 Mayuri TokunagaTakuya Imamura*
Mayuri TokunagaTakuya Imamura*- Program of Biomedical Science, Graduate School of Integrated Sciences for Life, Hiroshima University, Hiroshima, Japan
Microcephaly is characterized as a small head circumference, and is often accompanied by developmental disorders. Several candidate risk genes for this disease have been described, and mutations in non-coding regions are occasionally found in patients with microcephaly. Various non-coding RNAs (ncRNAs), such as microRNAs (miRNAs), SINEUPs, telomerase RNA component (TERC), and promoter-associated lncRNAs (pancRNAs) are now being characterized. These ncRNAs regulate gene expression, enzyme activity, telomere length, and chromatin structure through RNA binding proteins (RBPs)-RNA interaction. Elucidating the potential roles of ncRNA-protein coordination in microcephaly pathogenesis might contribute to its prevention or recovery. Here, we introduce several syndromes whose clinical features include microcephaly. In particular, we focus on syndromes for which ncRNAs or genes that interact with ncRNAs may play roles. We discuss the possibility that the huge ncRNA field will provide possible new therapeutic approaches for microcephaly and also reveal clues about the factors enabling the evolutionary acquisition of the human-specific “large brain.”
1 Introduction
Abnormal brain growth leads to aberrant brain size and developmental disorders. Microcephaly is defined as a head circumference < −2 standard deviations (SD) in humans (Whelan, 2010). Genetic mutations have been identified in half of such patients. Patients with severe microcephaly (<-3 SD) are more likely to be also have other developmental diseases such as epilepsy, cerebral palsy, autism, and intellectual disabilities simultaneously (Pirozzi et al., 2018). Numerous studies have revealed a variety of risk genes for microcephaly. For example, the assembly factor for spindle microtubules (ASPM) gene, which encodes a centrosomal protein, is one of the most frequent candidate genes for this symptom (Nicholas et al., 2009). Dysfunction of other centrosomal proteins such as WDR62, CEP135, CENPE, and MCHP1 also causes microcephaly, which indicates the importance of centrosomes for brain volume expansion in infants (Pirozzi et al., 2018). On the other hand, non-genetic factors (e.g., Zika virus infection, excessive maternal alcohol drinking, drug overdose, and malnutrition) can also be causes for such diseases (Whelan, 2010). In addition, epigenetic factors are known to be involved in abnormal brain growth phenotypes. For example, Rett syndrome, an epigenetic disease, was first described in 1966 (Rett, 1966). The syndrome appears in approximately 1 in 10,000 female births. Patients grow and develop normally until 6-8 months of age, and then gradually lose speech and hand skills and appear to have stereotypic hand movements. The head circumference growth decelerates and patients are diagnosed with microcephaly (Weng et al., 2011). This disorder is caused by mutation in X-linked methyl-CpG-binding protein 2 (MeCP2), whose protein product binds to methyl-CpG sites (Amir et al., 1999), affecting both genic and intergenic regions in the genome to modulate RNA transcription. The occurrence of the complex disease phenotypes is further supported by recent studies showing that many central nervous system (CNS) disorders are also associated with mutations in non-coding regions in the human genome (Simon-Sanchez and Singleton, 2008). Single nucleotide polymorphisms (SNPs) located within non-coding regions have occasionally been found in infants with microcephaly (Xia et al., 2017). A recent study indicated that ASPM is modulated by circular RNA and microRNA (miRNA), both of which are types of non-coding RNAs (ncRNAs) (Han et al., 2021). Therefore, better understanding of the involvement of non-coding regions in the pathogenesis of microcephaly through ncRNA transcription is needed. NcRNAs play important roles in genome transcription, RNA translation, RNA degradation, and protein scaffolding (Yan et al., 2021). For example, several miRNAs related to Feingold syndrome function in RNA interference in which the precursors of these miRNAs are transcribed by RNA polymerase Ⅱ, and then the miRNA is incorporated into the miRNA-induced silencing complex called miRISC to degrade the target mRNA (de Pontual et al., 2011), as described later. In addition to miRNA, long ncRNA (lncRNA) with size greater than 200 nt (Novikova et al., 2013) also seems to function for regulating brain size by forming a complex structure with chromatinic DNA to regulate gene expression (Chi et al., 2019). Here, we will introduce diseases with microcephaly candidate genes including those for RNA-binding proteins (RBP) and with intergenic mutations that affect the generation of ncRNAs and discuss how ncRNAs are involved in establishing the nature of human-specific “large-brain” and how RNA-involving epigenetic mechanisms can be therapeutic targets (Figure 1). In fact, there are several brain diseases that affect brain size but are not annotated as microcephaly. Since little information on ncRNAs contributing to microcephaly is available, we will also refer to ncRNAs known to be physically and/or functionally connected to brain-size-affecting diseases (e.g., autism spectrum disorder: ASD) other than known microcephaly-related diseases.
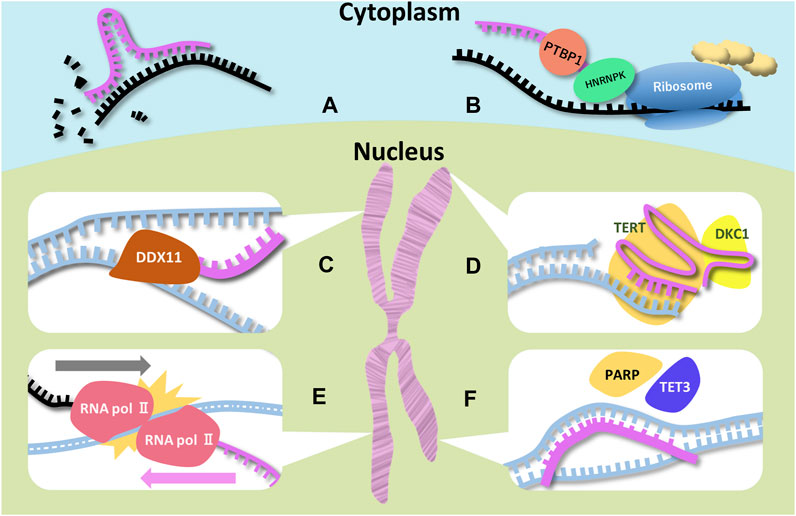
FIGURE 1. Schematic representations of function of ncRNAs and RBPs associated with microcephaly Six examples for ncRNAs (miRNA, SINEUP, DDX11-AS1 (CONCR), TERC, UBE3A-ATS, and pancRNA) are illustrated. Magenta, black, and blue strands indicate ncRNA, mRNA, and DNA respectively. (A) Degradation of the target mRNA is a miRNA function. MiRNAs have the complementary sequence of the target mRNAs. (B) Two RBPs (PTBP1 and HNRNPK) bind to SINEUP and recruit the ribosome. SINEUP upregulates translation through RBPs. (C) DDX-AS1 bind to DDX11 directly to promote its enzymatic activity. In addition, the lncRNA traps miRNA targeting DDX11 mRNA. (D) TERC is the template for telomere elongation. DKC1 is essential for the TERC stability. (E) RNA polymerases colliding is thought to lead to stopping elongation of UBE3A mRNA. (F) pancRNA recruits transcription and histone acetylation factors by changing DNA structure.
2 Cytosolic function of ncRNAs in brain diseases
2.1 Microcephaly-related inhibitory ncRNAs in the cytoplasm
Feingold syndrome is an autosomal dominant syndrome including microcephaly, short stature, and short mesophalanx of the fifth finger (brachymesophalangy). Several ncRNAs are involved in the pathogenesis of this syndrome. In many cases, the deletion of either MYCN (type 1) or MIR17HG (type 2) seems to cause this type of disease (de Pontual et al., 2011). MIR17HG generates six miRNAs, namely, miR-17, 18a, 19a, 20a, 19b-1, and 92a-1 (Mendell, 2008), which have been reported to be involved in proliferation of various tumors (Tan et al., 2022). MYCN protein seems to regulate the expression of these miRNAs by binding to the MIR17HG promoter region to upregulate miRNA expression (de Pontual et al., 2011). MiR-17-92 cluster is described as a human oncogene in several cancers (Hayashita et al., 2005) (Mu et al., 2009). The deletion of the cluster promotes apoptosis because the miRNAs target BIM (Ventura et al., 2008), which initiates the intrinsic apoptotic pathway (Sionov et al., 2015). Mice models for Feingold syndrome type 2 exhibit brachymesophalangy, small body (short stature), and microcephaly. The homozygous deletion of MIR17HG frequently leads to perinatal lethality (de Pontual et al., 2011). MIR17HG targets TGF-β receptor type 2 (TGFBR2) (Ma et al., 2016) (Mirzamohammadi et al., 2018), and cases deficient for MIR17HG are associated with excessive TGF-β signaling, which is supported by the fact that treatment with a TGF-β receptor inhibitor, LY364947, prevented the skeletal defect and microcephaly in the Feingold syndrome type 2 mouse model. GW788388, another TGF-β receptor inhibitor, and 1D11, a neutralizing antibody against TGF-β ligands, also caused similar effects (Mirzamohammadi et al., 2018).
