
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cell Dev. Biol. , 10 March 2023
Sec. Molecular and Cellular Pathology
Volume 11 - 2023 | https://doi.org/10.3389/fcell.2023.1142937
This article is part of the Research Topic Personalized Medicine for Neuromuscular Disorders View all 9 articles
 Sergi Cesar1,2,3*
Sergi Cesar1,2,3* Oscar Campuzano4,5,6
Oscar Campuzano4,5,6 Jose Cruzalegui1,2,3
Jose Cruzalegui1,2,3 Victori Fiol1,2,3Isaac Moll1,2,3
Victori Fiol1,2,3Isaac Moll1,2,3 Estefania Martínez-Barrios1,2,3Irene Zschaeck1,2,3,7,8
Estefania Martínez-Barrios1,2,3Irene Zschaeck1,2,3,7,8 Daniel Natera-de Benito7,8Carlos Ortez7,8Laura Carrera7,8
Daniel Natera-de Benito7,8Carlos Ortez7,8Laura Carrera7,8 Jessica Expósito7,8
Jessica Expósito7,8 Rubén Berrueco9
Rubén Berrueco9 Carles Bautista-Rodriguez10,11
Carles Bautista-Rodriguez10,11 Ivana Dabaj12Marta Gómez García-de-la-Banda12
Ivana Dabaj12Marta Gómez García-de-la-Banda12 Susana Quijano-Roy12Josep Brugada1,2,3,6,13Andrés Nascimento7,8,14Georgia Sarquella-Brugada1,2,3
Susana Quijano-Roy12Josep Brugada1,2,3,6,13Andrés Nascimento7,8,14Georgia Sarquella-Brugada1,2,3Introduction: LMNA-related muscular dystrophy is a rare entity that produce “laminopathies” such as Emery–Dreifuss muscular dystrophy (EDMD), limb–girdle muscular dystrophy type 1B (LGMD1B), and LMNA-related congenital muscular dystrophy (L-CMD). Heart failure, malignant arrhythmias, and sudden death may occur. No consensus exists on cardiovascular management in pediatric laminopathies. The aim was to perform an exhaustive cardiologic follow-up in pediatric patients diagnosed with LMNA-related muscular dystrophy.
Methods: Baseline cardiac work-up consisted of clinical assessment, transthoracic Doppler echocardiography, 12-lead electrocardiogram, electrophysiological study, and implantation of a long-term implantable cardiac loop recorder (ILR).
Results: We enrolled twenty-eight pediatric patients diagnosed with EDMD (13 patients), L-CMD (11 patients), LGMD1B (2 patients), and LMNA-related mild weakness (2 patients). Follow-up showed dilated cardiomyopathy (DCM) in six patients and malignant arrhythmias in five (four concomitant with DCM) detected by the ILR that required implantable cardioverter defibrillator (ICD) implantation. Malignant arrhythmias were detected in 20% of our cohort and early-onset EDMD showed worse cardiac prognosis.
Discussion: Patients diagnosed with early-onset EDMD are at higher risk of DCM, while potentially life-threatening arrhythmias without DCM appear earlier in L-CMD patients. Early onset neurologic symptoms could be related with worse cardiac prognosis. Specific clinical guidelines for children are needed to prevent sudden death.
LMNA-related muscular dystrophy is a very rare (0.5 per 100,000) disease caused by pathogenic alterations in the LMNA gene. The disorder is characterized by cervical–axial weakness, scapuloperoneal weakness, joint contractures, thoracic lordosis, a dystrophic muscle biopsy, and mildly elevated creatine kinase levels (Quijano-roy et al., 2008). Children with early-onset LMNA-related muscular dystrophy may show decreased fetal movement and early lack of motor development since the first months of life, or later develop a loss of head and trunk control and ability to walk or sit, followed by progressive loss of axial and limb motor function. As these patients age, there is an increased risk for respiratory insufficiency, appears joint and spinal deformities and cardiac involvement. Practically all patients exhibit heart disease in long follow up studies (Dubowitz, 1999; Quijano-roy et al., 2008).
LMNA encodes the nuclear envelope proteins lamins A and C, intermediate filaments that are required during development and cell differentiation (Bonne et al., 2003). Lamins facilitate signal transduction between the cytoskeleton and the nucleus (Aebi et al., 1986; Worman and Bonne, 2007), provide genome stability and modulation of chromatin organization and gene expression (Lammerding et al., 2004; Dechat et al., 2007; Andrés and González, 2009; Dauer and Worman, 2009; Gonzalez-Suarez et al., 2009; Hutchison, 2011). Lamins consist of a globular N-terminal head domain, a central coiled-coil rod domain implicated in protein dimerization, and a C-terminal tail domain that includes an immunoglobulin-like domain where various posttranslational modifications occur (Burke and Stewart, 2012; Ho and Lammerding, 2012). LMNA was first identified in 1986 in humans, but it was not until 1999 that a pathogenic rare variant in the LMNA gene was linked to Emery–Dreifuss muscular dystrophy (EDMD) (Bonne et al., 1999). To date, over 600 disease-causing rare LMNA variants are characterized (Worman and Bonne, 2007; Dittmer and Misteli, 2011), and these “laminopathies” are associated with heterogeneous clinical phenotypes, including neuromuscular, cardiac, and metabolic disorders (Bertrand et al., 2011; Worman, 2012; Maggi et al., 2014). However, there is no clear correlation between genotype and phenotype, including within the same muscle, suggesting the presence of genetic modifiers and representing an example of allelic heterogeneity (Granger et al., 2011).
Muscle laminopathies may associate cardiac disease at any age and range from congenital muscular dystrophy (LMNA-related congenital muscular dystrophy, or L-CMD) to late-onset manifestations (limb–girdle muscular dystrophy 1B, or LGMD1B; and autosomal-dominant EDMD). L-CMD is the most severe and early phenotype, and typically presents in the first 2 years of life, either by an arrest of motor milestones before sitting or walking are acquired, or as a later presentation with a characteristic loss of head support, while sitting and or walking are still maintained (dropped head syndrome). It is a very progressive and severe disease, and shares with EDMD a recognizable scapulo-humero-peroneal pattern of muscle weakness and atrophy (predominantly proximal in upper limbs and distal in lower limbs). The heart may be involved in all three entities, and the manifestation of heart disease may precede muscle weakness or be isolated. Globally, early-onset phenotypes before 5 years of age, and specially before 2 years of age, are related with worse motor and cardiac prognosis, although heart involvement is rarely observed initially (Granger et al., 2011; Carboni et al., 2013; Maggi et al., 2014; Ben Yaou et al., 2021). Dilated cardiomyopathy (DCM) with conduction disease and sudden cardiac death (SCD) can occur in LMNA-related muscular dystrophies, in children and adults (Finsterer et al., 2006; Finsterer et al., 2010; Lu et al., 2011; Groh, 2012; Quarta et al., 2012; Cattin et al., 2013; Hasselberg et al., 2014; Alastalo et al., 2015; Dobrzynska et al., 2016; Finsterer and St, 2016; Heller et al., 2017; Muscogiuri, 2017; Wang et al., 2017; Groh et al., 2022). These patients have a high incidence of malignant arrhythmias at early ages, worsening prognosis and posing a clinical challenge for cardiologists, neurologists, and genetic counselors (Meune et al., 2006; Groh, 2012; Rijsingen et al., 2012; Carboni et al., 2013; Rajdev and Groh, 2015; Kumar et al., 2016; Feingold et al., 2017; Peretto et al., 2019; Ben Yaou et al., 2021). Despite the risk prediction scores for life-threatening ventricular tachyarrhythmias (VT) have been defined in adults with laminopathies (Wahbi et al., 2019; Marchel et al., 2021a; Groh et al., 2022), there are no published guidelines for risk prediction scores in pediatric laminopathies to prevent early life-threatening VT. Given the rarity of the disease in the pediatric population, phenotype–genotype correlations are difficult to be established, and data on age of onset and course of the cardiac disease or the risk of malignant arrhythmias and SCD is scarce (Ben Yaou et al., 2021).
To bridge this knowledge gap, we performed a mid-to-long-term cardiovascular follow-up study focused on cardiomyopathies and arrhythmia during childhood and genotype–phenotype correlation in several families with LMNA-related muscular dystrophy.
We enrolled patients <18 years of age from the international reference center in neuromuscular diseases at our institution that were diagnosed with LMNA-related muscular dystrophy (carrying a rare pathogenic or likely pathogenic variant in the LMNA gene with L-CMD, EDMD, LGMD1B, and LMNA-related atypical phenotype with mild weakness) between 2014 and 2020. The study was motivated and promoted by the national patient association. Due to the severity of pediatric laminopathies, we analyzed in an independent category all the patients with early-onset neuromuscular phenotypes (before 2 years of age) regardless of the final diagnosed clinical phenotype (L-CMD and early-onset EDMD). At enrollment, we collected retrospective clinical data. Participants were prospectively followed from enrollment to the last follow-up evaluation according to a specific schedule. Written informed consent to participate was provided by the participants’ legal guardians. Non-LMNA-related neuromuscular dystrophies were excluded from this study.
Baseline cardiac work-up consisted of a clinical evaluation, DNA samples from the patient and first-degree relatives, transthoracic Doppler echocardiography (Philips, IE33, software Intellispace Cardiovascular for standard measurements and QLAB for offline strain analysis), a 12-lead electrocardiogram (ECG), electrophysiological study (EPS), and implantation of a long-term cardiac implantable loop recorder (ILR; Medtronic Reveal LINQ) with a home monitoring system. The definition of DCM and reference values of echocardiographic measurements were adopted from the most recent pediatric guidelines and normal values published for children (Lang et al., 2005; Pettersen et al., 2008; Lee et al., 2014; Lee et al., 2017; Lipshultz et al., 2019).
EPS was performed in all patients at the time of inclusion to exclude underlying arrhythmogenic conditions and arrhythmia inducibility and to describe cardiac conduction system characteristics. The procedure was performed under mild sedation via the brachial or femoral vein (depending on joint retractions and grade of hyperlordosis). Baseline neuromuscular evaluation consisted of a detailed and standardized neurologic examination with diagnostic tests based on clinical indications. Retrospective clinical data from the referral center were collected. During follow-up, cardiologic and neurologic data were collected at least once a year; arrhythmia events were reviewed daily by long-term ILR software using a home monitoring system. Medical therapy and device implantation (pacemaker [PM] and implantable cardioverter defibrillator [ICD]) were indicated according to current clinical evidence.
For the echocardiogram analysis, a single observer obtained the following measurements at enrollment and during the follow-up: M-mode-derived left ventricular end-diastolic and end-systolic dimensions, left ventricular ejection fraction (LVEF; %) measured by the Simpson method, tricuspid annular plane systolic excursion (TAPSE; mm), mitral annular plane systolic excursion (MAPSE; mm), tissue doppler values from lateral and septal mitral annulus (cm/s), and spectral doppler E and A waves from inflow mitral and tricuspid filling pattern (cm/s). Global longitudinal strain (GLS [%]) was obtained from four-chamber view, and reference values (mean and p5) were obtained from published pediatric data (Koopman et al., 2019).
Major cardiac events included cardiac death, heart transplant, and malignant arrhythmias, which were defined as sustained VT, ventricular fibrillation (VF), asystole, complete atrioventricular block (AVB), cardiac arrest from VT/VF (witnessed SCD occurring within 1 h of acute symptoms), or appropriate treatment (antitachycardia pacing or shock) by ICD. Minor cardiac events included worsening of heart failure, reaching DCM criteria, conduction system abnormalities (except complete AVB), supraventricular tachycardia, or any structural or functional echocardiographic abnormality according to updated definitions and references. The timing of each event was reported as age, years from clinical onset, and months from study enrollment.
