019). Analysis of spermatogenesis and fertility in adult mice with a hypomorphic mutation in the Mtrr gene. Reprod. Fertil. Dev. 31, 1730–1741. doi:10.1071/RD19064
Blake, G. E. T., Zhao, X., Yung, H. W., Burton, G. J., Ferguson-Smith, A. C., Hamilton, R. S., et al. (2021). Defective folate metabolism causes germline epigenetic instability and distinguishes Hira as a phenotype inheritance biomarker. Nat. Commun. 12, 3714. doi:10.1038/s41467-021-24036-5
Blake, G. E., and Watson, E. D. (2016). Unravelling the complex mechanisms of transgenerational epigenetic inheritance. Curr. Opin. Chem. Biol. 33, 101–107. doi:10.1016/j.cbpa.2016.06.008
Brielmaier, J., Matteson, P. G., Silverman, J. L., Senerth, J. M., Kelly, S., Genestine, M., et al. (2012). Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS One 7, e40914. doi:10.1371/journal.pone.0040914
Cheh, M. A., Millonig, J. H., Roselli, L. M., Ming, X., Jacobsen, E., Kamdar, S., et al. (2006). En2 knockout mice display neurobehavioral and neurochemical alterations relevant to autism spectrum disorder. Brain Res. 1116, 166–176. doi:10.1016/j.brainres.2006.07.086
Chen, Y. Y., Gupta, M. B., Grattton, R., Powell, T. L., and Jansson, T. (2018). Down-regulation of placental folate transporters in intrauterine growth restriction. J. Nutr. Biochem. 59, 136–141. doi:10.1016/j.jnutbio.2018.06.003
Chen, Z., Schwahn, B. C., Wu, Q., He, X., and Rozen, R. (2005). Postnatal cerebellar defects in mice deficient in methylenetetrahydrofolate reductase. Int. J. Dev. Neurosci. 23, 465–474. doi:10.1016/j.ijdevneu.2005.05.007
Christensen, K. E., Deng, L., Bahous, R. H., Jerome-Majewska, L. A., and Rozen, R. (2015). MTHFD1 formyltetrahydrofolate synthetase deficiency, a model for the MTHFD1 R653Q variant, leads to congenital heart defects in mice. Birth Defects Res. A Clin. Mol. Teratol. 103, 1031–1038. doi:10.1002/bdra.23451
Davis, C. A., Noble-Topham, S. E., Rossant, J., and Joyner, A. L. (1988). Expression of the homeo box-containing gene En-2 delineates a specific region of the developing mouse brain. Genes. Dev. 2, 361–371. doi:10.1101/gad.2.3.361
Deng, L., Elmore, C. L., Lawrance, A. K., Matthews, R. G., and Rozen, R. (2008). Methionine synthase reductase deficiency results in adverse reproductive outcomes and congenital heart defects in mice. Mol. Genet. Metab. 94, 336–342. doi:10.1016/j.ymgme.2008.03.004
Ducker, G. S., and Rabinowitz, J. D. (2017). One-carbon metabolism in health and disease. Cell. Metab. 25, 27–42. doi:10.1016/j.cmet.2016.08.009
Elmore, C. L., Wu, X., Leclerc, D., Watson, E. D., Bottiglieri, T., Krupenko, N. I., et al. (2007). Metabolic derangement of methionine and folate metabolism in mice deficient in methionine synthase reductase. Mol. Genet. Metab. 91, 85–97. doi:10.1016/j.ymgme.2007.02.001
Emery, A. E., Timson, J., and Watson-Williams, E. J. (1969). Pathogenesis of spina bifida. Lancet 2, 909–910. doi:10.1016/s0140-6736(69)92371-x
Ficz, G., Branco, M. R., Seisenberger, S., Santos, F., Krueger, F., Hore, T. A., et al. (2011). Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 473, 398–402. doi:10.1038/nature10008
Furness, D. L., Fenech, M. F., Khong, Y. T., Romero, R., and Dekker, G. A. (2008). One-carbon metabolism enzyme polymorphisms and uteroplacental insufficiency. Am. J. Obstet. Gynecol. 199, 276 e271–e8. doi:10.1016/j.ajog.2008.06.020