2.2 Gene-activating ncRNAs functioning in the cytoplasm
In recent years, patients with de novo mutation of RAB11B have been described. The symptoms include absent speech, epilepsy, hypotonia, and microcephaly (Lamers et al., 2017) (Jauss et al., 2022), in spite of the fact that, in mouse, Rab11b deficiency exhibits no phenotypes (Nassari et al., 2020). This suggests that human RAB11B has acquired human-specific functions. RAB11B is a small GTPase belonging to the Rab family. Rab forms and transfers vesicles, and fuses them with the cellular membrane (Stenmark and Olkkonen, 2001). RAB11B is expressed in the brain, heart, and testis (Lai et al., 1994). Mislocalization of abnormal RAB11B due to mutations at its GTP/GDP binding pocket causes disorganized brain structures and functions (Lamers et al., 2017). Interestingly, RAB11B-AS1 is transcribed from the bidirectional RAB11B promoter to modulate RAB11B functions. RAB11B-AS1 is expressed in humans including in the brain, and functions as a “SINEUP” RNA for RAB11B, that can promote RAB11B translation (Zarantonello et al., 2021). SINEUP is a category of lncRNAs that promote translation of partly overlapping mRNAs (Zucchelli et al., 2015). This mechanism involves Polypyrimidine tract-binding protein (PTBP1), which is also known to function in alternative splicing for Filamin A (FLNA) (Zhang et al., 2016), a causative gene for microcephaly in mice, and the deregulation of this alterative splicing leads to periventricular heterotopia (PH) in human (Lian et al., 2012). In addition, PTBP1 can function together with heterogeneous nuclear ribonucleoprotein K (HNRNPK) to bind to the SINEUP RNAs to target mRNAs. These two RBPs help to recruit ribosomal subunits for enhancing the translation of the target mRNAs (Toki et al., 2020). It is noteworthy that RAB11B and RAB11B-AS1 are downregulated by CDH8 (Zarantonello et al., 2021), and therefore, known CDH8 variants may modulate the cellular level of RAB11B, leading to macrocephaly and intellectual disability (Bernier et al., 2014). Although the molecular function of RAB11B-AS1 in brain contexts is still obscure, it is possible that evolutionary acquisition of RAB11B-AS1 was actively involved in enlarging the human brain because some studies have shown an association with cancer via oncogene effects such as cell proliferation (Li et al., 2020), migration and invasion (Niu et al., 2020).
3 Epigenetic function of ncRNAs in brain disease
3.1 Chromatinic ncRNAs acting on intergenic regions
Warsaw breakage syndrome is a recessive hereditary disease caused by mutation in DEAD/H-box helicase 11 (DDX11, also known as hChlR1) (van der Lelij et al., 2010). The clinical features include microcephaly, hearing loss, and facial dysmorphia. DDX11 regulates chromatin structure (Pisani et al., 2018). DDX11 also controls chromosome separation and sister chromatid condensation in mitosis. DDX11 is hypothesized to prevent abnormal DNA structure in the replication folk (Leman et al., 2010). In line with this idea, defective sister chromatid cohesion is frequently observed in Warsaw breakage syndrome patients’ cells (Pisani, 2019). Separation of the centromere and sister chromatid pairs observed in mitomycin C-induced chromosomal breakage is a remarkable feature of DDX11 mutations (van der Lelij et al., 2010). The mutations in conserved helicase motifs result in unwinding forked duplex DNA substrates. DDX11 destabilization occurs due to misfolding of the protein (Santos et al., 2021). In mouse models, Ddx11 is indispensable for mouse embryonic and placental development, and Ddx11 knock out causes embryonic lethality (Inoue et al., 2007). In zebrafish models, embryonic lethality was increased and craniofacial and vertebral abnormalities were observed. In addition, ddx11 dysfunction generated heterochromatic structures ectopically. This gene also affects histone epigenetic modifications (Sun et al., 2015). Interestingly, a lncRNA, DDX11 antisense RNA 1 (DDX11-AS1, also known as CONCR) is transcribed bidirectionally from the DDX11 promoter region. Although the molecular function of DDX11-AS1 in microcephaly contexts is still obscure, deletion of DDX11-AS1 causes a defect in sister chromatid condensation in mitosis like Warsaw breakage syndrome. Unlike SINEUP, the DDX11 protein level is not affected by the ncRNA knockdown. Levels of histone H3K9 acetylation at the DDX11 promoter region and DDX11 mRNA are also unchanged. Surprisingly, however, the ncRNA can bind DDX11 protein directly, and thus activates hydrolysis of ATP. DDX11-AS1 maintains proper chromatin structure through promoting the enzymatic activity of DDX11 (Marchese et al., 2016). Another report indicated that DDX11-AS1 also function to regulate DDX11 through sponging miR-873-5p, which can target DDX11 (Zhang et al., 2020).
3.2 ncRNA that maintains telomere length
Mutation of dyskerin pseudouridine synthase 1 (DKC1) frequently result in Hoyeraal-Hreidarsson syndrome, a microcephaly disease (Dehmel et al., 2016). DKC1 has TRUB (tRNA pseudouridine synthase B-like) and PUA (pseudouridine synthase and archaeosine transglycosylase) domains. The TRUB domain constitutes the catalytic core of DKC1, whereas the PUA domain seems to function as a RNA binding motif (Garus and Chantal, 2021). DKC1 plays an important role in pseudouridylation of rRNA and telomere extension. The telomerase complex is composed of telomerase reverse transcriptase (TERT), telomerase RNA component (TERC), and other protein factors including DKC1 (Czekay and Kothe, 2021). Apoptosis and chromosomal aberrations increase and proliferative potential decreases in Terc−/− mouse cells (Wong et al., 2003). Both Hoyeraal-Hreidarsson syndrome patients and mouse models for DKC1 dysfunction show reduced rRNA processing and telomerase activities (Mochizuki et al., 2004). In fact, loss of telomere length cause dyskeratosis congenita characterized by bone marrow failure, hyperpigmentation, nail dystrophy and leukoplakia (AlSabbagh, 2020). In particular, DKC1 is involved in the Xp28 and X-linked recessive dyskeratosis congenita, known as a profound type of dyskeratosis congenita, including growth retardation and microcephaly (Dehmel et al., 2016). In most cases of Hoyeraal-Hreidarsson syndrome, the variant A353V located in the PUA domain of DKC1 is observed (Knight et al., 1999). The same mutation attenuates the binding of DKC1 to the TERC, leading to TERC destabilization (Czekay and Kothe, 2021). Accordingly, some patients with mild dyskeratosis congenita also have telomere shortening (Vulliamy et al., 2001) (Yamaguchi et al., 2003). Because bone marrow failure also accompanies dyskeratosis congenita, the patients are frequently treated with hematopoietic cell transplantation (HCT) or androgen therapy (Savage and Niewisch, 2009). Considering the potential of ncRNAs as future therapeutic agents for curing such diseases, their physical association with EXOSC10, a component of the RNA exosome complex which eliminates TERC, may be notable, because its knockdown restored telomerase activity in DKC1 knockdown cells (Shukla et al., 2016).
