 Mia C. Borlongan1
Mia C. Borlongan1 Hongbin Wang1,2,3*
Hongbin Wang1,2,3*- 1Master Program of Pharmaceutical Science College of Graduate Studies, Elk Grove, CA, United States
- 2Department of Pharmaceutical and Biomedical Sciences College of Pharmacy, Elk Grove, CA, United States
- 3Department of Basic Science College of Medicine, California Northstate University, Elk Grove, CA, United States
Tumorigenic cancer stem cells (CSCs) represent a subpopulation of cells within the tumor that express genetic and phenotypic profiles and signaling pathways distinct from the other tumor cells. CSCs have eluded many conventional anti-oncogenic treatments, resulting in metastases and relapses of cancers. Effectively targeting CSCs’ unique self-renewal and differentiation properties would be a breakthrough in cancer therapy. A better characterization of the CSCs’ unique signaling mechanisms will improve our understanding of the pathology and treatment of cancer. In this paper, we will discuss CSC origin, followed by an in-depth review of CSC-associated signaling pathways. Particular emphasis is given on CSC signaling pathways’ ligand-receptor engagement, upstream and downstream mechanisms, and associated genes, and molecules. Signaling pathways associated with regulation of CSC development stand as potential targets of CSC therapy, which include Wnt, TGFβ (transforming growth factor-β)/SMAD, Notch, JAK-STAT (Janus kinase-signal transducers and activators of transcription), Hedgehog (Hh), and vascular endothelial growth factor (VEGF). Lastly, we will also discuss milestone discoveries in CSC-based therapies, including pre-clinical and clinical studies featuring novel CSC signaling pathway cancer therapeutics. This review aims at generating innovative views on CSCs toward a better understanding of cancer pathology and treatment.
1 Introduction
Although cancer death rates decreased by 27% from 200.8 deaths in 1999 to 146.2 deaths in 2019 per 100,000 population, cancer remains the second leading cause of death, after heart disease, in the United States (Siegel et al., 2021). In 2019, more male cancer patients (315,876) succumbed to death than females (283,725), with increased mortality rates in males (172.9 deaths per 100,000 population) than females (126.2 deaths per 100,000 population) (Siegel et al., 2021). The economic burden to the society of cancer care for patients stands at $21 billion in the United States in 2019 (Ma and Richardson, 2022). Approximately 16.9 million Americans displayed a history of cancer in 2019 and by 2050 it is predicted the total number of incident cases will increase by almost 50% (Weir et al., 2021). In the world, prostate, colorectum, and melanoma of the skin correspond to the top three prevalent cancers in males, while breast, uterine corpus, and colorectum represent the most prevalent cancers in females (Cao et al., 2021). The deadliest cancer types across all races include lung and bronchus, colorectal, pancreatic, and breast cancers, which account for nearly 50% of all cancer deaths (Cao et al., 2021). About one-third of cancer deaths implicate risk factors, such as tobacco use, high body mass index, alcohol use, low fruit and vegetable intake, and lack of physical activity. Unfortunately, these risk factors skew towards increased morbidity among low and middle income populations due to late-stage presentation and lack of access to diagnosis and treatment (Dieteren and Bonfrer, 2021). Despite significant advances in our scientific understanding of the disease and improved clinical care, cancer treatments remain a significant unmet need. Here, we explore the concept of cancer stem cells (CSCs), which exist as a small subpopulation of cells within tumors exhibiting self-renewal, differentiation, and tumorigenicity (Pattabiraman and Weinberg, 2014; Müller et al., 2020; Chen et al., 2021a). Noteworthy, CSCs elude conventional chemotherapy and radiation treatment, suggesting that CSCs may very well be the origin of cancer metastasis (Chen et al., 2021a). The structure of CSCs also varies based on type of cancer, genetics, and epigenetics, and can differentiate into multiple cell types (e.g., fibroblasts, pericytes, endothelial cells, etc.) (Pattabiraman and Weinberg, 2014; Chen et al., 2021a). Recognizing these unique properties of CSCs opens novel avenues for a better understanding of cancer pathology and its treatment. Indeed, recent innovative cancer treatments utilize a CSC-based treatment platform. To this end, we discuss new lines of investigations probing CSC origin and microenvironment, detailing CSC genotypic and phenotypic profiles and signaling pathways that directly target tumor-forming CSCs.
2 Defining cancer stem cells
Cancer is defined as a malignant growth or tumor resulting from the division of abnormal cells (Cowan et al., 2022). The cells within the tumor microenvironment (TME) display varying levels of proliferation and differentiation leading to functional heterogeneity of tumor cells (Pizer et al., 1998; Naruse et al., 2021). Such functional heterogeneity among tumor cells begs the question on the origin of these cells. The conventional concept of abnormal cell division stipulates that cancer is caused by changes to DNA or genetic mutations (Germain et al., 2022). A cell becomes cancerous via multiple alterations to the cell’s DNA sequence (Lasorsa et al., 2022; Valle-Mendiola et al., 2022), with the mutations capable of arising from any cell in the body, thereafter, leading to cancer (Noble, 2021). The stochastic model or clonal evolution model of cancer origin states that all cells exhibit the capacity of self-renewal or differentiation, which becomes uncontrolled through genetic mutations, propelling the growth of heterogenic tumor cells (Odoux et al., 2008; Greaves and Maley, 2012). A newly proposed model of cancer origin advances the notion that a small population of stem cells exists within the tumor that express tumorigenic characteristics, namely the cells’ enduring ability to self-renew and to differentiate into cancer cells, thereby specifically modulating cancer onset and progression. These cancer-driving cells are the CSCs, and fittingly enough this concept of cancer origin is called the CSC model (Kreso and Dick, 2014). Recognizing that CSCs and non-CSCs represent distinct compartments within the tumor provides the functional heterogeneity of the tumor.
In parallel to the aforementioned models of cancer origin (i.e., conventional model versus the stochastic model or clonal evolution model), there are multiple noteworthy theories about origins for CSCs that have garnered compelling evidence. CSCs were first discovered in 1994 in acute myeloid leukemia (AML) rodents displaying severe combined immunodeficient (SCID) (Pattabiraman and Weinberg, 2014; Chen et al., 2021a). These unique cells exhibit not only self-renewing and tumor initiating capabilities akin to stem cells, but they also possess features of asymmetric division (Pattabiraman and Weinberg, 2014). The theory of asymmetric division states that one lineage of CSCs has self-renewing capabilities while the other lineage will lose their stemness properties and become epithelial cells which form the bulk of the tumor (Pattabiraman and Weinberg, 2014). Moreover, a small population of CSCs can form the bulk of the tumor mass, highlighting CSC’s cancer initiating potential (Sancho et al., 2015). This theory was further supported via in vivo experimentation on squamous skin cancer cells that confirmed the existence of two distinct cell populations: one population had stem cell-like characteristics, and the other population had a slower cycling rate that generated terminally differentiated cells (Driessens et al., 2012). Conversely, another theory states that CSCs arise from spontaneous dedifferentiation. In this theory, tumor progenitor cells undergo tumorigenesis in response to external toxic exposure, which then induces mutations to create CSCs (Nimmakayala et al., 2019). Accordingly, non-CSCs can also revert to CSCs without having to alter their genetics, downstream (Tsuchiya and Shiota, 2021). Other factors contributing to CSC origin include a metabolic shift, cell fusion, or horizontal gene transfer that drive non-CSCs to reprogram into CSCs (Nimmakayala et al., 2019). Eliminating CSCs alone may not be sufficient to stop cancer progression. Since CSCs can spontaneously differentiate from non-CSCs within the tumor, a comprehensive therapy may be more effective by combination of conventional cancer therapy and new CSC-targeted treatment. Next, we will highlight CSCs’ phenotypic markers, which define the tumor-driving cells and stand as therapeutic targets.
These models of cancer origin suggest two approaches in our understanding not only of cancer origin but also of cancer treatment. Conventional cancer treatments are designed to cause tumor shrinkage assessed by the ablation fraction of tumor mass or fractional kill (Palmer et al., 2019). Although the bulk of the tumor, comprised of differentiated or differentiating cells, may show shrinkage, the CSC subpopulation may escape detection by such treatment and may trigger relapse and metastasis. Since CSCs account for a miniscule number of cells within the tumor, such fractional kill index may not reveal the CSC status after conventional chemotherapies. Targeting the tumor-driving CSC directly may prevent tumor relapse (Walcher et al., 2020) while circumventing the need to treat the entire tumor (Miyoshi et al., 2021). Thus, focusing on CSCs as the pivotal origin of cancer may pave the way for innovative views about cancer onset, progression, and its treatment.
2.1 Phenotypic markers associated with cancer stem cells
In-depth analysis of the distinct phenotypic markers on CSCs not only allows us to differentiate CSCs’ unique biological properties from its stem cell counterpart but also provides us with key insights into specific CSC-targeted therapies. Specific CSC markers which are upregulated on the surface of CSCs, referred to as the cluster of differentiation (CD), designate them as unique compared to non-tumorigenic stem cells (Kaur et al., 2021). Similarly, CSCs markers also include proteins unique to CSCs and ATP-binding cassette (ABC) efflux transporters (Pattabiraman and Weinberg, 2014). Among these CSC markers that have been widely characterized include: 1) CD44 routinely found in breast cancer, prostate cancer, and gastric cancer, Head and neck squamous cell (HNSCC) (Müller et al., 2020); 2) CD133 detected in glioblastoma, lung cancer, sarcomas, pancreas, and prostate (Pattabiraman and Weinberg, 2014; Müller et al., 2020); 3) CD90 in glioblastoma, breast cancer (Müller et al., 2020); 4) CD117 in glioblastoma, breast cancer, ovarian cancer, lung cancer (Müller et al., 2020); 5) CD29 in breast cancer, colon cancer (Müller et al., 2020); 6) CD47 found on majority of CSC types; engages signal regulatory protein alpha (SIRPα) on macrophages to inhibit their phagocytosis (Müller et al., 2020); 7) aldehyde dehydrogenase 1 (ALDH1) protein that is upregulated in the majority of CSCs (Pattabiraman and Weinberg, 2014); 8) increased expression of lysyl oxidase (LOX) in CSCs is associated with breast cancer (Tsuchiya and Shiota, 2021), and; 9) efflux pumps upregulated in the majority of CSCs, contributes to CSC resistance to conventional chemotherapy (Pattabiraman and Weinberg, 2014).
2.2 Cancer stem cells versus stem cells
Similar to CSCs, stem cells (SCs) have self-renewal and differentiation capabilities, and SCs are defined as either embryonic, germinal, or somatic (Kakarala and Wicha, 2007). Embryonic SCs, derived from the inner cell mast of a blastocyte, are totipotent or pluripotent and can generate into any cell type with unlimited replication potential (Kakarala and Wicha, 2007). Germinal SCs, harvested from the germinal layer of the embryo, differentiate into specific organs (Kakarala and Wicha, 2007). Somatic SCs are multipotent and have the capacity to self-renew and differentiate into many types of cells but are limited to a specialized tissue sub-type (Rossi et al., 2020).
Activation of Hh, Wnt, Notch, and TGF-β signaling pathways in CSCs lead to the induction of the embryonic signaling pathway, which is linked to epithelial-to-mesenchymal transition (EMT) activation that ultimately transforms adhesive cells into a mobile phenotype (Takebe et al., 2011). In fact, the embryonic signaling pathway activity is deregulated in various cancers, such as breast, pancreatic, and lung cancers (Takebe et al., 2011). Interestingly, the reverse process of mesenchymal-to-epithelial transition (MET) is also crucial in cancer progression (Yamamoto et al., 2017). In vitro studies evidence that both EMT and MET processes, which are governed by the vital proteins Zinc finger E-box-binding homeobox 1 (ZEB1) and Zinc finger protein SNAI2 (SLUG), are crucial for cellular metastasis (Yamamoto et al., 2017). The embryonic pathway and EMT process are associated with both CSC’s invasive metastatic potential as well as traditional SC’s plasticity properties (Scheel and Weinberg, 2011). A secondary theory to CSC origin is that CSCs retain the SC’s EMT infrastructure and reactivate the embryonic pathway, rather than curating CSC metastatic properties de novo (Scheel and Weinberg, 2011). In other words, SCs can mutate and evolve into a CSCs with EMT potential. The overlap between embryonic pathways in SCs and CSCs highlights the difficulty in targeted therapeutics that must ablate CSCs while preserving vital SCs. Therefore, future CSC research may need to delve into the intricacies of ZEB1 and SLUG proteins to grasp a better understanding of the embryonic pathway within various stem cell subtypes.
CSC-targeted therapeutics should highlight the differences in surface markers expressed on CSCs compared to normal stem cells. Despite our depiction of known CSC phenotypic markers above (see Section 2.1), there is still no sole ubiquitous marker that encompasses all CSC populations. The ability to differentiate CSCs from SCs poses as an overarching challenge, but certain markers are highly expressed among different CSC subpopulations, namely CD44 (Müller et al., 2020), CD133 (Müller et al., 2020), CD90 (Müller et al., 2020), CD47 (Müller et al., 2020), ALDH1 (Pattabiraman and Weinberg, 2014), LOX (Tsuchiya and Shiota, 2021), and efflux pumps (Pattabiraman and Weinberg, 2014). In comparison, embryonic SCs surface markers include CD9, CD24, CD29, CD90, CD117, and CD324 (Zhao et al., 2012). Somatic SCs are further subdivided into hematopoietic SCs, which give rise to blood cells during hematopoiesis, and mesenchymal stromal cells (MSCs), which spawn cells of mesenchymal origin (Rossi et al., 2020). Moreover, CD34, CD38, CD45, CD90, and CD133 are primary clinical marker for hematopoietic SCs, while CD73, CD90, and CD150 are primary markers of MSCs (Tweedell, 2017; Rossi et al., 2020). Since CD90 is depicted as a common CD marker across the majority of stem cell subtypes, it is more prudent to target CD44 and CD133 as targets for CSC directed therapy. In support of this theory, CD44 and CD133 have been the key targets of liver, pancreatic, gastric, breast, and urinary cancer subtypes (Huang et al., 2022). The most streamlined cancer treatment may also aim to target a combination of CD markers with monoclonal antibody therapy. Most notably combination therapy targeting CD133, CD44, and ALDH (aldehyde dehydrogenase) in breast cancer CSCs halted metastatic progression in pre-clinical models (Croker et al., 2009).
It is known that similar signaling pathways in regulating the self-renewal activity are shared in CSCs and SCs. For example, in regards to the Wnt pathway, Wnt5a, an activator of the non-canonical pathway, is involved in the regulation of CSCs and embryonic SCs, but is not activated in somatic SCs (Zhou et al., 2017). Notch signaling is also required both for SC differentiation and CSC generation via Notch regulated transcription factors (Wang et al., 2009). JAK-STAT signaling is also implicated in maintain somatic SCs’ critical homeostatic and regenerative processes, while STAT signaling also defines CSCs’ self-renewal ability (Herrera and Bach, 2019; Yang et al., 2020). TGF-β mostly partakes in tissue repair and maintenance in somatic SCs through Smad3 induction, and together with CSCs, TGF-β initiates various tumor subtypes (Rossi et al., 2020). Hh signaling activation induces stem cell proliferation via increased proliferation of Sox2+ and Sox9+ in adult pituitary stem cells and Hh signaling serves as vital factor in ovarian somatic SCs (Zhang and Kalderon, 2001; Pyczek et al., 2016). In the same token, Hh is linked with development of CSC formation (Takebe et al., 2015). Lastly, VEGF regulates both hematopoietic SC survival and CSC survival through a similar autocrine loop mechanism (Gerber et al., 2002; Mercurio, 2019a; Müller et al., 2020). Therefore, while CSCs and SCs act through similar signaling pathways, including Wnt, TGF-β, Notch, JAK-STAT, Hedgehog, and VEGF, the pathways fail to reach homeostasis in CSCs, contributing to CSC’s chemoresistance (Takebe et al., 2015). Our present review is to gather a better understanding of CSC’s signaling pathways in order to synthesize novel modes of cancer treatment.
3 Signaling pathways linked to cancer stem cells
The phenotypic markers and signaling pathways that are involved in CSC activation and maintenance stand as the same pathways that can be targeted for treating cancer. Because CSCs rely on multi-pronged molecular cues, in particular their signaling pathways, for their stemness, directly manipulating these pathways may abrogate their proliferative phenotype, thereby preventing CSC-mediated relapse (Correia et al., 2022). Among these potent signaling pathways that mediate CSCs include Wnt, Notch, TGFβ-SMAD, Hh, JAK-STAT, VEGF, IL-8, granulocyte macrophage colony-stimulating factor (GM-CSF), and bone morphogenic protein (BMP), and below we will discuss their unique therapeutic targets.
A postulated network of signaling pathways associated with CSC activation and maintenance can be deduced, with the overarching concept that CSCs communicate with the TME via paracrine and juxtracrine signaling pathways (Lau et al., 2017). Some major signaling pathways that have been examined to identify the localization of CSCs include: 1) Wnt found in pancreatic, breast, glioma, leukemia, colon, carcinoma and gastric cancer (Sharma et al., 2021); 2) TGFβ-SMAD observed in pancreatic, breast, and glioma (Peng et al., 2022); 3) Notch in breast, ovarian, glioma, pancreatic, and colon cancer (Zhdanovskaya et al., 2021); 4) Hedgehog detected in leukemia, myeloma, pancreatic, breast and glioma (Pattabiraman and Weinberg, 2014; Chen et al., 2021a); 5) JAK-STAT seen in glioblastoma, colon, prostate, and breast cancer (Hu et al., 2021); 6) platelet derived growth factor receptor (PDGFR) apparent in breast cancer (Farooqi and Siddik, 2015); 7) Nanog in glioma, colon, and gastric cancer (Chen et al., 2021a), and; 8) phosphatidylinositol-3-kinase (PI3K) in glioma, colon, and gastric cancer (Tran et al., 2016).
Based on the stemness feature of CSCs, signaling pathways have been postulated as key mechanisms that participate in CSC activation and maintenance. Principal signaling pathways that have been linked to CSC stemness include: 1) Wnt for CSC formation and maintenance (Kim and Kahn, 2014); 2) Notch for control of CSC replication, survival and differentiation, as well as renewal (Venkatesh et al., 2018); 3) TGFβ-SMAD promotes CSC self-renewal, migration, and invasion of the tumor by facilitating an inflammatory TME (Derynck et al., 2021; Yan et al., 2021); 4) Hh regulates CSC metabolism, thereby increasing CSC generation and maintenance (Statkiewicz et al., 2014), while promoting macrophage recruitment abetting the TME to become more conducive for CSC growth (Lee et al., 2018); 5) JAK-STAT regulates CSC growth, formation, and size (Dolatabadi et al., 2019), and contributes to sustained inflammation of the TME, further exacerbating CSC proliferation (Owen et al., 2019); 6) VEGF facilitate CSCs survival and self-renewal via an angiogenic system to support the cells (Mercurio, 2019b; Tsuchiya and Shiota, 2021); 7) IL-8 facilitates CSC proliferation and expansion by inducing an immunosuppressive TME (David et al., 2016; Kim et al., 2021a; Hirata et al., 2022); 8) GM-CSF contributes to macrophage recruitment to the TME, serving as an immune modulation and hematopoiesis platform for CSC to communicate with tumor cells (Hong, 2016; Liu et al., 2016; Li et al., 2020). Below we will delve into the intricacies of the major CSC signaling pathways (Figure 1).
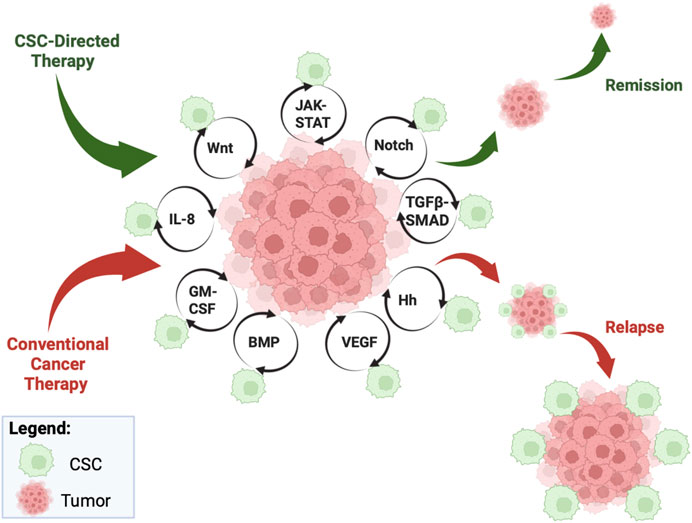
FIGURE 1. Overview of CSC pathways. Cancer stem cells (CSCs) escape conventional cancer therapy, resulting in an increase in non-CSC progression and maintenance via multi-pronged molecular pathways, such as JAK-STAT, Notch, TGFβ-SMAD, Hh, VEGF, BMP, GM-CSF, IL-8, and Wnt. While conventional cancer therapy will result in relapse (see red pathway), CSC-directed therapy can directly target the CSCs and ultimately afford tumor remission (see green pathway). Figure constructed via Biorender.com.
3.1 Wnt signaling pathway in CSCs
3.1.1 Wnt ligand-receptor engagement
Wnts comprise a family of nineteen glycoproteins, which act as ligands (Komiya and Habas, 2008). The Wnt ligand binds to one of the ten types of Frizzled (Fz) extracellular receptors, which is categorized as a seven-transmembrane G-protein coupled receptors (GPCRs) (Komiya and Habas, 2008). There are two main Wnt pathways: 1) the canonical pathway, which is dependent on a low-density-lipoprotein-related protein5/6 (LRP5/6) co-receptor, and 2) the non-canonical β-catenin-independent pathway, which features ROR1/ROR2/RYK co-receptors, which can be further subdivided into the Planar Cell Polarity (Wnt/PCP) and the Wnt/Ca2+ pathways (Komiya and Habas, 2008; Ackers and Malgor, 2018). Most notably, the canonical Wnt pathway features a central element, β-catenin, which upon translocating to the nucleus, regulates gene expression by recruiting CREB-binding protein (CBP) to form complexes with the transcription factors, T-cell/lymphoid enhancer (TCF/Lef) (Wiese et al., 2017). If the Wnt ligands fail to activate the receptor, β-catenin is directed to a degradation complex composed of the tumor suppressor Adenomatous Polyposis Coli (APC), AXIN1/2, and kinases CK1α/GSK-3β (Duchartre et al., 2016). Tankyrase (TNKS) is an enzyme that mediates the ubiquitination and degradation of AXIN1/2; most notably tankyrase inhibitors will upregulate the function of the β-catenin degradation complex, serving as a promising target for therapeutics (Wang et al., 2021). Under normal conditions, the Wnt pathway serves a vital role in embryogenesis and the cell cycle, in which β-catenin promotes differentiation and development of specific T-cells, dendritic cells, and tissue systems (Kaldis and Pagano, 2009; Zhan et al., 2017). However, CSCs will “hijack” the Wnt pathway, resulting in uncontrolled proliferation of cancer cells (Fang et al., 2016). Over time, Wnt remains constitutively active due to mutations in tumor suppressor genes and oncogenes in select cancer subtypes (Fang et al., 2016). The large number of Wnt ligand-receptor complexes trigger multiple complex cascades, which play a large role in various types of cancer. For example, Wnt pathway activation results in reduced survival rates in over 50% of breast cancer patients, Wnt signaling modulates gastrointestinal cancers, and the balance between canonical and non-canonical Wnt signaling is linked to melanoma progression (Zhan et al., 2017). However, due to the complexity of the cascade, current Wnt targeted therapeutics are typically halted at the preclinical stage or phase I/II stages of clinical trials (Katoh, 2017). Regardless, further exploration into combined Wnt therapeutics and targeted treatments will provide valuable insight on the intricacies of CSCs.
3.1.2 Wnt upstream and downstream mechanisms
Wnt pathways exhibit crosstalk with the Ras/MAP kinase, PI3/Akt, PLCγ, Notch, Hh and TGFβ/BMP pathways through various feedback loops (Katoh, 2017). Specifically, TGF-β pathway upregulates Wnt2B and Wnt3 expressions in turn activating the canonical Wnt pathway, while Wnt5A and Wnt11 genes are upstream activators of the non-canonical Wnt pathway (Katoh, 2017; Lecarpentier et al., 2019). Both Wnt signaling pathways require activation by the endoplasmic reticulum acyltransferase Porcupine5-7 (PORCN) (Liu et al., 2022). Specifically, PORCN is a membrane-bound O-acyltransferase that is required for the covalent attachment of fatty acids to Wnt ligands in a palmitoylation process, a vital step in Wnt ligand-receptor activation (Guan and Fierke, 2011; Liu et al., 2013a). Upon receptor activation, Dishevelled (Dsh/Dvl) is the first downstream activated intracellular signaling protein, which is involved in each branch of the Wnt cascade (Komiya and Habas, 2008). Activation of the canonical Wnt cascade upregulates the Hh cascade in rodent breast CSCs downstream, while the non-canonical Wnt cascade triggers PI3K-Akt activation promoting CSC survival (Katoh, 2017). Activation of Dsh also leads to β-catenin accumulation in the cytosol, which will then translocate to the nucleus to bind the TCF/Lef to induce cyclin D1, cMYC, monocarboxylate transporter 1 (MCT1), pyruvate dehydrogenase kinase (PDK), and fibronectin target genes. T cell factor 1 (TCF1) is also a downstream transcription factor of canonical Wnt signaling and plays a key role in the development on CD8+ memory and effector T cells (Lecarpentier et al., 2019).
