 Xingxing Xu†
Xingxing Xu† Nannan Sun
Nannan Sun Junbiao Chang
Junbiao Chang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cell Dev. Biol., 18 April 2023
Sec. Cancer Cell Biology
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1173432
This article is part of the Research TopicNew Targets and Strategies for Digestive System Tumor Drug Discovery and DevelopmentView all articles
Gastrointestinal malignancies are common digestive system tumor worldwide. Nucleoside analogues have been widely used as anticancer drugs for the treatment of a variety of conditions, including gastrointestinal malignancies. However, low permeability, enzymatic deamination, inefficiently phosphorylation, the emergence of chemoresistance and some other issues have limited its efficacy. The prodrug strategies have been widely applied in drug design to improve pharmacokinetic properties and address safety and drug-resistance issues. This review will provide an overview of the recent developments of prodrug strategies in nucleoside analogues for the treatment of gastrointestinal malignancies.
Gastrointestinal (GI) cancers include colorectal, gastric, oesophageal, pancreatic, and liver cancer, which continue to be a significant and common cause of mortality and morbidity worldwide. Among them, colorectal and gastric cancers rank second and fourth in global mortality respectively (Sung et al., 2021). In the United States, about 153,020 individuals will be diagnosed with colorectal cancer (Siegel et al., 2023) and 26,500 new cases of stomach cancer (American Cancer Society, 2023) in 2023. It is no doubt that radiotherapy, surgery, chemotherapy, targeted therapy, and immunotherapy all have been proven effectiveness in the treatment of GI malignancies, there still remains unmet need for new drugs and therapies to protect the patient from relapse and ensure long-term survival.
Nucleoside analogues (NAs, composed of nucleobases and ribose or deoxyribose) and nucleobases are invaluable components for various malignancies chemotherapy treatment. To induce cytotoxicity, these compounds act as antimetabolites, competing with physiological nucleosides, and interacting with a multitude of intracellular targets (Galmarini et al., 2001; Galmarini et al., 2002; Acharjee, 2020). After a series of phosphorylation, nucleoside-5′-triphosphate analogs are generated, and they can be incorporated into DNA as DNA polymerases substrates during DNA replication or excision repair synthesis. As a result, cell progression is terminated and that leads to apoptosis (Figure 1A) (Jackson and Bartek, 2009; Shelton et al., 2016). Due to the fact that cancer cells duplicate their genomes often more frequently than normal cells, it is possible for the enrichment of nucleoside-5′-triphosphate analogs in cancer cells (Parker, 2009; Counihan et al., 2018). The use of nucleoside analogue drugs, however, has some limitations: 1) NAs are structurally different from natural nucleosides, therefore, they are often inefficiently phosphorylated, which limits the active triphosphate metabolite production (Thornton et al., 2016; Fàbrega et al., 2021; Zhang et al., 2022). 2) As polar molecules, NAs and nucleobases also have poor oral bioavailability due to their low permeability (Li et al., 2008; Yan et al., 2023). 3) A number of complex mechanisms are involved in the development of NAs to avoid drug resistance and toxicity, such as the dephosphorylation of active NA metabolites by phosphatases, or the inactive metabolite of NAs by increased catabolic bioconversion. Facing the challenges of anticancer NAs research, a good way is to developing prodrugs of NAs which could circumvent some of the parent NA drug disadvantages.
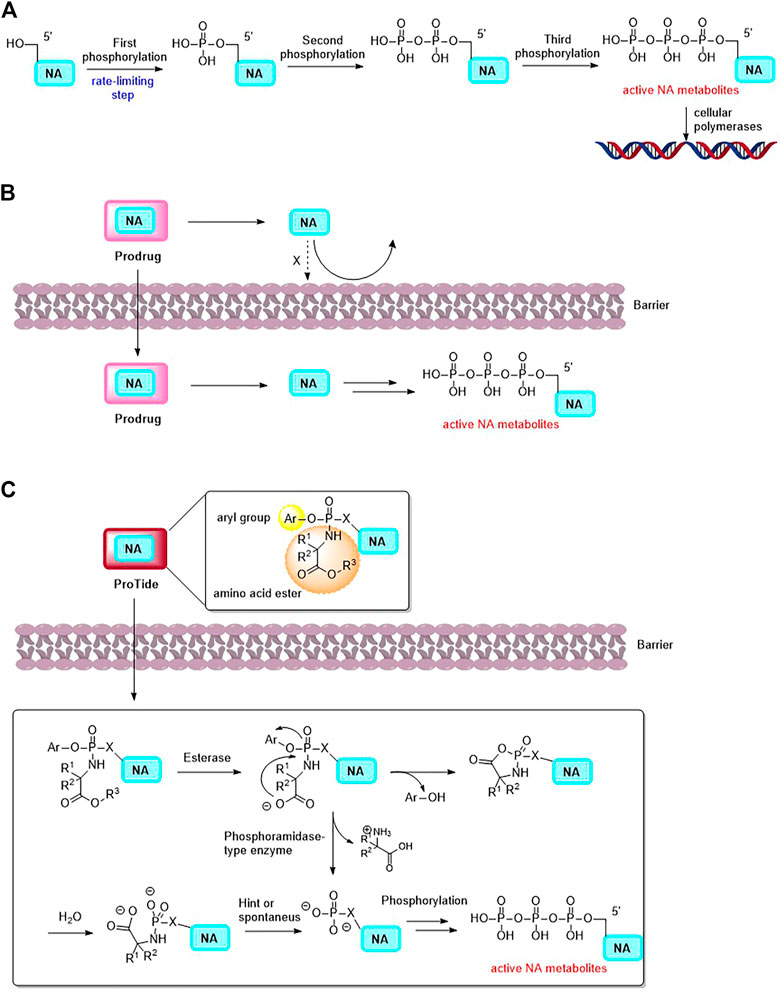
FIGURE 1. (A) The activation mechanism of NA (NA: nucleoside analogue). (B) The concept of Prodrug. Prodrug is able to deliver the parent drug (NA) to specific tissue. (C) Postulated mechanism of the mechanism of ProTides to release NA and its active metabolites.
