 Hannah M. Neuendorf
Hannah M. Neuendorf Jacinta L. Simmons1,2,3
Jacinta L. Simmons1,2,3 Glen M. Boyle
Glen M. Boyle- 1Cancer Drug Mechanisms Group, QIMR Berghofer Medical Research Institute, Herston, QLD, Australia
- 2School of Biomedical Sciences, Faculty of Health, Queensland University of Technology, Brisbane, QLD, Australia
- 3School of Biomedical Sciences, Faculty of Medicine, University of Queensland, Brisbane, QLD, Australia
The acquisition of resistance to anoikis, the cell death induced by loss of adhesion to the extracellular matrix, is an absolute requirement for the survival of disseminating and circulating tumour cells (CTCs), and for the seeding of metastatic lesions. In melanoma, a range of intracellular signalling cascades have been identified as potential drivers of anoikis resistance, however a full understanding of the process is yet to be attained. Mechanisms of anoikis resistance pose an attractive target for the therapeutic treatment of disseminating and circulating melanoma cells. This review explores the range of small molecule, peptide and antibody inhibitors targeting molecules involved in anoikis resistance in melanoma, and may be repurposed to prevent metastatic melanoma prior to its initiation, potentially improving the prognosis for patients.
1 Introduction
Anoikis is a type of apoptosis induced by detachment from the extracellular matrix (ECM) and surrounding cells (Meredith et al., 1993; Frisch and Francis, 1994). Under a physiological state, anoikis functions to maintain cell number equilibrium by triggering apoptosis in cells with inappropriate cell-to-ECM and cell-to-cell interactions (Frisch and Francis, 1994). Anoikis thereby functions in preventing cells from migrating, and subsequently proliferating at inappropriate sites within the body. However, as a hallmark of cancer and an absolute requirement for metastasis, tumour cells must acquire mechanisms to resist cell death by anoikis to detach from the primary lesion, persist in circulation and facilitate the seeding of metastases (Hanahan, 2022). In these cells, anoikis resistance is achieved through the oncogenic deregulation of survival and death signalling as a result of genetic, epigenetic and (micro-) environmental variation.
Cutaneous Malignant Melanoma, a cancer arising from melanocytes in the skin, is a highly aggressive and invasive cancer type that demonstrates early dissemination into lymphatic circulation from primary tumours less than 0.5 mm thick in at least one-third of patients (Werner-Klein et al., 2018). In addition, metastatic melanoma is a vastly complex and heterogeneous disease, with multiple, highly plastic subpopulations of cells contributing to disease progression and drug resistance (Tsoi et al., 2018; Rambow et al., 2019). Despite significant advancement in the treatment of metastatic melanoma since the advent of targeted and immune checkpoint inhibitors, three-quarters of patients who are diagnosed or relapse with an advanced stage of melanoma inevitably succumb to the disease (AIHW, 2021). With the incidence of melanoma predicted to increase into the future (Guy et al., 2015; Whiteman et al., 2016), the development or repurposing of existing drugs to target and prevent the progression of melanoma are urgently required. Anoikis resistance poses an attractive target for the therapeutic intervention of melanoma progression, with potential to prevent the dissemination of cells from the primary tumour and allow the targeting of circulating tumour cells (CTCs). However, the understanding of mechanisms involved in anoikis resistance in melanoma remains limited. This review details anoikis resistance mechanisms identified in cutaneous melanoma, examines therapeutic inhibitors with potential to be repurposed to target anoikis resistance mechanisms, and discusses the feasibility of targeting anoikis resistance to prevent the progression of melanoma to metastasis (Figure 1).
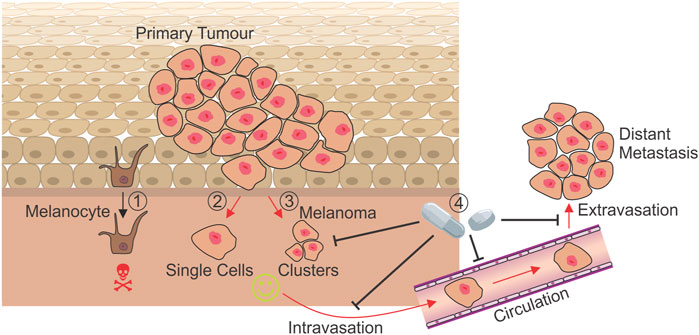
FIGURE 1. Mechanisms of anoikis resistance during metastasis. While the detachment of a non-transformed melanocyte results in cell death by anoikis (1), melanoma cells leaving the dermis as either single cells (2) or clusters (3) must develop mechanisms to resist anoikis in order to metastasise and form secondary tumours at distant sites. Treatments targeting currently known mechanisms of anoikis resistance have the potential to prevent cells leaving the primary tumour (4), and may target those in circulation, preventing the seeding of metastases and improving prognosis for patients. Red arrows: Processes where anoikis resistance is absolutely required.
1.1 General mechanisms of anoikis resistance
Anoikis occurs through a combination of both the intrinsic and extrinsic apoptosis pathways converging at the mitochondria and resulting in the activation of caspases, triggering DNA fragmentation and cell death (Paoli et al., 2013). The Bcl-2 family of proteins play a key role in both apoptosis pathways and is comprised of three subgroups: the anti-apoptotic proteins (Bcl-2, Bcl-XL, Bcl-w, Mcl-1 and A1) (Opferman and Kothari, 2018), the multi-domain pro-apoptotic members (Bax, Bak, Bok) (Reed, 2006), and the BH3-only pro-apoptotic proteins (Bim, Bad, Bid, Noxa, Puma) (Adams and Cory, 2007; Hao et al., 2007; Anvekar et al., 2011). Following cellular detachment, the pro-apoptotic members translocate from the cytosol to the mitochondria where they cause mitochondrial outer-membrane permeabilisation (MOMP) and cytochrome c release, triggering apoptosome assembly, caspase cleavage and cell death (Gilmore, 2005). As a result, it’s generally accepted that anoikis is marked by activation of caspase-3, -8 and -9, and PARP-1 cleavage upon anchorage loss (Zhang et al., 2011; Hasnat et al., 2015; Fanfone et al., 2022). However in the case of malignant cells, activation of oncogenic signalling results in deregulation of the pathways triggering caspase activation and PARP cleavage, inevitably resulting in abrogated apoptosis despite the presence of cell death-inducing stimuli.
Importantly, the “attach or die” phenotype is not true for all cells, and applies mostly to those of epithelial origin, while circulating blood and immune cells, for example, are intrinsically anoikis resistant (Zhu et al., 2001). As an exception to this rule, epithelial cells are able to overcome anoikis under certain conditions such as embryogenesis and wound healing, through the process of epithelial-to-mesenchymal transition (EMT). EMT allows epithelial cells to gain reversible migratory and invasive properties, as well as stem cell-ness and the ability to evade apoptosis with the loss of cell-to-cell adhesion in response to microenvironmental cues (Kalluri and Weinberg, 2009; Thiery et al., 2009). Despite residing in the skin, melanocytes are not derived from the epithelial lineage and therefore do not undergo true EMT (Kalluri and Weinberg, 2009). Rather, EMT is involved in the formation of melanoblasts (Figure 2, melanocyte precursor cells) from neural crest cells during embryonic development (Thomas and Erickson, 2008; Kalluri and Weinberg, 2009; Vandamme and Berx, 2014). As a result of their developmental lineage, melanoma cells are able to “hijack” EMT-like pathways to facilitate metastatic progression, including driving resistance to anoikis (Alonso et al., 2007). Subsequently, a model has been proposed that describes the intrinsic phenotypic plasticity observed in distinct subpopulations of melanoma cells; the phenotype switching model. Similar to the way epithelial cells are able to reversibly switch between epithelial and mesenchymal phenotypes, this model describes the ability of cells in the primary tumour to switch between proliferative and invasive transcriptional states subject to regulation by the proximal microenvironment (Hoek et al., 2006; Hoek et al., 2008). Subsequently, many of the molecules identified as drivers of EMT have likewise been identified to contribute to the acquisition of anoikis resistance in melanoma (Figure 2).
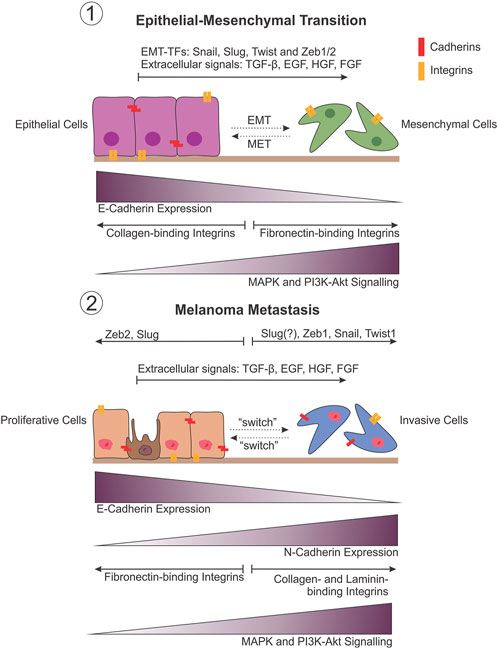
FIGURE 2. Comparison between EMT and melanoma metastasis. Phenotypic plasticity observed during melanoma metastasis draws striking similarities with the changes that occur during epithelial-mesenchymal transition (EMT) (1). This is believed to be the result of the developmental lineage of melanocytes in the neural crest, allowing melanoma cells to undergo an EMT-like transition to facilitate metastasis by varying the expression of adhesion molecules in particular, in response to shifts in transcriptional regulation (EMT-TFs) (2).
2 Mechanisms of anoikis resistance and potential therapies
2.1 Adhesion molecules
2.1.1 Integrins
Integrins are transmembrane cell surface receptors that are primarily responsible for mediating cell adhesion to the ECM (Paoli et al., 2013). In addition to their function in physical attachment, integrins transduce signals from the ECM to the inner machinery of the cell by directly binding components of the cytoskeleton. This results in actin filament rearrangement within the cell and activation of downstream signalling pathways that promote migration, proliferation and survival. In addition to their role in “outside-in” signalling, integrins can transduce “inside-out” signalling via the activation of extra-cellular ligands, particularly TGF-β (Barczyk et al., 2010). Integrins consist of two transmembrane glycoprotein subunits, α and β, which associate via non-covalent bonding interactions (Hynes, 2002). Depending on the cell type and composition of the ECM, different types of integrins may be present; those that bind fibronectin, laminin, vitronectin, or collagen (Friedl and Wolf, 2003).
During the process of EMT, epithelial cells undergo the phenomenon of ‘integrin switching’, where collagen-binding integrins are downregulated, and fibronectin-binding integrins upregulated to allow detachment from the basement membrane and mesenchymal cell migration (Maschler et al., 2005). As a hallmark of cancer, self-sufficiency in growth signalling is an absolute requirement for disease progression. A prominent mechanism employed by cancer cells to achieve this is integrin switching, as it favours receptors that transmit pro-growth or pro-survival signals, including those that prevent cell death upon detachment. As such, a distinct change in integrin expression has been observed between melanocytes and melanoma cells throughout progression. Studies analysing integrin expression demonstrate that melanocytes predominantly express the α3β1 and α6β1 laminin-binding integrins for adhesion to the basement membrane in vivo (Krengel et al., 2005). However, during the progression to malignant disease, melanoma cells upregulate expression of fibronectin- and collagen-binding integrins to facilitate vertical migration through the dermis, and attachment to vascular endothelial cells allowing dissemination into circulation.
To date, the most comprehensively studied integrin with links to anoikis resistance in melanoma metastasis is the αvβ3 integrin. Studies demonstrate αvβ3 binds multiple ECM components, including fibronectin, vitronectin, fibrinogen and osteopontin (Cheresh, 1987). It is best known for its role in angiogenesis, where blocking of the αvβ3 receptor in melanoma patients through the administration of combination high-dose tumour necrosis factor (TNF) and interferon γ (IFN-γ) inhibits tumour angiogenesis and growth due to the specific inhibitory effect of these cytokines on β3 subunit protein synthesis (Defilippi et al., 1991; Brooks et al., 1994; Rüegg et al., 1998). In addition, expression of αvβ3 on the surface of invasive cells allows the recruitment, localisation and activation of matrix metalloproteinases (MMP-1 and MMP-2), facilitating ECM remodelling through collagen degradation, promoting migration (Brooks et al., 1996; Brooks et al., 1998; Hofmann et al., 2000). As such, αvβ3 integrin is widely recognised as a molecular marker of metastasis, where its expression correlates with melanoma progression from radial growth phase (RGP) to the invasive vertical growth phase (VGP) (Albelda et al., 1990; Danen et al., 1994; Hsu et al., 1998; Johnson, 1999; Van Belle et al., 1999; Kageshita et al., 2000; Alonso et al., 2007). Expression of αvβ3 integrin in primary cutaneous melanoma is likewise associated with increased sentinel lymph node metastasis (Meves et al., 2015).
Importantly, αvβ3 integrin has been demonstrated to contribute to anoikis resistance through driving upregulation of anti-apoptotic proteins such as Bcl-2 (Montgomery et al., 1994; Petitclerc et al., 1999). A five-fold increase in the relative Bcl-2/Bax ratio conferred increased cellular survival (Petitclerc et al., 1999; Zhang et al., 2011). Recent studies have demonstrated that the αvβ3 integrin is able to induce partial EMT independent of TGF-β signalling (Kariya et al., 2021). This is important considering the recent emergence of evidence for distinct partial EMT-like phenotypes in melanoma that possess increased invasiveness and motility (Sahoo et al., 2022). However, an independent study demonstrated that αvβ3 integrin expression was not influenced by Snail, a transcription factor that promotes EMT (Kuphal et al., 2005).
Integrins containing the β1-subunit in conjunction with a range of alpha subunits (α2, α3, α4, α5, α6 or α7) have similarly been implicated in melanoma metastasis, invasion and anoikis resistance (Etoh et al., 1993; Danen et al., 1994; Ziober et al., 1999; Hegerfeldt et al., 2002; Toricelli et al., 2013; Kolli-Bouhafs et al., 2014; Kozlova et al., 2018; Kozlova et al., 2019; Kozlova et al., 2020). Kozlova and others demonstrated that inhibition of integrin α2β1 increased sensitivity to anoikis, while blocking α3β1 and α5β1 reduced invasion and activation of MMP-2 in vitro (Kozlova et al., 2019; Kozlova et al., 2020). Integrins containing the β1 subunit are thought to confer increased cell survival under anchorage-independent conditions through increased expression of Bcl-2 and c-Myc (Kozlova et al., 2019).
Integrins play an important role in the progression of melanoma, however data surrounding the activation and deactivation of integrin expression and clustering throughout the metastatic process, remains incomplete. Studies investigating integrins in metastasis focus on their expression levels and largely ignore their activity. It’s understood that integrin internalisation through endocytosis is important in enabling cell motility as it allows their recycling to the leading edge of migrating cells (Krengel et al., 2005). Therefore, future studies investigating integrin recycling throughout each stage of metastasis would provide valuable information on their functional role. Nevertheless, inhibitors targeting integrin clustering and expression have been used successfully in a range of non-malignant diseases (Slack et al., 2022). Given that integrins are strongly upregulated in a range of cancers including melanoma, the use of integrin inhibitors in this context has been extensively explored (Figure 3).
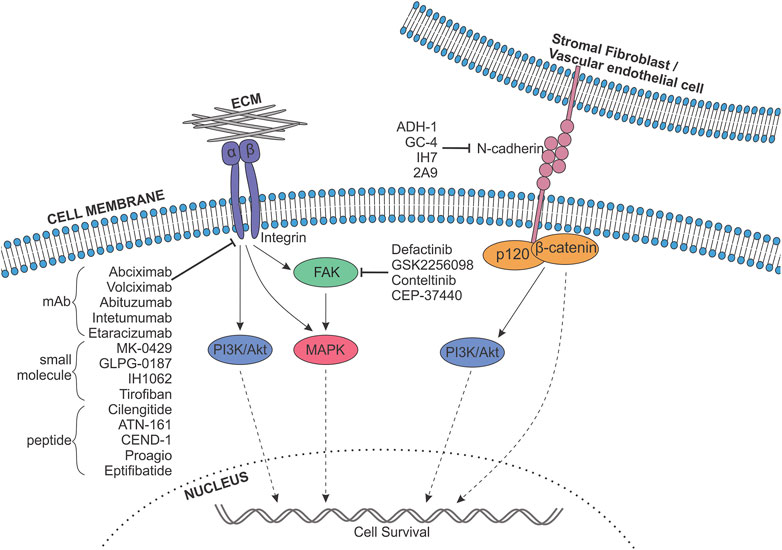
FIGURE 3. Targeting adhesion molecules in melanoma anoikis resistance. A range of integrins, as well as N-cadherin, and their downstream signalling pathways, including FAK (green), Mitogen-Activated Protein Kinase (MAPK; pink), PI3K-Akt (purple) and catenin pathways (orange), are known to contribute to anoikis resistance in melanoma. Pharmaceutical inhibitors targeting integrins, cadherins and FAK exist, and have the potential to be repurposed to block melanoma cell survival upon detachment. Different classes of integrin inhibitors are shown, mAb, monoclonal antibody; ECM, Extracellular matrix. Indicated inhibitors have been sourced from data from all cancers.
Eptifibatide (integrilin) and tirofiban (aggrastat) target β3 integrins, and were registered by the FDA (1998) for use in patients with heart conditions. While neither inhibitor has been examined in clinical trials against any cancer type, Kim et al. demonstrated that eptifibatide significantly reduced the ability of B16-F10 melanoma cells to form nodules on the lungs of mice following arsenic exposure (Kim et al., 2019).
A number of monoclonal antibody therapies targeting integrins have been evaluated in clinical trials (Table 1). However, none of these therapies have progressed past phase II trials. In addition, many small molecule and peptide compounds have demonstrated efficacy as integrin inhibitors. Cilengitide is a cyclic Arg-Gly-Asp (iRGD) peptide that inhibits αvβ3 and αvβ5 integrins, thus inhibiting tumour cell interactions with vascular endothelial cells, as well as cell-matrix interactions and angiogenesis. A phase III clinical trial examining its use in glioblastoma was completed (NCT00689221), in addition to multiple phase II studies in a range of cancers (NCT00093964, NCT00813943, NCT01124240, NCT00103337, NCT00121238, NCT00842712, NCT00679354). Despite preclinical evidence supporting its use in melanoma patients (Lode et al., 1999), a phase II trial in stage III/IV metastatic melanoma patients was terminated (NCT00082875) due to low Progression Free Survival (PFS) after 8 weeks, indicating the treatment was not effective (Kim et al., 2012). Nevertheless, more recent studies revealed the ability of cilengitide to inhibit invasion and vasculogenic mimicry of melanoma cells, and inhibit adhesion to vitronectin (Ruffini et al., 2015) indicating that further investigation into the use of cilengitide in metastatic melanoma patients may be warranted. While one study advised against the use of cilengitide in combination with paclitaxel due to decreased sensitivity to the chemotherapeutic agent (Stojanović et al., 2018), another revealed that combination therapy with doxorubicin synergistically suppressed tumour growth and reversed drug resistance in vivo, extending the survival of mice subcutaneously injected with A375 cells expressing β3 integrin (Zhu X. et al., 2019). Furthermore, combining cilengitide with anti-PD-L1 therapy significantly reduced tumour volume in a B16 murine melanoma model, and positively regulated anti-tumour immune responses (Pan et al., 2022).
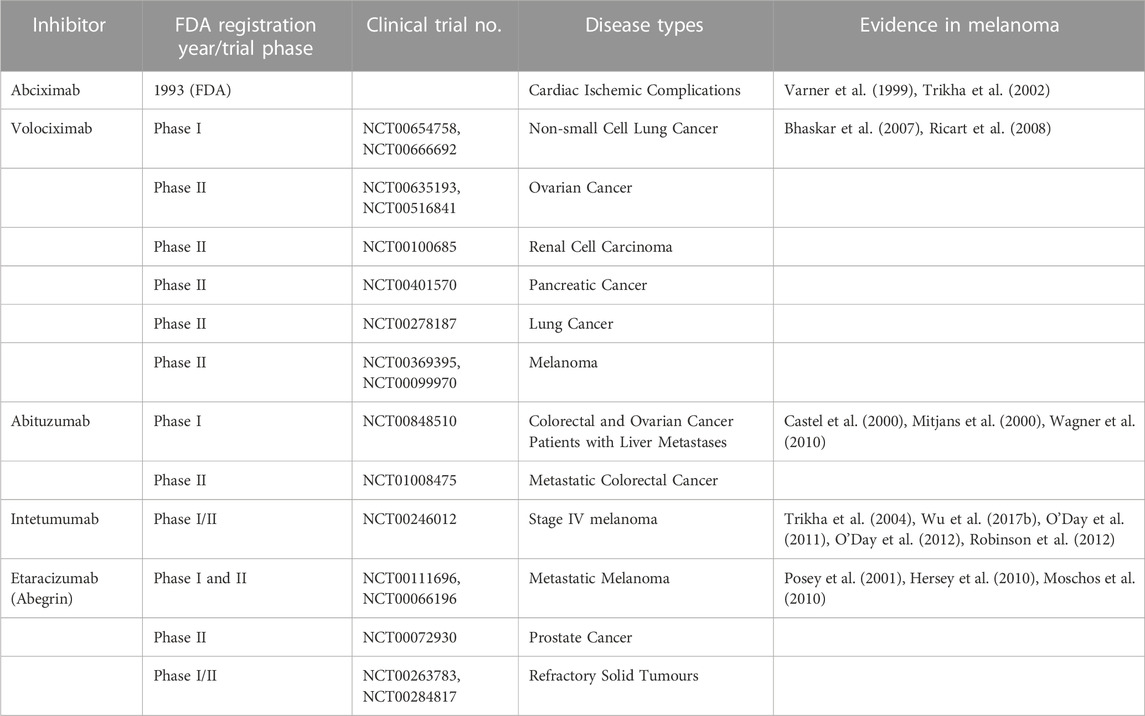
TABLE 1. Monoclonal antibody therapies targeting integrins.
A small peptide antagonist ATN-161, similarly targeting αvβ3 and αvβ5 integrins, has been tested in phase I/II trials with carboplatin (NCT00352313). Preclinical evidence utilising ATN-161 loaded reversibly cross-linked polymersomes for drug delivery into C57BL/6 mice bearing B16-F10 tumours demonstrated significant inhibition of tumour growth, and significantly improved survival rates (Zhang et al., 2017). Likewise, MK-0429 is a non-peptide antagonist compound initially examined in a phase II trial for the treatment of osteoporosis [NCT00533650 (Hutchinson et al., 2003)] for its ability to target the αvβ3 receptor. MK-0429 reduced the number of metastatic colonies in the lungs of mice injected with B16-F10 melanoma cells by 64% at 100 mg/kg (Pickarski et al., 2015). Similar to cilengitide, CEND-1 is an Arg-Gly-Asp cyclic peptide (iRGD) targeting αvβ3 and αvβ5 integrins in the tumour vasculature, promoting tumour penetration to enhance the efficacy and specificity of chemotherapy treatment. A phase I clinical trial in pancreatic ductal adenocarcinoma (PDAC) patients combining CEND-1 with nabpaclitaxel and gemcitabine was successfully completed [NCT03517176 (Dean et al., 2020)]. The use of CEND-1 in melanoma patients has not been examined in clinical trials. However preclinical evidence demonstrated that sterically-stabilised liposomes modified with iRGD peptides such as CEND-1, containing either paclitaxel or doxorubicin, significantly reduce volume of melanoma tumours and increase percentage survival in murine models (Yu et al., 2013; Du et al., 2014).
IH1062 is a novel small molecule inhibitor of αvβ3 integrins. Preclinical evidence in melanoma demonstrated its ability to induce anoikis and suppress metastasis in human melanoma cells, interrupting ECM attachment and FAK phosphorylation, and resulting in caspase activation through a decrease in the Bcl-2/Bax protein ratio (Zhang et al., 2011). Other molecules with therapeutic potential that are yet to be examined in melanoma are summarised in Table 2.

TABLE 2. Pharmacological Inhibitors yet to be examined in melanoma.
Caution must be used when considering integrin-targeting inhibitors for the treatment of melanoma patients. Natalizumab, for example, targets α4, β7 and β1 integrins, and was registered by the FDA in 2004 for multiple sclerosis patients. However, multiple studies have reported a significantly increased risk of developing cutaneous melanomas following natalizumab treatment (Mullen et al., 2008; Vavricka et al., 2011; Munguía-Calzada et al., 2017; Sabol et al., 2017; Kelm et al., 2019). As α4β1 integrin expression is thought to prevent melanoma metastasis formation, therapeutic inhibition with natalizumab was shown to increase invasive potential and cell migration in vitro, and resulted in the dissolution of clusters to single cells (Qian et al., 1994; Carbone et al., 2020). These concerns are further compounded by the findings that targeting orthosteric binding sites of integrins has the potential to induce a shift in integrin binding affinity to a higher binding state, promoting tumour growth and angiogenesis rather than inhibiting it (Slack et al., 2022).