3.1.3 Wnt associated genes, markers, and molecules
Many human cancers are associated with mutation or loss-of-function in both canonical and non-canonical Wnt genes: 1) abnormal catenin beta 1 gene (CTNNB1), a downstream co-activator of TCF/Lef, is associated with human breast cancer (van Schie and van Amerongen, 2020) 2) tumor suppressor APC mutations are mainly associated with colorectal cancers (Aghabozorgi et al., 2019), and 3) mutated AXIN1/2 genes, which encode the AXIN protein, a key component of the β-catenin degradation complex, are associated with gastrointestinal cancers (Mazzoni and Fearon, 2014; Zhan et al., 2017; Lecarpentier et al., 2019). The hallmark of canonical Wnt associated cancers is an upregulation of cyclin-D1 and C-Myc genes, which are associated with the cell cycle (Lecarpentier et al., 2019). Interestingly, the mutations in the canonical Wnt pathway are commonly accompanied by mutations in the circadian genes: CLOCK, BMAL1, PER (Lecarpentier et al., 2019). Notable canonical Wnt pathway associated CSC surface markers include: 1) LGR5, a Wnt target gene that encodes the receptor for the R-spondin (RSPO) ligand, which is associated with colorectal, pancreatic, endometrial, and intestinal CSCs (Barker and Clevers, 2010; Lin et al., 2015; Tomita et al., 2020), 2) epithelial cell adhesion molecule (EPCAM) upregulation associated with colon cancer (Zhou et al., 2015), 3) CD44+/CD133+ CSCs are positive regulators of the Wnt pathway associated with prostate (Acikgoz et al., 2021) and colorectal cancer (Tsuchiya and Shiota, 2021), and 5) CD44v6 associated with colorectal CSCs (Lin et al., 2015; Duchartre et al., 2016; Katoh, 2017; Zhan et al., 2017). TERT genes, which maintain CSC’s long telomeres, enhance binding of β-catenin to its promoter, thereby highlighting the correlation between telomerase activity and the Wnt pathway (Zhan et al., 2017). Overall genes associated for the canonical Wnt pathway initiate the EMT of CSCs, while non-canonical Wnt genes are responsible for the persistence and metastasis of CSCs (Katoh, 2017). CSCs undergo EMT to transition from their previous static epithelial cell state to gain migratory and anti-apoptotic abilities (Pattabiraman and Weinberg, 2014). Also of interest, the key genes, AXIN2, APC downregualted-1 gene (APCDD1), and Dickkopf Wnt pathway inhibitor 1 (DKK1) (Koinuma et al., 2006; Sato et al., 2007; de Sousa and Vermeulen, 2016), are negative regulators of the Wnt pathway, so silencing these genes will inhibit CSC expansion. Combined therapeutics should target both the canonical and non-canonical Wnt pathways to irradicate CSCs.
3.1.4 Wnt signaling and cancer: preclinical and clinical studies
The Wnt signaling pathway plays a critical role in stem cell biology and is implicated in CSC/tumor initiating cell population with aberrant Wnt signaling associated with tumor formation, suggesting that arresting Wnt signaling may block CSC maintenance (Kim and Kahn, 2014). Therapeutics which target the Wnt pathway have four general categories: 1) ligand/receptor-targeted drugs (ex. cirmtuzumab, rosmantuzumab and vantictumab): are currently in clinical trials and show anti-CSC effects, 2) PORCN inhibitors are under clinical trials targeting the small molecule PORCN, an O-acyltransferase that is required for Wnt ligand palmitoylation (ex. as IWP-2, WNT974, and ETC-159) (Liu et al., 2013b), 3) Tankyrase inhibitors, such as AZ1366, G007-LK, and JW55, upregulate the destruction complex via AXIN1/2 and are undergoing preclinical testing, 4) β-catenin inhibitors block CSC motility, most notably ICG-001 (and the clinical equivalent PRI-724) are in phase I/II clinical trials (Katoh, 2017; Wang et al., 2021). β-catenin inhibitors are challenging therapeutics due to the complexity of the Wnt pathway, but specific small molecule and monoclonal antibody treatments for select Wnt pathway proteins may also prove successful. For example, ICG-001, a small molecule inhibitor, specifically targets the CBP to arrest tumor growth in both animal and human cancer cell lines (Pattabiraman and Weinberg, 2014; Deng et al., 2020; Wiese et al., 2020). LF3 another small molecule inhibitor of the β-catenin/TCF4 interaction, which was found in an in vitro colon cancer mouse cell line, was shown to block CSC’s self-renewal, inhibit migration, and trigger differentiation into a benign tumor state, while not interfering with any other signaling pathway (Fang et al., 2016). Anti-FZD mAb, anti-ROR1 mAb, anti-RSPO3, and anti-LGR5 mAb therapeutics are currently in preclinical or clinical trials (Katoh, 2017). In another study, knockdown of mutated Wnt pathway members in chronic lymphocytic leukemia (CLL) cells showed that leukemia initiating cells (LIC) are dependent on the Wnt pathway for survival (Zhan et al., 2017). Studying patient specific CSC’s for their EMT and MET, immune editing, and metabolism through a genome sequencing process referred to as “omics monitoring” is vital for Wnt targeted therapy (Katoh, 2017). Through omics monitoring, Wnt pathway specific therapeutics can be optimized with combination therapy to target CSCs.
Despite extensive research on the Wnt signaling pathway, as of March 2023 there is still no FDA approved drug to treat cancer that specifically targets Wnt signaling. Notwithstanding the number of Wnt inhibitors in phase I/II clinical trials [NCT02413853, NCT02278133], overall safety and effectiveness remains a challenge as generalized Wnt inhibitors have detrimental effects on embryogenesis and overall cell homeostasis (Kaldis and Pagano, 2009; Zhan et al., 2017). For instance, the PORCN inhibitor WNT974 (aka LGK974) not only resulted in tumor growth inhibition in head and neck cancer cell lines but also decreased intestinal epithelium when administered at high doses (Liu et al., 2013a). Moreover, in human clinical trials, WNT974 treatment was generally well tolerated to treat advanced solid tumors with the most common side effect being dysgeusia, altered taste perception, in 50% of patients [NCT01351103], (Rodon et al., 2021). However, higher grade adverse events are expected to occur in clinical trials involving OMP-54F28, a Wnt recombinant fusion protein, as bone density loss had to be supplemented with vitamin D3 and calcium carbonate [NCT01608867]. Overall, concerns regarding Wnt pathway inhibition include effects on intestinal cells, bone density, dysgeusia, decreased appetite, fatigue, muscle spasms, and overall cell homeostasis (Jimeno et al., 2017). The questions as to whether the benefit of Wnt inhibitors outweighs the risks remains. However, despite diverging opinions regarding Wnt targeted signaling in cancer, as our understanding of the Wnt pathway increases, so does the growing enthusiasm for novel Wnt therapeutics (Figure 2).
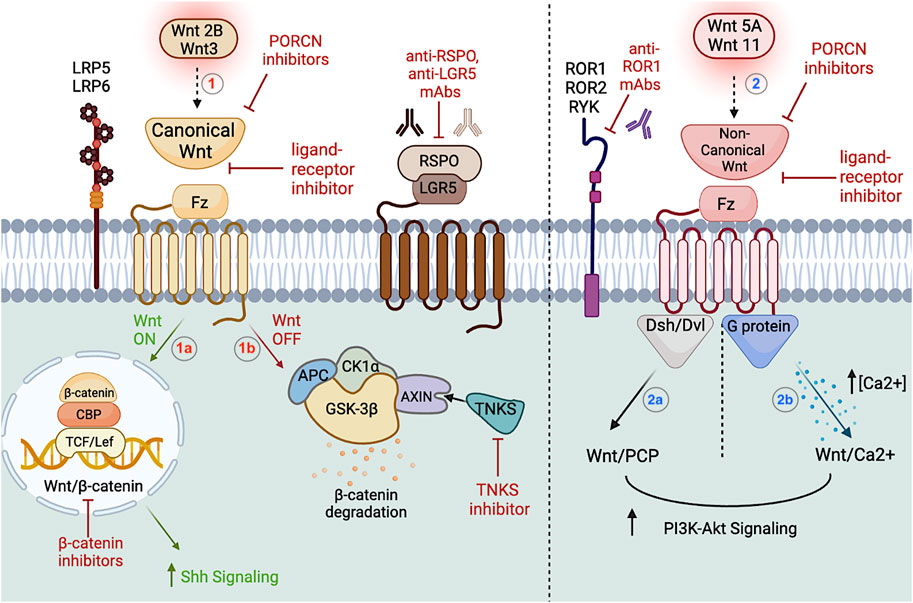
FIGURE 2. Wnt signaling pathway in CSCs. The left portion of the figure depicts the canonical pathway, which upon Wnt2B and Wnt3 activation, the ligand will bind to the Fz receptor in association with the LRP5/6 co-receptor (1). LGR5 is a membrane bound target GPCR encoding the RSPO protein associated with upregulating CSCs in the canonical pathway. Upon receptor activation (Wnt ON), β-catenin will translocate to the nucleus and form complexes with TCF/Lef to regulate CBP expression (1a). If the Wnt ligand fails to activate the receptor (Wnt OFF), β-catenin is directed to a degradation complex composed of APC, AXIN, and kinases CK1α/GSK-3β (1b). TNKS polymerase upregulates the destruction complex via AXIN1/2. Ultimately, the Wnt/β-catenin results in downstream Hh cascade upregulation. Featured inhibitors of the canonical pathway are PORCN inhibitors, ligand-receptor inhibitors, anti-RSPO and anti-LGR5 mAbs, TNKS inhibitors, and β-catenin inhibitors. The right portion of the figure depicts the non-canonical pathway, which upon Wnt5A and Wnt11 activation, the ligand will bind to the Fz receptor in association with the ROR1/ROR2/RYK co-receptors (2). The non-canonical pathway can be further subdivided into Wnt/PCP (2a) and the Wnt/Ca2+ pathways (2b), which upon activation of Dsh/Dvl and G proteins, will upregulate PI3-Akt signaling downstream. Featured inhibitors of the non-canonical pathway are PORCN inhibitors, ligand-receptor inhibitors, and anti-ROR1 mAbs. Figure constructed via Biorender.com.
3.2 Notch signaling pathway in CSCs
3.2.1 Notch ligand-receptor engagement
The Notch signaling pathway not only plays a critical role in embryogenesis, but also in regulating CSC proliferation, maintenance, and differentiation, with notable contribution to angiogenesis (Venkatesh et al., 2018). Classically, Notch serves a dual role acting as both a tumor suppressor and an oncogene, at times leading to tumorigenesis depending on tissue type or genetic mutation (Meisel et al., 2020). Depending on the microenvironment, Notch is typically downregulated in prostate, skin, lung, liver, and some breast cancers, while Notch is upregulated in gastric, colon, pancreatic, and some breast cancers (Yang et al., 2020). Under hypoxic conditions, the Notch pathway undergoes EMT, therefore the consensus is that CSCs typically upregulate the Notch pathway to enhance their stemness properties (Meisel et al., 2020). Notch signaling initiates when Delta/Serrate/Lag2 (DSL) transmembrane Notch ligands (i.e., Delta-like 1/3/4, or Jagged 1/2) bind to a Notch receptor (Notch1–4) in a juxtracrine fashion, triggering cleavage via ADAM-10 protease, followed by gamma-secretase (γ-secretase) intramembrane protease, which releases the intracellular Notch portion from the plasma membrane (Anders et al., 2006; Venkatesh et al., 2018). This cleavage results in Notch’s intracellular domain (NICD) translocating to the nucleus and binding to a CBF1/Suppressor of Hairless/Lag-1 (CSL) transcription factor, ultimately activating target genes (Venkatesh et al., 2018; Yang et al., 2020). Since activation of the Notch signal transduction pathway in CSCs, treatment directed at this pathway may control CSC replication, survival, and differentiation, thus regulating CSC renewal and modulating CSC-mediated tumor formation and its recurrence (Venkatesh et al., 2018).
3.2.2 Notch upstream and downstream mechanisms
The following upstream activators of the Notch pathway contribute to the self-renewal capacity of CSCs: 1) nitric oxide (NO) synthase increases the stemness of liver CSCs, and NO targeted therapy increases tamoxifen potency in breast CSCs (López-Sánchez et al., 2021), and 2) MAP17, a 17 kDa non-glycosylated membrane protein that is associated with an increase in reactive oxygen species (ROS), is upregulated in cervical, breast, colon, and lung CSCs (Guijarro et al., 2007; Garcia-Heredia et al., 2017; Yang et al., 2020). Hyperactivation of Notch can typically occur through a genetic mutation resulting in either increased expression of the cleaved portion of Notch’s intracellular domain, or ligand-independent receptor activation (Meisel et al., 2020). However, genetically independent receptor activation via ligand abundance or increased ligand-receptor affinity could also result in carcinogenesis (Meisel et al., 2020). For example, an increase in the expression of Notch2 and Jag1 are correlated with an increased incidence of medulloblastoma and prostate cancer, respectively (Meisel et al., 2020). Downstream, Notch communicates with a variety of immune cells in the TME, such as myeloid derived suppressor cells (MDSCs), tumor associated macrophages (TAMs), and T regulatory cells (Tregs) (Venkatesh et al., 2018). MDSCs are exceedingly dynamic cells that further promote CSC stemness via NO secretion via both the STAT-3 (Müller et al., 2020) and Notch pathway (Chen et al., 2021a). Notch signaling also regulates the downstream proto-oncogene C-Myc, cell cycle regulators (ex. cyclin, D1, and Cdkn1) and can act as a positive or negative regulator on itself via Deltex E3 Ubiquitin Ligase 1 (DTX1), a protein coding gene (Matsuno et al., 1995; Meisel et al., 2020). The Hh pathway also induces C-myc expression, which acts in parallel with the Notch pathway (Meisel et al., 2020). The Notch pathway also exhibits crosstalk with STAT3 signaling to promote EMT transition (Shan et al., 2021).
3.2.3 Notch associated genes, markers, and molecules
The Notch signaling pathway significantly contributes to CSC maintenance in several cancer types, paving the way for development of Notch inhibitors as an anti-cancer strategy (Purow, 2009). The following markers and molecules are known to enhance and maintain the stemness properties of CSCs via Notch signaling: 1) delta-like ligand 4 (DLL4) maintains gastric CSCs and regulates tumorigenesis (Segami et al., 2021), 2) tumor necrosis factor-α (TNFα) inhibition in liver CSCs decreases cancer metastasis (Renz et al., 2018), 3) BMP4 has a dual role - it is upregulated in breast CSCs (Bach et al., 2018) while BMP4 inhibited hepatic CSC self-renewal (Zhang et al., 2012a), 4) Jagged 2 is upregulated on breast and lung CSCs under hypoxic conditions, and 5) blocking VEGFR2 causes skin CSC pool size reduction (Beck et al., 2011; Takebe et al., 2014; Yang et al., 2020). Similarly, the following genes aid in CSC expression via Notch: 1) Gli3 in oral squamous cell carcinoma (SCC) CSCs, 2) Notch1 in ovarian CSCs under hypoxic conditions, 3) BRCA1 in breast CSCs, 4) Hairy enhancer of split genes (Hes1-7) in medulloblastoma CSCs (de Antonellis et al., 2013), 5) Hey (Hey1, Hey2 and HeyL) in HNSCC (Moon et al., 2019), 6) Notch-regulated ankyrin-repeat protein (NRARP) associates with non-small lung cancer (Liao et al., 2018), 7) cyclin D1 silencing suppresses liver CSC differentiation (Zhang, 2020), 8) DVL1 gene is upregulated on glioblastoma CSCs (Hsu et al., 2021), and 9) ADAM19 is elevated on breast CSCs (Takebe et al., 2014; Meisel et al., 2020; Yang et al., 2020). In a microarray study, the following genes: Notch1, Hes4/5, Hey1/L, and NRARP were inversely proportional to concentrations of the Notch inhibitors (i.e., MK-0752), confirming their effectiveness as biomarkers (Takebe et al., 2014). Strategies that target Notch genes, such as Notch 1, may prove beneficial for cancer treatment (Takebe et al., 2015; Gharaibeh et al., 2020). The following molecules are known to inhibit Notch signaling: 1) microRNA-34a is a potent tumor suppressor known to inhibit gastric CSCs (Jang et al., 2016), 2) PER3 polymorphisms are associated with colorectal CSCs, 3) miR-200b-3p expression is associated with pancreatic CSCs and downregulates colorectal cancer (CRC) cells (Feifei et al., 2019), and 4) miR-26a expression is found on osteosarcoma CSCs and inhibits ovarian (Gao et al., 2020) and colorectal cancers (Yang et al., 2020; Chen et al., 2021b). Inhibition of these Notch signaling molecules could also serve as candidate targets for developing potential cancer therapeutics (Zhang et al., 2017a; Zhang et al., 2017b).
3.2.4 Notch signaling and cancer: preclinical and clinical studies
The overall premise in targeting Notch signaling pathway is that combining a specific notch inhibitor with a traditional chemotherapeutic will result in a more potent anti-cancer treatment plan. For example, Notch1 inhibition in combination with a chemotherapeutic drug reduced CSC self-renewal in HNSCC CSCs in vitro and in vivo, confirming Notch1 as an ideal target for cancer treatments (Fukusumi and Califano, 2018; Venkatesh et al., 2018). Clinical agents which target Notch signaling fall under two categories: 1) γ-secretase inhibitors (GSIs) (ex. peptide isosteres, azepines, and sulfonamides) inhibit cleavage of Notch resulting in suppression of angiogenesis and apoptosis of tumor cells, and 2) monoclonal antibodies (mABs) that interfere with Notch ligand-receptor bonding or prevent the conformational change required for cleavage (Venkatesh et al., 2018; Yang et al., 2020). The following GSIs have emerged as promising therapeutics for inhibiting CSCs specifically by modulating the Notch pathway: 1) RO4929097 combined with bevacizumab decreased glioma CSCs in phase I clinical trials, and other RO4929097 combinations are known to combat advanced solid tumors [ NCT0113123], 2) PF-03084014 combined with gemcitabine decreased pancreatic CSCs in vivo and decreased desmoid tumors in phase II studies, 3) MRK003 decreased glioma CSCs in vivo, and 4) MK-0752 combined with docetaxel decreased metastatic breast cancer CSCs in vitro, MK-0752 combined with cisplatin treated ovarian cancer in vivo, MK-0752 combined with gemcitabine treated ductal adenocarcinoma of the pancreas in preclinical trials, and MK-0752 showed inhibition of pediatric central nervous system tumors in phase I clinical trials (Venkatesh et al., 2018; Yang et al., 2020). Interestingly, GSIs proved ineffective against in triple negative (ER, PgR and HER-2) breast cancer, which displayed elevated Notch signaling, emphasizing the unique complexity of CSCs and tumor types (Meisel et al., 2020).
In parallel, preclinical studies and clinical trials which feature mAbs that target the Notch pathway and inhibit CSCs have also shown encouraging results: 1) Tarextumab (OMP-59R5) decreased breast, small-cell lung, ovarian, and pancreatic CSCs by 40% after chemotherapy termination in vivo and effectively decreased solid tumors in phase Ib/II clinical trials by targeting Notch2/3 [NCT01277146], and 2) enoticumab (ex. REGN421, SAR153192) partially treated ovarian cancer and solid tumors by inhibiting DLL4 (Chiorean et al., 2015; Venkatesh et al., 2018; Smith et al., 2019) [NCT0087155]. Interestingly, through a combination of both a GSI and a mAb in early-stage cancers, there is a marked increase in chemotherapeutic effect, but a particular side-effect that commonly arises is gastrointestinal toxicities (Takebe et al., 2014; Venkatesh et al., 2018). Through identification of patient-specific Notch pathway pharmacodynamic biomarkers, the patient can be matched to the appropriate Notch inhibitor to create an appropriate treatment plan (Takebe et al., 2014; Venkatesh et al., 2018).
The discovery of effective, safe Notch inhibitors that predict a positive clinical outcome is essential. However, the Notch signaling pathway is not only vital for CSCs self-renewal but is also necessary for embryogenesis and angiogenesis (Venkatesh et al., 2018), posing as a hurdle for selective Notch-targeted therapies. Research is currently focused on Notch-selective GSIs, which inhibit angiogenesis and trigger tumor cell apoptosis, as well as inhibitory monoclonal antibodies that target Notch pathway molecules (Venkatesh et al., 2018). For example, in an in vivo study Notch pathway inhibition via delivery of a GSI reduced the percentage of CSCs within the tumor, while activation of the Notch pathway increased the percentage of CSCs within the tumor (Abel et al., 2014). Moreover, in a phase I clinical trial of Tarextumab, a mAb Notch2/3 inhibitor that targets solid tumors, GI toxicity was the most common adverse event, with patients experiencing diarrhea (81%), fatigue (48%), nausea (45%), anorexia (38%), vomiting (38%), abdominal pain (24%), and constipation (24%) [NCT01277146] (Smith et al., 2019). Tarextumab was also tolerated better at low doses <2.5 mg weekly [NCT01277146] (Smith et al., 2019). Despite intestinal toxicity being the most common side effect of Notch inhibitors, in vivo testing confirms that GI toxicity can be reduced by implementing a Notch3-selective antibody rather than Notch1/2 inhibitors (Wu et al., 2010). Therefore, in order to limit the toxicity of GSIs and mAbs doses must be selective, moderated, and taken intermittently. In conclusion, while drugs targeting the Notch pathway are typically halted at phase I/II clinical trials, an in-depth evaluation of Notch signaling has paved the way for novel CSC therapeutics with the ultimate goal of curing cancer (Figure 3).
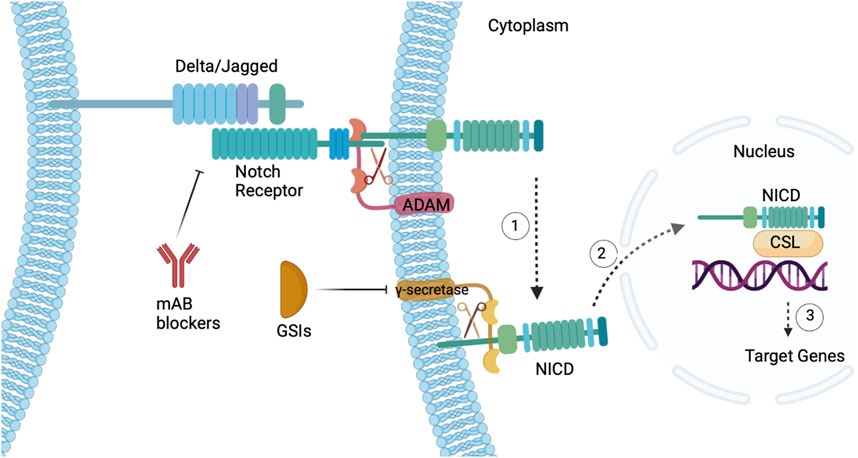
FIGURE 3. Notch signaling pathway in CSCs. (1) Delta/Jagged are notch ligands that bind to a Notch receptor in a juxtacrine fashion triggering cleavage via ADAM protease and γ-secretase. (2) Upon cleavage, the NICD translocates to the nucleus and binds to the CSL transcription factor, (3) ultimately activating target genes. Featured inhibitors of the Notch pathway are mAbs that target the ligand-receptor interaction and GSIs that target γ-secretase. Figure constructed via Biorender.com.
3.3 JAK-STAT signaling pathway in CSCs
3.3.1 JAK-STAT ligand-receptor engagement
The JAK-STAT signaling pathway is integral to the survival, self-renewal, maintenance, and metastasis of CSCs (Yang et al., 2020). The binding of ligands, typically cytokines or interferons (IFNs), to the cytokine cell-surface receptor results in receptor dimerization, which induces both positive and negative regulatory pathways (Kisseleva et al., 2002). After receptor-ligand engagement, the tyrosine kinase JAK (i.e., JAK1-3, and Tyk2), which is composed of seven domains, will phosphorylate the receptor, creating binding sites for proteins possessing an SH2 domain, such as STAT proteins (Kisseleva et al., 2002; Yang et al., 2020). STAT proteins (i.e., STAT1-4, STAT5a/b, STAT6) are transcription activators composed of N and C terminals, a DNA-binding region, and SH2/3 domains (Yang et al., 2020). Upon STAT binding to the receptor, it also undergoes tyrosine phosphorylation, causing the STAT to dissociate from its corresponding receptor, form an anti-parallel dimer, and translocate to the nucleus to affect target downstream molecules (Schindler et al., 2007; Yang et al., 2020).