It was successfully demonstrated that nucleotide prodrugs could overcome above issues. NA prodrugs decrease drug resistance by avoiding certain routes of catabolic bioconversion and increase nucleotides oral absorption (Thornton et al., 2016; Acharjee, 2020; Yan et al., 2023). Moreover, nucleotide prodrugs can deliver nucleotides to a specific tissue in vivo, changing their selectivity and reducing their toxicity (Figure 1B) (Li et al., 2008; Seley-Radtke and Deval, 2018). Phosphonate-containing NA prodrugs have also been investigated, as well as isosteric and isoelectronic phosphonates, to cross the monophosphorylation barrier and improve half-lives of the nucleoside monophosphonates as compared to their monophosphates (Thornton et al., 2016). Among these prodrug approaches, the ProTide (Pro-Nucleotide, the NA monophosphate or monophosphonate groups are masked by aromatic groups and amino acid esters, which could be cleaved off inside cells to release free nucleoside monophosphate and monophosphonate. Figure 1C) prodrug technology is notable, that was pioneered by Prof. Chris McGuigan (Cardiff, United Kingdom) (Mehellou et al., 2009; Mehellou, 2016; Mehellou et al., 2018). A number of anticancer NA prodrugs have been developed using ProTide technology. In this review, focusing on the chemical structures and the application of prodrug strategies, we discuss recent developments in NA prodrugs for GI malignancies treatment.
Chemotherapy is still the main treatment for GI malignancies. In current guidelines, perioperative chemotherapy or postoperative chemotherapy plus chemoradiation are listed as preferred approaches, although chemotherapy after surgery is also an option (Joshi and Badgwell, 2021). Nucleoside analogues, as 5-fluorouracil (5-FU) and gemcitabine, represent an integral component of recommended first-line GI malignancies treatment regimens. Some novel NAs also demonstrate potentials in GI cancer. Azvudine is one of these NAs, a notable cytidine analogue having both anti-HIV and anti-SARS-CoV-2 activities, but also having anticancer activities. The azvudine treated gastric cancer xenografts mouse (SGC7901) demonstrated considerable growth inhibition activity in a dose-dependent manner with lower toxicity (the antitumor effect of 2.0 mg/kg azvudine is comparable with 400 mg/kg capecitabine) (Wang et al., 2011; Fayzullina et al., 2022). Though NAs have showed their anticancer potency, the emerging issues, like drug resistance and low drug concentration at the tumor site, block the NAs application of anti-GI cancer. Prodrug strategies of NAs, particularly the ProTide approach, could address the emerging problems with promising future in oncology. The prodrug of 5-FU and gemcitabine are the most advanced NA prodrugs for the treatment of GI malignancies.
5-FU is an essential nucleobase which has been frequently administered to GI cancer patients since 1957 (Verma et al., 2022) (Table 1). Bearing a fluorine atom at position 5 of uracil, 5-FU is catalyzed by the corresponding enzyme as active metabolites that could incorporate into RNA (5-FU-5′-triphosphate, FUTP) and DNA (2′-deoxyribose-5-FU-5′-triphosphate, FdUTP) and inhibit the nucleotide synthetic enzyme thymidylate synthase (Longley et al., 2003; Sethy and Kundu, 2021). Although the efficacy of 5-FU makes it one of the most widely used chemotherapy agents, the phosphorylation of 5-FU in the digestive tract causing myelosuppression and gastrointestinal disorders, restricted cellular uptake and complex enzymatic hydrolysis processes directly contribute to drug resistance, the poor oral bioavailability limits the administration mode of 5-FU, it has a short half-life (t1/2: 8–20 min) and is rapidly eliminated from the plasma (Sethy and Kundu, 2021). Besides, intravenous injection brings the risk of venous thrombosis or infection round the catheter (Grem, 2000; More et al., 2021). Thus, developing prodrugs of 5-FU that diminish or circumvent some of these disadvantages is a major challenge.
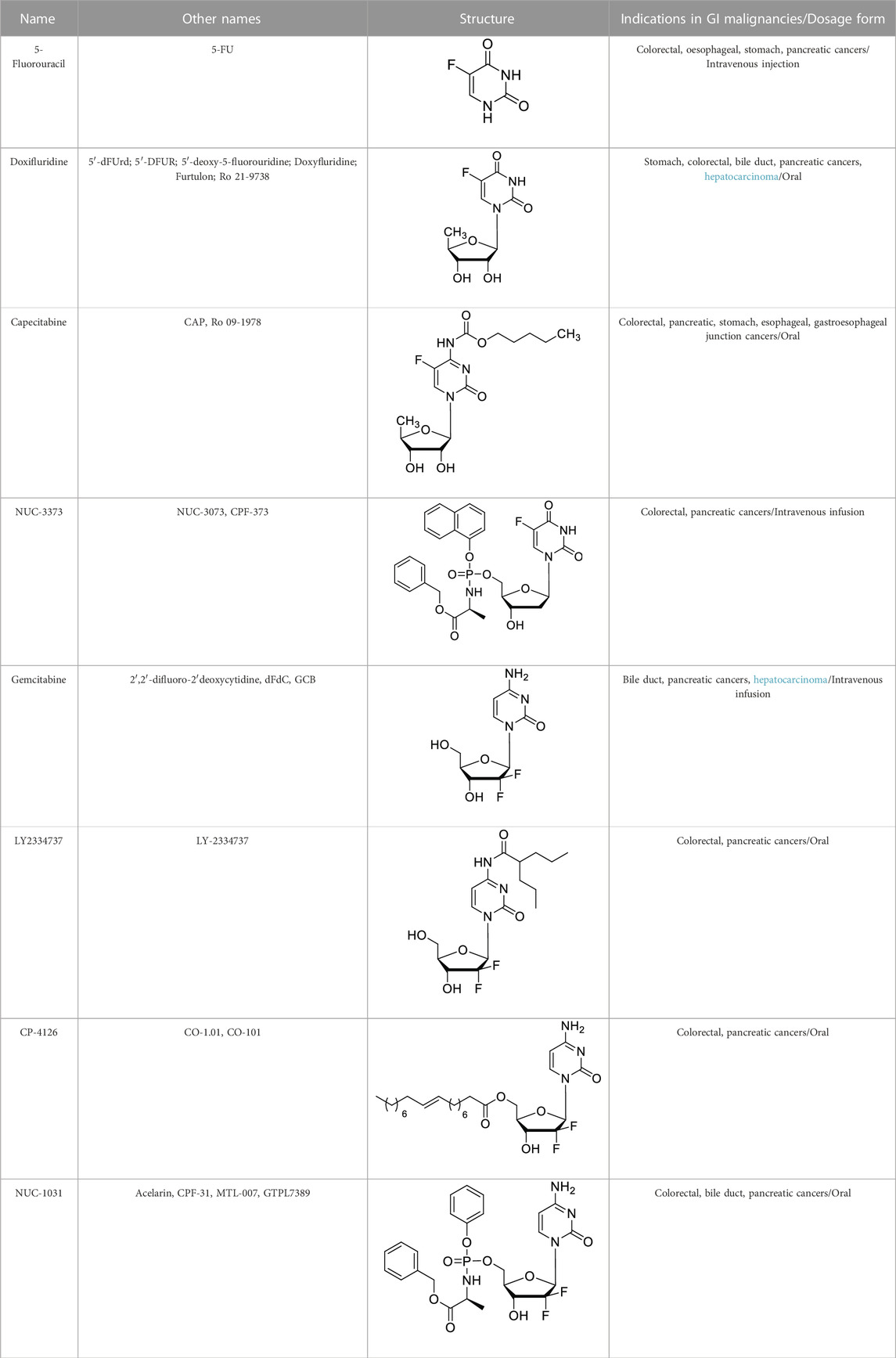
TABLE 1. The structures of NAs for the treatment of GI malignancies.