3.3 The convergent regulation of gene expression by ncRNA
Angelman syndrome, another microcephaly disease, was first described in 1965. It was characterized by unusual arm position and jerky movement (Kishino et al., 1997). Major characteristics include severe intellectual disability, lack of speech, sleep disruption, and microcephaly (Levin et al., 2022). Mouse models for Angelman syndrome frequently exhibit motor dysfunction and deficits in learning and memory. Abnormal electroencephalogram (EEG) is also observed (Miura et al., 2002). Mutation in the E6-AP ubiquitin-protein ligase gene (UBE3A) was found in chromosome 15 of many patients (Kishino et al., 1997) (Matsuura et al., 1997). Normally, UBE3A is expressed only from the maternal allele in the brain, while the paternal allele is silenced by genome imprinting. Some patients have a UBE3A mutation in the maternal allele, and others have paternal uniparental disomy (PUD) and/or imprinting defects (ID) (Saitoh et al., 2005) (Bai et al., 2014). The deletion patients have more profound effects than PUD and ID patients (Lossie et al., 2001). A UBE3A antisense transcript, UBE3A-ATS, suppresses UBE3A on the paternal chromosome (Meng et al., 2012). In the paternal chromosome, UBE3A and UBE3A-ATS are transcribed at the same time. However, in contrast to SINEUP, UBE3A-ATS prevents UBE3A transcription at the expressing paternal allele. It has been thought that 2 opposing RNA polymerases collide and stop the elongation of UBE3A mRNA (Wang et al., 2021) (Mabb et al., 2011). Although the molecular function of UBE3A-ATS in microcephaly contexts is still obscure, disrupting UBE3A-ATS transcription is noted as a potential therapy to increase UBE3A expression in the gene therapy field. For example, a clinical trial using antisense oligonucleotides is ongoing (Schmid et al., 2021). In a mouse model, such treatment can recover paternal UBE3A expression. Early treatment in mouse models (at postnatal day 1) is more effective compared with treatment of the adult (at 2 to 4 months of age). Partial improvement of motor deficiency and anxiety is observed only in young models. However, the behavioral phenotypes are hardly recovered. Nonetheless, both early and adult treatments ameliorate the memory impairment in fear conditioning tests (Milazzo et al., 2021) (Meng et al., 2015). Creation of indels located between the Ube3a 3’ UTR and Snord115 (Small Nucleolar RNA, C/D Box 115) by CRISPR/Cas9 rescued the behavioral phenotype (Schmid et al., 2021). Cas9 targeting the Snord115 cluster also prevent the motor deficiency (Wolter et al., 2020). Injection of the adeno-associated virus (AAV) expressing Zinc finger-based artificial transcription factors (ATFs), that repress Ube3a-ats to induce Ube3a expression (Bailus et al., 2016), also recovers the behavioral phenotype in mouse models (O'Geen et al., 2023). It is noteworthy that these targetings simultaneously truncate UBE3A-ATS, supporting the idea that allele-specific artificial removal of UBE3A-ATS is essential for ongoing therapies. How the lncRNA represses UBE3A expression is still uncertain, and elucidation of the mechanism will enable more effective therapy for Angelman syndrome.
3.4 ncRNAs leading to divergent transcription
We have illuminated various points of ncRNA actions in the above sections. Independently from convergent lncRNAs, we have found a different class composed of thousands of lncRNAs resulting from divergent transcription that originates from protein-coding gene promoters (Uesaka et al., 2014). Later on, we will introduce the functional mode and the potentials of such divergent ncRNAs based on our previous and other studies. As shown in the upper panel of Figures 2A lncRNA seems to be transcribed in the reverse direction to the partner gene. Comparison between RNA-seq reads from human neural stem cells (Brattas et al., 2017) and those from human cardiomyocyte cells (Lopacinski et al., 2021), revealed that aryl hydrocarbon receptor nuclear translocator 2 (ARNT2) is more highly expressed in brain than in cardiomyocytes. A variant of the gene causes Webb-Dattani syndrome, of which the features include microcephaly (Webb et al., 2013). Likewise, cyclin dependent kinase 6 (CDK6) is also microcephaly candidate gene (Naveed et al., 2018) and lncRNA expression was synchronized with that of mRNA. It would be interesting to see the possible effects of divergent lncRNAs on the pathogenesis of microcephaly-related diseases. As noted above, we have discovered a new type of divergent lncRNAs, called promoter-associated lncRNAs (pancRNAs) that are transcribed in the reverse direction to a set of tissue-specific genes (Uesaka et al., 2017). Approximately half of mammalian promoters show CpG-rich sequences and lack of TATA elements. In these CpG island-type promoters without TATA elements, TATA-binding protein (TBP) is recruited together with CpG-rich sequence-specific transcription factors such as Sp1 (Wu and Sharp, 2013) in both strands, thereby driving bidirectional transcription (Mahpour et al., 2018). Although enormous numbers of genes, including housekeeping genes, have CpG-rich promoters, the characteristics of the promoters for pancRNA-partnered genes include the acquisition of a G- and/or C-skewed motif, while such a skew cannot be seen in housekeeping genes. In addition, the lack of a poly(A) site sequence in the body of the pancRNAs has enabled then to get longer (An et al., 2021). Promoter-proximal Ser2 phosphorylation further reinforces a longer RNAPII dwell time at the start site, which may be beneficial for recruiting U1 snRNP upstream of the gene, thereby suppressing the recognition of poly(A) sites and the coupled termination of divergent transcription (Almada et al., 2013). In line with the concordant expression of pancRNAs and the partnered genes, as shown in Figures 2A,B, pancRNA production is associated with DNA demethylation, H3K4 trimethylation (Hamazaki et al., 2015), and H3K27 acetylation (Uesaka et al., 2017). In terms of the biological functions of pancRNAs, these are dependent on the roles of the downstream genes. For example, in rat PC12 cells, pancNusap1 functions in nucleolar and spindle associated protein 1 (Nusap1) activation through histone acetylation, accelerating the cell cycle since Nusap1 plays a role in spindle microtubule organization (Yamamoto et al., 2016). Another example is mouse pancIl17d, which enhances demethylation of the interleukin 17days (Il17d) promoter by recruiting ten-eleven translocation 3 (Tet3) and poly ADP-ribose polymerase (Parp). Silencing pancIl17d is embryonic lethal, probably because Il17d functions to support proliferation/differentiation of pluripotent stem cells, which has been evidenced by the fact that supplying Il17d protein rescues embryonic survival (Hamazaki et al., 2015). pancRNAs occasionally form a triple helix structure with the DNA duplex of promoters and/or enhancers, and interact with some regulatory proteins, such as histone modifiers and transcription factors, to regulate gene transcription in cis. A second mechanism is based on transcriptional activation via formation of a DNA-RNA hybrid (R-loop). In mammalian cells, the asymmetrical distribution of cytosine and guanine, one of the characteristics of CpG islands for tissue-specific genes as discussed above, makes it easy to form R-loops (Chen et al., 2017). Therefore, targeting these structures triggered by pancRNA expression might be a strategy to mitigate microcephaly-related diseases in the future.
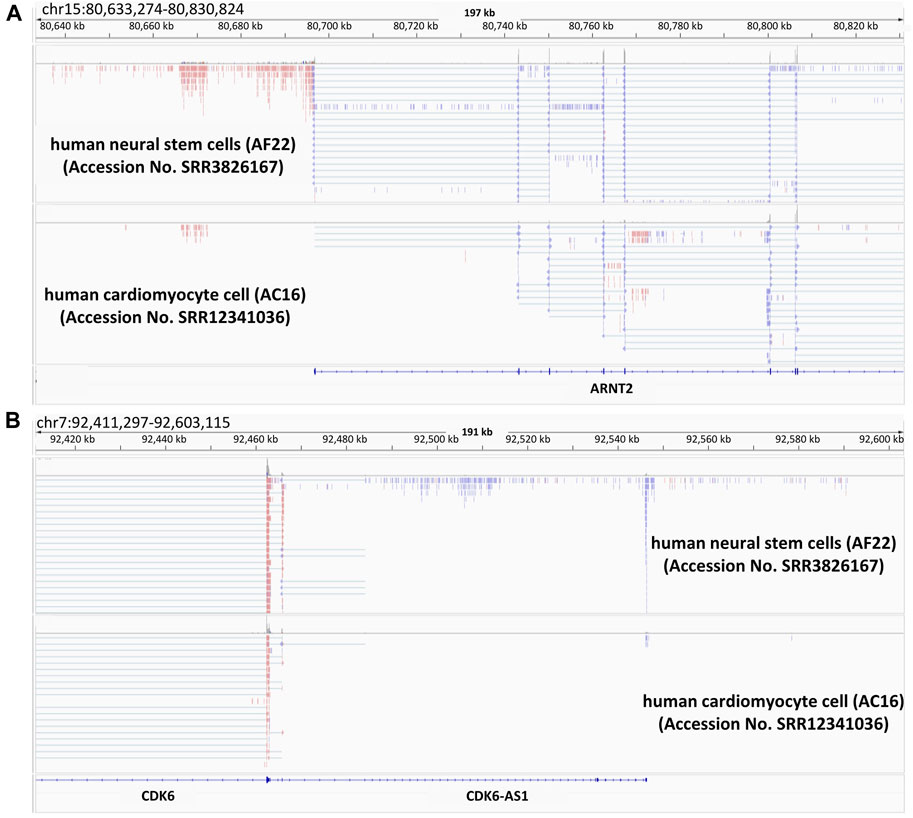
FIGURE 2. Presence of divergent lncRNAs in two examples of microcephaly-related genes Snapshots of the Integrative Genomics Viewer. Publicly available RNA-seq data of human-iPS cell-derived neural stem cell (AF22) and human hybrid cardiomyocyte (AC16) are shown. The data is from (Brattas et al., 2017; Lopacinski et al., 2021). ARNT2 (A) and CDK6 (B) are microcephaly related genes. In this figure, the colors indicate the differential strand usage.