3.3.2 JAK-STAT upstream and downstream mechanisms
Specifically, JAK-STAT pathway constitutive activation and mutation are associated with many tumors (Yang et al., 2020). The following JAK-STAT upstream activators contribute to the self-renewal capacity of CSCs: 1) IL-6 activates the JAK1/STAT3 pathway in endometrial CSCs, induces EMT in breast and colorectal CSCs (Zhang et al., 2018a), 2) IL-10, an immunosuppressive cytokine, induces stemness properties in lung CSCs (Yang et al., 2019), 3) GM-CSF will create a positive feedback loop between CSCs and TAMs within the TME (Kokubu et al., 2016), 4) PDGF increased stemness and metastatic potential in ovarian CSCs (Raghavan et al., 2020). Downstream, JAK-STAT upregulates PI3-Akt signaling and promotes activation of the MAPK/ERK pathway. The PI3K protein (similar to STAT) contains an SH2 domain, so it can bind to JAK phosphorylated tyrosine receptors and activate the PI3-Akt pathway (Rawlings et al., 2004). In a similar fashion Grb2, an integral protein for MAPK/ERK signaling, also features a SH2 domain, which allows it to bind JAK phosphorylated receptors, and activate the MAPK/ERK pathway (Rawlings et al., 2004). JAK-STAT signaling can integrate multiple signaling pathways, contributing to its complexity and therapeutic potential.
3.3.3 JAK-STAT-associated genes, markers, and molecules
JAK-STAT signaling behaves as a double-edged sword, whereby STAT3 is commonly linked to CSC’s immunosuppression capacity and aberrant TME, while STAT 1/2 activates an anti-tumor immune response via interferons, (i.e., IFN I/II) (Owen et al., 2019; Yang et al., 2020). Therefore, therapeutic avenues should navigate the complexity of JAK-STAT signaling with caution. The following genes, markers, and molecules are known to enrich the stemness properties of CSCs via JAK-STAT signaling: 1) Oct4 promotes ovarian CSCs, and lung CSCs through M2 macrophage polarization (Lu et al., 2020); 2) erythropoietin (Epo) upregulates breast and colorectal CSCs, as well as human gliomas via Epo-dependent constitutive activation of STAT-5 (Kondyli et al., 2010); 3) retinol-binding protein 4 (RBP4) promotes colon CSCs self-renewal via the JAK2-STAT3 pathway (Karunanithi et al., 2017); 4) Hypoxia inducible factor-1 alpha (HIF-1α), a key transcription factor in cancer progression (Rashid et al., 2021), is associated with glioma CSC upregulation (Yang et al., 2020); 5) miR-500a-3p in hepatocellular carcinoma CSCs leads to STAT3 constitutive activation (Yang et al., 2020); 6) TH17, an immunosuppressive cytokine, upregulates CSCs via the secretion of IL-17 via a STAT3-dependent signaling pathway in ovarian, colorectal, and gastric cancers (Chen et al., 2021a). In addition to STAT3, the MAPK (Mitogen Activated Protein Kinase) signaling pathway appears to also solicit the link between TH17 and CSCs in ovarian and pancreatic cancers (Chen et al., 2021a). The following molecules are known to inhibit JAK-STAT signaling: 1) Mir-218 inhibits the JAK-STAT3 pathway resulting in downregulation of lung CSC’s self-renewal capacity (Yang et al., 2017); 2) Ajuba, a LIM domain-containing scaffolding protein, promotes the proliferation of colorectal CSCs through suppression of JAK1/STAT1 (Jia et al., 2017); 3) Von Hippel–Lindau (VHL) cell surface proteins act via the JAK2/STAT3 pathway to suppresses self-renewal ability of glioma CSCs (Yang et al., 2020). Recognizing the dual properties of JAK-STAT, the activation and inhibition of these JAK-STAT signaling regulators represent potential candidate targets for cancer therapy.
3.3.4 JAK-STAT signaling and cancer: preclinical and clinical studies
Due to the integral contribution of the JAK-STAT signaling pathway in CSC preservation, JAK-STAT inhibitors serve as a notable approach to anti-cancer treatment. Targeted JAK-STAT pathway therapeutics fall into one of three categories: 1) cytokine or receptor antibodies, 2) JAK inhibitors, 3) STAT inhibitors (Hu et al., 2021).
In reference to the first category, antibody-cytokine novel fusion proteins (immunocytokines) exert an anti-cancer effect, as shown in reference to IL-2 in phase I/II clinical trials (Mortara et al., 2018). Ruxolitinib in combination tocilizumab, antibodies against IL-6, show improved survival ovarian cancer tumors in vivo (Qureshy et al., 2020). Ruxolitinib enhances cancer treatment in HNSCC, pancreatic, and glioblastoma (Qureshy et al., 2020).
Under the second category, JAK inhibitors typically result in immunosuppressive effects through decreasing proinflammatory cytokines. For example, pacritinib is a JAK2 inhibitor currently under clinical trials to treat AML, prostate, colon, rectal, and non-small cell lung cancer, but no response was observed in colorectal cancer patients (Hu et al., 2021). Cerdulatinib, a JAK1/2 inhibitor, is undergoing clinical trials for treatment of non-Hodgkin’s lymphoma [NCT04757259].
In the third category, STAT inhibitors include SMIs, peptide inhibitors, STAT-targeting small interfering RNAs (siRNA), ASOs that interfere with STAT mRNA, and decoy oligonucleotides (ODNs) (Hu et al., 2021). STAT3 inhibition can occur via the SMI drug, Napabucasin (Chen et al., 2021a). Napabucasin suppresses MDSC immunosuppressive capacity in melanoma-bearing mice (Bitsch et al., 2022) and is in phase I clinical trials for assessment of safety, tolerability, and pharmacokinetics in healthy volunteers (Dai et al., 2021) and patients with metastatic colorectal cancer (Taniguchi et al., 2021). A notable peptide inhibitor is PY*LKTK, which was shown to disrupt STAT3:STAT3 dimerization in vitro (Yue and Turkson, 2009). siRNA targeting STAT 5b enhances the chemosensitivity of gastric cancer cells to gefitinib in clinical trials (Sun et al., 2015). AZD9150, a STAT3 ASO, is undergoing phase 1 clinical trials for patients with advanced hepatocellular carcinoma (HCC) [NCT01839604] as well as advanced solid malignancies [NCT03394144]. ODNs bind to the DNA binding domain preventing STAT proteins from reacting with their appropriate DNA response element (Furqan et al., 2013). Specific ODN targeting STAT3/STAT5 (ex. K562, U251, A172, etc.) were studied in vitro and in vivo on a mice xenograft lung cancer model (Furqan et al., 2013).
JAK-STAT pathway dysregulation is a major contributor to cancer progression. Within the TME, JAK-STAT signaling controls cytokine secretion, inflammatory cascades, and regulates CSCs maintenance and proliferation via upstream and downstream mechanisms. In most cancer subtypes JAK-STAT pathway inhibition serves as a potential chemotherapeutic treatment, but selective targeting remains a challenge due to variabilities in patient genetics, epigenetics, and variations in tumor subtypes. Therefore, JAK-STAT pathway combined therapy targeted drugs should not only inhibit overactivation of the pathway but also delve into the intricacies of JAK-STAT pathway crosstalk as a potential means for novel therapeutics. Current JAK-STAT pathway therapeutics, which include antibody therapy, JAK-inhibitors, and STAT-inhibitors, are typically in phase I/II clinical trials and are focused on administering the drug alone or in combination with chemotherapy, rather than targeting multiple major CSC signaling pathways [NCT03421353, NCT04021082]. JAK-STAT is a complex, non-linear signaling pathway and in some cases crosstalk with JAK-STAT and downstream signaling pathways, such as MAPK/Erk and PI3K/Akt, can contribute to chemotherapy resistance. For example, in a colon cancer cell line, IL-6 secretion mediated the activation of JAK-STAT3 and MAPK/Erk pathways, which increased anti-apoptotic proteins, Bad and Bcl-2, thus leading to chemoresistance to 5-Fluorouracil (Li et al., 2018a). Moreover, JAK1/2 inhibitors, namely AZD1480 for the treatment of solid tumors and Momelotinib that treats non-small cell lung cancer, resulted in clinical trial termination due to neurotoxicity and neutropenia, respectively [NCT01112397, NCT02206763]. Conversely, Ruxolitinib showed improved overall survival rates in a subgroup of pancreatic ductal adenocarcinoma patients with inflammation [NCT01423604]. Despite a clinical trial depicting the benefit of Ruxolitinib in a select patient population, the majority of clinical trials testing the effects of Ruxolitinib resulted in termination, perhaps due to JAK inhibition resulting in decreased immune response, which hinders overall anti-tumor effects [NCT02117479, NCT02119663, NCT01562873], (Schwartz et al., 2017). Despite potential drawbacks in research, JAK-STAT targeted CSC therapy is a novel research avenue with the future direction of curating personalized medicine treatment plans based on variations in JAK-STAT signaling among different tumor subtypes (Figure 4).
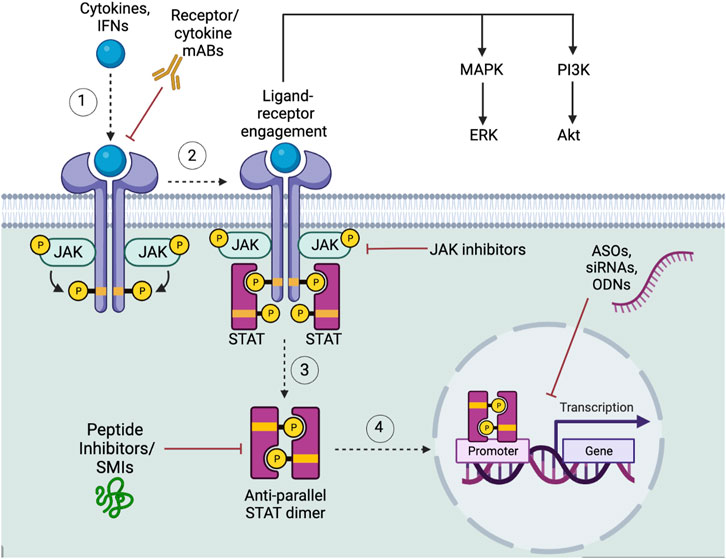
FIGURE 4. JAK-STAT signaling pathway in CSCs. (1) Upon the activation of the JAK-STAT receptor by the appropriate cytokines or IFNs (2) the tyrosine kinase JAK (i.e. JAK1-3, and Tyk2), will phosphorylate the receptor, followed by STAT proteins (i.e. STAT1-4, STAT5a/b, STAT6) tyrosine phosphorylation, (3) leading to STAT dissociation and formation of its anti-parallel dimer. (4) The STAT dimer will then translocate to the nucleus to affect downstream target molecules. Featured inhibitors of the JAK-STAT signaling pathway include receptor mAb inhibitors, targeted upstream cytokine mAb inhibitors, JAK inhibitors, and STAT inhibitors. STAT inhibitors can be subcategorized into SMIs, peptide inhibitors, siRNAs, ASOs, and ODNs. Downstream, JAK-STAT ligand-receptor engagement activates the MAPK/ERK and PI3K/Akt pathways. Figure constructed via Biorender.com.
3.4 Hh signaling pathway in CSCs
3.4.1 Hh ligand-receptor engagement
The hedgehog family is comprised of three homologs: Sonic hedgehog (Shh), Indian hedgehog (Ihh), and Desert hedgehog (Dhh), of which Shh is the best understood (Sasai et al., 2019). Under normal conditions, the hedgehog signaling pathway plays a unique role in embryonic development and cell differentiation (Yang et al., 2020). However, abnormal Hh pathway activation is linked to a variety of cancer types (Schulenburg et al., 2015), and CSCs uniquely express high levels of Hh activation (Yang et al., 2020). When extracellular Hh ligands bind to its downstream twelve-pass transmembrane receptor, Patched (PTCH), such engagement triggers signal transduction, most notably activating downstream GLI transcription factors (i.e., Gli1/2/3) (Pietrobono et al., 2019; Yang et al., 2020). In the absence of ligand-receptor activation, PTCH acts a negative regulator that will inhibit the seven-pass transmembrane GPCR Smoothened (SMO), preventing signal transduction (Pietrobono et al., 2019; Yang et al., 2020). Similarly, in the absence of a ligand, suppressor of fused (SUFU) binds to GLIs, anchors them in the cytoplasm, and prevents them from activating GLI target genes, thereby inhibiting Hh signaling (Rimkus et al., 2016). Selective inhibition of Hh signaling is an effective strategy for impeding cancer progression and halting CSC metastasis.
3.4.2 Hh upstream and downstream mechanisms
The following Hh upstream activators contribute to the maintenance, self-renewal, and regenerative capacity of CSCs: 1) IL-6 activates non-small-cell lung CSCs (Wei, 2019), 2) Gli1 initiates Hh transcription and notably promotes EMT and metastasis of non-small cell lung carcinoma (Jiang et al., 2020), and 3) Gli2 mainly inhibits transcription but in some instances also triggers Hh transcription due to variation in levels of phosphorylation (Pietrobono et al., 2019; Yang et al., 2020). Downstream, the Hh pathway also activates Gli3, a known transcription inhibitor, and Gli3 mutation has been linked to pancreatic adenocarcinoma (Yang et al., 2020). After translation of the Hh protein, the membrane-bound Hedgehog acyltransferase (Hhat) catalyzes a Hh-palmitate linkage and recycles the Hh ligand, so it may again bind to PTCH1 (Rimkus et al., 2016). Hh signaling also contributes to the metastasis of CSCs through upregulation of the following downstream markers: 1) ALDH1 a biomarker associated with early-stage non-small cell lung cancer via Hh signaling (Raz et al., 2012), 2) CD44 a biomarker associated with hepatocellular carcinoma via Hh signaling (Wang et al., 2020), 3) Twist1, Snail, and ZEB1 contribute to CSCs EMT capacity (Rimkus et al., 2016; Li et al., 2021), 4) C-Myc and cyclin-D1 are transcription factors involved in CSC proliferation and cell cycle progression (Rimkus et al., 2016; Elbadawy et al., 2019), 5) Nanog, Oct4, and Sox2 are transcription factors that contribute to CSC self-renewal (Shan et al., 2012; Yin et al., 2015; Rimkus et al., 2016; Yang et al., 2020) 6) PDGFR upregulation on glioblastoma CSCs (Schulenburg et al., 2015; Yang et al., 2020). The Hh pathway also displays crosstalk with the Wnt and Notch pathways via Hh upregulation of Wnt2 and Jagged1, respectively (Ding and Wang, 2017; Yang et al., 2020).
3.4.3 Hh-associated genes, markers, and molecules
Increased Hh signaling is observed in CSCs to support their maintenance, growth, and metastasis (Yang et al., 2020). The following genes, markers, and molecules are known to enrich the stemness properties of CSCs via Hh signaling: 1) CK2α, a protein kinase, supports lung CSCs via upregulation of Gli1 (Zhang et al., 2012b), 2) retinoic acid receptor α2 (RARα2) upregulates Hh and Wnt pathways to maintain myeloma CSCs (Yang et al., 2013), 3) PPM1D, a protein phosphatase, increased Hh signaling via target protein Gli1 in medulloblastoma CSCs (Wen et al., 2016), and 4) lncHDAC2 promotes liver CSCs through Hh signaling (Yang et al., 2020; Jahangiri et al., 2021). Speckle-type POZ (SPOP) is a unique protein in that it at times inhibits Hh signaling in gastric cancer through increasing the degradation of Gli2 (Zeng et al., 2014), while SPOP can also upregulate Hh signaling, decreasing CRC CSC’s rate of apoptosis (Zhi et al., 2016). Similarly, Vasohibin 2 (VASH2) protein suppresses pancreatic CSCs through decreasing SMO and Gli1/2 (Yang et al., 2020), while also functioning as a tumor promoter, stimulating resistance to doxorubicin (DOX) (Mirzaei et al., 2021) upregulating pancreatic CSCs via the Hh and Notch pathways (Liang et al., 2021a). The following molecules are known to inhibit Hh signaling thereby hindering the metastasis of CSCs: 1) BCL6, a transcriptional repressor, inhibits the positive Hh effectors, Gli1 and Gli2, in medulloblastoma CSCs (Yang et al., 2020), 2) RUNX3, a transcription factor, suppresses colorectal CSCs metastasis by blocking Gli1 (Kim et al., 2020), 3) miR-361-3p hinders retinoblastoma CSC self-renewal, by Gli1/3 inhibition and regulates liver CSCs through directly targeting SOX1 (Pietrobono et al., 2019; Qu et al., 2021), 4) miR-326, a scaffolding protein, impedes Gli2 and regulates SMO, thus creating a negative feedback loop for modulating Hh signal transduction (Jiang et al., 2014; Pietrobono et al., 2019), 5) β-arrestin1 (Arrb1), a scaffolding protein, acts as another negative modulator of Hh signaling through acetylation of Gli1, specifically identified in relation to medulloblastoma CSCs (Miele et al., 2017). Probing Hh-associated genes, markers, and molecules may reveal novel pathways or refine existing signaling pathways associated with CSC metastasis, cancer onset, and progression, paving the way for Hh-based cancer therapeutics.
3.4.4 Hh signaling and cancer: preclinical and clinical studies
The following molecules and proteins have emerged as promising therapeutics for inhibiting CSCs by controlling the Hh pathway. Targeted Hh pathway therapeutics fall into one of three categories: 1) SMO inhibitors, 2) Gli inhibitors, and 3) ligand/enzyme inhibitors (Rimkus et al., 2016).
Under the first category, SMO inhibitors prevent Gli activation downstream, leading to target gene inhibition and CSC suppression (Rimkus et al., 2016). LDE225 (also known as Erismodegib/Sonidegib) is a SMO inhibitor as evidenced in vitro and in vivo as a SMO antagonist (D’Amato et al., 2014), and is currently marketed as Odomzo® to treat advanced stage basal cell carcinoma (BCC) [NCT04066504]. The same SMO inhibitor, Sonidegib, also acts synergistically with JAK2 inhibitors (ex. ruxolitinib) to inhibit HER2-positive triple-negative breast CSCs in vitro (Doheny et al., 2020). In the same token, the Hh antagonist Vismodegib, a cyclopamine derivative that binds SMO preventing Gli activation and macrophage recruitment modifier, is a clinically approved therapy to treat BCC and is undergoing phase II clinical trials for medulloblastoma and other cancers (Gampala et al., 2021) [NCT00833417]. Another SMO inhibitor, steroidal alkaloid cyclopamine, is a natural plant product that directly antagonizes Hh in vivo (Incardona et al., 1998; Tremblay et al., 2009) and is currently under phase II clinical trials for medulloblastoma [ NCT01878617]. Due to the integral role of SMO in Hh signaling, it is the main target of many therapeutics.
Under the second category, Gli antagonists (GANTs) act downstream of the Hh signaling pathway and, therefore can be activated via Hh-independent pathways (Rimkus et al., 2016). Arsenic trioxide (ATO), a Gli1/2 suppressor, is the first FDA approved drug that targeted the Hh pathway to treat acute promyelocytic leukemia ATO and currently undergoing preclinical testing for a variety of cancers, including prostate and colon cancer (Rimkus et al., 2016; Carpenter and Ray, 2019). GANT-61 inhibits both Gli1/2 downstream effectors and decreases tumor growth in vivo and in vitro in a variety of cancers, including prostate and ovarian cancers (Rimkus et al., 2016; Carpenter and Ray, 2019). Interestingly, Balanophora polyandra Griff (BPPs) plant extract polysaccharides also suppress the Hh pathway through EMT inhibition downstream, in vivo and in vitro, but the specific mechanism remains largely unknown (Li et al., 2021). Combined SMO and Gli-targeted therapies are currently undergoing preclinical testing (Rimkus et al., 2016), as finding the balance between upstream and downstream regulation could be the key to Hh targeted therapeutics.
Under the third category, Shh ligand is the most abundant of the hedgehog ligands, and therefore is a promising therapeutic target (Rimkus et al., 2016). Shh monoclonal antibody 5E1 has been implicated to inhibit medulloblastoma (Coon et al., 2010) and to reduce pancreatic tumor size (Chang et al., 2013) in vivo, but 5E1 has not yet reached human trials (Carpenter and Ray, 2019). RU-SKI 43 inhibits the downstream enzyme Hhat and reduces proliferation of breast cancer and pancreatic cancer cells, as shown in preclinical trials (Carpenter and Ray, 2019). Through both upstream and downstream inhibitors, Hh targeted therapeutics serve as effective strategies to decrease tumor growth and metastasis.
The Hh pathway is necessary for the maintenance of CSCs in multiple cancer subtypes. Current research targeting the Hh pathway is focused on SMO inhibitors, GANTs, and ligand/enzyme inhibitors, of which select inhibitors are FDA approved to treat BCC while others are still undergoing pre-clinical and clinical trials. For example, GDC-0449 (Vismodegib), a SMO antagonist that suppresses the activation of downstream Hh target genes, displays anti-tumor activity in locally advanced and metastatic BCC [NCT00607724]. Moreover, Vismodegib, displayed good activity when treating BCC over long-term regimens (>3 months) (Ally et al., 2014; Dréno et al., 2017). However, a phase II clinical trial administering Vismodegib to treat BCC resulted in poor efficacy and multiple adverse events among patients, including muscle spasms (76%), alopecia (58%), and dysgeusia (50%) (Sofen et al., 2015). While there are several unresolved cases regarding the specific role of the Hh signaling pathway, it is known that Hh signaling plays a critical role in cancer progression, and a better understanding of Hh interactions in each tumor sub-type is vital. It is also important to highlight that the Hh pathway may be activated in response to chemotherapy (Merchant and Matsui, 2010) and can overcome chemotherapy resistance in medulloblastomas (Bar et al., 2007) and pancreatic cancer (Olive et al., 2009). Therefore, Hh inhibitors may be better served to combat early-stage tumors or should be administered following surgical removal of the tumor mass. Overall, Hh inhibitor administration during end-stage tumor progression typically correlates with poorer prognosis. In association with this claim, one established paradox in regard to CSC treatment states that positive patient response rates do necessarily correlate to patient survival rates, and the key to novel CSC treatments should be to develop new statistical methods to better asses changes in CSCs at multiple timepoints (Huff et al., 2006). Despite the vast array of clinical knowledge surrounding Hh signaling pathway (Figure 5), a greater understanding of Hh pathway’s role in CSCs is essential for designing combination therapy that utilizes selective Hh-inhibitors, traditional chemotherapeutics, and/or other major CSC signaling pathway inhibitors.
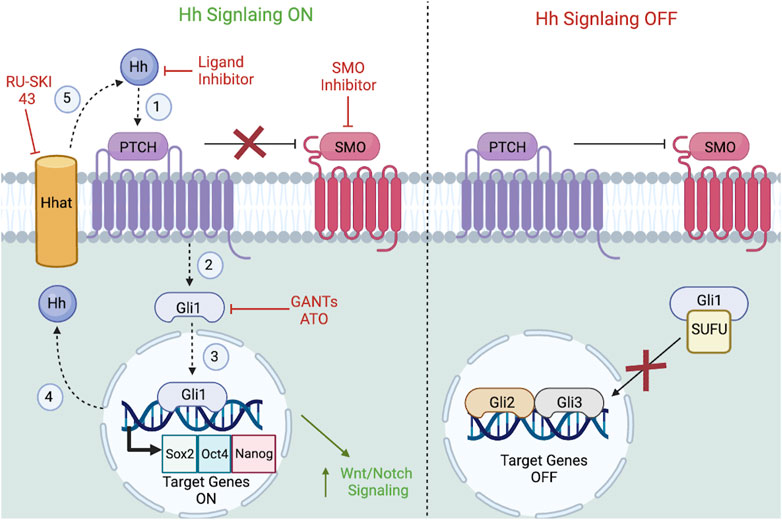
FIGURE 5. Hh signaling pathway in CSCs. Referring to the left side of the figure, Hh signaling is considered ON when (1) upon Hh ligands binding to the PTCH receptor, (2) signal transduction activates downstream GLI transcription factors, (3) which will translocate to the nucleus to transcribe target genes. (4) After the translation of the Hh protein in the nucleus, membrane-bound Hhat catalyzes palmitoylation of Hh ligands, (5) so that it can bind to PTCH1 in a cyclic fashion. Referring to the right side of the figure, PTCH acts a negative regulator of SMO in the absence of ligand-receptor activation. SUFU will also bind to GLIs and anchor them to the cytoplasm to prevent target gene activation. Featured inhibitors of the Hh signaling pathway include SMO inhibitors, GLI inhibitors, and ligand/enzyme inhibitors. Downstream, Hh ligand-receptor engagement interacts with the Wnt and Notch pathways. Figure constructed via Biorender.com.
3.5 VEGF signaling pathway in CSCs
3.5.1 VEGF ligand-receptor engagement
VEGF is not only necessary for angiogenesis and tumor progression but also acts independently of its angiogenic role to maintain CSCs via paracrine and autocrine signaling (Mercurio, 2019a; Müller et al., 2020). Inhibitors of VEGF may regulate CSCs by decreasing VEGF and subsequently suppressing tumor progression (Müller et al., 2020). VEGF ligands A-E and placental growth factor (PIGF) ligand each activate varying vascular endothelial growth factor receptors (VEGFRs), which each possess seven immunoglobulin-like extracellular domains and an intracellular tyrosine kinase domain (Shibuya, 2011; Takahashi, 2011). There are 3 types of VGFRs: VEGFR-1 (Flt-1), VEGFR-2 (Flk-1/KDR), and VEGFR-3 (Flt-4) (Shibuya, 2011). For instance, VEGF-A regulates angiogenesis through activating VEGFR-1/2, while ligands VEGF-C/D activate VEGFR-3 to initiate lymphangiogenesis, the formation of new lymphatic vessels from pre-existing ones (Shibuya, 2011). Therefore, VEGF-A and VEGFR-1/2 are the classical targets for anti-angiogenic therapeutics (Takahashi, 2011). Upon ligand-receptor engagement, the cell surface receptors will become activated and dimerized through transphosphorylation, resulting in a downstream signaling cascade (Holmes et al., 2007).