Data were anonymized and stored in local institutions. We used StatCrunch (Pearson Education Inc.) for the statistical analysis. Categorical variables were expressed as numbers and percentages, and continuous variables were expressed as median and interquartile range (IQR). When appropriate, echocardiographic data were analyzed comparing enrollment and last control measurements. Because the data were not normally distributed, a Mann–Whitney U non-parametric test was used, as appropriate, for analysis of quantitative variables between two groups. A p-value of <0.05 was considered statistically significant. Graphical presentations of the major and minor events that occurred during follow-up were generated with GraphPad Software (Prism, version 9.1.1).
Twenty-eight individuals (median age of 8.5 years at enrollment; IQR of 4–12.5 years) from 27 families were enrolled. Table 1 summarizes the main clinical and genetic features of our cohort (Bonne et al., 2000a; Quijano-roy et al., 2008; Chemla et al., 2010; Komaki et al., 2011; Pasqualin et al., 2014; Heller et al., 2017; Fan et al., 2020). The median age at last follow-up was 13 years (IQR of 8–17 years). All participants met clinical criteria and had confirmed LMNA-related neuromuscular disease. 13 (46.43%) had EDMD, 11 had L-CMD (39.28%), 2 (7.14%) had LGMD1B, and 2 (7.14%) had an LMNA-related atypical phenotype with mild weakness (Supplementary Figure S1). All had a pediatric onset of skeletal muscle symptoms, with most of them presenting before 2 years of age (23 of the 28 cases, 82%).

TABLE 1. Clinical and genetic data.
The median follow-up from study enrollment to last clinical evaluation was 4 years (IQR of 3–5 years) (Supplementary Figure S2). All living participants (except eight patients) had a comprehensive cardiac follow-up every 6 months that included echocardiography and remote cardiac rhythm monitoring. In the other eight patients, only clinical data and remote cardiac rhythm monitoring were available. Events by age and events by enrollment date are represented graphically in Figure 1 (Figures 1A, B).
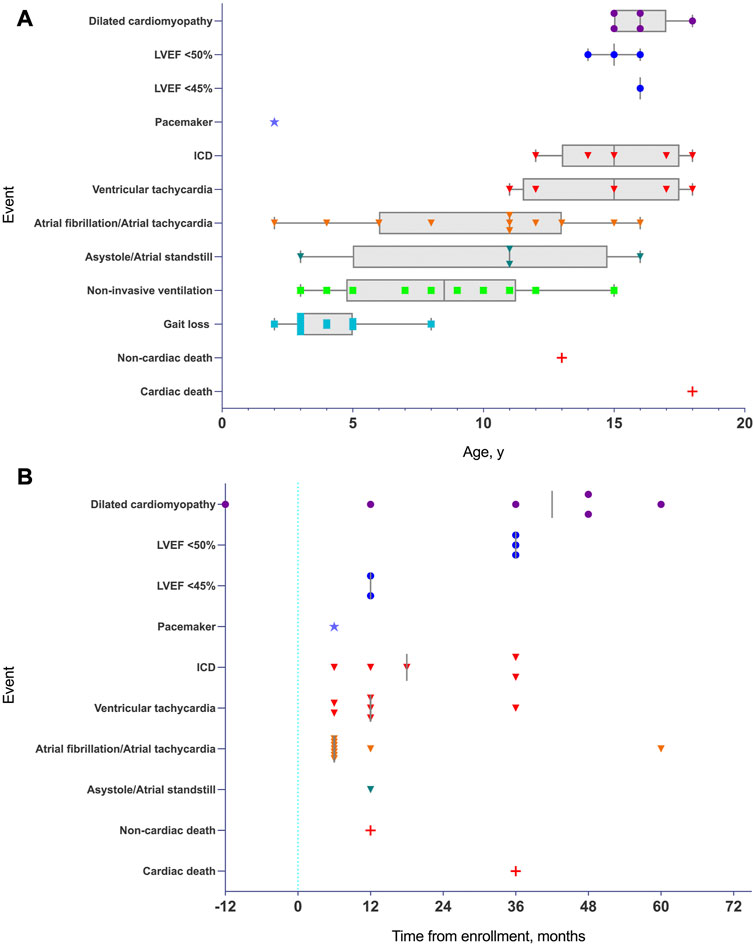
FIGURE 1. (A): Timeline of major and minor events in pediatric patients with lamin-related muscular dystrophy defined on the basis of age. Atrial arrhythmias, asystole/atrial standstill, and malignant arrhythmias preceded structural and cardiac functional abnormalities (DCM criteria or depressed LVEF). Median time values are represented by vertical grey line when required. DCM and LVEF are represented by a dot. Pacemaker are represented by a star. Arrhythmias and ICD are represented by an inverted triangle. Gait loss and NIMV are represented by a square, and death are represented by a cross. When required, minimum to maximum range age is represented in grey box and whiskers. Abbreviations: LVEF, left ventricular ejection fraction. (B): Timeline of major and minor events in pediatric patients with lamin-related muscular dystrophy defined on the basis of enrollment (time 0 is the time of enrollment represented by a vertical blue dotted line). Median time values are represented by a vertical grey line. DCM and LVEF are represented by a dot, pacemaker is represented by a star, arrhythmias and ICD are represented by an inverted triangle and death is represented by a cross. Abbreviations: LVEF, left ventricular ejection fraction.
A total of 57.1% of participants were male. Two of the cases enrolled were female monozygotic twins. Families were originally from Spain (Bonne et al., 1999), the United Kingdom (Bonne et al., 2003), the United States (Bonne et al., 2003), Australia (Dubowitz, 1999), Canada (Quijano-roy et al., 2008), France (Quijano-roy et al., 2008), Greece (Quijano-roy et al., 2008), Russia (Quijano-roy et al., 2008), and Venezuela (Quijano-roy et al., 2008). Eleven patients (39.29%) had L-CMD, 13 (46.43%) had EDMD, 2 (7.14%) had LGMD1B, and 2 (7.14%) had an LMNA-related atypical phenotype with mild weakness. Early-onset skeletal muscle impairment before 2 years of age was detected in 23 of the 28 cases (82%). Table 1 summarizes the main clinical and genetic features of our cohort (Bonne et al., 2000a; Quijano-roy et al., 2008; Chemla et al., 2010; Komaki et al., 2011; Pasqualin et al., 2014; Heller et al., 2017; Fan et al., 2020). Eleven patients (39.2%; seven males) required non-invasive mechanical ventilation (NIMV) either at inclusion or during follow-up. Patients requiring NIMV had early-onset EDMD and L-CMD phenotypes. For those with gait loss, the median age for gait loss was 6 years (IQR of 5–8 years). Supplementary Table S1 compares our cohort with previous publications not focused on pediatric patients.
Table 2 and Supplementary Table S2 summarize major and minor cardiac events and statistical analyses of the echocardiographic data. During follow-up, rapid progression to DCM was seen in six cases (21.4%). (patient 3 [died], 5, 9, 11, 13, and 17), all showing early neuromuscular impairment before 2 years of age; five were diagnosed with early-onset EDMD, and one was diagnosed with L-CMD (patient 11).
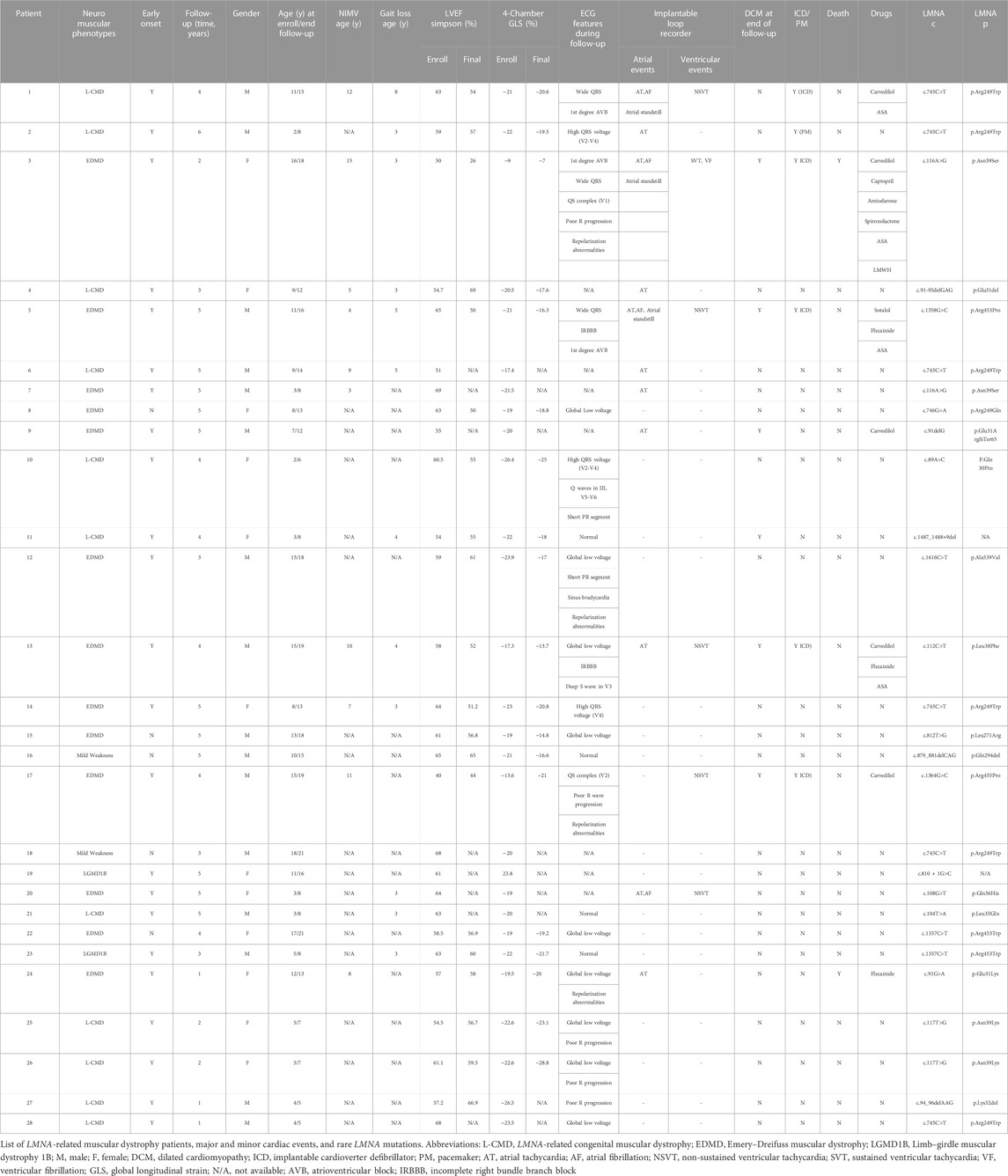
TABLE 2. Demographic data and overall follow-up.
Two of these patients showed LVEF values of less than 45% at last follow-up, and four showed DCM requiring an ICD. No right ventricle (RV) involvement was detected in our cohort, but advanced RV myocardial strain analysis could not be adequately performed because of suboptimal image quality from the RV related to chest wall deformities in the majority of patients.