Furthermore, integrin therapies induce the detachment of melanoma cells from the ECM, but do not result in tumour cell apoptosis in most cases. Consequently, integrin inhibitors may induce amoeboid migration, characterised by a rounded cell morphology with bleb-like protrusions, and weak cell-ECM interactions (Wu J. S. et al., 2021), by encouraging detachment of melanoma cells from the primary tumour. For instance, in a study investigating the αvβ3 targeting cyclic oligopeptide cRGDfV, cell adhesion was blocked however apoptosis was not induced (Allman et al., 2000). Rather, a change in cell morphology imitating amoeboid migration was observed following treatment. A similar phenomenon was detected following the treatment of cells with abituzumab (Castel et al., 2000). Amoeboid invasion has been associated with weakening of integrin adhesions (Carragher et al., 2006; Wolf et al., 2007). While previously shown that β1 integrin expression is essential for amoeboid migration of melanoma cells (Sanz-Moreno et al., 2008), other studies have revealed that blocking β1 expression cannot abolish amoeboid crawling or dissemination (Hegerfeldt et al., 2002). To overcome this, combining integrin inhibitors with urokinase-plasminogen activator (uPAR) inhibitors may be effective in blocking amoeboid invasion as uPAR, in association with integrins and the actin cytoskeleton, are believed to drive amoeboid invasion (Margheri et al., 2014).
2.1.2 Cadherins
Cadherins are calcium-dependent transmembrane glycoproteins that function in maintaining cell-to-cell adhesion through the formation of adherens junctions and contribute to intracellular signalling via p120 and β-catenin recruitment in the cytosol, influencing proliferation, cell survival and invasion (Venhuizen et al., 2020). There are several subtypes of cadherins, however type I cadherins are the most relevant to melanoma, and include: epithelial cadherin (E-cadherin, CDH1), present in epithelial cells, melanocytes and keratinocytes; neural cadherin (N-cadherin, CDH2); and placental cadherin (P-cadherin, CDH3) (Yu et al., 2019).
In the skin, E-cadherin functions to maintain the adhesion of melanocytes to keratinocytes through the formation of the epidermal melanin unit; one melanocyte bound to ten keratinocytes (Tang et al., 1994; Danen et al., 1996; D'Arcy and Kiel, 2021). However, loss of E-cadherin is an early event in the progression of most melanomas, believed to occur between radial and vertical growth phases (Silye et al., 1998; Kuphal et al., 2004), and results in the loss of interactions within the epidermal melanin unit (Hsu M. et al., 2000). Coinciding with the reduced levels of E-cadherin, melanoma cells gain expression of N-cadherin allowing them to interact with stromal fibroblasts and endothelial cells, promoting migration from the epidermis and dissemination into circulation (Li et al., 2001a; Nguyen and Mège, 2016; Murtas et al., 2017). This ‘switch’ in cadherin expression is driven by downregulation of the EMT transcription factors (EMT-TFs) Snail and Zeb1 (Poser et al., 2001; Kuphal et al., 2004; Zhu L. et al., 2019) and correlates with a low proliferation rate and acquisition of an invasive phenotype in melanoma cells (Kovacs et al., 2016).
As overexpression of MITF is shown to increase E-cadherin expression, while directly repressing CDH2 (N-cadherin), MITF downregulation results in cadherin switching in melanoma (Dilshat et al., 2021). Other molecules identified to regulate cadherin expression in melanoma cells include: STAT3 and MEK, demonstrated to induce expression of N-cadherin (Vultur et al., 2014); FAK, knockdown of which is shown to reduce expression of E-cadherin (Pei et al., 2017); and Integrin Linked Kinase (ILK) which regulates N-cadherin membranous localisation through regulating its’ endocytosis and recycling (Gil et al., 2020). Notch1 is likewise demonstrated to increase expression of N-cadherin in melanoma cells and coincides with the acquisition of an invasive phenotype (Murtas et al., 2017). Co-expression of these molecules within a patients’ lesion correlates with significantly poorer prognosis compared to those expressing either protein alone. Crucially, expression of N-cadherin facilitates metastasis by promoting the survival of cells under anchorage-independent conditions (Li et al., 2001a). This is achieved through the activation of anti-apoptotic proteins Akt and PKB, increased β-catenin levels, and the inactivation of pro-apoptotic protein Bad.
However, controversy arises between studies analysing the expression levels of E- and N-cadherin in ex vivo melanomas. Early studies indicated that the majority of melanoma tumours retain no expression of E-cadherin as a result of autocrine HGF secretion from melanoma cells activating c-Met and the MAPK and PI3K pathways resulting in E-cadherin downregulation (Li et al., 2001b). However, more recent studies demonstrated mixed expression of E- and N-cadherin in patient samples, trending towards an increase in N-cadherin expression as melanoma progresses (Yan et al., 2016). Granted, melanoma cells are known to be highly plastic in their protein expression and phenotype. Therefore, it’s possible that the E-to N-cadherin switch is highly transient and may occur only in a small subpopulation of cells with the propensity for metastasis, with E-cadherin re-expressed to promote adhesion and proliferation at the secondary tumour site. As a result the switch may not be identifiable through histological analysis of tumour sections. Furthermore, studies thus far fail to analyse the function and activity of the cadherins present. It is hypothesised that reduced activity, rather than reduced expression of E-cadherin may be sufficient to promote anoikis resistance in some patients. Nevertheless, the survival advantage conferred by cooperative dissemination and the formation of CTC clusters or circulating tumour microemboli (CTM) versus single cells suggests an important role for the expression of cell-to-cell adhesion molecules during metastasis and may explain why a partial switch in cadherin expression is observed in melanoma (Hou et al., 2011).
As a result of the switch in expression of E-to N-cadherin observed in a range of cancers driven by EMT-like transcriptional programs, therapies that promote a reversal of this transition have been utilised (Figure 3). ADH-1 (exherin) is a synthetic cyclic peptide containing the His-Ala-Val (HAV) sequence (N-Ac-CHAVC-NH2), designed to bind and inhibit N-cadherin clustering and interactions (Eslami et al., 2019), inhibiting angiogenesis and metastasis, and promoting apoptosis in multiple myeloma, neuroblastoma and pancreatic cancer (Shintani et al., 2008; Lammens et al., 2012; Sadler et al., 2013). Subsequently, the safety and efficacy of ADH-1 as a single-agent therapy against neoplasms has been examined in phase I/II trials (NCT00264433, NCT00265057), as well as in combination with carboplatin, docetaxel or capecitabine (NCT00390676), and melphalan (LPAM) (NCT00421811). In melanoma, pre-clinical evidence suggests combining systemic ADH-1 therapy with regionally infused LPAM has the potential to improve survival of melanoma patients with in-transit metastases. Studies investigating the combination in mouse models of melanoma demonstrate a synergistic reduction in tumour volume associated with suppression of N-cadherin expression, induction of apoptosis and changes in the levels of genes related to cell adhesion (Augustine et al., 2008; Turley et al., 2015). ADH-1 treatment further resulted in an increase in endothelial cell permeability, which was hypothesised to improve the delivery of chemotherapeutic agents to melanoma tumours. However, both studies demonstrate increased volume of specific tumours treated with ADH-1 alone, which appears to correlate with the PTEN expression status of the cell line employed. Additionally, examination of ADH-1 combined with temozolomide (TMZ) in vivo yielded conflicting results, demonstrating synergism in DM443 and DM366 xenograft models, but increased tumour volume in A375 xenografts. It is therefore clear that the mutation status of a patient is an important consideration for the administration of ADH-1 therapies. A phase I trial examining safety, pharmacokinetics and anti-tumour activity of ADH-1 + LPAM in stage IIIB/C melanoma patients with in-transit limb metastases demonstrated complete response in 50% of patients (N = 16) (Beasley et al., 2009), with a subsequent phase II study revealing the combination therapy resulted in significantly improved response rates when compared with standard-of-care isolated limb infusion (ILI) alone [NCT00421811 (Beasley et al., 2011)].
While yet to be examined in clinical trials, monoclonal antibody therapies designed to block N-cadherin activity have demonstrated preclinical efficacy in a range of cancers (Hazan et al., 2000; Zhang et al., 2007; Wallerand et al., 2010; Groen et al., 2011; Zhang et al., 2013; Eiring et al., 2015; Klymenko et al., 2017) (Table 2). GC-4 binds the EC1 domain of N-cadherin, blocking adhesion and intracellular signalling. Treatment of melanoma cells with GC-4 resulted in knockdown of N-cadherin, and subsequently blocked melanoma cell adhesion to endothelial cells, inhibiting transendothelial migration (Qi et al., 2005).
In addition to the use of therapies aimed at reducing N-cadherin activity, studies have investigated whether increasing E-cadherin expression within the primary tumour is a feasible method of reducing the loss of interactions within the epidermal melanin unit, thus preventing cells from entering circulation. Inducing expression of E-cadherin in melanoma cells is demonstrated to reduce colony formation, and restore keratinocyte-mediated inhibition of invasion, resulting in smaller tumours in vivo (Hsu M. Y. et al., 2000). Although no data has been presented to support this to date, it’s possible that the use of these agents may potentiate the attachment of CTCs to secondary sites that express E-cadherin, such as the liver, lungs and intestines, or may promote mesenchymal-to-epithelial transition (MET) of pre-existing micro-metastases resulting in E-cadherin re-expression, cellular proliferation and subsequent formation of clinically detectable metastases, as has been observed in breast cancer (Chao et al., 2010; Padmanaban et al., 2019).
2.2 Receptor tyrosine kinase (RTK) hyperactivation
Activated upon ligand binding or via ligand-independent mechanisms facilitated by integrins, tyrosine kinase receptors (RTKs) function as the upstream initiators of intracellular signalling cascades (Moro et al., 1998; Shen and Kramer, 2004; Mitra et al., 2011). Melanoma cells are demonstrated to significantly upregulate expression of a range of RTKs to maintain growth and survival signalling, avoid apoptosis and ultimately drive the processes of invasion and metastasis. RTK hyperactivation is also postulated to be one of the main mechanisms driving intrinsic and acquired resistance to treatment utilising targeted inhibitors against the MAPK signalling pathway (Manzano et al., 2016). In addition to the growth factor receptors discussed in this section, many others such as FGFR, PDGFR, IGFR and VEGFR, are known to be important in anoikis resistance in a range of other cancers (Hilmi et al., 2008; Molhoek et al., 2011; Paoli et al., 2013; Chen et al., 2016), and likely also have a role in melanoma. However, little evidence is currently available, and they are therefore outside the scope of this review.
2.2.1 EGFR
A switch in expression levels of epidermal growth factor receptors is demonstrated to occur during melanoma progression and correlates with the acquisition of invasive or proliferative cell behaviour (Tsoi et al., 2018). EGFR (ERBB1) is a neural crest-associated gene that is found to be highly expressed in subpopulations of melanoma tumour cells exhibiting an undifferentiated or neural crest stem cell-like (NCSC) gene expression signature, including upregulation of SOX9, NGFR and AXL, downregulation of SOX10, and a highly invasive phenotype (Sun et al., 2014; Tsoi et al., 2018). EGFR has been identified as a driver of resistance to therapeutic MAPK pathway inhibition (Abel et al., 2013; Dugo et al., 2015), where autocrine activation of EGFR stimulates activation of the downstream MAPK and PI3K-Akt signalling pathways (Oberst et al., 2008; Sun et al., 2014; Liu S. et al., 2021). Conversely, subpopulations of cells with a proliferative phenotype preferentially express ERBB3 (Tsoi et al., 2018); with ERBB1 and ERBB3 expression shown to be mutually exclusive in melanoma cell lines (Dugo et al., 2015). However, the ERBB3 molecule demonstrates no kinase activity except when dimerised with EGFR or ERBB2, while ERBB2 expression is reported to be very low or absent in melanoma (Liu S. et al., 2021, Ueno et al., 2008; Inman et al., 2003). This suggests that a switch in EGF receptor expression occurs, with ERBB3 slowly downregulated and EGFR upregulated during the transition to an invasive phenotype in order to drive MAPK and PI3K-Akt pathway activity and ultimately metastasis. However, these findings contradict a study utilising B16-BL6 melanoma cells indicating that ERBB3, when dimerised with EGFR, is essential for tumour metastasis both in vitro and in vivo, and upregulates mesenchymal genes downstream of MAPK, JNK and PI3K-Akt pathway activation (Ueno et al., 2008). Nevertheless, the study by Ueno and others is based on a single murine melanoma cell line which likely fails to recapitulate the intratumoural heterogeneity found in a patient tumour, and thus the switch observed.
In conjunction with overexpression of EGFR, metastatic melanoma cells are demonstrated to overproduce the epidermal growth factor (EGF) ligand. EGF can subsequently act in an autocrine signalling manner allowing melanoma cells to produce their own growth and survival signals, as well as in a paracrine manner acting on endothelial cells to drive neoangiogenesis (Bracher et al., 2013). The hyperactivated EGF signalling cascade promotes expression of MMPs, adhesion molecules and initiators of EMT, subsequently driving invasion and metastasis (Kajanne et al., 2007; Lafky et al., 2008; Hardy et al., 2010; Kim et al., 2011; Zuo et al., 2011).
Overexpression and hyperactivation/hyper-phosphorylation of EGFR has been linked to anoikis resistance in a range of cancers, including hepatocellular carcinoma (Lim et al., 2020), GBME (Talukdar et al., 2018), breast (Oberst et al., 2008), lung (Chunhacha et al., 2013) and prostate cancer (Giannoni et al., 2009). In epithelial cells, hyperactivation of EGFR results in abrogated activity of pro-apoptotic Bim through the maintenance of MAPK pathway signalling, allowing the survival of cells upon ECM detachment (Reginato et al., 2003). While investigating the effects of reduced pH on the survival and metastasis of human melanoma cells, Peppicelli and others identified that cells possessing an anoikis resistant phenotype expressed high levels of EGFR, and reduced levels of cleaved PARP-1 (Peppicelli et al., 2019). These cells additionally displayed enhanced motility and invasion through matrigel, and expressed markers of a mesenchymal-like phenotype including N-cadherin. Furthermore, CTCs from melanoma are demonstrated to express receptors from the EGFR family (Tsao et al., 2018). While the role for EGFR signalling in anoikis resistance in melanoma cells remains poorly defined, it is hypothesised that the sustained survival signalling provided by autocrine EGF/EGFR activation within clusters of melanoma cells contributes to the survival of disseminated cells.
A range of small molecule inhibitors and monoclonal antibody therapies have been designed to target EGFR hyperactivation in cancers of epithelial origin (Figure 4). Lapatinib is an antineoplastic small molecule kinase inhibitor registered by the FDA (2007) for the treatment of patients with advanced metastatic breast cancer (HER2/ERBB2+) in conjunction with the chemotherapeutic agent capecitabine (Ryan et al., 2008). Lapatinib prevents phosphorylation of multiple RTKs, including EGFR, ERBB2, ERK1/2 and Akt and has demonstrated efficacy in clinical trials covering other malignancies (NCT00486954, NCT00949455, NCT02230553, NCT00095940, NCT01184482, NCT04608409). Similarly, three monoclonal antibody therapies targeting EGFR have been registered by the FDA. Cetuximab (2004) was first approved for the treatment of metastatic colorectal carcinoma (CRC) and head and neck squamous cell carcinoma (HNSCC), while avelumab (2017, NCT03089658) and amivantamab (2021, NCT04599712) were approved for the treatment of metastatic Merkel cell carcinoma and EGFR-mutant NSCLC, respectively. A multitude of other ERBB inhibitors (ERBBi) have demonstrated efficacy in clinical trials, with the most notable examined in phase IV trials: osimertinib (NCT03853551), dacomitinib (NCT04511533), erlotinib (NCT01230710, NCT00949910, NCT01402089, NCT01609543, NCT01066884), gefitinib (NCT00770588, NCT01203917, NCT00608868, NCT01000740), afatinib (NCT02514174, NCT02208843).
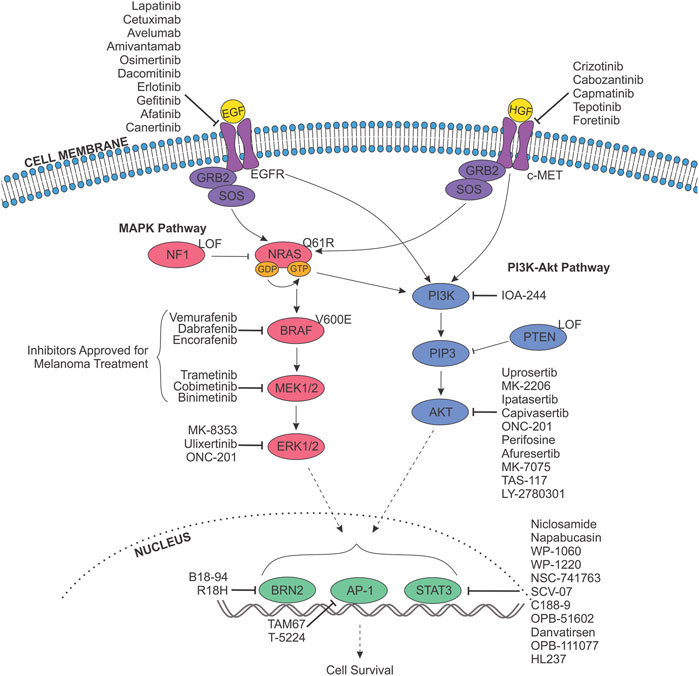
FIGURE 4. Targeting receptor tyrosine kinase signalling pathways in melanoma anoikis resistance. The MAPK (pink) and PI3K-Akt (purple) signalling pathways are constitutively activated in melanoma and stimulated by the binding of a growth factor (Epidermal Growth Factor (EGF)/Hepatocyte Growth Factor (HGF); yellow) to their respective receptor tyrosine kinase at the cell membrane (EGFR/c-MET). Oncogenic activation of these pathways is due to one of three proteins, whose genes are mutated with high frequently in melanoma; BRAF, NRAS and NF1. Most commonly, the BRAF gene harbours a mutation resulting in the V600E alteration, while NRAS usually possesses mutations leading to the Q61R variant, and NF1 is frequently effected by a genetic alteration resulting in loss of function (LOF). These variations result in abnormal signalling to downstream effectors, resulting in altered proliferation, differentiation, apoptosis, cell survival and metabolism; inevitably driving melanoma initiation and progression. As such, components of the MAPK pathway are targeted with small molecule inhibitors for the therapeutic treatment of melanoma, and have been approved as either single-agent therapies or in combination. Inhibitors approved for treatment of melanoma are indicated. In addition, inhibitors targeting EGFR, c-MET, the PI3K-Akt pathway, and downstream transcription factors (green) exist, and have the potential to block anoikis resistance. Indicated inhibitors have been sourced from data from all cancers.
Extensive preclinical studies have examined the effects of ERBBi in metastatic melanoma. A study by Girotti and others demonstrated that BRAFi-resistant melanoma cell lines express high levels of phosphorylated EGFR, and secrete increased levels of EGF (Girotti et al., 2013). Treatment of resistant A375 cells with gefitinib reduced in vitro proliferation and invasion. Subsequent in vivo treatment of BRAFi-resistant A375 tumour xenografts with combination gefitinib plus vemurafenib (PLX4720) synergistically reduced tumour volume, compared to vemurafenib or gefitinib alone. As BRAFi treatment has been demonstrated to promote SOX10-low/EGFR-high expressing cell populations, an independent study investigated the same inhibitor combination in the A375 cell line in the context of SOX10 knockout (Sun et al., 2014). However, the combination of gefitinib and vemurafenib did not lead to proliferation arrest in this setting, suggesting that further investigation is required to identify the specific cohort of patients that may benefit from ERBB inhibition. Interestingly, a study comparing single-agent treatment using gefitinib, erlotinib or lapatinib versus the pan-ERBB inhibitor (targeting EGFR, ERBB2,3 and 4), canertinib (CI-1033), revealed increased ability of the multi-kinase inhibitor to reduce in vitro proliferation of both BRAF-wild-type (WT) and BRAFV600E melanoma cell lines (Ng et al., 2014). Canertinib was additionally shown to synergise with vemurafenib in vitro, significantly reducing the IC50 of vemurafenib in 4 of 6 melanoma lines assessed.
ERBB4 is found to be somatically mutated in 19% of melanomas, with the majority of alterations occurring in the extracellular domain of the receptor, resulting in enhanced kinase activity (Prickett et al., 2009). Importantly, ERBB4 mutations increased the ability of melanoma cells to grow under anchorage-independent conditions, as assessed by the ability of SK-MEL-2 cells to form colonies in soft agar. Treatment with lapatinib was demonstrated to significantly reduce cell growth in ERBB4-mutant melanoma cells lines at 10- to 250- fold higher sensitivity (IC50) than ERBB4 WT cells, resulting in dose-dependent inhibition of ERBB4 auto-phosphorylation and downstream Akt signalling. However, a phase II clinical trial evaluating the safety and efficacy of lapatinib in treatment refractory stage IV melanoma patients carrying ERBB4 mutations (NCT01264081) was terminated with 30/34 patients failing to complete the trial. Nevertheless, the in vitro and in vivo evidence supporting the use of ERBBi in melanoma is strong, and further investigation into their use in melanoma patients with high levels of EGFR expression is warranted, particularly given the evidence for its role in driving anoikis resistance.
2.2.2 c-MET/HGF
c-MET (encoded by the proto-oncogene, MET) is a transmembrane RTK activated by its’ ligand, hepatocyte growth factor (HGF). The c-MET/HGF signalling pathway normally acts in a paracrine manner, with HGF secreted by mesenchymal cells and acting on surrounding epithelial cells (Li et al., 2001b). The conformation change induced by HGF binding, and the subsequent auto-phosphorylation of the homodimerised receptor subunits at Y1234 and Y1235, results in the recruitment of downstream effector proteins such as STAT3 and Grb2 (Organ and Tsao, 2011). Guanine nucleotide exchange factors (GEFs), including SOS, are subsequently recruited resulting in the stimulation of the MAPK pathway via the activation of Ras-GDP to Ras-GTP, as well as the initiation of PI3K-Akt pathway signalling (Zhang et al., 2018). During embryonic development, c-MET/HGF signalling plays a crucial role in the survival and migration of myogenic progenitor cells through stimulation of EMT (Bladt et al., 1995; Jeon and Lee, 2017).
Expression of c-MET is detected in both keratinocytes and melanocytes in the dermis and epidermis of the skin (Saitoh et al., 1994). However, following oncogenic transformation, melanoma cells gain expression of HGF, allowing autocrine c-MET/HGF signalling that functionally decouples melanoma cells from keratinocytes, facilitating invasion and migration (Natali et al., 1993; Otsuka et al., 1998; Rusciano et al., 1998; Li et al., 2001b). Studies subsequently established that melanoma cells with high c-MET/HGF autocrine signalling have an increased propensity for metastasis to the liver (Lin et al., 1998; Otsuka et al., 1998). Expression of c-MET in BRAFV600E melanoma cell lines, mouse xenografts and patient tumours is further demonstrated to contribute to vemurafenib resistance under hypoxic conditions (Qin et al., 2016). At a cellular level, c-MET is localised to sites of cellular adhesion (Kenessey et al., 2010), and co-immunoprecipitates with adhesion molecules such as E-cadherin and desmoglein 1 (Li et al., 2001b). A study by Koefinger and others demonstrated that expression of HGF in melanoma cells induces a switch in cadherin expression from E-to N-cadherin, driven by the downregulation of Slug and upregulation of Twist (Koefinger et al., 2011). Overexpression of c-MET also promotes proteolytic processes associated with invasion through the upregulation of urokinase-type plasminogen activator (uPA) and MMP expression (Rusciano et al., 1998; Tanaka et al., 2021).
Given the role of c-MET in melanoma invasion and migration, and its association with adhesion molecules and EMT processes, the involvement of c-MET/HGF signalling in anoikis resistance is unsurprising. An early study in hepatocytes revealed that c-MET functionally sequesters the Fas death receptor to prevent apoptosis, suggesting that a high c-MET to Fas ratio may be involved in cancer cell survival (Wang et al., 2002). In accordance with this hypothesis, c-MET is demonstrated to contribute to anoikis resistance in detached ovarian cancer cells through activation of MAPK and PI3K pathways (Tang et al., 2010), as well as in gastric cancer (Toiyama et al., 2012), HNSCC (Zeng et al., 2002) and prostate cancer (Dai and Siemann, 2012). In a study by Pierce and others demonstrating increased anoikis resistance in melanoma cells following overexpression of the BRN2 transcription factor, a significant increase in c-MET expression and phosphorylation were observed (Pierce et al., 2020). Comparable with autocrine EGFR signalling discussed previously, autocrine c-MET/HGF signalling in disseminated cells and CTC clusters is hypothesised to contribute to the survival signalling necessary to evade anoikis during melanoma metastasis.