3.5.2 VEGF upstream and downstream mechanisms
VEGF not only promotes CSC stemness by stimulating angiogenesis through paracrine signaling but also creates a microenvironment for CSCs through neuropilin1 (NRP1) via an autocrine signaling pathway (Beck et al., 2011). VEGF ligands can also act independently of the VEGFRs via neuropilins1/2 (NRPs), transmembrane glycoprotein receptors largely expressed on tumor cells, which function in tumor initiation and maintenance (Mercurio, 2019a). VEGF-NRP2 triggers the extracellular matrix (ECM) glycoprotein laminin’s engagement with α6β1 integrins on the cell membrane (Goel et al., 2012). Laminin-integrin engagement activates the FAK/Ras pathway, stimulates Hh target gene Gli1, and ultimately enhances the expression of a stem cell factor, BMI-1 (Mercurio, 2019a). The VEGF pathway also activates the Rac1GTPase, which in turn inhibits the Hippo kinase LATS, thus ultimately promoting the activation of TAZ, which can reprogram cancer cells into cancer stem cells (Piccolo et al., 2014; Elaimy et al., 2018). While the classically activated VEGF-VEGFR pathway contributes to angiogenesis and lymphangiogenesis to support the maintenance of the tumor, the VEGF-NRP loop facilitates the self-renewal and persistence of CSCs (Müller et al., 2020; Yang et al., 2020). Most notably, the VEGF-NRP loop results in a positive feedback loop with the Hh pathway, since Gli1 upregulates NRP2 expression (Mercurio, 2019a). Similarly, the crosstalk between the VEGF-NRP loop and Hh pathway is also crucial to Hh signaling that engages the PIGF ligand in the onset and progression of medulloblastomas (Snuderl et al., 2013).
3.5.3 VEGF-associated genes, markers, and molecules
The following genes, markers, and molecules enhance the stemness properties of CSCs via VEGF signaling: 1) C-Myc and Sox2 genes show a strong correlation with VEGFR-2 through activation of JAK2/STAT3 signaling in breast CSCs in vivo (Zhao et al., 2015); 2) Hypoxia-inducible factor-1α α (HIF-1α) is a known target gene of VEGF that interacts via the PI3K/Akt pathway (Choi et al., 2011); 3) CD133, a transmembrane protein biomarker found on CSCs, promotes tumor stemness and reoccurrence of HCC through a mechanism dependent on VEGFR2 and Nanog (Liu et al., 2017); 4) COUP transcription factor II (COUP-TFII) is a unique protein that stimulates lymphangiogenesis and tumorigenesis in prostate cancer, enhancing NRP-2 expression (Lin et al., 2010; Qin et al., 2013); and 5) Heparin and NRP1 are involved in VEGFR activation via VEGF-A mechanisms (Takahashi, 2011). Interestingly, some Notch ligands (ex. DLL4) display unique properties in that they may suppress VEGFR2 and NRP1 expression, while other Notch ligands, such as DLL1, stimulate VEGF signaling (Mercurio, 2019a). The following molecules are known to naturally inhibit VEGF signaling, thereby impeding CSCs metastasis: 1) Vitamin C inhibits VEGF signaling through degrading HIF-1α (Zhao et al., 2020); and 2) Melatonin (MLT), a natural pineal gland hormone, regulates neoangiogenesis and inhibits lung and breast cancer cells through the downregulation of HIF-1α/ROS/VEGF (Cheng et al., 2019). While VEGF activates the Raf/MEK/ERK and PI3K/Akt pathways downstream (Ferrara et al., 2003), additional studies are warranted to explore the distinct crosstalk between VEGF and its associated genes and molecules, such as Notch and JAK-STAT signaling components, to unravel novel CSC-targeted therapeutics.
3.5.4 VEGF signaling and cancer: preclinical and clinical studies
The following VEGF targeted therapeutics are promising therapeutics for inhibiting CSCs. Targeted VEGF pathway therapies fall into one of four categories: 1) VEGF ligand inhibitors, 2) receptor inhibitors, 3) VEGF decoy receptors, and 4) ribozymes targeting VEGF (Cardones and Banez, 2006).
Under the first category, bevacizumab (Avastin) is an anti-VEGFA ligand inhibitor approved by the FDA to treat metastatic CRC and renal cell carcinomas (RCC) in combination with traditional therapeutics (Cardones and Banez, 2006). Bevacizumab is currently undergoing phase II clinical trials to treat advanced stage prostate cancer in combination with traditional chemotherapeutic Docetaxel [NCT00574769]. Despite bevacizumab being the most common anti-VEGF drug by blocking VEGFR tyrosine kinases it also enhances VEGF/NRP signaling, thereby resulting in a “VEGF paradox”: bevacizumab cannot fully eradicate prostate CSCs since the drug will enrich CSCs while primarily targeting non-CSCs (Ferrara, 2005; Mercurio, 2019a). To this end, VEGFR therapy combined with NRP2 inhibitors is more effective than the stand-alone drug to treat prostate cancer (Mercurio, 2019a). Similar to bevacizumab, microRNA-140-5p is also an anti-VEGFA ligand inhibitor that hinders breast CSC tumor metastasis and angiogenesis both in vitro and in vivo (Lu et al., 2017). The most common anti-VEGF strategies have focused on molecules and ligands aimed at diminishing VEGFR action, but the downside to this approach is that VEGFRs bind to multiple ligands within their super-family (Ferrara, 2005). Ligand targeted therapeutics should not only diminish non-CSC tumor cells but also eradicate CSCs by hindering the VEGF-NRP loop.
Under the second category, VEGFR tyrosine kinase (TRK) inhibitors hinder angiogenesis and tumor growth (Takahashi, 2011). Sunitinib, a VEGFR tyrosine kinase inhibitor, is FDA approved in an oral tablet form to treat RCCs and gastrointestinal tumors (Takahashi, 2011). Sorafenib is also a tyrosine kinase VEFGR2/3 inhibitor and is FDA approved to treat advanced renal carcinoma and HCC (Takahashi, 2011). Similarly, ramucirumab (Cyramza) is a monoclonal antibody blocks VEGFR2 and is FDA approved to treat multiple solid cancers (Itatani et al., 2018). Vatalanib blocks kinase VEGFR activity and has completed phase II clinical trials with promising results to treat progressive meningioma [NCT00348790] and generalized advanced stage cancers [NCT00171587]. Furthermore, an in vivo study of CSC skin papillomas highlighted how anti-VEGFR2 antibodies in combination with NRP1 deletion prevented tumor metastasis and decreased tumor size through impairing CSC stemness properties (Beck et al., 2011). VEGFR inhibitors show promising effects in both clinical and non-clinical studies.
Under the third category of VEGF decoy receptors, Aflibercept (Zaltrap®) is a soluble recombinant fusion protein that acts as a decoy receptor by binding to VEGF-A ligand with high affinity, preventing VEGFR1/2 activation (Ciombor and Berlin, 2014). Aflibercept has been clinically approved to treat metastatic colorectal cancer in combination with traditional therapeutics (Ciombor and Berlin, 2014), and is currently undergoing phase II clinical trials to treat esophageal and gastric cancers [NCT01747551]. Although effective in the short-term, anti-VEGF therapy is not stable, thus new approaches involving VEGF splicing (Lee et al., 2018) and co-adjunctive treatment with anti-vascular mimics have been explored (Liang et al., 2021b).
Under the fourth category, Angiozyme, a synthetic ribozyme-based VEGFR1 inhibitor cleaves site-specific RNA molecules, which downregulates Flt-1 and KDR, paving the way for a new class of VEGF therapeutics (Weng and Usman, 2001; Weng et al., 2005). A phase I dose escalation study of Angiozyme on patients with refractory tumors showed promising results (Weng et al., 2005), and a phase II clinical trial on metastatic kidney cancer patients was also well tolerated [NCT00021021]. In in vitro studies, anti-VEGF165 ribozyme showed 90.7% efficacy in anti-angiogenesis gene therapy for the treatment of tumors (Gu et al., 2004). More research should be conducted to better understand the promising effects of this novel class of anti-VEGF ribozyme therapeutics.
Both the classical VEGF pathway and VEGF-NRP pathway promote the progression of CSCs within multiple cancer subtypes. Targeting VEGF signaling in combination with chemotherapeutic drugs that target non-CSC pathways can potentially overcome therapy resistance. While VEGF pathway inhibitors are typically well tolerated via multiple routes of administration, common toxicities include fatigue, hypertension, proteinuria, hypothyroidism, and deficient wound healing (Kieran et al., 2012). Further studies are ongoing to address the more serious adverse events that can occur as a result of anti-VEGF related therapies [NCT05108519, NCT04788381], namely thrombosis, encephalopathy, deficient growth plate formation, wound healing deficiencies, and death (Kieran et al., 2012). Current research targeting the VEGF pathway is focused on ligand inhibitors, receptor inhibitors, VEGF decoy receptors, and ribozymes targeting VEGF, of which select inhibitors are marketed to treat CRC, RCC, HCC, solid tumors, and gastrointestinal tumors. Clinical trials also highlight how VEGF combination therapy (i.e., anti-VEGF drug in combination with traditional chemotherapy) is more effective than standard therapy. For example, the most studied FDA approved anti-VEGF inhibitor, bevacizumab, is currently in phase III trials in combination with capecitabine to treat metastatic breast cancer and showed increased response rate from 9.1% to 9.8% (p = 0.001) (Miller et al., 2005). Similarly, another phase III study on bevacizumab and paclitaxel for the treatment of breast cancer provided further supporting evidence of improved objective response rates (Ho and Kuo, 2007). However, positive response rates to combination therapy may not necessarily transfer to overall survival rates of the patient. Moreover, although bevacizumab is widely used to treat CRC and RCC, as of 2010 bevacizumab’s breast cancer indication has been withdrawn (Sasich and Sukkari, 2012). The FDA withdrawing bevacizumab’s indication highlights the major hurdle of anti-angiogenic therapies at treating an array of heterogenic cancer subtypes. As stated above, perhaps the reason for bevacizumab’s inefficacy at treating breast cancer is the VEGF paradox; the paradox states that anti-VEGF drugs commonly target the classical pathway while simultaneously upregulating the VEGF-NRP pathway, which supports CSC maintenance. In accordance with this paradox, it may be more beneficial to treat early tumors with anti-angiogenic drugs that inhibit tumor vascularization, rather than to treat highly vascularized, late-stage tumors, but research on early-stage anti-VEGF treatments is currently limited. Clinical trials on the VEGF signaling pathway (Figure 6) should be expanded upon to encompass the unique signaling mechanisms involved in CSC function.
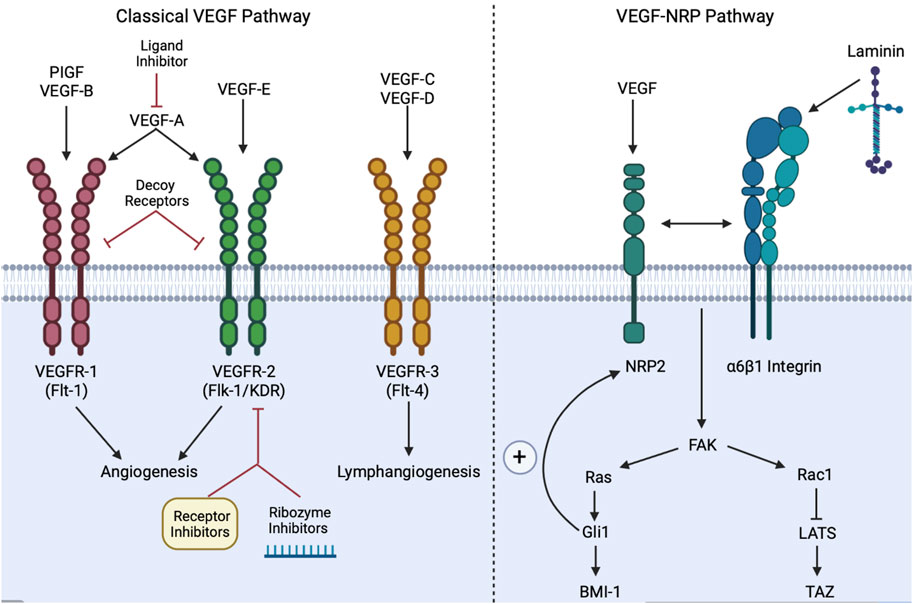
FIGURE 6. VEGF signaling pathway in CSCs. Referring to the left side of the figure, VEGF ligands A-E and PIGF ligand each activate varying VEGFRs, i.e., VEGFRs 1-3. Upon ligand-receptor engagement, tyrosing kinase receptors dimerize and transphosphorylate resulting in VEGFR-1/2 activating angiogenesis and VEGFR-3 initiating lymphangiogenesis. Referring to the right side of the figure, VEGF ligands can also act in an autocrine fashion via NRP1/2. Specifically, NRP2 triggers laminin engagement with α6β1 integrins, triggering the FAK/Ras pathway, which branches off into two downstream mechanisms: (1) FAK protein tyrosine kinase activates Ras, followed by Hh target gene Gli1, and ultimately enhances the expression of a stem cell factor, BMI-1; or (2) FAK activates Rac1 GTPase, which in turn inhibits the Hippo kinase LATS, thus ultimately promoting the activation of TAZ, permitting the turnover of non-cancer cells into cancer cells. Featured inhibitors of the VEGF signaling pathway include ligand inhibitors, receptor inhibitors, decoy receptors, and ribozyme inhibitors. Figure constructed via Biorender.com.
3.6 TGFβ-Smad signaling pathway in CSCs
3.6.1 TGFβ ligand-receptor engagement
Under normal conditions the TGF-β signaling pathway plays a vital role in cell differentiation, homeostasis, and organism development (Yang et al., 2020). However, when downstream signaling proteins are mutated, TGF-β suppresses the development of immune cells and allows for the metastasis and growth of prostate, lung, and breast cancer CSCs (Bellomo et al., 2016; Futakuchi et al., 2019). Upon ligand binding, a type II TGF-β ligand bound receptor will phosphorylate a type I receptor, and the TβRII–TβRI receptor complex will ultimately phosphorylate receptor-regulated Smads (R-Smads) to transcribe targeted genes in the nucleus (Yang et al., 2020). There are three categories of Smads: 1) receptor-regulated Smads are either activated by activin, a beta subunit dimer of the TGF-β protein superfamily, termed activin TGF-β (i.e., Smads 2/3) or are activated by BMP (i.e. Smads1/5/8/9), 2) coSmad (i.e. Smad4), is common to multiple TGF-β signaling pathways, and 3) i-Smads (i.e. Smad6/7) will inhibit the signal transduction of the TGF-β family (Yang et al., 2020). While the canonical TGF-β signaling pathway utilizes Smad2/3 to regulate gene transcription, the non-canonical TGF-β signaling pathways involves Smad 1/5 and can lead to the downstream activation of MAPK/ERK pathway proteins and transcription factors, which are involved in fibrosis and tumorigenesis (Clayton et al., 2020; Finnson et al., 2020; Guo et al., 2020). The non-canonical pathway also leads to the activation of the PI3K/Akt pathway, which is one of the most frequently over-activated downstream intracellular pathways that is involved in several human cancers (Rascio et al., 2021). Other molecules activated by the non-canonical TGF-β signaling pathways include NF-κB, Rho/Rac1, Cdc42, focal adhesion kinase (FAK), Src, and Abl (Neuzillet et al., 2013). The TβRII–TβRI receptor complex will also activate other non-Smad proteins signals, such as ubiquitin ligases, or small GTPases, resulting in a diverse molecular cascade (Bellomo et al., 2016). The TGFβ pathway is a potential avenue for cancer therapeutics due to its vital role in homoeostasis, induction of inflammatory cytokines in the ECM and regulation of T and B lymphocytes (Bellomo et al., 2016). Non-cellular components are mainly defined by the ECM, which make up the bulk of the stem cell niche (Pattabiraman and Weinberg, 2014). In this way, the TGF-β pathway monitors CSCs, and aberrant TGF-β signaling results in improper function of the immune response.
3.6.2 TGFβ upstream and downstream mechanisms
It is important to highlight TGFβ’s dual function—at times inhibiting and at other times promoting CSCs progression. Interestingly, in the early stages of cancer, TGF-β suppresses tumors, but in the later stages, TGF-β promotes tumor growth and survival (Futakuchi et al., 2019). On one hand, in the bone microenvironment, TGF-β leads to liver, gastric, melanoma, prostate, renal, glioblastoma, leukemia and bladder cancer proliferation via the downstream activation of the MAPK/ERK and PI3K/Akt, and induces breast cancer metastasis via activation of the Wnt pathway (Bellomo et al., 2016; Futakuchi et al., 2019). TGFβ features an autocrine loop via its downstream targets Sox4, Oct4, and Sox2, and disruption of the TGFβ pathway would hinder the progression of glioma CSCs (Ikushima et al., 2009). TGFβ is also implicated in cancer progression by inducing EMT and attenuating the anti-tumorigenic effects of dendritic cells, natural killer (NK) cells, CD8+ T cells, and CD4+ T cells, which can be further subdivided into pro-inflammatory TH1 and anti-inflammatory TH2 cells (Akhurst and Hata, 2012; Kim et al., 2021a; Kim et al., 2021b). On the other hand, TGFβ also inhibits ALDH1 on CSCs, thereby limiting their self-renewal capacity and halting tumor progression (Bellomo et al., 2016). Along these lines, TGFβ may act as a double-edged sword, wielding both pro- and anti-inflammatory effects by driving TH17 via secretion of IL-6 (Akhurst and Hata, 2012).
Moreover, Nodal and Activin are TGFβ ligands can both signal through the same receptors and in many cases the effects are indistinguishable from each other in that both Activin and Nodal and share the downstream effectors Smad2/3 (Pauklin and Vallier, 2015). Under normal conditions, Nodal ligand plays a vital role in embryogenesis, namely inducing epiblast implantation and guiding left-right axis neurulation (Pauklin and Vallier, 2015). Consistent with Nodal’s role in embryogenesis, Activin/Nodal signaling maintains pluripotency in stem cells through induction of Nanog and other cell cycle factors (Pauklin and Vallier, 2015). Cripto-1 is similar to Nodal in that it is an embryonic protein involved in TGF-β signaling (Arboretto et al., 2021). Both Cripto-1 and Nodal have minimal expression in terminally differentiated cells, but are often re-expressed in CSCs and in a dose-dependent manner can trigger more aggressive phenotypes and worsened prognosis for several cancer-subtypes (Arboretto et al., 2021).
Due to the complexity of the TGFβ’s highly context-dependent downstream mechanisms, developing therapeutics should first identify the stage of cancer progression optimal for a personalized, patient-specific treatment plan, which balances TGFβ’s pro-inflammatory and anti-inflammatory effects.
3.6.3 TGFβ associated genes, markers, and molecules
The following genes and markers are known to upregulate the stemness properties of CSCs via TGF-β/Smad signaling: 1) TGF-β1 in low concentration over short time periods upregulate breast CSCs (Hariyanto et al., 2021), 2) cancer upregulated gene (CUG) 2 promoters associated with YAP1 increase EMT in lung CSCs (Kaowinn et al., 2019), 3) cyclin D1 activation of Smad2/3/4 increases liver CSCs self-renewal and stemness (Xia et al., 2017), 4) CD51 increased migratory and invasive potential in colorectal CSCs (Wang et al., 2017), 5) CD133 acts via a Smad-dependent transcriptional mechanism in HCC, melanoma (Korn et al., 2021), and ovarian CSCs (Ikram et al., 2021), and 6) CD44/CD44v integrates with TME signals to upregulate HCC CSCs (Yan et al., 2015; Bellomo et al., 2016; Yang et al., 2020). Targeting these known upstream molecules, will result in inhibition of the TGF-β/Smad signaling pathway and could serve as potential therapeutics. Within the bone microenvironment, TGF-β is also known to induce the parathyroid thyroid hormone-related peptide (PTHrP) downstream, which stimulates osteoclast activity to destroy bone and promote tumor growth and metastasis (Futakuchi et al., 2019). Currently, the following molecules are known to inhibit TGF-β/Smad signaling: 1) miR-106 targeting Smad7 inhibits gastric CSCs and is implicated as a potential biomarker in CRC, and 2) Dkk-3 inhibits matrix metallopeptidases 9/13, which are downstream TGF-β-induced enzymes, implicated in preventing prostate CSC metastasis (Peng et al., 2020; Yang et al., 2020). Further investigation will uncover the distinct crosstalk between TGF-β and the TME, leading to breakthrough immunotherapeutics.
3.6.4 TGFβ signaling and cancer: preclinical and clinical studies
TGF-β is a valuable target in oncology that should be regulated accordingly. Current drugs on the market that inhibit TGFβ signaling include angiotensin type II receptor inhibitors (i.e. losartan and candesartan) (Akhurst and Hata, 2012). Drugs targeting the TGF-β pathway under clinical trials include anti-ligand antisense oligonucleotides (ASOs), competitive ligand/receptors, antibodies, kinase inhibitors, and small molecule inhibitors (SMIs) (Akhurst and Hata, 2012).
TGFβ antibody therapy has proven to be very effective; since TGFβ can induce an inflammatory TME thereby lowering the efficacy of cancer and radiotherapy (RT) treatments, inhibiting the TGFβ pathway is likely to directly enhance the efficacy of RT (Chen et al., 2021a). Indeed, TGFβ receptor 2 (TGFβR2)-neutralizing antibody MT1 and the small molecule TGFβR1 inhibitor LY3200882 in combination with RT results in increased anti-tumor efficacy against murine orthotopic models in HNSCC (Hamon et al., 2022). The TME is composed of an array of different immune cells and proteins that support the development of cancer cells, including CSCs (Arneth, 2019). The mechanism through which TGFβ interacts with the TME is closely connected to CSCs, in part providing the inflammatory microenvironment, which plays a critical role in tumorigenesis, tumor progression, and metastasis (Zhang et al., 2018b).
In preclinical studies, TGF-β can be directly inhibited. For example, through receptor kinase inhibitors, TGF-β signaling plays a vital role in the bone microenvironment, which harbors significantly more SOX2, CD44, and CD166 positive CSCs than those in the subcutaneous (subQ) microenvironment (Futakuchi et al., 2019). Treatment with TGF-β R1 kinase inhibitor (R1-Ki) in mouse mammary tumor cells reduced tumor volume in the bone microenvironment via TGF-β activation of ERK 1/2 and Akt pathways, but BMP signaling did not contribute to tumor growth (Futakuchi et al., 2019). Similarly, another R1-Ki, SD-208, suppressed the development of melanoma bone metastasis by blocking TGF-β induction of Smad3 phosphorylation (Mohammed et al., 2016). Other studies, which not only block the TGF-β Nodal/Activin Alk4/7 receptor but also simultaneously inhibit the Hh pathway, abolish CSC’s self-renewal capacity and render CSCs more susceptible to gemcitabine in vivo (Lonardo et al., 2011). Monoclonal antibodies that interfere with TGF-β proteins Cripto-1 or Nodal in vitro have also shown promising effects at abrogating CSCs (Arboretto et al., 2021). Moreover, SB431542, a SMI, partially reversed Nodal-induced chemoresistance in melanoma CSCs, in vitro (Li et al., 2018b). Therefore, the majority of TGF-β cancer therapeutics work to downregulate rather than upregulate TGF-β.
Rather than inhibiting TGF-β, treatment with TGF-β resulted in a significant decrease in proliferative CSC cytokines (i.e., cytokeratin-14, frizzled-7) and saw an increase in markers for slow proliferation (i.e., mucin-1 and cytokeratin-18) (Bellomo et al., 2016). Similarly, in vivo TGFβ’s activation of Smad2/3 inhibited ABCG2, a chemotherapy efflux transporter, leading to reduction of gastric CSCs and overall reduction in tumor size (Ehata et al., 2011). Therefore, due to the dual role of TGF-β in the immune response, at times benefiting and at times hampering the growth of CSCs, we must turn to personalized medicine.
In the clinic, ample consideration of the patient’s genetics and specific biomarkers may guide the development of effective TGF-β treatments. While many TGF-β treatments aim to discover patient-specific anti-TGF-β therapy, there is growing interest in assessing patients based on their unique TGF-β tumor response and relevant oncogenic pathways in cancer cells (Chen et al., 2021c). Such strategy requires probing germline genetic variation between individuals, which should further guide the identification of patients likely to respond to anti-TGF-β therapy (Chen et al., 2021c). Besides looking into patient genetics, TGF-β biomarkers include circulating levels of TGFβ, levels of P-SMAD2 levels in the peripheral blood mononuclear cells (PMNCs), and USP15 in glioblastoma breast and ovarian cancer (Akhurst and Hata, 2012; Eichhorn et al., 2012). Biomarkers serve as a non-invasive way of providing targeted therapeutic treatment. Moving forward, the goal of TGFβ drug development should be to enhance the tumor-suppressing effects of TGFβ while inhibiting TGFβ’s role in CSC progression.