5′-Deoxy-5-fluorouridine (5′dFUrd, doxifluridine, Table 1), an orally available prodrug of 5-FU, was synthesized by Cook et al. in 1979 (Cook et al., 1979). To avoid degradation in gut and phosphorylation in digestive tract, researchers at Roche used prodrug strategies by adding a pseudo pentose is bonded in position 1 of 5-FU to develop an orally available prodrug doxifluridine (Zampino et al., 1999). As missing the 5′-hydroxyl groups, doxifluridine cannot be phosphorylated and thus cannot be directly metabolized in DNA or RNA synthesis. In vivo, doxifluridine is converted to 5-FU by either thymidine phosphorylase or pyrimidine-nucleoside phosphorylase (Kono et al., 1983; Nishioka et al., 2007). Pyrimidine nucleoside phosphorylase is highly expressed in patients with colorectal cancer and esophageal squamous cell carcinoma, and especially in tumor tissues such as esophageal, pancreatic, and hepatic, indicating a higher tumor specificity and better safety of doxifluridine than that of 5-FU (Taguchi, 1987; Noguchi et al., 2003; Ogata et al., 2007).
At the beginning of developing doxifluridine, intravenous administration of doxifluridine can produce comparable tumor-free survival while the systemic exposure to the cytotoxic metabolite 5-fluorouracil is minimized. Then the first clinical trials of doxifluridine was by i.v. administration and has indicated good activity for colorectal cancers, however, toxic effects on the nervous system and the cardiovascular system prevents this mode of administration (Van Der Heyden et al., 1999). The oral bioavailability of doxifluridine was 34%–47%, which was more satisfactory than that of 5-FU, and with a comparably show elimination half-life (t1/2 of 32–45 min) (Van Der Heyden et al., 1999; Vivian and Polli, 2014). Besides, oral administration of doxifluridine can avoid the neurological and cardiac complications caused by i.v. administration. Because gastrointestinal tracts rich in thymidine phosphorylase, the dose-limiting toxicity of orally dosed doxifluridine was found in human intestinal tract (nausea, diarrhea) (Lamont and Schilsky, 1999). Doxifluridine is used as a cytostatic agent in chemotherapy in several Asian countries including Japan, China and South Korea. In clinical trials, doxifluridine has also been administered intravenously either as a monotherapy or in combination with mitomycin-C and cisplatin.
To circumvent the unacceptable gastrointestinal toxicity of doxifluridine, researchers at Roche continually developed a series of N4-substituted 5′-deoxy-5-fluorocytidine derivatives, which could be hydrolyzed to doxifluridine and 5-FU. As a result, N4-pentyloxycarbonyl-5′-deoxy-5-fluorocytidine (capecitabine, CAP, Table 1) stood out by such prodrug strategy as an orally chemotherapeutic drug that was FDA-approved for the treatment of colorectal cancer, pancreatic adenocarcinoma, stomach cancer, esophageal cancer, or gastroesophageal junction cancer (Siddiqui et al., 2019; Pouya et al., 2021; Ge et al., 2023). As mentioned above, the gastrointestinal toxicity of doxifluridine is attributed to the liberation of 5-FU in intestine tract under the action of thymidine phosphorylase, capecitabine was thus designed as a prodrug of doxifluridine that could not be the substrate for thymidine phosphorylase in the intestine with an oral bioavailability of about 100%, Cmax of 3.9 mg/L, tmax of 1.5–2 h, and AUC of 5.96 mg·h/L (Walko and Lindley, 2005; Rehman et al., 2022). Capecitabine is rapidly and intently absorbed after oral administration, then it is cleaved by liver carboxylesterases after passing through the intestinal mucosa and catalyzed by cytidine deaminase to form doxifluridine in either liver or tumor, then capecitabine is subsequently converted to 5-FU by thymidine phosphorylase (Ishitsuka, 2003). Diarrhea, the typical toxicity issue caused by doxifluridine, could be well avoid by capecitabine because of the intact across of the gastrointestinal barrier.
Comparing to doxifluridine or 5-FU, capecitabine was highly effective in CXF280, HCT116 and COLO205 human colon cancer xenograft models with higher levels of parent drug 5-FU in tumor than in plasma (114- to 209-fold) and muscle (21.6-fold) (Ishikawa et al., 1998). In colorectal cancer patients, after capecitabine was orally administered with 1,255 mg/m2 of twice daily over 5–7 days, the concentration of parent drug 5-FU was on average 3.2 times higher in primary colorectal tumors than in adjacent healthy tissue, 21 times greater than in plasma (Li et al., 2023). Clinical studies showed that a statistically significant improvement in the overall response rates was observed and the safety profile was in favor of capecitabine (Beenet, 2021; Bryson et al., 2021). Capecitabine has been approved by FDA to be used alone or with other drugs to treat GI cancers and is now commercialized in Europe as a monotherapy for first-line treatment of metastatic colorectal cancer. In brief, using prodrug strategy, capecitabine was developed with increased concentration at the tumor site and decreased concentration in healthy tissues with a consequent reduction in systemic toxicity.