4 RBPs as potential targets for brain diseases
Although the information on ncRNAs in microcephaly is still limited, we can learn more about ncRNAs in relation to brain diseases. In addition to the examples of functional ncRNAs noted above, several other ncRNAs that specify social interactions and behavior have been identified by using mouse models (Table 1). For example, brain cytoplasmic 1 (BC1), which has a motif for dendritic localization (Robeck et al., 2016), regulates neuronal activity-dependent translation in neurons (Eom et al., 2014). Memory and learning dysfunction were observed in some mouse knockout models of BC1 ncRNAs (Chung et al., 2017). The lncRNA nuclear paraspeckle assembly transcript 1 (Neat1) sponges various miRNAs (Azizidoost et al., 2022). The knockout model of Neat1 lost interest in a social interaction (Kukharsky et al., 2020). The ncRNA of synapsin2 (syn2) is decreased in mice with dominant behavior. The ncRNA modulates the social rank thorough binding syn2b pre-mRNA directly and protecting against its destabilization (Ma et al., 2020). In ASD and schizophrenia (SCZ), differentially expressed lncRNAs were detected (Ziats and Rennert, 2013) (Chen et al., 2016) (Table 2). One can hypothesize that most of the lncRNAs function together with RBPs. Although we still do not know of RBPs specifically functioning in the context of microcephaly, in some cases of brain diseases, detailed relationships between RBPs and ncRNAs have been revealed. Cyrano (OIP5-AS1), which is a schizophrenia candidate gene in females (Safari et al., 2019), sponges HuR (human antigen R) and inhibits the protein (Kim et al., 2016). Gomafu (RNCR2/MIAT) binds to the RNA-binding protein Celf3 and splicing factor SF1. The complex is speculated to control splicing and transcription (Ishizuka et al., 2014). TUNA (Tcl1 Upstream Neuron-Associated lincRNA) forms an RNA-RBP complex with three RBPs, PTBP1, HNRNPK, and nucleolin (NCL), and the complex binds to the sox2 promoter (Lin et al., 2014). Rhabdomyosarcoma 2-associated transcript (RMST) and SOX2 interaction plays an important role in neural stem cell fate specification (Ng et al., 2013). A recent study has shown that lncRNAs determine Sox2’s genomic localization (Hamilton et al., 2023). In another example, the interaction of the transcription factor POU3F3 and DNMT1-associated long intergenic (Dali) was described (Chalei et al., 2014). We believe that accumulating evidence further opens up the possibility of lncRNAs as therapeutic targets to artificially regulate their association with various RBPs.
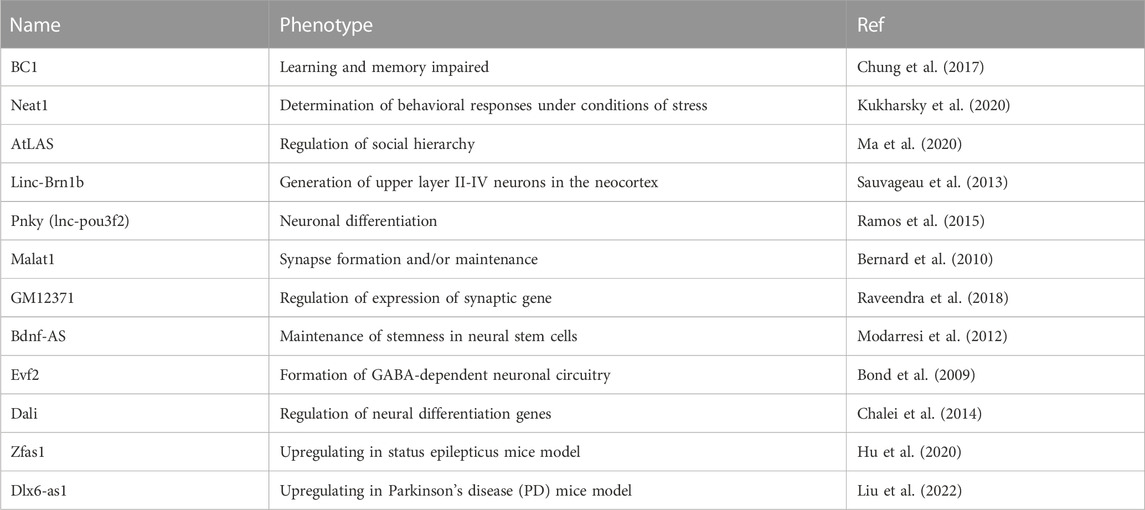
TABLE 1. NcRNA-involving phenotypes in mouse model.
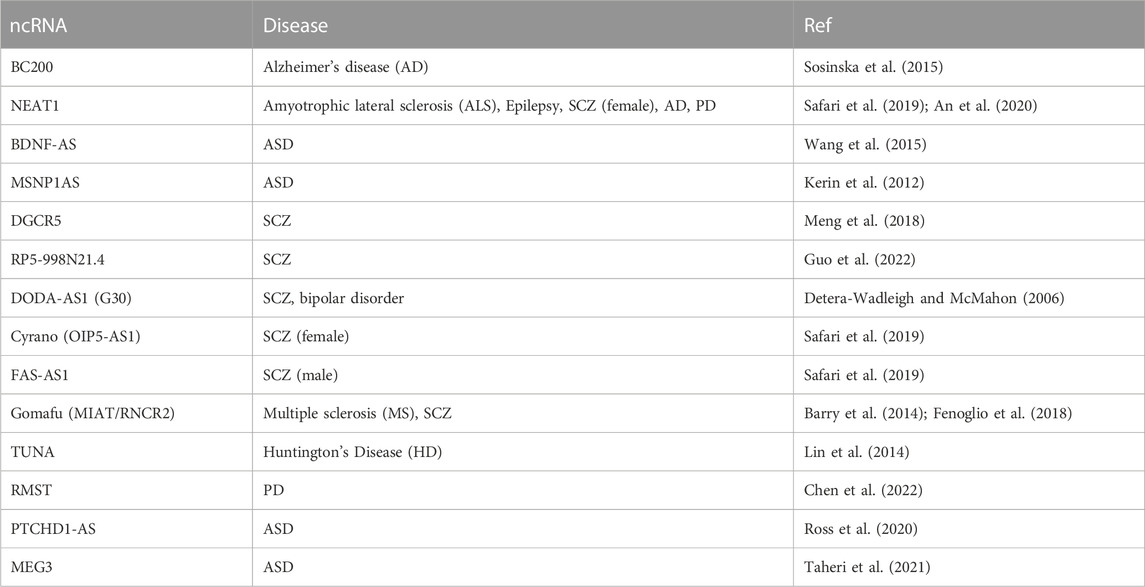
TABLE 2. NcRNAs related to human brain diseases.
5 Conclusion
In this review, we described five ncRNAs that regulate microcephaly-related genes. Although little information is available on ncRNAs responsible for microcephaly, multiple factors are known to provoke microcephaly. For example, 30% of case of ASD are accompanied by the features of diminishing brain size (Fombonne et al., 1999). Table 2 shows the ncRNAs known to be related to brain diseases. In fact, there are many ncRNAs related to ASD. Interestingly, most of the ncRNAs in this list are categorized as lncRNA species. Therefore, it would be interesting to confirm whether the lncRNA class rather than the small RNA class brain function tends to affect the determination of brain size by analyzing the lncRNAs listed in Table 2. Along with understanding of the human genome, tailor-made medicine is a center of attention these days. Acquisition of the sequences of individual genomes become easier and less expensive, revealing mutations that occur not only in coding genes but also in intergenic regions. In particular, accessible and affordable sequence reading enables us to find new intergenic mutations that could have been missed previously because of mild disease symptoms and poor sequencing technology. The resultant studies on intergenic regions allow us to highlight the potentials of ncRNAs for understanding human pathology in clinical research. Since the intergenic regions are poorly conserved among the enormous variety of organisms, and the large size and complicated functions of the brain are human-unique features, it is intriguing possibility that the intergenic regions contribute a big controlling center for determining such interesting traits. Considering the human-specific features of the brain structure and function, it seems likely that model animals such as mouse, zebra fish, and fruit fly would be of limited use for searching for human-specific ncRNAs. Leveraging human brain organoids, genome-wide association studies (GWASs), and massive annotation of human-specific ncRNA functions are essential for pioneering this vast ncRNA field. This field will lead us to new treatments for brain disease and understanding what makes us human.
Author contributions
Writing the manuscript, MT and TI; review and editing, MT and TI; supervision, TI; funding acquisition, TI. All authors contributed to the article and approved the submitted version.
Funding
This study was financially supported by the KAKENHI (JP22H02531), from the Japan Society for the Promotion of Sciences to TI.