Due to the high prevalence of c-MET hyperactivation in a variety of cancers, inhibitors have been designed to target the molecule for therapeutic purposes (Figure 4). Crizotinib is an ATP competitive inhibitor against c-MET, as well as ALK, FDA registered (2011) for the treatment of patients with advanced or metastatic NSCLC. Preclinical evidence in uveal melanoma revealed the feasibility of crizotinib treatment, demonstrating reduced in vitro viability and migration, and a significant reduction in vivo formation of macro-metastases (Surriga et al., 2013). Furthermore, multiple studies have investigated combining crizotinib with other inhibitors for the treatment of cutaneous melanoma. The combination of crizotinib with the ERBBi afatinib demonstrated synergistic cytotoxic effects, significantly reducing 2D and 3D invasion, migration and colony formation independent of BRAF/NRAS mutation status, and resulting in decreased tumour volume in vivo (Das et al., 2019; Das et al., 2020). Importantly, a phase I study investigating the use of crizotinib in combination with vemurafenib in advanced melanoma patients has demonstrated safety and efficacy [NCT01531361 (Janku et al., 2021)]. Single-agent treatment with crizotinib is under investigation in a phase II trial for the treatment of uveal melanoma patients (NCT02223819), while a phase I/II study is currently recruiting patients with solid tumours carrying GNAQ/11 mutations or PRKC gene (encoding PKC protein family members) fusions (NCT03947385), as these alterations are shown to result in HGF hypersecretion and hyperactivation of c-MET (Kermorgant et al., 2004; Surriga et al., 2013).
Cabozantinib is a small molecule inhibitor targeting a range of RTKs including c-MET, KIT, VEGFR-1/2/3, AXL and TRKB. It was first registered by the FDA (2012) for the treatment of patients with progressive and unresectable advanced medullary thyroid cancer (MTC), and has subsequently received FDA registration for use in treatment-refractory advanced renal cell carcinoma (2016), hepatocellular carcinoma (HCC) (2019), and as a first line treatment for differentiated thyroid cancer (DTC) (2021) and RCC (2021). A preclinical study investigating cabozantinib treatment of cell lines derived from melanoma brain metastases demonstrated the ability of the inhibitor to significantly reduce viability of cells in monolayer and 3D spheroid cultures, reduce migration and colony formation, and downregulate phosphorylation of Akt and MEK1/2 (Mannsåker et al., 2021). Phase I and II trials investigating the response of uveal and cutaneous melanoma patients to cabozantinib treatment have been completed [NCT01709435, NCT00940225 (Daud et al., 2017), NCT01835145 (Luke et al., 2020)].
Capmatinib is a c-MET inhibitor registered by the FDA (2020) for the treatment of metastatic NSCLC patients with MET exon 14 skipping mutations. Preclinical studies have evaluated the treatment of melanoma patient-derived xenografts (PDXs) with single-agent capmatinib, and in combination with encorafenib (BRAFi), binimetinib (MEKi) or both (Krepler et al., 2016). The study revealed complete tumour regression in 100% of mice treated with capmatinib + binimetinib + encorafenib, with no evidence of therapy resistance. A phase II clinical trial is currently recruiting advanced melanoma patients for treatment with the triple combination (NCT02159066). Similarly, tepotinib was FDA registered in 2021 for the treatment of NSCLC, and demonstrated evidence against melanoma in preclinical studies. Treatment of WM451 cells suppressed invasion and migration, and induced apoptosis by blocking PI3K-Akt signalling resulting in reduced Bcl-2, increased Bax expression and caspase-3 cleavage, accompanied by a switch in N- to E-cadherin expression suggesting suppression of EMT (Jing et al., 2022).
Foretinib (EXEL-2880, GSK1363089), a c-Met and VEGFR2 inhibitor, has demonstrated a high level of efficacy in clinical trials for other cancer types (NCT00742131, NCT00742261, NCT00743067), and in preclinical studies. Treatment of B16-F10 melanoma cells with foretinib blocked anchorage-independent growth under hypoxic conditions in vitro, and significantly reduced spontaneous metastasis to the lungs of mice in a dose-dependent manner (Qian et al., 2009). Similar to crizotinib, combining foretinib treatment with ERBBi gefitinib or lapatinib demonstrated synergistic cytotoxic effects on melanoma cells (Dratkiewicz et al., 2018; Simiczyjew et al., 2019), while combination treatment with vemurafenib produced comparable effects (Dratkiewicz et al., 2019).
In addition to the direct effect of c-MET inhibitors on tumour growth and invasion, treatment with c-MET inhibitors foretinib, crizotinib and cabozantinib are demonstrated to reduce the viability of vascular endothelial cells immortalised from melanoma tumours grown in immunocompetent mice (Jenkins et al., 2021), revealing the potential efficacy of c-METi on tumour cells, stromal cells and CTCs.
2.3 Signalling pathways
2.3.1 FAK signalling
First linked to anoikis resistance in mammary epithelial cells by Frisch et al. (1996), focal adhesion kinase (FAK/PTK2) plays a role in tyrosine kinase signalling where it interacts with the β subunit of integrins in the cytosol [reviewed in (Paoli et al., 2013)]. Cell adhesion to ECM components through integrin binding results in rapid activating phosphorylation of FAK (p-FAK) at tyrosine residues 397 and 576, resulting in the recruitment of proteins including Src kinase, the actin cytoskeleton-binding paxillin and adaptor proteins such as Grb2, activating multiple downstream pathways including MAPK and PI3k-Akt signalling (Goundiam et al., 2012). FAK also has a distinct role independent of focal adhesions as a result of a nuclear localisation signal within the FERM domain of the protein (Aplin et al., 1998; Parsons et al., 2000). Normally, interrupting focal adhesions through cellular detachment results in rapid dephosphorylation of FAK and its cleavage facilitated by caspase-3, -6 and -9, resulting in translocation to the nucleus where FAK prevents activation of wild-type p53 signalling, triggering intrinsic apoptosis (Del Mistro et al., 2022).
In melanoma, studies indicate that inappropriate phosphorylation of FAK upon cell detachment contributes to anoikis resistance by allowing persistent survival signalling (Hess et al., 2005). Studies utilising B16-F10 murine melanoma cells cultured on poly-HEMA (an ultra-low attachment coating) demonstrate increased expression of p-FAK, alongside increased expression of downstream molecule RhoA, and increased activating phosphorylation of Akt (p-Akt) and ERK1/2 (p-ERK1/2) when compared to the Swiss 3T3 anoikis sensitive cell line used as a control (Goundiam et al., 2010). Interestingly, a study demonstrated that stabilisation of p-FAK through interaction with the cell cycle regulator p14ARF and subsequent sumoylation contributes to anoikis resistance (Vivo et al., 2017). Mutant forms of p14ARF containing point mutations identified in melanoma were able to stabilise p-FAK more effectively than wild-type p14ARF. Comparably, FAK knockdown (siFAK) in B16-F10 cells was shown to suppress migration and metastasis in vivo, with decreased number of metastatic nodules present in the lungs of mice upon siFAK in comparison to controls (Pei et al., 2017). FAK knockdown decreased expression of p-ERK1/2, p-STAT3 and increase expression of E-cadherin, while siRNA against Akt and PI3K reduced p-FAK expression (Goundiam et al., 2012; Pei et al., 2017).
Small molecule inhibitors that target FAK activity have been explored in both phase I and II trials for use in a range of advanced solid and haematological malignancies, and may represent a viable option for repurposing to target melanoma CTCs. Defactinib (VS-6063, PF-04554878) is a specific inhibitor of FAK with demonstrated antioangiogenic and antineoplastic activities. Defactinib is shown to be safe in healthy subjects, and has demonstrated efficacy against advanced malignancies in phase I trials (NCT02913716, NCT02546531 and NCT01943292). Interestingly, in a study utilising melanoma cells from patients who relapsed following treatment with BRAF or MEK targeted inhibitors, treatment with the FAK inhibitor (FAKi) defactinib was able to resensitise cells to killing by MAPK pathway inhibition (Del Mistro et al., 2022). Other FAKi yet to be examined in melanoma are summarised in Table 2.
The literature suggests that clusters of tumour cells in circulation have greater metastatic potential and increased ability to evade anoikis than single tumour cells (Hou et al., 2011; Hou et al., 2012). A study revealed the potential efficacy of using FAKi to target anoikis resistant tumour cells in circulation (Au et al., 2016). It was demonstrated that CTC clusters are able to transit through capillaries with over 90% efficacy by unfolding into single-file chains of cells. Use of a FAK inhibitor tool compound (FAK I-14) resulted in a significantly greater likelihood of CTC clusters being disrupted when transiting through capillaries than untreated clusters (Figure 1).
2.3.2 MAPK signalling pathway
Melanomagenesis occurs primarily through the acquisition of pathogenic mutations in genes that encode key components of the Mitogen-Activated Protein Kinase (MAPK) signalling pathway (Guo et al., 2020). Mutations in BRAF, (N/H/K) RAS or NF-1 were detected in 52%, 30.9% and 14% of patients in the Cancer Genome Atlas Network (TCGA) dataset, respectively (Akbani et al., 2015). These mutations cause constitutive activation of the MAPK pathway, driving processes such as proliferation, differentiation and abrogated apoptosis via the activation of a kinase cascade resulting in downstream nuclear transcription factor phosphorylation and activation (Guo et al., 2020). MAPK signalling is demonstrated to be stimulated by microenvironmental growth factors, and result in activation of the EMT-TFs (Lamouille et al., 2014). The downregulation of Slug and Zeb2, and upregulation of Zeb1, Snail and Twist1 promote differentiation and metastasis in melanoma cells (Hoek et al., 2004; Kuphal et al., 2005; Caramel et al., 2013). However, the role of Slug in melanoma appears to be highly context dependent, with other studies demonstrating increased expression in invasive cell populations (Fenouille et al., 2012; Pearlman et al., 2017).
Accordingly, multiple small molecule inhibitors against key components of this pathway have been registered by the FDA for use in metastatic melanoma (Figure 4). Vemurafenib (approved in 2011) and dabrafenib (2013) target and inhibit mutant-BRAF (BRAFi) (Jenkins and Fisher, 2021). In addition, between 2014 and 2018, three regimens that combine the use of a BRAFi with an inhibitor against MEK (MEKi) were approved for use. The combinations of dabrafenib + trametinib, vemurafenib + cobimetinib, and encorafenib + binimetinib have demonstrated efficacy against melanomas with mutations in both the NRAS and BRAF genes, and are shown to improve therapy response and overall PFS (Flaherty et al., 2012; Ascierto et al., 2013; Larkin et al., 2014; Long et al., 2015; Robert et al., 2015). However, following treatment with such inhibitors, multiple distinct and drug resistant subpopulations of melanoma cells persist, and quickly drive relapse in a proportion of patients (Fedorenko et al., 2011; Rambow et al., 2018; Rambow et al., 2019).
Together with its role in driving melanoma formation and progression the MAPK signalling pathway contributes significantly to the acquisition of resistance to cell death by anoikis in melanoma cells. Unlike primary human epidermal melanocytes, melanoma cell lines carrying the BRAFV600E alteration demonstrate enhanced cellular survival and reduced apoptosis when plated on agar, as measured by caspase-3 cleavage (Boisvert-Adamo and Aplin, 2006). It was identified that mutant-BRAF drives anoikis resistance through constitutive activation of MAPK signalling, resulting in the depletion of two pro-apoptotic Bcl-2 family members; the Bcl-xl/Bcl-2-associated death promoter (Bad) and Bcl-2-interacting mediator of cell death (Bim), while simultaneously increasing expression of anti-apoptotic myeloid cell leukemia-1 (Mcl-1) (Boisvert-Adamo and Aplin, 2008; Boisvert-Adamo et al., 2009; Goldstein et al., 2009). Consistent with these findings, siRNA-mediated ablation of BRAF, or pharmacological inhibition of MEK, were demonstrated to induce susceptibility to anoikis in melanoma cells (Boisvert-Adamo and Aplin, 2006). However, it is clear from the vast number of studies detailing intrinsic and acquired resistance to single-agent and combination MAPKi therapy (Fedorenko et al., 2011; Winder and Virós, 2018), that the use of BRAF or MEK targeted inhibitors alone is insufficient to block anoikis resistance long-term in patients.
Reactivation of ERK1/2 is considered a primary mechanism of MAPKi therapy resistance (Lee et al., 2020). Given its role in cell cycle progression and evasion of apoptosis, inhibitors against ERK1/2 are being investigated in a range of advanced and metastatic malignancies (Figure 4). MK-8353 is a selective inhibitor that targets both phosphorylated and unphosphorylated ERK1/2. Preclinical evaluation in an in vivo human xenograft model derived from the BRAFV600E-mutant melanoma cell line SK-MEL-28, demonstrated significant reduction in tumour volume compared to untreated controls (Moschos et al., 2018). Despite reporting efficacy and safety in a phase I trial of patients with advanced solid tumours (NCT01358331), MK-8353 demonstrated an anti-tumour response in only 3 of 8 BRAFV600E melanoma patients, with all patients withdrawing from the trial as a result of disease progression or adverse effects. MK-8353 has also undergone testing in a phase 1b trial in combination with the ATP competitive MEK1/2 inhibitor Selumetinib in advanced or metastatic solid tumours in an attempt to suppress MAPK therapy resistance (NCT03745989), and is likewise currently under investigation in combination with the immunotherapy pembrolizumab (NCT02972034). Similarly, Ulixertinib (BVD-523) in an ATP-competitive inhibitor against ERK1/2 that has successfully completed phase I/II trials examining dosage and safety, displaying favourable pharmacokinetics in NRAS-mutant and BRAFV600E advanced solid malignancies, as well as those carrying BRAF mutations at non-V600 sites (NCT01781429) (Sullivan et al., 2018; Wu J. et al., 2021). Interestingly, ulixertinib was recently used in a phase II trial for the treatment of patients with metastatic uveal melanoma (NCT03417739). However, the therapy failed to demonstrate anti-tumour activity, with those treated demonstrating a median overall survival of 6.9 months (3.2–8.3), and 38.46% of participants reporting serious adverse events as a result of the treatment (Buchbinder et al., 2020). In addition to the use of ERKi, the current gold-standard treatment combining the use of targeted inhibitors with immunotherapies suggests there may be efficacy in further combining BRAF or MEK inhibitors with those against other known mediators of anoikis resistance described in this review to target CTCs, especially given the considerable level of crosstalk between MAPK, PI3K-Akt, EGFR, FAK and other key signalling pathways in melanoma.
2.3.3 PI3K-Akt signalling
The Phosphatidylinositol 3-Kinase Akt (PI3K-Akt) signalling pathway has been demonstrated to synergise with oncogenic MAPK signalling to increase proliferation and disease progression (Atefi et al., 2011; Greger et al., 2012). PI3K-Akt signalling is constitutively activated by mutant NRAS (Johnson and Puzanov, 2015), as well as the PTEN tumour suppressor which is found to be effected by loss of function mutations in approximately 20% of BRAF-mutant melanomas, resulting in Akt activation (Shull et al., 2012; Akbani et al., 2015). Overall, expression of Akt is elevated in approximately 70% of malignant melanomas resulting in the dysregulation of downstream effector molecules, such as mTOR (Pearlman et al., 2017). Studies propose that melanoma invasiveness may be regulated by a PI3K-PAX3-BRN2 axis, with inhibition of PI3K signalling shown to reduce invasion and downregulate expression of both PAX3 and BRN2 (Bonvin et al., 2012). In epithelial cells, activation of PI3K-Akt signalling drives the process of EMT through the activation of the EMT-TFs, protecting against death by anoikis in suspension culture conditions (Khwaja et al., 1997; Xu et al., 2015). Mechanistically, Akt activity is able to abrogate apoptosis via the phosphorylation of Bim and Bad pro-apoptotic proteins, inhibiting caspase-9 activity and transcription of the Fas death receptor ligand (Qi et al., 2006; del Peso et al., 1997; Brunet et al., 1999; Kennedy et al., 1999).
In melanoma, PI3K-Akt signalling is thought to contribute to anoikis resistance. Early research suggested the pathway may act as a secondary survival cue, protecting cells against apoptosis in the absence of mutant-BRAF (Boisvert-Adamo and Aplin, 2006). In the presence of mutant-BRAF, PTEN-deficient melanoma cells express constitutively active Akt3, which protects against apoptosis by upregulating Bim and Bmf (Shao and Aplin, 2010). When cultured on poly-HEMA, B16-F10 cells demonstrated a transient increase in Akt phosphorylation, while total protein level was unchanged (Goundiam et al., 2010). A subsequent study revealed that inhibition of Akt activation, as well as downstream RhoA and RhoC expression, resulted in induction of anoikis through the inactivation of the FAK signalling pathway (Goundiam et al., 2012). Similarly, two consecutive studies by Toricelli et al. validated these findings, revealing that inhibition of PI3K or Akt proteins reversed anoikis resistance in melanoma cells (Toricelli et al., 2013; Toricelli et al., 2017). Inhibition of Timp1, an MMP inhibitor whose overexpression activates PI3K-Akt signalling, induced sensitivity to anoikis in vitro and reduced tumour volume and metastatic colony formation in vivo. Subsequent studies have shown that anoikis resistance induced via the knockdown of α2, α3β1 or α5β1 integrins can be rescued via the specific inhibition of Akt (Kozlova et al., 2019; Kozlova et al., 2020). Furthermore, MIST1 and SNAI1 transcription factors are thought to contribute to anoikis resistance by directly repressing PTEN (Lee et al., 2018), while knockdown of the NCAM adhesion molecule induced apoptosis in melanoma cells by suppressing Akt activation (Li et al., 2020).
Given the frequency of Akt hyperactivation in melanoma, a range of inhibitors have been designed against its’ isoforms and may be repurposed in order to target anoikis resistance (Table 2; Figure 4). Uprosertib is an orally bioavailable inhibitor against Akt. Preclinical evidence demonstrated the potential efficacy of combining uprosertib with MEKi against a range of cancer cell lines with mutations in the BRAF or KRAS genes (NCT01935973, NCT01989598) (Dumble et al., 2014). However, a phase II study investigating the combination in BRAF/NRAS wild-type, and NRAS-mutant melanoma patients (NCT01941927) revealed no improvement to overall or progression-free survival (Algazi et al., 2018). A similar phase II trial in stage IV uveal melanoma patients (NCT01979523) revealed analogous results, with dose reductions required due to the frequency of adverse events (Shoushtari et al., 2016), as predicted in the phase I dose-escalation study [NCT01138085 (Tolcher et al., 2020)]. Despite this, a phase I/II trial into uprosertib plus dabrafenib and trametinib in stage III/IV metastatic solid cancers, including melanoma, is currently underway (NCT01902173).
MK-2206 inhibits Akt, blocking downstream PI3K-Akt signalling. Its’ anti-tumour activity has been assessed in clinical trials against a range of cancers (NCT01071018, NCT00670488, NCT00848718, NCT01283035, NCT01604772, NCT01349933, NCT01802320, NCT01333475, NCT01258998, NCT01481129, NCT01253447, NCT01231919, NCT01369849, NCT01277757) (Molife et al., 2014). Preclinical studies treating melanoma cells in vitro with MK-2206 resulted in a concentration-dependent downregulation of phosphorylated Akt, inhibition of cell growth and colony formation, and induced apoptosis through altered expression of Bax, and increased ROS generation (Quast et al., 2013; Petit et al., 2019). MK-2206 has further demonstrated efficacy in combination with binimetinib, where treatment resulted in a synergistic reduction in tumour volume (Petit et al., 2019), as well as in combination with the mTOR inhibitor everolimus (Ciołczyk-Wierzbicka et al., 2020), and vemurafenib (Su et al., 2012; Thang et al., 2015). Interestingly, a phase I trial investigating MK-2206 in combination with paclitaxel and carboplatin for the treatment of two patients with BRAF-wild-type stage IV melanoma reported long-term, enhanced responses to chemotherapy [NCT00848718 (Rebecca et al., 2014)]. However, a phase II clinical trial investigating MK-2206 plus selumetinib (MEK1/2 inhibitor) in stage III/IV melanoma patients who previously failed vemurafenib or dabrafenib treatment was terminated due to slow accrual (NCT01519427).
The small molecule Akt inhibitor Ipatasertib (GDC-0068) has been examined in phase I trials against breast, ovarian and prostate cancers (NCT03840200, NCT01562275, NCT01362374) (Yan et al., 2013), as well as in phase II trials for gastric cancers (NCT01896531) and in combination with paclitaxel for breast cancer treatment (NCT02301988, NCT02162719). While its efficacy in melanoma patients is yet to be examined, ipatasertib is demonstrated to prevent growth of the PTEN-null melanoma tumours in vivo (Saura et al., 2017). Similarly, capivasertib (AZD-5363) targets all isoforms of Akt, and has completed phase I trials against solid tumours (NCT04742036), haematological malignancies (NCT04944771) and prostate cancer (NCT04087174). In a study by Dinavahi and others, simultaneously administering capivasertib with the WEE1 inhibitor AZD-1775, synergistically reduced melanoma cell survival in vitro and tumour growth in vivo by driving increased expression of p53 and blocking Akt-mediated activation of FOXM1 (Dinavahi et al., 2018).
ONC-201 (TIC-10) is an orally available small molecule inhibitor with activity against both Akt and ERK. The efficacy of ONC-201 for the treatment of advanced malignancies has been investigated in phase I and II (NCT02324621, NCT02250781, NCT02609230, NCT03394027). Interestingly, as ONC-201 is water-soluble and able to cross the blood-brain barrier, phase II/III clinical trials are currently recruiting participants to examine its efficacy against gliomas (NCT05476939, NCT05009992). While yet to be trialled in melanoma patients, pre-clinical evidence demonstrated treatment of melanoma cell lines with ONC-201 reduced colony formation and migration in vitro, decreased expression of p-Akt and p-ERK, and resulted in a significant reduction in tumour volume (Wagner et al., 2017). Further, combination treatment with bortezomib, an inhibitor of the ubiquitin-proteasome pathway, demonstrated a synergistic ability to reduce cell viability and induce apoptosis (Takács et al., 2021). Although ONC-201 has been examined in clinical trials as a specific inhibitor against Akt and ERK, conjecture surrounding its mechanism of action remains, with studies suggesting it may indirectly modulate Akt activity as a downstream effect of dopamine receptor D2 inhibition (McSweeney et al., 2020; Prabhu et al., 2020).
Perifosine (KRX-0401) is a small molecule inhibitor against Akt that has been examined in phase II trials against a range of cancers (NCT00389077, NCT00873457, NCT00455559, NCT00590954, NCT00448721, NCT00498966, NCT00375791, NCT00398879, NCT00053924, NCT00059982). Despite evidence that perifosine treatment of melanoma cells reduces expression of p-Akt (Liu and Xing, 2012), a phase II trial in metastatic cutaneous melanoma patients failed to demonstrate efficacy [NCT00053781 (Ernst et al., 2005)]. Afuresertib (GSK-2110183) is an orally available small molecule inhibitor of Akt. A phase I/II dose escalation study combining afuresertib with trametinib in patients with advanced solid tumours observed a partial response in a patient with BRAF-wild-type melanoma, however the combination therapy was poorly tolerated with extensive adverse effects recorded [NCT01476137 (Tolcher et al., 2015)].
Furthermore, there has been growing investigation into the use of inhibitors targeting overexpression of PI3K directly in breast cancer and haematological malignancies [reviewed in (Vanhaesebroeck et al., 2021)], which is beginning to expand into other solid tumour types with a phase I trial examining the safety and tolerability of IOA-244 in uveal melanoma patients currently recruiting participants (NCT04328844). In addition, given that constitutive activation of the PI3K-Akt pathway activates downstream mTOR signalling to drive survival, motility, invasion and proliferation, multiple studies have investigated the use of mTOR inhibitors in combination with other therapies for the treatment of metastatic melanoma patients [recently reviewed in (Chamcheu et al., 2019)]. Such combination treatments may also be efficacious in the targeting of anoikis resistant melanoma cells.