Though the prodrugs of 5-FU have achieved clinical success as anticancer agents, acquired drug resistance have been observed because of reduced levels of the activating enzyme (phosphorylating FdUTP), overexpression of thymidylate synthase (the main target for 5-FU and FUDR), upregulation of the degradative enzyme thymidine phosphorylase (TP) and reduced transporter-mediated entry of nucleoside/nucleobase (5-FU or FdUTP) into cells (Parker, 2009; Kumar et al., 2022). Nucleotides could overcome these matters, but since nucleotides are poorly membrane soluble and readily dephosphorylated, a better approach for solving this drug resistance is to dissimulate the monophosphate as a phosphate prodrug. In 2011, McGuigan et al. reported phosphate prodrugs of 5-FU and FUDR by phosphorochloridate chemistry with variation in the aryl, ester, and amino acid regions (McGuigan et al., 2011). NUC-3373 is one of the phosphoramidate prodrugs (Table 1).
The cytotoxic activity of NUC-3373 can be achieved without thymidine kinase in TK-deficient tumor cells, and NUC-3373 remains resistant to degradation by catabolic enzymes including TP and dihydropyrimidine dehydrogenase (DPD) (Vande Voorde et al., 2011). NUC-3373 produced more FdUMP (active metabolite of 5-FU, 5-fluoro-2-deoxyuridine monophosphate) than 5-FU (366-fold) in human colorectal cancer cell line HT29 and NUC-3373 showed greater tumor reduced volume than 5-FU in HT29 xenograft studies (Ghazaly et al., 2017). PK studies demonstrated that NUC-3373 has a long plasma half-life (t1/2 = 9.7 h), which is significantly improved compared to 5-FU. A Phase II study of NUC-3373 in combination with standard agents used in colorectal cancer treatment (ClinicalTrials.gov Identifier: NCT05678257) is ongoing and a Phase I clinical trial for treatment of advanced solid tumors has been complete.
Gemcitabine is a pyrimidine nucleoside analog (2′,2′-difluoro-2′deoxycytidine, Table 1), which isoriginally synthesized by Hertel and colleagues at Eli Lilly (Hertel et al., 1990). Gemcitabine is another useful chemotherapy for GI cancer, especially is used as a first-line treatment alone for pancreatic cancer. Gemcitabine in combination with cisplatin is currently being investigated as an adjuvant therapy for biliary tract cancers (Oh et al., 2022; Beutel and Halbrook, 2023; Ioka et al., 2023). But there are still challenges associated with gemcitabine’s long-term use in cancer therapy. The main challenges include the inactive deaminated metabolite 2′,2′-difluoro-2′-deoxyuridine (dFdU), which is deaminated by cytidine deaminase at the N4 position of the cytidine base in the serum, and dFdU-5′-monophoshate, which is deaminated by deoxycytdine deaminase in the cell (Serdjebi et al., 2015; Shelton et al., 2016; Miao et al., 2020). Prodrug strategies have also been applied for developing novel NAs based on gemcitabine.
LY2334737 was designed by blocking the deamination site at the N4 position of gemcitabine (Table 1). In 2009, Bender and collaborators introduced a valproic acid to the N4 position of the cytosine to improve the orally bioavailability of gemcitabine (Bender et al., 2009; Zhou et al., 2018). The enzymatic stability of LY2334737 is remarkable. A study showed that LY2334737 is insensitive about many hydrolases, only carboxylesterase-2 hydrolyzed LY2334737 to gemcitabine and required long incubation periods of 0.5–2 h (Pratt et al., 2013). Prodrugs with an amide linkage to valproic acid, instead of ester linkages, are likely to exhibit slow cleavage rates. With oral dosing, this prodrug passes through the intestinal epithelium and is absorbed intact and gemcitabine is formed slowly in the body. As in a mice PK study, orally administered LY2334737, the systemic exposure (AUC) of gemcitabine was 778 ng·h/mL with tmax of 1 h, which was higher and longer compared with the value that was under the same molar dose of oral gemcitabine (AUC of 536 ng·h/mL, tmax = 0.5 h) (Bender et al., 2009). Additionally, mice treated with LY2334737 had a less than half ratio of dFdU to gemcitabine than mice given an equal dose of gemcitabine, demonstrating the possibility of N4-modification strategy for new nucleoside-based prodrug discovery.
CP-4126 (CO-1.01, Table 1) is a 5′-elaidic acid ester prodrug of gemcitabine developed by Clavis Pharma. The fatty acid ester chain could increase the ability to diffuse through the plasma membrane and avoid the resulting drug resistance. As a result of this modification, CP-4126 is a transporter-independent analog of gemcitabine, entering cells primarily without hENT1 (Galmarini et al., 2009; Alexander et al., 2016). Besides, the 5′-elaidic acid ester of CP-4126 could effectively protected gemcitabine from deamination (Bergman et al., 2011). Thus, it was suggested that CP-4126 had efficacy as a second-line treatment in gemcitabine-resistant pancreatic adenocarcinoma patients with negative tumor hENT1 expression. Safety and efficacy of CP-4126 have been evaluated in several Phase I/II clinical trials, including metastatic pancreatic adenocarcinoma (ClinicalTrials.gov Identifier: NCT01124786), gemcitabine-resistant pancreatic adenocarcinoma (ClinicalTrials.gov Identifier: NCT01233375) and other advance solid tumors (Stuurman et al., 2013). Whereas, the primary endpoint of this study was not met, and no efficacy signal was identified for CP-4126 in treating pancreatic adenocarcinoma with progressive metastatic disease (Li et al., 2014).