Acknowledgments
We appreciate Hiroto Akuta for his excellent guidance on visualizing the IGV genome browser snapshots from RNA-seq data, and Elizabeth Nakajima for the English editing on this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Almada, A. E., Wu, X. B., Kriz, A. J., Burge, C. B., and Sharp, P. A. (2013). Promoter directionality is controlled by U1 snRNP and polyadenylation signals. Nature 499 (7458), 360–363. doi:10.1038/nature12349
AlSabbagh, M. M. (2020). Dyskeratosis congenita: A literature review. J. Der Deutschen Dermatologischen Gesellschaft 25. doi:10.1111/ddg.14268
Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U., and Zoghbi, H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23 (2), 185–188. doi:10.1038/13810
An, B. Y., Kameda, T., and Imamura, T. (2021). “The evolutionary acquisition and mode of functions of promoter-associated non-coding RNAs (pancRNAs) for mammalian development,” in Non-coding genome. Essays in biochemistry 65 Editor C. C. Hon (London: Portland Press Ltd), 697–708. doi:10.1042/EBC20200143
An, H., Williams, N. G., and Shelkovnikova, T. A. (2020). NEAT1 and paraspeckles in neurodegenerative diseases: A missing lnc found? (vol 3, pg 243, 2018). Non-Coding Rna Res. 5 (4), 219.doi:10.1016/j.ncrna.2018.11.003
Azizidoost, S., Ghaedrahmati, F., Anbiyaee, O., Ali, R. A., Cheraghzadeh, M., and Farzaneh, M. (2022). Emerging roles for lncRNA-NEAT1 in colorectal cancer. Cancer Cell Int. 22 (1), 209. doi:10.1186/s12935-022-02627-6
Bai, J. L., Qu, Y. J., Jin, Y. W., Wang, H., Yang, Y. L., Jiang, Y. W., et al. (2014). Molecular and clinical characterization of Angelman syndrome in Chinese patients. Clin. Genet. 85 (3), 273–277. doi:10.1111/cge.12155
Bailus, B. J., Pyles, B., McAlister, M. M., O'Geen, H., Lockwood, S. H., Adams, A. N., et al. (2016). Protein delivery of an artificial transcription factor restores widespread Ube3a expression in an angelman syndrome mouse brain. Mol. Ther. 24 (3), 548–555. doi:10.1038/mt.2015.236
Barry, G., Briggs, J. A., Vanichkina, D. P., Poth, E. M., Beveridge, N. J., Ratnu, V. S., et al. (2014). The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 19 (4), 486–494. doi:10.1038/mp.2013.45
Bernard, D., Prasanth, K. V., Tripathi, V., Colasse, S., Nakamura, T., Xuan, Z. Y., et al. (2010). A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. Embo J. 29 (18), 3082–3093. doi:10.1038/emboj.2010.199
Bernier, R., Golzio, C., Xiong, B., Stessman, H. A., Coe, B. P., Penn, O., et al. (2014). Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158 (2), 263–276. doi:10.1016/j.cell.2014.06.017
Bond, A. M., VanGompel, M. J. W., Sametsky, E. A., Clark, M. F., Savage, J. C., Disterhoft, J. F., et al. (2009). Balanced gene regulation by an embryonic brain ncRNA is critical for adult hippocampal GABA circuitry. Nat. Neurosci. 12 (8), 1020–1027. doi:10.1038/nn.2371
Brattas, P. L., Jonsson, M. E., Fasching, L., Wahlestedt, J. N., Shahsavani, M., Falk, R., et al. (2017). TRIM28 controls a gene regulatory network based on endogenous retroviruses in human neural progenitor cells. Cell Rep. 18 (1), 1–11. doi:10.1016/j.celrep.2016.12.010
Chalei, V., Sansom, S. N., Kong, L. S., Lee, S., Montiel, J., Vance, K. W., et al. (2014). The long non-coding RNA Dali is an epigenetic regulator of neural differentiation. Elife 3, e04530. doi:10.7554/eLife.04530
Chen, C. L., Zhang, S. J., Wei, Y. H., and Sun, X. B. (2022). LncRNA RMST regulates neuronal apoptosis and inflammatory response via sponging miR-150-5p in Parkinson's disease. Neuroimmunomodulation 29 (1), 55–62. doi:10.1159/000518212
Chen, L., Chen, J. Y., Zhang, X., Gu, Y., Xiao, R., Shao, C. W., et al. (2017). R-ChIP using inactive RNase H reveals dynamic coupling of R-loops with transcriptional pausing at gene promoters. Mol. Cell 68 (4), 745–757. doi:10.1016/j.molcel.2017.10.008
Chen, S. D., Sun, X. Y., Niu, W., Kong, L. M., He, M. J., Li, W. S., et al. (2016). Aberrant expression of long non-coding RNAs in schizophrenia patients. Med. Sci. Monit. 22, 3340–3351. doi:10.12659/msm.896927
Chi, Y. J., Wang, D., Wang, J. P., Yu, W. D., and Yang, J. C. (2019). Long non-coding RNA in the pathogenesis of cancers. Cells 8 (9), 1015. doi:10.3390/cells8091015
Chung, A., Dahan, N., Alarcon, J. M., and Fenton, A. A. (2017). Effects of regulatory BC1 RNA deletion on synaptic plasticity, learning, and memory. Learn. Mem. 24 (12), 646–649. doi:10.1101/lm.045617.117
Czekay, D. P., and Kothe, U. (2021). H/ACA small ribonucleoproteins: Structural and functional comparison between archaea and eukaryotes. Front. Microbiol. 12, 654370. doi:10.3389/fmicb.2021.654370
de Pontual, L., Yao, E., Callier, P., Faivre, L., Drouin, V., Cariou, S., et al. (2011). Germline deletion of the miR-17∼92 cluster causes skeletal and growth defects in humans. Nat. Genet. 43 (10), 1026–1030. doi:10.1038/ng.915
Dehmel, M., Brenner, S., Suttorp, M., Hahn, G., Schutzle, H., Dinger, J., et al. (2016). Novel mutation in the DKC1 gene: Neonatal hoyeraal-hreidarsson syndrome as a rare differential diagnosis in pontocerebellar hypoplasia, primary microcephaly, and progressive bone marrow failure. Neuropediatrics 47 (3), 182–186. doi:10.1055/s-0036-1578799
Detera-Wadleigh, S. D., and McMahon, F. J. (2006). G72/G30 in schizophrenia and bipolar disorder: Review and meta-analysis. Biol. Psychiatry 60 (2), 106–114. doi:10.1016/j.biopsych.2006.01.019
Eom, T., Muslimov, I. A., Tsokas, P., Berardi, V., Zhong, J., Sacktor, T. C., et al. (2014). Neuronal BC RNAs cooperate with eIF4B to mediate activity-dependent translational control. J. Cell Biol. 207 (2), 237–252. doi:10.1083/jcb.201401005
Fenoglio, C., Oldoni, E., Serpente, M., De Riz, M. A., Arcaro, M., D'Anca, M., et al. (2018). LncRNAs expression profile in peripheral blood mononuclear cells from multiple sclerosis patients. J. Neuroimmunol. 324, 129–135. doi:10.1016/j.jneuroim.2018.08.008
Fombonne, E., Roge, B., Claverie, J., Courty, S., and Fremolle, J. (1999). Microcephaly and macrocephaly in autism. J. Autism Dev. Disord. 29 (2), 113–119. doi:10.1023/a:1023036509476
Garus, A., and Chantal, A. (2021). Dyskerin: An essential pseudouridine synthase with multifaceted roles in ribosome biogenesis, splicing, and telomere maintenance. New York, N.Y. RNA. doi:10.1261/rna.078953.121
Guo, B., Jiang, T. Y., Wu, F. C., Ni, H. Y., Ye, J. P., Wu, X. H., et al. (2022). LncRNA RP5-998N21.4 promotes immune defense through upregulation of IFIT2 and IFIT3 in schizophrenia. Schizophrenia 8 (1), 11. doi:10.1038/s41537-021-00195-8
Hamazaki, N., Uesaka, M., Nakashima, K., Agata, K., and Imamura, T. (2015). Gene activation-associated long noncoding RNAs function in mouse preimplantation development. Development 142 (5), 910–920. doi:10.1242/dev.116996