Gelineau-Van Waes, J., Heller, S., Bauer, L. K., Wilberding, J., Maddox, J. R., Aleman, F., et al. (2008). Embryonic development in the reduced folate carrier knockout mouse is modulated by maternal folate supplementation. Birth Defects Res. A Clin. Mol. Teratol. 82, 494–507. doi:10.1002/bdra.20453
Goodier, J. L., and Kazazian, H. H. (2008). Retrotransposons revisited: The restraint and rehabilitation of parasites. Cell. 135, 23–35. doi:10.1016/j.cell.2008.09.022
Hanna, C. W., Perez-Palacios, R., Gahurova, L., Schubert, M., Krueger, F., Biggins, L., et al. (2019). Endogenous retroviral insertions drive non-canonical imprinting in extra-embryonic tissues. Genome Biol. 20, 225. doi:10.1186/s13059-019-1833-x
Holven, K. B., Aukrust, P., Holm, T., Ose, L., and Nenseter, M. S. (2002). Folic acid treatment reduces chemokine release from peripheral blood mononuclear cells in hyperhomocysteinemic subjects. Arterioscler. Thromb. Vasc. Biol. 22, 699–703. doi:10.1161/01.atv.0000013288.35930.90
Howe, F. S., Fischl, H., Murray, S. C., and Mellor, J. (2017). Is H3K4me3 instructive for transcription activation? Bioessays 39, 1–12. doi:10.1002/bies.201600095
Joyner, A. L., Herrup, K., Auerbach, B. A., Davis, C. A., and Rossant, J. (1991). Subtle cerebellar phenotype in mice homozygous for a targeted deletion of the En-2 homeobox. Science 251, 1239–1243. doi:10.1126/science.1672471
Karahan, G., Chan, D., Shirane, K., Mcclatchie, T., Janssen, S., Baltz, J. M., et al. (2021). Paternal MTHFR deficiency leads to hypomethylation of young retrotransposons and reproductive decline across two successive generations. Development 148 (13), 199492. doi:10.1242/dev.199492
Kobayashi, H., Sakurai, T., Imai, M., Takahashi, N., Fukuda, A., Yayoi, O., et al. (2012). Contribution of intragenic DNA methylation in mouse gametic DNA methylomes to establish oocyte-specific heritable marks. PLoS Genet. 8, e1002440. doi:10.1371/journal.pgen.1002440
Krishnaswamy, K., and Madhavan Nair, K. (2001). Importance of folate in human nutrition. Br. J. Nutr. 85 (2), S115–S124. doi:10.1079/bjn2000303
Leclerc, D., Wilson, A., Dumas, R., Gafuik, C., Song, D., Watkins, D., et al. (1998). Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc. Natl. Acad. Sci. U. S. A. 95, 3059–3064. doi:10.1073/pnas.95.6.3059
Lee, S. M., Danielian, P. S., Fritzsch, B., and Mcmahon, A. P. (1997). Evidence that FGF8 signalling from the midbrain-hindbrain junction regulates growth and polarity in the developing midbrain. Development 124, 959–969. doi:10.1242/dev.124.5.959
Lin, J. G., Kourtis, S., Ghose, R., Pardo Lorente, N., Kubicek, S., and Sdelci, S. (2022). Metabolic modulation of transcription: The role of one-carbon metabolism. Cell. Chem. Biol. 29, P1664–P1679. doi:10.1016/j.chembiol.2022.11.009
Lismer, A., Siklenka, K., Lafleur, C., Dumeaux, V., and Kimmins, S. (2020). Sperm histone H3 lysine 4 trimethylation is altered in a genetic mouse model of transgenerational epigenetic inheritance. Nucleic Acids Res. 48, 11380–11393. doi:10.1093/nar/gkaa712
Ma, C., Liu, G., Liu, W., Xu, W., Li, H., Piao, S., et al. (2021). CXCL1 stimulates decidual angiogenesis via the VEGF-A pathway during the first trimester of pregnancy. Mol. Cell. Biochem. 476, 2989–2998. doi:10.1007/s11010-021-04137-x