2.4 Transcription factors
2.4.1 BRN2
BRN2, the POU domain transcription factor encoded by the gene POU3F2, has been linked to melanoma progression in the phenotype switching model as a potential driver of invasive behaviour (Cook and Sturm, 2008; Hoek and Goding, 2010) [reviewed in (Fane et al., 2019)]. BRN2 plays a role in the delineation of neural crest cells to the melanocytic lineage during embryonic development, similar to other factors such as SOX10 and the Microphthalmia-associated transcription factor (MITF) (Thomson et al., 1995; Cook et al., 2003). BRN2 is expressed in melanoma tissues (Sturm et al., 1991; Thomson et al., 1993), with 10-fold higher expression observed in melanoma cell lines in comparison to normal melanocytes (Eisen et al., 1995). Early research revealed that inhibition of BRN2 expression results in complete loss of tumour formation in mice, and the loss of melanocyte markers including MITF (Thomson et al., 1995). Subsequently, it was identified that BRN2 expression is inversely correlated with, and mutually exclusive to MITF expression in patient tumours and xenografts (Goodall et al., 2008). MITF and BRN2 therefore regulate opposing functions in the phenotype switching phenomenon by virtue of their ability to modulate each other; BRN2 represses expression of MITF by directly binding to its promoter (Goodall et al., 2008) as well as indirectly through upregulating NFIB (Fane et al., 2017); while MITF negatively regulates BRN2 via miR-211, a micro-RNA derived from the MITF-regulated gene melastatin (TRPM1) (Boyle et al., 2011). As a result, melanoma cells expressing low levels of MITF and high BRN2 (MITFlow/BRN2high) are demonstrated to be significantly more tumorigenic than MITFhigh/BRN2low cells when injected subcutaneously into mice (Goodall et al., 2008). In addition, BRN2 is shown to interact with the DNA damage response proteins PARP1 and Ku70/80 at sites of damage induced by UVB, chemotherapy or vemurafenib treatment, promoting error-prone repair via non-homologous end joining (NHEJ) and suppressing apoptosis (Herbert et al., 2019).
It’s therefore unsurprising given the role of BRN2 in driving invasion and metastasis that it has been similarly implicated in resistance to cell death by anoikis in melanoma. A recent study by our lab revealed that overexpression of BRN2 in human metastatic melanoma cell lines increased cellular viability when grown under non-adherent conditions (Pierce et al., 2020). The increased survival ability coincided with amplified expression of known markers of the anoikis-resistant and mesenchymal-like phenotypes; including the β1 integrin subunit (ITGB1), TWIST1 and MET, as well as increased STAT3 phosphorylation. The study further demonstrated that inhibition of c-MET was able to significantly reduce percentage viability in BRN2 overexpressing cells in ultra-low attachment conditions.
Currently, there are no FDA registered inhibitors against BRN2, nor are there any in clinical trials. However, a study in neuroendocrine prostate cancer recently emerged detailing the development of a potent and specific small molecule inhibitor against wild-type BRN2 (B18-94) (Thaper et al., 2022) (Figure 4). Inhibition of BRN2 using B18-94, or inducible CRISPR/Cas9 knockout as a control, was shown to result in the upregulation of pathways for epithelial development and apoptosis, suggesting that direct inhibition of BRN2 may be a feasible way of inducing anoikis in cancer cells that overexpress BRN2. While yet to be examined in humans, in vivo administration of B18-94 via both intra-peritoneal (IP) and oral routes in a murine model suggests the compound has efficacious therapeutic likeness. Furthermore, treatment of xenograft tumours reduced tumour volume and cell proliferation, and increased apoptosis. Similarly, a synthetic peptide derived from the POU domain of BRN2 (R18H) has been demonstrated to induce apoptosis in B16-F10-Nex2 melanoma cells in vitro, while IP administration in C57BL/6 mice resulted in a significant reduction in the formation of metastatic nodules in the lungs, compared to untreated mice (da Cunha et al., 2019).
2.4.2 STAT3
Signal transducer and activator of transcription 3 (STAT3) is activated downstream of receptor tyrosine kinase and growth factor receptors, particularly cMET/HGF, EGFR and PDGFR, as well as janus kinase (JAK) and Src Kinase pathways. Phosphorylation of STAT3 results in its dimerization and translocation to the nucleus where it promotes transcription. STAT3 has been extensively studied in the context of melanoma, where its constitutive activation is shown to promote melanoma cell proliferation, metastasis and invasion, and contribute to immune evasion by stimulating persistent expression of VEGF (Kortylewski et al., 2005; Lee et al., 2012; Swoboda et al., 2021; Graziani et al., 2022). Studies demonstrate that STAT3 drives melanoma cell survival through the upregulation of anti-apoptotic proteins Bcl-XL, Mcl-1, Cyclin D1 and survivin, and downregulation of p53 (Zhuang et al., 2007). As such, STAT3 has been identified as a contributor to the anoikis resistant phenotype in melanoma.
A study demonstrated increased STAT3 phosphorylation at Y705 in melanoma cell lines cultured in suspension conditions compared to adherent cells, driving upregulation of Bcl-2 and Mcl-1 (Fofaria and Srivastava, 2014). The subsequent increase in STAT3 activity increased migration and invasion of cells replated from suspension cultures in vitro, while knockout of STAT3 prevented the formation of tumours in vivo. An independent study confirmed these findings, revealing that p-STAT3 (Y705) stimulated anoikis resistance of B16-F10 melanoma cells as part of the FAK, p-ERK1/2 and PPARγ signalling pathways (Pei et al., 2017). Furthermore, STAT3 activity upregulated V-ATPase expression in B16-F10 cells to drive anoikis resistance, while pharmacological blockade of STAT3 repressed V-ATPase, inducing anoikis through ROS-mediated misfolded protein accumulation (Adeshakin et al., 2021b). An increase in STAT3 phosphorylation was similarly observed in melanoma cells cultured in ultra-low attachment conditions with an anoikis resistant phenotype induced by overexpression of BRN2 (Pierce et al., 2020).
The use of STAT3 inhibitors (STAT3i) for metastatic melanoma patients has been extensively investigated, given that combining STAT3i with anti-PD-1 immunotherapy has the potential to remodel the tumour microenvironment and resensitise treatment-refractory cells to vemurafenib treatment, while increasing CD8+ T cell infiltration into the tumour (Su et al., 2018; Zhao et al., 2020; Kim et al., 2022) (Figure 4). In vitro studies suggest the potential of utilising STAT3i to target anoikis resistance, with preclinical studies demonstrating that the synthetic inhibitor, AG-490, and the natural compound from black pepper, piplartine (PL), reduce anoikis resistance and induce PARP cleavage, while reducing the migratory potential of melanoma cells (Fofaria and Srivastava, 2014).
The FDA registered anti-helminthic drug niclosamide has potential inhibitory effects against STAT3. It’s unclear whether these effects are direct, however the molecule is predicted to bind to both the SH site and the Y705 phosphorylation site (Shi et al., 2017). Niclosamide may alternatively block STAT3 activation by targeting the androgen receptor variant V7 (AR-V7) upstream. Its use has been examined in phase I trials for castration resistant metastatic prostate cancer (NCT02532114) and refractory AML (NCT05188170). In melanoma, preclinical evidence revealed that niclosamide inhibited melanoma cell proliferation in vitro, independent of BRAF or NRAS mutation status, inhibiting tumour growth in xenograft models by uncoupling mitochondria and increasing metabolic stress (Figarola et al., 2018). Treatment of melanoma cells reduced STAT3 phosphorylation (Y705), inducing apoptosis via activation of Bax, reduction in Bcl-2 expression, and caspase-3 cleavage (Zhu Y. et al., 2019; Zheng et al., 2022). Niclosamide was further established to block melanoma cell migration and invasion, inhibiting activation of MMP-2 and 9, while inducing ROS generation in a dose-dependent manner. Other studies reveal improved delivery and efficacy of niclosamide against melanoma cells when the drug is packaged into liposomes (Hatamipour et al., 2019; Shah et al., 2022). A range of other STAT3 inhibitors have been examined in many cancers including melanoma, and are summarised in Table 3.
TABLE 3. STAT3 inhibitors.
Upstream, STAT3 is activated by the cytokine IL-6, and as such therapies blocking IL-6 are being investigated for their ability to prevent STAT3 activation (Table 4). In addition, as Janus kinases (JAK) are upstream mediators of STAT3 activation and signalling, a range of JAK inhibitors under investigation in clinical trials have demonstrated efficacy against melanoma in preclinical studies (Wu K. J. et al., 2017), and subsequently have the potential to be repurposed to block anoikis resistance by targeting JAK/STAT signalling in melanoma cells (Table 5).
TABLE 4. IL-6 inhibitors.
TABLE 5. JAK inhibitors.
2.4.3 AP-1 family
The activating protein-1 (AP-1) family of transcription factors are comprised of homodimers between Jun proteins (cJun, JunB and JunD) or heterodimers between Jun and Fos (c-Fos, FosB, Fra1 and Fra2) (Wisdom, 1999; Eferl and Wagner, 2003; Gurzov et al., 2008), and demonstrate a wide range of functions that differ depending on the composition of the complex, the target gene, and the cell type, influencing cell growth, proliferation and cell cycle progression (Chinenov and Kerppola, 2001; Jin et al., 2011). AP-1 transcription factors act as part of the immediate-early response initiated by MAPK pathway signalling. As such, during melanomagenesis the constitutive activation of this pathway results in over-activation of the AP-1 factors, driving cellular dedifferentiation and transcriptional heterogeneity (Comandante-Lou et al., 2022). In particular, Fra-1 (encoded by the gene FOSL1) is important for melanoma progression as its accumulation triggers the transcription factor switch that drives (partial)-EMT, downregulating Zeb2 and SNAI2 and directly upregulating Zeb1 at its promoter (Caramel et al., 2013; Casalino et al., 2022). Subsequently, Fra-1 drives changes in cytoskeletal organisation, polarisation, motility and invasion (Dhillon and Tulchinsky, 2015; Casalino et al., 2022). Critically, Maurus and others demonstrated that FOSL1 promotes anoikis resistant growth of melanoma cells on soft agar, and allows subcutaneous tumour growth in vivo via a Fra-1 target gene product, the chromatin modifier HMGA1 (Maurus et al., 2017).
Fra-1 was previously thought to be a non-viable target for pharmacological inhibition by small molecules, and as such most of the attempts to target the molecule focus on its destabilisation by inhibition of upstream kinases, or via targeting FOSL1 mRNA (Sobolev et al., 2022). Some metabolites and molecules are reported to non-specifically inhibit its expression and function, such as PARP1 inhibitors, glucocorticoid dexamethasone, ranpirnase and rosiglitazone, while vemurfanib and retinoic acid are shown to activate Fra-1 expression (Zeng et al., 2022). However, a few polypeptide and small molecule inhibitors against Fra-1 have demonstrated promise in recent years (reviewed in (Casalino et al., 2022) Table 6 and Figure 4).

TABLE 6. AP-1 family inhibitors.
2.5 Other contributing factors
Additional factors, including the microenvironment and ECM, cellular metabolism, and production of reactive oxygen species (ROS) and nitric oxide (NO) are believed to contribute to anoikis resistance in melanoma. However, we have chosen to focus on the contribution and targeting of intracellular signalling cascades, and as such detailed discussion of these topics are beyond the scope of this review.
Research in the field of cellular microenvironment and ECM have focused on the contribution of MMPs and TIMP-1 to anoikis resistance. TIMP-1, despite being described as an MMP inhibitor, is interestingly shown to increase the survival of melanoma cells in suspension, and increase metastatic potential by interacting with β1 integrins, resulting in PDK1 activation (Zhu et al., 2001; Ricca et al., 2009; Toricelli et al., 2013; Toricelli et al., 2017). Furthermore, adhesion to fibronectin is thought to activate PI3K-Akt signalling, and protect cells from apoptosis (Boisvert-Adamo and Aplin, 2006; Fedorenko et al., 2016). Drugs targeting these processes have the potential to block anoikis resistance in melanoma cells [as reviewed in (Winer et al., 2018)].
Studies investigating the cellular metabolism of CTCs have identified autophagy as a contributing factor to anoikis resistance driven by p53, PI3K-Akt, mTOR, STAT and EGFR signalling (Guadamillas et al., 2011; Liu M. et al., 2021). The contribution of autophagy, as well as ferroptosis and necroptosis to anoikis resistance in melanoma were recently reviewed (Ashrafizadeh et al., 2019). Small molecule inhibitors targeting these pathways have the potential to induce anoikis in melanoma cells [as reviewed in (Wu et al., 2022)]. Regulators of fatty acid oxidation (FAO), CROT and CRAT, are shown to regulate anoikis in melanoma cells (Lasheras-Otero et al., 2022), while inhibitors of FAO, such as thioridazine and ranolazine may have therapeutic potential [as reviewed in (Munir et al., 2022)]. Anoikis induction is additionally linked to increased ROS and reduced NO production (Adeshakin et al., 2021b; Zhu et al., 2020; Monteiro et al., 2019; Ribeiro-Pereira et al., 2014; Giannoni et al., 2008; da Costa et al., 2018), and as such therapies that target these processes may be a viable option against melanoma cells (Adeshakin et al., 2021a). These topics were recently reviewed in (Sattari Fard et al., 2022).
3 Discussion/concluding remarks
There is an opportunity to repurpose existing therapies for patients with a high risk of disseminating melanoma. For instance, patients with ulcerated disease may benefit from novel therapy due to early dissemination leading to worse prognosis than those with non-ulcerated, but similarly staged melanoma. Further, treatments targeting pathways involved in invasion and cellular migration may prevent cells leaving the primary tumour, while those impacting anoikis resistance mechanisms may potentially target melanoma cells in circulation prior to the seeding of metastases (Figure 1). Ultimately, it is clear that transcriptomic testing is required to administer personalised targeted therapies based on the expression signature of an individuals’ tumour. This personalised approach will hopefully culminate in better health outcomes for patients with malignant melanoma.
Author contributions
Conceptualisation, HN and GB; writing—original draft preparation, HN; writing—review and editing, JS and GB. All authors have read and agreed to the published version of the manuscript.
Funding
The authors wish to thank the National Health and Medical Research Council of Australia (APP1158283 Project Grant, GB).
Acknowledgments
The authors wish to thank the Queensland University of Technology for supporting this work through an Australian Government Research Training Program (RTP) Scholarship, and QIMR Berghofer Medical Research Institute for a Top-Up Scholarship.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abel, E. V., Basile, K. J., Kugel, C. H., Witkiewicz, A. K., Le, K., Amaravadi, R. K., et al. (2013). Melanoma adapts to RAF/MEK inhibitors through FOXD3-mediated upregulation of ERBB3. J. Clin. Invest 123, 2155–2168. doi:10.1172/JCI65780
Adams, J. M., and Cory, S. (2007). The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337. doi:10.1038/sj.onc.1210220
Adeshakin, F. O., Adeshakin, A. O., Liu, Z., Cheng, J., Zhang, P., Yan, D., et al. (2021a). Targeting oxidative phosphorylation-proteasome activity in extracellular detached cells promotes anoikis and inhibits metastasis. Life (Basel) 12, 42. doi:10.3390/life12010042
Adeshakin, F. O., Adeshakin, A. O., Liu, Z., Lu, X., Cheng, J., Zhang, P., et al. (2021b). Upregulation of V-ATPase by STAT3 activation promotes anoikis resistance and tumor metastasis. J. Cancer 12, 4819–4829. doi:10.7150/jca.58670
Akbani, R., Akdemir, K., Aksoy, B., Albert, M., Ally, A., Amin, S., et al. (2015). Genomic classification of cutaneous melanoma. Cell 161, 1681–1696. doi:10.1016/j.cell.2015.05.044
Albelda, S. M., Mette, S. A., Elder, D. E., Stewart, R., Damjanovich, L., Herlyn, M., et al. (1990). Integrin distribution in malignant melanoma: Association of the beta 3 subunit with tumor progression. Cancer Res. 50, 6757–6764.
Algazi, A. P., Esteve-Puig, R., Nosrati, A., Hinds, B., Hobbs-Muthukumar, A., Nandoskar, P., et al. (2018). Dual MEK/AKT inhibition with trametinib and GSK2141795 does not yield clinical benefit in metastatic NRAS-mutant and wild-type melanoma. Pigment. Cell Melanoma Res. 31, 110–114. doi:10.1111/pcmr.12644
Allman, R., Cowburn, P., and Mason, M. (2000). In vitro and in vivo effects of a cyclic peptide with affinity for the alpha(nu)beta3 integrin in human melanoma cells. Eur. J. Cancer 36, 410–422. doi:10.1016/s0959-8049(99)00279-8
Alonso, S. R., Tracey, L., Ortiz, P., Pérez-Gómez, B., Palacios, J., Pollán, M., et al. (2007). A high-throughput study in melanoma identifies epithelial-mesenchymal transition as a major determinant of metastasis. Cancer Res. 67 (7), 3450–3460. doi:10.1158/0008-5472.CAN-06-3481
Anvekar, R., Asciolla, J., Missert, D., and Chipuk, J. (2011). Born to be alive: A role for the BCL-2 family in melanoma tumor cell survival, apoptosis, and treatment. Front. Oncol. 1, 34. doi:10.3389/fonc.2011.00034
Aplin, A. E., Howe, A., Alahari, S. K., and Juliano, R. L. (1998). Signal transduction and signal modulation by cell adhesion receptors: The role of integrins, cadherins, immunoglobulin-cell adhesion molecules, and selectins. Pharmacol. Rev. 50, 197–263.
Ascierto, P. A., Schadendorf, D., Berking, C., Agarwala, S. S., Van Herpen, C. M., Queirolo, P., et al. (2013). MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 14, 249–256. doi:10.1016/S1470-2045(13)70024-X
Ashrafizadeh, M., Mohammadinejad, R., Tavakol, S., Ahmadi, Z., Roomiani, S., and Katebi, M. (2019). Autophagy, anoikis, ferroptosis, necroptosis, and endoplasmic reticulum stress: Potential applications in melanoma therapy. J. Cell Physiol. 234, 19471–19479. doi:10.1002/jcp.28740
Atefi, M., Von Euw, E., Attar, N., Ng, C., Chu, C., Guo, D., et al. (2011). Reversing melanoma cross-resistance to BRAF and MEK inhibitors by co-targeting the AKT/mTOR pathway. PLoS One 6, e28973. doi:10.1371/journal.pone.0028973
Au, S. H., Storey, B. D., Moore, J. C., Tang, Q., Chen, Y.-L., Javaid, S., et al. (2016). Clusters of circulating tumor cells traverse capillary-sized vessels. Proc. Natl. Acad. Sci. 113, 4947–4952. doi:10.1073/pnas.1524448113
Augustine, C. K., Yoshimoto, Y., Gupta, M., Zipfel, P. A., Selim, M. A., Febbo, P., et al. (2008). Targeting N-cadherin enhances antitumor activity of cytotoxic therapies in melanoma treatment. Cancer Res. 68, 3777–3784. doi:10.1158/0008-5472.CAN-07-5949
Barczyk, M., Carracedo, S., and Gullberg, D. (2010). Integrins. Cell Tissue Res. 339, 269–280. doi:10.1007/s00441-009-0834-6
Beasley, G. M., Mcmahon, N., Sanders, G., Augustine, C. K., Selim, M. A., Peterson, B., et al. (2009). A phase 1 study of systemic ADH-1 in combination with melphalan via isolated limb infusion in patients with locally advanced in-transit malignant melanoma. Cancer 115, 4766–4774. doi:10.1002/cncr.24509
Beasley, G. M., Riboh, J. C., Augustine, C. K., Zager, J. S., Hochwald, S. N., Grobmyer, S. R., et al. (2011). Prospective multicenter phase II trial of systemic ADH-1 in combination with melphalan via isolated limb infusion in patients with advanced extremity melanoma. J. Clin. Oncol. 29, 1210–1215. doi:10.1200/JCO.2010.32.1224
Beatty, G. L., Shahda, S., Beck, T., Uppal, N., Cohen, S. J., Donehower, R., et al. (2019). A phase ib/II study of the JAK1 inhibitor, itacitinib, plus nab-paclitaxel and gemcitabine in advanced solid tumors. Oncologist 24, 14–e10. doi:10.1634/theoncologist.2017-0665
Bhaskar, V., Zhang, D., Fox, M., Seto, P., Wong, M. H., Wales, P. E., et al. (2007). A function blocking anti-mouse integrin alpha5beta1 antibody inhibits angiogenesis and impedes tumor growth in vivo. J. Transl. Med. 5, 61. doi:10.1186/1479-5876-5-61
Bhoumik, A., Jones, N., and Ronai, Z. (2004). Transcriptional switch by activating transcription factor 2-derived peptide sensitizes melanoma cells to apoptosis and inhibits their tumorigenicity. Proc. Natl. Acad. Sci. U. S. A. 101, 4222–4227. doi:10.1073/pnas.0400195101
Bitsch, R., Kurzay, A., Özbay Kurt, F., De La Torre, C., Lasser, S., Lepper, A., et al. (2022). STAT3 inhibitor Napabucasin abrogates MDSC immunosuppressive capacity and prolongs survival of melanoma-bearing mice. J. Immunother. Cancer 10, e004384. doi:10.1136/jitc-2021-004384
Bladt, F., Riethmacher, D., Isenmann, S., Aguzzi, A., and Birchmeier, C. (1995). Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 376, 768–771. doi:10.1038/376768a0
Boisvert-Adamo, K., and Aplin, A. E. (2006). B-RAF and PI-3 kinase signaling protect melanoma cells from anoikis. Oncogene 25, 4848–4856. doi:10.1038/sj.onc.1209493
Boisvert-Adamo, K., and Aplin, A. E. (2008). Mutant B-RAF mediates resistance to anoikis via Bad and Bim. Oncogene 27, 3301–3312. doi:10.1038/sj.onc.1211003
Boisvert-Adamo, K., Longmate, W., Abel, E. V., and Aplin, A. E. (2009). Mcl-1 is required for melanoma cell resistance to anoikis. Mol. Cancer Res. 7, 549–556. doi:10.1158/1541-7786.MCR-08-0358
Bonvin, E., Falletta, P., Shaw, H., Delmas, V., and Goding, C. R. (2012). A phosphatidylinositol 3-kinase-Pax3 axis regulates Brn-2 expression in melanoma. Mol. Cell Biol. 32, 4674–4683. doi:10.1128/MCB.01067-12
Boyle, G. M., Woods, S. L., Bonazzi, V. F., Stark, M. S., Hacker, E., Aoude, L. G., et al. (2011). Melanoma cell invasiveness is regulated by miR-211 suppression of the BRN2 transcription factor. Pigment Cell and Melanoma Res. 24, 525–537. doi:10.1111/j.1755-148X.2011.00849.x
Bracher, A., Cardona, A. S., Tauber, S., Fink, A. M., Steiner, A., Pehamberger, H., et al. (2013). Epidermal growth factor facilitates melanoma lymph node metastasis by influencing tumor lymphangiogenesis. J. Invest Dermatol 133, 230–238. doi:10.1038/jid.2012.272
Brooks, P. C., Clark, R. A., and Cheresh, D. A. (1994). Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science 264, 569–571. doi:10.1126/science.7512751
Brooks, P. C., Silletti, S., Von Schalscha, T. L., Friedlander, M., and Cheresh, D. A. (1998). Disruption of angiogenesis by PEX, a noncatalytic metalloproteinase fragment with integrin binding activity. Cell 92, 391–400. doi:10.1016/s0092-8674(00)80931-9
Brooks, P. C., Strömblad, S., Sanders, L. C., Von Schalscha, T. L., Aimes, R. T., Stetler-Stevenson, W. G., et al. (1996). Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 85, 683–693. doi:10.1016/s0092-8674(00)81235-0
Brunet, A., Bonni, A., Zigmond, M. J., Lin, M. Z., Juo, P., Hu, L. S., et al. (1999). Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96, 857–868. doi:10.1016/s0092-8674(00)80595-4
Buchbinder, E. I., Cohen, J. V., Haq, R., Hodi, F. S., Lawrence, D. P., Giobbie-Hurder, A., et al. (2020). A phase II study of ERK inhibition by ulixertinib (BVD-523) in metastatic uveal melanoma. J. Clin. Oncol. 38, 10036. doi:10.1200/jco.2020.38.15_suppl.10036
Caramel, J., Papadogeorgakis, E., Hill, L., Browne, G. J., Richard, G., Wierinckx, A., et al. (2013). A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 24, 466–480. doi:10.1016/j.ccr.2013.08.018
Carbone, M. L., Lacal, P. M., Messinese, S., De Giglio, L., Pozzilli, C., Persechino, S., et al. (2020). Multiple sclerosis treatment and melanoma development. Int. J. Mol. Sci. 21, 2950. doi:10.3390/ijms21082950
Carragher, N. O., Walker, S. M., Scott Carragher, L. A., Harris, F., Sawyer, T. K., Brunton, V. G., et al. (2006). Calpain 2 and Src dependence distinguishes mesenchymal and amoeboid modes of tumour cell invasion: A link to integrin function. Oncogene 25, 5726–5740. doi:10.1038/sj.onc.1209582
Casalino, L., Talotta, F., Cimmino, A., and Verde, P. (2022). The fra-1/AP-1 oncoprotein: From the “undruggable” transcription factor to therapeutic targeting. Cancers 14, 1480. doi:10.3390/cancers14061480
Castel, S., Pagan, R., García, R., Casaroli-Marano, R. P., Reina, M., Mitjans, F., et al. (2000). Alpha v integrin antagonists induce the disassembly of focal contacts in melanoma cells. Eur. J. Cell Biol. 79, 502–512. doi:10.1078/0171-9335-00067
Chakraborty, B., Byemerwa, J., Shepherd, J., Haines, C. N., Baldi, R., Gong, W., et al. (2021). Inhibition of estrogen signaling in myeloid cells increases tumor immunity in melanoma. J. Clin. Invest 131, e151347. doi:10.1172/JCI151347
Chamcheu, J. C., Roy, T., Uddin, M. B., Banang-Mbeumi, S., Chamcheu, R. N., Walker, A. L., et al. (2019). Role and therapeutic targeting of the PI3K/Akt/mTOR signaling pathway in skin cancer: A review of current status and future trends on natural and synthetic agents therapy. Cells 8, 803. doi:10.3390/cells8080803
Chao, Y. L., Shepard, C. R., and Wells, A. (2010). Breast carcinoma cells re-express E-cadherin during mesenchymal to epithelial reverting transition. Mol. Cancer 9, 179. doi:10.1186/1476-4598-9-179
Chen, Y., Xie, X., Li, X., Wang, P., Jing, Q., Yue, J., et al. (2016). FGFR antagonist induces protective autophagy in FGFR1-amplified breast cancer cell. Biochem. Biophys. Res. Commun. 474, 1–7. doi:10.1016/j.bbrc.2016.03.017
Cheresh, D. A. (1987). Human endothelial cells synthesize and express an Arg-Gly-Asp-directed adhesion receptor involved in attachment to fibrinogen and von Willebrand factor. Proc. Natl. Acad. Sci. U. S. A. 84, 6471–6475. doi:10.1073/pnas.84.18.6471
Chinenov, Y., and Kerppola, T. K. (2001). Close encounters of many kinds: Fos-Jun interactions that mediate transcription regulatory specificity. Oncogene 20, 2438–2452. doi:10.1038/sj.onc.1204385
Chunhacha, P., Sriuranpong, V., and Chanvorachote, P. (2013). Epithelial-mesenchymal transition mediates anoikis resistance and enhances invasion in pleural effusion-derived human lung cancer cells. Oncol. Lett. 5, 1043–1047. doi:10.3892/ol.2013.1108
Ciołczyk-Wierzbicka, D., Gil, D., Zarzycka, M., and Laidler, P. (2020). mTOR inhibitor everolimus reduces invasiveness of melanoma cells. Hum. Cell 33, 88–97. doi:10.1007/s13577-019-00270-4
Cirkel, G. A., Kerklaan, B. M., Vanhoutte, F., Van Der Aa, A., Lorenzon, G., Namour, F., et al. (2016). A dose escalating phase I study of GLPG0187, a broad spectrum integrin receptor antagonist, in adult patients with progressive high-grade glioma and other advanced solid malignancies. Invest New Drugs 34, 184–192. doi:10.1007/s10637-015-0320-9
Comandante-Lou, N., Baumann, D. G., and Fallahi-Sichani, M. (2022). AP-1 transcription factor network explains diverse patterns of cellular plasticity in melanoma cells. Cell Rep. 40, 111147. doi:10.1016/j.celrep.2022.111147
Cook, A. L., Donatien, P. D., Smith, A. G., Murphy, M., Jones, M. K., Herlyn, M., et al. (2003). Human melanoblasts in culture: Expression of BRN2 and synergistic regulation by fibroblast growth factor-2, stem cell factor, and endothelin-3. J. Invest Dermatol 121, 1150–1159. doi:10.1046/j.1523-1747.2003.12562.x
Cook, A. L., and Sturm, R. A. (2008). POU domain transcription factors: BRN2 as a regulator of melanocytic growth and tumourigenesis. Pigment Cell and Melanoma Res. 21, 611–626. doi:10.1111/j.1755-148X.2008.00510.x
D'arcy, C., and Kiel, C. (2021). Cell adhesion molecules in normal skin and melanoma. Biomolecules 11, 1213. doi:10.3390/biom11081213
Da Costa, P. E., Batista, W. L., Moraes, M. S., Stern, A., and Monteiro, H. P. (2018). Src kinase activation by nitric oxide promotes resistance to anoikis in tumour cell lines. Free Radic. Res. 52, 592–604. doi:10.1080/10715762.2018.1455095
Da Cunha, F. F. M., Mugnol, K. C. U., De Melo, F. M., Nascimento, M., De Azevedo, R. A., Santos, R. T. S., et al. (2019). Peptide R18H from BRN2 transcription factor POU domain displays antitumor activity in vitro and in vivo and induces apoptosis in B16F10-nex2 cells. Anticancer Agents Med. Chem. 19, 389–401. doi:10.2174/1871520618666181109164246
Dai, Y., and Siemann, D. W. (2012). Constitutively active c-Met kinase in PC-3 cells is autocrine-independent and can be blocked by the Met kinase inhibitor BMS-777607. BMC Cancer 12, 198. doi:10.1186/1471-2407-12-198
Danen, E. H., Jansen, K. F., Klein, C. E., Smit, N. P., Ruiter, D. J., and Van Muijen, G. N. (1996). Loss of adhesion to basement membrane components but not to keratinocytes in proliferating melanocytes. Eur. J. Cell Biol. 70, 69–75.