Hamilton, D. J., Hein, A. E., Wuttke, D. S., and Batey, R. T. (2023). The DNA binding high mobility group box protein family functionally binds RNA. Wiley Interdiscip. Reviews-Rna 31. doi:10.1002/wrna.1778
Han, J., Thurnherr, T., Chung, A. Y. F., Goh, B. K. P., Chow, P. K. H., Chan, C. Y., et al. (2021). Clinicopathological-associated regulatory network of deregulated circRNAs in hepatocellular carcinoma. Cancers 13 (11), 2772. doi:10.3390/cancers13112772
Hayashita, Y., Osada, H., Tatematsu, Y., Yamada, H., Yanagisawa, K., Tomida, S., et al. (2005). A polycistronic microRNA cluster, miR-17-92, is overexpressed in human lung cancers and enhances cell proliferation. Cancer Res. 65 (21), 9628–9632. doi:10.1158/0008-5472.CAN-05-2352
Hu, F., Shao, L., Zhang, J., Zhang, H., Wen, A., and Zhang, P. (2020). Knockdown of ZFAS1 inhibits hippocampal neurons apoptosis and autophagy by activating the PI3K/AKT pathway via up-regulating miR-421 in epilepsy. Neurochem. Res. 45 (10), 2433–2441. doi:10.1007/s11064-020-03103-1
Inoue, A., Li, T. Y., Roby, S. K., Valentine, M. B., Inoue, M., Boyd, K., et al. (2007). Loss of ChlR1 helicase in mouse causes lethality due to the accumulation of aneuploid cells generated by cohesion defects and placental malformation. Cell Cycle 6 (13), 1646–1654. doi:10.4161/cc.6.13.4411
Ishizuka, A., Hasegawa, Y., Ishida, K., Yanaka, K., and Nakagawa, S. (2014). Formation of nuclear bodies by the lncRNA Gomafu-associating proteins Celf3 and SF1. Genes Cells 19 (9), 704–721. doi:10.1111/gtc.12169
Jauss, R. T., Schliesske, S., and Abou Jamra, R. (2022). Routine diagnostics confirm novel neurodevelopmental disorders. Genes 13 (12), 16. doi:10.3390/genes13122305
Kerin, T., Ramanathan, A., Rivas, K., Grepo, N., Coetzee, G. A., and Campbell, D. B. (2012). A noncoding RNA antisense to moesin at 5p14.1 in autism. Sci. Transl. Med. 4 (128), 128ra40. doi:10.1126/scitranslmed.3003479
Kim, J., Abdelmohsen, K., Yang, X. L., De, S., Grammatikakis, I., Noh, J. H., et al. (2016). LncRNA OIP5-AS1/cyrano sponges RNA-binding protein HuR. Nucleic Acids Res. 44 (5), 2378–2392. doi:10.1093/nar/gkw017
Kishino, T., Lalande, M., and Wagstaff, J. (1997). UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 15 (1), 70–73. doi:10.1038/ng0197-70
Knight, S. W., Heiss, N. S., Vulliamy, T. J., Greschner, S., Stavrides, G., Pai, G. S., et al. (1999). X-linked dyskeratosis congenita is predominantly caused by missense mutations in the DKC1 gene. Am. J. Hum. Genet. 65 (1), 50–58. doi:10.1086/302446
Kukharsky, M. S., Ninkina, N. N., An, H. Y., Telezhkin, V., Wei, W. B., de Meritens, C. R., et al. (2020). Long non-coding RNA Neat1 regulates adaptive behavioural response to stress in mice. Transl. Psychiatry 10 (1), 171. doi:10.1038/s41398-020-0854-2
Lai, F., Stubbs, L., and Artzt, K. (1994). Molecular analysis of mouse Rab11b - a new-type of mammalian ypt/rab protein. Genomics 22 (3), 610–616. doi:10.1006/geno.1994.1434
Lamers, I. J. C., Reijnders, M. R. F., Venselaar, H., Kraus, A., Jansen, S., de Vries, B. B. A., et al. (2017). Recurrent de novo mutations disturbing the GTP/GDP binding pocket of RAB11B cause intellectual disability and a distinctive brain phenotype. Am. J. Hum. Genet. 101 (5), 824–832. doi:10.1016/j.ajhg.2017.09.015
Leman, A. R., Noguchi, C., Lee, C. Y., and Noguchi, E. (2010). Human Timeless and Tipin stabilize replication forks and facilitate sister-chromatid cohesion. J. Cell Sci. 123 (5), 660–670. doi:10.1242/jcs.057984
Levin, Y., Hosamane, N. S., McNair, T. E., Kunnam, S. S., Philpot, B. D., Fan, Z., et al. (2022). Evaluation of electroencephalography biomarkers for Angelman syndrome during overnight sleep. Autism Res. 15 (6), 1031–1042. doi:10.1002/aur.2709
Li, T. G., Wu, D., Liu, Q., Wang, D. D., Chen, J. B., Zhao, H. J., et al. (2020). Upregulation of long noncoding RNA RAB11B-AS1 promotes tumor metastasis and predicts poor prognosis in lung cancer. Ann. Transl. Med. 8 (9), 582. doi:10.21037/atm.2020.04.52
Lian, G. W., Lu, J., Hu, J. J., Zhang, J. P., Cross, S. H., Ferland, R. J., et al. (2012). Filamin A regulates neural progenitor proliferation and cortical size through wee1-dependent Cdk1 phosphorylation. J. Neurosci. 32 (22), 7672–7684. doi:10.1523/JNEUROSCI.0894-12.2012
Lin, N. W., Chang, K. Y., Li, Z. H., Gates, K., Rana, Z. A., Dang, J. S., et al. (2014). An evolutionarily conserved long noncoding RNA TUNA controls pluripotency and neural lineage commitment. Mol. Cell 53 (6), 1005–1019. doi:10.1016/j.molcel.2014.01.021
Liu, L., Zhou, T. T., Li, T., Liang, Z. H., and Luo, X. G. (2022). LncRNA DLX6-AS1 promotes microglial inflammatory response in Parkinson?s disease by regulating the miR-223-3p/NRP1 axis. Behav. Brain Res. 431, 113923. doi:10.1016/j.bbr.2022.113923
Lopacinski, A. B., Sweatt, A. J., Smolko, C. M., Gray-Gaillard, E., Borgman, C. A., Shah, M., et al. (2021). Modeling the complete kinetics of coxsackievirus B3 reveals human determinants of host-cell feedback. Cell Syst. 12 (4), 304–323.e13. doi:10.1016/j.cels.2021.02.004
Lossie, A. C., Whitney, M. M., Amidon, D., Dong, H. J., Chen, P., Theriaque, D., et al. (2001). Distinct phenotypes distinguish the molecular classes of Angelman syndrome. J. Med. Genet. 38 (12), 834–845. doi:10.1136/jmg.38.12.834
Ma, H. B., Pan, J. S., Jin, L. X., Wu, J. F., Ren, Y. D., Chen, P. D., et al. (2016). MicroRNA-17∼92 inhibits colorectal cancer progression by targeting angiogenesis. Cancer Lett. 376 (2), 293–302. doi:10.1016/j.canlet.2016.04.011
Ma, M., Xiong, W., Hu, F., Deng, M. F., Huang, X., Chen, J. G., et al. (2020). A novel pathway regulates social hierarchy via lncRNA AtLAS and postsynaptic synapsin IIb. Cell Res. 30 (2), 105–118. doi:10.1038/s41422-020-0273-1
Mabb, A. M., Judson, M. C., Zylka, M. J., and Philpot, B. D. (2011). Angelman syndrome: Insights into genomic imprinting and neurodevelopmental phenotypes. Trends Neurosci. 34 (6), 293–303. doi:10.1016/j.tins.2011.04.001
Mahpour, A., Scruggs, B. S., Smiraglia, D., Ouchi, T., and Gelman, I. H. (2018). A methyl-sensitive element induces bidirectional transcription in TATA-less CpG island-associated promoters. Plos One 13 (10), e0205608. doi:10.1371/journal.pone.0205608
Marchese, F. P., Grossi, E., Marin-Bejar, O., Bharti, S. K., Raimondi, I., Gonzalez, J., et al. (2016). A long noncoding RNA regulates sister chromatid cohesion. Mol. Cell 63 (3), 397–407. doi:10.1016/j.molcel.2016.06.031
Matsuura, T., Sutcliffe, J. S., Fang, P., Galjaard, R. J., Jiang, Y. H., Benton, C. S., et al. (1997). De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat. Genet. 15 (1), 74–77. doi:10.1038/ng0197-74