Macrae, T. A., Fothergill-Robinson, J., and Ramalho-Santos, M. (2023). Regulation, functions and transmission of bivalent chromatin during mammalian development. Nat. Rev. Mol. Cell. Biol. 24, 6–26. doi:10.1038/s41580-022-00518-2
Maksakova, I. A., Romanish, M. T., Gagnier, L., Dunn, C. A., Van De Lagemaat, L. N., and Mager, D. L. (2006). Retroviral elements and their hosts: Insertional mutagenesis in the mouse germ line. PLoS Genet. 2, e2. doi:10.1371/journal.pgen.0020002
Mislanova, C., Martsenyuk, O., Huppertz, B., and Obolenskaya, M. (2011). Placental markers of folate-related metabolism in preeclampsia. Reproduction 142, 467–476. doi:10.1530/REP-10-0484
MRC Vitamin Study Research Group (1991). Prevention of neural tube defects: Results of the medical research council vitamin study MRC vitamin study research group. Lancet 338, 131–137. doi:10.1016/0140-6736(91)90133-A
Padmanabhan, N., Jia, D., Geary-Joo, C., Wu, X., Ferguson-Smith, A. C., Fung, E., et al. (2013). Mutation in folate metabolism causes epigenetic instability and transgenerational effects on development. Cell. 155, 81–93. doi:10.1016/j.cell.2013.09.002
Padmanabhan, N., Menelaou, K., Gao, J., Anderson, A., Blake, G. E. T., Li, T., et al. (2018). Abnormal folate metabolism causes age-sex- and parent-of-origin-specific haematological defects in mice. J. Physiol. 596, 4341–4360. doi:10.1113/JP276419
Padmanabhan, N., Rakoczy, J., Kondratowicz, M., Menelaou, K., Blake, G. E. T., and Watson, E. D. (2017). Multigenerational analysis of sex-specific phenotypic differences at midgestation caused by abnormal folate metabolism. Environ. Epigenet 3, dvx014. doi:10.1093/eep/dvx014
Parry, A., Rulands, S., and Reik, W. (2021). Active turnover of DNA methylation during cell fate decisions. Nat. Rev. Genet. 22, 59–66. doi:10.1038/s41576-020-00287-8
Provenzano, G., Pangrazzi, L., Poli, A., Pernigo, M., Sgado, P., Genovesi, S., et al. (2014). Hippocampal dysregulation of neurofibromin-dependent pathways is associated with impaired spatial learning in engrailed 2 knock-out mice. J. Neurosci. 34, 13281–13288. doi:10.1523/JNEUROSCI.2894-13.2014
Schoenfelder, S., Mifsud, B., Senner, C. E., Todd, C. D., Chrysanthou, S., Darbo, E., et al. (2018). Divergent wiring of repressive and active chromatin interactions between mouse embryonic and trophoblast lineages. Nat. Commun. 9, 4189. doi:10.1038/s41467-018-06666-4
Schuh, S., Rosenblatt, D. S., Cooper, B. A., Schroeder, M. L., Bishop, A. J., Seargeant, L. E., et al. (1984). Homocystinuria and megaloblastic anemia responsive to vitamin B12 therapy. An inborn error of metabolism due to a defect in cobalamin metabolism. N. Engl. J. Med. 310, 686–690. doi:10.1056/NEJM198403153101104
Senner, C. E., Krueger, F., Oxley, D., Andrews, S., and Hemberger, M. (2012). DNA methylation profiles define stem cell identity and reveal a tight embryonic-extraembryonic lineage boundary. Stem Cells 30, 2732–2745. doi:10.1002/stem.1249
Sgado, P., Genovesi, S., Kalinovsky, A., Zunino, G., Macchi, F., Allegra, M., et al. (2013). Loss of GABAergic neurons in the hippocampus and cerebral cortex of engrailed-2 null mutant mice: Implications for autism spectrum disorders. Exp. Neurol. 247, 496–505. doi:10.1016/j.expneurol.2013.01.021