Danen, E. H., Ten Berge, P. J., Van Muijen, G. N., Van 'T Hof-Grootenboer, A. E., Bröcker, E. B., and Ruiter, D. J. (1994). Emergence of alpha 5 beta 1 fibronectin- and alpha v beta 3 vitronectin-receptor expression in melanocytic tumour progression. Histopathology 24, 249–256. doi:10.1111/j.1365-2559.1994.tb00517.x
Das, I., Chen, H., Maddalo, G., Tuominen, R., Rebecca, V. W., Herlyn, M., et al. (2020). Inhibiting insulin and mTOR signaling by afatinib and crizotinib combination fosters broad cytotoxic effects in cutaneous malignant melanoma. Cell Death Dis. 11, 882. doi:10.1038/s41419-020-03097-2
Das, I., Wilhelm, M., Höiom, V., Franco Marquez, R., Costa Svedman, F., Hansson, J., et al. (2019). Combining ERBB family and MET inhibitors is an effective therapeutic strategy in cutaneous malignant melanoma independent of BRAF/NRAS mutation status. Cell Death Dis. 10, 663. doi:10.1038/s41419-019-1875-8
Daud, A., Kluger, H. M., Kurzrock, R., Schimmoller, F., Weitzman, A. L., Samuel, T. A., et al. (2017). Phase II randomised discontinuation trial of the MET/VEGF receptor inhibitor cabozantinib in metastatic melanoma. Br. J. Cancer 116, 432–440. doi:10.1038/bjc.2016.419
Dean, A., Gill, S., Mcgregor, M., Broadbridge, V., Jarvelainen, H. A., and Price, T. J. (2020). 1528P Phase I trial of the first-in-class agent CEND-1 in combination with gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer. Ann. Oncol. 31, S941. doi:10.1016/j.annonc.2020.08.2011
Defilippi, P., Truffa, G., Stefanuto, G., Altruda, F., Silengo, L., and Tarone, G. (1991). Tumor necrosis factor alpha and interferon gamma modulate the expression of the vitronectin receptor (integrin beta 3) in human endothelial cells. J. Biol. Chem. 266, 7638–7645. doi:10.1016/s0021-9258(20)89495-2
Del Mistro, G., Riemann, S., Schindler, S., Beissert, S., Kontermann, R. E., Ginolhac, A., et al. (2022). Focal adhesion kinase plays a dual role in TRAIL resistance and metastatic outgrowth of malignant melanoma. Cell Death Dis. 13, 54. doi:10.1038/s41419-022-04502-8
Del Peso, L., González-García, M., Page, C., Herrera, R., and Nuñez, G. (1997). Interleukin-3-induced phosphorylation of BAD through the protein kinase Akt. Science 278, 687–689. doi:10.1126/science.278.5338.687
Dhillon, A. S., and Tulchinsky, E. (2015). FRA-1 as a driver of tumour heterogeneity: A nexus between oncogenes and embryonic signalling pathways in cancer. Oncogene 34, 4421–4428. doi:10.1038/onc.2014.374
Dilshat, R., Fock, V., Kenny, C., Gerritsen, I., Lasseur, R. M. J., Travnickova, J., et al. (2021). MITF reprograms the extracellular matrix and focal adhesion in melanoma. Elife 10, e63093. doi:10.7554/eLife.63093
Dinavahi, S. S., Noory, M. A., Gowda, R., Drabick, J. J., Berg, A., Neves, R. I., et al. (2018). Moving synergistically acting drug combinations to the clinic by comparing sequential versus simultaneous drug administrations. Mol. Pharmacol. 93, 190–196. doi:10.1124/mol.117.110759
Dratkiewicz, E., Pietraszek-Gremplewicz, K., Simiczyjew, A., Mazur, A. J., and Nowak, D. (2018). Gefitinib or lapatinib with foretinib synergistically induce a cytotoxic effect in melanoma cell lines. Oncotarget 9, 18254–18268. doi:10.18632/oncotarget.24810
Dratkiewicz, E., Simiczyjew, A., Pietraszek-Gremplewicz, K., Mazurkiewicz, J., and Nowak, D. (2019). Characterization of melanoma cell lines resistant to vemurafenib and evaluation of their responsiveness to EGFR- and MET-inhibitor treatment. Int. J. Mol. Sci. 21, 113. doi:10.3390/ijms21010113
Du, R., Zhong, T., Zhang, W. Q., Song, P., Song, W. D., Zhao, Y., et al. (2014). Antitumor effect of iRGD-modified liposomes containing conjugated linoleic acid-paclitaxel (CLA-PTX) on B16-F10 melanoma. Int. J. Nanomedicine 9, 3091–3105. doi:10.2147/IJN.S65664
Dugo, M., Nicolini, G., Tragni, G., Bersani, I., Tomassetti, A., Colonna, V., et al. (2015). A melanoma subtype with intrinsic resistance to BRAF inhibition identified by receptor tyrosine kinases gene-driven classification. Oncotarget 6, 5118–5133. doi:10.18632/oncotarget.3007
Dumble, M., Crouthamel, M. C., Zhang, S. Y., Schaber, M., Levy, D., Robell, K., et al. (2014). Discovery of novel AKT inhibitors with enhanced anti-tumor effects in combination with the MEK inhibitor. PLoS One 9, e100880. doi:10.1371/journal.pone.0100880
Eferl, R., and Wagner, E. F. (2003). AP-1: A double-edged sword in tumorigenesis. Nat. Rev. Cancer 3, 859–868. doi:10.1038/nrc1209
Eiring, A. M., Khorashad, J. S., Anderson, D. J., Yu, F., Redwine, H. M., Mason, C. C., et al. (2015). β-Catenin is required for intrinsic but not extrinsic BCR-ABL1 kinase-independent resistance to tyrosine kinase inhibitors in chronic myeloid leukemia. Leukemia 29, 2328–2337. doi:10.1038/leu.2015.196
Eisen, T., Easty, D. J., Bennett, D. C., and Goding, C. R. (1995). The POU domain transcription factor brn-2: Elevated expression in malignant melanoma and regulation of melanocyte-specific gene expression. Oncogene 11, 2157–2164.
Ernst, D. S., Eisenhauer, E., Wainman, N., Davis, M., Lohmann, R., Baetz, T., et al. (2005). Phase II study of perifosine in previously untreated patients with metastatic melanoma. Invest New Drugs 23, 569–575. doi:10.1007/s10637-005-1157-4
Eslami, M., Nezafat, N., Khajeh, S., Mostafavi-Pour, Z., Bagheri Novir, S., Negahdaripour, M., et al. (2019). Deep analysis of N-cadherin/ADH-1 interaction: A computational survey. J. Biomol. Struct. Dyn. 37, 210–228. doi:10.1080/07391102.2018.1424035
Etoh, T., Thomas, L., Pastel-Levy, C., Colvin, R. B., Mihm, M. C., and Byers, H. R. (1993). Role of integrin alpha 2 beta 1 (VLA-2) in the migration of human melanoma cells on laminin and type IV collagen. J. Invest Dermatol 100, 640–647. doi:10.1111/1523-1747.ep12472299
Fane, M. E., Chhabra, Y., Hollingsworth, D. E. J., Simmons, J. L., Spoerri, L., Oh, T. G., et al. (2017). NFIB mediates BRN2 driven melanoma cell migration and invasion through regulation of EZH2 and MITF. EBioMedicine 16, 63–75. doi:10.1016/j.ebiom.2017.01.013
Fane, M. E., Chhabra, Y., Smith, A. G., and Sturm, R. A. (2019). BRN2, a POUerful driver of melanoma phenotype switching and metastasis. Pigment Cell and Melanoma Res. 32, 9–24. doi:10.1111/pcmr.12710
Fanfone, D., Wu, Z., Mammi, J., Berthenet, K., Neves, D., Weber, K., et al. (2022). Confined migration promotes cancer metastasis through resistance to anoikis and increased invasiveness. Elife 11, e73150. doi:10.7554/eLife.73150
Fedorenko, I. V., Abel, E. V., Koomen, J. M., Fang, B., Wood, E. R., Chen, Y. A., et al. (2016). Fibronectin induction abrogates the BRAF inhibitor response of BRAF V600E/PTEN-null melanoma cells. Oncogene 35, 1225–1235. doi:10.1038/onc.2015.188
Fedorenko, I. V., Paraiso, K. H., and Smalley, K. S. (2011). Acquired and intrinsic BRAF inhibitor resistance in BRAF V600E mutant melanoma. Biochem. Pharmacol. 82, 201–209. doi:10.1016/j.bcp.2011.05.015
Fenouille, N., Tichet, M., Dufies, M., Pottier, A., Mogha, A., Soo, J. K., et al. (2012). The epithelial-mesenchymal transition (EMT) regulatory factor SLUG (SNAI2) is a downstream target of SPARC and AKT in promoting melanoma cell invasion. PLoS One 7, e40378. doi:10.1371/journal.pone.0040378
Figarola, J. L., Singhal, J., Singhal, S., Kusari, J., and Riggs, A. (2018). Bioenergetic modulation with the mitochondria uncouplers SR4 and niclosamide prevents proliferation and growth of treatment-naïve and vemurafenib-resistant melanomas. Oncotarget 9, 36945–36965. doi:10.18632/oncotarget.26421
Flaherty, K. T., Infante, J. R., Daud, A., Gonzalez, R., Kefford, R. F., Sosman, J., et al. (2012). Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 367, 1694–1703. doi:10.1056/NEJMoa1210093
Fofaria, N. M., and Srivastava, S. K. (2014). Critical role of STAT3 in melanoma metastasis through anoikis resistance. Oncotarget 5, 7051–7064. doi:10.18632/oncotarget.2251
Friedl, P., and Wolf, K. (2003). Tumour-cell invasion and migration: Diversity and escape mechanisms. Nat. Rev. Cancer 3, 362–374. doi:10.1038/nrc1075
Frisch, S., and Francis, H. (1994). Disruption of epithelial cell-matrix interactions induces apoptosis. J. Cell Biol. 124, 619–626. doi:10.1083/jcb.124.4.619
Frisch, S. M., Vuori, K., Ruoslahti, E., and Chan-Hui, P. Y. (1996). Control of adhesion-dependent cell survival by focal adhesion kinase. J. Cell Biol. 134, 793–799. doi:10.1083/jcb.134.3.793
Giannoni, E., Fiaschi, T., Ramponi, G., and Chiarugi, P. (2009). Redox regulation of anoikis resistance of metastatic prostate cancer cells: Key role for Src and EGFR-mediated pro-survival signals. Oncogene 28, 2074–2086. doi:10.1038/onc.2009.77
Giannoni, E., Buricchi, F., Grimaldi, G., Parri, M., Cialdai, F., Taddei, M. L., et al. (2008). Redox regulation of anoikis: Reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 15, 867–878. doi:10.1038/cdd.2008.3
Gil, D., Zarzycka, M., Ciołczyk-Wierzbicka, D., and Laidler, P. (2020). Integrin linked kinase regulates endosomal recycling of N-cadherin in melanoma cells. Cell Signal 72, 109642. doi:10.1016/j.cellsig.2020.109642
Girotti, M. R., Pedersen, M., Sanchez-Laorden, B., Viros, A., Turajlic, S., Niculescu-Duvaz, D., et al. (2013). Inhibiting EGF receptor or SRC family kinase signaling overcomes BRAF inhibitor resistance in melanoma. Cancer Discov. 3, 158–167. doi:10.1158/2159-8290.CD-12-0386
Goldstein, N. B., Johannes, W. U., Gadeliya, A. V., Green, M. R., Fujita, M., Norris, D. A., et al. (2009). Active N-Ras and B-Raf inhibit anoikis by downregulating Bim expression in melanocytic cells. J. Invest Dermatol 129, 432–437. doi:10.1038/jid.2008.227
Goodall, J., Carreira, S., Denat, L., Kobi, D., Davidson, I., Nuciforo, P., et al. (2008). Brn-2 represses microphthalmia-associated transcription factor expression and marks a distinct subpopulation of microphthalmia-associated transcription factor–negative melanoma cells. Cancer Res. 68, 7788–7794. doi:10.1158/0008-5472.CAN-08-1053
Goundiam, O., Nagel, M. D., and Vayssade, M. (2012). Akt and RhoA inhibition promotes anoikis of aggregated B16F10 melanoma cells. Cell Biol. Int. 36, 311–319. doi:10.1042/CBI20110069
Goundiam, O., Nagel, M. D., and Vayssade, M. (2010). Growth and survival signalling in B16F10 melanoma cells in 3D culture. Cell Biol. Int. 34, 385–391. doi:10.1042/CBI20090147
Graziani, V., Rodriguez-Hernandez, I., Maiques, O., and Sanz-Moreno, V. (2022). The amoeboid state as part of the epithelial-to-mesenchymal transition programme. Trends Cell Biol. 32, 228–242. doi:10.1016/j.tcb.2021.10.004
Greger, J. G., Eastman, S. D., Zhang, V., Bleam, M. R., Hughes, A. M., Smitheman, K. N., et al. (2012). Combinations of BRAF, MEK, and PI3K/mTOR inhibitors overcome acquired resistance to the BRAF inhibitor GSK2118436 dabrafenib, mediated by NRAS or MEK mutations. Mol. Cancer Ther. 11, 909–920. doi:10.1158/1535-7163.MCT-11-0989
Groen, R. W., De Rooij, M. F., Kocemba, K. A., Reijmers, R. M., De Haan-Kramer, A., Overdijk, M. B., et al. (2011). N-cadherin-mediated interaction with multiple myeloma cells inhibits osteoblast differentiation. Haematologica 96, 1653–1661. doi:10.3324/haematol.2010.038133
Guadamillas, M. C., Cerezo, A., and Del Pozo, M. A. (2011). Overcoming anoikis-pathways to anchorage-independent growth in cancer. J. Cell Sci. 124, 3189–3197. doi:10.1242/jcs.072165
Guo, Y. J., Pan, W. W., Liu, S. B., Shen, Z. F., Xu, Y., and Hu, L. L. (2020). ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 19, 1997–2007. doi:10.3892/etm.2020.8454
Gurzov, E. N., Bakiri, L., Alfaro, J. M., Wagner, E. F., and Izquierdo, M. (2008). Targeting c-Jun and JunB proteins as potential anticancer cell therapy. Oncogene 27, 641–652. doi:10.1038/sj.onc.1210690
Guy, G. P., Thomas, C. C., Thompson, T., Watson, M., Massetti, G. M., Richardson, L. C., et al. (2015). Vital signs: Melanoma incidence and mortality trends and projections - United States, 1982-2030. MMWR Morb. Mortal. Wkly. Rep. 64, 591–596.
Hanahan, D. (2022). Hallmarks of cancer: New dimensions. Cancer Discov. 12, 31–46. doi:10.1158/2159-8290.CD-21-1059
Hao, H., Dong, Y., Bowling, M. T., Gomez-Gutierrez, J. G., Zhou, H. S., and Mcmasters, K. M. (2007). E2F-1 induces melanoma cell apoptosis via PUMA up-regulation and Bax translocation. BMC Cancer 7, 24. doi:10.1186/1471-2407-7-24
Hardy, K. M., Booth, B. W., Hendrix, M. J., Salomon, D. S., and Strizzi, L. (2010). ErbB/EGF signaling and EMT in mammary development and breast cancer. J. Mammary Gland. Biol. Neoplasia 15, 191–199. doi:10.1007/s10911-010-9172-2
Hasnat, M. A., Pervin, M., Lim, J. H., and Lim, B. O. (2015). Apigenin attenuates melanoma cell migration by inducing anoikis through integrin and focal adhesion kinase inhibition. Molecules 20, 21157–21166. doi:10.3390/molecules201219752
Hatamipour, M., Jaafari, M. R., Momtazi-Borojeni, A. A., Ramezani, M., and Sahebkar, A. (2019). Nanoliposomal encapsulation enhances in vivo anti-tumor activity of niclosamide against melanoma. Anticancer Agents Med. Chem. 19, 1618–1626. doi:10.2174/1871520619666190705120011
Hatiboglu, M. A., Kong, L. Y., Wei, J., Wang, Y., Mcenery, K. A., Fuller, G. N., et al. (2012). The tumor microenvironment expression of p-STAT3 influences the efficacy of cyclophosphamide with WP1066 in murine melanoma models. Int. J. Cancer 131, 8–17. doi:10.1002/ijc.26307
Hazan, R. B., Phillips, G. R., Qiao, R. F., Norton, L., and Aaronson, S. A. (2000). Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J. Cell Biol. 148, 779–790. doi:10.1083/jcb.148.4.779
Hegerfeldt, Y., Tusch, M., Bröcker, E. B., and Friedl, P. (2002). Collective cell movement in primary melanoma explants: Plasticity of cell-cell interaction, beta1-integrin function, and migration strategies. Cancer Res. 62, 2125–2130.