LVEF was compared by the Simpson method in all 28 pediatric patients from enrollment to the end of follow-up, and a global reduction in LVEF during follow-up was found (LVEF 60.75% [IQR of 56–63.5] versus 56.75% [IQR of 51.6–59.75], p < 0.05; Figure 2A). Patients requiring an ICD (patients 1, 3, 5, 13, and 17) showed worse LVEF values at last follow-up than the rest of the cohort and were older (median age at enrollment of 15 years [IQR of 11–15] and median age at last follow-up of 18 years [IQR of 16–19]) than the rest of the cohort (median age at enrollment of 7 years [IQR of 3–11] and median age at last follow-up of 12 years [IQR of 8–15]). The median time of follow-up was 4 years in the group requiring an ICD (IQR of 4 to 4) and in the rest of the cohort (IQR of 3–5). Five patients showed LVEF values between 45% and 55% (patients 1, 5, 8, 13, and 14). No significant differences in TAPSE were observed when comparing values at enrollment with those at last follow-up (TAPSE of 19.9 mm [IQR of 15.3–22]). The value of TAPSE was ≤17 mm at last follow-up in four of five patients requiring an ICD. GLS (%) analyzed in the apical four-chamber view showed significantly lower values at enrollment than at last follow-up (−21 [IQR of −19 to −22.6] versus −17.6 [IQR of −16.3 to −20.6], p = 0.01; Figure 2B). At enrollment, GLS values below the mean (<−20.6%) were detected despite normal LVEF values (>55%) in most participants. At last follow-up, these patients showed decreasing GLS and LVEF (Figures 2A, 3B). No other significant findings were detected in the analysis of MAPSE, lateral E/E′ ratio, and septal E/E′ ratio.
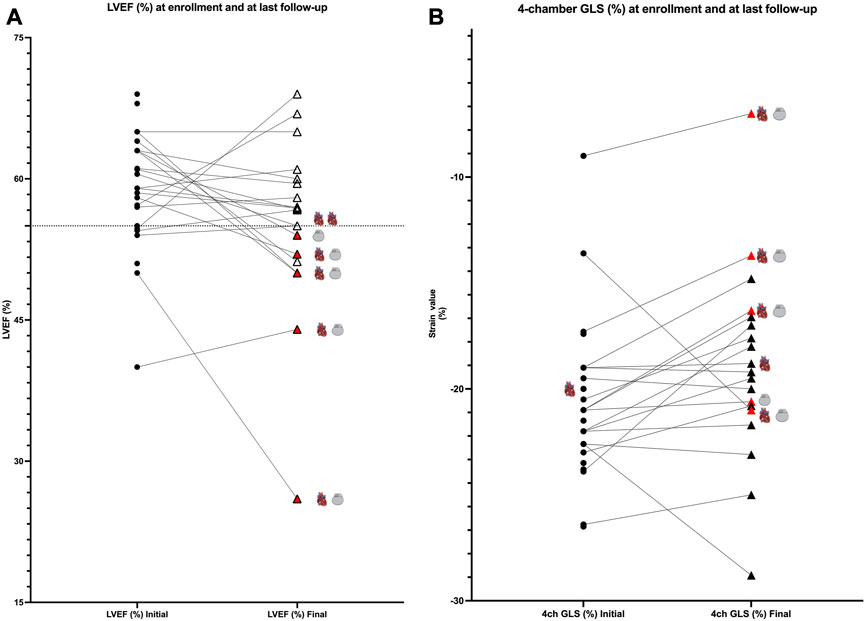
FIGURE 2. (A): comparison of values at enrollment and at last follow-up; LVEF values (%). Patients are represented as black dots at enrollment, and triangles represent patients at last follow-up. Red triangles show patients requiring an ICD (gray device represented in the graphic). Cardiomyopathy is represented by a heart. Black lines link the same patient in the two different moments of follow-up. (B): comparison of values at enrollment and at last follow-up; four-chamber GLS values (%). Patients are represented as black dots at enrollment and triangles at last follow-up. Red triangles show patients requiring an ICD (gray device represented in the graphic). Cardiomyopathy is represented by a heart. Black lines link the same patient in the two different moments of follow-up.
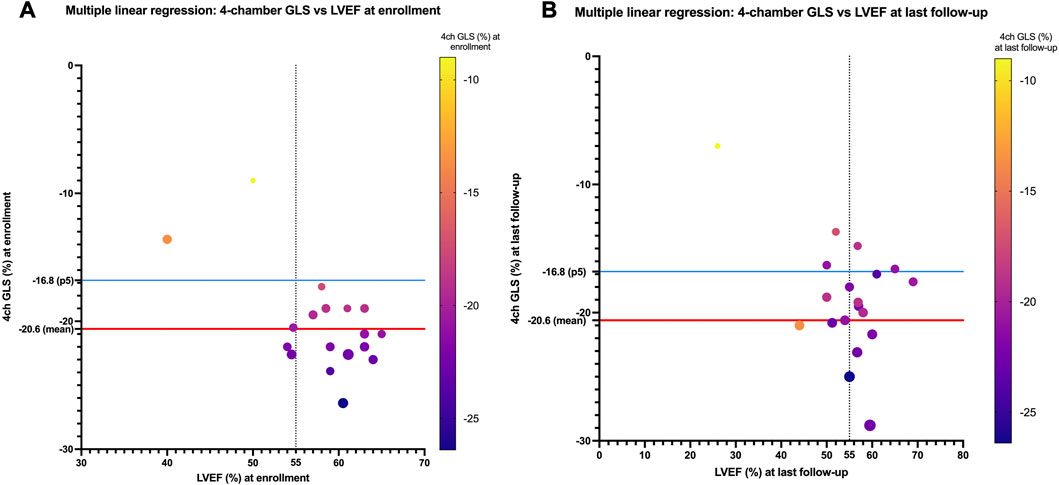
FIGURE 3. (A) Four-chamber GLS values (%) compared with LVEF values (%) for each patient at enrollment. At enrollment, despite normal LVEF (>55%), values below 20.6% are detected (20.6% is the mean normal value published in children). These LVEF values show a decreasing trend at last follow-up (B).
Patients that finally developed DCM showed their first neuromuscular symptoms before 2 years of age (early-onset EDMD and L-CMD phenotypes) suggesting an early-onset aggressive form of laminopathy.
The ECG analysis is summarized in Table 2. Minor ECG abnormalities were present in 16 patients. Within these minor abnormalities, the following were described: first-degree AVB, wide QRS complex, short PR interval, global QRS low voltage, high QRS voltage V1–V4, QS complex in V1–V2, poor R progression, incomplete right bundle-branch block (IRBBB), abnormal Q waves, sinus bradycardia, and repolarization abnormalities. The most frequent finding (28.5%, eight patients) was global low QRS voltage that presented early; seven of eight cases had neither ventricular dysfunction nor DCM (Supplementary Figures S3A–C). The early-onset group showed the following ECG abnormalities before 2 years of age: global QRS low voltage and poor R progression.
EPS at enrollment showed seven patients (25%) with atrial conduction disorders (short non-sustained atrial tachycardia [n = 6] and intermittent atrial standstill [n = 1]) that did not merit any further treatment at that moment. No other forms of supraventricular or ventricular tachycardia were induced, and no accessory pathways were detected. HV intervals (time from the proximal His bundle to the ventricular myocardium) were within normal values. When retrospectively analyzing the EPS data in patients who presented with ventricular arrhythmia during follow-up, no abnormalities were observed.
In the ILR monitoring device analysis, malignant arrhythmia (VT/VF) was detected in five cases. (patients 1, 3 [died], 5, 13, and 17). Supplementary Table S3 summarizes the indication and the median age for device implantation. All had normal EPS results at enrollment and were never diagnosed with malignant arrhythmias at our center or from a referring hospital. In all five cases, malignant arrhythmias were detected a few months after enrollment, and they were then considered candidates for ICD implantation (Supplementary Table S3). Two of these patients that were diagnosed with VT through the ILR monitoring device had histories of intermittent pallor and fainting episodes. After ICD implantation, two patients received appropriate shocks. No inappropriate shocks were detected during follow-up. All patients carrying an ICD showed their first neuromuscular symptoms before 2 years of age.
In one participant with an L-CMD phenotype, a premature PM implantation was required because of symptomatic prolonged asystole (patient 2, 2-year-old boy). Patients 1, 3, and 5 (median age of 11 years [IQR of 11–16]) showed intermittent atrial standstill detected in traces from the ILR. During follow-up, atrial fibrillation was diagnosed in four cases (patients 1, 3, 5, and 20; median age at atrial fibrillation diagnosis of 11 years [IQR of 7.5–13.5]), and three were later diagnosed with DCM. Atrial tachycardia was diagnosed in 11 patients (patients 1–7, 9, 13, 20, and 24; median age at atrial tachycardia diagnosis of 11 years [IQR of 6–12]), and 4 were later diagnosed with DCM. Both atrial tachycardia and atrial fibrillation were present simultaneously in four patients. Example electrocardiographic traces from an ILR monitoring device are represented in Figure 4. Neither arrhythmia nor major cardiac events were detected within the patients with mild muscular impairment and late onset after 2 years old of age (patients 8, 15, 16, 18, and 22).
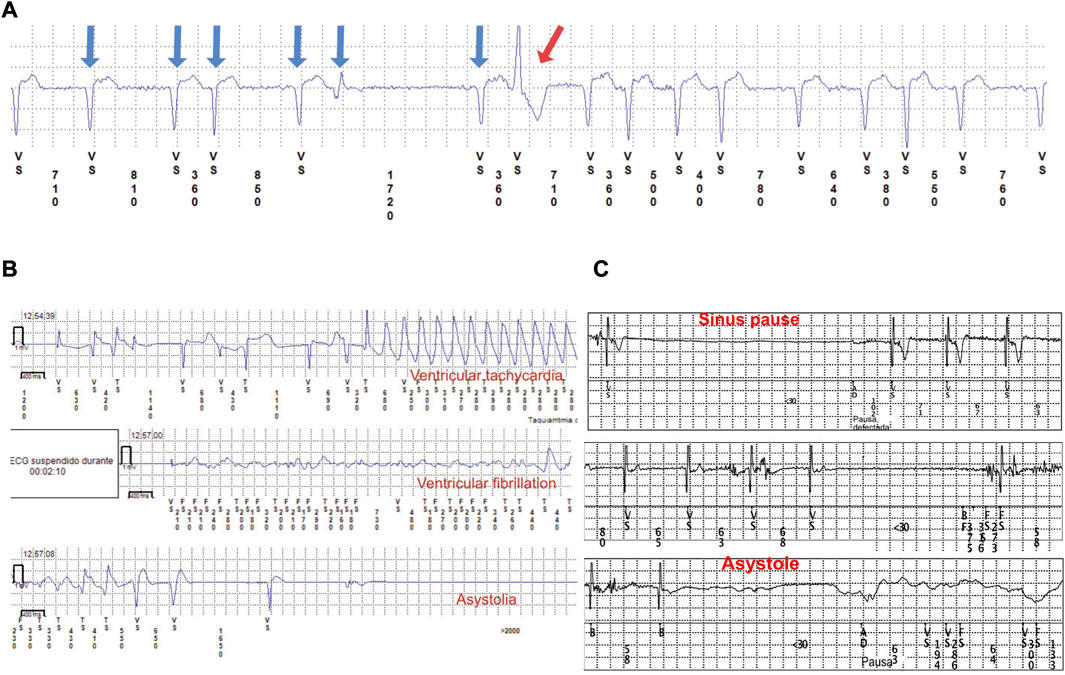
FIGURE 4. Three ECG traces from the ILR. (A) Atrial fibrillation (blue arrows) and a ventricular extrasystole (red arrow). (B) Ventricular tachycardia, VF, and asystole from a pediatric patient diagnosed with EDMD. (C) Sinus pause and asystole in a 3-year-old patient.