Mendell, J. T. (2008). miRiad roles for the miR-17-92 cluster in development and disease. Cell 133 (2), 217–222. doi:10.1016/j.cell.2008.04.001
Meng, L. Y., Person, R. E., and Beaudet, A. L. (2012). Ube3a-ATS is an atypical RNA polymerase II transcript that represses the paternal expression of Ube3a. Hum. Mol. Genet. 21 (13), 3001–3012. doi:10.1093/hmg/dds130
Meng, L. Y., Ward, A. J., Chun, S., Bennett, C. F., Beaudet, A. L., and Rigo, F. (2015). Towards a therapy for Angelman syndrome by targeting a long non-coding RNA. Nature 518 (7539), 409–412. doi:10.1038/nature13975
Meng, Q. T., Wang, K. L., Brunetti, T., Xia, Y., Jiao, C., Dai, R., et al. (2018). The DGCR5 long noncoding RNA may regulate expression of several schizophrenia-related genes. Sci. Transl. Med. 10 (472), eaat6912. doi:10.1126/scitranslmed.aat6912
Milazzo, C., Mientjes, E. J., Wallaard, I., Rasmussen, S. V., Erichsen, K. D., Kakunuri, T., et al. (2021). Antisense oligonucleotide treatment rescues UBE3A expression and multiple phenotypes of an Angelman syndrome mouse model. Jci Insight 6 (15), e145991. doi:10.1172/jci.insight.145991
Mirzamohammadi, F., Kozlova, A., Papaioannou, G., Paltrinieri, E., Ayturk, U. M., and Kobayashi, T. (2018). Distinct molecular pathways mediate Mycn and Myc-regulated miR-17-92 microRNA action in Feingold syndrome mouse models. Nat. Commun. 9, 1352. doi:10.1038/s41467-018-03788-7
Miura, K., Kishino, T., Li, E., Webber, H., Dikkes, P., Holmes, G. L., et al. (2002). Neurobehavioral and electroencephalographic abnormalities in Ube3a maternal-deficient mice. Neurobiol. Dis. 9 (2), 149–159. doi:10.1006/nbdi.2001.0463
Mochizuki, Y., He, J., Kulkarni, S., Bessler, M., and Mason, P. J. (2004). Mouse dyskerin mutations affect accumulation of telomerase RNA and small nucleolar RNA, telomerase activity, and ribosomal RNA processing. Proc. Natl. Acad. Sci. U. S. A. 101 (29), 10756–10761. doi:10.1073/pnas.0402560101
Modarresi, F., Faghihi, M. A., Lopez-Toledano, M. A., Fatemi, R. P., Magistri, M., Brothers, S. P., et al. (2012). Inhibition of natural antisense transcripts in vivo results in gene-specific transcriptional upregulation. Nat. Biotechnol. 30 (5), 453–459. doi:10.1038/nbt.2158
Mu, P., Han, Y. C., Betel, D., Yao, E., Squatrito, M., Ogrodowski, P., et al. (2009). Genetic dissection of the miR-17∼92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes and Dev. 23 (24), 2806–2811. doi:10.1101/gad.1872909
Nassari, S., Del Olmo, T., and Jean, S. (2020). Rabs in signaling and embryonic development. Int. J. Mol. Sci. 21 (3), 1064. doi:10.3390/ijms21031064
Naveed, M., Kazmi, S. K., Amin, M., Asif, Z., Islam, U., Shahid, K., et al. (2018). Comprehensive review on the molecular genetics of autosomal recessive primary microcephaly (MCPH). Genet. Res. 100, e7. doi:10.1017/S0016672318000046
Ng, S. Y., Bogu, G. K., Soh, B. S., and Stanton, L. W. (2013). The long noncoding RNA RMST interacts with SOX2 to regulate neurogenesis. Mol. Cell 51 (3), 349–359. doi:10.1016/j.molcel.2013.07.017
Nicholas, A. K., Swanson, E. A., Cox, J. J., Karbani, G., Malik, S., Springell, K., et al. (2009). The molecular landscape of ASPM mutations in primary microcephaly. J. Med. Genet. 46 (4), 249–253. doi:10.1136/jmg.2008.062380
Niu, Y. L., Bao, L., Chen, Y., Wang, C. L., Luo, M. W., Zhang, B., et al. (2020). HIF2-Induced long noncoding RNA RAB11B-AS1 promotes hypoxia-mediated angiogenesis and breast cancer metastasis. Cancer Res. 80 (5), 964–975. doi:10.1158/0008-5472.CAN-19-1532
Novikova, I. V., Hennelly, S. P., and Sanbonmatsu, K. Y. (2013). Tackling structures of long noncoding RNAs. Int. J. Mol. Sci. 14 (12), 23672–23684. doi:10.3390/ijms141223672
O'Geen, H., Beitnere, U., Garcia, M. S., Adhikari, A., Cameron, D. L., Fenton, T. A., et al. (2023). Transcriptional reprogramming restores UBE3A brain-wide and rescues behavioral phenotypes in an Angelman syndrome mouse model. Mol. Ther. 31 (4), 1088–1105. doi:10.1016/j.ymthe.2023.01.013
Pirozzi, F., Nelson, B., and Mirzaa, G. (2018). From microcephaly to megalencephaly: Determinants of brain size. Dialogues Clin. Neurosci. 20 (4), 267–282. doi:10.31887/DCNS.2018.20.4/gmirzaa
Pisani, F. M., Napolitano, E., Napolitano, L. M. R., and Onesti, S. (2018). Molecular and cellular functions of the Warsaw breakage syndrome DNA helicase DDX11. Genes 9 (11), 564. doi:10.3390/genes9110564
Pisani, F. M. (2019). Spotlight on Warsaw breakage syndrome. Appl. Clin. Genet. 12, 239–248. doi:10.2147/TACG.S186476
Ramos, A. D., Andersen, R. E., Liu, S. J., Nowakowski, T. J., Hong, S. J., Gertz, C. C., et al. (2015). The long noncoding RNA pnky regulates neuronal differentiation of embryonic and postnatal neural stem cells. Cell Stem Cell 16 (4), 439–447. doi:10.1016/j.stem.2015.02.007
Raveendra, B. L., Swarnkar, S., Avchalumov, Y., Liu, X. A., Grinman, E., Badal, K., et al. (2018). Long noncoding RNA GM12371 acts as a transcriptional regulator of synapse function. Proc. Natl. Acad. Sci. U. S. A. 115 (43), E10197–E205. doi:10.1073/pnas.1722587115
Rett, A. (1966). On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 116, 723–726.
Robeck, T., Skryabin, B. V., Rozhdestvensky, T. S., Skryabin, A. B., and Brosius, J. (2016). BC1 RNA motifs required for dendritic transport in vivo. Sci. Rep. 6, 28300. doi:10.1038/srep28300
Ross, P. J., Zhang, W. B., Mok, R. S. F., Zaslavsky, K., Deneault, E., D'Abate, L., et al. (2020). Synaptic dysfunction in human neurons with autism-associated deletions in PTCHD1-AS. Biol. Psychiatry 87 (2), 139–149. doi:10.1016/j.biopsych.2019.07.014
Safari, M. R., Komaki, A., Arsang-Jang, S., Taheri, M., and Ghafouri-Fard, S. (2019). Expression pattern of long non-coding RNAs in schizophrenic patients. Cell. Mol. Neurobiol. 39 (2), 211–221. doi:10.1007/s10571-018-0640-3
Saitoh, S., Wada, T., Okajima, M., Takano, K., Sudo, A., and Niikawa, N. (2005). Uniparental disomy and imprinting defects in Japanese patients with Angelman syndrome. Brain and Dev. 27 (5), 389–391. doi:10.1016/j.braindev.2003.12.013
Santos, D., Mahtab, M., Boavida, A., and Pisani, F. M. (2021). Role of the DDX11 DNA helicase in Warsaw breakage syndrome etiology. Int. J. Mol. Sci. 22 (5), 2308. doi:10.3390/ijms22052308
Sauvageau, M., Goff, L. A., Lodato, S., Bonev, B., Groff, A. F., Gerhardinger, C., et al. (2013). Multiple knockout mouse models reveal lincRNAs are required for life and brain development. Elife 2, e01749. doi:10.7554/eLife.01749
Savage, A. S., and Niewisch, M. R. (2009). “Dyskeratosis congenita and related telomere Biology disorders,” in GeneReviews®: Margaret P adam (Seattle (WA): University of Washington).