Though 5′-ester prodrugs CP-4126 improved metabolic properties, it is still failed to show superiority over gemcitabine during clinical trials. In 2014, McGuigan’s research team used the ProTide technology to design a series of 5′-phosphoramidate gemcitabine prodrugs in order to overcome the mechanisms of cancer cell resistance that limit the clinical treatment of gemcitabine, within these agents, NUC-1031 is the most potent one (Table 1) (Slusarczyk et al., 2014). NUC-1031 could avert gemcitabine resistance mainly benefit from its unique ProTide characteristic. First, NUC-1031 effectively avoid the deamination of cytidine deaminase because of the presence of the phosphoramidate moiety on the 5′-O-position (Kapacee et al., 2020). Besides, this ProTide was lipophilic enough for passive diffusion into cells and the cellular uptake of NUC-1031 was not dependent on nucleoside transporters (McNamara et al., 2020). In addition, the 5′-phosphoramidate structure of the ProTide is able to bypass the rate-limiting step of the monophosphorylation by deoxycytidine kinase which is downregulated in gemcitabine-resistant cancer cells (Boyd et al., 2020). The pharmacokinetic analysis of NUC-1031 revealed that it had a significantly longer half-life than gemcitabine, with a plasma t1/2 of 9.7 h (Kapacee et al., 2020). It was also observed that NUC-1031 was rapidly absorbed intracellularly by peripheral blood mononuclear cells. NUC-1031 (Cmax was 764 pmol/106 cells/h with a tmax of 20 min) will be successfully converted to the active moiety dFdCTP with a Cmax of 727.5 pmol/106 cells at the 500 mg/m2 equivalent dose after 30 min post end of infusion. NUC-1031 also demonstrated potent intracellular concentrations of the active gemcitabine triphosphate metabolite that was 13 times higher than achieved by gemcitabine (Mohanty et al., 2018). In terms of safety and tolerability, NUC-1031 was comparable to gemcitabine, with similar side effects and serious adverse events (SAEs) (Kapacee et al., 2020).
NUC-1031 was studied in vivo, particularly in gemcitabine resistant pancreatic cancers. After administration of NUC-1031 by 7 days, the tumor size reduced faster and more significantly than that in dosed gemcitabine, and NUC-1031 showed good tolerance with less than 4% reduction in body weight over the treatment (Slusarczyk et al., 2014). The clinical study of NUC-1031 in metastatic pancreatic carcinoma (a Phase III, open label, multicentre randomised clinical study comparing NUC-1031 with gemcitabine in patients with metastatic pancreatic carcinoma, ClinicalTrials.gov Identifier: NCT03610100) is attractive, but now, it is suspended to recruitment following a review on efficacy and toxicities.
Developing NAs is important for the discovery of chemotherapy agents. Prodrug strategies to discover new NAs are widely used, particularly the ProTide strategy. With the ProTide technology, novel nucleoside monophosphate analogues have been discovered and applied to the clinic in recent years, and there have been two FDA-approved ProTides (tenofovir alafenamide and sofosbuvir) as well-known antiviral drugs. This prodrug technology has already been proven effective in delivering the activated parental drug into cells avoiding drug resistance and toxicity. However, it is important to note that nucleoside monophosphate analogues with their stereocenter at the phosphorus atom may cause different biological activities and potencies of enantiomers or diastereomers (Slusarczyk et al., 2021). Developing diastereoselective synthetic and separation methods for ProTide is therefore necessary. In summary, in light of the applications of prodrug technology, it is possible that new NA ProTides will be developed for treating cancers, especially GI malignancies, with improved efficacy and avoidable drug resistance.
NS and JC conceived the idea. NS, XX, ZL, and XY drafted the manuscript and prepared the artworks. NS and JC supervised the whole work.
This work was supported by the Natural National Science Foundation of China (Grant No: 82003577; 82130103), China Postdoctoral Science Foundation (Grant No: 2022M722893), the Key Science and Technology Project of Henan Province (222102310275).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acharjee, N. (2020). Unravelling the regio-and stereoselective synthesis of bicyclic N, O-nucleoside analogues within the molecular electron density theory perspective. J. Struct. Chem. 31 (6), 2147–2160. doi:10.1007/s11224-020-01569-x
Alexander, P., Kucera, G., and Pardee, T. S. (2016). Improving nucleoside analogs via lipid conjugation: Is fatter any better? Crit. Rev. Oncol. Hematol. 100, 46–56. doi:10.1016/j.critrevonc.2016.01.015
American Cancer Society (2023). Key statistics about stomach cancer. Available at: https://www.cancer.org/cancer/stomach-cancer/about/key-statistics.html (Accessed April 1, 2023).