The development of cancer therapeutics that target the TGF-β pathway have demonstrated satisfactory safety and efficacy in cancer patients. However, TGF-β’s dual role in cancer progression should be further explored to develop superior patient-specific anti-TGF-β treatments. Moreover, since TGF-β is ubiquitously expressed in multiple cell types to regulate cell homeostasis and organism development, there is an inherent physiological barrier to treatment. Current anti-TGF-β treatments including ASOs, competitive ligand/receptors, antibodies, kinase inhibitors, and SMIs are limited to pre-clinical and phase I-II clinical trials. Due to the limited dual role of TGF-β functioning as both a tumor promoter and tumor suppressor, the development of safe and effective TGF-β antagonists remains a challenge. For example, in a phase II study the SMI, galunisertib, in combination with the chemotherapeutic lomustine, failed to efficiently treat glioma compared to the placebo [NCT01582269], and galunisertib must be closely monitored to avoid cardiac toxicity (Kovacs et al., 2015). Furthermore, selective TGF-β biomarkers are only in the initial stages of clinical development. While TGF-β treatment alone is limited, combination therapy has proven to be more effective at targeting not only non-CSCs but also ablating CSCs. For example, Vactosertib, a TGF-β kinase inhibitor, in combination with chemotherapy has proven to be well tolerated in phase I-II clinical trials to treat solid tumors [NCT02160106], metastatic colorectal or gastric cancers [NCT03724851, NCT03698825], non-small cell lung cancer [NCT03732274], and urothelial cancer [NCT04064190]. In conclusion, drugs that target the TGF-β pathway are highly valuable to the cancer research field, as they boast improved patient response when compared to classical cancer treatments. While there are still many hurdles that TGF-β therapies must overcome before reaching the market, expanding knowledge of targeted CSC treatments are enhanced by continued TGF-β research (Figure 7).
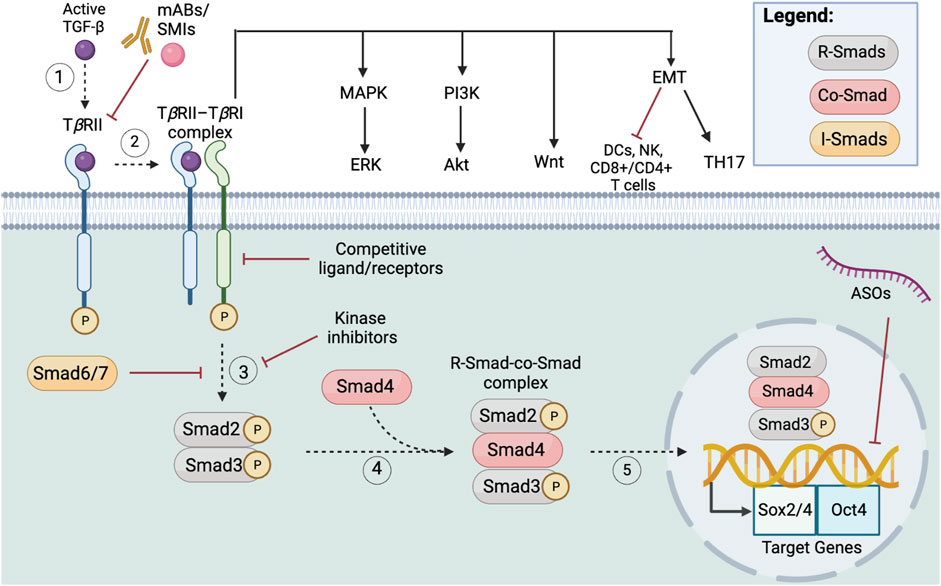
FIGURE 7. TGF-β-Smad signaling pathway in CSCs. (1) Upon the active TGF-β ligand binding to a type II receptor, (2) there is phosphorylation of the type I receptor, and (3) the TβRII–TβRI receptor complex will ultimately phosphorylate R-Smads. (4) The R-Smads will form a complex with co-Smad 4 and (5) translocate to the nucleus to transcribe the known targeted genes (Sox2/4, Oct4). Featured inhibitors of the TGF-β pathway are SMI mAbs, ligand-receptor inhibitors, kinase inhibitors, upregulation of the endogenous i-Smad6/7, and ASOs. Downstream of ligand-receptor engagement, TGF-β will upregulate the MAPK/ERK, PI3K/Akt, and Wnt pathways. In terms of the TME, TGF-β upregulates TH17 secretion while downregulating dendritic cells, NK cells, CD8+/CD4+ T cells. Figure constructed via Biorender.com.
3.7 Accessory CSC signaling therapeutics
CSCs have also been targeted by inhibition of IL-8 treatment, which prevents the recruitment of MSCs (Müller et al., 2020). IL-8 production is closely associated with the expansion of CSCs in the TME (Kim and Kahn, 2014) and its activation has been implicated in the proliferation of CSCs in the highly aggressive triple-negative breast cancer (Hirata et al., 2022), suggesting the use of IL-8 inhibitors to sequester CSCs. CSCs can also be regulated through blockade of GM-CSF, which suppresses the recruitment of TAMs (Müller et al., 2020). There are two main lineages of macrophages that are known to interact with CSCs. M1, which are classically activated, pro-inflammatory macrophages that induce and maintain CSCs through the production of IL-6. Conversely, M2 are alternatively activated, anti-inflammatory macrophages (Tsuchiya and Shiota, 2021). In leukemic mice, TAMs are shown to significantly contribute to the TME with bone marrow-derived macrophages becoming polarized into leukemic cells following injection of leukemic cells into the animals, indicating that targeting TAMs may retard the onset and progression of tumors (Li et al., 2020). Of note, the use of GM-CSF and TAM (tumor associated macrophages) inhibitors, like CSF-1R, have shown promising results in blocking the crosstalk between macrophages and tumor cells (Liu et al., 2016; Li et al., 2020).
Conversely, strategies designed to indirectly target CSCs by modulating non-CSCs have also demonstrated some success in reducing tumor growth. For example, BMP and Gremlin decrease TGFβ and target non-CSC state polarization (Pattabiraman and Weinberg, 2014). The BMP-antagonist, Gremlin 1 or GREM1, is closely linked with metastasis, specifically stemness of breast cancer cells (Ren et al., 2019), and accompanies the poor prognosis of patients with estrogen receptor-negative breast cancer (Neckmann et al., 2019), suggesting that Gremlin stands as a potent therapy for abrogating cancer cell (i.e., CSC) proliferation. Another approach for indirectly arresting CSCs is via specific CTLA-4 inhibitor, ipilimumab and anti-PD-1, nivolumab, both of which increase T cell cytotoxicity and shows success in treating melanoma and leukemia (Pattabiraman and Weinberg, 2014; Greiner et al., 2020). Targeting leukemic progenitor and stem cells by specific cytotoxic T lymphocytes can dampen leukemia-associated antigens that mediate immune responses against colony-forming cells including leukemic progenitor cells, which are thought to correspond to the source population of relapse of the disease (Greiner et al., 2020). Altogether, promising results in the laboratory and early limited clinical trials that target the proteins and signaling pathways implicated in CSC proliferation appear to sequester the aberrant self-renewal of CSCs with corresponding reduction in tumor size and prevention of relapse.
3.8 Crosstalk of CSC signaling pathways and combination therapy
The signaling pathways Wnt, TGFβ, Notch, JAK-STAT, Hh, and VEGF often interact with each other to maintain and regulate CSCs. Hh and VEGF targeted therapeutics have made great strides in the field of CSC-directed therapeutics as they boast multiple FDA approved drugs, namely Odomzo®, Avastin®, Cyramza®, and Zaltrap® [NCT04066504] (Cardones and Banez, 2006). Conversely, Wnt targeted therapeutics are typically halted at phase I/II due to the complexity of the Wnt pathway (Katoh, 2017). Additionally, it is important to synthesize the connection among the major CSC signaling pathways, as the best mode of treatment may be to target multiple signaling pathways.
The canonical Wnt pathway is known to display crosstalk with the Hh pathway and vice versa (Katoh, 2017; Yang et al., 2020). For example, β-catenin upregulates Hh signaling via stabilization of Gli mRNAs, but β-catenin can also lead to the proteasomal degradation of Gli (Noubissi et al., 2009; Zinke et al., 2015). In turn, Hh signaling can also suppress Wnt signaling via induction of Gli 1/2 mediated Hh target gene, soluble frizzled-related protein 1 (sFrP1) (He et al., 2006). Conversely, Gli1 also activates Wnt2b, Wnt4, Wnt7b genes, which upregulate Wnt signaling (Li et al., 2007). This leads to the question as to why Hh CSC targeted therapeutics are more effective than Wnt targeted therapeutics, given the intimate crosstalk between Wnt and Hh signaling. Perhaps, the interplay between Wnt and Hh signaling should also be viewed in accordance with Notch signaling, a key mediator between Wnt and Hh signaling (Koury et al., 2017). Specifically, in colorectal cancer cells, Notch’s Jagged 1 ligand elevates β-catenin activity to drive Wnt signaling (Pannequin et al., 2009). Wnt signaling also directly upregulates Notch signaling components, namely DLL1, Hes1, Notch 2, and Jagged 1 (Kumar et al., 2021). Moreover, Notch’s target gene, Hes 1, regulates Hh signaling in glioblastomas, and Hh signaling can also directly modulate Notch signaling through inducing the same Hes1 gene, as well (Wall et al., 2009; Schreck et al., 2010). Through inhibiting Notch and Hh signaling via combination therapy, prostate cancer CSCs were more sensitive to chemotherapy in vitro (Domingo-Domenech et al., 2012). In addition, phase II clinical trials are currently being carried out with Hh pathway inhibitor (Vismodegib) and Notch pathway inhibitor (RO4929097) that show promising results for the treatment of advanced stage sarcoma (Gounder et al., 2022) [NCT00833417]. Future studies should aim to unravel the unique relationship between Wnt, Hh, and Notch in order to develop novel CSC combination therapeutics.
Wnt signaling pathway is also displays crosstalk with TGF-β through a variety of mechanisms. TGF-β and Wnt signaling pathway transcription factors, namely Smad and Lef, respectively, synergistically regulate a set of shared target genes (Guo and Wang, 2009). TGF-β and Wnt also display protein-protein interaction in the cytoplasm, via the binding of Smad7-Axin, and can reciprocally regulate ligand production (Guo and Wang, 2009). The crosstalk between TGF-β and Wnt signaling serve as a potential cancer therapeutic. For example, SMI A83-01, which inhibits TGF-β-induced upregulation of Wnt3, enhances trastuzumab treatment for targeting breast cancer cells in vitro (Wu et al., 2017). Current clinical trials that combine Wnt and TGF-β inhibitors are limited.
JAK-STAT pathway displays multiple diverse mechanisms, which overlap with TGF-β and Notch pathways. For instance, in pancreatic ductal carcinoma TGF-β inhibited and IL-1 induced JAK/STAT signaling cascade in vitro (Biffi et al., 2019). In comparison, in hematopoietic SCs, TGF-β increased IL-6 mediated STAT3 activation (Tang et al., 2017). Moreover, Notch signaling activates IL-6 induced JAK/STAT signaling in breast cancer tumors (Jin et al., 2013). Current clinical trials featuring combination therapy aim to decipher variations in targeting JAK family members and STATs, rather than targeting multiple signaling pathways. For instance, one clinical trial in phase I for the treatment of lung cancer features Afatinib plus Ruxolitinib combination therapy, which targets IL-6 receptor and JAK1/2 [NCT02145637].
Lastly, VEGF signaling is also vital for Wnt and Notch pathways. VEGF induces Notch signaling, while Notch signaling also modulates the VEGF pathway. Specifically, in vivo and in vitro studies confirm that VEGF will upregulate Notch pathway modulators, namely DLL1/4, Notch 1, and ADAM-10 (Li and Harris, 2009). Notch signaling also regulates VEGF ligand expression (VEGF and PLGF) and VEGF receptor expression (NRP1/2 and VEGFR1/2/3) (Li and Harris, 2009). Moreover, another study in vitro proved that VEGFR-1 kinase activity is required for Wnt/β-catenin CRC cells (Zeitlin et al., 2009). Current VEGF combination therapeutics are in phase IV clinical trials and display promising results for ablating CRC using anti-VEGF drugs plus traditional chemotherapeutics [NCT01972490]. Other VEGF combination therapeutics are in phase II trials and feature anti-VEGF drugs (Bevacizumab) in combination with a DNA synthesis and cell cycle inhibitors (Gemcitabine, Paclitaxel) [NCT00403130]. Future VEGF therapeutics should target not only the VEGF pathway but also inhibit Notch signaling as a potential means for CSC directed therapeutics.
Wnt, TGFβ, Notch, JAK-STAT, Hh, and VEGF signaling are vital for CSC regulation and restoring the pathways to equilibrium is a potential novel avenue for cancer therapeutics. Moreover, a better understanding of these key pathways’ genes, markers, and molecules will allow us to better assess unique therapeutic targets (Table 1). While most clinical trials are focused on targeting one singular pathway in combination with traditional chemotherapy, a more effective approach would be to curate combination products that blocks multiple CSC signaling mechanisms (Table 2). Moreover, while the Wnt pathway is the most complex and centralized among each signaling pathway, targeting signaling pathways that are more downstream of Wnt will result in more specific therapeutics.
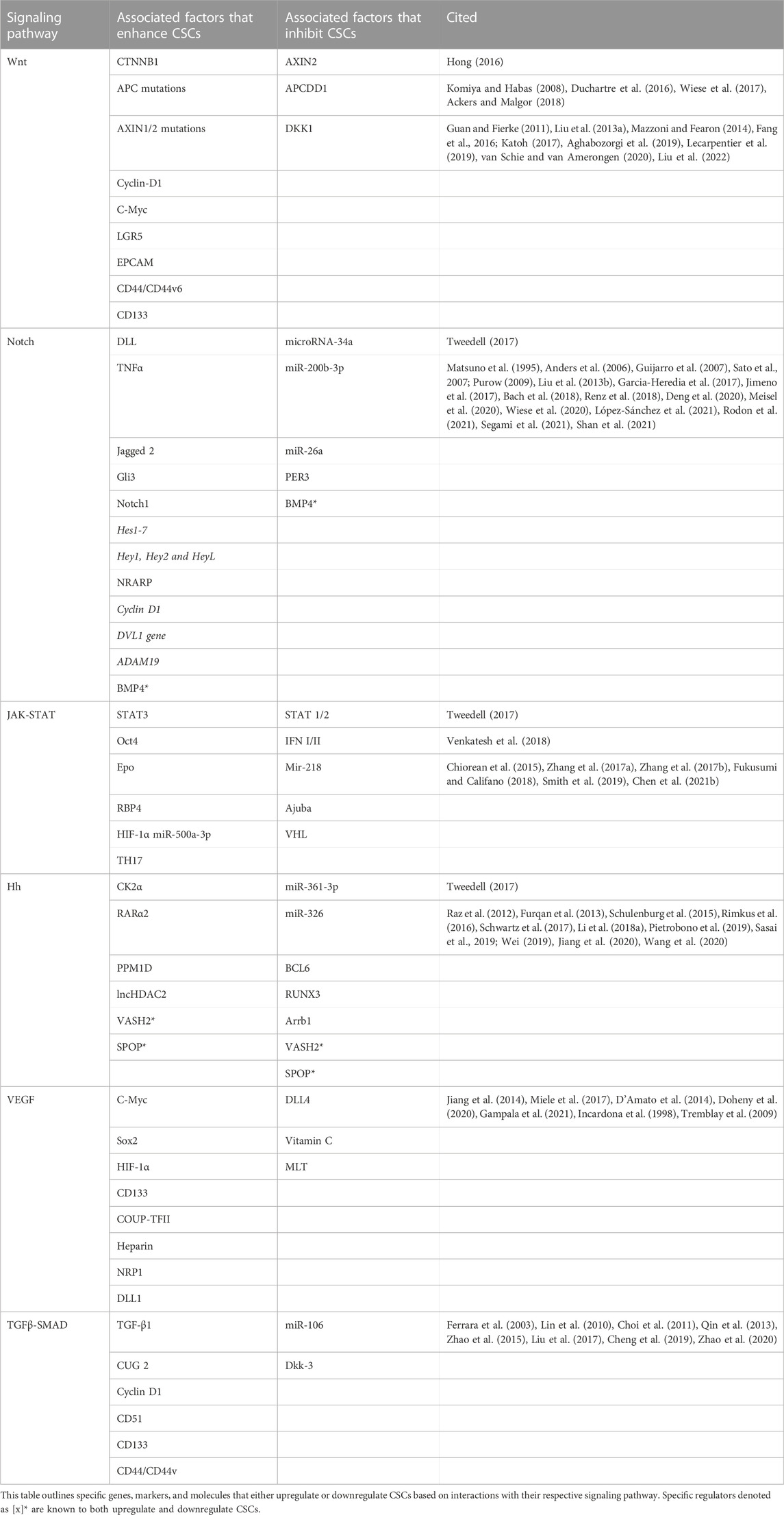
TABLE 1. CSC signaling pathways’ associated genes, markers, and molecules.
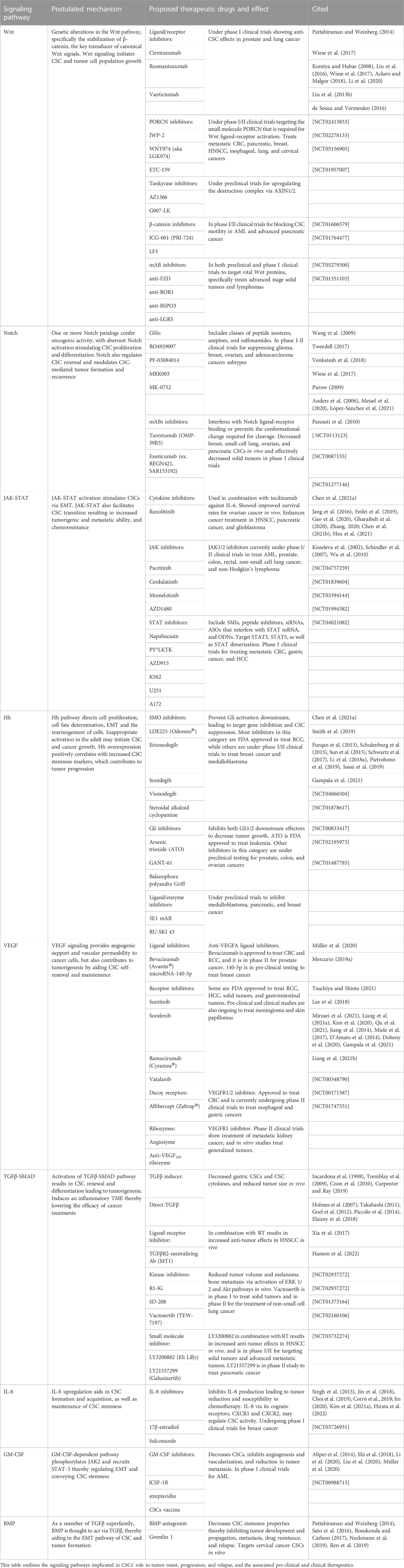
TABLE 2. CSC signaling pathways and associated therapeutics.
4 Conclusion and perspective
CSCs are a small subclass of cancer cells with self-renewal and metastatic capacity, resulting in resistance to traditional chemotherapeutics and multidrug therapies (Pattabiraman and Weinberg, 2014). Many clinical trials on CSCs have shown promising evidence for cancer therapy. Signaling pathways, including Wnt, TGFβ-SMAD, Notch, JAK-STAT, Hh, and VEGF, are essential regulators of CSCs. Notably, signaling pathway inhibitors include ligand-receptor inhibitors (Cardones and Banez, 2006), decoy receptors (Ciombor and Berlin, 2014), mAbs (Katoh, 2017), siRNA (Hu et al., 2021), and specific molecule (Akhurst and Hata, 2012) and enzyme inhibitors (Gu et al., 2004; Carpenter and Ray, 2019). Inhabiting these signaling pathways may prove beneficial against cancers. Cancer therapies must navigate the intricacies of CSC signaling pathways to eliminate CSC effectively. CSCs may act differently in different cancer sub-types (Yang et al., 2020), resulting in a less than satisfactory classification of CSC therapeutics. Under normal conditions certain signaling pathway may play an intrinsic role in cell homeostasis (Yang et al., 2020), but the same signaling proteins vital to cell development can result in metastasis and growth of CSCs (Bellomo et al., 2016; Futakuchi et al., 2019). CSCs also share signaling pathways with traditional stem cells (Pattabiraman and Weinberg, 2014), limiting therapeutic potential in cancer treatment. While there are multiple novel avenues for cancer therapeutics, the sheer volume of processes and pathways that contribute to CSC stemness can be daunting, and finding one cure-all treatment may be restrictive. Instead, patient-specific therapies that focus on the patient’s cancer profile and genotypic signal processes may prove promising (Katoh, 2017). Future CSC-targeted therapies should aim to create specific inhibitors rather than generalized signaling pathway inhibitors to decrease the number of side effects associated with traditional, indiscriminate cancer therapies. Natural products that target CSCs should also be studied in the future as they may be effective at targeting CSCs without impairing non-cancerous stem cell (Incardona et al., 1998; Cheng et al., 2019; Zhao et al., 2020). Future studies should also delve into the accumulating evidence implicating a crosstalk between CSCs and immune cells within the TME providing key concepts on signaling mechanisms mediating CSC-mediated tumor formation (Müller et al., 2020) as well as possible immunotherapeutics against cancer (Chen et al., 2021a). An integrated understanding of CSC signaling pathways and immune cell crosstalk factors is expected to provide significant improvements to the current knowledge of cancer pathology and treatment.