Two patients (2/28, 7.1%) died during follow-up. One (patient 3) was an 18-year-old female with intermittent atrial standstill diagnosed in the EPS and had an ICD due to VT/VF. The patient died because of rapidly progressive heart failure despite optimal treatment. Patient 24 was a 10-year-old female with no cardiovascular involvement who died due to respiratory infection. Both patients were early-onset EDMD, non-ambulant, required NIMV, and showed their first clinical manifestations of muscle weakness before 2 years of age.
All 28 patients carried one rare variant in LMNA classified as likely pathogenic or pathogenic following American College of Medical Genetics (ACMG) guidelines and according to currently available data (Supplementary Table S4). Family segregation showed that in 26 cases (92.85%) the rare variant in LMNA was de novo. Family history of cardiac disease or neurological impairment was present in only two cases (patients 11 [c.1487_1488+9del] and 20 [p.Gln36His]). In both cases, the parents carried the same variant in LMNA and had mild neuromuscular impairment and malignant arrhythmias.
Twenty rare LMNA variants were identified (18 exonic and 2 intronic). Of all exonic rare variants, 4 were deletions, and 14 were missense. Thirteen rare variants (65%) were classified as likely pathogenic and seven (35%) as definitively pathogenic. Twelve variants were novel (two intronic, two deletions, and eight missense; Supplementary Table S4).
The most prevalent variant identified was p. Arg249Trp (six patients, 21.4%) located in exon 4 of LMNA. These six patients were diagnosed with L-CMD (four patients), EDMD (one patient), or lamin-related mild weakness (one patient). One patient showed malignant arrhythmias and required an ICD, and another patient needed a PM due to asystole (both L-CMD with dropped head). No DCM was detected with this rare variant. Figure 5 shows the correlation of genotype to phenotype of all 28 patients. The LMNA mutations identified in the six patients with DCM (patient 3 (died), 5, 9, 11, 13 and 17) were p. Asn39Ser, p. Arg453Pro, p. Glu31ArgfsTer65, c.1487_1488+9del, p. Leu38Phe, and p. Arg455Pro. In the five patients with ICDs implanted due to malignant arrhythmias (patients 1, 3 [died], 5, 13, and 17), all carried rare variants often identified in pediatric LMNA-muscular dystrophy patients (p.Arg249Trp, p. Asn39Ser, p. Arg453Pro, p. Leu38Phe and p. Arg455Pro, respectively). Four cases were diagnosed with EDMD and one with L-CMD. The two cases who died (patients 3 [with an ICD] and 24) carried also a rare variant (p.Asn39Ser and p. Glu31Lys, respectively), both located in exon 1 of LMNA. Both were diagnosed with early-onset EDMD and showed their first neuromuscular symptoms before 2 years of age.
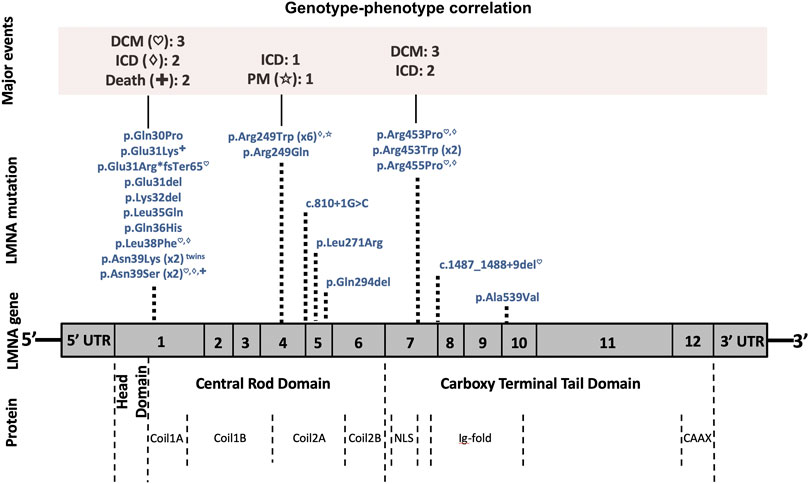
FIGURE 5. Phenotype–genotype graphic correlation. A schematic representation of the LMNA gene is shown. Top, major cardiac events, followed by rare pathogenic/likely pathogenic LMNA variants identified in our patients with the corresponding amino acid or nucleotide changes. Bottom, structural protein: the coil 1A–1B and coil 2A–2B constituting the ⍺-helical rod domain. Abbreviations: DCM, dilated cardiomyopathy; ICD, implantable cardioverter defibrillator; PM, pacemaker; NLS, nuclear location signal; Ig, immunoglobulin.
Laminopathies are a group of ultrarare genetic diseases attributable to pathogenic rare variants in the LMNA gene (Worman and Bonne, 2007; Bonne and Quijano-Roy, 2013). Due to its low prevalence, few cases have been diagnosed and reported so far, and there are not enough published data about its natural history. This fact, reinforces the importance of our prospective pediatric international registry including congenital phenotypes. Other multicenter laminopathy studies and expert consensus statement on arrhythmic risk have been published for adult populations (Pasotti et al., 2008; Rijsingen et al., 2012; Haas et al., 2014; Kumar et al., 2016; Peretto et al., 2019; Groh et al., 2022) but did not focus on cardiac involvement in children, as we describe here. Additionally, reports on cardiac impairment in pediatric laminopathies with neuromuscular involvement are lacking, despite that cardiac events can be present at early stages (Benedetti et al., 2007; Maggi et al., 2014; Fan et al., 2020; Groh et al., 2022). Our study offers longer follow-up in a pediatric population and valuable information about cardiac events, neuromuscular phenotypes, and genotype–phenotype correlations.
LMNA-related diseases cause cardiac symptoms in children and young adults that gradually worsen to bradyarrhythmias and tachyarrhythmias (Groh, 2012; Rijsingen et al., 2012; Rijsingen et al., 2013; Alexandra et al., 2015; Priori et al., 2015; Groh et al., 2022). Approximately half of patients need a PM or an ICD during adulthood (Taylor et al., 2003; Priori et al., 2015). Cardiac involvement occurs in a high percentage of cases, and implantation of preventative measures, such as an ICD, is recommended in patients with malignant arrhythmias (Pasotti et al., 2008; Rijsingen et al., 2012; Maggi et al., 2014; Groh et al., 2022). Pathogenesis of LMNA-related cardiomyopathy remains unclear, but lamin A/C haploinsufficiency may have negative effects on the heart (Wolf et al., 2008). Both fibroblasts from L-CMD patients and myoblasts from L-CMD mouse models demonstrate increased nucleoplasmic localization of lamin A/C compared to controls and EDMD. Mislocalization of nuclear envelope proteins leads to defects in myoblast differentiation, contributing to the more severe phenotype observed in L-CMD. (Bertrand et al., 2020). Clinical recommendations for these patients were identical to those for patients with other cardiomyopathies or heart failure: pharmacologic treatment with neurohormonal antagonists, diuretics, and vasodilators and non-pharmacological treatment with ventricular device therapy, such as early PM or resynchronization therapy for progressive conduction delays and an ICD to prevent SCD (Yancy et al., 2013; Priori et al., 2015; Bozkurt et al., 2016; Yancy et al., 2016; Atalaia et al., 2021). An expert consensus on evaluation and management of arrhythmic risk in neuromuscular disorders, published recently highlighted the especial recommendations for diagnostic testing and risk stratification in adults affected with EDMD or LGMD1B. Within the recommendations in adult population, implantable cardiac monitoring is reasonable even in the setting of a normal 12-lead ECG, ambulatory ECG monitoring and normal echocardiogram. Sustained arrhythmias, AVB and SCD are highly present in these patients and comprehensive risk stratification is needed, including EP study in select patients. Heart transplantation may be considered in certain cases with mild neuromuscular impairment (Taylor et al., 2003; Finsterer et al., 2006; Maggi et al., 2014; Pérez-Serra et al., 2016).
Six pediatric patients carrying a deleterious rare variant in LMNA concomitant with congenital heart disease (CHD) were previously found to have no major neuromuscular involvement (Baban et al., 2020) Most cases showed a family history of CHD and/or DCM associated with arrhythmias. Neither CHD nor aortic involvement were found in our cohort. Despite a rare deleterious variant in the LMNA gene, this previously reported phenotype is not similar to our study because most of our cases showed severe neuromuscular involvement, and rare LMNA variants are de novo. The wide range of phenotypes associated with rare pathogenic variants located in the LMNA gene are well known. During follow-up in a retrospective series of 15 pediatric patients carrying LMNA variants with early-onset neuromuscular symptoms, no major cardiac involvement was described (two cases of supraventricular arrhythmia, no malignant arrhythmias, no DCM, and no sudden death) or specific cardiovascular description (Jędrzejowska et al., 2021). In contrast to theses previous studies, our study characterizes the impact of LMNA variants in pediatric patients with both neuromuscular and cardiovascular involvement.
To our knowledge, no more than 10 reports have focused on pediatric laminopathies with both neuromuscular and cardiac involvement. In a retrospective study, 151 patients carrying a mutation in LMNA showed an early-onset phenotype, and the most frequent mutation was p. Arg249Trp; 63% of patients never acquired independent ambulation, and 37% died. Early cardiac interventions (heart medication and/or PM or ICD implantation) were usually associated with earlier respiratory interventions (intermittent positive pressure breathing, non-invasive ventilation of tracheostomy), and clinical severity was positively correlated across the triad of skeletal muscles, respiratory muscles, and myocardium. Correlation to cardiac device placement is less strongly linked with the timing of respiratory interventions and the progression of skeletal muscle weakness. Prospective natural history studies should therefore be conducted to further validate the stratification of L-CMD (Ben Yaou et al., 2021). We performed a prospective cardiac natural history of pediatric patients with LMNA-related muscular dystrophy.
Other studies included no more than eight patients, were retrospective, and had no adoption of preventive arrhythmogenic measures or follow-up (Bonne et al., 2000b; Komaki et al., 2011; Pasqualin et al., 2014; Parent et al., 2015; Petillo et al., 2015; Tan et al., 2015; Heller et al., 2017). In our cohort, patients 1, 2, 6, 14, 18, and 28 carried variant p. Arg249Trp, which is present in pediatric patients with arrhythmic complications without major ventricular dysfunction and could be related to neuromuscular severity (Komaki et al., 2011; Ben Yaou et al., 2021). Patient 3 and patient seven carried p. Asn39Ser, which was previously described in two patients (one died of cardiac arrest due to malignant arrhythmias, and one had premature ventricular contractions [PVCs]) (Pasqualin et al., 2014). Patient 8 carried p. Arg249Gln, which was previously reported in children with arrhythmias (Bonne et al., 2000b; Komaki et al., 2011; Heller et al., 2017).