Herbert, K., Binet, R., Lambert, J. P., Louphrasitthiphol, P., Kalkavan, H., Sesma-Sanz, L., et al. (2019). BRN2 suppresses apoptosis, reprograms DNA damage repair, and is associated with a high somatic mutation burden in melanoma. Genes Dev. 33, 310–332. doi:10.1101/gad.314633.118
Hersey, P., Sosman, J., O'day, S., Richards, J., Bedikian, A., Gonzalez, R., et al. (2010). A randomized phase 2 study of etaracizumab, a monoclonal antibody against integrin alpha(v)beta(3), + or - dacarbazine in patients with stage IV metastatic melanoma. Cancer 116, 1526–1534. doi:10.1002/cncr.24821
Hess, A. R., Postovit, L.-M., Margaryan, N. V., Seftor, E. A., Schneider, G. B., Seftor, R. E. B., et al. (2005). Focal adhesion kinase promotes the aggressive melanoma phenotype. Cancer Res. 65, 9851–9860. doi:10.1158/0008-5472.CAN-05-2172
Hilmi, C., Larribere, L., Giuliano, S., Bille, K., Ortonne, J. P., Ballotti, R., et al. (2008). IGF1 promotes resistance to apoptosis in melanoma cells through an increased expression of BCL2, BCL-X(L), and survivin. J. Invest Dermatol 128, 1499–1505. doi:10.1038/sj.jid.5701185
Hoek, K., Rimm, D. L., Williams, K. R., Zhao, H., Ariyan, S., Lin, A., et al. (2004). Expression profiling reveals novel pathways in the transformation of melanocytes to melanomas. Cancer Res. 64, 5270–5282. doi:10.1158/0008-5472.CAN-04-0731
Hoek, K. S., Eichhoff, O. M., Schlegel, N. C., Döbbeling, U., Kobert, N., Schaerer, L., et al. (2008). In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. 68, 650–656. doi:10.1158/0008-5472.CAN-07-2491
Hoek, K. S., and Goding, C. R. (2010). Cancer stem cells versus phenotype-switching in melanoma. Pigment. Cell Melanoma Res. 23, 746–759. doi:10.1111/j.1755-148X.2010.00757.x
Hoek, K. S., Schlegel, N. C., Brafford, P., Sucker, A., Ugurel, S., Kumar, R., et al. (2006). Metastatic potential of melanomas defined by specific gene expression profiles with no BRAF signature. Pigment. Cell Res. 19, 290–302. doi:10.1111/j.1600-0749.2006.00322.x
Hofmann, U. B., Westphal, J. R., Van Kraats, A. A., Ruiter, D. J., and Van Muijen, G. N. (2000). Expression of integrin alpha(v)beta(3) correlates with activation of membrane-type matrix metalloproteinase-1 (MT1-MMP) and matrix metalloproteinase-2 (MMP-2) in human melanoma cells in vitro and in vivo. Int. J. Cancer 87, 12–19. doi:10.1002/1097-0215(20000701)87:1<12::aid-ijc3>3.0.co;2-a
Hou, J. M., Krebs, M. G., Lancashire, L., Sloane, R., Backen, A., Swain, R. K., et al. (2012). Clinical significance and molecular characteristics of circulating tumor cells and circulating tumor microemboli in patients with small-cell lung cancer. J. Clin. Oncol. 30, 525–532. doi:10.1200/JCO.2010.33.3716
Hou, J. M., Krebs, M., Ward, T., Sloane, R., Priest, L., Hughes, A., et al. (2011). Circulating tumor cells as a window on metastasis biology in lung cancer. Am. J. Pathol. 178, 989–996. doi:10.1016/j.ajpath.2010.12.003
Hsu, M., Andl, T., Li, G., Meinkoth, J. L., and Herlyn, M. (2000a). Cadherin repertoire determines partner-specific gap junctional communication during melanoma progression. J. Cell Sci. 113 (9), 1535–1542. doi:10.1242/jcs.113.9.1535
Hsu, M. Y., Meier, F. E., Nesbit, M., Hsu, J. Y., Van Belle, P., Elder, D. E., et al. (2000b). E-cadherin expression in melanoma cells restores keratinocyte-mediated growth control and down-regulates expression of invasion-related adhesion receptors. Am. J. Pathol. 156, 1515–1525. doi:10.1016/S0002-9440(10)65023-7
Hsu, M. Y., Shih, D. T., Meier, F. E., Van Belle, P., Hsu, J. Y., Elder, D. E., et al. (1998). Adenoviral gene transfer of beta3 integrin subunit induces conversion from radial to vertical growth phase in primary human melanoma. Am. J. Pathol. 153, 1435–1442. doi:10.1016/s0002-9440(10)65730-6
Hutchinson, J. H., Halczenko, W., Brashear, K. M., Breslin, M. J., Coleman, P. J., Duong, L. T., et al. (2003). Nonpeptide alphavbeta3 antagonists. 8. in vitro and in vivo evaluation of a potent alphavbeta3 antagonist for the prevention and treatment of osteoporosis. J. Med. Chem. 46, 4790–4798. doi:10.1021/jm030306r
Hynes, R. O. (2002). Integrins: Bidirectional, allosteric signaling machines. Cell 110, 673–687. doi:10.1016/s0092-8674(02)00971-6
Inman, J. L., Kute, T., White, W., Pettenati, M., and Levine, E. A. (2003). Absence of HER2 overexpression in metastatic malignant melanoma. J. Surg. Oncol., 84, 82–8. doi:10.1002/cncr.33242
Janku, F., Sakamuri, D., Kato, S., Huang, H. J., Call, S. G., Naing, A., et al. (2021). Dose-escalation study of vemurafenib with sorafenib or crizotinib in patients with BRAF-mutated advanced cancers. Cancer 127, 391–402. doi:10.1002/cncr.33242
Jenkins, R. W., and Fisher, D. E. (2021). Treatment of advanced melanoma in 2020 and beyond. J. Invest Dermatol 141, 23–31. doi:10.1016/j.jid.2020.03.943
Jenkins, S. V., Alimohammadi, M., Terry, A. S., Griffin, R. J., Tackett, A. J., Leung, J. W., et al. (2021). Dysbiotic stress increases the sensitivity of the tumor vasculature to radiotherapy and c-Met inhibitors. Angiogenesis 24, 597–611. doi:10.1007/s10456-021-09771-z
Jeon, H.-M., and Lee, J. (2017). MET: Roles in epithelial-mesenchymal transition and cancer stemness. Ann. Transl. Med. 5, 5. doi:10.21037/atm.2016.12.67
Jin, J. Y., Ke, H., Hall, R. P., and Zhang, J. Y. (2011). c-Jun promotes whereas JunB inhibits epidermal neoplasia. J. Invest Dermatol 131, 1149–1158. doi:10.1038/jid.2011.1
Jing, G., Yu, F., and Xue, H. (2022). Tepotinib suppresses proliferation, invasion, migration, and promotes apoptosis of melanoma cells via inhibiting MET and PI3K/AKT signaling pathways. Oncol. Lett. 23, 170. doi:10.3892/ol.2022.13290
Johnson, D. B., and Puzanov, I. (2015). Treatment of NRAS-mutant melanoma. Curr. Treat. Options Oncol. 16, 15. doi:10.1007/s11864-015-0330-z
Johnson, J. P. (1999). Cell adhesion molecules in the development and progression of malignant melanoma. Cancer Metastasis Rev. 18, 345–357. doi:10.1023/a:1006304806799
Kageshita, T., Hamby, C. V., Hirai, S., Kimura, T., Ono, T., and Ferrone, S. (2000). Alpha(v)beta3 expression on blood vessels and melanoma cells in primary lesions: Differential association with tumor progression and clinical prognosis. Cancer Immunol. Immunother. 49, 314–318. doi:10.1007/s002620000124
Kajanne, R., Miettinen, P., Mehlem, A., Leivonen, S. K., Birrer, M., Foschi, M., et al. (2007). EGF-R regulates MMP function in fibroblasts through MAPK and AP-1 pathways. J. Cell Physiol. 212, 489–497. doi:10.1002/jcp.21041
Kalluri, R., and Weinberg, R. A. (2009). The basics of epithelial-mesenchymal transition. J. Clin. Invest 119, 1420–1428. doi:10.1172/JCI39104
Kamide, D., Yamashita, T., Araki, K., Tomifuji, M., Tanaka, Y., Tanaka, S., et al. (2016). Selective activator protein-1 inhibitor T-5224 prevents lymph node metastasis in an oral cancer model. Cancer Sci. 107, 666–673. doi:10.1111/cas.12914
Kariya, Y., Oyama, M., Suzuki, T., and Kariya, Y. (2021). αvβ3 Integrin induces partial EMT independent of TGF-β signaling. Commun. Biol. 4, 490. doi:10.1038/s42003-021-02003-6
Kelm, R. C., Hagstrom, E. L., Mathieu, R. J., Orrell, K. A., Serrano, L., Mueller, K. A., et al. (2019). Melanoma subsequent to natalizumab exposure: A report from the radar (research on adverse drug events and reports) program. J. Am. Acad. Dermatol 80, 820–821. doi:10.1016/j.jaad.2018.10.052
Kenessey, I., Keszthelyi, M., Krámer, Z., Berta, J., Adám, A., Dobos, J., et al. (2010). Inhibition of c-Met with the specific small molecule tyrosine kinase inhibitor SU11274 decreases growth and metastasis formation of experimental human melanoma. Curr. Cancer Drug Targets 10, 332–342. doi:10.2174/156800910791190184
Kennedy, S. G., Kandel, E. S., Cross, T. K., and Hay, N. (1999). Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell Biol. 19, 5800–5810. doi:10.1128/mcb.19.8.5800
Kermorgant, S., Zicha, D., and Parker, P. J. (2004). PKC controls HGF-dependent c-Met traffic, signalling and cell migration. Embo J. 23, 3721–3734. doi:10.1038/sj.emboj.7600396
Khwaja, A., Rodriguez-Viciana, P., Wennström, S., Warne, P. H., and Downward, J. (1997). Matrix adhesion and Ras transformation both activate a phosphoinositide 3-OH kinase and protein kinase B/Akt cellular survival pathway. Embo J. 16, 2783–2793. doi:10.1093/emboj/16.10.2783
Kim, H.-D., Meyer, A. S., Wagner, J. P., Alford, S. K., Wells, A., Gertler, F. B., et al. (2011). Signaling network state predicts twist-mediated effects on breast cell migration across diverse growth factor contexts. Mol. Cell. Proteomics 10, M111.008433. doi:10.1074/mcp.M111.008433
Kim, K. B., Prieto, V., Joseph, R. W., Diwan, A. H., Gallick, G. E., Papadopoulos, N. E., et al. (2012). A randomized phase II study of cilengitide (EMD 121974) in patients with metastatic melanoma. Melanoma Res. 22, 294–301. doi:10.1097/CMR.0b013e32835312e4
Kim, K., Heo, Y. K., Chun, S., Kim, C. H., Bian, Y., Bae, O. N., et al. (2019). Arsenic may act as a pro-metastatic carcinogen through promoting tumor cell-induced platelet aggregation. Toxicol. Sci. 168, 18–27. doi:10.1093/toxsci/kfy247
Kim, T. W., Kim, Y., Keum, H., Jung, W., Kang, M., and Jon, S. (2022). Combination of a STAT3 inhibitor with anti-PD-1 immunotherapy is an effective treatment regimen for a vemurafenib-resistant melanoma. Mol. Ther. Oncolytics 26, 1–14. doi:10.1016/j.omto.2022.06.001
Kirkwood, J. M., Iannotti, N., Cho, D., O'day, S., Gibney, G., Hodi, F. S., et al. (2018). Abstract CT176: Effect of JAK/STAT or PI3Kδ plus PD-1 inhibition on the tumor microenvironment: Biomarker results from a phase Ib study in patients with advanced solid tumors. Cancer Res. 78, CT176. doi:10.1158/1538-7445.AM2018-CT176
Klymenko, Y., Kim, O., Loughran, E., Yang, J., Lombard, R., Alber, M., et al. (2017). Cadherin composition and multicellular aggregate invasion in organotypic models of epithelial ovarian cancer intraperitoneal metastasis. Oncogene 36, 5840–5851. doi:10.1038/onc.2017.171
Koefinger, P., Wels, C., Joshi, S., Damm, S., Steinbauer, E., Beham-Schmid, C., et al. (2011). The cadherin switch in melanoma instigated by HGF is mediated through epithelial–mesenchymal transition regulators. Pigment Cell and Melanoma Res. 24, 382–385. doi:10.1111/j.1755-148X.2010.00807.x
Kolli-Bouhafs, K., Sick, E., Noulet, F., Gies, J. P., De Mey, J., and Rondé, P. (2014). FAK competes for Src to promote migration against invasion in melanoma cells. Cell Death Dis. 5, e1379–e. doi:10.1038/cddis.2014.329
Kong, L. Y., Abou-Ghazal, M. K., Wei, J., Chakraborty, A., Sun, W., Qiao, W., et al. (2008). A novel inhibitor of signal transducers and activators of transcription 3 activation is efficacious against established central nervous system melanoma and inhibits regulatory T cells. Clin. Cancer Res. 14, 5759–5768. doi:10.1158/1078-0432.CCR-08-0377
Kortylewski, M., Jove, R., and Yu, H. (2005). Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev. 24, 315–327. doi:10.1007/s10555-005-1580-1
Kovacs, D., Migliano, E., Muscardin, L., Silipo, V., Catricalà, C., Picardo, M., et al. (2016). The role of wnt/β-catenin signaling pathway in melanoma epithelial-to-mesenchymal-like switching: Evidences from patients-derived cell lines. Oncotarget 7, 43295–43314. doi:10.18632/oncotarget.9232
Kozlova, N. I., Morozevich, G. E., Gevorkian, N. M., and Berman, A. E. (2020). Implication of integrins α3β1 and α5β1 in invasion and anoikis of SK-Mel-147 human melanoma cells: Non-canonical functions of protein kinase akt. Aging (Albany NY) 12, 24345–24356. doi:10.18632/aging.202243
Kozlova, N. I., Morozevich, G. E., Ushakova, N. A., and Berman, A. E. (2019). Implication of integrin α2β1 in anoikis of SK-Mel-147 human melanoma cells: A non-canonical function of akt protein kinase. Oncotarget 10, 1829–1839. doi:10.18632/oncotarget.26746
Kozlova, N. I., Morozevich, G. E., Ushakova, N. A., and Berman, A. E. (2018). Implication of integrin α2β1 in proliferation and invasion of human breast carcinoma and melanoma cells: Noncanonical function of akt protein kinase. Biochem. (Mosc) 83, 738–745. doi:10.1134/S0006297918060111
Krengel, S., Stark, I., Geuchen, C., Knoppe, B., Scheel, G., Schlenke, P., et al. (2005). Selective down-regulation of the alpha6-integrin subunit in melanocytes by UVB light. Exp. Dermatol 14, 411–419. doi:10.1111/j.0906-6705.2005.00295.x
Krepler, C., Xiao, M., Sproesser, K., Brafford, P. A., Shannan, B., Beqiri, M., et al. (2016). Personalized preclinical trials in BRAF inhibitor-resistant patient-derived xenograft models identify second-line combination therapies. Clin. Cancer Res. 22, 1592–1602. doi:10.1158/1078-0432.CCR-15-1762
Kuphal, S., Palm, H. G., Poser, I., and Bosserhoff, A. K. (2005). Snail-regulated genes in malignant melanoma. Melanoma Res. 15, 305–313. doi:10.1097/00008390-200508000-00012
Kuphal, S., Poser, I., Jobin, C., Hellerbrand, C., and Bosserhoff, A. K. (2004). Loss of E-cadherin leads to upregulation of NFkappaB activity in malignant melanoma. Oncogene 23, 8509–8519. doi:10.1038/sj.onc.1207831
Lafky, J. M., Wilken, J. A., Baron, A. T., and Maihle, N. J. (2008). Clinical implications of the ErbB/epidermal growth factor (EGF) receptor family and its ligands in ovarian cancer. Biochim. Biophys. Acta 1785, 232–265. doi:10.1016/j.bbcan.2008.01.001
Lammens, T., Swerts, K., Derycke, L., De Craemer, A., De Brouwer, S., De Preter, K., et al. (2012). N-Cadherin in neuroblastoma disease: Expression and clinical significance. PLoS One 7, e31206. doi:10.1371/journal.pone.0031206
Lamouille, S., Xu, J., and Derynck, R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15, 178–196. doi:10.1038/nrm3758
Larkin, J., Ascierto, P. A., Dréno, B., Atkinson, V., Liszkay, G., Maio, M., et al. (2014). Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N. Engl. J. Med. 371, 1867–1876. doi:10.1056/NEJMoa1408868
Lasheras-Otero, I., Feliu, I., Maillo, A., Moreno, H., Redondo-Muñoz, M., Aldaz, P., et al. (2022). The regulators of peroxisomal acyl-carnitine shuttle CROT and CRAT promote metastasis in melanoma. J. Invest Dermatol 143, 305–316.e5. doi:10.1016/j.jid.2022.08.038
Lee, I., Fox, P. S., Ferguson, S. D., Bassett, R., Kong, L. Y., Schacherer, C. W., et al. (2012). The expression of p-STAT3 in stage IV melanoma: Risk of CNS metastasis and survival. Oncotarget 3, 336–344. doi:10.18632/oncotarget.475
Lee, J. B., Jung, M., Beom, S. H., Kim, G. M., Kim, H. R., Choi, H. J., et al. (2021). Phase 2 study of TAS-117, an allosteric akt inhibitor in advanced solid tumors harboring phosphatidylinositol 3-kinase/v-akt murine thymoma viral oncogene homolog gene mutations. Invest New Drugs 39, 1366–1374. doi:10.1007/s10637-021-01085-7
Lee, S., Rauch, J., and Kolch, W. (2020). Targeting MAPK signaling in cancer: Mechanisms of drug resistance and sensitivity. Int. J. Mol. Sci. 21, 1102. doi:10.3390/ijms21031102
Lee, Y., Yao, W., Yang, C., Li, Y., Ni, H., Wang, L., et al. (2018). MIST1 regulates SNAI1 and acts through the PTEN/AKT signaling axis to promote anoikisresistance in human melanoma cells. Exp. Ther. Med. 16, 695–703. doi:10.3892/etm.2018.6225
Li, G., Satyamoorthy, K., and Herlyn, M. (2001a). N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res. 61, 3819–3825.
Li, G., Schaider, H., Satyamoorthy, K., Hanakawa, Y., Hashimoto, K., and Herlyn, M. (2001b). Downregulation of E-cadherin and Desmoglein 1 by autocrine hepatocyte growth factor during melanoma development. Oncogene 20, 8125–8135. doi:10.1038/sj.onc.1205034
Li, J., Yang, R., Yang, H., Chen, S., Wang, L., Li, M., et al. (2020). NCAM regulates the proliferation, apoptosis, autophagy, EMT, and migration of human melanoma cells via the Src/Akt/mTOR/cofilin signaling pathway. J. Cell Biochem. 121, 1192–1204. doi:10.1002/jcb.29353
Lim, W.-C., Kim, H., Kim, Y.-J., Jeon, B.-N., Kang, H.-B., and Ko, H. (2020). Catechol inhibits epidermal growth factor-induced epithelial-to-mesenchymal transition and stem cell-like properties in hepatocellular carcinoma cells. Sci. Rep. 10, 7620. doi:10.1038/s41598-020-64603-2
Lin, S., Rusciano, D., Lorenzoni, P., Hartmann, G., Birchmeier, W., Giordano, S., et al. (1998). C-met activation is necessary but not sufficient for liver colonization by B16 murine melanoma cells. Clin. Exp. Metastasis 16, 253–265. doi:10.1023/a:1006596909948
Liu, M., Yang, J., Xu, B., and Zhang, X. (2021a). Tumor metastasis: Mechanistic insights and therapeutic interventions. MedComm 2, 587–617. doi:10.1002/mco2.100
Liu, R., Tan, J., Shen, X., Jiang, K., Wang, C., Zhu, G., et al. (2021b). Therapeutic targeting of FOS in mutant TERT cancers through removing TERT suppression of apoptosis via regulating survivin and TRAIL-R2. Proc. Natl. Acad. Sci. U. S. A. 118, e2022779118. doi:10.1073/pnas.2022779118
Liu, S., Geng, R., Lin, E., Zhao, P., and Chen, Y. (2021c). ERBB1/2/3 expression, prognosis, and immune infiltration in cutaneous melanoma. Front. Genet. 12, 602160. doi:10.3389/fgene.2021.602160
Liu, Z., and Xing, M. (2012). Induction of sodium/iodide symporter (NIS) expression and radioiodine uptake in non-thyroid cancer cells. PLoS One 7, e31729. doi:10.1371/journal.pone.0031729
Lode, H. N., Moehler, T., Xiang, R., Jonczyk, A., Gillies, S. D., Cheresh, D. A., et al. (1999). Synergy between an antiangiogenic integrin alphav antagonist and an antibody-cytokine fusion protein eradicates spontaneous tumor metastases. Proc. Natl. Acad. Sci. U. S. A. 96, 1591–1596. doi:10.1073/pnas.96.4.1591
Long, G. V., Stroyakovskiy, D., Gogas, H., Levchenko, E., De Braud, F., Larkin, J., et al. (2015). Dabrafenib and trametinib versus dabrafenib and placebo for Val600 BRAF-mutant melanoma: A multicentre, double-blind, phase 3 randomised controlled trial. Lancet 386, 444–451. doi:10.1016/S0140-6736(15)60898-4
Luke, J. J., Olson, D. J., Allred, J. B., Strand, C. A., Bao, R., Zha, Y., et al. (2020). Randomized phase II trial and tumor mutational spectrum analysis from cabozantinib versus chemotherapy in metastatic uveal melanoma (alliance A091201). Clin. Cancer Res. 26, 804–811. doi:10.1158/1078-0432.CCR-19-1223
Madireddi, M. T., Dent, P., and Fisher, P. B. (2000). AP-1 and C/EBP transcription factors contribute to mda-7 gene promoter activity during human melanoma differentiation. J. Cell Physiol. 185, 36–46. doi:10.1002/1097-4652(200010)185:1<36::AID-JCP3>3.0.CO;2-V
Mannsåker, T. A., Hoang, T., Aasen, S. N., Bjørnstad, O. V., Parajuli, H., Sundstrøm, T., et al. (2021). Cabozantinib is effective in melanoma brain metastasis cell lines and affects key signaling pathways. Int. J. Mol. Sci. 22, 12296. doi:10.3390/ijms222212296
Manzano, J. L., Layos, L., Bugés, C., De Los Llanos Gil, M., Vila, L., Martínez-Balibrea, E., et al. (2016). Resistant mechanisms to BRAF inhibitors in melanoma. Ann. Transl. Med. 4, 237. doi:10.21037/atm.2016.06.07
Margheri, F., Luciani, C., Taddei, M. L., Giannoni, E., Laurenzana, A., Biagioni, A., et al. (2014). The receptor for urokinase-plasminogen activator (uPAR) controls plasticity of cancer cell movement in mesenchymal and amoeboid migration style. Oncotarget 5, 1538–1553. doi:10.18632/oncotarget.1754
Maschler, S., Wirl, G., Spring, H., Bredow, D. V., Sordat, I., Beug, H., et al. (2005). Tumor cell invasiveness correlates with changes in integrin expression and localization. Oncogene 24, 2032–2041. doi:10.1038/sj.onc.1208423
Matthews, C. P., Birkholz, A. M., Baker, A. R., Perella, C. M., Beck, G. R., Young, M. R., et al. (2007). Dominant-negative activator protein 1 (TAM67) targets cyclooxygenase-2 and osteopontin under conditions in which it specifically inhibits tumorigenesis. Cancer Res. 67, 2430–2438. doi:10.1158/0008-5472.CAN-06-0522
Maurus, K., Hufnagel, A., Geiger, F., Graf, S., Berking, C., Heinemann, A., et al. (2017). The AP-1 transcription factor FOSL1 causes melanocyte reprogramming and transformation. Oncogene 36, 5110–5121. doi:10.1038/onc.2017.135
Mcsweeney, K. R., Gadanec, L. K., Qaradakhi, T., Gammune, T. M., Kubatka, P., Caprnda, M., et al. (2020). Imipridone enhances vascular relaxation via FOXO1 pathway. Clin. Exp. Pharmacol. Physiol. 47, 1816–1823. doi:10.1111/1440-1681.13377
Meredith, J. E., Fazeli, B., and Schwartz, M. A. (1993). The extracellular matrix as a cell survival factor. Mol. Biol. Cell 4, 953–961. doi:10.1091/mbc.4.9.953
Meves, A., Nikolova, E., Heim, J. B., Squirewell, E. J., Cappel, M. A., Pittelkow, M. R., et al. (2015). Tumor cell adhesion as a risk factor for sentinel lymph node metastasis in primary cutaneous melanoma. J. Clin. Oncol. 33, 2509–2515. doi:10.1200/JCO.2014.60.7002
Mitjans, F., Meyer, T., Fittschen, C., Goodman, S., Jonczyk, A., Marshall, J. F., et al. (2000). In vivo therapy of malignant melanoma by means of antagonists of αv integrins. Int. J. Cancer 87, 716–723. doi:10.1002/1097-0215(20000901)87:5<716::aid-ijc14>3.0.co;2-r
Mitra, A. K., Sawada, K., Tiwari, P., Mui, K., Gwin, K., and Lengyel, E. (2011). Ligand-independent activation of c-Met by fibronectin and α(5)β(1)-integrin regulates ovarian cancer invasion and metastasis. Oncogene 30, 1566–1576. doi:10.1038/onc.2010.532
Mo, X., Kazmi, H. R., Preston-Alp, S., Zhou, B., and Zaidi, M. R. (2022). Interferon-gamma induces melanogenesis via post-translational regulation of tyrosinase. Pigment. Cell Melanoma Res. 35, 342–355. doi:10.1111/pcmr.13036
Molhoek, K. R., Shada, A. L., Smolkin, M., Chowbina, S., Papin, J., Brautigan, D. L., et al. (2011). Comprehensive analysis of receptor tyrosine kinase activation in human melanomas reveals autocrine signaling through IGF-1R. Melanoma Res. 21, 274–284. doi:10.1097/CMR.0b013e328343a1d6
Molife, L. R., Yan, L., Vitfell-Rasmussen, J., Zernhelt, A. M., Sullivan, D. M., Cassier, P. A., et al. (2014). Phase 1 trial of the oral AKT inhibitor MK-2206 plus carboplatin/paclitaxel, docetaxel, or erlotinib in patients with advanced solid tumors. J. Hematol. Oncol. 7, 1. doi:10.1186/1756-8722-7-1
Monteiro, H. P., Rodrigues, E. G., Amorim Reis, A. K. C., Longo, L. S., Ogata, F. T., Moretti, A. I. S., et al. (2019). Nitric oxide and interactions with reactive oxygen species in the development of melanoma, breast, and colon cancer: A redox signaling perspective. Nitric Oxide 89, 1–13. doi:10.1016/j.niox.2019.04.009
Montgomery, A. M., Reisfeld, R. A., and Cheresh, D. A. (1994). Integrin alpha v beta 3 rescues melanoma cells from apoptosis in three-dimensional dermal collagen. Proc. Natl. Acad. Sci. U. S. A. 91, 8856–8860. doi:10.1073/pnas.91.19.8856
Moro, L., Venturino, M., Bozzo, C., Silengo, L., Altruda, F., Beguinot, L., et al. (1998). Integrins induce activation of EGF receptor: Role in MAP kinase induction and adhesion-dependent cell survival. Embo J. 17, 6622–6632. doi:10.1093/emboj/17.22.6622
Moschos, S. J., Sander, C. A., Wang, W., Reppert, S. L., Drogowski, L. M., Jukic, D. M., et al. (2010). Pharmacodynamic (phase 0) study using etaracizumab in advanced melanoma. J. Immunother. 33, 316–325. doi:10.1097/CJI.0b013e3181c1f216
Moschos, S. J., Sullivan, R. J., Hwu, W. J., Ramanathan, R. K., Adjei, A. A., Fong, P. C., et al. (2018). Development of MK-8353, an orally administered ERK1/2 inhibitor, in patients with advanced solid tumors. JCI Insight 3, e92352. doi:10.1172/jci.insight.92352
Mullen, J. T., Vartanian, T. K., and Atkins, M. B. (2008). Melanoma complicating treatment with natalizumab for multiple sclerosis. New Engl. J. Med. 358, 647–648. doi:10.1056/NEJMc0706103
Munguía-Calzada, P., Vivanco, B., Oliva-Nacarino, P., and Santos-Juanes, J. (2017). Melanoma, eruptive naevi and natalizumab: Causal relation or coincidence? Australas. J. Dermatol 58, 330–331. doi:10.1111/ajd.12724
Munir, R., Lisec, J., Swinnen, J. V., and Zaidi, N. (2022). Too complex to fail? Targeting fatty acid metabolism for cancer therapy. Prog. Lipid Res. 85, 101143. doi:10.1016/j.plipres.2021.101143
Murtas, D., Maxia, C., Diana, A., Pilloni, L., Corda, C., Minerba, L., et al. (2017). Role of epithelial-mesenchymal transition involved molecules in the progression of cutaneous melanoma. Histochem Cell Biol. 148, 639–649. doi:10.1007/s00418-017-1606-0
Natali, P. G., Nicotra, M. R., Di Renzo, M. F., Prat, M., Bigotti, A., Cavaliere, R., et al. (1993). Expression of the c-met/HGF receptor in human melanocytic neoplasms: Demonstration of the relationship to malignant melanoma tumour progression. Br. J. Cancer 68, 746–750. doi:10.1038/bjc.1993.422
Ng, Y. K., Lee, J. Y., Supko, K. M., Khan, A., Torres, S. M., Berwick, M., et al. (2014). Pan-erbB inhibition potentiates BRAF inhibitors for melanoma treatment. Melanoma Res. 24, 207–218. doi:10.1097/CMR.0000000000000060
Nguyen, T., and Mège, R. M. (2016). N-Cadherin and Fibroblast Growth Factor Receptors crosstalk in the control of developmental and cancer cell migrations. Eur. J. Cell Biol. 95, 415–426. doi:10.1016/j.ejcb.2016.05.002
Niu, G., Heller, R., Catlett-Falcone, R., Coppola, D., Jaroszeski, M., Dalton, W., et al. (1999). Gene therapy with dominant-negative Stat3 suppresses growth of the murine melanoma B16 tumor in vivo. Cancer Res. 59, 5059–5063.