Author contributions
HW: Conceptualization, data curation, analysis, methodology, projection administration, resources, supervision, validation, writing, review, and editing. MB: Conceptualization, methodology, data curation, analysis, resources, validation, writing, review, and editing. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abel, E. V., Kim, E. J., Wu, J., Hynes, M., Bednar, F., Proctor, E., et al. (2014). The Notch pathway is important in maintaining the cancer stem cell population in pancreatic cancer. PLoS One 9 (3), e91983. doi:10.1371/journal.pone.0091983
Acikgoz, E., Soner, B. C., Ozdil, B., and Guven, M. (2021). CD133+/CD44+ prostate cancer stem cells exhibit embryo-like behavior patterns. Acta histochem. 123 (5), 151743. doi:10.1016/j.acthis.2021.151743
Ackers, I., and Malgor, R. (2018). Interrelationship of canonical and non-canonical Wnt signalling pathways in chronic metabolic diseases. Diab Vasc. Dis. Res. 15 (1), 3–13. doi:10.1177/1479164117738442
Aghabozorgi, A. S., Bahreyni, A., Soleimani, A., Bahrami, A., Khazaei, M., Ferns, G. A., et al. (2019). Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie 157, 64–71. doi:10.1016/j.biochi.2018.11.003
Akhurst, R. J., and Hata, A. (2012). Targeting the TGFβ signalling pathway in disease. Nat. Rev. Drug Discov. 11 (10), 790–811. doi:10.1038/nrd3810
Aliper, A. M., Frieden-Korovkina, V. P., Buzdin, A., Roumiantsev, S. A., and Zhavoronkov, A. (2014). A role for G-CSF and GM-CSF in nonmyeloid cancers. Cancer Med. 3 (4), 737–746. doi:10.1002/cam4.239
Ally, M. S., Aasi, S., Wysong, A., Teng, C., Anderson, E., Bailey-Healy, I., et al. (2014). An investigator-initiated open-label clinical trial of vismodegib as a neoadjuvant to surgery for high-risk basal cell carcinoma. J. Am. Acad. Dermatol 71 (5), 904–911. doi:10.1016/j.jaad.2014.05.020
Anders, L., Mertins, P., Lammich, S., Murgia, M., Hartmann, D., Saftig, P., et al. (2006). Furin-ADAM 10-and gamma-secretase-mediated cleavage of a receptor tyrosine phosphatase and regulation of beta-catenin's transcriptional activity. Mol. Cell Biol. 26 (10), 3917–3934. doi:10.1128/MCB.26.10.3917-3934.2006
Arboretto, P., Cillo, M., and Leonardi, A. (2021). New insights into cancer targeted therapy: Nodal and cripto-1 as attractive candidates. Int. J. Mol. Sci. 22 (15), 7838. doi:10.3390/ijms22157838
Bach, D. H., Park, H. J., and Lee, S. K. (2018). The dual role of bone morphogenetic proteins in cancer. Mol. Ther. Oncolytics 8, 1–13. doi:10.1016/j.omto.2017.10.002
Bar, E. E., Chaudhry, A., Farah, M. H., and Eberhart, C. G. (2007). Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am. J. Pathol. 170 (1), 347–355. doi:10.2353/ajpath.2007.060066
Barker, N., and Clevers, H. (2010). Leucine-rich repeat-containing G-protein-coupled receptors as markers of adult stem cells. Gastroenterology 138 (5), 1681–1696. doi:10.1053/j.gastro.2010.03.002
Beck, B., Driessens, G., Goossens, S., Youssef, K. K., Kuchnio, A., Caauwe, A., et al. (2011). A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature 478 (7369), 399–403. doi:10.1038/nature10525
Bellomo, C., Caja, L., and Moustakas, A. (2016). Transforming growth factor β as regulator of cancer stemness and metastasis. Br. J. Cancer 115 (7), 761–769. doi:10.1038/bjc.2016.255
Biffi, G., Spielman, B., Hao, Y., Elyada, E., and Park, Y. (2019). IL1-Induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov. 9 (2), 282–301. doi:10.1158/2159-8290.CD-18-0710
Bitsch, R., Kurzay, A., Özbay Kurt, F., De La Torre, C., Lasser, S., Lepper, A., et al. (2022). STAT3 inhibitor Napabucasin abrogates MDSC immunosuppressive capacity and prolongs survival of melanoma-bearing mice. J. Immunother. Cancer 10 (3), e004384. doi:10.1136/jitc-2021-004384
Bosukonda, A., and Carlson, W. (2017). Harnessing the BMP signaling pathway to control the formation of cancer stem cells by effects on epithelial-to-mesenchymal transition. Biochem. Soc. Trans. 45, 223–228. doi:10.1042/BST20160177
Cao, W., Chen, H. D., Yu, Y. W., and Chen, W. Q. (2021). Changing profiles of cancer burden worldwide and in China: A secondary analysis of the global cancer statistics 2020. Chin. Med. J. Engl. 134 (7), 783–791. doi:10.1097/CM9.0000000000001474
Cardones, A. R., and Banez, L. L. (2006). VEGF inhibitors in cancer therapy. Curr. Pharm. Des. 12 (3), 387–394. doi:10.2174/138161206775201910
Carpenter, R. L., and Ray, H. (2019). Safety and tolerability of sonic hedgehog pathway inhibitors in cancer. Drug Saf. 42 (2), 263–279. doi:10.1007/s40264-018-0777-5
Chang, Q., Foltz, W. D., Chaudary, N., Hill, R. P., and Hedley, D. W. (2013). Tumor-stroma interaction in orthotopic primary pancreatic cancer xenografts during hedgehog pathway inhibition. Int. J. Cancer 133 (1), 225–234. doi:10.1002/ijc.28006
Chen, B., Deng, Y. N., Wang, X., Xia, Z., He, Y., Zhang, P., et al. (2021). miR-26a enhances colorectal cancer cell growth by targeting RREB1 deacetylation to activate AKT-mediated glycolysis. Cancer Lett. 521, 1–13. doi:10.1016/j.canlet.2021.08.017
Chen, C., Hou, J., Yu, S., Li, W., Wang, X., Sun, H., et al. (2021). Role of cancer-associated fibroblasts in the resistance to antitumor therapy, and their potential therapeutic mechanisms in non-small cell lung cancer. Oncol. Lett. 21 (5), 413. doi:10.3892/ol.2021.12674
Chen, P., Hsu, W. H., Han, J., Xia, Y., and DePinho, R. A. (2021). Cancer stemness meets immunity: From mechanism to therapy. Cell Rep. 34 (1), 108597. doi:10.1016/j.celrep.2020.108597
Cheng, J., Yang, H. L., Gu, C. J., Liu, Y. K., Shao, J., Zhu, R., et al. (2019). Melatonin restricts the viability and angiogenesis of vascular endothelial cells by suppressing HIF-1α/ROS/VEGF. Int. J. Mol. Med. 43 (2), 945–955. doi:10.3892/ijmm.2018.4021
Chiorean, E. G., LoRusso, P., Strother, R. M., Diamond, J. R., Younger, A., Messersmith, W. A., et al. (2015). A phase I first-in-human study of enoticumab (REGN421), a fully human delta-like ligand 4 (Dll4) monoclonal antibody in patients with advanced solid tumors. Clin. Cancer Res. 21 (12), 2695–2703. doi:10.1158/1078-0432.CCR-14-2797
Choi, H. S., Kim, J. H., Kim, S. L., and Lee, D. S. (2019). Disruption of the NF-κB/IL-8 signaling Axis by sulconazole inhibits human breast cancer stem cell formation. Cells 8 (9), 1007. doi:10.3390/cells8091007
Choi, S. B., Park, J. B., and Song, T. J. (2011). Molecular mechanism of HIF-1-independent VEGF expression in a hepatocellular carcinoma cell line. Int. J. Mol. Med. 28 (3), 449–454. doi:10.3892/ijmm.2011.719
Ciombor, K. K., and Berlin, J. (2014). Aflibercept--a decoy VEGF receptor. Curr. Oncol. Rep. 16 (2), 368. doi:10.1007/s11912-013-0368-7
Clayton, S. W., Ban, G. I., Liu, C., and Serra, R. (2020). Canonical and noncanonical TGF-β signaling regulate fibrous tissue differentiation in the axial skeleton. Sci. Rep. 10 (1), 21364. doi:10.1038/s41598-020-78206-4
Coon, V., Laukert, T., Pedone, C. A., Laterra, J., Kim, K. J., and Fults, D. W. (2010). Molecular therapy targeting Sonic hedgehog and hepatocyte growth factor signaling in a mouse model of medulloblastoma. Mol. Cancer Ther. 9 (9), 2627–2636. doi:10.1158/1535-7163.MCT-10-0486
Correia, C., Weiskittel, T. M., Ung, C. Y., Villasboas Bisneto, J. C., Billadeau, D. D., Kaufmann, S. H., et al. (2022). Uncovering pharmacological opportunities for cancer stem cells-A systems biology view. Front. Cell Dev. Biol. 10, 752326. doi:10.3389/fcell.2022.752326
Corrò, C., Healy, M. E., Engler, S., Bodenmiller, B., Li, Z., Schraml, P., et al. (2019). IL-8 and CXCR1 expression is associated with cancer stem cell-like properties of clear cell renal cancer. J. Pathol. 248 (3), 377–389. doi:10.1002/path.5267
Cowan, A. J., Green, D. J., Kwok, M., Lee, S., Coffey, D. G., Holmberg, L. A., et al. (2022). Diagnosis and management of multiple myeloma: A review. Jama 327 (5), 464–477. doi:10.1001/jama.2022.0003
Croker, A. K., Goodale, D., Chu, J., Postenka, C., Hedley, B. D., Hess, D. A., et al. (2009). High aldehyde dehydrogenase and expression of cancer stem cell markers selects for breast cancer cells with enhanced malignant and metastatic ability. J. Cell Mol. Med. 13 (8), 2236–2252. doi:10.1111/j.1582-4934.2008.00455.x
Dai, X., Karol, M. D., Hitron, M., Hard, M. L., Goulet, M. T., McLaughlin, C. F., et al. (2021). Napabucasin drug-drug interaction potential, safety, tolerability, and pharmacokinetics following oral dosing in healthy adult volunteers. Clin. Pharmacol. Drug Dev. 10 (8), 824–839. doi:10.1002/cpdd.961
D’Amato, C., Rosa, R., Marciano, R., D'Amato, V., Formisano, L., Nappi, L., et al. (2014). Inhibition of Hedgehog signalling by NVP-LDE225 (Erismodegib) interferes with growth and invasion of human renal cell carcinoma cells. Br. J. Cancer 111 (6), 1168–1179. doi:10.1038/bjc.2014.421
David, J. M., Dominguez, C., Hamilton, D. H., and Palena, C. (2016). The IL-8/IL-8R Axis A double agent in tumor immune resistance. Vaccines (Basel) 4 (3), 22. doi:10.3390/vaccines4030022
de Antonellis, P., Liguori, L., Falanga, A., Carotenuto, M., Ferrucci, V., Andolfo, I., et al. (2013). MicroRNA 199b-5p delivery through stable nucleic acid lipid particles (SNALPs) in tumorigenic cell lines. Naunyn Schmiedeb. Arch. Pharmacol. 386 (4), 287–302. doi:10.1007/s00210-013-0837-4
de Sousa, E. M. F., and Vermeulen, L. (2016). Wnt signaling in cancer stem cell biology. Cancers (Basel) 8 (7), 60. doi:10.3390/cancers8070060
Deng, J., Hua, L., Han, T., Tian, M., Wang, D., Tang, H., et al. (2020). The CREB-binding protein inhibitor ICG-001: A promising therapeutic strategy in sporadic meningioma with NF2 mutations. Neurooncol Adv. 2 (1), vdz055. doi:10.1093/noajnl/vdz055
Derynck, R., Turley, S. J., and Akhurst, R. J. (2021). TGFβ biology in cancer progression and immunotherapy. Nat. Rev. Clin. Oncol. 18 (1), 9–34. doi:10.1038/s41571-020-0403-1
Dieteren, C., and Bonfrer, I. (2021). Socioeconomic inequalities in lifestyle risk factors across low- and middle-income countries. BMC Public Health 21 (1), 951. doi:10.1186/s12889-021-11014-1
Ding, M., and Wang, X. (2017). Antagonism between Hedgehog and Wnt signaling pathways regulates tumorigenicity. Oncol. Lett. 14 (6), 6327–6333. doi:10.3892/ol.2017.7030
Doheny, D., Sirkisoon, S., Carpenter, R. L., Aguayo, N. R., Regua, A. T., Anguelov, M., et al. (2020). Combined inhibition of JAK2-STAT3 and SMO-GLI1/tGLI1 pathways suppresses breast cancer stem cells, tumor growth, and metastasis. Oncogene 39 (42), 6589–6605. doi:10.1038/s41388-020-01454-1
Dolatabadi, S., Jonasson, E., Lindén, M., Fereydouni, B., Bäcksten, K., Nilsson, M., et al. (2019). JAK-STAT signalling controls cancer stem cell properties including chemotherapy resistance in myxoid liposarcoma. Int. J. cancer 145 (2), 435–449. doi:10.1002/ijc.32123
Domingo-Domenech, J., Vidal, S. J., Rodriguez-Bravo, V., Castillo-Martin, M., Quinn, S. A., Rodriguez-Barrueco, R., et al. (2012). Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell 22 (3), 373–388. doi:10.1016/j.ccr.2012.07.016
Dréno, B., Kunstfeld, R., Hauschild, A., Fosko, S., Zloty, D., Labeille, B., et al. (2017). Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): A randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 18 (3), 404–412. doi:10.1016/S1470-2045(17)30072-4
Driessens, G., Beck, B., Caauwe, A., Simons, B. D., and Blanpain, C. (2012). Defining the mode of tumour growth by clonal analysis. Nature 488 (7412), 527–530. doi:10.1038/nature11344
Duchartre, Y., Kim, Y. M., and Kahn, M. (2016). The Wnt signaling pathway in cancer. Crit. Rev. Oncol. Hematol. 99, 141–149. doi:10.1016/j.critrevonc.2015.12.005
Ehata, S., Johansson, E., Katayama, R., Koike, S., Watanabe, A., Hoshino, Y., et al. (2011). Transforming growth factor-β decreases the cancer-initiating cell population within diffuse-type gastric carcinoma cells. Oncogene 30 (14), 1693–1705. doi:10.1038/onc.2010.546
Eichhorn, P. J., Rodón, L., Gonzàlez-Juncà, A., Dirac, A., Gili, M., Martínez-Sáez, E., et al. (2012). USP15 stabilizes TGF-β receptor I and promotes oncogenesis through the activation of TGF-β signaling in glioblastoma. Nat. Med. 18 (3), 429–435. doi:10.1038/nm.2619
Elaimy, A. L., Guru, S., Chang, C., Ou, J., Amante, J. J., Zhu, L. J., et al. (2018). VEGF-neuropilin-2 signaling promotes stem-like traits in breast cancer cells by TAZ-mediated repression of the Rac GAP β2-chimaerin. Sci. Signal 11 (528), eaao6897. doi:10.1126/scisignal.aao6897
Elbadawy, M., Usui, T., Yamawaki, H., and Sasaki, K. (2019). Emerging roles of C-myc in cancer stem cell-related signaling and resistance to cancer chemotherapy: A potential therapeutic target against colorectal cancer. Int. J. Mol. Sci. 20 (9), 2340. doi:10.3390/ijms20092340
Fang, L., Zhu, Q., Neuenschwander, M., Specker, E., Wulf-Goldenberg, A., Weis, W. I., et al. (2016). A small-molecule antagonist of the β-catenin/TCF4 interaction blocks the self-renewal of cancer stem cells and suppresses tumorigenesis. Cancer Res. 76 (4), 891–901. doi:10.1158/0008-5472.CAN-15-1519
Farooqi, A. A., and Siddik, Z. H. (2015). Platelet-derived growth factor (PDGF) signalling in cancer: Rapidly emerging signalling landscape. Cell Biochem. Funct. 33 (5), 257–265. doi:10.1002/cbf.3120
Feifei, W., Hui, G., Ruiqiang, Z., Qunxiang, J., and Yu'an, X. (2019). MAGP2, a component of extracellular matrix, is upregulated in colorectal cancer and negatively modulated by miR-200b-3p. Technol. Cancer Res. Treat. 18, 1533033819870777. doi:10.1177/1533033819870777
Ferrara, N., Gerber, H.-P., and LeCouter, J. (2003). The biology of VEGF and its receptors. Nat. Med. 9 (6), 669–676. doi:10.1038/nm0603-669
Ferrara, N. (2005). VEGF as a therapeutic target in cancer. Oncology 69 (3), 11–16. doi:10.1159/000088479
Finnson, K. W., Almadani, Y., and Philip, A. (2020). Non-canonical (non-SMAD2/3) TGF-β signaling in fibrosis: Mechanisms and targets. Seminars Cell & Dev. Biol. 101, 115–122. doi:10.1016/j.semcdb.2019.11.013
Fukusumi, T., and Califano, J. A. (2018). The NOTCH pathway in head and neck squamous cell carcinoma. J. Dent. Res. 97 (6), 645–653. doi:10.1177/0022034518760297
Furqan, M., Akinleye, A., Mukhi, N., Mittal, V., Chen, Y., and Liu, D. (2013). STAT inhibitors for cancer therapy. J. Hematol. Oncol. 6, 90. doi:10.1186/1756-8722-6-90
Futakuchi, M., Lami, K., Tachibana, Y., Yamamoto, Y., Furukawa, M., and Fukuoka, J. (2019). The effects of TGF-β signaling on cancer cells and cancer stem cells in the bone microenvironment. Int. J. Mol. Sci. 20 (20), 5117. doi:10.3390/ijms20205117
Gampala, S., Zhang, G., Chang, C. J., and Yang, J. Y. (2021). Activation of AMPK sensitizes medulloblastoma to Vismodegib and overcomes Vismodegib-resistance. FASEB bioAdvances 3 (6), 459–469. doi:10.1096/fba.2020-00032
Gao, S., Bian, T., Su, M., Liu, Y., and Zhang, Y. (2020). miR-26a inhibits ovarian cancer cell proliferation, migration and invasion by targeting TCF12. Oncol. Rep. 43 (1), 368–374. doi:10.3892/or.2019.7417
Garcia-Heredia, J. M., Lucena-Cacace, A., Verdugo-Sivianes, E. M., Pérez, M., and Carnero, A. (2017). The cargo protein MAP17 (PDZK1IP1) regulates the cancer stem cell pool activating the notch pathway by abducting NUMB. Clin. Cancer Res. 23 (14), 3871–3883. doi:10.1158/1078-0432.CCR-16-2358
Gerber, H. P., Malik, A. K., Solar, G. P., Sherman, D., Liang, X. H., Meng, G., et al. (2002). VEGF regulates haematopoietic stem cell survival by an internal autocrine loop mechanism. Nature 417 (6892), 954–958. doi:10.1038/nature00821
Germain, S., Commodore, P., Sinha, V., Sanz, L. A., Chédin, F., and Barlow, J. H. (2022). Genomic patterns of transcription–replication interactions in mouse primary B cells. Nucleic Acids Res. 50 (4), 2051–2073. doi:10.1093/nar/gkac035
Gharaibeh, L., Elmadany, N., Alwosaibai, K., and Alshaer, W. (2020). Notch1 in cancer therapy: Possible clinical implications and challenges. Mol. Pharmacol. 98 (5), 559–576. doi:10.1124/molpharm.120.000006
Goel, H. L., Pursell, B., Standley, C., Fogarty, K., and Mercurio, A. M. (2012). Neuropilin-2 regulates α6β1 integrin in the formation of focal adhesions and signaling. J. Cell Sci. 125 (2), 497–506. doi:10.1242/jcs.094433
Gounder, M. M., Rosenbaum, E., Wu, N., Dickson, M. A., Sheikh, T. N., D'Angelo, S. P., et al. (2022). A phase Ib/II randomized study of RO4929097, a gamma-secretase or notch inhibitor with or without vismodegib, a hedgehog inhibitor, in advanced sarcoma. Clin. Cancer Res. 28 (8), 1586–1594. doi:10.1158/1078-0432.CCR-21-3874
Greaves, M., and Maley, C. C. (2012). Clonal evolution in cancer. Nature 481 (7381), 306–313. doi:10.1038/nature10762
Greiner, J., Götz, M., Hofmann, S., Schrezenmeier, H., Wiesneth, M., Bullinger, L., et al. (2020). Specific T-cell immune responses against colony-forming cells including leukemic progenitor cells of AML patients were increased by immune checkpoint inhibition. Cancer Immunol. Immunother. 69 (4), 629–640. doi:10.1007/s00262-020-02490-2
Gu, Z. P., Wang, Y. J., Wu, Y., Li, J. G., and Chen, N. A. (2004). Synthesis of ribozyme against vascular endothelial growth factor165 and its biological activity in vitro. World J. Gastroenterol. 10 (10), 1495–1498. doi:10.3748/wjg.v10.i10.1495
Guan, X., and Fierke, C. A. (2011). Understanding protein palmitoylation: Biological significance and enzymology. Sci. China Chem. 54 (12), 1888–1897. doi:10.1007/s11426-011-4428-2
Guijarro, M. V., Leal, J. F. M., Blanco-Aparicio, C., Alonso, S., Fominaya, J., Lleonart, M., et al. (2007). MAP17 enhances the malignant behavior of tumor cells through ROS increase. Carcinogenesis 28 (10), 2096–2104. doi:10.1093/carcin/bgm124
Guo, X., and Wang, X.-F. (2009). Signaling cross-talk between TGF-beta/BMP and other pathways. Cell Res. 19 (1), 71–88. doi:10.1038/cr.2008.302
Guo, Y. J., Pan, W. W., Liu, S. B., Shen, Z. F., Xu, Y., and Hu, L. L. (2020). ERK/MAPK signalling pathway and tumorigenesis. Exp. Ther. Med. 19 (3), 1997–2007. doi:10.3892/etm.2020.8454
Hamon, P., Gerbé De Thoré, M., Classe, M., Signolle, N., Liu, W., Bawa, O., et al. (2022). TGFβ receptor inhibition unleashes interferon-β production by tumor-associated macrophages and enhances radiotherapy efficacy. J. Immunother. Cancer 10 (3), e003519. doi:10.1136/jitc-2021-003519
Hariyanto, N. I., Purwandhita, R. P., Syahrani, R. A., Louisa, M., and Wanandi, S. I. (2021). Role of TGF-β1 in human breast cancer stem cells. J. Pak Med. Assoc. 71 (2), S84–s89.
He, J., Sheng, T., Stelter, A. A., Li, C., Zhang, X., Sinha, M., et al. (2006). Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. J. Biol. Chem. 281 (47), 35598–35602. doi:10.1074/jbc.C600200200
Herrera, S. C., and Bach, E. A. (2019). JAK/STAT signaling in stem cells and regeneration: From Drosophila to vertebrates. Development 146 (2), dev167643. doi:10.1242/dev.167643
Hirata, N., Yamada, S., Yanagida, S., Ono, A., Yasuhiko, Y., Nishida, M., et al. (2022). Lysophosphatidic acid promotes the expansion of cancer stem cells via TRPC3 channels in triple-negative breast cancer. Int. J. Mol. Sci. 23 (4), 1967. doi:10.3390/ijms23041967
Ho, Q. T., and Kuo, C. J. (2007). Vascular endothelial growth factor: Biology and therapeutic applications. Int. J. Biochem. Cell Biol. 39 (7-8), 1349–1357. doi:10.1016/j.biocel.2007.04.010
Holmes, K., Roberts, O. L., Thomas, A. M., and Cross, M. J. (2007). Vascular endothelial growth factor receptor-2: Structure, function, intracellular signalling and therapeutic inhibition. Cell. Signal. 19 (10), 2003–2012. doi:10.1016/j.cellsig.2007.05.013
Hong, I. S. (2016). Stimulatory versus suppressive effects of GM-CSF on tumor progression in multiple cancer types. Exp. Mol. Med. 48 (7), e242. doi:10.1038/emm.2016.64
Hsu, J. B., Lee, T. Y., Cheng, S. J., Lee, G. A., Chen, Y. C., Le, N. Q. K., et al. (2021). Identification of differentially expressed genes in different glioblastoma regions and their association with cancer stem cell development and temozolomide response. J. Pers. Med. 11 (11), 1047. doi:10.3390/jpm11111047
Hu, X., Li, J., Fu, M., Zhao, X., and Wang, W. (2021). The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target Ther. 6 (1), 402. doi:10.1038/s41392-021-00791-1
Huang, B., Miao, L., Liu, J., Zhang, J., and Li, Y. (2022). A promising antitumor method: Targeting CSC with immune cells modified with CAR. Front. Immunol. 13, 937327. doi:10.3389/fimmu.2022.937327
Huff, C. A., Matsui, W., Smith, B. D., and Jones, R. J. (2006). The paradox of response and survival in cancer therapeutics. Blood 107 (2), 431–434. doi:10.1182/blood-2005-06-2517
Ikram, D., Masadah, R., Nelwan, B. J., Zainuddin, A. A., Ghaznawie, M., and Wahid, S. (2021). CD133 act as an essential marker in ovarian carcinogenesis. Asian Pac J. Cancer Prev. 22 (11), 3525–3531. doi:10.31557/APJCP.2021.22.11.3525
Ikushima, H., Todo, T., Ino, Y., Takahashi, M., Miyazawa, K., and Miyazono, K. (2009). Autocrine TGF-beta signaling maintains tumorigenicity of glioma-initiating cells through Sry-related HMG-box factors. Cell Stem Cell 5 (5), 504–514. doi:10.1016/j.stem.2009.08.018
Incardona, J. P., Gaffield, W., Kapur, R. P., and Roelink, H. (1998). The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 125 (18), 3553–3562. doi:10.1242/dev.125.18.3553
Itatani, Y., Kawada, K., Yamamoto, T., and Sakai, Y. (2018). Resistance to anti-angiogenic therapy in cancer-alterations to anti-VEGF pathway. Int. J. Mol. Sci. 19 (4), 1232. doi:10.3390/ijms19041232
Jahangiri, L., Ishola, T., Pucci, P., Trigg, R. M., Pereira, J., Williams, J. A., et al. (2021). The role of autophagy and lncRNAs in the maintenance of cancer stem cells. Cancers (Basel) 13 (6), 1239. doi:10.3390/cancers13061239
Jang, E., Kim, E., Son, H. Y., Lim, E. K., Lee, H., Choi, Y., et al. (2016). Nanovesicle-mediated systemic delivery of microRNA-34a for CD44 overexpressing gastric cancer stem cell therapy. Biomaterials 105, 12–24. doi:10.1016/j.biomaterials.2016.07.036
Jia, H., Song, L., Cong, Q., Wang, J., Xu, H., Chu, Y., et al. (2017). The LIM protein AJUBA promotes colorectal cancer cell survival through suppression of JAK1/STAT1/IFIT2 network. Oncogene 36 (19), 2655–2666. doi:10.1038/onc.2016.418
Jiang, L., Huang, J., Hu, Y., Lu, P., Luo, Q., and Wang, L. (2020). Gli promotes tumor progression through regulating epithelial-mesenchymal transition in non-small-cell lung cancer. J. Cardiothorac. Surg. 15 (1), 18. doi:10.1186/s13019-020-1049-x
Jiang, Z., Cushing, L., Ai, X., and Lü, J. (2014). miR-326 is downstream of Sonic hedgehog signaling and regulates the expression of Gli2 and smoothened. Am. J. Respir. Cell Mol. Biol. 51 (2), 273–283. doi:10.1165/rcmb.2013-0127OC
Jimeno, A., Gordon, M., Chugh, R., Messersmith, W., Mendelson, D., Dupont, J., et al. (2017). A first-in-human phase I study of the anticancer stem cell agent ipafricept (OMP-54F28), a decoy receptor for wnt ligands, in patients with advanced solid tumors. Clin. Cancer Res. 23 (24), 7490–7497. doi:10.1158/1078-0432.CCR-17-2157
Jin, F., Miao, Y., Xu, P., and Qiu, X. (2018). IL-8 regulates the stemness properties of cancer stem cells in the small-cell lung cancer cell line H446. OncoTargets Ther. 11, 5723–5731. doi:10.2147/OTT.S161760
Jin, S., Mutvei, A. P., Chivukula, I. V., Andersson, E. R., Ramsköld, D., Sandberg, R., et al. (2013). Non-canonical Notch signaling activates IL-6/JAK/STAT signaling in breast tumor cells and is controlled by p53 and IKKα/IKKβ. Oncogene 32 (41), 4892–4902. doi:10.1038/onc.2012.517
Jin, W. (2020). Role of JAK/STAT3 signaling in the regulation of metastasis, the transition of cancer stem cells, and chemoresistance of cancer by epithelial-mesenchymal transition. Cells 9 (1), 217. doi:10.3390/cells9010217
Kakarala, M., and Wicha, M. S. (2007). Cancer stem cells: Implications for cancer treatment and prevention. Cancer J. 13 (5), 271–275. doi:10.1097/PPO.0b013e318156da4e
Kaldis, P., and Pagano, M. (2009). Wnt signaling in mitosis. Dev. Cell 17 (6), 749–750. doi:10.1016/j.devcel.2009.12.001
Kaowinn, S., Yawut, N., Koh, S. S., and Chung, Y. H. (2019). Cancer upregulated gene (CUG)2 elevates YAP1 expression, leading to enhancement of epithelial-mesenchymal transition in human lung cancer cells. Biochem. Biophys. Res. Commun. 511 (1), 122–128. doi:10.1016/j.bbrc.2019.02.036
Karunanithi, S., Levi, L., DeVecchio, J., Karagkounis, G., Reizes, O., Lathia, J. D., et al. (2017). RBP4-STRA6 pathway drives cancer stem cell maintenance and mediates high-fat diet-induced colon carcinogenesis. Stem Cell Rep. 9 (2), 438–450. doi:10.1016/j.stemcr.2017.06.002
Katoh, M. (2017). Canonical and non-canonical WNT signaling in cancer stem cells and their niches: Cellular heterogeneity, omics reprogramming, targeted therapy and tumor plasticity (Review). Int. J. Oncol. 51 (5), 1357–1369. doi:10.3892/ijo.2017.4129
Kaur, S., Isenberg, J. S., and Roberts, D. D. (2021). CD47 (cluster of differentiation 47). Atlas Genet. Cytogenet Oncol. Haematol. 25 (2), 83–102.