Two groups are distinguished depending of the severity of onset, a very early form with arrest of motor development before the age of 6 months (no sitting or walking acquisition) and another with initial normal or subnormal motor milestones and subsequent loss, beginning by a characteristic presentation of loss of head support (dropped head syndrome) (Quijano-roy et al., 2008). All children have a progressive course with an initial rapid decline in cervical and axial tonus strength followed by a period of slower progression or plateau. Respiratory insufficiency is a major complication in the course of both entities, being responsible for early death in the early severe group, as early as the first 2 years of age (if no adequate ventilatory support is provided), or leading to mechanical ventilation which can evolve from non-invasive to invasive ventilation via tracheostomy in few years. Cardiac involvement is rarely observed initially in these children and is often subclinical in very young patients, but SCD can occur (Quijano-roy et al., 2008); therefore, routine cardiac follow-up is recommended (Quijano-roy et al., 2008; Lu et al., 2011). Our study revealed only one case (dropped head) with mild DCM with borderline LVEF values diagnosed before 10 years of age. During our follow-up, malignant arrhythmia was detected in one case (patient 1, dropped head) and an ICD was implanted at 12 years of age. Four patients showed atrial tachycardia, and one patient (patient 1, dropped head and non-ambulant) showed simultaneous atrial fibrillation and atrial tachycardia starting at 11 years of age. An ambulant child with dropped head syndrome (patient 2) had asystole and needed a PM. Both patients carried the same pathogenic rare LMNA variant p. Arg249Trp, which is the most frequent in pediatric squeletal laminopathies and is associated with worse clinical prognosis (Ben Yaou et al., 2021). These results support the need for close follow-up, which could show subclinical cardiac involvement in pediatric patients diagnosed with L-CMD. In a recent publication, echocardiography abnormalities were identified in 22% of L-CMD patients, and device implantation (ICD and PM) was described in 9% and 7% of the cohort, respectively (Ben Yaou et al., 2021); however, there is a lack information about the cause of death and reason for device implantation (Heller et al., 2017; Ben Yaou et al., 2021).
EDMD is the third most prevalent muscular dystrophy, and most patients present with autosomal-dominant EDMD due to LMNA (with higher risk of VT and DCM). EMD (emerin) gene is less frequent, with X-linked transmission (Bonne et al., 1999; Bonne and Quijano-Roy, 2013). However, more than 60% of EDMD cases have no deleterious variant in EMD or LMNA genes. Cardiac complications in EDMD patients can be life-threatening and lead to progressive cardiac failure, cardiac conduction disease, and SCD. DCM may occur at an advanced stage, but conduction disease is frequent (complete heart block, silent atria, atrial tachycardia, atrial fibrillation, and VT) (Figures 4B, C). Patients with EDMD are also at risk of cerebral emboli and sudden death (Bonne et al., 1999; Lu et al., 2011; Bonne and Quijano-Roy, 2013; Cattin et al., 2013; Finsterer et al., 2015; Priori et al., 2015; Marchel et al., 2021a; Marchel et al., 2021b; Groh et al., 2022). In our study, early presentation of atrial tachycardia (seven patients), atrial fibrillation (three patients), and different grades of ventricular dysfunction (six patients) that led to heart failure and malignant arrhythmias (four patients) were detected in early-onset EDMD children. Four patients required an ICD because of malignant arrhythmias (between 14 and 18 years old at ICD implantation). Surprisingly, all showed an EPS with no ventricular arrhythmia Inducibility, and the arrythmias were instead detected with the ILR. These malignant arrhythmias corresponded to sustained and non-sustained VT, and all were asymptomatic except patient 3 who exhibited pallor and seizures during VT/VF episodes. As previously described (Steckiewicz et al., 2016; Marchel et al., 2021a), the low rate of symptoms and presentation of atypical symptoms could make the global management of patients with EDMD difficult, worsening the prognosis. Antiaggregant therapy was needed in four patients because of atrial standstill, and one patient (patient 3) presented with an episode of cerebral emboli at 17 years of age, likely of multifactorial origin, before starting treatment. LMNA carriers have an increased risk of thromboembolic events, with an inherent risk independent of heart condition (Rijsingen et al., 2012; Rijsingen et al., 2013; Van Rijsingen et al., 2013; Groh et al., 2022) This thromboembolic risk could be related to cardiac dysfunction secondary to DCM and/or cardiac conduction disease, such as silent atria, sinus node dysfunction, sinus pauses, and atrial fibrillation (Groh et al., 2022). Long-term systemic anticoagulation therapy with a vitamin K antagonist and international normalized ratio (INR) target of 2.0–3.0 would be reasonable in DCM patients with arrhythmias, previous thromboembolic events, thrombophilic conditions, or an ejection fraction of ≤25%, but there is no evidence in pediatric laminopathies and traditional algorithms, such as CHA2DS2-VASc, are inappropriate for children. When silent atria or atrial fibrillation are present in children, long-term anticoagulation therapy is only indicated when there are other risk factors (previous stroke, transient ischemic attack, hypertension, or heart failure). As atrial arrhythmias, immobilization, and heart failure are usually concomitant findings in LMNA-related muscular dystrophy patients, long-term systemic anticoagulation therapy may benefit higher-risk patients, for example, those with a LVEF of ≤25% and atrial arrhythmias. If patients have any contraindication to receive vitamin K antagonist or low-molecular-weight heparin, aspirin should be recommended (Monagle, 2012; Giglia et al., 2015). We observed that patients diagnosed with early-onset EDMD are at higher risk of severe cardiac involvement, mainly DCM in those older patients of our cohort, while life-threatening arrhythmias without DCM appear earlier in L-CMD patients. These findings are in agreement with previous studies (Maggi et al., 2014; Fan et al., 2020; Marchel et al., 2021b), but more studies should be performed. Arrhythmic events predict myocardial involvement in pediatric patients carrying a deleterious LMNA variant (Bonne et al., 2000b; Komaki et al., 2011; Pasqualin et al., 2014; Parent et al., 2015; Petillo et al., 2015; Tan et al., 2015; Heller et al., 2017; Groh et al., 2022) reinforcing the importance of investigating cardiac involvement in childhood laminopathy cohorts, even in the absence of clear neuromuscular involvement (Baban et al., 2020). Our results showed a high rate of arrhythmias at pediatric age, so early detection and treatment for each case according to guidelines is critical to preventing complications, improving prognosis, and avoiding death.
LGMD1B is a subtype of limb–girdle muscular dystrophy that presents with progressive shoulder and hip gridle weakness with prior effects on the inferior limbs versus the upper limbs. LGMD1B is autosomal dominant and associated with AV conduction defects, supraventricular arrhtyhmias, and ventricular arrthythmias, but late-onset DCM (Muchir et al., 2000; Lu et al., 2011) and SCD (Finsterer et al., 2015; Groh et al., 2022) have been reported. Evaluation and management of arrhythmic risk are similar to EDMD patients as published in expert consensus statement in adult population with neuromuscular disorders (Groh et al., 2022). We saw no patients of pediatric age (8 and 16 years old at last control) diagnosed with DCM and no ventricular dysfunction, and the ILR registries showed no arrhythmias.
Patients with LMNA-related atypical or ‘undefined’ phenotype were identified in this study because they did not reach phenotypic criteria for L-CMD, EDMD, and LGMD1B phenotypes. They showed an intermediate phenotype between ‘dropped head’ and ‘early EDMD’ (mild weakness, selective hypotrophy and weakness in quadriceps and elbow flexors, mild retractions in elbows, hamstrings, or paraspinal muscles, and mild weakness in neck flexors and foot extensors) as it has been showed in the literature, confirming that these phenotypes are not different entities but probably a continuum (Maggi et al., 2016). Some patients have shown DCM in the literature (Maggi et al., 2014). Our study included two patients (10 and 18 years old at enrollment, 5 and 3 years of follow-up respectively) that showed late-onset muscular manifestations, and none presented with cardiovascular involvement during follow-up at pediatric age. These findings support the idea that early-onset, severe muscular dystrophy is associated with earlier and more severe cardiac involvement.
Patients with early-onset neuromuscular impairment could show early cardiac manifestations, as published recently in a retrospective review (Ben Yaou et al., 2021). Twenty-three of the 28 patients included in our cohort showed early-onset muscle impairment before 2 years of age. Those patients diagnosed with DCM during follow-up had an early-onset phenotype (L-CMD and early-onset EDMD), and no patients with late-onset phenotypes were candidates for an ICD during follow-up. These findings suggest that earlier onset neuromuscular impairment may lead to a worse cardiac phenotype and higher risk of SCD. It would therefore be reasonable to implant an ILR device in LMNA children presenting inf the first 2 years of life with muscle weakness.
Despite a preserved LVEF, longitudinal myocardial strain analysis speckle tracking could show different patterns indicative of early abnormal segmental strain deformation (especially septal strain compared with non-septal strain), post-systolic deformation, and mechanical dispersion (Hasselberg et al., 2014; Haugaa et al., 2015; Boer et al., 2017). (Hasselberg et al., 2014; Haugaa et al., 2015; Boer et al., 2017). This myocardial strain analysis is well reported in other neuromuscular diseases, such as cardiomyopathy-related Duchenne muscular dystrophy. In our study, echocardiographic analysis during follow-up suggested that pediatric laminopathies with neuromuscular involvement are linked to relatively rapid progression to DCM and low LVEF values in a few years. Despite well-preserved LVEF values, four-chamber GLS values at an early age could identify patients that may need an ICD at pediatric age. To our knowledge, this is the first publication that suggests this relationship early during childhood (Supplementary Figure S4). The RV may be involved in LMNA-related cardiomyopathy with or without neuromuscular disease (Quarta et al., 2012; Forleo et al., 2015; Peretto et al., 2019; Marchel et al., 2021b). The lower TAPSE values found in patients that eventually needed an ICD could be associated with RV involvement described previously in adult series with an EDMD phenotype, (Buckley et al., 1999; Marchel et al., 2021a; Marchel et al., 2021b), but the suboptimal acoustic window did not allow for an exhaustive analysis to describe a strong trend or relationship.
Typical early manifestations of arrhythmia in an ECG are flat P wave, AVB, and supraventricular and ventricular arrhythmias (Maggi et al., 2016; Finocchiaro et al., 2020). Other published ECG features include LV hypertrophy data, ST depression, wide QRS complex, and P terminal force. Septal remodeling data in leads V1–V3 seem to be frequent (Ollila et al., 2017), such as Q waves in V1–V2, fragmented QRS in V2–V3, RV1>RV2, RV2>RV3, and poor R wave progression. During follow-up, 12-lead ECG and 24-h Holter monitoring are recommended at least yearly. In our study, first degree AVB, flat P wave, LV hypertrophy data, fragmented and wide QRS complex, poor R wave progression, global low QRS voltage (Supplementary Figure S3C), IRBBB, RBBB, and LBBB were detected during follow-up, in agreement with previous reports (Maggi et al., 2016; Ollila et al., 2017; Finocchiaro et al., 2020). According to our data, ILR with home monitoring is useful for early detection of potential life-threatening arrhythmias, which are usually asymptomatic (Heller et al., 2017).
LMNA carriers may experience DCM and SCD before they experience overt heart failure, as ∼30% of patients will have SCD and another 30% will develop congestive heart failure. Males carrying a deleterious LMNA variant have worse prognosis because of malignant arrhythmias and heart failure. Laminopathies are the third neuromuscular disease in which SCD is frequently reported (Finsterer and St, 2016). In our pediatric study, and in agreement with previously published reports in adults, malignant arrhythmias are related in four/five cases to male sex, but death was related to female sex in early-onset EDMD (Marchel et al., 2021b). Our study may not be large enough to make conclusions about these trends. Atrial conduction disease and malignant arrhythmias are detected during pediatric follow-up that may lead to thromboembolism, heart failure, and SCD, especially in pediatric patients diagnosed with early-onset EDMD and L-CMD. Because there are no detailed recommendations on cardiovascular management in consensus guidelines (Wang et al., 2010), a specific protocol to detect arrhythmias is needed in this pediatric population because SCD may occur before heart failure (Berlo et al., 2005; Lu et al., 2011). We propose a tentative protocol (Supplementary Protocol S1).