Beenet, L. (2021). Capecitabine treatment: A safe and effective therapy in the field of oncology. Clin. Colorectal Cancer 20 (3), e194. doi:10.1016/j.clcc.2021.04.003
Bender, D. M., Bao, J., Dantzig, A. H., Diseroad, W. D., Law, K. L., Magnus, N. A., et al. (2009). Synthesis, crystallization, and biological evaluation of an orally active prodrug of gemcitabine. J. Med. Chem. 52 (22), 6958–6961. doi:10.1021/jm901181h
Bergman, A. M., Adema, A. D., Balzarini, J., Bruheim, S., Fichtner, I., Noordhuis, P., et al. (2011). Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Invest. New Drugs 29 (3), 456–466. doi:10.1007/s10637-009-9377-7
Beutel, A. K., and Halbrook, C. J. (2023). Barriers and opportunities for gemcitabine in pancreatic cancer therapy. Am. J. Physiol. Cell. Physiol. 324 (2), C540–c552. doi:10.1152/ajpcell.00331.2022
Boyd, L. N. C., Peters, G. J., Kazemier, G., and Giovannetti, E. (2020). NUC-1031 in biliary tract cancer: From bench to bedside and back? Cancer Chemother. Pharmacol. 85 (6), 1011–1014. doi:10.1007/s00280-020-04080-6
Bryson, E., Sakach, E., Patel, U., Watson, M., Hall, K., Draper, A., et al. (2021). Safety and efficacy of 7 Days on/7 Days off versus 14 Days on/7 Days off schedules of capecitabine in patients with metastatic colorectal cancer: A retrospective review. Clin. Colorectal Cancer 20 (2), 153–160. doi:10.1016/j.clcc.2020.12.002
Cook, A. F., Holman, M. J., Kramer, M. J., and Trown, P. W. (1979). Fluorinated pyrimidine nucleosides. 3. Synthesis and antitumor activity of a series of 5'-deoxy-5-fluoropyrimidine nucleosides. J. Med. Chem. 22 (11), 1330–1335. doi:10.1021/jm00197a010
Counihan, J. L., Grossman, E. A., and Nomura, D. K. (2018). Cancer metabolism: Current understanding and therapies. Chem. Rev. 118 (14), 6893–6923. doi:10.1021/acs.chemrev.7b00775
Fàbrega, C., Clua, A., Eritja, R., and Aviñó, A. (2021). Oligonucleotides carrying nucleoside antimetabolites as potential prodrugs. Curr. Med. Chem. 30, 1304–1319. doi:10.2174/0929867328666211129124039
Fayzullina, D., Kharwar, R. K., Acharya, A., Buzdin, A., Borisov, N., Timashev, P., et al. (2022). FNC: An advanced anticancer therapeutic or just an underdog? Front. Oncol. 12, 820647. doi:10.3389/fonc.2022.820647
Galmarini, C. M., Mackey, J. R., and Dumontet, C. (2002). Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 3 (7), 415–424. doi:10.1016/s1470-2045(02)00788-x
Galmarini, C. M., Mackey, J. R., and Dumontet, C. (2001). Nucleoside analogues: Mechanisms of drug resistance and reversal strategies. Leukemia 15 (6), 875–890. doi:10.1038/sj.leu.2402114
Galmarini, C. M., Myhren, F., and Sandvold, M. L. (2009). CP-4055 and CP-4126 are active in ara-C and gemcitabine-resistant lymphoma cell lines. Br. J. Haematol. 144 (2), 273–275. doi:10.1111/j.1365-2141.2008.07467.x
Ge, C., Huang, X., Zhang, S., Yuan, M., Tan, Z., Xu, C., et al. (2023). In vitro co-culture systems of hepatic and intestinal cells for cellular pharmacokinetic and pharmacodynamic studies of capecitabine against colorectal cancer. Cancer Cell. Int. 23 (1), 14. doi:10.1186/s12935-023-02853-6
Ghazaly, E., Woodcock, V. K., Spilipoulou, P., Spiers, L., Moschandreas, J., Griffiths, L., et al. (2017). Interim pharmacokinetic (PK) and pharmacodynamic (PD) data from the first-in-human study of NUC-3373, a pyrimidine nucleotide analogue, in patients with advanced solid tumors. Ann. Oncol. 28, v128. doi:10.1093/annonc/mdx367.019
Grem, J. L. (2000). 5-Fluorouracil: Forty-plus and still ticking. A review of its preclinical and clinical development. Invest. New Drugs 18 (4), 299–313. doi:10.1023/a:1006416410198
Hertel, L. W., Boder, G. B., Kroin, J. S., Rinzel, S. M., Poore, G. A., Todd, G. C., et al. (1990). Evaluation of the antitumor activity of gemcitabine (2',2'-difluoro-2'-deoxycytidine). Cancer Res. 50 (14), 4417–4422.
Ioka, T., Kanai, M., Kobayashi, S., Sakai, D., Eguchi, H., Baba, H., et al. (2023). Randomized phase III study of gemcitabine, cisplatin plus S-1 versus gemcitabine, cisplatin for advanced biliary tract cancer (KHBO1401- MITSUBA). J. Hepatobiliary Pancreat. Sci. 30 (1), 102–110. doi:10.1002/jhbp.1219
Ishikawa, T., Utoh, M., Sawada, N., Nishida, M., Fukase, Y., Sekiguchi, F., et al. (1998). Tumor selective delivery of 5-fluorouracil by capecitabine, a new oral fluoropyrimidine carbamate, in human cancer xenografts. Biochem. Pharmacol. 55 (7), 1091–1097. doi:10.1016/s0006-2952(97)00682-5
Ishitsuka, H. (2003). “Discovery and preclinical pharmacology of capecitabine,” in Fluoropyrimidines in cancer therapy (New Jersey: Humana Totowa), 249–259. doi:10.1007/978-1-59259-337-8_20
Jackson, S. P., and Bartek, J. (2009). The DNA-damage response in human biology and disease. Nature 461 (7267), 1071–1078. doi:10.1038/nature08467
Joshi, S. S., and Badgwell, B. D. (2021). Current treatment and recent progress in gastric cancer. CA Cancer J. Clin. 71 (3), 264–279. doi:10.3322/caac.21657
Kapacee, Z. A., Knox, J. J., Palmer, D., Blagden, S. P., Lamarca, A., Valle, J. W., et al. (2020). NUC-1031, use of ProTide technology to circumvent gemcitabine resistance: Current status in clinical trials. Med. Oncol. 37 (7), 61. doi:10.1007/s12032-020-01386-6
Kono, A., Hara, Y., Sugata, S., Karube, Y., Matsushima, Y., and Ishitsuka, H. (1983). Activation of 5'-deoxy-5-fluorouridine by thymidine phosphorylase in human tumors. Chem. Pharm. Bull. (Tokyo) 31 (1), 175–178. doi:10.1248/cpb.31.175
Kumar, A., Singh, A. K., Singh, H., Thareja, S., and Kumar, P. (2022). Regulation of thymidylate synthase: An approach to overcome 5-FU resistance in colorectal cancer. Med. Oncol. 40 (1), 3. doi:10.1007/s12032-022-01864-z
Lamont, E. B., and Schilsky, R. L. (1999). The oral fluoropyrimidines in cancer chemotherapy. Clin. Cancer Res. 5 (9), 2289–2296.