Kieran, M. W., Kalluri, R., and Cho, Y. J. (2012). The VEGF pathway in cancer and disease: Responses, resistance, and the path forward. Cold Spring Harb. Perspect. Med. 2 (12), a006593. doi:10.1101/cshperspect.a006593
Kim, B.-G., Malek, E., Choi, S. H., Ignatz-Hoover, J. J., and Driscoll, J. J. (2021). Novel therapies emerging in oncology to target the TGF-β pathway. J. Hematol. Oncol. 14 (1), 55. doi:10.1186/s13045-021-01053-x
Kim, B. R., Na, Y. J., Kim, J. L., Jeong, Y. A., Park, S. H., Jo, M. J., et al. (2020). RUNX3 suppresses metastasis and stemness by inhibiting Hedgehog signaling in colorectal cancer. Cell Death Differ. 27 (2), 676–694. doi:10.1038/s41418-019-0379-5
Kim, B., Seo, Y., Kwon, J. H., Shin, Y., Kim, S., Park, S. J., et al. (2021). IL-6 and IL-8, secreted by myofibroblasts in the tumor microenvironment, activate HES1 to expand the cancer stem cell population in early colorectal tumor. Mol. Carcinog. 60 (3), 188–200. doi:10.1002/mc.23283
Kim, Y. M., and Kahn, M. (2014). The role of the wnt signaling pathway in cancer stem cells: Prospects for drug development. Res. Rep. Biochem. 4, 1–12. doi:10.2147/RRBC.S53823
Kisseleva, T., Bhattacharya, S., Braunstein, J., and Schindler, C. W. (2002). Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 285 (1), 1–24. doi:10.1016/s0378-1119(02)00398-0
Koinuma, K., Yamashita, Y., Liu, W., Hatanaka, H., Kurashina, K., Wada, T., et al. (2006). Epigenetic silencing of AXIN2 in colorectal carcinoma with microsatellite instability. Oncogene 25 (1), 139–146. doi:10.1038/sj.onc.1209009
Kokubu, Y., Tabu, K., Muramatsu, N., Wang, W., Murota, Y., Nobuhisa, I., et al. (2016). Induction of protumoral CD11c(high) macrophages by glioma cancer stem cells through GM-CSF. Genes cells. 21 (3), 241–251. doi:10.1111/gtc.12333
Komiya, Y., and Habas, R. (2008). Wnt signal transduction pathways. Organogenesis 4 (2), 68–75. doi:10.4161/org.4.2.5851
Kondyli, M., Gatzounis, G., Kyritsis, A., Varakis, J., and Assimakopoulou, M. (2010). Immunohistochemical detection of phosphorylated JAK-2 and STAT-5 proteins and correlation with erythropoietin receptor (EpoR) expression status in human brain tumors. J. Neurooncol 100 (2), 157–164. doi:10.1007/s11060-010-0156-2
Korn, P., Kampmann, A., Spalthoff, S., Jehn, P., Tavassol, F., Lentge, F., et al. (2021). Suitability of CD133 as a marker for cancer stem cells in melanoma. Asian Pac J. Cancer Prev. 22 (5), 1591–1597. doi:10.31557/APJCP.2021.22.5.1591
Koury, J., Zhong, L., and Hao, J. (2017). Targeting signaling pathways in cancer stem cells for cancer treatment. Stem Cells Int. 2017, 2925869. doi:10.1155/2017/2925869
Kovacs, R. J., Maldonado, G., Azaro, A., Fernández, M. S., Romero, F. L., Sepulveda-Sánchez, J. M., et al. (2015). Cardiac safety of TGF-β receptor I kinase inhibitor LY2157299 monohydrate in cancer patients in a first-in-human dose study. Cardiovasc Toxicol. 15 (4), 309–323. doi:10.1007/s12012-014-9297-4
Kreso, A., and Dick, J. E. (2014). Evolution of the cancer stem cell model. Cell Stem Cell 14 (3), 275–291. doi:10.1016/j.stem.2014.02.006
Kumar, V., Vashishta, M., Kong, L., Wu, X., Lu, J. J., Guha, C., et al. (2021). The role of notch, hedgehog, and wnt signaling pathways in the resistance of tumors to anticancer therapies. Front. Cell Dev. Biol. 9, 650772. doi:10.3389/fcell.2021.650772
Lasorsa, V. A., Montella, A., Cantalupo, S., Tirelli, M., de Torres, C., Aveic, S., et al. (2022). Somatic mutations enriched in cis-regulatory elements affect genes involved in embryonic development and immune system response in neuroblastoma. Cancer Res. 82 (7), 1193–1207. doi:10.1158/0008-5472.CAN-20-3788
Lau, E. Y., Ho, N. P., and Lee, T. K. (2017). Cancer stem cells and their microenvironment: Biology and therapeutic implications. Stem Cells Int. 2017, 3714190. doi:10.1155/2017/3714190
Lecarpentier, Y., Schussler, O., Hébert, J. L., and Vallée, A. (2019). Multiple targets of the canonical WNT/β-Catenin signaling in cancers. Front. Oncol. 9, 1248. doi:10.3389/fonc.2019.01248
Lee, E. Q., Duda, D. G., Muzikansky, A., Gerstner, E. R., Kuhn, J. G., Reardon, D. A., et al. (2018). Phase I and biomarker study of plerixafor and bevacizumab in recurrent high-grade glioma. Clin. cancer Res. 24 (19), 4643–4649. doi:10.1158/1078-0432.CCR-18-1025
Li, H., Chen, J., Wang, X., Zhang, Z., and Cen, Y. (2018). Nodal induced by hypoxia exposure contributes to dacarbazine resistance and the maintenance of stemness in melanoma cancer stem-like cells. Oncol. Rep. 39 (6), 2855–2864. doi:10.3892/or.2018.6387
Li, J.-L., and Harris, A. L. (2009). Crosstalk of VEGF and notch pathways in tumour angiogenesis: Therapeutic implications. FBL 14 (8), 3094–3110. doi:10.2741/3438
Li, K., Xu, W., Lu, K., Wen, Y., Xin, T., Shen, Y., et al. (2020). CSF-1R inhibition disrupts the dialog between leukaemia cells and macrophages and delays leukaemia progression. J. Cell. Mol. Med. 24 (22), 13115–13128. doi:10.1111/jcmm.15916
Li, L., Zhou, G., Fu, R., He, Y., Xiao, L., Peng, F., et al. (2021). Polysaccharides extracted from balanophora polyandra Griff (BPP) ameliorate renal Fibrosis and EMT via inhibiting the Hedgehog pathway. J. Cell Mol. Med. 25 (6), 2828–2840. doi:10.1111/jcmm.16313
Li, S., Tian, J., Zhang, H., Zhou, S., Wang, X., Zhang, L., et al. (2018). Down-regulating IL-6/GP130 targets improved the anti-tumor effects of 5-fluorouracil in colon cancer. Apoptosis Int. J. Program. Cell death 23 (5-6), 356–374. doi:10.1007/s10495-018-1460-0
Li, X., Deng, W., Lobo-Ruppert, S. M., and Ruppert, J. M. (2007). Gli1 acts through Snail and E-cadherin to promote nuclear signaling by beta-catenin. Oncogene 26 (31), 4489–4498. doi:10.1038/sj.onc.1210241
Liang, Y., Yang, L., and Xie, J. (2021). The role of the hedgehog pathway in chemoresistance of gastrointestinal cancers. Cells 10 (8), 2030. doi:10.3390/cells10082030
Liang, Z., Liu, H., Zhang, Y., Xiong, L., Zeng, Z., He, X., et al. (2021). Cyr61 from adipose-derived stem cells promotes colorectal cancer metastasis and vasculogenic mimicry formation via integrin α(V) β(5). Mol. Oncol. 15 (12), 3447–3467. doi:10.1002/1878-0261.12998
Liao, Y., Chen, J., Ma, J., Mao, Q., Wei, R., and Zheng, J. (2018). Notch-regulated ankyrin-repeat protein is a novel tissue biomarker that predicts poor prognosis in non-small cell lung cancer. Oncol. Lett. 16 (2), 1885–1891. doi:10.3892/ol.2018.8826
Lin, F. J., Chen, X., Qin, J., Hong, Y. K., Tsai, M. J., and Tsai, S. Y. (2010). Direct transcriptional regulation of neuropilin-2 by COUP-TFII modulates multiple steps in murine lymphatic vessel development. J. Clin. Invest. 120 (5), 1694–1707. doi:10.1172/JCI40101
Lin, Y. U., Wu, T., Yao, Q., Zi, S., Cui, L., Yang, M., et al. (2015). LGR5 promotes the proliferation of colorectal cancer cells via the Wnt/β-catenin signaling pathway. Oncol. Lett. 9 (6), 2859–2863. doi:10.3892/ol.2015.3144
Liu, C. J., Kuo, F. C., Wang, C. L., Kuo, C. H., Wang, S. S. W., Chen, C. Y., et al. (2016). Suppression of IL-8-Src signalling axis by 17β-estradiol inhibits human mesenchymal stem cells-mediated gastric cancer invasion. J. Cell Mol. Med. 20 (5), 962–972. doi:10.1111/jcmm.12786
Liu, J., Pan, S., Hsieh, M. H., Ng, N., Sun, F., Wang, T., et al. (2013). Targeting wnt-driven cancer through the inhibition of porcupine by LGK974. Proc. Natl. Acad. Sci. U. S. A. 110 (50), 20224–20229. doi:10.1073/pnas.1314239110
Liu, J., Pan, S., Hsieh, M. H., Ng, N., Sun, F., Wang, T., et al. (2013). Targeting wnt-driven cancer through the inhibition of porcupine by LGK974. Proc. Natl. Acad. Sci. 110 (50), 20224–20229. doi:10.1073/pnas.1314239110
Liu, K., Hao, M., Ouyang, Y., Zheng, J., and Chen, D. (2017). CD133(+) cancer stem cells promoted by VEGF accelerate the recurrence of hepatocellular carcinoma. Sci. Rep. 7, 41499. doi:10.1038/srep41499
Liu, Q., Wu, H., Li, Y., Zhang, R., Kleeff, J., Zhang, X., et al. (2020). Combined blockade of TGf-β1 and GM-CSF improves chemotherapeutic effects for pancreatic cancer by modulating tumor microenvironment. Cancer Immunol. Immunother. 69 (8), 1477–1492. doi:10.1007/s00262-020-02542-7
Liu, Y., Qi, X., Donnelly, L., Elghobashi-Meinhardt, N., Long, T., Zhou, R. W., et al. (2022). Mechanisms and inhibition of porcupine-mediated wnt acylation. Nature 607 (7920), 816–822. doi:10.1038/s41586-022-04952-2
Lonardo, E., Hermann, P. C., Mueller, M. T., Huber, S., Balic, A., Miranda-Lorenzo, I., et al. (2011). Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell 9 (5), 433–446. doi:10.1016/j.stem.2011.10.001
López-Sánchez, L. M., Mena, R., Guil-Luna, S., Mantrana, A., Peñarando, J., Toledano-Fonseca, M., et al. (2021). Nitric oxide-targeted therapy inhibits stemness and increases the efficacy of tamoxifen in estrogen receptor-positive breast cancer cells. Lab. Invest. 101 (3), 292–303. doi:10.1038/s41374-020-00507-z
Lu, C. S., Shiau, A. L., Su, B. H., Hsu, T. S., Wang, C. T., Su, Y. C., et al. (2020). Oct4 promotes M2 macrophage polarization through upregulation of macrophage colony-stimulating factor in lung cancer. J. Hematol. Oncol. 13 (1), 62. doi:10.1186/s13045-020-00887-1
Lu, Y., Qin, T., Li, J., Wang, L., Zhang, Q., Jiang, Z., et al. (2017). MicroRNA-140-5p inhibits invasion and angiogenesis through targeting VEGF-A in breast cancer. Cancer Gene Ther. 24 (9), 386–392. doi:10.1038/cgt.2017.30
Ma, Z. Q., and Richardson, L. C. (2022). Cancer screening prevalence and associated factors among US adults. Prev. Chronic Dis. 19, E22. doi:10.5888/pcd19.220063
Matsuno, K., Diederich, R. J., Go, M. J., Blaumueller, C. M., and Artavanis-Tsakonas, S. (1995). Deltex acts as a positive regulator of Notch signaling through interactions with the Notch ankyrin repeats. Development 121 (8), 2633–2644. doi:10.1242/dev.121.8.2633
Mazzoni, S. M., and Fearon, E. R. (2014). AXIN1 and AXIN2 variants in gastrointestinal cancers. Cancer Lett. 355 (1), 1–8. doi:10.1016/j.canlet.2014.09.018
Meisel, C. T., Porcheri, C., and Mitsiadis, T. A. (2020). Cancer stem cells, quo vadis? The notch signaling pathway in tumor initiation and progression. Cells 9 (8), 1879. doi:10.3390/cells9081879
Merchant, A. A., and Matsui, W. (2010). Targeting Hedgehog--a cancer stem cell pathway. Clin. Cancer Res. 16 (12), 3130–3140. doi:10.1158/1078-0432.CCR-09-2846
Mercurio, A. M. (2019). VEGF/Neuropilin signaling in cancer stem cells. Int. J. Mol. Sci. 20 (3), 490. doi:10.3390/ijms20030490
Mercurio, A. M. (2019). VEGF/Neuropilin signaling in cancer stem cells. Int. J. Mol. Sci. 20 (3), 490. doi:10.3390/ijms20030490
Miele, E., Po, A., Begalli, F., Antonucci, L., Mastronuzzi, A., Marras, C. E., et al. (2017). β-arrestin1-mediated acetylation of Gli1 regulates Hedgehog/Gli signaling and modulates self-renewal of SHH medulloblastoma cancer stem cells. BMC Cancer 17 (1), 488. doi:10.1186/s12885-017-3477-0
Miller, K. D., Chap, L. I., Holmes, F. A., Cobleigh, M. A., Marcom, P. K., Fehrenbacher, L., et al. (2005). Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J. Clin. Oncol. 23 (4), 792–799. doi:10.1200/JCO.2005.05.098
Mirzaei, S., Zarrabi, A., Hashemi, F., Zabolian, A., Saleki, H., Azami, N., et al. (2021). Nrf2 signaling pathway in chemoprotection and doxorubicin resistance: Potential application in drug discovery. Antioxidants (Basel) 10 (3), 349. doi:10.3390/antiox10030349
Miyoshi, N., Haraguchi, N., Mizushima, T., Ishii, H., Yamamoto, H., and Mori, M. (2021). Targeting cancer stem cells in refractory cancer. Regen. Ther. 17, 13–19. doi:10.1016/j.reth.2021.01.002
Mohammed, J., Beura, L. K., Bobr, A., Astry, B., Chicoine, B., Kashem, S. W., et al. (2016). Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-β. Nat. Immunol. 17 (4), 414–421. doi:10.1038/ni.3396
Moon, J. H., Rho, Y. S., Lee, S. H., Koo, B. S., Lee, H. J., Do, S. I., et al. (2019). Role of integrin β1 as a biomarker of stemness in head and neck squamous cell carcinoma. Oral Oncol. 96, 34–41. doi:10.1016/j.oraloncology.2019.07.001
Mortara, L., Balza, E., Bruno, A., Poggi, A., Orecchia, P., and Carnemolla, B. (2018). Anti-cancer therapies employing IL-2 cytokine tumor targeting: Contribution of innate, adaptive and immunosuppressive cells in the anti-tumor efficacy. Front. Immunol. 9, 2905. doi:10.3389/fimmu.2018.02905
Müller, L., Tunger, A., Plesca, I., Wehner, R., Temme, A., Westphal, D., et al. (2020). Bidirectional crosstalk between cancer stem cells and immune cell subsets. Front. Immunol. 11, 140. doi:10.3389/fimmu.2020.00140
Naruse, M., Ochiai, M., Sekine, S., Taniguchi, H., Yoshida, T., Ichikawa, H., et al. (2021). Re-expression of REG family and DUOXs genes in CRC organoids by co-culturing with CAFs. Sci. Rep. 11 (1), 2077. doi:10.1038/s41598-021-81475-2
Neckmann, U., Wolowczyk, C., Hall, M., Almaas, E., Ren, J., Zhao, S., et al. (2019). GREM1 is associated with metastasis and predicts poor prognosis in ER-negative breast cancer patients. Cell Commun. Signal 17 (1), 140. doi:10.1186/s12964-019-0467-7
Neuzillet, C., de Gramont, A., Tijeras-Raballand, A., de Mestier, L., Cros, J., Faivre, S., et al. (2013). Perspectives of TGF-? Inhibition in pancreatic and hepatocellular carcinomas. Oncotarget 5, 78–94. doi:10.18632/oncotarget.1569
Nimmakayala, R. K., Batra, S. K., and Ponnusamy, M. P. (2019). Unraveling the journey of cancer stem cells from origin to metastasis. Biochim. Biophys. Acta Rev. Cancer 1871 (1), 50–63. doi:10.1016/j.bbcan.2018.10.006
Noble, D. (2021). Cellular Darwinism: Regulatory networks, stochasticity, and selection in cancer development. Prog. Biophys. Mol. Biol. 165, 66–71. doi:10.1016/j.pbiomolbio.2021.06.007
Noubissi, F. K., Goswami, S., Sanek, N. A., Kawakami, K., Minamoto, T., Moser, A., et al. (2009). Wnt signaling stimulates transcriptional outcome of the Hedgehog pathway by stabilizing GLI1 mRNA. Cancer Res. 69 (22), 8572–8578. doi:10.1158/0008-5472.CAN-09-1500
Odoux, C., Fohrer, H., Hoppo, T., Guzik, L., Stolz, D. B., Lewis, D. W., et al. (2008). A stochastic model for cancer stem cell origin in metastatic colon cancer. Cancer Res. 68 (17), 6932–6941. doi:10.1158/0008-5472.CAN-07-5779
Olive, K. P., Jacobetz, M. A., Davidson, C. J., Gopinathan, A., McIntyre, D., Honess, D., et al. (2009). Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 324 (5933), 1457–1461. doi:10.1126/science.1171362
Owen, K. L., Brockwell, N. K., and Parker, B. S. (2019). JAK-STAT signaling: A double-edged sword of immune regulation and cancer progression. Cancers (Basel) 11 (12), 2002. doi:10.3390/cancers11122002
Palmer, A. C., Chidley, C., and Sorger, P. K. (2019). A curative combination cancer therapy achieves high fractional cell killing through low cross-resistance and drug additivity. Elife 8, e50036. doi:10.7554/eLife.50036
Pannequin, J., Bonnans, C., Delaunay, N., Ryan, J., Bourgaux, J. F., Joubert, D., et al. (2009). The wnt target jagged-1 mediates the activation of notch signaling by progastrin in human colorectal cancer cells. Cancer Res. 69 (15), 6065–6073. doi:10.1158/0008-5472.CAN-08-2409
Pannuti, A., Foreman, K., Rizzo, P., Osipo, C., Golde, T., Osborne, B., et al. (2010). Targeting notch to target cancer stem cells. Clin. Cancer Res. 16 (12), 3141–3152. doi:10.1158/1078-0432.CCR-09-2823
Pattabiraman, D. R., and Weinberg, R. A. (2014). Tackling the cancer stem cells - what challenges do they pose? Nat. Rev. Drug Discov. 13 (7), 497–512. doi:10.1038/nrd4253
Pauklin, S., and Vallier, L. (2015). Activin/Nodal signalling in stem cells. Development 142 (4), 607–619. doi:10.1242/dev.091769
Peng, D., Fu, M., Wang, M., Wei, Y., and Wei, X. (2022). Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol. Cancer 21 (1), 104. doi:10.1186/s12943-022-01569-x
Peng, Q., Shen, Y., Zhao, P., Cheng, M., Zhu, Y., and Xu, B. (2020). Biomarker roles identification of miR-106 family for predicting the risk and poor survival of colorectal cancer. BMC Cancer 20 (1), 506. doi:10.1186/s12885-020-06863-9
Piccolo, S., Dupont, S., and Cordenonsi, M. (2014). The biology of YAP/TAZ: Hippo signaling and beyond. Physiol. Rev. 94 (4), 1287–1312. doi:10.1152/physrev.00005.2014
Pietrobono, S., Gagliardi, S., and Stecca, B. (2019). Non-canonical hedgehog signaling pathway in cancer: Activation of GLI transcription factors beyond smoothened. Front. Genet. 10, 556. doi:10.3389/fgene.2019.00556
Pizer, E. S., Lax, S. F., Kuhajda, F. P., Pasternack, G. R., and Kurman, R. J. (1998). Fatty acid synthase expression in endometrial carcinoma: Correlation with cell proliferation and hormone receptors. Cancer 83 (3), 528–537. doi:10.1002/(sici)1097-0142(19980801)83:3<528::aid-cncr22>3.0.co;2-x
Purow, B. (2009). Notch inhibitors as a new tool in the war on cancer: A pathway to watch. Curr. Pharm. Biotechnol. 10 (2), 154–160. doi:10.2174/138920109787315060
Pyczek, J., Buslei, R., Schult, D., Hölsken, A., Buchfelder, M., Heß, I., et al. (2016). Hedgehog signaling activation induces stem cell proliferation and hormone release in the adult pituitary gland. Sci. Rep. 6 (1), 24928. doi:10.1038/srep24928
Qin, J., Wu, S. P., Creighton, C. J., Dai, F., Xie, X., Cheng, C. M., et al. (2013). COUP-TFII inhibits TGF-β-induced growth barrier to promote prostate tumorigenesis. Nature 493 (7431), 236–240. doi:10.1038/nature11674
Qu, S., Zhang, X., Wu, Y., Li, H., Zhai, J., and Wu, D. (2021). miR-361-3p regulates liver tumor-initiating cells expansion and chemo-resistance. J. Cancer 12 (5), 1483–1492. doi:10.7150/jca.52395
Qureshy, Z., Johnson, D. E., and Grandis, J. R. (2020). Targeting the JAK/STAT pathway in solid tumors. J. Cancer Metastasis Treat. 6, 27. doi:10.20517/2394-4722.2020.58
Raghavan, S., Snyder, C. S., Wang, A., McLean, K., Zamarin, D., Buckanovich, R. J., et al. (2020). Carcinoma-associated mesenchymal stem cells promote chemoresistance in ovarian cancer stem cells via PDGF signaling. Cancers (Basel) 12 (8), 2063. doi:10.3390/cancers12082063
Rascio, F., Spadaccino, F., Rocchetti, M. T., Castellano, G., Stallone, G., Netti, G. S., et al. (2021). The pathogenic role of PI3K/AKT pathway in cancer onset and drug resistance: An updated review. Cancers (Basel) 13 (16), 3949. doi:10.3390/cancers13163949
Rashid, M., Zadeh, L. R., Baradaran, B., Molavi, O., Ghesmati, Z., Sabzichi, M., et al. (2021). Up-down regulation of HIF-1α in cancer progression. Gene 798, 145796. doi:10.1016/j.gene.2021.145796
Rawlings, J. S., Rosler, K. M., and Harrison, D. A. (2004). The JAK/STAT signaling pathway. J. Cell Sci. 117 (8), 1281–1283. doi:10.1242/jcs.00963
Raz, G., Allen, K. E., Kingsley, C., Cherni, I., Arora, S., Watanabe, A., et al. (2012). Hedgehog signaling pathway molecules and ALDH1A1 expression in early-stage non-small cell lung cancer. Lung Cancer 76 (2), 191–196. doi:10.1016/j.lungcan.2011.10.015
Ren, J., Smid, M., Iaria, J., Salvatori, D. C. F., van Dam, H., Zhu, H. J., et al. (2019). Cancer-associated fibroblast-derived Gremlin 1 promotes breast cancer progression. Breast Cancer Res. 21 (1), 109. doi:10.1186/s13058-019-1194-0
Renz, B. W., Tanaka, T., Sunagawa, M., Takahashi, R., Jiang, Z., Macchini, M., et al. (2018). Cholinergic signaling via muscarinic receptors directly and indirectly suppresses pancreatic tumorigenesis and cancer stemness. Cancer Discov. 8 (11), 1458–1473. doi:10.1158/2159-8290.CD-18-0046
Rimkus, T. K., Carpenter, R. L., Qasem, S., Chan, M., and Lo, H. W. (2016). Targeting the sonic hedgehog signaling pathway: Review of smoothened and GLI inhibitors. Cancers 8 (2), 22. doi:10.3390/cancers8020022