The role of EPS in pediatric neuromuscular disease has not been reported; however, it would be reasonable to include it during follow-up childhood and adulthood if any symptoms (typical or atypical) occur because it could be related to potentially malignant arrhythmia. As suggested in adult population with EDMD or LGMD1B, when individuals exhibits symptoms consistent with bradycardia or VT-related symptoms or ECG shows conduction disorder, EP study may be considered for risk stratification for sustained arrhythmias, AVB and SCD (Groh et al., 2022). Our cohort includes early-onset phenotypes in the majority of patients. In our prospective cohort, early-onset patients characterized by their first neuromuscular impairment before 2 years of age seem to have a direct relationship with major and minor cardiac events, regardless of the final neuromuscular phenotype, as previously suggested (Ben Yaou et al., 2021). We therefore propose a clinical protocol for a comprehensive cardiac assessment of pediatric patients with LMNA-related muscular dystrophy.
Our study has some limitations. Our hospital is an international reference center for neuromuscular diseases, and the most severe patients are referred to our institution. We had missing echocardiographic data because some patients were recruited internationally and there was no follow-up. LMNA-related muscular dystrophy is a very rare, often underdiagnosed, disease; therefore, all collected data are of great value.
Additionally, cardiac magnetic resonance imaging (CMRI) was not consistently available for all patients during follow-up. CMRI with late gadolinium enhancement imaging is recommended to study the presence and distribution of myocardial fibrosis. Fibrosis is frequently located along the interventricular septum, according to the worst values in the myocardial strain analysis by speckle tracking. Underlying septal fibrosis could explain the high incidence of ventricular arrhythmias, cardiac conduction delays, and ventricular dysfunction (Holmström et al., 2011; Muscogiuri, 2017). Biomarkers may help during cardiac follow-up, such as N-terminal probrain natriuretic peptide (NT proBNP), a well-recognized biomarker that increases when ventricular function worsens (Yancy et al., 2016; Marchel et al., 2021b). In our series, NT proBNP was not homogeneously analyzed, and it might help in some cases with a high risk of ventricular dysfunction. As published previously, proteomic analysis of plasma samples could be used in the future to identify individuals with a high risk of sudden death with LMNA-related cardiomyopathy (Izquierdo et al., 2016). Our study also lacks analysis of other genes that may be implicated in laminopathies or related muscular diseases. In the future, whole-exome sequencing and/or whole-genome sequencing can be used to identify new alterations in any region of genome.
Malignant arrhythmias and SCD in LMNA-related muscular dystrophy occurs frequently, but no comprehensive studies focused on early identification, adoption of preventative measures, and follow-up have been performed. ILR with home monitoring identified five cases (17%) with malignant arrhythmias, and ICDs were implanted for prevention. Two cases with an ICD showed appropriate shocks. ILR may be critical for early diagnosis of life-threatening arrhythmias in laminopathies with an early-onset neuromuscular phenotype. Remote home monitoring helps for close follow-up. Echocardiographic follow-up, including myocardial strain analysis, might be helpful to identify worse prognosis in patients because DCM is present early before adulthood. Specific clinical guidelines that include management in children and emphasize the use of ILR are needed to standardize treatment and mitigate the risk of SCD, especially in those with early-onset phenotypes.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by Hospital Sant Joan de Déu (Identification code: PIC-59-14). Written informed consent to participate in this study was provided by the participants and legal guardian/next of kin.
GS-B, SC, OC, JB, and AN developed the concept and prepared the manuscript. SC, JC, VF, CB-R, and EM-B acquired, pre-processed, and analyzed the data. GS-B, SC, OC, and JB supervised the study. IZ, DN-dB, CB-R, LC, JE, RB, ID, MG, and SQ-R contributed to manuscript revision. All authors have read and agreed to the published version of the manuscript.
This work has also been supported by Obra Social “La Caixa Foundation” (LCF/PR/GN19/50320002) and Instituto de Salud Carlos III, Fondo Investigación Sanitaria -FIS- (PI21/00094). CIBERCV is an initiative of the ISCIII, Spanish Ministry of Economy and Competitiveness.
This is the result of a hard work from patients, families and professionals working together to find answers. This long-term work could not have been possible without the unconditional support of “Fundación Andrés Marcio–Niños contra la Laminopatía” to who we are deeply thankful.
We would like to truly thank all our patients and their families, especially to Carlota who inspired us to become better doctors.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer AF declared a shared affiliation with the authors ID, MG, SQR to the handling editor at the time of review.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2023.1142937/full#supplementary-material
AV, atrioventricular; AVB, atrioventricular block; CHD, congenital heart disease; CMRI, cardiac magnetic resonance imaging; DCM, dilated cardiomyopathy; EDMD, Emery–Dreifuss muscular dystrophy; ECG, electrocardiogram; EPS, electrophysiological study; GLS, global longitudinal strain; HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter defibrillator; ILR, implantable loop recorder; IQR, interquartile range; IRBBB, incomplete right bundle-branch block; LBBB, left bundle-branch block; LGMD1B, limb–girdle muscular dystrophy 1B; L-CMD, LMNA-related congenital muscular dystrophy; LV, left ventricle; LVEF, left ventricular ejection fraction; MAPSE, mitral annular plane systolic excursion; NIMV, non-invasive mechanical ventilation; NT proBNP, N-terminal probrain natriuretic peptide; PVC, premature ventricular contractions; RBBB, right bundle-branch block; RV, right ventricle; SCD, sudden cardiac death; TAPSE, tricuspid annular plane systolic excursion; VT, ventricular tachycardia; VF, ventricular fibrillation.
Aebi, U., Cohn, J., Buhle, L., and Gerace, L. (1986). The nuclear lamina is a meshwork of intermediate-type filaments. Nature 323 (6088), 560–564. Internet.
Alastalo, T-P., West, G., Li, S-P., Keinänen, A., Helenius, M., Tyni, T., et al. (2015). LMNA mutation c.917T>G (p.L306R) leads to deleterious hyper-assembly of lamin A/C and associates with severe right ventricular cardiomyopathy and premature aging. Hum. Mutat. 36 (7), 694–703. doi:10.1002/humu.22793
Alexandra, P., Rocío, T., Oscar, C., Paola, B., Iglesias, A., Alipio, M., et al. (2015). A novel mutation in lamin A/C causing familial dilated cardiomyopathy associated with sudden cardiac death. J. Card. Fail 21, 217–225. Internet. doi:10.1016/j.cardfail.2014.12.003
Andrés, V., and González, J. M. (2009). Role of A-type lamins in signaling, transcription, and chromatin organization. J. Cell Biol. 187 (7), 945–957. doi:10.1083/jcb.200904124
Atalaia, A., Ben Yaou, R., Wahbi, K., De Sandre-Giovannoli, A., Vigouroux, C., and Bonne, G. (2021). Laminopathies’ treatments systematic review: A contribution towards a ‘treatabolome. ’ J. Neuromuscul. Dis. 8 (3), 419–439. doi:10.3233/JND-200596
Baban, A., Cicenia, M., Magliozzi, M., Gnazzo, M., Cantarutti, N., Silvetti, M. S., et al. (2020). Cardiovascular involvement in pediatric laminopathies. Report of six patients and literature revision. Front. Pediatr. 8, 374–379. doi:10.3389/fped.2020.00374
Ben Yaou, R., Yun, P., Dabaj, I., Norato, G., Donkervoort, S., Xiong, H., et al. (2021). International retrospective natural history study of LMNA -related congenital muscular dystrophy. Brain Commun. 3 (3), fcab075–15. doi:10.1093/braincomms/fcab075
Benedetti, S., Menditto, I., Degano, M., Rodolico, C., Merlini, L., D’Amico, A., et al. (2007). Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurol. [Internet] 69 (12), 1285–1292.
Berlo, J. H. V., Voogt, W. G. D., Rabah, B., Yaou, B., Duboc, D., Rossenbacker, T., et al. (2005). Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death. J. Mol. Med. 83, 79–83. doi:10.1007/s00109-004-0589-1
Bertrand, A. T., Brull, A., Azibani, F., Benarroch, L., Chikhaoui, K., Stewart, C. L., et al. (2020). Lamin A/C assembly defects in LMNA-congenital muscular dystrophy is responsible for the increased severity of the disease compared with emery-dreifuss muscular dystrophy. Cells 9 (4), 844. doi:10.3390/cells9040844
Bertrand, A. T., Chikhaoui, K., Yaou, R. B., and Bonne, G. (2011). Clinical and genetic heterogeneity in laminopathies. Biochem. Soc. Trans. 39 (6), 1687–1692. doi:10.1042/BST20110670
Boer, S. L. D., Gideon, J., Sarvaas, M., Klitsie, L. M., Iperen, G. G. V., Tanke, R. B., et al. (2017). Distribution of strain patterns in children with dilated cardiomyopathy. Echocardiography 34, 881–887. doi:10.1111/echo.13548
Bonne, G., Di Barletta, M. R., Varnous, S., Bécane, H. M., Hammouda, E. H., Merlini, L., et al. (1999). Mutations in the gene encoding lamin A/C cause autosomal dominant Emery-Dreifuss muscular dystrophy. Nat. Genet. 21, 285–288. doi:10.1038/6799
Bonne, G., Mercuri, E., Muchir, A., Urtizberea, A., Bécane, H. M., Recan, D., et al. (2000). Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 48 (2), 170–180. doi:10.1002/1531-8249(200008)48:2<170::aid-ana6>3.0.co;2-j
Bonne, G., Mercuri, E., Muchir, A., Urtizberea, A., Bécane, H. M., Recan, D., et al. (2000). Clinical and molecular genetic spectrum of autosomal dominant Emery-Dreifuss muscular dystrophy due to mutations of the lamin A/C gene. Ann. Neurol. 48 (2), 170–180. doi:10.1002/1531-8249(200008)48:2<170::aid-ana6>3.0.co;2-j
Bonne, G., and Quijano-Roy, S. (2013). Emery-Dreifuss muscular dystrophy, laminopathies, and other nuclear envelopathies. Handb. Clin. Neurol. 113, 1367–1376. doi:10.1016/B978-0-444-59565-2.00007-1
Bonne, G., Yaou, R. B., Béroud, C., Boriani, G., Brown, S., de Visser, M., et al. (2003). 108th ENMC international workshop, 3rd workshop of the MYO-CLUSTER project: EUROMEN, 7th international emery-dreifuss muscular dystrophy (EDMD) workshop, 13–15 september 2002, naarden, The Netherlands. Neuromuscul. Disord. 13 (6), 508–515. doi:10.1016/s0960-8966(03)00063-4
Bozkurt, B., Colvin, M., Cook, J., Cooper, L. T., Deswal, A., Fonarow, G. C., et al. (2016). Current diagnostic and treatment strategies for specific dilated cardiomyopathies: A scientific statement from the American heart association. Circulation 134, 579–646.