Schmid, R. S., Deng, X. F., Panikker, P., Msackyi, M., Breton, C., and Wilson, J. M. (2021). CRISPR/Cas9 directed to the Ube3a antisense transcript improves Angelman syndrome phenotype in mice. J. Clin. Investigation 131 (5), 7. doi:10.1172/jci142574
Shukla, S., Schmidt, J. C., Goldfarb, K. C., Cech, T. R., and Parker, R. (2016). Inhibition of telomerase RNA decay rescues telomerase deficiency caused by dyskerin or PARN defects. Nat. Struct. Mol. Biol. 23 (4), 286–292. doi:10.1038/nsmb.3184
Simon-Sanchez, J., and Singleton, A. (2008). Genome-wide association studies in neurological disorders. Lancet Neurol. 7 (11), 1067–1072. doi:10.1016/S1474-4422(08)70241-2
Sionov, R. V., Vlahopoulos, S. A., and Granot, Z. (2015). Regulation of bim in health and disease. Oncotarget 6 (27), 23058–23134. doi:10.18632/oncotarget.5492
Sosinska, P., Mikula-Pietrasik, J., and Ksiazek, K. (2015). The double-edged sword of long non-coding RNA: The role of human brain-specific BC200 RNA in translational control, neurodegenerative diseases, and cancer. Mutat. Research-Reviews Mutat. Res. 766, 58–67. doi:10.1016/j.mrrev.2015.08.002
Stenmark, H., and Olkkonen, V. M. (2001). The Rab GTPase family. Genome Biol. 2 (5), 3007. doi:10.1186/gb-2001-2-5-reviews3007
Sun, X. L., Chen, H. B., Deng, Z. A., Hu, B., Luo, H., Zeng, X. B., et al. (2015). The Warsaw breakage syndrome-related protein DDX11 is required for ribosomal RNA synthesis and embryonic development. Hum. Mol. Genet. 24 (17), 4901–4915. doi:10.1093/hmg/ddv213
Taheri, M., Tamizkar, K. H., Omrani, S., Arsang-Jang, S., Ghafouri-Fard, S., and Omrani, M. D. (2021). MEG3 lncRNA is over-expressed in autism spectrum disorder. Metab. Brain Dis. 36 (8), 2235–2242. doi:10.1007/s11011-021-00764-x
Tan, F. S., Chen, J. L., Du, Z. Y., Zhao, F. N., Liu, Y. L., Zhang, Q., et al. (2022). MIR17HG: A cancerogenic long-noncoding RNA in different cancers. Curr. Pharm. Des. 28 (15), 1272–1281. doi:10.2174/1381612828666220310144500
Toki, N., Takahashi, H., Sharma, H., Valentine, M. N. Z., Rahman, F. U. M., Zucchelli, S., et al. (2020). SINEUP long non-coding RNA acts via PTBP1 and HNRNPK to promote translational initiation assemblies. Nucleic Acids Res. 48 (20), 11626–11644. doi:10.1093/nar/gkaa814
Uesaka, M., Agata, K., Oishi, T., Nakashima, K., and Imamura, T. (2017). Evolutionary acquisition of promoterassociated non-coding RNA (pancRNA) repertoires diversifies species-dependent gene activation mechanisms in mammals. Bmc Genomics 18, 285. doi:10.1186/s12864-017-3662-1
Uesaka, M., Nishimura, O., Go, Y., Nakashima, K., Agata, K., and Imamura, T. (2014). Bidirectional promoters are the major source of gene activation-associated non-coding RNAs in mammals. Bmc Genomics 15, 35. doi:10.1186/1471-2164-15-35
van der Lelij, P., Chrzanowska, K. H., Godthelp, B. C., Rooimans, M. A., Ostra, A. B., Stumm, M., et al. (2010). Warsaw breakage syndrome, a cohesinopathy associated with mutations in the XPD helicase family member DDX11/ChlR1. Am. J. Hum. Genet. 86 (2), 262–266. doi:10.1016/j.ajhg.2010.01.008
Ventura, A., Young, A. G., Winslow, M. M., Lintault, L., Meissner, A., Erkeland, S. J., et al. (2008). Targeted deletion reveals essential and overlapping functions of the miR-17 through 92 family of miRNA clusters. Cell 132 (5), 875–886. doi:10.1016/j.cell.2008.02.019
Vulliamy, T., Marrone, A., Goldman, F., Dearlove, A., Bessler, M., Mason, P. J., et al. (2001). The RNA component of telomerase is mutated in autosomal dominant dyskeratosis congenita. Nature 413 (6854), 432–435. doi:10.1038/35096585
Wang, T. X., Li, J. J., Yang, L. Y., Wu, M. Y., and Ma, Q. (2021). The role of long non-coding RNAs in human imprinting disorders: Prospective therapeutic targets. Front. Cell Dev. Biol. 9, 730014. doi:10.3389/fcell.2021.730014
Wang, Y., Zhao, X., Ju, W., Flory, M., Zhong, J., Jiang, S., et al. (2015). Genome-wide differential expression of synaptic long noncoding RNAs in autism spectrum disorder. Transl. Psychiatry 5, e660. doi:10.1038/tp.2015.144
Webb, E. A., AlMutair, A., Kelberman, D., Bacchelli, C., Chanudet, E., Lescai, F., et al. (2013). ARNT2 mutation causes hypopituitarism, post-natal microcephaly, visual and renal anomalies. Brain 136, 3096–3105. doi:10.1093/brain/awt218
Weng, S. M., Bailey, M. E. S., and Cobb, S. R. (2011). Rett syndrome: From bed to bench. Pediatr. Neonatol. 52 (6), 309–316. doi:10.1016/j.pedneo.2011.08.002
Whelan, M. A. (2010). Practice parameter: Evaluation of the child with microcephaly(an evidence based review): Report of the quality standards subcommittee of the American Academy of neurology and the practice committee of the child neurology society. Neurology 74, 1079. doi:10.1212/WNL.0b013e3181d5dfca
Wolter, J. M., Mao, H. Q., Fragola, G., Simon, J. M., Krantz, J. L., Bazick, H. O., et al. (2020). Cas9 gene therapy for Angelman syndrome trapsUbe3a-ATSlong non-coding RNA. Nature 587(7833), 281. doi:10.1038/s41586-020-2835-2
Wong, K. K., Maser, R. S., Bachoo, R. M., Menon, J., Carrasco, D. R., Gu, Y. S., et al. (2003). Telomere dysfunction and Atm deficiency compromises organ homeostasis and accelerates ageing. Nature 421 (6923), 643–648. doi:10.1038/nature01385
Wu, X. B., and Sharp, P. A. (2013). Divergent transcription: A driving force for new gene origination? Cell 155 (5), 990–996. doi:10.1016/j.cell.2013.10.048
Xia, K., Zhang, J., Ahn, M., Jha, S., Crowley, J. J., Szatkiewicz, J., et al. (2017). Genome-wide association analysis identifies common variants influencing infant brain volumes. Transl. Psychiatry 7, e1188. doi:10.1038/tp.2017.159
Yamaguchi, H., Baerlocher, G. M., Lansdorp, P. M., Chanock, S. J., Nunez, O., Sloand, E., et al. (2003). Mutations of the human telomerase RNA gene (TERC) in aplastic anemia and myelodysplastic syndrome. Blood 102 (3), 916–918. doi:10.1182/blood-2003-01-0335
Yamamoto, N., Agata, K., Nakashima, K., and Imamura, T. (2016). Bidirectional promoters link cAMP signaling with irreversible differentiation through promoter-associated non-coding RNA (pancRNA) expression in PC12 cells. Nucleic Acids Res. 44 (11), 5105–5122. doi:10.1093/nar/gkw113
Yan, H. W., and Bu, P. C. (2021). “Non-coding RNA in cancer,” in Non-coding genome. Essays in biochemistry 65 Editor C. C. Hon (London: Portland Press Ltd), 625–639. doi:10.1042/EBC20200032
Zarantonello, G., Arnoldi, M., Filosi, M., Tebaldi, T., Spirito, G., Barbieri, A., et al. (2021). Natural SINEUP RNAs in autism spectrum disorders: RAB11B-AS1 dysregulation in a neuronal CHD8 suppression model leads to RAB11B protein increase. Front. Genet. 12, 12. doi:10.3389/fgene.2021.745229
Zhang, H., Lin, J. T., Chen, J. J., Gu, W. Q., Mao, Y. J., Wang, H. X., et al. (2020). DDX11-AS1 contributes to osteosarcoma progression via stabilizing DDX11. Life Sci. 254, 117392. doi:10.1016/j.lfs.2020.117392
Zhang, X. C., Chen, M. H., Wu, X. B., Kodani, A., Fan, J., Doan, R., et al. (2016). Cell-type-specific alternative splicing governs cell fate in the developing cerebral cortex. Cell 166 (5), 1147–1162. doi:10.1016/j.cell.2016.07.025
Ziats, M. N., and Rennert, O. M. (2013). Aberrant expression of long noncoding RNAs in autistic brain. J. Mol. Neurosci. 49 (3), 589–593. doi:10.1007/s12031-012-9880-8
Keywords: microcephaly, non-coding RNA, RNA-binding protein, human medicine, epigenetic, brain diversity
Citation: Tokunaga M and Imamura T (2023) Emerging concepts involving inhibitory and activating RNA functionalization towards the understanding of microcephaly phenotypes and brain diseases in humans. Front. Cell Dev. Biol. 11:1168072. doi: 10.3389/fcell.2023.1168072
Received: 17 February 2023; Accepted: 12 June 2023;
Published: 20 June 2023.
Edited by:
Masako Suzuki, Texas A&M University, United StatesReviewed by:
Masato Yano, Niigata University, JapanPanagiotis Politis, Biomedical Research Foundation of the Academy of Athens (BRFAA), Greece
Copyright © 2023 Tokunaga and Imamura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Takuya Imamura, timamura@hiroshima-u.ac.jp