Li, D., Pant, S., Ryan, D. P., Laheru, D., Bahary, N., Dragovich, T., et al. (2014). A phase II, open-label, multicenter study to evaluate the antitumor efficacy of CO-1.01 as second-line therapy for gemcitabine-refractory patients with stage IV pancreatic adenocarcinoma and negative tumor hENT1 expression. Pancreatology 14 (5), 398–402. doi:10.1016/j.pan.2014.07.003
Li, F., Maag, H., and Alfredson, T. (2008). Prodrugs of nucleoside analogues for improved oral absorption and tissue targeting. J. Pharm. Sci. 97 (3), 1109–1134. doi:10.1002/jps.21047
Li, X., Cui, D., Xiong, J., Dang, Q., Wen, Q., Yan, M., et al. (2023). Pharmacokinetics and comparative bioavailability of test or reference capecitabine and discrepant pharmacokinetics among various tumors in Chinese solid cancer patients. Clin. Pharmacol. Drug Dev. 12 (3), 324–332. doi:10.1002/cpdd.1202
Longley, D. B., Harkin, D. P., and Johnston, P. G. (2003). 5-fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 3 (5), 330–338. doi:10.1038/nrc1074
McGuigan, C., Murziani, P., Slusarczyk, M., Gonczy, B., Vande Voorde, J., Liekens, S., et al. (2011). Phosphoramidate ProTides of the anticancer agent FUDR successfully deliver the preformed bioactive monophosphate in cells and confer advantage over the parent nucleoside. J. Med. Chem. 54 (20), 7247–7258. doi:10.1021/jm200815w
McNamara, M. G., Goyal, L., Doherty, M., Springfeld, C., Cosgrove, D., Sjoquist, K. M., et al. (2020). NUC-1031/cisplatin versus gemcitabine/cisplatin in untreated locally advanced/metastatic biliary tract cancer (NuTide:121). Future Oncol. 16 (16), 1069–1081. doi:10.2217/fon-2020-0247
Mehellou, Y., Balzarini, J., and McGuigan, C. (2009). Aryloxy phosphoramidate triesters: A technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 4 (11), 1779–1791. doi:10.1002/cmdc.200900289
Mehellou, Y., Rattan, H. S., and Balzarini, J. (2018). The ProTide prodrug technology: From the concept to the clinic. J. Med. Chem. 61 (6), 2211–2226. doi:10.1021/acs.jmedchem.7b00734
Miao, H., Chen, X., and Luan, Y. (2020). Small molecular gemcitabine prodrugs for cancer therapy. Curr. Med. Chem. 27 (33), 5562–5582. doi:10.2174/0929867326666190816230650
Mohanty, A. J., Sishc, B. J., Falls, K. C., Heer, C. D., Spitz, D. R., and Story, M. D. (2018). Abstract 2929: GC4419 enhances the response of non-small cell lung carcinoma cell lines to cisplatin and cisplatin plus radiation through a ROS-mediated pathway. Cancer Res. 78 (13), 2929. doi:10.1158/1538-7445.Am2018-2929
More, L. A., Lane, S., and Asnani, A. (2021). 5-FU cardiotoxicity: Vasospasm, myocarditis, and sudden death. Curr. Cardiol. Rep. 23 (3), 17. doi:10.1007/s11886-021-01441-2
Nishioka, M., Miyamoto, H., Kurita, N., Higashijima, J., Yoshikawa, K., Miyatani, T., et al. (2007). Pyrimidine nucleoside phosphorylase and dihydropyrimidine dihydrogenase activities as predictive factors for the efficacy of doxifluridine together with mitomycin C as adjuvant chemotherapy in primary colorectal cancer. Hepato-gastroenterology 54 (76), 1089–1093.
Noguchi, T., Moriyama, H., Wada, S., Takeno, S., Kimura, Y., Uchida, Y., et al. (2003). High level concentration of pyrimidine nucleoside phosphorylase in esophageal squamous cell carcinoma but no correlation with clinicopathological parameters. Dis. Esophagus 16 (4), 307–311. doi:10.1111/j.1442-2050.2003.00352.x
Ogata, Y., Sasatomi, T., Mori, S., Matono, K., Ishibashi, N., Akagi, Y., et al. (2007). Significance of thymidine phosphorylase in metronomic chemotherapy using CPT-11 and doxifluridine for advanced colorectal carcinoma. Anticancer Res. 27 (4), 2605–2611.
Oh, D. Y., Lee, K. H., Lee, D. W., Yoon, J., Kim, T. Y., Bang, J. H., et al. (2022). Gemcitabine and cisplatin plus durvalumab with or without tremelimumab in chemotherapy-naive patients with advanced biliary tract cancer: An open-label, single-centre, phase 2 study. Lancet Gastroenterol. Hepatol. 7 (6), 522–532. doi:10.1016/s2468-1253(22)00043-7
Parker, W. B. (2009). Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 109 (7), 2880–2893. doi:10.1021/cr900028p
Pouya, F. D., Rasmi, Y., Camci, I. Y., Tutar, Y., and Nemati, M. (2021). Performance of capecitabine in novel combination therapies in colorectal cancer. J. Chemother. 33 (6), 375–389. doi:10.1080/1120009x.2021.1920247
Pratt, S. E., Durland-Busbice, S., Shepard, R. L., Heinz-Taheny, K., Iversen, P. W., and Dantzig, A. H. (2013). Human carboxylesterase-2 hydrolyzes the prodrug of gemcitabine (LY2334737) and confers prodrug sensitivity to cancer cells. Clint. Cancer Res. 19 (5), 1159–1168. doi:10.1158/1078-0432.CCR-12-1184
Rehman, U., Sarfraz, R. M., Mahmood, A., Akbar, S., Altyar, E. A., Khinkar, R. M., et al. (2022). pH responsive hydrogels for the delivery of capecitabine: Development, optimization and pharmacokinetic studies. Gels 8 (12), 775. doi:10.3390/gels8120775
Seley-Radtke, K., and Deval, J. (2018). Advances in antiviral nucleoside analogues and their prodrugs. Antivir. Chem. Chemother. 26, 2040206618781410. doi:10.1177/2040206618781410
Serdjebi, C., Gagnière, J., Desramé, J., Fein, F., Guimbaud, R., François, E., et al. (2015). FFCD-1004 clinical trial: Impact of cytidine deaminase activity on clinical outcome in gemcitabine-monotherapy treated patients. PLoS One 10 (8), e0135907. doi:10.1371/journal.pone.0135907