Buckley, A. E., Dean, J., and Mahy, I. R. (1999). Cardiac involvement in Emery dreifuss muscular dystrophy: A case series. Heart 82 (1), 105–108. doi:10.1136/hrt.82.1.105
Burke, B., and Stewart, C. L. (2012). The nuclear lamins: Flexibility in function. Nat. Rev. Mol. Cell Biol. [Internet] 14, 13–24. doi:10.1038/nrm3488
Carboni, N., Politano, L., Floris, M., Mateddu, A., Solla, E., Olla, S., et al. (2013). Overlapping syndromes in laminopathies: A meta-analysis of the reported literature. Acta Myol. 32 (5), 7–17.
Cattin, M-E., Muchir, A., and Bonne, G. (2013). State-of-the-heart” of cardiac laminopathies. Curr. Opin. Cardiol. 28 (3), 297–304. doi:10.1097/HCO.0b013e32835f0c79
Chemla, J. C., Kanter, R. J., Carboni, M. P., and Smith, E. C. (2010). Two children with “dropped head” syndrome due to lamin A/C mutations. Muscle Nerve 42 (5), 839–841. doi:10.1002/mus.21820
Dauer, W. T., and Worman, H. J. (2009). The nuclear envelope as a signaling node in development and disease. Dev. Cell [Internet] 17 (5), 626–638. doi:10.1016/j.devcel.2009.10.016
Dechat, T., Shimi, T., Adam, S. A., Rusinol, A. E., Andres, D. A., Spielmann, H. P., et al. (2007). Alterations in mitosis and cell cycle progression caused by a mutant lamin A known to accelerate human aging. Proc. Natl. Acad. Sci. U. S. A. [Internet] 104 (12), 4955–4960. doi:10.1073/pnas.0700854104
Dittmer, T., and Misteli, T. (2011). The lamin protein family. Genome Biol. 12 (5), 222. doi:10.1186/gb-2011-12-5-222
Dobrzynska, A., Gonzalo, S., Shanahan, C., and Askjaer, P. (2016). The nuclear lamina in health and disease. Nucleus 7 (3), 233–248. Internet. doi:10.1080/19491034.2016.1183848
Dubowitz, V. (1999). 68th ENMC international workshop (5th inter-national workshop) on congenital muscular dystrophy, 9-11 april 1999, naarden, The Netherlands. Neuromuscul. Disord. 9, 446–454. doi:10.1016/s0960-8966(99)00074-7
Fan, Y., Tan, D., Song, D., Zhang, X., Chang, X., Wang, Z., et al. (2020). Clinical spectrum and genetic variations of LMNA -related muscular dystrophies in a large cohort of Chinese patients. J. Med. Genet. 58, 326–333. doi:10.1136/jmedgenet-2019-106671
Feingold, B., Mahle, W. T., Auerbach, S., Clemens, P., Domenighetti, A. A., Jefferies, J. L., et al. (2017). Management of cardiac involvement associated with neuromuscular diseases: A scientific statement from the American heart association. Circulation 136, 200–231. doi:10.1161/CIR.0000000000000526
Finocchiaro, G., Merlo, M., Sheikh, N., De Angelis, G., Papadakis, M., Olivotto, I., et al. (2020). The electrocardiogram in the diagnosis and management of patients with dilated cardiomyopathy. Eur. J. Heart Fail 22 (7), 1097–1107. doi:10.1002/ejhf.1815
Finsterer, J., Ramaciotti, C., Wang, C. H., Wahbi, K., Rosenthal, D., Duboc, D., et al. (2010). Cardiac findings in congenital muscular dystrophies. Pediatrics 126 (3), 538–545. doi:10.1542/peds.2010-0208
Finsterer, J., and St, C. (2016). Heart disease in disorders of muscle, neuromuscular transmission, and the nerves. Korean Circ. J. 46, 117–134. doi:10.4070/kcj.2016.46.2.117
Finsterer, J., Sto, C., and Blazek, G. (2006). Neuromuscular implications in left ventricular hypertrabeculation/noncompaction. Int. J. Cardiol. 110, 288–300. doi:10.1016/j.ijcard.2005.10.028
Finsterer, J., Stöllberger, C., and Maeztu, C. (2015). Sudden cardiac death in neuromuscular disorders. Int. J. Cardiol. [Internet] 203, 508–515. doi:10.1016/j.ijcard.2015.10.176
Forleo, C., Carmosino, M., Resta, N., Rampazzo, A., Valecce, R., Sorrentino, S., et al. (2015). Clinical and functional characterization of a novel mutation in lamin a/C gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS One 10 (4), e0121723. doi:10.1371/journal.pone.0121723
Giglia, T. M., Dinardo, J., Ghanayem, N. S., Ichord, R., Niebler, R. A., Odegard, K. C., et al. (2015). Bleeding and thrombotic emergencies in pediatric cardiac intensive care: Unchecked balances. World J. Pediatr. Congenit. Hear Surg. 3 (4), 470–491. doi:10.1177/2150135112460866
Gonzalez-Suarez, I., Redwood, A. B., Perkins, S. M., Vermolen, B., Lichtensztejin, D., Grotsky, D. A., et al. (2009). Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J. [Internet 28 (16), 2414–2427. doi:10.1038/emboj.2009.196
Granger, B., Gueneau, L., Drouin-Garraud, V., Pedergnana, V., Gagnon, F., Ben Yaou, R., et al. (2011). Modifier locus of the skeletal muscle involvement in Emery-Dreifuss muscular dystrophy. Hum. Genet. 129 (2), 149–159. doi:10.1007/s00439-010-0909-1
Groh, W. J. (2012). Arrhythmias in the muscular dystrophies. HRTHM 9 (11), 1890–1895. Internet. doi:10.1016/j.hrthm.2012.06.038
Groh, W. J., Bhakta, D., Vice-chair, C., Tomaselli, G. F., Vice-chair, F., Aleong, R. G., et al. 2022 HRS expert consensus statement on evaluation and management of arrhythmic risk in neuromuscular disorders. Hear Rhythm [Internet]; 19, e61-e120. doi:10.1016/j.hrthm.2022.04.022
Haas, J., Frese, K. S., Peil, B., Kloos, W., Keller, A., Kayvanpour, E., et al. (2014). Atlas of the clinical genetics of human dilated cardiomyopathy. Eur. Heart J. 36, 1123–35a. doi:10.1093/eurheartj/ehu301
Hasselberg, N. E., Edvardsen, T., Petri, H., Berge, K. E., Leren, T. P., Bundgaard, H., et al. (2014). Risk prediction of ventricular arrhythmias and myocardial function in Lamin A/C mutation positive subjects. Europace 16 (4), 563–571. doi:10.1093/europace/eut291
Haugaa, K. H., Hasselberg, N. E., and Edvardsen, T. (2015). Mechanical dispersion by strain echocardiography: A predictor of ventricular arrhythmias in subjects with lamin A/C mutations. JACC Cardiovasc Imaging 8 (1), 104–106. doi:10.1016/j.jcmg.2014.04.029
Heller, F., Dabaj, I., Mah, J. K., Bergounioux, J., Essid, A., Bönnemann, C. G., et al. (2017). Cardiac manifestations of congenital LMNA-related muscular dystrophy in children: Three case reports and recommendations for care. Cardiol. Young 27, 1076–1082. doi:10.1017/S1047951116002079
Ho, C. Y., and Lammerding, J. (2012). Lamins at a glance. J. Cell Sci. 125 (9), 2087–2093. Internet. doi:10.1242/jcs.087288
Holmström, M., Kivistö, S., Heliö, T., Jurkko, R., Kaartinen, M., Antila, M., et al. (2011). Late gadolinium enhanced cardiovascular magnetic resonance of lamin A/C gene mutation related dilated cardiomyopathy. J. Cardiovasc Magn. Reson [Internet 13 (1), 30. doi:10.1186/1532-429X-13-30
Hutchison, C. J. (2011). The role of DNA damage in laminopathy progeroid syndromes. Biochem. Soc. Trans. [Internet 39 (6), 1715–1718. doi:10.1042/BST20110700
Izquierdo, I., Rosa, I., Bravo, S. B., Guitián, E., Pérez-Serra, A., Campuzano, O., et al. (2016). Proteomic identification of putative biomarkers for early detection of sudden cardiac death in a family with a LMNA gene mutation causing dilated cardiomyopathy. J. Proteomics 148, 75–84. Internet. doi:10.1016/j.jprot.2016.07
Jędrzejowska, M., Potulska-Chromik, A., Gos, M., Gambin, T., Dębek, E., Rosiak, E., et al. (2021). Floppy infant syndrome as a first manifestation of LMNA-related congenital muscular dystrophy. Eur. J. Paediatr. Neurol. 32, 115–121. doi:10.1016/j.ejpn.2021.04.005
Komaki, H., Hayashi, Y. K., Tsuburaya, R., Sugie, K., Kato, M., Nagai, T., et al. (2011). Inflammatory changes in infantile-onset lmna-associated myopathy. Neuromuscul. Disord. [Internet] 21 (8), 563–568. doi:10.1016/j.nmd.2011.04.010
Koopman, L. P., Rebel, B., Gnanam, D., Menting, M. E., Helbing, W. A., and Boersma, E. (2019). Reference values for two-dimensional myocardial strain echocardiography of the left ventricle in healthy children. Cardiol. Young 29 (3), 325–337. doi:10.1017/S1047951118002378
Kumar, S., Androulakis, A. F. A., Sellal, J. M., Maury, P., Gandjbakhch, E., Waintraub, X., et al. (2016). Multicenter experience with catheter ablation for ventricular tachycardia in lamin A/C cardiomyopathy. Circ. Arrhythm. Electrophysiol. 9, e004357. doi:10.1161/CIRCEP.116.004357
Lammerding, J., Schulze, P. C., Takahashi, T., Kozlov, S., Sullivan, T., Kamm, R. D., et al. (2004). Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest. 113 (3), 370–378. doi:10.1172/JCI19670
Lang, R. M., Bierig, M., Devereux, R. B., Flachskampf, F. A., Foster, E., Pellikka, P. A., et al. (2005). Recommendations for chamber quantification: A report from the American society of echocardiography's guidelines and standards committee and the chamber quantification writing group, developed in conjunction with the European association of echocardiography, a branch of the European society of cardiology. J. Am. Soc. Echocardiogr. 18 (12), 1440–1463. doi:10.1016/j.echo.2005.10.005
Lee, C. K., Margossian, R., Sleeper, L. A., Canter, C. E., Chen, S., Tani, L. Y., et al. (2014). Variability of M-mode versus two-dimensional echocardiography measurements in children with dilated cardiomyopathy. Pediatr. Cardiol. 35 (4), 658–667. doi:10.1007/s00246-013-0835-9
Lee, T. M., Hsu, D. T., Kantor, P., Towbin, J. A., Ware, S. M., Colan, S. D., et al. (2017). Pediatric cardiomyopathies. Circ. Res. 121 (7), 855–873. doi:10.1161/CIRCRESAHA.116.309386
Lipshultz, S. E., Law, Y. M., Asante-Korang, A., Austin, E. D., Dipchand, A. I., Everitt, M. D., et al. (2019). Cardiomyopathy in children: Classification and diagnosis: A scientific statement from the American heart association. Circulation 140, 9–68. doi:10.1161/CIR.0000000000000682
Lu, J. T., Muchir, A., Nagy, P. L., and Worman, H. J. (2011). LMNA cardiomyopathy: Cell biology and genetics meet clinical medicine. Dis. Model Mech. [Internet 4 (5), 562–568. doi:10.1242/dmm.006346