Rodon, J., Argilés, G., Connolly, R. M., Vaishampayan, U., de Jonge, M., Garralda, E., et al. (2021). Phase 1 study of single-agent WNT974, a first-in-class Porcupine inhibitor, in patients with advanced solid tumours. Br. J. Cancer 125 (1), 28–37. doi:10.1038/s41416-021-01389-8
Rossi, F., Noren, H., Jove, R., Beljanski, V., and Grinnemo, K. H. (2020). Differences and similarities between cancer and somatic stem cells: Therapeutic implications. Stem Cell Res. Ther. 11 (1), 489. doi:10.1186/s13287-020-02018-6
Sancho, P., Burgos-Ramos, E., Tavera, A., Bou Kheir, T., Jagust, P., Schoenhals, M., et al. (2015). MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell metab. 22 (4), 590–605. doi:10.1016/j.cmet.2015.08.015
Sasai, N., Toriyama, M., and Kondo, T. (2019). Hedgehog signal and genetic disorders. Front. Genet. 10, 1103. doi:10.3389/fgene.2019.01103
Sasich, L. D., and Sukkari, S. R. (2012). The US FDAs withdrawal of the breast cancer indication for Avastin (bevacizumab). Saudi Pharm. J. 20 (4), 381–385. doi:10.1016/j.jsps.2011.12.001
Sato, H., Suzuki, H., Toyota, M., Nojima, M., Maruyama, R., Sasaki, S., et al. (2007). Frequent epigenetic inactivation of DICKKOPF family genes in human gastrointestinal tumors. Carcinogenesis 28 (12), 2459–2466. doi:10.1093/carcin/bgm178
Sato, M., Kawana, K., Fujimoto, A., Yoshida, M., Nakamura, H., Nishida, H., et al. (2016). Clinical significance of Gremlin 1 in cervical cancer and its effects on cancer stem cell maintenance. Oncol. Rep. 35 (1), 391–397. doi:10.3892/or.2015.4367
Scheel, C., and Weinberg, R. A. (2011). Phenotypic plasticity and epithelial-mesenchymal transitions in cancer and normal stem cells? Int. J. Cancer 129 (10), 2310–2314. doi:10.1002/ijc.26311
Schindler, C., Levy, D. E., and Decker, T. (2007). JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 282 (28), 20059–20063. doi:10.1074/jbc.R700016200
Schreck, K. C., Taylor, P., Marchionni, L., Gopalakrishnan, V., Bar, E. E., Gaiano, N., et al. (2010). The notch target Hes1 directly modulates Gli1 expression and hedgehog signaling: A potential mechanism of therapeutic resistance. Clin. Cancer Res. 16 (24), 6060–6070. doi:10.1158/1078-0432.CCR-10-1624
Schulenburg, A., Blatt, K., Cerny-Reiterer, S., Sadovnik, I., Herrmann, H., Marian, B., et al. (2015). Cancer stem cells in basic science and in translational oncology: Can we translate into clinical application? J. Hematol. Oncol. 8, 16. doi:10.1186/s13045-015-0113-9
Schwartz, D. M., Kanno, Y., Villarino, A., Ward, M., Gadina, M., and O'Shea, J. J. (2017). JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 16 (12), 843–862. doi:10.1038/nrd.2017.201
Segami, K., Aoyama, T., Hiroshima, Y., Komori, K., Hashimoto, I., Watanabe, H., et al. (2021). Clinical significance of TAP1 and DLL4 expression in patients with locally advanced gastric cancer. Vivo 35 (5), 2771–2777. doi:10.21873/invivo.12562
Shan, J., Shen, J., Liu, L., Xia, F., Xu, C., Duan, G., et al. (2012). Nanog regulates self-renewal of cancer stem cells through the insulin-like growth factor pathway in human hepatocellular carcinoma. Hepatology 56 (3), 1004–1014. doi:10.1002/hep.25745
Shan, N. L., Shin, Y., Yang, G., Furmanski, P., and Suh, N. (2021). Breast cancer stem cells: A review of their characteristics and the agents that affect them. Mol. Carcinog. 60 (2), 73–100. doi:10.1002/mc.23277
Sharma, A., Mir, R., and Galande, S. (2021). Epigenetic regulation of the wnt/β-catenin signaling pathway in cancer. Front. Genet. 12, 681053. doi:10.3389/fgene.2021.681053
Shi, X., Zhang, X., Li, J., Mo, L., Zhao, H., Zhu, Y., et al. (2018). PD-1 blockade enhances the antitumor efficacy of GM-CSF surface-modified bladder cancer stem cells vaccine. Int. J. Cancer 142 (10), 2106–2117. doi:10.1002/ijc.31219
Shibuya, M. (2011). Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2 (12), 1097–1105. doi:10.1177/1947601911423031
Siegel, R. L., Miller, K. D., Fuchs, H. E., and Jemal, A. (2021). Cancer statistics, 2021. CA Cancer J. Clin. 71 (1), 7–33. doi:10.3322/caac.21654
Singh, J. K., Simões, B. M., Clarke, R. B., and Bundred, N. J. (2013). Targeting IL-8 signalling to inhibit breast cancer stem cell activity. Expert Opin. Ther. Targets 17 (11), 1235–1241. doi:10.1517/14728222.2013.835398
Smith, D. C., Chugh, R., Patnaik, A., Papadopoulos, K. P., Wang, M., Kapoun, A. M., et al. (2019). A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Invest. New Drugs 37 (4), 722–730. doi:10.1007/s10637-018-0714-6
Snuderl, M., Batista, A., Kirkpatrick, N. D., Ruiz de Almodovar, C., Riedemann, L., Walsh, E. C., et al. (2013). Targeting placental growth factor/neuropilin 1 pathway inhibits growth and spread of medulloblastoma. Cell 152 (5), 1065–1076. doi:10.1016/j.cell.2013.01.036
Sofen, H., Gross, K. G., Goldberg, L. H., Sharata, H., Hamilton, T. K., Egbert, B., et al. (2015). A phase II, multicenter, open-label, 3-cohort trial evaluating the efficacy and safety of vismodegib in operable basal cell carcinoma. J. Am. Acad. Dermatol 73 (1), 99–105. doi:10.1016/j.jaad.2015.03.013
Statkiewicz, M., Maryan, N., Lipiec, A., Grecka, E., Grygorowicz, M. A., Omiotek, M., et al. (2014). The role of the SHH gene in prostate cancer cell resistance to paclitaxel. Prostate 74 (11), 1142–1152. doi:10.1002/pros.22830
Sun, T., Jia, Y., and Xiao, D. (2015). Interference of STAT 5b expression enhances the chemo-sensitivity of gastric cancer cells to gefitinib by promoting mitochondrial pathway-mediated cell apoptosis. Oncol. Rep. 34 (1), 227–234. doi:10.3892/or.2015.3994
Takahashi, S. (2011). Vascular endothelial growth factor (VEGF), VEGF receptors and their inhibitors for antiangiogenic tumor therapy. Biol. Pharm. Bull. 34 (12), 1785–1788. doi:10.1248/bpb.34.1785
Takebe, N., Miele, L., Harris, P. J., Jeong, W., Bando, H., Kahn, M., et al. (2015). Targeting notch, hedgehog, and wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 12 (8), 445–464. doi:10.1038/nrclinonc.2015.61
Takebe, N., Nguyen, D., and Yang, S. X. (2014). Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol. Ther. 141 (2), 140–149. doi:10.1016/j.pharmthera.2013.09.005
Takebe, N., Warren, R. Q., and Ivy, S. P. (2011). Breast cancer growth and metastasis: Interplay between cancer stem cells, embryonic signaling pathways and epithelial-to-mesenchymal transition. Breast Cancer Res. 13 (3), 211. doi:10.1186/bcr2876
Tang, L. Y., Heller, M., Meng, Z., Yu, L. R., Tang, Y., Zhou, M., et al. (2017). Transforming growth factor-β (TGF-β) directly activates the JAK1-STAT3 Axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem. 292 (10), 4302–4312. doi:10.1074/jbc.M116.773085
Taniguchi, H., Masuishi, T., Kawazoe, A., Muro, K., Kadowaki, S., Bando, H., et al. (2021). Phase I study of napabucasin in combination with FOLFIRI + bevacizumab in Japanese patients with metastatic colorectal cancer. Int. J. Clin. Oncol. 26 (11), 2017–2024. doi:10.1007/s10147-021-01987-9
Tomita, H., and Hara, A. (2020). “Chapter 4 - serrated lesions and stem cells on drug resistance and colon cancer,” in Drug resistance in colorectal cancer: Molecular mechanisms and therapeutic strategies. Editors C. H. Cho, and T. Hu (Cambridge: Academic Press), 75–82.
Tran, P., Nguyen, C., and Klempner, S. J. (2016). Targeting the phosphatidylinositol-3-kinase pathway in gastric cancer: Can omics improve outcomes? Int. Neurourol. J. 20 (2), S131–S140. doi:10.5213/inj.1632740.370
Tremblay, M. R., Lescarbeau, A., Grogan, M. J., Lin, G., and Austad, B. C. (2009). Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926). J. Med. Chem. 52 (14), 4400–4418. doi:10.1021/jm900305z
Tsuchiya, H., and Shiota, G. (2021). Immune evasion by cancer stem cells. Regen. Ther. 17, 20–33. doi:10.1016/j.reth.2021.02.006
Tweedell, K. S. (2017). The adaptability of somatic stem cells: A review. J. Stem Cells Regen. Med. 13 (1), 3–13. doi:10.46582/jsrm.1301002
Valle-Mendiola, A., Bustos-Rodríguez, R., Domínguez-Melendez, V., Zerecero-Carreón, O., Gutiérrez-Hoya, A., Weiss-Steider, B., et al. (2022). Mutations in the helix αC of the catalytic domain from the EGFR affect its activity in cervical cancer cell lines. Oncol. Lett. 23 (2), 71. doi:10.3892/ol.2022.13191
van Schie, E. H., and van Amerongen, R. (2020). Aberrant WNT/CTNNB1 signaling as a therapeutic target in human breast cancer: Weighing the evidence. Front. Cell Dev. Biol. 8, 25. doi:10.3389/fcell.2020.00025
Venkatesh, V., Nataraj, R., Thangaraj, G. S., Karthikeyan, M., Gnanasekaran, A., Kaginelli, S. B., et al. (2018). Targeting Notch signalling pathway of cancer stem cells. Stem Cell Investig. 5, 5. doi:10.21037/sci.2018.02.02
Walcher, L., Kistenmacher, A. K., Suo, H., Kitte, R., Dluczek, S., Strauß, A., et al. (2020). Cancer stem cells-origins and biomarkers: Perspectives for targeted personalized therapies. Front. Immunol. 11, 1280. doi:10.3389/fimmu.2020.01280
Wall, D. S., Mears, A. J., McNeill, B., Mazerolle, C., Thurig, S., Wang, Y., et al. (2009). Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J. Cell Biol. 184 (1), 101–112. doi:10.1083/jcb.200805155
Wang, J., Zhang, B., Wu, H., Cai, J., Sui, X., Wang, Y., et al. (2017). CD51 correlates with the TGF-beta pathway and is a functional marker for colorectal cancer stem cells. Oncogene 36 (10), 1351–1363. doi:10.1038/onc.2016.299
Wang, S., Wang, Y., Xun, X., Zhang, C., Xiang, X., Cheng, Q., et al. (2020). Hedgehog signaling promotes sorafenib resistance in hepatocellular carcinoma patient-derived organoids. J. Exp. Clin. Cancer Res. 39 (1), 22. doi:10.1186/s13046-020-1523-2
Wang, W., Liu, P., Lavrijsen, M., Li, S., Zhang, R., Li, S., et al. (2021). Evaluation of AXIN1 and AXIN2 as targets of tankyrase inhibition in hepatocellular carcinoma cell lines. Sci. Rep. 11 (1), 7470. doi:10.1038/s41598-021-87091-4
Wang, Z., Li, Y., Banerjee, S., and Sarkar, F. H. (2009). Emerging role of Notch in stem cells and cancer. Cancer Lett. 279 (1), 8–12. doi:10.1016/j.canlet.2008.09.030
Wei, H. (2019). Interleukin 6 signaling maintains the stem-like properties of bladder cancer stem cells. Transl. Cancer Res. 8 (2), 557–566. doi:10.21037/tcr.2019.03.16
Weir, H. K., Thompson, T. D., Stewart, S. L., and White, M. C. (2021). Cancer incidence projections in the United States between 2015 and 2050. Prev. Chronic Dis. 18, E59. doi:10.5888/pcd18.210006
Wen, J., Lee, J., Malhotra, A., Nahta, R., Arnold, A. R., Buss, M. C., et al. (2016). WIP1 modulates responsiveness to Sonic Hedgehog signaling in neuronal precursor cells and medulloblastoma. Oncogene 35 (42), 5552–5564. doi:10.1038/onc.2016.96
Weng, D. E., Masci, P. A., Radka, S. F., Jackson, T. E., Weiss, P. A., Ganapathi, R., et al. (2005). A phase I clinical trial of a ribozyme-based angiogenesis inhibitor targeting vascular endothelial growth factor receptor-1 for patients with refractory solid tumors. Mol. Cancer Ther. 4 (6), 948–955. doi:10.1158/1535-7163.MCT-04-0210
Weng, D. E., and Usman, N. (2001). Angiozyme: A novel angiogenesis inhibitor. Curr. Oncol. Rep. 3 (2), 141–146. doi:10.1007/s11912-001-0014-7
Wiese, M., Hamdan, F. H., Kubiak, K., Diederichs, C., Gielen, G. H., Nussbaumer, G., et al. (2020). Combined treatment with CBP and BET inhibitors reverses inadvertent activation of detrimental super enhancer programs in DIPG cells. Cell Death Dis. 11 (8), 673. doi:10.1038/s41419-020-02800-7
Wiese, M., Walther, N., Diederichs, C., Schill, F., Monecke, S., Salinas, G., et al. (2017). The β-catenin/CBP-antagonist ICG-001 inhibits pediatric glioma tumorigenicity in a Wnt-independent manner. Oncotarget 8 (16), 27300–27313. doi:10.18632/oncotarget.15934
Wu, Y., Cain-Hom, C., Choy, L., Hagenbeek, T. J., de Leon, G. P., Chen, Y., et al. (2010). Therapeutic antibody targeting of individual Notch receptors. Nature 464 (7291), 1052–1057. doi:10.1038/nature08878
Wu, Y., Tran, T., Dwabe, S., Sarkissyan, M., Kim, J., Nava, M., et al. (2017). A83-01 inhibits TGF-β-induced upregulation of Wnt3 and epithelial to mesenchymal transition in HER2-overexpressing breast cancer cells. Breast Cancer Res. Treat. 163 (3), 449–460. doi:10.1007/s10549-017-4211-y
Xia, W., Lo, C. M., Poon, R. Y. C., Cheung, T. T., Chan, A. C. Y., Chen, L., et al. (2017). Smad inhibitor induces CSC differentiation for effective chemosensitization in cyclin D1-and TGF-β/Smad-regulated liver cancer stem cell-like cells. Oncotarget 8 (24), 38811–38824. doi:10.18632/oncotarget.16402
Yamamoto, M., Sakane, K., Tominaga, K., Gotoh, N., Niwa, T., Kikuchi, Y., et al. (2017). Intratumoral bidirectional transitions between epithelial and mesenchymal cells in triple-negative breast cancer. Cancer Sci. 108 (6), 1210–1222. doi:10.1111/cas.13246
Yan, G., Dai, M., Zhang, C., Poulet, S., Moamer, A., Wang, N., et al. (2021). TGFβ/cyclin D1/Smad-mediated inhibition of BMP4 promotes breast cancer stem cell self-renewal activity. Oncogenesis 10 (3), 21. doi:10.1038/s41389-021-00310-5
Yan, Y., Zuo, X., and Wei, D. (2015). Concise review: Emerging role of CD44 in cancer stem cells: A promising biomarker and therapeutic target. Stem Cells Transl. Med. 4 (9), 1033–1043. doi:10.5966/sctm.2015-0048
Yang, L., Dong, Y., Li, Y., Wang, D., Liu, S., Wang, D., et al. (2019). IL-10 derived from M2 macrophage promotes cancer stemness via JAK1/STAT1/NF-κB/Notch1 pathway in non-small cell lung cancer. Int. J. Cancer 145 (4), 1099–1110. doi:10.1002/ijc.32151
Yang, L., Shi, P., Zhao, G., Xu, J., Peng, W., Zhang, J., et al. (2020). Targeting cancer stem cell pathways for cancer therapy. Signal Transduct. Target Ther. 5 (1), 8. doi:10.1038/s41392-020-0110-5
Yang, Y., Ding, L., Hu, Q., Xia, J., Sun, J., Wang, X., et al. (2017). MicroRNA-218 functions as a tumor suppressor in lung cancer by targeting IL-6/STAT3 and negatively correlates with poor prognosis. Mol. Cancer 16 (1), 141. doi:10.1186/s12943-017-0710-z
Yang, Y., Shi, J., Tolomelli, G., Xu, H., Xia, J., Wang, H., et al. (2013). RARα2 expression confers myeloma stem cell features. Blood 122 (8), 1437–1447. doi:10.1182/blood-2013-02-482919
Yin, X., Zhang, B. H., Zheng, S. S., Gao, D. M., Qiu, S. J., Wu, W. Z., et al. (2015). Coexpression of gene Oct4 and Nanog initiates stem cell characteristics in hepatocellular carcinoma and promotes epithelial-mesenchymal transition through activation of Stat3/Snail signaling. J. Hematol. Oncol. 8, 23. doi:10.1186/s13045-015-0119-3
Yue, P., and Turkson, J. (2009). Targeting STAT3 in cancer: How successful are we? Expert Opin. Investig. Drugs 18 (1), 45–56. doi:10.1517/13543780802565791
Zeitlin, B. D., Ellis, L. M., and Nör, J. E. (2009). Inhibition of vascular endothelial growth factor receptor-1/wnt/{beta}-catenin crosstalk leads to tumor cell death. Clin. Cancer Res. 15 (24), 7453–7455. doi:10.1158/1078-0432.CCR-09-2578
Zeng, C., Wang, Y., Lu, Q., Chen, J., Zhang, J., Liu, T., et al. (2014). SPOP suppresses tumorigenesis by regulating Hedgehog/Gli2 signaling pathway in gastric cancer. J. Exp. Clin. Cancer Res. 33 (1), 75. doi:10.1186/s13046-014-0075-8
Zhan, T., Rindtorff, N., and Boutros, M. (2017). Wnt signaling in cancer. Oncogene 36 (11), 1461–1473. doi:10.1038/onc.2016.304
Zhang, F., Sun, H., Zhang, S., Yang, X., Zhang, G., and Su, T. (2017). Overexpression of PER3 inhibits self-renewal capability and chemoresistance of colorectal cancer stem-like cells via inhibition of notch and β-catenin signaling. Oncol. Res. 25 (5), 709–719. doi:10.3727/096504016X14772331883976
Zhang, H. (2020). CCND1 silencing suppresses liver cancer stem cell differentiation through inhibiting autophagy. Hum. Cell 33 (1), 140–147. doi:10.1007/s13577-019-00295-9
Zhang, L., Sun, H., Zhao, F., Lu, P., Ge, C., Li, H., et al. (2012). BMP4 administration induces differentiation of CD133+ hepatic cancer stem cells, blocking their contributions to hepatocellular carcinoma. Cancer Res. 72 (16), 4276–4285. doi:10.1158/0008-5472.CAN-12-1013
Zhang, S., Wang, Y., Mao, J. H., Hsieh, D., Kim, I. J., Hu, L. M., et al. (2012). Inhibition of CK2α down-regulates Hedgehog/Gli signaling leading to a reduction of a stem-like side population in human lung cancer cells. PLoS One 7 (6), e38996. doi:10.1371/journal.pone.0038996
Zhang, S., Yang, X., Wang, L., and Zhang, C. (2018). Interplay between inflammatory tumor microenvironment and cancer stem cells. Oncol. Lett. 16 (1), 679–686. doi:10.3892/ol.2018.8716
Zhang, X., Ai, F., Li, X., Tian, L., Wang, X., Shen, S., et al. (2017). MicroRNA-34a suppresses colorectal cancer metastasis by regulating Notch signaling. Oncol. Lett. 14 (2), 2325–2333. doi:10.3892/ol.2017.6444
Zhang, X., Hu, F., Yang, X., and Liu, L. (2018). Human colorectal cancer-derived mesenchymal stem cells promote colorectal cancer progression through IL-6/JAK2/STAT3 signaling. Cell Death Dis. 9 (2), 25. doi:10.1038/s41419-017-0176-3
Zhang, Y., and Kalderon, D. (2001). Hedgehog acts as a somatic stem cell factor in the Drosophila ovary. Nature 410 (6828), 599–604. doi:10.1038/35069099
Zhao, D., Pan, C., Sun, J., Gilbert, C., Drews-Elger, K., Azzam, D. J., et al. (2015). VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene 34 (24), 3107–3119. doi:10.1038/onc.2014.257
Zhao, L., Wang, J., Zhang, Y., Wang, L., Yu, M., and Wang, F. (2020). Vitamin C decreases VEGF expression levels via hypoxia‑inducible factor‑1α dependent and independent pathways in lens epithelial cells. Mol. Med. Rep. 22 (1), 436–444. doi:10.3892/mmr.2020.11103
Zhao, W., Ji, X., Zhang, F., and Li, L. (2012). Embryonic stem cell markers. Molecules 17 (6), 6196–6236. doi:10.3390/molecules17066196
Zhdanovskaya, N., Firrincieli, M., Lazzari, S., Pace, E., Scribani Rossi, P., Felli, M. P., et al. (2021). Targeting notch to maximize chemotherapeutic benefits: Rationale, advanced strategies, and future perspectives. Cancers (Basel) 13 (20), 5106. doi:10.3390/cancers13205106
Zhi, X., Tao, J., Zhang, L., Tao, R., Ma, L., and Qin, J. (2016). Silencing speckle-type POZ protein by promoter hypermethylation decreases cell apoptosis through upregulating Hedgehog signaling pathway in colorectal cancer. Cell Death Dis. 7 (12), e2569. doi:10.1038/cddis.2016.435
Zhou, F. Q., Qi, Y. M., Xu, H., Wang, Q. Y., Gao, X. S., and Guo, H. G. (2015). Expression of EpCAM and Wnt/β-catenin in human colon cancer. Genet. Mol. Res. 14 (2), 4485–4494. doi:10.4238/2015.May.4.6
Zhou, Y., Kipps, T. J., and Zhang, S. (2017). Wnt5a signaling in normal and cancer stem cells. Stem Cells Int. 2017, 5295286. doi:10.1155/2017/5295286
Zinke, J., Schneider, F. T., Harter, P. N., Thom, S., Ziegler, N., Toftgård, R., et al. (2015). β-Catenin-Gli1 interaction regulates proliferation and tumor growth in medulloblastoma. Mol. Cancer 14 (1), 17. doi:10.1186/s12943-015-0294-4
Glossary

Keywords: cancer stem cells (CSCs), Wnt, TGFβ, Notch, JAK-STAT, hedgehog, VEGF, therapeutics
Citation: Borlongan MC and Wang H (2023) Profiling and targeting cancer stem cell signaling pathways for cancer therapeutics. Front. Cell Dev. Biol. 11:1125174. doi: 10.3389/fcell.2023.1125174
Received: 16 December 2022; Accepted: 15 May 2023;
Published: 25 May 2023.
Edited by:
Lauren Gollahon, Texas Tech University, United StatesReviewed by:
Maria Cristina Rangel, University of São Paulo, BrazilHira Lal Goel, UMass Chan Medical School, United States
Copyright © 2023 Borlongan and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongbin Wang, aG9uZ2Jpbi53YW5nQGNuc3UuZWR1