Nummela, P., Yin, M., Kielosto, M., Leaner, V., Birrer, M. J., and Hölttä, E. (2006). Thymosin beta4 is a determinant of the transformed phenotype and invasiveness of S-adenosylmethionine decarboxylase-transfected fibroblasts. Cancer Res. 66, 701–712. doi:10.1158/0008-5472.CAN-05-2421
O'day, S. J., Pavlick, A. C., Albertini, M. R., Hamid, O., Schalch, H., Lang, Z., et al. (2012). Clinical and pharmacologic evaluation of two dose levels of intetumumab (CNTO 95) in patients with melanoma or angiosarcoma. Invest New Drugs 30, 1074–1081. doi:10.1007/s10637-011-9639-z
O'day, S., Pavlick, A., Loquai, C., Lawson, D., Gutzmer, R., Richards, J., et al. (2011). A randomised, phase II study of intetumumab, an anti-αv-integrin mAb, alone and with dacarbazine in stage IV melanoma. Br. J. Cancer 105, 346–352. doi:10.1038/bjc.2011.183
Oberst, M. D., Beberman, S. J., Zhao, L., Yin, J. J., Ward, Y., and Kelly, K. (2008). TDAG51 is an ERK signaling target that opposes ERK-mediated HME16C mammary epithelial cell transformation. BMC Cancer 8, 189. doi:10.1186/1471-2407-8-189
Opferman, J. T., and Kothari, A. (2018). Anti-apoptotic BCL-2 family members in development. Cell Death Differ. 25, 37–45. doi:10.1038/cdd.2017.170
Organ, S. L., and Tsao, M. S. (2011). An overview of the c-MET signaling pathway. Ther. Adv. Med. Oncol. 3, S7–s19. doi:10.1177/1758834011422556
Otsuka, T., Takayama, H., Sharp, R., Celli, G., Larochelle, W. J., Bottaro, D. P., et al. (1998). c-Met autocrine activation induces development of malignant melanoma and acquisition of the metastatic phenotype. Cancer Res. 58, 5157–5167.
Padmanaban, V., Krol, I., Suhail, Y., Szczerba, B. M., Aceto, N., Bader, J. S., et al. (2019). E-cadherin is required for metastasis in multiple models of breast cancer. Nature 573, 439–444. doi:10.1038/s41586-019-1526-3
Pan, X., Yi, M., Liu, C., Jin, Y., Liu, B., Hu, G., et al. (2022). Cilengitide, an αvβ3-integrin inhibitor, enhances the efficacy of anti-programmed cell death-1 therapy in a murine melanoma model. Bioengineered 13, 4557–4572. doi:10.1080/21655979.2022.2029236
Paoli, P., Giannoni, E., and Chiarugi, P. (2013). Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 1833, 3481–3498. doi:10.1016/j.bbamcr.2013.06.026
Parsons, J. T., Martin, K. H., Slack, J. K., Taylor, J. M., and Weed, S. A. (2000). Focal adhesion kinase: A regulator of focal adhesion dynamics and cell movement. Oncogene 19, 5606–5613. doi:10.1038/sj.onc.1203877
Pearlman, R. L., Montes De Oca, M. K., Pal, H. C., and Afaq, F. (2017). Potential therapeutic targets of epithelial-mesenchymal transition in melanoma. Cancer Lett. 391, 125–140. doi:10.1016/j.canlet.2017.01.029
Pei, G., Lan, Y., Chen, D., Ji, L., and Hua, Z. C. (2017). FAK regulates E-cadherin expression via p-SrcY416/p-ERK1/2/p-Stat3Y705 and PPARγ/miR-125b/Stat3 signaling pathway in B16F10 melanoma cells. Oncotarget 8, 13898–13908. doi:10.18632/oncotarget.14687
Peppicelli, S., Ruzzolini, J., Bianchini, F., Andreucci, E., Nediani, C., Laurenzana, A., et al. (2019). Anoikis resistance as a further trait of acidic-adapted melanoma cells. J. Oncol. 2019, 8340926. doi:10.1155/2019/8340926
Petit, V., Raymond, J., Alberti, C., Pouteaux, M., Gallagher, S. J., Nguyen, M. Q., et al. (2019). C57BL/6 congenic mouse NRAS(Q61K) melanoma cell lines are highly sensitive to the combination of Mek and Akt inhibitors in vitro and in vivo. Pigment. Cell Melanoma Res. 32, 829–841. doi:10.1111/pcmr.12807
Petitclerc, E., Strömblad, S., Von Schalscha, T. L., Mitjans, F., Piulats, J., Montgomery, A. M., et al. (1999). Integrin alpha(v)beta3 promotes M21 melanoma growth in human skin by regulating tumor cell survival. Cancer Res. 59, 2724–2730.
Pickarski, M., Gleason, A., Bednar, B., and Duong, L. T. (2015). Orally active αvβ3 integrin inhibitor MK-0429 reduces melanoma metastasis. Oncol. Rep. 33, 2737–2745. doi:10.3892/or.2015.3910
Pierce, C. J., Simmons, J. L., Broit, N., Karunarathne, D., Ng, M. F., and Boyle, G. M. (2020). BRN2 expression increases anoikis resistance in melanoma. Oncogenesis 9, 64. doi:10.1038/s41389-020-00247-1
Poser, I., Domínguez, D., De Herreros, A. G., Varnai, A., Buettner, R., and Bosserhoff, A. K. (2001). Loss of E-cadherin expression in melanoma cells involves up-regulation of the transcriptional repressor Snail. J. Biol. Chem. 276, 24661–24666. doi:10.1074/jbc.M011224200
Posey, J. A., Khazaeli, M. B., Delgrosso, A., Saleh, M. N., Lin, C. Y., Huse, W., et al. (2001). A pilot trial of Vitaxin, a humanized anti-vitronectin receptor (anti alpha v beta 3) antibody in patients with metastatic cancer. Cancer Biother Radiopharm. 16, 125–132. doi:10.1089/108497801300189218
Prabhu, V. V., Morrow, S., Rahman Kawakibi, A., Zhou, L., Ralff, M., Ray, J., et al. (2020). ONC201 and imipridones: Anti-cancer compounds with clinical efficacy. Neoplasia 22, 725–744. doi:10.1016/j.neo.2020.09.005
Prickett, T. D., Agrawal, N. S., Wei, X., Yates, K. E., Lin, J. C., Wunderlich, J. R., et al. (2009). Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat. Genet. 41, 1127–1132. doi:10.1038/ng.438
Qi, J., Chen, N., Wang, J., and Siu, C. H. (2005). Transendothelial migration of melanoma cells involves N-cadherin-mediated adhesion and activation of the beta-catenin signaling pathway. Mol. Biol. Cell 16, 4386–4397. doi:10.1091/mbc.e05-03-0186
Qi, X. J., Wildey, G. M., and Howe, P. H. (2006). Evidence that Ser87 of BimEL is phosphorylated by Akt and regulates BimEL apoptotic function. J. Biol. Chem. 281, 813–823. doi:10.1074/jbc.M505546200
Qian, F., Engst, S., Yamaguchi, K., Yu, P., Won, K.-A., Mock, L., et al. (2009). Inhibition of tumor cell growth, invasion, and metastasis by EXEL-2880 (XL880, GSK1363089), a novel inhibitor of HGF and VEGF receptor tyrosine kinases. Cancer Res. 69, 8009–8016. doi:10.1158/0008-5472.CAN-08-4889
Qian, F., Vaux, D. L., and Weissman, I. L. (1994). Expression of the integrin alpha 4 beta 1 on melanoma cells can inhibit the invasive stage of metastasis formation. Cell 77, 335–347. doi:10.1016/0092-8674(94)90149-x
Qin, Y., Roszik, J., Chattopadhyay, C., Hashimoto, Y., Liu, C., Cooper, Z. A., et al. (2016). Hypoxia-driven mechanism of vemurafenib resistance in melanoma. Mol. Cancer Ther. 15, 2442–2454. doi:10.1158/1535-7163.MCT-15-0963
Quast, S. A., Berger, A., and Eberle, J. (2013). ROS-dependent phosphorylation of Bax by wortmannin sensitizes melanoma cells for TRAIL-induced apoptosis. Cell Death Dis. 4, e839. doi:10.1038/cddis.2013.344
Ramasamy, T., Chen, X., Qin, B., Johnson, D. E., Grandis, J. R., and Villanueva, F. S. (2020). STAT3 decoy oligonucleotide-carrying microbubbles with pulsed ultrasound for enhanced therapeutic effect in head and neck tumors. PLoS One 15, e0242264. doi:10.1371/journal.pone.0242264
Rambow, F., Marine, J. C., and Goding, C. R. (2019). Melanoma plasticity and phenotypic diversity: Therapeutic barriers and opportunities. Genes Dev. 33, 1295–1318. doi:10.1101/gad.329771.119
Rambow, F., Rogiers, A., Marin-Bejar, O., Aibar, S., Femel, J., Dewaele, M., et al. (2018). Toward minimal residual disease-directed therapy in melanoma. Cell 174, 843–855. doi:10.1016/j.cell.2018.06.025
Rebecca, V. W., Massaro, R. R., Fedorenko, I. V., Sondak, V. K., Anderson, A. R., Kim, E., et al. (2014). Inhibition of autophagy enhances the effects of the AKT inhibitor MK-2206 when combined with paclitaxel and carboplatin in BRAF wild-type melanoma. Pigment. Cell Melanoma Res. 27, 465–478. doi:10.1111/pcmr.12227
Reed, J. C. (2006). Proapoptotic multidomain bcl-2/bax-family proteins: Mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 13, 1378–1386. doi:10.1038/sj.cdd.4401975
Reginato, M. J., Mills, K. R., Paulus, J. K., Lynch, D. K., Sgroi, D. C., Debnath, J., et al. (2003). Integrins and EGFR coordinately regulate the pro-apoptotic protein Bim to prevent anoikis. Nat. Cell Biol. 5, 733–740. doi:10.1038/ncb1026
Reilley, M. J., Mccoon, P., Cook, C., Lyne, P., Kurzrock, R., Kim, Y., et al. (2018). STAT3 antisense oligonucleotide AZD9150 in a subset of patients with heavily pretreated lymphoma: Results of a phase 1b trial. J. Immunother. Cancer 6, 119. doi:10.1186/s40425-018-0436-5
Ribeiro-Pereira, C., Moraes, J. A., Souza Mde, J., Laurindo, F. R., Arruda, M. A., and Barja-Fidalgo, C. (2014). Redox modulation of FAK controls melanoma survival-role of NOX4. PLoS One 9, e99481. doi:10.1371/journal.pone.0099481
Ricart, A. D., Tolcher, A. W., Liu, G., Holen, K., Schwartz, G., Albertini, M., et al. (2008). Volociximab, a chimeric monoclonal antibody that specifically binds alpha5beta1 integrin: A phase I, pharmacokinetic, and biological correlative study. Clin. Cancer Res. 14, 7924–7929. doi:10.1158/1078-0432.CCR-08-0378
Ricca, T. I., Liang, G., Suenaga, A. P., Han, S. W., Jones, P. A., and Jasiulionis, M. G. (2009). Tissue inhibitor of metalloproteinase 1 expression associated with gene demethylation confers anoikis resistance in early phases of melanocyte malignant transformation. Transl. Oncol. 2, 329–340. doi:10.1593/tlo.09220
Robert, C., Karaszewska, B., Schachter, J., Rutkowski, P., Mackiewicz, A., Stroiakovski, D., et al. (2015). Improved overall survival in melanoma with combined dabrafenib and trametinib. New Engl. J. Med. 372, 30–39. doi:10.1056/NEJMoa1412690
Robinson, D. W., Cormier, J. N., Zhao, N., Uhlar, C. M., Revicki, D. A., and Cella, D. (2012). Health-related quality of life among patients with metastatic melanoma: Results from an international phase 2 multicenter study. Melanoma Res. 22, 54–62. doi:10.1097/CMR.0b013e32834d3da0
Rüegg, C., Yilmaz, A., Bieler, G., Bamat, J., Chaubert, P., and Lejeune, F. J. (1998). Evidence for the involvement of endothelial cell integrin alphaVbeta3 in the disruption of the tumor vasculature induced by TNF and IFN-gamma. Nat. Med. 4, 408–414. doi:10.1038/nm0498-408
Ruffini, F., Graziani, G., Levati, L., Tentori, L., D'atri, S., and Lacal, P. M. (2015). Cilengitide downmodulates invasiveness and vasculogenic mimicry of neuropilin 1 expressing melanoma cells through the inhibition of αvβ5 integrin. Int. J. Cancer 136, E545–E558. doi:10.1002/ijc.29252
Rusciano, D., Lin, S., Lorenzoni, P., Casella, N., and Burger, M. M. (1998). Influence of hepatocyte growth factor/scatter factor on the metastatic phenotype of B16 melanoma cells. Tumour Biol. 19, 335–345. doi:10.1159/000030026
Ryan, Q., Ibrahim, A., Cohen, M. H., Johnson, J., Ko, C. W., Sridhara, R., et al. (2008). FDA drug approval summary: Lapatinib in combination with capecitabine for previously treated metastatic breast cancer that overexpresses HER-2. Oncologist 13, 1114–1119. doi:10.1634/theoncologist.2008-0816
Sabol, R. A., Noxon, V., Sartor, O., Berger, J. R., Qureshi, Z., Raisch, D. W., et al. (2017). Melanoma complicating treatment with natalizumab for multiple sclerosis: A report from the southern network on adverse reactions (sonar). Cancer Med. 6, 1541–1551. doi:10.1002/cam4.1098
Sadler, N. M., Harris, B. R., Metzger, B. A., and Kirshner, J. (2013). N-cadherin impedes proliferation of the multiple myeloma cancer stem cells. Am. J. Blood Res. 3, 271–285.
Sahoo, S., Ashraf, B., Duddu, A. S., Biddle, A., and Jolly, M. K. (2022). Interconnected high-dimensional landscapes of epithelial–mesenchymal plasticity and stemness in cancer. Clin. Exp. Metastasis 39, 279–290. doi:10.1007/s10585-021-10139-2
Saitoh, K., Takahashi, H., Sawada, N., and Parsons, P. G. (1994). Detection of the c-met proto-oncogene product in normal skin and tumours of melanocytic origin. J. Pathology 174, 191–199. doi:10.1002/path.1711740308
Sanz-Moreno, V., Gadea, G., Ahn, J., Paterson, H., Marra, P., Pinner, S., et al. (2008). Rac activation and inactivation control plasticity of tumor cell movement. Cell 135, 510–523. doi:10.1016/j.cell.2008.09.043
Sattari Fard, F., Jalilzadeh, N., Mehdizadeh, A., Sajjadian, F., and Velaei, K. (2022). Understanding and targeting anoikis in metastasis for cancer therapies. Cell Biol. Int. 47, 683–698. doi:10.1002/cbin.11970
Sau, S., Mondal, S. K., Kashaw, S. K., Iyer, A. K., and Banerjee, R. (2017). Combination of cationic dexamethasone derivative and STAT3 inhibitor (WP1066) for aggressive melanoma: A strategy for repurposing a phase I clinical trial drug. Mol. Cell Biochem. 436, 119–136. doi:10.1007/s11010-017-3084-z
Saura, C., Roda, D., Roselló, S., Oliveira, M., Macarulla, T., Pérez-Fidalgo, J. A., et al. (2017). A first-in-human phase I study of the ATP-competitive AKT inhibitor ipatasertib demonstrates robust and safe targeting of AKT in patients with solid tumors. Cancer Discov. 7, 102–113. doi:10.1158/2159-8290.CD-16-0512
Sen, M., Thomas, S. M., Kim, S., Yeh, J. I., Ferris, R. L., Johnson, J. T., et al. (2012). First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: Implications for cancer therapy. Cancer Discov. 2, 694–705. doi:10.1158/2159-8290.CD-12-0191
Shah, S., Famta, P., Fernandes, V., Bagasariya, D., Charankumar, K., Kumar Khatri, D., et al. (2022). Quality by design steered development of niclosamide loaded liposomal thermogel for melanoma: In vitro and ex vivo evaluation. Eur. J. Pharm. Biopharm. 180, 119–136. doi:10.1016/j.ejpb.2022.09.024
Shao, Y., and Aplin, A. E. (2010). Akt3-mediated resistance to apoptosis in B-RAF-targeted melanoma cells. Cancer Res. 70, 6670–6681. doi:10.1158/0008-5472.CAN-09-4471
Sharma, M., Turaga, R. C., Yuan, Y., Satyanarayana, G., Mishra, F., Bian, Z., et al. (2021). Simultaneously targeting cancer-associated fibroblasts and angiogenic vessel as a treatment for TNBC. J. Exp. Med. 218, e20200712. doi:10.1084/jem.20200712
Shen, H., Huang, F., Zhang, X., Ojo, O. A., Li, Y., Trummell, H. Q., et al. (2022). Selective suppression of melanoma lacking IFN-γ pathway by JAK inhibition depends on T cells and host TNF signaling. Nat. Commun. 13, 5013. doi:10.1038/s41467-022-32754-7
Shen, X., and Kramer, R. H. (2004). Adhesion-mediated squamous cell carcinoma survival through ligand-independent activation of epidermal growth factor receptor. Am. J. Pathol. 165, 1315–1329. doi:10.1016/S0002-9440(10)63390-1
Shi, L., Zheng, H., Hu, W., Zhou, B., Dai, X., Zhang, Y., et al. (2017). Niclosamide inhibition of STAT3 synergizes with erlotinib in human colon cancer. Onco Targets Ther. 10, 1767–1776. doi:10.2147/OTT.S129449
Shintani, Y., Fukumoto, Y., Chaika, N., Grandgenett, P. M., Hollingsworth, M. A., Wheelock, M. J., et al. (2008). ADH-1 suppresses N-cadherin-dependent pancreatic cancer progression. Int. J. Cancer 122, 71–77. doi:10.1002/ijc.23027
Shoushtari, A. N., Kudchadkar, R. R., Panageas, K., Murthy, R. K., Jung, M., Shah, R., et al. (2016). A randomized phase 2 study of trametinib with or without GSK2141795 in patients with advanced uveal melanoma. J. Clin. Oncol. 34, 9511. doi:10.1200/jco.2016.34.15_suppl.9511
Shull, A. Y., Latham-Schwark, A., Ramasamy, P., Leskoske, K., Oroian, D., Birtwistle, M. R., et al. (2012). Novel somatic mutations to PI3K pathway genes in metastatic melanoma. PLoS One 7, e43369. doi:10.1371/journal.pone.0043369
Silye, R., Karayiannakis, A. J., Syrigos, K. N., Poole, S., Van Noorden, S., Batchelor, W., et al. (1998). E-cadherin/catenin complex in benign and malignant melanocytic lesions. J. Pathology 186, 350–355. doi:10.1002/(SICI)1096-9896(199812)186:4<350::AID-PATH181>3.0.CO;2-K
Simiczyjew, A., Pietraszek-Gremplewicz, K., Dratkiewicz, E., Podgórska, M., Matkowski, R., Ziętek, M., et al. (2019). Combination of selected MET and EGFR inhibitors decreases melanoma cells' invasive abilities. Front. Pharmacol. 10, 1116. doi:10.3389/fphar.2019.01116
Slack, R. J., Macdonald, S. J. F., Roper, J. A., Jenkins, R. G., and Hatley, R. J. D. (2022). Emerging therapeutic opportunities for integrin inhibitors. Nat. Rev. Drug Discov. 21, 60–78. doi:10.1038/s41573-021-00284-4
Sobolev, V. V., Khashukoeva, A. Z., Evina, O. E., Geppe, N. A., Chebysheva, S. N., Korsunskaya, I. M., et al. (2022). Role of the transcription factor FOSL1 in organ development and tumorigenesis. Int. J. Mol. Sci. 23, 1521. doi:10.3390/ijms23031521
Sonbol, M. B., Ahn, D. H., Goldstein, D., Okusaka, T., Tabernero, J., Macarulla, T., et al. (2019). CanStem111P trial: A phase III study of napabucasin plus nab-paclitaxel with gemcitabine. Future Oncol. 15, 1295–1302. doi:10.2217/fon-2018-0903
Stojanović, N., Dekanić, A., Paradžik, M., Majhen, D., Ferenčak, K., Ruščić, J., et al. (2018). Differential effects of integrin αv knockdown and cilengitide on sensitization of triple-negative breast cancer and melanoma cells to microtubule poisons. Mol. Pharmacol. 94, 1334–1351. doi:10.1124/mol.118.113027
Sturm, R. A., Bisshop, F., Takahashi, H., and Parsons, P. G. (1991). A melanoma octamer binding protein is responsive to differentiating agents. Cell Growth Differ. 2, 519–524.
Su, F., Bradley, W. D., Wang, Q., Yang, H., Xu, L., Higgins, B., et al. (2012). Resistance to selective BRAF inhibition can Be mediated by modest upstream pathway activation. Cancer Res. 72, 969–978. doi:10.1158/0008-5472.CAN-11-1875
Su, Y., Yu, Y., He, D., Zhang, J., Wang, Z., Sun, P., et al. (2018). Targeting STAT3 restores BRAF inhibitor sensitivity through miR-759-3p in human cutaneous melanoma cells. Int. J. Clin. Exp. Pathol. 11, 2550–2560.