Maggi, L., Amico, A. D., Pini, A., Sivo, S., Ambrosio, P. D., Sala, S., et al. (2014). LMNA-Associated myopathies: The Italian experience in a large cohort of patients. Neurology 83, 1634–1644. doi:10.1212/WNL.0000000000000934
Maggi, L., Carboni, N., and Bernasconi, P. (2016). Skeletal muscle laminopathies: A review of clinical and molecular features. Cells 5, 33. doi:10.3390/cells5030033
Marchel, M., Madej-Pilarczyk, A., Steckiewicz, R., Stolarz, P., Peller, M., Tymińska, A., et al. (2021). Predictors of mortality and cardiovascular outcomes in Emery-Dreifuss muscular dystrophy in a long-term follow-up. Kardiol. Pol. 79 (12), 1335–1342. doi:10.33963/KP.a2021.0159
Marchel, M., Madej-Pilarczyk, A., Tymí Nska, A., Steckiewicz, R., Ostrowska, E., Wysí Nska, J., et al. (2021). Cardiac arrhythmias in muscular dystrophies associated with emerinopathy and laminopathy: A cohort study. J. Clin. Med. 10, 732. doi:10.3390/jcm10040732
Meune, C., Berlo, J. H. V., Anselme, F., Bonne, G., Pinto, Y. M., and Duboc, D. (2006). Primary prevention of sudden death in patients with Lamin A/C gene mutations. N. Engl. J. Med. 354 (2), 209–210. doi:10.1056/NEJMc052632
Monagle, P. (2012). Diagnosis and management of deep venous thrombosis and pulmonary embolism in neonates and children. Semin. Thromb. Hemost. 38, 683–690. doi:10.1055/s-0032-1326784
Muchir, A., Bonne, G., Kooi, A. J. V. D., Meegen, M. V., Baas, F., Bolhuis, P. A., et al. (2000). Identification of mutations in the gene encoding lamins A/C in autosomal dominant limb girdle muscular dystrophy with atrioventricular conduction disturbances (LGMD1B). Hum. Mol. Genet. 9 (9), 1453–1459. doi:10.1093/hmg/9.9.1453
Muscogiuri, G. (2017). Results of late gadolinium enhancement in children affected by dilated cardiomyopathy. Front. Prediator 5, 13. doi:10.3389/fped.2017.00013
Ollila, L., Nikus, K., Holmström, M., Jalanko, M., Jurkko, R., Kaartinen, M., et al. (2017). Clinical disease presentation and ECG characteristics of LMNA mutation carriers. Open Hear [Internet] 4 (1), e000474. doi:10.1136/openhrt-2016-000474
Parent, J. J., Towbin, J. A., and Jefferies, J. L. (2015). Left ventricular noncompaction in a family with lamin A/C gene mutation. Tex. Hear Inst. J. 42 (1), 73–76. doi:10.14503/THIJ-13-3843
Pasotti, M., Klersy, C., Pilotto, A., Marziliano, N., Rapezzi, C., Serio, A., et al. (2008). Long-term outcome and risk stratification in dilated cardiolaminopathies. J. Am. Coll. Cardiol. 52 (15), 1250–1260. doi:10.1016/j.jacc.2008.06.044
Pasqualin, L. M. A., Reed, U. C., Costa, T. V. M. M., Quedas, E., Albuquerque, M. A. V., Resende, M. B. D., et al. (2014). Congenital muscular dystrophy with dropped head linked to the LMNA gene in a Brazilian cohort. Pediatr. Neurol. [Internet] 50 (4), 400–406. doi:10.1016/j.pediatrneurol.2013.11.010
Peretto, G., Di Resta, C., Perversi, J., Forleo, C., Maggi, L., Politano, L., et al. (2019). Cardiac and neuromuscular features of patients with LMNA-related cardiomyopathy. Ann. Intern Med. 171 (7), 458–463. doi:10.7326/M18-2768
Pérez-Serra, A., Toro, R., Sarquella-Brugada, G., de Gonzalo-Calvo, D., Cesar, S., Carro, E., et al. (2016). Genetic basis of dilated cardiomyopathy. Int. J. Cardiol. 224, 461–472. Internet. doi:10.1016/j.ijcard.2016.09.068
Petillo, R., Ambrosio, P. D., Torella, A., Taglia, A., Picillo, E., Testori, A., et al. (2015). Novel mutations in LMNA A/C gene and associated phenotypes. Acta Myol. 34, 116–119.
Pettersen, M. D., Du, W., Skeens, M. E., and Humes, R. A. (2008). Regression equations for calculation of Z scores of cardiac structures in a large cohort of healthy infants, children, and adolescents: An echocardiographic study. J. Am. Soc. Echocardiogr. 21 (8), 922–934. doi:10.1016/j.echo.2008.02.006
Priori, S., Blomstrom-Lundqvist, C., Mazzanti, A., Blom, N., Borggrefe, M., Camm, J., et al. 2015 2015 ESC guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European society of cardiology (ESC). Endorsed by: Association for European paediatric and congenital cardiology (AEPC). Eur. Heart J.; 36(41):2793–2867. doi:10.1093/eurheartj/ehv316
Quarta, G., Syrris, P., Ashworth, M., Jenkins, S., Zuborne Alapi, K., Morgan, J., et al. (2012). Mutations in the Lamin A/C gene mimic arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 33 (9), 1128–1136. doi:10.1093/eurheartj/ehr451
Quijano-roy, S., Mbieleu, B., Bo, C. G., Jeannet, P., Colomer, J., Clarke, N. F., et al. (2008). De novo LMNA mutations cause a new form of congenital muscular dystrophy. Ann. Neurol. 64, 177–186. doi:10.1002/ana.21417
Rajdev, A., and Groh, W. (2015). Arrhythmias in the muscular dystrophies. Card. Electrophysiol. Clin. 7 (2), 303–308. doi:10.1016/j.ccep.2015.03.011
Rijsingen, I. A. W. V., Arbustini, E., Elliott, P. M., Mogensen, J., Ast, J. F. H., Jenkins, S., et al. (2012). Risk factors for malignant ventricular arrhythmias in lamin A/C mutation carriers A European cohort study. JAC [Internet] 59 (5), 493–500. doi:10.1016/j.jacc.2011.08.078
Rijsingen, I. A. W. V., Nannenberg, E. A., Arbustini, E., Elliott, P. M., Mogensen, J., Ast, J. F. H., et al. (2013). Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. Eur. J. Heart Fail 15, 376–384. doi:10.1093/eurjhf/hfs191
Steckiewicz, R., Stolarz, P., Świȩtoń, E., Madej-Pilarczyk, A., Grabowski, M., Marchel, M., et al. (2016). Cardiac pacing in 21 patients with emery-dreifuss muscular dystrophy: A single-centre study with a 39-year follow-up. Kardiol. Pol. 74 (6), 576–583. doi:10.5603/KP.a2015.0218
Tan, D., Yang, H., Yuan, Y., Bonnemann, C., Chang, X., Wang, S., et al. (2015). Phenotype-genotype analysis of Chinese patients with early-onset LMNA-related muscular dystrophy. PLoS One 10 (6), e0129699. doi:10.1371/journal.pone.0129699
Taylor, M. R. G., Fain, P. R., Sinagra, G., Robinson, M. L., Robertson, A. D., Carniel, E., et al. (2003). Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J. Am. Coll. Cardiol. [Internet] 41 (5), 771–780. doi:10.1016/S0735-1097(02)02954-6
Van Rijsingen, I. A. W., Bakker, A., Azim, D., Hermans-Van Ast, J. F., Van Der Kooi, A. J., Van Tintelen, J. P., et al. (2013). Lamin A/C mutation is independently associated with an increased risk of arterial and venous thromboembolic complications. Int. J. Cardiol. [Internet] 168 (1), 472–477. doi:10.1016/j.ijcard.2012.09.118
Wahbi, K., Ben Yaou, R., Gandjbakhch, E., Anselme, F., Gossios, T., Lakdawala, N. K., et al. (2019). Development and validation of a new risk prediction score for life- threatening ventricular tachyarrhythmias in laminopathies. Circulation 140, 293–302. doi:10.1161/CIRCULATIONAHA.118.039410
Wang, C. H., Bonnemann, C. G., Rutkowski, A., Sejersen, T., Bellini, J., Battista, V., et al. (2010). Consensus statement on standard of care for congenital muscular dystrophies. J. Child. Neurol. 25 (12), 1559–1581. doi:10.1177/0883073810381924
Wang, X., Zabell, A., and Koh, W. (2017). Lamin A/C cardiomyopathies: Current understanding and novel treatment strategies. Curr. Treat. Options Cardiovasc Med. 19, 21. doi:10.1007/s11936-017-0520-z
Wolf, C. M., Wang, L., Alcalai, R., Pizard, A., Burgon, P. G., Ahmad, F., et al. (2008). Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. J. Mol. Cell Cardiol. 44 (2), 293–303. doi:10.1016/j.yjmcc.2007.11.008
Worman, H. J., and Bonne, G. (2007). Laminopathies:” a wide spectrum of human diseases. Exp. Cell Res. 313 (10), 2121–2133. doi:10.1016/j.yexcr.2007.03.028
Worman, H. J. (2012). Nuclear lamins and laminopathies. J. Pathology 226, 316–325. doi:10.1002/path.2999
Yancy, A. C. W., Jessup, M., Bozkurt, B., Butler, J., Jr, D. E. C., Colvin, M. M., et al. 2016 2016 ACC/AHA/HFSA focused update on new pharmacological therapy for heart failure: An update of the 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of cardiology/American heart association task force on clinical practice guidelines and the heart failure society of America. J. Card. Fail [Internet]; 22(9):659–669. doi:10.1016/j.cardfail.2016.07.001
Yancy, C., Jessup, M., Bozkurt, B., Masoudi, F. F. A., Casey, E., Mcmurray, J. J. V., et al. 2013 2013 ACCF/AHA guideline for the management of heart failure: A report of the American College of cardiology foundation/American heart association task force on practice guidelines. J. Am. Coll. Cardiol.; 62(16):e147-e239. doi:10.1016/j.jacc.2013.05.019
Keywords: laminopathies, sudden cardiac death, cardiomyopathy, A/C lamins, LMNA-related diseases, LMNA-related cardiomyopathy, long-term implantable loop recorder
Citation: Cesar S, Campuzano O, Cruzalegui J, Fiol V, Moll I, Martínez-Barrios E, Zschaeck I, Natera-de Benito D, Ortez C, Carrera L, Expósito J, Berrueco R, Bautista-Rodriguez C, Dabaj I, Gómez García-de-la-Banda M, Quijano-Roy S, Brugada J, Nascimento A and Sarquella-Brugada G (2023) Characterization of cardiac involvement in children with LMNA-related muscular dystrophy. Front. Cell Dev. Biol. 11:1142937. doi: 10.3389/fcell.2023.1142937
Received: 12 January 2023; Accepted: 27 February 2023;
Published: 10 March 2023.
Edited by:
Marc Bartoli, Aix Marseille Université, FranceReviewed by:
Masamichi Ito, The University of Tokyo, JapanCopyright © 2023 Cesar, Campuzano, Cruzalegui, Fiol, Moll, Martínez-Barrios, Zschaeck, Natera-de Benito, Ortez, Carrera, Expósito, Berrueco, Bautista-Rodriguez, Dabaj, Gómez García-de-la-Banda, Quijano-Roy, Brugada, Nascimento and Sarquella-Brugada. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sergi Cesar, c2VyZ2kuY2VzYXJAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.