Sethy, C., and Kundu, C. N. (2021). 5-Fluorouracil (5-FU) resistance and the new strategy to enhance the sensitivity against cancer: Implication of DNA repair inhibition. Biomed. Pharmacother. 137, 111285. doi:10.1016/j.biopha.2021.111285
Shelton, J., Lu, X., Hollenbaugh, J. A., Cho, J. H., Amblard, F., and Schinazi, R. F. (2016). Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs. Chem. Rev. 116 (23), 14379–14455. doi:10.1021/acs.chemrev.6b00209
Siddiqui, N. S., Godara, A., Byrne, M. M., and Saif, M. W. (2019). Capecitabine for the treatment of pancreatic cancer. Expert Opin. Pharmacother. 20 (4), 399–409. doi:10.1080/14656566.2018.1560422
Siegel, R. L., Wagle, N. S., Cercek, A., Smith, R. A., and Jemal, A. (2023). Colorectal cancer statistics, 2023. CA Cancer J. Clin., 1–22. doi:10.3322/caac.21772
Slusarczyk, M., Lopez, M. H., Balzarini, J., Mason, M., Jiang, W. G., Blagden, S., et al. (2014). Application of ProTide technology to gemcitabine: A successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 57 (4), 1531–1542. doi:10.1021/jm401853a
Slusarczyk, M., Serpi, M., Ghazaly, E., Kariuki, B. M., McGuigan, C., and Pepper, C. (2021). Single diastereomers of the clinical anticancer ProTide agents NUC-1031 and NUC-3373 preferentially target cancer stem cells in vitro. J. Med. Chem. 64 (12), 8179–8193. doi:10.1021/acs.jmedchem.0c02194
Stuurman, F. E., Voest, E. E., Awada, A., Witteveen, P. O., Bergeland, T., Hals, P. A., et al. (2013). Phase I study of oral CP-4126, a gemcitabine derivative, in patients with advanced solid tumors. Invest. New Drugs 31 (4), 959–966. doi:10.1007/s10637-013-9925-z
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71 (3), 209–249. doi:10.3322/caac.21660
Thornton, P. J., Kadri, H., Miccoli, A., and Mehellou, Y. (2016). Nucleoside phosphate and phosphonate prodrug clinical candidates: Miniperspective. J. Med. Chem. 59 (23), 10400–10410. doi:10.1021/acs.jmedchem.6b00523
Van Der Heyden, S. A., Highley, M. S., De Bruijn, E. A., Tjaden, U. R., Reeuwijk, H. J., Van Slooten, H., et al. (1999). Pharmacokinetics and bioavailability of oral 5'-deoxy-5-fluorouridine in cancer patients. Br. J. Clin. Pharmacol. 47 (4), 351–356. doi:10.1046/j.1365-2125.1999.00899.x
Vande Voorde, J., Liekens, S., McGuigan, C., Murziani, P. G., Slusarczyk, M., and Balzarini, J. (2011). The cytostatic activity of NUC-3073, a phosphoramidate prodrug of 5-fluoro-2'-deoxyuridine, is independent of activation by thymidine kinase and insensitive to degradation by phosphorolytic enzymes. Biochem. Pharmacol. 82 (5), 441–452. doi:10.1016/j.bcp.2011.05.024
Verma, H., Narendra, G., Raju, B., Singh, P. K., and Silakari, O. (2022). Dihydropyrimidine dehydrogenase-mediated resistance to 5-fluorouracil: Mechanistic investigation and solution. ACS Pharmacol. Transl. Sci. 5 (11), 1017–1033. doi:10.1021/acsptsci.2c00117
Vivian, D., and Polli, J. E. (2014). Synthesis and in vitro evaluation of bile acid prodrugs of floxuridine to target the liver. Int. J. Pharm. 475 (1-2), 597–604. doi:10.1016/j.ijpharm.2014.09.014
Walko, C. M., and Lindley, C. (2005). Capecitabine: A review. Clin. Ther. 27 (1), 23–44. doi:10.1016/j.clinthera.2005.01.005
Wang, Q., Liu, X., Wang, Q., Zhang, Y., Jiang, J., Guo, X., et al. (2011). FNC, a novel nucleoside analogue inhibits cell proliferation and tumor growth in a variety of human cancer cells. Biochem. Pharmacol. 81 (7), 848–855. doi:10.1016/j.bcp.2011.01.001
Yan, V. C., Barekatain, Y., Lin, Y.-H., Satani, N., Hammoudi, N., Arthur, K., et al. (2023). Comparative pharmacology of a bis-pivaloyloxymethyl phosphonate prodrug inhibitor of enolase after oral and parenteral administration. ACS Pharmacol. Transl. Sci. 6, 245–252. doi:10.1021/acsptsci.2c00216
Zampino, M. G., Colleoni, M., Bajetta, E., Stampino, C. G., Guenzi, A., and de Braud, F. (1999). Pharmacokinetics of oral doxifluridine in patients with colorectal cancer. Tumori J. 85 (1), 47–50. doi:10.1177/030089169908500110
Zhang, Q., Peng, Y., Hou, J., Chen, Y., Liu, B., Zhang, P., et al. (2022). An O-benzyl phosphonamidate prodrug of tenofovir for the treatment of hepatitis B virus infection. J. Med. Chem. 65 (13), 9493–9505. doi:10.1021/acs.jmedchem.2c00869
Zhou, Y., Chang, Q., Wang, W., Zhang, X., Zhou, F., Sun, J., et al. (2018). Sensitive analysis and pharmacokinetic study of a novel gemcitabine carbamate prodrug and its active metabolite gemcitabine in rats using LC-ESI-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 1083, 249–257. doi:10.1016/j.jchromb.2018.03.015
Keywords: gastrointestinal malignancies, anticancer, nucleoside analogue, prodrug strategy, ProTide
Citation: Xu X, Li Z, Yao X, Sun N and Chang J (2023) Advanced prodrug strategies in nucleoside analogues targeting the treatment of gastrointestinal malignancies. Front. Cell Dev. Biol. 11:1173432. doi: 10.3389/fcell.2023.1173432
Received: 24 February 2023; Accepted: 07 April 2023;
Published: 18 April 2023.
Edited by:
Dingding Gao, Shanghai University of Traditional Chinese Medicine, ChinaReviewed by:
Jinsong Han, China Pharmaceutical University, ChinaCopyright © 2023 Xu, Li, Yao, Sun and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nannan Sun, c3VuX25uQHp6dS5lZHUuY24=; Junbiao Chang, Y2hhbmdqdW5iaWFvQHp6dS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.