Sullivan, R. J., Infante, J. R., Janku, F., Wong, D. J. L., Sosman, J. A., Keedy, V., et al. (2018). First-in-Class ERK1/2 inhibitor ulixertinib (BVD-523) in patients with MAPK mutant advanced solid tumors: Results of a phase I dose-escalation and expansion study. Cancer Discov. 8, 184–195. doi:10.1158/2159-8290.CD-17-1119
Sun, C., Wang, L., Huang, S., Heynen, G. J., Prahallad, A., Robert, C., et al. (2014). Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122. doi:10.1038/nature13121
Surriga, O., Rajasekhar, V. K., Ambrosini, G., Dogan, Y., Huang, R., and Schwartz, G. K. (2013). Crizotinib, a c-Met inhibitor, prevents metastasis in a metastatic uveal melanoma model. Mol. Cancer Ther. 12, 2817–2826. doi:10.1158/1535-7163.MCT-13-0499
Swoboda, A., Soukup, R., Eckel, O., Kinslechner, K., Wingelhofer, B., Schörghofer, D., et al. (2021). STAT3 promotes melanoma metastasis by CEBP-induced repression of the MITF pathway. Oncogene 40, 1091–1105. doi:10.1038/s41388-020-01584-6
Takács, A., Szász, Z., Kalabay, M., Bárány, P., Csámpai, A., Hegyesi, H., et al. (2021). The synergistic activity of bortezomib and TIC10 against A2058 melanoma cells. Pharm. (Basel) 14, 820. doi:10.3390/ph14080820
Talukdar, S., Pradhan, A. K., Bhoopathi, P., Shen, X. N., August, L. A., Windle, J. J., et al. (2018). MDA-9/Syntenin regulates protective autophagy in anoikis-resistant glioma stem cells. Proc. Natl. Acad. Sci. U. S. A. 115, 5768–5773. doi:10.1073/pnas.1721650115
Tanaka, H., Kono, E., Tran, C. P., Miyazaki, H., Yamashiro, J., Shimomura, T., et al. (2010). Monoclonal antibody targeting of N-cadherin inhibits prostate cancer growth, metastasis and castration resistance. Nat. Med. 16, 1414–1420. doi:10.1038/nm.2236
Tanaka, R., Terai, M., Londin, E., and Sato, T. (2021). The role of HGF/MET signaling in metastatic uveal melanoma. Cancers (Basel) 13, 5457. doi:10.3390/cancers13215457
Tang, A., Eller, M. S., Hara, M., Yaar, M., Hirohashi, S., and Gilchrest, B. A. (1994). E-cadherin is the major mediator of human melanocyte adhesion to keratinocytes in vitro. J. Cell Sci. 107 (4), 983–992. doi:10.1242/jcs.107.4.983
Tang, M. K., Zhou, H. Y., Yam, J. W., and Wong, A. S. (2010). c-Met overexpression contributes to the acquired apoptotic resistance of nonadherent ovarian cancer cells through a cross talk mediated by phosphatidylinositol 3-kinase and extracellular signal-regulated kinase 1/2. Neoplasia 12, 128–138. doi:10.1593/neo.91438
Tang, Q., Li, J., Zhu, H., Li, P., Zou, Z., and Xiao, Y. (2013). Hmgb1-IL-23-IL-17-IL-6-Stat3 axis promotes tumor growth in murine models of melanoma. Mediat. Inflamm. 2013, 713859. doi:10.1155/2013/713859
Thang, N. D., Nghia, P. T., Kumasaka, M. Y., Yajima, I., and Kato, M. (2015). Treatment of vemurafenib-resistant SKMEL-28 melanoma cells with paclitaxel. Asian Pac J. Cancer Prev. 16, 699–705. doi:10.7314/apjcp.2015.16.2.699
Thaper, D., Munuganti, R., Aguda, A., Kim, S., Ku, S., Sivak, O., et al. (2022). Discovery and characterization of a first-in-field transcription factor BRN2 inhibitor for the treatment of neuroendocrine prostate cancer. bioRxiv, 2022.05.04.490172.
Thiery, J. P., Acloque, H., Huang, R. Y., and Nieto, M. A. (2009). Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890. doi:10.1016/j.cell.2009.11.007
Thomas, A. J., and Erickson, C. A. (2008). The making of a melanocyte: The specification of melanoblasts from the neural crest. Pigment. Cell Melanoma Res. 21, 598–610. doi:10.1111/j.1755-148X.2008.00506.x
Thomson, J. A., Murphy, K., Baker, E., Sutherland, G. R., Parsons, P. G., Sturm, R. A., et al. (1995). The brn-2 gene regulates the melanocytic phenotype and tumorigenic potential of human melanoma cells. Oncogene 11, 691–700.
Thomson, J. A., Parsons, P. G., and Sturm, R. A. (1993). In vivo and in vitro expression of octamer binding proteins in human melanoma metastases, brain tissue, and fibroblasts. Pigment. Cell Res. 6, 13–22. doi:10.1111/j.1600-0749.1993.tb00576.x
Toiyama, Y., Yasuda, H., Saigusa, S., Matushita, K., Fujikawa, H., Tanaka, K., et al. (2012). Co-expression of hepatocyte growth factor and c-Met predicts peritoneal dissemination established by autocrine hepatocyte growth factor/c-Met signaling in gastric cancer. Int. J. Cancer 130, 2912–2921. doi:10.1002/ijc.26330
Tolcher, A. W., Kurzrock, R., Valero, V., Gonzalez, R., Heist, R. S., Tan, A. R., et al. (2020). Phase I dose-escalation trial of the oral AKT inhibitor uprosertib in combination with the oral MEK1/MEK2 inhibitor trametinib in patients with solid tumors. Cancer Chemother. Pharmacol. 85, 673–683. doi:10.1007/s00280-020-04038-8
Tolcher, A. W., Patnaik, A., Papadopoulos, K. P., Rasco, D. W., Becerra, C. R., Allred, A. J., et al. (2015). Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 75, 183–189. doi:10.1007/s00280-014-2615-5
Toricelli, M., Melo, F. H. M., Hunger, A., Zanatta, D., Strauss, B. E., and Jasiulionis, M. G. (2017). Timp1 promotes cell survival by activating the PDK1 signaling pathway in melanoma. Cancers (Basel) 9, 37. doi:10.3390/cancers9040037
Toricelli, M., Melo, F. H., Peres, G. B., Silva, D. C., and Jasiulionis, M. G. (2013). Timp1 interacts with beta-1 integrin and CD63 along melanoma Genesis and confers anoikis resistance by activating PI3-K signaling pathway independently of Akt phosphorylation. Mol. Cancer 12, 22. doi:10.1186/1476-4598-12-22
Trikha, M., Zhou, Z., Nemeth, J. A., Chen, Q., Sharp, C., Emmell, E., et al. (2004). CNTO 95, a fully human monoclonal antibody that inhibits alphav integrins, has antitumor and antiangiogenic activity in vivo. Int. J. Cancer 110, 326–335. doi:10.1002/ijc.20116
Trikha, M., Zhou, Z., Timar, J., Raso, E., Kennel, M., Emmell, E., et al. (2002). Multiple roles for platelet GPIIb/IIIa and alphavbeta3 integrins in tumor growth, angiogenesis, and metastasis. Cancer Res. 62, 2824–2833.
Tsao, S. C., Wang, J., Wang, Y., Behren, A., Cebon, J., and Trau, M. (2018). Characterising the phenotypic evolution of circulating tumour cells during treatment. Nat. Commun. 9, 1482. doi:10.1038/s41467-018-03725-8
Tsoi, J., Robert, L., Paraiso, K., Galvan, C., Sheu, K. M., Lay, J., et al. (2018). Multi-stage differentiation defines melanoma subtypes with differential vulnerability to drug-induced iron-dependent oxidative stress. Cancer Cell 33, 890–904. doi:10.1016/j.ccell.2018.03.017
Turaga, R. C., Sharma, M., Mishra, F., Krasinskas, A., Yuan, Y., Yang, J. J., et al. (2021). Modulation of cancer-associated fibrotic stroma by an integrin αvβ3 targeting protein for pancreatic cancer treatment. Cell Mol. Gastroenterol. Hepatol. 11, 161–179. doi:10.1016/j.jcmgh.2020.08.004
Turaga, R. C., Yin, L., Yang, J. J., Lee, H., Ivanov, I., Yan, C., et al. (2016). Rational design of a protein that binds integrin αvβ3 outside the ligand binding site. Nat. Commun. 7, 11675. doi:10.1038/ncomms11675
Turley, R. S., Tokuhisa, Y., Toshimitsu, H., Lidsky, M. E., Padussis, J. C., Fontanella, A., et al. (2015). Targeting N-cadherin increases vascular permeability and differentially activates AKT in melanoma. Ann. Surg. 261, 368–377. doi:10.1097/SLA.0000000000000635
Tuthill, C. W., and Sonis, S. T. (2012). Biomarkers to track Response to SCV-07. US patent application WO2012040656A2. Geneva, Switzerland: WORLD INTELLECTUAL PROPERTY ORGANIZATION.
Ueno, Y., Sakurai, H., Tsunoda, S., Choo, M. K., Matsuo, M., Koizumi, K., et al. (2008). Heregulin-induced activation of ErbB3 by EGFR tyrosine kinase activity promotes tumor growth and metastasis in melanoma cells. Int. J. Cancer, 123, 340–7.
Van Belle, P. A., Elenitsas, R., Satyamoorthy, K., Wolfe, J. T., Guerry, D. T., Schuchter, L., et al. (1999). Progression-related expression of beta3 integrin in melanomas and nevi. Hum. Pathol. 30, 562–567. doi:10.1016/s0046-8177(99)90202-2
Vandamme, N., and Berx, G. (2014). Melanoma cells revive an embryonic transcriptional network to dictate phenotypic heterogeneity. Front. Oncol. 4, 352. doi:10.3389/fonc.2014.00352
Vanhaesebroeck, B., Perry, M. W. D., Brown, J. R., André, F., and Okkenhaug, K. (2021). PI3K inhibitors are finally coming of age. Nat. Rev. Drug Discov. 20, 741–769. doi:10.1038/s41573-021-00209-1
Varner, J. A., Nakada, M. T., Jordan, R. E., and Coller, B. S. (1999). Inhibition of angiogenesis and tumor growth by murine 7E3, the parent antibody of c7E3 Fab (abciximab; ReoPro). Angiogenesis 3, 53–60. doi:10.1023/a:1009019223744
Vavricka, B. M., Baumberger, P., Russmann, S., and Kullak-Ublick, G. A. (2011). Diagnosis of melanoma under concomitant natalizumab therapy. Mult. Scler. 17, 255–256. doi:10.1177/1352458510389629
Venhuizen, J.-H., Jacobs, F. J. C., Span, P. N., and Zegers, M. M. (2020). P120 and E-cadherin: Double-edged swords in tumor metastasis. Seminars Cancer Biol. 60, 107–120. doi:10.1016/j.semcancer.2019.07.020
Vivo, M., Fontana, R., Ranieri, M., Capasso, G., Angrisano, T., Pollice, A., et al. (2017). p14ARF interacts with the focal adhesion kinase and protects cells from anoikis. Oncogene 36, 4913–4928. doi:10.1038/onc.2017.104
Vultur, A., Villanueva, J., Krepler, C., Rajan, G., Chen, Q., Xiao, M., et al. (2014). MEK inhibition affects STAT3 signaling and invasion in human melanoma cell lines. Oncogene 33, 1850–1861. doi:10.1038/onc.2013.131
Wagner, J., Kline, C. L., Ralff, M. D., Lev, A., Lulla, A., Zhou, L., et al. (2017). Preclinical evaluation of the imipridone family, analogs of clinical stage anti-cancer small molecule ONC201, reveals potent anti-cancer effects of ONC212. Cell Cycle 16, 1790–1799. doi:10.1080/15384101.2017.1325046
Wagner, S., Rothweiler, F., Anhorn, M. G., Sauer, D., Riemann, I., Weiss, E. C., et al. (2010). Enhanced drug targeting by attachment of an anti alphav integrin antibody to doxorubicin loaded human serum albumin nanoparticles. Biomaterials 31, 2388–2398. doi:10.1016/j.biomaterials.2009.11.093
Wallerand, H., Cai, Y., Wainberg, Z. A., Garraway, I., Lascombe, I., Nicolle, G., et al. (2010). Phospho-Akt pathway activation and inhibition depends on N-cadherin or phospho-EGFR expression in invasive human bladder cancer cell lines. Urol. Oncol. 28, 180–188. doi:10.1016/j.urolonc.2008.09.041
Wang, X., Defrances, M. C., Dai, Y., Pediaditakis, P., Johnson, C., Bell, A., et al. (2002). A mechanism of cell survival: Sequestration of Fas by the HGF receptor met. Mol. Cell 9, 411–421. doi:10.1016/s1097-2765(02)00439-2
Werner-Klein, M., Scheitler, S., Hoffmann, M., Hodak, I., Dietz, K., Lehnert, P., et al. (2018). Genetic alterations driving metastatic colony formation are acquired outside of the primary tumour in melanoma. Nat. Commun. 9, 595. doi:10.1038/s41467-017-02674-y
Whiteman, D. C., Green, A. C., and Olsen, C. M. (2016). The growing burden of invasive melanoma: Projections of incidence rates and numbers of new cases in six susceptible populations through 2031. J. Invest Dermatol 136, 1161–1171. doi:10.1016/j.jid.2016.01.035
Winder, M., and Virós, A. (2018). Mechanisms of drug resistance in melanoma. Handb. Exp. Pharmacol. 249, 91–108. doi:10.1007/164_2017_17
Winer, A., Adams, S., and Mignatti, P. (2018). Matrix metalloproteinase inhibitors in cancer therapy: Turning past failures into future successes. Mol. Cancer Ther. 17, 1147–1155. doi:10.1158/1535-7163.MCT-17-0646
Wisdom, R. (1999). AP-1: One switch for many signals. Exp. Cell Res. 253, 180–185. doi:10.1006/excr.1999.4685
Wolf, K., Wu, Y. I., Liu, Y., Geiger, J., Tam, E., Overall, C., et al. (2007). Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat. Cell Biol. 9, 893–904. doi:10.1038/ncb1616
Wong, A. L., Soo, R. A., Tan, D. S., Lee, S. C., Lim, J. S., Marban, P. C., et al. (2015). Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann. Oncol. 26, 998–1005. doi:10.1093/annonc/mdv026
Wu, J., Liu, D., Offin, M., Lezcano, C., Torrisi, J. M., Brownstein, S., et al. (2021a). Characterization and management of ERK inhibitor associated dermatologic adverse events: Analysis from a nonrandomized trial of ulixertinib for advanced cancers. Invest New Drugs 39, 785–795. doi:10.1007/s10637-020-01035-9
Wu, J. S., Jiang, J., Chen, B. J., Wang, K., Tang, Y. L., and Liang, X. H. (2021b). Plasticity of cancer cell invasion: Patterns and mechanisms. Transl. Oncol. 14, 100899. doi:10.1016/j.tranon.2020.100899
Wu, J., Ye, J., Xie, Q., Liu, B., and Liu, M. (2022). Targeting regulated cell death with pharmacological small molecules: An update on autophagy-dependent cell death, ferroptosis, and necroptosis in cancer. J. Med. Chem. 65, 2989–3001. doi:10.1021/acs.jmedchem.1c01572
Wu, K. J., Huang, J. M., Zhong, H. J., Dong, Z. Z., Vellaisamy, K., Lu, J. J., et al. (2017a). A natural product-like JAK2/STAT3 inhibitor induces apoptosis of malignant melanoma cells. PLoS One 12, e0177123. doi:10.1371/journal.pone.0177123
Wu, Y. J., Pagel, M. A., Muldoon, L. L., Fu, R., and Neuwelt, E. A. (2017b). High αv integrin level of cancer cells is associated with development of brain metastasis in athymic rats. Anticancer Res. 37, 4029–4040. doi:10.21873/anticanres.11788
Xu, W., Yang, Z., and Lu, N. (2015). A new role for the PI3K/Akt signaling pathway in the epithelial-mesenchymal transition. Cell Adh Migr. 9, 317–324. doi:10.1080/19336918.2015.1016686
Yan, S., Holderness, B. M., Li, Z., Seidel, G. D., Gui, J., Fisher, J. L., et al. (2016). Epithelial-mesenchymal expression phenotype of primary melanoma and matched metastases and relationship with overall survival. Anticancer Res. 36, 6449–6456. doi:10.21873/anticanres.11243
Yan, Y., Serra, V., Prudkin, L., Scaltriti, M., Murli, S., Rodríguez, O., et al. (2013). Evaluation and clinical analyses of downstream targets of the Akt inhibitor GDC-0068. Clin. Cancer Res. 19, 6976–6986. doi:10.1158/1078-0432.CCR-13-0978
Yang, H., Grossniklaus, H. E., Harris, W., Zhang, Q., Waller, E. K., Chakra, R., et al. (2015). Effect of Anti-angiogenic Agents Targeting Integrins ανβ3 on human uveal melanoma and vascular endothelia. Investigative Ophthalmol. Vis. Sci. 56, 5324.
Yang, S., Mcnulty, S., and Meyskens, F. L. (2004). During human melanoma progression AP-1 binding pairs are altered with loss of c-Jun in vitro. Pigment. Cell Res. 17, 74–83. doi:10.1046/j.1600-0749.2003.00114.x
Yu, K. F., Zhang, W. Q., Luo, L. M., Song, P., Li, D., Du, R., et al. (2013). The antitumor activity of a doxorubicin loaded, iRGD-modified sterically-stabilized liposome on B16-F10 melanoma cells: In vitro and in vivo evaluation. Int. J. Nanomedicine 8, 2473–2485. doi:10.2147/IJN.S46962
Yu, W., Yang, L., Li, T., and Zhang, Y. (2019). Cadherin signaling in cancer: Its functions and role as a therapeutic target. Front. Oncol. 9, 989. doi:10.3389/fonc.2019.00989
Zhang, Y., Yang, M., Ji, Q., Fan, D., Peng, H., Yang, C., et al. (2011). Anoikis induction and metastasis suppression by a new integrin αvβ3 inhibitor in human melanoma cell line M21. Invest New Drugs 29, 666–673. doi:10.1007/s10637-010-9616-y
Zaki, M. H., Nemeth, J. A., and Trikha, M. (2004). CNTO 328, a monoclonal antibody to IL-6, inhibits human tumor-induced cachexia in nude mice. Int. J. Cancer 111, 592–595. doi:10.1002/ijc.20270
Zeng, F., He, J., Jin, X., Liao, Q., Chen, Z., Peng, H., et al. (2022). FRA-1: A key factor regulating signal transduction of tumor cells and a potential target molecule for tumor therapy. Biomed. Pharmacother. 150, 113037. doi:10.1016/j.biopha.2022.113037
Zeng, Q., Mccauley, L. K., and Wang, C. Y. (2002). Hepatocyte growth factor inhibits anoikis by induction of activator protein 1-dependent cyclooxygenase-2. Implication in head and neck squamous cell carcinoma progression. J. Biol. Chem. 277, 50137–50142. doi:10.1074/jbc.M208952200
Zhang, B., Groffen, J., and Heisterkamp, N. (2007). Increased resistance to a farnesyltransferase inhibitor by N-cadherin expression in Bcr/Abl-P190 lymphoblastic leukemia cells. Leukemia 21, 1189–1197. doi:10.1038/sj.leu.2404667
Zhang, B., Li, M., Mcdonald, T., Holyoake, T. L., Moon, R. T., Campana, D., et al. (2013). Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-β-catenin signaling. Blood 121, 1824–1838. doi:10.1182/blood-2012-02-412890
Zhang, N., Xia, Y., Zou, Y., Yang, W., Zhang, J., Zhong, Z., et al. (2017). ATN-161 peptide functionalized reversibly cross-linked polymersomes mediate targeted doxorubicin delivery into melanoma-bearing C57bl/6 mice. Mol. Pharm. 14, 2538–2547. doi:10.1021/acs.molpharmaceut.6b00800
Zhang, Y., Xia, M., Jin, K., Wang, S., Wei, H., Fan, C., et al. (2018). Function of the c-Met receptor tyrosine kinase in carcinogenesis and associated therapeutic opportunities. Mol. Cancer 17, 45. doi:10.1186/s12943-018-0796-y
Zhao, K., Lu, Y., Chen, Y., Cheng, J., and Zhang, W. (2020). Dual inhibition of MAPK and JAK2/STAT3 pathways is critical for the treatment of BRAF mutant melanoma. Mol. Ther. Oncolytics 18, 100–108. doi:10.1016/j.omto.2020.06.004
Zhao, Q., Zhang, K., Li, Y., Ren, Y., Shi, J., Gu, Y., et al. (2021). OLFML2A is necessary for anti-triple negative breast cancer effect of selective activator protein-1 inhibitor T-5224. Transl. Oncol. 14, 101100. doi:10.1016/j.tranon.2021.101100
Zheng, X., Zhang, J., Li, S., Gao, X., Zhang, Y., Wang, M., et al. (2022). Low doses of niclosamide and quinacrine combination yields synergistic effect in melanoma via activating autophagy-mediated p53-dependent apoptosis. Transl. Oncol. 21, 101425. doi:10.1016/j.tranon.2022.101425
Zhong, S., Wu, B., Li, J., Wang, X., Jiang, S., Hu, F., et al. (2019). T5224, RSPO2 and AZD5363 are novel drugs against functional pituitary adenoma. Aging (Albany NY) 11, 9043–9059. doi:10.18632/aging.102372
Zhu, G., Xu, P., Guo, S., Yi, X., Wang, H., Yang, Y., et al. (2020). Metastatic melanoma cells rely on Sestrin2 to acquire anoikis resistance via detoxifying intracellular ROS. J. Invest Dermatol 140, 666–675. doi:10.1016/j.jid.2019.07.720
Zhu, L., Liu, Z., Dong, R., Wang, X., Zhang, M., Guo, X., et al. (2019a). MicroRNA-3662 targets ZEB1 and attenuates the invasion of the highly aggressive melanoma cell line A375. Cancer Manag. Res. 11, 5845–5856. doi:10.2147/CMAR.S200540
Zhu, X., Tao, X., Lu, W., Ding, Y., and Tang, Y. (2019b). Blockade of integrin β3 signals to reverse the stem-like phenotype and drug resistance in melanoma. Cancer Chemother. Pharmacol. 83, 615–624. doi:10.1007/s00280-018-3760-z
Zhu, Y., Zuo, W., Chen, L., Bian, S., Jing, J., Gan, C., et al. (2019c). Repurposing of the anti-helminthic drug niclosamide to treat melanoma and pulmonary metastasis via the STAT3 signaling pathway. Biochem. Pharmacol. 169, 113610. doi:10.1016/j.bcp.2019.08.012
Zhu, Z., Sanchez-Sweatman, O., Huang, X., Wiltrout, R., Khokha, R., Zhao, Q., et al. (2001). Anoikis and metastatic potential of cloudman S91 melanoma cells. Cancer Res. 61, 1707–1716.
Zhuang, L., Lee, C. S., Scolyer, R. A., Mccarthy, S. W., Zhang, X. D., Thompson, J. F., et al. (2007). Mcl-1, Bcl-XL and Stat3 expression are associated with progression of melanoma whereas Bcl-2, AP-2 and MITF levels decrease during progression of melanoma. Mod. Pathol. 20, 416–426. doi:10.1038/modpathol.3800750
Ziober, B. L., Chen, Y. Q., Ramos, D. M., Waleh, N., and Kramer, R. H. (1999). Expression of the alpha7beta1 laminin receptor suppresses melanoma growth and metastatic potential. Cell Growth Differ. 10, 479–490.
Zuo, J. H., Zhu, W., Li, M. Y., Li, X. H., Yi, H., Zeng, G. Q., et al. (2011). Activation of EGFR promotes squamous carcinoma SCC10A cell migration and invasion via inducing EMT-like phenotype change and MMP-9-mediated degradation of E-cadherin. J. Cell Biochem. 112, 2508–2517. doi:10.1002/jcb.23175
Keywords: melanoma, anoikis, therapy, metastasis, repurposing, targeting
Citation: Neuendorf HM, Simmons JL and Boyle GM (2023) Therapeutic targeting of anoikis resistance in cutaneous melanoma metastasis. Front. Cell Dev. Biol. 11:1183328. doi: 10.3389/fcell.2023.1183328
Received: 10 March 2023; Accepted: 14 April 2023;
Published: 26 April 2023.
Edited by:
Peter Hart, Roosevelt University College of Pharmacy, United StatesReviewed by:
Marcelo Yudi Icimoto, Federal University of São Paulo, BrazilMarika Quadri, University of Modena and Reggio Emilia, Italy
Copyright © 2023 Neuendorf, Simmons and Boyle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hannah M. Neuendorf, aGFubmFoLm5ldWVuZG9yZkBxaW1yYmVyZ2hvZmVyLmVkdS5hdQ==
†ORCID: Hannah M. Neuendorf, orcid.org/0000-0001-5238-5529; Jacinta L. Simmons, orcid.org/0000-0002-9311-1046; Glen M. Boyle, orcid.org/0000-0002-1385-529X