Shane, B., and Stokstad, E. L. (1985). Vitamin B12-folate interrelationships. Annu. Rev. Nutr. 5, 115–141. doi:10.1146/annurev.nu.05.070185.000555
Shirane, K., Miura, F., Ito, T., and Lorincz, M. C. (2020). NSD1-deposited H3K36me2 directs de novo methylation in the mouse male germline and counteracts Polycomb-associated silencing. Nat. Genet. 52, 1088–1098. doi:10.1038/s41588-020-0689-z
Sun, W., Dong, H., Becker, A. S., Dapito, D. H., Modica, S., Grandl, G., et al. (2018). Cold-induced epigenetic programming of the sperm enhances Brown adipose tissue activity in the offspring. Nat. Med. 24, 1372–1383. doi:10.1038/s41591-018-0102-y
Takahashi, Y., Morales Valencia, M., Yu, Y., Ouchi, Y., Takahashi, K., Shokhirev, M. N., et al. (2023). Transgenerational inheritance of acquired epigenetic signatures at CpG islands in mice. Cell. 186, 715–731.e19. doi:10.1016/j.cell.2022.12.047
Tanaka, S., Kunath, T., Hadjantonakis, A. K., Nagy, A., and Rossant, J. (1998). Promotion of trophoblast stem cell proliferation by FGF4. Science 282, 2072–2075. doi:10.1126/science.282.5396.2072
Vento-Tormo, R., Efremova, M., Botting, R. A., Turco, M. Y., Vento-Tormo, M., Meyer, K. B., et al. (2018). Single-cell reconstruction of the early maternal-fetal interface in humans. Nature 563, 347–353. doi:10.1038/s41586-018-0698-6
Wilkinson, A. L., Menelaou, K., Rakoczy, J., Tan, X. S., and Watson, E. D. (2021). Disruption of folate metabolism causes poor alignment and spacing of mouse conceptuses for multiple generations. Front. Cell. Dev. Biol. 9, 723978. doi:10.3389/fcell.2021.723978
Wilson, A., Platt, R., Wu, Q., Leclerc, D., Christensen, B., Yang, H., et al. (1999). A common variant in methionine synthase reductase combined with low cobalamin (vitamin B12) increases risk for spina bifida. Mol. Genet. Metab. 67, 317–323. doi:10.1006/mgme.1999.2879
Wolf, G., De Iaco, A., Sun, M. A., Bruno, M., Tinkham, M., Hoang, D., et al. (2020). KRAB-zinc finger protein gene expansion in response to active retrotransposons in the murine lineage. Elife 9, e56337. doi:10.7554/eLife.56337
Wu, X., Yang, K., Tang, X., Sa, Y., Zhou, R., Liu, J., et al. (2015). Folate metabolism gene polymorphisms MTHFR C677T and A1298C and risk for preeclampsia: A meta-analysis. J. Assist. Reprod. Genet. 32, 797–805. doi:10.1007/s10815-014-0408-8
Yamada, K., Gravel, R. A., Toraya, T., and Matthews, R. G. (2006). Human methionine synthase reductase is a molecular chaperone for human methionine synthase. Proc. Natl. Acad. Sci. U. S. A. 103, 9476–9481. doi:10.1073/pnas.0603694103
Keywords: DNA methylation, folate, histone methylation, MTRR, sperm, transposable elements, trophoblast, epigenetic inheritance
Citation: Senner CE, Dong Z, Prater M, Branco MR and Watson ED (2023) One-carbon metabolism is required for epigenetic stability in the mouse placenta. Front. Cell Dev. Biol. 11:1209928. doi: 10.3389/fcell.2023.1209928
Received: 21 April 2023; Accepted: 02 June 2023;
Published: 27 June 2023.
Edited by:
Mellissa Mann, University of Pittsburgh, United StatesReviewed by:
Julie Brind’Amour, Université de Montréal, CanadaCopyright © 2023 Senner, Dong, Prater, Branco and Watson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Erica D. Watson, ZWR3MjNAY2FtLmFjLnVr; Claire E. Senner, Y2VzMjA3QGNhbS5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.